
Investigations Concerning the Laser Ignition of Energetic ...
Upload
independentCategory
view
0download
0
2685
Twenty-Sixth Symposium (International) on Combustion/The Combustion Institute, 1996/pp. 2685–2692
EXTENTS OF ALKANE COMBUSTION DURING RAPID COMPRESSIONLEADING TO SINGLE- AND TWO-STAGE IGNITION
A. COX, J. F. GRIFFITHS and C. MOHAMEDSchool of Chemistry, The University
Leeds LS2 9JT, UK
H. J. CURRAN, W. J. PITZ and C. K. WESTBROOKLawrence Livermore National Laboratory
Livermore, CA 94350, USA
Extents of reactant consumption have been measured during the course of spontaneous ignition follow-ing rapid compression of n-pentane and n-heptane and also of PRF 60 (n-heptane ` i-octane, 2,2,4-trimethylpentane) in stoichiometric mixtures with air. Compressed gas temperatures of 720–750 K and845–875 K were studied at reactant densities of 131 mol m13. At the lower gas temperature, there wasno evidence of reactant consumption during the course of the compression stroke. Two-stage ignitionoccurred at these temperatures, but only modest proportions of n-pentane were consumed during the firststage (,15%), whereas about 40% of n-heptane reacted under the same conditions. At the higher com-pressed gas temperature, the oxidation of n-pentane began only after the piston had stopped, whereasmore than 30% of the n-heptane had already been consumed in the final stage of the compression stroke.The first stage of a two-stage ignition was evident during the compression of the n-heptane mixture underthese conditions. The behavior of the PRF 60 mixture differed somewhat from that of n-pentane despitethe similarity of the research octane numbers. Although there was a preferential oxidation of n-heptane atTc 4 850 K, which persisted throughout the early development of spontaneous ignition during the post-compression period, oxidation of both components of the PRF 60 mixture began before the piston hadstopped. Numerical simulations of the spontaneous ignition under conditions resembling those of the rapidcompression experiments show that the predicted reactivity from detailed kinetics is consistent with theobserved features. The modeling results also substantiate the characterization of single and two-stageignition in compression machines.
Introduction
The spontaneous ignition (or autoignition) char-acteristics of hydrocarbons have been of interest tochemists and engineers for many decades. The cur-rent interest relates predominantly to combustion inreciprocating engines and the occurrence of knockin spark-ignition (SI) engines in particular. For thetemperatures that are reached during compressionin engines or rapid compression machines (RCM),spontaneous ignition often occurs in two stages. Atsufficiently high compression temperatures, the ig-nition occurs in a single stage. However, for highlyreactive fuels, the two-stage character may persist inthe higher temperature regime because the firststage chemistry can occur during the course of thecompression stroke. The extent to which this takesplace is governed by the reactivity of the fuel andthe pressure and temperature to which the reactantcharge is finally compressed. This chemistry is ig-nored completely in experimental or numerical stud-ies that are not begun until the conditions corre-
spond to those of the postcompression period. Forexample, the extent of reaction and its effect on au-toignition cannot be measured in shock tubes orwhen reactants are admitted very rapidly to pre-heated vessels. It has been taken into account insome numerical simulations of combustion in en-gines [1].
In previous studies in an RCM, it was shown fromthe pressure–time records that during the combus-tion of n-C7H16 and n-C6H14 at compressed gas tem-peratures above 750 K, a sufficiently rapid exother-mic reaction occurred in the final stages of pistonmotion that there was an enhancement of the gastemperature beyond that reached by adiabatic com-pression alone [2]. This arose because the first (orcool flame) stage of a two-stage ignition coincidedwith the end of the compression stroke. The corol-lary is that spontaneous ignition that is observed asa single-stage ignition in a shock tube at gas tem-peratures above 850 K may continue to evolve in twostages in a compression machine (engine or RCM),so the measured ignition delays would not be the

2686 SPARK IGNITION ENGINES
same. It was also shown in the earlier studies [2] thatlittle or no oxidation occurred during the compres-sion stroke when a less-reactive fuel, such as n-C4H10, was involved. In such circumstances, the ig-nition delays for gas temperatures above 750 K in ashock tube or RCM are likely to be in reasonableagreement [3]. In RCM studies, the ignition delay(ti), measured from the pressure records, is definedas the time from the end of compression to the max-imum rate of pressure rise in ignition [2–6].
In the present work, we report direct experimentalmeasurement of the extents of reactant consumptionduring the development of spontaneous ignition ofalkanes in an RCM, with particular emphasis onreactivity during the compression stroke. Three fuelshave been studied in stoichiometric mixtures in air,namely n-C5H10, n-C7H16, and the PRF 60 mixture0.4 n-C7H16 ` 0.6 i-C8H18 (2,2,4-trimethylpen-tane). PRF 60 was chosen because its research oc-tane number is close to that of n-C5H12 (RON 462.5).
