
Quality control despite mistranslation caused by an ambiguous genetic code
Upload
independentCategory
view
1download
0
A
FSowB
rH©
K
1
aleshva
0d
Virus Research 123 (2007) 72–85
Dynamics of a hepatitis B virus e antigen minus population ascribed togenotype F during the course of a chronic infection despite
the presence of anti-HBs antibodies
V.L. Mathet a, J.L. Lopez b, V. Ruiz a, D.O. Sanchez c,G. Carballal d, R.H. Campos b, J.R. Oubina a,∗
a Department of Microbiology, Faculty of Medicine, University of Buenos Aires,Paraguay 2155, Piso 11, (1121) Buenos Aires, Argentina
b Faculty of Pharmacy and Biochemistry, University of Buenos Aires, Junin 956,Piso 4, (1113) Buenos Aires, Argentina
c IIB, University of San Martın, Av. Gral, Paz y Albarellos Parque Tecnologico Miguelet,INTI – Edificio 24, (1650) San Martın, Pcia, de Buenos Aires, Argentina
d CEMIC, Av. E. Galvan 4102, (1431) Buenos Aires, Argentina
Received 16 May 2006; received in revised form 7 August 2006; accepted 8 August 2006Available online 18 September 2006
bstract
The in vivo evolution of genotype F HBV variants was recorded in a chronically infected patient throughout a 3-year observation period.luctuating levels of HBs Ag and anti-HBs antibodies were recorded, both of them cocirculating in peripheral blood samples at given times. Fiftygene derived clones were sequenced and phylogenetically analyzed. As expected, some amino acid replacements within the S ORF were also
bserved within the P ORF while others were silent for the former. Such change was statistically significant for both S and P overlapping genes,hich clearly indicates the appearance of a positive selection pressure. Supporting this notion, amino acid replacements were documented at bothand T cell epitopes in samples from 1997 and 1998.
Several mutations were documented within and outside the “a” determinant in the major hydrophilic region. Such substitutions might haveesulted from the attempt of HBV to evade both humoral and/or cellular immune response. To the best of our knowledge this unusual profile ofBV variants in presence of usually “neutralizing” anti-HBs antibodies was examined in vivo for the first time.2006 Published by Elsevier B.V.
irp(t1u
eywords: HBV variants evolution
. Introduction
Hepatitis B virus (genus Orthohepadnavirus, family Hep-dnaviridae) is one of the main causes of chronic hepatitis,iver cirrhosis and hepatocellular carcinoma. A conservativestimate assumes that more than 350 million individuals are per-istently infected worldwide. Both host- and viral-related factors
ave been associated with viral persistence. Among the latter,iral variability is considered a key strategy in the attempt tovoid both humoral and cellular immune responses (reviewed∗ Corresponding author. Tel.: +54 11 59509500x2175; fax: +54 11 45083705.E-mail address: [email protected] (J.R. Oubina).
p1ieoca
168-1702/$ – see front matter © 2006 Published by Elsevier B.V.oi:10.1016/j.virusres.2006.08.004
n Rehermann and Nascimbeni, 2005). In this regard, severaleports have documented the appearance of escape mutants torotective anti-HBs antibodies as well as to CTL specific clonesantagonism for T cell receptor), though the clinical relevance ofhe latter appears to be limited (reviewed in Chisari and Ferrari,995). The epidemiological importance of such HBs mutants isnderlined by reports from Taiwan, where the HBs Ag mutants’revalence was 7.8% in 1984, 19.6% in 1989, and 28.1% in994, concurrent with a 10-fold decrease in the HBs carrier raten children (Hsu et al., 1999). In contrast to the vaccine-induced
scape mutations predominantly detected in the aa 137–149 loopf the a determinant, the most frequent mutations in patientshronically infected with genotype C, were observed within thea 107–137 loop (Ogura et al., 1999). Moreover, mutations in
Rese
t(ac1
tcaartMdIstaHpsa
vacfIi
pwt
2
2
isaHkAa
f(s2HttRiw
dpr
t
2o
ptt
R
n
wd(arr
tAa
wcDEwoNTusl
(AA(AA
2s
o
V.L. Mathet et al. / Virus
he CTL epitope encompassing the 28–51 amino acid regionLiu et al., 2002) mainly at positions 40 and 47, were recordedmong chronic patients, suggesting that they might have alsoontributed to viral persistence (Chen and Oon, 1999; Tai et al.,997).
In a previous study carried out by the authors, several muta-ions have been described within the S gene in a patient withhronic active hepatitis and cirrhosis, despite the presence ofnti-HBs antibodies. These mutations were located at both thedeterminant, and outside of it, involving some crucial Cys
esidues (Mathet et al., 2003). This isolate was ascribed to geno-ype F, as frequently observed in Argentina (Franca et al., 2004;
bayed et al., 1998, 2001; Telenta et al., 1997). To date, longitu-inal studies on HBV quasispecies evolution are almost lacking.n this connection, to our knowledge there is only one studyhowing the emergence of naturally occurring S gene muta-ions in six HBV chronically infected patients (Yamamoto etl., 1994). Since the variants evolution was only analyzed in oneBe Ag positive patient after sequencing 6 clones from 3 timeoints – the last one corresponding to the anti-HBe seroconver-ion –, the evolution of variants in an HBe Ag(−) strain, despitenti-HBs antibodies is still unknown at present.
Studies on viral quasispecies and their evolution may pro-ide important hints about pathogenetic issues of infection, suchs adaptation to a new host (including its immune response),ompartmentalization in different cells and organs, pathologiceatures, infection prognosis and resistance to antiviral agents.t may also be related to the viral ability to establish a persistentnfection and disease progression.
In this report, we are addressing the issue of HBV quasis-ecies molecular evolution of the same patient mentioned aboveithin a 3-year observation period (Mathet et al., 2003) without
he influence of antiviral treatment.
. Materials and methods
.1. Serological and virological profile
This study depicts the evolution of HBV variants in a chron-cally infected patient (named “RI”) who has not received anypecific antiviral treatment throughout the studied period. HBsntigen (Ag) and HBe Ag, as well as both anti-HBs and anti-Be antibodies were measured by using a commercial ELISAit (Abbott Laboratories, IL, USA). HBs Ag was measured withuszyme (Abbott) which uses a monoclonal antibody directed
gainst the a determinant.S gene PCR products spanning nucleotide positions 256–796
rom samples obtained in 1995 (RI95), 1997 (RI97) and 1998RI98) were initially typed by RFLP (Lindh et al., 1997), andubsequently sequenced for further analysis (Mathet et al.,003). Viral DNA had been measured as pg/ml (by using theepatitis B viral DNA kit, [a radiological molecular hybridiza-
ion assay], Abbott Diagnostic Division, Abbot Park, IL) at the
ime of sample collection, i.e. RI95 = 60 pg/ml, RI97 = 30 pg/ml;I98 = 85 pg/ml. Thus, these levels of HBV DNA were not lim-ting the PCR amplification procedure, and consequently theyere not considered as an influencing factor for a bottleneck
s
pt
arch 123 (2007) 72–85 73
uring the in vitro amplification. Phylogenetic analysis, entropylot and the hydrophilicity/antigenicity pattern were focused onegions depicted in Fig. 1.
This study received approval from CEMIC Ethics Commit-ee. The patient provided written informed consent.
.2. HBV population dynamics throughout a 3-yearbservation period
PCR products from RI95, RI97, and RI98 were cloned intoGEMR-T Easy Vector (Promega, Madison, WI, USA.). Eigh-een, 14 and 18 clones were, respectively, analyzed by bidirec-ionally performed DNA sequencing.
Direct sequences from PCR products derived from RI95,I97, and RI98 samples were also recorded.
Consensus sequences were obtained as an average of the totalumber of clones analyzed from each time point.
The potential Taq-dependent DNA misincorporation rateas investigated by bi-directionally sequencing PCR productserived from a GBV-C/HGV clone as previously describedMathet et al., 2003). Briefly, a c-DNA fragment of 325 pb wasmplified in triplicate and sequenced with specific forward andeverse primers, without detecting any nucleotidic misincorpo-ation.
