
Decomposition-Based Assembly Synthesis for Structural Modularity

Efficient synthesis and structural analysis of newdioxopiperazine isoquinolines
Nuria Cabedo,a Noureddine El Aouad,a Inmaculada Berenguer,a Miguel Zamora,b
M. Carmen Ramırez de Arellano,c Fernando Suvire,b Almudena Bermejo,a,†
Daniel Enrizb and Diego Cortesa,*
aLaboratorio de Farmacognosia, Departamento de Farmacologıa, Facultad de Farmacia,
Universidad de Valencia, 46100 Burjassot, Valencia, SpainbDepartamento de Quımica, Universidad Nacional de San Luis, Chacabuco 917, 5700 San Luis, Argentina
cDepartamento de Quımica Organica, Facultad de Farmacia, Universidad de Valencia, 46100 Burjassot, Valencia, Spain
Received 22 September 2005; revised 21 February 2006; accepted 21 February 2006
Available online 20 March 2006
Abstract—We report herein the synthesis of new dioxopiperazine isoquinolines using the Pictet–Spengler cyclisation. Our synthetic strategyfor the preparation of two new compounds (5, 6), with a tetrahydro-6H-pyrazino[1,2-b]isoquinoline-1,4-dione moiety was developed in onlyfour steps. To understand better the crucial step of the synthesis reported here, theoretical calculations using semiempirical (PM3), ab initioand DFT computations were carried out on a reduced system model. The structure of chlorohydrate water solvate of tetrahydro(2-piperidinylethyl)-6H-pyrazino [1,2-b]isoquinoline-1,4-dione (6$HCl$2H2O) was determined by X-ray diffraction. Theoreticalcalculations (RHF/3-21G and RB3LYP/6-31G(d)) were also performed for compound 6 neutralised with a chloride ion.q 2006 Elsevier Ltd. All rights reserved.
1. Introduction
Substituted 1,2,3,4 isoquinolines represent a class of naturaland synthetic compounds that has received considerableattention because of their significant and powerful biologi-cal activities, including antitumor, antibiotic and anti-convulsant properties.1–4 In many studies aimed atdeveloping simple and efficient syntheses of polyfunctionalheteroaromatic fused isoquinolines,5 it has been reportedthat the exact structure of the reaction compounds could notbe established unequivocally, because several closelysimilar isomeric products could be formed.6,7
In the present paper, as a part of our search for newpolyfunctional heterocycles,8,9 we report the synthesis oftwo new tetrahydro-6H-pyrazino[1,2-b]isoquinoline-1,4-diones. The rigid framework of these triclyclic compoundsincorporates both L-phenylalanine and piperazine-2,5-dionebones using Pictet–Spengler cyclisation. We have introduced
0040–4020/$ - see front matter q 2006 Elsevier Ltd. All rights reserved.doi:10.1016/j.tet.2006.02.065
Keywords: Dioxopiperazine isoquinolines; Synthesis; Pictet–Spengler;X-ray; Semiempirical (PM3); Ab initio and DFT calculations.* Corresponding author. Tel.: C34 963 54 49 75; fax: C34 963 54 49 43;e-mail: [email protected]
† Current address: Departamento de Citricultura, IVIA, CarreteraMoncada-Naquera Km 4.5, 46113 Moncada, Valencia, Spain.
a flexible linker into the molecule bearing basic nitrogen as ameans of improving aqueous solubility. We describe a shortand convenient method in the asymmetric total synthesis ofthis type of compounds that may be of potential use for theconstruction of several functionalised piperazine isoquinolinederivatives and will serve as building blocks for naturalproducts.3 To know better the alkylation–cyclisation step ofthe synthesis reported here, theoretical calculations usingsemiempirical (PM3), ab initio and density functional theory(DFT) computations were carried out on a reduced systemmodel. Structure elucidation of the new products wasestablished by spectroscopy and X-ray crystallography.In addition, geometrical optimisations using both RHF/3-21G and RB3LYP/6-31G(d) calculations were performedfor tetrahydro (2-piperidinylethyl)-6H-pyrazino[1,2-b]isoqui-noline-1,4-dione (compound 6) neutralisedwith a chloride ionwith the aim to compare these computations with theexperimental results.
2. Results and discussion
2.1. Synthesis
Our approach is based on the use of Pictet–Spenglercyclisation, reaction that was readily adaptable to preparing
Tetrahedron 62 (2006) 4408–4418
NH2
CO2H
NH. HCl
CO2R
NNHO
N
CO2CH3
N
CO2CH3
BrO
L-Phenylalanine (1)
a
2: R= H
3: R= CH3
b
n
c
4
N
N
O
O
N1119
8
7 65 4
3
210
1'2'
3'
4'
7'
6'
5'
5
11a
H
N
N
O
O
N111
65 4
2
1'2'
6
H3'
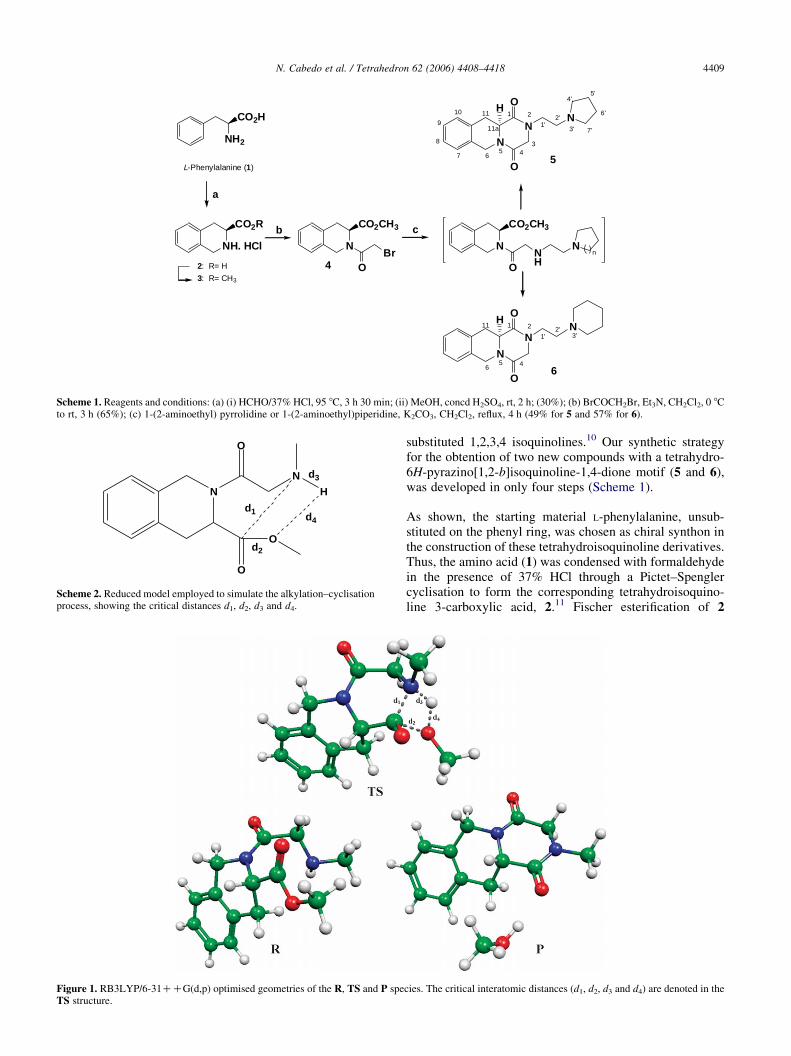
Scheme 1. Reagents and conditions: (a) (i) HCHO/37% HCl, 95 8C, 3 h 30 min; (ii) MeOH, concd H2SO4, rt, 2 h; (30%); (b) BrCOCH2Br, Et3N, CH2Cl2, 0 8Cto rt, 3 h (65%); (c) 1-(2-aminoethyl) pyrrolidine or 1-(2-aminoethyl)piperidine, K2CO3, CH2Cl2, reflux, 4 h (49% for 5 and 57% for 6).
N
O
N
O
H
O
d3
d4
d2
d1
Scheme 2. Reduced model employed to simulate the alkylation–cyclisationprocess, showing the critical distances d1, d2, d3 and d4.
Figure 1. RB3LYP/6-31CCG(d,p) optimised geometries of the R, TS and P speTS structure.
N. Cabedo et al. / Tetrahedron 62 (2006) 4408–4418 4409
substituted 1,2,3,4 isoquinolines.10 Our synthetic strategyfor the obtention of two new compounds with a tetrahydro-6H-pyrazino[1,2-b]isoquinoline-1,4-dione motif (5 and 6),was developed in only four steps (Scheme 1).
