
Expert system for solving problems in carbon-13 nuclear magnetic resonance spectroscopy
13
Analytica Chimica Acta, 200 (1987) 333-345 Elsevier Science Publishers B.V., Amsterdam -Printed in The Netherlands EXPERT SYSTEM FOR SOLVING PROBLEMS IN CARBON-13 NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY J. ZUPAN*, M. NOVIC, S. BOHANEC and M. RAZINGER Kemijski Idtitut “Boris KidrZ”, Llubljana (Yugoslavia) L. LAH, M. TUSAR and I. KOSIR Department of Chemistry, University of Ljubljana, Ljubljana (Yugoslavia) (Received 8th April 1987) SUMMARY The expert system CARBON is built around a knowledge base consisting of spectra/ structure correlations, tables of data, mathematical formulae and graph-theory pro- cedures and on a data base of 2500 assigned “C-NMR spectra. The built-in knowledge enables the user to obtain suggestions for solutions to problems of different types arising in “C-NMR spectroscopy. Use of the system is facilitated by appropriate command files, large on-line help files, and user-friendly dialogue. The system can be used with spectrom- etries other than ‘“C-NMR and in other fields concerned with correlations between chemi- cal structures and properties. New improved instrumentation linked with better computers requires better and more powerful software for instant and diverse use by spectros- copists. Simple search systems are no longer regarded as satisfactory and more problem-solving power is increasingly sought in the software packages applicable to different problems [l-4]. This means that better and more refined information is required rather than simple answers in the form of a retrieved structure or a spectrum, so that as much information as possible can be retrieved from available data. The case of 13C-NMR spectroscopy is very illustrative. The user does not necessarily start the inquiry with a 13C- *Jure Zupan is the Professor of Chemometrics at the Chemistry Department of the University of Ljubljana. He graduated in 1966 and obtained his Ph.D. in 1972 in Chem- istry at the University of Ljubljana. From 1966 to 1973 he worked on ceramic and quantum chemistry problems at Institute Josef Stefan. In 1974 he joined the Department for Structural Chemistry lead by Prof. D. Had% at the Boris KidriE Institute of Chemistry in order to form a group for research on applications of computers in chemistry. The group is presently involved in the development of different spectroscopies based on chemical expert systems. His research interests are algorithms and methods for hierarchi- cal clustering, and feature extractions from complex data. He has worked at ETH Ziirich and EPA, Washington, DC, and in 1982 was a visiting professor in the Department of Chemistry at Arizona State University in Tempe. He is the author and editor of several books in the field. 0003-2670/87/$03.50 o 1987 Elsevier Science Publishers B.V.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Expert system for solving problems in carbon-13 nuclear magnetic resonance spectroscopy

Analytica Chimica Acta, 200 (1987) 333-345 Elsevier Science Publishers B.V., Amsterdam -Printed in The Netherlands
EXPERT SYSTEM FOR SOLVING PROBLEMS IN CARBON-13 NUCLEAR MAGNETIC RESONANCE SPECTROSCOPY
J. ZUPAN*, M. NOVIC, S. BOHANEC and M. RAZINGER
Kemijski Idtitut “Boris KidrZ”, Llubljana (Yugoslavia)
L. LAH, M. TUSAR and I. KOSIR
Department of Chemistry, University of Ljubljana, Ljubljana (Yugoslavia)
(Received 8th April 1987)
SUMMARY
The expert system CARBON is built around a knowledge base consisting of spectra/ structure correlations, tables of data, mathematical formulae and graph-theory pro- cedures and on a data base of 2500 assigned “C-NMR spectra. The built-in knowledge enables the user to obtain suggestions for solutions to problems of different types arising in “C-NMR spectroscopy. Use of the system is facilitated by appropriate command files, large on-line help files, and user-friendly dialogue. The system can be used with spectrom- etries other than ‘“C-NMR and in other fields concerned with correlations between chemi- cal structures and properties.
