
Antigen receptor signalling: a distinctive role for the p110δ isoform of PI3K

Cardiovascular pharmacology
Evidence that the MEK/ERK but not the PI3K/Akt pathway is requiredfor protection from myocardial ischemia–reperfusion injuryby 30,40-dihydroxyflavonol
Colleen J. Thomas a,d,n, Nicholas R. Lim b, Alphious Kedikaetswe a, Yvonne Y. Yeap b,Owen L. Woodman c, Dominic C.H. Ng b,1, Clive N. May a
a Florey Institute of Neuroscience and Mental Health, Parkville, Victoria, Australiab Department of Biochemistry and Molecular Biology, Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville,Victoria, Australiac School of Medical Sciences, RMIT University, Bundoora, Victoria, Australiad Department of Physiology, Anatomy and Microbiology, La Trobe University, Bundoora, Victoria, 3086, Australia
a r t i c l e i n f o
Article history:Received 29 August 2014Received in revised form13 March 2015Accepted 17 March 2015Available online 25 March 2015
Keywords:FlavonolMyocardial ischemia/reperfusion injuryOxidative stressSurvival kinasesERK1/2AktPD98059LY294002
a b s t r a c t
The novel pro-drug of 3040-dihydroxyflavonol, NP202, potently reduces myocardial infarct size resultingfrom ischemia–reperfusion (I/R) through mechanisms that remain to be fully defined. In this study, weinvestigated whether cardioprotection induced by NP202 depended on activation of the reperfusioninjury survival kinase (RISK) pathways. We therefore examined the effects of PD98059 and LY294002,specific inhibitors of the MEK/ERK1/2 and PI3K/Akt pathways, respectively. In isolated cardiomyocytes,H2O2induced oxidative stress activated ERK1/2 and this was further enhanced by DiOHF, the activeparent compound of NP202. Although oxidative stress did not stimulate Akt in cardiomyocytes, co-treatment with DiOHF substantially increased Akt phosphorylation. This suggests that DiOHF is a potentmodulator of RISK pathways specifically in the context of stress stimulation. In anesthetised sheep,following 1 h ischemia and 3 h reperfusion, the contribution of the RISK pathways to NP202-mediatedcardioprotection was determined by treating the animals with PD98059, LY294002 or vehicle prior toNP202 administration and reperfusion. Infarct size, as a percentage of the area-at-risk, was substantiallyreduced by NP202 (from 7876 to 4674%, Po0.05). Inhibition of MEK/ERK1/2 abolished thecardioprotective effects of NP202 (infarct size 8174%), whereas inhibition of PI3K/Akt had no effect(infarct size 5374%). Our combined cellular and animal studies indicate that NP202 potently protectsagainst myocardial I/R injury through complex mechanisms that involved augmentation of MEK/ERK1/2signaling, but not PI3K/Akt signaling.
& 2015 Elsevier B.V. All rights reserved.
1. Introduction
The primary therapeutic strategy to reduce infarct size followingmyocardial infarction (MI) is coronary reperfusion by means ofangioplasty or thrombolysis. Paradoxically, blood flow restorationcauses death of vulnerable, but still viable, myocardial tissue, leadingto so-called ‘reperfusion injury’ (Braunwald and Kloner, 1985).Among the key contributors to lethal myocardial reperfusion injuryare oxidative stress due to generation of reactive oxygen species
(ROS), neutrophil accumulation, cytokine release, an increase in pHfrom an acidic to a normal environment, a rise in intracellular Ca2þ ,
microvascular dysfunction and altered myocardial metabolism(Hausenloy and Yellon, 2013; Yellon and Hausenloy, 2007).
Ischemic preconditioning (IPC), consisting of brief periods ofrepetitive ischemia–reperfusion (I/R), recruits endogenous mechan-isms that protect against subsequent prolonged periods of ischemia.Currently IPC is the most effective way to inhibit the pathologicalprocesses initiated by myocardial reperfusion injury (Kharbanda,2010). The Reperfusion Injury Survival Kinase (RISK) pathways havebeen identified as critical mediators of the cardioprotective effect ofIPC against I/R injury (Hausenloy and Yellon, 2007). These pathwaysconsist of a group of pro-survival, anti-apoptotic kinases, includingextracellular signal regulated kinase (ERK1/2, also referred to as p44/p42) and phosphoinositide-3 kinase (PI3K)/Akt (Hausenloy and Yellon,2007; Zhang et al., 2010). The identification of a cardioprotective agent
Contents lists available at ScienceDirect
journal homepage: www.elsevier.com/locate/ejphar
European Journal of Pharmacology
http://dx.doi.org/10.1016/j.ejphar.2015.03.0540014-2999/& 2015 Elsevier B.V. All rights reserved.
n Corresponding author at: Department of Physiology, Anatomy and Microbiol-ogy, La Trobe University, Bundoora, Victoria 3086, Australia. Tel.: þ61 3 9479 5593;fax: þ61 3 9479 5784.
E-mail address: [email protected] (C.J. Thomas).1 Current address: School of Biomedical Sciences, University of Queensland,
Australia.
European Journal of Pharmacology 758 (2015) 53–59

that can mimic IPC and prevent reperfusion injury has great clinicalappeal, but has thus far proved elusive.
We have previously demonstrated that the synthetic flavonol30,40-dihydroxyflavonol (DiOHF) provided a similar level of protec-tion from myocardial reperfusion injury to IPC in sheep (Wanget al., 2004a). DiOHF has multiple beneficial actions, including apotent antioxidant effect, reduction of superoxide-induced NOinactivation, preservation of endothelial NOS, enhancement ofpost-ischemic recovery of coronary endothelial function, reducedCa2þ utilization and inhibition of inflammatory responses, cardi-omyocyte apoptosis and opening of the mitochondrial permeabil-ity transition pore (mPTP) (Wang et al., 2004a, 2004b, 2009;Woodman and Chan, 2004; Woodman et al., 2014). Furtherexamination of the mechanism of action of NP202, a pro-drug ofDiOHF, indicated that it inhibited activation of pro-injury p38-MAPK and c-Jun N-terminal kinase (JNK) that occurred at 30 minof reperfusion in anesthetised sheep (Lim et al., 2013). We alsoobserved increased activation of cardioprotective ERK1/2 and Aktpathways in sheep, although their levels were not substantiallyaltered by NP202 at early stages of reperfusion (Lim et al., 2013).Thus, it remains unclear whether the cardioprotective effect ofDiOHF involves the kinase cascades of the RISK pathway.
