
Gene Expression Similarity with Polygonal Chain Alignment.
12
Elsevier Editorial System(tm) for Scientia Iranica Manuscript Draft Manuscript Number: SCIENTIA-D-11-00115 Title: Gene Expression Similarity with Polygonal Chain Alignment Article Type: Full Article Section/Category: Transactions D - Computer Science & Engineering Keywords: gene expression time series; polygonal chain matching; global sequence alignment Corresponding Author: Mr Hadi Banaee, Corresponding Author's Institution: Amirkabir University of Technology, Tehran, Iran First Author: Hadi Banaee Order of Authors: Hadi Banaee; Ali Mohades, associate professor; Fatemeh Zare, Assistant Professor Abstract: In this paper, we propose a new method to evaluate and find the similarity between two gene expression time series and regulatory gene pairs, respectively. In this method, each gene expression time series is modeled by a polygonal chain. Then, a polygonal chain sequence is defined based on the polygonal chain's geometric parameters. Finally, two polygonal chain sequences are aligned. This algorithm finds optimal similar sub-chains of two polygonal chains with gaps. We perform our approach on the known gene expression time series datasets and compare our results with some common methods. The experimental results show that our method finds regulatory gene pairs more efficient than the other methods. Suggested Reviewers: Abbas Nowzari Dalini Associate Professor Computer Science, University of Tehran [email protected] Mehdi Sadeghi Associate Professor National Institute of Genetic Engineering and Biotechnology [email protected] Mohammad Ganjtabesh Assistant Professor Computer Science, University of Tehran [email protected]
Transcript of Gene Expression Similarity with Polygonal Chain Alignment.

Elsevier Editorial System(tm) for Scientia Iranica Manuscript Draft Manuscript Number: SCIENTIA-D-11-00115 Title: Gene Expression Similarity with Polygonal Chain Alignment Article Type: Full Article Section/Category: Transactions D - Computer Science & Engineering Keywords: gene expression time series; polygonal chain matching; global sequence alignment Corresponding Author: Mr Hadi Banaee, Corresponding Author's Institution: Amirkabir University of Technology, Tehran, Iran First Author: Hadi Banaee Order of Authors: Hadi Banaee; Ali Mohades, associate professor; Fatemeh Zare, Assistant Professor Abstract: In this paper, we propose a new method to evaluate and find the similarity between two gene expression time series and regulatory gene pairs, respectively. In this method, each gene expression time series is modeled by a polygonal chain. Then, a polygonal chain sequence is defined based on the polygonal chain's geometric parameters. Finally, two polygonal chain sequences are aligned. This algorithm finds optimal similar sub-chains of two polygonal chains with gaps. We perform our approach on the known gene expression time series datasets and compare our results with some common methods. The experimental results show that our method finds regulatory gene pairs more efficient than the other methods. Suggested Reviewers: Abbas Nowzari Dalini Associate Professor Computer Science, University of Tehran [email protected] Mehdi Sadeghi Associate Professor National Institute of Genetic Engineering and Biotechnology [email protected] Mohammad Ganjtabesh Assistant Professor Computer Science, University of Tehran [email protected]

SCIENTIA IRANICA International Journal of Science and Technology
Journal
CICIS'11 Manuscript ID
Manuscript Type
Complete List of Authors
Keywords
Title
Cover Letter

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
Gene Expression Similarity with Polygonal Chain
Alignment
Hadi Banaee, Ali Mohades∗, Fatemeh Zare
Laboratory of Algorithms and Computational Geometry
Department of Mathematics and Computer Science
Amirkabir University of Technology, Tehran, Iran
{h.banaee, mohades, f.zare}@aut.ac.ir
Abstract
In this paper, we propose a new method to evaluate and find the similaritybetween two gene expression time series and regulatory gene pairs, respec-tively. In this method, each gene expression time series is modeled by apolygonal chain. Then, a polygonal chain sequence is defined based on thepolygonal chain’s geometric parameters. Finally, two polygonal chain se-quences are aligned. This algorithm finds optimal similar sub-chains of twopolygonal chains with gaps.
