
Gilthead sea bream (Sparus auratus) and European sea bass (Dicentrarchus labrax) expressed sequence...
13
Gilthead sea bream (Sparus auratus) and European sea bass (Dicentrarchus labrax) expressed sequence tags: Characterization, tissue-specific expression and gene markers Bruno Louro a,j , Ana Lúcia S. Passos a , Erika L. Souche b,1 , Costas Tsigenopoulos c , Alfred Beck d , Jacques Lagnel c , François Bonhomme e , Leonor Cancela a , Joan Cerdà f , Melody S. Clark g , Esther Lubzens h , Antonis Magoulas c , Josep V. Planas i , Filip A.M. Volckaert b , Richard Reinhardt d , Adelino V.M. Canario a, ⁎ a Centre of Marine Sciences, University of Algarve, Building 7, Gambelas, 8000-139 Faro, Portugal b Laboratory of Animal Diversity and Systematics, Katholieke Universiteit Leuven, Charles Deberiotstraat 32, B-3000 Leuven, Belgium c Hellenic Centre for Marine Research, Institute of Marine Biology and Genetics, Thalassocosmos, Ex-US base at Gournes, P.O. Box 2214, Gournes Pediados, 715 00 Heraklion, Crete, Greece d MPI Molecular Genetics, Ihnestrasse 63-73, D-14195 Berlin-Dahlem, Germany e Département Biologie Intégrative, Institut des Sciences de l'Evolution, UMR 5554 Université de Montpellier 2, cc 63 — Pl. E Bataillon, F34095 Montpellier Cedex 5, France f Laboratory of Institut de Recerca i Tecnologia Agroalimentaries (IRTA)-Institut de Ciencies del Mar (Consejo Superior de Investigaciones Científicas, CSIC), Passeig Marítim 37-49, 08003-Barcelona, Spain g British Antarctic Survey, Natural Environment Research Council, High Cross, Madingley Road, Cambridge CB3 0ET, UK h National Institute of Oceanography, Israel Oceanographic & Limnological Research, P.O. Box 8030, Haifa 31080, Israel i Departament de Fisiologia, Facultat de Biologia, Universitat de Barcelona, Av. Diagonal 645, 08028 Barcelona, Spain j Division of Genetics and Genomics, Roslin Institute and Royal (Dick) School of Veterinary Sciences, University of Edinburgh, Roslin, Midlothian, EH25 9PS, United Kingdom abstract article info Article history: Received 16 May 2010 Received in revised form 17 September 2010 Accepted 21 September 2010 Keywords: Expressed Sequence Tag Microsatellite Single nucleotide polymorphism Annotation Aquaculture Teleost fish The gilthead sea bream, Sparus auratus, and the European sea bass, Dicentrarchus labrax, are two of the most important marine species cultivated in Southern Europe. This study aimed at increasing genomic resources for the two species and produced and annotated two sets of 30,000 expressed sequence tags (EST) each from 14 normalized tissue-specific cDNA libraries from sea bream and sea bass. Clustering and assembly of the ESTs formed 5268 contigs and 12,928 singletons for sea bream and 4573 contigs and 13,143 singletons for sea bass, representing 18,196 and 17,716 putative unigenes, respectively. Assuming a similar number of genes in sea bass, sea bream and in the model fish Gasterosteus aculeatus genomes, it was estimated that approximately two thirds of the sea bream and the sea bass transcriptomes were covered by the unigene collections. BLAST sequence similarity searches (using a cut off of e-value b 10 -5 ) against fully the curated SwissProt (and TrEMBL) databases produced matches of 28%(37%) and 43%(53%) of the sea bream and sea bass unigene datasets respectively, allowing some putative designation of function. A comparative approach is described using human Ensembl peptide ID homolog's for functional annotation, which increased the number of unigenes with GO terms assigned and resulted in more GO terms assigned per unigene. This allowed the identification of tissue-specific genes using enrichment analysis for GO pathways and protein domains. The comparative annotation approach represents a good strategy for transferring more relevant biological information from highly studied species to genomic resource poorer species. It was possible to confirm by interspecies mRNA-to-genomic alignments 25 and 21 alternative splice events in sea bream and sea bass genes, respectively. Even using normalized cDNA from relatively few pooled individuals it was possible to identify 1145 SNPs and 1748 microsatellites loci for genetic marker development. The EST data are being applied to a range of projects, including the development microarrays, genetic and radiation hybrid maps and QTL genome scans. This highlights the important role of ESTs for generating genetic and genomic resources of aquaculture species. © 2010 Elsevier B.V. All rights reserved. Marine Genomics 3 (2010) 179–191 ⁎ Corresponding author. E-mail addresses: [email protected] (B. Louro), [email protected] (A.L.S. Passos), [email protected] (E.L. Souche), [email protected] (C. Tsigenopoulos), [email protected] (A. Beck), [email protected] (J. Lagnel), [email protected] (F. Bonhomme), [email protected] (L. Cancela), [email protected] (J. Cerdà), [email protected] (M.S. Clark), [email protected] (E. Lubzens), [email protected] (A. Magoulas), [email protected] (J.V. Planas), [email protected] (F.A.M. Volckaert), [email protected] (R. Reinhardt), [email protected] (A.V.M. Canario). 1 Current address: Institut Pasteur, Plate-Forme Intégration et Analyse Génomiques, 28 Rue du Docteur Roux, F-75724 Paris Cedex 15, France. 1874-7787/$ – see front matter © 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.margen.2010.09.005 Contents lists available at ScienceDirect Marine Genomics journal homepage: www.elsevier.com/locate/margen
Transcript of Gilthead sea bream (Sparus auratus) and European sea bass (Dicentrarchus labrax) expressed sequence...

Marine Genomics 3 (2010) 179–191
Contents lists available at ScienceDirect
Marine Genomics
j ourna l homepage: www.e lsev ie r.com/ locate /margen
Gilthead sea bream (Sparus auratus) and European sea bass (Dicentrarchus labrax)expressed sequence tags: Characterization, tissue-specific expression andgene markers
Bruno Louro a,j, Ana Lúcia S. Passos a, Erika L. Souche b,1, Costas Tsigenopoulos c, Alfred Beck d,Jacques Lagnel c, François Bonhomme e, Leonor Cancela a, Joan Cerdà f, Melody S. Clark g, Esther Lubzens h,Antonis Magoulas c, Josep V. Planas i, Filip A.M. Volckaert b, Richard Reinhardt d, Adelino V.M. Canario a,⁎a Centre of Marine Sciences, University of Algarve, Building 7, Gambelas, 8000-139 Faro, Portugalb Laboratory of Animal Diversity and Systematics, Katholieke Universiteit Leuven, Charles Deberiotstraat 32, B-3000 Leuven, Belgiumc Hellenic Centre for Marine Research, Institute of Marine Biology and Genetics, Thalassocosmos, Ex-US base at Gournes, P.O. Box 2214, Gournes Pediados, 715 00 Heraklion, Crete, Greeced MPI Molecular Genetics, Ihnestrasse 63-73, D-14195 Berlin-Dahlem, Germanye Département Biologie Intégrative, Institut des Sciences de l'Evolution, UMR 5554 Université de Montpellier 2, cc 63 — Pl. E Bataillon, F34095 Montpellier Cedex 5, Francef Laboratory of Institut de Recerca i Tecnologia Agroalimentaries (IRTA)-Institut de Ciencies del Mar (Consejo Superior de Investigaciones Científicas, CSIC),Passeig Marítim 37-49, 08003-Barcelona, Spaing British Antarctic Survey, Natural Environment Research Council, High Cross, Madingley Road, Cambridge CB3 0ET, UKh National Institute of Oceanography, Israel Oceanographic & Limnological Research, P.O. Box 8030, Haifa 31080, Israeli Departament de Fisiologia, Facultat de Biologia, Universitat de Barcelona, Av. Diagonal 645, 08028 Barcelona, Spainj Division of Genetics and Genomics, Roslin Institute and Royal (Dick) School of Veterinary Sciences, University of Edinburgh, Roslin, Midlothian, EH25 9PS, United Kingdom
⁎ Corresponding author.E-mail addresses: [email protected] (B. Louro), ccmar@u
(A. Beck), [email protected] (J. Lagnel), bonhomme@[email protected] (E. Lubzens), [email protected](R. Reinhardt), [email protected] (A.V.M. Canario).
1 Current address: Institut Pasteur, Plate-Forme Intég
1874-7787/$ – see front matter © 2010 Elsevier B.V. Aldoi:10.1016/j.margen.2010.09.005
a b s t r a c t
a r t i c l e i n f oArticle history:Received 16 May 2010Received in revised form 17 September 2010Accepted 21 September 2010
Keywords:Expressed Sequence TagMicrosatelliteSingle nucleotide polymorphismAnnotationAquacultureTeleost fish
The gilthead sea bream, Sparus auratus, and the European sea bass, Dicentrarchus labrax, are two of the mostimportant marine species cultivated in Southern Europe. This study aimed at increasing genomic resources forthe two species and produced and annotated two sets of 30,000 expressed sequence tags (EST) each from 14normalized tissue-specific cDNA libraries from sea bream and sea bass. Clustering and assembly of the ESTsformed 5268 contigs and 12,928 singletons for sea bream and 4573 contigs and 13,143 singletons for sea bass,representing 18,196 and 17,716 putative unigenes, respectively. Assuming a similar number of genes in seabass, sea bream and in the model fish Gasterosteus aculeatus genomes, it was estimated that approximatelytwo thirds of the sea bream and the sea bass transcriptomes were covered by the unigene collections. BLASTsequence similarity searches (using a cut off of e-value b10−5) against fully the curated SwissProt (andTrEMBL) databases produced matches of 28%(37%) and 43%(53%) of the sea bream and sea bass unigenedatasets respectively, allowing some putative designation of function. A comparative approach is describedusing human Ensembl peptide ID homolog's for functional annotation, which increased the number ofunigenes with GO terms assigned and resulted in more GO terms assigned per unigene. This allowed theidentification of tissue-specific genes using enrichment analysis for GO pathways and protein domains. Thecomparative annotation approach represents a good strategy for transferring more relevant biologicalinformation from highly studied species to genomic resource poorer species. It was possible to confirm byinterspecies mRNA-to-genomic alignments 25 and 21 alternative splice events in sea bream and sea bassgenes, respectively. Even using normalized cDNA from relatively few pooled individuals it was possible toidentify 1145 SNPs and 1748 microsatellites loci for genetic marker development.The EST data are being applied to a range of projects, including the development microarrays, genetic andradiation hybridmaps and QTL genome scans. This highlights the important role of ESTs for generating geneticand genomic resources of aquaculture species.
alg.pt (A.L.S. Passos), [email protected] (E.L. Souchit.univ-montp2.fr (F. Bonhomme), [email protected] (L. Car (A. Magoulas), [email protected] (J.V. Planas), Filip.Volck
ration et Analyse Génomiques, 28 Rue du Docteur Roux,
l rights reserved.
