
Direct potential fit analysis of the X 1Sigmag+ state of Rb2: Nothing else will do
10
Direct potential fit analysis of the X 1 S g ¿ state of Rb 2 : Nothing else will do! Jenning Y. Seto and Robert J. Le Roy a) Guelph-Waterloo Centre for Graduate Work in Chemistry and Biochemistry, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada Jean Verge ` s and Claude Amiot b) Laboratoire Aime ´ Cotton, Ba ˆtiment 505, Campus d’Orsay, 91405 Orsay Cedex, France ~Received 8 May 2000; accepted 23 May 2000! High resolution A-X emission data involving vibrational levels of the ground X 1 S g 1 electronic state up to v 9 5113, spanning 99.8% of the potential well, have been acquired for three isotopomers of Rb 2 . While a good fit ( s ¯ f 51.03) to the 12 148 transition frequencies ~with uncertainties 60.001 cm 21 ! is obtained from an unconstrained combined-isotopomer Dunham-type analysis, it requires a large number ~62! of expansion parameters, and the resulting empirical centrifugal distortion constants ~CDCs! are unreliable for extrapolation to higher-J. Moreover, Dunham expansion fits using constrained theoretical values of the first six CDCs ~up to O v ! fail to properly represent the data, as even higher-order CDCs are required. However, a direct fit of these data to an analytical ‘‘modified Lennard-Jones’’ potential energy function involving only 16 fitted parameters yields essentially the same quality of fit as did the unconstrained Dunham fit, and should be reliable for extrapolation to arbitrarily high J. This potential form incorporates the proper R 26 asymptotic behavior of the potential, and is constrained to have the theoretically predicted C 6 dispersion coefficient. Although the dataset involves the three isotopomers 85,85 Rb 2 , 85,87 Rb 2 , and 87,87 Rb 2 , none of the present analyses was able to determine any Born–Oppenheimer breakdown effects. © 2000 American Institute of Physics. @S0021-9606~00!01932-2# I. INTRODUCTION The Rb 2 molecule has been studied since the beginning of the century, and a comprehensive review of spectroscopic studies up to 1990 was presented by one of us a decade ago. 1 In that paper the observation of ground state vibrational lev- els up to v 9 5112 obtained by recording laser-induced fluo- rescence with a Fourier transform spectrometer yielding data with an accuracy of 60.005 cm 21 was reported. A potential energy curve spanning 99.7% of the potential energy well was obtained from those data. In recent years Rb 2 has figured prominently in applica- tions of the powerful new technique of photoassociation spectroscopy ~PAS! of ultracold atoms. Rubidium has been widely used in laser cooling experiments, and was the me- dium for the first demonstration of a Bose–Einstein conden- sate in a dilute medium. 2 An accurate knowledge of the ground-state potential energy curve of this system is neces- sary for an understanding of ultracold collisions and for the determination of the ‘‘scattering length,’’ which is an impor- tant parameter in the prediction of Bose–Einstein condensa- tion. To model the potential energy curve satisfactorily, it is important to have accurate observations of very highly ex- cited vibrational levels lying as close as possible to the dis- sociation limit. Using two-color PAS, Tsai et al. 3 observed 12 levels lying within 20 GHz of the dissociation limit of 85 Rb 2 , and a coupled-states analysis of those results using the potential of Ref. 1 yielded an accurate new ground-state dis- sociation energy of D e 53993.53( 60.06) cm 21 . Other appli- cations of the potential curve of Ref. 1 include ~i! the pre- diction of Feshbach resonances in collisions of ultracold Rb atoms, 4 ~ii! the calculation of magnetic-dipolar decay-rate constants in Bose–Einstein condensation experiments, 5 ~iii! the prediction of binary collision parameters for ultracold mixed-isotope collisions and the prospect of mixed isotope Bose–Einstein condensation, 6 ~iv! the calculation of scatter- ing lengths for ultracold collisions of the short-lived radio- active Rb isotopes, 7 and ~v! the formation of cold rubidium molecules in a magneto-optical trap. 8 The work of Ref. 1 was based on a less extensive and less accurate dataset than that obtained here, and shortcom- ings of the analysis techniques available at that time required the dataset used in the final analysis to be truncated further. In particular, since the then available computer codes for the calculation of centrifugal distortion constants ~CDCs! from a given potential 9 would only generate the first four CDCs, several hundred high-J 9 lines had to be omitted from the final ‘‘mechanically consistent’’ fits with constrained CDC constants. Moreover, the differences between experiment and the vibrational energies calculated from the final potential showed a disturbingly regular oscillatory behavior ~see Fig. 7 of Ref. 1!, which suggests the presence of some systematic error. In view of the considerable interest in this system, the present work therefore set out to improve our knowledge of the ground state of Rb 2 , both by making new, more accurate measurements, and by applying a more sophisticated method of analysis. In the following, in Sec. II we describe the experimental a! Electronic mail: [email protected] b! Electronic mail: [email protected] JOURNAL OF CHEMICAL PHYSICS VOLUME 113, NUMBER 8 22 AUGUST 2000 3067 0021-9606/2000/113(8)/3067/10/$17.00 © 2000 American Institute of Physics Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Direct potential fit analysis of the X 1Sigmag+ state of Rb2: Nothing else will do

JOURNAL OF CHEMICAL PHYSICS VOLUME 113, NUMBER 8 22 AUGUST 2000
Direct potential fit analysis of the X 1Sg¿ state of Rb 2: Nothing else will do!
Jenning Y. Seto and Robert J. Le Roya)
Guelph-Waterloo Centre for Graduate Work in Chemistry and Biochemistry, University of Waterloo,Waterloo, Ontario N2L 3G1, Canada
Jean Verges and Claude Amiotb)
Laboratoire Aime´ Cotton, Batiment 505, Campus d’Orsay, 91405 Orsay Cedex, France
~Received 8 May 2000; accepted 23 May 2000!
High resolutionA-X emission data involving vibrational levels of the groundX 1Sg1 electronic state
up to v95113, spanning 99.8% of the potential well, have been acquired for three isotopomers ofRb2. While a good fit (s f51.03) to the 12 148 transition frequencies~with uncertainties60.001cm21! is obtained from an unconstrained combined-isotopomer Dunham-type analysis, it requires alarge number~62! of expansion parameters, and the resulting empirical centrifugal distortionconstants~CDCs! are unreliable for extrapolation to higher-J. Moreover, Dunham expansion fitsusing constrained theoretical values of the first six CDCs~up to Ov! fail to properly represent thedata, as even higher-order CDCs are required. However, a direct fit of these data to an analytical‘‘modified Lennard-Jones’’ potential energy function involving only 16 fitted parameters yieldsessentially the same quality of fit as did the unconstrained Dunham fit, and should be reliable forextrapolation to arbitrarily highJ. This potential form incorporates the properR26 asymptoticbehavior of the potential, and is constrained to have the theoretically predictedC6 dispersioncoefficient. Although the dataset involves the three isotopomers85,85Rb2,
85,87Rb2, and 87,87Rb2,none of the present analyses was able to determine any Born–Oppenheimer breakdown effects.© 2000 American Institute of Physics.@S0021-9606~00!01932-2#
inopagevo-a
le
-ion
enecethr-sis
exis
ft
di
Rbte
ldpe
-o-
andcom-iredher.the
,
Cand
tial
atictheof
tethod
tal
I. INTRODUCTION
The Rb2 molecule has been studied since the beginnof the century, and a comprehensive review of spectroscstudies up to 1990 was presented by one of us a decade1
In that paper the observation of ground state vibrational lels up tov95112 obtained by recording laser-induced flurescence with a Fourier transform spectrometer yielding dwith an accuracy of60.005 cm21 was reported. A potentiaenergy curve spanning 99.7% of the potential energy wwas obtained from those data.