The aim of this study was to determine the role of both arms ofthe RISK pathway in mediating the cardioprotective effect ofNP202, a pro-drug of DiOHF designed to be water soluble forhuman use (Williams et al., 2011). Specifically, we examined, inprimary cultures of neonatal rat cardiac myocytes, the effects ofDiOHF on oxidative stress-stimulated phosphorylation of kinasesin the RISK pathways. Subsequently, we determined whetherpharmacological blockade of the PI3K/Akt or the MEK/ERK1/2kinase cascades, with LY294002 or PD98059, respectively, inhib-ited NP202 cardioprotection following myocardial I/R injury inanesthetized sheep.
2. Materials and methods
The present studies were approved by the Animal EthicsCommittees of (i) the Florey Institute of Neuroscience and MentalHealth and (ii) Biochemistry and Molecular Biology, Dental Science,Medicine, Microbiology and Immunology, University of Melbourne,in accordance with the guidelines laid down by the National Healthand Medical Research Council of Australia's Code of Practice for theCare and Use of Animals for Experimental Purposes.
2.1. Primary cardiac myocyte isolation and treatment
Primary neonatal cardiac myocytes were isolated from 1–2 dayold Sprague–Dawley rats (Ng et al., 2001). Briefly, ventricular cellswere isolated by collagenase digestion and then pre-plated to depletenon-myocyte cells. Cardiac myocytes were then plated on gelatin-coated 60 mm dishes in Dulbecco's modified Eagle's medium/Med-ium 199 (4:1 v/v) containing 10% (v/v) horse serum, 5% (v/v) fetal calfserum, and penicillin/streptomycin (100 Units/ml). The serum wasremoved after 18 h, and cells starved for 24 h prior to experimentaltreatment. Cells were then pre-treated with DiOHF – the pharmaco-logically active parent drug of NP202 (10 mM, 30 min, IndofineChemical Co. New Jersey, U.S.A.) or an equivalent volume of vehicle(0.1% [v/v] DMSO) as a control, as we have described previously (Limet al., 2013). Simulated I/R was achieved using a ROS treatment.Oxidative stress was induced in primary cell cultures by treating withhydrogen peroxide (H2O2; 1 mM, 60 min), as we have describedpreviously (Lim et al., 2013). Neonatal myocytes remained contractileand actively beating during stress stimulation. In separate experi-ments, cells were pre-treated with PD 98059 (20 mM, 30 min;blocking MEK/ERK1/2) or LY 294002 (20 mM, 30 min; blocking Akt)
alone or concomitant with DiOHF, prior to stress treatment. Theseconcentrations were based on previously established data indicatingeffective dose ranges for in vitro studies (Lash, 2005).
2.2. Protein extraction and immunoblotting from cardiac cells
Protein lysates were prepared and immunoblotted as we havepreviously described (Lim et al., 2013). Briefly, cell lysates were preparedin radioimmune precipitation assay (RIPA) buffer [50mM Tris/HCl, pH7.3, 150mM NaCl, 0.1mM EDTA, 1% (w/v) sodium deoxycholate, 1% (v/v)Triton X-100, 0.2% (w/v) NaF and 100 μM Na3VO4] supplemented withprotease inhibitors (Ng et al., 2010). After 10min on ice, cell debris wasremoved by centrifugation (14,000 g, 10min). Protein concentrationswere then determined by Bradford assay. Protein lysates were resolvedby SDS/PAGE, transferred onto polyvinylidene fluoride membranes,blocked with 5% (w/v) non-fat dried skimmed milk powder in TBST(10mM Tris, pH 7.5, 150mM NaCl and 0.1% Tween 20) and then blottedwith primary antibodies (1/250 to 1/1000 dilution, depending on theantibody and determined experimentally) in the block solution. Thephospho-specific MEK1/2 antibody was obtained from Cell SignalingTechnology (Massachusetts, USA). Horseradish peroxidase-conjugatedsecondary antibodies were diluted 1/20,000 in 1% (w/v) non-fat milkpowder in TBST. ECL detection was with SuperSignal West Pico (PIERCE)or Lumi-Light Plus (Roche) according to the manufacturers' instructions.Chemiluminescence was imaged on a Chemidoc MP system (Bio-RadLaboratories) and densitometric band quantitation performed usingImageJ (NIH).
2.3. Surgical instrumentation for myocardial ischemia–reperfusion
Myocardial I/R was performed in anesthetised adult ewes (30–50 kg) as described previously (Wang et al., 2004a). Briefly, anesthe-sia was induced with intravenous sodium thiopentone (15 mg/kg)and, after endotracheal intubation, was maintained with 1.5–2.0%isoflurane–O2/air mixture. Following a left thoracotomy and openingof the pericardium, the second branch of the left anterior descendingcoronary artery (D2) was isolated and a vascular snare was placedproximal to a transit-time flow probe (2 mm). Animals were instru-mented to record hemodynamics (arterial blood pressure and heartrate) and cardiac function (dP/dt), according to our previouslypublished methodology (Wang et al., 2004a).
2.4. Experimental Protocol
Following stabilization after surgery, the experimental protocolconsisted of 30 min baseline recording of systemic hemodynamicsand coronary flow, followed by 1 h ischemia and 3 h reperfusion.Studies were performed on 35 sheep randomized into 7 groups.Group 1 (n¼6), infusion of vehicle used for kinase inhibitorsstarting 10 min before reperfusion followed at 5 min beforereperfusion by NP202 (6.6 mg/kg IV bolus, NeuProtect Pty, Ltd,Australia), dissolved in 10 ml 0.1 M Na2CO3 (Sigma-Aldrich Inc., St.Louis, MO, U.S.A.) given over 1 min, as described previously(Thomas et al., 2011; Williams et al., 2011). Groups 2 and 3 (bothn¼5), administration at 10 min before reperfusion of either thePI3K/Akt inhibitor LY294002 (4 mg IV bolus followed by 10 mg/12 ml IV infusion over 1 h, Sapphire Bioscience, Redfern, N.S.W.Australia) or the MEK1/2-ERK1/2 inhibitor PD98059 (4 mg IVbolus followed by 8 mg/12 ml IV infusion over 1 h, SapphireBioscience, Redfern, N.S.W. Australia). For the bolus, drugs weredissolved in 1 ml dimethyl sulphoxide (DMSO, Ajax Finechem,Seven Hill, NSW, Australia) and 4 ml polyethylene glycol (PEG,Sigma-Aldrich Inc., St. Louis, MO, USA) and for the infusions, at12 ml/h, drugs were dissolved in 2 ml DMSO and 12 ml PEG. Inboth groups NP202 was given 5 min before reperfusion. Group 4(n¼5), vehicle for kinase inhibitors started at 10 min before
C.J. Thomas et al. / European Journal of Pharmacology 758 (2015) 53–5954
reperfusion was followed by vehicle for NP202 at 5 min beforereperfusion. Groups 5 and 6 (both n¼3), LY294002 or PD98059 at10 min before reperfusion was followed by vehicle for NP202 at5 min before reperfusion. Group 7 (n¼3), a combination ofPD98059 and LY294002 was administered 5 min before reperfu-sion followed by vehicle for NP202. In all groups, hearts werecollected and analyzed as described below.