© 2010 Elsevier B.V. All rights reserved.
e), [email protected] (C. Tsigenopoulos), [email protected]), [email protected] (J. Cerdà), [email protected] (M.S. Clark),[email protected] (F.A.M. Volckaert), [email protected]
F-75724 Paris Cedex 15, France.

180 B. Louro et al. / Marine Genomics 3 (2010) 179–191
1. Introduction
The gilthead sea bream, Sparus auratus, and the European sea bass,Dicentrarchus labrax, are two of themainmarine fish species cultured inSouthern Europe. Both species are economically valuable and, therefore,over the last two decades efforts have been made to increase theknowledge of their physiology, reproduction, immunology, ecology,quantitative genetics and population genetics (Power et al., 2001;Scapigliati et al., 2002; Piferrer et al., 2005; Pawson et al., 2007; Lemaireet al., 2005; Laiz-Carrion et al., 2005; Arends et al., 1999; Senger et al.,2006; Meiri et al., 2002). The fact that these are not designated modelfish species has contributed to the shortage of genomic resources.However, recently there has been growing awareness of the value ofgenomic information in the efficient implementation of strategies forselective breeding, disease prevention, treatment and quality control incultured fish species. Therefore, the EU Network of Excellence MarineGenomics Europe (MGE; http://www.marine-genomics-europe.org/)set out todevelopEST(expressed sequence tags; single-pass sequencingof cDNA libraries)projects on aquaculturedmarinefish species, focusingon the sea bream and the sea bass. These EST projects represent animportant contribution to the genomic resources of these two species.The generation and analysis of ESTs is a valuable approach for theidentification and characterization of new genes and provide a platformfor developing functional genomics methods (Gong, 1999; Douglaset al., 1999; 2007; Gonzalez et al., 2007; Cerdà et al., 2008; Canario et al.,2008; Koop et al., 2008).
Representation of transcript abundance and diversity in ESTcollections is dependent on the method of cDNA library construction.If the objective is to obtain thewidest possible representation of cDNAs,then it is a common procedure to normalize the cDNA libraries prior tosequencing in order to increase the presence of rare transcripts,however the gene expression profiles are lost (Bonaldo et al., 1996;Coblentz et al., 2006;Govoroun et al., 2006). In contrast, nonnormalizedcDNA libraries may be deficient in rare transcripts, unless deepsequencing coverage is achieved, something that is nowmore accessiblewith Next Generation pyrosequencing (Morozova and Marra, 2008).Fish EST projects started in the late 90s with the model fishes zebrafish,Danio rerio (Gong, 1999; Gong et al., 1997) and medaka, Oryzias latipes(Hirono and Aoki, 1997). These are still the species with the largestcollections of published ESTs. These were followed by another twomodel fishes, the Japanese puffer fish, Takifugu rubripes (Clark et al.,2003) and themummichogkillifish, Fundulus heteroclitus (Paschall et al.,2004). Since then, large scale EST projects from the cultured salmonids,Atlantic salmon, Salmo salar (Adzhubei et al., 2007; Hagen-Larsen et al.,2005) and rainbow trout, Onchorynchus mykiss (Govoroun et al., 2006),and other cultured fish, such as the catfish, Ictalurus spp (Li et al., 2007;Wang et al., 2010), and the common carp, Cyprinus carpio (Gonzalezet al., 2007), Nile tilapia, Oreochromis niloticus (Lee et al., 2010) havebeen developed. Currently, there are more than a dozen teleost ESTcollections comprising more than 50,000 ESTs, of which eight arecultured fish species. Prior to the study described in this article, therewere less that 2,500 transcript sequences published for each of seabream and sea bass, which included a randommix from directed gene-based studies and small scale EST collections (Chini et al., 2006).
The objective of this work was to increase the knowledge base ofsea bream and sea bass genomics through the generation and analysisof two medium-scale EST collections. This study has producedapproximately 30,000 ESTs using Sanger sequencing from 14 tissue-specific normalized cDNA libraries for each species. These nowrepresent 44% of sea bream and 54% sea bass entries in the NCBIGenBank dbEST (accessed 24 April 2010). These EST collections havealready had a significant impact in the development of additionalgenomics tools and applications such as microarrays (Ferraresso et al.,2008; Calduch-Giner et al., 2010; Ferraresso et al., 2010),microsatellitemarkers and linkage maps (Vogiatzi et al., submitted for publication),SNP development and population studies (Souche et al., 2007),
assembly and annotation of draft genome sequence (Kuhl et al.,2010)], sea bass radiation hybrid map and QTL mapping (Massault etal., 2010).
2. Methods
2.1. cDNA libraries construction
The sea breamand sea basswere obtained from localfish farms andmaintained at the Ramalhete marine station (University of Algarve,Faro, Portugal). The sea bream cDNA libraries were made from tissuepools of 9 animals, ranging in weight between 0.3 and 4 kg andincluded males (2), females (4) and hermaphrodites (3). The sea basscDNA libraries weremade from tissue pools from 5male individuals ofapproximately 450 g except for the ovarian tissue which came from asingle individual from a separate stock. Animals were sacrificed withan excess of 2-phenoxyethanol, tissues were dissected and stored inliquid nitrogen at −80°C. Total RNA was extracted using the acidguanidinium thiocyanate-phenol-chloroform method (Chomczynskiand Sacchi, 1987).
Fourteen normalized tissue-specific cDNA libraries were con-structed and normalized from liver; ovary; testis; bone/cartilage;brain/pituitary; heart/vessels; adipose; head/kidney; trunk/kidney;gill; intestine; spleen; muscle; and skin of each species. cDNA libraryconstruction and sequencing was carried out at the Max PlanckInstitute of Molecular Genetics (Berlin, Germany). mRNA purificationsand concentration were performed using the Dynal's oligo-dTmagnetic beads (Invitrogen Dynal AS, Olso, Norway). cDNA librarieswere constructed using the cDNA SMART-kit (Clontech, Palo Alto, CA,USA) (Zhu et al., 2001) and normalized using thermostable duplex-specific nuclease (Zhulidov et al., 2004).
2.2. Sequencing: editing and cluster assembly
Sequencing was performed using Capillary Sequencer systems (ABI3730 XL and GEHealthcareMegaBace 4500) at theMax Planck Instituteof Molecular Genetics (Berlin, Germany). For both sequencing systems,the sequencing kit used was the ABI BigDye Terminator v.3.1 (AppliedBiosystems, Foster City, CA, USA). For each library between 2304and 2688 ESTs were sequenced from the 5´ ends for both species.
The resulting ESTs were quality-trimmed (Nq20) and vector-clipped using Phred (Ewing et al., 1998); http://www.phrap.org,(Ewing and Green, 1998) and LUCY (Chou and Holmes, 2001); http://www.tigr.org, and all sequences shorter than 100 bp were discarded.Cross-match (http://www.phrap.org) was then used to screen thesequences for contaminant vector sequences. Repeatsweremasked byRepeatMasker (Smit et al., 1996–2010) before the ESTs were clusteredand assembled, according to the default parameters, using TGICL(Pertea et al., 2003). Consensus sequences originating from clustersof ESTs representing the same transcripts were termed contigs andunique sequences were classified as singletons. All sequences weredeposited in EMBL with accession numbers AM950553 to AM980446(S. auratus) for and FM178562 to FM178778 (D. labrax).
2.3. Unigene identification using sequence similarity searching
The BLASTX algorithm (Altschul et al., 1990; 1997) was used toquery for sequence similarity of the unigenes against the Swissprotand Trembl databases using a local blast engine, blastall (ftp://ftp.ncbi.nih.gov/blast/executables/). The fully manually curated and annotat-ed Swissprot database (ftp://ftp.expasy.org/databases/uniprot/knowledgebase/uniprot_sprot.fasta.gz, version 10/04/2008) was que-ried in order to obtain putative functional identifications for theunigenes. BLASTX was also used to query the Trembl database (ftp://ftp.expasy.org/databases/uniprot/knowledgebase/uniprot_trembl.fasta.gz, version 10/04/2008) to improve the unigene identifications.

181B. Louro et al. / Marine Genomics 3 (2010) 179–191
Both BLASTX queries were performed using the blosum62 matrixwith 1e−5 e-value, 10 open gap penalty, 1 extended gap penalty and3 word size as cut-off values.
In order to identify novel transcripts, Genbank dbEST was alsoqueried using BLASTN (with a cut-off value of E-value of b 1e−5 and abit score N40). The sequence similarity searches were performed bothon the unigenes resulting from all tissues clustered together into aspecies-specific EST database and also on each tissue-specific ESTdatabase (data not shown). Spreadsheet tables were produced withthe BLAST results, cluster consensus sequences and links to the EMBLUniProt (Supplementary file 1 and 2).
2.4. Unigene GO annotation
Two methods were used to obtain Gene Ontology (GO) terms(Ashburner et al., 2000). The first approach was to directly retrieve theGO terms, if available, from the first match of the BLASTX query againstTrembl. The second approachwas to obtain the GO terms for sea breamand sea bass unigenes separately, via a comparative approach to thestickleback, Gasterosteus aculeatus, Ensembl transcripts (http://www.ensembl.org/Gasterosteus_aculeatus) and human, Homo sapiens,Ensembl peptides (http://www.ensembl.org/Homo_sapiens) (Fig. 1).The unigenes were queried against the human Ensembl peptides (Fig. 1step 1) (Homo_sapiens.NCBI36.47.pep.all.fa, 26/10/2007) and stickle-back Ensembl transcripts (Fig. 1 step 2) (Gasterosteus_aculeatus.BROADS1.46.cdna.all.fa, 26/10/2007) via local blast using BLASTX andBLASTN, respectively. Blast parameters were as described previously.BLAST results against the human and stickleback were merged andoverlapping clones were eliminated. For this purpose sticklebackEnsembl transcripts identifiers retrieved from the blastn output wereused as input filters via biomart martview (http://www.biomart.org/martview) to retrievehumanpeptidesorthologs (Fig. 1 step3)asoutputattributes. Because several homology relationships exist (one-to-one,one-to-many, many-to-many, and many-to-one), a Microsoft OfficeAccess 2003 database was created and queried to filter maximum
Fig. 1. Flowchart of Gene Ontology annotation methodology carried out for the two species a(step 1) and stickleback Ensembl transcripts (step 2) using BLASTX and BLASTN, respectivtranscripts identifiers retrieved from the blastn output were used as input filters via biomarthomology human peptides were filtered in step 4. Ensembl human peptide identifiers retrievcomparison were merged and overlapping identifiers for sea bream or sea bass unigenes elimquerying the “Ensembl 50 genes (Sanger UK), Homo sapiens gene (NCBI36)” datasets with
homology human peptides (Fig. 1 step 4). Ensembl human peptidesidentifiers retrieved via blastp (Fig. 1 step 1) and unique Ensemblhuman peptides identifiers retrieved via stickleback comparison weremerged and overlapping identifiers for sea bream or sea bass unigeneswere eliminated. In order to do this the Ensembl human identifiers datawere uploaded to the Microsoft Office Access 2003 database and cross-queriedusing sea breamor sea bass unigenes asprimary key link tofilterfor redundancy (Fig. 1 step 5). A unique file with unigenes and theirrespective human peptide orthologous Ensembl IDs was obtained forboth sea bass and sea bream unigenes separately. The procedure wasalso carried out separately for each tissue library and also for all librariescombined. The output files were used to retrieve all the human GOannotations by querying the “Ensembl 50 genes (Sanger UK), Homosapiens gene (NCBI36)” datasets with biomart martview (Fig. 1 step 6).The retrieved GO terms were submitted to the GO term classificationcounter tool from theGeneOntologyConsortium(Zhi-Liang et al., 2008)to produce a representative GO set (GOA-slim set plus the direct childterms of binding and cellular processes) of the two ancestral GO terms(Biological process and Molecular function) separately. All GO classifi-cation data output was integrated in spreadsheet tables with all ofthe BLAST results usingMicrosoft Office Access 2003 with sea bream orsea bass unigenes defined as primary key to link all incremental datain one file (Supplementary file 1 and 2).