As shown, the starting material L-phenylalanine, unsub-stituted on the phenyl ring, was chosen as chiral synthon inthe construction of these tetrahydroisoquinoline derivatives.Thus, the amino acid (1) was condensed with formaldehydein the presence of 37% HCl through a Pictet–Spenglercyclisation to form the corresponding tetrahydroisoquino-line 3-carboxylic acid, 2.11 Fischer esterification of 2
cies. The critical interatomic distances (d1, d2, d3 and d4) are denoted in the
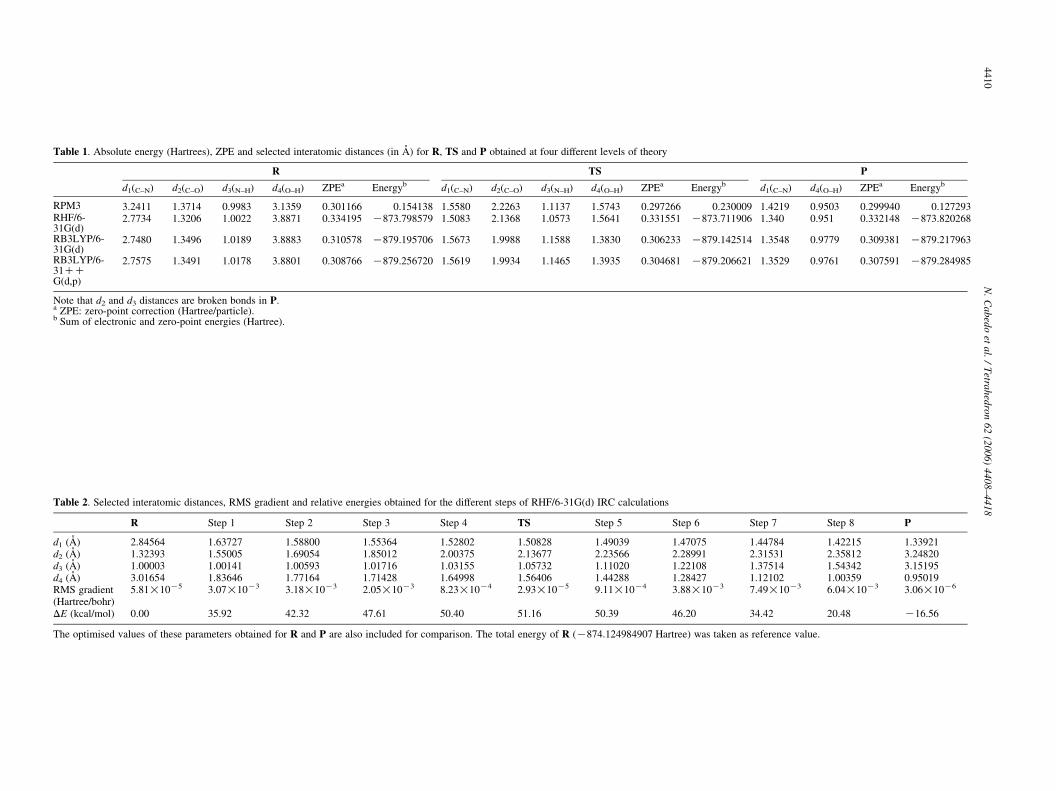
Table 1. Absolute energy (Hartrees), ZPE and selected interatomic distances (in A) for R, TS and P obtained at four different levels of theory
R TS P
d1(C–N) d2(C–O) d3(N–H) d4(O–H) ZPEa Energyb d1(C–N) d2(C–O) d3(N–H) d4(O–H) ZPEa Energyb d1(C–N) d4(O–H) ZPEa Energyb
RPM3 3.2411 1.3714 0.9983 3.1359 0.301166 0.154138 1.5580 2.2263 1.1137 1.5743 0.297266 0.230009 1.4219 0.9503 0.299940 0.127293RHF/6-31G(d)
2.7734 1.3206 1.0022 3.8871 0.334195 K873.798579 1.5083 2.1368 1.0573 1.5641 0.331551 K873.711906 1.340 0.951 0.332148 K873.820268
RB3LYP/6-31G(d)
2.7480 1.3496 1.0189 3.8883 0.310578 K879.195706 1.5673 1.9988 1.1588 1.3830 0.306233 K879.142514 1.3548 0.9779 0.309381 K879.217963
RB3LYP/6-31CCG(d,p)
2.7575 1.3491 1.0178 3.8801 0.308766 K879.256720 1.5619 1.9934 1.1465 1.3935 0.304681 K879.206621 1.3529 0.9761 0.307591 K879.284985
Note that d2 and d3 distances are broken bonds in P.a ZPE: zero-point correction (Hartree/particle).b Sum of electronic and zero-point energies (Hartree).
Table 2. Selected interatomic distances, RMS gradient and relative energies obtained for the different steps of RHF/6-31G(d) IRC calculations
R Step 1 Step 2 Step 3 Step 4 TS Step 5 Step 6 Step 7 Step 8 P
d1 (A) 2.84564 1.63727 1.58800 1.55364 1.52802 1.50828 1.49039 1.47075 1.44784 1.42215 1.33921d2 (A) 1.32393 1.55005 1.69054 1.85012 2.00375 2.13677 2.23566 2.28991 2.31531 2.35812 3.24820d3 (A) 1.00003 1.00141 1.00593 1.01716 1.03155 1.05732 1.11020 1.22108 1.37514 1.54342 3.15195d4 (A) 3.01654 1.83646 1.77164 1.71428 1.64998 1.56406 1.44288 1.28427 1.12102 1.00359 0.95019RMS gradient(Hartree/bohr)
5.81!10K5 3.07!10K3 3.18!10K3 2.05!10K3 8.23!10K4 2.93!10K5 9.11!10K4 3.88!10K3 7.49!10K3 6.04!10K3 3.06!10K6
DE (kcal/mol) 0.00 35.92 42.32 47.61 50.40 51.16 50.39 46.20 34.42 20.48 K16.56
The optimised values of these parameters obtained for R and P are also included for comparison. The total energy of R (K874.124984907 Hartree) was taken as reference value.
N.Cabedoet
al./Tetra
hedron62(2006)4408–4418
4410
–3.0 –2.5 –2.0 –1.5 –1.0 –0.5 0.0 0.5 1.0 1.5 2.0 2.5 3.0–20
–15
–10
–5
0
5
10
15
20
25
30
35
40
45
50
55(a)
(b)
(c)
To ward P
To ward R 8
5
6
7
43
P
TS
R
1
2
Rel
ativ
e E
nerg
y (k
cal/m
ol)
IRC Reaction Coordinate (amu1/2 Bohr)
IRC Reaction Coordinate (amu1/2 Bohr)
–1.4 –1.2 –1.0 –0.8 –0.6 –0.4 –0.2 0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.40.91.01.11.21.31.41.51.6
1.71.81.92.02.12.22.32.42.5
TSd
1
d2
d3
d4
Dis
tanc
e (Å
)
IRC Reaction Coordinate (amu1/2 Bohr)
–1.4 –1.2 –1.0 –0.8 –0.6 –0.4 –0.2 0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4–0.001
0.000
0.001
0.002
0.003
0.004
0.005
0.006
0.007
0.008 7
8
6
54
3
21
TS
RM
S G
radi
ent (
Har
tree
/Boh
r)
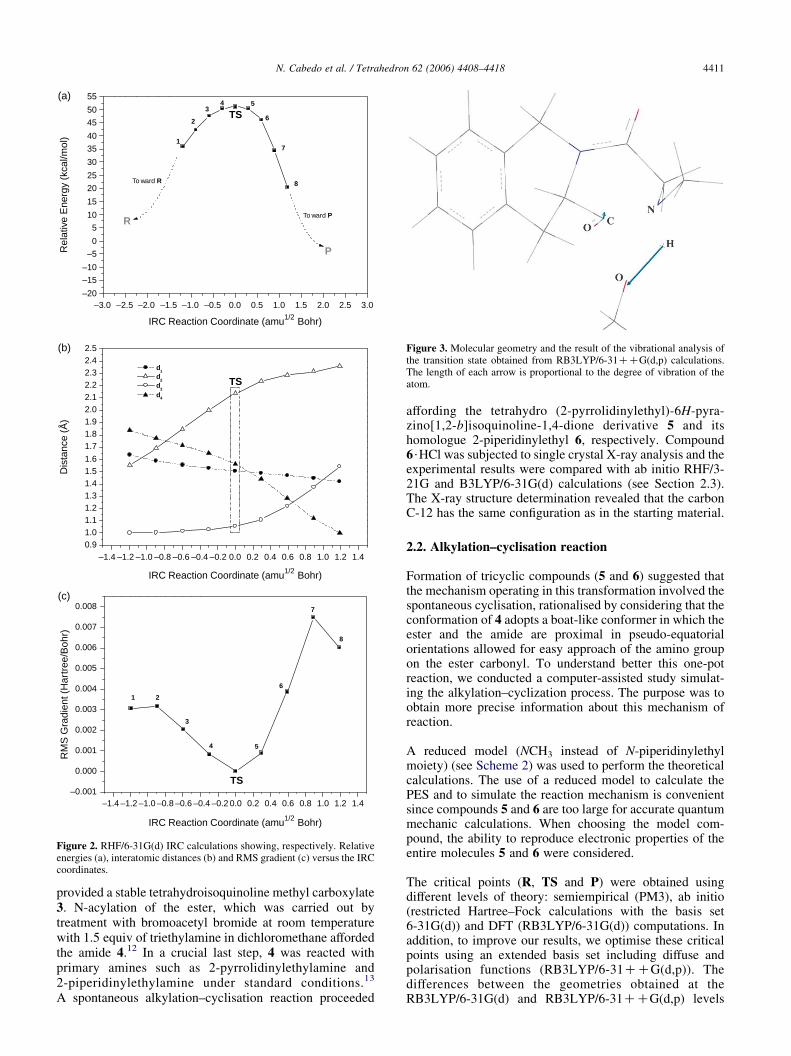
Figure 2. RHF/6-31G(d) IRC calculations showing, respectively. Relativeenergies (a), interatomic distances (b) and RMS gradient (c) versus the IRCcoordinates.
