
Corrigendum: Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting...
9
1;' -- îiîëdicine ARTICLES Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptoslS GI C U :0 GI E f ::1 1ii c E 0 u I!! ::1 ... t'CI j -..; ë. := .c D.. :1 E! C) CI c :c .! :ë :1 Q. CI) ... :1 - ca Z co 0 0 N @ Adriano G Rossi1, Deborah A Sawatzky1.3, Annemieke WaIker1,3, Carol Ward1, Tara A Sheldrake1, Nicola A Riley1, Alison Caldicott1, Magdalena Martinez-Losa1, Trevor R WaIkerl, Rodger Duffinl, Mohini Gray1, Elvira Crescenzi2, Morag C MartinI, Hugh J Bradf, John S Savill1,lan Dransfield1& Christopher Haslett1 Apoptosis is essential for clearance of potentially injurious inflammatory cells and subsequent efficient resolution of inflammation. Here we report that human neutrophils contain functionally active cyclin-dependent kinases (CDKs),and that structurally diverse CDK inhibitors induce caspase-dependent apoptosis and override powerful anti-apoptosis signais from survival factors such as granulocyte-macrophage colony-stimulating factor (GM-CSF).We show that the CDKinhibitor R-roscovitine (Seliciclib or CYC202) markedly enhances resolution of established neutrophil-dependent inflammation in carrageenan-elicited acute pleurisy, bleomycin- induced lung injury,and passively induced arthritis in mice. ln the pleurisy model, the caspase inhibitor zVAD-fmkprevents R-roscovitine-enhanced resolution of inflammation, indicating that this CDKinhibitor augments inflammatory cell apoptosis. We also provide evidence that R-roscovitine promotes apoptosis by reducing concentrations of the anti-apoptotic protein Md-l. Thus, CDK inhibitors enhance the resolution of established inflammation by promoting apoptosis of inflammatory cells, thereby demonstrating a hitherto unrecognized potential for the treatment of inflammatory disorders. Neutrophils have a central role in innate immunity and are rapidly recruited to sites of infection and injury; however, their many defense mechanisms that destroy and digest invading microorganisms are potentially deleterious to tissuesi. Thus, it is vital that, once their physiological function has been achieved, these inflammatory cells and their potentially histotoxic contents are eleared rapidly. During spontaneous resolution of inflammation, neutrophils undergo apoptosis: a pre-programmed and highly regulated cell death process that results in shutdown of secretory capacity and allows recognition and removal by macrophages2,3. Neutrophil survival and apoptosis are profoundly influenced by the inflammatory milieu: inflammatory mediators such as GM-CSF or lipopolysaccharide (LP5), environ- mental conditions such as hypoxia, and the presence of pro-apoptotic stimuli such as tunIor necrosis factor-a; or Fas ligand markedly alter neutrophillongevity4,S. Neutrophil apoptosis is controlled by a complex network of sigrtal- ing pathways that regulate both the turnover of key molecules, ineluding the anti-apoptotic protein myeloid cell leukemia 1 (Mel-1) and the pro-apoptotic Bel-2 farnily member Bax, and activation of the caspase family of proteasess. Once apoptosis has been engaged, the neutrophil secretory activity is shutdown; the cells remain intact and are phagocytosed by macrophages using recognition mechanisms that Et fail to elicit a pro-infiammatory response2,6. If macrophage phago- cytosis or neutrophil apoptosis is irnpaired, however, chronic inflam- mation may ensuéS,7. Consequently, the mechanisms involved in regulating inflammatory cell survival and apoptosis are the subject of considerable research endeavor. Cell division ofeukaryotic cells occurs in four phases (Gl, 5, G2, M) and in some circumstances, for example where growth factors are withdrawn or the cell is terminally differentiated, the cell will rest in GO phase. The CDKs have been traditionally described as key regulators of the cell cyele, whereby different CDKs become activated during cell-cyele progression when complexed with their associated cyclin partners8. For this reason, targeting CDKs by specific inhibitors may prevent or limit tunIor progression. Indeed, CDK inhibitors are under clinical trial for esophageal, lung, prostate and non-small-cell lung cancers9. ln contrast, inhibition ofCDKs attenuates apoptosis of terrninally differentiated neuronslO. The precise mechanisrns that deterrnine the effects of CDK inhibitors on apoptosis remain unelear, although these inhibitors downregulate Mel-1 (relS. 11,12), a key protein regulating apoptosisI3 ineluding neutrophil apoptosisI4. Neu- trophils are terrninally differentiated cells and therefore CDK inhibi- tors such as R-roscovitine wouId be predicted either to have no effect or, as in neurons, to inhibit apoptosislO. IMRC Centre for Inflammafion Research, Queen's Medical Research Institute, University of Edinburgh, 47 Little France Crescent, Edinburgh, EH16 4TJ, UK. 2Molecular Haematology and Cancer Biology Unit, Institute of Child Health, Great Ormond Street Hospital for Children NHS Trust, University College London, 30 Guilford Street, London WC1N 1EH, UK. 3These authors contributed equally to this article. Correspondence should be addressed to A.G.R. ([email protected]). Received 26 October 2005; accepted 24 July 2006; published online 3 September 2006; doi:1O.1038/nm1468 NATURE MEDICINE ADVANCE ON LINE PUBLICATION
Transcript of Corrigendum: Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting...

1;'--
îiîëdicine
ARTICLES
Cyclin-dependent kinase inhibitors enhance theresolution of inflammation by promotinginflammatory cell apoptoslS
GICU:0GIEf::11iicE0uI!!::1...t'CI
j-..;ë.:=.cD..:1E!
C)CIc:c.!:ë:1Q.CI)...:1-ca
Zco00N
@
Adriano G Rossi1, Deborah A Sawatzky1.3, Annemieke WaIker1,3, Carol Ward1, Tara A Sheldrake1,Nicola A Riley1, Alison Caldicott1, Magdalena Martinez-Losa1, Trevor R WaIkerl, Rodger Duffinl,Mohini Gray1, Elvira Crescenzi2, Morag C MartinI, Hugh J Bradf, John S Savill1,lan Dransfield1&Christopher Haslett1
Apoptosis is essential for clearance of potentially injurious inflammatory cells and subsequent efficient resolution of inflammation.Here we report that human neutrophils contain functionally active cyclin-dependent kinases (CDKs),and that structurally diverseCDK inhibitors induce caspase-dependent apoptosis and override powerful anti-apoptosis signais from survival factors such asgranulocyte-macrophage colony-stimulating factor (GM-CSF).We show that the CDKinhibitor R-roscovitine (Seliciclib or CYC202)markedly enhances resolution of established neutrophil-dependent inflammation in carrageenan-elicited acute pleurisy, bleomycin-induced lung injury, and passively induced arthritis in mice. ln the pleurisy model, the caspase inhibitor zVAD-fmkpreventsR-roscovitine-enhanced resolution of inflammation, indicating that this CDKinhibitor augments inflammatory cell apoptosis.We also provide evidence that R-roscovitine promotes apoptosis by reducing concentrations of the anti-apoptotic protein Md-l.Thus, CDK inhibitors enhance the resolution of established inflammation by promoting apoptosis of inflammatory cells, therebydemonstrating a hitherto unrecognized potential for the treatment of inflammatory disorders.
Neutrophils have a central role in innate immunity and are rapidlyrecruited to sites of infection and injury; however, their many defensemechanisms that destroy and digest invading microorganismsare potentially deleterious to tissuesi. Thus, it is vital that, oncetheir physiological function has been achieved, these inflammatorycells and their potentially histotoxic contents are eleared rapidly.During spontaneous resolution of inflammation, neutrophils undergoapoptosis: a pre-programmed and highly regulated cell death processthat results in shutdown of secretory capacity and allows recognitionand removal by macrophages2,3. Neutrophil survival and apoptosis areprofoundly influenced by the inflammatory milieu: inflammatorymediators such as GM-CSF or lipopolysaccharide (LP5), environ-mental conditions such as hypoxia, and the presence of pro-apoptoticstimuli such as tunIor necrosis factor-a; or Fas ligand markedly alterneutrophillongevity4,S.
Neutrophil apoptosis is controlled by a complex network of sigrtal-ing pathways that regulate both the turnover of key molecules,ineluding the anti-apoptotic protein myeloid cell leukemia 1 (Mel-1)and the pro-apoptotic Bel-2 farnily member Bax, and activation of thecaspase family of proteasess. Once apoptosis has been engaged, theneutrophil secretory activity is shutdown; the cells remain intact andare phagocytosed by macrophages using recognition mechanisms that
Et
fail to elicit a pro-infiammatory response2,6. If macrophage phago-cytosis or neutrophil apoptosis is irnpaired, however, chronic inflam-mation may ensuéS,7. Consequently, the mechanisms involved inregulating inflammatory cell survival and apoptosis are the subject ofconsiderable research endeavor.
Cell division ofeukaryotic cells occurs in four phases (Gl, 5, G2, M)and in some circumstances, for example where growth factors arewithdrawn or the cell is terminally differentiated, the cell will restin GO phase. The CDKs have been traditionally described as keyregulators of the cell cyele, whereby different CDKs become activatedduring cell-cyele progression when complexed with their associatedcyclin partners8. For this reason, targeting CDKs by specific inhibitorsmay prevent or limit tunIor progression. Indeed, CDK inhibitors areunder clinical trial for esophageal, lung, prostate and non-small-celllung cancers9. ln contrast, inhibition ofCDKs attenuates apoptosis ofterrninally differentiated neuronslO. The precise mechanisrns thatdeterrnine the effects of CDK inhibitors on apoptosis remain unelear,although these inhibitors downregulate Mel-1 (relS. 11,12), a keyprotein regulating apoptosisI3 ineluding neutrophil apoptosisI4. Neu-trophils are terrninally differentiated cells and therefore CDK inhibi-tors such as R-roscovitine wouId be predicted either to have no effector, as in neurons, to inhibit apoptosislO.
