
Determinants for transformation induced by the Axl receptor ...
11
Determinants for transformation induced by the Axl receptor tyrosine kinase Andreas Burchert 1 , Eyal C Attar 1 , Patrick McCloskey 3 , Yih-Woei C Fridell 1 and Edison T Liu 1,2,3,4 Departments of 1 Medicine, 2 Biochemistry and Biophysics, 3 Curriculum in Genetics and Molecular Biology, Lineberger Comprehensive Cancer Center, the University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, 27599-7295, USA The Axl receptor tyrosine kinase is a transforming oncogene in NIH3T3 cells. In order to define structural requirements of the Axl receptor necessary for transfor- mation we passaged recombinant retroviruses carrying the axl cDNA in NIH3T3 cells, generating randomly mutated axl variants. Using this strategy, we have isolated three axl viral strains (1B1, SV8, and FFa4) that show augmented 3T3 cell transforming capacity associated with elevated p140 Axl . Upon sequencing, the 1B1 and SV8 proviruses possessed only silent mutations, making p140 Axl overexpression the most likely explana- tion for their increased transformation activity. However, the characterization of FFa4 revealed a deletion of sequences encoding the carboxy-terminal 45 amino acids leading to the generation of a chimeric transcript comprised of a truncated Axl receptor with a segment of the 3’ UTR region. Mutational analysis revealed that the transforming activity of FFa4 was specific to the formation of the chimeric receptor rather than to the carboxyl-terminal truncation. Intriguingly, none of the viral strains were able to transform the murine cell lines NR-6 and 32D despite equivalent expression of surface p140 Axl protein. Further analysis showed that Axl’s transforming potential is dependent on the host cell type, the presence of a putative pp190 as a facilitator for transformation, and the level of p140 Axl expression. Taken together, these results underscore the complexity of Axl biology which is dependent on receptor stoichiometry and the cellular background. Keywords: receptor tyrosine kinase; retroviruses, mutations; transformations; Axl Introduction The Axl/Ufo receptor is the founding member of a family of receptor tyrosine kinases (RTKs) character- ized by their unique extracellular composition consist- ing of immunglobulin-like and fibronectin type Ill-like domains. This domain architecture is also found in adhesion molecules of the cadherin- and immunoglo- bulin superfamily and in receptor protein tyrosine phosphatases such as LAR or PTPm and PTPk (Edelmann and Crossin, 1991; Fischer et al., 1991; Charbonneau and Tonks, 1992). In addition, unique sequences within the kinase domain distinguish axl and its related kinases (sky and mer) from other type I RTKs. These structural findings suggest that Axl family members may mediate both cell adhesion and intracellular signaling (Bellosta et al., 1995; Pulido et al., 1992). Axl’s role in normal and malignant cell biology has not been completely elucidated but appears to be complex. We and others have found that Axl is a transforming gene that is overexpressed in some metastatic epithelial cancers (Quong et al., 1994; Craven et al., 1995) and is overexpressed in approxi- mately 25% of primary breast cancers (Attar and Liu, manuscript in preparation). All members of the Axl/ Ufo family can transform NIH3T3 cells. sky and mer/ eyk are rendered transforming by 5’ truncation or substitution leading to an RTK with an altered extracellular domain (Taylor et al., 1995; Jia et al., 1992). When transfected, axl transforms by over- expression with no activating mutations reported. The transforming activity, however, is weak in that transfection followed by short term passaging of 3T3 cells was necessary to uncover the transforming activity. Using a chimeric receptor construct contain- ing the EGFR extracellular and transmembrane domains and Axl kinase domain (called EAK for EGFR- Axl- Kinase) introduced into the murine mye- loid cell line 32D, we found that prolonged stimulation of the kinase by the foster ligand, EGF, resulted in the generation of factor independent 32D sublines able to grow in the absence of IL-3. Factor independent growth is a hallmark of transformation in this cell system and appeared to be correlated with dramatic overexpression of the EAK chimeric receptor leading to ligand independent signaling (McCloskey et al., 1994). Thus, in several systems, Axl is capable of cellular transformation through constitutive activation of its kinase function achieved through receptor overexpression. The same results have been seen in other type I RTKs: HER-2/neu is primarily transform- ing through amplification and overexpression, though amino acid substitutions in the transmembrane domain will activate the RTK by enhancing receptor dimeriza- tion (Bargmann et al., 1986; Slamon et al., 1989). Surprisingly, however, the Axl ligand, GAS-6, is only a weak mitogen in a restricted range of tissue such as 3T3, Schwann, and vascular smooth muscle cells (Goruppi et al., 1996; Gasic et al., 1992; Li et al., 1996) and appears to play a greater role in modulating cellular responses to other factors such as thrombin and angiotensin II (Nakano et al., 1995). That a ligand Correspondence: ET Liu 4 Current address: Division of Clinical Sciences, National Cancer Institute, National Institutes of Health, 31/3A11, 31 Center Drive, MSC 2440, Bethesda, MD 20892 – 2440 The first two authors contributed equally to this work Received 16 June 1997; revised 23 January 1998; accepted 26 January 1998 Oncogene (1998) 16, 3177 – 3187 1998 Stockton Press All rights reserved 0950 – 9232/98 $12.00 http://www.stockton-press.co.uk/onc
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of Determinants for transformation induced by the Axl receptor ...

Determinants for transformation induced by the Axl receptor tyrosinekinase
Andreas Burchert1, Eyal C Attar1, Patrick McCloskey3, Yih-Woei C Fridell1 andEdison T Liu1,2,3,4
Departments of 1Medicine, 2Biochemistry and Biophysics, 3Curriculum in Genetics and Molecular Biology, LinebergerComprehensive Cancer Center, the University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, 27599-7295, USA
The Axl receptor tyrosine kinase is a transformingoncogene in NIH3T3 cells. In order to de®ne structuralrequirements of the Axl receptor necessary for transfor-mation we passaged recombinant retroviruses carryingthe axl cDNA in NIH3T3 cells, generating randomlymutated axl variants. Using this strategy, we haveisolated three axl viral strains (1B1, SV8, and FFa4)that show augmented 3T3 cell transforming capacityassociated with elevated p140Axl. Upon sequencing, the1B1 and SV8 proviruses possessed only silent mutations,making p140Axl overexpression the most likely explana-tion for their increased transformation activity. However,the characterization of FFa4 revealed a deletion ofsequences encoding the carboxy-terminal 45 amino acidsleading to the generation of a chimeric transcriptcomprised of a truncated Axl receptor with a segmentof the 3' UTR region. Mutational analysis revealed thatthe transforming activity of FFa4 was speci®c to theformation of the chimeric receptor rather than to thecarboxyl-terminal truncation. Intriguingly, none of theviral strains were able to transform the murine cell linesNR-6 and 32D despite equivalent expression of surfacep140Axl protein. Further analysis showed that Axl'stransforming potential is dependent on the host celltype, the presence of a putative pp190 as a facilitator fortransformation, and the level of p140Axl expression.Taken together, these results underscore the complexityof Axl biology which is dependent on receptorstoichiometry and the cellular background.
Keywords: receptor tyrosine kinase; retroviruses,mutations; transformations; Axl
Introduction
The Axl/Ufo receptor is the founding member of afamily of receptor tyrosine kinases (RTKs) character-ized by their unique extracellular composition consist-ing of immunglobulin-like and ®bronectin type Ill-likedomains. This domain architecture is also found inadhesion molecules of the cadherin- and immunoglo-bulin superfamily and in receptor protein tyrosinephosphatases such as LAR or PTPm and PTPk
(Edelmann and Crossin, 1991; Fischer et al., 1991;Charbonneau and Tonks, 1992). In addition, uniquesequences within the kinase domain distinguish axl andits related kinases (sky and mer) from other type IRTKs. These structural ®ndings suggest that Axlfamily members may mediate both cell adhesion andintracellular signaling (Bellosta et al., 1995; Pulido etal., 1992).
Axl's role in normal and malignant cell biology hasnot been completely elucidated but appears to becomplex. We and others have found that Axl is atransforming gene that is overexpressed in somemetastatic epithelial cancers (Quong et al., 1994;Craven et al., 1995) and is overexpressed in approxi-mately 25% of primary breast cancers (Attar and Liu,manuscript in preparation). All members of the Axl/Ufo family can transform NIH3T3 cells. sky and mer/eyk are rendered transforming by 5' truncation orsubstitution leading to an RTK with an alteredextracellular domain (Taylor et al., 1995; Jia et al.,1992). When transfected, axl transforms by over-expression with no activating mutations reported. Thetransforming activity, however, is weak in thattransfection followed by short term passaging of 3T3cells was necessary to uncover the transformingactivity. Using a chimeric receptor construct contain-ing the EGFR extracellular and transmembranedomains and Axl kinase domain (called EAK forEGFR-Axl-Kinase) introduced into the murine mye-loid cell line 32D, we found that prolonged stimulationof the kinase by the foster ligand, EGF, resulted in thegeneration of factor independent 32D sublines able togrow in the absence of IL-3. Factor independentgrowth is a hallmark of transformation in this cellsystem and appeared to be correlated with dramaticoverexpression of the EAK chimeric receptor leadingto ligand independent signaling (McCloskey et al.,1994). Thus, in several systems, Axl is capable ofcellular transformation through constitutive activationof its kinase function achieved through receptoroverexpression. The same results have been seen inother type I RTKs: HER-2/neu is primarily transform-ing through ampli®cation and overexpression, thoughamino acid substitutions in the transmembrane domainwill activate the RTK by enhancing receptor dimeriza-tion (Bargmann et al., 1986; Slamon et al., 1989).