Kwok and Higuchi (1989) rules were followed strictlyhroughout the whole procedures to avoid DNA contamination.ppropriate negative controls (SDW) were included in all PCR
mplification events.Direct sequences from both full length HBV PCR products as
ell as S gene derived amplicons from the above mentioned 50lones were used for alignment and phylogenetic analysis. TheNA alignments were generated with the Clustal X program.volutionary distances between sequences were determinedith the DNADIST program (Kimura two-parameter method)f the PHYLIP package, version 3.5c (Felsenstein, 1993). Theeighbor Joining phylogenetic tree was constructed with theREEVIEW program. Bootstrap analysis was performed bysing the programs SEQBOOT (to generate 1000 reshuffledequences), CONSENSE, and RETREE at the midpoint of theongest branch for comparative purposes.
Sequences used to construct the phylogenetic treeFig. 2) belonged to genotypes A–H (AY128092, AY217370,Y217376, AB090270, AB091256, X75658, AF160501 andY090460, respectively) and to genotype F from Argentina
AF1–AF10: AF288628, AF288627, AF288626, AF288625,F288624, AF288623, AF043578, AF043577, AF043573 andF043571, respectively).
.3. HBV mutations as compared with the ancestralequence
All mutations were referred to an ancestor HBV RI sequencebtained after SPLIT decomposition of all 50 clones’ derived
equences by using the Split Tree Program.A mathematical model for clustering referred as Split Decom-osition proved useful for viral evolution analysis, especially forhose cases where viral quasispecies are involved (Bandelt and
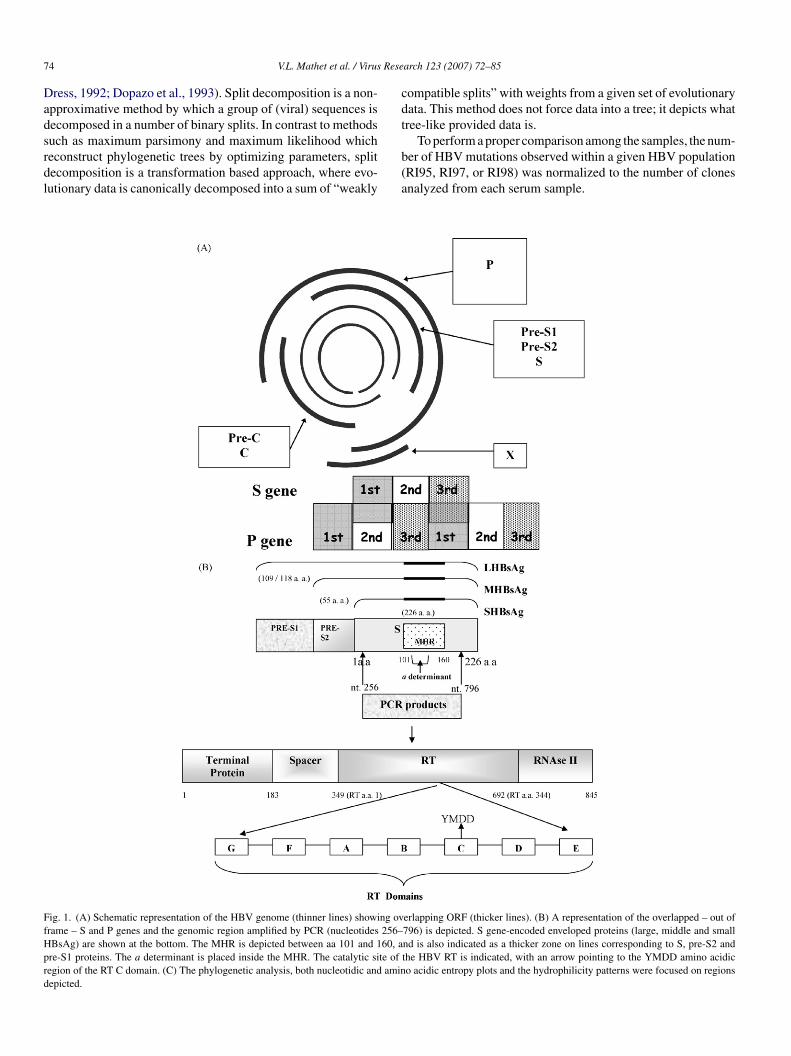
7 Rese
Dadsrdl
cdt
FfHprd
4 V.L. Mathet et al. / Virus
ress, 1992; Dopazo et al., 1993). Split decomposition is a non-pproximative method by which a group of (viral) sequences isecomposed in a number of binary splits. In contrast to methods
uch as maximum parsimony and maximum likelihood whicheconstruct phylogenetic trees by optimizing parameters, splitecomposition is a transformation based approach, where evo-utionary data is canonically decomposed into a sum of “weaklyb(a
ig. 1. (A) Schematic representation of the HBV genome (thinner lines) showing ovrame – S and P genes and the genomic region amplified by PCR (nucleotides 256–BsAg) are shown at the bottom. The MHR is depicted between aa 101 and 160, are-S1 proteins. The a determinant is placed inside the MHR. The catalytic site ofegion of the RT C domain. (C) The phylogenetic analysis, both nucleotidic and aminepicted.
arch 123 (2007) 72–85
ompatible splits” with weights from a given set of evolutionaryata. This method does not force data into a tree; it depicts whatree-like provided data is.
To perform a proper comparison among the samples, the num-er of HBV mutations observed within a given HBV populationRI95, RI97, or RI98) was normalized to the number of clonesnalyzed from each serum sample.
erlapping ORF (thicker lines). (B) A representation of the overlapped – out of796) is depicted. S gene-encoded enveloped proteins (large, middle and smallnd is also indicated as a thicker zone on lines corresponding to S, pre-S2 andthe HBV RT is indicated, with an arrow pointing to the YMDD amino acidico acidic entropy plots and the hydrophilicity patterns were focused on regions

V.L. Mathet et al. / Virus Research 123 (2007) 72–85 75
(Cont
2
secQtmcstqPa
2
atblcgb
2
uBe
2n
sSi
3
3
so
Fig. 1.
.4. Quasispecies heterogeneity
The extent of HBV quasispecies heterogeneity was mea-ured in all three samples by two ways. In the first, the het-rogeneity index was calculated as the proportion of HBVlones not bearing the dominant (majority) sequences, i.e.H = 1 − [proportion of clones bearing majority sequences]. In
he second, Shannon entropy analysis was applied. This entropyeasure was calculated as S = −∑
(p, lnp) which takes intoonsideration the frequency of each sequence (p) in a given qua-ispecies. Each entropy value was then normalized as Sn = S/lnN,o consider the total number of sequences (N) studied in eachuasispecies. An Entropy Plot was built by using the BioEditrogram (Hall, 1999) and applied for both nucleotide and aminocid sequences.
.5. Nucleotide substitution rate
To study the dynamics of this population during the course ofchronic infection despite the presence of anti-HBs antibodies,
he nucleotide substitution rate per site per year was determinedy using the most likely ancestral sequence. This rate was calcu-
ated by correlating the number of nucleotide substitutions perlone and the number of analyzed nucleotides (358 nt of the Sene) taking into account the elapsed time of infection (2 yearsetween RI95 and RI97, and 1.5 years between RI97 and RI98).aeid
inued ).
.6. Hydrophilicity/antigenicity pattern
The hydrophilicity/antigenicity pattern was investigated bysing the general method of Hopp and Woods by means of theioEdit Program (Hall, 1999). The window’s size used to gen-rate this pattern was set up at one amino acid.
.7. Transitions/Transversions andon-synonymous/synonymous substitutions
The number of both transitions/transversions and non-ynonymous/synonymous substitutions within the overlapping
and P genes was determined manually by analysis of eachndividual codon.
. Results
.1. Serological and virological profile
Fig. 3 shows the virological follow up of patient RI. Fifteenerum samples were obtained between 1995 and 1999. Through-ut this elapsed time fluctuating levels of HBs Ag and anti-HBs
ntibodies were recorded, both of them cocirculating in periph-ral blood samples at given times. HBe Ag proved negativen all 15 samples, while anti-HBe antibodies were consistentlyetected.
76 V.L. Mathet et al. / Virus Research 123 (2007) 72–85
F ic infl e eighA ce fro
gtt1is
3i
ig. 2. Phylogenetic analysis of HBV quasispecies throughout a 3-year chronength represents the specified genetic distance. A–H capital letters represent thrgentina. PCR and CON represent direct PCR sequence and consensus sequen
Six of the 15 samples proved DNA positive for the Sene by PCR amplification (Mathet et al., 2003). Two restric-ion patterns corresponding to genotype F were ascribed
o all samples by amplified S-gene RFLP (Lindh et al.,997). Three (RI95, RI97, and RI98) of these 6 DNA pos-tive samples were selected for HBV molecular evolutiontudies.pah
ection. Fifty clones were analyzed from samples RI95, RI97, and RI98. Bart known HBV genotypes. AF1–AF10 represent ten genotype F sequences fromm RI95, RI97, and RI98, respectively. Bootstrap value is shown.
