
Discovery of Inhibitors of Protein-Protein Interactions from Combinatorial Libraries
34
1 Discovery of Inhibitors of Protein-Protein Interactions from Combinatorial Libraries María J. Vicent 1 , Enrique Pérez-Payá 1,2 and Mar Orzáez 1,* 1 Dept. Química Médica. Centro de Investigación Príncipe Felipe. E-46013 Valencia, Spain. 2 Instituto de Biomedicina de Valencia (CSIC). E-46010 Valencia, Spain. • Corresponding author: Dr. Mar Orzáez. Centro de Investigación Príncipe Felipe, Avda. Autopista del Saler, 16, E-46013 Valencia, Spain. Phone: +34-963289680; Fax: +34-963289701; E-mail: [email protected]
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Discovery of Inhibitors of Protein-Protein Interactions from Combinatorial Libraries

1
Discovery of Inhibitors of Protein-Protein Interactions from
Combinatorial Libraries
María J. Vicent1, Enrique Pérez-Payá1,2 and Mar Orzáez1,*
1 Dept. Química Médica. Centro de Investigación Príncipe Felipe. E-46013 Valencia,
Spain.
2 Instituto de Biomedicina de Valencia (CSIC). E-46010 Valencia, Spain.
• Corresponding author: Dr. Mar Orzáez. Centro de Investigación Príncipe Felipe,
Avda. Autopista del Saler, 16, E-46013 Valencia, Spain. Phone: +34-963289680;
Fax: +34-963289701; E-mail: [email protected]

2
Abstract: Protein-protein interactions play a central role within numerous processes in the cell.
The relevance of the processes in which this type of interactions are implicated make
them responsible of many pathological situations. In the last decade protein-protein
interfaces have shown their potential as new drug targets, and combinatorial chemistry
has been defined as an useful tool in this line. This review gives a global vision of the
actual situation of combinatorial chemistry, highlighting its applicability to high-
throughput drug discovery and giving some crucial examples of its contribution to find
modulators of protein-protein interactions.
Abbreviations:
Acquired immunodeficiency syndrome (AIDS), conformationally restricted libraries
(CRL), hightroughput methods of screening (HTS), human immunodeficiency virus
type 1 (HIV-1), nuclear magnetic resonance (NMR), rabies virus Nucleoprotein (N),
rabies virus Phosphoprotein (P), structure activity relationship (SAR).

3
PROTEIN-PROTEIN INTERACTIONS AS DRUG TARGETS.
The most generalised approach used to develop new therapeutic agents has been
mostly based on the choice of a single gene product of clinical relevance as target.
However, as it was early postulated and more recently experimentally confirmed, a
large number of proteins inside the cell rarely stays alone. Molecular signalling and
signal transduction pathways at cellular level require massive protein usage in order to
transmit signals through protein-protein interactions producing cell-answerings in
response to different environmental changes or modifications. Not only proteins in
cellular pathways are busy seeking for interactions but structural proteins are also
forming part of macromolecular structures and enzymes have regulatory and
catalytically active subunits. In this crowded scenario, in which protein-protein
interactions are the main actors, it is reasonable to speculate that the modulation of
protein-protein interactions could open new alternatives to the development of
molecules that could modulate cellular pathways that in turn could have relevant future
therapeutic applications. Special attention is devoted to the drawing of the protein
interaction maps. In fact, there are now available cellular-protein interaction maps of
model organisms[1], human[2,3], and some cellular pathways and, in addition, many
genomes have been completely sequenced (see http://www.ensembl.org). However,
these maps represent only a very partial picture of the complete set of real interactions.
Because detecting and analysing all protein-protein interactions by classical
experimental methods is not as fast as genome sequencing, there is an unbalance
between our knowledge on the basic pieces of the puzzle (the proteins) and the higher
order relationships between them. Thus, it would be useful for the identification of new
potential targets the use of predictive bioinformatics methods based on different types
of evidences such as sequence features [4] or different indirect experimental evidences

4
such as co-expression [5], domain-domain co-occurrence [6] and others methods.
These approaches are becoming more and more accurate and now it is reasonable to
take them as a starting point in absence of previous experimental evidence. In addition,
systematic whole-cell mass spectrometry approaches are currently used to identify
protein complexes [7,8], which includes strong, transient and weak interactions between
proteins that will increase our knowledge of interacting networks [9,10]. Nevertheless,
data derived from all these methodologies clearly point to the need of taking into
consideration protein-protein interactions as targets for the development of new drugs.
In fact a study of the pairwise interactions among 8100 human open reading frames
(that represents only about 10% of the total protein-coding genes), showed that at least
in 424 of them one of the partners is implicated in human diseases [3] .
Figure 1
The development of modulators for protein-protein interactions is not exempt of
difficulties. Among others, one of the first identified problems is related to the nature of
the surfaces that define these interactions. While the ‘classical’ protein sites targeted by
drug discovery programs are ligand binding sites, defined by small-concave
cartographic spots, the protein-protein interaction surfaces are flat and large. An
extensive network of weak molecular interactions maintains the structure of the
complex, then at a first glance, they were classified as extremely difficult target for
small-molecules, that in turn were the favourite choice for drugs. Years of investigation
are now providing information on the detailed nature of these interactions that for sure
will provide the hints to afford disruption of this type of complexes. For example, now
we know that the packing between molecules is not as tight as the core packing of

5
proteins, probably due to the less hydrophobic interaction surfaces and the presence of
interfacial water molecules [11]. This flexibility allows multiple forms of protein-
protein complexes that can be initially classified as permanent, with no individual
components found in the cell during protein lifetime and, transient complexes that are
formed and disrupted in response to different cell situations. The total extension of the
interactive surface, considered a key parameter of the discovery strategy, would then
probably vary when facing a small (close to 1000 Å2) or a middle to large (> 4000 Å2)
interacting interface [11]. In addition, it is well established now that when comparing
the folding of an individual protein versus the structural arrangements needed for a
protein-protein interaction there are differences on the role of the structural elements. A
correct ‘hydrophobic-driven’ single protein packing tends to collect hydrophobic
elements of secondary structure inside the protein core regardless of their secondary
structure. However, it has been observed that in protein-protein interaction surfaces
there is a ranking of preferences on packing, from high to low, between β-sheet/β-sheet,
loop/loop, α-helix/α-helix while α-helix/β-sheet interactions are less favoured [11].
Furthermore, statistical data also suggests that some defined amino acids have tendency
to be more present than others at the protein-protein interfaces. Methionine, tryptophan,
cysteine, phenylalanine, tyrosine, arginine and histidine are overrepresented at transient
molecular interfaces opposed to hydrophobic residues that are in general
underrepresented in this type of surfaces [12]. Some amino acid pairs are frequently
found in association like tryptophan and proline or phenylalanine and isoleucine. It
should be also noted that exposed hydrophobic interfaces tend to be small and although
hydrophobic residues contribute to the binding affinity, these types of interactions are
less specific than those composed of polar or charged residues.

6
Taking all this information together it seems that there is an infinite diversity in the
universe of protein-protein interactions difficult to cope with from the point of view of
the design of new therapeutics. Nevertheless proteins are often constructed of modular
domains that are used repeatedly in distinct molecules to mediate their interactions [13].
For example, proline rich motifs are recognized by conserved interaction modules such
as SH3, WW and EVH1 domains or phosphotyrosine motifs are recognized by SH2 or
PTB domains. These common features facilitate the development of general strategies
for the inhibition of different molecular targets, which share an interaction motif
although their cellular function could be completely different.
In any case the remaining question is whether or not relatively small molecules, a
desirable characteristic to consider a molecule as a potential drug candidate, could or
not be capable of disrupting protein-protein interaction surfaces. Recent progress in the
field has provided a large number of examples [14,15] in which this strategy of
modulation has prospered opening a new field for drug discovery. Relevant and
difficult to target diseases such as acquired immunodeficiency syndrome (AIDS),
Alzheimer or cancer are being favoured with recent discoveries from methodologies
using this strategy.
A second key question is related to the methodological strategy that could be used to
ensure the identification of active compounds able to inhibit these protein-protein
interactions. Is it necessary to use traditional searching or it is maybe better to use a
combinatorial approach?. The limiting factor for rational design is the requirement of
an exhaustive knowledge of the targets in order to identify ‘hot spots’ along the
interaction surface of proteins where small drugs could disrupt the complexes.
Unfortunately, our current knowledge about protein-protein surfaces, even with enough
structural data available, is extremely limited to permit the use of this rational strategy

7
as a general approach to modulate interactions. Moreover in this new technological era
where the information about protein interactions emerge as global interactome maps [3]
in which thousands of proteins are inter-connected, it seems necessary to find a global
strategy to perform the search of modulators for all these interactions. It is in these
situations where combinatorial approaches could be extremely beneficial and profitable,
offering a great diversity of compounds to test protein-protein disruption activity and
therefore overcoming the problem of lack of information. From the technological point
of view the use of combinatorial chemistry requires the development of hightroughput
methods of screening (HTS) for protein-protein interactions in order to increase the
probability of finding a lead compound. There are many techniques available to
identify protein partners such as yeast two hybrid system, mass spectrometry, protein
chips (reviewed in [16]), POSSYCCAT for the study of interactions between
transmembrane fragments [17], etc. Once an active product has been identified, deeper
studies could permit the location of the interaction site with its target and give the
necessary information to improve the drug-protein interaction surface. This site could
probably be considered as a ‘hot spot’ on the interaction surface between the proteins.
Furthermore, the discovery of a new inhibitor could facilitate the identification of the
cellular processes in which this interaction is implicated. In the cases where enough
information about the target is available to initiate an adequate drug rational design,
combinatorial chemistry could also contribute offering the possibility of improving its
inhibition capabilities through structural modifications of the defined lead compound
[18,19].
This review will try to illustrate in a first stage the principles and types of
combinatorial chemistry and how with this combinatorial approach it is possible to
identify inhibitors for key protein-protein interactions.