New improved instrumentation linked with better computers requires better and more powerful software for instant and diverse use by spectros- copists. Simple search systems are no longer regarded as satisfactory and more problem-solving power is increasingly sought in the software packages applicable to different problems [l-4]. This means that better and more refined information is required rather than simple answers in the form of a retrieved structure or a spectrum, so that as much information as possible can be retrieved from available data. The case of 13C-NMR spectroscopy is very illustrative. The user does not necessarily start the inquiry with a 13C-
*Jure Zupan is the Professor of Chemometrics at the Chemistry Department of the University of Ljubljana. He graduated in 1966 and obtained his Ph.D. in 1972 in Chem- istry at the University of Ljubljana. From 1966 to 1973 he worked on ceramic and quantum chemistry problems at Institute Josef Stefan. In 1974 he joined the Department for Structural Chemistry lead by Prof. D. Had% at the Boris KidriE Institute of Chemistry in order to form a group for research on applications of computers in chemistry. The group is presently involved in the development of different spectroscopies based on chemical expert systems. His research interests are algorithms and methods for hierarchi- cal clustering, and feature extractions from complex data. He has worked at ETH Ziirich and EPA, Washington, DC, and in 1982 was a visiting professor in the Department of Chemistry at Arizona State University in Tempe. He is the author and editor of several books in the field.
0003-2670/87/$03.50 o 1987 Elsevier Science Publishers B.V.

334
NMR spectrum. The query could be any entity from the field of 13C-NMR spectroscopy. Hence, an expert system in this field must accept spectra, subspectra, single chemical shifts, structures, substructures, sets of frag- ments, etc. as input data. Using input data of different types, the expert system should be able to handle various tasks from spectral and structural searches to simulation of spectra, prediction of structures and substructures, detection of structural locations where conformational isomerism can arise, pinpointing the positions for substituents, etc. The more such problems a system can handle, the more it deserves the term “expert”.
Unfortunately, the present chemical expert systems are far from being able to solve really difficult problems. However, they can provide assistance with a number of valuable suggestions and hints deduced from the built-in knowledge, obtained by brute-force algorithms, or retrieved from data bases 151. In the present paper, the CARBON system, which is designed for solving different problems raised in 13C-NMR spectroscopy is described.
PHILOSOPHY OF THE CARBON SYSTEM
When the CARBON system is used, work is started on the problem by selecting one of the following seven routines: identification of entire and/or partial spectra, identification of structures and/or substructures, prediction of possible structural features, simulation of 13C-NMR spectra, assignment of 13C-NMR chemical shifts to selected carbon atoms, generation of all isomers from a given set of fragments, and various combinations of these options.
In addition to the manipulation of spectroscopic knowledge, handling and maintenance of intermediate results are also provided. The utility features are easy to deal with, hence even users having little experience with com- puters can work successfully.
At present, the data base contains 2536 13C-NMR spectra of various chemical compounds with about 30 000 assigned chemical shifts. All shifts m the data base are given with an accuracy of kO.1 ppm, relative to TMS. The data base serves primarily as a source of spectra/structure correlations (needed for predictions of structural and spectral features) and shifts of different substituents (needed for assignments and spectra simulation), and secondarily, as a reference collection for different searches.
The quality of an expert system is determined by the quality of the knowledge built into it. In the CARBON system, knowledge is incorporated in the following four forms: (1) as a hierarchical tree of clusters of similar spectra, which serves for the prediction of structural fragments and struc- tural types of queries, (2) as tables of chemical shifts of different substit- uents in different steric positions and conformation states, which can be used for simulation of r3C-NMR spectra; (3) as mathematical formulae and statistical procedures on groups of data of different kinds and sizes; and (4) as graph-theory algorithms implemented in substructure search, assign- ments, and generation of constitutional isomers.

335
During the application of the system, it is possible to select any option offered and to use the intermediate results obtained in previous sessions as a new data base for further work. For example, the user can generate interactively a molecular graph and then choose one of the possible con- tinuations: these can be either the simulation of a 13C-NMR spectrum, a substructure search, or building a data base of structures by writing the generated structures on external file. In all cases, the results can be stored permanently or temporarily for further use. In another example, the user can begin the work with a single chemical shift, with a group of shifts, or with a whole ‘“C-NMR spectrum. Having this type of input data, the user can select the search in the hierarchical tree, shift-by-shift search in the data base, or assignment of shifts. There are other features m the CARBON sys- tem which are valuable for spectroscopists and organic chemists, e.g., inter- active generation of chemical structures and generation of all constitutional isomers from a given set of fragments.