IMRC Centre for Inflammafion Research, Queen's Medical Research Institute, University of Edinburgh, 47 Little France Crescent, Edinburgh, EH16 4TJ, UK.2Molecular Haematology and Cancer Biology Unit, Institute of Child Health, Great Ormond Street Hospital for Children NHS Trust, University College London,30 Guilford Street, London WC1N 1EH, UK. 3These authors contributed equally to this article. Correspondence should be addressed to A.G.R. ([email protected]).
Received 26 October 2005; accepted 24 July 2006; published online 3 September 2006; doi:1O.1038/nm1468
NATURE MEDICINE ADVANCE ON LINE PUBLICATION
QIC'ü'5QIEQI:;aï.eE0uai...::J+'lU
i:::::ii:t:s:.a.:J0...~C)1::c.!!!:ë::J
Go
e::J1üzCI)00C\I
@.
ARTICLES
a b100
C 103
83' F2
",,102
2 10' F3 -F'10°
100 10' 102 103FL110g
g>go'g 80:E:;- 70
.~ 60ê 50m'0 40"g> 30~ 20~if 10
00
~0.1 1 10
Log[conc] Ù1M)
--- R-Roscovitine (20 !lM)
-+- NG75 (10 !lM)
-+- HO (10 !lM)
8 12Time(h)
16 20
d 103
~'o2~ 10'1J.
100
r. .
.,..t. /# fi_.
"tf---","..~ :.'"~
e 103m
",,1020
~ lO'lF3"-10°
##
#
-. ....f g> 100
i :> 70.~ 60
:g 50
:ii 40'0 30" 20g> 10
~ 0~0.
#
il#
i#
i0 10 1520
1
0 1015201
0 1015201
0 101520Control dbcAMP GM-GSF LPS
R-Roscovitine (j1M)
Here we have investigated whether CDK inlulJitors can influenceneutrophil apoptosis in vitro and consequently the resolution ofneutrophilic inflammation in vivo. We show that human neutrophilsexpress functional CDKs and that different CDK inhibitors directiyinduce caspase-dependent neutrophil apoptosis and inhibit ceIlsurvival induced by several biologically important powerful anti-apoptotic agents. We also show that the CDK inlulJitor R-roscovitinedownregulates Md-1 expression induced by survival fàctors in neu-trophils. ln addition, we demonstrate in vivo that R-roscovitinemarkedly enhances resolution of inflammation in mouse modelsof carrageenan-induced acute pleurisy, bleomycin-induced lunginflammation and passively induced arthritis. The R-roscovitine-enhanced resolution of established pleurisy is driven by a caspase-mediated pro-apoptotic effect. These findings suggest that CDKinhibitors may provide a therapeutic strategy to promote resolutionof inflammatory diseases.
RESULTS
CDK inhibitors induce human neutrophil apoptosisTo investigate whether CDK inhibitors can affect apoptosis directiy,human neutrophils were incubated over a 20-h period with thestructurally diverse CDK inhibitors R-roscovitine15-17, NG75 (refs.
g g> 100'g go:E 80> 70.~ 60:g 50Iii 40'0 30& 20J'! 10B 0:f
10° 10' 102 103
FU log
##
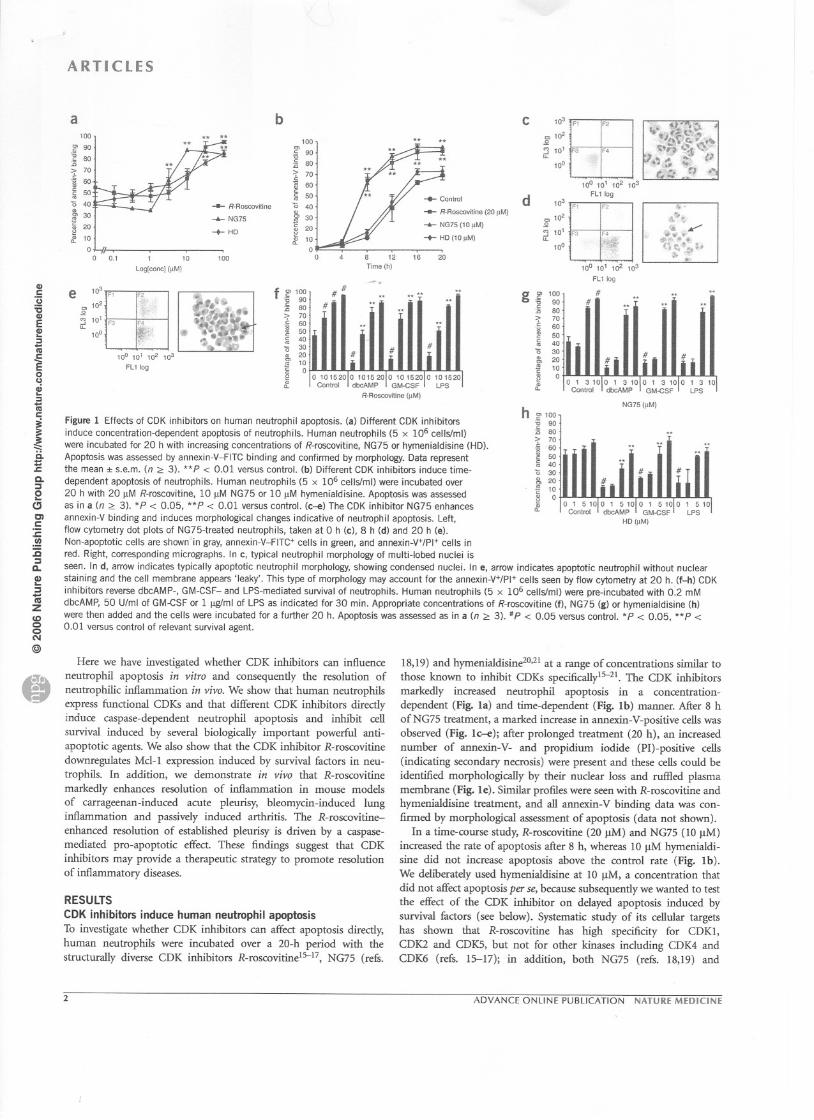
Figure 1 Effects of CDK inhibitors on hum an neutrophil apoptosis. (a) Different CDK inhibitorsinduce concentration-dependent apoptosis of neutrophils. Human neutrophils (5 x 106 cellsfm!)were incubated for 20 h with increasing concentrations of R-roscovitine, NG75 or hymenialdisine (HD).Apoptosis was assessed by annexin-V-FITC binding and confirmed by morphology. Data representthe mean :1:s.e.m. (n 2:: 3). **P < 0.01 versus control. (b) Different CDK inhibitors induce time-dependent apoptosis of neutrophils. Human neutrophils (5 x 106 cellsfm!) were incubated over20 h with 20 IlM R-roscovitine, 10 IlM NG75 or 10 IlM hymenialdisine. Apoptosis was assessedas in a (n 2::3). *P < 0.05, **P < 0.01 versus controL (c-e) The CDK inhibitor NG75 enhances
annexin-V binding and induces morphological changes indicative of neutrophil apoptosis. Left,flow cytometry dot plots of NG75-treated neutraphils, taken at 0 h (c), 8 h (d) and 20 h (e).Non-apoptotic cells are shown in gray, annexin-V-FITC+ cells in green, and annexin-V+/PI+ cells inred. Right, corresponding micrographs. ln c, tYpical neutrophil morphology of multi-Iobed nuclei isseen. ln d, arrow indicates tYpically apoptotic neutrophil morphology, showing condensed nuclei. ln e, arrow indicates apoptotic neutrophil without nuclearstaining and the cell membrane appears 'Ieaky'. This tYpe of morphology may account for the annexin-V+/PI+ cells seen by flow cytometry at 20 h. (f-h) CDKinhibitors reverse dbcAMP-, GM-CSF- and LPS-mediated survival of neutrophils. Human neutrophils (5 x 106 cellsfm!) were pre-incubated with 0:2 mMdbcAMP, 50 Ulml of GM-CSF or 1 ~ml of LPS as indicated for 30 min. Appropriate concentrations of R-roscovitine (f), NG75 (g) or hymenialdisine (h)were then added and the cells were incubated for a further 20 h. Apoptosis was assessed as in a (n 2:: 3). IlP < 0.05 versus control. *P < 0.05, **P <0.01 versus contrai of relevant survival agent.
0 1 3 101
0 1 3 101
0 1 3 101
0 1 3 10Control dbcAMP GM-GSF LPS
NG75 (!lM)
h go 100
i ::;- 70.~ 60~ 50:ii 40'0 30& 20i!! 10" 0~0. 0 c~;.,:OI °oo:,~~OI 0G~_~s~OI 0 ~P~ 10
HO Ù1M)
18,19) and hymenialdisine2°,21 at a range of concentrations similar tothose known to inhibit CDKs specificallyl5-21. The CDK inhibitorsmarkedly increased neutrophil apoptosis in a concentration-dependent (Fig. la) and time-dependent (Fig. 1b) manner. After 8 hofNG75 treatrnent, a marked increase in annexin-V-positive cells wasobserved (Fig. 1c--e); after prolonged treatrnent (20 h), an increasednumber of annexin-V- and propidium iodide (PI)-positive cells(indicating secondary necrosis) were present and these cells could beidentified morphologically by their nuclear loss and ruffled plasmamembrane (Fig. le). Similar profiles were seen with R-roscovitine andhymenialdisine treatrnent, and all annexin-V binding data was con-finned by morphological assessment of apoptosis (data not shown).
ln a time-course study, R-roscovitine (20 vM) and NG75 (10 !-lM)increased the rate of apoptosis after 8 h, whereas 10 vM hymenialdi-sine did not increase apoptosis above the control rate (Fig. Ib).We deliberately used hymenialdisine at 10 !lM, a concentration thatdid not affect apoptosis per se, because subsequently we wanted to testthe effect of the CDK inhibitor on delayed apoptosis induced bysurvival fàctors (see below). Systematic study of its cellular targetshas shown that R-roscovitine has high specificity for CDK1,CDK2 and CDKS, but not for other kinases including CDK4 andCDK6 (refs. 15--17); in addition, both NG75 (refs. 18,19) and
2 ADVANCE ONLINE PUBLICATION NATURE MEDICINE
** *.100
""." 90."1j 80
;;; 70. 60
:ii 50--- R-Roscovitine 40
-+- NG75 30
-+- HO Iii 20
if 100
100 0 4
F' F2
F3 F.f
.
