
FLAVIN OXYGEN CHEMISTRY BROUGHT TO DATE Thomas ...
12
FLAVIN OXYGEN CHEMISTRY BROUGHT TO DATE Thomas C. Bruice Department of Chemistry University of California, Santa Barbara Santa Barbara, California USA 93106 In this contribution the author provides a summary of the research accomplishments of his laboratory which deal with chemistry related to the flavoenzyme mixed function oxidases. Studies which are discussed are those which have come to fruition since the 7th Symposium on Flavins and flavoenzymes. When added to the chapter published in the volume edited by Williams and Massey (1) the salient features of our contri- butions to this area of research are reviewed. The reader is also directed to a recent review in the Israeli Journal of Chemistry (2). For the reader who may wish to gain an understanding of the important aspects of our investigations, with little invest- ment of time, the following statements can be made: 1) 1,5-Dihydrof lavins ( F l rec j) react with molecular oxygen by way of a le transfer to yield as intermediate a radical pair consisting of flavin radical (Fl ) and superoxide (0,-). 3 sem c 2 This radical pair can partition back to 1,5-dihydroflavin and C>2 or forward to a 4a, 5-dihydrof lavin substituted by a hydro- peroxy anion at the 4a-position. This flavin hydroperoxy anion becomes protonated to yield 4a-hydroperoxy flavin which may then eliminate hydrogen peroxide to yield flavin ( F 1 QX )• This step-wise mechanism is shown in eq. 1. (Note: Scheme II in ref. 1 is an alternate mechanism which is now known to be Flavins and Flavoproteins © 1984 Walter de Gruyter & Co., Berlin • New York - Printed in Germany
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of FLAVIN OXYGEN CHEMISTRY BROUGHT TO DATE Thomas ...

FLAVIN OXYGEN CHEMISTRY BROUGHT TO DATE
Thomas C. Bruice Department of Chemistry University of California, Santa Barbara Santa Barbara, California USA 93106
In this contribution the author provides a summary of the research accomplishments of his laboratory which deal with chemistry related to the flavoenzyme mixed function oxidases. Studies which are discussed are those which have come to fruition since the 7th Symposium on Flavins and flavoenzymes. When added to the chapter published in the volume edited by Williams and Massey (1) the salient features of our contri-butions to this area of research are reviewed. The reader is also directed to a recent review in the Israeli Journal of Chemistry (2).
For the reader who may wish to gain an understanding of the important aspects of our investigations, with little invest-ment of time, the following statements can be made:
1) 1,5-Dihydrof lavins (Flrecj) react with molecular oxygen by way of a le transfer to yield as intermediate a radical pair consisting of flavin radical (Fl ) and superoxide (0,-). 3 sem c 2
This radical pair can partition back to 1,5-dihydroflavin and C>2 or forward to a 4a, 5-dihydrof lavin substituted by a hydro-peroxy anion at the 4a-position. This flavin hydroperoxy anion becomes protonated to yield 4a-hydroperoxy flavin which may then eliminate hydrogen peroxide to yield flavin ( F1 Q X)• This step-wise mechanism is shown in eq. 1. (Note: Scheme II in ref. 1 is an alternate mechanism which is now known to be
Flavins and Flavoproteins © 1984 Walter de Gruyter & Co., Berlin • New York - Printed in Germany
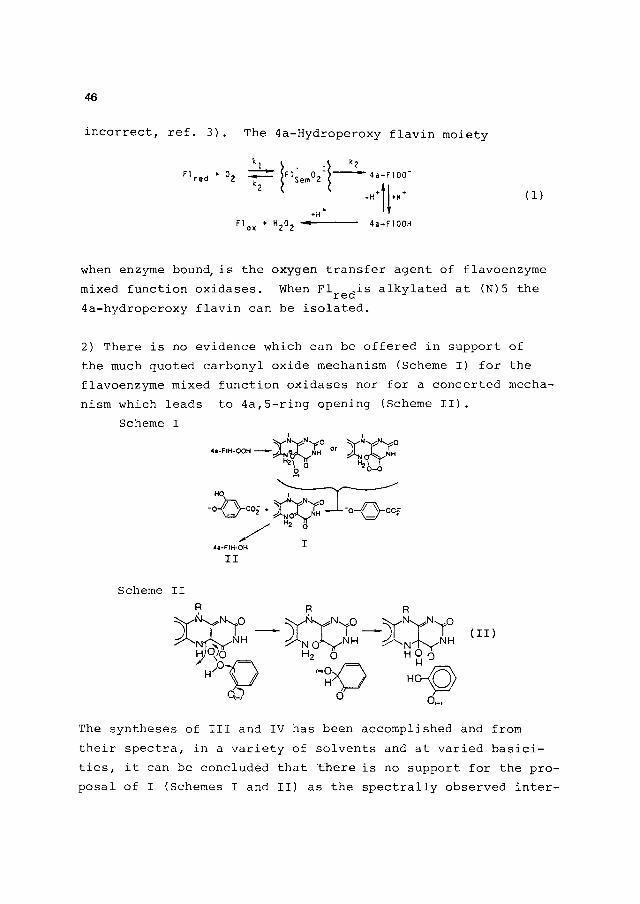
46
incorrect, réf. 3). The 4a-Hydroperoxy flavin moiety
Fl r e d 4 a - F 1 0 0
+ ( 1 )
4 a - F 1 0 0 H
when enzyme bound, is the oxygen transfer agent of flavoenzyme mixed function oxidases. When Flre(jis alkylated at (N) 5 the 4a-hydroperoxy flavin can be isolated.
2) There is no evidence which can be offered in support of the much quoted carbonyl oxide mechanism (Scheme I) for the flavoenzyme mixed function oxidases nor for a concerted mecha-nism which leads to 4a,5-ring opening (Scheme II).
The syntheses of III and IV has been accomplished and from their spectra, in a variety of solvents and at varied basici-ties, it can be concluded that there is no support for the pro-posal of I (Schemes I and II) as the spectrally observed inter-
Scheme I
II
Scheme II
(II)