The general idea was to link different programs together so that the CARBON system could be used for attacking many different problems, and partial results of one option could be taken as the input data for another option.
DESCRIPTION OF FEATURES OFFERED BY THE SYSTEM
Search by chemical shifts This is the simplest and, for beginners, the most used part of the system.
It enables those spectra having all the chemical shifts entered consecutively by the user to be retrieved from the data base of 2536 items. The default tolerance for each shift is kO.5 ppm, but the user can alter this value pro- vided that the number of retrieved spectra does not exceed 1000. The search procedure is shown in Fig. 1. The user can inspect or store the “good list” of structures at any point during the search. The possibility of selecting and storing permanently a small data base from which the work can be continued later is very useful. The number of data bases created in this manner is limi- ted only by the external computer space available.
Shift-by-shift search is important for another reason. Selecting the answer “A” (for assignment) in the above search procedure (Fig. la) causes the system to check all retrieved spectra if the entered chemical shifts are assigned to carbon atoms linked together. The system acknowledges the connections between the carbon atoms even if they are separated by a number of consecutive heteroatoms. For example, two carbon atoms in a chain -C-O-C(=O)- having chemical shifts of 64.0 and 173.2 ppm res- pectively, are treated as connected. In the next step, the system searches for different fragments (up to the preselected size of the neighbourhood) having in the centre the carbon atom with the chemical shift given first. At present, the system displays the structures containing the selected fragments and writes their connection tables to a separate file for further use.

336
Search with complete spectrum There are two reasons why the shift-by-shift search cannot satisfy all
needs. First, an inherent problem in the shift-by-shift search, i.e., the very large number of retrieved references in certain regions of the 13C-NMR spec- trum, prevents fast, precise, and thus reliable, answers; secondly, very often a holistic approach to the retrieval of spectra is desired (i.e., the user is interested in the information about the type of structures that fit best the entire query spectrum not just an arbitrary part of it. In the case of a full spectrum search, the system starts with the preprocessing of the query spectrum into a 40dimensional vector [ 6, 71 and inputs this representation at the root of the hierarchical tree (Fig. 2a). The choice of the path through the tree is based at each node on the comparison of three distances: two distances between both descending clusters and the query spectrum, and the third one between the descending clusters themselves. After the query traverses an assigned node, the assigned structural property is associated with the unknown structure. The reliability of the prediction is inversely
Fig. 1. Search procedure using single peaks. The default tolerance is kO.5 ppm. The user can display the retrieved references at any point by typing “I”, or save them on a perma- nent file using the “F” command. The command “A” prompts the system to check if the given chemical shifts belong to carbon atoms that are linked together in the reference structures. As seen in example (a), the resulting list of 5 compounds was reduced to only two compounds, (b) and (c), in which both shifts 180.0 f 1.0 ppm and 48.0 * 0.5 ppm belong to neighbouring atoms.

337
Fig. 2. Search in the hierarchical tree. First, the complete spectrum (213.6, 36.7, 22.0 ppm) is entered; secondly, the nodes on the main path are displayed together with the most interesting parameters (number of spectra in the node, omitted nodes, distances, the relative difference in distances in the percentage with the respect to the smallest one, and the identification number of predicted structural fragments). Thirdly, the ID num- bers of the members of the final cluster to which the query spectrum was linked are shown (Nos. 908 and 1122 in this case). The omitted nodes at which the relative differ- ence in the distances was small are potential candidates for the feed-back search (i.e., nodes 6 and 1715, 495, etc.). In node 1544, fragment no. 4 (-CH,-) was predicted and in node 235 fragment no. 9 (-C!(K))-) was predicted. To clarify the procedure, a small part of the top of the decision tree is shown in detail in (a). Both compounds from cluster 977 are shown as (b) and (c). In fact, the query spectrum belonged to the same compound with spectra recorded under different conditions.