We perform our approach on the known gene expression time series datasetsand compare our results with some common methods. The experimental re-sults show that our method finds regulatory gene pairs more efficient thanthe other methods.
Keywords: gene expression time series, polygonal chain matching, andglobal sequence alignment
1. Introduction
Microarray is a high throughput technique to investigate the expressionof thousands of genes simultaneously [1]. Comparison of gene expressiontime series has a substantial role for identification of regulatory relationships
∗Corresponding authorEmail address: [email protected] (Ali Mohades)
Preprint submitted to Elsevier July 15, 2011

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
among various genes. The comparison of gene expression time series leads tofind the similar functionalities of genes.
Several measurement methods have been proposed to compare gene ex-pression time series. The most common measure is the Pearson correlationcoefficient. This measure compares the expression levels at each time point[2, 3].
Edge detection [4], Event method [5], and Spectral component correlationmethod [6] extract regulatory gene pairs. Cho et al. [7] compare gene expres-sion patterns by hypothetical sine wave patterns using correlation coefficient.Qian et al. [3] apply a local alignment algorithm to study time-shifted andinvert gene expression profiles.
Aach and Church [8] use linear interpolation to align cell-cycle expressionexperiments. Baysian similarity metric is applied in GEST [9] algorithm tosearch an individual profile. Horton et al. [10] built web based gene expres-sion similarity search system, called CellMontage. Lee et al. [11] presenta new similarity metric, DTWT-SSIM Index, based on wavelet transform,to solve the time shift in gene expressions. These measures are not able todetect the best correspondent intervals between two time series.
Most of these methods are applied on interpolated gene expression datawhere some part of the knowledge is lost. Biological experiments have demon-strated amplitude scaling, vertical shift, time delay, and missing value are sig-nificant to detect similarity between each gene expression pair [11]. However,these methods do not consider these observations.
In this research, a new similarity metric is proposed to analyze gene ex-pression time series considering vertical shift, time delay and noisy data.Each Gene expression time series is modeled by a polygonal chain. Then,some features are defined on each edge of the polygonal chain based on thegeometric parameters. In following a polygonal chain sequence is made fromthese parameters. After generating two polygonal chain sequences, they arealigned to find the best similarity between them.
In geometry, some algorithms have applied to find similarity betweenpolygonal chains called matching. In matching polygonal chains, translationor rotation are considered. Hausdorf distance and Frechet distance are twocommon functions to measure the similarity of polygonal chains [9]. Thematching methods under translation can solve vertical shift and horizontalshift (time delay). However, none of them can ignore noisy data or splitchains to sub-chains for finding the best matching. Alignment algorithmsare suitable approaches for splitting chains to similar sub-chains. The part
2

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
of our method, polygonal chain alignment, finds similar sub-chains betweentwo polygonal chains. We use global sequence alignment, Needleman-Wunsch[13], to align two polygonal chain sequences, In Section 2, we define polygo-nal chain sequences and describe the global alignment on these sequences. InSection 3, we apply our method, polygonal chain alignment, on the knownregulated datasets and compare the results with some gene expression simi-larity methods.
2. Method
In this section, our algorithm is described. Two Gene expression timeseries are given as inputs to the algorithm as follows. Each gene expressiontime series G, with n+1 time point levels, is modeled by a polygonal chain Pwith n+1 vertices (set V ) in the plane and vi : (x, y) ∈ V where x and y arespecified by the time value and expression level on G, respectively. Polygonalchain P contains n edges (set E) that each edge pi ∈ E connects two verticesvi and vi+1 (1 ≤ i ≤ n).
This algorithm contains two main steps. In the first step, a polygonalchain sequence is made and normalized from each polygonal chain. In thesecond step, two polygonal chain sequences are aligned to find similaritybetween them.