Over the last decade, there has been considerabledevelopment of numerical approaches to the predic-tion of spontaneous ignition phenomena by model-ing the detailed chemistry and its associated heatrelease [1,7]. Ignition delays have been a primaryfocus of attention [7], with additional validation ofthe models against experimental studies of the for-mation of intermediate and final molecular products[1,5,6,8,9]. Thus, it is both timely and opportune toapply numerical modeling to the experimental re-sults reported here for reactant consumption.
The purpose of the computation here is not to testthe present state of numerical modeling; the goal isto substantiate the characterization of single andtwo-stage ignition and the conditions in which thefirst stage reaction is subsumed into the compressionso that ignition appears as a single stage in the post-compression period. The initial, numerical studies ofthe binary PRF 60 proved to be less satisfactory thanthose for the modeling of n-C5H10 and n-C7H16 ox-idation, possibly through difficulties associated withthe i-octane oxidation model. A spatial uniformity oftemperature and concentration of species is assumedin the kinetic analysis to restrict the numerical in-tegration to that of ordinary differential equations,as is normally the case. However, spatial inhomo-geneities of temperature and concentration areknown to exist to varying extents throughout thecourse of piston motion of rapid compression ma-chines and throughout the postcompression intervalin which spontaneous ignition develops fully [10–12].
Experimental and Numerical Methods
Experimental StudiesFull details of the apparatus and experimental pro-
cedures have been described previously [2,13–16].
In summary, fuel vapor was premixed with air con-sisting of 21% oxygen and a 79% composite of non-reactive components comprising argon or nitrogen.The proportions of the inerts were varied to alter theoverall heat capacity of each mixture and hence theratio Cp/Cv (4 c). Thus, a range of compressed gastemperatures could be covered at a fixed compres-sion ratio of the machine (CR 4 11.00 5 0.15:1).Experiments were performed at the compressed gastemperatures of 720–750 K and 845–875 K.Mixtures, stored in a Pyrex glass vacuum line, weretransferred at an initial pressure of 33.3 kPa to thecombustion chamber and then compressed into acylindrical combustion chamber by a piston that wasdriven by compressed air and that stopped at topdead center (TDC), the stroke taking 22 ms. Thecylinder and combustion chamber temperature (Ti)were maintained at 323–348 (51) K. The com-pressed gas density was 131.0 5 1.5 mol m13 in allexperiments.
Pressure–time data during the compression andthroughout the postcompression period were mea-sured by Kistler transducer and recorded digitally ona PC at a sampling rate of 200 ls per point via an A/D converter, 200 ms in total. Chemiluminescentemission accompanying reaction was measured byphotomultiplier in selected experiments (ThornEMI 9924B). In general, the ignition delays, mea-sured in three to six experiments at each reactantcomposition and compressed gas temperature, werereproducible to 52.0 ms at long durations and 51.0ms at short durations. The compressed gas temper-ature, Tc, was derived from the measured pressuresat the start and end of compression, pi and pc, andtaking into account the temperature dependence ofc, using the relationship
Tc c pcdlnT 4 ln# 1 2T c 1 1 pi i
To analyze the reactant and product compositionsat intermediate stages, the reaction was quenchedby the bursting of a diaphragm (99% Al, 0.1 mmthickness) in the end wall of the combustion cham-ber, which formed the cylinder head. This was fol-lowed by the expansion of the reactant mixture intoa hemispherical dump tank (1.5 dm3), as describedpreviously [13]. The time at which the diaphragmburst was controlled by a time-delayed signal gen-erated at a threshold pressure during the compres-sion stroke that was used to actuate an electromag-netically driven needle. The condensablecomponents were then pumped to a cold trap cou-pled to the vacuum line. To obtain time-dependentprofiles for the reactant consumption, many experi-ments were performed under the same compressionconditions but with quenching after different inter-vals. The results for each set of experimental con-ditions were obtained from a number of separate

ALKANE COMBUSTION DURING RAPID COMPRESSION 2687
cyclic ether ` OH
or b scission ` HO2
O O ↑ O2 2 2
RH → R s RO s QO H s O QO H s OQ8O H ` OH → branching2 2 2 2 2X fO f2
alkene ` HO alkene ` HO2 2
mixtures made up to the same composition. To stan-dardize the extents of reaction in a series of experi-ments, the sealed cold trap was allowed to warm tolaboratory temperature and a standard volume of n-C4H10 was injected into the trap via a septum cap.After complete mixing, a gaseous sample was with-drawn from the trap via a microsyringe and injectedinto the analytical system. A separation of the mix-ture was performed by gas chromatography (CarloErbe) on a fused silica, Poraplot Q-coated, capillarycolumn (25 m 2 0.32 mm), and a quantitative anal-ysis was obtained from the integrated peak areas ofthe eluting compounds. The proportions of the fuelremaining in each experiment were obtained from adetermination of the peak area of the primary fuelpeak relative to that of n-butane. This result wasthen normalized to a constant peak area that repre-sented the n-butane signal.
