
Cross-link network of polydimethylsiloxane via addition and condensation (RTV) mechanisms. Part I:...
7
Cross-link network of polydimethylsiloxane via addition and condensation (RTV) mechanisms. Part I: Synthesis and thermal properties Mohamad Riduwan Ramli, Muhammad Bisyrul Hafi Othman, Azlan Arifin, Zulkifli Ahmad * School of Materials and Mineral Resources Engineering, Engineering Campus, Universiti Sains Malaysia, Seri Ampangan, 14300 Nibong Tebal, Penang, Malaysia article info Article history: Received 24 August 2011 Accepted 2 October 2011 Available online 6 October 2011 Keywords: Polysiloxane Cross-link network Thermal Crystallinity Fractional free volume X-ray diffraction abstract A series of highly cross-linked polysiloxane was synthesised via hydrosilylation and condensation reaction. Structural identification using Fourier Transform Infrared (FTIR) and 1 H-NMR confirmed their chemical structures. Their thermal and, mechanical properties, and crystallinity, were analysed and related to the level of cross-link density. These systems displayed elevated thermal and hardness properties at an increased cross-link density. Furthermore, the level of crystallinity was reduced as displayed by XRD analysis. Along with this observation, the calculated fractional free volume (FFV) showed a decreasing trend leading to the ‘densification’ effect. It was envisaged that the linear poly- siloxane chain segments aligned parallel to each other in a triclinic crystal system to generate a crys- talline domain. The spacing between these stacking chains was found to be about 7.2 Å as measured from simulated XRD pattern. Ó 2011 Elsevier Ltd. All rights reserved. 1. Introduction Cross-linked polydimethylsiloxane (PDMS), or silicone elas- tomer, is of major interest to researchers because of its unique properties. It is generally produced by synthesising reactive PDMS prepolymers, which are subsequently cross-linked to give a highly tough and durable elastomeric material. Crosslinking or network formation of silicone polymer can be obtained by three typical routes: condensation reaction (moisture cure), addition reaction (hydrosilylation cure), or radical reaction, which is normally per- formed at higher temperatures [1e3]. Crosslinking of polysiloxane via condensation reaction affording thermally stable materials as been reported recently [4e7]. Han et al. [6] studied the effect of cross-link density towards the thermal stability of room temperature vulcanization PDMS by incorporating it with polymethylmethoxysiloxane (PMOS) via hydrolytic condensation reaction. Highly cross-linked PMOS phases were formed in situ and the average cross-link density increased as the loading of PMOS was increased. They suggested that dense PMOS phases could reduce the pyrolysis of PDMS at elevated temperature. Chen et al. [7] investigated the thermal degradation, thermo- oxidative stability and mechanical properties of hybrid PDMS with an octavinyl-Polyhedral oligomeric silsesquioxanes (POSS) derivative prepared by hydrolytic condensation. They reported that the improvement in thermal properties could be attributed to the effective three-dimensional network structures resulting from the structure of the octavinyl-POSS derivative. Furthermore, the improvement of mechanical properties could be attributed to the synergistic effect of the structure of three-dimensional multi-arm cross-linker vinyl-POSS derivative and the perfect distribution of the vinyl-POSS derivative. Meanwhile the addition reaction of polysiloxane provides a more stable material at elevated temper- atures, and was developed for rapid processing and fast curing rate of deep section part [8,9]. The two-part heat curable system, which consists of the vinyl and hydride reactive functional groups, and the addition of a platinum catalyst hydrosilylation cure, provides a fast cure system without any by-product. Furthermore, a longer shelf time, one-component system was developed by premixing a scav- enging agent such as oximatosilanes, carbamatosilane or amino- silanes [10]. These scavenging agents react with excess hydroxyl groups whether from methanol, silanol, or water, and would not react with the alkoxy groups to prematurely cross-link the PDMS system. A high flame retardancy effect could be accomplished by a cross-linked structure performed under platinum catalysed hydrosilylation reaction as suggested by Hayashida et al. [11] by suppressing the thermal decomposition. The condensation reaction of PDMS however, requires a longer curing time [12,13]. On the other hand, the addition reaction of PDMS in a one-component system requires less mixing and provide a problem-free production material but requires an elevated curing temperature [1,3]. * Corresponding author. Tel.: þ604 599 5099; fax: þ604 594 1011. E-mail addresses: [email protected] (M.R. Ramli), zulkifl[email protected]. my (Z. Ahmad). Contents lists available at SciVerse ScienceDirect Polymer Degradation and Stability journal homepage: www.elsevier.com/locate/polydegstab 0141-3910/$ e see front matter Ó 2011 Elsevier Ltd. All rights reserved. doi:10.1016/j.polymdegradstab.2011.10.001 Polymer Degradation and Stability 96 (2011) 2064e2070
-
Upload
independent -
Category
Documents
-
view
9 -
download
0
Transcript of Cross-link network of polydimethylsiloxane via addition and condensation (RTV) mechanisms. Part I:...