.2. Phylogenetic studies: genotype assignment andntra-genotypic clustering
Phylogenetic analysis of 50 clones from serum samples fromatient RI (RI95, RI97, and RI98) allowed all the clones to bessigned to Ib cluster of genotype F (Fig. 2), with an identityigher than 96%.

V.L. Mathet et al. / Virus Research 123 (2007) 72–85 77
F les wa anti-H
3
ttgRtRt2
copsfo
sfnsp
(aaetaAaitlot
tRto(a(
3
mpisRnRwcRwoct
3
i
nwaw1
ig. 3. HBV serological and virological profile from patient RI. Fifteen sampntibody is observed in several samples obtained in 1995, 1996 and 1999. IgM
.3. Nucleotide sequence analysis
As shown in Fig. 2, the phylogenetic analysis demonstratedhat nucleotide sequences from HBV clones corresponding tohe 3-year observation period were intermingled placed. Higherenetic distances were observed among several sequences fromI98, as compared with those from RI95 and RI97. Nevertheless,
he most divergent sequence was observed in one clone fromI97 (RI97.1), where 17 mutations were recorded. In contrast,
he remaining 13 clones derived from RI97 exhibited only up tonucleotide changes per clone.The above mentioned unusual sequence exhibited also a stop
odon at the 3′ end of the S gene (aa position 156), together withther mutations not found among the remaining clones (analysiserformed bidirectionally by duplicate from two independentequencing events). Such stop codon within the S gene resultedrom a transition, which – in turn – was synonymous for theverlapping P gene.
The sequence obtained from RI95 PCR products (directequencing of PCR products) was different from that derivedrom the consensus of RI95 clone sequences (Fig. 2). Likewise,one of the RI95 clone sequences was identical to such consen-us sequence. In contrast, both PCR and consensus sequencesroved, respectively, identical in samples RI97 and RI98.
Variants’ analysis detected at the beginning of this studyRI95 sample), showed the coexistence of different nucleotidest several analyzed positions, which was reflected by the appear-nce of indeterminations when PCR products were directlyxamined. Gene cloning was carried out taking into accounthat some nucleotide positions could not be properly assigned togiven base due to the quasispecies composition of RI sample.fter analyzing the respective contribution of each nucleotide atgiven position throughout the observation period, it was real-
zed that some minor populations became predominant by fixing
he corresponding change, while others disappeared below theimits of detection (less than 5.5%; <1/18 clones) after 3 yearsf viral evolution (Table 1). In this connection, it was observedhat some nucleotide substitutions (as compared with the ances-aamc
ere analyzed between 1995 and 1999. Cocirculation of HBsAg and anti-HBsBc was positive only in 1997.
ral sequence) already present in several clones from sampleI95, were missed throughout the course of the chronic infec-
ion (as observed in Fig. 5, i.e. A575C/G). In sharp contrast,ther changes were fixed at the end of the observation periodsample RI98). Only some of these substitutions were associ-ted with amino acid changes for HBs Ag and/or the Pol proteinsee below and Section 4; data available upon request).
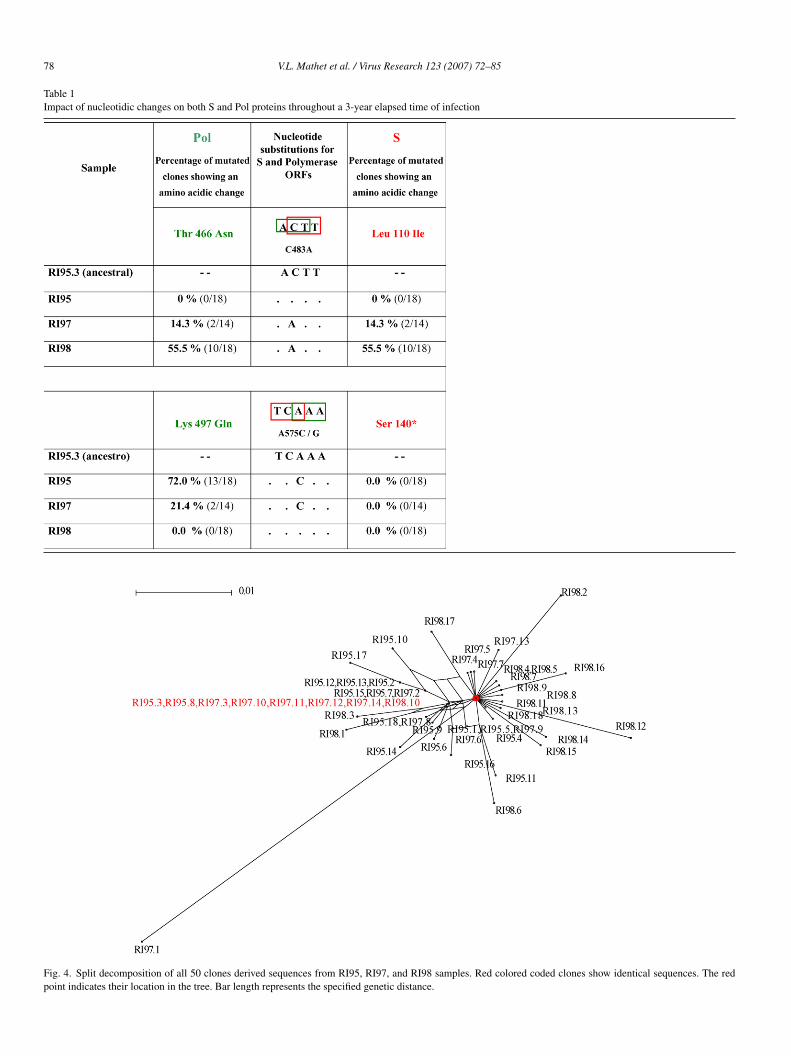
.3.1. Determining the ancestral nucleotidic sequenceThe Split decomposition method was used to establish the
ost probable ancestral sequence for the studied clones. Toerform this analysis all the above mentioned 50 clones werencluded. It was established that the most likely ancestralequence corresponded to that observed in clones RI95.3 andI95.8, which are 100% identical (Fig. 4). Likewise, suchucleotide sequence was also detected in 5 out of 14 clones fromI97 and from 1 out of 18 from RI98. When sequence alignmentas performed comparing both the ancestral sequence with the
onsensus sequence obtained from the 18 clones from sampleI95, only 1 nt change was recorded (99.7% identity). Togetherith the above mentioned results, it is inferred that the sequencebtained by using the Split decomposition method – but not theonsensus from the 18 clones analyzed from sample RI95 – ishe most appropriate for subsequent analysis.
.3.2. Analysis of the genetic heterogeneityHeterogeneity was determined by both the heterogeneity
ndex and the Shanon’s entropy index (Table 2).A greater genetic distance was observed among the
ucleotide sequences from clones derived from sample RI98,ith regard to that observed from the remaining samples (RI95
nd RI97). Concomitantly, the greatest genetic heterogeneityas observed among the 18 clones from samples collected in998 (RI98), since its HI was equal to 1 (as compared with either
HI = 0.89 in 1995, or a HI = 0.64 in 1997). In 1995, three vari-nts – one of them exhibiting the ancestral sequence – were theost predominant (each of them represented by 2 clones). Inontrast, in 1997 a single variant was significantly represented
78 V.L. Mathet et al. / Virus Research 123 (2007) 72–85
Table 1Impact of nucleotidic changes on both S and Pol proteins throughout a 3-year elapsed time of infection
Fig. 4. Split decomposition of all 50 clones derived sequences from RI95, RI97, and RI98 samples. Red colored coded clones show identical sequences. The redpoint indicates their location in the tree. Bar length represents the specified genetic distance.