In our acute myocardial I/R sheep model, arrhythmic episodesof up to 15 min develop about 20 min after the start of ischemiaand again early in reperfusion. During these times we administer2% lignocaine (�7 and �4 ml, respectively) into the left atria.Lignocaine has a transient mild hypotensive action, but this effectis controlled for by its use in the control groups. A total of 35animals were instrumented and commenced the acute myocardialI/R protocol. Five sheep died due to irreversible ventricularfibrillation which occurred between 20–25 min after the start ofthe coronary ischemic period. In each animal, this was beforetreatment; these animals were excluded from further analysis.
2.5. Ischemic Preconditioning (IPC) experiments
To confirm effectiveness of the dose of LY294002 used, weexamined the capacity of LY294002 to inhibit the cardioprotectiveeffect of IPC, which is mediated by the both the ERK1/2 and Aktpathways of the RISK cascade (Hausenloy and Yellon, 2007). Group1 (n¼3), induction of IPC by 3 cycles of 5-min regional coronaryischemia separated by 5-min reperfusion, followed by 1 h sus-tained ischemia and 30 min reperfusion. Group 2 (n¼2), treat-ment with LY294002 (4 mg IV bolus followed by 10 mg/h over 2 h)started at 10 min before IPC followed by 1 h sustained ischemiaand 30 min reperfusion. Hearts were collected and analyzed asdescribed below.
2.6. Area-at-risk and infarct size determination
The area of myocardium at risk of infarction and infarct size weredelineated by Evan's blue and triphenyltetrazolium chloride (TTC)staining, respectively, as we have previously described (Wang et al.,2004a). Briefly, after 3 h reperfusion, the D2 artery was re-occludedwith the vascular snare. Immediately prior to arresting the heart withan intravenous injection of pentobarbitone (100 mg/kg; Virbac, Aus-tralia), Evan's blue dye (1.5% in 60 ml saline, Sigma, Australia) wasinjected into the left atrial catheter to define the myocardium madeischemic, i.e., the area at risk of infarction. The heart was rapidlyexcised and the left ventricle was sliced into six 1-cm thick transverserings. These were photographed and the unstained areas were tracedonto transparencies. The rings were incubated in 0.1 M sodiumphosphate buffer containing 1% TTC (Sigma, Australia) for 20 min at37 1C, pH 7.4, re-photographed and traced onto transparencies withthe infarcted area outlined. The area of myocardium at risk ofinfarction and the infarct size were measured by computerizedplanimetry (MCID-M 2, Imaging Research Inc., Canada). Analysis wasperformed blinded to treatment group. The myocardium-at-risk wasexpressed as a percentage of total left ventricular volume and infarctsize was expressed as a percentage of the area of myocardium at risk.
2.7. Statistical analysis
All results are expressed as mean7standard error of mean(S.E.M.), and data were analyzed using SigmaStat (Version 2.03,SPSS, USA). Effects of different treatments on MEK1/2, ERK1/2 andAkt in isolated cardiomyocytes were compared by 1-way ANOVA.The effects of time and treatment on hemodynamic and cardiacfunction parameters were analyzed by 2-way repeated measures
ANOVA followed by the Dunnett's post-hoc test for multiplecomparisons. Effects of different treatments on infarct size werecompared by 1-way ANOVA. Statistically significant differenceswere reported when Po0.05.
3. Results
3.1. Effect of DiOHF on activation of ERK1/2 and Akt in cardiacmyocytes
Previously, we had observed in cultured myoblasts a trendtowards increased ERK activation in the presence of DiOHF, theactive parent compound of NP202 (Lim et al., 2013). We extendedour investigation of the effects of DiOHF on RISK pathway kinasesto include primary culture of neonatal rat cardiomyocytes during
