
Epstein-Barr virus and carcinomas: rare association of the virus with gastric adenocarcinomas
Upload
independentCategory
view
1download
0
JOURNAL OF VIROLOGY, Apr. 2005, p. 4415–4424 Vol. 79, No. 70022-538X/05/$08.00�0 doi:10.1128/JVI.79.7.4415–4424.2005Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Epstein-Barr Virus Transforming Protein LMP1 Plays aCritical Role in Virus Production
Nazmul Ahsan,1 Teru Kanda,2 Kazuo Nagashima,3 and Kenzo Takada1*Department of Tumor Virology1 and Center for Virus Vector Development,2 Institute for Genetic Medicine,
Hokkaido University, and Laboratory of Molecular and Cellular Pathology, Hokkaido UniversityGraduate School of Medicine,3 Sapporo, Japan
Received 23 July 2004/Accepted 9 November 2004
The Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1), which is critical for EBV-induced B-celltransformation, is also abundantly expressed during the lytic cycle of viral replication. However, the biologicalsignificance of this strong LMP1 induction remains unknown. We engineered a bacterial artificial chromosomeclone containing the entire genome of Akata strain EBV to specifically disrupt the LMP1 gene. Akata cell clonesharboring the episomes of LMP1-deleted EBV were established, and the effect of LMP1 loss on virus produc-tion was investigated. We found that the degree of viral DNA amplification and the expression levels of virallate gene products were unaffected by LMP1 loss, demonstrating that the LMP1-deleted EBV entered the lyticreplication cycle as efficiently as the wild-type counterpart. This was confirmed by our electron microscopicobservation that nucleocapsid formation inside nuclei occurred even in the absence of LMP1. By contrast, lossof LMP1 severely impaired virus release into culture supernatants, resulting in poor infection efficiency. Theexpression of truncated LMP1 in Akata cells harboring LMP1-deleted EBV rescued the virus release into theculture supernatant and the infectivity, and full-length LMP1 partially rescued the infectivity. These resultsindicate that inducible expression of LMP1 during the viral lytic cycle plays a critical role in virus production.
Epstein-Barr virus (EBV) is a human herpesvirus that es-tablishes a life-long persistence in the host (33). The virus in-fects the vast majority of the world’s adult population and iswell known for its association with a broad spectrum of benignand malignant diseases, including infectious mononucleosis,Burkitt’s lymphoma, nasopharyngeal carcinoma, B-cell lym-phoma in immunocompromised patients, and gastric carci-noma (33). In vitro EBV infection of peripheral B lymphocytesresults in their immortalization and continuous proliferation(39). In immortalized lymphoblastoid cell lines, EBV expressessix nuclear proteins (EBNAs), three integral membrane pro-teins (LMP1, -2A, and -2B), two small nuclear RNAs (EBER1and -2), and a transcript from the BamHI-A region (21).
Latent membrane protein 1 (LMP1) is one of the viral geneproducts that are essential for B-cell transformation (19). LMP1is an integral membrane protein composed of a short cytoplas-mic amino-terminal domain, six hydrophobic transmembranedomains, and a cytoplasmic carboxy-terminal domain (10).It has been demonstrated that LMP1 acts as a constitutivelyactive receptor that mimics activated CD40, a member of thetumor necrosis factor receptor family (12, 26). The cytoplasmiccarboxy terminus of LMP1 plays a critical role in EBV-inducedB-cell transformation through its binding to a tumor necrosisfactor receptor-associated factor (TRAF) and a tumor necrosisfactor receptor-associated death domain (TRADD) protein (6,21).
The LMP1 (BNLF1) gene contains three exons that arelocated within the BamHI-N region of the EBV genome (10).Two open reading frames (ORFs) have been identified based
on nucleotide sequences (2, 10) and mRNA mapping (16) ofB95-8 strain EBV. A transcript starting from the ED-L1 pro-moter, which is located upstream of the first exon (16), encodesthe first ORF. This ORF encodes full-length LMP1 (386 aminoacids) that is abundantly expressed in lymphoblastoid cell lines.Another transcript starting from the EDL1A promoter, whichis located within the first intron of the LMP1 gene, encodes thesecond ORF. The translation initiation site of this second ORFis methionine-129 of full-length LMP1 (16), and the ORF thusencodes an amino-terminally truncated form of the LMP1 pro-tein. The truncated LMP1 (258 amino acids) consists of thefifth and sixth transmembrane domains and the cytoplasmiccarboxy terminus of full-length LMP1.
The B95-8 cell line, an in vitro-immortalized lymphoblastoidcell line, was initially used to characterize truncated LMP1. Anumber of studies demonstrated that expression of truncatedLMP1 was specifically upregulated during the lytic phase of vi-ral replication (4, 5, 16, 28, 34, 44). Therefore, truncated formof LMP1 is frequently referred to as lytic LMP1 (8, 43). Incontrast to full-length LMP1, truncated LMP1 does not trans-form rodent cells (3, 44) or alter the phenotypes of human Blymphocytes (45). The only biological activity of truncatedLMP1 identified to date is its ability to negatively regulateLMP1 signaling pathways (9), although truncated LMP1 re-tains the carboxy terminus of LMP1.
Recent studies revealed that Met-129 of the B95-8 LMP1ORF is not common in many virus isolates (7, 42). For exam-ple, the LMP1 gene of Akata strain EBV encodes isoleucine atcodon 129, resulting in loss of the translation initiation codonfor the ORF of truncated LMP1. The Akata cell line is derivedfrom an EBV-positive Burkitt’s lymphoma from a Japanesepatient (41). LMP1 expression is not detected in unstimulatedAkata cells (42), as Akata cells retain the Burkitt’s-type viral
* Corresponding author. Mailing address: Department of TumorVirology, Institute for Genetic Medicine, Hokkaido University, N15W7, Kita-ku, Sapporo 060-0815, Japan. Phone: 81-11-706-5071. Fax:81-11-706-7540. E-mail: [email protected].
4415
on January 4, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from

gene expression (36). A unique feature of Akata cells is thatcross-linking of cell surface immunoglobulin with anti-immu-noglobulin antibodies can efficiently induce the lytic cycle ofviral replication (40). Interestingly, expression of full-lengthLMP1 is strongly upregulated after anti-immunoglobulin treat-ment of Akata cells (42). The induction of full-length LMP1from Met-129-negative EBV was also observed in Raji andBL74 cells after tetradecanoyl phorbol acetate and butyratetreatment (5, 7). Therefore, the induction of full-length LMP1after stimulating virus production is not peculiar to Akata cells.
These observations prompted us to investigate the biologicalsignificance of LMP1 upregulation during the lytic cycle ofviral replication in Akata cells. We therefore set out to gener-ate a recombinant Akata EBV containing the disrupted LMP1gene. We recently reported the establishment of a bacterialartificial chromosome (BAC) system for engineering the ge-nome of Akata strain EBV and used the system to producehigh-titer recombinant viruses (18). We employed the systemto disrupt the LMP1 gene of Akata strain EBV and investi-gated whether Akata cells harboring the episomes of LMP1-deleted EBV (d.LMP1-EBV) could produce infectious viruses.We found that the d.LMP1-EBV entered the lytic replicationcycle as efficiently as the wild-type counterpart. However, weunexpectedly found that loss of LMP1 severely impaired virusrelease into the culture supernatant, resulting in poor infectionefficiency. We expressed either truncated LMP1 or full-lengthLMP1 in Akata cells harboring d.LMP1-EBV to rescue the in-fectivity of the culture supernatants. The results revealed thatloss of LMP1 is truly responsible for the impaired virus release.
MATERIALS AND METHODS
Cell culture. Akata, a human Burkitt’s lymphoma-derived cell line carryingEBV as episomes (41) and EBV-negative Akata cells (36) were maintained inRPMI 1640 medium (Sigma-Aldrich Fine Chemicals, St. Louis, Mo.) containing10% fetal bovine serum at 37°C in a 5% humidified CO2 atmosphere.
Plasmids. A plasmid, pBS246 (Life Technologies), having two loxP sites andmultiple cloning sites, was used for excising a loxP cassette. Two loxP sites of pBS246,one located between BspEI and SpeI and the other between EcoRI and Acc65I,were replaced with mutated loxP sites (5171 loxP) with 43-bp synthetic oligonucle-otides to construct pBS246-mloxp5171-EXA. The 5171 loxP site has the substitutionof two nucleotide pairs within the asymmetric 8-nucleotide spacer region of the loxPsequence, and therefore it barely recombines with the wild-type loxP site (23).
A synthetic oligonucleotide containing triple stop codons and restriction en-zyme sites (BamHI, NotI, and SalI) was inserted into the SfiI site (after codon 9of the LMP1 gene) of pUC119-Nhet (pUC119 containing the BamHI-Nhetfragment of the Akata EBV genome) to construct pUC119-Nhet-del.LMP1.