Surprisingly, however, the Axl ligand, GAS-6, isonly a weak mitogen in a restricted range of tissue suchas 3T3, Schwann, and vascular smooth muscle cells(Goruppi et al., 1996; Gasic et al., 1992; Li et al., 1996)and appears to play a greater role in modulatingcellular responses to other factors such as thrombinand angiotensin II (Nakano et al., 1995). That a ligand
Correspondence: ET Liu4Current address: Division of Clinical Sciences, National CancerInstitute, National Institutes of Health, 31/3A11, 31 Center Drive,MSC 2440, Bethesda, MD 20892 ± 2440The ®rst two authors contributed equally to this workReceived 16 June 1997; revised 23 January 1998; accepted 26 January1998
Oncogene (1998) 16, 3177 ± 3187 1998 Stockton Press All rights reserved 0950 ± 9232/98 $12.00
http://www.stockton-press.co.uk/onc

shows little mitogenic activity when interacting with aRTK with transforming function has been seen in theEph receptor tyrosine kinase family (Brambilla et al.,1995). Inducing mitogenesis is therefore only a limitedaspect of RTK signaling as Eph family membersdemonstrate the ability to mediate fasciculation andcollapse of axons of the retinal ganglion and therebyregulate targeting of axons.
Developmentally, Axl is temporally expressed inembryonic mesenchymal cells immediately prior toorganogenesis (Sadler and Liu, manuscript in prepara-tion). Recently, we have determined that Axl cane�ectively mediate cell-cell adhesion in a GAS-6dependent manner (McCloskey and Liu, in press).Taken together, these results raise the possibility thatAxl is potentially involved in cell recruitment duringembryogenesis through its adhesion and signalingfunctions.
Given the complexity of Axl biology, we sought toidentify the structural requirements for Axl kinaseactivation using 3T3 transformation as the biologicalassay. Our screening strategy employed the error-pronenature of the reverse transcriptase during the retrovirallife cycle to generate functionally transforming mutantsof axl; the in vivo forward point mutation rate of theretroviral reverse transcriptase varies between 1075 and1073 per base per replication cycle, whereas insertionsand deletions are less common (161077/b/replicationcycle) (Monk et al., 1992; Dougherty and Temin,1988). A similar approach of randomly mutatinggenetic sequences through retroviral replication wasused for the derivation of an oncogenic variant of theerythropoietin (EPO) receptor (Yoshimura et al., 1990).After selection for retroviral strains containing viralparticles with an augmented ability to transform 3T3cells, we analysed the structural correlates to theheightened transforming action. We found thatthough overexpression remained the most potentmeans to render Axl transforming, C-terminal trunca-tion resulting in the formation of a chimeric receptorcomprised of appended sequences from the 3'untranslated region (UTR) could also activate theAxl kinase. Furthermore, we found that NR6 and 32Dcells were resistant to Axl transformation whencompared to 3T3 cells. This resistance appeared to beassociated with the absence of a pp190 phosphoproteinfound in passaged 3T3 cells and could be overcome bya 30 ± 60-fold further increase in surface Axl expres-sion.
Taken together, our results suggest that Axl'stransforming activity is dependent not only onstructural changes within the Axl protein, but also onthe level of surface p140Axl expression in particularcellular contexts.
Results
Generation of transforming axl mutants
In order to develop infectious axl-retroviruses, thewild-type ax16-2 cDNA was cloned into the retroviralexpression vector pLXSN (thereafter called pLaxlSN)and subsequently transfected into NIH3T3 ®broblasts.After G418 selection, axl-transfected 3T3 cells wereinfected with conditioned media (CM) containing the
wild-type Moloney Murine Leukernia Virus (MoMLV)as described in Materials and methods. MoMLV thusserved as the helper virus in packaging pLaxlSNtranscripts. The generation of replication competentretroviruses in the CM of these transduced 3T3 cellswas con®rmed by the presence of reverse transcriptaseactivity as previously described (data not shown) (Fanet al., 1988).
Conditioned media derived from MoMLV infectedpLaxlSN cells was then tested for the presence of axl-containing retroviruses by Western blot analysis of thehuman p140Axl protein in newly infected 3T3 cells. Axlexpression could be found only after infection withsupernatant obtained from MoMLV-pLaxlSN cells.Despite detectable levels of Axl expression, MoMLV-pLaxlSN infected cells were not transformed (data notshown). This is in accordance with our earliertransfection results using the pLaxlSN constructshowing that axl is a weak oncogene requiring cellpassage for transformation to become manifest(O'Bryan et al., 1991).
In order to generate axl-containing viral strains withaugmented transforming activity, we serially passagedthe MuLV-pLaxlSN viral supernatant in naive 3T3cells. Initial infections were performed using theMuLV-pLaxlSN virus stock, and cells were selectedin G418 for incorporation of the viral construct.Conditioned media from these G418 resistant popula-tions was then used to further infect naive 3T3 cells.After a total of three rounds of viral passage, a viralstrain emerged capable of transforming 3T3 cellswithin 12 days post infection.
Seven transformed foci from this infection wereclonally expanded and found to exhibit di�erent levelsof p140Axl overexpression as seen by Western analysisemploying a C-terminal anti-Axl antibody (Figure 1).Of these, three transformed clones showing the highestlevel of Axl tyrosine phosphorylation (SV8, 1B1 andFFa) were isolated for further study. Each was foundto release axl-containing virions capable of limitedtransformation in an NIH3T3 cell focus formationassay; transformation was observed within 12 days ofinfection, with approximately 20 foci per plate. Thissuggested that within each viral strain, a subpopulationexisted with higher transforming potential.
SV
8/1
FFa/
1
1B1/
1
SV
8/2
SV
8/3
SV
8/6
Par
enta
l
1A1
NIH3T3
p140AXL
p120AXL
Blotting: α–COOH AXL
Figure 1 Axl expression in various 3T3 cell lines generated fromexpansion of transformed foci that emerged after the thirdpassage of the axl containing retroviral strain. Equal amounts ofprotein (50 mg) were separated by 9% SDS±PAGE and analysedby immunoblotting with anti-COOH Axl antiserum 17-4(1 : 5000). The positions of p120Axl and p140Axl are indicated.These data demonstrate expression of Axl in the transformed 3T3sublines
Axl overexpression in 3T3 transformationA Burchert et al
3178

To select for these transforming retroviral sub-lines, CM from SV8, 1B1 and FFa infected cellswere subjected to further viral passage. We screenedfor new viral phenotypes by systematically assessingthe extent of and time required for focus formation.In addition, Axl expression and tyrosine phosphor-ylation were analysed after every completed cycle ofviral passage. Interestingly, after the fourth passageof FFa-CM, the FFa virus (termed FFa4) trans-formed an entire 100 mm dish of NIH3T3 cellswithin a period of 6 days. By contrast, comparablepassage of the 1B1 and SV8 viral strains resulted inviruses that still could induce only 30 and 26 fociper plate, respectively. This di�erence was not dueto a greater amount of virions in the FFa-CM sincequantitation of the viral concentration as assessed bythe number of G418 resistant colonies after limitingdilution showed comparable levels between the FFa,SV8, and 1B1 viral sublines. Furthermore, thetransforming potential resided in the recombinantvirus and was not due to proviral insertion sincesubsequent passages of the FFa4 viral sublines(FFa5 and FFa6) demonstrated an equally hightransforming capacity.
Biochemical characterization of transforming axlmutants
Western blot analysis was used to characterize the3T3 cells transformed by the three viral sublineswith respect to Axl expression and tyrosinephosphorylation. Immunoblotting with an anti-phosphotyrosine antibody (a-pTyr) demonstratedhighly phosphorylated proteins at 120 and 140 kDconsistent with the human Axl protein found in 1B1,SV8 and FFa4 strains but not in parental 3T3 cells.When the same lysates were analysed using apolyclonal antibody speci®c for the Axl carboxy-terminus (17-4), cell lines 1B1 and SV8 demonstratedhigh levels of Axl expression. However, the sameantibody failed to detect the presence of Axl inFFa4 cell lysates either by immunoblotting orimmunoprecipitation (Figure 2). To further investi-gate these ®ndings, lysates were also blotted with anNH2-terminal Axl antibody. This, in contrast,revealed signi®cant Axl expression in all cell lines,including FFa4 (Figure 2a). Taken together, thesedata indicated that the most transforming virus,FFa4, may contain alterations which alter thecarboxyl terminus of Axl, thus rendering itundetectable by the 17-4 antibody.