H2O2(1 mM, 60 min)Control
DiOHF (10 M)- - - - + + PD98059 (20 M)- + - + - +
phospho-ERK1/2
total-ERK1/2
05
10152025303540
Arb
itrar
y U
nits
(pho
spho
-ER
K/to
tal-E
RK
)ns
*
- 50 kD
- 37 kD- 50 kD
- 37 kD
H2O2(1 mM, 60 min)Control
- + - + DiOHF (10 M)
phospho-MEK1/2
total-MEK1/2
- 50 kD
- 37 kD- 50 kD
- 37 kD
0
5
10
15
20
25 *
0
5
10
15
20
25
Arb
itrar
y U
nits
(pho
spho
-ME
K/to
tal-M
EK
)
µµ
µ
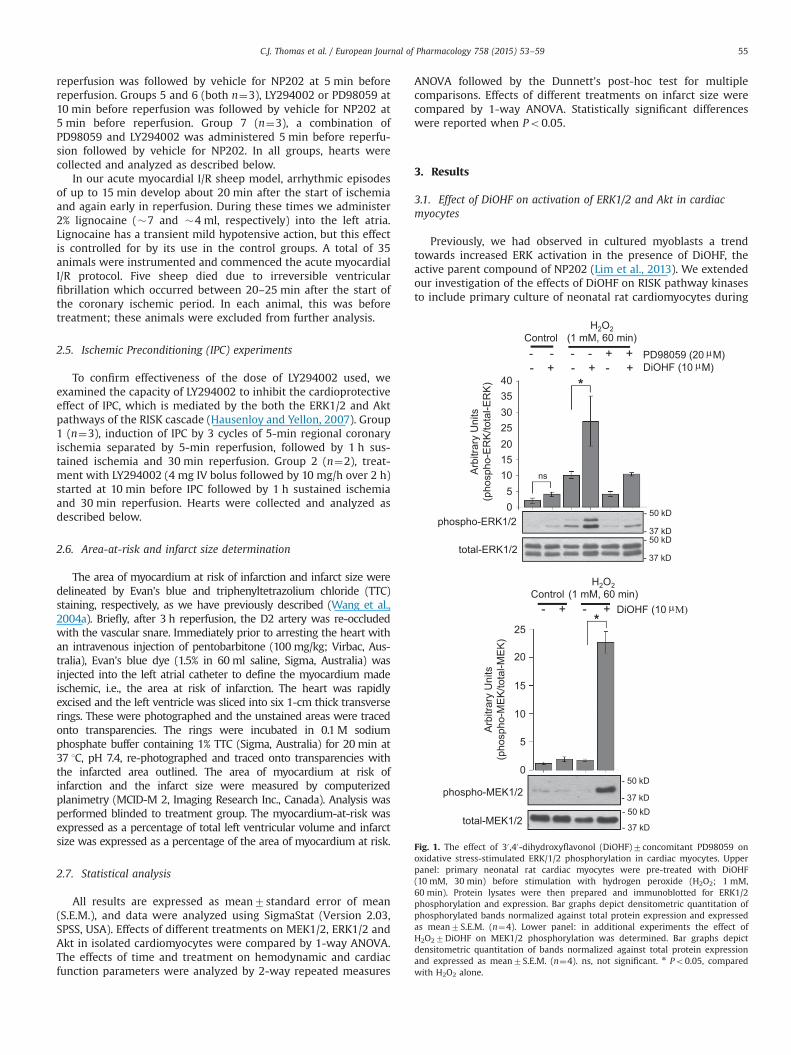
Fig. 1. The effect of 30 ,40-dihydroxyflavonol (DiOHF)7concomitant PD98059 onoxidative stress-stimulated ERK/1/2 phosphorylation in cardiac myocytes. Upperpanel: primary neonatal rat cardiac myocytes were pre-treated with DiOHF(10 mM, 30 min) before stimulation with hydrogen peroxide (H2O2; 1 mM,60 min). Protein lysates were then prepared and immunoblotted for ERK1/2phosphorylation and expression. Bar graphs depict densitometric quantitation ofphosphorylated bands normalized against total protein expression and expressedas mean7S.E.M. (n¼4). Lower panel: in additional experiments the effect ofH2O27DiOHF on MEK1/2 phosphorylation was determined. Bar graphs depictdensitometric quantitation of bands normalized against total protein expressionand expressed as mean7S.E.M. (n¼4). ns, not significant. n Po0.05, comparedwith H2O2 alone.
C.J. Thomas et al. / European Journal of Pharmacology 758 (2015) 53–59 55
oxidative stress stimulated with hydrogen peroxide (H2O2). ERKphosphorylation, indicative of activation, was increased in cardi-omyocytes following H2O2 treatment (Fig. 1, upper panel). We
observed a trend of increased ERK phosphorylation in cardiomyo-cytes in the presence of DiOHF alone (Fig. 1, upper panel). More-over, during oxidative stress, DiOHF significantly enhanced ERKphosphorylation and this was abolished by PD98059, a smallmolecule inhibitor targeting MEK specifically (Fig. 1, upper panel).Consistent with these findings, DiOHF treatment significantlyenhanced MEK phosphorylation stimulated by H2O2 (Fig. 1, lowerpanel). Similarly, DiOHF induced activation of Akt following H2O2
treatment in cardiomyocytes and this was abolished by LY294002(Fig. 2), whereas in the absence of oxidative stress DiOHF did notalter phosphorylated AKT levels in cardiomyocytes (Fig. 2). Takentogether, our studies in cultured cardiomyocytes indicate thatDiOHF substantially augments MEK/ERK and Akt pathway activa-tion, particularly in the context of cellular stress.
3.2. Hemodynamic effects of RISK pathway blockers
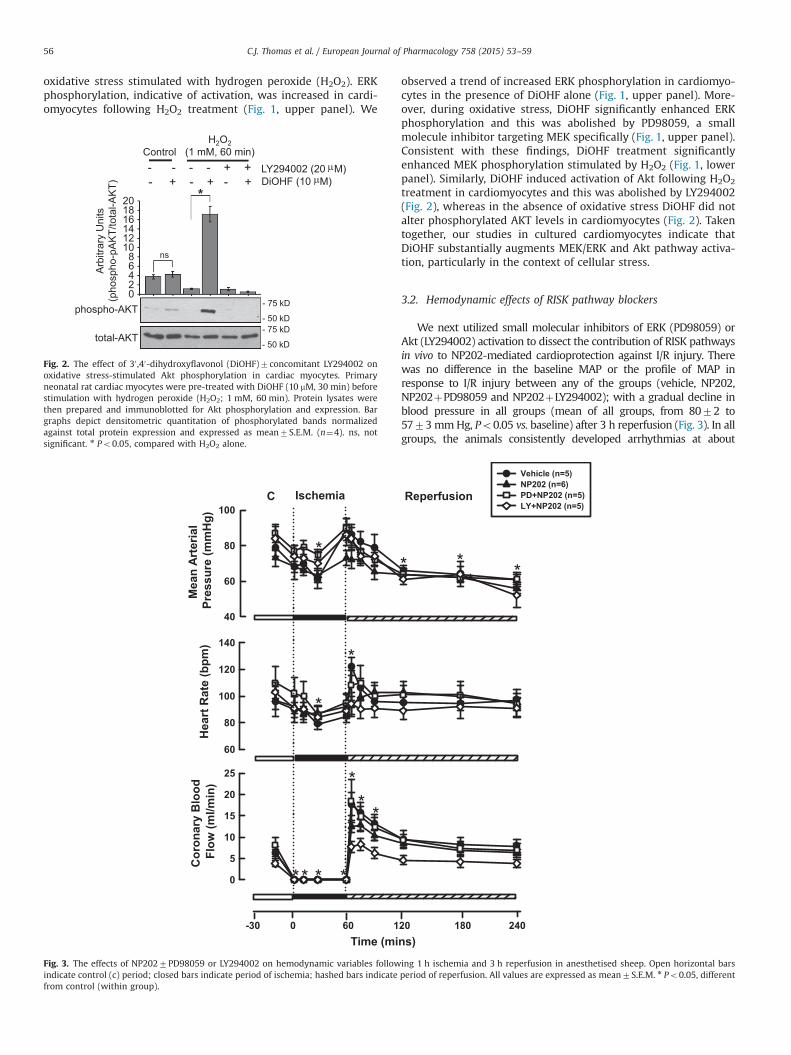
We next utilized small molecular inhibitors of ERK (PD98059) orAkt (LY294002) activation to dissect the contribution of RISK pathwaysin vivo to NP202-mediated cardioprotection against I/R injury. Therewas no difference in the baseline MAP or the profile of MAP inresponse to I/R injury between any of the groups (vehicle, NP202,NP202þPD98059 and NP202þLY294002); with a gradual decline inblood pressure in all groups (mean of all groups, from 8072 to5773mmHg, Po0.05 vs. baseline) after 3 h reperfusion (Fig. 3). In allgroups, the animals consistently developed arrhythmias at about
40
60
80
100
Vehicle (n=5)NP202 (n=6)PD+NP202 (n=5)LY+NP202 (n=5)
0
5
10
15
20
25
Cor
onar
y B
lood
Flow
(ml/m
in)
Time (mins)
Mea
n Ar
teria
l Pr
essu
re (m
mH
g)
ReperfusionIschemiaC
Hea
rt R
ate
(bpm
)
0 60-30 240120 180
60
80
100
120
140
**
* *
*
* ** *
** *
*
*
Fig. 3. The effects of NP2027PD98059 or LY294002 on hemodynamic variables following 1 h ischemia and 3 h reperfusion in anesthetised sheep. Open horizontal barsindicate control (c) period; closed bars indicate period of ischemia; hashed bars indicate period of reperfusion. All values are expressed as mean7S.E.M. n Po0.05, differentfrom control (within group).