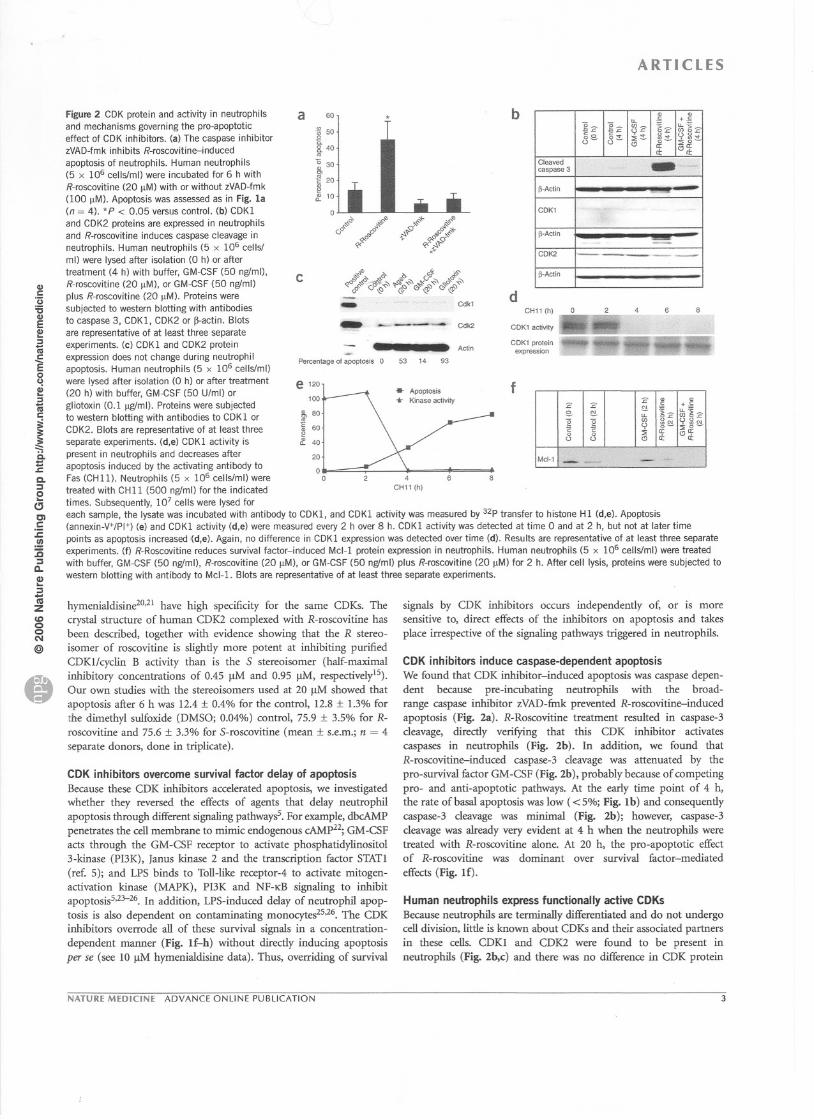
Figure 2 CDK protein and activity in neutrophils
and mechanisms governing the pro-apoptoticeffect of CDK inhibitors. (a) The caspase inhibitorzVAD-fmk inhibits R-roscovitine-induced
apoptosis of neutrophils. Human neutrophils
(5 x 106 cellslml) were incubated for 6 h with
R-roscovitine (20 !lM) with or without zVAD-fmk
(100 !lM). Apoptosis was assessed as in Fig. la(n = 4). *P < 0.05 versus control. (b) CDKI
and CDK2 proteins are expressed in neutrophils
and R-roscovitine induces caspase cleavage inneutrophils. Human neutrophils (5 x 106 cellsl
ml) were Iysed after isolation (0 h) or aftertreatment (4 h) with buffer, GM-CSF (50 ngfml),
R-roscovitine (20 !lM), or GM-CSF (50 ngfml)
plus R-roscovitine (20 !lM). Proteins were
subjected to western blotting with antibodies
to caspase 3, CDKl, CDK2 or ~actin. Blots
are representative of at least three separate
experiments. (c) CDKI and CDK2 protein
expression does not change during neutrophilapoptosis. Human neutrophils (5 x 106 cellslml)
were Iysed after isolation (0 h) or after treatment
(20 h) with buffer, GM-CSF (50 U/ml) or
gliotoxin (0.1 !lgfmi). Proteins were subjected
to western blotting with antibodies to CDK 1 or
CDK2. Blots are representative of at least three
separate experiments. (d,e) CDKI activity is
present in neutrophils and decreases after
apoptosis induced by the activating antibody to
Fas (CHll). Neutrophils (5 x 106 cellslml) were
treated with CHII (500 ngfml) for the indicated
times. Subsequently, 107 cells were Iysed for
each sam pie, the Iysate was incubated with antibody to CDKl, and CDKI activity was measured by 32p transfer to histone Hl (d,e). Apoptosis(annexin-V+/PI+j (e) and CDKl activity (d,e) were measured every 2 h over 8 h. CDKl activity was detected at time 0 and at 2 h, but not at later time
points as apoptosis increased (d,e). Again, no difference in CDKl expression was detected over time (d). Results are representative of at least three separateexperiments. (f) R-Roscovitine reduces survival factor-induced Md-l protein expression in neutrophils. Human neutrophils (5 x 106 cellslml) were treated
with buffer, GM-CSF (50 ngfml), R-roscovitine (20 !lM), or GM-CSF (50 ngfml) plus R-roscovitine (20 !lM) for 2 h. After cell Iysis, proteins were subjected to
western blotting with antibody to Md-!. Blots are representative of at least three separate experiments.
a 60
'"! 508.40..
~30'"j!! 208:f 10
0
ARTICLES
b
if"" ;,.{>,,0 #" $,,0(,0 ,,00 "".? ,,00~~..,
C«~o C«~:~x""
C ."-"',, "" b 4 cf"~~<Jf'!~ ~<r."'~0~~"'~&~~d' ~>Œ >Œ~- d
CI)&::
'ü:sCI)ECI):ï1ii
~0uai:ï1ii
i:::::S.1:0..::J0(;CIc:c:.!!:a::Ja..e::J-ni
ZCD00N
@
--Percentage of apoptosis 0
e120
100
Cdk1CHII (h) 0 2 - 4
CDKI activity I~ ~----CDK1 prot.,;n """. ""'0"-""expression- - - --
6Cdk2- . Actin
53 9314
... Apoptosis
... Kinase activity
f
&80..ë 60~a. 40
20
00
hymenialdisine2°,21 have high specificity for the same CDKs. Thecrystal structure of human CDK2 complexed with R-roscovitine hasbeen described, together with evidence showing that the R stereo-isomer of roscovitine is slightly more potent at inhibiting purifiedCDKl/cyclin B activity than is the S stereoisomer (half-maximalinhibitory concentrations of 0.45 ~ and 0.95 ~, respectively15).Our own studies with the stereoisomers used at 20 /lM showed thatapoptosis after 6 h was 12.4 :t 0.4% for the control, 12.8 :t 1.3% forthe dimethyl sulfoxide (DMSO; 0.04%) control, 75.9 :t 3.5% for R-roscovitine and 75.6 :t 3.3% for S-roscovitine (mean :t s.e.m.; n = 4
separate donors, done in triplicate).
CDK inhibitors overcome survival factor delay of apoptosisBecause these CDK inhibitors acœlerated apoptosis, we investigatedwhether they reversed the effects of agents that delay neutrophilapoptosis through different signaling pathwaysS. For example, dbcAMPpenetrates the celI membrane to mimic endogenous cAMP22;GM-CSFacts through the GM-CSF receptor to activate phosphatidylinositol3-kinase (PI3K), Janus kinase 2 and the transcription factor STAT!
(ref. 5); and LPS binds to TolI-like receptor-4 to activate mitogen-activation kinase (MAPK), PI3K and NF-KB signaling to inhibitapoptosis5,23-26. ln addition, LPS-induced delay of neutrophil apop-tosis is also dependent on contaminating monocytes25,26. The CDKinhibitors overrode all of these survival signals in a concentration-dependent manner (Fig. lf-h) without directIy inducing apoptosisper se (see 10 ~ hymenialdisine data). Thus, overriding of survival
4CH11(h)
6
signals by CDK inhibitors occurs independently of, or is moresensitive to, direct effects of the inhibitors on apoptosis and takesplace irrespective of the signaling pathways triggered in neutrophils.
CDK inhibitors induce caspase-dependent apoptosisWe found that CDK inhibitor-induced apoptosis was caspase depen-dent because pre-incubating neutrophils with the broad-range caspase inhibitor zVAD-fmk prevented R-roscovitine-inducedapoptosis (Fig. 2a). R-Roscovitine treatment resulted in caspase-3cleavage, directIy verifying that this CDK inhibitor activatescaspases in neutrophils (Fig. 2b). ln addition, we found thatR-roscovitine-induced caspase-3 cleavage was attenuated by thepro-survival factor GM -CSF (Fig. 2b), probably because of competingpro- and anti-apoptotic pathways. At the early time point of 4 h,the rate ofbasal apoptosis was low «5%; Fig. Ib) and consequentlycaspase-3 cleavage was minimal (Fig. 2b); however, caspase-3cleavage was already very evident at 4 h when the neutrophils weretreated with R-roscovitine alone. At 20 h, the pro-apoptotic effectof R-roscovitine was dominant over survival factor-mediated
effects (Fig. If).