In recent years Rb2 has figured prominently in applications of the powerful new technique of photoassociatspectroscopy~PAS! of ultracold atoms. Rubidium has beewidely used in laser cooling experiments, and was the mdium for the first demonstration of a Bose–Einstein condsate in a dilute medium.2 An accurate knowledge of thground-state potential energy curve of this system is nesary for an understanding of ultracold collisions and fordetermination of the ‘‘scattering length,’’ which is an impotant parameter in the prediction of Bose–Einstein condention. To model the potential energy curve satisfactorily, itimportant to have accurate observations of very highlycited vibrational levels lying as close as possible to the dsociation limit. Using two-color PAS, Tsaiet al.3 observed12 levels lying within 20 GHz of the dissociation limit o85Rb2, and a coupled-states analysis of those results usingpotential of Ref. 1 yielded an accurate new ground-state
a!Electronic mail: [email protected]!Electronic mail: [email protected]
3060021-9606/2000/113(8)/3067/10/$17.00
Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to A
gico.-
ta
ll
n
e--
s-e
a-
--
hes-
sociation energy ofDe53993.53(60.06) cm21. Other appli-cations of the potential curve of Ref. 1 include~i! the pre-diction of Feshbach resonances in collisions of ultracoldatoms,4 ~ii ! the calculation of magnetic-dipolar decay-raconstants in Bose–Einstein condensation experiments,5 ~iii !the prediction of binary collision parameters for ultracomixed-isotope collisions and the prospect of mixed isotoBose–Einstein condensation,6 ~iv! the calculation of scattering lengths for ultracold collisions of the short-lived radiactive Rb isotopes,7 and ~v! the formation of cold rubidiummolecules in a magneto-optical trap.8
The work of Ref. 1 was based on a less extensiveless accurate dataset than that obtained here, and shortings of the analysis techniques available at that time requthe dataset used in the final analysis to be truncated furtIn particular, since the then available computer codes forcalculation of centrifugal distortion constants~CDCs! from agiven potential9 would only generate the first four CDCsseveral hundred high-J9 lines had to be omitted from thefinal ‘‘mechanically consistent’’ fits with constrained CDconstants. Moreover, the differences between experimentthe vibrational energies calculated from the final potenshowed a disturbingly regular oscillatory behavior~see Fig. 7of Ref. 1!, which suggests the presence of some systemerror. In view of the considerable interest in this system,present work therefore set out to improve our knowledgethe ground state of Rb2, both by making new, more accurameasurements, and by applying a more sophisticated meof analysis.
In the following, in Sec. II we describe the experimen
7 © 2000 American Institute of Physics
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

inded
aet
pt
-
-
do
mv
-atio
w
,eioum
deipri
imerwthrr-0
2hesu
in
-we
-al
ofaser
lli-ine
al
148sly
ethe
2.erses onset
onsti-
or,
e
s
edlet
3068 J. Chem. Phys., Vol. 113, No. 8, 22 August 2000 Seto et al.
methodology and measurements, while the results obtausing various methods of analysis and our recommennew ground-state potential energy function are presenteSec. III.
II. EXPERIMENT
As in previous work,1 Rb2 molecules were produced instainless steel heat pipe oven by heating rubidium m~99.95% purity from the Aldrich Chemical Company! with10 Torr of argon as a buffer gas. The temperature was ke600 K, where the vapor pressures of Rb and Rb2 are, respec-tively, 2.4 and 0.002 Torr.10 The molecules were then excited with various fixed frequencies from an Ar1 laser~Spectra-Physics 171-19! and a Kr1 laser~Coherent Radia-tion Innova K-3000!, both oscillating under multimode conditions, and by a linear single mode dye laser~Coherent Ra-diation 599-21! operating with DCM or LD700 dyes pumpeby an Ar1 laser. The emission excited in this way consiststransitions from the three systemsC 1Pu→X 1Sg
1 , B 1Pu
→X 1Sg1 , andA 1Su
1→X 1Sg1 .
As with all other homo- and heteronuclear alkali diatoics, the best way to obtain fluorescence into the highestbrational levels of the ground state of Rb2 is to pump theupper levels of the first excited1Su
1 electronic state. In particular, due to the relative positions of their potential minimthe best way to obtain spectra nearly up to the dissocialimit of the groundX 1Sg
1 state Rb2 is through the excitationof the A 1Su
1 state. In the present work, isolatedA 1Su1
←X 1Sg1 rovibrational transitions were excited by a c
single frequency titanium sapphire laser~Coherent 899-21!pumped by an argon–ion laser~Spectra Physics 2080-155all lines power: 18 W!. The Ti:Sa laser was operated with thshorter- to the longer-wavelength optics, so its emissrange extended from 750 to 1000 nm, with the maximdelivered laser power being 2.3 W.
The experimental arrangement was similar to thatscribed in Ref. 1: backward fluorescence from the heat pwas collected and focused on the entrance iris of the Foutransform spectrometer constructed in the Laboratoire A´Cotton. The frequency and intensity stability of the laswas sufficient to allow spectra recording times of about thours. The wave numbers were calibrated relative tofixed frequency reference line~the Xe atomic transition nea3.5 mm! used to monitor the path difference of the interfeometer. Spectra were recorded between 5 000 and 13cm21 with a resolution limit ranging from 0.005 to 0.01cm21, corresponding to the range of Doppler widths in tspectral range studied. The absolute wave number meament uncertainty varied from 131023 cm21 for the strongestlines to 331023 cm21 for the weakest ones; these uncertaties are some 3–5 times smaller than those of Ref. 1.
A typical A 1Su1→X 1Sg
1 transition, obtained by excitation with the 785.843 nm output of the Ti:Sa laser, is shoin the upper part of Fig. 1. This series originates in an uppstate level withJ8514; $P(15),R(13)% doublets are clearlyobserved for ground-state vibrational levels up tov95113.Fainter lines around theseP-R doublets correspond to transitions from neighboring collisionally populated rotation
Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to A
eddin
al
at
f
-i-
,n
n
-eeresoe
00
re-
-
nr-
levels. The lower portion of Fig. 1 shows a small portionthe fluorescence spectrum induced by the 869.440 nm lline, which generates aJ8554 series of the85,87Rb2 isoto-pomer, together with an additional structure due to cosional transfer of an upper-state population. Such multilpatterns, also observed for NaLi and RbCs,11,12 occur allalong thisJ8554 series, and are probably due to collisiontransfer from the initially excitedA 1Su
1 level into levels ofthe nearbyb 3Pu state.
The dataset generated in this way consists of 12transitions in 424 fluorescence series. Use of the previoureported ground-state constants1 made their assignment quitstraightforward. Of the observed lines, 7009 belonged tomost abundant~52.1%! isotopomer85,85Rb2, 5044 to themixed isotopomer85,87Rb2 ~40.2% abundance!, and 95 to87,87Rb2 ~7.8% abundance!. For each vibrational level, therange of associated rotational levels is indicated in Fig.This dataset includes levels with rotational quantum numbranging fromJ958 to 243, which considerably extends thpreviously reported dataset and places severe demandour analysis procedure. A complete listing of this datamay be obtained from the Journal’s electronic archive.13
III. ANALYSIS
In all of the fits reported herein, the observed transitienergies were weighted by the inverse square of their emated uncertainties of 0.001 cm21, and the quality of fit ischaracterized by the value of dimensionless standard err14
FIG. 1. Two examples of Ti:Sa laser-induced spectra.Upper segment: withthe laser set at 785.843 nm, vibrational levels are observed up tov95113 ina series withJ8514; lower segment: with the laser set at 869.440 nm, thdoublet linked by solid lines areP(55) andR(53) transitions originating inan A-state level withJ8554. TheJ8554 doublet joined with dashed linecorresponds to transitions from a collisionally populatedb 3Pu-state leveldown to the samev9534 ground state level; the additional doublets label56 and 52 arise from collisional rotational energy transfer in that tripstate.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

eriem
topasal
stedvebb
ca
alf teer
s
-m
artsip
a-re
sed
Eq.
andta-
rstnod to
forthe
ttsately,entialheed
so-ar,
424
m-aly-to-
n-
and
3069J. Chem. Phys., Vol. 113, No. 8, 22 August 2000 Potential of Rb2(X1Sg
1)
s f5F 1
N2M (i 51
N S ycalc~ i !2yobs~ i !
u~ i ! D 2G1/2
, ~1!
where each of theN experimental datayobs( i ) has an uncer-tainty of u( i ), andycalc( i ) is the value of datumi predictedby theM-parameter model being fitted. All parameter unctainties quoted herein are 95% confidence limit uncertaintand the atomic masses used in the combined-isotopoanalysis were taken from the 1993 mass table.15 In order tosimplify the representation of these datasets, to extractmost physically meaningful molecular parameters and prerties, and to allow a search for Born–Oppenheimer bredown effects, all of the analyses reported below were baon combined-isotopomer fits in which all of the data forthree isotopomers were treated simultaneously.