H2O2(1 mM, 60 min)Control
DiOHF (10 M)- - - - + + LY294002 (20 M)- + - + - +
phospho-AKT
total-AKT
02468
101214161820
Arb
itrar
y U
nits
(pho
spho
-pA
KT/
tota
l-AK
T)
ns
*
- 75 kD
- 50 kD- 75 kD
- 50 kD
µµ
Fig. 2. The effect of 30 ,40-dihydroxyflavonol (DiOHF)7concomitant LY294002 onoxidative stress-stimulated Akt phosphorylation in cardiac myocytes. Primaryneonatal rat cardiac myocytes were pre-treated with DiOHF (10 mM, 30 min) beforestimulation with hydrogen peroxide (H2O2; 1 mM, 60 min). Protein lysates werethen prepared and immunoblotted for Akt phosphorylation and expression. Bargraphs depict densitometric quantitation of phosphorylated bands normalizedagainst total protein expression and expressed as mean7S.E.M. (n¼4). ns, notsignificant. n Po0.05, compared with H2O2 alone.
C.J. Thomas et al. / European Journal of Pharmacology 758 (2015) 53–5956

20min into the ischemic period. A significant fall in HR was observedin all groups after �30min coronary ischemia, generally coincidingwith the arrhythmic disturbances (Fig. 3). Otherwise, there wereminimal overall changes in HR during the I/R protocol. The zerocoronary flow measurements recorded during the ischemic periodconfirmed complete occlusion of the D2 artery (Fig. 3). A hyperemicresponse to reperfusion (CBF increased to 14.072.0 ml/min, Po0.05)was recorded in all animal groups. CBF gradually returned to baseline,reaching 6.270.6 ml/min at 3 h reperfusion. Overall, there was nosignificant difference in coronary hemodynamics between the differ-ent treatment groups. During myocardial I/R there was no significantchange in LV dP/dt from baseline and this was not altered by any ofthe treatments (data not shown). In sheep treated with LY294002 andPD98059 alone, the hemodynamic responses followed a similarpattern to the other groups (resting MAP, 7373mm Hg and9075mm Hg; resting HR, 9676 bpm and 9375 bpm; resting CBF,6.370.9 ml/min and 6.971.4 ml/min, LY alone and PD alone,respectively).
3.3. Effect of selective RISK pathway blockers on infarct size aftermyocardial I/R injury
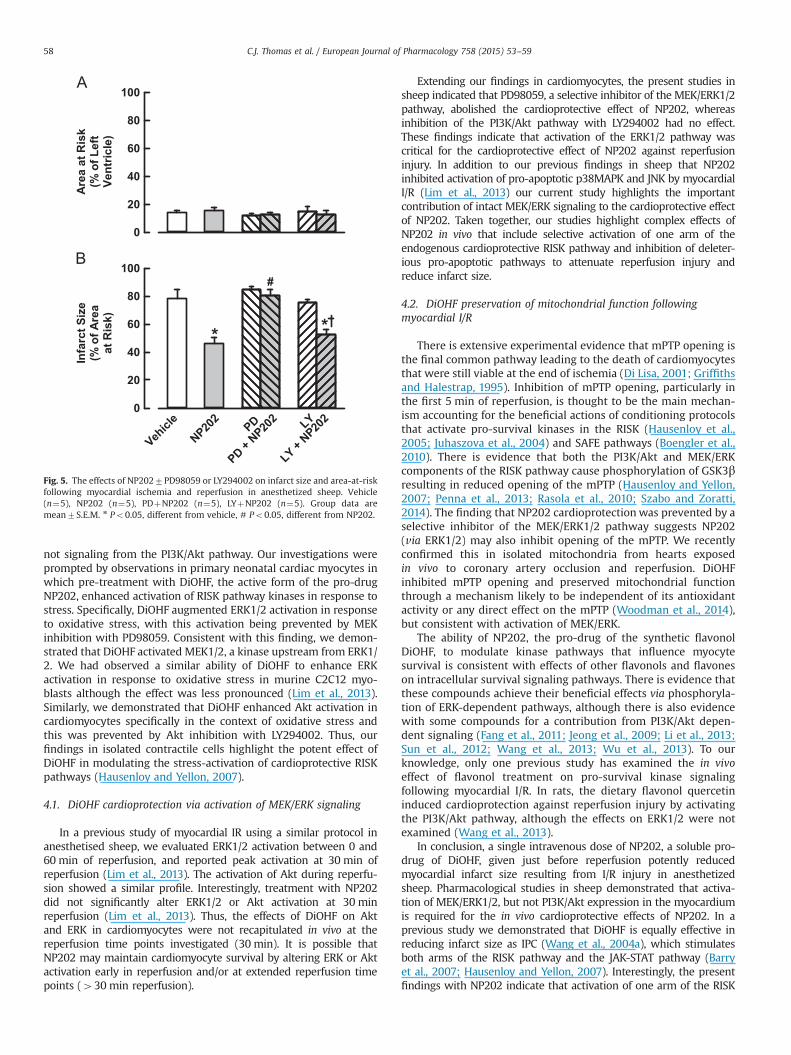
The area of myocardium at risk and infarct size were delineated byEvans blue and TTC staining following 1 h D2 coronary arteryocclusion and 3 h reperfusion (Fig. 4). The area-at-risk, expressed asa percentage of total LV area, was not significantly different betweenvehicle (14.271.0%) and drug treated groups of sheep (average13.771.3%) (Fig. 5). Infarct area was reduced from 7876% of the area
at risk with vehicle treatment, to 4674% with NP202 (Po0.05)(Fig. 5). Treatment with PD98059 abolished the cardioprotective effectof NP202; infarct size was similar to that with vehicle treatment(Fig. 5). In contrast, treatment with LY294002 did not prevent theNP202-induced reduction in infarct size, with infarct size being similarto that with NP202 alone (Fig. 5). Infarct size following myocardial IRwas not reduced by treatment with LY294002 alone (7572%),PD98059 alone (8572%) or a combination of both drugs (8572%),compared with vehicle (7876%).