8
COMBINATORIAL CHEMISTRY: A VALID APPROACH FOR TARGET
VALIDATION AND DRUG DISCOVERY
Combinatorial vs. traditional chemistry. Principles of combinatorial chemistry
Traditionally, natural product extracts or industrial collections of randomly
synthesised organic molecules (synthesised one at a time) were screened to discover
“hits”, which were then optimised in an iterative process by the synthesis of
derivatives. Many important drugs were identified following this approach however
the ratio of novel to previously discovered compounds started to diminish with time.
This fact together with the cost constraints found on pharmaceutical research forced
the investigation of methods in the pharmaceutical industry that could offer higher
productivity at lower expenses. By that time, genomics and proteomics had
experienced an exponential growth and more potent and efficient technological devices
for biological screening had also been achieved. Altogether, triggered the appearance
of a new discipline in the early 1980s: Combinatorial Chemistry [20].
Combinatorial chemistry could be defined as “the generation of large collections,
or ‘libraries’, of compounds by synthesising all possible combinations of a set of
smaller chemical structures, or ‘building blocks’, in a time and submitted for
pharmacological assay” [19-28]. In this way, the chemist can synthesise up to
thousands of compounds at once instead of preparing only a few by simple
methodology. The traditional approach of organic synthesis to sequentially synthesise
a logically designed set of analogues based on a lead compound has been surpassed by
the ability to screen whole compound libraries accumulated over the years by large
pharmaceutical companies. Therefore, with the combinatorial strategy the productivity
has been amplified beyond the levels obtained as routine in the last century [29,30].

9
Solid-phase and solution-phase combinatorial chemistry
Solid phase peptide synthesis methodology, introduced by Merrifield in 1963,
paved the way for solid-phase combinatorial approaches (extensively reviewed in
[27,28,31-35]). Geysen [36,37] was the pioneer in this field synthesising peptides on
pin-shaped solid supports followed by Houghten with his “teabag” approach to epitope
mapping [38]. Solid-phase combinatorial chemistry has been widely implemented for
hit discovery as well as lead optimisation due to its advantages and simplicity of use,
advantages such as: (i) the compounds can be isolated once they are attached to solid
supports, (ii) the isolation of the immobilised product by simple filtration permits the
use of large reagent excesses to get high-yield conversions for each of the steps, (iii)
pseudo-dilution effects are present and (iv) the use of split-and-mix synthesis
simplifies the preparation of large libraries [15].
The split-and-mix solid–phase synthesis on beads was introduced by Furka et
al.[39,40]. This technique has been enthusiastically exploited by many others since its
first disclosure. For example, Houghten has used split-and-mix on a macro scale in a
"teabag" approach for the generation of large libraries of peptides [41]. Lam et al. [42-
44] was able to enhance the production and rapid evaluation of random libraries of
millions of peptides in a way that acceptor-binding ligands of high affinity could be
rapidly identified and sequenced, on the basis of a 'one-bead, one-peptide' approach.
Consequently to the large acceptance achieved by the split-and-mix strategy, several
identification techniques have been also developed, such as nucleotide- [45,46],
peptide- [47,48], chemical- [49], radiofrecuency- [50], color- [51], and shape- [52-54]
encoded or iterative [55-57] and recursive [58,59] deconvolution.
Unfortunately, there is not an ideal general technique, therefore, among the
limitations of solid-phase combinatorial synthesis should be mentioned the restriction

10
of being only suitable in linear synthetic strategies and the requirements for linkage or
cleavage steps that may restrict the possible chemical reactions. A hydroxyl, amine,
carboxyl, or other polar group must be present on a molecule to be able to attach it to a
support. This is a potentially undesirable constraint on the structure of compounds
synthesised on solid phase, as products retain the polar group even after they are
cleaved from the support. Several groups have been devoted to diminish these
drawbacks in order to continue improving the implementation of solid-phase synthesis.
The use of ‘Traceless linkers’ [12,60-62] have been one of the most remarkable
implementations. For instance, Mori and coworkers [60,63] have developed an aniline
linker for solid-phase synthesis of azomethines or Ellman et al. [64] have reported an
acylsulfonamide linker able to add diversity to a library when displaced by various
nucleophiles at the same time that is completely removed from products during the
cleavage process. Furthermore, other innovative techniques for solid-phase synthesis
have been developed, including: react-and-release type chemistry [65], in which
reagents used to cleave products from the solid support are incorporated into the
product; use of scavenger resins [66]; the introduction of safety-catch linkers [67-69]
or the improvement of resin properties and loading capacities [70-72].
Although the solid-phase combinatorial chemistry is still the most widely used,
several groups have been applying an alternative solution-phase approach to
complement solid-phase techniques. Solution-phase synthesis offers the possibility of
expanding the repertoire of chemical reactions and allows the application of
convergent synthetic strategies, the synthesis of mixture libraries or the use of dynamic
libraries [73,74].
Dynamic combinatorial chemistry [29,73,74] is based on continuous
interconversion between the library constituents, it uses reversible connections

11
between the initial building blocks producing flexible and adaptive libraries. This
approach is driven by the interactions of the library constituents with the target site.
This strategy allows a target-driven generation or amplification of active constituents
by performing a self-screening process where the active species are preferentially
expressed and retrieved from the library [73]. A similar concept is found in Phage
displayed libraries (biologically displayed libraries) [75-77] first published on 1985 by
Smith et al.[78]. These offer a strategy to isolate peptide ligands to target proteins and
to define interaction sites between proteins [79]. In phage displayed libraries, instead
of using inter-convertible building blocks as it is done in dynamic libraries, possible
active peptides or oligonucleotides (phage population) are screened by incubation with
the target molecule adsorbed to a solid support. Active phages bind the target, then
target-bound phages are isolated and propagated by infection of E. coli and subjected
to an additional round of adsorption to the immobilised target. The use of biological
displayed libraries for the isolation of peptide ligands is an interesting alternative to
chemical libraries.
Both, solid-phase and solution-phase strategies can be easily automated, however,
the major limitation to solution-phase is the isolation and purification of the library
compounds [80,81]. To date, most of the isolation/purification steps are based on acid-
base chemistry and sequestration-enabling reagents [82-84]. The pioneering acid-base
extraction approach of Boger et al. [85], clearly showed the possibility of generate
large libraries of pure molecules. His work has been mainly focused on the main
concept raised in this review, the discovery of small compounds (mainly
peptidomimetics) inhibitors of protein-protein interactions [15]. Mixture synthesis
provides for solution-phase synthesis what split-and-mix synthesis provides for solid-
phase approaches (reviewed in [86,87]).

12
Analogous to encoding approaches for split-and-mix synthesis of large libraries
(also known as iterative approach), there are two key and complementary
deconvolution strategies that permit the immediate identification of active lead
compounds from large mixture libraries: positional scanning[88,89] and deletion
synthesis [90,91] techniques. The positional scanning approach was first described by
Houghten et al. [88,89] and was developed as an alternative to the iterative strategy
where the lead identification was achieved through successive steps of mixture
selection with the active compound, synthesis of sublibrary and subsequent evaluation,
that is, only one residue can be identified at a time by a synthesis-assay cycle. When
using a positional scanning approach [92,93] several sublibraries can be synthesised at
a time, in each sublibrary an individual component is placed in one position but
mixtures are used in all others. The most active sublibrary in each set indicates the
optimal substituent for that position so, an increase in activity is measured that allows
the identification of active lead structures. On the other hand, in deletion synthesis
deconvolution libraries [90], an individual component is missing in each sublibrary but
full mixtures are used in all other positions. The least active sublibrary in each set
indicates the optimal substituent for that position as the missing component that in
turn, will be the most active. This strategy provides less global information that
positional-scanning but is better at identifying a uniquely potent library member. It is
important to note that, with either of both techniques, the lead identification comes out
from a single round of screening.
SUITABILITY OF COMBINATORIAL APPROACH TO TARGET PROTEIN-
PROTEIN INTERACTIONS

13
Despite the already described inherent difficulties in the process to identify small
compounds modulators of protein-protein interactions, there are several important
modulators of this type of complexes that have been defined using combinatorial
approaches. Some of the more representative examples are described in more detail
below (Table 1).
Apoptosis inhibitors
Programmed cell death or apoptosis is a highly regulated process of cell deletion
that plays crucial roles in development and maintenance of tissue homeostasis in
multicellular organisms. Due to the relevance of this cellular mechanism its
deregulation is a key issue in the pathogenesis of several human diseases such as cancer
or neurodegenerative disorders [94]. This type of cellular death can be triggered by a
variety of extrinsic and intrinsic signals [95].
Inhibitors of apoptosome formation.
In the apoptosis pathway in mammals the formation of a multiprotein complex
called apoptosome between cytochrome c, Apaf-1 (apoptotic protease-activating factor)
and procaspase-9 links mitochondria dysfunction with the activation of the effector
caspases starting the cascade of cell death [8,96-98]. Our laboratory focused on the
identification of compounds that inhibit the apoptosome-mediated activation of
procaspase-9 from the screening of a positional scanning diversity-oriented
combinatorial library of trimers of N-alkylglycines [99]. These type of molecules also
known as peptoids present a series of characteristics that make them good candidates to
cope with the disruption of protein-protein interactions. Its main chain is quite similar
to a polypeptide but with the lateral side chain on the nitrogen atom instead of the