Figure 3. Molecular geometry and the result of the vibrational analysis ofthe transition state obtained from RB3LYP/6-31CCG(d,p) calculations.The length of each arrow is proportional to the degree of vibration of theatom.
N. Cabedo et al. / Tetrahedron 62 (2006) 4408–4418 4411
provided a stable tetrahydroisoquinoline methyl carboxylate3. N-acylation of the ester, which was carried out bytreatment with bromoacetyl bromide at room temperaturewith 1.5 equiv of triethylamine in dichloromethane affordedthe amide 4.12 In a crucial last step, 4 was reacted withprimary amines such as 2-pyrrolidinylethylamine and2-piperidinylethylamine under standard conditions.13
A spontaneous alkylation–cyclisation reaction proceeded
affording the tetrahydro (2-pyrrolidinylethyl)-6H-pyra-zino[1,2-b]isoquinoline-1,4-dione derivative 5 and itshomologue 2-piperidinylethyl 6, respectively. Compound6$HCl was subjected to single crystal X-ray analysis and theexperimental results were compared with ab initio RHF/3-21G and B3LYP/6-31G(d) calculations (see Section 2.3).The X-ray structure determination revealed that the carbonC-12 has the same configuration as in the starting material.
2.2. Alkylation–cyclisation reaction
Formation of tricyclic compounds (5 and 6) suggested thatthe mechanism operating in this transformation involved thespontaneous cyclisation, rationalised by considering that theconformation of 4 adopts a boat-like conformer in which theester and the amide are proximal in pseudo-equatorialorientations allowed for easy approach of the amino groupon the ester carbonyl. To understand better this one-potreaction, we conducted a computer-assisted study simulat-ing the alkylation–cyclization process. The purpose was toobtain more precise information about this mechanism ofreaction.
A reduced model (NCH3 instead of N-piperidinylethylmoiety) (see Scheme 2) was used to perform the theoreticalcalculations. The use of a reduced model to calculate thePES and to simulate the reaction mechanism is convenientsince compounds 5 and 6 are too large for accurate quantummechanic calculations. When choosing the model com-pound, the ability to reproduce electronic properties of theentire molecules 5 and 6 were considered.
The critical points (R, TS and P) were obtained usingdifferent levels of theory: semiempirical (PM3), ab initio(restricted Hartree–Fock calculations with the basis set6-31G(d)) and DFT (RB3LYP/6-31G(d)) computations. Inaddition, to improve our results, we optimise these criticalpoints using an extended basis set including diffuse andpolarisation functions (RB3LYP/6-31CCG(d,p)). Thedifferences between the geometries obtained at theRB3LYP/6-31G(d) and RB3LYP/6-31CCG(d,p) levels
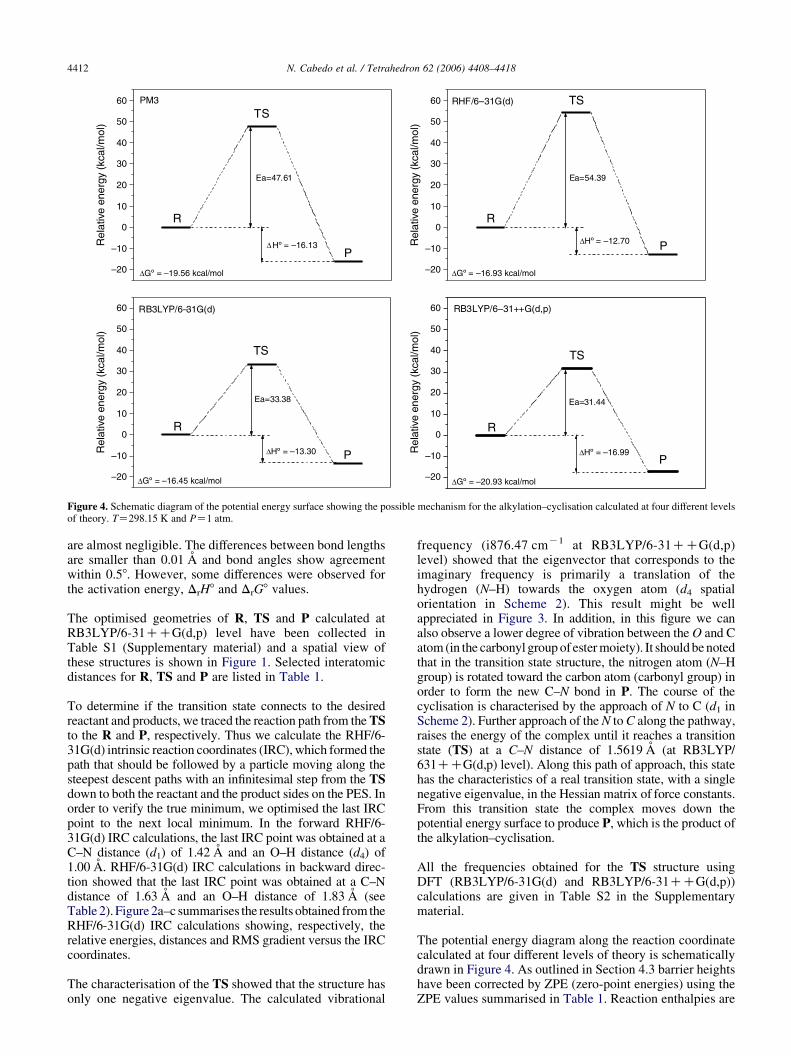
Figure 4. Schematic diagram of the potential energy surface showing the possible mechanism for the alkylation–cyclisation calculated at four different levelsof theory. TZ298.15 K and PZ1 atm.
N. Cabedo et al. / Tetrahedron 62 (2006) 4408–44184412
are almost negligible. The differences between bond lengthsare smaller than 0.01 A and bond angles show agreementwithin 0.58. However, some differences were observed forthe activation energy, DrH8 and DrG8 values.
The optimised geometries of R, TS and P calculated atRB3LYP/6-31CCG(d,p) level have been collected inTable S1 (Supplementary material) and a spatial view ofthese structures is shown in Figure 1. Selected interatomicdistances for R, TS and P are listed in Table 1.
To determine if the transition state connects to the desiredreactant and products, we traced the reaction path from theTSto the R and P, respectively. Thus we calculate the RHF/6-31G(d) intrinsic reaction coordinates (IRC), which formed thepath that should be followed by a particle moving along thesteepest descent paths with an infinitesimal step from the TSdown to both the reactant and the product sides on the PES. Inorder to verify the true minimum, we optimised the last IRCpoint to the next local minimum. In the forward RHF/6-31G(d) IRC calculations, the last IRC point was obtained at aC–N distance (d1) of 1.42 A and an O–H distance (d4) of1.00 A. RHF/6-31G(d) IRC calculations in backward direc-tion showed that the last IRC point was obtained at a C–Ndistance of 1.63 A and an O–H distance of 1.83 A (seeTable 2). Figure2a–c summarises the results obtained from theRHF/6-31G(d) IRC calculations showing, respectively, therelative energies, distances and RMS gradient versus the IRCcoordinates.