The extents of reaction were measured relative tothe amount of reactant measured following com-pression under nonreactive conditions. Productcompositions were not analyzed in the present work.
Numerical Analysis and Kinetic Model
The numerical modeling calculations were carriedout using the HCT program [17], which solves thecoupled nonlinear differential equations for conser-vation of mass, momentum, energy, and each chem-ical species in finite-difference form. The compres-sion stroke was simulated ab initio from the initialconditions and reactant compositions for the exper-imental results, adiabatic conditions being assumed.The reaction mechanism included more than 2000elementary chemical reactions with their reversesteps. This reaction mechanism includes subme-chanisms for oxidation of C1–C8 hydrocarbon spe-cies, validated against a variety of experimental datafor ignition and combustion environments [1]. Thesemechanisms include reactions governed by forma-tion and consumption of alkylperoxy radical speciesthat are appropriate to low temperatures.
Distinctions were made between the rates of ab-straction of H atoms from primary, secondary, andtertiary sites in each alkane and between equilibriumconstants for addition of molecular oxygen to differ-ent types of alkyl radicals. The most significant, sub-sequent reactions of the alkylperoxy radicals(CnH2n`1OO) so formed are their isomerizations via
internal H-atom abstraction, at rates that dependprimarily on the type of C—H bond being brokenand the ring strain energy of the intermediate tran-sition state [8]. The many distinct CnH2nOOH rad-icals can decompose thermally, producing hydroxylradicals and cyclic ethers. Depending on their spe-cific structure, some alkylperoxy radicals can also de-compose to produce stable alkene or oxygenatedspecies and HO2 or OH radicals. All of these reac-tions lead to chain propagation. The only reactionsof alkylperoxy radicals that lead to vigorous chainbranching involve addition of a second oxygen mol-ecule producing ketohydroperoxide intermediatesthat then decompose to produce radicals. Rates andequilibrium constants for these reaction paths havebeen developed in recent studies [1,8,9]. The kineticstructure to represent alkane oxidation at low tem-peratures may be summarised in the equation above[9,18,19].
Results
Combustion at Tc 4 720–750 KMeasured pressure change and the measured and
computed proportions of reactants consumed duringthe spontaneous ignition of n-C5H12 and n-C7H16 atf 4 1 are shown in Figs. 1 and 2. The records beginat 2 ms before the end of piston motion and continuethroughout the development of ignition. The pres-sure record shown in Fig. 1 also includes the sharpfall that occurred when the quenching diaphragmwas punctured. This illustrates the rate of expansionof the products into the dump tank. In this case, thepressure did not reach the maximum that was nor-mally attained at ignition (.3.0 MPa).