14
carbon thus giving the advantage of the similarity at the same time that conferring
flexibility to the scaffold which could favour the interaction with the target surface
[100]. Moreover, due to this chemical difference with proteins, cellular proteases are
not able to degrade this type of substrates increasing the lifetime of the drug inside the
organism. The library consisted in 52 controlled mixtures and a total of 5120
compounds. Mixtures 1 to 20 (O1XX) contain as defined position one of 20 selected
commercially available primary amines, while at the ‘X’ positions (mixture positions) a
set of 16 primary amines was present. Mixtures 21 to 36 (XO2X) and 37 to 52 (XXO3)
contain at the defined position only a set of 16 amines. The mixtures making up each
sublibrary were screened for their ability to prevent the apoptosome-dependent
activation of procaspase-9. For this goal the apoptosome was assembled in vitro by
incubating rApaf-1, cytochrome c, dATP and [35S]-Met procaspase-9.
After the identification of a lead compound (peptoid 1) rescued from the library a
rational strategy was used to improve its solubility as well as its cellular internalisation
capability, obtaining finally a drug with antiapoptotic activity in different cell lines, see
Fig (2).
Figure 2
Inhibitors of interaction between pro- and anti-apoptotic proteins.
The response of mitochondria to apoptotic signals is a key point of regulation of
apoptosis that is controlled by the Bcl-2 family of proteins. In particular by the balance
between pro- and anti-apoptotic proteins in the mitochondrial membrane. Anti-
apoptotic Bcl-2 proteins (for example, Bcl-2 and Bcl-XL) are the target of pro-apoptotic
proteins (like Bad, Bax, Bak) that through their BH3 domain bind to a hydrophobic cleft
on the surface of Bcl-2 or Bcl-XL. Several lines of evidence indicate that BH3-mediated

15
binding has a key role in regulating apoptotic functions. In fact, short peptides derived
from BH3 domains of various pro-apoptotic Bcl-2 family members are sufficient to
induce apoptosis in cells [101]. To identify small-molecule inhibitors of Bcl-XL-BH3
domain interaction, Degterev et al. [102] screened a commercially available library of
16320 chemicals and found hit compounds that after chemical optimisation rendered
biologically active compounds that inhibited the interaction between Bcl-XL and BH3
domains and induced apoptosis in Jurkat cells. The same protein-protein complex was
selected as target by Oltersdorf et al. [103]. However in this study they applied the
methodology of SAR by NMR. In this methodology a library of small chemical
fragments is screened against the protein target to identify low-affinity binding
compounds. Then the chemical linkage of proximal fragments would render a new
molecule with high binding affinity to the target. Using this technique a small
molecule, named ABT-737, with high affinity (Ki< 1 nM) to Bcl-XL, was identified, see
Fig (3). ABT-737 displayed synergism with chemotherapeutics and radiation.
Furthermore, the compound showed in vivo anti-tumor activity causing complete
regression of established tumor xenografts.
Figure 3
Viral inhibitors
Combinatorial libraries have also contributed to the inhibition of the life cycle of
certain viruses such as the human immunodeficiency virus or the rabies virus.
HIV protease dimerisation inhibitors
Human immunodeficiency virus type 1 (HIV-1) is a retrovirus with a very complex
life cycle in which at least 15 protein are implicated [104]. Several drugs targeting

16
different steps of this cycle have been developed. One of the main pharmacological
targets is the virus protease (PR), a pivotal enzyme in viral maturation responsible of
processing the polyproteins encoded by the virus rendering active proteins needed for
the assembly and the infection steps of the virus particle [105]. PR is an homodimeric
aspartil protease with the active site located at the interface of the dimmer [106]. This
dimerisation interface is a highly conserved region which makes it a relevant target for
drug discovery. The main dimerisation region is folded as an interdigitating N-and C-
terminal four stranded β-sheet. The group of Chmielewski using rational design
developed inhibitors of the PR dimerisation derived from crosslinked interfacial
peptides corresponding to this conserved β-sheet region and identified the minimal
structure necessary for the activity of these peptides [107]. Moreover in an attempt to
improve its activity a focused library on the basis of this minimized molecule was
designed (compound 6) in which certain side chains were substituted by different
natural and non natural amino acid side chains, see Fig (4). The analysis of the library
not only gave information about the groups that confer better characteristics in each
position but also rendered a molecule (compound 36, Fig (3)) with improved activity
respect to the initial scaffold.
Figure 4
Rabies virus infection inhibition
Infection by rabies virus is the cause of thousands of deaths by encephalomyelitis
per year in the world. The clear need to have a post-exposure treatment led the group of
Yves Jacob to work on the development of a new virucidal drug [108]. Rabies virus is a
member of the lyssaviruses family. Among other viral constituents the Phosphoprotein
(P) is crucial for the formation of the transcription-replication complex, in which

17
interacts with the RNA-polymerase and the nucleoprotein (N). Moreover P also
interacts in the cell with Dynein LC8 a protein implicated in retrograde transport. These
characteristics made P a good target to develop a new virucidal drug, although there was
no structural information about the complexes in which it participates. Ybes Jacob and
coworkers designed two genetically encoded combinatorial peptide libraries, called
coactamer libraries. In these libraries the peptide scaffold is structurally constrained
due to the presence of either prolines or cysteines. This structure mimics conotoxins
and insect antimicrobial peptides, which easily reach the cellular interior, in an attempt
to increase drug bioavailability. A two hybrid assay was used to identify peptides
interacting with phosphoproteins from two different lyssaviruses. Peptides selected
from the library were tested for its viral transcription-replication complex interference
activity. To be sure that their activity were related with an alteration of the N-P
interaction a ProteinChip with an anti-Flag M2 monoclonal antibody cross-linked was
used to immunoprecipitate P and its associated proteins from cell extracts in the
presence or absence of the co-transfected inhibitor peptides. Peaks corresponding to
the interaction between P and N were analysed by SELDI-TOF, and four peptides
modulators of the ratio P/N (indicator of an altered interaction) were identified.
Table 1
Inhibitors of cell migration
Integrin αvβ3 / MMP-2
Integrin αvβ3 is an heterodimeric cell surface receptor that promotes cell attachment
to the extracellular matrix. MMP-2 (gelatinase A) a protein secreted by vascular
endothelial cells is crucial for the degradation of collagen matrix which permits new
vessels to proliferate. Moreover, the interaction between integrin αvβ3 and MMP-2

18
seems to be responsible for the activation of MMP-2 in invasive endothelial cells [109].
Disruption of this protein-protein interaction is one of therapeutic strategies that are
being exploited for tumor growth inhibition. Screening of a library composed of ten
mixtures of 60 small organic compounds designed to mimic potential protein–protein
interactive moieties have led to the identification of a lead compound with in vitro
activity. Posterior refinement experiments permitted to obtain a more water-soluble
molecule with antitumor and antiangiogenic in vivo activity [110]. One of the active
compounds, termed TSRI265, was shown to prevent collagen IV degradation in hamster
CS-1 melanoma cells transfected with the human 3-integrin, see Fig (3). Furthermore,
TSRI265 almost completely inhibited FGF-stimulated angiogenesis in 10-day-old
chick chorioallantoic membrane. The data obtained from this study set the basis to
consider inhibition of MMP-2/ v 3 binding as a crucial molecular target to block the
angiogenic process [111].
Integrin α4β1 / Paxillin
The interaction between the signal adaptor protein Paxillin and the cytoplasmic tail
of integrin α4β1, a cell surface receptor, has been implicated in several pathogenic
situations including enhanced rates of cell migration, reduced rates of cell spreading,
focal adhesion, and stress fibber formation [112]. All these processes contribute to
leukocyte migration into tissues and to the expression of genes involved in chronic
inflammation. From the screening of a combinatorial library in an ELISA assay using
immobilised His-tagged α4 and examining the binding of Paxillin, Boger and coworkers
obtained a lead molecule that inhibits this interaction [92]. The library consisted of a
scaffold composed of three different variable subunits (X,Y and Z) linked by amide
bonds, and a basic side chain 4-(dimethylamino) butyric acid linked to the X subunit,

