
DNA methyltransferase 3B mutations linked to the ICF syndrome cause dysregulation of lymphogenesis...
16
© 2001 Oxford University Press Human Molecular Genetics, 2001, Vol. 10, No. 25 2917–2931 DNA methyltransferase 3B mutations linked to the ICF syndrome cause dysregulation of lymphogenesis genes Melanie Ehrlich*, Kent L. Buchanan 1 , Fern Tsien, Guanchao Jiang, Baodong Sun, William Uicker 1 , Corry M.R. Weemaes 2 , Dominique Smeets 3 , Karl Sperling 4 , Bernd H. Belohradsky 5 , Niels Tommerup 6 , David E. Misek 7 , Jean-Marie Rouillard 7 , Rork Kuick 7 and Samir M. Hanash 7 Human Genetics Program and Department of Biochemistry, Tulane Medical School, New Orleans, LA 70112, USA, 1 Department of Microbiology and Immunology, Tulane Medical School, New Orleans, LA 70112, USA, 2 Department of Pediatrics and 3 Department of Human Genetics, University Medical Center St. Radboud, Nijmegen, The Netherlands, 4 Institute of Human Genetics, Charitéé, Berlin, Germany, 5 Ludwig-Maximilians-Universitat, Munchen, Germany, 6 Wilhelm Johannsen Centre for Functional Genome Research, Department of Medical Genetics, Institute of Medical Biochemistry and Genetics, University of Copenhagen, Denmark and 7 Department of Pediatric Hematology Oncology, Box 0656, MSRB I, Room A520C, University of Michigan, Ann Arbor, MI 48109, USA Received August 17, 2001; Revised and Accepted October 8, 2001 ICF (immunodeficiency, centromeric region instability and facial anomalies) is a recessive disease caused by mutations in the DNA methyltransferase 3B gene (DNMT3B). Patients have immunodeficiency, chromo- some 1 (Chr1) and Chr16 pericentromeric anomalies in mitogen-stimulated lymphocytes, a small decrease in overall genomic 5-methylcytosine levels and much hypomethylation of Chr1 and Chr16 juxtacentromeric heterochromatin. Microarray expression analysis was done on B-cell lymphoblastoid cell lines (LCLs) from ICF patients with diverse DNMT3B mutations and on control LCLs using oligonucleotide arrays for approximately 5600 different genes, 510 of which showed a lymphoid lineage-restricted expression pattern among several different lineages tested. A set of 32 genes had consistent and significant ICF-specific changes in RNA levels. Half of these genes play a role in immune function. ICF-specific increases in immunoglobulin (Ig) heavy constant µ and δ RNA and cell surface IgM and IgD and decreases in Igγ and Igα RNA and surface IgG and IgA indicate inhibition of the later steps of lymphocyte maturation. ICF-specific increases were seen in RNA for RGS1, a B-cell specific inhibitor of G-protein signaling implicated in negative regulation of B-cell migration, and in RNA for the pro- apoptotic protein kinase C eta gene. ICF-associated decreases were observed in RNAs encoding proteins involved in activation, migration or survival of lymphoid cells, namely, transcription factor negative regulator ID3, the enhancer-binding MEF2C, the iron regulatory transferrin receptor, integrin β7, the stress protein heme oxygenase and the lymphocyte-specific tumor necrosis factor receptor family members 7 and 17. No differences in promoter methylation were seen between ICF and normal LCLs for three ICF upregulated genes and one downregulated gene by a quantitative methylation assay [combined bisulfite restriction analysis (COBRA)]. Our data suggest that DNMT3B mutations in the ICF syndrome cause lymphogenesis-associated gene dysregulation by indirect effects on gene expression that interfere with normal lymphocyte signaling, maturation and migration. INTRODUCTION ICF (immunodeficiency, centromeric region instability and facial anomalies) is a rare chromosome instability syndrome that is autosomal recessive. It is the only disease shown to be associated with mutations in a DNA methyltransferase gene (1,2). Chromosomal anomalies are targeted specifically to the vicinity of the centromere (pericentromeric region) of chromo- some 1 (Chr1) or Chr16 in mitogen-stimulated lymphocytes and B lymphoblastoid cell lines (LCLs) from these patients (3,4). The only other types of chromosomal aberrations associated with ICF are rare abnormalities in the pericentromeric region of Chr9 (3) and telomeric associations, which have been reported so far only in two ICF LCLs (4). Severe immunodeficiency and high frequencies of peri- centromeric decondensation and rearrangements in Chr1 and Chr16 upon karyotype analysis of blood samples are diagnostic for this syndrome (3). Also, in all studied ICF tissue samples, hypomethylation has been found in satellite 2 (Sat2) DNA, the main component of centromere-adjacent (juxtacentromeric) heterochromatin, where the ICF-specific decondensation is seen (4–6). Sat2 DNA is highly methylated in normal postnatal *To whom correspondence should be addressed. Tel: +1 504 584 2449; Fax: +1 504 584 1763; Email: [email protected]
Transcript of DNA methyltransferase 3B mutations linked to the ICF syndrome cause dysregulation of lymphogenesis...

© 2001 Oxford University Press Human Molecular Genetics, 2001, Vol. 10, No. 25 2917–2931
DNA methyltransferase 3B mutations linked to the ICFsyndrome cause dysregulation of lymphogenesis genesMelanie Ehrlich*, Kent L. Buchanan1, Fern Tsien, Guanchao Jiang, Baodong Sun,William Uicker1, Corry M.R. Weemaes2, Dominique Smeets3, Karl Sperling4,Bernd H. Belohradsky5, Niels Tommerup6, David E. Misek7, Jean-Marie Rouillard7,Rork Kuick7 and Samir M. Hanash7
Human Genetics Program and Department of Biochemistry, Tulane Medical School, New Orleans, LA 70112, USA,1Department of Microbiology and Immunology, Tulane Medical School, New Orleans, LA 70112, USA, 2Department ofPediatrics and 3Department of Human Genetics, University Medical Center St. Radboud, Nijmegen, The Netherlands,4Institute of Human Genetics, Charitéé, Berlin, Germany, 5Ludwig-Maximilians-Universitat, Munchen, Germany,6Wilhelm Johannsen Centre for Functional Genome Research, Department of Medical Genetics, Institute of MedicalBiochemistry and Genetics, University of Copenhagen, Denmark and 7Department of Pediatric HematologyOncology, Box 0656, MSRB I, Room A520C, University of Michigan, Ann Arbor, MI 48109, USA
Received August 17, 2001; Revised and Accepted October 8, 2001
ICF (immunodeficiency, centromeric region instabilityand facial anomalies) is a recessive disease causedby mutations in the DNA methyltransferase 3B gene(DNMT3B). Patients have immunodeficiency, chromo-some 1 (Chr1) and Chr16 pericentromeric anomaliesin mitogen-stimulated lymphocytes, a small decreasein overall genomic 5-methylcytosine levels and muchhypomethylation of Chr1 and Chr16 juxtacentromericheterochromatin. Microarray expression analysiswas done on B-cell lymphoblastoid cell lines (LCLs)from ICF patients with diverse DNMT3B mutationsand on control LCLs using oligonucleotide arrays forapproximately 5600 different genes, 510 of whichshowed a lymphoid lineage-restricted expressionpattern among several different lineages tested. A setof 32 genes had consistent and significant ICF-specificchanges in RNA levels. Half of these genes play arole in immune function. ICF-specific increases inimmunoglobulin (Ig) heavy constant µ and δ RNA andcell surface IgM and IgD and decreases in Igγ and IgαRNA and surface IgG and IgA indicate inhibition ofthe later steps of lymphocyte maturation. ICF-specificincreases were seen in RNA for RGS1, a B-cell specificinhibitor of G-protein signaling implicated in negativeregulation of B-cell migration, and in RNA for the pro-apoptotic protein kinase C eta gene. ICF-associateddecreases were observed in RNAs encoding proteinsinvolved in activation, migration or survival oflymphoid cells, namely, transcription factor negativeregulator ID3, the enhancer-binding MEF2C, the ironregulatory transferrin receptor, integrin β7, the stressprotein heme oxygenase and the lymphocyte-specific
tumor necrosis factor receptor family members 7 and17. No differences in promoter methylation wereseen between ICF and normal LCLs for three ICFupregulated genes and one downregulated gene by aquantitative methylation assay [combined bisulfiterestriction analysis (COBRA)]. Our data suggest thatDNMT3B mutations in the ICF syndrome causelymphogenesis-associated gene dysregulation byindirect effects on gene expression that interferewith normal lymphocyte signaling, maturation andmigration.
INTRODUCTION
ICF (immunodeficiency, centromeric region instability andfacial anomalies) is a rare chromosome instability syndromethat is autosomal recessive. It is the only disease shown to beassociated with mutations in a DNA methyltransferase gene(1,2). Chromosomal anomalies are targeted specifically to thevicinity of the centromere (pericentromeric region) of chromo-some 1 (Chr1) or Chr16 in mitogen-stimulated lymphocytesand B lymphoblastoid cell lines (LCLs) from these patients(3,4). The only other types of chromosomal aberrations associatedwith ICF are rare abnormalities in the pericentromeric regionof Chr9 (3) and telomeric associations, which have beenreported so far only in two ICF LCLs (4).
Severe immunodeficiency and high frequencies of peri-centromeric decondensation and rearrangements in Chr1 andChr16 upon karyotype analysis of blood samples are diagnosticfor this syndrome (3). Also, in all studied ICF tissue samples,hypomethylation has been found in satellite 2 (Sat2) DNA, themain component of centromere-adjacent (juxtacentromeric)heterochromatin, where the ICF-specific decondensation isseen (4–6). Sat2 DNA is highly methylated in normal postnatal
*To whom correspondence should be addressed. Tel: +1 504 584 2449; Fax: +1 504 584 1763; Email: [email protected]