Both n-C5H12 and n-C7H16 exhibited a two-stageignition after the end of compression, which was ev-ident both in the pressure record and in the accom-panying chemiluminescent emission from the reac-tion (Figs. 1 and 2). The first stage of two-stageignition is interpreted as the development from theend of compression to the point of inflexion in thepressure rise [4–6]. There was very little or negligi-ble consumption of either reactant before the pistonhad stopped. Whereas nearly 40% of the n-C7H16had reacted by the end of the first stage (Fig. 2), lessthan 15% of the n-C5H12 had been consumed (Fig.1). There was a particularly sharp acceleration in therate of consumption of n-C5H12 during the devel-opment of the second stage, and 80% of it had been
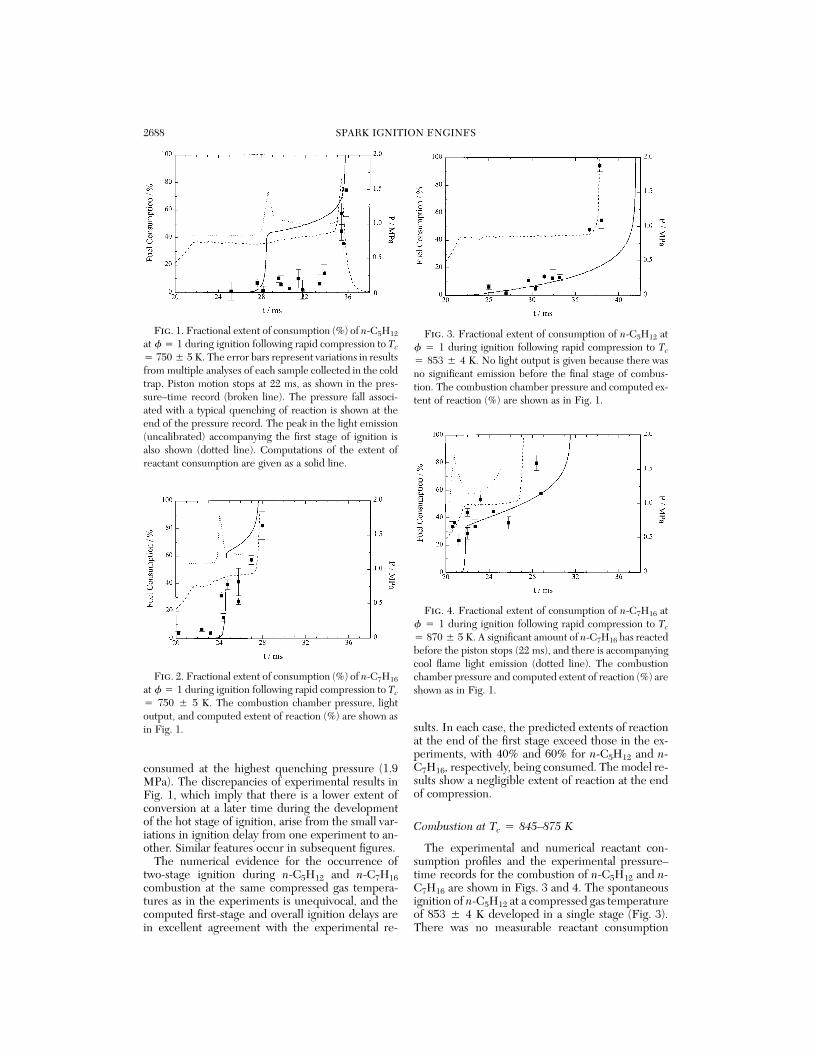
2688 SPARK IGNITION ENGINES
Fig. 1. Fractional extent of consumption (%) of n-C5H12
at f 4 1 during ignition following rapid compression to Tc
4 750 5 5 K. The error bars represent variations in resultsfrom multiple analyses of each sample collected in the coldtrap. Piston motion stops at 22 ms, as shown in the pres-sure–time record (broken line). The pressure fall associ-ated with a typical quenching of reaction is shown at theend of the pressure record. The peak in the light emission(uncalibrated) accompanying the first stage of ignition isalso shown (dotted line). Computations of the extent ofreactant consumption are given as a solid line.
Fig. 2. Fractional extent of consumption (%) of n-C7H16
at f 4 1 during ignition following rapid compression to Tc
4 750 5 5 K. The combustion chamber pressure, lightoutput, and computed extent of reaction (%) are shown asin Fig. 1.
Fig. 3. Fractional extent of consumption of n-C5H12 atf 4 1 during ignition following rapid compression to Tc
4 853 5 4 K. No light output is given because there wasno significant emission before the final stage of combus-tion. The combustion chamber pressure and computed ex-tent of reaction (%) are shown as in Fig. 1.
Fig. 4. Fractional extent of consumption of n-C7H16 atf 4 1 during ignition following rapid compression to Tc
4 870 5 5 K. A significant amount of n-C7H16 has reactedbefore the piston stops (22 ms), and there is accompanyingcool flame light emission (dotted line). The combustionchamber pressure and computed extent of reaction (%) areshown as in Fig. 1.
consumed at the highest quenching pressure (1.9MPa). The discrepancies of experimental results inFig. 1, which imply that there is a lower extent ofconversion at a later time during the developmentof the hot stage of ignition, arise from the small var-iations in ignition delay from one experiment to an-other. Similar features occur in subsequent figures.
The numerical evidence for the occurrence oftwo-stage ignition during n-C5H12 and n-C7H16combustion at the same compressed gas tempera-tures as in the experiments is unequivocal, and thecomputed first-stage and overall ignition delays arein excellent agreement with the experimental re-
sults. In each case, the predicted extents of reactionat the end of the first stage exceed those in the ex-periments, with 40% and 60% for n-C5H12 and n-C7H16, respectively, being consumed. The model re-sults show a negligible extent of reaction at the endof compression.