To validate the efficacy of LY294002 treatment in vivo, as apositive control we investigated the effect of the LY294002 dosewe used on the cardioprotective effect of IPC. We induced IPCwith three 5 min periods of ischemia of the 2D coronary arteryinterspersed with 5 min reperfusion, prior to 60 min ischemiaand 3 h reperfusion. We found that LY294002 markedly atte-nuated the cardioprotective effect of IPC. Infarct size wasincreased from 3876% (n¼3) in the IPC treated group to 67%(n¼2) in the IPC group treated with LY294002. Since IPC ismediated via activation of both the ERK1/2 and Akt kinasepathways (Hausenloy and Yellon, 2007), our data provide strongevidence that the dose of LY294002 used effectively inhibitedPI3K/Akt activation in vivo.
4. Discussion
The main finding was that the cardioprotective effect of NP202in a large animal model of I/R injury required MEK/ERK activity, but
NP202
NP202+LY
Vehicle
NP202+PDFig. 4. Cross section of sheep left ventricle rings following 1 h ischemia and 3 h reperfusion. Top panel, representative digital photos showing effect of NP202 treatment inreducing infarct size (dotted line, following TTC staining) relative to the area-at-risk (solid white outline, following Evans Blue staining) compared to vehicle. Bottom panel,representative photos showing differential effect of NP2027PD98059 or LY294002 on infarct size. TTC, triphenyltetrazolium chloride.
C.J. Thomas et al. / European Journal of Pharmacology 758 (2015) 53–59 57
not signaling from the PI3K/Akt pathway. Our investigations wereprompted by observations in primary neonatal cardiac myocytes inwhich pre-treatment with DiOHF, the active form of the pro-drugNP202, enhanced activation of RISK pathway kinases in response tostress. Specifically, DiOHF augmented ERK1/2 activation in responseto oxidative stress, with this activation being prevented by MEKinhibition with PD98059. Consistent with this finding, we demon-strated that DiOHF activated MEK1/2, a kinase upstream from ERK1/2. We had observed a similar ability of DiOHF to enhance ERKactivation in response to oxidative stress in murine C2C12 myo-blasts although the effect was less pronounced (Lim et al., 2013).Similarly, we demonstrated that DiOHF enhanced Akt activation incardiomyocytes specifically in the context of oxidative stress andthis was prevented by Akt inhibition with LY294002. Thus, ourfindings in isolated contractile cells highlight the potent effect ofDiOHF in modulating the stress-activation of cardioprotective RISKpathways (Hausenloy and Yellon, 2007).
4.1. DiOHF cardioprotection via activation of MEK/ERK signaling
In a previous study of myocardial IR using a similar protocol inanesthetised sheep, we evaluated ERK1/2 activation between 0 and60 min of reperfusion, and reported peak activation at 30 min ofreperfusion (Lim et al., 2013). The activation of Akt during reperfu-sion showed a similar profile. Interestingly, treatment with NP202did not significantly alter ERK1/2 or Akt activation at 30 minreperfusion (Lim et al., 2013). Thus, the effects of DiOHF on Aktand ERK in cardiomyocytes were not recapitulated in vivo at thereperfusion time points investigated (30 min). It is possible thatNP202 may maintain cardiomyocyte survival by altering ERK or Aktactivation early in reperfusion and/or at extended reperfusion timepoints (430 min reperfusion).
Extending our findings in cardiomyocytes, the present studies insheep indicated that PD98059, a selective inhibitor of the MEK/ERK1/2pathway, abolished the cardioprotective effect of NP202, whereasinhibition of the PI3K/Akt pathway with LY294002 had no effect.These findings indicate that activation of the ERK1/2 pathway wascritical for the cardioprotective effect of NP202 against reperfusioninjury. In addition to our previous findings in sheep that NP202inhibited activation of pro-apoptotic p38MAPK and JNK by myocardialI/R (Lim et al., 2013) our current study highlights the importantcontribution of intact MEK/ERK signaling to the cardioprotective effectof NP202. Taken together, our studies highlight complex effects ofNP202 in vivo that include selective activation of one arm of theendogenous cardioprotective RISK pathway and inhibition of deleter-ious pro-apoptotic pathways to attenuate reperfusion injury andreduce infarct size.
4.2. DiOHF preservation of mitochondrial function followingmyocardial I/R
There is extensive experimental evidence that mPTP opening isthe final common pathway leading to the death of cardiomyocytesthat were still viable at the end of ischemia (Di Lisa, 2001; Griffithsand Halestrap, 1995). Inhibition of mPTP opening, particularly inthe first 5 min of reperfusion, is thought to be the main mechan-ism accounting for the beneficial actions of conditioning protocolsthat activate pro-survival kinases in the RISK (Hausenloy et al.,2005; Juhaszova et al., 2004) and SAFE pathways (Boengler et al.,2010). There is evidence that both the PI3K/Akt and MEK/ERKcomponents of the RISK pathway cause phosphorylation of GSK3βresulting in reduced opening of the mPTP (Hausenloy and Yellon,2007; Penna et al., 2013; Rasola et al., 2010; Szabo and Zoratti,2014). The finding that NP202 cardioprotectionwas prevented by aselective inhibitor of the MEK/ERK1/2 pathway suggests NP202(via ERK1/2) may also inhibit opening of the mPTP. We recentlyconfirmed this in isolated mitochondria from hearts exposedin vivo to coronary artery occlusion and reperfusion. DiOHFinhibited mPTP opening and preserved mitochondrial functionthrough a mechanism likely to be independent of its antioxidantactivity or any direct effect on the mPTP (Woodman et al., 2014),but consistent with activation of MEK/ERK.