The blunted FokI-BclI fragment of pCDNA4/HisMax (Invitrogen, Carlsbad,Calif.) containing the zeocin resistance marker gene (zeo) was cloned into theEcoRV site of pBS246-mloxp5171-EXA to make pBS246-mloxp-zeo. The NotIfragment of pBS246-mloxp-zeo containing the zeocin resistance marker flankedby two 5171 loxP sites was then cloned into the NotI site of pUC119-Nhet-del.LMP1 to construct pUC119-Nhet-del.LMP1-zeo.
To construct a plasmid expressing full-length LMP1, an LMP1 cDNA of Akatastrain EBV was cloned into the pGEM-T easy vector (Promega) and verified byDNA sequencing. The LMP1 cDNA was subcloned into the pSG5 vector (Strat-agene), which was then transferred to a vector equipped with a hygromycin re-sistance marker gene to construct pSG5-AK-LMP1-Hyg.
The sequences of oligonucleotides used for constructions are available uponrequest.
Electrotransformation of E. coli. Electrocompetent Escherichia coli DH10Bbacterial cells were prepared as described previously (35). The Bio-Rad GenePulser II electroporation system (0.1-cm cuvette, 1.25 kV, 25 �F, 100 �, 40 �l ofcells) was used for electrotransformation.
Construction of d.LMP1-EBV. Construction of d.LMP1-EBV was performedin E. coli with Red�, RecE, RecT (GET) recombination (29, 31) as described(18). Plasmid pUC119-Nhet-del.LMP1-zeo was PCR amplified with primers
NA03 (5�-AGAGCAAGGCCTATGGAAGAGGAG-3�) and NA04 (5�-CCTCAGTTGCCTTGCTCCTGCCAC-3�) to obtain a linear targeting construct re-quired for GET recombination. The resultant PCR product (912 bp long) had60-bp (5� end) and 62-bp (3� end) sequences homologous to the target region ofAK-BAC-GFP (18). The PCR product obtained was gel purified and DpnIdigested, and 200 ng of the linear PCR product was electroporated into recom-binase-induced DH10B electrocompetent E. coli (harboring AK-BAC-GFP andpGETrec). Colonies of recombinant BAC clones were identified by plating cellson Luria-Bertani (LB) plates containing chloramphenicol (12.5 �g/ml) and zeo-cin (20 �g/ml). The modified BAC clone (d.LMP1-EBV-zeo) was purified frompGETrec by miniprep DNA isolation and electroporation into E. coli DH10B.
To remove the zeocin resistance marker, the miniprep DNA of d.LMP1-EBV-zeo was treated by Cre recombinase (Novagen) in vitro according to the manu-facturer’s instructions. One microliter of the reaction mixture was then used totransform electrocompetent E. coli DH10B, followed by selection on LB platescontaining chloramphenicol. Bacterial clones of d.LMP1-EBV which had thezeocin marker gene deleted were chosen by checking their sensitivity to zeocin.
BACmid preparation and DNA analysis. For minipreparations, each BACplasmid (BACmid) was isolated from 1.5 ml of a bacterial culture with analkaline lysis protocol (Current Protocols in Human Genetics, Unit 5.15. Con-struction of Bacterial Artificial Chromosome BAC/PAC libraries, found at http://murdoch.rch.unimelb.edu.au/). For maxipreparations, BACmid DNA was iso-lated from each 500-ml bacterial culture with the Nucleobond BAC 100 kit(Macherey-Nagel, Duren, Germany). BACmid DNAs digested with restrictionenzymes were resolved by 0.8% agarose gel electrophoresis for 16 h at 40 V andvisualized by ethidium bromide staining.
Transfection of BACmids into Akata cells. Akata cells (5.0 � 106) weretransfected with 10 �g of d.LMP1-EBV DNA (isolated by maxipreparation) viaelectroporation (Bio-Rad Gene Pulser II; 0.190 kV, 950 �F). Transfected cellswere resuspended in 5 ml of culture medium and plated into six-well dishes. At2 days posttransfection, cells were plated at 104 cells per well in 96-well tissueculture plates in medium containing 375 �g of G418 (Sigma) per ml. Half of theculture medium was replaced with fresh G418-containing medium every 5 daysuntil G418-resistant cell clones emerged (3 to 4 weeks after plating). EpisomalDNA fractions were prepared from G418-resistant cell clones by an alkaline lysisprocedure as described previously (18, 38). The purified episomal DNAs wereused to transform electrocompetent E. coli DH10B to rescue episomally main-tained BACmids as bacterial clones. Out of 39 G418-resistant cell clones, 21 cellclones contained episomally maintained BACmids, and two cell clones containedBACmids identical to the transfected d.LMP1-EBV.
Southern blot analysis. Genomic DNAs were extracted by the standard pro-teinase K-sodium dodecyl sulfate method, followed by phenol-chloroform ex-traction and ethanol precipitation. Southern blotting was performed as previ-ously described (27). The BamHI X and EcoRI K fragments of the Akata strainEBV genome were used as probes for Southern blotting.
Immunofluorescence. Cells were smeared on glass slides and fixed with ace-tone for 2 min. Indirect immunofluorescence was performed with either mono-clonal antibody Cl.50-1 (specific to gp110) or monoclonal antibody C-1 (specificto gp350) as primary antibodies and a Cy3-conjugated anti-mouse immunoglob-ulin G (IgG) (Jackson ImmunoResearch) as a secondary antibody.
Immunoblots. Cells (1.0 � 106) were harvested before and after inducing virusproduction (48 h after starting anti-IgG treatment) and suspended in 200 �l oflow-salt buffer. The cell suspensions were sonicated and centrifuged at 10,000rpm at 4°C for 5 min, and the supernatant fractions were collected. Ten micro-liters of each protein sample was resolved in sodium dodecyl sulfate–8.0%polyacrylamide gels and blotted onto nitrocellulose membranes. The expressionof LMP1 was detected with monoclonal antibody S12 (specific to LMP1) (25, 45)as a primary antibody and horseradish peroxidase-conjugated anti-mouse IgG asa secondary antibody. Signals were detected by an enhanced chemiluminescencemethod (Amersham) according to the manufacturer’s protocol.
Rescue experiments via LMP1 expression. For transient expression, Akatacells harboring d.LMP1-EBV episomes (d.LMP1-EBV cells) (5.0 � 106 cells)were transfected with 50 �g of plasmid pSG5-�XLMP1 with a BTX electropo-rator (BTX, San Diego, Calif.) at 1,000 �F and 200 V. To get cell clones withconstitutive expression of truncated LMP1, cells were cotransfected with 50 �gof pSG5-�XLMP1 (24) and 5 �g of the hygromycin B phosphotransferaseexpression vector. For constitutively expressing full-length LMP1, cells weretransfected with 30 �g of plasmid pSG5-AK-LMP1-Hyg by electroporation. At 2days posttransfection, transfected cells were plated at 104 cells per well in 96-welltissue culture plates in 200 �l of complete medium containing 250 �g of hygro-mycin B (Calbiochem) and 700 �g of G418 per ml. Cells were fed with freshselective medium every 5 days until hygromycin B-resistant cell clones emerged(3 to 4 weeks after plating).
4416 AHSAN ET AL. J. VIROL.
on January 4, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from

Virus production, infection, and preparation of viral DNA. Akata cells har-boring EBV episomes (2.0 � 106) were resuspended in 1 ml of fresh mediumcontaining 0.5% rabbit anti-human IgG (DakoCytomation, Carpinteria, Calif.)and incubated for 8 to 12 h. The culture medium was replaced with fresh medium,and 48 h later, the culture supernatant was harvested. The culture supernatant wasfiltered through a 0.45-�m-pore-size membrane and used as a virus solution. Forinfection, EBV-negative Akata cells (1.0 � 106) were suspended in 1 ml of filteredvirus solution and incubated at 37°C for 90 min with continuous gentle mixing.
For quantification of virions in the culture supernatants, 8 ml of virus solutionwas prepared as described above and ultracentrifuged at 15,000 rpm with anSW41 rotor (L8-80 M ultracentrifuge, Beckman) at 4°C for 1.5 h. Pellets wereresuspended in 400 �l of phosphate-buffered saline, and 200 �l of lysis buffer(3.0% sodium dodecyl sulfate, 75 mM Tris-HCl [pH 8.0], 25 mM EDTA) and1 �l of proteinase K (10 mg/ml) were added. After incubating the lysates at 37°Cfor 1 h, they were extracted twice with phenol and once with a chloroform-iso-amyl alcohol (24:1) mixture. The viral DNAs were then ethanol precipitated, dis-solved in Tris-EDTA, digested with EcoRI, and used for Southern blot analysis.
Flow cytometry analysis. Green fluorescent protein (GFP) expression in in-fected EBV-negative Akata cells was analyzed by fluorescence-activated cellsorting (FACS) (20,000 events) with a FACScalibur flow cytometer (BectonDickinson Co., San Jose, Calif.). Statistical differences in infection efficiencybetween groups were determined by Welch’s t test.