19
see Fig (5). For each variable position ten aromatic amino acids were used. The library
was prepared in solution using two different formats, as 100 mixtures of 10 compounds
and using a positional scanning approach in which 30 sublibraries were generated.
Results using the two types of library formats were quite similar. The lead molecule
(11 X7-Y7-Z7 ), see Fig (3), obtained from the screening not only disrupted Paxillin/
Integrin α4β interaction but also demonstrated a potent inhibition activity of human
Jurkat T cell migration.
Figure 5
Cell cycle inhibitors
Cell growth in eukaryotic cells is under control of a series of concerted molecular
mechanisms defined as the cell cycle. In cell life cycle there are strict checkpoints
controlling cells prepared to enter mitosis. The p16-cyclinD-pRB-E2F pathway
controls the G1/S transition of the mammalian cell cycle. E2Fs are a family of
transcription factors (E2F1 to E2F7 in mammalian cells) that require heterodimerization
with proteins of the DP family to bind DNA and exert its regulatory function over genes
implicated in DNA replication [113,114]. Activation of E2F dependent transcription
promotes progression from G1 to S and conversely its inhibition arrests cells in G1
[115]. Sardet and coworkers [116] identified, from the screening of a combinatorial
library of thiorredoxin-20mer peptide (called aptamers), a molecule that interacts with
E2F1 dimerization domain. For this purpose, a two hybrid assay with E2F1
dimerization domain as a bait, was used. Among the molecules identified there was one
peptide (Apt5) that shared notable sequence similarity with a region of DP1. More
extensive studies demonstrated that this molecule interferes with the interaction between
E2F/DP. Moreover, deeper studies in mammalian cells demonstrated that Apt5 is

20
capable of blocking fibroblasts in G1 reinforcing the hypothesis that this interaction
could be considered as a key target for the development of pharmaceutical
antiproliferative agents.
Inhibitors of G-proteins
Heterotrimeric G-proteins (Gαβγ) transform the signal produced by G-protein
coupled receptors in an intracellular signal. In humans the wide diversity of receptors
together with the great number of isoforms of G-proteins generate a pool of complexes
implicated in a wide variety of cellular processes ranging from neurotransmission or
embryonic development [117] to respiratory control [118]. Drug discovery efforts that
initially were directed to G-coupled receptors now have been focused in G-proteins and
its intracellular partners.
In an attempt of interfere with these interactions the group of Scott JK used different
phage displayed peptide libraries (Table 1) that were screened against immobilized βγ
subunits as target [119]. Selected peptides were grouped into different families and one
of the groups identified was shown to share notable homology with peptides derived
from Phospholipase C β2 (PLC β2). These peptides in fact prevented activation of PLC
β2 by Gβγ subunits but did no block Gβγ-mediated of voltage-gated calcium channels
which remarks the pathway specificity of the molecules. Moreover all peptides selected
were predicted to bind to the same site of the Gβγ-subunits. In this sense the screening
of the library not only rendered active molecules but also gave information about a ‘hot
spot’ site in Gβγ for binding interaction [120]. This information could be of great
interest for the posterior refinement of the peptide/protein interactions as well as for the
development of new pharmaceuticals using a traditional strategy.

21
Conformationally restricted libraries (CRL).
The main characteristic of these libraries is that the random sequences (the
diversity) are grafted onto a rigid natural protein domain or into stable secondary
structural motifs usually named as the scaffold of the CRL [121,122]. The sequence of
the scaffold is kept in its major part and only a few positions are combinatorialised.
Hence, the library presents dual diversity, namely of sequences and structures. An
adequate selection of the combinatorialised positions in the scaffold allows the
generation of molecules that virtually populate the conformational space between the
random and the fully folded conformation which, in turn, depends on the selected
scaffold. The aims of the design of a monomeric α-helix CRL was based on its
potential use as source for the identification of molecules that would modulate protein-
protein interactions. In this context, the α-helical based CRL was screened in two
different biological assays. In particular, we were interested in the modulation of
protein-protein interactions that mediate membrane fusion. The mechanism of
virus-cell fusion of some enveloped viruses and in particular for rhabdoviruses
revealed that, to fuse with the cellular membranes, the G protein trimeric spikes find
and bind to their target cells. However, the molecular mechanisms involved in
rhabdovirus fusion are not well understood and it might involve new proteins or
principles yet to be discovered. The screening of the library allowed the identification
of peptides that enhanced the infectivity of rhabdoviruses [8,123]. These peptides are
currently being used in studies addressed to understand the molecular mechanism that
control the fusion of the rhabdovirus to the target cell. In a different biological assay
we addressed the identification of novel modulators of the SNARE complex as
inhibitors of regulated exocytosis. Calcium-dependent exocytosis in excitable cells is
mediated by the precise docking and fusion of neurotransmitter-loaded cargo vesicles

22
[124,125]. Mechanistically, neuronal exocytosis is an orchestrated cascade of protein-
protein interactions that involve several proteins. At the centre of the process are
found the so-called SNARE proteins that assemble into a highly stable, ternary
complex known as the SNARE core complex. [124-127]. Structurally, the SNARE
complex is a highly stable parallel four helix bundle formed by coiled coil
arrangements. The high stability of the SNARE complex has hampered the discovery
of small molecules that modulate the assembly of the proteins and only clostridial
neurotoxins and peptides patterned after protein domains of SNARE proteins have
been reported to have an effect on the assembly [128-130]. However, the discovery of
amino acid sequences unrelated to the SNARE proteins capable to inhibit the assembly
of the core complex remained elusive. From the screening of the α-helix CRL we
identified up to 8 peptides that inhibited in vitro the formation of the SDS resistant
SNARE complex. The most active 17-mer peptide abrogated the Ca2+-dependent
release of L-glutamate in intact hippocampal neurons [130].
Conclusion
The network of protein interactions that defines the full interactome of proteins of
cellular regulatory mechanisms is far broader than the subject matter reviewed here.
Consequently, the identification of molecules that inhibit protein-protein interactions
reviewed here should be viewed as a starting point upon which to build the concept
that such protein-protein interactions are pharmacologically accessible by small
molecules. The actual and future molecules that will modulate interactions between
proteins, not only would have the chance to be promote to ‘more promising hits to
drugs’ but also will define valuable tools for the study of cell biology. The use of
collections of molecules or compound libraries together with the availability of

23
structural and bioinformatics information for key protein-protein complexes have
significantly open new views to the understanding of how modulators of protein-
protein interaction could be developed. In turn, these findings open important
questions and perspectives that will be the driving force for future research. Questions
ahead will look upon the ‘strength’ of the interactions between proteins and how many
transient interactions (difficult to identify in the cellular context) that are still
undiscovered, will be classified as key points of regulation in cellular pathways.
However, combinatorial chemistry in its different formats will be always a valuable
and useful tool to face research programs for the identification of modulators of
protein-protein interactions, specially when structural information of the complex is
not available but a biological activity could be evaluated.
References [1] Giot, L.; Bader, J. S.; Brouwer, C.; Chaudhuri, A.; Kuang, B.; Li, Y.; Hao, Y.
L.; Ooi, C. E.; Godwin, B.; Vitols, E.; Vijayadamodar, G.; Pochart, P.; Machineni, H.; Welsh, M.; Kong, Y.; Zerhusen, B.; Malcolm, R.; Varrone, Z.; Collis, A.; Minto, M.; Burgess, S.; McDaniel, L.; Stimpson, E.; Spriggs, F.; Williams, J.; Neurath, K.; Ioime, N.; Agee, M.; Voss, E.; Furtak, K.; Renzulli, R.; Aanensen, N.; Carrolla, S.; Bickelhaupt, E.; Lazovatsky, Y.; DaSilva, A.; Zhong, J.; Stanyon, C. A.; Finley, R. L.; White, K. P.; Braverman, M.; Jarvie, T.; Gold, S.; Leach, M.; Knight, J.; Shimkets, R. A.; McKenna, M. P.; Chant, J.; Rothberg, J. M. A protein interaction map of Drosophila melanogaster. Science 2003, 302, 1727-1736.
[2] Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F. H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; Timm, J.; Mintzlaff, S.; Abraham, C.; Bock, N.; Kietzmann, S.; Goedde, A.; Toksoz, E.; Droege, A.; Krobitsch, S.; Korn, B.; Birchmeier, W.; Lehrach, H.; Wanker, E. E. A human protein-protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957-968.
[3] Rual, J. F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G. F.; Gibbons, F. D.; Dreze, M.; Ayivi-Guedehoussou, N.; Klitgord, N.; Simon, C.; Boxem, M.; Milstein, S.; Rosenberg, J.; Goldberg, D. S.; Zhang, L. V.; Wong, S. L.; Franklin, G.; Li, S.; Albala, J. S.; Lim, J.; Fraughton, C.; Llamosas, E.; Cevik, S.; Bex, C.; Lamesch, P.; Sikorski, R. S.; Vandenhaute, J.; Zoghbi, H. Y.; Smolyar, A.; Bosak, S.; Sequerra, R.; Doucette-Stamm, L.; Cusick, M. E.; Hill, D. E.; Roth, F. P.; Vidal, M. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 28, 28.