The ability of NP202, the pro-drug of the synthetic flavonolDiOHF, to modulate kinase pathways that influence myocytesurvival is consistent with effects of other flavonols and flavoneson intracellular survival signaling pathways. There is evidence thatthese compounds achieve their beneficial effects via phosphoryla-tion of ERK-dependent pathways, although there is also evidencewith some compounds for a contribution from PI3K/Akt depen-dent signaling (Fang et al., 2011; Jeong et al., 2009; Li et al., 2013;Sun et al., 2012; Wang et al., 2013; Wu et al., 2013). To ourknowledge, only one previous study has examined the in vivoeffect of flavonol treatment on pro-survival kinase signalingfollowing myocardial I/R. In rats, the dietary flavonol quercetininduced cardioprotection against reperfusion injury by activatingthe PI3K/Akt pathway, although the effects on ERK1/2 were notexamined (Wang et al., 2013).
In conclusion, a single intravenous dose of NP202, a soluble pro-drug of DiOHF, given just before reperfusion potently reducedmyocardial infarct size resulting from I/R injury in anesthetizedsheep. Pharmacological studies in sheep demonstrated that activa-tion of MEK/ERK1/2, but not PI3K/Akt expression in the myocardiumis required for the in vivo cardioprotective effects of NP202. In aprevious study we demonstrated that DiOHF is equally effective inreducing infarct size as IPC (Wang et al., 2004a), which stimulatesboth arms of the RISK pathway and the JAK-STAT pathway (Barryet al., 2007; Hausenloy and Yellon, 2007). Interestingly, the presentfindings with NP202 indicate that activation of one arm of the RISK
0
20
40
60
80
100
0
20
40
60
80
100
Area
at R
isk
(% o
f Lef
tVe
ntric
le)
Infa
rct S
ize
(% o
f Are
aat
Ris
k)
Vehicl
e
NP202
PD + NP20
2PD
LY + NP20
2LY
* *
#
Fig. 5. The effects of NP2027PD98059 or LY294002 on infarct size and area-at-riskfollowing myocardial ischemia and reperfusion in anesthetized sheep. Vehicle(n¼5), NP202 (n¼5), PDþNP202 (n¼5), LYþNP202 (n¼5). Group data aremean7S.E.M. n Po0.05, different from vehicle, # Po0.05, different from NP202.
C.J. Thomas et al. / European Journal of Pharmacology 758 (2015) 53–5958

pathway is sufficient to induce a similarly high level of cardioprotec-tion. The ability of NP202 to activate this endogenous pro-survivalsignaling cascade in the heart and reduce infarct size indicates itstherapeutic potential as an adjunct treatment for myocardial I/Rinjury.
Funding
This work was supported by a Grant-in Aid from the NationalHeart Foundation of Australia (G 08M 3761). CNM is supported bya Research Fellowship from the National Health and MedicalResearch Council of Australia NHMRC ID, (566819). DCHN issupported by an Australian Research Council Future Fellowship(FT120100193).
Disclosures
O.L. Woodman and C.N. May are shareholders in NeuProtectPty. Ltd., a company which holds a patent for use of flavonols as atreatment for myocardial ischaemia–reperfusion injury.
Acknowledgments
We thank Alan McDonald and Tony Dornom for their experttechnical assistance. NeuProtect Pty. Ltd. generously provided NP202.
References
Barry, S.P., Townsend, P.A., Latchman, D.S., Stephanou, A., 2007. Role of the JAK–STAT pathway in myocardial injury. Trends Mol. Med. 13, 82–89.
Boengler, K., Hilfiker-Kleiner, D., Heusch, G., Schulz, R., 2010. Inhibition of perme-ability transition pore opening by mitochondrial STAT3 and its role inmyocardial ischemia/reperfusion. Basic Res. Cardiol. 105, 771–785.
Braunwald, E., Kloner, R.A., 1985. Myocardial reperfusion: a double-edged sword?J. Clin. Investig. 76, 1713–1719.
Di Lisa, F., 2001. Mitochondrial contribution in the progression of cardiac ischemicinjury. IUBMB Life 52, 255–261.
Fang, F., Li, D., Pan, H., Chen, D., Qi, L., Zhang, R., Sun, H., 2011. Luteolin inhibitsapoptosis and improves cardiomyocyte contractile function through the PI3K/Akt pathway in simulated ischemia/reperfusion. Pharmacology 88, 149–158.
Griffiths, E.J., Halestrap, A.P., 1995. Mitochondrial non-specific pores remain closedduring cardiac ischaemia, but open upon reperfusion. Biochem. J. 307 (Pt 1),93–98.
Hausenloy, D.J., Tsang, A., Yellon, D.M., 2005. The reperfusion injury salvage kinasepathway: a common target for both ischemic preconditioning and postcondi-tioning. Trends Cardiovasc. Med. 15, 69–75.
Hausenloy, D.J., Yellon, D.M., 2007. Reperfusion injury salvage kinase signaling:taking a RISK for cardioprotection. Heart Fail. Rev. 12, 217–234.
Hausenloy, D.J., Yellon, D.M., 2013. Myocardial ischemia–reperfusion injury: aneglected therapeutic target. J. Clin. Investig. 123, 92–100.
Jeong, J.J., Ha, Y.M., Jin, Y.C., Lee, E.J., Kim, J.S., Kim, H.J., Seo, H.G., Lee, J.H., Kang, S.S.,Kim, Y.S., Chang, K.C., 2009. Rutin from Lonicera japonica inhibits myocardialischemia/reperfusion-induced apoptosis in vivo and protects H9c2 cells againsthydrogen peroxide-mediated injury via ERK1/2 and PI3K/Akt signals in vitro.Food Chem. Toxicol. 47, 1569–1576.