The data used in the analyses reported below consionly of the new A-X emission measurements describabove. The PAS study of Ref. 3 did observe a dozen lelying much closer to dissociation, but they are perturbedtriplet states sharing the same dissociation limit and splithyperfine coupling interactions, and the coupled-channelculations required for their analysis3 are not readily com-bined with the more conventional single-channel data ansis techniques required to accurately describe the rest odata for this state. As a result, the PAS measurements thselves were not used here, although the dissociation endetermined in Ref. 3 was utilized in the~recommended!‘‘direct-potential-fit’’ analysis of Sec. III C.
A. Unconstrained combined-isotopomer Dunham-typeanalysis
The observed transitions for isotopomera of speciesA–B formed from atoms of massMA
a and MBa were ex-
pressed as differences between level energies, written a16
Ea~v,J!5 (~ l ,m!Þ~0,0!
Yl ,m1 S m1
maD m1 l /2
~v11/2! l@J~J11!#m
1 (~ l ,m!>~0,0!
H DMAa
MAa d l ,m
A 1DMB
a
MBa d l ,m
B J3S m1
maD m1 l /2
~v11/2! l@J~J11!#m, ~2!
whereDMAa5MA
a2MA1, anda51 identifies a selected ref
erence species, in this case the most abundant isotopo85,85Rb2. This expression is equivalent to the familiRoss–Eng–Kildal–Bunker–Watson17–19 expansion, excepthat the Born–Oppenheimer and JWKB breakdown termthe second sum are included as additive rather than multcative corrections, and the reference species~isotopomera51, 85,85Rb2! is a real physical molecule. The conventionDunham constants for other (aÞ1) isotopomers are therefore not independent, but may be generated from the expsion
Yl ,ma 5S Yl ,m
l 1DMA
a
MAa d l ,m
A 1DMB
a
MBa d l ,m
B D S m1
maD m1 l /2
. ~3!
Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to A
-s,er
he-
k-edl
ed
lsyyl-
y-hem-gy
er,
inli-
l
s-
Other advantages of this expansion are discuselsewhere.16
Our complete three-isotopomer dataset was fitted to~2! using program ‘‘DSParFit,’’20 which simplifies the re-sulting parameters by applying the sequential roundingrefitting procedure described in Ref. 14. After experimention to determine the optimum lengthl max(m) for the vibra-tional expansion associated with each power~m! of @J(J11)#, this yielded the molecular constants given in the ficolumn of Table I. Within the experimental uncertainties,Born–Oppenhiemer breakdown parameters were requirefit this multi-isotopomer dataset.
For the user’s convenience, the Dunham parametersthe minority isotopomers, as generated by substitutingfitted parameters of column 1 into Eq.~3! and rounded at thefirst digit of the parameter sensitivity,14 are shown in the lastwo columns of Table I. Relatively more significant digiare required to represent these derived constants adequas the compensating changes associated with the sequrounding and refitting procedure do not come into play. Tresults in the last row of this table show that these derivconstants reproduce the data for the individual minority itopomers with no significant loss of precision. In particulthe value ofs f under the85,85Rb2 column is that for the fullcombined-isotopomer fit to 62 Dunham parameters andfluorescence origins, while the values for85,87Rb2 and87,87Rb2 are associated with fits in which the Dunham paraeters were fixed at the values derived from the global ansis and only the fluorescence origin energies for that isopomer were free parameters.
B. Constrained-CDC combined-isotopomer Dunham-type analysis
It has long been known that centrifugal distortion costants~CDCs! of diatomic molecules,
Km~v !5 (l 50
l max~m!
Yl ,m~v11/2! l@J~J11!#m, ~4!
for m>2 @K2(v)52Dv , K3(v)5Hv ,..., etc.#, are not in-dependent physical parameters, but are determined by
FIG. 2. The data field$v,J% for the X 1Sg1 ground state of Rb2.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
ces as
3070 J. Chem. Phys., Vol. 113, No. 8, 22 August 2000 Seto et al.
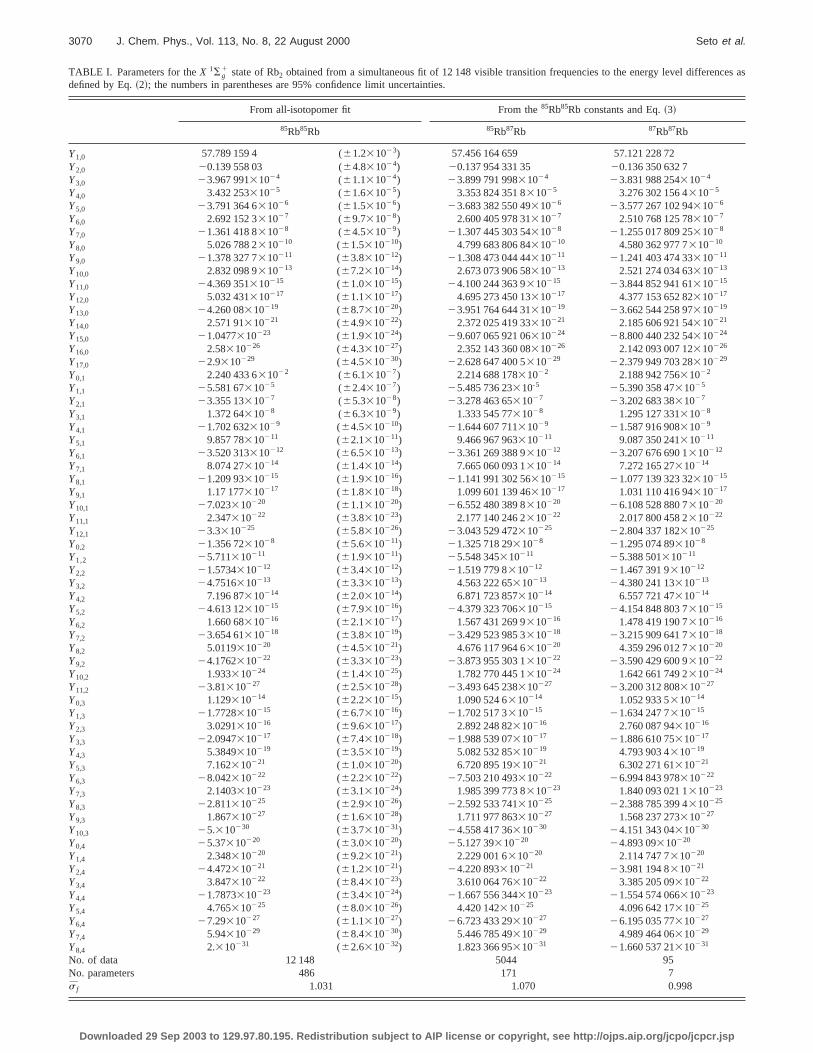
TABLE I. Parameters for theX 1Sg1 state of Rb2 obtained from a simultaneous fit of 12 148 visible transition frequencies to the energy level differen
defined by Eq.~2!; the numbers in parentheses are 95% confidence limit uncertainties.
From all-isotopomer fit From the85Rb85Rb constants and Eq.~3!