Human neutrophils express functionally active CDKsBecause neutrophils are terminally differentiated and do not undergocelI division, little is known about CDKs and their associated partnersin these celIs. CDK1 and CDK2 were found to be present inneutrophils (Fig. 2b,c) and there was no difference in CDK protein
NATURE MEDICINE ADVANCE ONLINE PUBLICATION 3
" .."- ,s +.EO
g:2 ë- "'- 15:2 1:2E"" 0"":Se. '..,. ::!- 1J:!0"" ::;;-0 0- C) cr: C)a::
ci: ci:
Cleaved - ..
caspase 3
.Actin
CDK1
.AClin
CDK2 -j!-Actin
:2 " ":2 :2 c +.Se. '" ü
:2g,:2ILë '" '" y",E 0 0- ::;;0-
::1; cr: C)0 0ci:0 0 CJ
Mcl-1I...,... -.
CI)r:'ü:eCI)E!!!::11iir:E0u
2!::11ii
j--ii:=.r:Q.~!:!CJCIr::c.!!!:c~Q.CI)..~1iizCO00('II
@
e
ARTICLES
a .,. 350
S30
Ji! 25q;~ 200
t 15""E 10::i-= 5$t52 0
b 7
Saline Vehicle 10 mg/kg 100 mg/kg
control R-Roscovitine
Treatment24 h post-carrageenan
Saline Vehicle 10 mglkg 100 mglkg
control R-Roscovltine
Treatment 24 h post-carrageenan
C 0.20
0.15
Ê-;; 0.10E"."w 0.05
* ~6S. 5"'~ 4"(; 31J 2E~ 1
25
ln1Lu- 20 .-.Ë'" 15
f10 .. *
- 5 .0
Saline Vehicle 10 mglkg 100 mglkgcontrai R-Roscovitine
Treatment24 h post-carrageenan
0.00
Saline Vehicle 10 mg/kg 100 mg/kg
control R-Roscovitine
Treatment24 h post-carrageenan
f40
~'[30S>~ 20
~ 1: ** **Saline Vehicle 10 mg/kg 100 mglkg
control R-Roscovitine
Treatment 24 h post-carrageenan
100 mg/kg R-Roscovitine orvehicle control (i.p.)
t'~11
11 .
" ~ Acute inflammation
1 *** H R-Roscovitine**
0 1 . . ,If-,0 24 48 72 168
Ouralion of pleurisy post-carrageenan(h)
quantities in isolated cells feU rapidly within 2 h of culture, an effect
that was prevented by GM-CSF treatment. R-Roscovitine inhibitedGM-CSF-mediated upregulation of Md-l (Fig. 2f).
Roscovitine-induced apoptosis enhances inflammatory resolutionHaving shown in vitro that neutrophil apoptosis was markedlyinfluenced by CDK inhibitors, we determined the effects ofR-roscovitine on the resolution of neutrophil-dominant inflammation
in vivo. We used a weU-established acute resolving mouse modelof carrageenan-induced pleural inflammation28-31 to assess theeffects of the CDK inhibitor on the kinetics of inflammatoryceU recruitment and to investigate inflammatory mechanisms.R-Roscovitine accelerated the resolution of established inflammation
when administered intraperitoneally (i.p.) 24 h after intrapleuralinjection of 1% carrageenan (Fig. 3). Thus. treatment with 10 mgper kg body weight of R-roscovitine (i.p.) inhibited the totalinHammatory cell number by more than 50% as compared withvehide control (Fig. 3a), and also reduced the number of monocytes!macrophages and neutrophils (Fig. 3b). Of note, R-roscovitineat 100 mg per kg reduced the inflammatory cells to near basalnumbers normally found in the naive mouse pleural cavity.Anti-inflammatory effects of R-roscovitine were also evident because
edema formation (total exudate volume obtained by pleural
.-l--1***
0
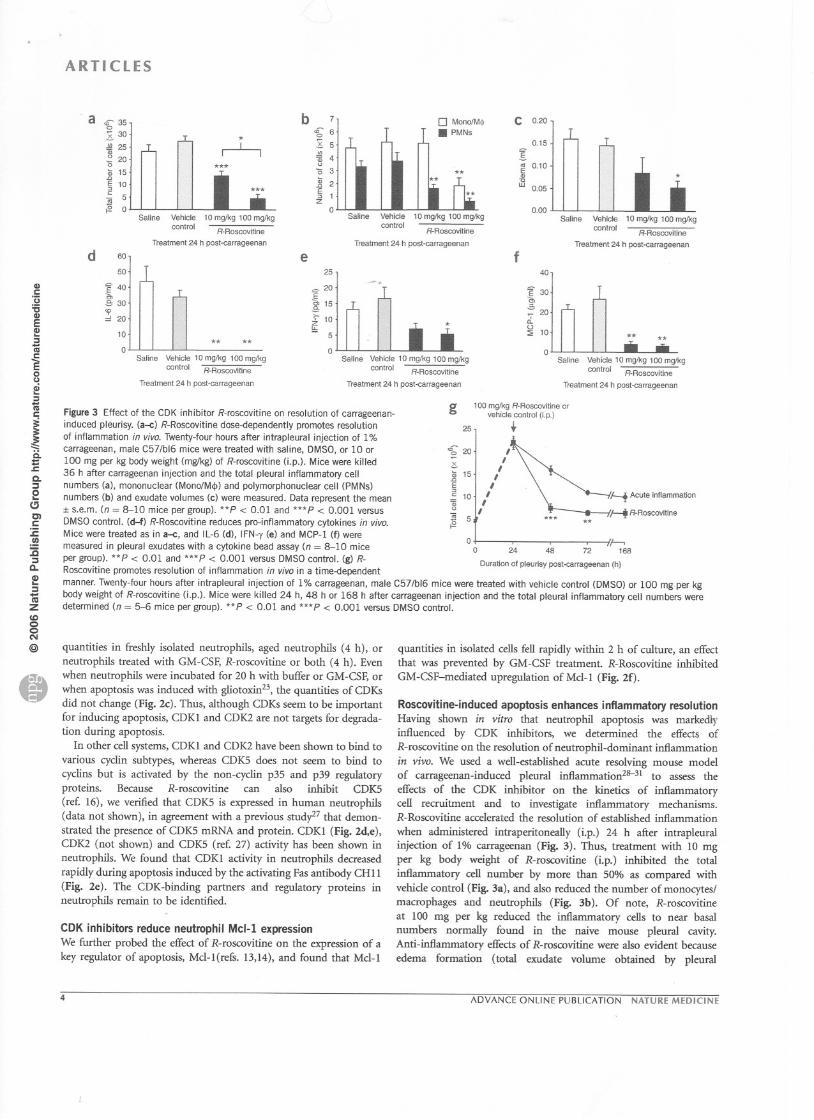
Figure 3 Effect of the CDK inhibitor R-roscovitine on resolution of carrageenan-induced pleurisy. (a--c) R-Roscovitine dose-dependently promotes resolution
of inflammation in vivo. Twenty-four hours after intrapleural injection of 1%
carrageenan, male C57/b16 mice were treated with saline, DMSO, or 10 or
100 mg per kg body weight (mg/kg) of R-roscovitine (j.p.). Mice were killed
36 h after carrageenan injection and the total pleural inflammatory cell
numbers (a), mononuclear (MonofM<!» and polymorphonuclear cell (PMNs)
numbers (b) and exudate volumes (c) were measured. Data represent the mean:1:s.e.m. (n = 8-10 mice per group). **P < 0.01 and ***P < 0.001 versus
DMSO control. (d-f) R-Roscovitine reduces pro-inflammatory cytokines in vivo.
Mice were treated as in a--c, and IL-6 (d), IFN-y (e) and MCP-l (f) were
measured in pleural exudates with a cytokine bead assay (n = 8-10 mice
per group). **P < 0.01 and ***P < 0.001 versus DMsa control. (g) R-Roscovitine promotes resolution of inflammation in vivo in a time-dependent
manner. Twenty-four hours after intrapleural injection of 1% carrageenan, male C57/b16 mice were treated with vehicle control (DMSO) or 100 mg.per kg
body weight of R-roscovitine (i.p.). Mice were killed 24 h, 48 h or 168 h after carrageenan injection and the total pleural inflammatory cell numbers weredetermined (n = 5-6 mice per group). **P < 0.01 and ***P < 0.001 versus DMSO control.
e
Saline Vehicle 10 mg/kg 100 mglkg
control R-RoscoVinne
Treatment 24 h post-carrageenan
quantities in freshly isolated neutrophils, aged neutrophils (4 h), orneutrophils treated with GM-CSF, R-roscovitine or both (4 h). Evenwhen neutrophils were incubated for 20 h with buffer or GM-CSF, orwhen apoptosis was induced with gliotoxin23, the quantities of CDKsdid not change (Fig. 2c). Thus, although CDKs seem to be importantfor inducing apoptosis, CDKI and CDK2 are not targets for degrada-tion during apoptosis.
ln other ceil systems, CDKI and CDK2 have been shown to bind tovarious cyclin subtypes, whereas CDKS does not seem to bind tocyclins but is activated by the non-cyclin p35 and p39 regulatorypro teins. Because R-roscovitine can also inhibit CDK5
(ref. 16), we verified that CDK5 is expressed in human neutrophils(data not shown), in agreement with a previous study27 that demon-
strated the presence of CDK5 mRNA and protein. CDKI (Fig. 2d,e),CDK2 (not shown) and CDKS (ref. 27) activity has been shown inneutrophils. We found that CDKI activity in neutrophils decreasedrapidly during apoptosis induced by the activating Fas antibody CH II(Fig. 2e). The CDK-binding partners and regulatory proteins inneutrophils remain to be identified.