Combustion at Tc 4 845–875 K
The experimental and numerical reactant con-sumption profiles and the experimental pressure–time records for the combustion of n-C5H12 and n-C7H16 are shown in Figs. 3 and 4. The spontaneousignition of n-C5H12 at a compressed gas temperatureof 853 5 4 K developed in a single stage (Fig. 3).There was no measurable reactant consumption
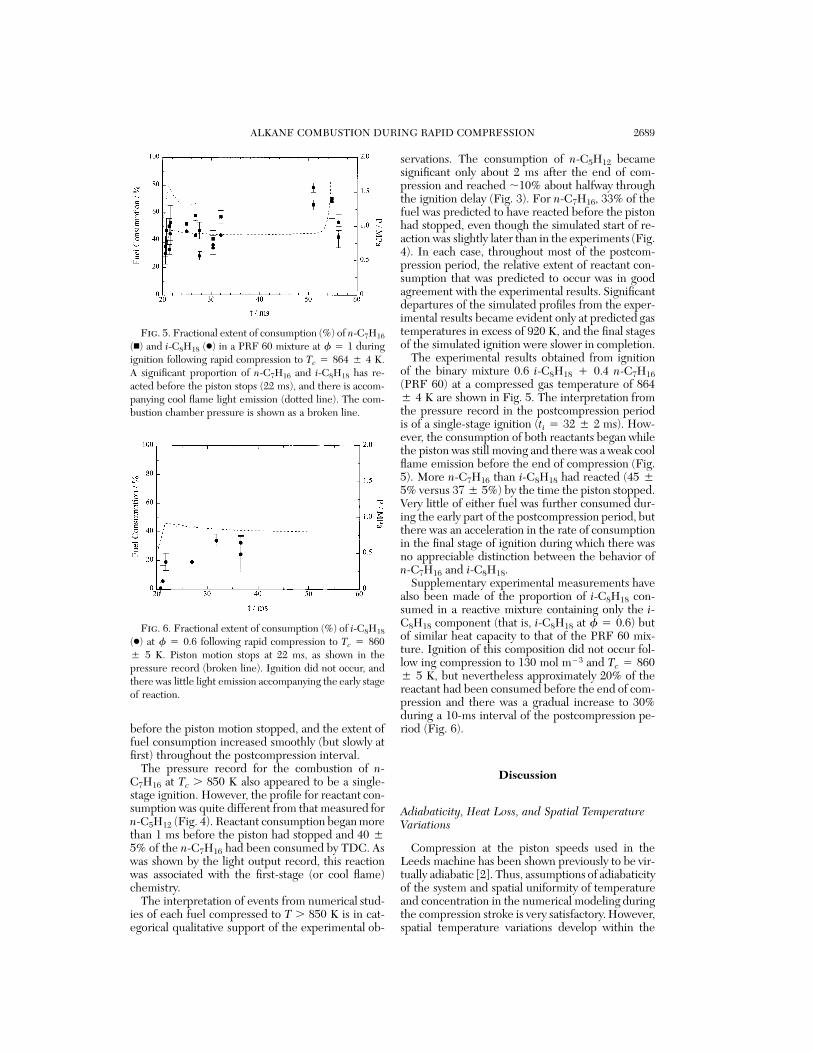
ALKANE COMBUSTION DURING RAPID COMPRESSION 2689
Fig. 5. Fractional extent of consumption (%) of n-C7H16
(n) and i-C8H18 (●) in a PRF 60 mixture at f 4 1 duringignition following rapid compression to Tc 4 864 5 4 K.A significant proportion of n-C7H16 and i-C8H18 has re-acted before the piston stops (22 ms), and there is accom-panying cool flame light emission (dotted line). The com-bustion chamber pressure is shown as a broken line.
Fig. 6. Fractional extent of consumption (%) of i-C8H18
(●) at f 4 0.6 following rapid compression to Tc 4 8605 5 K. Piston motion stops at 22 ms, as shown in thepressure record (broken line). Ignition did not occur, andthere was little light emission accompanying the early stageof reaction.
before the piston motion stopped, and the extent offuel consumption increased smoothly (but slowly atfirst) throughout the postcompression interval.
The pressure record for the combustion of n-C7H16 at Tc . 850 K also appeared to be a single-stage ignition. However, the profile for reactant con-sumption was quite different from that measured forn-C5H12 (Fig. 4). Reactant consumption began morethan 1 ms before the piston had stopped and 40 55% of the n-C7H16 had been consumed by TDC. Aswas shown by the light output record, this reactionwas associated with the first-stage (or cool flame)chemistry.
The interpretation of events from numerical stud-ies of each fuel compressed to T . 850 K is in cat-egorical qualitative support of the experimental ob-
servations. The consumption of n-C5H12 becamesignificant only about 2 ms after the end of com-pression and reached ;10% about halfway throughthe ignition delay (Fig. 3). For n-C7H16, 33% of thefuel was predicted to have reacted before the pistonhad stopped, even though the simulated start of re-action was slightly later than in the experiments (Fig.4). In each case, throughout most of the postcom-pression period, the relative extent of reactant con-sumption that was predicted to occur was in goodagreement with the experimental results. Significantdepartures of the simulated profiles from the exper-imental results became evident only at predicted gastemperatures in excess of 920 K, and the final stagesof the simulated ignition were slower in completion.