24
[4] Fariselli, P.; Pazos, F.; Valencia, A.; Casadio, R. Prediction of protein-protein interaction sites in heterocomplexes with neural networks. Eur. J. Biochem. 2002, 269, 1356-1361.
[5] Deng, M. H.; Mehta, S.; Sun, F. Z.; Chen, T. Inferring domain-domain interactions from protein-protein interactions. Genome Res. 2002, 12, 1540-1548.
[6] Bader, J. S.; Chaudhuri, A.; Rothberg, J. M.; Chant, J. Gaining confidence in high-throughput protein interaction networks. Nat. Biotechnol. 2004, 22, 78-85.
[7] Gavin, A. C.; Bosche, M.; Krause, R.; Grandi, P.; Marzioch, M.; Bauer, A.; Schultz, J.; Rick, J. M.; Michon, A. M.; Cruciat, C. M.; Remor, M.; Hofert, C.; Schelder, M.; Brajenovic, M.; Ruffner, H.; Merino, A.; Klein, K.; Hudak, M.; Dickson, D.; Rudi, T.; Gnau, V.; Bauch, A.; Bastuck, S.; Huhse, B.; Leutwein, C.; Heurtier, M. A.; Copley, R. R.; Edelmann, A.; Querfurth, E.; Rybin, V.; Drewes, G.; Raida, M.; Bouwmeester, T.; Bork, P.; Seraphin, B.; Kuster, B.; Neubauer, G.; Superti-Furga, G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141-147.
[8] Acehan, D.; Jiang, X.; Morgan, D. G.; Heuser, J. E.; Wang, X.; Akey, C. W. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002, 9, 423-432.
[9] Vasilescu, J.; Guo, X.; Kast, J. Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics. 2004, 4, 3845-3854.
[10] Guerrero, C.; Tagwerker, C.; Kaiser, P.; Huang, L. An Integrated Mass Spectrometry-based Proteomic Approach: Quantitative Analysis of Tandem Affinity-purified in vivo Cross-linked Protein Complexes (qtax) to Decipher the 26 s Proteasome-interacting Network. Mol Cell Proteomics. 2006, 5, 366-378.
[11] Lo Conte, L.; Chothia, C.; Janin, J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999, 285, 2177-2198.
[12] Ansari, S.; Helms, V. Statistical analysis of predominantly transient protein-protein interfaces. Proteins 2005, 61, 344-355.
[13] Pawson, T.; Raina, M.; Nash, P. Interaction domains: from simple binding events to complex cellular behavior. FEBS Lett. 2002, 513, 2-10.
[14] Archakov, A. I.; Govorun, V. M.; Dubanov, A. V.; Ivanov, Y. D.; Veselovsky, A. V.; Lewi, P.; Janssen, P. Protein-protein interactions as a target for drugs in proteomics. Proteomics 2003, 3, 380-391.
[15] Boger, D. L.; Desharnais, J.; Capps, K. Solution-phase combinatorial libraries: modulating cellular signaling by targeting protein-protein or protein-DNA interactions. Angew. Chem. Int. Ed. 2003, 42, 4138-4176.
[16] Xenarios, I.; Eisenberg, D. Protein interaction databases. Curr. Opin. Biotechnol. 2001, 12, 334-339.
[17] Gurezka, R.; Langosch, D. In vitro selection of membrane-spanning leucine zipper protein-protein interaction motifs using POSSYCCAT. J. Biol. Chem. 2001, 276, 45580-45587.
[18] Pacofsky, G. J.; Lackey, K.; Alligood, K. J.; Berman, J.; Charifson, P. S.; Crosby, R. M.; Dorsey, G. F., Jr.; Feldman, P. L.; Gilmer, T. M.; Hummel, C. W.; Jordan, S. R.; Mohr, C.; Shewchuk, L. M.; Sternbach, D. D.; Rodriguez, M. Potent dipeptide inhibitors of the pp60c-src SH2 domain. J. Med. Chem. 1998, 41, 1894-1908.
[19] Curran, D. P.; Wipf, P. Combinatorial definitions. Chem. & Eng. News 1997, 75, 6-7.

25
[20] Gold, L.; Alper, J. Drug discovery - Keeping pace with genomics through combinatorial chemistry. Nat. Biotechnology 1997, 15, 297-297.
[21] Several web pages are dedicated to give state-of the-art information about development of combinatorial chemistry techniques and its applications, s. a., http://www.combichemistry.com, http://www.combi-web.com or http://www.chemsoc.org/networks/cnn/ (from the Royal Society of Chemistry (RSC)).
[22] Baum, R. M. Combinatorial Approaches Provide Fresh Leads for Medicinal Chemistry. Chem.l & Eng. News 1994, 72, 20-26.
[23] Borman, S. Combinatorial chemists focus on small molecules, molecular recognition, and automation. Chemical & Engineering News 1996, 74, 29-.
[24] Service, R. F. Chemistry - Combinatorial chemistry hits the drug market. Science 1996, 272, 1266-1268.
[25] Plunkett, M. J.; Ellman, J. A. Combinatorial chemistry and new drugs. Sci. Am. 1997, 276, 68-73.
[26] Lazo, J. S.; Wipf, P. Combinatorial chemistry and contemporary pharmacology. Journal of Pharmacology and Experimental Therapeutics 2000, 293, 705-709.
[27] Choong, I. C.; Ellman, J. A. Solid-phase synthesis: Applications to combinatorial libraries. Annu. Rep. Med. Chem., Vol 31, 1996; pp 309-318.
[28] Obrecht, D.; Villalgordo, J. M. Solid-supported combinatorial and parallel synthesis of small-molecular-weigth compound libraries. Pergamon Press Ltd., Oxford, UK 1998.
[29] Sanchez-Martin, R. M.; Mittoo, S.; Bradley, M. The impact of combinatorial methodologies on medicinal chemistry. Curr. Top. Med. Chem. 2004, 4, 653-669.
[30] Bradley, M. The combinatorial centre of excellence - A unique industrial & academic partnership. Current Medicinal Chemistry 2002, 9, 2173-2177.
[31] Chabala, J. C. Solid-Phase Combinatorial Chemistry and Novel Tagging Methods for Identifying Leads. Curr. Opin. Biotechnol. 1995, 6, 632-639.
[32] Kamal, A.; Reddy, K. L.; Devaiah, V.; Shankaraiah, N.; Reddy, D. R. Recent advances in the solid-phase combinatorial synthetic strategies for the benzodiazepine based privileged structures. Mini-Rev. Med. Chem. 2006, 6, 53-69.
[33] Whitehead, D. M.; McKeown, S. C.; Routledge, A. Recent advances in analytical construct resins. Comb. Chem. High Throughput Screen. 2005, 8, 361-371.
[34] Nefzi, A.; Ostresh, J. M.; Houghten, R. A. The current status of heterocyclic combinatorial libraries. Chem. Rev. 1997, 97, 449-472.
[35] Gordon, E. M.; Gallop, M. A.; Patel, D. V. Strategy and tactics in combinatorial organic synthesis. Applications to drug discovery. Acc. Chem. Res. 1996, 29, 144-154.
[36] Geysen, H. M.; Meloen, R. H.; Barteling, S. J. Use of Peptide-Synthesis to Probe Viral-Antigens for Epitopes to a Resolution of a Single Amino-Acid. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 3998-4002.
[37] Geysen, H. M.; Barteling, S. J.; Meloen, R. H. Small Peptides Induce Antibodies with a Sequence and Structural Requirement for Binding Antigen Comparable to Antibodies Raised against the Native Protein. Proc. Natl. Acad. Sci. U.S.A 1985, 82, 178-182.

26
[38] Houghten, R. A. General-Method for the Rapid Solid-Phase Synthesis of Large Numbers of Peptides - Specificity of Antigen-Antibody Interaction at the Level of Individual Amino-Acids. Proc. Natl. Acad. Sci. U.S.A 1985, 82, 5131-5135.
[39] Furka, A. History of Combinatorial Chemistry. Drug Dev.Res. 1995, 36, 1-12. [40] Sebestyen, F.; Dibo, G.; Kovacs, A.; Furka, A. Chemical Synthesis of Peptide
Libraries. Bioorg. Med. Chem. Lett. 1993, 3, 413-418. [41] Houghten, R. A.; Pinilla, C.; Blondelle, S. E.; Appel, J. R.; Dooley, C. T.;
Cuervo, J. H. Generation and Use of Synthetic Peptide Combinatorial Libraries for Basic Research and Drug Discovery. Nature 1991, 354, 84-86.
[42] Lam, K. S.; Lebl, M.; Krchnak, V. The ''one-bead-one-compound'' combinatorial library method. Chem. Rev. 1997, 97, 411-448.
[43] Salmon, S. E.; Lam, K. S.; Lebl, M.; Kandola, A.; Khattri, P. S.; Wade, S.; Patek, M.; Kocis, P.; Krchnak, V.; Thorpe, D.; Felder, S. Discovery of Biologically-Active Peptides in Random Libraries - Solution-Phase Testing after Staged Orthogonal Release from Resin Beads. Proc. Natl. Acad. Sci. U.S.A 1993, 90, 11708-11712.
[44] Lam, K. S.; Salmon, S. E.; Hersh, E. M.; Hruby, V. J.; Kazmierski, W. M.; Knapp, R. J. A New Type of Synthetic Peptide Library for Identifying Ligand-Binding Activity. Nature 1991, 354, 82-84.
[45] Brenner, S.; Lerner, R. A. Encoded Combinatorial Chemistry. Proc. Natl. Acad. Sci. U.S.A 1992, 89, 5381-5383.
[46] Yingyongnarongkul, B. E.; How, S. E.; Diaz-Mochon, J. J.; Muzerelle, M.; Bradley, M. Parallel and multiplexed bead-based assays and encoding strategies. Com. Chem. High Throughput Screen. 2003, 6, 577-587.
[47] Franz, A. H.; Liu, R. W.; Song, A. M.; Lam, K. S.; Lebrilla, C. B. High-throughput one-bead-one-compound approach to peptide-encoded combinatorial libraries: MALDI-MS analysis of single TentaGel beads. Journal of Comb. Chem. 2003, 5, 125-137.
[48] Vagner, J.; Barany, G.; Lam, K. S.; Krchnak, V.; Sepetov, N. F.; Ostrem, J. A.; Strop, P.; Lebl, M. Enzyme-mediated spatial segregation on individual polymeric support beads: Application to generation and screening of encoded combinatorial libraries. Proc. Natl. Acad. Sci. U.S.A 1996, 93, 8194-8199.
[49] Still, W. C. Discovery of sequence-selective peptide binding by synthetic receptors using encoded combinatorial libraries. Acc. Chem. Res. 1996, 29, 155-163.
[50] Parandoosh, Z.; Knowles, S. K.; Xiao, X. Y.; Zhao, C.; David, G. S.; Nova, M. P. Encoded chemical synthesis coupled to screening: "Pot Assay". Com.Chem. High Throughput Screen. 1998, 1, 135-142.
[51] Guiles, J. W.; Lanter, C. L.; Rivero, R. A. A visual tagging process for mix and sort combinatorial chemistry. Angew. Chem. Int. Ed. 1998, 37, 926-928.
[52] Meiring, J. E.; Schmid, M. J.; Grayson, S. M.; Kirby, R.; Manthiram, K.; Hsia, B. I.; Ellington, A. D.; Willson, C. G. Development of a shape-encoded self-assembled hydrogel biosensor array. Abstr. Pap. Am. Chem. Soc. 2004, 227, U515-U515.
[53] Chen, Z.; Tsai, J.; Merriman, B.; Chen, J.; Kim, C.; Nelson, S. Shape encoded particles for biological probes. Am. J. Hum. Genet. 2003, 73, 434-434.
[54] Vaino, A. R.; Janda, K. D. Euclidean shape-encoded combinatorial chemical libraries. Proc. Natl. Acad. Sci. U.S.A 2000, 97, 7692-7696.