CDK inhibitors reduce neutrophil Mcl-! expressionWe further probed the effèct of R-roscovitine on the expression of akey reguiator of apoptosis. Mel-l(refs. 13,14), and found that Mel-l
4
g25
'f2O~t 15""E"-= 10q;(J
]i 5t52
ADVANCE ONLINE PUBLICATION NATURE MEDICINE
d 60
50
= 40
.e, 30<0
20
10
0

a b 0.35
0.30
ARTICLES
C-30g>~ 25
",:0~t 20c.'X0"ê' § 15~~~ ~ 10gc>~S
~ 5~~ 0
60
€ 50xi 40
~30"'~ 20'iij
.2 10
0.25
IO.20"g 0.15'"w
0.10
0.05
~<?t if''<-,~",,0.,:'t' s..{)~0~6'~
"''«'?'
;,,""° ii>Q"';. ;,,;,>""°",,'" .0.°<11'- <3'
<,0 <S''«'?'
~~ ,$''<-''1;>''''°
.,:'t' s..{) O~"O~"''«'?'
0 0.00
.-. f g
° ~0 .;:.°
""~'" .o.i-~(y i'",,0 O~
'«'?'
~,<?t if''<-';s:.,>",,0
.,:'r' s..{):~6'"'Q:<'?'
""~,,,,0 ii>Q~ p~.0.° "" ,,0
,,0 0"Q:<'?'
Figure 4 Role of caspase-dependent apoptosis in R-roscovitine-enhanced resolution of carrageenan-induced pleurisy. (a,b) The caspase inhibitor zVAD-fmkprevents R-roscovitine-induced resolution of carrageenan-induced inflammation. Twenty-four hours after intrapleural injection of 1% carrageenan, maleC57/b16 mice were treated with 10 mg per kg of R-roscovitine (Lp.) and/or 1 ~ per kg of zVAD-fmk (j.p. at 4-h intervals). Mice were killed 36 h aftercarrageenan injection and total pleural inflammatory cell numbers (a) and exudate volumes (b) were measured. Data represent the mean :!:s.e.m.(8-10 mice per group). 'p < 0.05, **P < 0.01 and *'*p < 0.001 versus DMSOcontrol. (c) zVAD-fmkprevents apoptosis during R-roscovitine-inducedresolution of carrageenan-induced inflammation. Mice were treated as in a,b, and apoptosis was analyzed in pleural inflammatory cells by annexin-V labelingand flowcytometry(n = 8-10 mice per group). 'p < 0.05 versus DMSOcontrol. Inflammatorycell apoptosis was also assessed by morphologyusingcyto-œntrifuge preparations stained with hematoxylin and eosin. (d-g) R-Roscovitine reduces inflammation in pleural lavage exudates and Jung tissue, anddecreases the numbers of macrophages containing apoptotic bodies in pleural lavage. Twenty four hours after intrapleural injection of 1% carrageenan,male C571b16 mice were treated i.p. with vehicle control (d,t) or 100 mg per kg of R-roscovitine (e,g). Mice were killed 36 h after carrageenan injection.Cyto-centrifuge preparations stained with hematoxylin and eosin (d,e) and tissue sections of pleural lavage and lungs (f,g) were assessed morphologicallyby microscopy (original magnification x40). Asterisks indicate viable neutrophils (dJ; arrows indicate phagocytosed apoptotic neutrophils (e).
.
CIlC
Ü
'6CIlEI!!:J1iicE0u~:J1ii
jë.:=.s::.
Q.~2~C1c.:c.!!:c~Q.CI)...~'IiizCI)00N
@
d
e "r ~ï,
;:' ~,'1..,,,...~, '
iif.
'
,
' ""''',
" :Mi'
,
:,
"
~.,~ .~.,~~t '~, . - ""'~. '
i',
""
'"
"". ., t'Ii<,Y;;. " ,,'#
'~,'"
,
',~
* "1
~.J. ~
i'
'/'''''''''
,
-,
"!l'" *, ",
",
",
tf ... J,,,,,!'
*
"':'
6*
washouts) decreased markedly with R-roscovitine treatment ascompared with saline control (*P < 0.05; Fig. 3c). ln parallel, releaseof the pro-inflarnmatory mediators IL-6 (Fig. 3d), IFN-y (Fig. 3e)and MCP-1 (Fig. 3f) in the exudate was also diminishedby R-roscovitine.
We next determined the effect, over a period of 7 d, of adminis-
tering R-roscovitine (100 mg per kg i.p.) at the peak of inflammation(24 h). We observed a reduction in total cell numbers in the pleuralcavity at ail time points (Fig. 3g) and in neutrophil and monocytes!macrophages numbers at 36 h after carrageenan injection (Fig.3a,b),providing further evidence that this CDK inhibitor enhances inflam-matory resolution. Because R-roscovitine induced caspase-dependentapoptosis of human neutrophils in vitro (Fig. 2a,b), we establishedwhether the enhanced resolution of inflammation by R-roscovitinewas mediated by a similar induction of caspase-dependentinflammatory cell apoptosis in vivo. Systemic administration ofthe broad-spectrum caspase inhibitor zVAD-fmk prevented theR-roscovitine-induced decrease in inflarnmatory cells and edemaformation in the pleural cavity in carrageenan-induced pleuralinflammation (Fig. 4). Administration of zVAD-fmk prevented theresolution of inflammation in vivo by increasing the total inflamma-tory cell number (Fig. 4a) and edema formation (Fig. 4b) ascompared with saline or vehicle controls. ln addition, the increasein inflarnmatory cell apoptosis (Fig. 4c) and macrophages containingapoptotic bodies (see Fig.4d,e) observed with R-roscovitine treatmentin the pleural cavity was inhibited markedly by treatment with zVAD-
fmk. We also noted that lungs of mice treated intrapleurally withcarrageenan had a consequent marked inflarnmatory infiltrate(Fig. 4f), which was reduced by R-roscovitine treatment (Fig. 4g).
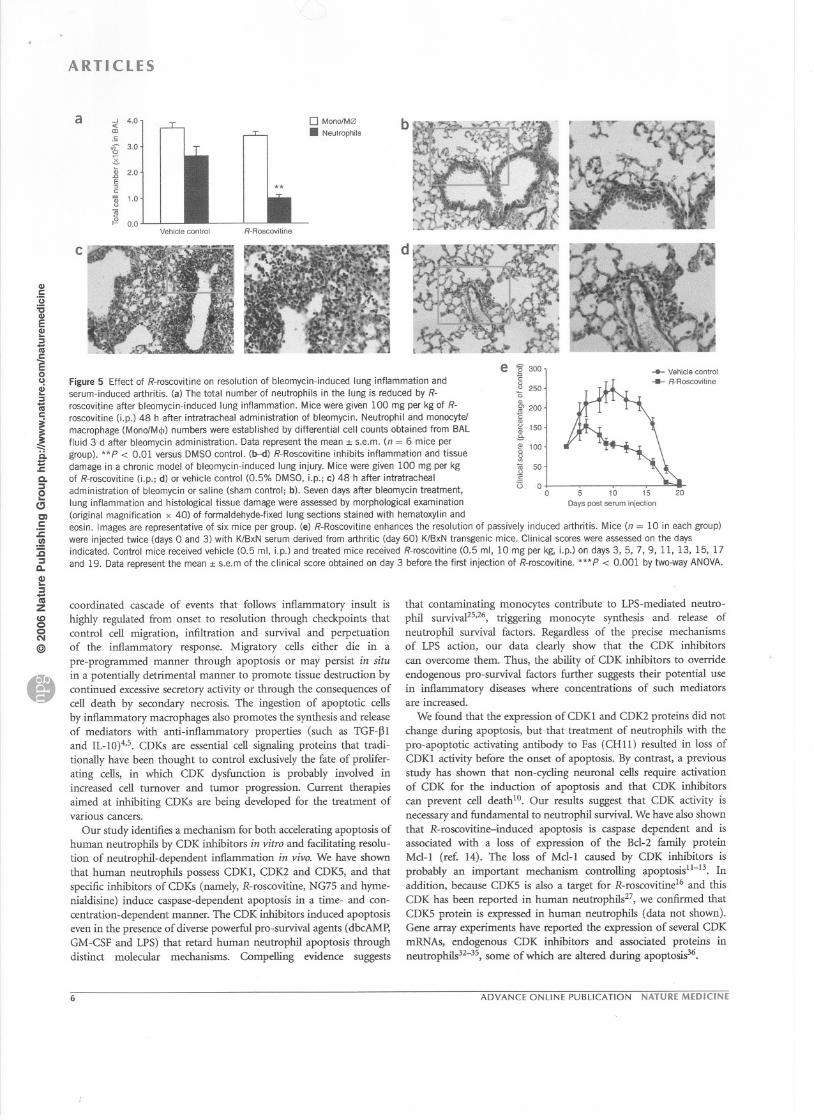
Roscovitine enhances inflammatory resolution in other modelsThe anti-inf1arnmatory and pro-resolving effects of R-roscovitine werefurther confirmed in another two inflarnmatory models. ln thebleomycin-induced lung injury mode!, R-roscovitine (100 mg per kgLp.), given after inflammation was established, reduced the totalnumber of neutrophils in the bronchoalveolar fluid 3 d after bleomy-cin administration (**P < 0.01; Fig. Sa). Histological examinationshowed that, when bleomycin-induced lung injury was aIlowed topersist for 7 d, R-roscovitine reduced bleomycin-induced inflamma-tion (Fig. Sb-d). R-Roscovitine, when administered with the shamintratracheal saline control, had no obvious detrimental effects on the
lung pathology (Fig. Sb). R-Roscovitine also reduced bleomycin-induced lethality (3 out of 8 mice treated with bleomycin alone diedby day 12, as compared with none of the R-roscovitine- and bleo-mycin-treated mice). ln addition, in a mode! of passively inducedarthritis, we observed that arthritis, as assessed by clinical scores,resolved more quickly in R-roscovitine-treated than in controlmice (Fig. Se).