Sequence analysis of the axl transforming mutants
In order to determine the structural basis for Axltransformation, the three derived axl proviruses (1B1,SV8 and FFa4) were subcloned and subjected tosequence analysis. Using axl primers spanning approxi-mately 500 ± 600 base pairs, various segments of theentire FFa4-axl cDNA were ampli®ed from the FFa4infected 3T3 cells. Interestingly, using a primer pair(PJA18 and PJA20) that ampli®es the 3' portion of theaxl cDNA between bp 2534 (end of codon 792) and bp3023 (in the 3' UTR), we detected two fragments withdistinct mobilities when resolved on a 1% agarose gel.One PCR fragment was of the expected 489 bp length
and the other, much stronger band, measured 294 bps.This 294 bp band was not seen using DNA from otheraxl transduced 3T3 cells as template. These observa-tions suggested that FFa4 CM contains two distinctviral subclones: one missing sequences in the 3' domainof the axl cDNA (which we designate FFa4short), andthe other carrying an axl cDNA of the appropriatelength.
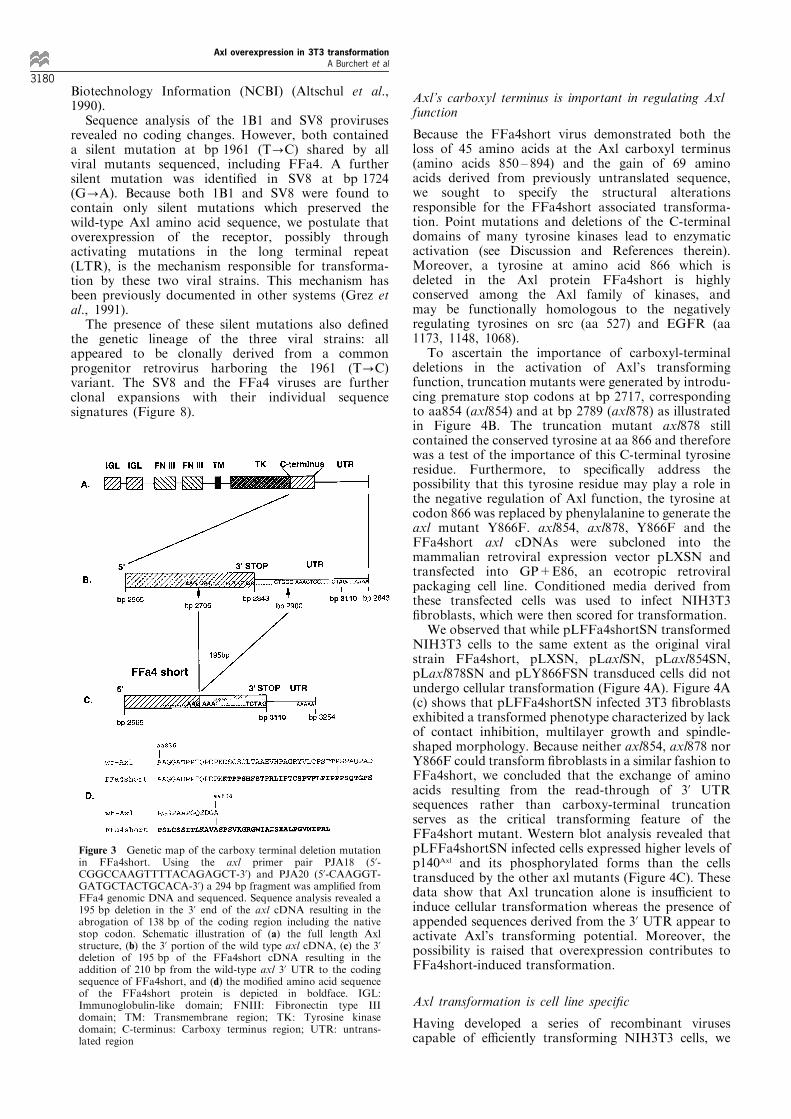
Sequence analysis of the smaller fragment revealeda 195 bp deletion in the 3' end of the axl cDNA frombp 2706 to bp 2900, removing 138 bps of the codingregion. Because the normal axl stop codon is involvedin this deletion, FFa4short would be predicted togenerate a read through transcript adding 210 bpsfrom the wild-type axl 3' UTR to the truncated FFa4sequence (Figure 3a, b and c). Thus, the FFa4shortvirus expresses an Axl protein that consists of the®rst 849 amino acids of the native protein followedby 69 amino acid residues derived from sequencesfrom the 3' UTR before terminating at a new stopcodon at bp 3110 (Figure 3d). This con®rms ourearlier ®ndings suggesting the absence of C-terminalAxl peptides in FFa4, since the anti-Axl antibody 17-4 recognizes epitopes located within the deleted138 bp 3' region. We used the University ofWisconsin Sequence Analysis Software Package byGenetics Computer, Inc. (GCG) and determined thatthe new C-terminus does not contain known proteinpatterns de®ned in the PROSITE Dictionary ofProtein Sites and Patterns (MOTIFS). In addition,no homology to known proteins in the PIR andSwissProt protein databases was seen using theBLAST network service at the National Center for
AF6295 AF6295FFa4 FFa4FFa6 FFa6
p140AXL
p140AXL
p120AXL
p120AXL
Blotting: α-COOH AXL Blotting: α-NH2 AXL
SV8 1B1 FFa4
NIH3T3 NR6
IP: α–COOH AXLBlotting: α-NH2 AXL
a
b
Figure 2 Analysis of Axl expression using both amino andcarboxy terminal directed antibodies. (a) Immunoblot analysis ofFFa4 and FFa6 transduced NIH3T3 cells and of AF6295 cells.Equal amounts of total cell lysates from each cell line wereimmunoblotted using either anti-COOH Axl or anti-NH2 Axlantibodies. AF6295 serves as a control cell line overexpressinghuman wild-type Axl protein. (b) Immunoprecipitation of Axlcomplexes from NIH3T3 and NR6 cells transduced with di�erentviral strains using anti-COOH Axl antiserum 17-4. Immunoblot-ting was performed with the anti-NH2 terminal Axl antibody. Thepositions of p120Axl and p140Axl are indicated. These data suggestthat FFa4 and FFa6 express an Axl protein with C-terminalabnormalities
Axl overexpression in 3T3 transformationA Burchert et al
3179
Biotechnology Information (NCBI) (Altschul et al.,1990).
Sequence analysis of the 1B1 and SV8 provirusesrevealed no coding changes. However, both containeda silent mutation at bp 1961 (T?C) shared by allviral mutants sequenced, including FFa4. A furthersilent mutation was identi®ed in SV8 at bp 1724(G?A). Because both 1B1 and SV8 were found tocontain only silent mutations which preserved thewild-type Axl amino acid sequence, we postulate thatoverexpression of the receptor, possibly throughactivating mutations in the long terminal repeat(LTR), is the mechanism responsible for transforma-tion by these two viral strains. This mechanism hasbeen previously documented in other systems (Grez etal., 1991).
The presence of these silent mutations also de®nedthe genetic lineage of the three viral strains: allappeared to be clonally derived from a commonprogenitor retrovirus harboring the 1961 (T?C)variant. The SV8 and the FFa4 viruses are furtherclonal expansions with their individual sequencesignatures (Figure 8).
Axl's carboxyl terminus is important in regulating Axlfunction
Because the FFa4short virus demonstrated both theloss of 45 amino acids at the Axl carboxyl terminus(amino acids 850 ± 894) and the gain of 69 aminoacids derived from previously untranslated sequence,we sought to specify the structural alterationsresponsible for the FFa4short associated transforma-tion. Point mutations and deletions of the C-terminaldomains of many tyrosine kinases lead to enzymaticactivation (see Discussion and References therein).Moreover, a tyrosine at amino acid 866 which isdeleted in the Axl protein FFa4short is highlyconserved among the Axl family of kinases, andmay be functionally homologous to the negativelyregulating tyrosines on src (aa 527) and EGFR (aa1173, 1148, 1068).
To ascertain the importance of carboxyl-terminaldeletions in the activation of Axl's transformingfunction, truncation mutants were generated by introdu-cing premature stop codons at bp 2717, correspondingto aa854 (axl854) and at bp 2789 (axl878) as illustratedin Figure 4B. The truncation mutant axl878 stillcontained the conserved tyrosine at aa 866 and thereforewas a test of the importance of this C-terminal tyrosineresidue. Furthermore, to speci®cally address thepossibility that this tyrosine residue may play a role inthe negative regulation of Axl function, the tyrosine atcodon 866 was replaced by phenylalanine to generate theaxl mutant Y866F. axl854, axl878, Y866F and theFFa4short axl cDNAs were subcloned into themammalian retroviral expression vector pLXSN andtransfected into GP+E86, an ecotropic retroviralpackaging cell line. Conditioned media derived fromthese transfected cells was used to infect NIH3T3®broblasts, which were then scored for transformation.
We observed that while pLFFa4shortSN transformedNIH3T3 cells to the same extent as the original viralstrain FFa4short, pLXSN, pLaxlSN, pLaxl854SN,pLaxl878SN and pLY866FSN transduced cells did notundergo cellular transformation (Figure 4A). Figure 4A(c) shows that pLFFa4shortSN infected 3T3 ®broblastsexhibited a transformed phenotype characterized by lackof contact inhibition, multilayer growth and spindle-shaped morphology. Because neither axl854, axl878 norY866F could transform ®broblasts in a similar fashion toFFa4short, we concluded that the exchange of aminoacids resulting from the read-through of 3' UTRsequences rather than carboxy-terminal truncationserves as the critical transforming feature of theFFa4short mutant. Western blot analysis revealed thatpLFFa4shortSN infected cells expressed higher levels ofp140Axl and its phosphorylated forms than the cellstransduced by the other axl mutants (Figure 4C). Thesedata show that Axl truncation alone is insu�cient toinduce cellular transformation whereas the presence ofappended sequences derived from the 3' UTR appear toactivate Axl's transforming potential. Moreover, thepossibility is raised that overexpression contributes toFFa4short-induced transformation.