27
[55] Xue, F. T.; Seto, C. T. Selective inhibitors of the serine protease plasmin: Probing the S3 and S3 ' subsites using a combinatorial library. J. Med. Chem. 2005, 48, 6908-6917.
[56] Vilaivan, T.; Saesaengseerung, N.; Jarprung, D.; Kamchonwongpaisan, S.; Sirawaraporn, W.; Yuthavong, Y. Synthesis of solution-phase combinatorial library of 4,6-diamino-1,2-dihydro-1,3,5-triazine and identification of new leads against A16V+S108T mutant dihydrofolate reductase of Plasmodium falciparum. Bioorg. Med. Chem. 2003, 11, 217-224.
[57] Dostmann, W. R. G.; Taylor, M. S.; Nickl, C. K.; Brayden, J. E.; Frank, R.; Tegge, W. J. Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase I alpha inhibit NO-induced cerebral dilation. Proc. Natl. Acad. Sci. U.S.A 2000, 97, 14772-14777.
[58] Lawrence, R. N. Recursive deconvolution for real-time lead identification. Drug Discov. Today 2001, 6, S2-S3.
[59] Erb, E.; Janda, K. D.; Brenner, S. Recursive Deconvolution of Combinatorial Chemical Libraries. Proc. Natl. Acad. Sci. U.S.A 1994, 91, 11422-11426.
[60] Hioki, H.; Fukutaka, M.; Takahashi, H.; Kubo, M.; Matsushita, K.; Kodama, M.; Kubo, K.; Ideta, K.; Mori, A. Development of a new traceless aniline linker for solid-phase synthesis of azomethines. Application to parallel synthesis of a rod-shaped liquid crystalline library. Tetrahedron 2005, 61, 10643-10651.
[61] Rombouts, F. J. R.; Fridkin, G.; Lubell, W. D. Deazapurine solid-phase synthesis: Construction of 3-substituted pyrrolo 3,2-d pyrimidine-6-carboxylates on cross-linked polystyrene bearing a cysteamine linker. J. Comb. Chem. 2005, 7, 589-598.
[62] Brase, S.; Dahmen, S. Traceless linkers - Only disappearing links in solid-phase organic synthesis? Chemistry European J. 2000, 6, 1899-1905.
[63] Hioki, H.; Fukutaka, M.; Takahashi, H.; Kodama, M.; Kubo, K.; Ideta, K.; Mori, A. Development of a new traceless aniline linker for combinatorial solid-phase parallel synthesis of rod-shaped liquid crystals with an azomethine linkage. Tetrahedron Lett. 2004, 45, 7591-7594.
[64] Backes, B. J.; Ellman, J. A. Solid support linker strategies. Curr. Opin. Chem. Biol. 1997, 1, 86-93.
[65] De Luca, L.; Giacomelli, G.; Porcheddu, A. Synthesis of 1-alkyl-4-imidazolecarboxylates: A catch and release strategy. J. Comb. Chem. 2005, 7, 905-908.
[66] Cho, J. K.; White, P. D.; Klute, W.; Dean, T. W.; Bradley, M. Self-indicating amine scavenger resins. Chem.l Commun. 2004, 502-503.
[67] Porcheddu, A.; Giacomelli, G.; De Luca, L.; Ruda, A. M. A "catch and release" strategy for the parallel synthesis of 2,4,5-trisubstituted pyrimidines. J. Comb. Chem.2004, 6, 105-111.
[68] McAllister, L. A.; McCormick, R. A.; Procter, D. J. Sulfide- and selenide-based linkers in phase tag-assisted synthesis. Tetrahedron 2005, 61, 11527-11576.
[69] Ravn, J.; Bourne, G. T.; Smythe, M. L. A safety catch linker for Fmoc-based assembly of constrained cyclic peptides. J. Pept. Sci. 2005, 11, 572-578.
[70] Masquelin, T.; Sprenger, D.; Baer, R.; Gerber, F.; Mercadal, Y. A novel solution- and solid-phase approach to 2,4,5-tri- and 2,4,5,6-tetrasubstituted pyrimidines and their conversion into condensed heterocycles. Helv. Chim. Acta 1998, 81, 646-660.
[71] Kappe, C. O. Synthesis and reactions of Biginelli compounds, part 17 - Highly versatile solid phase synthesis of biofunctional 4-aryl-3,4-dihydropyrimidines

28
using resin-bound isothiourea building blocks and multidirectional resin cleavage. Bioorg. Med. Chem. Lett. 2000, 10, 49-51.
[72] Yu, Z. R.; Bradley, M. Solid supports for combinatorial chemistry. Current Opinion in Chemical Biology 2002, 6, 347-352.
[73] Ramstrom, O.; Lehn, J. M. Drug discovery by dynamic combinatorial libraries. Nat. Rev. Drug Discov. 2002, 1, 26-36.
[74] Huc, I.; Nguyen, R. Dynamic combinatorial chemistry. Comb. Chem. & High Throughput Screening 2001, 4, 53-74. [1] Giot, L.; Bader, J. S.; Brouwer, C.;. 2005, 48, 6908-6917.
[75] Rivas, L.; Riveiro, O. M. Andreu y Rivas Eds. Péptidos en biología y medicina, . Col. Nuevas Tendencias, CSIC, Madrid. 1997, pp. 238-254.
[76] Scholle, M. D.; Kehoe, J. W.; Kay, B. K. Efficient construction of a large collection of phage-displayed combinatorial peptide libraries Comb. Chem. High Throughput Screen. 2005, 8, 545-551.
[77] Kay, B. K.; Hamilton, P. T. Identification of enzyme inhibitors from phage-displayed combinatorial peptide libraries. Comb. Chem. High Throughput Screen. 2001, 4, 535-543.
[78] Smith, G. P. Filamentous Fusion Phage - Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 1985, 228, 1315-1317.
[79] Uppala, A.; Koivunen, E. Targeting of phage display vectors to mammalian cells. Comb. Chem. High Throughput Screen. 2000, 3, 373-392.
[80] Ripka, W. C.; Barker, G.; Krakover, J. High-throughput purification of compound libraries. Drug Discov. Today 2001, 6, 471-477.
[81] Diaz-Mochon, J. J.; Bialy, L.; Keinicke, L.; Bradley, M. Combinatorial libraries - from solution to 2D microarrays. Chem. Commun. 2005, 1384-1386.
[82] Gayo, L. M.; Suto, M. J. Ion-exchange resins for solution phase parallel synthesis of chemical libraries. Tetrahedron Lett. 1997, 38, 513-516.
[83] Parlow, J. J.; Naing, W.; South, M. S.; Flynn, D. L. In situ chemical tagging: Tetrafluorophthalic anhydride as a ''sequestration enabling reagent'' (SER) in the purification of solution-phase combinatorial libraries. Tetrahedron Lett. 1997, 38, 7959-7962.
[84] Siegel, M. G.; Hahn, P. J.; Dressman, B. A.; Fritz, J. E.; Grunwell, J. R. Kaldor, S. W et al. Rapid purification of small molecule libraries by ion exchange chromatography. Tetrahedron Lett. 1997, 38, 3357-3360.
[85] Boger, D. L.; Tarby, C. M.; Myers, P. L.; Caporale, L. H. Generalized dipeptidomimetic template: Solution phase parallel synthesis of combinatorial libraries. J. Am. Chem. Soc. 1996, 118, 2109-2110.
[86] Houghten, R. A.; Pinilla, C.; Appel, J. R.; Blondelle, S. E.; Dooley, C. T. Eichler, J., Nefzi, A., Ostresh, J. M.. Mixture-based synthetic combinatorial libraries. J. Med.Chem. 1999, 42, 3743-3778.
[87] Pinilla, C.; Rubio-Godoy, V.; Dutoit, V.; Guillaume, P.; Simon, R. Zhao, Y. D. Houghten, R. A., Cerottini, J. C, Romero, P, Valmori, D Combinatorial peptide libraries as an alternative approach to the identification of ligands for tumor-reactive cytolytic T lymphocytes. Cancer Res. 2001, 61, 5153-5160.
[88] Pinilla, C.; Appel, J. R.; Blanc, P.; Houghten, R. A. Rapid Identification of High-Affinity Peptide Ligands Using Positional Scanning Synthetic Peptide Combinatorial Libraries. Biotechniques 1992, 13, 901-&.
[89] Dooley, C. T.; Houghten, R. A. The Use of Positional Scanning Synthetic Peptide Combinatorial Libraries for the Rapid-Determination of Opioid Receptor Ligands. Life Sci. 1993, 52, 1509-1517.