2.1. Polygonal Chain Sequence and Normalization
Assume that a polygonal chain P has n edges, [p1, p2, · · · , pn]. Each edgepi is explained by four parameters:
• The length of pi: len(pi),
• Angle between pi, and the x−axis: ϕb(pi),
• Angle difference between pi+1 and pi: ϕn(pi) = ϕb(pi+1)− ϕb(pi), and
• Angle difference between pi−1 and pi: ϕp(pi) = ϕb(pi)− ϕb(pi−1).
These parameters represent geometric properties of the edge pi. So, eachedge pi is represented by a quadruple Kpi :< len(pi), ϕb(pi), ϕn(pi), ϕp(pi) >.
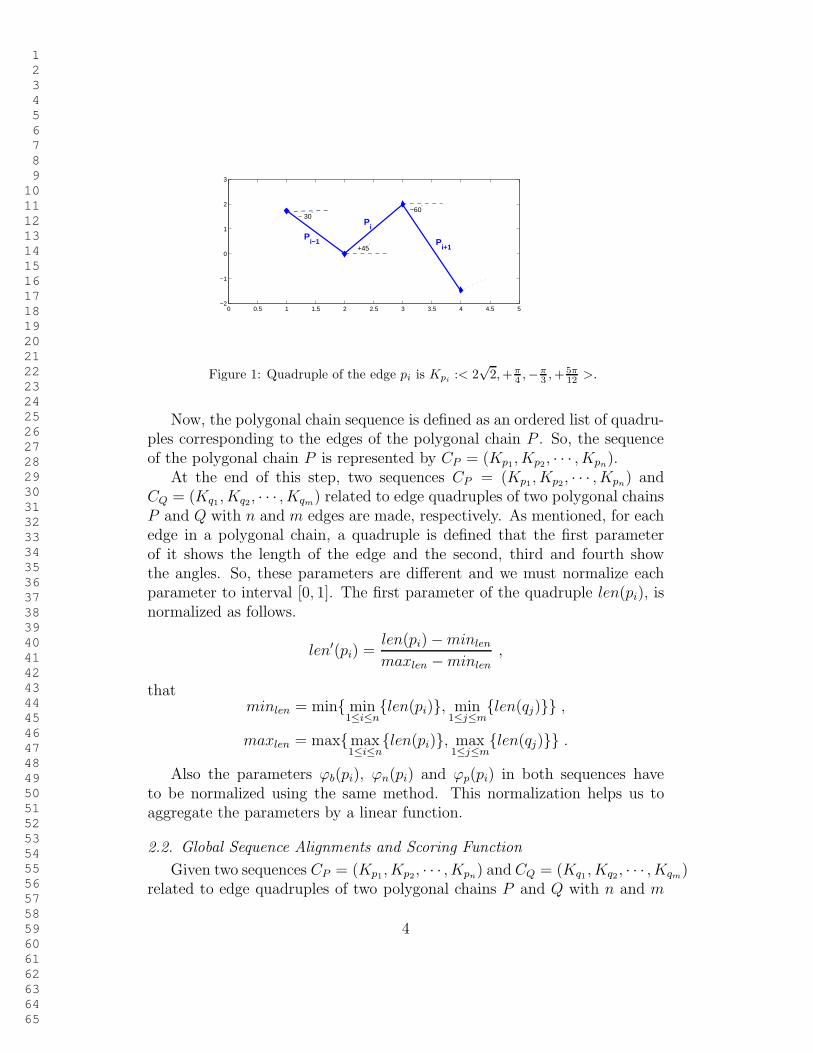
For example, figure 1 illustrates the geometric parameters of the edge piin the polygonal chain P . The corresponding quadruple of the edge pi isequal to Kpi :< 2
√2,+π
4,−π
3,+5π
12>.