Axl transformation is cell line speci®c
Having developed a series of recombinant virusescapable of e�ciently transforming NIH3T3 cells, we
Figure 3 Genetic map of the carboxy terminal deletion mutationin FFa4short. Using the axl primer pair PJA18 (5'-CGGCCAAGTTTTACAGAGCT-3') and PJA20 (5'-CAAGGT-GATGCTACTGCACA-3') a 294 bp fragment was ampli®ed fromFFa4 genomic DNA and sequenced. Sequence analysis revealed a195 bp deletion in the 3' end of the axl cDNA resulting in theabrogation of 138 bp of the coding region including the nativestop codon. Schematic illustration of (a) the full length Axlstructure, (b) the 3' portion of the wild type axl cDNA, (c) the 3'deletion of 195 bp of the FFa4short cDNA resulting in theaddition of 210 bp from the wild-type axl 3' UTR to the codingsequence of FFa4short, and (d) the modi®ed amino acid sequenceof the FFa4short protein is depicted in boldface. IGL:Immunoglobulin-like domain; FNIII: Fibronectin type IIIdomain; TM: Transmembrane region; TK: Tyrosine kinasedomain; C-terminus: Carboxy terminus region; UTR: untrans-lated region
Axl overexpression in 3T3 transformationA Burchert et al
3180

wished to determine whether this transformingpotential could be assessed in other cell systems. Tothis end, we infected the IL-3 dependent myeloid
precursor cell line 32D (gift of Dr J Pierce) with FFa4,1B1 and SV8 viral stocks giving rise to 32D cellsublines: FFa4.1, harboring mainly the truncated axlvariant (not shown), and 1B1 and SV8, expressing therespective axl cDNAs. Transformation was scored asthe ability of infected 32D cells to grow in a factorindependent manner (McCloskey et al., 1994).
Intriguingly, none of the axl viruses transformed32D cells, despite the presence of comparable orgreater surface levels of Axl than in 3T3 cells asassessed by ¯ow cytometry and con®rmed byWestern blot analysis (Figures 5a, b and 6a). Thedi�erence, however, was that whereas the humanAxl expressed in transduced 3T3 cells was tyrosinephosphorylated, there was no detectable Axlphosphorylation on tyrosine in the infected 32Dcells (Figure 5a and b). To prove that the axl-transduced 32D cells harbored a transforming axlvirus, supernatants from infected 32D cells wereapplied to naive 3T3 cells which were then scoredfor transformation. All 32D supernatants e�cientlytransformed 3T3 cells within 10 ± 12 days, showingthat passage through 32D cells did not a�ect thetransforming capability of the axl viruses.
The observation that 32D cells cannot be trans-formed when expressing comparable levels of Axloncoprotein to Axl-transformed 3T3 cells indicatedthat cell lines may vary in their requirements fortransformation by Axl. Alternatively, the biology ofthese transforming viruses may di�er in cells grown insuspension (e.g. 32D cells) compared to adherent cells(i.e. 3T3). To test this latter possibility, the ®broblastcell line NR-6, a modi®ed NIH3T3 cell line devoid ofEGF receptors, was similarly infected with the FFa4and SV8 viruses and observed for phenotypic changes.Following G418 selection, the infected NR-6 cellsappeared morphologically normal despite signi®cantAxl expression detectable on Western blot analysis(Figure 5c). Flow cytometry demonstrated comparablelevels of surface Axl expression in the axl-transducedbut untransformed NR-6 cells and in the axl virustransformed 3T3 cell lines (data not shown). Again,like the 32D cells, the axl-infected NR-6 cell lysatefailed to demonstrate tyrosine phosphorylation of thetransduced Axl protein (Figure 5c).
We were intrigued by the inability of the FFa4 andSV8 viral strains to transform 32D and NR-6 cells andasked whether we could generate axl viruses that couldtransform these apparently resistant cells. To this end,we continued passaging the FFa4.1 and SV8/FFa4.1viruses in naive 32D and NR6 cells and observed forthe emergence of IL-3 independent cell lines (32D) orfor transformed foci (NR6). Even after four viralpassages, we were not able to induce 32D or NR6 celltransformation under these conditions. Thus, 32D andNR6 cells appear to be relatively resistant totransformation by Axl as compared to 3T3 cells.
In investigating the mechanism of this resistance, weexamined the phosphoprotein pro®les of axl-trans-duced 3T3, NR6 and 32D cells. As mentioned before,whereas p140Axl is phosphorylated on tyrosine intransformed 3T3 cells, it is present but not phosphory-lated in transduced NR6 and 32D cells (Figure 5a, band c). Moreover, transformation of 3T3 cells by Axl isassociated with the appearance of a 190 kD phospho-protein which is not present in any axl virus infected
A
B
pLX
SN
pLa
z6-2
SN
pLF
Fa4s
ho
rtS
N
pLa
x854
SN
pLa
x878
SN
pLY
866F
SN
pp140Axl
pp120Axl
pp140Axl
pp120Axl
Blotting: α-NH2AXL
Blotting: α-pTYR
C
Figure 4 Morphology of NIH3T3 cells infected with axlrecombinant mutants. (A) Morphology of con¯uent cultures ofNIH3T3 cells 30 days after infection with replication de®cientretroviruses containing (a) pLXSN, (b) pLaxl6-2SN, (c)pLFFa4shortSN, (d) pLaxl854SN, (e) pLaxl878SN, (f)pLY866FSN cDNAs and selection in G418 (1006magnifica-tion). (B) Schematic representation of the axl recombinantmutants (c-f) and wild-type axl (b). (C) Western blot analysis ofAxl expression and tyrosine phosphorylation of the axlrecombinant mutants showing that all constructs are expressedafter transduction. The positions of p120Axl and p140Axl areindicated. These results demonstrate that transformation byFFa4short is due to gain of sequences derived from the 3' UTRrather than truncation of the Axl C-terminus
Axl overexpression in 3T3 transformationA Burchert et al
3181
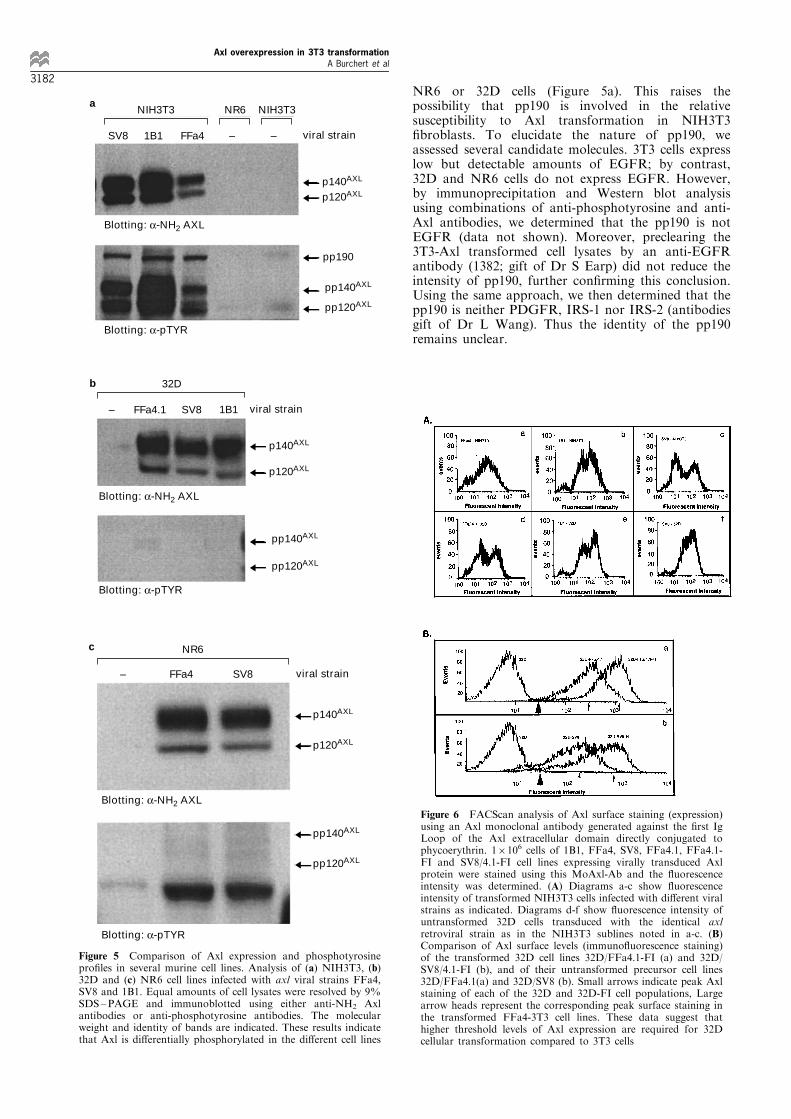
NR6 or 32D cells (Figure 5a). This raises thepossibility that pp190 is involved in the relativesusceptibility to Axl transformation in NIH3T3®broblasts. To elucidate the nature of pp190, weassessed several candidate molecules. 3T3 cells expresslow but detectable amounts of EGFR; by contrast,32D and NR6 cells do not express EGFR. However,by immunoprecipitation and Western blot analysisusing combinations of anti-phosphotyrosine and anti-Axl antibodies, we determined that the pp190 is notEGFR (data not shown). Moreover, preclearing the3T3-Axl transformed cell lysates by an anti-EGFRantibody (1382; gift of Dr S Earp) did not reduce theintensity of pp190, further con®rming this conclusion.Using the same approach, we then determined that thepp190 is neither PDGFR, IRS-1 nor IRS-2 (antibodiesgift of Dr L Wang). Thus the identity of the pp190remains unclear.