85Rb85Rb 85Rb87Rb 87Rb87Rb
Y1,0 57.789 159 4 (61.231023) 57.456 164 659 57.121 228 72Y2,0 20.139 558 03 (64.831024) 20.137 954 331 35 20.136 350 632 7Y3,0 23.967 99131024 (61.131024) 23.899 791 99831024 23.831 988 25431024
Y4,0 3.432 25331025 (61.631025) 3.353 824 351 831025 3.276 302 156 431025
Y5,0 23.791 364 631026 (61.531026) 23.683 382 550 4931026 23.577 267 102 9431026
Y6,0 2.692 152 331027 (69.731028) 2.600 405 978 3131027 2.510 768 125 7831027
Y7,0 21.361 418 831028 (64.531029) 21.307 445 303 5431028 21.255 017 809 2531028
Y8,0 5.026 788 2310210 (61.5310210) 4.799 683 806 84310210 4.580 362 977 7310210
Y9,0 21.378 327 7310211 (63.8310212) 21.308 473 044 44310211 21.241 403 474 33310211
Y10,0 2.832 098 9310213 (67.2310214) 2.673 073 906 58310213 2.521 274 034 63310213
Y11,0 24.369 351310215 (61.0310215) 24.100 244 363 9310215 23.844 852 941 61310215
Y12,0 5.032 431310217 (61.1310217) 4.695 273 450 13310217 4.377 153 652 82310217
Y13,0 24.260 08310219 (68.7310220) 23.951 764 644 31310219 23.662 544 258 97310219
Y14,0 2.571 91310221 (64.9310222) 2.372 025 419 33310221 2.185 606 921 54310221
Y15,0 21.0477310223 (61.9310224) 29.607 065 921 06310224 28.800 440 232 54310224
Y16,0 2.58310226 (64.3310227) 2.352 143 360 08310226 2.142 093 007 12310226
Y17,0 22.9310229 (64.5310230) 22.628 647 400 5310229 22.379 949 703 28310229
Y0,1 2.240 433 631022 (66.131027) 2.214 688 17831022 2.188 942 75631022
Y1,1 25.581 6731025 (62.431027) 25.485 736 23310-5 25.390 358 4731025
Y2,1 23.355 1331027 (65.331028) 23.278 463 6531027 23.202 683 3831027
Y3,1 1.372 6431028 (66.331029) 1.333 545 7731028 1.295 127 33131028
Y4,1 21.702 63231029 (64.5310210) 21.644 607 71131029 21.587 916 90831029
Y5,1 9.857 78310211 (62.1310211) 9.466 967 963310211 9.087 350 241310211
Y6,1 23.520 313310212 (66.5310213) 23.361 269 388 9310212 23.207 676 690 1310212
Y7,1 8.074 27310214 (61.4310214) 7.665 060 093 1310214 7.272 165 27310214
Y8,1 21.209 93310215 (61.9310216) 21.141 991 302 56310215 21.077 139 323 32310215
Y9,1 1.17 177310217 (61.8310218) 1.099 601 139 46310217 1.031 110 416 94310217
Y10,1 27.023310220 (61.1310220) 26.552 480 389 8310220 26.108 528 880 7310220
Y11,1 2.347310222 (63.8310223) 2.177 140 246 2310222 2.017 800 458 2310222
Y12,1 23.3310225 (65.8310226) 23.043 529 472310225 22.804 337 182310225
Y0,2 21.356 7231028 (65.6310211) 21.325 718 2931028 21.295 074 8931028
Y1,2 25.711310211 (61.9310211) 25.548 345310211 25.388 501310211
Y2,2 21.5734310212 (63.4310212) 21.519 779 8310212 21.467 391 9310212
Y3,2 24.7516310213 (63.3310213) 4.563 222 65310213 24.380 241 13310213
Y4,2 7.196 87310214 (62.0310214) 6.871 723 857310214 6.557 721 47310214
Y5,2 24.613 12310215 (67.9310216) 24.379 323 706310215 24.154 848 803 7310215
Y6,2 1.660 68310216 (62.1310217) 1.567 431 269 9310216 1.478 419 190 7310216
Y7,2 23.654 61310218 (63.8310219) 23.429 523 985 3310218 23.215 909 641 7310218
Y8,2 5.0119310220 (64.5310221) 4.676 117 964 6310220 4.359 296 012 7310220
Y9,2 24.1762310222 (63.3310223) 23.873 955 303 1310222 23.590 429 600 9310222
Y10,2 1.933310224 (61.4310225) 1.782 770 445 1310224 1.642 661 749 2310224
Y11,2 23.81310227 (62.5310228) 23.493 645 238310227 23.200 312 808310227
Y0,3 1.129310214 (62.2310215) 1.090 524 6310214 1.052 933 5310214
Y1,3 21.7728310215 (66.7310216) 21.702 517 3310215 21.634 247 7310215
Y2,3 3.0291310216 (69.6310217) 2.892 248 82310216 2.760 087 94310216
Y3,3 22.0947310217 (67.4310218) 21.988 539 07310217 21.886 610 75310217
Y4,3 5.3849310219 (63.5310219) 5.082 532 85310219 4.793 903 4310219
Y5,3 7.162310221 (61.0310220) 6.720 895 19310221 6.302 271 61310221
Y6,3 28.042310222 (62.2310222) 27.503 210 493310222 26.994 843 978310222
Y7,3 2.1403310223 (63.1310224) 1.985 399 773 8310223 1.840 093 021 1310223
Y8,3 22.811310225 (62.9310226) 22.592 533 741310225 22.388 785 399 4310225
Y9,3 1.867310227 (61.6310228) 1.711 977 863310227 1.568 237 273310227
Y10,3 25.310230 (63.7310231) 24.558 417 36310230 24.151 343 04310230
Y0,4 25.37310220 (63.0310220) 25.127 39310220 24.893 09310220
Y1,4 2.348310220 (69.2310221) 2.229 001 6310220 2.114 747 7310220
Y2,4 24.472310221 (61.2310221) 24.220 893310221 23.981 194 8310221
Y3,4 3.847310222 (68.4310223) 3.610 064 76310222 3.385 205 09310222
Y4,4 21.7873310223 (63.4310224) 21.667 556 344310223 21.554 574 066310223
Y5,4 4.765310225 (68.0310226) 4.420 142310225 4.096 642 17310225
Y6,4 27.29310227 (61.1310227) 26.723 433 29310227 26.195 035 77310227
Y7,4 5.94310229 (68.4310230) 5.446 785 49310229 4.989 464 06310229
Y8,4 2.310231 (62.6310232) 1.823 366 95310231 21.660 537 21310231
No. of data 12 148 5044 95No. parameters 486 171 7s f 1.031 1.070 0.998
Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

rga
hricDuelerxg
A.
d.
ses
in--lew
vedofeen
t ofdb-ntseic-
ode
ocu-st
r ofhisid-
-sis.te
C’s
li-rstheto
r--her
atae-
thethe
II.
3071J. Chem. Phys., Vol. 113, No. 8, 22 August 2000 Potential of Rb2(X1Sg
1)
can be calculated from a knowledge of the potential enefunction.21–25 They are therefore implicitly determined byknowledge of the vibrational energies@Gv5K0(v)# and in-ertial rotational constants@Bv5K1(v)# whose use in theRKR inversion procedure26 yields the potential function. Asa result, while unconstrained Dunham-type analyses sucthat described above can usually provide accurate empirepresentations of experimental datasets, the resulting Ctend to be ‘‘effective’’ parameters incorporating effects dto perturbations, interparameter correlation, and the negof undetermined higher-order CDCs. This means that thesulting term-value expressions will not be reliable for etrapolation to higherJ values, and that small compensatinerrors are introduced into the fittedGv and Bv values, andhence into the potential function determined from them.an illustration of this problem for the present system, Figcompares CDCs for 85,85Rb2 generated from theunconstrained-fit empirical parameters listed in Table I~solidcurves! with values calculated numerically27 from the RKRpotential generated28 from the Gv and Bv expansions pre-sented there~dashed curves!, and also with CDCs calculatefrom the final recommended ‘‘MLJ’’ potential of Sec. III C
FIG. 3. A comparison of empirical CDCs from unconstrained fit~solidcurves! with values calculated from the initial RKR potential~dashedcurves! and from the final recommended analytic MLJ potential of Table
Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to A
y
asalCs
cte--
s3
The error in the unconstrained-fit CDCs clearly increawith order ~from Dv to Hv to Lv!; for Lv (m54) it is typi-cally much greater than a factor of 2, and for ordersm>5empirical CDCs could not even be determined.