3
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 5−2
−1
0
1
2
3
Pi−1
Pi
Pi+1
− 30’
+45’
−60’
Figure 1: Quadruple of the edge pi is Kpi:< 2
√2,+π
4,−π
3,+ 5π
12>.
Now, the polygonal chain sequence is defined as an ordered list of quadru-ples corresponding to the edges of the polygonal chain P . So, the sequenceof the polygonal chain P is represented by CP = (Kp1, Kp2, · · · , Kpn).
At the end of this step, two sequences CP = (Kp1, Kp2, · · · , Kpn) andCQ = (Kq1 , Kq2, · · · , Kqm) related to edge quadruples of two polygonal chainsP and Q with n and m edges are made, respectively. As mentioned, for eachedge in a polygonal chain, a quadruple is defined that the first parameterof it shows the length of the edge and the second, third and fourth showthe angles. So, these parameters are different and we must normalize eachparameter to interval [0, 1]. The first parameter of the quadruple len(pi), isnormalized as follows.
len′(pi) =len(pi)−minlen
maxlen −minlen
,
thatminlen = min{ min
1≤i≤n{len(pi)}, min
1≤j≤m{len(qj)}} ,
maxlen = max{max1≤i≤n
{len(pi)}, max1≤j≤m
{len(qj)}} .
Also the parameters ϕb(pi), ϕn(pi) and ϕp(pi) in both sequences haveto be normalized using the same method. This normalization helps us toaggregate the parameters by a linear function.
2.2. Global Sequence Alignments and Scoring Function
Given two sequences CP = (Kp1, Kp2, · · · , Kpn) and CQ = (Kq1 , Kq2, · · · , Kqm)related to edge quadruples of two polygonal chains P and Q with n and m
4

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
edges, respectively. The similarity of two sequences is defined as the bestscore among all possible alignments between them. The global alignment be-tween sequences CP and CQ is computed by Needleman-Wunsch algorithm.This algorithm needs a proper scoring function to find the optimal alignmentof sequences CP and CQ.
Similarity is specified by a scoring function S, in which S(Kpi, Kqj) isthe similarity of two edges pi and qj using the quadruples Kpi and Kqj . Thescoring function is defined by the weighted aggregation of similarities betweencorresponded parameters of the edge pi and edge qj , as follows:
S(Kpi, Kqj) =ωlen × ( 1− |len′(pi)− len′(qj)| )+ ωϕb
× ( 1− |ϕ′b(pi)− ϕ′
b(qj)| )+ ωϕn
× ( 1− |ϕ′n(pi)− ϕ′
n(qj)| )+ ωϕp
× ( 1− |ϕ′p(pi)− ϕ′
p(qj)| )
where, weights ωlen, ωϕb, ωϕn
and ωϕpare the weights of their corresponded
parts in the quadruples. These weights explain the influence of the corre-sponding parts on the alignment.