DISCUSSION
Specific induction of inflammatory cell apoptosis represents anew approach for future treatment of inflammatory diseases. The
NATURE MEDICINE ADVANCE ONLINE PUBLICATION 5
.
Figure 5 Effect of R-roscovitine on resolution of bleomycin-induced lung inflammation andserum-induced arthritis. (a) The total number of neutrophils in the lung is reduced by R-
roscovitine after bleomycin-induced lung inflammation. M ice were given 100 mg per kg of R-
roscovitine (j.p.) 48 h after intratracheal administration of bleomycin. Neutrophil and monocyte!
macrophage (MonolMcjJ) numbers were established by differential cell counts obtained from BAL
fluid 3 d after bleomycin administration. Data represent the mean :1:s.e.m. (n = 6 mice per
group). ** P < 0.01 versus DMSO control. (b--d) R-Roscovitine inhibits inflammation and tissue
damage in a chronic model of bleomycin-induced lung injury. Mice were given 100 mg per kg
of R-roscovitine (j.p.; d) or vehicle control (0.5% DMSO, i.p.; c) 48 h after intratracheal
administration of bleomycin or saline (sham control; b). Seven days after bleomycin treatrnent,
lung inflammation and histological tissue damage were assessed by morphological examination
(original magnification x 40) of formaldehyde-fixed lung sections stained with hematoxylin and
eosin. Images are representative of six mice per group. (e) R-Roscovitine enhances the resolution of passively induced arthritis. Mice (n = 10 in each group)
were injected twice (days 0 and 3) with KlBxN serum derived from arthritic (day 60) K/BxN transgenic mice. Clinical scores were assessed on the daysindicated. Control mice received vehicle (0.5 ml, i.p.) and treated mice received R-roscovitine (0.5 ml, 10 mg per kg, i.p.) on days 3,5, 7, 9, Il, 13, 15, 17
and 19. Data represent the mean :1:s.e.m of the clinical score obtained on day 3 before the first injection of R-roscovitine. ***P < 0.001 by two-way ANOVA.
ARTICLES
a 0 Mono/M0. Neutrophils;i. 4.0ro.5,;) 3.002$.:n 2.0.cE"c:~ 1.0<J<ü;§ 0.0
Vehide control R-Roscovitine
c
CIlc:::ë:ï:sCIlE2!~..lU
~0()
ai:sRi
i:::::ë.:t:.cCo::J0
(;CIc:ë.!Il::ë::J0..CI)...::J'Iiizco00(101@
coordinated cascade of events that follows inflammatory insult ishighly regulated from onset to resolution through checkpoints thatcontrol cell migration, infiltration and survival and perpetuationof the inflammatory response. Migratory cells either die in apre-programmed manner through apoptosis or may persist in situin a potentially detrimental manner to promote tissue destruction bycontinued excessive secretory activity or through the consequences ofcell death by secondary necrosis. The ingestion of apoptotic censby inflammatory macrophages also promotes the synthesis and releaseof mediators with anti-inflammatory properties (such as TGF-~land 1L-1O)4,5.CDKs are essential cell signaling proteins that tradi-tionally have been thought to control exclusively the fate of prolifer-ating cells, in which CDK dysfunction is probably involved inincreased cell turnover and tumor progression. Current therapiesaimed at inhibiting CDKs are being developed for the treatrnent ofvarious cancers.
Our study identifies a mechanism for both accelerating apoptosis ofhuman neutrophils by CDK inhibitors in vitro and facilitating resolu-tion of neutrophil-dependent inflammation in vivo. We have shownthat human neutrophils possess CDK1, CDK2 and CDKS, and thatspecific inhibitors of CDKs (namely, R-roscovitine, NG75 and hyme-nialdisine) induce caspase-dependent apoptosis in a time- and con-centration-dependent manner. The CDK inhibitors induced apoptosiseven in the presence of diverse powerful pro-survival agents (dbcAMP,GM-CSF and LPS) that retard human neutrophil apoptosis throughdistinct molecular mechanisms. Compelling evidence suggests
b
d
e'8300E0<J 250
ë
N 200c:"~ 150.s,~ 100~<ü 50<J:si3. 0
0
~ VehiclecontrolR-Roscovitine
5 10 15Days post serum injection
20
that contaminating monocytes contribute to LPS-mediatedneutro-phil survival25,26, triggering monocyte synthesis and release ofneutrophil survival factors. Regardless of the precise mechanismsof LPS action, our data clearly show that the CDK inhibitorscan overcome them. Thus, the ability of CDK inhibitors to overrideendogenous pro-survival factors further suggests their potential usein inflammatory diseases where concentrations of such mediatorsare increased.
We found that the expression of CDK1 and CDK2 proteins did notchange during apoptosis, but that treatrnent of neutrophils with thepro-apoptotic activating antibody to Fas (CHU) resulted in loss ofCDK1 activitybefore the onset of apoptosis. Bycontrast, a previousstudy has shown that non-cycling neuronal cells require activationof CDK for the induction of apoptosis and that CDK inhibitorscan prevent cell deathlO. Our results suggest that CDK activity isnecessary and fundamental to neutrophil survival. We have also shownthat R-roscovitine-induced apoptosis is caspase dependent and isassociated with a loss of expression of the Bcl-2 family proteinMcl-1 (ref. 14). The loss of Mcl-1 caused by CDK inhibitors is
probably an important mechanism controlling apoptosisll-13. lnaddition, because CDKS is also a target for R-roscovitinel6 and thisCDK has been reported in human neutrophils27, we confirrned thatCDK5 protein is expressed in human neutrophils (data not shown).Gene array experiments have reported the expression of several CDKmRNAs, endogenous CDK inhibitors and associated proteins inneutropMs32-35, $Orne of which are altered during apoptosis36.
6 ADVANCE ONLINE PUBLICATION NATURE MEDICINE

CIl1:U'6CIlEI!!:11;j1:E0u2!:11;j
j......
ii.=J:.Q.:JeCJCIc:c.!:a:JQ.G)...:J-10ZCO00N@)
Cf»
We previously showed that inhibiting pro-survival molecules fromthe MAPK and Bel-2 families (namely, ERKl/2 and Bel-xL>promotesresolution of inflammation by inducing inflammatory ceIl apoptosisin an acute rat model of carrageenan-induced pleurisy, whereasinhibition of the pro-apoptotic molecule Bax prevents resolution ofinflammation30. We have now shown that R-roscovitine promotesresolution of inflammation in carrageenan-induced pleurisy in adose-dependent manner. R-Roscovitine induced apoptosis in vivo,
as assessed by annexin-V labeling and morphological analysis.R-Roscovitine also inhibited the release of pro-inflammatorycytokines, ineluding IL-6, MCP-I and IFN-y, further supporting itspro-resolution role. ln addition, R-roscovitine, administered atthe peak of inflammation, enhanced the resolution of bleomyciiî:induced lung inflammation. This model was used for its clinicalrelevance37 and because progression of the marked acute inflamma-tory response induced by bleomycin leads to chronic inflammationand fibrosis38,39.Early accumulation of neutrophils in the BAL fluidand subsequent lung inflammation and injury were attenuated byR-roscovitine treatrnent. R-Roscovitine also reduced bleomycin-induced lethality, indicating that CDK inhibition attenuates prolongedinflammation, leading to lung damage and consequent death.R-Roscovitine also caused enhanced inflammatory resolution inserum-induced arthritis as assessed by improved clinical scores.
Our in vitro work showing that the CDK inhibitors promoteneutrophil apoptosis adds to the evidence indicating that neutrophilsare essential in regulating the inflammatory responses in vivo,ineluding models of carrageenan-induced pleurisro, bleomycininduced-Iung inj~8,39 and arthritis7. We also showed that inhibitionof apoptosis with zVAD-fink prevents the enhanced resolutionof inflammation by R-roscovitine in carrageenan-induced pleurisy.The dosing regime for zVAD-fink administration is paramount toelicit an effect in vivo and was based on studies where injections wereadministered every 3 h (ref. 40). Notably, R-roscovitine-inducedapoptosis and its anti-inflammatory effects were reversed in vivo bythe caspase inhibitor zVAD-fmk, providing further evidence thatthe anti-inflammatory mechanism of the CDK inhibitor is due tothe induction of caspase-dependent inflammatory ceIl apoptosis.
Although neutrophils are terminally differentiated, evidence indi-cates that during the early stages of resolution of inflammation theyare capable of phenotypic alteration, switching from a phenotype thatgenerates pro-inflammatory mediators to one generating mediatorsthat are anti-inflammatory and/or pro-resolving. For example, neu-trophils can produce and respond to agents, such as lipoxins andresolvins, that have anti-inflammatory and pro-resolution proper-ties41--43.ln addition, accelerated resolution of inflammation has been
reported for endogenous lipid mediators such as protectin Dl (ref.44), suggesting that control of inflammation can be achieved physio-logically through production of endogenous mediators. Our hypo-thesis that manipulation of pro-resolution mechanisms can haveprofound effects on established inflammation is strongly supportedby data showing that pharrnacological manipulation of apoptosisand/or macrophage clearance can be achieved3o,45 and our presentfinding that modulation of granulocyte apoptosis with CDK inhibitorspromotes resolution of inflammation.