at SciVerse ScienceDirect
Polymer Degradation and Stability 96 (2011) 2064e2070
Contents lists available
Polymer Degradation and Stability
journal homepage: www.elsevier .com/locate/polydegstab
Cross-link network of polydimethylsiloxane via addition and condensation(RTV) mechanisms. Part I: Synthesis and thermal properties
Mohamad Riduwan Ramli, Muhammad Bisyrul Hafi Othman, Azlan Arifin, Zulkifli Ahmad*
School of Materials and Mineral Resources Engineering, Engineering Campus, Universiti Sains Malaysia, Seri Ampangan, 14300 Nibong Tebal, Penang, Malaysia
a r t i c l e i n f o
Article history:Received 24 August 2011Accepted 2 October 2011Available online 6 October 2011
Keywords:PolysiloxaneCross-link networkThermalCrystallinityFractional free volumeX-ray diffraction
* Corresponding author. Tel.: þ604 599 5099; fax:E-mail addresses: [email protected] (M
my (Z. Ahmad).
0141-3910/$ e see front matter � 2011 Elsevier Ltd.doi:10.1016/j.polymdegradstab.2011.10.001
a b s t r a c t
A series of highly cross-linked polysiloxane was synthesised via hydrosilylation and condensationreaction. Structural identification using Fourier Transform Infrared (FTIR) and 1H-NMR confirmed theirchemical structures. Their thermal and, mechanical properties, and crystallinity, were analysed andrelated to the level of cross-link density. These systems displayed elevated thermal and hardnessproperties at an increased cross-link density. Furthermore, the level of crystallinity was reduced asdisplayed by XRD analysis. Along with this observation, the calculated fractional free volume (FFV)showed a decreasing trend leading to the ‘densification’ effect. It was envisaged that the linear poly-siloxane chain segments aligned parallel to each other in a triclinic crystal system to generate a crys-talline domain. The spacing between these stacking chains was found to be about 7.2 Å as measured fromsimulated XRD pattern.
� 2011 Elsevier Ltd. All rights reserved.
1. Introduction
Cross-linked polydimethylsiloxane (PDMS), or silicone elas-tomer, is of major interest to researchers because of its uniqueproperties. It is generally produced by synthesising reactive PDMSprepolymers, which are subsequently cross-linked to give a highlytough and durable elastomeric material. Crosslinking or networkformation of silicone polymer can be obtained by three typicalroutes: condensation reaction (moisture cure), addition reaction(hydrosilylation cure), or radical reaction, which is normally per-formed at higher temperatures [1e3].
Crosslinking of polysiloxane via condensation reaction affordingthermally stable materials as been reported recently [4e7]. Hanet al. [6] studied the effect of cross-link density towards the thermalstability of room temperature vulcanization PDMS by incorporatingit with polymethylmethoxysiloxane (PMOS) via hydrolyticcondensation reaction. Highly cross-linked PMOS phases wereformed in situ and the average cross-link density increased as theloading of PMOS was increased. They suggested that dense PMOSphases could reduce the pyrolysis of PDMS at elevated temperature.Chen et al. [7] investigated the thermal degradation, thermo-oxidative stability and mechanical properties of hybrid PDMSwith an octavinyl-Polyhedral oligomeric silsesquioxanes (POSS)
þ604 594 1011..R. Ramli), [email protected].
All rights reserved.
derivative prepared by hydrolytic condensation. They reported thatthe improvement in thermal properties could be attributed to theeffective three-dimensional network structures resulting from thestructure of the octavinyl-POSS derivative. Furthermore, theimprovement of mechanical properties could be attributed to thesynergistic effect of the structure of three-dimensional multi-armcross-linker vinyl-POSS derivative and the perfect distribution ofthe vinyl-POSS derivative. Meanwhile the addition reaction ofpolysiloxane provides a more stable material at elevated temper-atures, and was developed for rapid processing and fast curing rateof deep section part [8,9]. The two-part heat curable system, whichconsists of the vinyl and hydride reactive functional groups, and theaddition of a platinum catalyst hydrosilylation cure, provides a fastcure system without any by-product. Furthermore, a longer shelftime, one-component system was developed by premixing a scav-enging agent such as oximatosilanes, carbamatosilane or amino-silanes [10]. These scavenging agents react with excess hydroxylgroups whether from methanol, silanol, or water, and would notreact with the alkoxy groups to prematurely cross-link the PDMSsystem. A high flame retardancy effect could be accomplished bya cross-linked structure performed under platinum catalysedhydrosilylation reaction as suggested by Hayashida et al. [11] bysuppressing the thermal decomposition. The condensation reactionof PDMS however, requires a longer curing time [12,13]. On theother hand, the addition reaction of PDMS in a one-componentsystem requires less mixing and provide a problem-free productionmaterial but requires an elevated curing temperature [1,3].

Table 1Feed molar ratio of hydride terminated PDMS which have been synthesised.