The characterisation of the TS showed that the structure hasonly one negative eigenvalue. The calculated vibrational
frequency (i876.47 cmK1 at RB3LYP/6-31CCG(d,p)level) showed that the eigenvector that corresponds to theimaginary frequency is primarily a translation of thehydrogen (N–H) towards the oxygen atom (d4 spatialorientation in Scheme 2). This result might be wellappreciated in Figure 3. In addition, in this figure we canalso observe a lower degree of vibration between theO and Catom(in the carbonyl group of estermoiety). It shouldbenotedthat in the transition state structure, the nitrogen atom (N–Hgroup) is rotated toward the carbon atom (carbonyl group) inorder to form the new C–N bond in P. The course of thecyclisation is characterised by the approach of N to C (d1 inScheme 2). Further approach of theN toC along the pathway,raises the energy of the complex until it reaches a transitionstate (TS) at a C–N distance of 1.5619 A (at RB3LYP/631CCG(d,p) level). Along this path of approach, this statehas the characteristics of a real transition state, with a singlenegative eigenvalue, in the Hessian matrix of force constants.From this transition state the complex moves down thepotential energy surface to produce P, which is the product ofthe alkylation–cyclisation.
All the frequencies obtained for the TS structure usingDFT (RB3LYP/6-31G(d) and RB3LYP/6-31CCG(d,p))calculations are given in Table S2 in the Supplementarymaterial.
The potential energy diagram along the reaction coordinatecalculated at four different levels of theory is schematicallydrawn in Figure 4. As outlined in Section 4.3 barrier heightshave been corrected by ZPE (zero-point energies) using theZPE values summarised in Table 1. Reaction enthalpies are
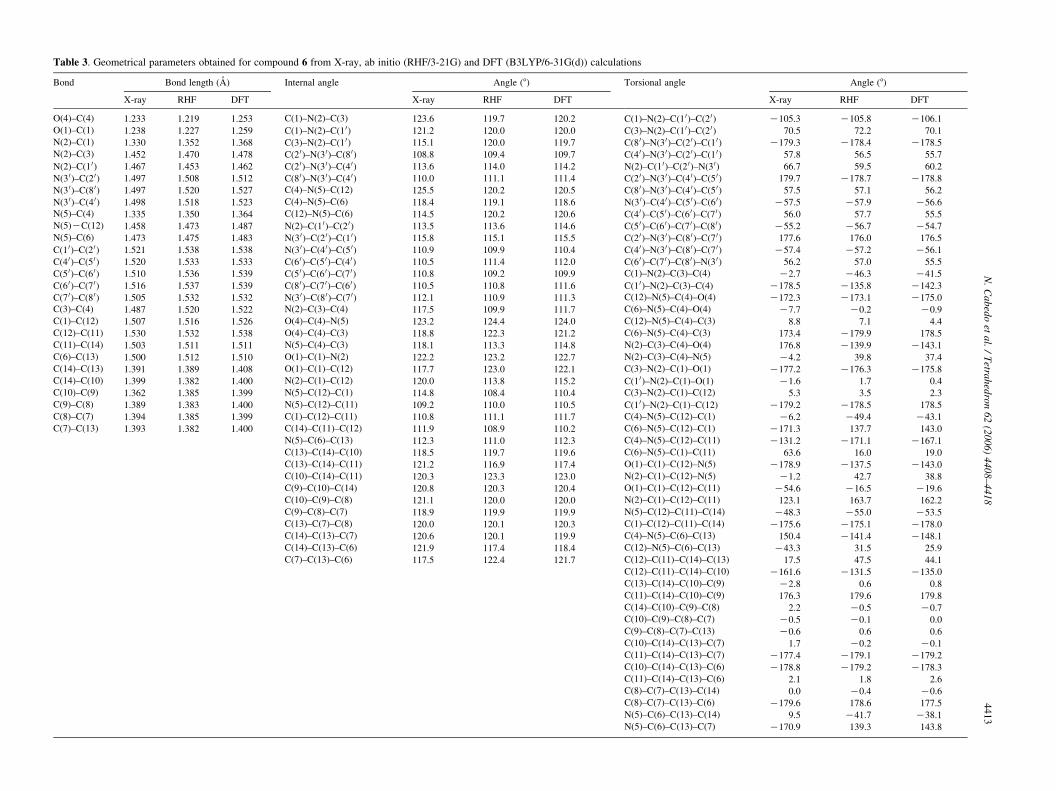
Table 3. Geometrical parameters obtained for compound 6 from X-ray, ab initio (RHF/3-21G) and DFT (B3LYP/6-31G(d)) calculations
Bond Bond length (A) Internal angle Angle (8) Torsional angle Angle (8)
X-ray RHF DFT X-ray RHF DFT X-ray RHF DFT
O(4)–C(4) 1.233 1.219 1.253 C(1)–N(2)–C(3) 123.6 119.7 120.2 C(1)–N(2)–C(1 0)–C(2 0) K105.3 K105.8 K106.1
O(1)–C(1) 1.238 1.227 1.259 C(1)–N(2)–C(1 0) 121.2 120.0 120.0 C(3)–N(2)–C(1 0)–C(2 0) 70.5 72.2 70.1
N(2)–C(1) 1.330 1.352 1.368 C(3)–N(2)–C(1 0) 115.1 120.0 119.7 C(8 0)–N(3 0)–C(2 0)–C(1 0) K179.3 K178.4 K178.5
N(2)–C(3) 1.452 1.470 1.478 C(2 0)–N(3 0)–C(8 0) 108.8 109.4 109.7 C(4 0)–N(3 0)–C(2 0)–C(1 0) 57.8 56.5 55.7
N(2)–C(1 0) 1.467 1.453 1.462 C(2 0)–N(3 0)–C(4 0) 113.6 114.0 114.2 N(2)–C(1 0)–C(2 0)–N(3 0) 66.7 59.5 60.2
N(3 0)–C(2 0) 1.497 1.508 1.512 C(8 0)–N(3 0)–C(4 0) 110.0 111.1 111.4 C(2 0)–N(3 0)–C(4 0)–C(5 0) 179.7 K178.7 K178.8
N(3 0)–C(8 0) 1.497 1.520 1.527 C(4)–N(5)–C(12) 125.5 120.2 120.5 C(8 0)–N(3 0)–C(4 0)–C(5 0) 57.5 57.1 56.2
N(3 0)–C(4 0) 1.498 1.518 1.523 C(4)–N(5)–C(6) 118.4 119.1 118.6 N(3 0)–C(4 0)–C(5 0)–C(6 0) K57.5 K57.9 K56.6
N(5)–C(4) 1.335 1.350 1.364 C(12)–N(5)–C(6) 114.5 120.2 120.6 C(4 0)–C(5 0)–C(6 0)–C(7 0) 56.0 57.7 55.5
N(5)KC(12) 1.458 1.473 1.487 N(2)–C(1 0)–C(2 0) 113.5 113.6 114.6 C(5 0)–C(6 0)–C(7 0)–C(8 0) K55.2 K56.7 K54.7
N(5)–C(6) 1.473 1.475 1.483 N(3 0)–C(2 0)–C(1 0) 115.8 115.1 115.5 C(2 0)–N(3 0)–C(8 0)–C(7 0) 177.6 176.0 176.5
C(1 0)–C(2 0) 1.521 1.538 1.538 N(3 0)–C(4 0)–C(5 0) 110.9 109.9 110.4 C(4 0)–N(3 0)–C(8 0)–C(7 0) K57.4 K57.2 K56.1
C(4 0)–C(5 0) 1.520 1.533 1.533 C(6 0)–C(5 0)–C(4 0) 110.5 111.4 112.0 C(6 0)–C(7 0)–C(8 0)–N(3 0) 56.2 57.0 55.5
C(5 0)–C(6 0) 1.510 1.536 1.539 C(5 0)–C(6 0)–C(7 0) 110.8 109.2 109.9 C(1)–N(2)–C(3)–C(4) K2.7 K46.3 K41.5
C(6 0)–C(7 0) 1.516 1.537 1.539 C(8 0)–C(7 0)–C(6 0) 110.5 110.8 111.6 C(1 0)–N(2)–C(3)–C(4) K178.5 K135.8 K142.3
C(7 0)–C(8 0) 1.505 1.532 1.532 N(3 0)–C(8 0)–C(7 0) 112.1 110.9 111.3 C(12)–N(5)–C(4)–O(4) K172.3 K173.1 K175.0
C(3)–C(4) 1.487 1.520 1.522 N(2)–C(3)–C(4) 117.5 109.9 111.7 C(6)–N(5)–C(4)–O(4) K7.7 K0.2 K0.9
C(1)–C(12) 1.507 1.516 1.526 O(4)–C(4)–N(5) 123.2 124.4 124.0 C(12)–N(5)–C(4)–C(3) 8.8 7.1 4.4
C(12)–C(11) 1.530 1.532 1.538 O(4)–C(4)–C(3) 118.8 122.3 121.2 C(6)–N(5)–C(4)–C(3) 173.4 K179.9 178.5
C(11)–C(14) 1.503 1.511 1.511 N(5)–C(4)–C(3) 118.1 113.3 114.8 N(2)–C(3)–C(4)–O(4) 176.8 K139.9 K143.1
C(6)–C(13) 1.500 1.512 1.510 O(1)–C(1)–N(2) 122.2 123.2 122.7 N(2)–C(3)–C(4)–N(5) K4.2 39.8 37.4
C(14)–C(13) 1.391 1.389 1.408 O(1)–C(1)–C(12) 117.7 123.0 122.1 C(3)–N(2)–C(1)–O(1) K177.2 K176.3 K175.8
C(14)–C(10) 1.399 1.382 1.400 N(2)–C(1)–C(12) 120.0 113.8 115.2 C(1 0)–N(2)–C(1)–O(1) K1.6 1.7 0.4
C(10)–C(9) 1.362 1.385 1.399 N(5)–C(12)–C(1) 114.8 108.4 110.4 C(3)–N(2)–C(1)–C(12) 5.3 3.5 2.3
C(9)–C(8) 1.389 1.383 1.400 N(5)–C(12)–C(11) 109.2 110.0 110.5 C(1 0)–N(2)–C(1)–C(12) K179.2 K178.5 178.5
C(8)–C(7) 1.394 1.385 1.399 C(1)–C(12)–C(11) 110.8 111.1 111.7 C(4)–N(5)–C(12)–C(1) K6.2 K49.4 K43.1
C(7)–C(13) 1.393 1.382 1.400 C(14)–C(11)–C(12) 111.9 108.9 110.2 C(6)–N(5)–C(12)–C(1) K171.3 137.7 143.0