47
mediate in the reaction of phenol mono oxygeneses. No evidence for the intermediacy of I has been offered for any other flavin mixed function oxidase.
i
III IV
3) The mechanism for the microsomal flavin mixed function oxi-dase involves the direct conversion of the flavin hydroperox-ide to its pseudo-base (eq. 2).
4a-FIEt -CL o — 4a-FIEtOH *• XO , £ 0 =X < 2 >
The reaction occurs by nucleophilic attack of the substrate (:X) upon the terminal oxygen of the 4a-hydroperoxy flavin. The second order rate constant for reaction of nucleophiles (:X) with hydroperoxides and percarboxylie acids depends upon the pK of the generated hydroxyl compound (ROH in eq. 2). a The large oxygen donation potential of 4a-flavin hydroperoxide is due to the low (for an alcohol) pK of II.
l pKa 9.2 I
H
II
4) There are at present no model systems that duplicate the spectral observations made with the various phenolate hydrox-ylases. Perhaps suitable mechanisms will evolve for the enzym-atic reactions which will be based on our observations that numerous phenolate and indole anion substrates are dioxygen-ated by the anion of 4a-hydroperoxyflavin. A suitable example is provided in Scheme III.

48
Scheme III
o* 4a-FlEt02" + F1Et" +
0 0 FT Et" + Fl! ox Et •
0 OH
In what follows there is provided in greater detail a descrip-tion of our recent contributions which have led to a portion of the conclusions just presented. The mechanism of eq. 1 for the reduction of oxygen to hydrogen peroxide by 1,5-dihydro-flavins was reached by a combination of electrochemical and kinetic experiments (3). From the one-electron reduction potential for both oxidized flavin — f l a v i n radical and flavin radical 1,5-reduced flavin plus the one-electron reduction potentials for 0 2 °2 ' ant^ °2* t^lere
were calculated the standard free energies for the reactions:
1,5-reduced flavin + C>2 »-flavin radical + 02~(AG-l°) (4)
Flavin radical + 0 2~ »-flavin + f^O^AG^ 0) (5)
Plots of AG1° vs. pH were found to parallel in shape plots of AG* vs. pH for the bimolecular oxidation of 1,5-reduced--5-ethyl-3-methyllumiflavin (Figure 1). In the plot of Figure 1 the free energy content of the transition state (AG* ) exceeds the free energy content of the 0^' + flavin radical pairs (AG^0 ) by 15 kJM-1. In eq.l, AG* pertains to k ^ The values of AG* - AG^° equals the free energy of activation for reduction of flavin radicals by 0 ( A G * ). The free energy of activation AG.* pertains to of eq. 1. Since AG^ >> AG * , the critical transition state for the reaction of 0-, r ' 2 with 1,5-dihydro-5-ethyl-3-methyllumiflavin must closely resemble an (Fl 0,") pair (pH range 3 to 10.5). A reaction