Electron microscopy. Cells were pelleted 48 h after starting anti-IgG treat-ment, and the pellets were washed with washing buffer (0.1 M phosphate buffer[pH 7.4], 7% sucrose) three times. The pellets were then immersed in 2.5%glutaraldehyde (TAAB) in 0.1 M phosphate buffer (pH 7.4) for 24 h at roomtemperature. The samples were then washed in the same washing buffer, post-fixed in 1% osmium tetroxide (Merck) in 0.1 M phosphate buffer for 1.5 h atroom temperature, dehydrated in graded acetones, and embedded in Epon
(TAAB). Ultrathin sections (0.1 �m) were stained with 3% uranyl acetate for 20min and 0.1% lead citrate for 15 min, and they were examined under a HitachiH-7100 electron microscope.
RESULTS
Construction of d.LMP1-EBV via homologous recombina-tion in E. coli. We recently cloned the genome of Akata strainEBV as a BAC clone (designated AK-BAC) and used deriva-tives of AK-BAC to generate recombinant EBVs (18). One ofthe derivatives of AK-BAC, designated AK-BAC-GFP, thathad a GFP gene and neomycin and kanamycin resistance genesinserted into the region deleted in B95-8 strain EBV producedprogeny virus in the absence of wild-type EBV. The recombi-nant virus AK-BAC-GFP exhibited high B-cell-transformingability and expressed all of the latently expressed viral genes inthe established lymphoblastoid cell lines (18). Therefore, weused AK-BAC-GFP as the starting material for mutagenesis.
A homologous recombination method called GET recombi-nation (29, 31) was employed in order to specifically disrupt theLMP1 gene of AK-BAC-GFP. First, a nonsense linker (encodingtriple stop codons) and a zeocin resistance gene were introducedafter codon 9 of the LMP1 gene via GET recombination to gen-erate d.LMP1-EBV-Zeor (see Materials and Methods and Fig.1). Subsequently, the zeocin resistance gene, which was flanked
FIG. 1. Construction of d.LMP1-EBV. (A) Schematic maps of AK-BAC-GFP (18) and d.LMP1-EBV. The locations of the BamHI fragmentsof Akata strain EBV are shown on top. Triple stop codons (indicated by crosses) were inserted after codon 9 of the LMP1 gene of AK-BAC-GFPvia GET recombination (29, 31). A zeocin resistance gene (Zeor), which served as a marker gene in GET recombination, was subsequently removedby in vitro Cre recombinase treatment. The positions of wild-type and 5171 loxP sites (mloxP) are indicated by open arrowheads. The chloram-phenicol resistance gene (Cmr), neomycin resistance gene (Neor), kanamycin resistance gene (Kmr), right origin of lytic replication (oriLyt), andinternal repeat 4 (IR4) are also indicated. (B) Partial sequence of the PCR product that was used for GET recombination to generated.LMP1-EBV. The coding sequence of LMP1 is in the complementary strand of this sequence, and an arrow indicates its translation start site. Thesequences homologous to the LMP1 gene are in bold. The positions of the primer sequences (NA03 and NA04) are underlined, the artificial stopcodons (which appear in the complementary strand) are in italic, and the mutated loxP sites (23) are indicated by parentheses. Mutated loxP siteswere used to avoid unwanted recombination, as AK-BAC-GFP had one wild-type loxP site located upstream of the BAC vector sequence (18).Stars indicate base substitution mutations created in 5171 loxP sites.
VOL. 79, 2005 ROLE OF LMP1 IN VIRUS PRODUCTION 4417
on January 4, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
by a pair of 5171 loxP sites (23), was removed by an in vitro Crerecombinase reaction. The resultant BAC clone, designatedd.LMP1-EBV, had triple stop codons and one residual 5171loxP site (with short flanking sequences) inserted after co-don 9 of the LMP1 gene. The insertion of triple stop codonsafter codon 9 of the LMP1 gene is expected to completelyabolish LMP1 expression, as only full-length LMP1 protein isexpressed from the LMP1 gene of Akata strain EBV, whichlacks the Met-129 in the LMP1 gene of B95-8 strain EBV (7,42). The modified genomic locus of d.LMP1-EBV was PCRamplified, and the modification was verified by DNA sequenc-ing (data not shown). Restriction enzyme digestion analysesrevealed the expected changes in band sizes corresponding tothe modified LMP1 locus, while the bands of unmodified lociremained unchanged (data not shown).
Generation of Akata cells harboring d.LMP1-EBV. We in-troduced the DNA of d.LMP1-EBV into Akata cells in orderto establish cell lines producing the LMP1-deleted recombi-nant viruses. We previously showed that transfected AK-BAC-GFP efficiently formed episomes in EBV-positive Akata cellsbut not in EBV-negative Akata cells (18). Accordingly, EBV-positive Akata cells were transfected with d.LMP1-EBV, andthe transfected cells were selected with G418. Screening of 39G418-resistant cell clones resulted in obtaining two cell clones(clones 28 and 31) that harbored intact d.LMP1-EBV epi-somes (data not shown).
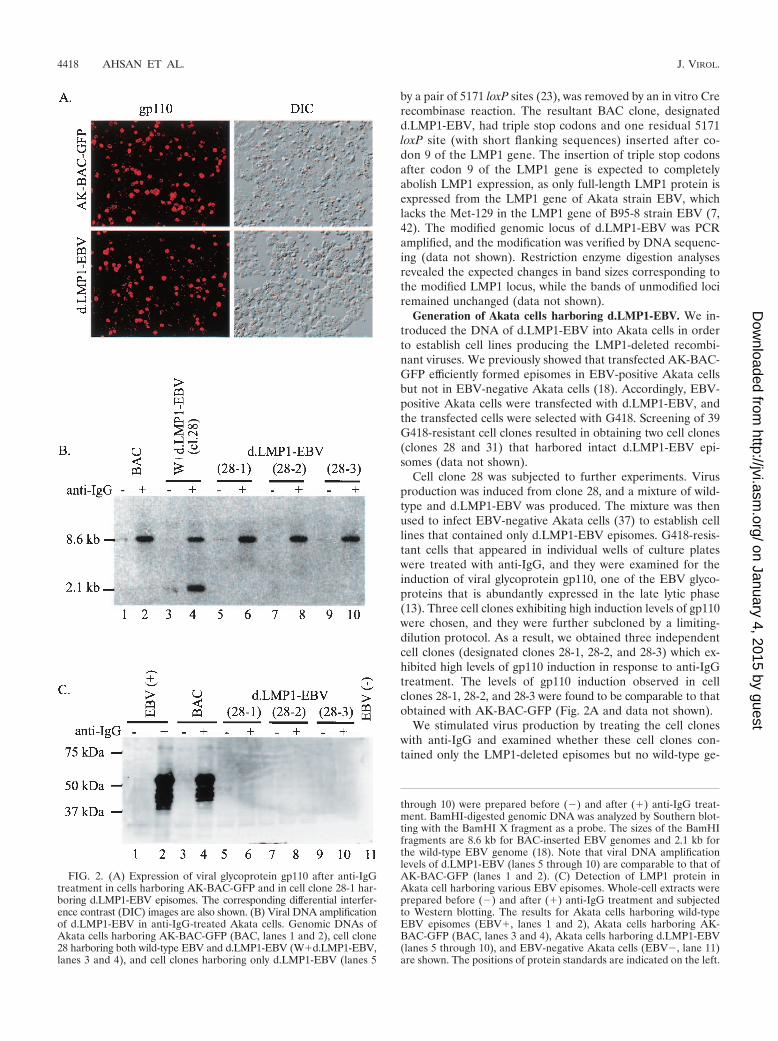
Cell clone 28 was subjected to further experiments. Virusproduction was induced from clone 28, and a mixture of wild-type and d.LMP1-EBV was produced. The mixture was thenused to infect EBV-negative Akata cells (37) to establish celllines that contained only d.LMP1-EBV episomes. G418-resis-tant cells that appeared in individual wells of culture plateswere treated with anti-IgG, and they were examined for theinduction of viral glycoprotein gp110, one of the EBV glyco-proteins that is abundantly expressed in the late lytic phase(13). Three cell clones exhibiting high induction levels of gp110were chosen, and they were further subcloned by a limiting-dilution protocol. As a result, we obtained three independentcell clones (designated clones 28-1, 28-2, and 28-3) which ex-hibited high levels of gp110 induction in response to anti-IgGtreatment. The levels of gp110 induction observed in cellclones 28-1, 28-2, and 28-3 were found to be comparable to thatobtained with AK-BAC-GFP (Fig. 2A and data not shown).