338
proportional to the distance measure between the query spectrum and the assigned node at which the prediction was made. The search can stop either at the end spectrum in the tree (at the leaf) or in the middle of the tree (at a cluster of spectra). The distance of the query spectrum to the leaf or to all members of the cluster at which the search stops, gives the user the information how similar the unknown structure and the reference structures are. A case in which the search stopped at node no. 977 is shown in Fig. Z(a). This node represents two identical compounds with very similar spectra. In the 40dimensional representation, both spectra in node 977 are identical, not only with each other but also with the query. Both reference compounds found in node 977 are shown in Fig. 2(b and c). When the two spectra are compared witlithe chemical shifts in the query, the advantage of the method is evident: the differences in corresponding shifts (reference-to-query) are too large for a retrieval on the shift-by-shift basis to be successful. In fact, there are more than 1500 spectra in the data base for the peak 22.0 in the interval of 50.5 ppm. Additionally, the mentioned interval does not even cover both reference spectra having 23.5 and 23.4 ppm shifts, respectively.
The assigned nodes (clusters of three or more spectra) in the hierarchical tree represent spectra of compounds having at least one structural feature in common. On the search path shown in Fig. 2, nodes 1544 and 235 are assigned to fragments 4 (-CH,--) and 9 (--C(e)-), respectively. The traverse of the query spectrum through the tree can, in principle, start at any node in the tree. The default start implemented in the first pass is at the root. After the first pass is complete, the user can redirect the next start of the query spectrum at any chosen node in the tree. The potential candidates for such feed-back searches [8] are the nodes at which the decision about the continuation was not a clear-cut one (i.e., the differ- ences were approximately equal). In Fig. 2, nodes 4 and 1715 are the most appropriate candidates for the feed-back search.
Generation of molecular graphs For many purposes (simulation of spectra, substructure search, update,
etc.), the user must have a utility for simple and flexible generation of molecular graphs (i.e., connection tables). The set of commands for this purpose consists of the standard ones (for generation and linking of differ- ent structural parts together like RING, CHAIN, BRIDGE, ATOM, BOND, DELETE, OPEN, INSERT, etc.) and utility commands for making the generation and manipulation of structures more convenient (MENU, SAVE, DROP, TERMINAL, HELP, CT for connection table, etc.). With the option SAVE, for example, up to 20 userdefined structures can be saved for instant recall (with MENU, n command) when needed.
When a fragment is built, the atoms that could be connected to other fragments, substituents, or structures should be clearly marked as such. The atoms where a substituent could be attached are marked with an A, while the atoms where a substituent must be connected are marked with an X.

339
After a structure (or fragment) has been generated, it is written on a tem- porary file accessible from any option that uses connection tables as input (substructure search, assignment, simulation, update, etc.).
Substructure search In the 13C-NMR and other spectroscopies, structurally similar compounds
are frequently studied. In the CARBON system, the substructure search is done in four\ steps: (1) generation of the query substructure; (2) fast pruning of the existing structural da& base using the inverted files of atomic centred fragments; (3) atom-by-atom comparison of connection tables on the short remaining file with the connection table of the query fragment, and (4) search for all number-to-number correspondences between the number- ing of atoms in the query and the numbering of atoms in the retrieved structures.
Because a reference structure can contain the query fragment in more than one structural position, more than one number-to-number correspon- dences can result. Because all carbon atoms in the data base are numbered and assigned to chemical shifts, the distribution of shifts at the equivalent positions is easily obtained. Figure 3 shows the query, the retrieved list of ID numbers of compounds containing the query fragment, the distribution of shifts for atom no. 4 in the query fragment, and two structures from the good-list.
Simulation of ‘%-NMR spectra For any structure currently generated by the CARBON system, its 13C-
NMR spectrum can be simulated. The values of the chemical shifts associ- ated with carbon atoms are calculated by adding partial contributions of neighbours to the standard chemical shift of each carbon atom. The partial contributions of substituents and standard shifts are taken from the liter- ature [9, lo] . The system checks each atom, its (Y, p, y and 6 neighbours, and the substituents on these positions. The system identifies the type of the particular carbon atom (sp, sp2, sp3 hybridization, carbonyl-type, aro- matic, pyridine ring, etc.), type and number of the substituents, and the position of the substituent with the respect to the central atom. Steric con- tributions (depending on whether the central atom is primary, secondary, tertiary or quaternary) to the chemical shifts are also considered. Addition- ally, the system detects the positions where isomerism can arise and asks for specification of the conformation (synperiplanar, synclinal, anticlinal, antiperiplanar, or free rotation). The corresponding contribution is then added to the total chemical shift.