A widely used approach for addressing this problemvolves calculating CDCs from an initial RKR potential computed fromGv and Bv expressions obtained from an initiaunconstrained fit. These CDCs are then held fixed in a nfit to the original dataset to obtain improvedGv andBv ex-pansions, which, in turn, are used to generate an improRKR potential and then improved CDCs. In a wide varietyapplications this iterative self-consistent procedure has bfound to converge after very few cycles.1,25,29–31However,when applied to Rb2 X 1Sg
1 in Ref. 1, it was found wanting,since the computational tools then available9 could only cal-culate the first four CDCs~Dv , Hv , Lv , andM v!, and thiswas inadequate to describe the existing dataset. Neglecseveral hundred high-J data circumvented this problem anallowed a fairly reliable potential energy curve to be otained, but the resulting molecular band consta$Gv ,Bv ,Dv ,...,M v% could not accurately describe all of thexisting data, and certainly would not yield accurate predtions for even higherJ values.
Since that time a generally accessible computer cable to calculate the first six CDCs@up to K7(v)5Ov# hasbecome available.27 However, even the inclusion of the twadditional calculated CDCs does not explain all of our acrate high-J data for this system. In particular, with the firsix CDCs fixed at values calculated27 from the RKR poten-tial generated28 from the Gv and Bv expansions~$Yl ,m% form50 and 1! in Table I, a fit to the full~12 148 data! three-isotopomer dataset gave a dimensionless standard erros f55.9, and iterating to self-consistency only reduced tvalue to 3.0. Reducing the range of rotational levels consered from the dataset maximum ofJ5243 toJmax5150 ~ex-cluding 1584 data! reducess f to 1.26, and truncating therange further by settingJmax5100 ~excluding an additional3021 data! reduceds f to 1.07, which is the level of agreement attained in the full unconstrained-parameter analyThis indicates that the problem in the fit to the compledataset was due to the lack of even higher-order CD@Km(v) for m.7#.32 While the RKR potential obtained fromthe Jmax5100 reduced dataset would have been fairly reable ~see Sec. IV!, the associated molecular parametewould be unable to represent a substantial fraction ofavailable data, and would be unreliable for extrapolationhigherJ. The computation of yet higher-order CDC’s is cetainly possible~see, e.g., Ref. 33!, but the numerical procedure tends to become increasingly unstable for the higorders, and it is not even clear that the expansion in@J(J11)# truly converges. Thus, it seems clear that the danalysis approach of constraining the CDCs at their ‘‘mchanically consistent’’ calculated values and iteratingparameter-fit analysis to convergence is not adequate forpresent system.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

aysri-
ac
tstia
te–
n–a
pln
vet
ns
te
e
rg
too
iah
-
un
foreder-e-
in-
aseto
sta-lly
setlly
-ent
ver,
ate.con-
ofrela-is-
ed
ial-
e-
-
dd
ff76-
3072 J. Chem. Phys., Vol. 113, No. 8, 22 August 2000 Seto et al.
C. Combined-isotopomer direct-potential-fit „DPF…analysis
A more compact and more physically significant alterntive to the traditional parameter-fit approach to data analis to fit the data directly to eigenvalue differences numecally calculated from the Schro¨dinger equation for some parametrized analytical potential energy function.34–36 For di-atomic molecules, most early applications of this appro~called the ‘‘IPA’’ method! involved fitting to determineanalytic corrections to a pointwise RKR potential,35–37 andthat was the method used in the Rb2 study of Ref. 1. Morerecently, the preferred approach has been to use direct fidetermine parameters characterizing fully analytic potenenergy functions, and~where possible! to determine simulta-neously atomic-mass-dependent radial and centrifugal potial corrections due to the breakdown of the BornOppenheimer approximation.38–44
Since Rb2 has a relatively large reduced mass, BorOppenheimer breakdown effects are expected to be smBoth the parameter-fit analyses described above and exatory direct-potential-fit~DPF! analyses showed that withithe precision of the available data for Rb2 (X 1Sg
1), theireffect is negligible. As a result, the observed pattern of leenergies for all three isotopomers can be described byeigenvalues of the simple radial Schro¨dinger equation,
S 2\2
2ma
d2
dR2 1V~R!1\2J~J11!
2maR2 2Ev,Ja Dcv,J~R!50,
~5!
whereV(R) is the parametrized analytic potential functiowe wish to determine,ma is the reduced mass of the atomforming isotopomera, and$Ev,J
a % are its eigenvalues.In the present work the potential energy was represen
by the ‘‘Modified Lennard-Jones’’~MLJ! function,45
V~R!5De@12~Re /R!ne2b~z!z#2, ~6!
whereDe is the well depth,Re the equilibrium internucleardistance,z[(R2Re)/(R1Re), andb(z) is a simple powerseries inz,
b~z!5(j 50
b j zj , ~7!
whose expansion parameters$b j% are dimensionless. Sincthe groundX 1Sg
1 state of Rb2 dissociates to yield two1Sstate atoms, the asymptotic behavior of its potential enecurve is V(R);D2C6 /R6, so the powern in our modelMLJ potential for this state isn56.46–48Our numerical treat-ment of Eq. ~5! was performed on the interval 2.6>R>42.0 Å with a radial mesh of 0.001 Å. This is sufficientensure that all of the eigenvalues used in the fits were cverged to better than 0.0002 cm21.
Optimization of the parameters of our model potentenergy function started from an initial trial potential in whicDe was taken from Ref. 3, andRe and the first three exponent parameters~b0 , b1 , andb2! were calculated from thelow-order molecular constants~Y1,1, Y1,0, and Y2,0; seeTable I! using Taylor series expansions matched to the Dham relations.49 In most of our fitsDe was held fixed at the
Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to A
-is-
h
tol
n-
ll.or-
lhe
d
y
n-
l
-
value determined in Ref. 3, because it was based on datalevels lying much closer to dissociation than those includin the present analysis. Initial trial values of the higher-ordexponent parametersb j were estimated by a ‘‘bootstrapping’’ method that started by obtaining a good fit to a rduced dataset spanning a modest range ofv, with all b j
values for j >3 set to zero. In stages, the range of datacluded in the fit was then expanded and one additionalb j
parameter~at a time! set free. Our practice was to initiallyfree only this one additional parameter, and to then relethe lower-order coefficients one by one in an attemptminimize the occurrence of large changes that might debilize the fit; however, other strategies may work equawell.
As the highest vibrational levels included in our dataare bound by only a few wave numbers, it was initiahoped that the value ofDe could be treated as a free parameter and the fit used to determine an improved independestimate of the ground-state dissociation energy. Howethe values obtained from fits in whichDe was also set freediffered from one another and from the current best estim3
of 3993.53~60.06! cm21 by a number of wave numbersThe magnitude of these discrepancies was somewhat discerting. Further inspection showed that the associatedb(z)functions tended to grow very quickly beyond the rangethe data, causing the potential to approach an asymptotetively abruptly, which led to an underestimation of the dsociation energy. This rapid growth inb(z) beyond therange of R spanned by the data is shown by the dashcurves~labeled ‘‘unconstrained’’! in Fig. 4. One implicationof this behavior is that although the resulting MLJ potentwill be ‘‘well behaved,’’ smoothly approaching its asymptote with the theoretically predictedD2C6 /R6 behavior, thevalue of theC6 coefficient, defined within the MLJ form bythe expression
C652De~Re!6e2b`, ~8!
where b`[b(z51)5b@z(R5`)#, is unconstrained, andwhile finite, it is ~in this case! unrealistically large.
Our initial attempt to address this problem involved dfining the highest-order coefficient (b j5b last) in the powerseries expansion forb(z) in terms of both the other coefficients and a fixed limiting value of
b`5 ln@2De~Re!6/C6#, ~9!
determined by the theoretically known50 C6 constant for thisspecies:
b last5b`2 (j 50
last21
b j5 ln@2De~Re!6/C6#2 (
j 50
last21
b j . ~10!