We stimulated virus production by treating the cell cloneswith anti-IgG and examined whether these cell clones con-tained only the LMP1-deleted episomes but no wild-type ge-
FIG. 2. (A) Expression of viral glycoprotein gp110 after anti-IgGtreatment in cells harboring AK-BAC-GFP and in cell clone 28-1 har-boring d.LMP1-EBV episomes. The corresponding differential interfer-ence contrast (DIC) images are also shown. (B) Viral DNA amplificationof d.LMP1-EBV in anti-IgG-treated Akata cells. Genomic DNAs ofAkata cells harboring AK-BAC-GFP (BAC, lanes 1 and 2), cell clone28 harboring both wild-type EBV and d.LMP1-EBV (W�d.LMP1-EBV,lanes 3 and 4), and cell clones harboring only d.LMP1-EBV (lanes 5
through 10) were prepared before (�) and after (�) anti-IgG treat-ment. BamHI-digested genomic DNA was analyzed by Southern blot-ting with the BamHI X fragment as a probe. The sizes of the BamHIfragments are 8.6 kb for BAC-inserted EBV genomes and 2.1 kb forthe wild-type EBV genome (18). Note that viral DNA amplificationlevels of d.LMP1-EBV (lanes 5 through 10) are comparable to that ofAK-BAC-GFP (lanes 1 and 2). (C) Detection of LMP1 protein inAkata cell harboring various EBV episomes. Whole-cell extracts wereprepared before (�) and after (�) anti-IgG treatment and subjectedto Western blotting. The results for Akata cells harboring wild-typeEBV episomes (EBV�, lanes 1 and 2), Akata cells harboring AK-BAC-GFP (BAC, lanes 3 and 4), Akata cells harboring d.LMP1-EBV(lanes 5 through 10), and EBV-negative Akata cells (EBV�, lane 11)are shown. The positions of protein standards are indicated on the left.
4418 AHSAN ET AL. J. VIROL.
on January 4, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
nomes. Genomic DNA of clone 28 (harboring wild-type andd.LMP1-EBV) and cell clones harboring the LMP1-deletedepisomes (28-1, 28-2, and 28-3) were prepared before and afteranti-IgG treatment, and they were subjected to Southern blotanalysis. The result revealed that clone 28 exhibited bandsderived from both wild-type and d.LMP1-EBV episomes,while clones 28-1, 28-2, and 28-3 exhibited only the d.LMP1-EBV bands (Fig. 2B). The result also demonstrated that, afterstimulating virus production, the genomes of d.LMP1-EBVamplified significantly in the absence of wild-type episomes.The degrees of viral DNA amplification of the LMP1-deletedepisomes were comparable to that of its wild-type counterpart(AK-BAC-GFP) (Fig. 2B).
We next examined whether insertion of the triple stop co-dons into the LMP1 ORF abolished the expression of LMP1during the lytic cycle of viral replication. The expression ofLMP1 was examined before and after stimulating virus pro-duction by Western blot analysis. Akata cells retain the Bur-kitt’s-type viral gene expression, and LMP1 expression is un-detectable unless virus production is stimulated (42). EBV-positive Akata cells and the cell clones with LMP1-deletedepisomes (28-1, 28-2, and 28-3) were then treated with anti-IgG, and the expression of LMP1 protein was examined.Strong inductions of full-length LMP1 protein (55 kDa insodium dodecyl sulfate-polyacrylamide gel electrophoresis)and its associated faster-migrating products were observed inAkata cells harboring either wild-type EBV or AK-BAC-GFP.By contrast, no LMP1 expression was detected in the cell clonesharboring d.LMP1-EBV (Fig. 2C). Taken together, these datademonstrate the successful knockout of LMP1 protein expres-sion as well as the establishment of cell clones harboring onlyLMP1-deleted episomes.
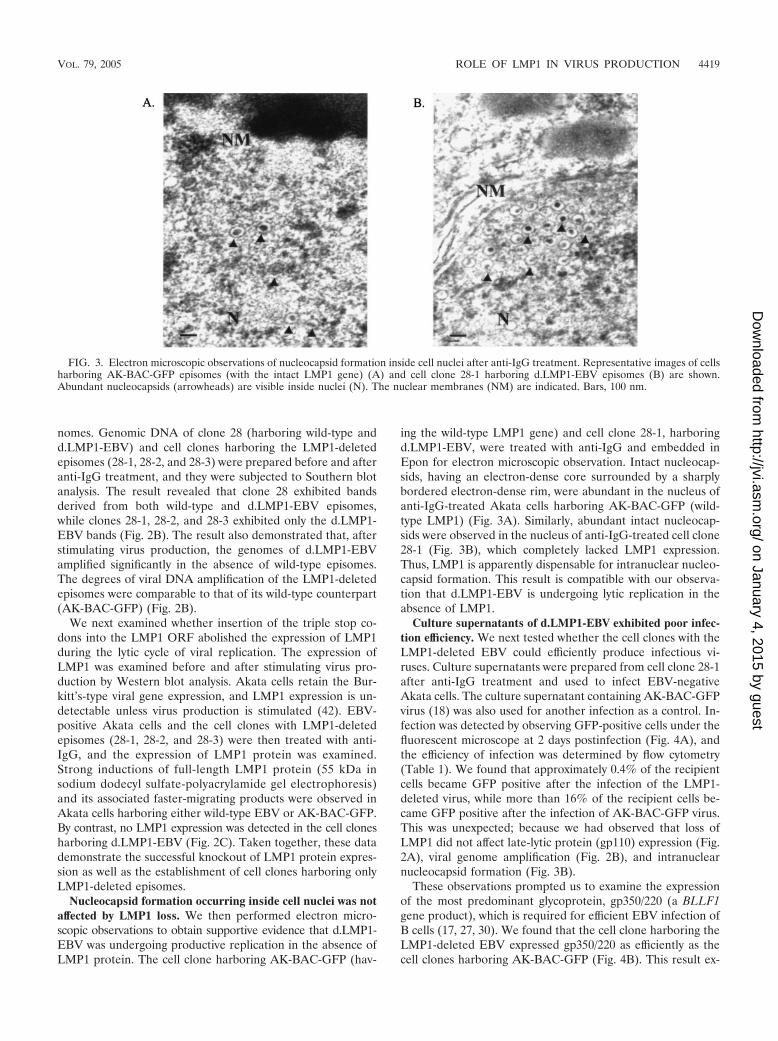
Nucleocapsid formation occurring inside cell nuclei was notaffected by LMP1 loss. We then performed electron micro-scopic observations to obtain supportive evidence that d.LMP1-EBV was undergoing productive replication in the absence ofLMP1 protein. The cell clone harboring AK-BAC-GFP (hav-
ing the wild-type LMP1 gene) and cell clone 28-1, harboringd.LMP1-EBV, were treated with anti-IgG and embedded inEpon for electron microscopic observation. Intact nucleocap-sids, having an electron-dense core surrounded by a sharplybordered electron-dense rim, were abundant in the nucleus ofanti-IgG-treated Akata cells harboring AK-BAC-GFP (wild-type LMP1) (Fig. 3A). Similarly, abundant intact nucleocap-sids were observed in the nucleus of anti-IgG-treated cell clone28-1 (Fig. 3B), which completely lacked LMP1 expression.Thus, LMP1 is apparently dispensable for intranuclear nucleo-capsid formation. This result is compatible with our observa-tion that d.LMP1-EBV is undergoing lytic replication in theabsence of LMP1.