If a contribution for a found substituent is not present in the tables, the system informs the user about the missing data. The user then inputs the appropriate value or just notes the insufficiency. A comparison between the simulated 13C-NMR spectrum of a given structure and the experimental spectrum of the same compound found in the data base is given in Fig. 4.

Fig. 3. The substructure search. In the query fragment (a), the sites where substitutions can take place are marked with A or X, depending whether the user wants a possible (A) or mandatory (X) substituent at a particular position. The substructure search starts with the command Q,S (for quit the structure generator and start searching). The hst of retrieved compounds is accompanied by the number of times that the query fragment is encountered in each of them. After the resulting reference have been dlsplayed, the user can inspect distributions of chemical shifts for any selected carbon atom m the query or look at the full reference (c and d). In all intervals of the distribution of chemical shifts, the ID numbers of the compounds together with the number of the particular carbon atom are shown. For example, compound 1236 (c) has 2 carbon atoms (1 and 9) that correspond to the atom 4 in the query structure. Both shifts (169.4 ppm) assigned to these 2 atoms are placed in the first interval of the distribution shown.
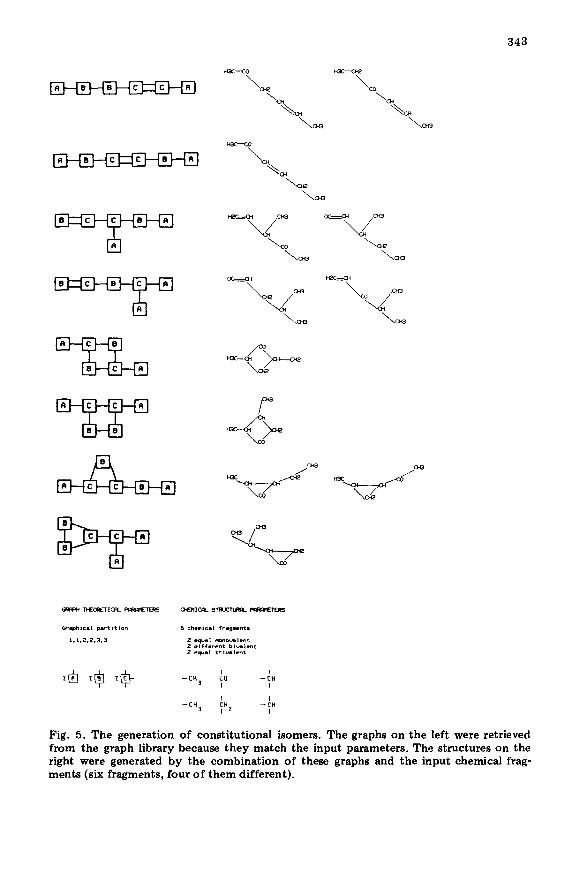
Generation of all constitutional isomers A routine part of the structure elucidation process is assembling the
structure from the set of fragments known to be present in the unknown compound. The list of possible fragments can be acquired and/or updated from various sources: the similarity search, the separate peak-position search, or from any other analytical method. Several isomer generators

341
Fig. 4. Simulation of the ‘%NMR spectrum. First, the structure is generated (a), then in an interactive dialogue between the system and the user, questions about the confor- mational positions are resolved (b), and the simulated spectrum is displayed. For com- parison, the experimental spectrum of the same compound is also displayed (c). The OK entries in the fourth column of the simulated spectrum (labelled STATUS) refer to the fact that all partial contributions to all chemical shifts were found.
are implemented in different, systems (e.g., DENDRAL [ll] and CASE [12] ). With the possibility of implementation of the designed system on personal computers, it was decided to try a new approach to the isomer generation problem.