This constraint did successfully forceb(z) to reach the the-oretical limit of b`'0.676. However, as sometimes founby Hajigeorgiou and Le Roy,45 beyond the region governeby the data, the resultingb(z) function had an implausiblylarge maximum, following the ‘‘unconstrained’’ curves oscale in Fig. 4, and only dropping back toward the 0.6limit at very largeR, and the fit still underestimated the dissociation energy by a number of wave numbers.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
oftta
eo
at
rsu-frio
mfo
hluulg
apes in
gpo-
ain,sing14.
LJat
fit to
tial
e’’uc-ofldus
the
onfi-the
3073J. Chem. Phys., Vol. 113, No. 8, 22 August 2000 Potential of Rb2(X1Sg
1)
Our final solution was simply to follow the approachRef. 45 and introduce a switching function parametrizedmake b(z) beyond the range of the experimental dasmoothly approach the asymptotic limit defined by the thretical C6 value. This switching function has the form45
bS~z!5 f S~R!S (j 50
last
b j zj2b`D 1b` , ~11!
f S~R!51/@eaS~R2RS!11#, ~12!
where the parametersaS and RS are chosen to ensure thb(z) ‘‘looks’’ smooth.
To determine optimum switching-function parametewe performed global fits to the data for a range of ‘‘plasible’’ trial sets of$aS ,RS% values. In all cases the quality ofit to the data was essentially the same, and the criteapplied was a desire to obtain abS(R) function that wouldapproach the theoretically known limiting value (b`
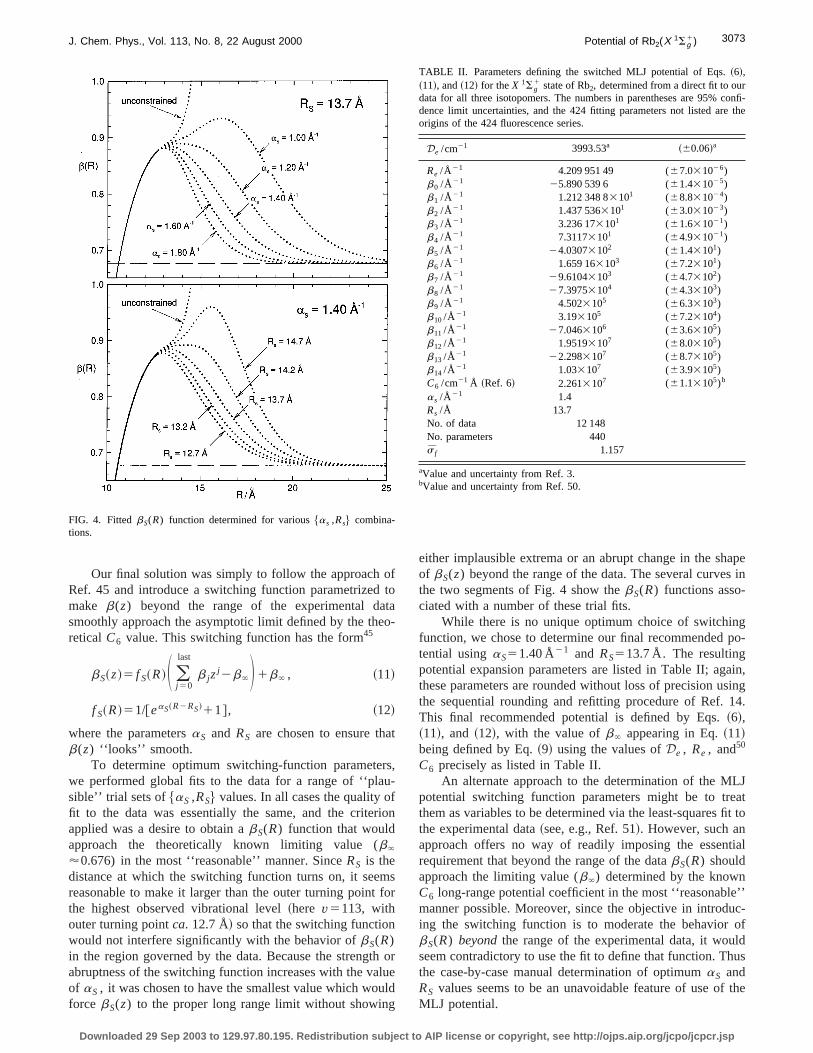
'0.676) in the most ‘‘reasonable’’ manner. SinceRS is thedistance at which the switching function turns on, it seereasonable to make it larger than the outer turning pointthe highest observed vibrational level~here v5113, withouter turning pointca. 12.7 Å! so that the switching functionwould not interfere significantly with the behavior ofbS(R)in the region governed by the data. Because the strengtabruptness of the switching function increases with the vaof aS , it was chosen to have the smallest value which woforce bS(z) to the proper long range limit without showin
FIG. 4. FittedbS(R) function determined for various$as ,Rs% combina-tions.
Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to A
o
-
,
n
sr
ored
either implausible extrema or an abrupt change in the shof bS(z) beyond the range of the data. The several curvethe two segments of Fig. 4 show thebS(R) functions asso-ciated with a number of these trial fits.
While there is no unique optimum choice of switchinfunction, we chose to determine our final recommendedtential usingaS51.40 Å21 and RS513.7 Å. The resultingpotential expansion parameters are listed in Table II; agthese parameters are rounded without loss of precision uthe sequential rounding and refitting procedure of Ref.This final recommended potential is defined by Eqs.~6!,~11!, and ~12!, with the value ofb` appearing in Eq.~11!being defined by Eq.~9! using the values ofDe , Re , and50
C6 precisely as listed in Table II.An alternate approach to the determination of the M
potential switching function parameters might be to trethem as variables to be determined via the least-squaresthe experimental data~see, e.g., Ref. 51!. However, such anapproach offers no way of readily imposing the essenrequirement that beyond the range of the databS(R) shouldapproach the limiting value (b`) determined by the knownC6 long-range potential coefficient in the most ‘‘reasonablmanner possible. Moreover, since the objective in introding the switching function is to moderate the behaviorbS(R) beyondthe range of the experimental data, it wouseem contradictory to use the fit to define that function. Ththe case-by-case manual determination of optimumaS andRS values seems to be an unavoidable feature of use ofMLJ potential.
TABLE II. Parameters defining the switched MLJ potential of Eqs.~6!,~11!, and~12! for theX 1Sg
1 state of Rb2, determined from a direct fit to ourdata for all three isotopomers. The numbers in parentheses are 95% cdence limit uncertainties, and the 424 fitting parameters not listed areorigins of the 424 fluorescence series.
De /cm21 3993.53a ~60.06!a
Re /Å 21 4.209 951 49 (67.031026)b0 /Å 21 25.890 539 6 (61.431025)b1 /Å 21 1.212 348 83101 (68.831024)b2 /Å 21 1.437 5363101 (63.031023)b3 /Å 21 3.236 173101 (61.631021)b4 /Å 21 7.31173101 (64.931021)b5 /Å 21 24.03073102 (61.43101)b6 /Å 21 1.659 163103 (67.23101)b7 /Å 21 29.61043103 (64.73102)b8 /Å 21 27.39753104 (64.33103)b9 /Å 21 4.5023105 (66.33103)b10 /Å 21 3.193105 (67.23104)b11 /Å 21 27.0463106 (63.63105)b12 /Å 21 1.95193107 (68.03105)b13 /Å 21 22.2983107 (68.73105)b14 /Å 21 1.033107 (63.93105)C6 /cm21 Å ~Ref. 6! 2.2613107 (61.13105)b
as /Å 21 1.4Rs /Å 13.7No. of data 12 148No. parameters 440s f 1.157
aValue and uncertainty from Ref. 3.bValue and uncertainty from Ref. 50.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

ete
lyeltin
tod
ch
he
e
eltiapabct
a-onllyl
r-
ncalvethfiis
ronlla
DPl o
-
hePFlar,
sis148pa-
ultsls orntsof
b-a
e IIsy-atecesle-n
c-tialmrst-ncan-or-
nts
li-
ose
al-estnsen
tersve
e-
-
ec.-
e ofthe-
thew
i--sisur
3074 J. Chem. Phys., Vol. 113, No. 8, 22 August 2000 Seto et al.
D. Fitting to determine the dissociation energy
A slightly disappointing feature of the DPF analysis dscribed above is the fact that the fits seem unable to demine a reliable value for the dissociation energyDe . Thisseems to be a general problem associated with most anamodel potentials in direct fits to data for many-levsystems.40,41,44,51 For ground-state Rb2 the independencoupled-channel analysis of the PAS data for levels lyextremely close to dissociation yielded a reliableDe that washeld fixed in our analysis,3 but that is a special situation thadoes not usually arise. Moreover, unconstrained-constrained-CDC Dunham-expansion fits such as thosescribed in Secs. III A and III B are of no help, since suempirical polynomials in (v11/2) are generally unreliablefor vibrational extrapolation. For example, although thighest vibrational level used in our analysis (v5113) isonly bound by 7 cm21, a vibrational extrapolation using thDunham parameters of Table I predictsvD'118.1, andhence misses almost half of the (9210) levels lying abovev5113.3
The ideal solution to this problem would be the devopment and application of a better analytic model potenenergy function that is more robust and stable in the extralation region. In the meantime, however, we note thatalternate type of parameter-fit analysis can provide reliavalues ofDe that can then be held fixed in the global direpotential fit to the data.