SV8 1B1 FFa4 – –
NIH3T3 NR6 NIH3T3
viral strain
viral strain
p140AXL
p140AXL
p120AXL
p120AXL
pp190
pp140AXL
pp120AXL
pp140AXL
pp120AXL
Blotting: α-NH2 AXL
Blotting: α-NH2 AXL
Blotting: α-pTYR
Blotting: α-pTYR
– FFa4.1 SV8 1B1
32D
a
b
– FFa4 SV8 viral strain
NR6
p140AXL
p120AXL
pp140AXL
pp120AXL
Blotting: α-NH2 AXL
Blotting: α-pTYR
c
Figure 5 Comparison of Axl expression and phosphotyrosinepro®les in several murine cell lines. Analysis of (a) NIH3T3, (b)32D and (c) NR6 cell lines infected with axl viral strains FFa4,SV8 and 1B1. Equal amounts of cell lysates were resolved by 9%SDS±PAGE and immunoblotted using either anti-NH2 Axlantibodies or anti-phosphotyrosine antibodies. The molecularweight and identity of bands are indicated. These results indicatethat Axl is di�erentially phosphorylated in the di�erent cell lines
Figure 6 FACScan analysis of Axl surface staining (expression)using an Axl monoclonal antibody generated against the ®rst IgLoop of the Axl extracellular domain directly conjugated tophycoerythrin. 16106 cells of 1B1, FFa4, SV8, FFa4.1, FFa4.1-FI and SV8/4.1-FI cell lines expressing virally transduced Axlprotein were stained using this MoAxl-Ab and the ¯uorescenceintensity was determined. (A) Diagrams a-c show ¯uorescenceintensity of transformed NIH3T3 cells infected with di�erent viralstrains as indicated. Diagrams d-f show ¯uorescence intensity ofuntransformed 32D cells transduced with the identical axlretroviral strain as in the NIH3T3 sublines noted in a-c. (B)Comparison of Axl surface levels (immuno¯uorescence staining)of the transformed 32D cell lines 32D/FFa4.1-FI (a) and 32D/SV8/4.1-FI (b), and of their untransformed precursor cell lines32D/FFa4.1(a) and 32D/SV8 (b). Small arrows indicate peak Axlstaining of each of the 32D and 32D-FI cell populations, Largearrow heads represent the corresponding peak surface staining inthe transformed FFa4-3T3 cell lines. These data suggest thathigher threshold levels of Axl expression are required for 32Dcellular transformation compared to 3T3 cells
Axl overexpression in 3T3 transformationA Burchert et al
3182
Suprathreshold expression of the axl gene is associatedwith 32D cell transformation
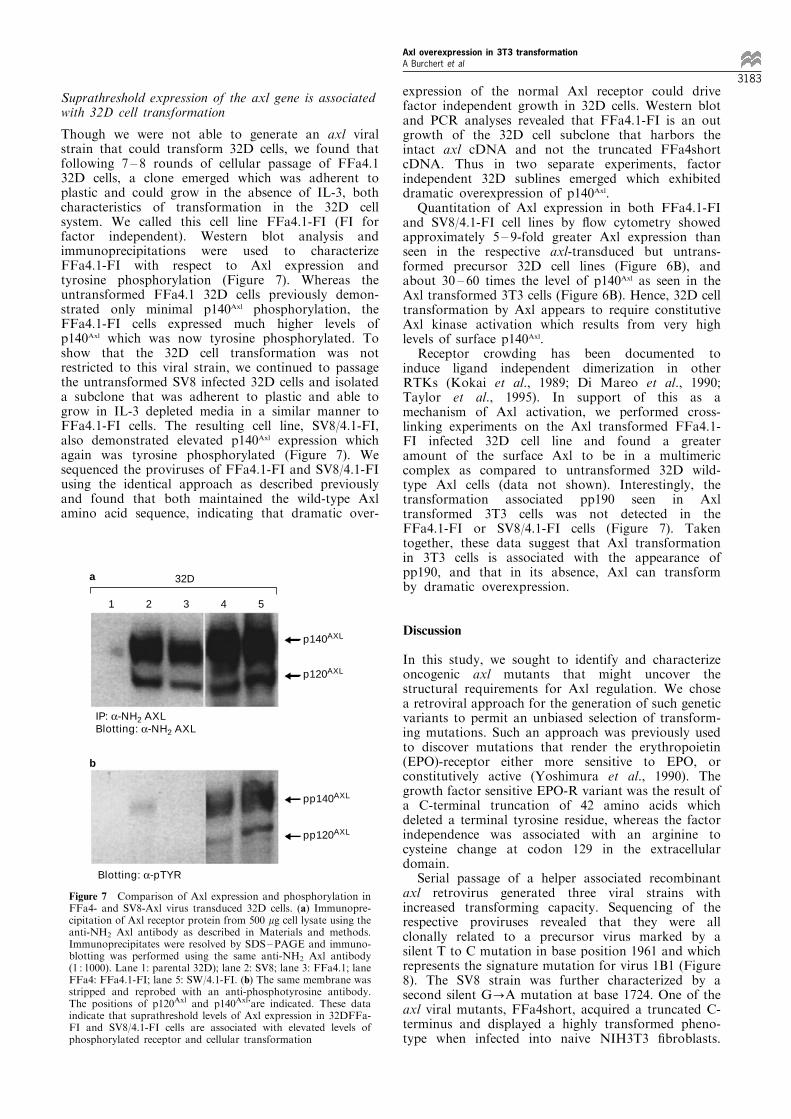
Though we were not able to generate an axl viralstrain that could transform 32D cells, we found thatfollowing 7 ± 8 rounds of cellular passage of FFa4.132D cells, a clone emerged which was adherent toplastic and could grow in the absence of IL-3, bothcharacteristics of transformation in the 32D cellsystem. We called this cell line FFa4.1-FI (FI forfactor independent). Western blot analysis andimmunoprecipitations were used to characterizeFFa4.1-FI with respect to Axl expression andtyrosine phosphorylation (Figure 7). Whereas theuntransformed FFa4.1 32D cells previously demon-strated only minimal p140Axl phosphorylation, theFFa4.1-FI cells expressed much higher levels ofp140Axl which was now tyrosine phosphorylated. Toshow that the 32D cell transformation was notrestricted to this viral strain, we continued to passagethe untransformed SV8 infected 32D cells and isolateda subclone that was adherent to plastic and able togrow in IL-3 depleted media in a similar manner toFFa4.1-FI cells. The resulting cell line, SV8/4.1-FI,also demonstrated elevated p140Axl expression whichagain was tyrosine phosphorylated (Figure 7). Wesequenced the proviruses of FFa4.1-FI and SV8/4.1-FIusing the identical approach as described previouslyand found that both maintained the wild-type Axlamino acid sequence, indicating that dramatic over-
expression of the normal Axl receptor could drivefactor independent growth in 32D cells. Western blotand PCR analyses revealed that FFa4.1-FI is an outgrowth of the 32D cell subclone that harbors theintact axl cDNA and not the truncated FFa4shortcDNA. Thus in two separate experiments, factorindependent 32D sublines emerged which exhibiteddramatic overexpression of p140Axl.
Quantitation of Axl expression in both FFa4.1-FIand SV8/4.1-FI cell lines by ¯ow cytometry showedapproximately 5 ± 9-fold greater Axl expression thanseen in the respective axl-transduced but untrans-formed precursor 32D cell lines (Figure 6B), andabout 30 ± 60 times the level of p140Axl as seen in theAxl transformed 3T3 cells (Figure 6B). Hence, 32D celltransformation by Axl appears to require constitutiveAxl kinase activation which results from very highlevels of surface p140Axl.
Receptor crowding has been documented toinduce ligand independent dimerization in otherRTKs (Kokai et al., 1989; Di Mareo et al., 1990;Taylor et al., 1995). In support of this as amechanism of Axl activation, we performed cross-linking experiments on the Axl transformed FFa4.1-FI infected 32D cell line and found a greateramount of the surface Axl to be in a multimericcomplex as compared to untransformed 32D wild-type Axl cells (data not shown). Interestingly, thetransformation associated pp190 seen in Axltransformed 3T3 cells was not detected in theFFa4.1-FI or SV8/4.1-FI cells (Figure 7). Takentogether, these data suggest that Axl transformationin 3T3 cells is associated with the appearance ofpp190, and that in its absence, Axl can transformby dramatic overexpression.