Juhaszova, M., Zorov, D.B., Kim, S.H., Pepe, S., Fu, Q., Fishbein, K.W., Ziman, B.D.,Wang, S., Ytrehus, K., Antos, C.L., Olson, E.N., Sollott, S.J., 2004. Glycogen
synthase kinase-3beta mediates convergence of protection signaling to inhibitthe mitochondrial permeability transition pore. J. Clin. Investig. 113, 1535–1549.
Kharbanda, R.K., 2010. Cardiac conditioning: a review of evolving strategies toreduce ischemia-reperfusion injury. Heart 96, 1179–1186.
Lash, L.H. (Ed.), 2005. Drug Metabolism and Transport: Molecular Methods andMechanisms. Humana Press Inc., Portland USA.
Li, Z.L., Hu, J., Li, Y.L., Xue, F., Zhang, L., Xie, J.Q., Liu, Z.H., Li, H., Yi, D.H., Liu, J.C.,Wang, S.W., 2013. The effect of hyperoside on the functional recovery of theischemic/reperfused isolated rat heart: potential involvement of the extracel-lular signal-regulated kinase 1/2 signaling pathway. Free Radic. Biol. Med. 57,132–140.
Lim, N.R., Thomas, C.J., Silva, L.S., Yeap, Y.Y., Yap, S., Bell, J.R., Delbridge, L.M.,Bogoyevitch, M.A., Woodman, O.L., Williams, S.J., May, C.N., Ng, D.C., 2013.Cardioprotective 30 ,40-dihydroxyflavonol attenuation of JNK and p38(MAPK)signaling involves CaMKII inhibition. Biochem. J. 456, 149–161.
Ng, D.C., Long, C.S., Bogoyevitch, M.A., 2001. A role for the extracellular signal-regulated kinase and p38 mitogen-activated protein kinases in interleukin-1beta-stimulated delayed signal tranducer and activator of transcription 3 acti-vation, atrial natriuretic factor expression, and cardiac myocyte morphology.J. Biol. Chem. 276, 29490–29498.
Ng, D.C., Zhao, T.T., Yeap, Y.Y., Ngoei, K.R., Bogoyevitch, M.A., 2010. c-Jun N-terminalkinase phosphorylation of stathmin confers protection against cellular stress.J. Biol. Chem. 285, 29001–29013.
Penna, C., Perrelli, M.G., Pagliaro, P., 2013. Mitochondrial pathways, permeabilitytransition pore, and redox signaling in cardioprotection: therapeutic implica-tions. Antioxid. Redox Signal. 18, 556–599.
Rasola, A., Sciacovelli, M., Pantic, B., Bernardi, P., 2010. Signal transduction to thepermeability transition pore. FEBS Lett. 584, 1989–1996.
Sun, B., Sun, G.B., Xiao, J., Chen, R.C., Wang, X., Wu, Y., Cao, L., Yang, Z.H., Sun, X.B.,2012. Isorhamnetin inhibits H(2)O(2)-induced activation of the intrinsic apop-totic pathway in H9c2 cardiomyocytes through scavenging reactive oxygenspecies and ERK inactivation. J. Cell. Biochem. 113, 473–485.
Szabo, I., Zoratti, M., 2014. Mitochondrial channels: ion fluxes and more. Physiol.Rev. 94, 519–608.
Thomas, C.J., Ng, D.C., Patsikatheodorou, N., Limengka, Y., Lee, M.W., Darby, I.A.,Woodman, O.L., May, C.N., 2011. Cardioprotection from ischaemia–reperfusioninjury by a novel flavonol that reduces activation of p38 MAPK. Eur.J. Pharmacol. 658, 160–167.
Wang, S., Dusting, G.J., May, C.N., Woodman, O.L., 2004a. 30 ,40-Dihydroxyflavonolreduces infarct size and injury associated with myocardial ischaemia andreperfusion in sheep. Br. J. Pharmacol. 142, 443–452.
Wang, S., Dusting, G.J., Woodman, O.L., May, C.N., 2004b. Selective vasodilator andchronotropic actions of 30 ,40-dihydroxyflavonol in conscious sheep. Eur.J. Pharmacol. 491, 43–51.
Wang, S., Fei, K., Xu, Y.W., Wang, L.X., Chen, Y.Q., 2009. Dihydroxyflavonol reducespost-infarction left ventricular remodeling by preventing myocyte apoptosis inthe non-infarcted zone in goats. Chin. Med. J. 122, 61–67.
Wang, Y., Zhang, Z.Z., Wu, Y., Ke, J.J., He, X.H., Wang, Y.L., 2013. Quercetinpostconditioning attenuates myocardial ischemia/reperfusion injury in ratsthrough the PI3K/Akt pathway. Braz. J. Med. Biol. Res. 46, 861–867.
Williams, S.J., Thomas, C.J., Boujaoude, M., Gannon, C.T., Zanatta, S.D., Jarrott, B.,May, C.N., Woodman, O.L., 2011. Water soluble flavonol prodrugs protectagainst reperfusion injury. Med. Chem. Comm. 2, 321–324.
Woodman, O.L., Chan, E., 2004. Vascular and anti-oxidant actions of flavonols andflavones. Clin. Exp. Pharmacol. Physiol. 31, 786–790.
Woodman, O.L., Long, R., Pons, S., Eychenne, N., Berdeaux, A., Morin, D., 2014. Thecardioprotectant 30 ,40-dihydroxyflavonol inhibits opening of the mitochondiralpermeability transition pore after myocardial ischemia and reperfusion in rats.Pharmacol. Res. 81, 26–33.
Wu, X., Xu, T., Li, D., Zhu, S., Chen, Q., Hu, W., Pan, D., Zhu, H., Sun, H., 2013. ERK/PP1a/PLB/SERCA2a and JNK pathways are involved in luteolin-mediated pro-tection of rat hearts and cardiomyocytes following ischemia/reperfusion. PLoSOne 8, e82957.
Yellon, D.M., Hausenloy, D.J., 2007. Myocardial reperfusion injury. N. Engl. J. Med.357, 1121–1135.
Zhang, J., Bian, H.J., Li, X.X., Liu, X.B., Sun, J.P., Li, N., Zhang, Y., Ji, X.P., 2010. ERK-MAPK signaling opposes rho-kinase to reduce cardiomyocyte apoptosis in heartischemic preconditioning. Mol. Med. 16, 307–315.
C.J. Thomas et al. / European Journal of Pharmacology 758 (2015) 53–59 59
Copyright © 2022 FDOKUMEN




