V.L. Mathet et al. / Virus Research 123 (2007) 72–85 79
Table 2HBV RI variability throughout a 3-year observation period
Evaluated parameter RI95 (18 clones) RI97 (14 clones) RI98 (18 clones)
Heterogeneity index 0.89 0.64 1
No. of clones of the predominant quasispecies 2a (clones 3 and 8) 5a (clones 3, 10, 11, 12 and 14) 0a
2 (clones 12 and 13)2 (clones 1 and 5)
N −3 −3 −3
es. Inq
a[sst
3
Fn
ucleotide substitution rate/site/year –
a Two clones in 1995, five in 1997 and one in 1998 shared identical sequencuasispecies could be demonstrated.
s a master sequence (5 clones out of 14 exhibited the same
ancestral] sequence: 37.7%; Fig. 4 and Table 2). Thus, suchequence was identical to that obtained from two clones corre-ponding to one of the 3 predominant quasispecies in 1995, ando a single clone detected in 1998.pvl
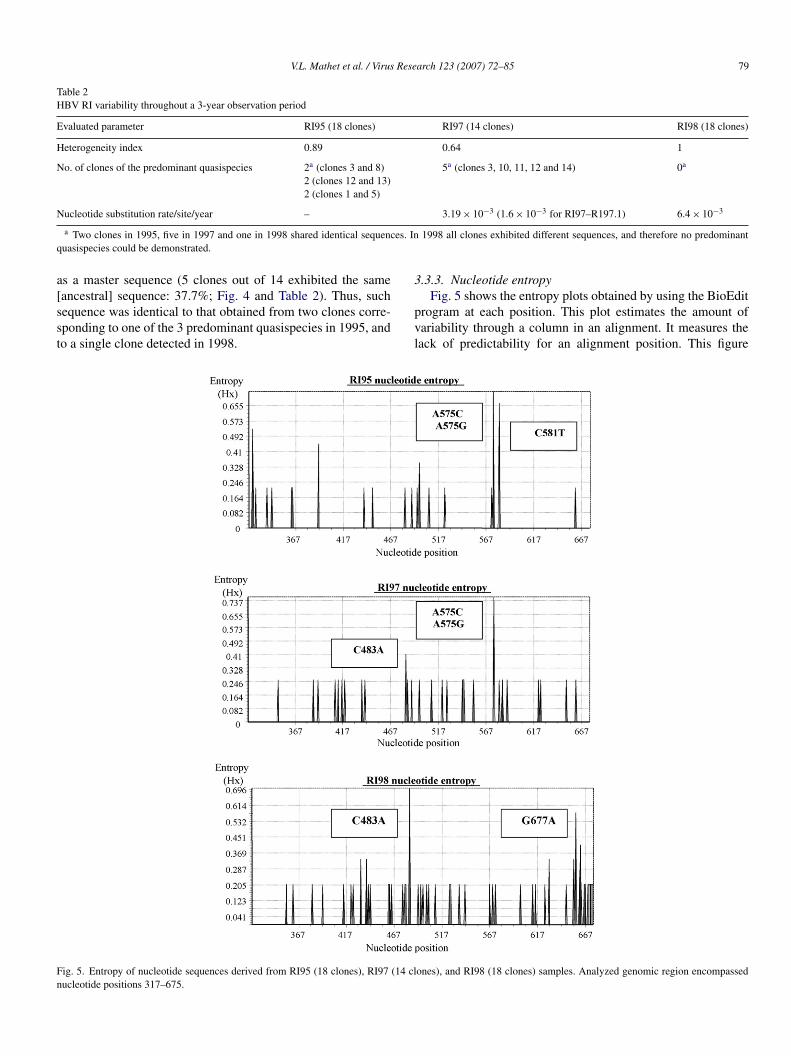
ig. 5. Entropy of nucleotide sequences derived from RI95 (18 clones), RI97 (14 clucleotide positions 317–675.
3.19 × 10 (1.6 × 10 for RI97–R197.1) 6.4 × 10
1998 all clones exhibited different sequences, and therefore no predominant
.3.3. Nucleotide entropy
Fig. 5 shows the entropy plots obtained by using the BioEditrogram at each position. This plot estimates the amount ofariability through a column in an alignment. It measures theack of predictability for an alignment position. This figure
ones), and RI98 (18 clones) samples. Analyzed genomic region encompassed
8 Rese
s(wmafit
3
prptwnffi(q
3
pcfpii
arToR
Fa
0 V.L. Mathet et al. / Virus
hows the entropy of nucleotide sequences derived from RI9518 clones), RI97 (14 clones) and RI98 (18 clones) samples,hich occasionally represented changes for both S and Poly-erase protein. Some polymorphisms were gained or missing
t specific nucleotide positions, due to the loss (i.e. A575C/G) orxation (i.e. C483A) of mutations which – in turn – determined
he entropy level (see also Table 1).
.3.4. Nucleotide substitution rateInitially, nucleotide sequences from directly sequenced PCR
roducts were aligned to visualize nucleotide changes as aeflection of the viral evolution. As a consequence of severalolymorphisms, clones’ sequences from the S gene were fur-her studied. Such analysis showed a higher rate as comparedith that reportedly known for both e+ and e− strains. Theucleotide substitution rate per site per year was determined
or both elapsed periods: 1995–1997 and 1997–1998. For therst period, the nucleotide substitution rate was 3.19 × 10−31.6 × 10−3 without considering RI97.1) while for the subse-uent period it was 6.4 × 10−3 (Table 2).
3
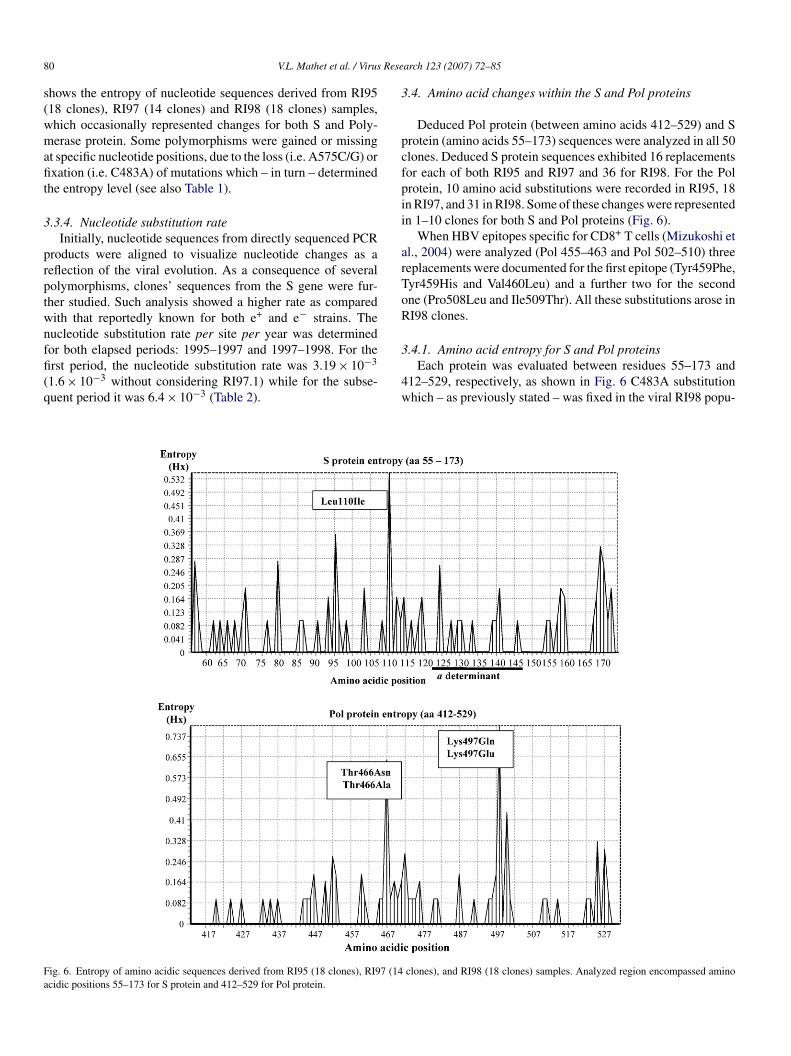
4w
ig. 6. Entropy of amino acidic sequences derived from RI95 (18 clones), RI97 (14cidic positions 55–173 for S protein and 412–529 for Pol protein.
arch 123 (2007) 72–85
.4. Amino acid changes within the S and Pol proteins
Deduced Pol protein (between amino acids 412–529) and Srotein (amino acids 55–173) sequences were analyzed in all 50lones. Deduced S protein sequences exhibited 16 replacementsor each of both RI95 and RI97 and 36 for RI98. For the Polrotein, 10 amino acid substitutions were recorded in RI95, 18n RI97, and 31 in RI98. Some of these changes were representedn 1–10 clones for both S and Pol proteins (Fig. 6).