Samples Concentration (mmol) aMolarRatio, r
bMn
(g/mol)
bMw
(g/mol)Mw/Mn
cIntrinsicViscosities(dL/g)
Monomer I(D4)
Monomer II(EC)
A 67.6 20.0 0.30 3858 4697 1.22 0.0122B 67.6 10.1 0.15 5866 8206 1.40 0.0404C 67.6 5.4 0.08 11,104 14,838 1.34 0.0673D 67.6 2.7 0.04 18,971 28,672 1.51 0.1495
a Calculated based on molar ratio monomer I and monomer II, r ¼ [EC/D4].b Measured using gel permeation chromatography (GPC).c Obtained from Ubbelohde solution viscometer using toluene as the solvent.
Fig. 1. Repeat unit for polysiloxane network used to determine the FFV. The n valuesfor samples RTV-A ¼ 51, RTV-B ¼ 78, RTV-C ¼ 149, RTV-D ¼ 256.
M.R. Ramli et al. / Polymer Degradation and Stability 96 (2011) 2064e2070 2065
This work attempted to incorporate both mechanisms to yielda novel elastomeric product having synergistic thermal andmechanical properties. It comprised of investigation into thethermal and physical properties of cured PDMS at varying cross-link densities. The degree of cross-link density was designedbased on a series of linear prepolymers prepared as the startingmaterials. The effect of cross-link density on the level of crystal-linity was evaluated and the simulated crystal structure of thepolysiloxane was generated based on the XRD pattern.
2. Experimental
2.1. Materials
Octamethylcyclotetrasiloxane (D4), trifluoromethane sulfonic acid(triflic acid), Platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxanecomplex (Karstedt’s catalyst) and vinyltrimethoxysilane 98% wereobtained fromALDRICH.1,1,3,3-tetramethyldisiloxanewas purchasedfrom Fluka. The solvents such as the toluene and diethyl ether wereobtained from J.T. Baker and Merck respectively. All materials wereused as received.
2.2. Synthesis of a,u-dihydrido-polydimethylsiloxane (PDMS-1)
Octamethylcyclotetrasiloxane, I (D4) (20 g) and 1,1,3,3-tetramethyldisiloxane II (2.7 g, 20.0 mmol) were charged intoa round bottom flask and purged with nitrogen gas. The tempera-ture was increased to 55 �C and triflic acid (0.2 g) was slowly addedinto the reaction. The reaction was equilibrated at 55 �C for 72 h.The mixture was dissolved in diethyl ether then neutralized byrepeatedly being washed with deionized water. The solution wasdried over anhydrous magnesium sulphate for 1 h and subse-quently filtered through glass wool. Then, diethyl ether andunreacted monomer were distilled out under reduced pressure at80 �C for 2 h. A clear liquid form was obtained with over 90 wt%yield. Four pre-polymer with different molar ratio of I and II weresynthesised (Table 1). 1H-NMR (CDCl3): d ¼ 4.7 (m, 2H, SiH);d¼ 0.1e0.05 (m, 6H, SiCH3). IR y: 2962, 2907, 2127, 1412, 1258, 1079,1011, 911, 864, 788, 699 cm�1.
Table 2Some physical properties of RTV samples.
Samples Molar Ratio, r aDensity, re(g/cm3)
Hardness(Shore A)
Optimum Q
RTV-A 0.30 0.985 67 1.99RTV-B 0.15 0.982 63 2.79RTV-C 0.08 0.975 60 3.44RTV-D 0.04 0.971 59 4.08
a Were taken using pycnometer.
2.3. Synthesis of a,u-bis-(trimethoxysilane)-polydimethylsiloxane(PDMS-2)
Vinyltrimethoxysilane IV (4.58 ml, 30 mmol), toluene (10 ml)and Karstedt’s catalyst (0.075 ml) were charged into a 50 ml two-neck round bottom flask equipped with a dropping funnel.PDMS-1 III (10 g) was charged into the dropping funnel and thetemperature was increased to 50 �C. PDMS-1 was slowly droppedinto the reaction mixture, which was maintained at a temperatureof 50 �C. The temperature was then increased to 75 �C for 2 h. Aftercompletion of the reaction, the solution was cooled to roomtemperature and the remaining unreacted monomers wereremoved using rotary evaporator at 90 �C for 3 h to give a clearliquid formwith over 90 wt% yield. 1H-NMR (CDCl3): d¼ 3.5 (s, 18H,eSi(OCH3)3); 1.1 (m, 4H, (CH3O)3SiCH2e); 0.6 (d, 4H,e(CH3)2SiCH2e); 0.1e0.05 (6H, eSi(CH3)2e). IR y: 2962, 2907, 2839,1412, 1258, 1191, 1079, 1013, 788, 699 cm�1.