Thus, systemic administration of a specific inhibitor of CDKs inducesapoptosis of inflammatorycells in situ and promotes inflammatoryresolution. ln vivo studies investigating the effect of CDK inhibition incancer have shown that -systemic administration of R-roscovitinereduces tumor Size46and that R-roscovitine (even at high doses of2 g per kg) is well tolerated. It is widely acknowledged that tumor cellshave defective CDK pathways that promote ceIl proliferation and
ARTICLES
prevent ceIl apoptosis8,9, suggesting that CDK inhibitors specificallytarget these pathways to elicit their therapeutic action. Our study hasshown that CDK inhibitors also promote apoptosis in non-proliferatinginflammatory cells in vitro and accelerate inflammatory resolutionby promoting apoptosis and subsequent safe elearance of neutrophilsby macrophages in vivo. ln conclusion, given the renewed interest inthe elose association between inflammatory processes and cancer47,48and the potential problems with using monoelonal antibodies andother biological agents, this newly identified role of the small-moleculeanti-cancer CDK inhibitors may have potential for the treatrnent ofdiseases associated with increased or persistent inflammatory responses.
METHODSNeutrophil isolation and assessment of apoptosis. Weisolated hwnan bloodneutrophiJs23,49, a proœss that takes - 2 h, and culture<! the cells (5 x 106/ml,
37°C) in Iscove's modified Dulbecco's mediwn (IMDM) containing 10%aulologous serum. The ceIIs were incubated with the following reagents:
R-roscovitine, (R)- 2-[ [9-( 1-methylethyl)-6-[ (phenylmethyl)amino] -9H-purin-2-yl]amino ]-l-butanol (A.G. Scientific); hymenialdisine, 4-(2-amino-4-oxo-2-imidazolidin-5-ylidene )-2-bromo-4,5,6,7 -tetrahydropyrrolo[ 2,3-c] azepin-8-one(from L. Meijer, Roscoff, France); NG75, 2-(lR-isopropyl-2-hydroxyethyl-amino)-6-(4-metoxy-benzylamino)-9-isopropylpurine (from N. Gray, Univer-sity of Califomia, Berkeley); db-cAMP (Sigma); GM-CSF (R&D SySlems); andLPS (Escherichia coli 0127:B8, Sigma). We assessed apoptosis by flow cytometryusing annexin-V-conjugated fluorescein isothiocynate (frIC; Roche) in com-bination with PI (Sigma) and used light microscopy to confirm the presence ofmorphological changes characteristic of apoptosïsU3.
Western blotting. We lysed cells (5 x 106) for 15 min with 0.1% Nonidet P40
in 1 ml of TBS containing a protease inhibitor cocktail23 before centrifugation(23,100g, 4°C, 20 min). Protein samples (40 I1gor the equivalent of 1.5 x 106ceIIs per Jane) were resolved by SDS-PAGE and then transferred to polyvinyli-dene difluoride (PVDF) membranes. Blots were blocked with 5% skimmed
miIk powder in TES plus Tween before probing with antI"bodies to CDK1(Transduction Laboratories), CDK2 (Transduction Laboratories), CDlC5 (San-
ta-Cruz Biotechnology), caspase-3 (Cell Signaling Technologies), Mcl-1 (Santa-Cruz Biotechnology) or IJ-actin (Sigma).
Kinase assays. We assayed kinase activity by immunoprecipitation of CDK1(A17 antibody; Phanningen) or CDK2 (Ml antibody; Santa Cruz Biotechno-logy), followed by incubation with histone Hl and [y-32p]ATP (ref. 50).1mmunoprecipitates were reso1ved by SDS-PAGE, stained with Coomassie blueto visuaIize the histone Hl bands, dried and exposed for auloradiography.
Carrageenan-induœd pleurisy. Male C57BU6J miœ (aged 8-12 weeks) wereinjected intrapleurally with 0.1 ml of 1% Â.-carrageenan (Marine Colloids),followed 24 hlater byO.5 ml of saline contro~ 0.5 ml ofO.5% DMSO vehicle con-trol, or 10 or 100 mg per kg of R-roscovitine (i.p.). Mice were killed and pleuralcavities were lavaged with 1 ml of 3.15% (w/v) sodiwn citrate in PBS. Edema
formation was measured by pleural exudate weight. CelI counts, apoptosis and
macrophage phagocytosis in pleural cell samples were assessed microscopically.
Administration of zVAD-fmk. Twenty-four hOUTSafter intrapleural injection
of carrageenan, mice were injected i.p. with 0.5 ml of 10 mg per kg ofR-roscovitine and/or 0.5 ml of 1 J1gper kg of zVAD-fmk (z-Val-Ala-DL-Asp-fluoromethylketone; Bachem) at a final concentration of 1 mg/ml in saline, orwith appropriate amounts of DMSO vehicle control. Two additional doses ofzVAD-fmk were given i.p. 4 and 8 h later, and aIl mice were killed after 12 h.
Cytokine analysis. We measured cytokine leveJsby using a mouse inflammatÎoncytokine bead kit (BD Biosciences) according to the manufacturer's instructions.
Bleomycin-inducedlung injury. MaleC57BU6Jmice (aged8-12 weeks)weregiven either 0.05 ml of 0.1 U of bleomycin (Apollo Scientific) or salineintratracheally(sham control), and then treated 48 h later with either 0.5 mlof 0.5% DMSO vehiclecontrol or 100mg per kg of R-roscovitine.The micewere killed72 h or 7 d after bleomycinor saline administration. ln the acute
NATURE MEDICINE ADVANCE ON LINE PUBLICATION 7

.
ARTICLES
cu1:U:scuEcu:s1ii1:E0(,)ai:s1ii
i~ii:t:J:C.~e~CIc:c.!:ë~
0..
~~-alZco00N@)
72-h experiments, bronchoalveolar lavage were performed with three sequentialwashes with 0.8 ml of iœ-cold saline before perfusion with 4% formaldehydefor tissue histological analysis. Differentiai œil counts were done on cytoœn-trifuge preparations with eosin and hemotoxylin staining. Histological analysisof the 7-d experiments was done without bronchoalveolar lavage to maintaintissue integrity. Experiments were done on untreated control, sham control(saline and DMSO treatment), R-roscovitine control (saline and 100 mg per kg
of R-roscovitine), bleomycin plus vehicle (DMSO) control, and bleomycin plusR-roscovitine treatment groups, with six miœ per group. Lungs were inflated,fixed with 1 ml of 4% formaldehyde and then decalcified with 5% nitric acid
for 3 h, and lung injury was assessed by histological examination of paraffin-embedded lung sections stained with hernatoxylin and eosin.
Induction and assessment of arthritis. Female C57BU6J mice (aged (HI
weeks) reœived two i.p. injections at 48-h intervals of sera (100 J.Ll)ttomarthritic KlBxN miœ (aged 60 d) and R-roscovitine or vehicle was administeredi.p. every 48 h. Arthritis was scored by clinical examination: a score of 1represents erythema alone or swelling of 1 digit; 2 represents erythema plus
swelling of the tarsal joints, or swelling of the hock or wrist joint alone;3 represents swelling in both tarsal and hock joints, both wrist and digits, ormore than two digits and the tarsal joints. The mean cumulative limb scores for
each mouse were calculated for each group, and data are expressed as theperœntage of the clinical score obtained on day 3 before the first injectionof R-roscovitine.
Statistical analysis. We analyzed in vitro experiments by analysis of variance
(ANOVA) and Student-Newman-Keuls comparison tests. AIl in vivo experi-ments included at least six miœ per group and experiments were repeated to
verify the original findings. Statistical analysis was done by a one-way ANOVAwith a Bonferroni multiple comparison post hoc test with a 95% confidenceintervaL Data are expressed as the mean :!:s.e.m. and values of P < 0.05 wereconsidered significant.
ACKNOWLEDGMENTS
We thank M. Clay and K. Miles for help with the in vivo models. We thank theArthritis Research Campaign, the Medical Research Council, UK, the NormanSalvesen Emphysema Trust, Asthma UK, and the Juan Esplugues Foundation forfinanciai support.
AUTHOR CONTRIBUTIONS
A.G.R. initiated, designed, directed and performed experiments and took overalIresponsibility for planning and writing the manuscript. O.A.S. helped design,perform and analyze the in vivo experiments and contributed to the writing ofthe in vivo component. A.W. helped design, perform and analyze the in vitroexperiments and contributed to the writing of the in vitro component. c.w.contributed intellectually to the manuscript and helped with the in vitro work.T.A.S.performed some in vitro apoptosis and western blotting experiments.N.A.R. performed the McI-1 western blot experiments. A.c. and MM.-Lperformed some in vitro apoptosis experiments. T.R.W.helped in the designof the western blotting and kinase experiments. R.D. helped in the bleomycinexperiments. M.G. helped in the design and execution of the arthritisexperiments. E.C. helped in the design and execution of the kinase experiments.M.C.M. performed preliminary apoptosis experiments with the CDK inhibitors.H.J.B. provided intellectual input and provided significant input to the designand execution of the kinase experiments. J.S.S.provided intellectual input. LD.provided intellectual input and contributed to the design of the experiments andthe writing of the manuscript. C.H. provided intellectual input and helped in thedesign and coordination of the project.
COMPETING INTERESTS STATEMENT
The authors dedare that they have no competing financial interests.
Published online at http://www.nature.com/naturemedicinel
Reprints and permissions information is available online at httpHnpg.nature.com/œprintsandperm5s~nsI
1. Nathan, C. Points of control in inflammation. Nature 420, 846-852 (2002).2. Savill, J.S. et al. Macrophage phagocytosis of aging neutrophils in inflammation.
Programmed cell death in the neutrophil leads to its recognition by macrophages.J. Clin. Invest. 83, 865-875 (1989).
3. Savill, J., Dransfield, 1., Hogg, N. & Haslett, C. Vitronectin receptor-mediated phago-cytosis of cells undergoing apoptosis. Nature 343, 170--173 (1990).
4. Gilroy, D.w., Lawrence, T., Perretti, M. & Rossi, A.G. Inflammatory resolution: newopportunities for drug discovery. Nat. Rev. Orug Olscov. 3, 401-416 (2004).
5. Riley, NA et al. Granulocyte apoptosis and macrophage clearance of apoptotic cells astargets for pharmacological intervention in inflammatory diseases. Antl-Inflamm. Antl-AI/ergy Agents Medicinal Chem. 5, 3-12 (2006).