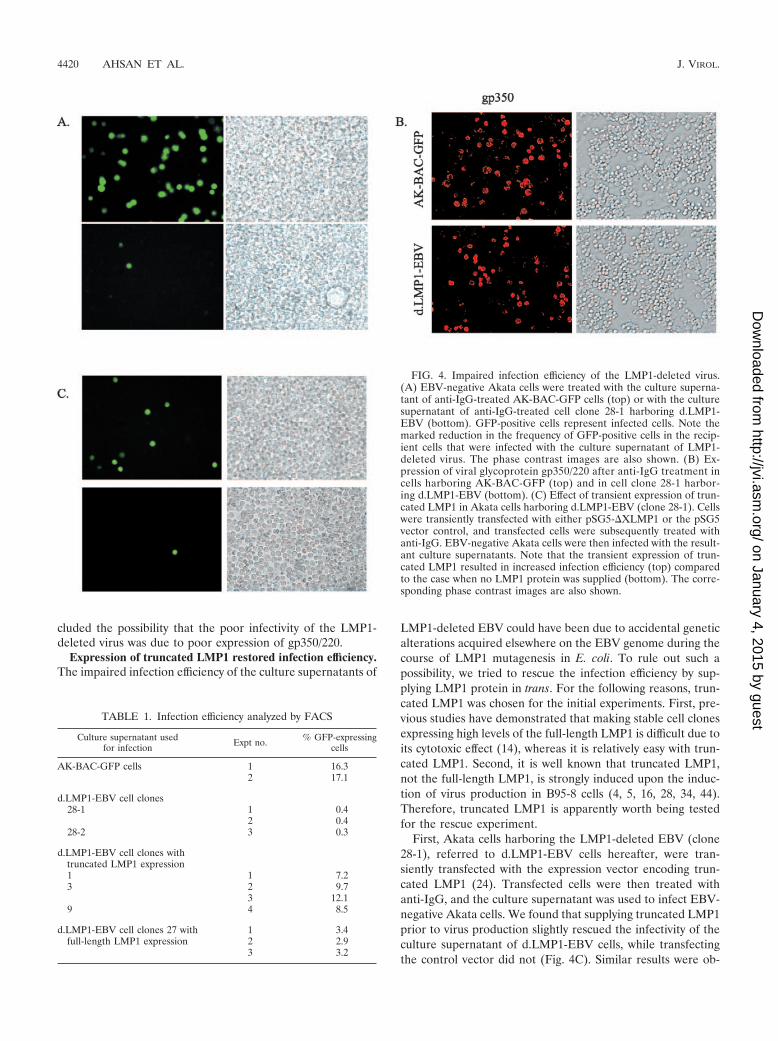
Culture supernatants of d.LMP1-EBV exhibited poor infec-tion efficiency. We next tested whether the cell clones with theLMP1-deleted EBV could efficiently produce infectious vi-ruses. Culture supernatants were prepared from cell clone 28-1after anti-IgG treatment and used to infect EBV-negativeAkata cells. The culture supernatant containing AK-BAC-GFPvirus (18) was also used for another infection as a control. In-fection was detected by observing GFP-positive cells under thefluorescent microscope at 2 days postinfection (Fig. 4A), andthe efficiency of infection was determined by flow cytometry(Table 1). We found that approximately 0.4% of the recipientcells became GFP positive after the infection of the LMP1-deleted virus, while more than 16% of the recipient cells be-came GFP positive after the infection of AK-BAC-GFP virus.This was unexpected; because we had observed that loss ofLMP1 did not affect late-lytic protein (gp110) expression (Fig.2A), viral genome amplification (Fig. 2B), and intranuclearnucleocapsid formation (Fig. 3B).
These observations prompted us to examine the expressionof the most predominant glycoprotein, gp350/220 (a BLLF1gene product), which is required for efficient EBV infection ofB cells (17, 27, 30). We found that the cell clone harboring theLMP1-deleted EBV expressed gp350/220 as efficiently as thecell clones harboring AK-BAC-GFP (Fig. 4B). This result ex-
FIG. 3. Electron microscopic observations of nucleocapsid formation inside cell nuclei after anti-IgG treatment. Representative images of cellsharboring AK-BAC-GFP episomes (with the intact LMP1 gene) (A) and cell clone 28-1 harboring d.LMP1-EBV episomes (B) are shown.Abundant nucleocapsids (arrowheads) are visible inside nuclei (N). The nuclear membranes (NM) are indicated. Bars, 100 nm.
VOL. 79, 2005 ROLE OF LMP1 IN VIRUS PRODUCTION 4419
on January 4, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
cluded the possibility that the poor infectivity of the LMP1-deleted virus was due to poor expression of gp350/220.
Expression of truncated LMP1 restored infection efficiency.The impaired infection efficiency of the culture supernatants of
LMP1-deleted EBV could have been due to accidental geneticalterations acquired elsewhere on the EBV genome during thecourse of LMP1 mutagenesis in E. coli. To rule out such apossibility, we tried to rescue the infection efficiency by sup-plying LMP1 protein in trans. For the following reasons, trun-cated LMP1 was chosen for the initial experiments. First, pre-vious studies have demonstrated that making stable cell clonesexpressing high levels of the full-length LMP1 is difficult due toits cytotoxic effect (14), whereas it is relatively easy with trun-cated LMP1. Second, it is well known that truncated LMP1,not the full-length LMP1, is strongly induced upon the induc-tion of virus production in B95-8 cells (4, 5, 16, 28, 34, 44).Therefore, truncated LMP1 is apparently worth being testedfor the rescue experiment.
First, Akata cells harboring the LMP1-deleted EBV (clone28-1), referred to d.LMP1-EBV cells hereafter, were tran-siently transfected with the expression vector encoding trun-cated LMP1 (24). Transfected cells were then treated withanti-IgG, and the culture supernatant was used to infect EBV-negative Akata cells. We found that supplying truncated LMP1prior to virus production slightly rescued the infectivity of theculture supernatant of d.LMP1-EBV cells, while transfectingthe control vector did not (Fig. 4C). Similar results were ob-
FIG. 4. Impaired infection efficiency of the LMP1-deleted virus.(A) EBV-negative Akata cells were treated with the culture superna-tant of anti-IgG-treated AK-BAC-GFP cells (top) or with the culturesupernatant of anti-IgG-treated cell clone 28-1 harboring d.LMP1-EBV (bottom). GFP-positive cells represent infected cells. Note themarked reduction in the frequency of GFP-positive cells in the recip-ient cells that were infected with the culture supernatant of LMP1-deleted virus. The phase contrast images are also shown. (B) Ex-pression of viral glycoprotein gp350/220 after anti-IgG treatment incells harboring AK-BAC-GFP (top) and in cell clone 28-1 harbor-ing d.LMP1-EBV (bottom). (C) Effect of transient expression of trun-cated LMP1 in Akata cells harboring d.LMP1-EBV (clone 28-1). Cellswere transiently transfected with either pSG5-�XLMP1 or the pSG5vector control, and transfected cells were subsequently treated withanti-IgG. EBV-negative Akata cells were then infected with the result-ant culture supernatants. Note that the transient expression of trun-cated LMP1 resulted in increased infection efficiency (top) comparedto the case when no LMP1 protein was supplied (bottom). The corre-sponding phase contrast images are also shown.
TABLE 1. Infection efficiency analyzed by FACS
Culture supernatant usedfor infection Expt no. % GFP-expressing
cells
AK-BAC-GFP cells 1 16.32 17.1
d.LMP1-EBV cell clones28-1 1 0.4
2 0.428-2 3 0.3
d.LMP1-EBV cell clones withtruncated LMP1 expression1 1 7.23 2 9.7
3 12.19 4 8.5
d.LMP1-EBV cell clones 27 withfull-length LMP1 expression
1 3.42 2.93 3.2
4420 AHSAN ET AL. J. VIROL.
on January 4, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
tained when two other d.LMP1-EBV cell clones (28-2 and28-3) were subjected to the same experiment (data not shown).
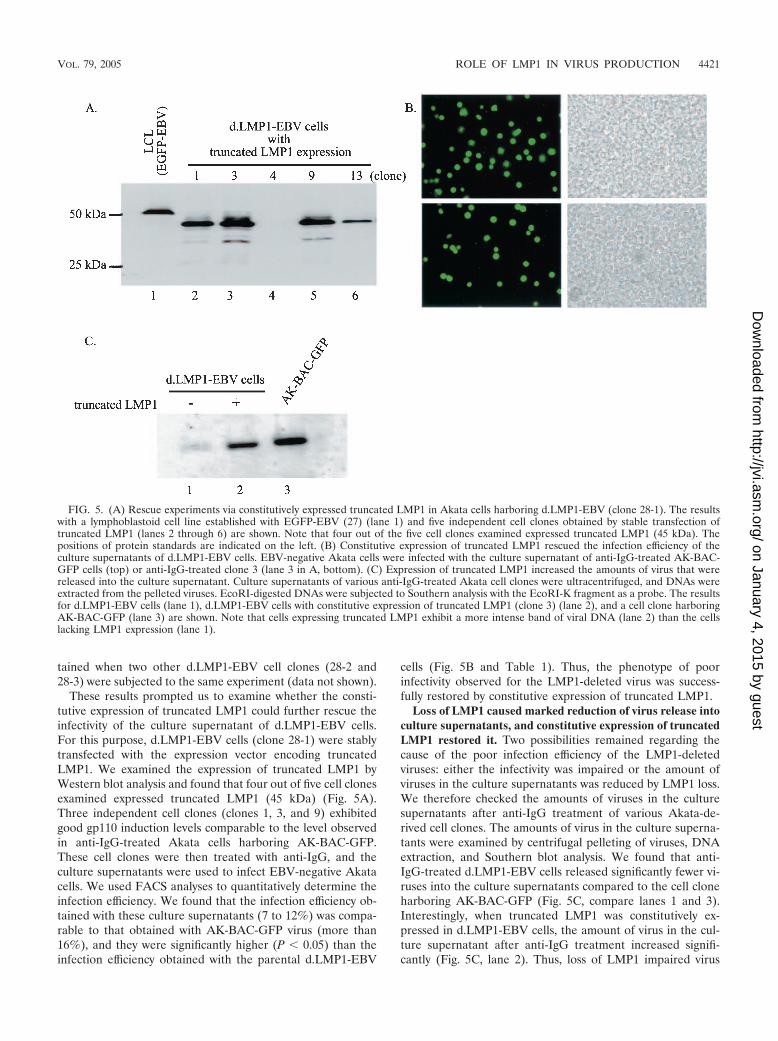
These results prompted us to examine whether the consti-tutive expression of truncated LMP1 could further rescue theinfectivity of the culture supernatant of d.LMP1-EBV cells.For this purpose, d.LMP1-EBV cells (clone 28-1) were stablytransfected with the expression vector encoding truncatedLMP1. We examined the expression of truncated LMP1 byWestern blot analysis and found that four out of five cell clonesexamined expressed truncated LMP1 (45 kDa) (Fig. 5A).Three independent cell clones (clones 1, 3, and 9) exhibitedgood gp110 induction levels comparable to the level observedin anti-IgG-treated Akata cells harboring AK-BAC-GFP.These cell clones were then treated with anti-IgG, and theculture supernatants were used to infect EBV-negative Akatacells. We used FACS analyses to quantitatively determine theinfection efficiency. We found that the infection efficiency ob-tained with these culture supernatants (7 to 12%) was compa-rable to that obtained with AK-BAC-GFP virus (more than16%), and they were significantly higher (P 0.05) than theinfection efficiency obtained with the parental d.LMP1-EBV
cells (Fig. 5B and Table 1). Thus, the phenotype of poorinfectivity observed for the LMP1-deleted virus was success-fully restored by constitutive expression of truncated LMP1.