The experimental results obtained from ignitionof the binary mixture 0.6 i-C8H18 ` 0.4 n-C7H16(PRF 60) at a compressed gas temperature of 8645 4 K are shown in Fig. 5. The interpretation fromthe pressure record in the postcompression periodis of a single-stage ignition (ti 4 32 5 2 ms). How-ever, the consumption of both reactants began whilethe piston was still moving and there was a weak coolflame emission before the end of compression (Fig.5). More n-C7H16 than i-C8H18 had reacted (45 55% versus 37 5 5%) by the time the piston stopped.Very little of either fuel was further consumed dur-ing the early part of the postcompression period, butthere was an acceleration in the rate of consumptionin the final stage of ignition during which there wasno appreciable distinction between the behavior ofn-C7H16 and i-C8H18.
Supplementary experimental measurements havealso been made of the proportion of i-C8H18 con-sumed in a reactive mixture containing only the i-C8H18 component (that is, i-C8H18 at f 4 0.6) butof similar heat capacity to that of the PRF 60 mix-ture. Ignition of this composition did not occur fol-low ing compression to 130 mol m13 and Tc 4 8605 5 K, but nevertheless approximately 20% of thereactant had been consumed before the end of com-pression and there was a gradual increase to 30%during a 10-ms interval of the postcompression pe-riod (Fig. 6).
Discussion
Adiabaticity, Heat Loss, and Spatial TemperatureVariations
Compression at the piston speeds used in theLeeds machine has been shown previously to be vir-tually adiabatic [2]. Thus, assumptions of adiabaticityof the system and spatial uniformity of temperatureand concentration in the numerical modeling duringthe compression stroke is very satisfactory. However,spatial temperature variations develop within the

2690 SPARK IGNITION ENGINES
combustion chamber during the postcompressionperiod as a result of heat transport to the combustionchamber walls. The heat-loss rates are highest justafter compression ceases because the residual gasmotion is greatest at this stage [11]. Thereafter, therate of heat loss decreases as the gas motion decays.In the present calculations, adiabaticity was assumedalso throughout the postcompression period. Thesimulation of ignition delay thus implies that the be-havior is dominated by conditions in the adiabaticcore [21], but there are complexities of the interac-tion between the kinetics and a nonhomogeneoustemperature field in a temperature range where anegative temperature dependence of reaction rateexists [11,12].
The calculated extent of reaction at any time afterthe end of compression, with adiabatic conditionsassumed, may be expected to be an overestimate rel-ative to an experimental measurement that is ob-tained from the amount of reactant remaining withina nonuniform temperature regime comprisingcooler regions surrounding an adiabatic core. Theheat-loss rate throughout the postcompression pe-riod may be characterized by a Newtonian heat-transfer coefficient in a zero-dimensional numericalmodel [22]. However, its application can adverselyaffect the predicted reactivity because there is ahighly nonlinear response of the chemical kinetics toreactant temperature. Rates and extents of reactioncannot be averaged in a simple way throughout acombustion chamber in which spatial variations intemperature exist, and so the correction has notbeen attempted in this work.
Extents of Reaction in Single- and Two-StageIgnition and during Compression
The greater reactivity of n-C7H16 over that of n-C5H12 at Tc ; 750 K is not only reflected in theshorter ignition delay but also is manifest directly inthe extents of reaction measured in the first stage oftwo-stage ignition (Fig. 1 and 2). In previous rapidcompression studies, Fish et al. [23] measured ap-proximately 25% consumption of 2-methylpentaneduring the first stage of two-stage ignition at Tc 4808 K. For n-C4H10, Minetti et al. [5] showed thatthere was only 10–15% reactant consumption duringthe first stage of two-stage ignition at Tc 4 708 K,whereas approximately 25% consumption of n-C7H16occurred during the first stage of its two-stage igni-tion at Tc 4 667 K [6].
The activity of the alkanes at the reactant temper-atures associated with the first stage may be attrib-uted to the generalized sequence shown in the nu-merical background. Each alkane is capable ofgenerating a number of different isomeric forms ofthe alkyl (R), alkylperoxy (RO2), hydroalkylperoxy(QO2H), or other species. The slow development ofthe first stage may be attributed to the chain-thermal
interaction in which the thermal feedback is re-quired to accelerate the degenerate branching route[19]. Its rate is controlled largely by the activationenergies associated with the RO2/QO2H isomeriza-tions, but the properties of each of the other stepsplay a part in the quantitative behavior. The morevigorous acceleration in rate of n-C7H16 consump-tion in the first stage of two-stage ignition, which wasfound both experimentally and numerically, testifiesto its much more facile chain-branching route thanthat of n-C5H12. The limit on chain branching is setby the displacement of the R/RO2 equilibrium to-ward dissociation as the reactant temperature in-creases beyond 800 K. This is the primary kineticorigin of the negative temperature dependence ofreaction rate since the main (nonbranching) propa-gation is taken up with relatively low exothermicityby HO2 radicals [19].
There are both chemical and thermal conse-quences of oxidation in the final stages of compres-sion. The thermal contribution is an augmentationof the gas temperature beyond that which would bereached by rapid compression alone, and the effectcan be seen in the difference between the pressuresattained under reactive and nonreactive conditions[2]. The temperature increase predicted in the nu-merical simulations for n-C7H16 combustion is 30 K.