49
Figure 1. Plots of the experimen-tally determined free energies of activation (AG+) and calculated standard free energies (AG0), for formation of flavin radical plus superoxide on reaction of 1,5-di-hydro-5-ethyl-3-methylluminflavin with 02(30 0 C; H20) VS. pH. Lines a and b represent the pH dependen-cies of the E for 0„ + le m 2 and FlEt + le sem respectively.
FlEt red
coordinate cartoon is shown in Figure 2. The pH dependence of the apparent second-order rate constant for reaction of 1,5-dihydro-5-ethyl-3-methyllumiflavin with 0 2 is quantita-tively explained by the assumption of second-order reactions of neutral (k ) and anionic (k, ) dihydroflavin species with 0,
Figure 2. Reaction coordinate cartoon for the oxidation of N(5)-ethyl-1,5-dihydro-3-methyllumifla-vin by 0 2 at 30 'C in H 20 at pH 4.6.
Reac t i on C o o r d i n a t e
The numerical values of k and k are such that the fraction cl O HC>2"/®2~ Pr°duced a s intermediates are as predicted from the pK of HO ' ^ a 2 (Scheme IV).

50
Scheme IV
^ o J O (l.S-FlEtH) (FI Et")
-H+it+H + PK. 6.7 T I
J o J o
(1,5-FlEt")
The mechanisms of the reaction of a series of 1,5-dihydro-flavins, possessing various substituents, with was studied
5 A at pH 4.6 (pK H0-). As in the instance of N -ethyl-3-methyl-
^ o lumiflavins, values of AG-̂ and isQ̂ were calculated from one-electron reduction potentials of oxygen and flavin species, AG,* by kinetic studies, and AG* as the difference AG* - AG,0, f 1 ' r f 1 In all instances AG* >> AG.„ so that {Fl 0^} pairs serve as f r sem 2 r
reasonable intermediates. These oxidations are neither gen-eral nor specific-acid catalyzed and since the rate constant
can be as large as 10 s it follows that further reactions of must occur by radical coupling (k2 in eq. 1) to yield flavin 4a-hydroperoxide anion, which may then be proton-ated to yield the 4a-hydroperoxyflavin. These results have been obtained from studies of 1-blocked, 5-blocked and 1,5-di-blocked flavins. From the kinetics for reaction of C^ with normal unblocked flavins we show that the second-order reaction of 1,5-dihydroflavin with 0^ cannot be derived from initial rates due to the very early onset of autocatalytic oxidation.
The synthesis of III and IV were carried out by percarboxylic acid oxidation of the appropriate N(5)-diethylated lumiflavin

51
as shown in eq. 6 (4,5).
<l)Na2S 204 or V ^ L N M , H2/Pd-C ' * m.CIC4H4C03H ,g
0 NaOH / \ 0
In Figure 3 there is compared the UV-VIS spectrum of III to that of an absorbing species formed, in time, between the 4a-hydroperoxyflavin (4a-FlH00H) and 4a-hydroxyflavin (II) with p-hydroxybenzoate hydroxylase. This absorbing species was assumed (6) to be the 6-amino-5-oxo-3H, 5H-uricil (Species I in Schemes I and II).
nm
Examination of Figure 3 shows that the spectrum of III does not resemble at all that of the enzyme bound intermediate (both in X and most importantly in e, 7,000 vs. 15,000 cm ^ max — M ). Compound IV shows the same spectral characteristics as III. Further, the spectrum of IV is not changed by the dielec-tric constant of the media or upon solution in t-BuOH contain-ing t-BuO K+. The latter observation shows that the long-wave length absorbance of the enzyme intermediate cannot be due to