6. Whyte, M.K., Meagher, L.C., MacDermot, J. & Haslett, C. Impairment of function inaging neutrophils is associated with apoptosis. J. Immuno/. 150, 5124-5134 (1993).
7. Jonsson, H., Allen, P. & Peng, S.L. Inflammatory arthritis requires Fox03a to preventFas ligand-induced neutrophil apoptosis. Nat. Med. Il, 666-671 (2005).
8. Vermeulen, K., Van Bockstaele, D.R. & Berneman, Z.N. The cell cycle: a review of regu-lation, deregulation and therapeutic targets in cancer. Cel/ Profif. 36,131-149 (2003).
9. Senderowicz, A.M. Small-molecule cyclin-dependent kinase modulators. Oncogene 22,6609-6620 (2003).
10. Monaco, LA., III. &Vallano, M.L. Cyclin-dependent kinase inhibitors: cancer killers toneuronal guardians. Curr. Med. Chem. 10, 367-379 (2003).
Il. MacCallum, D.E. et al. Seliciclib (CYC202, R-Roscovitine) induces cell death inmultiple myeloma cells by inhibition of RNA polymerase II-dependent transcriptionand down-regulation of Mcl-1. Cancer Res. 65, 5399-5407 (2005).
12. Raje, N. et al. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependentkinase inhibitor, mediates activity via down-regulatîon of McI-1 in multiple myeloma.Blood 106, 1042-1047 (2005).
13. Michels, J., Johnson, P.W. & Packham, G. McI-1. Int. J. BIochem. Cell BIol. 37,267-271 (2005).
14. Moulding, DA, Quayle, J.A., Hart, C.A. & Edwards, S.W. McI-1 expression inhuman neutrophils: regulation by cytokines and correlation with cell survival. Blood92, 2495-2502 (1998).
15. De Azevedo, W.F. et al. Inhibition of cyclin-dependent kinases by purine analogues:crystal structure of human cdk2 complexed with roscovitine. Eur. J. BIochem. 243,518-526 (1997).
16. Meijer, L. et al. Biochemical and cellular effects of roscovitine, a potent and selectiveinhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. BIochem. 243,527-536 (1997).
17. Bach, S. et al. Roscovitine targets, protein kinases and pyridoxal kinase. J. BIol. Chem.280, 31208-31219 (2005).
18. Gray, N.S. et al. Exploiting chemicallibraries, structure, and genomics in the search forkinase inhibitors. Science 281, 533-538 (1998).
19. Chang, Y.T.et al. Synthesis and application of functionally diverse 2,6,9-trisubstitutedpurine libraries as CDK inhibitors. Chem. BIol. 6, 361-375 (1999).
20. Meijer, L. et al. Inhibition of cyclin-dependent kinases, GSK-3j} and CK1 by hyme-nialdisine, a marine sponge constituent. Chem. BIol.7, 51-63 (2000).
21. Williams, O., Gil-Gomez, G., Norton, T., Kioussis, D. & Brady, H.J. Activation ofCdk2 isa requirement for antigen-mediated thymic negative selection. Eur. J. Immunol. 3D,709-713 (2000).
22. Martin, M.C., Dransfield, 1., Haslett, C. & Rossi, A.G. Cyclic AMP regulation ofneutrophil apoptosis occurs via a novel protein kinase A-independent signaling path-way. J. BIol. Chem. 276, 45041-45050 (2001).
23. Ward, C. et al. NF-KBactivation is a critical regulator of human granulocyte apoptosisln vitro. J. BIol. Chem. 274, 4309-4318 (1999).
24. Ward, C. et al. Interleukin-lO inhibits lipopolysaccharide-induced survival and extra-cellular signal-regulated kinase activation in human neutrophils. Eur. J. Immunol. 35,2728-2737 (2005).
25. Sabroe, 1., Jones, E.C., USher, L.R., Whyte, M.K. & Dower, S.K. Toll-likereceptor <TlR)2 and TlR4 in human peripheral blood granulocytes: a critical role formonocytes in leukocyte lipopolysaccharide responses. J. Immunol. 168, 4701-4710(2002).
26. Sabroe, 1. et al. Selective roles for TolI-like receptor (TlR)2 and TlR4 in the regulationof neutrophil activation and life span. J. Immunol. 170, 5268-5275 (2003).
27. Rosales, J.L., Ernst, J.D., Hallows, J. & Lee, K.Y. GTP-dependent secretion fromneutrophils is regulated by Cdk5. J. BIol. Chem.279, 53932-53936 (2004).
28. Gilroy, D.W. et al. Inducible cyclooxygenase may have anti-inflammatory properties.Nat. Med. 5, 698-701 (1999).
29. Gilroy, D.W. et al. Inducible cyclooxygenase-derived 15-<Jeoxy(À)l2-14PGJ2 bringsabout acute inflammatory resolulion in rat pleurisy by inducing neutrophil andmacrophage apoptosis. FASEB J. 17, 2269-2271 (2003).
30. Sawatzky, DA, Willoughby, DA, Calville-Nash, P.R. & Rossi, A.G. The involvement ofthe apoptosis-modulating proteins ERK 1/2, BcI-Xi.and Bax in the resolution of acuteinflammation ln vivo. Am. J. Pathol. 168, 33-41 (2006).
31. Cailhier, J.F. et al. Resident pleural macrophages are key orchestrators of neutrophilrecruitment in pleural inflammation. Am. J. Resplr. Crit. Care Med. 173, 540--547(2006).
32. Klausen, P., Bjerregaard, M.D., Borregaard, N. & Cowland, J.B. End-stage differentia-tion of neutrophil granulocytes ln vivo is accompanied by up-regulation of p27kipland down-regulation of CDK2, CDK4, and CDK6. J. Leukoc. BIol. 75, 569-578(2004).
33. Tsukahara, Y. et al. Gene expression in human neutrophils during activation andpriming by bacterial lipopolysaccharide. J. Cel/. BIochem. 89, 848-861 (2003).
34. Zhang, X. et al. Gene expression in mature neutrophils: early responses to inflammatorystimuli. J. Leukoc. BIo/. 75, 358-372 (2004).
35. Subrahmanyam, Y.V.et al. RNA expression patterns change dramatîcally in humanneutrophils exposed to bacteria. Blood 97, 2457-2468 (2001).
36. Kobayashi, S.D., Voyich, J.M., Whitney, A.R. & Deleo, F.R. Spontaneous neutrophilapoptosis and regulation of cell survival by granulocyte macrophage-colony stimulatingfactor. J. Leukoc. BIol.78, 1408-1418 (2005).
8 ADVANCE ON LINE PUBLICATION NATURE MEDICINE

CD1:'ü=ëCDECD..::1-;1:E0uf!::1...III1:
~~:::::ii=.t:.c..~2CJenc:ë.!!!:ci~
a..
!!!~-cuZte)00N
@.
37. Azambuja, E., Fleck, J.E, Batista, R.G. & Menna Barreto,S.S. Bleomycin lung toxicity:who are the patients with increasedrisk? Pulm. Pharmacol. Ther.18,363-366 (2005).
38. Nagase, T. et al. A pivotai role of cytosolic phospholipase A2 in bleomycin-inducedpulmonary fibrosis. Nat. Med. 8, 480-484 (2002).
39. Teder, P. et al. Resolution of lung intlammation by CD44. Science 296, 155--158(2002).
40. De Paepe, M.E., Mao, Q., Embree-Ku, M., Rubin, L.P. & Luks, El. FaslFasL-mediatedapoptosis in perinatal murine lungs. Am. J. Physiol. Lung Cel/. Mol. Physiol. 287,L730-L742 (2004).
41. Levy, B.D., Clish, C.B., Schmidt, B., Gronert, K. & Serhan, C.N. Lipid mediator classswitchingduringacute inflammation: signais in resolution. Nat.lmmunol. 2,612-619(2001).
42. Serhan, C.N. et al. Resolvins: a family of bioactive products of omega-3 fatty acidtransformation circuits initiated by aspirin treatment that counter proinflammationsignais.J. Exp.Med.196,1025--1037(2002).
43. Serhan, C.N. & Savill, J. Resolution of inflammation: the beginning programsthe end.Nat. Immuno/. 6, 1191-1197 (2005).
ARTICLES
.-.
44. Serhan, C.N.et al. Anti-inflammatory actions of neuroprotectin D1/protectin Dl and its
natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J. Immu-nol. 176, 1848-1859 (2006).
45. Pinho, V. et al. Phosphoinositide-3 kinases critically regulate the recruitment andsurvival of eosinophils in vivo: importance for the resolution of allergie inflammation.J. Leukoc. Biol. 77, 800-810 (2005).
46. McClue,S.J. et al. ln vitroand in vivoantitumar properties of the cyclin dependentkinase inhibitor CYC202(R-roscovitine).Int. J. Cancer102, 463-468 (2002).
47. Caussens, L.M. & Werb, Z. Inflammation and cancer. Nature 420, 860-867 (2002).48. Karin, M. & Greten, ER. NF-ICB: linking inflammation and immunity to cancer
development and progression. Nat. Rev. Immunol.5, 749-759 (2005).49. Haslett, C., Guthrie, L.A., Kopaniak, M.M., Johnston, R.B., Jr. & Henson, P.M.
Modulation of multiple neutrophil functions by preparative methods or trace concen-trations of bacteriallipopolysaccharide. Am. J. Pathol.119, 101-110 (1985).
50. Gil-Gornez,G., Bems, A. & Brady, H.J. A link between cell cycle and cell death:Bax and BcI-2 modulate Cdk2 activation during thymocyte apoptosis. EMBOJ. 17,7209-7218 (1998).
NATURE MEDICINE ADVANCE ONLINE PUBLICATION 9




