2918 Human Molecular Genetics, 2001, Vol. 10, No. 25
somatic tissues (4). Various other abnormalities are often seenin ICF patients, such as, facial anomalies, intestinal problems,growth retardation and neurological dysfunction (3). Also, werecently reported that ICF LCLs are hypersensitive to the lethaleffects of γ radiation but without deficient cell cycle checkpointresponses (7).
In order to elucidate the changes in gene expression thataccount for the symptoms of ICF and to understand therelationships between targeted DNA hypomethylation andgene expression, we compared RNA from LCLs of ICF patientsand normal controls using an oligonucleotide microarray.Among the genes that showed significant ICF-specificdecreases or increases in RNA are five Ig genes and nine genesinvolved in the activation or late maturation of lymphocytes.The results of our study can help explain the heretoforeenigmatic nature of the immunodeficiency in ICF.
RESULTS
Confirming the ICF-specific phenotype of the analyzedB-cell lines
For the microarray analysis of ICF-specific gene expression,six ICF and five control polyclonal B-cell LCLs were used.The choice of B-cell lines was predicated upon the central role ofB cells in abnormal Ig production in ICF and the inaccessibilityof uncultured B cells from multiple patients’ blood samplesbecause only slightly more than 30 ICF patients have beenreported in the last few decades and few reach adulthood.Despite their maintenance in cell culture for extended periodsof time, the LCLs exhibited highly significant ICF-specificdifferences in RNA levels as described below. Moreover, theICF LCLs displayed the diagnostic karyotype of mitogen-stimulated ICF lymphocytes, namely, much higher levels ofthe ICF-specific chromosomal abnormalities in the pericentro-meric regions of Chr1 and Chr16 than control LCLs (Table 1)and hypomethylation of Sat2 DNA in somatic cells (seebelow). The ICF LCLs were from five unrelated ICF patientswith various mutations in DNMT3B (Table 1). The five controlLCLs were from unaffected unrelated individuals (Normal Aand D) or phenotypically normal parents of ICF patients(Normal B, G and C). The LCLs from the ICF patients’ parentshad a similar passage history as the patients’ LCLs. All the ICFLCLs exhibited telomeric associations previously reported intwo examined ICF LCLs (4).
We also assayed these LCLs for hypomethylation of themajor DNA component of juxtacentromeric heterochromatinof Chr1, Sat2 DNA, which is characteristic of ICF. BstBIdigests of the LCL DNAs were hybridized in Southern blotswith a Chr1 Sat2-specific probe. All of the ICF LCLs exhibitedsimilarly hypomethylated Sat2 DNA compared with thecontrol LCLs, as previously demonstrated for ICF B and ICF Crelative to the LCLs from their parents and to Normal A andNormal D LCLs (4). ICF B and ICF C LCLs had been checkedfor overall genomic 5-methylcytosine (m5C) levels by analysisof DNA digested to deoxynucleosides and shown not to bedetectably m5C-deficient relative to normal LCLs (4).However, we had demonstrated that DNA from the oneassayed uncultured ICF tissue, ICF brain DNA, had ∼7% lessm5C than normal brain DNA samples (m5C to C ratios of 0.043and 0.0455, respectively) (4).
Overview of altered gene expression in ICF LCLs
RNA from ICF and control LCLs was compared using micro-arrays containing oligonucleotide probes for approximately5600 different human genes. In analyzing the data from thearrays, we looked for genes whose RNAs were significantlyover- or under-represented in ICF versus control LCLs. Werequired that at least three of the LCLs give significant expressionof that transcript (P < 0.01 comparing the perfect match tomismatch probes). There were 3881 of such probe sets, whichindicates transcription of more than half of the genes on thearray by at least some of the LCLs in this work. Forty-five ofthese displayed differences between the ICF and control LCLsat a significance level of P < 0.05 and a fold change (FC) of>2.0 or an FC > 1.5 and a P < 0.01. We also compared RNAsfrom five normal LCLs and three non-LCL cell lines (triplicateRNA samples from a lung carcinoma cell line, A549; a braintumor cell line, U118; and a colorectal cancer cell line, Lovo).In an ANOVA of these samples there were 510 probe sets thatwere restricted in their expression to LCLs, with an FC ≥ 1.5and P < 0.05 for all three LCL versus non-LCL comparisons.Out of the 45 probe sets displaying ICF versus normal LCLdifferences, 18 were among the 510 LCL-specific probe sets.That an excess of the probe sets which show up- or down-regulation in ICF are specifically expressed in LCLs relative tonon-lymphoid cell lines is significant at P < 0.00001 forFisher’s exact test (two-sided).
In Tables 2–4, we show data for 32 genes with significantICF-specific differences in RNA levels. The remaining 13 geneswere not included in the tables because their expression levelswere not sufficiently high (>200 arbitrary units as the averageexpression for the ICF or control LCLs) or reproducible amongthe ICF or control LCLs (at least three of the five patientsshowing an FC in the same direction and the average for theICF LCLs or control LCLs not strongly influenced by one ortwo outliers). These 32 genes were selected using just theseparameters without regard to functionality or tissue specificity.Many of these genes had functions that could be related to theICF phenotype and several of these showed consistent resultsfrom replicate or related probe sets. For example, the transferrinreceptor gene, TFRC, which is involved in lymphocyte activationas well as the general regulation of iron metabolism, showedan average of 2.0–2.4 times less RNA signal in ICF than innormal LCLs for three different TFRC probe sets in the arrays(Table 3). The tumor necrosis factor receptor superfamily member17 (TNFR17) gene, which is implicated in B-cell activation, and itsoverlapping antisense (AS) gene (8,9) gave concordant ICF-specific differences in their RNAs (Table 3; Fig. 1) consistentwith the finding that LCLs which express TNFR17 RNAusually also express TNFR17 AS RNA (8). GUCY1B3 andGUCY1A3, both of which exhibited higher RNA levels in ICFthan normal LCLs (Table 4; Fig. 1), encode two subunits of thesame enzyme and are generally coordinately expressed (10).Moreover, the inverse relationship between ICF-associatedincreases in RNA levels for the Ig heavy chains IgHM andIgHD and decreases for IgHG3, IgHA1 and IgHA2 areexpected for these classes of Ig genes given the genomicrecombination in Ig class switch regions which deletes IgHMand IgHD exons during generation of mature heavy-chain IgGand IgA genes.

Human Molecular Genetics, 2001, Vol. 10, No. 25 2919
We controlled for differences among the LCLs other thantheir status as ICF or control cell lines. Neither differences inploidy (Table 1) nor growth rate could explain ICF-associatedincreases or decreases in RNA levels and X-linked genes didnot show ICF-specific differences in expression. We checkedthat the ICF-specific expression of genes in Tables 3 and 4 wasnot just a consequence of the cells overexpressing IgHM andIgHD RNA. Because Normal A had a similar pattern of IgRNA expression to that of the ICF LCLs (Table 5), we deter-mined whether this control cell line had RNA levels similar tothose of the ICF LCLs for any of the genes in Tables 3 or 4.The only genes for which this was found to be the case werethose encoding GUCY1B3 and GUCY1A3 subunits (Table 4;Fig. 1). The mean RNA signals for GUCY1B3 among the ICFLCLs, Normal A, and the other control LCLs were 1316, 813and 162 and the analogous values for GUCY1A1 were 233,179 and 81, respectively. Therefore, in B cells, there might bea previously unreported association of expression of soluble
guanylate cyclase 1 subunit genes with the immunoglobulinstatus.
ICF-specific changes in immunoglobulin RNAs andsurface immunoglobulins
The reported immune problems in most of the known ICFpatients (30 patients; C.M.R.Weemaes and D.Smeets, unpublisheddata) are usually severe but occasionally slight. The patientsmay have no or low amounts of IgG, IgA and/or IgM (Table 5),and they usually have normal levels of B cells. The ICF LCLshad very low signals for IgG3 and IgA1 and IgA2 heavy-chainRNAs compared to most of the control LCLs (Tables 2 and 5).However, these ICF cell lines had high levels of IgM and IgDheavy-chain RNAs. For example, the ICF LCLs had IgD RNAsignals ranging from approximately 2400 to 5500 (mean 3383)compared to signals of 400–1700 (mean 808) for the normalLCLs. No significant difference in expression of λ or κ light-chainRNA was seen in the ICF versus normal LCLs.
Table 1. Frequency of different types of chromosomal abnormalities in ICF and control LCLs
aICF LCLs were from unrelated individuals but ICF B and ICF B′ were derived from the same culture which was subsequently propagated in different laborato-ries. Normal B, C, and G were LCLs from completely unaffected parents of ICF patients B, C, and G. Normal B and G have a mutant allele in common with thecorresponding patient but Normal C does not because one of the two mutant alleles in the ICF patient C apparently arose de novo (30). Normal A and D wereestablished from blood cultures of normal individuals not in ICF families.bMultiradials, delns, chrb and chtb are chromosomes with more than two arms fused in the pericentromeric region, whole-arm deletions, chromosome breaks andchromatid breaks, respectively. Fifty metaphases were examined for each LCL. Only ∼10% of the ICF cells with 1qh or 16qh decondensation had more than oneChr1 or Chr16 displaying this anomaly and none of the metaphases with rearrangements of Chr1 or Chr16 had both homologs with the same anomaly. Approxi-mately 80% of ICF B metaphases were tetraploid and the rest were diploid but the frequencies of 1qh decondensation, 16qh decondensation and pericentromericrearrangements were similar for the tetraploid and diploid ICF B cells. The percentage tetraploid cells in ICF S, Normal A, Normal D and the other LCLs were100, 80, 25 and <2.cNo evidence for centromeric decondensation was seen in ICF or normal chromosomes.
Cell typea Percent of metaphases with a given Chr1 or Chr16pericentromeric aberrationb
Percent of metaphases with the indicatedabnormalities
Mutations in DNMT3B (reference)
Decondensationc
Multiradials Delns, chrb,chtb
Telomereassociations
Other abnormalitiesc
(type of abnormality)
1qh 16qh
ICF B 22 16 4 14 10 4 [del(4p); chrb(6q)] Homozygous, exon 20, 2177T→G, V726G (30)
ICF B′ 14 4 0 6 2 0 Homozygous, exon 20, 2177T→G, V726G (30)
ICF C 16 6 6 6 2 0 Exon 16, 1807G→A, A603T and IVS22–11G→A, STP807ins (2,30)
ICF G 4 2 2 2 4 4 [chrb(3p);chrb(12q)]
Exon 2, ins 1bp codon 53, term 158 and IVS21G65452A, T65903C;IVS22 G67577A, alt. splice ∆737–798 (1)
ICF K 10 14 2 4 2 6 [t(2q;4q); chrb(8q);del(15q)]
IVS13 1G→A, skipping exon 13; exon 23, C→Gat R823G; variants:IVS2-22delC; 3′-UTR: 69127delT; K.Sperling,K.Uhlmann andM.Karbasiyan, this work
ICF S 8 2 8 4 8 0 Exon 17, 1817T→C, V606A; exon 6, 610C→T,Q204X (3)
Normal B 0 0 0 0 0 2 [chrb(7p)] –a
Normal C 2 0 0 0 0 0 –a
Normal G 0 0 0 0 0 2 [decond(6q10)] –a
Normal A 0 0 0 0 0 2 [chrb(10q)] –
Normal D 2 0 0 0 0 2 [chrb(2q)] –