N(5)–C(6)–C(13) 112.3 111.0 112.3 C(4)–N(5)–C(12)–C(11) K131.2 K171.1 K167.1
C(13)–C(14)–C(10) 118.5 119.7 119.6 C(6)–N(5)–C(1)–C(11) 63.6 16.0 19.0
C(13)–C(14)–C(11) 121.2 116.9 117.4 O(1)–C(1)–C(12)–N(5) K178.9 K137.5 K143.0
C(10)–C(14)–C(11) 120.3 123.3 123.0 N(2)–C(1)–C(12)–N(5) K1.2 42.7 38.8
C(9)–C(10)–C(14) 120.8 120.3 120.4 O(1)–C(1)–C(12)–C(11) K54.6 K16.5 K19.6
C(10)–C(9)–C(8) 121.1 120.0 120.0 N(2)–C(1)–C(12)–C(11) 123.1 163.7 162.2
C(9)–C(8)–C(7) 118.9 119.9 119.9 N(5)–C(12)–C(11)–C(14) K48.3 K55.0 K53.5
C(13)–C(7)–C(8) 120.0 120.1 120.3 C(1)–C(12)–C(11)–C(14) K175.6 K175.1 K178.0
C(14)–C(13)–C(7) 120.6 120.1 119.9 C(4)–N(5)–C(6)–C(13) 150.4 K141.4 K148.1
C(14)–C(13)–C(6) 121.9 117.4 118.4 C(12)–N(5)–C(6)–C(13) K43.3 31.5 25.9
C(7)–C(13)–C(6) 117.5 122.4 121.7 C(12)–C(11)–C(14)–C(13) 17.5 47.5 44.1
C(12)–C(11)–C(14)–C(10) K161.6 K131.5 K135.0
C(13)–C(14)–C(10)–C(9) K2.8 0.6 0.8
C(11)–C(14)–C(10)–C(9) 176.3 179.6 179.8
C(14)–C(10)–C(9)–C(8) 2.2 K0.5 K0.7
C(10)–C(9)–C(8)–C(7) K0.5 K0.1 0.0
C(9)–C(8)–C(7)–C(13) K0.6 0.6 0.6
C(10)–C(14)–C(13)–C(7) 1.7 K0.2 K0.1
C(11)–C(14)–C(13)–C(7) K177.4 K179.1 K179.2
C(10)–C(14)–C(13)–C(6) K178.8 K179.2 K178.3
C(11)–C(14)–C(13)–C(6) 2.1 1.8 2.6
C(8)–C(7)–C(13)–C(14) 0.0 K0.4 K0.6
C(8)–C(7)–C(13)–C(6) K179.6 178.6 177.5
N(5)–C(6)–C(13)–C(14) 9.5 K41.7 K38.1
N(5)–C(6)–C(13)–C(7) K170.9 139.3 143.8
N.Cabedoet
al./Tetra
hedron62(2006)4408–4418
4413

N. Cabedo et al. / Tetrahedron 62 (2006) 4408–44184414
moreover corrected for thermal energy calculated at298.15 K. Inspection of the collected data in Figure 4indicates that the barrier heights are sensitive tothe employed theory level. Thus, PM3 andRHF/6-31G(d) calculations predict an activation energy of47.61 and 54.39 kcal/mol, whereas RB3LYP/6-31G(d) andRB3LYP/6-31CCG(d,p) calculations suggest 33.38 and31.44 kcal/mol, respectively. Analysis of the DFT dataindicates that enlarging the basis set (from 6-31G(d) to6-31CCG(d,p)) increases the stability of the transitionstate structure by 1.94 kcal/mol. It is interesting to note thatthe free energy of activation of the reaction at the highestlevel of theory is 31.44 kcal/mol, an indicative value ofwhich the reaction can be carried out at room temperature ata reasonable rate.
The reaction enthalpies (DrH8) for the alkylation–cyclisa-tion calculated within the different approaches employed inthis study are summarised in Figure 3. In contrast with thefeatures found in the barrier height analysis, the calculatedvalues are quite similar at the different levels of theory. Ourtheoretical results strongly suggest that the alkylation–cyclisation is an exothermic process (DrH8ZK17.13 kcal/mol at RB3LYP/6-31CCG(d,p) level), which is inagreement with our experimental data. It occurs if thereagent can assume the proper orientation. The proposedreaction mechanism with four-centre TS that has beencharacterised, as described previously, is a true reaction paththat shows to be reasonable according to the estimatedactivation energy.
The Ea, DrH8 and DrG8 values obtained from DFTcalculations for the alkylation–cyclisation could explainthe spontaneous nature of this process. However, it shouldbe noted that our theoretical calculations had beenperformed for a reduced model system in vacuum; thereforecaution is needed in these interpretations, because thecomplete molecular systems as well as solvent effects couldchange somewhat these results.14
2.3. Crystal structure analysis
The solid state structure of 6$HCl$2H2O was determined bysingle crystal X-ray diffraction (Table 3, Fig. 5). Crystalstructure confirms the absolute configuration (S) on C(12)which is in accordance with the starting material,L-phenylalanine. The three fused rings are not coplanar
Figure 5. X-ray ellipsoid plot of the cation of 6$HCl (H2O molecules havebeen removed for a better view of the crystal structure).
and present a half-folded conformation. The plane formedby the aromatic ring C7–C8–C9–C10–C13–C14 and C6 andC11 (mean deviation 0.019 A) makes a dihedral angle of33.7(2)8with the C1–N2–C3–C4–N5–C12 ring plane (meandeviation 0.029 A). The piperidine ring adopts the chairconformation with the flexible connecting chain in theequatorial position. The bond lengths of N(3 0)–C(2 0)(1.497(5) A), C(2 0)–C(1 0) (1.519(6) A) and C(1 0)–N(2)(1.469(5) A) show the C–N and C–C single bond character.The ammonium hydrogen atom is acting as hydrogen bonddonor to the chloride atom (N3 0/Cl1 3.159(4) A, H3 0/Cl12.26 A, N3 0-H3 0/Cl1 168.38). Other hydrogen bonds havebeen found involving the two water solvated moleculeshydrogen atoms and the carbonyl or the water oxygen atomsor the chloride ion (O5–H5A/O1 163(5)8, O5/O12.793(5) A, H5A/O1 1.92(2); O5–H5B/O4i 166(5)8,(i) KxC1, yK1/2, KzC1/2, O5/O4i 2.812(5) A,H5B/O4i 1.94(2) A; O6–H6D/Cl1 131(5)8, O6/Cl13.249(6) A, H6D/Cl1 2.60(5) A; O6–H6C/O5ii 161(6),(ii)KxC1, yC1/2,KzC1/2, O6/O5ii 2.874(7) A, H6C/O5ii 2.02(4) A).