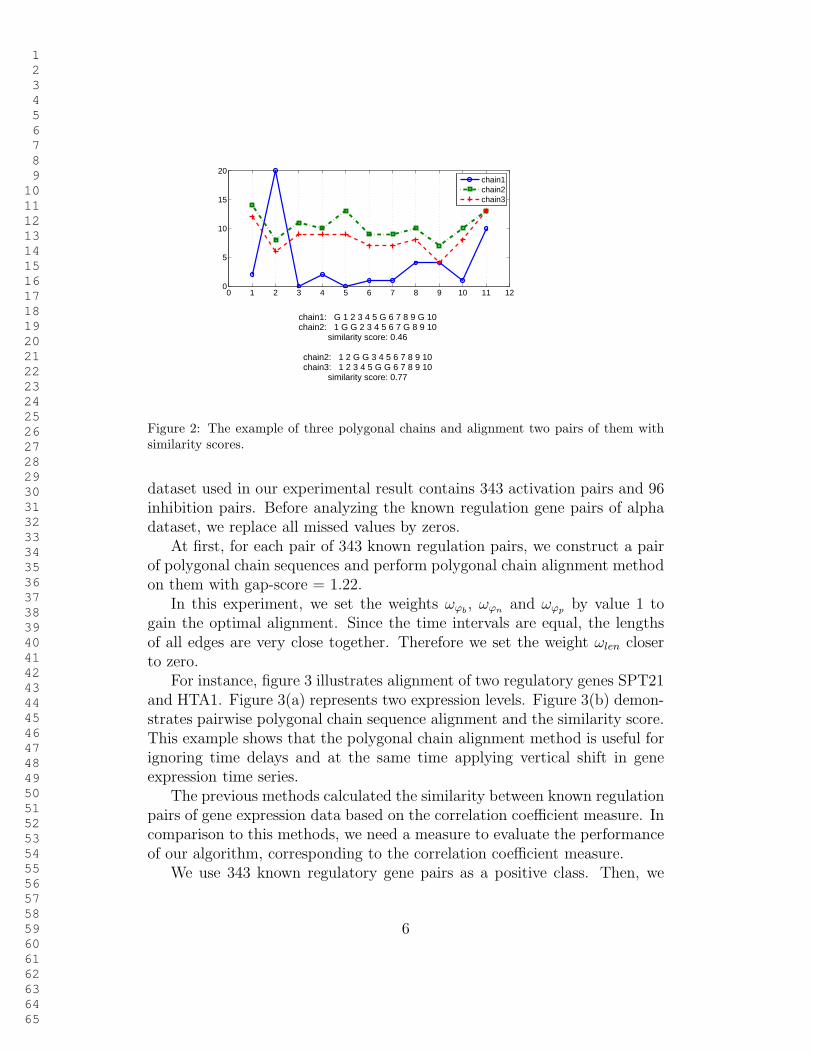
In figure 2, we align three polygonal chains two by two. Chain 1 andchain 2 are aligned with three gaps (gap is shown with ”G”) on the chain1 and three gaps on the chain 2. Figure 2, shows that the similarity of thechain 1 and the chain 2 is 0.46 using gap-score=0.4.
3. Results
We test the ability of the polygonal chain alignment method amongreal regulatory gene expression datasets. The gene pairs have been ex-tracted manually from the Cho and Spellman alpha and cdc28 datasets[14, 15]. These datasets are available from the web site at http://cellcycle-
www.stanford.edu. The alpha dataset contains gene expression time series pro-files of the characterization of mRNA transcription levels during the yeastcell cycle. This dataset includes the expression levels of the 6220 yeast genesduring the cell cycle.
These experiments have measured at 18 time points, at every 7-minutestime interval, from 0 to 119 minutes. Filkov et al. [4] constructed a subsetof 888 known regulation pairs from alpha and cdc28 datasets, containing 647activation gene pairs and 241 inhibition gene pairs. This dataset is availablefrom the web site at http://www.cs.sunysb.edu/∼skiena/gene/jizu/. The alpha
5
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
0 1 2 3 4 5 6 7 8 9 10 11 120
5
10
15
20
chain1chain2chain3
chain1: G 1 2 3 4 5 G 6 7 8 9 G 10chain2: 1 G G 2 3 4 5 6 7 G 8 9 10
similarity score: 0.46
chain2: 1 2 G G 3 4 5 6 7 8 9 10chain3: 1 2 3 4 5 G G 6 7 8 9 10
similarity score: 0.77
Figure 2: The example of three polygonal chains and alignment two pairs of them withsimilarity scores.
dataset used in our experimental result contains 343 activation pairs and 96inhibition pairs. Before analyzing the known regulation gene pairs of alphadataset, we replace all missed values by zeros.
At first, for each pair of 343 known regulation pairs, we construct a pairof polygonal chain sequences and perform polygonal chain alignment methodon them with gap-score = 1.22.
In this experiment, we set the weights ωϕb, ωϕn
and ωϕpby value 1 to
gain the optimal alignment. Since the time intervals are equal, the lengthsof all edges are very close together. Therefore we set the weight ωlen closerto zero.