2.5. Tissue specific gene-enrichment analysis
To obtain specific biological annotations for each sea bass and seabream tissue library, a gene-enrichment analysis was carried outusing the DAVID functional annotation tool (http://www.david.abcc.ncifcrf.gov/summary.jsp). Human Ensembl gene IDs obtained viabiomart, corresponding to human Ensembl peptides IDs homologs ofthe sea bream and sea bass unigenes present only in one given specifictissue were uploaded and submitted to the DAVID functionalannotation tool (http://www.david.abcc.ncifcrf.gov/summary.jsp).Batch annotations for all tissue-specific biological clusters containing
nd for each cDNA library. Unigenes were queried against the human Ensembl peptidesely. The two BLAST results were merged and overlapping clones eliminated. For thismartview to retrieve human peptides orthologs (step 3) as output attributes. Maximumed via blastp (step 1) and unique human peptide identifiers retrieved via the sticklebackinated (step 5). The output files were used to retrieve all the human GO annotations bybiomart martview (step 6). See also Section 2.4 for details.

182 B. Louro et al. / Marine Genomics 3 (2010) 179–191
molecular function and biological process GO terms, Interpro proteinfunctional domains, KEGG and Biocarta pathways were retrievedusing a false discovery rate of 10% as function of the EASE Score and amodified Fisher Exact P-value as a cut-off score. The same procedurewas repeated for all the unigenes in the different tissue-specificlibraries irrespective of their coverage within the libraries.
2.6. Alternative splice form analysis
In order to identify sequence insertions/gaps or partially dissimilarsequences, potentially indicative of alternative splicing events,“supracontigs” were created using a more relaxed clustering gappenalty parameter and aligned using ClustalX (Thompson et al., 1997).Contigs containing inserts or partially dissimilar sequences were usedas queries against the green spotted pufferfish (Tetraodon nigroviridis)genome (http://www.genoscope.cns.fr/externe/tetranew/) using Blat(Kent, 2002) in order to retrieve the respective gene loci necessary forthe mRNA-to-genomic alignments. To confirm the genomic organiza-tion and identify the type of alternative splicing present in each gene,the Spidey software (Wheelan et al., 2001) was used selecting thedivergent sequences option necessary for the interspecies alignment.
2.7. SNP detection
ESTs are single-pass reads and therefore they contain base callingerrors on average every 100 bp (Edwards, 2007) and are prone totranscriptional errors and misalignments (Garg et al., 1999). In order tolimit the number of false SNP candidates, only mismatches appearingtwice in an alignment (redundant mismatches) are usually consideredSNP candidates (Garg et al., 1999; Batley et al., 2003; Le Dantec et al.,2004). The software MiraEST [version 2.4; (Chevreux et al., 2004) wasconsidered to be themost efficient (data not shown) formining sea bass
Table 1Number of total sequences per tissue and their respective contigs, singletons and redundan
A
cDNA library Total ESTs EST Contigs
Sea breamLiver 2181 733 923Ovary 2107 670 845Testis 2141 718 913Bone/cartilage 2163 656 936Brain/pituitary 2104 618 889Heart/vessels 2210 726 992Adipose 2125 648 799Head/kidney 2161 679 937Trunk/kidney 2060 686 931Gill 2096 591 729Intestine 2088 671 930Spleen 2185 725 1021Muscle 2168 741 1050Skin 2105 601 823All libraries 29895 677 1049
Sea bassLiver 2139 699 909Ovary 2389 526 659Testis 2127 697 881Bone/cartilage 2045 645 839Brain/pituitary 2118 579 739Heart/vessels 2296 541 715Adipose 2295 631 818Head/kidney 1935 535 638Trunk/kidney 2203 680 890Gill 2101 701 873Intestine 1989 743 868Spleen 2277 608 817Muscle 1776 694 828Skin 1570 668 711All libraries 29260 636 928
ESTs for SNPs when redundant SNP candidates were selected. Defaultparameters and tissue information were used. Insertions/deletions(indels) of several base pairs, initially detected as several indels of onebase pair, were considered single indels of several base pairs.
2.8. Microsatellite detection
Both sea bass and sea bream unigenes were used to identify andcharacterize SSRs (Simple Sequence Repeats) using a Perl script basedon the algorithm of the MISA script (http://www.pgrc.ipk-gatersleben.de/misa). Perfect tandem repeats were defined in sequences with aminimum number of repeats of 9 for dinucleotide, 6 for trinucleotide, 5for tetranucleotide, and 4 for penta- and hexanucleotide repeats. SSR-ESTs were analyzed for redundancy by BLASTN and/or Cap3 software.SSR results were retrieved in a tabular form using a pipeline of SQLqueries and Perl scripts.
3. Results and discussion
3.1. cDNA library construction, sequencing and clustering
The aim of this project was to obtain a comprehensive overview ofthe sea bream and sea bass transcriptomes. Hence, 14 normalizedtissue-specific cDNA libraries were constructed for each species andapproximately 2000 clones from each library were single-passsequenced from their 5´ ends. The normalization procedure resultedin an average of 10% and 12% redundancy per tissue for the sea breamand sea bass, respectively, with the exception of the sea bass skinlibrary (60%; Table 1). Normalization success was confirmed by thefact that most contigs contained only two ESTs.
A total of 29,895 and 29,260 high quality ESTs were obtained fromthe sea bream and sea bass, respectively (Table 1). The average length
cy obtained by clustering in sea bream and sea bass.
verage length (bp)
Singletons Unigenes (contigs+singletons) Redundancy (%)
735 1799 (251+1548) 18670 1913 (155+1758) 9717 1999 (128+1871) 7654 2012 (141+1862) 7616 1957 (127+1830) 7722 1921 (217+1704) 13644 1864 (218+1646) 12675 1900 (206+1694) 12683 1907 (108+1799) 7591 1871 (197+1674) 11671 1881 (175+1706) 10721 2014 (141+1873) 8736 1858 (225+1633) 14601 1959 (131+1828) 7665 18196 (5268+12928) 39
697 1858 (213+1645) 13525 2069 (264+1805) 13692 1940 (153+1787) 9645 1842 (96+1746) 10578 1997 (95+1902) 6536 2082 (157+1925) 9629 2127 (123+2004) 7536 1687 (116+1571) 13672 1917 (161+1756) 13696 1877 (119+1758) 11738 1628 (97+1531) 18607 2074 (136+1938) 9697 1249 (102+1147) 30695 632 (62+570) 60619 17716 (4573+13143) 39

Table 2Summary of BLAST query results for sea bream and sea bass unigenes.
Unigenes hits
Database Sea bream Sea bass
Swissprot 5166 (28%) 7640 (43%)Trembl 6 716 (37%) 9 320 (53%)Genebank ESTdb 10470 (57%) 12104 (68%)Human Ensembl peptides 5126 (28%) 7595 (43%)Stickleback Ensembl transcripts 6056 (33%) 8544 (48%)Stickleback Ensembl peptides 5811 (32%) 8519 (48%)
Fig. 2. Gene Ontology annotation of the sea bream total ESTs according to Biological Procerepresented in their respective stacked bars.
Table 3Summary of GO annotation and method comparison. GO TrEMBL refers to annotationwith GO terms retrieved directly from the first match of Blastx query against theTrEMBL database. GO biomart refers to annotation with human GO terms retrieved viaan integrated and comparative approach (see Fig. 1).
Unigenes GO TrEMBL (A) GO biomart (B) B/A
Sea breamWith GO annotation (C) 3272 (18%) 5203 (29%) 1.6×Total GO terms (D) 9408 29,834 3.2×Average GO terms (D/C) 2.9 5.7 2×
Sea bassWith GO annotation (E) 4658 (26%) 7528 (43%) 1.6×Total GO terms (F) 13,435 41,511 3.1×Average GO terms (F/E) 2.9 5.5 1.9×
183B. Louro et al. / Marine Genomics 3 (2010) 179–191
of high quality unassembled ESTs was 676 bp for sea bream and636 bp for sea bass, with, respectively, 86% and 80% sequences longerthan 500 bp after quality trimming.
Clustering of all tissues for each species yielded 5,268 contigs and12,928 singletons for the sea bream and 4,573 contigs and 13,143singletons for the sea bass (Table 1). The contigs plus the singletonsputatively corresponded to different transcripts and were designatedunigenes.
Stickleback is, among the genome sequenced fish species, the onewith highest representation of Ensembl predicted transcripts (27,629;(http://www.ensembl.org/Gasterosteus_aculeatus/Info/StatsTable) atthe same time is the most phylogeneticaly closely related to seabreamand sea bass. Assuming the number of stickleback transcripts as anear representation of the whole transcriptome, the ratio of sea breamand sea bass unigenes with stickleback predicted transcripts indicate,that 66% and 64% transcriptomes were covered by the unigenescollections, respectively. The average length of the sea bream unigeneswas 776 bp,with amedian of 750 bp and amode of 782 bp. The sea bassunigenes comprised an average length of 698 bp, a median of 699 bpand a mode of 770 bp. The redundancy of the unigenes for the wholespecies datasets was 39% for both sea bream and sea bass. This washigher than the within tissue average redundancy due to two factors:redundancy is expected to increase with a higher transcriptomerepresentation and normalization procedures were carried out sepa-rately for each tissue library and not for the pool of tissues.
ss and Molecular Function. Direct child GO terms of cellular process and binding are

184 B. Louro et al. / Marine Genomics 3 (2010) 179–191
3.2. Unigene identification using sequence similarity
For the sea bream, 28% and 37% positive matches were obtained inSwissprot and Trembl databases, respectively using the unigenes(Table 2). The corresponding values for sea bass were 43% and 53%(Table 2). Although a smaller percentage of positive matches wereobtainedbyquerying the fully curatedSwissprot database, thesematchesproduced more reliable sequence similarity identification with addedbiological annotation. The Trembl BLASTX query outputs producedmorematches, with almost 80% of themost significantmatches to the proteinsof fish species. However the biological annotation of these matcheswas very limited. For both the sea bream and sea bass Trembl BLASTXoutputs, approximately half of the most significant matches were tothe green spotted pufferfish genome predicted proteins and aboutone fifth were to the zebrafish genome automatically predicted, non-curated, proteins. The sea bass Trembl BLASTXqueries had amuchhigherpercentage of positive matches compared with those of the sea bream(Table 2). This, together with the higher number of matches againstGenbank dbEST using BLASTN, indicated that the sea bass unigenesweremore representative of coding region sequences (CDS) with comparisonto sea bream. Additional evidence in support of this conclusion was the
Fig. 3. Gene Ontology annotation of the sea bass total ESTs according to Biological Procerepresented in their respective stacked bars.
higher sea bass unigene GC content (data not shown) and higherpercentage of trinucleotide microsatellites (see below), which aremore abundant in exons in all taxa (Tóth et al., 2000).