We were also interested to learn whether the experimentallyobtained structure of 6$HCl$2H2O could also be reproducedwith reasonable agreement by standard computationalmethods. Thus we calculate compound 6 neutralised with achloride (ClK) ion. To perform these optimisations we choseas starting geometry the conformation obtained from X-raywhere the chloride was located at an arbitrary C–NH distanceof 3.07 A. The z-matrix of the optimised geometry at theB3LYP/6-31G(d) level is given inTable S3 the supplementarymaterial. In order to obtain information about the electronicaspects, we calculate the molecular electrostatic potential(MEP) using RB3LYP/6-31G(d) calculations. A spatial viewof the MEP obtained for compound 6 neutralised with achloride ClK is shown in Figure 6.
The geometry optimisations have been carried out at therestricted Hartree–Fock (RHF) level with 3-21G basis setand RB3LYP/6-31G(d) level of theory. Table 3 gives themain geometrical parameters for the RHF/3-21G and DFToptimised geometries. Both levels of calculations wellreproduce the bond distances and angles observed at X-rayeven when periodical conditions were not considered in ourcalculations. Agreements between the calculated structuresand the experimentally determined X-ray crystal structurewere excellent; a strikingly significant correlation RZ0.99904 (RHF/3-21G) and RZ0.99911 (B3LYP/6-31G(d))was found between the bond lengths optimised at bothlevels of theory and those obtained from X-ray. Theexperimentally determined bond lengths are slightly shorterthan those observed in the DFT calculated geometry. Thesedifferences in bond lengths may be due to the shortintermolecular contacts in the crystal. Inspection of Figure 7reveals the similarity between the X-ray, RHF/3-21G andRB3LYP/6-31G(d) geometries. Specifically, only minutedeviation was found between torsion angle values found atX-ray when compared to those found at RHF/3-21G or atRB3LYP/6-31G(d) levels; being the main difference thespatial position of the piperidine ring (see Table 2). Thedifferent environments, that is, crystal versus isolatedmolecule may yield differences between both experimentaland theoretical structures due to the role played by
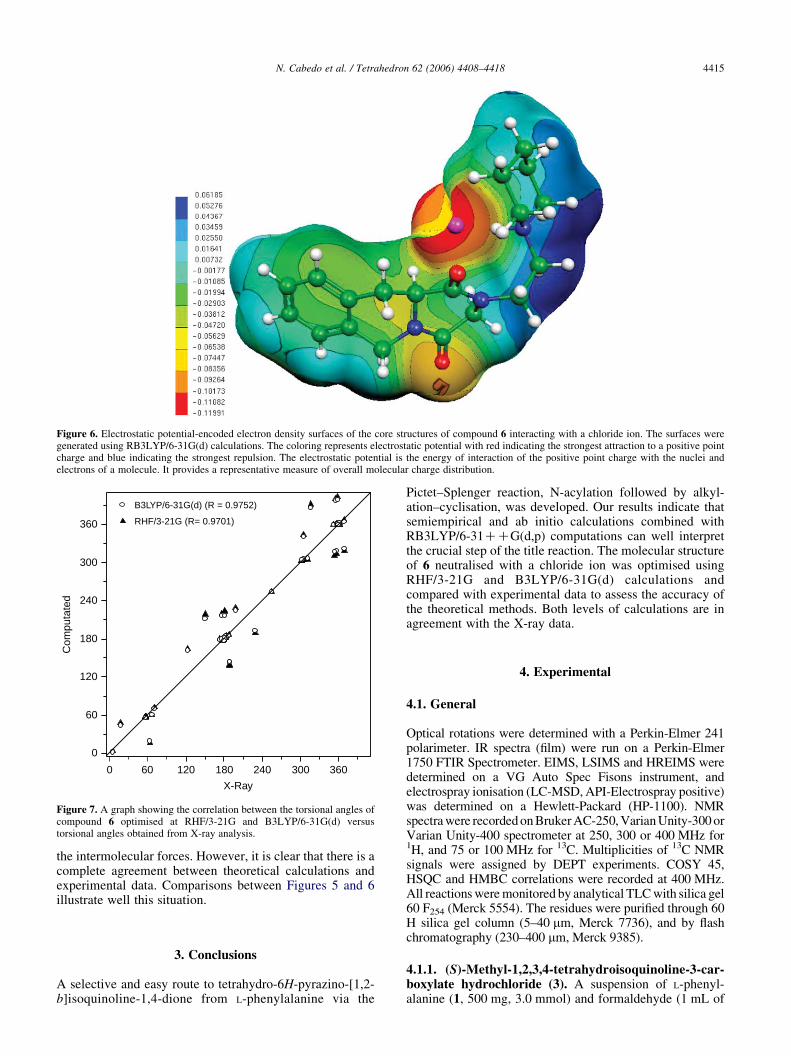
Figure 6. Electrostatic potential-encoded electron density surfaces of the core structures of compound 6 interacting with a chloride ion. The surfaces weregenerated using RB3LYP/6-31G(d) calculations. The coloring represents electrostatic potential with red indicating the strongest attraction to a positive pointcharge and blue indicating the strongest repulsion. The electrostatic potential is the energy of interaction of the positive point charge with the nuclei andelectrons of a molecule. It provides a representative measure of overall molecular charge distribution.
0 60 120 180 240 300 360
0
60
120
180
240
300
360 RHF/3-21G (R= 0.9701)
Com
puta
ted
X-Ray
B3LYP/6-31G(d) (R = 0.9752)
Figure 7. A graph showing the correlation between the torsional angles ofcompound 6 optimised at RHF/3-21G and B3LYP/6-31G(d) versustorsional angles obtained from X-ray analysis.
N. Cabedo et al. / Tetrahedron 62 (2006) 4408–4418 4415
the intermolecular forces. However, it is clear that there is acomplete agreement between theoretical calculations andexperimental data. Comparisons between Figures 5 and 6illustrate well this situation.
3. Conclusions
A selective and easy route to tetrahydro-6H-pyrazino-[1,2-b]isoquinoline-1,4-dione from L-phenylalanine via the
Pictet–Splenger reaction, N-acylation followed by alkyl-ation–cyclisation, was developed. Our results indicate thatsemiempirical and ab initio calculations combined withRB3LYP/6-31CCG(d,p) computations can well interpretthe crucial step of the title reaction. The molecular structureof 6 neutralised with a chloride ion was optimised usingRHF/3-21G and B3LYP/6-31G(d) calculations andcompared with experimental data to assess the accuracy ofthe theoretical methods. Both levels of calculations are inagreement with the X-ray data.
4. Experimental
4.1. General
Optical rotations were determined with a Perkin-Elmer 241polarimeter. IR spectra (film) were run on a Perkin-Elmer1750 FTIR Spectrometer. EIMS, LSIMS and HREIMS weredetermined on a VG Auto Spec Fisons instrument, andelectrospray ionisation (LC-MSD,API-Electrospray positive)was determined on a Hewlett-Packard (HP-1100). NMRspectrawere recordedonBrukerAC-250,VarianUnity-300orVarian Unity-400 spectrometer at 250, 300 or 400 MHz for1H, and 75 or 100 MHz for 13C. Multiplicities of 13C NMRsignals were assigned by DEPT experiments. COSY 45,HSQC and HMBC correlations were recorded at 400 MHz.All reactionsweremonitored by analytical TLCwith silica gel60 F254 (Merck 5554). The residues were purified through 60H silica gel column (5–40 mm, Merck 7736), and by flashchromatography (230–400 mm, Merck 9385).
4.1.1. (S)-Methyl-1,2,3,4-tetrahydroisoquinoline-3-car-boxylate hydrochloride (3). A suspension of L-phenyl-alanine (1, 500 mg, 3.0 mmol) and formaldehyde (1 mL of

N. Cabedo et al. / Tetrahedron 62 (2006) 4408–44184416
a 37% aqueous solution) in concd HCl (5 mL) was heatedfor 30 min at 95 8C. Then concd HCl (1 mL) andformaldehyde (0.5 mL) were added and the mixture washeated at 95 8C for another 3 h. The mixture reaction wasallowed to cool to room temperature overnight and the solidwas filtered and washed with H2O. Then, the residue ofTHIQ 2 hydrochloride was dissolved in hot MeOH (10 mL),treated with concd H2SO4 (0.2 mL) and shaken at roomtemperature for 2 h. The solvent was concentrated underreduced pressure to give 204 mg of the methyl esterhydrochloride 3 (0.9 mmol, 30%) as a white solid, whichwas used directly in the next step.