Discussion
In this study, we sought to identify and characterizeoncogenic axl mutants that might uncover thestructural requirements for Axl regulation. We chosea retroviral approach for the generation of such geneticvariants to permit an unbiased selection of transform-ing mutations. Such an approach was previously usedto discover mutations that render the erythropoietin(EPO)-receptor either more sensitive to EPO, orconstitutively active (Yoshimura et al., 1990). Thegrowth factor sensitive EPO-R variant was the result ofa C-terminal truncation of 42 amino acids whichdeleted a terminal tyrosine residue, whereas the factorindependence was associated with an arginine tocysteine change at codon 129 in the extracellulardomain.
Serial passage of a helper associated recombinantaxl retrovirus generated three viral strains withincreased transforming capacity. Sequencing of therespective proviruses revealed that they were allclonally related to a precursor virus marked by asilent T to C mutation in base position 1961 and whichrepresents the signature mutation for virus 1B1 (Figure8). The SV8 strain was further characterized by asecond silent G?A mutation at base 1724. One of theaxl viral mutants, FFa4short, acquired a truncated C-terminus and displayed a highly transformed pheno-type when infected into naive NIH3T3 ®broblasts.
1 2 3 4 5
32D
p140AXL
p120AXL
pp140AXL
pp120AXL
IP: α-NH2 AXLBlotting: α-NH2 AXL
Blotting: α-pTYR
a
b
Figure 7 Comparison of Axl expression and phosphorylation inFFa4- and SV8-Axl virus transduced 32D cells. (a) Immunopre-cipitation of Axl receptor protein from 500 mg cell lysate using theanti-NH2 Axl antibody as described in Materials and methods.Immunoprecipitates were resolved by SDS±PAGE and immuno-blotting was performed using the same anti-NH2 Axl antibody(1 : 1000). Lane 1: parental 32D); lane 2: SV8; lane 3: FFa4.1; laneFFa4: FFa4.1-FI; lane 5: SW/4.1-FI. (b) The same membrane wasstripped and reprobed with an anti-phosphotyrosine antibody.The positions of p120Axl and p140Axl'are indicated. These dataindicate that suprathreshold levels of Axl expression in 32DFFa-FI and SV8/4.1-FI cells are associated with elevated levels ofphosphorylated receptor and cellular transformation
Axl overexpression in 3T3 transformationA Burchert et al
3183

That these viruses induced transformation uponpassage using comparable viral titers suggests thattheir transforming potential is an intrinsic property ofthe individual retroviruses rather than a simpleconsequence of multiplicity of infection or of inser-tional activation.
The FFa4 transforming viral strain was found tobe a composite of two viruses, one identical to the1B1 in sequence, and the other (FFa4short)harboring a deletion of 195 base pairs in the 3'end of the axl cDNA that encompasses the TAGstop codon. This deletion results in the truncation of45 amino acids in the C-terminus of Axl and a readthrough that adds back 69 amino acids due to thetranslation of sequences within native 3' UTR. Toprove that FFa4short carries an axl cDNA withincreased oncogenic potential, we subcloned thetruncated axl into pLXSN and subsequentlyinfected the resultant helper free virus into 3T3indicator cells. Our results con®rm that FFa4shorthas augmented 3T3 transforming ability. Through aseries of deletion mutants, we determined that theincreased transforming activity of FFa4short did notresult from Axl carboxy-terminal truncation alonebut involved the addition of sequences encoded bythe 3' UTR. In addition, these transformed cellsoverexpressed p140Axl.
The presence of an appended carboxy terminusappeared to promote receptor activation and abrogatethe need for secondary passaging to induce transforma-tion. Whether this increased transforming abilityresulted from higher steady state levels of the Axlprotein or a functional di�erence of the chimericreceptor is unclear. Possible mechanisms for thisaugmented transforming ability include enhancedprotein stability, altered substrate selection, perturba-tion of receptor turnover, and altered intramolecularinteractions (Kornilova et al., 1996; Lorenzi et al.,1996; O'Connor et al., 1997). Nevertheless, it remainsconsistent that transformation by any form of Axl isassociated with receptor overexpression.
We furthermore observed that changing the tyrosineat codon 866 to phenylalanine (Y866F) was by itselfinsu�cient to render the axl cDNA transforming(Figure 4). By contrast, studies on other tyrosinekinases have implicated the terminal tyrosine in thenegative control of kinase signaling functions (i.e. theinsulin receptor, CSF-1 receptor, Src nonreceptortyrosine kinase; Roussel et al., 1987; Ellis et al., 1986;Thies et al., 1989; Cantley et al., 1991; Herbst et al.,1995; Cooper et al., 1986). For example, an insulinreceptor mutant lacking C-terminal Tyr1316 andTyr1322 demonstrates increased insulin inducedmitogenesis and mitogen activated protein kinaseactivity (Ando et al., 1992). The kinase activity of Srcis inhibited by the phosphorylation of the C-terminalTyr527. Intramolecular binding of the phosphorylatedtyrosine to the SH-2 domain of SRC inhibitsexogenous substrate interactions and downstreamsignaling, whereas deletion of this residue leads toconstitutive activation (Cooper and King, 1986). In thecase of the EGF receptor, the C-terminal tyrosineresidues (i.e. Tyr1173, Tyr1148, Tyr1068) may serve asintrinsic phosphorylation sites that compete withexogenous substrates for binding to the active domainof the receptor. Ligand induced autophosphorylationor deletion of these terminal residues are thought toincrease access of exogenous substrates to the receptor,resulting in augmented signaling capacity (Ullrich andSchlessinger, 1990; Honnegger et al., 1988a,b; Bertics etal., 1988; Khazaie et al., 1988; Velu et al., 1989). In thecase of EGFR, progressive C-terminal deletion mutantsincreased the phosphorylation of non-SH2 containingsubstrates (eps8 and eps15) which correlates well withenhanced transforming potential of these deletionmutants (Alvarez et al., 1995). Though conservationof Axl's terminal tyrosine 866 among members of theAxl family of receptor tyrosine kinases (forming theconsensus sequence RY(V/I)(L/F)) suggests functionalsigni®cance to this residue, mutation analysis of thistyrosine did not a�ect Axl's transforming ability in 3T3cells. Thus, the exact function of this motif remainsunclear.
Other members of the Axl/UFO family of receptortyrosine kinases can also induce transformationemploying di�erent structural mechanisms. v-eyk (v-ryk), the avian retroviral homologue of human c-mer,was discovered as the transduced oncogene in anacutely transforming retrovirus, RPL30, isolated froma variety of chicken mesenchymally derived tumors.v-eyk appears to be transforming through substitutionof the extracellular domain of its protooncogene, c-eyk/mer, with the viral gp37 envelope gene (Jia et al., 1992).The neural speci®c axl homologue, sky/rse, cantransform murine ®broblast cell lines either whenoverexpressed or when the extracellular domain isdeleted, resulting in an activated cytoplasmic version ofsky (also called brt) (Taylor et al., 1995; Lai et al.,1994). Recently, replacement of the axl ECD with viralgag sequences was found to enhance Axl's transform-ing potential in NIH3T3 cells and suggests thatmutations within the receptor ectodomain mayincrease Axl's transforming activity (Zhang et al.,1996). None of our transforming retroviruses har-bored ectodomain alterations. Moreover, an engineeredtruncation of the Axl ectodomain using the endogen-ous Axl initiating methionine did not render Axl
Figure 8 Viral lineage illustrating the etiology of axl retroviralmutants based on the sequence analysis. All transformed cell linesacquired a silent T?C mutation at bp 1961. Further passaging ofFFa resulted in a silent C?T transition and 195 bp loss in thecarboxy terminus resulting in a read-through mutation. FFa4.1-FI did not acquire further mutations. SV8/4.1 harbored a silentG?A transition at bp 1724, while further passaging resulted inthe development of another silent G?A transition at bp 2831 inSV8/4.1-FI. UTR: untranslated region
Axl overexpression in 3T3 transformationA Burchert et al
3184

transforming (Isaacs and Liu, unpublished results).Thus, N-terminal alterations of Axl appear activatingonly under speci®c conditions. Our investigations,however, suggest that receptor overexpression and C-terminal alterations identi®ed by a process of retroviralevolutionary selection may be the primary pathways toaugment Axl's transforming activity.
Despite the ®nding of a transforming C-terminalmutation, the vast majority of events leading to Axltransformation involve simple overexpression. We havepreviously described that transfection of axl into 3T3cells transforms through ampli®cation of the wild-typeaxl cDNA leading to overexpression (O'Bryan et al.,1991). In the error-prone retroviral system used here,transformation by two of three viral strains (SV8 and1B1) was primarily caused by dramatic overexpressionof Axl rather than through activating mutations, sinceboth viral sublines transmit the intact axl sequence.Activating promoter mutations of the MoMLV-LTRcan account for upregulated gene expression and havealso been documented to change the host expressionspeci®city of the retrovirus (Hanecak et al., 1991). Thepossibility that viral infection induced cellular trans-formation by insertional mutagenesis is unlikely sincethe acutely transforming nature of the viral strain wascarried with each successive viral passage. That bothtransfection and infection approaches lead almostexclusively to variants overexpressing a normal axlcDNA strongly suggest that overexpression is thesimplest and most potent mechanism by which Axlcan induce cellular transformation.