The difference in reactivity shown by n-C7H16 andn-C5H12 also accounts for the ability of n-C7H16 toreact during the compression stroke when compres-sion occurs beyond Tc 4 800 K (Figs. 3 and 4). Al-though thermal feedback has some effect on gastemperature, the reactant temperature is forcedthrough the most reactive temperature regime (750–800 K) at a rate determined by the compressionstroke. At a piston speed of 12 m s11 in the Leedsrapid compression machine, there is sufficient timefor n-C7H16 to react. Although this is not the casefor n-C5H12 under the present conditions, it couldbe so for n-C5H12 or other fuels of similar reactivityat higher reactant densities.
Schlieren imaging of spontaneous ignition in therapid compression machine shows that the hot stageoften begins at a single center, and the bulk of thereactants are consumed in a propagating combustionwave before many (and sometimes any) other cen-ters are initiated [24]. Consequently, a reactionquenched on the rising pressure profile of the finalstage of ignition gives rise to a product compositiondrawn from segregated burned gas and partially ox-idized reactants. Thus, the extents of reaction in thelate stage of reaction that are predicted numericallyfrom a zero-dimensional model cannot be expectedto match quantitatively the experimental behavior.Differences in the duration of the overall ignitiondelay, which in the present numerical analyses tendto be slightly longer than the experimental measure-ments, may be attributed to the same marked

ALKANE COMBUSTION DURING RAPID COMPRESSION 2691
departures in the experiments from the idealizationin the final stages of combustion.
Combustion of a PRF 60 Mixture
Although there were significant quantitative dif-ferences in the form of the predicted profiles for thePRF 60 mixture that still have to be addressed, thepredicted ratio of extents of reaction throughout theignition delay at temperatures below 900 K, d[n-C7H16]/d[i-C8H18] was 1.24. This is in excellentagreement with the experimental measurements atthe end of compression (1.2 5 0.1). It follows thatthe relative concentration of alkyl radicals, d[n-C7H15]/d[i-C8H17], generated in the PRF 60 mixturein this environment is (40/60) 2 1.2 4 0.83 5 0.02.This ratio is consistent with an almost exclusive roleof H-atom abstraction by OH radicals, as can be de-rived from the ratio for k(n-C7H16 ` OH)/k(i-C8H18` OH) in the range 800–900 K, taking into accountthe numbers of primary, secondary, or tertiary C—H bonds in n-C7H16 and i-C8H18 and the respectiveArrhenius parameters [25]. The relatively weak se-lectivity in favor of C8H17 radical formation in themixture 0.4 n-C7H16 ` 0.6 i-C8H18 would shift verysubstantially toward C8H17 radical formation in PRFmixtures containing higher proportions of i-C8H18.
Oxidation of n-C7H16 generates a rate of chainbranching that is much more intense than that of i-C8H18, since the structure of the C7H15 radicalsleads to higher rates of peroxy radical isomerization[1], ultimately causing chain branching from keto-hydroperoxide decomposition. Increasing fractionsof i-C8H18 in PRF mixtures would therefore reducethe overall rate of combustion because the rate ofchain branching would decrease. This is apparent ina qualitative way in a comparison between the reac-tivity at high compressed gas temperatures betweenn-C7H16 and PRF 60 (Figs. 4 and 5) and betweenPRF 60 and i-C8H18 at f 4 0.6 (Figs. 5 and 6). The“net branching factor” cannot be quantified fromthese global measurements in the rapid compressionmachine. However, the proportion of alkyl radicalsfrom each reactant that eventually contributes to thelow-temperature chain-branching channels can bederived from numerical simulations [26], althoughat present it appears that the branching character-istics included in the model for i-C8H18 oxidation arenot yet as vigorous as is observed in practice.
The Effect on Ignition Delay of ReactantConsumption in the Compression Stroke
It must be expected that reaction in the compres-sion stroke has a significant effect on the subsequentdevelopment of ignition. Although not amenable toexperimental test in an RCM, this can be assessedquantitatively by a numerical analysis in which acomparison is made between the computation in a
fully reactive compression simulation and when thechemical calculation is started only at the tempera-ture and pressure attained at the end of compres-sion.
Such calculations for n-C7H16 combustion at Tc .850 K show that when 33% or more of the fuel hasalready been consumed in the compression strokeand has conditioned the system accordingly, the ig-nition delay is about half of that predicted from anonreacted compressed gas state [26]. The modelcalculations also show clearly that, even in conditionsin which no identifiable fuel consumption has beenobserved at the end of the compression stroke, asignificant extent of chemical activity may have al-ready developed. For example, in the high-temper-ature compression of n-C5H12, at the time compres-sion is complete, although no n-C5H12 consumptionis calculated (,0.1%) or observed, C5 radicals, suchas RO2 and QOOH, as well as C5 alkenes and cyclicethers, are all present in excess of ppm concentra-tions. In this respect, the fuel ` air mixture is pre-conditioned at the end of the compression strokeand the subsequent ignition delay is 10% shorterthan would be predicted if a truly unreacted mixturewere assumed.