52
formation of a quinoid species by isomerization (eq. 7) or ionization of the N(3)-H. The steric influence of the di-methyl substituents of the dimethylamino moiety of III and IV
in preventing sufficient planarity, to allow internal charge transfer to occur and thereby change the spectra of the synthe-tic 6-amino-5-oxo-3 H ,5H-uracil to that of the enzymatic inter-mediate, has been considered (7). However, there is no evidence that such structures are subject to charge transfer nor could structure I (Scheme I and II) become coplanar in the coiled conformation shown.
The microsomal flavomonooxygenase of animals is responsible for the N-oxygenation of tertiary- and secondary amines as well as hydroxylamines and the S-oxygenation of organic sul-fides (eq. 2). That this mechanism involves a direct nucleo-philic displacement by substrate at the terminal oxygen of the hydroperoxyflavin (eq. 2) is shown by the following observa-tions. Displacements of this nature are common in hydroper-oxide chemistry where there is no possibility of special effects as ring opening to give a carbonyl oxide (as postu-lated in Scheme I and II). In the reaction of eq. 2 there is expected to be a relationship between the basicity of RO and the second order rate constant for 0-0 bond scission accompa-nying attack of X:. In Figure 4 there is plotted the log of the second order rate constant for reaction of the organic sulfide thioxane k vs. the pK of ROH (8). Figure 4 repre-S — a sents a correlation between the free energy of activation for thioxane sulfur displacement upon hydroperoxides (R00H) and the standard free energy for proton dissociation from ROH. Inspection of Figure 4 shows that a linear free energy correl-ation exists for thirteen powers of ten in the proton bicicity of RO- and eight and a half powers of ten in rate.
(7)

53
C02Me
Figure 4. Plot of the log of the second-order rate constants (relative to the rate constant for 4a-FlEt00H)for sulfoxidation of thioxane (k ) by the ROOH species vs. the pK of ROH species (solvent absolute t-BuOH for rate constants and H O for pK values, 30 °C). ¿. a
The slope of the plot is -0.6. Very similar plots may be made for the N-oxidation of N,N-dimethylbenzylamine and the oxida-tion of I by the same series of hydroperoxides and percarb-oxylic acid. The slope for these plots is also -0.6. The slope of these plots tell us that 0-0 bond breaking has pro-ceeded to about 60% in the transition states. In an earlier study (9) we found that, for the reaction of secondary-, and tertiary amines and hydroxylamines, a plot of amine pK vs. a — the log of the second order rate constant for attack of these nitrogen bases upon peroxides was of slope 0.2. A general picture emerges for the reaction of equation 2 where bond making has not proceeded as far as bond breaking. Note that

54
in figure 4 the point for the 4a-hydroperoxyflavin fits the correlation line.
So there it is, 4a-hydroperoxyflavin possesses a rather re-markable monooxygen donation potential because the pK of a 4a-hydroxyflavin (II) is about 9.2. This is low for an alcohol and is attributable to the electronegativity of the nitrogen substituents at the 1, 5 and 10 positions and the carbonyl group at the 4-position of the isoaloxazine ring structure. The pK of 9.2 for 4a-hydroxyflavin was calculated employing a the p,o, method of Charton (10). A kinetic pK value for I I aap the dissociation of the hydroxylic proton has been experimen-tally determined as 9.9 in a study of the solvolysis of 4a-hydroxy-5-ethyl-4a,5-dihydrolumiflavin (11).
Acknowledgement: The author thanks the National Institutes of Health and the National Science Foundation for continuing support of his laboratory in its studies of flavin chemistry.
References
1. Bruice, T.C. in "Flavins and Flavoproteins" (Massey, V. , Williams, C.H., eds.), Elsivier/North-Holland, 1982, ch.44.
2. Bruice, T.C.: Is. J. Chem., (1984). 3. Ebertein, G., Bruice, T.C.: J. Am. Chem. Soc., 105, 6685
(1983) . 4. Wessiak, A., Bruice, T.C.: J. Am. Chem. Soc., 105, 4809-
4825 (1983). 5. Wessiak, A., Noar, J.B., Bruice, T.C.: Proc. Natl. Acad.
Sci. (USA), 8_1, 332-336 (1984). 6. Entsch, B., Ballou, D.P., Massey, V. : J. Biol. Chem., 251,
2550, 7369 (1976). 7. Wessiak, A., Schoyfer, L.M., Yuan, L.-C, Bruice, T.C.,
Massey, V. : Proc. Natl. Acad. Sci. (USA), 8JL, July (1984).

55
8. Bruice, T.C.: J. Chem. Soc. Chem. Comm., (1983). Bruice, T.C., Noar, J.B., Ball, S.S., Vankataram, U.V.: J. Am. Chem. Soc., 105, 2452 (1983).
9. Ball, S.S., Bruice, T.C.: J. Amer. Chem. Soc., 102, 6498-6503 (1980).
10. Charton, M.: J. Org. Chem., 29, 1222 (1964). See also Fox, J.F., Jencks, W.P.: J. Am. Chem. Soc., 96̂ , 1436 (1974)
11. Venkataram, U.V., Bruice, T.C.: J. Chem. Soc. Chem. Comm. In Press.





