The main difficulty of this problem lies in the fact that the isomer list, has to be exhaustive but without redundancies. Because of this, each generated isomer has to be tested for possible graph isomorphism against all previously generated isomers. Because with a growing number of fragments (i.e., graph nodes) testing for isomorphism leads to a combinatorial explosion [ 131, it
is obviously unsuitable for implementation on small computers or personal computers. In our solution, this slow testing for isomorphism is replaced by a quick comparison of each generated isomer (i.e., chemical graph) with all relevant entries in a special graph library. The graphs in the library are repre- sented by their graphical partitions, i.e., sequences of valencies of all graph nodes. Because the input information has the same structure (set of frag- ments of known valency), the comparison can be done by simple matching and proceeds very quickly. The actual library contains graphs with 2-10 nodes.
At first glance, it would seem that such an approach is of very limited use because of the small number of nodes in a graph that can be precalculated

342
exhaustively and maintained efficiently later. But because the building blocks can also be larger fragments, rings, ring systems, etc., and not just single atoms, quite large molecules can be treated in this way. Compared with the brute-force approach, of course, the proposed scheme means that much of the work that could be done by the computer is left to the oper- ator but it is still far better than to do everything by hand.
The generation of isomers is done in two steps. In the first step, the input distribution of fragments in a form of a set of numbers is checked in the files for the identical set. In the second step, the nodes of the matching graphs are substituted with all possible chemical fragments (Fig. 5). During this step (which can be more time-consuming), the user can discard some structural types on the basis of chemical knowledge about the problem.
Isomer generation can be of use in various fields of structure elucida- tion where information on the identity of fragments is gathered (infrared, mass spectrometry, etc.) and thus the option can be used by different specialists.
File manipulation As has been shown, the user can start working on the system with queries
of very different form, combine partial results from different options, save intermediate or final results on permanent files, change or update files, etc. In order to accomplish these tasks, the system must have a uniform and strict file organization; data files containing the same type of information (i.e., connection tables, ID numbers, set‘of shifts, etc.) should always be written in the same format regardless of the option in which they are created. Further, the file-manipulation capability must be flexible. The file-manipula- tion option is designed to help in these steps and to meet the condition of minimal user knowledge about computers. Because all options (program modules) are linked via the command files, the appropriate HELP infor- mation is included in these files and is thus always at hand if a wrong com- mand is issued by the user. At present, there are about 100 kbytes of on- line help information available to assist users in different situations.
FUTURE DEVELOPMENT OF THE CARBON SYSTEM
Like any other expert system, the CARBON system can and will be further developed and improved in many areas. The following improvements are already in progress. (1) prediction of more structural features during the traverse of the hierarchical data base; (2) addition of chemical shifts of more substituents for simulation of 13C-NMR spectra; (3) addition of new graph files for structure generator; and (4) automation of assignment of chemical shifts.
The prediction of structural features is improved by generating new trees (based on new representations of ‘%!-NMR spectra such as Hadamard trans- forms [ 141. For every node in the new tree, an analysis of structural features
343
hoc---oe
\
‘“\, Nzk4
\CX3
Fig. 5. The generation of constitutional isomers. The graphs on the left were retrieved from the graph library because they match the input parameters. The structures on the right were generated by the combination of these graphs and the input chemical frag- ments (six fragments, four of them different).

344
of linked compounds is made. After a tree with better prediction ability and with a larger assortment of predicted structural features has been found, it replaces the old one.
The problem of simulation of more complex compounds is tackled from two sides: first, by using the existing data base of assigned spectra in order to extract chemical shifts for substituents not found in the literature, and second, by introducing a set of skeletons with known chemical shifts. The latter approach is connected with the so-called super-structure search. Its purpose is to identify all members of a given set of structures (templates) that are substructures of a query structure; in contrast, a substructure search tests all structures from a data base if they contain the query struc- ture as a substructure. Although the core algorithm for the comparison of structures is the same for both searches, the selection, exit conditions, and file organization for the super-structure search is quite different from that of the substructure search.
At present, the files of graphs for a given number of nodes (from 2 to 10) contain all acyclic, monocyclic and bicyclic graphs with single bonds and all acyclic graphs with double and triple bonds. The files of polycyclic graphs and cyclic graphs with multiple bonds are under development.
Assignment will be improved by the introduction of automatic assign- ment [4] and error detection. In the present version, the system shows structural fragments consisting of atoms assigned to a given set of chemical shifts and the user must choose the most appropriate fragment. In the next step, the user will input the set of shifts and the structure while the system will try to optimize the assignment.