2920 Human Molecular Genetics, 2001, Vol. 10, No. 25
To determine the percentage of cells in each LCL expressingIgG, IgA, IgM or IgD, these and several additional polyclonalB-cell lines were analyzed by flow cytometry with fluorochrome-labeled antibodies to human Ig. Most of the cells in six of eightexamined ICF LCLs were positive for cell surface IgM(sIgM+) and many of these cells were sIgD+ (Table 5 and datanot shown). In contrast, all the ICF LCLs displayed negligiblenumbers of sIgG+ or sIgA+ cells. Also consistent with themicroarray data, the normal LCLs gave very different results.Six of the seven normal LCLs had sIgG or sIgA on a largefraction (usually most) of their cells. The lack of cell-surface Igfor ICF B′ and ICF K probably reflects a post-transcriptionaldeficiency (such as, the loss of Ig-specific chaperone-assistedfolding of the heavy chain in the endoplasmic reticulum)resulting from changes during prolonged cell culture. ICF B′differed from ICF B, which was sIgM+ and sIgD+, by beingpassaged in a different laboratory after establishment from asingle blood sample. From the highly consistent results on thelevels of Ig RNA and the flow cytometry analysis at the levelof protein (Table 5 plus results from the four additional LCLsmentioned above), we conclude that the B cells in the peripheralblood of ICF patients are probably mostly IgM+ (or IgD+) withextremely low percentages of IgG+ or IgA+ cells, in contrast tothe cells from normal individuals which are mostly IgG+ orIgA+. The differences in the results from clinical analyses ofserum Ig in ICF patients and microarray or flow cytometricanalyses of ICF LCLs, notably for IgM and IgD, could beexplained by defects in B-cell maturation and/or activationafter V(D)J recombination in the patients.
ICF-specific changes in RNAs encoding proteinsregulating lymphocyte migration, activation or homing
Nine of the genes showing ICF-specific differences in RNAlevels are implicated in lymphoid cell differentiation orspecific lymphoid function after V(D)J recombination andmost of them are involved in signal transduction or in thecontrol of transcription (Table 3). The genes with increasedlevels of RNA in ICF LCLs are the regulator of G-proteinsignaling gene 1 (RGS1; Fig. 1) and protein kinase C eta(PRKCH). RGS1 is a B-cell- and monocyte-specific protein thatnegatively regulates chemokine signaling through hetero-trimeric guanine-nucleotide binding proteins (11) and controlsB-cell migration within lymphoid organs in response tochemokines (12). PKC eta is present at high levels in pro-Bcells and early-stage thymocytes and was shown to be pro-apoptotic in the former cells (13). The pro-B cells in ICFpatients could have yet higher levels of PKC eta RNA relativeto their normal counterparts than do ICF LCLs because onlythe ICF pro-B cells with less of an increase in this RNA mightsurvive selection in the bone marrow.
Lower RNA levels in ICF than in control LCLs were seen forthe TNFR17 and its overlapping AS gene (Table 3; Fig. 1).TNFR17, also known as B cell maturation factor, is amembrane receptor whose protein ligand (zTNF4) is importantfor antibody production (9). TNFR7, another TNFR super-family member assigned roles in antibody production (14),also had significantly lower RNA levels in ICF LCLs than innormal LCLs (Table 3). RNA for B lymphoid tyrosine kinase(BLK), which might function downstream of heterotrimeric Gproteins (15), was downregulated in ICF LCLs (Table 3). BLK
Table 2. Immunoglobulin or T-cell receptor genes which displayed ICF-specific changes in RNA levelsa
aIn Tables 2–4, we show data for 32 genes with significant and consistent differences in mean trimmed RNA levels between ICF LCLs and normal LCLs as described inResults. These genes were selected without regard to functionality or tissue-specific expression. The quantile-normalized RNA signals, trimmed means and P-values forall the probe sets in the microarray analysis of ICF and normal LCLs are publicly available at http://dot.ped.med.umich.edu:2000/pub/ICF/index.htmlbPositive FC values denote genes with higher mean values for ICF LCL RNAs than control LCL RNAs and are the mean of the RNAICF divided by the mean ofthe RNAcontrol for a given probe set. Negative FC values indicate genes with less RNA in ICF than control LCLs and are the negative of the mean of the RNAcontroldivided by that of the RNAICF for the indicated probe set. FC for IGHG3 is a minimum estimate because of the very high level of expression in the control LCLs.cP-values are for two-sample t-tests to evaluate the significance of ICF-associated decreases or increases in mean RNA levels relative to the controls.dOut of the six examined ICF LCLs, the number of ICF LCLs which showed a change in RNA levels similar to the average FC for ICF versus control LCLs.
Abbreviatedgenename
Accessionno.
Chromosomallocation
Description RNA signal for six ICF versus five control LCLs No. of ICFLCLs showingsimilar FCd
Mean forICF
Mean forcontrols
FCb P-value fromt-testc
IGHG3 M87789 14q32.33 Immunoglobulin heavy constant γ3 291 >18600 –64.0 0.004 6
IGHA2 S71043 14q32.33 Immunoglobulin heavy constant α2 459 8467 –18.5 0.009 6
IGHA1,membrane
M62628 14q32.33 Immunoglobulin heavy constant α1membrane region
287 709 –2.5 0.01 6
IGHD K02882 14q32.33 Immunoglobulin heavy constant δ 3383 808 +4.2 0.002 6
IGHM X58529 14q32.33 Immunoglobulin heavy constant µ 21701 10552 +2.1 0.08 6
IGHM V00563 14q32.33 Immunoglobulin heavy constant µ;duplicate probe set
7189 3353 +2.1 0.05 6
TRB@ M12886 7q35 T-cell receptor β locus 821 2885 –3.5 0.005 5

Human Molecular Genetics, 2001, Vol. 10, No. 25 2921
RNA has been proposed to be involved in signal transductionin the B-cell and thymocyte lineages (16).
Two RNAs downregulated in ICF LCLs are needed fornormal immunity as determined by mice knockout studies(Table 3; Fig. 1). They encode integrin β7 , which facilitateshoming to lymphoid tissue in the gut (17), and a transdominantnegative protein (ID3), which inhibits the activity and expressionof tissue-restricted bHLH proteins by sequestering differentiation-inducing E proteins (18). ICF-associated decreases in RNA
levels were also seen for the transcription enhancer MEF2C(19,20) (Table 3; Fig. 1) and the transferrin receptor (21)(Table 3), both of which are associated with immune function.Some of the noteworthy genes that did not display ICF-specificdifferences in RNA levels are the B-cell antigen receptorcomplex components CD79a and CD79b; the lymphocytesurface protein, CD81; the B-cell surface protein CD19, a panB-cell marker; and various members of the integrin familyother than ITGB7, of the protein kinase C family other than the
Table 3. Genes involved in leukocyte activation, survival or homing, which displayed ICF-specific changes in RNA levels
aFC, P-values and the numbers of ICF LCLs showing similar FC are as defined for Table 2. References for the description of the gene are in the text.
Abbreviatedgenename
Accessionno.
Chromosomallocation
Description RNA signal for six ICF versus five control LCLs No. of ICFLCLsshowingsimilar FCa
Mean forICF
Mean forcontrols
FCa P-valuefrom t-testa
RGS1 orBL34
S59049 1q31 Regulator of G-protein signaling 1; inhibitsB-cell migration from germinal centersto the peripheral blood in response to cytokines;inhibits downstream signaling byGiα and Gqα
2610 958 +2.7 0.005 5
PRKCH M55284 14q22–q23 Protein kinase C, eta; abundant in lung andinduces terminal differentiation in keratinocytes;also implicated in cell cycle arrest and apoptosisin pro-B cells
379 100 +3.8 0.04 5
TNFR17or BCMA
Z29574 16p13.1 TNFRSF17; B-cell maturation (BCMA); B-cellsurvival, activation, and proliferation in responseto antigens
2040 4040 –2.0 0.0006 6
AS TNFR17 Z29572 16p13.1 Antisense transcript of tumor necrosis factorreceptor 17; often coexpressed with TNFR17;has ORF; unknown function
191 358 –1.9 0.0006 6
TNFR7or CD27
M63928 12p13 Tumor necrosis factor receptor family, member 7(TNFRSF7); lymphoid cells;T-cell and B-cell proliferation response toantigens
1439 2890 –2.0 0.04 4
ID3 X69111 1p36.13–p36.12
Inhibitor of DNA binding 3; dominant negativetranscription controls; B-cell proliferationresponse to antigens; thymocyte positiveselection; embryogenesis
1663 3675 –2.2 0.01 5
ITGB7 S80335 12q13.13 Integrin, β7; homing of B and T cells tointestinal mucosa and to a lesser extent to lungs;maturation of immature monocyte-deriveddendritic cells
2217 5710 –2.1 0.03 4
BLK S76617 8p23–p22 B lymphoid tyrosine kinase in B-cell lineage andthymocytes but not mature T cells
465 1264 –2.7 0.02 4
MEF2C S57212 5q14 MADS box transcription enhancer factor 2,polypeptide C; high expression inmuscle and cerebrocortical neurons; alsoexpressed in B cells; implicated in stimulation ofJ chain expression
656 1063 –1.6 0.009 5
TFRC M11507 3q26.2-qter Transferrin receptor (p90, CD71) 5′ region 386 893 –2.3 0.004 4
TFRC M11507 3q26.2-qter Transferrin receptor (p90, CD71); middle region 278 657 –2.4 0.008 4
TFRC M11507 3q26.2-qter Transferrin receptor (p90, CD71); 3′ region;ubiquitous; regulation of ironmetabolism; upregulated during B-cell activationand involved in T-cell activation
938 1866 –2.0 0.01 4
2922 Human Molecular Genetics, 2001, Vol. 10, No. 25
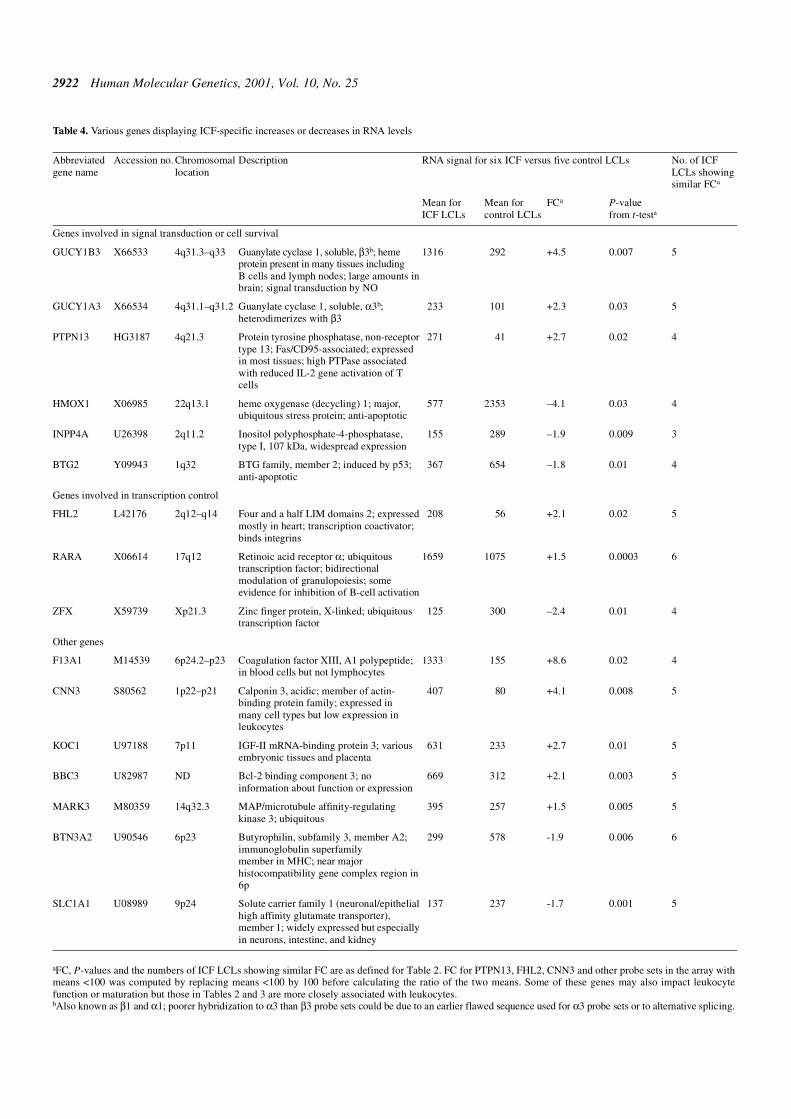
Table 4. Various genes displaying ICF-specific increases or decreases in RNA levels
aFC, P-values and the numbers of ICF LCLs showing similar FC are as defined for Table 2. FC for PTPN13, FHL2, CNN3 and other probe sets in the array withmeans <100 was computed by replacing means <100 by 100 before calculating the ratio of the two means. Some of these genes may also impact leukocytefunction or maturation but those in Tables 2 and 3 are more closely associated with leukocytes.bAlso known as β1 and α1; poorer hybridization to α3 than β3 probe sets could be due to an earlier flawed sequence used for α3 probe sets or to alternative splicing.
Abbreviatedgene name
Accession no. Chromosomallocation
Description RNA signal for six ICF versus five control LCLs No. of ICFLCLs showingsimilar FCa
Mean forICF LCLs
Mean forcontrol LCLs
FCa P-valuefrom t-testa
Genes involved in signal transduction or cell survival
GUCY1B3 X66533 4q31.3–q33 Guanylate cyclase 1, soluble, β3b; hemeprotein present in many tissues includingB cells and lymph nodes; large amounts inbrain; signal transduction by NO
1316 292 +4.5 0.007 5
GUCY1A3 X66534 4q31.1–q31.2 Guanylate cyclase 1, soluble, α3b;heterodimerizes with β3
233 101 +2.3 0.03 5
PTPN13 HG3187 4q21.3 Protein tyrosine phosphatase, non-receptortype 13; Fas/CD95-associated; expressedin most tissues; high PTPase associatedwith reduced IL-2 gene activation of Tcells
271 41 +2.7 0.02 4
HMOX1 X06985 22q13.1 heme oxygenase (decycling) 1; major,ubiquitous stress protein; anti-apoptotic
577 2353 –4.1 0.03 4
INPP4A U26398 2q11.2 Inositol polyphosphate-4-phosphatase,type I, 107 kDa, widespread expression
155 289 –1.9 0.009 3
BTG2 Y09943 1q32 BTG family, member 2; induced by p53;anti-apoptotic
367 654 –1.8 0.01 4
Genes involved in transcription control
FHL2 L42176 2q12–q14 Four and a half LIM domains 2; expressedmostly in heart; transcription coactivator;binds integrins
208 56 +2.1 0.02 5
RARA X06614 17q12 Retinoic acid receptor α; ubiquitoustranscription factor; bidirectionalmodulation of granulopoiesis; someevidence for inhibition of B-cell activation
1659 1075 +1.5 0.0003 6
ZFX X59739 Xp21.3 Zinc finger protein, X-linked; ubiquitoustranscription factor
125 300 –2.4 0.01 4
Other genes
F13A1 M14539 6p24.2–p23 Coagulation factor XIII, A1 polypeptide;in blood cells but not lymphocytes
1333 155 +8.6 0.02 4
CNN3 S80562 1p22–p21 Calponin 3, acidic; member of actin-binding protein family; expressed inmany cell types but low expression inleukocytes
407 80 +4.1 0.008 5
KOC1 U97188 7p11 IGF-II mRNA-binding protein 3; variousembryonic tissues and placenta
631 233 +2.7 0.01 5
BBC3 U82987 ND Bcl-2 binding component 3; noinformation about function or expression
669 312 +2.1 0.003 5
MARK3 M80359 14q32.3 MAP/microtubule affinity-regulatingkinase 3; ubiquitous
395 257 +1.5 0.005 5
BTN3A2 U90546 6p23 Butyrophilin, subfamily 3, member A2;immunoglobulin superfamilymember in MHC; near majorhistocompatibility gene complex region in6p
299 578 -1.9 0.006 6
SLC1A1 U08989 9p24 Solute carrier family 1 (neuronal/epithelialhigh affinity glutamate transporter),member 1; widely expressed but especiallyin neurons, intestine, and kidney
137 237 -1.7 0.001 5