For instance, figure 3 illustrates alignment of two regulatory genes SPT21and HTA1. Figure 3(a) represents two expression levels. Figure 3(b) demon-strates pairwise polygonal chain sequence alignment and the similarity score.This example shows that the polygonal chain alignment method is useful forignoring time delays and at the same time applying vertical shift in geneexpression time series.
The previous methods calculated the similarity between known regulationpairs of gene expression data based on the correlation coefficient measure. Incomparison to this methods, we need a measure to evaluate the performanceof our algorithm, corresponding to the correlation coefficient measure.
We use 343 known regulatory gene pairs as a positive class. Then, we
6

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18−3
−2
−1
0
1
2
(a)
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22−4
−3
−2
−1
0
1
2
3
(b)
SPT21
HTA1
SPT21
HTA1
G
G G
G GG
SPT21: G 1 2 G G 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17HTA1 : 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 G G G 17
Similarity score is 0.54
Figure 3: Aligned two polygonal chains sequences of SPT21 and HTA1 gene expressionlevels and similarity score of this pair.
utilize 6220 genes from alpha dataset to construct some random gene pairsets as a negative class. Our results show high similarity score for regulatorygene pairs and low similarity score for non-regulatory gene pairs.
Matthews correlation coefficient (MCC), is a measure of the evaluation inclassifying or predicting algorithms [16]. MCC value is between -1 and +1,that the +1 represents the perfect correlation. MCC is defined as follows:
MCC =(TP × TN − FP × FN)
√
(TP + FP )× (FP + TN)× (TN + FN)× (FN + TP )
where, TP (true positives) is the number of gene pairs in the positive classand is predicted by the algorithm. TN (true negatives) is the number of genepairs in the negative class and is not predicted by the algorithm. FP (falsepositives) is the number of gene pairs in the negative class, but is predictedby the algorithm. FN (false negatives) is the number of gene pairs in the
7
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
positive class, but is not predicted by the algorithm. At the end of algorithm,we need a cutoff to regulatory gene pair prediction. So, the similarity scoresare normalized between [0, 1]. Experimentally, we define cutoff = 0.615.
Our method is applied on 343 known regulatory gene pairs. The resultsindicate that 239 pairs of 343 pairs (69% of data) have similarity score greaterthan the cutoff (TP = 239). Our results show more TP gene pairs comparedto Pearson correlation and two other methods, Component-wise correlation[6] and DTWT-SSIM index [11].
The previous results demonstrated that less than 11% (36/343) of geneexpression pairs had Pearson coefficient greater than 0.5. In Component-wiseCorrelation and DTWT-SSIM Index methods, the ratios of the similarityscores greater than 0.5 are 65% (223/343) and 57% (198/343), respectively.
Table 1 shows the results of similarity methods among the 343 knownregulatory pairs and the number of TP in previous methods and our method.
Table 1: The similarity methods among the 343 known regulatory pairs.
Methods TP (true positives) Ratio
Pearson Correlation 36/343 11%
DTWT-SSIM 198/343 57%
Component-wise correlation 223/343 65%
Polygonal chain alignment 239/343 69%
4. Conclusion
In this paper, we presented a new method to measure the similarity be-tween gene expression time series. In the method we converted each geneexpression time series to a polygonal chain. In following, a polygonal chainsequence was defined. Then, two polygonal chain sequences were aligned andobtained a similarity score between them.
For testing our approach, we used the known regulation gene expressiontime series. Then, we compared our results with some common methods. Theexperimental results showed that the polygonal chain alignment method wasmore efficient than previous methods. Our method can predict regulationgene pairs with strong similar oscillatory, however they have time-shifted inexpression levels.
8

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
References
[1] Chee, M., Yang, R., Hubbell, E., Berno, A., Huang, X.C., Stern, D.,Winkler, J., Lockhart, D.J., Morris, M.S. and Fodor, S.P.A. ”Accessinggenetic information with high-density DNA arrays”, Science, 274, pp467-470 (1996).