2.4. Preparation of highly cross-linked PDMS
Room temperature vulcanization PDMS (RTV-PDMS) wasprepared via hydrolysis followed by condensation reaction ofmethoxy groups using triflic acid as catalyst (0.3 phr) underambient conditions for 24 h. The alcohol that was produced asa side product was eliminated from the reaction system usingDean-Stark apparatus. For comparison, a control sample wasprovided by Penchem Industry Sdn. Bhd. The control was PDMSsystem curedwith the platinum catalyst at 120 �C for 2 h. IR y: 2962,2907, 1412, 1258, 1079, 1013, 788, 699 cm�1.
2.5. Analytical techniques
1H-NMR spectra were recorded with a Bruker 400 UltraShieldlinked to a computer running WIN-NMR software. The frequencyused was 400 MHz for 1H. Experiments were performed in CDCl3with tetramethylsilane (TMS) as an internal reference. Infraredspectra were recorded using a PerkineElmer System 2000 Atten-uated Total Reflectance (ATR-FTIR) spectroscopy. The spectra wererecorded in 4000e500 cm�1 region using 16 scans.
Fig. 2. Synthetic route of highly cross-linked RTV-PDMS.
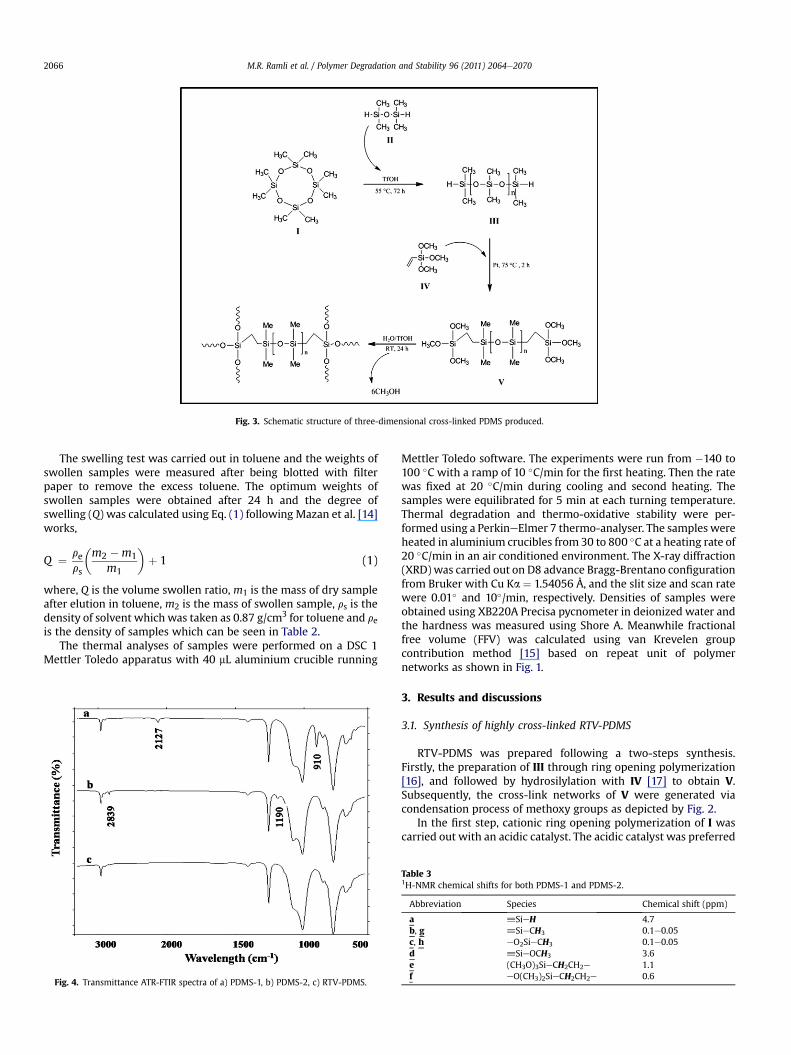
Fig. 3. Schematic structure of three-dimensional cross-linked PDMS produced.
M.R. Ramli et al. / Polymer Degradation and Stability 96 (2011) 2064e20702066
The swelling test was carried out in toluene and the weights ofswollen samples were measured after being blotted with filterpaper to remove the excess toluene. The optimum weights ofswollen samples were obtained after 24 h and the degree ofswelling (Q) was calculated using Eq. (1) following Mazan et al. [14]works,
Q ¼ rers
�m2 �m1
m1
�þ 1 (1)
where, Q is the volume swollen ratio, m1 is the mass of dry sampleafter elution in toluene, m2 is the mass of swollen sample, rs is thedensity of solvent which was taken as 0.87 g/cm3 for toluene and reis the density of samples which can be seen in Table 2.
The thermal analyses of samples were performed on a DSC 1Mettler Toledo apparatus with 40 mL aluminium crucible running
Fig. 4. Transmittance ATR-FTIR spectra of a) PDMS-1, b) PDMS-2, c) RTV-PDMS.