29
[90] Boger, D. L.; Lee, J. K.; Goldberg, J.; Jin, Q. Two comparisons of the performance of positional scanning and deletion synthesis for the identification of active constituents in mixture combinatorial libraries. J. Org. Chem. 2000, 65, 1467-1474.
[91] Boger, D. L.; Chai, W. Y.; Jin, Q. Multistep convergent solution-phase combinatorial synthesis and deletion synthesis deconvolution. J. Am. Chem. Soc. 1998, 120, 7220-7225.
[92] Ambroise, Y.; Yaspan, B.; Ginsberg, M. H.; Boger, D. L. Inhibitors of cell migration that inhibit intracellular paxillin/alpha4 binding: a well-documented use of positional scanning libraries. Chem Biol. 2002, 9, 1219-1226.
[93] Kacprzak, M. M.; Than, M. E.; Juliano, L.; Juliano, M. A.; Bode, W. Lindberg, I.. Mutations of the PC2 substrate binding pocket alter enzyme specificity. J. Biol. Chem. 2005, 280, 31850-31858.
[94] Reed, J. C. Apoptosis-regulating proteins as targets for drug discovery. Trends Mol. Med. 2001, 7, 314-319.
[95] Strasser, A.; O'Connor, L.; Dixit, V. M. Apoptosis signaling. Annu. Rev. Biochem. 2000, 69, 217-245.
[96] Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S. M.; Ahmad, M. Alnemri, E. S., Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997, 91, 479-489.
[97] Rodriguez, J.; Lazebnik, Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999, 13, 3179-3184.
[98] Srinivasula, S. M.; Ahmad, M.; Fernandes-Alnemri, T.; Alnemri, E. S. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell. 1998, 1, 949-957.
[99] Malet, G.; Martin, A. G.; Orzaez, M.; Vicent, M. J.; Masip, I. Sanclimens, G.,Ferrer-Montiel, A.,Mingarro, I.,Messeguer, A.,Fearnhead, H. O., Perez-Paya, E.. Small molecule inhibitors of Apaf-1-related caspase- 3/-9 activation that control mitochondrial-dependent apoptosis. Cell Death Differ. 2005, 9, 9.
[100] Simon, R. J.; Kania, R. S.; Zuckermann, R. N.; Huebner, V. D.; Jewell, D. A Banville, S., Ng, S.,Wang, L., Rosenberg, S., Marlowe, C. K. Peptoids: a modular approach to drug discovery. Proc. Natl. Acad. Sci. U S A. 1992, 89, 9367-9371.
[101] Holinger, E. P.; Chittenden, T.; Lutz, R. J. Bak BH3 peptides antagonize Bcl-x(L) function and induce apoptosis through cytochrome c-independent activation of caspases. J. Biol. Chem. 1999, 274, 13298-13304.
[102] Degterev, A.; Lugovskoy, A.; Cardone, M.; Mulley, B.; Wagner, G. Mitchison, T., Yuan, J. Y. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-x(L). Nat. Cell. Biol. 2001, 3, 173-182.
[103] Oltersdorf, T.; Elmore, S. W.; Shoemaker, A. R.; Armstrong, R. C.; Augeri, D. J. Belli, B. A.Bruncko, M, Deckwerth, T. L, Dinges, J., Hajduk, P. J., Joseph, M. K., Kitada, S., Korsmeyer, S. J., Kunzer, A. R., Letai, A., Li, C., Mitten, M. J., Nettesheim, D. G., Ng, S., Nimmer, P. M., O'Connor, J. M., Oleksijew, A., Petros, A. M., Reed, J. C. Shen, W., Tahir, S. K., Thompson, C. B., Tomaselli, K. J., Wang, B. L., Wendt, M. D.Zhang, H. C., Fesik, S. W., Rosenberg, S. H. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677-681.
[104] Frankel, A. D.; Young, J. A. HIV-1: fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1-25.

30
[105] Wu, J.; Adomat, J. M.; Ridky, T. W.; Louis, J. M.; Leis, J. Harrison, R. W., Weber, I. T. Structural basis for specificity of retroviral proteases. Biochemistry. 1998, 37, 4518-4526.
[106] Navia, M. A.; Fitzgerald, P. M.; McKeever, B. M.; Leu, C. T.; Heimbach, J. C. Herber, W. K., Sigal, I. S., Darke, P. L., Springer, J. P. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature. 1989, 337, 615-620.
[107] Shultz, M. D.; Bowman, M. J.; Ham, Y. W.; Zhao, X.; Tora, G. Chmielewski, J. Small-Molecule Inhibitors of HIV-1 Protease Dimerization Derived from Cross-Linked Interfacial Peptides This work was supported by NIH (GM52739) and NSF (9457372-CHE). Angew. Chem. In.t Ed. 2000, 39, 2710-2713.
[108] Jacob, Y.; Badrane, H.; Ceccaldi, P. E.; Tordo, N. Cytoplasmic dynein LC8 interacts with lyssavirus phosphoprotein. J. Virol. 2000, 74, 10217-10222.
[109] Brooks, P. C.; Stromblad, S.; Sanders, L. C.; von Schalscha, T. L.; Aimes, R. T. Stetler-Stevenson, W. G., Quigley, J. P., Cheresh, D. A. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 1996, 85, 683-693.
[110] Boger, D. L.; Goldberg, J.; Silletti, S.; Kessler, T.; Cheresh, D. A. Identification of a novel class of small-molecule antiangiogenic agents through the screening of combinatorial libraries which function by inhibiting the binding and localization of proteinase MMP2 to integrin alpha(V)beta(3). J. Am. Chem. Soc. 2001, 123, 1280-1288.
[111] Silletti, S.; Kessler, T.; Goldberg, J.; Boger, D. L.; Cheresh, D. A. Disruption of matrix metalloproteinase 2 binding to integrin alpha vbeta 3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 119-124.
[112] Han, J.; Rose, D. M.; Woodside, D. G.; Goldfinger, L. E.; Ginsberg, M. H. Integrin alpha 4 beta 1-dependent T cell migration requires both phosphorylation and dephosphorylation of the alpha 4 cytoplasmic domain to regulate the reversible binding of paxillin. J. Biol. Chem. 2003, 278, 34845-34853. Epub 32003 Jun 34830.
[113] Fahraeus, R.; Paramio, J. M.; Ball, K. L.; Lain, S.; Lane, D. P. Inhibition of pRb phosphorylation and cell-cycle progression by a 20-residue peptide derived from p16CDKN2/INK4A. Curr. Biol. 1996, 6, 84-91.
[114] Frolov, M. V.; Moon, N. S.; Dyson, N. J. dDP is needed for normal cell proliferation. Mol. Cell Biol. 2005, 25, 3027-3039.
[115] Asano, M.; Nevins, J. R.; Wharton, R. P. Ectopic E2F expression induces S phase and apoptosis in Drosophila imaginal discs. Genes Dev. 1996, 10, 1422-1432.
[116] Fabbrizio, E.; Le Cam, L.; Polanowska, J.; Kaczorek, M.; Lamb, N., Brent, R. Sardet, C. Inhibition of mammalian cell proliferation by genetically selected peptide aptamers that functionally antagonize E2F activity. Oncogene. 1999, 18, 4357-4363.
[117] Malbon, C. C. G proteins in development. Nat. Rev. Mol. Cell Biol. 2005, 6, 689-701.
[118] Hollmann, M. W.; Strumper, D.; Herroeder, S.; Durieux, M. E. Receptors, G proteins, and their interactions. Anesthesiology. 2005, 103, 1066-1078.
[119] Scott, J. K.; Huang, S. F.; Gangadhar, B. P.; Samoriski, G. M.; Clapp, P. Gross, R. A., Taussig, R., Smrcka, A. V. Evidence that a protein-protein interaction 'hot