3.3. EST GO annotations
The sea bream and sea bass unigene associated GO terms wereretrieved from two BLASTX IDs, either directly from the first BLASThit Trembl ID, or from the merged stickleback transcripts and humanpeptide Ensembl IDs (described in Fig. 1 and Section 2.4). Thestickleback was chosen because it is the most phylogenetically relatedteleost fish to the species under study for which genome data andannotation is available. Human was used as the final anchor species,since it is the species with the most GO term annotated genes. BLASTNqueries against the stickleback transcript database enabled identifica-tion of the ESTs consisting mainly of UTR regions, which had producedno significant results on the BLASTX queries to the Swissprot, Trembland human peptide databases.
A higher than threefold increase in total GO terms retrieved for thetwo species was obtained using human peptides IDs in comparison tousing Trembl IDs (Table 3). This was because of the greater depth of
ss and Molecular Function. Direct child GO terms of cellular process and binding are
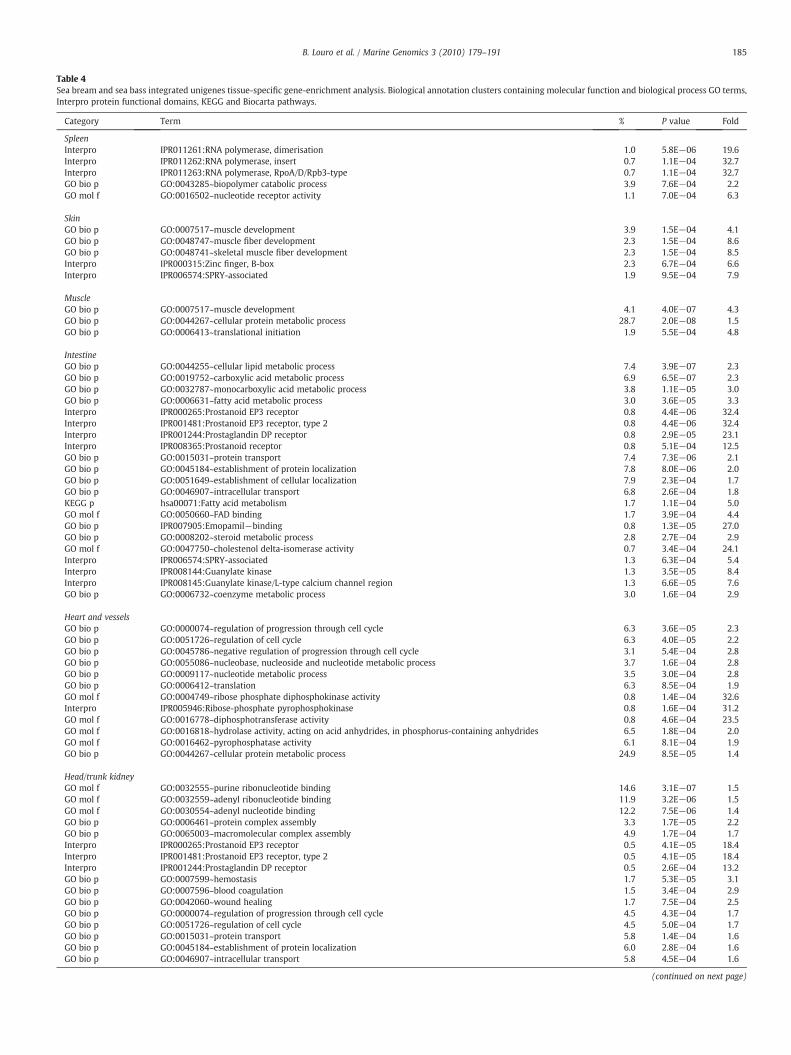
Table 4Sea bream and sea bass integrated unigenes tissue-specific gene-enrichment analysis. Biological annotation clusters containing molecular function and biological process GO terms,Interpro protein functional domains, KEGG and Biocarta pathways.
Category Term % P value Fold
SpleenInterpro IPR011261:RNA polymerase, dimerisation 1.0 5.8E−06 19.6Interpro IPR011262:RNA polymerase, insert 0.7 1.1E−04 32.7Interpro IPR011263:RNA polymerase, RpoA/D/Rpb3-type 0.7 1.1E−04 32.7GO bio p GO:0043285~biopolymer catabolic process 3.9 7.6E−04 2.2GO mol f GO:0016502~nucleotide receptor activity 1.1 7.0E−04 6.3
SkinGO bio p GO:0007517~muscle development 3.9 1.5E−04 4.1GO bio p GO:0048747~muscle fiber development 2.3 1.5E−04 8.6GO bio p GO:0048741~skeletal muscle fiber development 2.3 1.5E−04 8.5Interpro IPR000315:Zinc finger, B-box 2.3 6.7E−04 6.6Interpro IPR006574:SPRY-associated 1.9 9.5E−04 7.9
MuscleGO bio p GO:0007517~muscle development 4.1 4.0E−07 4.3GO bio p GO:0044267~cellular protein metabolic process 28.7 2.0E−08 1.5GO bio p GO:0006413~translational initiation 1.9 5.5E−04 4.8
IntestineGO bio p GO:0044255~cellular lipid metabolic process 7.4 3.9E−07 2.3GO bio p GO:0019752~carboxylic acid metabolic process 6.9 6.5E−07 2.3GO bio p GO:0032787~monocarboxylic acid metabolic process 3.8 1.1E−05 3.0GO bio p GO:0006631~fatty acid metabolic process 3.0 3.6E−05 3.3Interpro IPR000265:Prostanoid EP3 receptor 0.8 4.4E−06 32.4Interpro IPR001481:Prostanoid EP3 receptor, type 2 0.8 4.4E−06 32.4Interpro IPR001244:Prostaglandin DP receptor 0.8 2.9E−05 23.1Interpro IPR008365:Prostanoid receptor 0.8 5.1E−04 12.5GO bio p GO:0015031~protein transport 7.4 7.3E−06 2.1GO bio p GO:0045184~establishment of protein localization 7.8 8.0E−06 2.0GO bio p GO:0051649~establishment of cellular localization 7.9 2.3E−04 1.7GO bio p GO:0046907~intracellular transport 6.8 2.6E−04 1.8KEGG p hsa00071:Fatty acid metabolism 1.7 1.1E−04 5.0GO mol f GO:0050660~FAD binding 1.7 3.9E−04 4.4GO bio p IPR007905:Emopamil−binding 0.8 1.3E−05 27.0GO bio p GO:0008202~steroid metabolic process 2.8 2.7E−04 2.9GO mol f GO:0047750~cholestenol delta-isomerase activity 0.7 3.4E−04 24.1Interpro IPR006574:SPRY-associated 1.3 6.3E−04 5.4Interpro IPR008144:Guanylate kinase 1.3 3.5E−05 8.4Interpro IPR008145:Guanylate kinase/L-type calcium channel region 1.3 6.6E−05 7.6GO bio p GO:0006732~coenzyme metabolic process 3.0 1.6E−04 2.9
Heart and vesselsGO bio p GO:0000074~regulation of progression through cell cycle 6.3 3.6E−05 2.3GO bio p GO:0051726~regulation of cell cycle 6.3 4.0E−05 2.2GO bio p GO:0045786~negative regulation of progression through cell cycle 3.1 5.4E−04 2.8GO bio p GO:0055086~nucleobase, nucleoside and nucleotide metabolic process 3.7 1.6E−04 2.8GO bio p GO:0009117~nucleotide metabolic process 3.5 3.0E−04 2.8GO bio p GO:0006412~translation 6.3 8.5E−04 1.9GO mol f GO:0004749~ribose phosphate diphosphokinase activity 0.8 1.4E−04 32.6Interpro IPR005946:Ribose-phosphate pyrophosphokinase 0.8 1.6E−04 31.2GO mol f GO:0016778~diphosphotransferase activity 0.8 4.6E−04 23.5GO mol f GO:0016818~hydrolase activity, acting on acid anhydrides, in phosphorus-containing anhydrides 6.5 1.8E−04 2.0GO mol f GO:0016462~pyrophosphatase activity 6.1 8.1E−04 1.9GO bio p GO:0044267~cellular protein metabolic process 24.9 8.5E−05 1.4
Head/trunk kidneyGO mol f GO:0032555~purine ribonucleotide binding 14.6 3.1E−07 1.5GO mol f GO:0032559~adenyl ribonucleotide binding 11.9 3.2E−06 1.5GO mol f GO:0030554~adenyl nucleotide binding 12.2 7.5E−06 1.4GO bio p GO:0006461~protein complex assembly 3.3 1.7E−05 2.2GO bio p GO:0065003~macromolecular complex assembly 4.9 1.7E−04 1.7Interpro IPR000265:Prostanoid EP3 receptor 0.5 4.1E−05 18.4Interpro IPR001481:Prostanoid EP3 receptor, type 2 0.5 4.1E−05 18.4Interpro IPR001244:Prostaglandin DP receptor 0.5 2.6E−04 13.2GO bio p GO:0007599~hemostasis 1.7 5.3E−05 3.1GO bio p GO:0007596~blood coagulation 1.5 3.4E−04 2.9GO bio p GO:0042060~wound healing 1.7 7.5E−04 2.5GO bio p GO:0000074~regulation of progression through cell cycle 4.5 4.3E−04 1.7GO bio p GO:0051726~regulation of cell cycle 4.5 5.0E−04 1.7GO bio p GO:0015031~protein transport 5.8 1.4E−04 1.6GO bio p GO:0045184~establishment of protein localization 6.0 2.8E−04 1.6GO bio p GO:0046907~intracellular transport 5.8 4.5E−04 1.6
(continued on next page)
185B. Louro et al. / Marine Genomics 3 (2010) 179–191

Table 4 (continued)
Category Term % P value Fold
Head/trunk kidneyGO bio p GO:0006886~intracellular protein transport 3.7 5.3E−04 1.8GO bio p GO:0051649~establishment of cellular localization 6.7 8.1E−04 1.5GO bio p GO:0007067~mitosis 2.4 8.0E−04 2.1GO bio p GO:0000087~M phase of mitotic cell cycle 2.4 9.1E−04 2.0GO bio p GO:0044267~cellular protein metabolic process 22.2 5.9E−05 1.2GO bio p GO:0016192~vesicle-mediated transport 4.4 7.1E−04 1.7Interpro IPR013128:Peptidase C1A, papain 0.7 2.6E−04 7.2GO bio p GO:0044265~cellular macromolecule catabolic process 3.7 2.7E−05 2.1GO bio p GO:0044275~cellular carbohydrate catabolic process 1.6 4.0E−04 2.8GO bio p GO:0016052~carbohydrate catabolic process 1.6 7.1E−04 2.6Interpro IPR013833:Cytochrome c oxidase, subunit III, 4-helical bundle 0.5 4.1E−05 18.4Interpro IPR000298:Cytochrome c oxidase, subunit III 0.5 4.1E−05 18.4Interpro IPR014014:RNA helicase, DEAD-box type, Q motif 0.8 8.3E−04 4.4GO bio p GO:0008219~cell death 6.5 6.7E−04 1.5GO bio p GO:0012501~programmed cell death 6.1 8.4E−04 1.5GO bio p GO:0006915~apoptosis 6.1 9.0E−04 1.5Interpro IPR000568:ATPase, F0 complex, subunit A 0.6 2.7E−06 18.4GO bio p GO:0008632~apoptotic program 1.3 6.0E−04 3.0