Mettler Toledo software. The experiments were run from �140 to100 �C with a ramp of 10 �C/min for the first heating. Then the ratewas fixed at 20 �C/min during cooling and second heating. Thesamples were equilibrated for 5 min at each turning temperature.Thermal degradation and thermo-oxidative stability were per-formed using a PerkineElmer 7 thermo-analyser. The samples wereheated in aluminium crucibles from 30 to 800 �C at a heating rate of20 �C/min in an air conditioned environment. The X-ray diffraction(XRD) was carried out on D8 advance Bragg-Brentano configurationfrom Bruker with Cu Ka ¼ 1.54056 Å, and the slit size and scan ratewere 0.01� and 10�/min, respectively. Densities of samples wereobtained using XB220A Precisa pycnometer in deionized water andthe hardness was measured using Shore A. Meanwhile fractionalfree volume (FFV) was calculated using van Krevelen groupcontribution method [15] based on repeat unit of polymernetworks as shown in Fig. 1.
3. Results and discussions
3.1. Synthesis of highly cross-linked RTV-PDMS
RTV-PDMS was prepared following a two-steps synthesis.Firstly, the preparation of III through ring opening polymerization[16], and followed by hydrosilylation with IV [17] to obtain V.Subsequently, the cross-link networks of V were generated viacondensation process of methoxy groups as depicted by Fig. 2.
In the first step, cationic ring opening polymerization of I wascarried out with an acidic catalyst. The acidic catalyst was preferred
Table 31H-NMR chemical shifts for both PDMS-1 and PDMS-2.
Abbreviation Species Chemical shift (ppm)
a ^SieH 4.7b, g ^SieCH3 0.1e0.05c, h eO2SieCH3 0.1e0.05d ^SieOCH3 3.6e (CH3O)3SieCH2CH2e 1.1f eO(CH3)2SieCH2CH2e 0.6

Fig. 5. 1H-NMR spectrum of PDMS-1 (above) and PDMS-2 (below).
Fig. 6. The swelling ratio of RTV in toluene at 25 �C.
M.R. Ramli et al. / Polymer Degradation and Stability 96 (2011) 2064e2070 2067
over a basic catalyst due to the sensitivity of hydride (SieH) reactivegroups to the latter which might lead to O-silylation reaction [18].Siloxane cation was first generated in this system by the reactivetriflic acid which propagates with the other cyclic siloxanes. Thepropagation of linear PDMS-I was then terminated by II to obtaina hydride terminal group. Themolecular weight of this linear PDMSchain was tailored by controlling the mole ratio of the 1,3-tetramethyldisiloxane as the end-capper. The molecular weightfor this series is shown as in Table 1. Some physical properties areshown as in Table 2.
The hydrosilylation process that occurred in the second stepinvolves the hydrogen of the hydrosilyl group and the vinyl groupfrom the trimethoxy silane in the presence of Karstedt’s catalyst;this introduced the tri-methoxysilane group to the terminal of thechain. The condensation step was finally performed with the activemethoxy groups reacting under atmospheric moisture in thepresence of 0.3 phr of triflic acid. During the first two hours of thereaction, the methanol-like smell was detected suggesting that thecondensation and hydrolysis of the methoxy groups took place.Three methoxy groups attached at the terminal end of PDMS-2affording a three dimensionally cross-linked PDMS structure(Fig. 3).
3.2. FTIR spectroscopy
Complete formation of SieH bonds in PDMS-I was beenobserved after purification based on FTIR spectrum. Fig. 4a showsthe FTIR spectrum of SieH terminated PDMS with two distinctivebands at 2127 cm�1 and 910 cm�1 stretching represented by A andB respectively [19e21]. These bands can be used as the preliminaryevidence of the appearance of hydride groups in III.
The disappearance of SieH peaks at 2127 cm�1 and 910 cm�1
indicate that the hydrosilylation process was accomplished in thesystem (Fig. 4b). This is reiterated further by the appearance ofsymmetrical CH3eO stretching and SieOeCH3 peaks at 2839 cm�1
and 1190 cm�1 respectively. The peak at 2840 cm�1 (sharp) and1190 cm�1 can be assigned as SieOCH3 groups as has been reportedelsewhere [21,22].
The disappearance of methoxy groups represented by CH3eOand SieOeCH3 peaks at 2839 cm�1 and 1190 cm�1 respectivelyunambiguously proves the formation of crosslinking networks asshown in Fig. 4c.
3.3. Nuclear magnetic resonance
NMR analysis for PDMS-1 and PDMS-2 are given as in Table 3. InPDMS-1 spectrum, the proton from SieH groups occurred atd 4.7 ppm (assigned as a in Fig. 5) meanwhile siloxane groups(proton from methyl attached to siloxane groups) represented byb and c were assigned mostly upfield at (d 0.1ed 0.05 ppm)respectively. The chemical shifts for SieH and SieCH3 wereconsistent with the previous study [17,23]. The integration ratios ofthese two peaks were taken to determine the value of repeatingunit n.
In PDMS-2 spectrum, methoxy proton peak occurred mostlydownfield at d 3.6 ppm as a sharp singlet represented by d (see

Fig. 9. TGA curves for RTV samples compared to control sample measured in airatmosphere condition.