Human Molecular Genetics, 2001, Vol. 10, No. 25 2923
PKC eta gene, of the ID family other than ID3, and of the RGSfamily other than RGS1.
ICF-specific changes in RNA levels of other genes
Several genes encoding proteins involved in signal trans-duction or apoptosis, but not specifically associated withlymphogenesis, exhibited ICF-specific differences in RNAlevels. GUCY1B3 and GUCY1A3 (GUCY1B1 and GUCY1A1)(22) are ∼0.1 Mb apart and encode the subunits of a solubleguanylate cyclase. Both RNAs were overexpressed in ICFLCLs (Table 4; Fig. 1). Their hemoprotein mediates NO signalingand is involved in many biological processes (23). Alsoupregulated in the ICF LCLs was the RNA for protein tyrosinephosphatase 13, which can associate with the apoptosis-inducing Fas/CD95 (Table 4). Overexpression of proteinphosphatases may help maintain a resting, non-activatedphenotype of circulating T cells (24). Two anti-apoptotic genesthat are expressed in various tissues and were downregulatedin the ICF LCLs were the cell cycle-related BTG2 (25) and thestress response gene heme oxygenase 1 (HMOX1) (26).
Several genes displaying ICF-specific differences in RNAlevels are transcription factors or coactivators, e.g. MEF2C(Table 3), which is implicated in IgM secretion, and theubiquitous transcription factor retinoic acid receptor α (RARα;Table 4). RARα has been reported to inhibit activation ofresting peripheral blood B cells; to induce or inhibit apoptosis
depending on the concentration of retinoid ligand; and tomodulate granulopoeisis (27,28). Lastly, some RNAs that wereupregulated specifically in ICF LCLs are from poorly characterizedhuman genes, namely, BBC3 (BCL-2 binding component 3),KOC1, MARK3 CNN3, or a gene whose expression is notassociated with leukocytes, namely, F13A1 (Table 4).
Methylation analysis of promoter regions of four geneswith ICF-specific RNA levels
ICF-linked genomic hypomethylation includes Sat2 and Sat3,D4Z4 repeats, NBL2 repeats and some X-linked genes(5,29,30). We tested the LCLs for ICF-specific hypomethyl-ation of the promoter or 5′ region of three genes that displayedICF-linked upregulation of expression (RGS1, GUCY1B3 andF13A1; Tables 3 and 4). Because DNA hypomethylation andhypermethylation are established in different parts of thegenome during embryogenesis and often during carcino-genesis (31), we also analyzed an ICF-downregulated gene(TNFR17) for possible hypermethylation of its promoter. CpGislands overlapping vertebrate promoters and 5′ transcribedregions are usually constitutively unmethylated; therefore, twoof the four genes for this methylation analysis were chosenpartly because they did not have CpG islands in the examinedregion (RGS1 and TNFR17). For one of the two studied geneswhose promoter region is in a CpG island (GUCY1B3), weanalyzed an upstream DNA sequence with a low CpG density
Figure 1. Mean trimmed RNA levels for individual ICF and control LCLs for six probe sets. The data are for individual LCLs for probe sets from Tables 3 and 4and are quantile-normalized RNA signals.

2924 Human Molecular Genetics, 2001, Vol. 10, No. 25
as well as the CpG-rich 5′ region of the gene. These analyseswere done by combined bisulfite restriction analysis(COBRA), a quantitative assay of C methylation in whichrestriction sites can be destroyed or created because of C→Tconversions only at unmethylated C residues upon bisulfitemodification, alkaline treatment and PCR (32). We usedCOBRA rather than genomic sequencing because the latter ismore difficult to quantitate, especially when trying to explain2–4-fold differences of expression. With COBRA, we cananalyze methylation at individual CpG sites, as in Southernanalysis, but COBRA allows more rapid examination ofmultiple genes and displays methylation by a positive resultrather than by enzyme resistance. We analyzed two to eightCpG 5′ or promoter region sites for each of the four examinedgenes (Table 6) and, as described below, the results obtained atdifferent sites for a given DNA region were consistent.
Two of the studied genes, RGS1 and TNFR17, are preferen-tially expressed in mature B cells with little or no expression in
T-cell lines and mitogen-activated T cells (8,12,33,34). RGS1has not been detected in pre-B-cell lines (12). TNFR17 tran-scripts have been observed in pre-B-cell lines but not inlymphoid precursor nor myeloid cell lines (8). The RGS1promoter region has a very low CpG content. The 900 bpupstream of the beginning of TNFR17 has almost 2% CpG andthe predicted CpG frequency from its base composition wouldbe ∼4.5% if there were no under-representation of CpGs.Therefore, it does not display the 4–5-fold under-representationof CpGs typical of bulk vertebrate DNA but it is not as rich inCpGs as a CpG island. Two TNFR17 CpG sites, one located55 bp downstream of the transcription start site (+55; allpositions given relative to the beginning of the gene; Fig. 2)and one at –128 and two RGS1 CpG sites at –83 and –2282were amenable to COBRA (Fig. 2D and E). The –83 RGS1, –128TNFR17 and +55 TFNR17 CpG sites were unmethylated in allLCLs and in the B-cell fraction of blood (<5% digestion;Fig. 2A and C and data not shown). They were ∼40–70%
Table 5. ICF versus control LCLs: immunoglobulin RNA synthesis and surface immunoglobulin expression
aUnits for Ig RNA levels from microarray data are described in Materials and Methods and the legend to Table 2. For IgG and IgA RNA, results are for the IgG3and IgA2 heavy-chain RNAs. Fluorescence-activated cell sorting (FACS) data are given as the percentage of cells with the given antibody on the surface.bMicroarray and flow cytometry results on LCLs are summarized as: ++, moderate to high levels of RNA and cell surface Ig (sIg); +, low levels of RNA and sIg.Where IgG, IgA or IgM levels were insignificant or undetectable, that Ig is not listed in this summary.cClinical data on Ig levels prior to IgG supplementation are described relative to age-matched normal children; Ig–, no detectable Ig. No Ig levels in serum aregiven for the non-immunodeficient donors of the control LCLs.dAt this age, the IgG and IgM might be maternal.
LCL Analysis Ig heavy-chain RNA levels and % of cells with theindicated surface Iga
sIg status of LCLb Patient’s serum Ig (age at analysis)c
IgM IgD IgG IgA
ICF B Microarraya 8256 2,687 294 32 IgM++ IgD++ Ig– (1 year)
FACS (%)a 86 21 0 0
ICF B′ Microarray 7083 5455 180 203 RNA: IgM++ IgD++ Same patient as above
FACS (%) 0 0 0 0 Null for surface Ig
ICF C Microarray 7204 3341 275 780 IgM++ IgD++ IgG and IgM, low (6 months)d
FACS (%) 43 17 0 0
ICF G Microarray 7892 2916 245 100 IgM++ IgD++ IgG, normal; IgA and IgM, low (1.5 years)
FACS (%) 60 14 0 0
ICF K Microarray 7137 2417 197 1028 RNA: IgM++ IgD++ IgG–; IgA–; IgM, low (1.5 years)
FACS (%) 0 0 0 0 Null for surface Ig
ICF S Microarray 5561 3484 557 321 IgM++ IgD++ IgG very low; IgA and IgM, low (1.5 years)
FACS (%) 69 22 <1 0
Normal B Microarray 2632 484 8136 6440 IgG++ IgA++ IgM+ –
FACS (%) 0 0 29 16 IgG++ IgA++
Normal C Microarray 1009 133 27 121 376 IgG++ –
FACS (%) 1.6 <1 86 0
Normal G Microarray 5977 1728 24 289 18 614 IgG++ IgA++ IgM++ IgD+ –
FACS (%) 4 0 49 7 IgG++ IgA+ IgM+
Normal A Microarray 6971 1438 172 1916 IgA+ IgM++ IgD+ –
FACS (%) 46 9 0 <1 IgM++ IgD+
Normal D Microarray 175 258 33 617 14 981 IgG++ IgA++ –
FACS (%) 1 <1 63 29