GillInterpro IPR014745:MHC class II, alpha/beta chain, N-terminal 9.8 2.0E−43 11.0Interpro IPR000353:MHC class II, beta chain, N-terminal 9.6 2.6E−43 11.3Biocarta h_mhcPathway:Antigen Processing and Presentation 5.6 1.3E−20 6.7Biocarta h_eosinophilsPathway:The Role of Eosinophils in the Chemokine Network of Allergy 5.4 1.8E−20 6.9Biocarta h_bbcellPathway:Bystander B Cell Activation 5.4 2.9E−20 6.8Biocarta h_il5Pathway:IL 5 Signaling Pathway 5.4 4.7E−20 6.8Biocarta h_asbcellPathway:Antigen Dependent B Cell Activation 5.4 1.9E−19 6.5Biocarta h_blymphocytePathway:B Lymphocyte Cell Surface Molecules 5.4 1.9E−19 6.5Biocarta h_tcraPathway:Lck and Fyn tyrosine kinases in initiation of TCR Activation 5.4 2.4E−18 6.0Biocarta H_th1th2Pathway:Th1/Th2 Differentiation 5.4 5.3E−18 5.9Biocarta h_CSKPathway:Activation of Csk by cAMP-dependent Protein Kinase Inhibits Signaling through the T Cell Receptor 5.4 3.5E−17 5.6Biocarta h_inflamPathway:Cytokines and Inflammatory Response 5.4 7.2E−17 5.5Biocarta h_ctla4Pathway:The Co-Stimulatory Signal During T-cell Activation 5.4 1.0E−16 5.4GO mol f GO:0032555~purine ribonucleotide binding 13.9 8.8E−05 1.5GO mol f GO:0032555~purine ribonucleotide binding 13.9 8.8E−05 1.5Interpro IPR003006:Immunoglobulin/major histocompatibility complex motif 3.2 1.3E−05 3.4Interpro IPR003597:Immunoglobulin C1-set 3.1 2.0E−05 3.4GO bio p GO:0009889~regulation of biosynthetic process 2.9 2.5E−04 2.9Interpro IPR011769:Adenylate/cytidine kinase, N-terminal 0.9 1.5E−04 16.6GO bio p GO:0045184~establishment of protein localization 6.4 5.6E−04 1.8GO mol f GO:0019001~guanyl nucleotide binding 4.4 2.7E−04 2.2GO mol f GO:0032561~guanyl ribonucleotide binding 4.4 3.1E−04 2.2Interpro IPR001404:Heat shock protein Hsp90 1.0 1.5E−04 11.1
Bone and cartilageGO mol f IPR002048:Calcium-binding EF-hand 4.2 4.1E−07 3.2GO mol f GO:0005509~calcium ion binding 9.1 2.0E−05 1.8GO mol f IPR011992:EF-Hand type 3.5 5.3E−05 2.8GO bio p GO:0006941~striated muscle contraction 1.7 2.9E−07 8.7Interpro IPR001978:Troponin 0.9 7.9E−07 25.8GO bio p GO:0006936~muscle contraction 3.3 9.0E−07 3.7GO bio p GO:0006937~regulation of muscle contraction 1.6 2.8E−06 8.0Interpro IPR000169:Peptidase, cysteine peptidase active site 1.4 3.5E−05 6.9GO bio p GO:0006767~water-soluble vitamin metabolic process 1.4 1.6E−04 5.7Interpro IPR001101:Plectin repeat 0.8 3.9E−05 21.5Interpro IPR002017:Spectrin repeat 1.1 7.9E−04 6.2GO bio p GO:0051129~negative regulation of cellular component organization and biogenesis 1.4 1.3E−04 5.8GO bio p GO:0051128~regulation of cellular component organization and biogenesis 1.7 8.2E−04 3.7GO bio p GO:0006732~coenzyme metabolic process 3.0 1.2E−04 2.8GO bio p GO:0006767~water-soluble vitamin metabolic process 1.4 1.6E−04 5.7Interpro IPR008937:Ras guanine nucleotide exchange factor 1.3 5.6E−05 7.8Interpro IPR002396:Selectin (CD62E/L/P antigen) 0.8 1.3E−04 16.7
AdiposeInterpro IPR011261:RNA polymerase, dimerisation 1.1 2.5E−07 21.0Interpro IPR014001:DEAD-like helicase, N-terminal 2.0 2.6E−04 3.6Interpro IPR014021:Helicase, superfamily 1 and 2, ATP-binding 2.0 3.3E−04 3.5Interpro IPR001650:DNA/RNA helicase, C-terminal 2.0 3.9E−04 3.4GO mol f GO:0003899~DNA-directed RNA polymerase activity 1.5 2.2E−05 6.3GO bio p GO:0001570~vasculogenesis 0.9 7.9E−04 7.9
Ovary and testisKEGG p hsa00500:Starch and sucrose metabolism 1.5 5.1E−06 3.0KEGG p hsa00860:Porphyrin and chlorophyll metabolism 1.0 6.4E−06 4.1Biocarta h_arenrf2Pathway:Oxidative Stress Induced Gene Expression Via Nrf2 0.8 2.4E−05 4.4KEGG p hsa00040:Pentose and glucuronate interconversions 0.8 2.9E−05 4.9
186 B. Louro et al. / Marine Genomics 3 (2010) 179–191
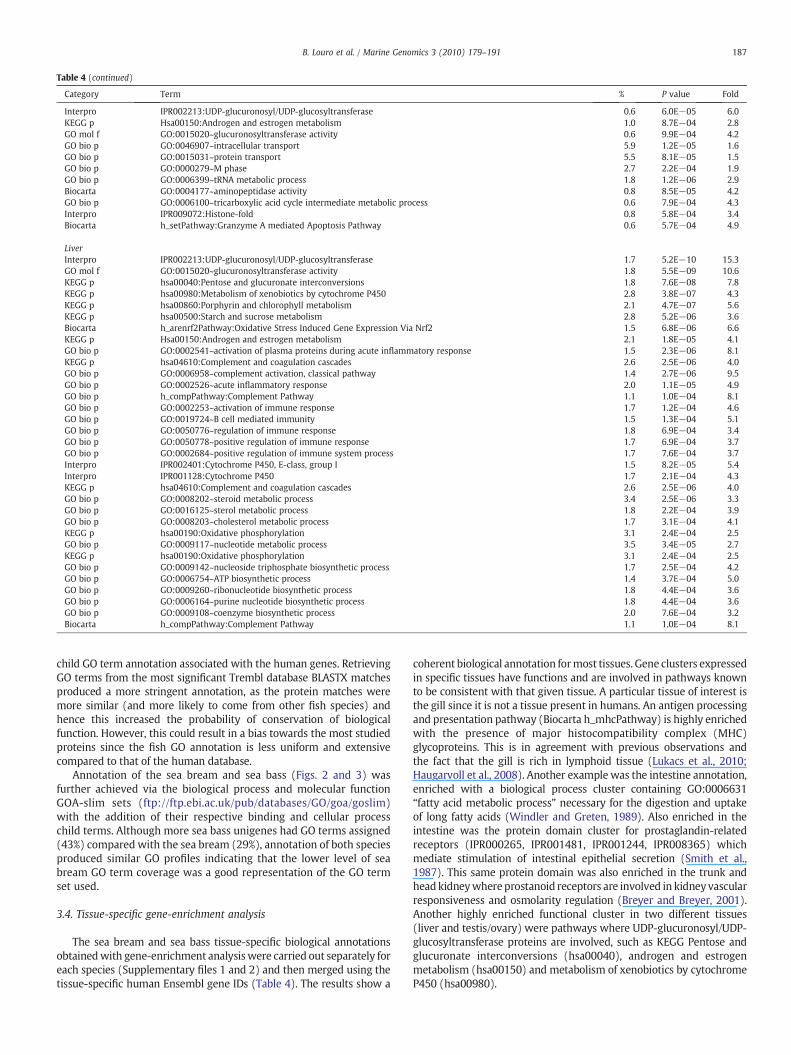
Table 4 (continued)
Category Term % P value Fold
Interpro IPR002213:UDP-glucuronosyl/UDP-glucosyltransferase 0.6 6.0E−05 6.0KEGG p Hsa00150:Androgen and estrogen metabolism 1.0 8.7E−04 2.8GO mol f GO:0015020~glucuronosyltransferase activity 0.6 9.9E−04 4.2GO bio p GO:0046907~intracellular transport 5.9 1.2E−05 1.6GO bio p GO:0015031~protein transport 5.5 8.1E−05 1.5GO bio p GO:0000279~M phase 2.7 2.2E−04 1.9GO bio p GO:0006399~tRNA metabolic process 1.8 1.2E−06 2.9Biocarta GO:0004177~aminopeptidase activity 0.8 8.5E−05 4.2GO bio p GO:0006100~tricarboxylic acid cycle intermediate metabolic process 0.6 7.9E−04 4.3Interpro IPR009072:Histone-fold 0.8 5.8E−04 3.4Biocarta h_setPathway:Granzyme A mediated Apoptosis Pathway 0.6 5.7E−04 4.9
LiverInterpro IPR002213:UDP-glucuronosyl/UDP-glucosyltransferase 1.7 5.2E−10 15.3GO mol f GO:0015020~glucuronosyltransferase activity 1.8 5.5E−09 10.6KEGG p hsa00040:Pentose and glucuronate interconversions 1.8 7.6E−08 7.8KEGG p hsa00980:Metabolism of xenobiotics by cytochrome P450 2.8 3.8E−07 4.3KEGG p hsa00860:Porphyrin and chlorophyll metabolism 2.1 4.7E−07 5.6KEGG p hsa00500:Starch and sucrose metabolism 2.8 5.2E−06 3.6Biocarta h_arenrf2Pathway:Oxidative Stress Induced Gene Expression Via Nrf2 1.5 6.8E−06 6.6KEGG p Hsa00150:Androgen and estrogen metabolism 2.1 1.8E−05 4.1GO bio p GO:0002541~activation of plasma proteins during acute inflammatory response 1.5 2.3E−06 8.1KEGG p hsa04610:Complement and coagulation cascades 2.6 2.5E−06 4.0GO bio p GO:0006958~complement activation, classical pathway 1.4 2.7E−06 9.5GO bio p GO:0002526~acute inflammatory response 2.0 1.1E−05 4.9GO bio p h_compPathway:Complement Pathway 1.1 1.0E−04 8.1GO bio p GO:0002253~activation of immune response 1.7 1.2E−04 4.6GO bio p GO:0019724~B cell mediated immunity 1.5 1.3E−04 5.1GO bio p GO:0050776~regulation of immune response 1.8 6.9E−04 3.4GO bio p GO:0050778~positive regulation of immune response 1.7 6.9E−04 3.7GO bio p GO:0002684~positive regulation of immune system process 1.7 7.6E−04 3.7Interpro IPR002401:Cytochrome P450, E-class, group I 1.5 8.2E−05 5.4Interpro IPR001128:Cytochrome P450 1.7 2.1E−04 4.3KEGG p hsa04610:Complement and coagulation cascades 2.6 2.5E−06 4.0GO bio p GO:0008202~steroid metabolic process 3.4 2.5E−06 3.3GO bio p GO:0016125~sterol metabolic process 1.8 2.2E−04 3.9GO bio p GO:0008203~cholesterol metabolic process 1.7 3.1E−04 4.1KEGG p hsa00190:Oxidative phosphorylation 3.1 2.4E−04 2.5GO bio p GO:0009117~nucleotide metabolic process 3.5 3.4E−05 2.7KEGG p hsa00190:Oxidative phosphorylation 3.1 2.4E−04 2.5GO bio p GO:0009142~nucleoside triphosphate biosynthetic process 1.7 2.5E−04 4.2GO bio p GO:0006754~ATP biosynthetic process 1.4 3.7E−04 5.0GO bio p GO:0009260~ribonucleotide biosynthetic process 1.8 4.4E−04 3.6GO bio p GO:0006164~purine nucleotide biosynthetic process 1.8 4.4E−04 3.6GO bio p GO:0009108~coenzyme biosynthetic process 2.0 7.6E−04 3.2Biocarta h_compPathway:Complement Pathway 1.1 1.0E−04 8.1
187B. Louro et al. / Marine Genomics 3 (2010) 179–191
child GO term annotation associated with the human genes. RetrievingGO terms from the most significant Trembl database BLASTX matchesproduced a more stringent annotation, as the protein matches weremore similar (and more likely to come from other fish species) andhence this increased the probability of conservation of biologicalfunction. However, this could result in a bias towards the most studiedproteins since the fish GO annotation is less uniform and extensivecompared to that of the human database.