When HBV epitopes specific for CD8+ T cells (Mizukoshi etl., 2004) were analyzed (Pol 455–463 and Pol 502–510) threeeplacements were documented for the first epitope (Tyr459Phe,yr459His and Val460Leu) and a further two for the secondne (Pro508Leu and Ile509Thr). All these substitutions arose inI98 clones.
.4.1. Amino acid entropy for S and Pol proteinsEach protein was evaluated between residues 55–173 and
12–529, respectively, as shown in Fig. 6 C483A substitutionhich – as previously stated – was fixed in the viral RI98 popu-
clones), and RI98 (18 clones) samples. Analyzed region encompassed amino
Rese
l(nr(S
3
1d
uettrHastto
cmc(ip
3a
mtwwti
3
Fp
V.L. Mathet et al. / Virus
ation produced non-synonymous changes in both the S proteinLeu110Ile) and the Pol protein (Thr466Asn/Ala). In contrast,ucleotide substitutions observed at position 575 (A575C/G)endered amino acid changes only within the Pol proteinLys497Gln/Glu), since it was a synonymous change for theprotein (Table 1).
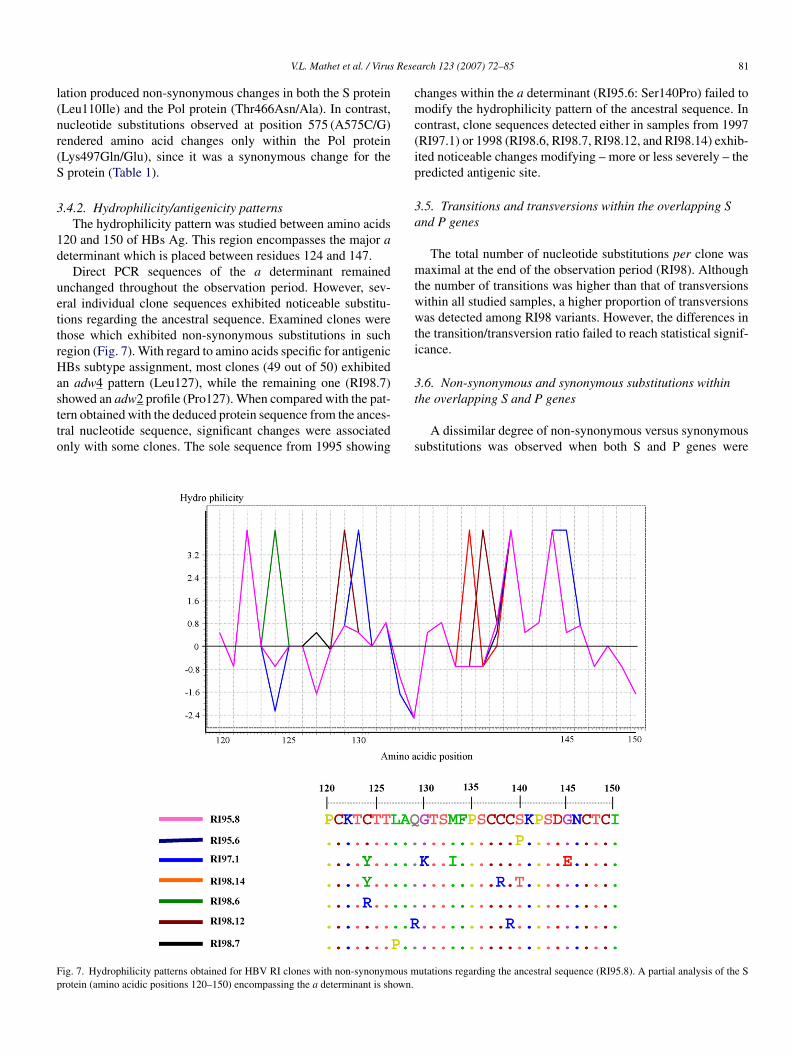
.4.2. Hydrophilicity/antigenicity patternsThe hydrophilicity pattern was studied between amino acids
20 and 150 of HBs Ag. This region encompasses the major aeterminant which is placed between residues 124 and 147.
Direct PCR sequences of the a determinant remainednchanged throughout the observation period. However, sev-ral individual clone sequences exhibited noticeable substitu-ions regarding the ancestral sequence. Examined clones werehose which exhibited non-synonymous substitutions in suchegion (Fig. 7). With regard to amino acids specific for antigenicBs subtype assignment, most clones (49 out of 50) exhibited
n adw4 pattern (Leu127), while the remaining one (RI98.7)
howed an adw2 profile (Pro127). When compared with the pat-ern obtained with the deduced protein sequence from the ances-ral nucleotide sequence, significant changes were associatednly with some clones. The sole sequence from 1995 showingt
s
ig. 7. Hydrophilicity patterns obtained for HBV RI clones with non-synonymous mrotein (amino acidic positions 120–150) encompassing the a determinant is shown.
arch 123 (2007) 72–85 81
hanges within the a determinant (RI95.6: Ser140Pro) failed toodify the hydrophilicity pattern of the ancestral sequence. In
ontrast, clone sequences detected either in samples from 1997RI97.1) or 1998 (RI98.6, RI98.7, RI98.12, and RI98.14) exhib-ted noticeable changes modifying – more or less severely – theredicted antigenic site.
.5. Transitions and transversions within the overlapping Snd P genes
The total number of nucleotide substitutions per clone wasaximal at the end of the observation period (RI98). Although
he number of transitions was higher than that of transversionsithin all studied samples, a higher proportion of transversionsas detected among RI98 variants. However, the differences in
he transition/transversion ratio failed to reach statistical signif-cance.
.6. Non-synonymous and synonymous substitutions within
he overlapping S and P genesA dissimilar degree of non-synonymous versus synonymousubstitutions was observed when both S and P genes were
utations regarding the ancestral sequence (RI95.8). A partial analysis of the S

8 Rese
csw(san
ttgfsrR
tngttl
SnSaanRtti(tc
grcnPRtscWput
4
uca
1dHaetcwqe
tdvpd
4
igett(
tbgosaHeaot2Bvr2
tftatsc
2 V.L. Mathet et al. / Virus
ompared throughout the observation period. A statisticallyignificant higher proportion of non-synonymous replacementsas detected within the sequence of the S (p = 0.009) and P
p = 0.001) genes after the 3-year elapsed time. Interestingly,uch increased number of non-synonymous replacements waslready statistically significant from RI97 sample in P gene butot in S gene.
A mathematical analysis performed manually showedhat Ka/Ks (non-synonymous versus synonymous substitu-ions) ratio was approximately 1 for the RI95 sample, (HBsene = 1.11; Pol gene = 0.89), while it was approximately 2or the RI98 sample (HBs gene = 1.83; Pol gene = 2.19). Whenuch analysis was focused on the a determinant the Ka/Ksatio was 0.25 for the first sample (RI95) and 2.33 forI98.
With regard to the P gene, no mutations were observed withinhe A or B domain of RI95 sample, as compared with severalon-synonymous substitutions observed throughout the sameene of RI98 sample. No mutations were observed throughouthe observation period at positions 528 and 552 of the Pol pro-ein, showing a lack of known resistance to lamivudine (3TC) ateast for these sites.
Total and non-synonymous nucleotide substitutions for bothand P genes were also examined by taking into account the
ucleotide codon position. Total nucleotide substitutions for thegene were higher at the third position within samples RI95
nd RI97. In contrast, in RI98 the highest value was recordedt the first codon position. Likewise, higher values of totalucleotide substitutions at P gene were observed in RI95 andI97 at the first codon position (third nucleotide position for
he S gene). Subsequently, higher numbers of nucleotide substi-utions were recorded at the second nucleotide codon positionn RI98. When clone RI97.1 was excluded from the analysisRI97–RI97.1) a similar codon location of nucleotide substitu-ions was observed, although the absolute number of them wasonsiderably reduced.
With regard to non-synonymous substitutions within the Sene, a statistically significant increase in the number of changesecorded at the first codon position in RI98 was observed, asompared with RI95 and RI97. Likewise, an increase in theumber of nucleotide non-synonymous substitutions within thegene was documented at the second nucleotide position in
I98 as compared with RI95 and RI97 (p < 0.05). A concomi-ant statistically significant decrease in the number of non-ynonymous substitutions was detected at the first nucleotideodon position when samples RI95 and RI98 were analyzed.
hen the analysis excluded RI97.1 clone, the nucleotide codonosition exhibiting non-synonymous substitutions remainednchanged for both S and P genes (second and first base, respec-ively).