Loss of LMP1 caused marked reduction of virus release intoculture supernatants, and constitutive expression of truncatedLMP1 restored it. Two possibilities remained regarding thecause of the poor infection efficiency of the LMP1-deletedviruses: either the infectivity was impaired or the amount ofviruses in the culture supernatants was reduced by LMP1 loss.We therefore checked the amounts of viruses in the culturesupernatants after anti-IgG treatment of various Akata-de-rived cell clones. The amounts of virus in the culture superna-tants were examined by centrifugal pelleting of viruses, DNAextraction, and Southern blot analysis. We found that anti-IgG-treated d.LMP1-EBV cells released significantly fewer vi-ruses into the culture supernatants compared to the cell cloneharboring AK-BAC-GFP (Fig. 5C, compare lanes 1 and 3).Interestingly, when truncated LMP1 was constitutively ex-pressed in d.LMP1-EBV cells, the amount of virus in the cul-ture supernatant after anti-IgG treatment increased signifi-cantly (Fig. 5C, lane 2). Thus, loss of LMP1 impaired virus
FIG. 5. (A) Rescue experiments via constitutively expressed truncated LMP1 in Akata cells harboring d.LMP1-EBV (clone 28-1). The resultswith a lymphoblastoid cell line established with EGFP-EBV (27) (lane 1) and five independent cell clones obtained by stable transfection oftruncated LMP1 (lanes 2 through 6) are shown. Note that four out of the five cell clones examined expressed truncated LMP1 (45 kDa). Thepositions of protein standards are indicated on the left. (B) Constitutive expression of truncated LMP1 rescued the infection efficiency of theculture supernatants of d.LMP1-EBV cells. EBV-negative Akata cells were infected with the culture supernatant of anti-IgG-treated AK-BAC-GFP cells (top) or anti-IgG-treated clone 3 (lane 3 in A, bottom). (C) Expression of truncated LMP1 increased the amounts of virus that werereleased into the culture supernatant. Culture supernatants of various anti-IgG-treated Akata cell clones were ultracentrifuged, and DNAs wereextracted from the pelleted viruses. EcoRI-digested DNAs were subjected to Southern analysis with the EcoRI-K fragment as a probe. The resultsfor d.LMP1-EBV cells (lane 1), d.LMP1-EBV cells with constitutive expression of truncated LMP1 (clone 3) (lane 2), and a cell clone harboringAK-BAC-GFP (lane 3) are shown. Note that cells expressing truncated LMP1 exhibit a more intense band of viral DNA (lane 2) than the cellslacking LMP1 expression (lane 1).
VOL. 79, 2005 ROLE OF LMP1 IN VIRUS PRODUCTION 4421
on January 4, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
release into the culture supernatant, and the constitutive ex-pression of truncated LMP1 significantly restored it. This re-sult indicates that the impaired infection efficiency observedfor the LMP1-deleted EBV is most likely due to the reductionof virus release into the culture supernatant rather than toimpaired infectivity of the viruses.
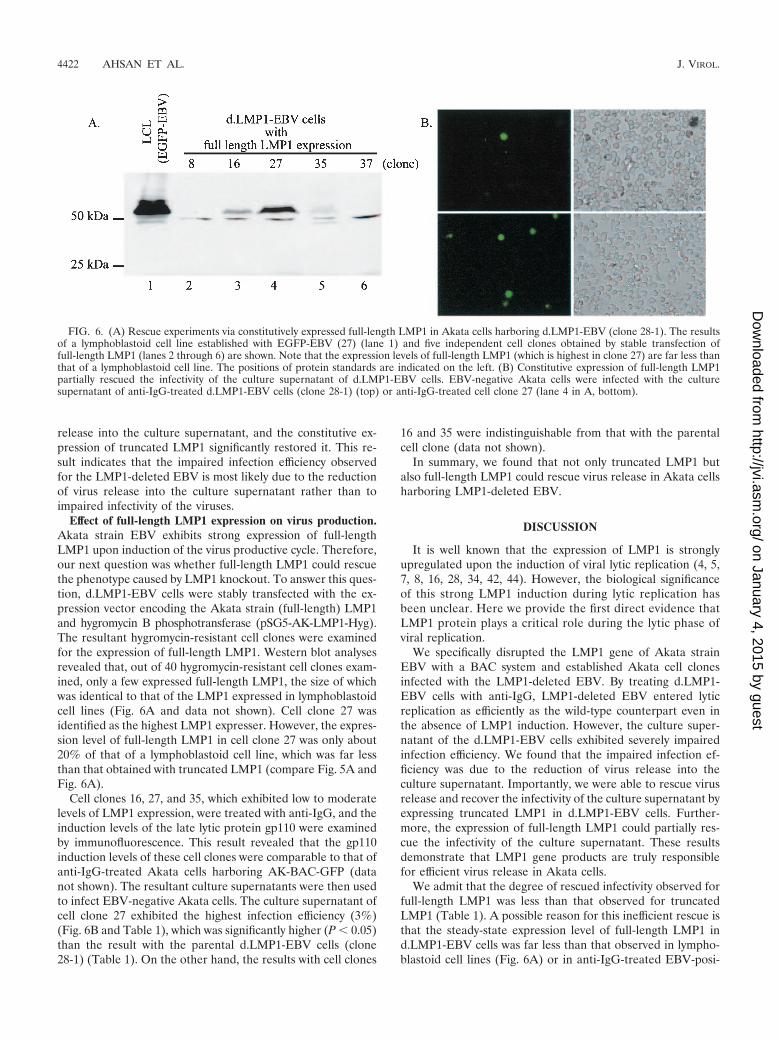
Effect of full-length LMP1 expression on virus production.Akata strain EBV exhibits strong expression of full-lengthLMP1 upon induction of the virus productive cycle. Therefore,our next question was whether full-length LMP1 could rescuethe phenotype caused by LMP1 knockout. To answer this ques-tion, d.LMP1-EBV cells were stably transfected with the ex-pression vector encoding the Akata strain (full-length) LMP1and hygromycin B phosphotransferase (pSG5-AK-LMP1-Hyg).The resultant hygromycin-resistant cell clones were examinedfor the expression of full-length LMP1. Western blot analysesrevealed that, out of 40 hygromycin-resistant cell clones exam-ined, only a few expressed full-length LMP1, the size of whichwas identical to that of the LMP1 expressed in lymphoblastoidcell lines (Fig. 6A and data not shown). Cell clone 27 wasidentified as the highest LMP1 expresser. However, the expres-sion level of full-length LMP1 in cell clone 27 was only about20% of that of a lymphoblastoid cell line, which was far lessthan that obtained with truncated LMP1 (compare Fig. 5A andFig. 6A).
Cell clones 16, 27, and 35, which exhibited low to moderatelevels of LMP1 expression, were treated with anti-IgG, and theinduction levels of the late lytic protein gp110 were examinedby immunofluorescence. This result revealed that the gp110induction levels of these cell clones were comparable to that ofanti-IgG-treated Akata cells harboring AK-BAC-GFP (datanot shown). The resultant culture supernatants were then usedto infect EBV-negative Akata cells. The culture supernatant ofcell clone 27 exhibited the highest infection efficiency (3%)(Fig. 6B and Table 1), which was significantly higher (P 0.05)than the result with the parental d.LMP1-EBV cells (clone28-1) (Table 1). On the other hand, the results with cell clones
16 and 35 were indistinguishable from that with the parentalcell clone (data not shown).
In summary, we found that not only truncated LMP1 butalso full-length LMP1 could rescue virus release in Akata cellsharboring LMP1-deleted EBV.