31
spot' on heterotrimeric G protein betagamma subunits is used for recognition of a subclass of effectors. Embo J. 2001, 20, 767-776.
[120] Davis, T. L.; Bonacci, T. M.; Sprang, S. R.; Smrcka, A. V. Structural and molecular characterization of a preferred protein interaction surface on G protein beta gamma subunits. Biochemistry. 2005, 44, 10593-10604.
[121] Pastor, M. T.; Mora, P.; Ferrer-Montiel, A.; Perez-Paya, E. Design of bioactive and structurally well-defined peptides from conformationally restricted libraries. Biopolymers 2004, 76, 357-365.
[122] Ho, Y.; Gruhler, A.; Heilbut, A.; Bader, G. D.; Moore, L., Adams, S. L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., Yang, L. Y., Wolting, C., Donaldson, I., Schandorff, S., Shewnarane, J., Vo, M., Taggart, J., Goudreault, M., Muskat, B., Alfarano, C., Dewar, D., Lin, Z., Michalickova, K., Willems, A. R., Sassi, H., Nielsen, P. A., Rasmussen, K. J., Andersen, J. R., Johansen, L. E., Hansen, L. H., Jespersen, H., Podtelejnikov, A., Nielsen, E., Crawford, J., Poulsen, V., Sorensen, B. D., Matthiesen, J., Hendrickson, R. C., Gleeson, F., Pawson, T., Moran, M. F., Durocher, D., Mann, M., Hogue, C. W. V., Figeys, D., Tyers, M.. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002, 415, 180-183.
[123] Mas, V.; Perez, L.; Encinar, J. A.; Pastor, M. T.; Rocha, A., Perez-Paya, E., Ferrer-Montiel, A., Ros, J. M. G., Estepa, A., Coll, J. M. Salmonid viral haemorrhagic septicaemia virus: fusion-related enhancement of virus infectivity by peptides derived from viral glycoprotein G or a combinatorial library. J. Gen. Virol. 2002, 83, 2671-2681.
[124] Brunger, A. T. Structure of proteins involved in synaptic vesicle fusion in neurons. Annu Rev Biophys Biomol Struct. 2001, 30, 157-171.
[125] Chen, Y. A.; Scheller, R. H. SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2001, 2, 98-106.
[126] Sutton, R. B.; Fasshauer, D.; Jahn, R.; Brunger, A. T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998, 395, 347-353.
[127] Xiao, W.; Poirier, M. A.; Bennett, M. K.; Shin, Y. K. The neuronal t-SNARE complex is a parallel four-helix bundle. Nat. Struct. Biol. 2001, 8, 308-311.
[128] Schiavo, G.; Matteoli, M.; Montecucco, C. Neurotoxins affecting neuroexocytosis. Physiol Rev. 2000, 80, 717-766.
[129] Blanes-Mira, C.; Pastor, M. T.; Valera, E.; Fernandez-Ballester, G.; Merino, J. M. Gutierrez, L. M., Perez-Paya, E., Ferrer-Montiel, A.. Identification of SNARE complex modulators that inhibit exocytosis from an alpha-helix-constrained combinatorial library. Biochem. J. 2003, 375, 159-166.
[130] Ferrer-Montiel, A. V.; Gutierrez, L. M.; Apland, J. P.; Canaves, J. M.; Gil, A. Viniegra, S., Biser, J. A., Adler, M., Montal, M. The 26-mer peptide released from SNAP-25 cleavage by botulinum neurotoxin E inhibits vesicle docking. FEBS Lett. 1998, 435, 84-88.
[131] Ambroise, Y.; Yaspan, B.; Ginsberg, M. H.; Boger, D. L. Inhibitors of cell migration that inhibit intracellular paxillin/alpha A binding: A well-documented use of positional scanning libraries. Chem. Biol. 2002, 9, 1219-1226.
[132] Qin, H. X.; Srinivasula, S. M.; Wu, G.; Fernandes-Alnemri, T.; Alnemri, E. S. Shi, Y. G. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 1999, 399, 549-557.
[133] Rutenber, E.; Fauman, E. B.; Keenan, R. J.; Fong, S.; Furth, P. S. Demontellano, P. R. O., Meng, E., Kuntz, I. D., Decamp, D. L., Salto, R., Rose, J. R., Craik, C.

32
S., Stroud, R. M.. Structure of a Nonpeptide Inhibitor Complexed with Hiv-1 Protease - Developing a Cycle of Structure-Based Drug Design. J Biol. Chem. 1993, 268, 15343-15346.
[134] Emsley, J.; Knight, C. G.; Farndale, R. W.; Barnes, M. J.; Liddington, R. C. Structural basis of collagen recognition by integrin alpha 2 beta 1. Cell 2000, 101, 47-56.
33
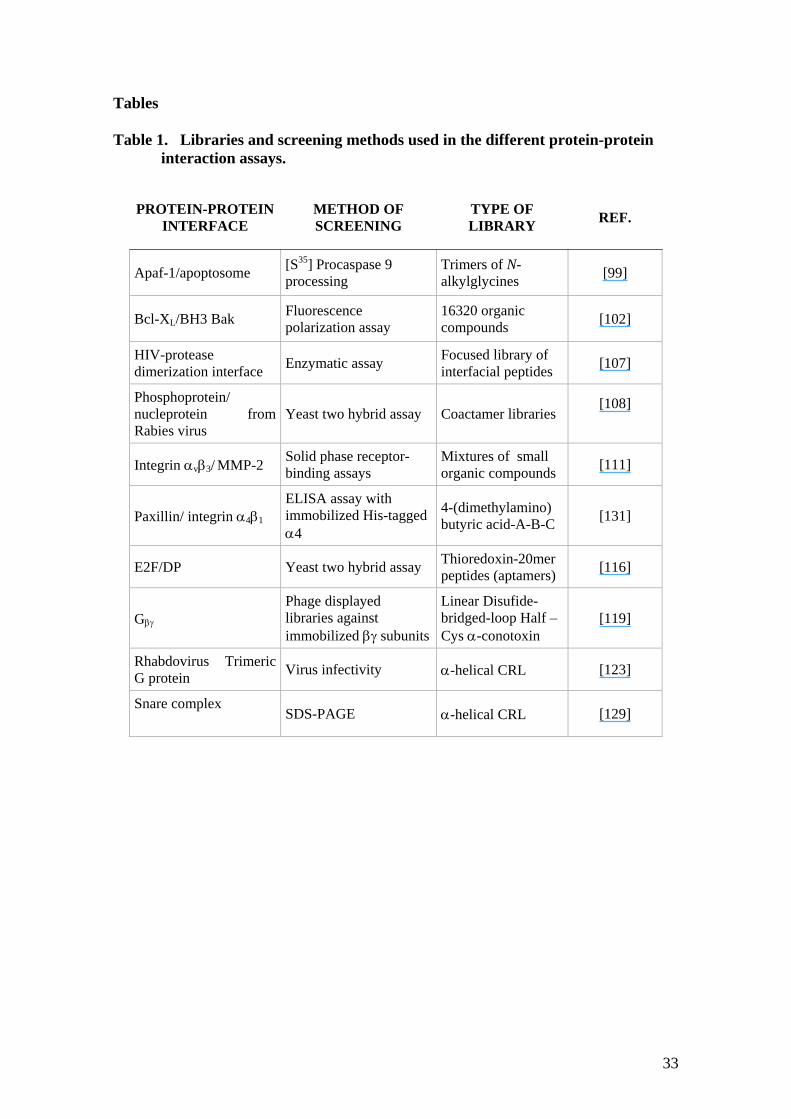
Tables Table 1. Libraries and screening methods used in the different protein-protein
interaction assays.
PROTEIN-PROTEIN INTERFACE
METHOD OF SCREENING
TYPE OF LIBRARY REF.
Apaf-1/apoptosome [S35] Procaspase 9 processing
Trimers of N-alkylglycines [99]
Bcl-XL/BH3 Bak Fluorescence polarization assay
16320 organic compounds [102]
HIV-protease dimerization interface Enzymatic assay Focused library of
interfacial peptides [107]
Phosphoprotein/ nucleprotein from Rabies virus
Yeast two hybrid assay Coactamer libraries [108]
Integrin αvβ3/ MMP-2 Solid phase receptor-binding assays
Mixtures of small organic compounds [111]
Paxillin/ integrin α4β1 ELISA assay with immobilized His-tagged α4
4-(dimethylamino) butyric acid-A-B-C [131]
E2F/DP Yeast two hybrid assay Thioredoxin-20mer peptides (aptamers) [116]
Gβγ Phage displayed libraries against immobilized βγ subunits
Linear Disufide-bridged-loop Half –Cys α-conotoxin
[119]
Rhabdovirus Trimeric G protein Virus infectivity α-helical CRL [123]
Snare complex
SDS-PAGE α-helical CRL [129]

34
Legend to figures
Figure 1. Representative examples of solved strutures of known protein-protein
interfaces. (A) CARD domain of Apaf-1/prodomain of caspase (3YGS) [132] (B)
Dimeric HIV protease (1AID) [133] (C) Integrin alpha2 I domain/collagen complex
(1DZI) [134]
Figure 2. Apaf 1 inhibition by identified peptoids. (A) Structure of the lead compound
obtained from the screening, peptoid 1, and a more soluble derivative peptoid 1a. (B)
Apoptosome-dependent activation of procaspase-9 followed by incubating in vitro
transcribed-translated [35S]-Met procaspase-9 and rApaf-1 in the presence cytochrome
at different concentrations of peptoid 1a. (C) Structures peptoid 1 third generation
derivatives: penetratin-GG-peptoid 1, cyclo-peptoid 1a and PGA-GG-peptoid 1.
Figure 3. Structure of active compounds obtained from the screening of different
libraries in protein-protein interaction assays.
Figure 4. Structure of the focused library designed to inhibit HIV protease dimerisation.
The library consisted of 49 single modifications of the parent compound in the four
marked different positions.
Figure 5. General structure of the library designed to interfere with the Paxillin/α4
interaction. The positions marked as X, Y and Z were substituded by ten different
aromatic amino acids.




