. Discussion
This study longitudinally examines the in vivo HBV e− pop-lation evolution in a patient with chronic active hepatitis asso-iated to cirrhosis despite the fluctuating detection of anti-HBsntibodies.
sRRu
arch 123 (2007) 72–85
Several researchers (Carman et al., 1995; Karthigesu et al.,994; Lee et al., 1997; Ogata et al., 1997) have previouslyemonstrated the cocirculation of mutated HBs Ag and anti-Bs antibodies in patients that have received an appropriate
ctive/passive immunization. In addition, some studies (Asahinat al., 1996; Ogura et al., 1997) directly correlated the Hepati-is B exacerbation with some HBs Ag mutations. Moreover, ahronic HBV infection has been recently shown to be associatedith a “wild type” adr strain which escaped from the patient’suasimonoclonal immune response directed to the y (but not d)pitope (Margeridon et al., 2005).
Results obtained provide new interesting insights into twoopics: (i) HBV population complexity; (ii) HBV populationynamic evolution. Although the pathogenic importance of indi-idual mutations was not the major target of this work, theutative biological significance of several nucleotide changeseserves to be highlighted.
.1. Population complexity
The viral population complexity observed in patient RI wasnitially inferred when two restriction patterns ascribed to the Fenotype and numerous variants were recorded in RI98 (Mathett al., 2003). Taq DNA polymerase induced error was consideredo be highly unlikely, taking into account that several con-rol experiments had been carried out, as previously describedMathet et al., 2003).
In this study, initial results were confirmed and retrospec-ively extended throughout a 3-year observation period whenoth the phylogenetic analysis from 50 clones and the hetero-eneity index (HI) were determined (Fig. 2 and Table 2). Asbserved in Fig. 2, an intermingled pattern was obtained whenequences from RI95, RI97, and RI98 were analyzed, indicatinglack of sequence clustering from a given time. As regards theBV RI population, this pattern might be interpreted as a unique
volved quasispecies structure (Holmes and Moya, 2002) whosencestral sequence was modulated. The dynamic distributionf mutants observed in this study throughout a 3-year observa-ion period is consistent with the quasispecies theory (Domingo,002; Domingo, 2003; Domingo and Holland, 1997; Eigen andiebricher, 1988). Moreover, a crucial cooperative role amongiral variants within a quasispecies provides a rationale for theole of quasispecies diversity in pathogenesis (Vignuzzi et al.,006).
Statistical analysis of the genetic distance comparisons (twoailed t Test for X ± S.D. for each time) among sequencesrom RI95, RI97, and RI98 confirmed the intermingled pat-ern observed in the phylogenetic study, since values obtainedt any given time failed to show significant differences amonghem (i.e. genetic distances between clones derived from theame sample exhibited similar values to those observed betweenlones derived from different samples).
The HI documented the diversity of all population samples,
ince values of 0.89, 0.65, and 1 were, respectively, recorded inI95, RI97, and RI98. Interestingly, only 1 out of 18 sequences inI98 was identical to the ancestral sequence (see below) lastingp to the end of the observation period.
Rese
4
4d
mesMpopuswm
vfisws
4
segossonsthefpp
iRc(tstee
4
rstw
siwmst
4
Vt(1eaiiei
srpoa1
nipp
swIaRctattcattbtaaco
V.L. Mathet et al. / Virus
.2. Population dynamics
.2.1. Determination of the ancestral sequence by the splitecomposition method
The most commonly used strategies to determine genomicutations are comparison of the analyzed sequence with ref-
rence sequences (Wang et al., 2002), or with a consensusequence derived from clones’ sequences (Hannoun et al., 2000;
anzini et al., 1998). However, both methods might exhibitotential drawbacks, such as erroneous mutation assignmentsr “creation” of an unreal sequence, respectively. In order toroperly assign genuine mutations, the ancestral sequence wassed for proper comparisons. To find out the most likely ancestralequence for HBV RI clones, the split decomposition methodas applied. This methodology allows to determining putativeutations as compared with the ancestral sequence.The ancestral sequence was observed throughout the obser-
ation period (as represented in two clones from RI95, otherve clones from RI97 and one from RI98). This was the uniqueequence showing this pattern (Fig. 4), which suggests that thehole constellation of variants at a given time might arise from
uch ancestor.
.2.2. Nucleotide substitution rate and nucleotide entropyIt is already known that the rate of HBV synonymous sub-
titution is about 104-fold faster than that of eukaryotes (Oritot al., 1989). A high substitution rate was observed in the HBVenome analyzed from RI patient. Although it has been previ-usly reported that e− strains roughly Exhibit 12 times higherubstitution rates than e+ strains (Simmonds, 2001), RI strainhowed approximately one order of magnitude above the levelf previously reported e− strains (Table 2). The high rate ofucleotide substitutions observed with HBV RI clone-derivedequences, suggests that a “cloud” of potentially beneficial muta-ions at the population level allowed the viral quasispecies aigher probability to evolve and adapt (Vignuzzi et al., 2006)ven in presence of circulating anti-HBs antibodies. When theull length HBV nucleotide sequences from RI95 were com-ared with RI98, such substitution rate reached 2.7 × 10−3 nter site per year (Lopez et al., submitted for publication).
Entropy profiles at nucleotide level demonstrated a signif-cant increase in RI98 sample, as compared with RI95 andI97. This result might reflect a viral attempt to adaptation tohanging environmental conditions. Newly generated variantsFig. 5) might contribute differentially to quasispecies struc-ure and function. It has been reported that such quasispeciestructure may account for the pathogenicity of a given virus, itsransmission capability to another host, its replication at differ-nt tissues or even as a decoy for the immune response (Vignuzzit al., 2006).
.2.3. Transitions/tranversionsPrevious studies on the S and P genes had documented the
atio of transitions/transversions replacements. As previouslyhown by Brunetto et al. (1999), our work demonstrated thatransitions were more frequently observed than transversionsithin the whole population studied at each time.
dH(1
arch 123 (2007) 72–85 83
The transitions/transversion ratio failed to show statisticallyignificant differences when comparisons among the three stud-ed time points were carried out, although their absolute numbersere higher at the end of the observation period. These dataight suggest a continuous – although not necessarily equally
trong – involvement of selective pressures throughout the infec-ion.
.2.4. Synonymous and non-synonymous mutationsOne of the most commonly used procedures in Evolutionary
irology to determine what sort of evolution process contributeso the fixation of a given mutation is to analyze synonymousS) and non-synonymous (NS) substitutions (Argentini et al.,999; Holmes et al., 1992; Kils-Hutten et al., 2001; Manzinit al., 1998; Plikat et al., 1997; Yang et al., 2000; Zhang etl., 1997). If S substitutions are neutral, their fixation velocitys equal to their production velocity. Likewise, if NS evolven a neutral manner, its velocity of substitutions should bequal to S sites (dNS = dS), as observed for pseudogenes andntrons.
It is reportedly known that the number of synonymous sub-titutions per nucleotide site is 5.5 times lower at overlappedegions of P and S genes as compared with the non-overlappingarts of the HBV genome. In this context, the number of syn-nymous substitutions per nucleotide in the S gene is quite lownd reflects a strongly constrained evolution (Mizokami et al.,997).
Since some synonymous substitutions in one gene may causeon-synonymous replacements in the overlapped gene locatedn a different frame, mutations that affect one protein (i.e. Polrotein) may or may not change the other one (herein, the Srotein) as shown in Table 1 and Fig. 6.
In our study, the initial dNS < dS pattern (RI95) was sub-equently replaced by a dNS > dS profile (RI98). Such changeas statistically significant for both S and P overlapping genes.
nterestingly, this switch of the dNS/dS ratio pattern firstppeared in RI97 at the P gene, and became noticeable inI98 at both P and S genes. This observation clearly indi-ates the appearance of a positive selection pressure, duringhe course of the infection. To support this notion, aminocid replacements were documented at both B and T cell epi-opes in RI97 and RI98 samples. Such substitutions might behe consequence of both anti-HBs antibodies and LT CD8+
ells, respectively. Indeed, it has been recently shown thatnti-HBs antibodies may facilitate the emergence and secre-ion of mutated variants, after such specific IgG is endocy-osed by hepatocytes (Schilling et al., 2003). In fact, it haseen recently reported that in chronic hepatitis B patients,he coexistence of HBs Ag and anti-HBs antibodies is associ-ted with an increase of a determinant variability, suggestingselection of HBV immune escape mutants during chronic
arriage (Lada et al., 2006). Furthermore, it has been previ-usly shown that some of the mutated variants which arose
uring the infection are poorly recognized by anti (wild type)-Bs antibodies, and even some of them fail to bind to themColeman et al., 1999; Coleman, 2006; Chong-Jin et al.,999).