A weak point in the system is the updating of new spectra and structures. The update is not intended for general public access because all the internal knowledge (hierarchical tree, assignments, structure prediction, etc.) is very sensitive to errors in the data base. Each addition of a new item changes many files and data on different levels (inverted files of shifts and structural fragments, hierarchical tree with assigned nodes, targets for assignments, etc.) and so the updated structures and spectra must be thoroughly checked. For these reasons, updating is done in blocks of 500 new entries at a time. Our main concern is to make the CARBON system a real expert system based on a good knowledge base rather than on a large data base. It is true that more knowledge can be extracted from a larger data base, but it is considered here to be more useful to make improvements on the basis of existing data and to add more features which will enable the CARBON system to interpret 13C-NMR spectra intelligently, rather than to focus in adding new spectra to improve obvious tasks such as retrieval of similar spectra and structures.
Conclusion The CARBON system was designed to help in solving problems related to
13C-NMR spectroscopy. At present, it finds spectra similar or identical to

345
that of the query, predicts structural features on the basis of a single chemi- cal shift, group of shifts, or entire 13C-NMR spectrum, simulates 13C-NMR spectra, helps in assignment problems, generates isomers from a given set of fragments, runs structure and substructure searches, and performs com- binations of all these tasks. Additionally, the CARBON system can help in other fields besides 13C-NMR and thus can be helpful to users interested in problems related to chemical structures. Substructure search and clustering of structures is applicable in many areas of structure elucidation while the isomer generator can be used successfully in mass spectrometry. The inter- active structure generator can serve as a stand-alone program for input of connection tables if a chemical structure-oriented data base is required. Finally, the system can serve very well for educational purposes.
The system consists of approximately 20 command files governing about 60 programs written in either Fortran or Pascal and of about 20 different permanent data files. The entire system uses slightly less than 20 Mbyte of memory.
The authors thank many friends, co-workers and users who contributed programs, comments, ideas and criticism, helping to improve the perfor- mance of the system considerably. Among these, special thanks are due to Prof. Dus’an Hadii, Prof. Morton E. Munk, Prof. Branko Stanovnik, Dr. Jurka KidriE, Mr. Duhn Turk and Mr. Milan Hodos’Eek. The financial sup- port of Research Community of Slovenia is gratefully acknowledged.
REFERENCES
1 W. Bremser, L. Ernst, B. Franke, R. Gerhards and A. Hardt, Carbon-13 NMR Spectral Data, 3rd edn., Verlag Chemie, Weinheim, 1981.
2 J. Zupan, (Ed.), Computer-Supported Spectroscopic Data bases, Horwood, Chichester, 1986.
3 N. A. B. Gray, Prog. Nucl. Magn. Res. Spectrosc., 15 (1985) 201. 4 H. Kalchhauser and W. Robien, J. Chem. Inf. Comput. Sci., 25 (1985) 103. 5 R. Dessy, Anal. Chem., 56 (1984) 1200A, 1312A. 6 J. Zupan, Clustering of Large Data Sets, Research Studies Press, Wiley, Chichester,
1982. 7 M. NoviE and J. Zupan, Anal. Chim. Acta, 177 (1985) 23. 8 J. Zupan and M. E. Munk, Anal. Chem., 58 (1986) 3219. 9 E. Pretsch, J. T. Clerc, J. Seibel and W. Simon, Tabellen zur Strukturaufkliirung
organischer Verbindungen mit spektroskopischen Methoden, Springer, Berlin, 1976. 10 D. W. Brown, A Short Set of Carbon-13 NMR Correlation Tables, J. Chem. Educ.,
62(3) (1985) 209. 11 R. E. Carhart, D. H. Smith, N. A. B. Gray, J. G. Nourse and C. Djerassi, J. Org. Chem.,
46 (1981) 1708. 12 C. A. Shelley and M. E. Munk, Anal. Chim. Acta, 133 (1981) 607. 13 R. C. Read, in B. Harris, Ed., Graph Theory Algorithms, Graph Theory and its Appli-
cations, Academic, New York, 1970, pp. 51-78. 14 R. Kaiser, J. Magn. Res., 15 (1974) 44.




