Human Molecular Genetics, 2001, Vol. 10, No. 25 2925
methylated in various normal tissues with the exception ofRGS1’s site in sperm, which displayed >99% methylation.That the examined RGS1 promoter’s CpG was also hypo-methylated in a non-B-cell leukocyte fraction, where this gene isnot expected to be expressed, suggests that hypomethylation ofthis region precedes expression rather than being a consequenceof it. Further upstream at the –2282 RGS1 CpG site, there was40–90% methylation in all tested cell populations but, again,there was no correlation of methylation status with the ICFphenotype (Fig. 2).
Four CpG sites within the 300 bp upstream of the beginningof the widely expressed guanylate cyclase β3 gene(GUCY1B3) were amenable to COBRA using TaqI (threesites) and RsaI (one site). This amplified promoter region wasundigested (<5% digestion) and, therefore, unmethylated in alltested LCLs and tissues with two exceptions (liver and NormalD, ∼10–50% digestion with either of the above enzymes). Thispromoter region is part of a 1.7 kb CpG island extending intothe beginning of the gene and has CpG and C + G contents of4 and 57%, respectively. Further upstream, in the –380 to –650region, which has a typical, low CpG content (1%), there wasmuch methylation of all tested LCL and somatic tissue samplesat four COBRA sites assayed with HpyCH4IV. In contrast,sperm was completely unmethylated at these sites. The levelsof methylation in the somatic cell samples were variable but,again, no correlation was seen between the ICF status and theextent of methylation. COBRA assays of three CpG sites (oneeach for HpyCH4IV, BstUI, and TaqI) from +53 to +144 ofF13A1, which encodes a subunit of the coagulation factor XIII,revealed a complete lack of methylation (<5%) in all the LCLsand tissues despite this protein’s specific association withmonocytes/macrophages, megakaryocytes/platelets and placenta(35). From approximately the transcription start point to ∼800 bpdownstream is a CpG island, which has CpG and C + G contentsof 5 and 64%, respectively.
DISCUSSION
ICF is usually discovered in early childhood by the repeatedoccurrence of severe infections and by the finding of highfrequencies of rearrangements in the pericentromeric regionsof Chr1 and Chr16 in mitogen-stimulated lymphocytes (3).ICF leukocytes always display hypomethylation in Chr1 andChr16 within the Sat2-containing heterochromatin adjacent tothe centromere, i.e. the juxtacentromeric heterochromatin (1qhand 16qh), as well as in other DNA repeats (29). ICF DNAhypomethylation, which is restricted to a small percentage ofthe genome (7% in brain) (4), is usually the result of mutationsin both alleles of DNMT3B (36). It could be due to a lack ofde novo methylation of these sequences early in embryo-genesis after most of the DNA methylation is lost and methyl-ation patterns are re-established and/or to a deficiency inDNMT3B activity in postnatal cell populations. Adult cells,including ICF LCLs (37), contain DNMT3B RNA althoughDNMT3B expression appears to be especially high in embryoniccells (2).
We studied differential expression of genes in ICF versusnormal LCLs using B-cell lines (LCLs) because the mostconsistent and important symptom of this syndrome involvesB-cell dysfunction, namely, decreased Ig levels in serum. Thesix ICF LCLs that we studied retained the cellular phenotype
of Sat2 hypomethylation and chromosomal abnormalities inthe Sat2-rich 1qh and 16qh (Table 1). Validation of themicroarray RNA analysis on LCLs was established in severalways. Among the genes showing ICF-specific over- or under-expression, lymphogenesis-associated genes were significantlyover-represented compared to all LCL-expressed sequences(see Results), and these differences correlate with the immuno-deficiency of ICF. For five of the genes displaying ICF-specific differences in RNA levels (TFRC, IGHM, IGHA1,GUCY1B3 and TNFR17), we had independent, closely relatedprobe sets on the microarray that confirmed the ICF-associatedchanges (Tables 2–4). Lastly, we verified the ICF-specificexpression from five genes (IGHG3, IGHA1, IGHA2, IGHDand IGHM) at the protein level (Table 5).
Whereas many of the genes that showed significant ICF-specificunderexpression or overexpression in the LCLs can accountfor much of the immunodeficiency that characterizes ICF (seebelow), none is a good candidate to explain the chromosomeinstability targeted to the pericentromeric regions of Chr1 andChr16 in mitogen-stimulated lymphocytes and in LCLs fromICF patients. Therefore, cis effects of hypomethylation at 1qhand 16qh might suffice in the context of the correct cell type(lymphocytes stimulated with mitogen in vitro or B-cell lines)(3,4,38) for predisposing to these chromosomal anomalies (4).Caveats are that the microarray that was used in this workrepresents only ∼20% of the estimated number of human genesand some of the genes that are involved in transcription controlor signal transduction and display ICF-specific differences inRNA levels (Tables 3 and 4) might indirectly contribute to theICF chromosome abnormalities.
The present work clarifies the previously unknown nature ofthe immunodeficiency in ICF. Sometimes, the defectiveresponse of ICF patients to antigens is seen as low in vitrostimulation indices for B or T cells upon exposure to mitogensor antigens (39–41). Infections usually affect the pulmonary orgastrointestinal tract. Serum Ig levels in blood samples arehighly variable in ICF patients, but the typical finding is lowserum Ig coupled with normal B- and T-cell numbers in peripheralblood (C.M.R.Weemaes and D.Smeets, unpublished data),indicating that the early stages of lymphocyte differentiationare usually not abnormal. In some patients, deficiencies are seen inonly certain, variable subclasses of IgG (41; B.H.Belohradsky,unpublished data). The variability in the phenotype of ICFpatients is probably partly attributable to the varied nature ofDNMT3B mutations. To date, such mutant alleles in ICFpatients always include at least one allele that is not a nullallele and so may specify different amounts of residualDNMT3B activity (36). This is consistent with the finding thathomozygous Dnmt3b knockout in mice is lethal duringembryogenesis (2).
Our analysis of ICF B-cell lines for surface antigens showedthat high percentages of these cells have membrane-boundIgM and/or IgD, indicating that V(D)J recombination wasnormal for the precursors of these cells. No surface IgG or IgAwas detected in these LCLs, in contrast to the control LCLs,and none had appreciable levels of IgG or IgA RNA. Despitethe unusually high percentages of cells containing surface IgMin six of the eight examined ICF LCLs, the patients from whomthese were derived were moderately to severely deficient inserum IgM (Table 5 and data not shown). These findingssuggest that a block at class switching is not the immediate

2926 Human Molecular Genetics, 2001, Vol. 10, No. 25
problem in the immune system of ICF patients but rather thatthey have other deficiencies in lymphocyte maturation oractivation.
In the microarray analysis, we found highly promising geneswhose dysregulation may be largely responsible for theimmunodeficiency in ICF. None of the genes exhibiting ICF-
specific differences in RNA levels correspond to genes shownby microarray analysis to be dysregulated in a human colonadenocarcinoma cell line treated with the DNA demethylating(and DNA damaging) agent 5-azadeoxycytidine (42). Also,none are the same as those whose RNAs are abnormallyregulated in murine fibroblasts containing a conditional
Table 6. Parameters for COBRA analysis of DNA methylation for six regions of genes displaying ICF-specific differences in RNA levels
aThe sequences (5′–3′) of the PCR primers for the bisulfite-treated strand (the sense strand) are given followed in parenthesis by the sequence of the sense strandin the primer region before bisulfite treatment.bTen micrograms of DNA was used for bisulfite treatment, purified and 1/20 of the DNA was subject to PCR as described previously (63). The annealing tem-perature and time for annealing are given. The indicated regions were amplified by one or two rounds of PCR of 30 or 32 cycles each with a 1:100 dilution of thefirst-round product and freshly added primers, buffer and enzyme in the case of two rounds of PCR. The necessity of more amplification with some of the primer-pairs than with others is due to the non-optimal nature of many primers for bisulfite-treated DNA, e.g. their enrichment in A + T and runs of T residues from C→Tconversions.cRestriction sites followed by an asterisk do not pre-exist in the untreated genomic DNA, but rather, are generated by C→T conversions at non-CpG C residuesin the sequence upon bisulfite treatment and PCR. Cleavage at all the given restriction sites was obtained only if the CpG-containing C residue in the sequencewas methylated.dThe sizes in base pairs of the restriction fragments that would be obtained from the PCR products if there were complete CpG methylation at the site are given. Frag-ments of >90 bp could be detected in the 1% electrophoresis gel.
Gene region Primer sequences (sequence in theamplifiedstrand before bisulfite modification)a
Annealing conditionsandcycle numberb
PCR product size andGenBank coordinates
Enzymec, positionof site in GeneBanksequence
Sizes (bp) ofdigestion fragmentsd
TNFR17 promoter and5′ region of gene
AAAGAATAGTTTGGAAGGA 42°C, 55 s, 32 cycles 412 bp; HSU95742:31931–32342
TaqI, 32246* 96, 316
(AAAGAACAGCCTGGAAGGA) BstUI, 32063 133, 279
AAACACAACACAAACCTT
(AAGGTCTGTGCTGTGCCC)
RGS1 promoter TTAGAATTAAAGATTGATGTT 36°C, 55 s, 30/30cycles
258 bp; AL136987:90890–91147
HpyCH4IV, 91051 96, 162
(CCAGAACCAAAGACTGATGCT)
AAATACAAACAAACACTTACT
(AGCAAGTGCCTGTCTGCATTC)
RGS1 upstream of promoter TGTATGGTATTATAATGATGG 40°C, 55 s, 32 cycles 652 bp; AL136987:88435–89086
HpyCH4IV, 88851 235, 417
(TGTATGGTACTATAATGATGG)
CTATTTACTCATCCACCC
(GGGTGGATGAGCAAACAG)
GUCY1B3 promoter TTGTTTGTGTTAATTATTGAGG 44°C, 55 s, 30/30cycles
302 bp; NT_006129:165088–165389
RsaI, 165188* 100, 202
(TTGCTTGTGTTAACCACTGAGG) TaqI, 165289*,165335*, 165354
19, 35, 46, 202
CTAATTACCACCTCTTCCCA
(TGGGAAGAGGTGGCAATCAG)
GUCY1B3 upstream ofpromoter and CpG island
AAGTTGGAGGTAGAGTAGTT 48°C, 55 s, 32 cycles 553 bp; NT_006129:164602–165154
HpyCH4IV, 164825,165050, 165056*,165073*
6, 17, 81, 224, 225
(AAGCTGGAGGTAGAGTAGTT)
CAACTCTTATAAACCCACT
(AGTGGGTCCACAAGAGCTG)
F13A1 5′ region of gene AGGATTGGGTAAGTGGAG 44°C, 55 s, 32 cycles 235 bp; AL133326:96301–96535
BstUI, 96470 65, 170
(AGGATTGGGCAAGTGGAG)
CCAACTATCTTATCTTTCCC TaqI, 96378* 78, 157
(GGGAAAGACAAGACAGTTGG) HpyCH4IV, 96396* 96, 139