8 Rese
4
vsae(npaa
apssttmmtsess
4tP
aSiRiomehwa2
ddCCotdrP
tb
gS
a(kt
A
As1iNf
R
A
A
B
B
C
C
C
C
C
C
DD
4 V.L. Mathet et al. / Virus
.3. Mutations as variability reservoirs
The emergence of escape mutants is a biologically importantiral strategy to evade the immune response. In this regard, thistudy undoubtedly showed that a majority of mutations arisingt the beginning of the study (RI95) were synonymous. How-ver, such synonymous replacements could mean a potentialintermediate) source for subsequent drastic non-synonymousucleotide changes which might rapidly appear when selectionressures are evidently operative (RI98), as the case was withnti-HBs antibodies’ reappearance (measured as free “in excess”ntibodies since 1999; Fig. 3).
When overlapped S and P genes’ evolution were compared,dissimilar profile was apparent, since most changes initially
laced at the third nucleotide of the codons for the S gene wereubsequently observed at the first position. Likewise, most sub-titutions from P gene turned their location from the first tohe second codon position. It is reportedly known that substi-utions at the second nucleotide position in a given codon are
ore restricted than changes at the first position, and these onesore than those at the third location. Consequently, it appears
hat P gene evolved more drastically than S gene. As a result ofuch variability, two analyzed Pol derived CD8 specific T cellpitopes exhibited five replacements from RI98 clones derivedequences, reinforcing the notion of a positive selection pres-ure.
.4. Biological significance of mutations within and outsidehe a determinant and its relationship with the overlapping
gene
Several mutations have been documented within (Fig. 6)nd outside the a determinant in the major hydrophilic region.ome of them are reportedly known to affect the antigenic-
ty of the S protein, as exemplified by RI97.1, RI98.6, andI98.14 clones, which drastically modified their hydrophilic-
ty profiles. Such substitutions might result from the attemptf HBV to evade both humoral and/or cellular responses, asentioned above. It has been recently postulated that S gene
scape mutants (but not wild type variants) may be secreted fromepatocytes after wild type HBV virus interacts intracellularlyith endocytosed specific anti-HBs antibodies, thus suggestingnovel mechanism for S mutant(s) emergence (Schilling et al.,003).
Most of these amino acid replacements have been previouslyetailed (Mathet et al., 2003). Briefly, key changes have beenocumented at cystein residues within the S protein (Cys69Tyr,ys76Arg, Cys90Tyr, Cys124Arg or Tyr, Cys138Arg, andys139Arg) which could affect either the S protein secretionr its conformational structure (Mangold and Streeck, 1993). Inhis connection, replacements along the S protein arise from aelicate balance between the lack of proof reading of the HBVeverse transcriptase and constraints exerted by the overlapped
gene to keep its functional activity.The current lack of the crystallographic structure of the wildype S protein together with the reportedly known transmem-rane topology precludes the authors to use bioinformatics pro-
D
D
arch 123 (2007) 72–85
rams to predict faithfully a secondary structure of the reportedmutants.In summary, we have shown HBV quasispecies evolution in
patient infected with the genotype F of HBV in presence ofusually) neutralizing anti-HBs antibodies. To the best of ournowledge this unusual profile has been examined for the firstime.
GenBank accession numbers:
Sample RI95: PCR direct sequence (DQ306924); clones 1–18(DQ306925 to DQ306942).Sample RI97: PCR direct sequence (DQ306943); clones 1–14(DQ306944 to DQ306957).Sample RI98: PCR direct sequence (DQ306958); clones 1–18,as depicted in Manzini et al., 1998.
cknowledgements
We are indebted to Miss M.L. Cuestas, M.A. Oubina and Mrs..M. Andreetta for their excellent technical assistance. This
tudy was partly supported by grants: BID 1201/OC-AR-PICT0871 from the National Agency for Scientific and Technolog-cal Promotion (ANPCyT), PIP 842 and PIP 6065 from theational Research Council (CONICET) and UBACYT M059
rom the University of Buenos Aires.
eferences
rgentini, C., La Sorsa, V., Bruni, R., D’Ugo, E., Giuseppetti, R., Rapicetta, M.,1999. Hepadnavirus evolution and molecular strategy of adaptation in a newhost. J. Gen. Virol. 80, 617–626.
sahina, Y., Enomoto, N., Ogura, Y., Kurosaki, M., Sakuma, I., Izumi, N.,Marumo, F., Sato, C., 1996. Sequential changes in full-length genomes ofhepatitis B virus accompanying acute exacerbation of chronic hepatitis B. J.Hepatol. 25, 787–794.
andelt, H.J., Dress, A.W.M., 1992. A canonical decomposition theory for met-rics on a finite set. Adv. Math. 92, 47–105.
runetto, M., Rodriguez, U.A., Bonino, F., 1999. Hepatitis B virus mutants.Intervirology 42, 69–80.
arman, W.F., Korula, J., Wallace, L., MacPhee, R., Mimms, L., Decker,R., 1995. Fulminant reactivation of hepatitis B due to envelope proteinmutant that escaped detection by monoclonal HBsAg ELISA. Lancet 345,1406–1407.
hen, W.N., Oon, C.J., 1999. Mutation “hot spot” in HLA class I-restricted T-cellepitope on hepatitis B surface antigen in chronic carriers and hepatocellularcarcinoma. Biochem. Biophys. Res. Commun. 262, 757–761.
hisari, F.V., Ferrari, C., 1995. Hepatitis B virus immunopathogenesis. Ann.Rev. Immunol. 13, 29–60.
hong-Jin, O., Ning, C.W., Shiuan, K., Keow, L.G., 1999. Identification of Hep-atitis B surface antigen variants with alterations outside the “a” determinantin immunized Singapore infants. J. Infect. Dis. 179, 259–263.
oleman, P.F., Jack Chen, Y.C., Mushahwar, I., 1999. Immunoassay detectionof Hepatitis B surface antigen mutants. J. Med. Virol. 59, 19–29.
oleman, P.F., 2006. Surveillance for hepatitis B surface antigen mutants. J.Med. Virol. 78 (Suppl. 1), 56–58.
omingo, E., 2002. Quasispecies theory in Virology. J. Virol. 76, 463–465.omingo, E., 2003. Quasispecies and the development of new antiviral strate-
gies. Prog. Drug Res. 60, 133–158.omingo, E., Holland, J.J., 1997. RNA virus mutations and fitness for survival.
Ann. Rev. Microbiol. 51, 151–178.opazo, J., Dress, A., von Haeseler, A., 1993. Split decomposition: a technique
to analyze viral evolution. Proc. Natl. Acad. Sci. U.S.A. 90, 10320–10324.

Rese
E
F
F
H
H
H
H
H
K
K
K
L
L
L
L
M
M
M
M
M
M
M
M
O
O
O
P
R
S
S
T
T
V
W
Y
Y
V.L. Mathet et al. / Virus
igen, M., Biebricher, C.K., 1988. Sequence space and quasispecies distribution.In: Domingo, Holland and Ahlquist’s RNA genetics: Variability of RNAGenomes, vol. III. CRC Press, Boca Raton, FL, pp. 211–245.
elsenstein, J., 1993. PHYLIP: Phylogeny Interference Package, Version 3.5c.Department of Genetics, University of Washington, Seattle, WA, USA.
ranca, P.H.C., Gonzalez, J.E., Munne, M.S., Brandao, L.H., Gouvea, V.S.,Sablon, E., Vanderborght, B.O.M., 2004. Strong association between geno-type F and Hepatitis B virus e antigen-negative variants among HBV-infectedArgentinean blood donors. J. Clin. Microbiol. 42, 5015–5021.
all, T.A., 1999. BioEdit: a user-friendly biological sequence alignment editorand analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 41,95–98.
announ, C., Horal, P., Lindh, M., 2000. Long-term mutation rates in the hep-atitis B virus genome. J. Gen. Virol. 81, 75–83.