It has long been known that particularly reliable vibrtional energy extrapolations may be obtained from fitsobserved level energies to ‘‘near-dissociation expansio~NDE! expressions that incorporate the theoreticaknown52,53 limiting near-dissociation behavior of vibrationalevel spacings.54,55 We therefore fitted the vibrational enegies of the 20 highest levels in the present dataset (v5942113) to such expansions,56 while fixing the values of thetwo longest-range inverse-power potential coefficients~C6
andC8! at their known theoretical values.50,57This approachpredicts De53993.47(60.18) cm21 and vD5123.01(60.19), values remarkably close to the PAS results3 De
53993.53(60.06) cm21 and vD5122.994(60.012), re-spectively. The PAS results should be more reliable, sithe binding energies of the highest levels used in that ansis are orders of magnitude smaller than those for the leused in our NDE fits. However, the results obtained fromlatter are clearly very good, and provide confidence thatto NDE functions should be able to provide the reliable dsociation energies that the DPF method is not~yet! able toprovide independently.
IV. DISCUSSION AND CONCLUSIONS
In the present paper we present new high-resolutionsolved fluorescence data characterizing the ground electrstate of Rb2 over essentially the whole potential energy weThese data are analyzed using three distinctly differentproaches. Of these, the unconstrained Dunham fit andanalyses are of equivalent quality, both representing althe available data within their uncertainties~of ca. 60.001cm21!, while the ‘‘mechanically consistent’’ constrained
Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to A
-r-
tic
g
re-
-l
o-nle
f’’
ey-lsets-
e-ic
.p-Ff
CDC Dunham-type fits cannot properly account for thigh-J data. Of the two successful fitting schemes, the Danalysis is preferred for a number of reasons. In particuwe have the following.
~i! The 16 fitted potential parameters of the DPF analyprovide a much more compact representation of the 12experimental data than do the 62 independent Dunhamrameters of Table I. It might be argued that the DPF resare less convenient to use for predicting unobserved levetransitions. However, because of the numbers of coefficieand significant digits involved, the Dunham parametersTable I would only practically be utilized through their sustitution into some computer program. If one is to usecomputer program, the MLJ potential parameters of Tablmay be equally readily input into one of the available eato-use Schro¨dinger-solver programs that can readily generarbitrary numbers of eigenvalues or eigenvalue differen~together with any desired expectation values or matrix ements! for any specified analytic potential energy functio~see, e.g., Ref. 27!.
~ii ! The resulting DPF-determined potential energy funtion is quantum mechanically accurate, while a potenfunction determined by the RKR method from the Dunhaconstants of Table I would only be as accurate as the fiorder JWKB approximation on which the RKR inversioprocedure is based. Moreover, the RKR-based approachnot take full account of Born–Oppenheimer breakdown crections. While these are not serious drawbacks for Rb2 ~seebelow!, they could introduce substantial errors in treatmeof small-reduced-mass species.
~iii ! The recommended MLJ potential should yield reable predictions for arbitrarily largeJ, while longJ extrapo-lations with unconstrained molecular constants such as thof Table I are quite unreliable.
~iv! The recommended MLJ potential should yield reistic predictions for most vibrational levels above the highone used in our analysis, while vibrational extrapolatiowith unconstrained high-order Dunham polynomials oftbehave very badly~see, e.g., Fig. 4 of Ref. 31!. In the presentcase vibrational extrapolation with the Dunham parameof Table I suggests that only five vibrational levels lie abothe highest level used in our analysis (v5113), while ourMLJ potential supports all of the nine additional levels prdicted by the PAS analysis of Ref. 3, and agrees~within theuncertainties! with their prediction for the effective vibrational index at dissociationvD .
The constrained-CDC Dunham-type analysis of SIII B is certainly unsatisfactory, in that it could not quantitatively account for the experimental data at highJ. On theother hand, it seems appropriate to ask whether that typapproach could yield a reliable potential energy curve ifanalysis simply neglected any high-J data that cannot be accounted for by the limited number~six! of CDCs one canreadily calculate. As a test of this question we appliediterative self-consistent procedure of Sec. III B to our nedataset for Rb2, while omitting the 4605 transitions assocated withJ9.100.32 In the plots of Fig. 3, the CDCs calculated from the RKR curve generated from this analywould be essentially identical to those calculated from o
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

oca
Puais
teieen–reba
uison
ateninevierrer-
elfaseri
as
eluar
A
. J
.
Le
ni,
ng
on
byme-
al for://
l.
o
codeite
erec-chmay
l.
osc.
le-
ia,
Le
l-iver-
hys.
3075J. Chem. Phys., Vol. 113, No. 8, 22 August 2000 Potential of Rb2(X1Sg
1)
final recommended MLJ potential function. However, a fitour full 3-isotopomer 12 148-line dataset to eigenvaluesculated from that RKR curve yieldeds f51.235, which isdistinctly worse than the agreement obtained from the Dor unconstrained parameter fit. Moreover, even had the qity of the predictions been essentially equivalent, a pointwRKR potential whose turning points must be interpolaover and extrapolated beyond is a distinctly less convenway to describe the system than our analytical MLJ pottial, which is valid at all distances. In addition, while BornOppenheimer breakdown corrections are expected to betively small for a large-reduced-mass system such as R2,this will not be true, in general, and the conventionconstrained-CDC parameter-fit approach cannot distingbetween adiabatic and nonadiabatic corrections to rotatienergies, while the DPF method can.16
Our final comment draws attention to two slightly unsisfactory features of the present direct fits to the MLJ pottial. These are~1! the need to include a switching functionthe exponent expansion to ensure that the MLJ potentialponent function does not have spurious unphysical behain the interval between the region governed by the expmental data and the asymptotic value defined by the theocally knownC6 constant, and~2! the associated need to peform a manual search to determineaS and RS values thatensure the ‘‘most reasonable’’ behavior there. Unfortunatwe have not devised a solution to these deficiencies. Thethat the MLJ potential form incorporates the correct inverpower asymptotic behavior does make it much more apppriate than most of the other potential models introducedrecent years.40,41 However, the somewhat unsatisfactory nture of this manual search requirement should spur effortdevelop even more effective potential function models.
ACKNOWLEDGMENTS
We are pleased to thank Professor P. F. Bernath for hful comments on the manuscript. This research was sported by the Natural Sciences and Engineering ReseCouncil of Canada.
1C. Amiot, J. Chem. Phys.93, 8591~1990!.2M. Anderson, J. R. Ensher, M. R. Matthews, C. E. Wieman, and E.Cornell, Science269, 198 ~1995!.
3C. C. Tsai, R. S. Freeland, J. M. Vogels, H. M. J. M. Boesten, BVerhaar, and D. J. Heinzen, Phys. Rev. Lett.79, 1245~1997!.
4J. M. Vogels, C. C. Tsai, R. S. Freeland, S. J. J. M. F. Kokkelmans, BVerhaar, and D. Heinzen, Phys. Rev. A56, R1067~1997!.
5H. M. J. M. Boesten, A. J. Moerdijk, and B. J. Verhaar, Phys. Rev. A54,R29 ~1996!.