Human Molecular Genetics, 2001, Vol. 10, No. 25 2927
knockout of Dnmt1 that causes massive demethylation (43),unlike the very limited demethylation in ICF cells. Our studyrevealed nine genes with ICF-specific dysregulation whoseprotein products are involved in lymphocyte migration,activation or survival consistent with abnormal lymphocytematuration or function (Table 3). For example, the ICF LCL-linkedoverexpression of RGS1 (Table 3; Fig. 1), a B-cell-associated
negative regulator of heterotrimeric G-protein signaling, probablyreflects a decreased ability of B cells in ICF patients to respondto chemokines (12,44). Also, RGS1 strongly inhibits thedirected migration of lymphoid cells (11,12). RGS1 expressionhas been postulated to lead to the retention of B cells in thegerminal center of lymphoid organs (12). That significantlymore RGS1 RNA was present in ICF LCLs than in the controlLCLs is consistent with the hypothesis that B-cell migrationwithin lymphoid organs is defective in ICF patients, in turn,preventing the normal final stages of maturation.
A gene that displayed ICF-specific changes in RNA levels,the integrin β7 gene (ITGB7; Table 3; Fig. 1), has been demon-strated to be important for promoting intra-organ movement oflymphocytes (45). Whereas RGS1 is involved in negative controlof lymphocyte migration and had an increased concentration of itsRNA in ICF LCLs, ITGB7 promotes lymphocyte migrationand, accordingly, had lower levels of RNA in ICF LCLs.Another RNA whose downregulation in ICF LCLs suggestsinvolvement in ICF immune dysfunction encodes TNFR17, aB-cell-associated member of the tumor necrosis receptorfamily without a death domain (8,34) (Table 3; Fig. 1). It is thereceptor for zTNF4 (also known as BLyS, BAFF, TALL-1 orTHANK), which plays an important role in B-cell expansionand antibody responses (9). Functional redundancy betweenTnfr17 and Taci, another zTNF4 receptor, may be responsiblefor the lack of a phenotype in Tnfr17 (Bcma) homozygousknockout mice (46), but such redundancy might not exist inhumans, especially because there is only 56% identity betweenthe human and murine proteins. TNFR17 has been proposed totransduce signals for cell survival and proliferation throughactivation of the MAP kinase pathway (47), which is an essentialpart of B- and T-cell development.
Two other genes that are also underexpressed in ICF LCLsand can be upregulated in stimulated B cells are transferrinreceptor (21) and B-MEF-2 (apparently encoded by MEF2C)(20). MEF2C is primarily expressed in skeletal muscle, brain,and spleen, and within the lymphoid lineage, it is restricted toB cells (20,48). In B cells, MEF2C is a positive regulator ofJ chain expression (20,49). Because the J chain is required foroptimal production of secreted pentameric IgM and dimericIgA, decreased expression of MEF2C (Table 3; Fig. 1) couldaffect the ability of ICF B cells to produce secreted forms ofIgM while maintaining normal expression of IgM on thesurface of the B cells, consistent with the phenotype of the ICFpatients in this microarray study. The product of the othergene, transferrin receptor, is critical for transport of iron intocells. Increased transferrin receptor in B cells is a marker foractivation (21) and has a role in antigen trafficking in B cells(50). Thus, the decrease in transferrin receptor RNA observedin ICF LCLs in comparison to normal LCLs (Table 3) mayindicate a diminished activation state of B cells in ICF patients.
A gene that functions in differentiation during embryo-genesis as well as during lymphogenesis and displayed ICF-specific decreases in RNA levels is ID3. Underexpression ofID3 can help explain defective immune function withoutdecreases in B- or T-cell numbers, as seen in the majority ofICF patients. ID3 inhibits the DNA binding of positive-actingtranscription factors (especially E factors) by heterodimerizingwith them. Several of these E factors play central roles in B- andT-cell maturation. Despite B and T cells being phenotypicallynormal and in normal numbers in Id3 homozygous knockout
Figure 2. Analysis of DNA methylation by COBRA in upstream or 5′ regionsof genes displaying ICF-specific changes in RNA levels. RepresentativeCOBRA electrophoresis gels stained with ethidium bromide and visualized for flu-orescent bands are shown. First, the DNA samples had been modified withbisulfite for differentiation between methylated and unmethylated CpGs atgiven restriction sites. Then, the samples were amplified by PCR and the prod-ucts digested with the appropriate enzyme. The undigested PCR products areshown by an asterisk and had unmethylated CpGs making them resistant todigestion during COBRA. The digested PCR products, which are shown by aplus sign, are indicative of methylation at the tested size. (A) PCR from – 245to +13 relative to the transcription start site of RGS1 followed by digestion withHpyCH4IV. The transcription start site [broken arrow in (D)] is approximateand derived from GenBank sequence AL136987 (position 91 134). (B) PCRfrom –2699 to –2048 of RGS1 followed by digestion with HpyCH4IV. (C) PCRfrom +188 to –224 relative to the transcription start site of TNFR17 with subse-quent digestion with BstUI. The transcription start site had been determinedexperimentally (8) and is at position 32 118 in GenBank sequence HSU95742.DNA from the total white blood cell (WBC) fraction from a normal individual,the B-cell fraction obtained by cell sorting of leukocytes, or the cells other thanB cells obtained by the cell sorting was analyzed in the last three lanes of (A–C).(D) Distribution of CpGs in the promoter region and further upstream of RGS1.(E) Distribution of CpGs in the 5′ region of TNFR17; the Taq I site was ana-lyzed in gels not shown. For (D and E), positions given are relative to the Gen-Bank sequences.

2928 Human Molecular Genetics, 2001, Vol. 10, No. 25
mice, these mice have reduced IgG1 and IgG2a levels andlarge decreases in production of various subclasses of serumIg, including IgM, in response to T-cell dependent or T-cellindependent antigens (51). Part of their deficient humoralimmunity may be due to a defective B-cell proliferationresponse to activation of the B-cell receptor complex (51).Furthermore, in another Id3–/– mouse model, Id3 was shown tobe necessary for thymocyte positive selection, possiblyoperating through T-cell receptor signaling (18). Importantlyfor elucidating the immunodeficiency associated with ICF,Id3+/– mice, which should have approximately half the normalamount of Id3 RNA, also had perturbed thymocyte maturation(18). This phenotype for the heterozygotes could be a result ofthe loss of a delicate balance between levels of Id3/ID3 and ofa thymocyte-specific transcription activator E protein. ICFLCLs also had approximately half the level of ID3 RNAcompared to controls (Table 3; Fig. 1).
Some of the changes in RNA levels in ICF versus controlLCLs may be secondary to changes in RNA levels specified byother genes. For example, the increase in RGS1 RNA in ICFLCLs (Table 3; Fig. 1) should lead to decreased Giα and Gqαsignaling resulting in downregulation of MAP kinase activityin response to platelet activation factor (PAF) (12). Effects ofmodest changes in RGS1 protein levels might be dramaticbecause RGS proteins generally have low Km values (52).Negative regulation of MAP kinase activity might be respon-sible for the ICF-specific decreases in ID3 RNA levels becauseID3 is regulated at the transcription level by MAP kinaseactivity (53). Also, a lack of maturity of B cells in ICF patients’peripheral blood could account for the low levels of some ofthe RNAs in ICF LCLs, e.g. integrin β7, TNFR17 and TFRCRNAs, because those proteins are associated with leukocyte orB-cell maturation (21,34,54,55).
However, the postulated immaturity of the B cells from whichthe LCLs were established would not explain the ICF-linkedincreases in RGS1 RNA. Higher levels of transcripts fromRGS1 are associated with mitogen activation of tonsil B cellsor induction of G-protein signaling in a B-cell line by PAF(12,33). RGS proteins have been proposed to be necessary forfeedback desensitization of G-protein activated pathways (11)and so the increase in RGS1 levels in B cells from ICF patients,who appear to be deficient in the activation of B cells, indicatesa premature induction of this feedback control. We proposethat RGS1 and a few other of the genes showing ICF-specificdifferences in RNA levels (as well as genes not represented inthis array) are proximal targets of the deficiency in DNMT3Bactivity in ICF patients. Most of the other genes with ICF-specificchanges in their RNA levels may be subject to changes intranscription or post-transcriptional processing or stabilizationof their mRNA secondary to proximally affected genes thatcontrol signal transduction, other types of feedback pathwaysor transcription.
The DNMT3B mutations in ICF patients are often missensemutations in the catalytic domain, suggesting that it is the lossof DNA methyltransferase activity and not some other functionof the protein that is responsible for the syndrome. Forvertebrate DNA sequences subject to transcription control byDNA methylation, this control is almost always downregulation.Furthermore, DNA methylation is often associated withheterochromatinization, which is generally repressive totranscription. Therefore, we hypothesize that it is some of the
upregulated genes in ICF LCLs whose RNA levels are mostdirectly affected by the DNMT3B mutations and that this is theresult of ICF-specific DNA hypomethylation. Nevertheless, noICF-specific differences in promoter methylation were seen inthree of the genes that had higher RNA levels in ICF than in thecontrol LCLs. COBRA analysis of promoter regions of the twoB-cell-specific genes did reveal B-cell- or leukocyte-specifichypomethylation, but this occurred irrespective of whether thecells were derived from ICF patients or unaffected individuals(Fig. 2A and C). In ICF lymphoid cells, hypomethylation ofregions of the genome that are normally constitutively hetero-chromatic (e.g. 1qh and 16qh) and consequent decreases inheterochromatinization (3,4) might affect regulation ofexpression of genes elsewhere in the genome. This could occurby altering the nuclear compartmentalization of a smallpercentage of the gene-containing euchromatin or modulatingthe sequestration of certain transcription regulatory proteins atspecific constitutive heterochromatin regions (56,57).Evidence suggests that, during murine lymphogenesis, centro-meric heterochromatin is involved in the heritable repressionof TdT and several other genes through an interaction witheuchromatin regions containing these genes (56,58). Ikaros, atranscription regulatory factor that binds specifically to TdTand λ5 promoters and to centromeric heterochromatin,mediates this repression of TdT and λ5 transcription (57,59).Similarly, centromeric heterochromatin seems to have arepressive interaction with the human β-globin locus (60).Different heterochromatin-rich domains, such as the humanSatα-rich centromeric heterochromatin and Sat2-rich1qh and16qh, might act independently of one another (56). Furtherstudies will be necessary to determine whether, in normallymphocytes, association of highly methylated DNA inconstitutive heterochromatin negatively controls expression ofgenes that exhibit upregulation of their RNA in ICF LCLs andwhether this control is abrogated in ICF lymphocytes by DNAhypomethylation.
MATERIALS AND METHODS
Cell lines and their characterization
All LCLs were Epstein–Barr virus-transformed B-cell lines.LCLs ICF B/B′, ICF C, ICF S, ICF K and ICF G have beendescribed previously (3,6,39,40,61); the first three were frompatients P1, P6 and P14, respectively, in Wijmenga et al. (36).Normal B, C and G were from phenotypically normal parentsof patients who were donors for ICF B/B′, C and G LCLs.Normal A and D were from phenotypically normal individuals(4). ICF B, ICF B′, ICF G, ICF K, Normal A, D and C werefrom males whereas the other LCLs were from females. Fiftymetaphases were examined for gross chromosomal aberrationsin each LCL from the same cell passage used for harvesting forthe microarray expression analysis. To prepare cell pellets forRNA extraction for microarray analysis, LCLs were split 1:3.3when they were at 5 × 105 cells/ml, incubated, and thenapproximately 10 × 106 cells were harvested when the culturesreached a concentration of 3 × 105 cells/ml. Chromosomalanomalies and Chr1 Sat2 DNA methylation was assessed asdescribed by Tuck-Muller et al. (4). ICF LCLs which wereused just for flow cytometry were from patient B in Xu et al.(1) and his affected brother.