olmes, E.C., Moya, A., 2002. Is the quasispecies concept relevant to RNAviruses? J. Virol. 76, 460–462.
olmes, E.C., Zhang, L.Q., Simmonds, P., Ludlam, C.A., Leighbrown, A.J.,1992. Convergent and divergent sequence evolution in the surface enve-lope glycoprotein of human immunodeficiency virus type 1 within a singleinfected patient. Proc. Natl. Acad. Sci. U.S.A. 89, 4835–4839.
su, H.Y., Chang, M.H., Liaw, S.H., Ni, Y.H., Chen, H.L., 1999. Changes of hep-atitis B surface antigen variants in carrier children before and after universalvaccination in Taiwan. Hepatology 30, 1312–1317.
arthigesu, V.D., Allison, L.M., Fortuin, M., Mendy, M., Whittle, H.C., Howard,C.R., 1994. A novel hepatitis B virus variant in the sera of immunized chil-dren. J. Gen. Virol. 75, 443–448.
ils-Hutten, L., Cheynier, R., Wain-Hobson, S., Meyerhans, A., 2001. Phyloge-netic reconstruction of intrapatient evolution of human immunodeficiencyvirus type 1: predominance of drift and purifying selection. J. Gen. Virol.82, 1621–1627.
wok, S., Higuchi, R., 1989. Avoding false positives with PCR. Nature 339,237–238.
ada, O., Benhamon, Y., Poynard, T., Thibault, V., 2006. Coexistence of Hepati-tis B surface antigen/HBs Ag) and anti-HBs antibodies in chronic HepatitisB virus carriers: influence of “a” determinant variants. J. Virol. 80, 2968–2975.
ee, P.I., Chang, L.Y., Lee, C.Y., Huang, L.M., Chang, M.H., 1997. Detectionof hepatitis B surface gene mutation in carrier children with or withoutimmunoprophylaxis at birth. J. Infect. Dis. 176, 427–430.
indh, M., Andersson, A.S., Gusdal, A., 1997. Genotypes, nt 1858 variants,and geographic origin of hepatitis B virus: Large-scale analysis using a newgenotyping method. J. Infect. Dis. 175, 1285–1293.
iu, C.J., Kao, J.H., Shau, W.Y., Chen, P.J., Lai, M.Y., Chen, D.S., 2002. Natu-rally occurring hepatitis B surface gene variants in chronic hepatitis B virusinfection: correlation with viral serotypes and clinical stages of liver disease.J. Med. Virol. 68, 50–59.
angold, C.M.T., Streeck, R.E., 1993. Mutational analysis of the cysteineresidues in the hepatitis B virus small envelope protein. J. Virol. 67,4588–4597.
anzini, A., Solforosi, L., Petrelli, E., Macarri, G., Tosone, G., Piazza, M.,Clementi, M., 1998. Evolution of hypervariable region 1 of hepatitis C virusin primary infection. J. Virol. 72, 6271–6276.
argeridon, S., Lachaux, A., Trepo, C., Zoulim, F., Kay, A., 2005. A quasi-monoclonal anti-HBs response can lead to immune escape of ‘wild-type’
hepatitis B virus. J. Gen. Virol. 86, 1687–1693.athet, V.L., Feld, M., Espinola, L., Sanchez, D.O., Ruiz, V., Mando, O., Car-ballal, G., Quarleri, J.F., D’Mello, F., Howard, C.R., Oubina, J.R., 2003.Hepatitis B virus S gene mutants in a patient with chronic active hepatitiswith circulating anti-HBs antibodies. J. Med. Virol. 69, 18–26.
Z
arch 123 (2007) 72–85 85
bayed, V.A., Barbini, L., Lopez, J.L., Campos, R.H., 2001. Phylogenetic anal-ysis of the hepatitis B virus (HBV) genotype F including Argentine isolates.Arch. Virol. 146, 1803–1810.
bayed, V.A., Lopez, J.L., Telenta, P.F.S., Palacios, G., Badıa, I., Ferro, A.,Galoppo, C., Campos, R.H., 1998. Distribution of Hepatitis B virus geno-types in two different pediatric populations from Argentina. J. Clin. Micro-biol. 36, 3362–3365.
izokami, M., Orito, E., Ohba, K., Ikeo, K., Lau, J.Y.N., Gojobori, T., 1997.Constrained evolution with respect to gene overlap of Hepatitis B virus. J.Mol. Evol. 44, 83–90.
izukoshi, E., Sidney, J., Livingston, B., Ghany, M., Hoofnagle, J.H., Sette, A.,Rehermann, B., 2004. Constrained evolution with respect to gene overlap ofHepatitis B virus. J. Mol. Evol. 44, 83–90.
gata, N., Zanetti, A.R., Yu, M., Miller, R.H., Purcell, R.H., 1997. Infectivityand pathogenicity in chimpanzees of a surface gene mutant of hepatitis Bvirus that emerged in a vaccinated infant. J. Infect. Dis. 175, 511–523.
gura, Y., Kurosaki, M., Asahina, Y., Enomoto, N., Maruno, F., Sato, C., 1999.Prevalence and significance of naturally occuring mutations in the surfaceand polymerase genes of hepatitis B virus. J. Infect. Dis. 180, 1444–1451.
rito, E., Mizokami, M., Ina, Y., Moriyama, N.E., Kameshima, N., Yamamoto,M., Gojobori, T., 1989. Host-independent evolution and genetic classifica-tion of the hepadnavirus family based on nucleotide sequences. Proc. Natl.Acad. Sci. U.S.A. 86, 7059–7062.
likat, U., Nieselt-Struwe, K., Meyerhans, A., 1997. Genetic drift can dominateshort-term human immunodeficiency virus type 1 nef quasispecies evolutionin vivo. J. Virol. 71, 4233–4240.
ehermann, B., Nascimbeni, M., 2005. Immunology of hepatitis B virus andHepatitis C virus infection. Nat. Immunol. 5, 215–218.
chilling, R., Ijaz, S., Davidoff, M., Yee Lee, J., Locarnini, S., Williams, R.,Naoumov, N.V., 2003. Endocytosis of Hepatitis B immune globulin intohepatocytes inhibits the secretion of Hepatitis B virus surface antigen andvirions. J. Virol. 77, 8882–8892.
immonds, P., 2001. The origin and evolution of hepatitis viruses in human. J.Gen. Virol. 82, 693–712.
ai, P.C., Banik, D., Lin, G.I., Pai, S., Pai, K., Lin, M.H., Yuoh, G., Che, S.,Hsu, S.H., Chen, T.C., Kuo, T.T., Lee, C.S., Yang, C.S., Shih, C., 1997.Novel and frequent mutations of hepatitis B virus coincide with a majorhistocompatibility complex class I-restricted T-cell epitope of the surfaceantigen. J. Virol. 71, 4852–4856.
elenta, P.F., Poggio, G.P., Lopez, J.L., Gonzalez, J., Lamberg, A., Campos,R.H., 1997. Increased prevalence of genotype F hepatitis B virus isolates inBuenos Aires, Argentina. J. Clin. Microbiol. 35, 1873–1875.
ignuzzi, M., Stone, J.K., Arnold, J.J., Cameron, C.E., Andino, R., 2006. Qua-sispecies diversity determines pathogenesis through cooperative interactionin a viral population. Nature 439, 344–348.
ang, H., Bian, T., Merrill, S.J., Eckels, D.D., 2002. Sequence variation in thegene encoding the nonstructural 3 protein of hepatitis C virus: evidence forimmune selection. J. Mol. Evol. 54, 465–473.
amamoto, K., Horikita, M., Tsuda, F., Itoh, K., Akahane, Y., Yotsumoto, S.,Okamoto, H., Miyakawa, Y., Mayumi, M., 1994. Naturally occurring escapemutants of hepatitis B virus with various mutations in the S gene in carri-ers seropositive for antibody to Hepatitis B surface antigen. J. Virol. 68,2671–2676.
ang, Z., Nielsen, R., Goldman, N., Pedersen, A.M., 2000. Codon-substitution
models for heterogeneous selection pressure al amino acid sites. Genetics155, 431–449.hang, L., Diaz, R.S., Ho, D.D., Mosley, J.W., Busch, M.P., Mayer, A., 1997.Host-specific driving force in human immunodeficiency virus type 1 evolu-tion in vivo. J. Virol. 71, 2555–2561.
Copyright © 2022 FDOKUMEN




