1H NMR (300 MHz, CDCl3) d: 2.86 (br s, 1H, NH), 2.91(dd, JZ16.2, 10.2 Hz, 1H, CH2-4a), 3.05 (dd, JZ16.2,4.8 Hz, 1H, CH2-4b), 3.71 (dd, JZ10.2, 4.8 Hz, 1H, CH-3),3.73 (s, 3H, OCH3), 4.05 (s, 2H, CH2-1), 6.95–7.14 (m, 4H,ArH); 13C NMR (300 MHz, CDCl3) d: 31.0 (CH2-4), 46.6(CH2-1), 52.0 (OCH3), 55.3 (CH-3), 125.9 (CH), 126.1(CH), 126.2 (CH), 128.8 (CH), 132.4 (2C), 172.9(COOCH3); APIES positive m/z (%): 214 (100) [MCCNaC], 192 (52) [MHC].
4.1.2. (S)-Methyl-2-(2-bromoacetyl)-1,2,3,4-tetrahydro-isoquinoline-3-carboxylate (4a,4b). Under N2, bromoace-tyl bromide (0.045 mL, 0.52 mmol, 1 equiv) was added to astirred solution of THIQ methyl ester hydrochloride, 3(118 mg, 0.52 mmol, 1 equiv) and Et3N (0.11 mL,0.78 mmol, 1.5 equiv) in anhydrous CH2Cl2 (4 mL) at0 8C and then, stirred at room temperature for 3 h. Thereaction mixture was washed with saturated NaHCO3
solution, H2O and brine, dried over Na2SO4, filtered andevaporated under reduced pressure. The residue waspurified by flash chromatography (CH2Cl2/EtOAc, 9.6:0.4)to afford 105 mg of bromoacetamide-THIQ derivative 4(65%) as a mixture of rotamers in a 2:1 ratio of 4a:4brotamers.
1H NMR (400 MHz, CDCl3) d: 3.15 (dd, JZ16.0, 6.0 Hz,1H, CH2a-4, rotamer-4a), 3.26 (dd, JZ16.0, 4.0 Hz, 1H,CH2b-4, rotamer-4a), 3.29–3.42 (m, 2H, CH2-4, rotamer-4b), 3.62 (s, 3H, OCH3, rotamer-4b), 3.63 (s, 3H, OCH3,rotamer-4a), 3.93 (m, 2H, CH2Br, rotamer-4b), 4.0 (d, JZ10.8 Hz, 1H, CH2a–Br, rotamer-4a), 4.06 (d, JZ10.8 Hz,1H, CH2b–Br, rotamer-4a), 4.53 (d, JZ17.6 Hz, 1H, CH2a-1, rotamer-4b), 4.78 (s, 2H, CH2-1, rotamer-4a), 4.95 (d, JZ17.6 Hz, 2H, CHb-1 and CH-3, rotamer-4b), 5.37 (dd, JZ6.0, 4.0 Hz, 1H, CH-3, rotamer-4a), 7.10–7.30 (m, 4H,ArH); 13C NMR (100 MHz, CDCl3) d: 26.1 (CH2Br), 30.5(CH2-4), 46.2 (CH2-1), 52.0 (OCH3), 52.4 (CH-3), 126.0(CH), 126.8 (CH), 127.0 (CH), 128.3 (CH), 131.4 (C), 131.8(C), 166.6 (CO), 170.8 (COOCH3); EIMS m/z (%): 312 (5)[MC], 232 (100) [MCKBr], 190 (75) [MCKCOCH2Br],146 (88) [MCKCOCH2Br–CH3], 130 (79).
4.1.3. (S)-2,3,11,11a-Tetrahydro-2-(2 0-(pyrrolidin-3 0-yl)ethyl)-6H-pyrazino-[1,2-b]isoquinoline-1,4-dione (5)and its 2-(2 0-piperidin-3 0-yl)ethyl homologue (6). UnderN2, a stirred suspension of (S)-methyl-2-(2-bromoacetyl)-1,2,3,4-tetrahydro-isoquinoline-3-carboxylate, 4 (100 mg,0.32 mmol, 1 equiv), 1-(2-aminoethyl)piperidine(0.046 mL, 0.32 mmol, 1 equiv) and anhydrous K2CO3
(70 mg) in dry CH2Cl2 (6 mL) was refluxed for 4 h. Then,
the reaction mixture was cool to room temperature, andwashed with H2O, brine, dried over NaSO4, filtered andevaporated under reduced pressure. The residue waspurified by flash chromatography (CH2Cl2/MeOH/NH4OH, 9.6:0.4:1 drop) to obtain 60 mg of compound 6(57%). The same procedure using 1-(2-aminoethyl)pyrroli-dine was accomplished for compound 5 (49 mg, 49%).
Compound 5. [a]DZK1428 (c 0.9, EtOH); IR (dry film)nmax: 3470, 2944, 1673, 1456, 1325 cmK1; 1H NMR(300 MHz, CDCl3) d: 1.70–1.84 (m, 4H, CH2-5
0 and CH2-6 0), 2.50–2.60 (m, 4H, CH2-4
0 and CH2-70), 2.71 (t, JZ
6.5 Hz, 2H, CH2-20), 3.01 (dd, JZ15.9, 3.4 Hz, 1H, CH2a-
11), 3.43 (dd, JZ15.9, 12.4 Hz, 1H, CH2b-11), 3.50–3.66(m, 2H, CH2-1
0), 4.19 (s, 2H, CH2-3), 4.16–4.25 (m, 1H,CH-11a), 4.33 (d, JZ17.1 Hz, 1H, CH2a-6), 5.29 (d, JZ17.1 Hz, 1H, CH2b-6), 7.10–7.30 (m, 4H, ArH); 13C NMR(75 MHz, CDCl3) d: 23.4 (CH2-5
0 and CH2-60), 33.5 (CH2-
11), 43.89 (CH2-6), 44.9 (CH2-10), 50.3 (CH2-3), 53.0 (CH2-
4 0and CH2-70), 54.1 (CH2-2
0), 55.5 (CH-11a), 126.2 (CH),126.9 (CH), 128.6 (2CH), 131.2 (C-10a), 132.4 (C-6a),162.6 (C-1), 164.8 (C-4); LSIMS m/z 314 [MHC].
Compound 6. [a]D25K1568 (c 0.8, EtOH); IR (dry film) nmax:
3486, 2934, 1661, 1470, 1325 cmK1; 1H NMR (300 MHz,CDCl3) d: 1.34–1.45 (m, 1H, H-6 0), 1.50–1.60 (m, 4H, CH2-5 0and CH2-7
0), 2.35–2.48 (m, 4H, CH2-40 and CH2-8
0), 2.54(t, JZ6.3 Hz, 2H, CH2-2
0), 3.01 (dd, JZ16.0, 12.4 Hz, 1H,CH2a-11), 3.40 (dd, JZ16.0, 3.7 Hz, 1H, CH2b-11), 3.44–3.54 (m, 1H, CH2a-1
0), 3.58–3.68 (m, 1H, CH2b-10), 4.15 (s,
2H, CH2-3), 4.18–4.24 (m, 1H, CH-11a), 4.30 (d, JZ17 Hz,1H, CH2a-6), 5.25 (d, JZ17 Hz, 1H, CH2b-6), 7.12–7.30(m, 4H, ArH); 13C NMR (75 MHz, CDCl3) d: 24.1 (CH2-6
0),25.8 (CH2-5
0 and CH2-70), 33.4 (CH2-11), 43.1 (CH2-1
0),43.7 (CH2-6), 50.3 (CH2-3), 54.5 (CH2-4
0 and CH2-80), 55.5
(CH-11a), 55.7 (CH2-20), 126.1 (CH), 126.8 (CH), 128.6
(2CH), 131.3 (C-10a), 132.4 (C-6a), 162.6 (C-4), 164.73(C-1); HRMS (EI) m/z calcd for C19H25N3O2 [MC]327.1947, found 327.1943.
4.2. Crystal structure determination of 6$HCl$2H2O
A colourless lath of 0.79!0.23!0.07 mm size wasmounted on a glass fibre and transferred to the diffracto-meter (orthorhombic, P212121, aZ7.9742(16), bZ12.875(3), cZ19.791(4) A, VZ2032.0(7) A3, ZZ4, rcalcd-Z1.307 g cmK3, qmaxZ24.98, Mo Ka, lZ0.71073 A,u-scan, diffractometer Nonius CAD4, TZ293(2) K, 5953reflections collected of which 3557 were independent,RintZ0.052). The structure was solved by direct methodsand refined anisotropically on F2 (SHELXS-97 andSHELXL-97, Sheldrick, University of Gottingen, 1997).The water hydrogen atoms were located in a differenceFourier synthesis and refined with restrained O–H bondlength. Other hydrogen atoms were included using a ridingmodel. The absolute structure was determined (Flackparameter 0.03(13); Flack, H. D. Acta Crystallogr., Sect. A,1983, 39, 876).