DISCUSSION
It is well known that the expression of LMP1 is stronglyupregulated upon the induction of viral lytic replication (4, 5,7, 8, 16, 28, 34, 42, 44). However, the biological significanceof this strong LMP1 induction during lytic replication hasbeen unclear. Here we provide the first direct evidence thatLMP1 protein plays a critical role during the lytic phase ofviral replication.
We specifically disrupted the LMP1 gene of Akata strainEBV with a BAC system and established Akata cell clonesinfected with the LMP1-deleted EBV. By treating d.LMP1-EBV cells with anti-IgG, LMP1-deleted EBV entered lyticreplication as efficiently as the wild-type counterpart even inthe absence of LMP1 induction. However, the culture super-natant of the d.LMP1-EBV cells exhibited severely impairedinfection efficiency. We found that the impaired infection ef-ficiency was due to the reduction of virus release into theculture supernatant. Importantly, we were able to rescue virusrelease and recover the infectivity of the culture supernatant byexpressing truncated LMP1 in d.LMP1-EBV cells. Further-more, the expression of full-length LMP1 could partially res-cue the infectivity of the culture supernatant. These resultsdemonstrate that LMP1 gene products are truly responsiblefor efficient virus release in Akata cells.
We admit that the degree of rescued infectivity observed forfull-length LMP1 was less than that observed for truncatedLMP1 (Table 1). A possible reason for this inefficient rescue isthat the steady-state expression level of full-length LMP1 ind.LMP1-EBV cells was far less than that observed in lympho-blastoid cell lines (Fig. 6A) or in anti-IgG-treated EBV-posi-
FIG. 6. (A) Rescue experiments via constitutively expressed full-length LMP1 in Akata cells harboring d.LMP1-EBV (clone 28-1). The resultsof a lymphoblastoid cell line established with EGFP-EBV (27) (lane 1) and five independent cell clones obtained by stable transfection offull-length LMP1 (lanes 2 through 6) are shown. Note that the expression levels of full-length LMP1 (which is highest in clone 27) are far less thanthat of a lymphoblastoid cell line. The positions of protein standards are indicated on the left. (B) Constitutive expression of full-length LMP1partially rescued the infectivity of the culture supernatant of d.LMP1-EBV cells. EBV-negative Akata cells were infected with the culturesupernatant of anti-IgG-treated d.LMP1-EBV cells (clone 28-1) (top) or anti-IgG-treated cell clone 27 (lane 4 in A, bottom).
4422 AHSAN ET AL. J. VIROL.
on January 4, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from

tive Akata cells (Fig. 2C). Full-length LMP1 could have res-cued the infectivity more efficiently if we had establishedhigher LMP1 expressers. However, it is well known that mak-ing stable cell clones expressing high levels of full-length LMP1is difficult due to its cytotoxic effect (14).
Our model is that strong induction of LMP1 expression iscritical for efficient virus release, but previous studies haveproposed an apparently conflicting model in which LMP1 caninhibit lytic cycle progression (1, 32). One study demonstratedthat constitutive LMP1 expression in lymphoblastoid cell lines(while EBNA2 expression was shut off) could inhibit EBVreactivation (1), and another study showed that transient over-expression of LMP1 could inhibit the virus-productive cycle ina derivative of the P3HR1 cell line (32). However, in ourrescue experiments, we saw no significant reduction in latelytic protein expression (gp110 and gp350) after constitutivelyexpressing low to moderate levels of full-length LMP1 ind.LMP1-EBV cells (data not shown). The discrepancy betweenour study and the previous studies could be due to the differentexperimental systems and different expression levels of LMP1proteins.
We still do not know where and how LMP1 can augment theprocess of virus release. A working model of intracellular nu-cleocapsid transport involves a deenvelopment process fromnucleus to cytoplasm and a reenvelopment process either atpost-Golgi-derived cytoplasmic vesicles or at the plasma mem-brane (13). It is possible that the LMP1 enhances the deen-velopment and reenvelopment processes either at the nuclearmembrane or at the cytoplasm during the lytic phase. Full-length LMP1 is known to localize to lipid microdomains, des-ignated lipid rafts, located on the plasma membrane (15, 20).However, recent studies reinvestigated the intracellular local-ization of LMP1 protein and found that a substantial fractionof LMP1 protein localized to intracellular compartments (11,22). Therefore, it is possible that full-length LMP1 expressedshortly after virus induction localizes to intracellular compart-ments and augments virus production.
Another open question is how full-length LMP1 induction instimulated Akata cells can be reconciled with truncated LMP1induction in stimulated B95-8 cells (43). We found that intro-ducing triple stop codons after codon 9 of the LMP1 gene ofAkata strain EBV abolished the induction of not only full-length LMP1 but also smaller LMP1-immunoreactive proteins(Fig. 2C). This result clearly indicates that proteolytic cleavageof full-length LMP1 generates these smaller products. As theepitope of the monoclonal antibody used for the Western anal-yses resides in the C terminus of LMP1 protein (25), they areapparently N-terminally truncated. Therefore, the possibilityremains that these N-terminally truncated LMP1 products ac-tually play a role in augmenting virus release in Akata cells.Whether these N-terminally truncated LMP1 products corre-spond to the lytic LMP1 in stimulated B95-8 cells remains to beclarified.
In summary, we propose a model in which EBV utilizesdifferentially expressed LMP1 gene products for dual purposesduring its life cycle. First, in latently infected B lymphocytes,steady-state expression of full-length LMP1 is critical for main-taining transformation status. Second, at the onset of the virusproduction cycle, the strong induction of LMP1 enables effi-cient virus release from cells. In B95-8 cells, it is probably a
truncated LMP1 that plays a critical role for virus release,fitting well with our finding that truncated LMP1 can restorethe defect of LMP1-deleted virus. In Akata cells, it is full-length LMP1 or its associated N-terminally truncated LMP1that plays a critical role during virus production.
LMP1 is apparently one of the most important viral proteinsabsolutely required for EBV’s life cycle. Choosing such animportant protein for augmenting virus release may be a mat-ter of importance for virus evolution.
ACKNOWLEDGMENTS
We thank P. Ioannou (Murdoch Institute, Melbourne, Australia) forthe generous gift of plasmid pGETrec, M. Sato for expertise withelectron microscopy, and S. Maruo for FACS analysis. We also thankJ. M. Martin (Boulder University), D. Iwakiri, and M. Tanaka forhelpful suggestions.
This work was supported by a Grant-in-Aid for Scientific Researchfrom the Ministry of Education, Culture, Sports, Science and Tech-nology, Japan (T.K. and K.T.), Akiyama Memorial Foundation (T.K.),and Uehara Memorial Foundation (K.T.).
REFERENCES
1. Adler, B., E. Schaadt, B. Kempkes, U. Zimber-Strobl, B. Baier, and G. W.Bornkamm. 2002. Control of Epstein-Barr virus reactivation by activatedCD40 and viral latent membrane protein 1. Proc. Natl. Acad. Sci. USA 99:437–442.
2. Baer, R., A. T. Bankier, M. D. Biggin, P. L. Deininger, P. J. Farrell, T. J.Gibson, G. Hatfull, G. S. Hudson, S. C. Satchwell, C. Seguin, et al. 1984.DNA sequence and expression of the B95-8 Epstein-Barr virus genome.Nature 310:207–211.
3. Baichwal, V. R., and B. Sugden. 1989. The multiple membrane-spanningsegments of the BNLF-1 oncogene from Epstein-Barr virus are required fortransformation. Oncogene 4:67–74.
4. Baichwal, V. R., and B. Sugden. 1987. Posttranslational processing of anEpstein-Barr virus-encoded membrane protein expressed in cells trans-formed by Epstein-Barr virus. J. Virol. 61:866–875.
5. Boos, H., R. Berger, C. Kuklik-Roos, T. Iftner, and N. Mueller-Lantzsch.1987. Enhancement of Epstein-Barr virus membrane protein (LMP) expres-sion by serum, TPA, or n-butyrate in latently infected Raji cells. Virology159:161–165.
6. Cahir McFarland, E. D., K. M. Izumi, and G. Mosialos. 1999. Epstein-barrvirus transformation: involvement of latent membrane protein 1-mediatedactivation of NF-kappaB. Oncogene 18:6959–6964.
7. Erickson, K. D., C. Berger, W. F. Coffin, 3rd, E. Schiff, D. M. Walling, andJ. M. Martin. 2003. Unexpected absence of the Epstein-Barr virus (EBV)lyLMP-1 open reading frame in tumor virus isolates: lack of correlationbetween Met129 status and EBV strain identity. J. Virol. 77:4415–4422.
8. Erickson, K. D., and J. M. Martin. 1997. Early detection of the lytic LMP-1protein in EBV-infected B-cells suggests its presence in the virion. Virology234:1–13.
9. Erickson, K. D., and J. M. Martin. 2000. The late lytic LMP-1 protein ofEpstein-Barr virus can negatively regulate LMP-1 signaling. J. Virol. 74:1057–1060.