Annotation of the sea bream and sea bass (Figs. 2 and 3) wasfurther achieved via the biological process and molecular functionGOA-slim sets (ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/goslim)with the addition of their respective binding and cellular processchild terms. Although more sea bass unigenes had GO terms assigned(43%) compared with the sea bream (29%), annotation of both speciesproduced similar GO profiles indicating that the lower level of seabream GO term coverage was a good representation of the GO termset used.
3.4. Tissue-specific gene-enrichment analysis
The sea bream and sea bass tissue-specific biological annotationsobtainedwith gene-enrichment analysiswere carried out separately foreach species (Supplementary files 1 and 2) and then merged using thetissue-specific human Ensembl gene IDs (Table 4). The results show a
coherent biological annotation formost tissues. Gene clusters expressedin specific tissues have functions and are involved in pathways knownto be consistent with that given tissue. A particular tissue of interest isthe gill since it is not a tissue present in humans. An antigen processingand presentation pathway (Biocarta h_mhcPathway) is highly enrichedwith the presence of major histocompatibility complex (MHC)glycoproteins. This is in agreement with previous observations andthe fact that the gill is rich in lymphoid tissue (Lukacs et al., 2010;Haugarvoll et al., 2008). Another example was the intestine annotation,enriched with a biological process cluster containing GO:0006631“fatty acid metabolic process” necessary for the digestion and uptakeof long fatty acids (Windler and Greten, 1989). Also enriched in theintestine was the protein domain cluster for prostaglandin-relatedreceptors (IPR000265, IPR001481, IPR001244, IPR008365) whichmediate stimulation of intestinal epithelial secretion (Smith et al.,1987). This same protein domain was also enriched in the trunk andheadkidneywhereprostanoid receptors are involved in kidney vascularresponsiveness and osmolarity regulation (Breyer and Breyer, 2001).Another highly enriched functional cluster in two different tissues(liver and testis/ovary) were pathways where UDP-glucuronosyl/UDP-glucosyltransferase proteins are involved, such as KEGG Pentose andglucuronate interconversions (hsa00040), androgen and estrogenmetabolism (hsa00150) and metabolism of xenobiotics by cytochromeP450 (hsa00980).

188 B. Louro et al. / Marine Genomics 3 (2010) 179–191
Fish are increasingly recognised as an important model species forbone disease related studies, hence it was noted that in the bone/cartilage tissues results that the GO molecular function cluster“calcium ion binding” was highly enriched. The parent GO term ”ionbinding” is represented in Figs. 2 and 3. Proteins tagged with this GOterm interact selectively and non-covalentlywith calcium ions (Ca2+),with obvious roles in the chemical dynamics of bone mineralization,e.g., the secreted “Cartilage Acidic protein 1” that contains an EGF-likedomain and is detected in humans cartilage, bone, cultured chon-drocytes and lung (Benz et al., 2002). Another example is “matrixgla 1” which associates with the organic matrix of bone and cartilage,in principle acting as an inhibitor of cartilage calcification (Luo et al.,1997).
3.5. Alternative splice form analysis
Single alignments of the 311 and 193 sea bream and sea basssupracontigs (see Section 2.6) containing 767 and 400 contigsrespectively identified89and 54 inserts, or partially different sequences.This indicated alternative splice events in 79 and 46 sea bream and seabass genes respectively. It was possible to confirm by interspeciesmRNA-to-genomic alignments that 25 and 21 alternative splice eventswere present in sea bream (Supplemental file 3, Fig. 4) and sea bass(Supplemental file 4, Fig. 4) genes, respectively. No alternative selectionof promoters was identified in either species (Fig. 4) due to themethodology used rather than because they are not present in thetranscriptomes. This latter type of alternative transcript isoform leads tovariation of thefirst exon present in the transcript containingmost if notall the 5' UTR region, and these tend not to align well in mRNA-to-genomic alignments between different species (Wheelan et al., 2001)and hence are difficult to detect in comparative analyses.
The remaining supracontigs for which it was not possible to confirmgenomic organization and the respective alternative splice event, waseither due to no retrieval of the homologous green spotted pufferfishgene loci, no alignment at the target splice site region, or no alignmentat all.
Several alternative transcripts were identified in specific tissues,although the possibility of overlap of expression cannot be ruled out.
Fig. 4. Alternative splice events identified in
Sea bass ornithine decarboxylase antizyme isoform a (SeabassC-CL86Contig1) was present in the trunk kidney library while isoform b(SeabassC-CL86Contig2) was present in muscle and brain/pituitarylibraries. Similarly, sea bream Prefoldin subunit 3 isoform a (Sea-bassC-CL86Contig1) was present in the adipose, liver, head kidney,ovary, gill, and trunk kidney libraries and isoform b (SeabassC-CL86Contig2) in ovary library.
Highly similar gene products within the sea bream and sea bassunigenes, numbering 57 and 33 respectively, were also identified (datanot shown) using multiple contig alignment. Future detailed phyloge-netic analysis will be needed to confirm these putative paralogousgenes.
3.6. SNP detection
All contigs with a minimum depth of four overlapping sequenceswere selected for SNPmining. For both sea bass and sea bream a singlecontig containingmore than50 ESTwas present and removed from thedata set to minimize discovery of false positive candidate SNPs (LeDantec et al., 2004). Both contigs (2,006 ESTs and 1026 bp long in seabass and 111 ESTs and 913 bp long in sea bream) mapped to thehuman aldolase A, fructose-bisphosphate (ALDOA) transcript variant2, mRNA. A total of 974 (21.3%) sea bass contigs of 4 to 37 ESTsand 1136 (21.6%) sea bream contigs of 4 to 22 ESTs were mined forSNPs. In sea bass, 570 SNP candidates were discovered in 477,224 bp,representing one SNP candidate every 837 bp. In sea bream, 575 SNPcandidates were discovered in 583,397 bp, representing one SNPcandidate every 1014 bp. Although more contigs (and more ESTs)were screened for SNPs in the sea bream, the proportion ofpolymorphic contigs was lower in sea bream (31.3%) than in seabass (36.4%). This could be due to fish originating from differentaquaculture facilitieswith different degrees of kinship and/or differentgeographical origins. Fewer transitions (49%) and transversions (25%)were detected in the sea bream than the sea bass (60% of transitionsand 30% of transversions). The proportion of detected indels wasmorethan double in sea bream (26%) compared with the sea bass (10%)possibly as a result of the fact that sea bream contigs include lessprotein-coding regions (data not shown). Most of the indels (59%
sea bream and sea bass gene products.

Table 5Detection of microsatellites (SSRs) found in sea bream and sea bass ESTs (SSR-ESTs). Number of repeats is considered 9, 6, 5, 4 and 4 for di-, tri, tetra-, penta- and hexanucleotides,respectively. Data for other fish are from (Ju et al., 2005).
Species Unigenes SSR-ESTs Number of SSRs / motif (%) SSRs total
n % Di- Tri- Tetra- Penta- Hexa- n
Sea bream 18,196 899 4.94 474 (47.6) 325 (32.6) 115 (11.5) 66 (6.6) 17 (1.7) 997Sea bass 17,716 795 3.97 267 (37.6) 312 (41.5) 109 (14.5) 47 (6.3) 16 (2.1) 751Zebrafish 24,003 1.749 7.3 1,497 (64) 579 (25) 202 (8.6) 55 (2.4) – 2,333Medaka 8,158 209 2.6 105 (47) 81 (36) 27 (12) 10 (4.5) – 223Killifish 16,726 369 2.2 237 (52) 155 (34) 43 (9.5) 18 (4.0) – 453
189B. Louro et al. / Marine Genomics 3 (2010) 179–191
for sea bass and 72% for sea bream) were only one nucleotide long. Inthe sea bass, 25% and 8% of the indels were two and three nucleotideslong, while in the sea bream, theywere 21% and 6%, respectively. Theirimpact varies depending on their position in the genome. For instance,SNPs located in the promoter region of a gene may influence geneexpression while non-synonymous SNPs may alter gene function(Barnes, 2009). Some polymorphic microsatellites can be detected asindels by SNP discovery tools and this could explain the high number ofindels longer than one nucleotide detected in the sea bream since thisspecies contains more microsatellites than sea bass. The proportion oftransitions, transversions and indels detected in the sea bass wasconsistent with the proportion found while sequencing 80 sea bassloci (Souche et al, unpublished). Sequencing of a few sea bream lociwould also clarify if theproportion of detected transitions, transversionsand indels (and the high number of one nucleotide long indels) iscorrect.