6J. P. Burke, Jr., J. L. Bohn, B. D. Esry, and C. H. Greene, Phys. Rev.80, 2097~1998!.
7J. P. Burke, Jr. and J. L. Bohn, Phys. Rev. A59, 1303~1999!.8C. Gabbanini, A. Fioretti, A. Lucchesini, S. Gozzini, and M. MazzoPhys. Rev. Lett.84, 2814~2000!.
9~a! J. M. Hutson, J. Phys. B14, 851 ~1981!; ~b! J. M. Hutson, QCPEBulletin, 2, No. 2, Program #435, Quantum Chemistry Program ExchaIndiana University, Bloomington, Indiana.
10A. N. Nesmeyanov,Vapor Pressure of the Chemical Elements~Elsevier,New York, 1963!.
11C. E. Fellows, C. Amiot, and J. Verge`s, J. Chem. Phys.93, 6281~1990!.12C. E. Fellows, R. F. Gutterres, A. P. C. Campos, J. Verge`s, and C. Amiot,
J. Mol. Spectrosc.197, 19 ~1999!.13See EPAPS Document No. E-JCPSA6-113-019032 for ASCII files c
Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to A
fl-
Fl-
ednt-
la-
lhal
--
x-ori-ti-
y,ct-
o-n-to
p-p-ch
.
.
J.
tt.
e,
-
taining listings of the data used and of all the fitted parameters yieldedthe present work. This document may be retrieved via the EPAPS hopage~http://www.aip.org/pubservs/epaps.html! or from ftp.aip.org in thedirectory /epaps/. See the EPAPS homepage for more information.
14R. J. Le Roy, J. Mol. Spectrosc.191, 223 ~1998!.15G. Audi and A. H. Wapstra, Nucl. Phys. A565, 1 ~1993!.16R. J. Le Roy, J. Mol. Spectrosc.194, 189 ~1999!.17A. H. M. Ross, R. S. Eng, and H. Kildal, Opt. Commun.12, 433 ~1974!.18P. R. Bunker, J. Mol. Spectrosc.68, 367 ~1977!.19J. K. G. Watson, J. Mol. Spectrosc.80, 411 ~1980!.20R. J. Le Roy,DSParFit 1.0. A Computer Program for Fitting Multi-
Isotopomer Diatomic Molecule Spectra, University of Waterloo ChemicalPhysics Research Report CP-646, 2000. The source code and manuthis program may be obtained from the www site httptheochem.uwaterloo.ca/;leroy.
21J. L. Dunham, Phys. Rev.41, 721 ~1932!.22S. Z. Moody and C. L. Beckel, Int. J. Quantum Chem., Symp.3, 469
~1970!.23D. L. Albritton, W. J. Harrop, A. L. Schmeltekopf, and R. N. Zare, J. Mo
Spectrosc.46, 25 ~1973!.24J. Tellinghuisen, Chem. Phys. Lett.18, 544 ~1973!.25J. D. Brown, G. Burns, and R. J. Le Roy, Can. J. Phys.51, 1664~1973!.26~a! R. Rydberg, Z. Phys.73, 376 ~1931!; ~b! O. Klein, ibid. 76, 226
~1932!; ~c! R. Rydberg,ibid. 80, 514 ~1933!; ~d! A. L. G. Rees, Proc.Phys. Soc. London59, 998 ~1947!.
27R. J. Le Roy,LEVEL 7.0. A Computer Program Solving the Radial Schr¨-dinger Equation for Bound and Quasibound Levels, University of Water-loo Chemical Physics Research Report CP-642, 2000. The sourceand manual for this program may be obtained from the www shttp://theochem.uwaterloo.ca/;leroy.
28R. J. Le Roy,RKR1. A Computer Program Implementing the First-OrdRKR Method for Determining Diatom Potential Energy Curves from Sptroscopic Constants, University of Waterloo Chemical Physics ResearReport CP-425, 1992. The source code and manual for this programbe obtained from the www site http://theochem.uwaterloo.ca/;leroy.
29D. L. Albritton, W. J. Harrop, A. L. Schmeltekopf, and R. N. Zare, J. MoSpectrosc.46, 89 ~1973!.
30J. M. Hutson, S. Gerstenkorn, P. Luc, and J. Sinzelle, J. Mol. Spectr96, 266 ~1982!.
31D. R. T. Appadoo, R. J. Le Roy, P. F. Bernathet al., J. Chem. Phys.104,903 ~1996!.
32A more sophisticated truncation procedure that cuts off theJ, range atdifferent points for different vibrational levels could also have been impmented here~Ref. 1!, but it would not have changed this conclusion.
33C. Boone, ‘‘Magnetic rotation in theA 3P1u2X 1Sg1 system of79Br2,’’
Ph.D. thesis, Department of Physics, University of British Columb1999.
34R. J. Le Roy, Can. J. Phys.52, 246 ~1974!.35W. M. Kosman and J. Hinze, J. Mol. Spectrosc.56, 93 ~1975!.36C. R. Vidal and H. Scheingraber, J. Mol. Spectrosc.65, 46 ~1977!.37J. A. Coxon, J. Mol. Spectrosc.117, 361 ~1986!.38M. Gruebele, Mol. Phys.69, 475 ~1990!.39R. Bruhl, J. Kapetanakis, and D. Zimmermann, J. Chem. Phys.94, 5865
~1991!.40J. A. Coxon and P. G. Hajigeorgiou, J. Mol. Spectrosc.150, 1 ~1991!.41H. G. Hedderich, M. Dulick, and P. F. Bernath, J. Chem. Phys.99, 8363
~1993!.42J. Tellinghuisen, J. Mol. Spectrosc.173, 223 ~1995!.43A. Surkus, Chem. Phys. Lett.279, 236 ~1997!.44J. Y. Seto, Z. Morbi, F. Charron, S. K. Lee, P. F. Bernath, and R. J.
Roy, J. Chem. Phys.110, 11 756~1999!.45P. G. Hajigeorgiou and R. J. Le Roy, J. Chem. Phys.112, 3949~2000!.46J. O. Hirschfelder, C. F. Curtiss, and R. B. Bird,Molecular Theory of
Gases and Liquids~Wiley, New York, 1964!.47J. O. Hirschfelder and W. J. Meath, inIntermolecular Forces, Vol. 12 of
Advances Chemical Physics, edited by J. O. Hirschfelder~Interscience,New York, 1967!, pp. 3–106.
48R. J. Le Roy and R. B. Bernstein, J. Chem. Phys.52, 3869~1970!.49J. Y. Seto, Direct fitting of analytic potential functions to diatomic mo
ecule spectroscopic data, M.Sc. thesis, Department of Chemistry, Unsity of Waterloo, 2000.
50A. Derevianko, W. R. Johnson, M. S. Safronova, and J. F. Babb, PRev. Lett.82, 3589~1999!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

-r-g a
3076 J. Chem. Phys., Vol. 113, No. 8, 22 August 2000 Seto et al.
51J. A. Coxon and P. G. Hajigeorgiou, J. Mol. Spectrosc.193, 306 ~1999!.52~a! R. J. Le Roy and R. B. Bernstein, Chem. Phys. Lett.5, 42 ~1970!; ~b!
R. J. Le Roy and R. B. Bernstein, J. Chem. Phys.52, 3869~1970!.53R. J. Le Roy, J. Chem. Phys.73, 6003~1980!.54R. J. Le Roy and W.-H. Lam, Chem. Phys. Lett.71, 544 ~1980!.55R. J. Le Roy, J. Chem. Phys.101, 10 217~1994!.
Downloaded 29 Sep 2003 to 129.97.80.195. Redistribution subject to A
56R. J. Le Roy,GvNDE. A Computer Program for Performing ‘‘NearDissociation-Expansion’’ Fits to Diatomic Molecule Vibrational Enegies. The source code for this program may be obtained by sendinrequest by e-mail to the author at ‘‘[email protected].’’
57M. Marinescu, H. R. Sadeghpour, and A. Dalgarno, Phys. Rev. A49, 982~1994!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp




