Fig. 7. Effect of molar ratio (end capper to monomer ratio) on cross-link density anddegree of swelling of RTV samples.
M.R. Ramli et al. / Polymer Degradation and Stability 96 (2011) 2064e20702068
Table 3) due to the presence of neighbouring electronegativeoxygen. The methylene proton e and f occur at d 1.1 and 0.6 ppmrespectively. For e, the silicon atom bonded to three methoxygroups which contain three oxygen atoms. The chemical shifttherefore, is slightly downfield compared to f of which the adjacentsilicone atom is attached to two methyl groups and one oxygenatom.
Comparison between spectra PDMS-1 and PDMS-2 shows thatpeak a in PDMS-1 completely disappeared in the PDMS-2 spectrumsuggesting a complete hydrosilylation process had occurred. Theappearance of peak d at d 3.6 ppm indicates the successful incor-poration of methoxy groups in the PDMS-2 structure. The forma-tion of methylene at peak e and f in PDMS-2 fully support thecomplete hydrosilylation process.
3.4. Cross-link density
Cross-link density of samples were obtained after curing processat room temperature and were determined using Flory-Rehner’sEq. (2),
Fig. 8. Glass transition temperature (2nd scan) of RTV-PDMS samples run at 20 �C/min.
n ¼�hlnð1� V2Þ þ V2 þ xpsV2
2
i
V1
V1=32 � V2
2
! (2)
where, V1 is molar volume of solvent (106.85 cm3/mol was takenfor toluene [24]), V2 is reciprocal of volume swelling ratio Q, xps ispolymeresolvent interaction coefficient and was taken as 0.465[24] for siloxane elastomer and toluene interaction.
In Fig. 6, the optimum degrees of swelling for RTV-B, RTV-C, andRTV-D were achieved after 23 h of swelling test. RTV-A attained itsoptimum degree of swelling after only 6 h of swelling test. Degreeof swelling is inversely proportional to the cross-link density sinceat higher cross-link density, solvent diffusion in the polymer matrixwould be less efficient. The order of cross-link density would beRTV-A > RTV-B > RTV-C > RTV-D.
It can be seen that, the cross-link density of RTV samplesincreased correspondingly with the increase of molar ratio of end-capper to the monomer as depicted in Fig. 7. At high molar ratio ofend-capper, the molecular weights of the chain will decrease andsubsequently provide a higher bulk concentration for the conden-sation reaction site with the silane cross-linker. The highest cross-link density was obtained at 0.30 ratio while the lowest value is at0.04 ratio. The end-capper was subsequently replaced by threeactive methoxy groups during the hydrosilylation process. Thereare six active methoxy groups per molecule, (i.e. three at each end-chain of the molecule), and it was these groups which formed thecross-link network through the condensation reaction betweenchains.
The effect of cross-link densities on hardness were shown inTable 2. The hardness of samples decreased with decreasing cross-
Table 4Effect of cross-link density on free volume and glass transition temperature.
Samples Cross-link density(mol/cm3)
aFractional freevolume (FFV)
Tg (�C)
RTV-A 13.48 � 10�4 0.186 �108RTV-B 4.52 � 10�4 0.187 �112RTV-C 2.43 � 10�4 0.192 �114RTV-D 1.51 � 10�4 0.195 �119
a Calculated using Krevelen method [15].

Fig. 10. XRD spectrum for sample RTV-A until RTV-D and the simulated pattern.
M.R. Ramli et al. / Polymer Degradation and Stability 96 (2011) 2064e2070 2069
link densities. Hardness values of samples produced in this studywere superior to the commercial RTV samples. The commercialhardness was in the range of 22e40 Shore A [12].
3.5. Thermal properties
Differential Scanning Calorimetry (DSC) was performed on RTV-PDMS to reveal the thermal transition in the polymer. The transi-tions in Fig. 8 were attributed to the glass transition (Tg) of RTV-PDMS samples in the range of �108 to �119 �C. This is in closeagreement with early studies [25] which reported the Tg of analo-gous RTV silicone elastomers cured with POSS structures wasbetween �118 and �120 �C. The Tg decreased progressively withthe decreasing cross-link density of the samples. Thus, sample RTV-A displayed the highest Tg compared to other samples due to chainrestriction as the result of a high cross-link density. All samplesshows a gradual step drop during the glass transition as due to theplasticizing effect of the linear siloxane chains which adjoined thecrosslinking points in the network.
Fractional free volume (FFV) of RTV samples were calculatedusing group contribution of van der Waals’ volume method asestablished by van Krevelen [15]. The values in the range 0.15e0.19are typical for polymeric systems. RTV-A contains the highestcross-link density when compared to other three samples, thus,exhibiting the lowest free volume. It was established from Positron
Fig. 11. Simulated crystal structure of crystallite domain of the cured RTV material s
Annihilation Lifetime Study that a high cross-link density inducesa lower fractional free volume [26]. This is because a high cross-linkdensity would affect a ‘densification’ between chains.