Conclusions
In both the experimental results and the supple-mentary modeling analysis, it has been shown thatconsiderable fuel consumption can take place beforecompletion of the compression stroke when suffi-ciently high compressed gas temperatures arereached. By implication, this will occur in other ma-chines with comparable or slower compression rates.When knock occurs in a spark-ignition engine (at,2000 rpm11, say, at an average piston speed of 4–6 m s11 with a stroke of 6–10 cm), there is a similartime interval during which spontaneous oxidation ofthe fuel ` air mixture to develop appreciably, es-pecially at high gas densities.
Acknowledgments
The authors thank EPSRC for financial support.
REFERENCES
1. Westbrook, C. K., Pitz, W. J., and Leppard, W. R., SAEPaper No. 91-2314, 1991.
2. Griffiths, J. F., Halford-Maw, P. A., and Rose, D. J.,Combust. Flame 95:291 (1993).
3. Ohta, Y., Hayashi, A. K., Fujiwara, T., and Takahashi,H., Prog. Astro. Aeronaut. 113:225 (1988).
4. Halstead, M. P., Kirsch, L. J., Prothero, A., and Quinn,C. P., Proc. R. Soc. London, Ser. A 346:515 (1975).
5. Minetti, R., Ribaucour, M., Carlier, M., Fittschen, C.,and Sochet, L. R., Combust. Flame 96:201 (1994).

2692 SPARK IGNITION ENGINES
6. Minetti, R., Carlier, M., Ribaucour, M., Therssen, E.,and Sochet, L. R., Combust. Flame 102:298 (1995).
7. Chevalier, C., Pitz, W. J., Warnatz, J., Westbrook,C. K., and Melenk, H., Twenty-Fourth Symposium(International) on Combustion, The Combustion In-stitute, Pittsburgh, 1992, p. 1293.
8. Curran, H. J., Pitz, W. J., Westbrook, C. K., Hisham,H. W. M., and Walker, R. W., submitted for publica-tion (1996).
9. Curran, H. J., Gaffuri, P., Pitz, W. J., Westbrook, C. K.,and Leppard, W. R., SAE Paper No. 95-2406, 1995.
10. Park, P. and Keck, J. C., SAE Paper No. 90-0027, 1990.11. Griffiths, J. F., Jiao, Q., Schreiber, M., Meyer, J., and
Knoche, K. F., Twenty-Fourth Symposium (Interna-tional) on Combustion, The Combustion Institute,Pittsburgh, 1992, p. 1809.
12. Schreiber, M., Poppe, C., Sadat Sakak, A., Griffiths,J. F., Rose, D. J., and Halford-Maw, P. A., SAE PaperNo. 93-2758, 1993.
13. Beeley, P., Gray, P., and Griffiths, J. F., Combust. Flame39:269 (1980).
14. Griffiths, J. F. and Hasko, S. M., Proc. R. Soc. London,Ser. A 393:371 (1984).
15. Griffiths, J. F. and Perche, A., Eighteenth Symposium
(International) on Combustion, The Combustion In-stitute, Pittsburgh, 1981, p. 893.
16. Griffiths, J. F., Jiao, Q., Schreiber, M., Meyer, J., andKnoche, K. F., Combust. Flame 91:209 (1992).
17. Lund, C. M., Lawrence Livermore National Labora-tory report UCRL-52504, 1978.
18. Morley, C., Combust. Sci. Technol. 55:115(1987).
19. Griffiths, J. F., Prog. Energy Combust. Sci. 21:25(1995).
20. Cartlidge, J. and Tipper, C. F. H., Combust. Flame5:187 (1961).
21. Hu, H. and Keck, J. C., Twenty-First Symposium (In-ternational) on Combustion, The Combustion Insti-tute, Pittsburgh, 1986, p. 521.
22. Griffiths, J. F., Hughes, K. J., Schreiber, M., andPoppe, C., Combust. Flame 99:533 (1994).
23. Fish, A., Haskell, W. W., and Read, I. A., Proc. R. Soc.London, Ser. A 313:261 (1969).
24. Mohamed, C. and Pan, J., 1996, unpublished results.25. Kwok, E. S. C. and Atkinson, R., Atmospheric Envi-
ron. 29:1685 (1995).26. Curran, H. J., Pitz, W. J., and Westbrook, C. K., to be
published.
Copyright © 2022 FDOKUMEN




