3.7. Microsatellite detection
A total of 899 and 795 sea bream and sea bass unigenes wereidentified that harboured 997 and 751 SSRs respectively (Table 5). Thefrequency of EST-SSRs was 3.97% for the sea bass and 4.94% for the seabream and is in the range reported for model fish species such asmedaka (2.6%), mummichog killifish (2.2%), zebrafish (7.3%) and11.2% in channel catfish, Ictalurus punctatus (Ju et al., 2005; Serapionet al., 2004). The dinucleotide repeat motifs were the most abundantSSRs in sea bream accounting for 47.6% of the SSRs as observed inother species (47%, 52%, and 64% for medaka, mummichog killifish,and zebrafish, respectively), followed by the tri-, tetra-, and penta-and hexanucleotide SSRs (Table 5). In contrast, the most abundantrepeats in the sea bass were the trinucleotides (Table 5). In the seabass, the greater abundance of trinucleotides together with thereduced number of EST-SSR % frequency might reflect the fact thatsea bass unigenes in comparison to sea bream unigenes representedfewer UTR and more coding regions (data not shown) wheretrinucleotides are more abundant (Tóth et al., 2000). Among thedinucleotide motifs, AC/TG was the most abundant type in bothspecies. This was also observed in the other species exceptmummichog killifish in which AT/TA was the most abundant motif(Ju et al., 2005). No CG/GC motif was found in the sea bass and seabream ESTs. Ten trinucleotide SSR motifs were identified in sea bassand sea bream; the AAC and AAGmotifs were themost frequent in seabass and sea bream, respectively, whereas the other 8 trinucleotidemotifs (AAT, ACC, ACG, ACT, AGC, AGG, ATC, and CCG) wereapproximately equally frequent. The length and location of an SSRmotif is an important factor in determining its usefulness as a marker,the longer the repeats, the higher the probability a marker will bepolymorphic. For subsequent marker development for linkagemapping and population genetics studies it is advisable to prioritizescreening of EST-SSRs with relatively high repeat number for eachtype, e.g. dinucleotides with more than 20 repeats (11.7% ofdinucleotides), tri- and tetranucleotides with more than 15 repeats(5.7 and 6.5%, respectively), and penta- and hexanucleotides withmore than 6 repeats (11.3% and 7.1, respectively).
4. Conclusions
We reported the characterisation and analysis of approximately30,000 ESTs from each of the sea bream and the sea bass ESTs. Thesewere generated from a range of tissues. The comparative annotationapproach described here shows the utility of retrieving useful biologicalannotation from less-related model species and in particular, fromhuman, as this increases the amount of biological annotation. Theincreasing democratization of genomics through the use of new,cheaper and higher throughput sequencing technologies (such asNext Gen sequencing) will produce a large increase in the number oftranscriptomes from new species. These will lack phylogeneticallyclosely related well-annotated species for assignment of putativefunctionality to genes and therefore the comparative annotationapproach described here is useful and easy to adapt. The primarygoal of this project was production of the EST datasets for theaquaculture community, with the aim of these acting as a cornerstoneresource for the development of new genomic tools with broodstockand culturing applications. As a result, the sea bass and the sea breamhave become part of the small group of genomic resource richaquaculture species.
Supplementarymaterials related to this article can be found onlineat doi:10.1016/j.margen.2010.09.005.
Acknowledgements
The authors acknowledge funding by the European Commission ofthe European Union through the Network of Excellence MarineGenomics Europe (contract GOCE-CT-2004-505403). BL benefitedfrom the Portuguese National Science Foundation (SFRH/BD/29171/2006) and SABRETRAIN Marie Curie EST fellowships. The authorsthankDr. Juan Fuentes andCarla Viegas (Centre ofMarine Sciences) forthe tissue collection and RNA extraction, respectively, and Sven Klages(MPI Molecular Genetics) for the bioinformatic support.
References
Adzhubei, A.A., Vlasova, A.V., Hagen-Larsen, H., Ruden, T.A., Laerdahl, J.K., Hoyheim, B.,2007. Annotated Expressed Sequence Tags (ESTs) from pre-smolt atlantic salmon(Salmo salar) in a searchable data resource. BMC Genomics 8, 209.
Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J., 1990. Basic local alignmentsearch tool. J. Mol. Biol. 215, 403–410.
Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., Lipman, D.J.,1997. Gapped BLAST and PSI-BLAST: a new generation of protein database searchprograms. Nucleic Acids Res. 25, 3389–3402.
Arends, R., Mancera, J., Munoz, J., Wendelaar Bonga, S., Flick, G., 1999. The stressresponse of gilthead sea bream (Sparus aurata L) to air exposure and confinement.J. Endocrinol. 143, 23–31.
Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P.,Dolinski, K., Dwight, S.S., Eppig, J.T., Harris, M.A., Hill, D.P., Issel-Tarver, L., Kasarskis,A., Lewis, S., Matese, J.C., Richardson, J.E., Ringwald, M., Rubin, G.M., Sherlock, G.,2000. Gene ontology: tool for the unification of biology. The Gene OntologyConsortium. Nat. Genet. 25, 25–29.
Barnes, M.R. Genetic variation analysis for biomedical researchers: A primer, in, 2009,pp. 1-20.
Batley, J., Barker, G., O'Sullivan, H., Edwards, K.J., Edwards, D., 2003. Mining for singlenucleotide polymorphisms and insertions/deletions in maize expressed sequencetag data. Plant Physiol. 132, 84–91.

190 B. Louro et al. / Marine Genomics 3 (2010) 179–191
Benz, K., Breit, S., Lukoschek, M., Mau, H., Richter, W., 2002. Molecular analysis ofexpansion, differentiation, and growth factor treatment of human chondrocytesidentifies differentiation markers and growth-related genes. Biochem. Biophys.Res. Commun. 293, 284–292.
Bonaldo, M.F., Lennon, G., Soares, M.B., 1996. Normalization and subtraction: twoapproaches to facilitate gene discovery. Genome Res. 6, 791–806.
Breyer, M.D., Breyer, R.M., 2001. G protein-coupled prostanoid receptors and thekidney. Annu. Rev. Physiol. 63, 579–605.
Calduch-Giner, J., Davey, G., Saera-Vila, A., Houeix, B., Talbot, A., Prunet, P., Cairns, M.,Perez-Sanchez, J., 2010. Use of microarray technology to assess the time course ofliver stress response after confinement exposure in gilthead sea bream (Sparusaurata L.). BMC Genomics 11, 193.
Canario, A.V.M., Bargelloni, L., Volckaert, F., Houston, R.D., Massault, C., Guiguen, Y.,2008. Genomics toolbox for farmed fish. Rev. Fish. Sci. 16 (S1), 1–13.
Cerdà, J., Mercade, J., Lozano, J., Manchado, M., Tingaud-Sequeira, A., Astola, A.,Infante, C., Halm, S., Vinas, J., Castellana, B., Asensio, E., Canabate, P., Martinez-Rodriguez, G., Piferrer, F., Planas, J., Prat, F., Yufera, M., Durany, O., Subirada, F.,Rosell, E., Maes, T., 2008. Genomic resources for a commercial flatfish, theSenegalese sole (Solea senegalensis): EST sequencing, oligo microarray design,and development of the Soleamold bioinformatics platform. BMC Genomics 9,508.
Chevreux, B., Pfisterer, T., Drescher, B., Driesel, A.J., Muller, W.E., Wetter, T., Suhai,S., 2004. Using the miraEST assembler for reliable and automated mRNAtranscript assembly and SNP detection in sequenced ESTs. Genome Res. 14,1147–1159.
Chini, V., Rimoldi, S., Terova, G., Saroglia, M., Rossi, F., Bernardini, G., Gornati, R., 2006.EST-based identification of genes expressed in the liver of adult seabass(Dicentrarchus labrax, L.). Gene 376, 102–106.
Chomczynski, P., Sacchi, N., 1987. Single-step method of RNA isolation by acidguanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162,156–159.
Chou, H.H., Holmes, M.H., 2001. DNA sequence quality trimming and vector removal.Bioinformatics 17, 1093–1104.
Clark, M.S., Edwards, Y.J., Peterson, D., Clifton, S.W., Thompson, A.J., Sasaki, M., Suzuki,Y., Kikuchi, K., Watabe, S., Kawakami, K., Sugano, S., Elgar, G., Johnson, S.L., 2003.Fugu ESTs: new resources for transcription analysis and genome annotation.Genome Res. 13, 2747–2753.
Coblentz, F.E., Towle, D.W., Shafer, T.H., 2006. Expressed sequence tags fromnormalized cDNA libraries prepared from gill and hypodermal tissues of the bluecrab, Callinectes sapidus. Comp. Biochem. Physiol. Part D Genomics Proteomics 1,200–208.
Douglas, S., Gallant, J., Bullerwell, C., Wolff, C., Munholland, J., Reith, M., 1999. Winterflounder expressed sequence tags: establishment of an EST database andidentification of novel fish genes. Mar. Biotechnol. 1, 458–464.
Douglas, S.E., Knickle, L.C., Kimball, J., Reith, M.E., 2007. Comprehensive EST analysis ofAtlantic halibut (Hippoglossus hippoglossus), a commercially relevant aquaculturespecies. BMC Genomics 8, 144.
Edwards, N.J., 2007. Novel peptide identification from tandem mass spectra using ESTsand sequence database compression. Mol. Syst. Biol. 3, 102.
Ewing, B., Green, P., 1998. Base-calling of automated sequencer traces using phred. II.Error probabilities. Genome Res. 8, 186–194.
Ewing, B., Hillier, L., Wendl, M.C., Green, P., 1998. Base-calling of automated sequencertraces using phred. I. Accuracy assessment. Genome Res. 8, 175–185.
Ferraresso, S., Vitulo, N., Mininni, A., Romualdi, C., Cardazzo, B., Negrisolo, E., Reinhardt,R., Canario, A., Patarnello, T., Bargelloni, L., 2008. Development and validation of agene expression oligo microarray for the gilthead sea bream (Sparus aurata). BMCGenomics 9, 580.
Ferraresso, S., Milan, M., Pellizzari, C., Vitulo, N., Reinhardt, R., Canario, A.V.M.,Patarnello, T., Bargelloni, L., 2010. Development of an oligo DNA microarray for theEuropean sea bass and its application to expression profiling of jaw deformity. BMCGenomics 11, 354.
Garg, K., Green, P., Nickerson, D.A., 1999. Identification of candidate coding regionsingle nucleotide polymorphisms in 165 human genes using assembled expressedsequence tags. Genome Res. 9, 1087–1092.
Gong, Z., 1999. Zebrafish expressed sequence tags and their applications. Methods CellBiol. 60, 213–233.
Gong, Z., Yan, T., Liao, J., Lee, S., He, J., Hew, C., 1997. Rapid identification and isolation ofzebrafish cDNA clones. Gene 201, 87–98.