A control from a commercial source was used in determinationof thermal stability to compare with other RTV samples syn-thesised in this work (Fig. 9). It can be seen that, RTV-A starts todegrade at 475 �C along with the control sample. Meanwhile RTV-Bwas degraded at around 450 �C followed by RTV-C and RTV-D ataround 375 �C. Thus, the thermo-oxidative stability of RTV samplesincreased with the cross-link density. Interestingly, the thermalstability of RTV samples was comparable with the control sample,although the control sample is the addition system, which is knownto be thermally stabile. This might be due to the high cross-linknetwork achievable in this system. Further, RTV-A showed thehighest char yield. It has been shown that char yield is indirectlyproportional to the cross-link density in a polysiloxane system. Alinear, uncross-linked polydimethylsiloxane heated in an Aratmospherewas completely degraded at 600 �Cwith no residue left[27].
The decreasing trend of Tg (Table 4) was further investigated bystudying the X-ray diffraction (XRD) spectra. Essentially the XRDspectra (Fig. 10) show broad halos representing the highly amor-phous nature of all the samples. It can be seen however, that theRTV-A shows broad peaks while RTV-D shows the sharpest peaks.This observation shows that the highly cross-linked sample of RTV-A is less crystalline than the low cross-linked sample of RTV-D. Thiscan be explained that as the RTV-A has more cross-link network,chain flexibility is immobilised to certain degree which restrict anypossible chain alignment to form crystalline domains. Alternatively,as the cross-link densities decreased, the molecular chains haveadequate space to align and rearrange into crystalline domainsthus, giving sharper peaks in XRD. However, the peak’s positions donot vary much between samples with the change in the cross-linkdensities implying that crystal system remains intact. The XRDanalysis, therefore, corroborate further the observed degree ofcross-link density as discuss in the preceding sections.
Simulated crystal structures based on the experimental XRDpattern were generated using Material Studio v4.4 software usingForcite module whose energy minimisation was perform underDreiding Forcefield. One chain per unit cell was generated andminimised in an initially constrained triclinic crystal system.Towards the end of the simulation cycle, this unit cell was relaxedand no apparent change was detected in either the crystal lattice orthe chain configuration. Thus, it was established that the obtainedunit cell configuration should be correct. Fig. 11 shows the simu-lated diffraction pattern as compared to the experimental data. Thelaterally adjacent chain distance was found to be 7.2 Å. These values
howing the stereo projection (a) and laterally (b) adjacent chain in a unit cell.

M.R. Ramli et al. / Polymer Degradation and Stability 96 (2011) 2064e20702070
corresponded very well to the main peak positions at 2q ¼ 12.0�
from the experimental data. This distance represents the d-spacingof the linear polysiloxane chain backbone. It is envisaged that thecrystalline domain is formed between the linear polysiloxanechains which are at close proximity and undergoes chainarrangement. The polysiloxane region resulting from condensationis unlikely to form any crystalline structure as expected froma highly cross-link network.
4. Conclusion
Synthesis of RTV siloxane by a combination of addition andcondensation systems has been presented in this paper. Structureidentifications were established using FTIR and H-NMR methods.The results showed significant improvement of RTV samplescompared with a high temperature vulcanization (control) sample.The greatest contribution to this improvement was from highcontent of cross-link density and synergistically having combinedthermal properties from both the addition and condensationsystem. The swelling test showed that the lower the chain length ofthe linear polysiloxane segment, the higher the cross-link density.A high cross-link network affects an improvement in hardness andthermal properties. The free volume is also reduced leading to theeffect of ‘densification’ of polymeric chains in the bulk system. Thehigh cross-link density of this system, however, reduced the level ofcrystallinity, which owed much to the chain immobilisation.Simulated crystalline structure based from XRD analysis estab-lished that the crystalline domain was generated from the parallelalignment of linear polysiloxane backbone in triclinic crystalsystem whose inter-chain spacing is about 7.2 Å.
Acknowledgement
The authors thank Universiti Sains Malaysia and the USMFellowship Scheme for the financial support of this work byResearch University Grant (No. 814069).
References
[1] Brook M. Silicon in organic, organometallic, and polymer chemistry. J. Wiley;2000.
[2] Gonzaga F, Yu G, Brook M. Polysiloxane elastomers via room temperature,metal-free click chemistry. Macromolecules 2009;42(23):9220e4.
[3] de Buyl F. Silicone sealants and structural adhesives* 1. Int J Adhes Adhes2001;21(5):411e22.
[4] Prado LASA, Sforça ML, de Oliveira AG, Yoshida IVP. Poly(dimethylsiloxane)networks modified with poly(phenylsilsesquioxane)s: synthesis, structuralcharacterisation and evaluation of the thermal stability and gas permeability.Eur Polym J 2008;44(10):3080e6.
[5] Chen D, Yi S, Wu W, Zhong Y, Liao J, Huang C, et al. Synthesis and charac-terization of novel room temperature vulcanized (RTV) silicone rubbers using
Vinyl-POSS derivatives as cross linking agents. Polymer 2010;51(17):3867e78.