Human Molecular Genetics, 2001, Vol. 10, No. 25 2929
Microarray analysis
Total RNA was isolated by standard methods (Trizol reagent,Invitrogen, Gibco Life Technology, Carlsbad, CA; RNeasyspin column, Qiagen, Valencia, CA). Preparation of cRNAfrom 5 µg of total RNA, hybridization and scanning of theoligonucleotide arrays (HuGeneFL, Affymetrix, Sanat Clara,CA) were performed as described by Le Naour et al. (62) on5 µg of total RNA reverse transcribed with an oligo(dT)24primer containing a T7 RNA polymerase promoter site. Then,labeled cRNA was generated by in vitro transcription. Fifteenmicrograms was fragmented at 94°C for 35 min in 40 mMTris–acetate, 100 mM potassium acetate, 30 mM magnesiumacetate pH 8.1 and used to prepare 300 µl of hybridizationmixture [100 mM 4-morpholineethanesulfonic acid (MES) pH6.7, 1 M NaCl, 20 mM EDTA, 0.01% Tween 20, 0.1 mg/mlherring sperm DNA, 500 µg/ml acetylated bovine serumalbumin] containing a mixture of control cRNAs for hybridiza-tion efficiency and quantitation of transcript levels. Thehybridization mixtures were heated to 94°C for 5 min, equili-brated at 45°C for 5 min, then clarified by centrifugation. Tenmicrograms of fragmented cRNA was hybridized to the arraysat 45°C for 16 h. The arrays were washed with 6× saline/sodium phosphate/EDTA (SSPE) at 25°C and then with100 mM MES, 0.1 M NaCl, 0.01% Tween 20 pH 6.7, at 50°C.They were stained with streptavidin-phycoerythrin, washedwith 6× SSPE, stained with biotinylated anti-streptavidin IgG,stained again with streptavidin-phycoerythrin, washed with 6×SSPE, then scanned, and subjected to image analysis (Gene-Array scanner and GeneChip 4.0 software, Affymetrix).
Statistical analyses
Each probe set on the microarrays typically consists of20 oligonucleotides complementary to a specific cDNA[perfect match (PM) features], and 20 identical oligonucleo-tides except for a central mismatch (MM features). Probe-pairsfor which the PM – MM intensities were less than –1000 onone normal LCL that was selected as a standard were excludedfrom the analysis, as were probe-pairs displaying saturation inthe image of the standard for either the PM or MM. For asaturated PM (or MM) value on other chips, the values wereimputed by considering the ratio of non-saturating PM (or MM)values compared to the standard chip. A one-sided signed-ranktest on the PM – MM differences was used to test expression forthe probe set. The average intensity for each probe set wascomputed as the mean of PM – MM differences after trimmingaway the 25% highest and lowest PM – MM differences (seehttp://dot.ped.med.umich.edu:2000/pub/ICF/index.html). Theaverage intensities for each chip were normalized using apiece-wise linear function that makes 99 evenly spaced quantileswhich have the same values as the corresponding quantiles ofthe standard. In order to fit linear models, the data were thenlog-transformed by adding 100 to each trimmed-mean,converting still negative values to 0, and then adding 1 andtaking logarithms. ICF and normal LCLs were compared usingtwo-sample t-tests on the log-transformed data. For compar-ison of non-LCL cell lines with LCLs, a one-way ANOVAmodel was fit and the resulting t-tests for comparing pairs ofgroups used to judge the significance of LCL versus non-LCLdifferences.
DNA methylation analysis (COBRA)
COBRA was done as described by Xiong and Laird (32) andSun et al. (63). Oligonucleotides and PCR conditions are givenin Table 6. Only the expected sized PCR products wereobtained. Ethidium bromide-induced fluorescence in the bandsrepresenting the cleaved versus the uncleaved PCR productswas quantitated by digital analysis. Internal controls forcomplete digestion that were included in each digest indicatedthat no inhibitors of digestion were present. We demonstratedthat the primer-pairs, which were designed to amplify onlybisulfite-treated DNA, could amplify the genomic DNA onlywhen it was bisulfite modified or that the PCR product fromthe bisulfite-modified DNA was unable to be cleaved by anon-CpG restriction endonuclease whose restriction siteshould be destroyed by a C→T conversion in the modifiedDNA whereas it could be cleaved by enzymes whose restrictionsite should be retained after bisulfite modification.
Flow cytometric analysis of LCLs of surface markerexpression
LCL cells (106) were incubated with monoclonal FITC-antihuman IgD, PE-anti human IgM, biotinylated-anti human IgG,biotinylated-anti human IgA and FITC-anti human CD19 (BDPharmingen, San Diego, CA) for 40 min on ice. The cells werewashed and incubated with streptavidin-CyChrome on ice for40 min. Appropriate conjugated isotype control antibodieswere used to quantify non-specific staining. Followingstaining, the LCLs were washed, fixed in cold 1% para-formaldehyde and analyzed by flow cytometry. Sorting for Blymphocytes from peripheral blood was done from a leukocytefraction obtained by lysing red blood cells and stainingremaining cells for 45 min with FITC-conjugated anti-humanCD19 and the subset of interest sorted.
ACKNOWLEDGEMENTS
We are very grateful to G.K.Hinkel for generously sharing ICFLCLs with us and to K.Uhlmann and M.Karbasiyan forunpublished data on the ICF K mutation. We are also verythankful to Alan Tucker for preparative sorting of the B-cellfraction. This research was supported in part by NIH grantsCA81506 (to M.E.) and CA26803 (to S.H.) from the NationalInstitutes of Health.
REFERENCES
1. Xu, G., Bestor, T.H., Bourc’his, D., Hsieh, C., Tommerup, N., Hulten, M.,Qu, S., Russo, J.J. and Viegas-Péquignot, E. (1999) Chromosomeinstability and immunodeficiency syndrome caused by mutations in aDNA methyltransferase gene. Nature, 402, 187–191.
2. Okano, M., Bell, D.W., Haber, D.A. and Li, E. (1999) DNA methyltransferasesDnmt3a and Dnmt3b are essential for de novo methylation and mammaliandevelopment. Cell, 98, 247–257.
3. Smeets, D.F.C.M., Moog, U., Weemaes, C.M.R., Vaes-Peeters, G.,Merkx, G.F.M., Niehof, J.P. and Hamers, G. (1994) ICF syndrome:a new case and review of the literature. Hum. Genet., 94, 240–246.
4. Tuck-Muller, C.M., Narayan, A., Tsien, F., Smeets, D., Sawyer, J.,Fiala, E.S., Sohn, O. and Ehrlich, M. (2000) DNA hypomethylation andunusual chromosome instability in cell lines from ICF syndrome patients.Cytogenet. Cell Genet., 89, 121–128.
5. Jeanpierre, M., Turleau, C., Aurias, A., Prieur, M., Ledeist, F., Fischer, A.and Viegas-Pequignot, E. (1993) An embryonic-like methylation pattern