10. Fennewald, S., V. van Santen, and E. Kieff. 1984. Nucleotide sequence of anmRNA transcribed in latent growth-transforming virus infection indicatesthat it may encode a membrane protein. J. Virol. 51:411–419.
11. Flanagan, J., J. Middeldorp, and T. Sculley. 2003. Localization of the Ep-stein-Barr virus protein LMP 1 to exosomes. J. Gen. Virol. 84:1871–1879.
12. Gires, O., U. Zimber-Strobl, R. Gonnella, M. Ueffing, G. Marschall, R.Zeidler, D. Pich, and W. Hammerschmidt. 1997. Latent membrane protein1 of Epstein-Barr virus mimics a constitutively active receptor molecule.EMBO J. 16:6131–6140.
13. Gong, M., and E. Kieff. 1990. Intracellular trafficking of two major Epstein-Barr virus glycoproteins, gp350/220 and gp110. J. Virol. 64:1507–1516.
14. Hammerschmidt, W., B. Sugden, and V. R. Baichwal. 1989. The transform-ing domain alone of the latent membrane protein of Epstein-Barr virus istoxic to cells when expressed at high levels. J. Virol. 63:2469–2475.
15. Higuchi, M., K. M. Izumi, and E. Kieff. 2001. Epstein-Barr virus latent-infection membrane proteins are palmitoylated and raft-associated: protein1 binds to the cytoskeleton through TNF receptor cytoplasmic factors. Proc.Natl. Acad. Sci. USA 98:4675–4680.
16. Hudson, G. S., P. J. Farrell, and B. G. Barrell. 1985. Two related but dif-ferentially expressed potential membrane proteins encoded by the EcoRIDhet region of Epstein-Barr virus B95-8. J. Virol. 53:528–535.
17. Janz, A., M. Oezel, C. Kurzeder, J. Mautner, D. Pich, M. Kost, W. Ham-merschmidt, and H. J. Delecluse. 2000. Infectious Epstein-Barr virus lacking
VOL. 79, 2005 ROLE OF LMP1 IN VIRUS PRODUCTION 4423
on January 4, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from

major glycoprotein BLLF1 (gp350/220) demonstrates the existence of addi-tional viral ligands. J. Virol. 74:10142–10152.
18. Kanda, T., M. Yajima, N. Ahsan, M. Tanaka, and K. Takada. 2004. Produc-tion of high-titer Epstein-Barr virus recombinants derived from Akata cellsby using a bacterial artificial chromosome system. J. Virol. 78:7004–7015.
19. Kaye, K. M., K. M. Izumi, and E. Kieff. 1993. Epstein-Barr virus latentmembrane protein 1 is essential for B-lymphocyte growth transformation.Proc. Natl. Acad. Sci. USA 90:9150–9154.
20. Kaykas, A., K. Worringer, and B. Sugden. 2001. CD40 and LMP-1 bothsignal from lipid rafts but LMP-1 assembles a distinct, more efficient signal-ing complex. EMBO J. 20:2641–2654.
21. Kieff, E., and A. B. Rikinson. 2001. Epstein-Barr virus and its replication, p.2511-2573. In M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed.Lippincott Williams & Wilkins, Philadelphia, Pa.
22. Lam, N., and B. Sugden. 2003. LMP1, a viral relative of the TNF receptorfamily, signals principally from intracellular compartments. EMBO J. 22:3027–3038.
23. Lee, G., and I. Saito. 1998. Role of nucleotide sequences of loxP spacerregion in Cre-mediated recombination. Gene 216:55–65.
24. Liebowitz, D., J. Mannick, K. Takada, and E. Kieff. 1992. Phenotypes ofEpstein-Barr virus LMP1 deletion mutants indicate transmembrane andamino-terminal cytoplasmic domains necessary for effects in B-lymphomacells. J. Virol. 66:4612–4616.
25. Mann, K. P., D. Staunton, and D. A. Thorley-Lawson. 1985. Epstein-Barrvirus-encoded protein found in plasma membranes of transformed cells.J. Virol. 55:710–720.
26. Martin, J., and B. Sugden. 1991. The latent membrane protein oncoproteinresembles growth factor receptors in the properties of its turnover. CellGrowth Differ. 2:653–700.
27. Maruo, S., L. Yang, and K. Takada. 2001. Roles of Epstein-Barr virusglycoproteins gp350 and gp25 in the infection of human epithelial cells.J. Gen. Virol. 82:2373–2383.
28. Modrow, S., and H. Wolf. 1986. Characterization of two related Epstein-Barrvirus-encoded membrane proteins that are differentially expressed in Burkittlymphoma and in vitro-transformed cell lines. Proc. Natl. Acad. Sci. USA 83:5703–5707.
29. Narayanan, K., R. Williamson, Y. Zhang, A. F. Stewart, and P. A. Ioannou.1999. Efficient and precise engineering of a 200 kb beta-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homol-ogous recombination system. Gene Ther. 6:442–447.
30. Nemerow, G. R., R. A. Houghten, M. D. Moore, and N. R. Cooper. 1989.Identification of an epitope in the major envelope protein of Epstein-Barrvirus that mediates viral binding to the B lymphocyte EBV receptor (CR2).Cell 56:369–377.
31. Orford, M., M. Nefedov, J. Vadolas, F. Zaibak, R. Williamson, and P. A.
Ioannou. 2000. Engineering EGFP reporter constructs into a 200 kb humanbeta-globin BAC clone using GET Recombination. Nucleic Acids Res. 28:E84.
32. Prince, S., S. Keating, C. Fielding, P. Brennan, E. Floettmann, and M. Rowe.2003. Latent membrane protein 1 inhibits Epstein-Barr virus lytic cycleinduction and progress via different mechanisms. J. Virol. 77:5000–5007.
33. Rickinson, A. B., and E. Kieff. 2001. Epstein-Barr virus, p. 2575-2627. In M.Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams& Wilkins, Philadelphia, Pa.
34. Rowe, M., H. S. Evans, L. S. Young, K. Hennessy, E. Kieff, and A. B.Rickinson. 1987. Monoclonal antibodies to the latent membrane protein ofEpstein-Barr virus reveal heterogeneity of the protein and inducible expres-sion in virus-transformed cells. J. Gen. Virol. 68:1575–1586.
35. Sharma, R. C., and R. T. Schimke. 1996. Preparation of electrocompetent E.coli using salt-free growth medium. BioTechniques 20:42–44.
36. Shimizu, N., A. Tanabe-Tochikura, Y. Kuroiwa, and K. Takada. 1994. Iso-lation of Epstein-Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt’s lymphoma (BL) line Akata: malignant phenotypes of BLcells are dependent on EBV. J. Virol. 68:6069–6073.
37. Shimizu, N., H. Yoshiyama, and K. Takada. 1996. Clonal propagation ofEpstein-Barr virus (EBV) recombinants in EBV-negative Akata cells. J. Vi-rol. 70:7260–7263.
38. Simpson, K., and C. Huxley. 1996. A shuttle system for transfer of YACsbetween yeast and mammalian cells. Nucleic Acids Res. 24:4693–4699.
39. Sugden, B. 1989. An intricate route to immortality. Cell 57:5–7.40. Takada, K. 1984. Cross-linking of cell surface immunoglobulins induces
Epstein-Barr virus in Burkitt lymphoma lines. Int. J. Cancer 33:27–32.41. Takada, K., K. Horinouchi, Y. Ono, T. Aya, T. Osato, M. Takahashi, and S.
Hayasaka. 1991. An Epstein-Barr virus-producer line Akata: establishmentof the cell line and analysis of viral DNA. Virus Genes 5:147–156.
42. Torii, T., K. Konishi, J. Sample, and K. Takada. 1998. The truncated formof the Epstein-Barr virus LMP-1 is dispensable or complementable by full-length form in virus infection and replication. Virology 251:273–278.
43. Vazirabadi, G., T. R. Geiger, W. F. Coffin, 3rd, and J. M. Martin. 2003.Epstein-Barr virus latent membrane protein-1 (LMP-1) and lytic LMP-1localization in plasma membrane-derived extracellular vesicles and intracel-lular virions. J. Gen. Virol. 84:1997–2008.
44. Wang, D., D. Liebowitz, and E. Kieff. 1988. The truncated form of theEpstein-Barr virus latent-infection membrane protein expressed in virusreplication does not transform rodent fibroblasts. J. Virol. 62:2337–2346.
45. Wang, D., D. Liebowitz, F. Wang, C. Gregory, A. Rickinson, R. Larson, T.Springer, and E. Kieff. 1988. Epstein-Barr virus latent infection membraneprotein alters the human B-lymphocyte phenotype: deletion of the aminoterminus abolishes activity. J. Virol. 62:4173–4184.
4424 AHSAN ET AL. J. VIROL.
on January 4, 2015 by guesthttp://jvi.asm
.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