[6] Han Y, Zhang J, Shi L, Qi S, Cheng J, Jin R. Improvement of thermal resistance ofpolydimethylsiloxanes with polymethylmethoxysiloxane as crosslinker.Polym Degrad Stab 2008;93(1):242e51.
[7] Chen D, Nie J, Yi S, Wu W, Zhong Y, Liao J, et al. Thermal behaviour andmechanical properties of novel RTV silicone rubbers using divinyl-hexa[(trimethoxysilyl)ethyl]-POSS as cross-linker. Polym Degrad Stab 2010;95(4):618e26.
[8] Cai D, Neyer A. Cost-effective and reliable sealing method for PDMS (Poly-DiMethylSiloxane)-based microfluidic devices with various substrates.Microfluid Nanofluid; 2010:1e10.
[9] Parbhoo B, O’Hare LA, Leadley SR. Fundamental aspects of adhesion tech-nology in silicones. In: Chaudhury M, Pocius AV, editors. Surfaces, chemistryand applications. Amsterdam: Elsevier Science B.V.; 2002. p. 677e709.
[10] White MA, Beers, Melvin D, Lucas, Gary M, Smith, Robert A, Swiger, Roger T.One package, stable, moisture curable, polyalkoxy-terminated organo-polysiloxane compositions and method for making. United States: GeneralElectric Company (Waterford, NY); 1983.
[11] Hayashida K, Tsuge S, Ohtani H. Flame retardant mechanism of poly-dimethylsiloxane material containing platinum compound studied byanalytical pyrolysis techniques and alkaline hydrolysis gas chromatography.Polymer 2003;44(19):5611e6.
[12] Jerschow P. Silicone elastomers. Rapra Technology Ltd; 2002.[13] Reilly B, Bruner S. Silicones as a material of choice for drug delivery appli-
cations; 2004.[14] Mazan J, Leclerc B, Galandrin N, Couarraze G. Diffusion of free poly-
dimethylsiloxane chains in polydimethylsiloxane elastomer networks. EurPolym J 1995;31(8):803e7.
[15] Van Krevelen DW, Te Nijenhuis K. Volumetric properties. Properties of poly-mers. 4th ed. Amsterdam: Elsevier; 2009. pp. 71e108.
[16] Desmurs J, Ghosez L, Martins J, Deforth T, Mignani G. Bis (trifluoromethane)sulfonimide initiated ring-opening polymerization of octamethylcyclote-trasiloxane. J Organomet Chem 2002;646(1e2):171e8.
[17] Cai G, Weber W. Synthesis of terminal Si-H irregular tetra-branched starpolysiloxanes. Pt-catalyzed hydrosilylation with unsaturated epoxides. Poly-siloxane films by photo-acid catalyzed cross-linking. Polymer 2004;45(9):2941e8.
[18] Pawlenko S. Organosilicon chemistry. W. de Gruyter; 1986.[19] Cai D, Neyer A, Kuckuk R, Heise HM. Raman, mid-infrared, near-infrared
and ultraviolet-visible spectroscopy of PDMS silicone rubber for charac-terization of polymer optical waveguide materials. J Mol Struct 2010;976(1e3):274e81.
[20] Satoh T, Hiraki A. Detailed study of SieH stretching modes in mc-Si: H filmthrough second derivative IR spectra. Jpn J Appl Phys 2 1985;24(7):L491e4.
[21] Smith A. Infrared spectra-structure correlations for organosilicon compounds.Spectrochim Acta 1960;16(1e2):87e105.
[22] Crompton T. Analysis of organosilicon compounds. The Chemistry of organicsilicon compounds. New York: Wiley; 1989. pp. 393e444.
[23] Louis E, Jussofie I, Kühn F, Herrmann W. Karstedt catalyst-catalyzed step-growth co-polyaddition of 1,9-decadiene and 1,1,3,3-tetramethyldisiloxane.J Organomet Chem 2006;691(9):2031e6.
[24] Hauser RL, Walker C, Kilbourne FL. Swelling of silicone elastomers. Ind EngChem 1956;48(7):1202e8.
[25] Chen D, Yi S, Fang P, Zhong Y, Huang C, Wu X. Synthesis and characterizationof novel room temperature vulcanized (RTV) silicone rubbers using octa[(trimethoxysilyl)ethyl]-POSS as cross-linker. React Funct Polym 2011;71(4):502e11.
[26] Xu J, Chen B, Zhang Q, Guo B. Prediction of refractive indices of linear poly-mers by a four-descriptor QSPR model. Polymer 2004;45(26):8651e9.
[27] Schiavon M, Redondo S, Pina S, Yoshida I. Investigation on kinetics of thermaldecomposition in polysiloxane networks used as precursors of silicon oxy-carbide glasses. J Non-Cryst Solids 2002;304(1e3):92e100.




