Gonzalez, S., Chatziandreou, N., Nielsen, M., Li, W., Rogers, J., Taylor, R., Santos, Y.,Cossins, A., 2007. Cutaneous immune responses in the common carp detected usingtranscript analysis. Mol. Immunol. 44, 1675–1690.
Govoroun, M., Le Gac, F., Guiguen, Y., 2006. Generation of a large scale repertoire ofExpressed Sequence Tags (ESTs) from normalised rainbow trout cDNA libraries.BMC Genomics 7, 196.
Hagen-Larsen, H., Laerdahl, J.K., Panitz, F., Adzhubei, A., Høyheim, B., 2005. An EST-based approach for identifying genes expressed in the intestine and gills of pre-smolt atlantic salmon (Salmo salar). BMC Genomics 6, 171.
Haugarvoll, E., Bjerkas, I., Nowak, B.F., Hordvik, I., Koppang, E.O., 2008. Identificationand characterization of a novel intraepithelial lymphoid tissue in the gills ofAtlantic salmon. J. Anat. 213, 202–209.
Hirono, I., Aoki, T., 1997. Expressed sequence tags of medaka (Oryzias latipes) livermRNA. Mol. Mar. Biol. Biotechnol. 6, 345–350.
Ju, Z., Wells, M.C., Martinez, A., Hazlewood, L., Walter, R.B., 2005. An in silico mining forsimple sequence repeats from expressed sequence tags of zebrafish, medaka,Fundulus, and Xiphophorus. In Silico Biol. 5, 439–463.
Kent, W.J., 2002. BLAT- the BLAST-like alignment tool. Genome Res. 12, 656–664.
Koop, B., von Schalburg, K., Leong, J., Walker, N., Lieph, R., Cooper, G., Robb, A., Beetz-Sargent, M., Holt, R., Moore, R., Brahmbhatt, S., Rosner, J., Rexroad, C., McGowan, C.,Davidson, W., 2008. A salmonid EST genomic study: genes, duplications, phylogenyand microarrays. BMC Genomics 9, 545.
Kuhl, H., Beck, A., Wozniak, G., Canario, A.V.M., Volckaert, F.A.M., Reinhardt, R., 2010.The European sea bass Dicentrarchus labrax genome puzzle: comparative BAC-mapping and low coverage shotgun sequencing. BMC Genomics 11, 68.
Laiz-Carrion, R., Guerreiro, P.M., Fuentes, J., Canario, A.V.M., Del Rio, M.P.M., Mancera,J.M., 2005. Branchial osmoregulatory response to salinity in the gilthead seabream, Sparus auratus. J. Exp. Zool. Comp. Exp. Biol. 303A, 563–576.
Le Dantec, L., Chagné, D., Pot, D., Cantin, O., Garnier- Géré, P., Bedon, F., Frigerio, J.-M.,Chaumeil, P., Léger, P., Garcia, V., Laigret, F., de Daruvar, A., Plomion, C., 2004.Automated SNP detection in expressed sequence tags: statistical considerationsand application to maritime pine sequences. Plant Mol. Biol. 54, 461–470.
Lee, B.Y., Howe, A.E., Conte, M.A., D'Cotta, H., Pepey, E., Baroiller, J.F., di Palma, F.,Carleton, K.L., Kocher, T.D., 2010. An EST resource for tilapia based on 17normalized libraries and assembly of 116, 899 sequence tags. BMC Genomics 11,278.
Lemaire, C., Versini, J.J., Bonhomme, F., 2005. Maintenance of genetic differentiationacross a transition zone in the sea: discordance between nuclear and cytoplasmicmarkers. J. Evolution Biol. 18, 70–80.
Li, P., Peatman, E., Wang, S., Feng, J., He, C., Baoprasertkul, P., Xu, P., Kucuktas, H., Nandi,S., Somridhivej, B., Serapion, J., Simmons, M., Turan, C., Liu, L., Muir, W., Dunham, R.,Brady, Y., Grizzle, J., Liu, Z., 2007. Towards the ictalurid catfish transcriptome:generation and analysis of 31, 215 catfish ESTs. BMC Genomics 8, 177.
Lukacs, M.F., Harstad, H., Bakke, H.G., Beetz-Sargent, M., McKinnel, L., Lubieniecki, K.P.,Koop, B.F., Grimholt, U., 2010. Comprehensive analysis of MHC class I genes fromthe U-, S-, and Z-lineages in Atlantic salmon. BMC Genomics 11, 154.
Luo, G., Ducy, P., McKee, M.D., Pinero, G.J., Loyer, E., Behringer, R.R., Karsenty, G., 1997.Spontaneous calcification of arteries and cartilage in mice lacking matrix GLAprotein. Nature 386, 78–81.
Massault, C., Hellemans, B., Louro, B., Batargias, C., Houdt, J.K.J.V., Canario, A., Volckaert,F.A.M., Bovenhuis, H., Haley, C., Koning, D.J.d., 2010. QTL for body weight,morphometric traits and stress response in European sea bass Dicentrarchuslabrax. Anim. Genet. 41, 337–345.
Meiri, I., Gothilf, Y., Zohar, Y., Elizur, A., 2002. Physiological changes in the spawninggilthead seabream, Sparus aurata, succeeding the removal of males. J. Exp. Zool.292, 555–564.
Morozova, O., Marra, M.A., 2008. Applications of next-generation sequencingtechnologies in functional genomics. Genomics 92, 255–264.
Paschall, J., Oleksiak, M., VanWye, J., Roach, J., Whitehead, J., Wyckoff, G., Kolell, K., DL,C., 2004. FunnyBase: a systems level functional annotation of Fundulus ESTs for theanalysis of gene expression. BMC Genomics 5, 96.
Pawson, M.G., Pickett, G.D., Leballeur, J., Brown, M., Fritsch, M., 2007. Migrations, fisheryinteractions, and management units of sea bass (Dicentrarchus labrax) inNorthwest Europe. ICES J. Mar. Sci. 64, 332–345.
Pertea, G., Huang, X., Liang, F., Antonescu, V., Sultana, R., Karamycheva, S., Lee, Y., White,J., Cheung, F., Parvizi, B., Tsai, J., Quackenbush, J., 2003. TIGR Gene Indices clusteringtools (TGICL): a software system for fast clustering of large EST datasets.Bioinformatics 19, 651–652.
Piferrer, F., Blazquez, M., Navarro, L., Gonzalez, A., 2005. Genetic, endocrine, andenvironmental components of sex determination and differentiation in theEuropean sea bass (Dicentrarchus labrax L.). Gen. Comp. Endocrinol. 142, 102–110.
Power, D., Llewellyn, L., Faustino, M., Nowell, M., Bjornsson, B., Einarsdottir, I., Canario,A., Sweeney, G., 2001. Thyroid hormones in growth and development of fish. Comp.Biochem. Physiol. C Toxicol. Pharmacol. 130, 447–459.
Scapigliati, G., Romano, N., Buonocore, F., Picchietti, S., Baldassini, M., Prugnoli, D.,Galice, A., Meloni, S., Secombes, C., Mazzini, M., Abelli, L., 2002. The immune systemof sea bass, Dicentrarchus labrax, reared in aquaculture. Dev. Comp. Immunol. 26,151–160.
Senger, F., Priat, C., Hitte, C., Sarropoulou, E., Franch, R., Geisler, R., Bargelloni, L., Power,D., Galibert, F., 2006. The first radiation hybrid map of a perch-like fish: the giltheadseabream (Sparus aurata L). Genomics 87, 793–800.
Serapion, J., Kucuktas, H., Feng, J., Liu, Z., 2004. Bioinformatic mining of type I micro-satellites from expressed sequence tags of channel catfish (Ictalurus punctatus).Mar. Biotechnol. 6, 364–377.
Smit, A.F.A., Hubley, R., Green, P. RepeatMasker Open-3.0, 1996-2010 http://www.repeatmasker.org.
Smith, G., Warhurst, G., Lees, M., Turnberg, L., 1987. Evidence that PGE2 stimulatesintestinal epithelial cell adenylate cyclase by a receptor-mediated mechanism. Dig.Dis. Sci. 32, 71–75.
Souche, E.L., Hellemans, B., Houdt, J.K.J.V., Canario, A., Klages, S., Reinhardt, R., Volckaert,F.A.M., 2007. Mining for single nucleotide polymorphisms in expressed sequencetags of European sea bass. J. Integr. Bioinform. 4, 73.
Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., Higgins, D.G., 1997. TheCLUSTAL_X windows interface: flexible strategies for multiple sequence alignmentaided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882.
Tóth, G., Gáspári, Z., Jurka, J., 2000. Microsatellites in different eukaryotic genomes:survey and analysis. Genome Res. 10, 967–981.
Vogiatzi, E., Lagnel, J., Pakaki, V., Louro, B., Canario, A.V.M., Reinhardt, R., Kotoulas, G.,Magoulas, A., Tsigenopoulos, C.S. In silico mining and characterization of simplesequence repeats from gilthead sea bream (Sparus aurata) expressed sequence tags(EST-SSRs); PCR amplification, polymorphism evaluation and multiplexing andcross-species assays, Marine Genomics, (submitted).
Wang, S., Peatman, E., Abernathy, J., Waldbieser, G., Lindquist, E., Richardson, P., Lucas,S., Wang, M., Li, P., Thimmapuram, J., Liu, L., Vullaganti, D., Kucuktas, H., Murdock,

191B. Louro et al. / Marine Genomics 3 (2010) 179–191
C., Small, B.C., Wilson, M., Liu, H., Jiang, Y., Lee, Y., Chen, F., Lu, J., Wang, W., Xu, P.,Somridhivej, B., Baoprasertkul, P., Quilang, J., Sha, Z., Bao, B., Wang, Y., Wang, Q.,Takano, T., Nandi, S., Liu, S., Wong, L., Kaltenboeck, L., Quiniou, S., Bengten, E., Miller,N., Trant, J., Rokhsar, D., Liu, Z., 2010. Assembly of 500,000 inter-specific catfishexpressed sequence tags and large scale gene-associated marker development forwhole genome association studies. Genome Biol. 11, R8.
Wheelan, S.J., Church, D.M., Ostell, J.M., 2001. Spidey: a tool for mRNA-to-genomicalignments. Genome Res. 11, 1952–1957.
Windler, E., Greten, H., 1989. Intestinal lipid and lipoprotein metabolism, in. W.Zuckschwerdt Verlag, München.
Zhi-Liang, H., Bao, J., Reecy, J.M., 2008. CateGOrizer: A web-based program to batchgene ontology classification categories, in. Online J. Bioinformatics 5.
Zhu, Y.Y., Machleder, E.M., Chenchik, A., Li, R., Siebert, P.D., 2001. Reverse transcriptasetemplate switching: a SMART approach for full-length cDNA library construction.Biotechniques 30, 892–897.
Zhulidov, P.A., Bogdanova, E.A., Shcheglov, A.S., Vagner, L.L., Khaspekov, G.L.,Kozhemyako, V.B., Matz, M.V., Meleshkevitch, E., Moroz, L.L., Lukyanov, S.A.,Shagin, D.A., 2004. Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 32, e37.




