2930 Human Molecular Genetics, 2001, Vol. 10, No. 25
of classical satellite DNA is observed in ICF syndrome. Hum. Mol.Genet., 2, 731–735.
6. Schuffenhauer, S., Bartsch, O., Stumm, M., Buchholz, T., Petropoulou, T.,Kraft, S., Belohradsky, B., Meitinger, T. and Wegner, R. (1995) DNA, FISHand complementation studies in ICF syndrome; DNA hypomethylation ofrepetitive and single copy loci and evidence for a trans acting factor.Hum. Genet., 96, 562–571.
7. Narayan, A., Tuck-Muller, C., Weissbecker, K., Smeets, D. and Ehrlich, M.(2000) Hypersensitivity to radiation-induced non-apoptotic and apoptoticdeath in cell lines from patients with the ICF chromosome instabilitysyndrome. Mutat. Res., 456, 1–15.
8. Laabi, Y., Gras, M.P., Brouet, J.C., Berger, R., Larsen, C.J. and Tsapis, A.(1994) The BCMA gene, preferentially expressed during B lymphoidmaturation, is bidirectionally transcribed. Nucleic Acids Res., 22, 1147–1154.
9. Gross, J.A., Johnston, J., Mudri, S., Enselman, R., Dillon, S.R., Madden, K.,Xu, W., Parrish-Novak, J., Foster, D., Lofton-Day, C. et al. (2000) TACIand BCMA are receptors for a TNF homologue implicated in B-cellautoimmune disease. Nature, 404, 995–999.
10. Budworth, J., Meillerais, S., Charles, I. and Powell, K. (1999) Tissuedistribution of the human soluble guanylate cyclases. Biochem. Biophys.Res. Commun., 263, 696–701.
11. Berman, D.M. and Gilman, A.G. (1998) Mammalian RGS proteins:barbarians at the gate. J. Biol. Chem., 273, 1269–1272.
12. Moratz, C., Kang, V.H., Druey, K.M., Shi, C.S., Scheschonka, A.,Murphy, P.M., Kozasa, T. and Kehrl, J.H. (2000) Regulator of G proteinsignaling 1 (RGS1) markedly impairs Gi α signaling responses ofB lymphocytes. J. Immunol., 164, 1829–1838.
13. Morrow, T.A., Muljo, S.A., Zhang, J., Hardwick, J.M. and Schlissel, M.S.(1999) Pro-B-cell-specific transcription and proapoptotic function ofprotein kinase Ceta. Mol. Cell. Biol., 19, 5608–5618.
14. Prasad, K.V., Ao, Z., Yoon, Y., Wu, M.X., Rizk, M., Jacquot, S. andSchlossman, S.F. (1997) CD27, a member of the tumor necrosis factorreceptor family, induces apoptosis and binds to Siva, a proapoptoticprotein. Proc. Natl Acad. Sci. USA, 94, 6346–6351.
15. Deehan, M.R., Klaus, G.G., Holman, M.J., Harnett, W. and Harnett, M.M.(1998) MAPkinase: a second site of G-protein regulation of B-cellactivation via the antigen receptors. Immunology, 95, 169–177.
16. Islam, K.B., Rabbani, H., Larsson, C., Sanders, R. and Smith, C.I. (1995)Molecular cloning, characterization and chromosomal localization of ahuman lymphoid tyrosine kinase related to murine Blk. J. Immunol., 154,1265–1272.
17. Artis, D., Humphreys, N.E., Potten, C.S., Wagner, N., Muller, W.,McDermott, J.R., Grencis, R.K. and Else, K.J. (2000) β7 integrin-deficientmice: delayed leukocyte recruitment and attenuated protective immunityin the small intestine during enteric helminth infection. Eur. J. Immunol.,30, 1656–1664.
18. Rivera, R.R., Johns, C.P., Quan, J., Johnson, R.S. and Murre, C. (2000)Thymocyte selection is regulated by the helix–loop–helix inhibitorprotein, Id3. Immunity, 12, 17–26.
19. Glynne, R., Ghandour, G., Rayner, J., Mack, D.H. and Goodnow, C.C.(2000) B-lymphocyte quiescence, tolerance and activation as viewed byglobal gene expression profiling on microarrays. Immunol. Rev., 176,216–246.
20. Rao, S., Karray, S., Gackstetter, E.R. and Koshland, M.E. (1998) Myocyteenhancer factor-related B-MEF2 is developmentally expressed in B cellsand regulates the immunoglobulin J chain promoter. J. Biol. Chem., 273,26123–26129.
21. Futran, J., Kemp, J.D., Field, E.H., Vora, A. and Ashman, R.F. (1989)Transferrin receptor synthesis is an early event in B cell activation.J. Immunol., 143, 787–792.
22. Zabel, U., Weeger, M., La, M. and Schmidt, H.H. (1998) Human solubleguanylate cyclase: functional expression and revised isoenzyme family.Biochem. J., 335, 51–57.
23. Lee, Y.C., Martin, E. and Murad, F. (2000) Human recombinant solubleguanylyl cyclase: expression, purification and regulation. Proc. NatlAcad. Sci. USA, 97, 10763–10768.
24. Gjorloff-Wingren, A., Saxena, M., Han, S., Wang, X., Alonso, A.,Renedo, M., Oh, P., Williams, S., Schnitzer, J. and Mustelin, T. (2000)Subcellular localization of intracellular protein tyrosine phosphatases in Tcells. Eur. J. Immunol., 30, 2412–2421.
25. Guardavaccaro, D., Corrente, G., Covone, F., Micheli, L., D’Agnano, I.,Starace, G., Caruso, M. and Tirone, F. (2000) Arrest of G(1)-S progression bythe p53-inducible gene PC3 is Rb dependent and relies on the inhibition ofcyclin D1 transcription. Mol. Cell. Biol., 20, 1797–1815.
26. Elbirt, K.K. and Bonkovsky, H.L. (1999) Heme oxygenase: recentadvances in understanding its regulation and role. Proc. Assoc. Am.Physicians, 111, 438–447.
27. Lomo, J., Smeland, E.B., Ulven, S., Natarajan, V., Blomhoff, R., Gandhi, U.,Dawson, M.I. and Blomhoff, H.K. (1998) RAR, not RXR, ligands inhibitcell activation and prevent apoptosis in B-lymphocytes. J. Cell Physiol.,175, 68–77.
28. Kastner, P., Lawrence, H.J., Waltzinger, C., Ghyselinck, N.B., Chambon, P.and Chan, S. (2001) Positive and negative regulation of granulopoiesis byendogenous RARα. Blood, 97, 1314–1320.
29. Kondo, T., Comenge, Y., Bobek, M.P., Kuick, R., Lamb, B., Zhu, X.,Narayan, A., Bourc’his, D., Viegas-Pequinot, E., Ehrlich, M. and Hanash, S.(2000) Whole-genome methylation scan in ICF syndrome: hypomethylationof non-satellite DNA repeats D4Z4 and NBL2. Hum. Mol. Genet., 9, 597–604.
30. Hansen, R.S., Stoger, R., Wijmenga, C., Stanek, A.M., Canfield, T.K.,Luo, P., Matarazzo, M.R., D’Esposito, M., Feil, R., Gimelli, G. et al.(2000) Escape from gene silencing in ICF syndrome: evidence foradvanced replication time as a major determinant. Hum. Mol. Genet., 9,2575–2587.
31. Ehrlich, M. (2000) DNA Alterations in Cancer: Genetic and EpigeneticChanges. Biotechniques Books, Eaton Publishing, Natick.
32. Xiong, Z. and Laird, P.W. (1997) COBRA: a sensitive and quantitativeDNA methylation assay. Nucleic Acids Res., 25, 2532–2534.
33. Hong, J.X., Wilson, G.L., Fox, C.H. and Kehrl, J.H. (1993) Isolation andcharacterization of a novel B cell activation gene. J. Immunol., 150, 3895–3904.
34. Gras, M.P., Laabi, Y., Linares-Cruz, G., Blondel, M.O., Rigaut, J.P.,Brouet, J.C., Leca, G., Haguenauer-Tsapis, R. and Tsapis, A. (1995)BCMAp: an integral membrane protein in the Golgi apparatus of humanmature B lymphocytes. Int. Immunol., 7, 1093–1106.
35. Kida, M., Souri, M., Yamamoto, M., Saito, H. and Ichinose, A. (1999)Transcriptional regulation of cell type-specific expression of theTATA-less A subunit gene for human coagulation factor XIII. J. Biol.Chem., 274, 6138–6147.
36. Wijmenga, C., Hansen, R.S., Gimelli, G., Bjorck, E.J., Davies, E.G.,Valentine, D., Belohradsky, B.H., van Dongen, J.J., Smeets, D.F.,van den Heuvel, L.P. et al. (2000) Genetic variation in ICF syndrome:evidence for genetic heterogeneity. Hum. Mutat., 16, 509–517.
37. Hansen, R.S., Wijmenga, C., Luo, P., Stanek, A.M., Canfield, T.K.,Weemaes, C.M. and Gartler, S.M. (1999) The DNMT3B DNAmethyltransferase gene is mutated in the ICF immunodeficiencysyndrome. Proc. Natl Acad. Sci. USA, 96, 14412–14417.
38. Fasth, A., Forestier, E., Holmberg, E., Holmgren, G., Nordenson, I.,Soderstrom, T. and Wahlstrom, J. (1990) Fragility of the centromeric regionof chromosome 1 associated with combined immunodeficiency in siblings:a recessively inherited entity? Acta Paediatr. Scand., 79, 605–612.
39. Carpenter, N.J., Fillpovich, A., Blaese, R.M., Carey, T.L. and Berkel, A.I.(1988) Variable immunodeficiency with abnormal condensation of theheterochromatin of chromosomes 1, 9 and 16. J. Pediatr., 112, 757–760.
40. Turleau, C., Cabanis, M.-O., Girault, D., Ledeist, F., Mettey, R., Puissant, H.,Marguerite, P. and de Grouchy, J. (1989) Multibranched chromosomes inthe ICF syndrome: immunodeficiency, centromeric instability and facialanomalies. Am. J. Med. Genet., 32, 420–424.
41. Franceschini, P., Martino, S., Ciocchini, M., Ciuti, E., Vardeu, M.P.,Guala, A., Signorile, F., Camerano, P., Franceschini, D. and Tovo, P.A.(1995) Variability of clinical and immunological phenotype inimmunodeficiency–centromeric instability–facial anomalies syndrome.Report of two new patients and review of the literature. Eur. J. Pediatr.,154, 840–846.
42. Karpf, A.R., Peterson, P.W., Rawlins, J.T., Dalley, B.K., Yang, Q.,Albertsen, H. and Jones, D.A. (1999) Inhibition of DNA methyltransferasestimulates the expression of signal transducer and activator of transcription 1,2 and 3 genes in colon tumor cells. Proc. Natl Acad. Sci. USA, 96,14007–14012.
43. Jackson-Grusby, L., Beard, C., Possemato, R., Tudor, M., Fambrough, D.,Csankovszki, G., Dausman, J., Lee, P., Wilson, C., Lander, E. andJaenisch, R. (2001) Loss of genomic methylation causes p53-dependentapoptosis and epigenetic deregulation. Nat. Genet., 27, 31–39.
44. Reif, K. and Cyster, J.G. (2000) RGS molecule expression in murine Blymphocytes and ability to down-regulate chemotaxis to lymphoidchemokines. J. Immunol., 164, 4720–4729.
45. Lim, S.P., Leung, E. and Krissansen, G.W. (1998) The β7 integrin gene(Itgb-7) promoter is responsive to TGF-β1: defining control regions.Immunogenetics, 48, 184–195.

Human Molecular Genetics, 2001, Vol. 10, No. 25 2931
46. Xu, S. and Lam, K.P. (2001) B-cell maturation protein, which binds thetumor necrosis factor family members BAFF and APRIL, is dispensablefor humoral immune responses. Mol. Cell. Biol., 21, 4067–4074.
47. Hatzoglou, A., Roussel, J., Bourgeade, M.F., Rogier, E., Madry, C.,Inoue, J., Devergne, O. and Tsapis, A. (2000) TNF receptor familymember BCMA (B cell maturation) associates with TNF receptor-associatedfactor (TRAF) 1, TRAF2 and TRAF3 and activates NF-κ B, elk-1, c-JunN-terminal kinase and p38 mitogen-activated protein kinase. J. Immunol.,165, 1322–1330.
48. Swanson, B.J., Jack, H.M. and Lyons, G.E. (1998) Characterization ofmyocyte enhancer factor 2 (MEF2) expression in B and T cells: MEF2Cis a B cell-restricted transcription factor in lymphocytes. Mol. Immunol.,35, 445–458.
49. Wallin, J.J., Rinkenberger, J.L., Rao, S., Gackstetter, E.R., Koshland, M.E.and Zwollo, P. (1999) B cell-specific activator protein prevents twoactivator factors from binding to the immunoglobulin J chain promoteruntil the antigen-driven stages of B cell development. J. Biol. Chem., 274,15959–15965.
50. Cheng, P.C., Steele, C.R., Gu, L., Song, W. and Pierce, S.K. (1999) MHCclass II antigen processing in B cells: accelerated intracellular targeting ofantigens. J. Immunol., 162, 7171–7180.
51. Pan, L., Sato, S., Frederick, J.P., Sun, X.H. and Zhuang, Y. (1999)Impaired immune responses and B-cell proliferation in mice lacking theId3 gene. Mol. Cell. Biol., 19, 5969–5980.
52. Wang, J., Ducret, A., Tu, Y., Kozasa, T., Aebersold, R. and Ross, E.M.(1998) RGSZ1, a Gz-selective RGS protein in brain. Structure, membraneassociation, regulation by G αz phosphorylation and relationship to a Gzgtpase-activating protein subfamily. J. Biol. Chem., 273, 26014–26025.
53. Bain, G., Cravatt, C.B., Loomans, C., Alberola-Ila, J., Hedrick, S.M. andMurre, C. (2001) Regulation of the helix–loop–helix proteins, E2A andId3, by the Ras-ERK MAPK cascade. Nat. Immunol., 2, 165–171.
54. Puig-Kroger, A., Sanz-Rodriguez, F., Longo, N., Sanchez-Mateos, P.,Botella, L., Teixido, J., Bernabeu, C. and Corbi, A.L. (2000)Maturation-dependent expression and function of the CD49d integrin onmonocyte-derived human dendritic cells. J. Immunol., 165, 4338–4345.
55. Jacquot, S., Kobata, T., Iwata, S., Morimoto, C. and Schlossman, S.F.(1997) CD154/CD40 and CD70/CD27 interactions have different andsequential functions in T cell-dependent B cell responses: enhancement ofplasma cell differentiation by CD27 signaling. J. Immunol., 159, 2652–2657.
56. Gasser, S.M. (2001) Positions of potential: nuclear organization and geneexpression. Cell, 104, 639–642
57. Sabbattini, P., Lundgren, M., Georgiou, A., Chow, C., Warnes, G. andDillon, N. (2001) Binding of Ikaros to the λ5 promoter silences transcriptionthrough a mechanism that does not require heterochromatin formation.EMBO J., 20, 2812–2822.
58. Brown, K.E., Baxter, J., Graf, D., Merkenschlager, M. and Fisher, A.G.(1999) Dynamic repositioning of genes in the nucleus of lymphocytespreparing for cell division. Mol. Cell, 3, 207–217.
59. Trinh, L.A., Ferrini, R., Cobb, B.S., Weinmann, A.S., Hahm, K., Ernst, P.,Garraway, I.P., Merkenschlager, M. and Smale, S.T. (2001) Down-regulationof TDT transcription in CD4+CD8+ thymocytes by Ikaros proteins indirect competition with an Ets activator. Genes Dev., 15, 1817–1832.
60. Schubeler, D., Francastel, C., Cimbora, D.M., Reik, A., Martin, D.I. andGroudine, M. (2000) Nuclear localization and histone acetylation: apathway for chromatin opening and transcriptional activation of thehuman β-globin locus. Genes Dev., 14, 940–950.
61. Kieback, P., Wendisch, H., Lorenz, P. and Hinkel, K. (1992) ICFsyndrome. Immunodeficiency, chromosomal centromere instability, facialanomalies. Case report and literature review. Monatsschr. Kinderheilkd.,140, 91–94.
62. Le Naour, F., Hohenkirk, L., Grolleau, A., Misek, D.E., Lescure, P.,Geiger, J.D., Hanash, S. and Beretta, L. (2001) Profiling changes in geneexpression during differentiation and maturation of monocyte-deriveddendritic cells using both oligonucleotide microarrays and proteomics.J. Biol. Chem., 276, 17920–17931.
63. Sun, B., Jiang, G., Zaydin, M., LaRussa, V., Safah, H. and Ehrlich, M.(2001) ABL promoter methylation can exist independently of BCR-ABLtranscription in chronic myeloid leukemia hematopoietic progenitors.Cancer Res., 61, 6931–6937.

2932 Human Molecular Genetics, 2001, Vol. 10, No. 25




