This conclusion is supported by our observations inthe cell line speci®city of Axl transformation. Our threetransforming viral strains were all able to e�cientlytransform 3T3 cells, yet were ine�ective when appliedto 32D and NR6 cells. Tissue speci®city for oncogenetransformation has been previously documented. Aninsertionally activated c-erbB (1Ac-erbB), which con-sists of gag sequences replacing the c-erbB ectodomain,can transform erythroblasts but not ®broblasts (Carteret al., 1995). The presence of an unde®ned transforma-tion inhibitory factor in ®broblasts absent in erythro-blasts was speculated. Further investigations into v-erbB variants displaying diverse tissue speci®c patternsof transformation revealed the engagement of speci®csignaling pathways associated with the transformationof di�erent tissue types (McManus et al., 1995). Ourdata, however, suggest that the tissue or cell linespeci®city for Axl transformation may be due to hostfactors that facilitate p140Axl tyrosine phosphorylation.In investigating the mechanism of this resistance totransformation, we found that although the levels ofp140Axl expression were equivalent in infected 3T3,NR6 and 32D cells, the inability to transform wascorrelated with the absence of tyrosine phosphorylationof the transduced Axl (Figure 5): Axl-transformed 3T3cells (FFa4-3T3, 1B1-3T3, and SW-3T3) all showedligand independent tyrosine phosphorylation of p140Axl
compared to the absence of phosphorylation in the32D cell equivalents. Notably, the lack of transformingpotential is not a result of inactive viral mutants sinceviral recovery experiments showed that the resistant32D and NR6 cells produced axl viruses that could stille�ciently transform naive NIH3T3 cells. Uponspontaneous transformation, however, the FFa4.1-FIand SV8/FFa4.1-FI 32D cells showed up to a ninefold
greater increase in surface Axl expression relative totheir infected but untransformed precursor 32Dsublines, which in turn express up to sixfold higherp140Axl levels than the Axl-transformed 3T3 cells(Figure 6b). Thus, the surface levels of p140Axl
required for NIH3T3 cell transformation are 1/60 ±1/30 that for 32D cell transformation.
The components mediating a lower threshold forp140Axl phosphorylation and transformation in 3T3cells remain unclear. However, the absence of an Axlspeci®c phosphatase or a role for the transformationassociated pp190 found speci®cally in 3T3 cells mayaccount for the di�erences in transforming suscept-ibility seen between 3T3, 32D and NR6 cells. Takentogether, our results suggest that the biological e�ectsof Axl expression are determined by the cellularcontext, and that extrapolation of expression dataand function should be made with caution.
Materials and methods
Generation and infection of replication competent recombinantaxl retroviruses
The axl cDNA 6-2 was cloned into the expression vectorpLXSN (pLaxlSN) as previously described (Miller andRosman, 1989; O'Bryan et al., 1991). Recombinant axlretroviruses were generated by transfection of 10 mg ofpLaxlSN into N1H3T3 cells using Lipofectamine (BRL,Grand Island, NY) followed by selection of resistantcolonies in G418 (BRL) at 800 mg/ml and subsequentinfection of these cells with conditioned media (CM)containing ecotropic MoMLV (kindly provided by Dr JOlsen, UNC-Chapel Hill). Cells were then reseeded andgrown to con¯uency. The axl retrovirus containing CM washarvested and ®ltered through a 0.2 mm membrane(Millipore). The viral titer was estimated by infectingparental NIH3T3 cells with serial dilutions of theMoMLV-pLaxlSN supernatant. For infections, cells wereseeded at a density of 16105 cells per 60 mm plate andincubated for 2 h with 46105 ± 16106 c.f.u. (colony formingunits) of the axl/MoMLV retrovirus-containing CM in thepresence of 8 mg/ml Polybrene (Sigma). Twenty-four hoursafter infection, G418 selection was applied and the culturewas observed for focus formation and transformation over a30 day period. Following selection, CM of the G418resistant cell lines was harvested, ®ltered and used for thenext round of infection of parental 3T3 cells.
Cell culture and cell lines
The murine IL-3 dependent 32D cell lines producedthrough viral infection with 1B1, FFa4, and SV8 CM,the factor-independent (FI) 32D cells, and the ecotropicmurine retroviral packaging cell line GP+E86 weremaintained as previously described (McCloskey et al.,1994; Fridell et al., 1996). Early-passage NIH3T3 fibro-blasts and viral-infected NIH3T3 cell lines were cultured inDulbecco's Modi®cation of Eagle's Medium (DMEM-H)(Gibco, BRL) containing 10% fetal bovine serum andpenicillin/streptomycin (Gibco, BRL). NR6 cells werepropagated in DMEM-L supplemented with 10% FBS,1% L-Glutamine and penicillin/streptomycin.
Immunoprecipitation and Western blot analysis
Cells were lysed in lysis bu�er containing 1% Triton X-100and protease inhibitors as previously described (McCloskeyet al., 1994). For immunoprecipitation, 350 ± 600 mg of celllysates were precleared with 5 mg per ml of Immunopure
Axl overexpression in 3T3 transformationA Burchert et al
3185

Rabbit Gamma Globulin (Pierce) followed by Protein A/GPLUS-Agarose beads (Santa Cruz) for 1 h at 48C.Precleared lysates were subsequently incubated with acarboxyl terminal antibody raised against the peptidesequence CGADRSPAAPGQEDGA (anti-COOH Axlantibody 17-4; 1 : 1000) or with an a�nity puri®edantibody raised against amino acids 33 ± 136 (anti-NH2
Axl; 1 : 250) overnight at 48C (McCloskey et al., 1994;O'Bryan et al., 1995). Immune complexes were precipitatedby adding 40 mL of Protein A/G PLUS-Agarose beads.Immune complexes were washed with 361 ml lysis bu�erand sample bu�er was added to the washed beads. Sampleswere heat denatured and eluted proteins were loaded on a9% SDS polyacrylamide gel. Resolved proteins wereelectrophoretically transferred onto Immobilon-P TransferMembranes (Millipore). Western blot analysis was per-formed as previously described (Fridell et al., 1996) withprimary antibodies indicated in the ®gure legends.
PCR cloning of proviral DNA, sequence analysis and generationof recombinant axl mutants
Genomic DNA (gDNA) was extracted from 66106 cellsusing a Genepure DNA extractor (341 Nucleic AcidPuri®cation System, Applied Biosystems). Each polymerasechain reaction (PCR) consisted of 100 ± 300 ng gDNA astemplate . For sequencing of FFa4-axl, primer pairs JB3/7-2-3; JB3/JB6; JB5/PJ6; PJA23/PJA3; PJA14/PJA17; PJA18/PJA20 were used to amplify FFa4-axl proviral cDNAfragments as previously described (O'Bryan et al., 1991).PCR was carried out in a 50 ml total volume, with 0.025 nmolof each primer, 0.01 mmol dNTPs, 16GeneAmp 106PCRBu�er (contains 15 mM MgCl2), and 0.25 ml AmpliTaq DNAPolymerase (Roche, Perkin-Elmer). PCR products weresubcloned into TA-cloning vector using TA Cloning Kit(Invitrogen, San Diego, CA) according to the manufacturer'sprotocol, and sequencing of FFa4-axl plasmid clones wasperformed using the Sequenase Version 2.0 Kit (USB,Cleveland, OH). Mutant proviral strains FFa4, FFa4short,SV8, 1B1, SV8/4.1-FI and FFa4.1-FI were ampli®ed fromgDNA using the Expand Long Template PCR System(Boehringer Mannheim, Indianapolis, IN) with 100 ±300 ng template gDNA, forward primer EcoJB3 (5'-CGGAATTCAGCCCAGCAACTTCTGAGGA-3') whichincludes an EcoRI site, and reverse primer 3'pLXSN (5'-GCGGATCCTCGAGTTAACG-3') which anneals 3' to anEcoRI site located in the pLXSN polylinker. PCR productswere electrophoretically separated, gel puri®ed, and digestedwith EcoRI prior to cloning into the EcoRI site of pLXSN.Plasmid sequencing was performed, and suspected mutationswere con®rmed using asymmetric PCR from gDNA andsequencing of single stranded products as described else-where (Medori et al., 1992).
Axl mutants axl854 and axl878 were generated by PCRwith the Expand Long Template PCR System using pLaxl6-
2SN as template with EcoJB3 as the forward primer andaxl854 (5'-CGGAATTCGCAGTGAGTCAGCTACAG-GAA-3') and axl878 (5'-CGGAATTCATCAGCAGGCT-TAGCGGGGCTA-3') as reverse primers, respectively.Amplicons were digested with EcoRI and cloned intopLXSN. Axl mutant Y866F was created with the AlteredSites in vitro Mutagenesis System (Promega, Madison, WI)using mutagenesis primer Y866F (5'-GCAGAGGA-CAAAGCGTCCAG-3' and was then cloned into the EcoRIsite of pLXSN.
Biologic activity of axl mutants
To con®rm the transforming potential of the viral andrecombinant axl mutants, a standard NIH3T3 cell focusformation assay was performed by infecting parental cellswith equivalent amounts of titered virus and selecting withG418. The GP+E86 retroviral packaging cell line was usedto generate viruses by transfecting with 10 mg of cDNAfrom pLXSN, pLaxl6-2SN, pLaxl854SN, pLaxl878SN,pLFFa4shortSN and pLY866HN constructs. CM fromthese transfected cell lines were then used to infect naiveNIH3T3 cells as described above. The extent of transfor-mation in NIH3T3 cells following G418 selection wasassessed both by observing the time required to achievefocus formation and by counting the number of foci after a30 day period. Transformation of 32D cells was determinedby withdrawing IL-3 and assessing for factor-independentgrowth.