4.3. Theoretical calculations
All the calculations have been performed with theGAUSSIAN 03 suit of programs.15 The geometries of all

N. Cabedo et al. / Tetrahedron 62 (2006) 4408–4418 4417
the relevant points along the reaction path for thealkylation–cyclisation (R, TS and P), have been optimisedat PM3, RHF/6-31G(d), RB3LYP/6-31G(d) and RB3LYP/6-31CCG(d,p) levels of theory. Vibrational frequenciesfor the optimised structures were computed to evaluate thezero-point energies (ZPE) as well as to confirm the nature ofthe singular points along the potential energy surface. Thestationary points have been identifies as a minimum with noimaginary frequencies, or as a maximum characterised bythe existence of only one imaginary frequency in the normalmode coordinate analysis. Transition state structures werelocated until the Hessian matrix had only one imaginaryeigenvalue, and the transition states were also confirmed byanimating the negative eigenvectors coordinate with avisualisation program and internal reaction coordinate(IRC) calculations.16,17 RHF/6-31G(d) IRC calculationswere performed on the transition state (TS) structure tocheck that the TS structure leads to reactants and to thereaction product (forward and reverse directions of thereaction path). IRC calculations steps 4 points in cartesianscoordiantes in the forward direction and 4 points in thereverse direction, in steps of 0.3 amu1/2 bohr along the pathwere carried out. Correlation effects were included usingdensity functional theory (DFT) with the Becke3-Lee-Yang-Parr (B3LYP)18 functional. The B3LYP method gavereasonable relative energies with diffuse basis sets, and, forexample, 6-31CG(d,p) was suggested by Csonka,19 andHoffman and Rychlewsky20 as sufficient but still computa-tionally economical choice. However, regarding the basisset superimposition errors (BSSEs), the basis set should beextended to 6-31CCG(d,p).20
Molecular geometry optimisations for the conformationobtained from X-ray neutralised with a chloride wereperformed at two levels of theory, RHF/3-21G andRB3LYP/6-31G(d). The electronic study of this confor-mation was carried out using molecular electrostaticpotentials (MEPs).21,22 MEPs were calculated usingRB3LYP/6-31G(d) wave function from Gaussian 03program. The Molekel program23 was used as the graphicinterface to visualise the MEPs.
Acknowledgements
The authors want to thank the Fundacion Ana y Jose Royofor postdoctoral fellowship to N.C. This work was partiallysupported by grants from Universidad Nacional de SanLuis-Argentina. R.D.E. is member of the Scientific staff ofCONICET (Argentina). Lic M.F. Massman originallycarried out some calculations reported here and wegratefully acknowledge his contribution to this research.
Supplementary data
Supplementary data associated with this article can befound, in the online version, at doi:10.1016/j.tet.2006.02.065. Crystallographic data (excluding structure factors) forthe structure in this paper have been deposited with theCambridge Crystallographic Data Centre as supplementarypublication number CCDC 284187. Copies of the data can
be obtained, free of charge, on application to CCDC, 12Union Road, Cambridge CB2 1EZ, UK [fax: C1 44 1223336033 or e-mail: [email protected]]. Depositedinformation necessary to guarantee reproducibility.
Table S2 gives the analytical frequencies of TS obtainedfrom RB3LYP/6-31G(d) and RB3LYP/6-31CCg(d,p)calculations.
Table S3 gives the RB3LYP/6-31G(d) optimised geometryof compound 6 neutralised with a chloride.
References and notes
1. Bentley, K. W. Nat. Prod. Rep. 2005, 22, 249.
2. Hamburger, M. Phytochem. Rev. 2002, 1, 333.
3. Hatzelt, A.; Laschat, S.; Jones, P. G.; Grunenberg, J. Eur.
J. Org. Chem. 2002, 3936.
4. Gitto, R.; Caruso, R.; Orlando, V.; Quartarone, S.; Barreca,
M. L.; Ferreri, G.; Russo, E.; De Sarro, G.; Chimirri, A. Il
Farmaco 2004, 59, 7.
5. Awad, E. M.; Elwan, N. M.; Hassaneen, H. M.; Linden, A.;
Heimgartner, H. Helv. Chim. Acta 2001, 84, 1172.
6. Elwan, N. M.; Abdelhadi, H. A.; Abdellah, T. A.; Hassaneen,
H. M. Tetrahedron 1996, 52, 3451.
7. Herberich, B.; Kinugawa, M.; Vazquez, A.; Williams, R. M.
Tetrahedron Lett. 2001, 42, 543.
8. Cabedo, N.; Protais, P.; Cassels, B. K.; Cortes, D. J. Nat. Prod.
1998, 61, 709.
9. Cabedo, N.; Andreu, I.; Ramırez de Arellano, M. C.;
Chagraoui, A.; Serrano, A.; Bermejo, A.; Protais, P.; Cortes,
D. J. Med. Chem. 2001, 44, 1794.
10. (a) Pictet, A.; Spengler, T. Ber. 1911, 44, 2030. (b) Cox, E. D.;
Cook, J. M. Chem. Rev. 1995, 95, 1797.
11. (a) Julian, P.-L.; Karpel, W.-J.; Magnani, A.; Meyer, E.-W.
J. Am. Chem. Soc. 1948, 70, 180. (b) Monsees, A.; Laschat, S.;
Kotila, S.; Fox, T.; Wurthwein, E.-U. Liebigs Ann./Recueil.
1997, 533.
12. Gois, P. M. P.; Afonso, C. A. M. Eur. J. Org. Chem. 2003,
3798.
13. Chiou, C.-M.; Kang, J.-J.; Lee, S.-S. J. Nat. Prod. 1998, 61, 46.
14. Suvire, F. D.; Rodriguez, A. M.; Mak, M. L.; Papp, J.; Enriz,
R. D. J. Mol. Struct. (Theochem) 2001, 540, 257.
15. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;
Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.;
Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar,
S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.;
Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada,
M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida,
M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.;
Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.;
Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.;
Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala,
P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg,
J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain,
M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.;
Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.;
Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.;
Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.
L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.;
Nanayakkara, A.; Challacombe, M.; Gill, P. M.W.; Johnson, B.;

N. Cabedo et al. / Tetrahedron 62 (2006) 4408–44184418
Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian
03, Revision B.05, Gaussian, Inc.: Pittsburgh PA, 2003.
16. Gonzalez, C.; Schlegel, H. B. J. Chem. Phys. 1989, 90,
2154.
17. Gonzalez, C.; Schlegel, H. B. J. Chem. Phys. 1990, 94,
5523.
18. Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
19. Csonka, G. I. J. Mol. Struct. (Theochem) 2002, 584, 1.
20. Hoffman, M.; Rychlewski, J. Comput. Meth. Sci. Technol.
2000, 6, 61.
21. Polilzer, P.; Truhlar, D. G. Chemical Applications of Atomic
and Molecular Eletrostatic Potentials; Plenum: New York,
1991.
22. Greeling, P.; Langenaeker, W.; De Proft, F.; Baeten, A.
Molecular Electrostatic Potentials: Concepts and Aplications;
Theoretical and Computational Chemistry; Elsevier Science
B.V.: Amsterdam, 1996; Vol. 3; pp 587–617.
23. Flukiger, P.; Luthi, H. P.; Portmann, S.; Weber, J. MOLEKEL
4.0; Swiss Center for Scientific Computing: Manno, Switzer-
land, 2000.
Copyright © 2022 FDOKUMEN










![Efficient synthesis of 2- and 3-substituted-2,3-dihydro [1,4]dioxino[2,3- b]pyridine derivatives](https://static.fdokumen.com/doc/165x107/6324e7974643260de90d793b/efficient-synthesis-of-2-and-3-substituted-23-dihydro-14dioxino23-bpyridine.jpg)







![Fast and Efficient Synthesis of Pyrano[3,2- c ]quinolines Catalyzed by Niobium(V) Chloride](https://static.fdokumen.com/doc/165x107/6337c72f65077fe2dd044087/fast-and-efficient-synthesis-of-pyrano32-c-quinolines-catalyzed-by-niobiumv.jpg)