[2] Eisen, M.B., Spellman, P.T. and Brown, P.O. ”Cluster analysis anddisplay of genome-wide expression patterns”, Proc Natl. Acad. Sci. USA,96, pp. 13118-13123. (1999).
[3] Qian, J., Dolled-Filhart, M., Lin, J., Yu, H. and Gerstein, M. ”BeyondSynexpression relationships: local clustering of time-shifted and invertedgene expression profiles identifies new, biologically relevant interactions”J. Mol. Biol, 314, pp. 1053-1066 (2001).
[4] Filkov, V., Skiena, S. and Zhi, J. ”Identifying gene regulatory networksfrom experiomental data” Proceeding of RECOMB, pp. 124-131 (2001).
[5] Kwon, A.T., Hoos, H.H. and Ng, R. ”Inference of transcriptional regu-lation relationships from gene expression data” Bioinformatics, 19, pp.905-912 (2003).
[6] Liew, A.W.C., Hong, Y. and Mengsu, Y. ”Pattern recognition tech-niques for the emerging field of bioinformatics: A review” Pattern Recog-nition 33, pp. 2055-2073 (2005).
[7] Cho, R. J., Huang, M., Campbell, M.J., Dong, H., Steinmetz, L.,Sapinoso, L., Hampton, G., Elledge, S.,J., Davis, R.W. and Lockhart,D. J. ”Transcriptional regulation and function during the human cellcycle” Nature Genetics, 27, pp. 48-54 (2001).
[8] Aach, J. and Church, G.M. ”Aligning gene expression time series withtime warping algorithms” Bioinformatics, 17, pp. 495-508 (2001).
[9] Hunter, L., Taylor, R.C., Lech, S.M. and Simon, R. ”GEST: a geneexpression search tool based on a novel besian similarity metric” Bioin-formatics, 17, pp. 115-122 (2001).
[10] Fujibuchi, W., Kiseleva, L., Taniguchi, T., Harada, H. and Horton, P.”CellMontage: similar expression profile search server” Bioinformatics,23, pp. 3103-3104 (2007).
9

1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65
[11] Lee, M., Chen, M. and Liu, L. ”Similarity matches of gene expressiondata based on wavelet transform” Lecture notes in computer science,Springer, 5575, pp. 540-549 (2009).
[12] Bereg, S., Buchin, K., Buchin, M., Gavrilova, M. nad Zhu, B ”VoronoiDiagram of Polygonal Chains under the Discrete Frechet Distance” CO-COON ’08, pp. 352-362. (2008).
[13] Needleman, S.B. and Wunsch, C.D. ”A general method applicable tothe search for similarities in the amino acid sequence of two proteins”Journal of Molecular Biology, 48, pp. 443-453 (1970).
[14] Cho, R.J., Campbell, M.J., Winzeler, E.A., Steinmetz, L., Conway, A.,Wodicka, L., Wolfsberg, T.G., Gabrielian, A.E., Landsman, D., Lock-hart, D.J. and Davis, R.W. ”A genome-wide transcriptional analysis ofthe mitotic cell cycle” Molecular Cell, 2, pp. 65-73 (1998).
[15] Spellman, P., Sherlock, G., Zhang, M.Q., Iyer, V.R., Anders, K., Eisen,M.B., Brown, P.O., Botstein, D. and Futcher, B. ”Comprehensive iden-tification of cell cycle-regulated genes of the yeast Saccharomyces cere-visiae by microarray hybridization” Molecular Biology of the Cell, 9, pp.3273-3297 (1998).
[16] Matthews, B.W. ”Comparison of the predicted and observed secondarystructure of T4 phage lysozyme” Biochim. Biophys, 405, pp. 442451(1975).
10




