Flow cytometry
FACS analysis was performed in a Becton DickinsonFACScan. An Axl monoclonal antibody (Axl MoAb #9)directed against the ®rst Ig Loop of Axl (Fridell et al., 1996)was used to assay surface Axl expression in virallytransduced murine cell lines. The MoAb was directlyconjugated to phycoerythrin (PE). For FACS analysis,800 ng of Axl-PE antibody was mixed with 16106 cells andincubated on ice for 20 min. Stained cells were then washedtwice with PBS containing 0.1% BSA. The reported valuerepresents the mode of ¯uorescence intensity determinedusing the Cyclops ¯ow cytometry software package.
AcknowledgementsThe authors wish to acknowledge the excellent technicalassistance of Dr Yan Jin. We also wish to thank Drs JOlsen, S Earp, J Pierce and L Wang for their contibutions.The work in this manuscript was supported by NCI RO1-CA49240 (ETL) and SPORE CA58223-04 (ETL). ETL is aLeukemia Society Scholar, AB is a recipient of a grantfrom the Deutscher Akademischer AustauschdienstBiomedical Exchange Program (BMEP), and ECA is aHoward Hughes Medical Institute Predoctoral Fellow.
References
Altschul SF, Gish W, Miller W, Myers EW and Lipman DJ.(1990). J. Mol. Biol., 215, 403 ± 410.
Alvarez CV, Shon KJ, Miloso M and Beguinot L. (1995). J.Biol. Chem., 270, 16271 ± 16276.
Ando A, Momomura K, Tobe K, Yamamoto-Honda R,Sakura H, Tamori Y, Kaburagi Y, Koshio O, AkanumaY, Yawki Y, Kasuga M and Kadowaki T. (1992). J. Biol.Chem., 267, 12788 ± 12796.
Bargmann CI, Hung MC and Weinberg RI. (1986). Cell, 45,649 ± 657.
Bellosta P, Costa M, Lin DA and Basilico C. (1995). Mol.Cell. Biol., 15, 614 ± 625.
Bertics PJ, William SC, Hubler L, Lazar CS, Rosenfeld MGand Gill GN. (1988). J. Biol. Chem., 8, 3610 ± 3617.
Brambilla R, Schnapp A, Casagranda F, Labrador JP,Bergemann AD, Flanagan JG, Pasquale EB and Klein R.(1995). EMBO J., 14, 3116 ± 3126.
Cantley LC, Auger KR, Carpenter C, Duckworth B,Graziani A, Kapeller R and Solto� S. (1991). Cell, 64,281 ± 302.
Carter TH, Dominguez N, Zeng L and Kung HI. (1995). J.Cell. Biol., 212, 277 ± 283.
Charbonneau H and Tonks NK. (1992). Annu. Rev. Cell.Biol., 8, 463 ± 493.
Axl overexpression in 3T3 transformationA Burchert et al
3186

Cooper JA, Gould KL, Cartwright CA and Hunter T.(1986). Science, 231, 1431 ± 1434.
Cooper JA and King CS. (1986). Mol. Cell. Biol., 12, 4467 ±4477.
Craven RJ, Xu LH, Weiner TM, Fridell YW, Dent GA,Srivastava S, Vamum B, Liu ET and Cance WG. (1995).Int. J. Cancer, 60, 791 ± 797.
DiMareo El, Pierce JH, Knickley CL and DiFiore PP.(1990). Mol. Cell. Biol., 10, 3247 ± 3252.
Dougherty JP and Temin HM. (1988). J. Virol., 62, 2817 ±2822.
Edelmann GM and Crossin U. (1991). Annu. Rev. Biochem.,60, 155 ± 190.
Ellis L, Clauser E, Morgan DO, Edery M, Roth RA andRutter WJ. (1986). Cell, 45, 721 ± 731.
Fan H, Chute H, Chao E and Pattengale PK. (1988).Virology, 166, 58 ± 65.
Fischer EH, Charbonneau H and Tonks NK. (1991).Science, 253, 401 ± 406.
Fridell YC, Jin Y, Quilliam LA, Burchert A, McCloskey P,Spizz G, Varnum B, Der C and Liu ET. (1996). Mol. Cell.Biol., 16, 135 ± 145.
Gasic GP, Arenas CP, Gasic TB and Gasic GJ. (1992). Proc.Nat. Acad. Sci. USA, 89, 2317 ± 2320.
Goruppi S, Ruaro E and Schneider C. (1996). Oncogene, 12,471 ± 480.
Grez M, Zornig M, Nowock J and Ziegler M. (1991). J.Virol., 65, 4691 ± 4698.
Hanecak R, Pattengale PK and Fan H. (1991). J. Virol., 65,5357 ± 5363.
Herbst R, Munemitsu S and Ullrich A. (1995). Oncogene, 10,369 ± 379.
Honegger A, Dull TJ, Bellot F, Van Obberghen E, SzaparyD, Schmidt A and Schlessinger J. (1988a). EMBO J., 7,3045 ± 3052.
Honegger A, Dull TJ, Szapary D, Kornoriya A, Kris R,Ullrich A and Schlessinger J. (1988b). EMBO J., 7, 3053 ±3061.
Jia R, Mayer BJ, Hanafusa T and Hanafusa H. (1992). J.Virol., 66, 5975 ± 5987.
Khazaie K, Dull TJ, Graf T, Schlessinger J, Ullrich A, BeugH and Venstroem B. (1988). EMBO J., 7, 3061 ± 3071.
Kokai Y, Meyers JN, Wada T, Brown VI, Leva CM, DavisJG, Dobashi K and Greene M. (1989). Cell, 58, 287 ± 292.
Kornilova E, Sorkina T, Beguinot L and Sorkin A. (1996). J.Biol. Chem., 271, 30340 ± 30346.
Lai C, Gore M and Lernke G. (1994). Oncogene, 9, 2567 ±2578.
Li R, Chen J, Hammonds G, Phillips H, Armanini M, WoodP, Bunge R, Godowski PJ, SliwIcowski NIX and MatherJP. (1996). J. Neurosci., 16, 2012 ± 2019.
Lorenzi MV, Horii Y, Yamanaka R, Sakaguchi K and MikiT. (1996). Proc. Nat. Acad. Sci. USA., 93, 8956 ± 8961.
McCloskey P, Pierce J, Koski RA, Varnum B and Liu ET.(1994). Cell Growth Di�er., 5, 1105 ± 1117.
Medori R, Tritschler H and Gambetti P. (1992). Biotechni-ques. 12, 346 ± 350.
Monk RJ, Malik FG, Stokesberry D and Evans LH. (1992).J. Virol., 66, 3683 ± 3689.
McManus J, Connolly DC andMai�e NT. (1995).Mol. Cell.Biol., 69, 3631 ± 3638.
Miller AD and Rosman GJ. (1989). Biotechniques, 7, 980 ±990.
Nakano T, Higashino K, Kikuchi N, Kishino J, Nomura K,Fujita H, Ohara O and Arita H. (1995). J. Biol. Chem.,270, 5702 ± 5705.
O'Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B,Prokop C, Espinosa III R, Le Beau MM, Earp HS and LiuET. (1991). Mol. Cell. Biol., 11, 5016 ± 5031.
O'Bryan JP, Fridell Y, Koski R, Varnum B and Liu ET.(1995). J. Biol. Chem., 270, 551 ± 557.
O'Connor R, Kau¯mann-Zeh A, Liu Y, Lehar S, Evan GI,Baserga R and Blattler WA. (1997). Mol. Cell. Biol., 17,427 ± 435.
Pulido D, Campuzano S, Koda T, Modolell J and BarbacidM. (1992). EMBO J, 11, 391 ± 404.
Quong RY, Bickford ST, Ing YL, Terman B, Herlyn M andLassam NJ. (1994). Melanoma Res., 4, 313 ± 319.
Roussel MF, Dull TJ, Rettenmier CW, Ralph P, Ullrich Aand Sherr CL. (1987). Nature, 325, 549 ± 552.
Slamon DJ, Godolphin W, Jones LA, Wong SG, Keith DE,Levin WJ, Stuart SG, Udove J, Ullrich A and Press MF.(1989). Science, 244, 707 ± 712.
Taylor IC, Roy S and Varmus HE. (1995). Oncogene, 11,2619 ± 2626.
Thies RS, Ullrich A and McClain DA. (1989). J. Biol. Chem.,264, 12820 ± 12825.
Velu TJ, Vass WC, Lowy DR and Beguinot L. (1989). Mol.Cell. Biol., 9, 1772 ± 1778.
Ullrich A and Schlessinger J. (1990). Cell, 61, 203 ± 212.Yoshimura A, Longmore G and Lodish HF. (1990). Nature,
348, 647 ± 649.Zhang QK, Boast S, De Los Santos K, Begemann M and
Go� SP. (1996). J. Virol., 70, 8089 ± 8097.
Axl overexpression in 3T3 transformationA Burchert et al
3187




















