
Conserved Amino Acid Residues within the Amino-terminal Domain of ClpB are Essential for the...
10
Conserved Amino Acid Residues within the Amino-terminal Domain of ClpB are Essential for the Chaperone Activity Zhonghua Liu, Vekalet Tek, Vladimir Akoev and Michal Zolkiewski * Department of Biochemistry Kansas State University, 104 Willard Hall, Manhattan, KS 66506, USA ClpB from Escherichia coli is a member of a protein-disaggregating multi- chaperone system that also includes DnaK, DnaJ, and GrpE. The sequence of ClpB contains two ATP-binding domains that are enclosed between the amino-terminal and carboxyl-terminal regions. The N-terminal sequence region does not contain known functional sequence motifs. Here, we performed site-directed mutagenesis of four polar residues within the N-terminal domain of ClpB (Thr7, Ser84, Asp103 and Glu109). These residues are conserved in several ClpB homologs. We found that the mutations, T7A, S84A, D103A, and E109A did not significantly affect the secondary structure and thermal stability of ClpB, nor did they inhibit the self-association of ClpB, its basal ATPase activity, or the enhanced rate of the ATP hydrolysis by ClpB in the presence of poly-L-lysine. We observed, however, that three mutations, T7A, D103A, and E109A, reduced the casein-induced activation of the ClpB ATPase. The same three mutant ClpB variants also showed low chaperone activity in the luciferase reactivation assay. We found, however, that the four ClpB mutants, as well as the wild-type, bound similar amounts of inactivated luciferase. In summary, we have identified three essential amino acid residues within the N-terminal region of ClpB that participate in the coupling between a protein-binding signal and the ATP hydrolysis, and also support the chaperone activity of ClpB. q 2002 Elsevier Science Ltd. All rights reserved Keywords: ClpB; molecular chaperone; site-directed mutagenesis; protein folding; protein aggregation *Corresponding author Introduction The protein family of Clp ATPases, also referred to as Hsp100, is involved in protein quality control in prokaryotic and eukaryotic organisms. 1,2 The best characterized Clp ATPases are bacterial proteins: ClpA, ClpB, ClpX, and ClpY (HslU). ClpA, ClpX, and ClpY form stable complexes with peptidase subunits (ClpP, ClpQ) and are involved in ATP-dependent protein degradation. The func- tion of Clp ATPases in such complexes is recog- nition and active unfolding of protein substrates, and their delivery to the peptidases for degradation. 3–5 In contrast to ClpA, ClpX, and ClpY, ClpB does not associate with the peptidase subunits and it is not involved in cellular protein degradation. 6 Instead, a protein disaggregation activity has been observed for ClpB 7–9 and its yeast homolog, Hsp104. 10 ClpB is a member of a highly-efficient multi-chaperone system (with DnaK, DnaJ, and GrpE) that reactivates strongly aggregated proteins. Other Clp ATPases (ClpA and ClpX) do not cooperate with the DnaK/J system and do not show a ClpB-like protein- reactivation activity in vitro (M. Zolkiewski, unpublished results). Thus, Clp ATPases play two distinct roles within the cellular protein-turnover machinery: Whereas ClpB assists other chaperones in rescuing misfolded and aggregated proteins, ClpA, ClpX, and ClpY participate in degradation of those proteins that cannot be rescued. All Clp ATPases contain one or two nucleotide binding domains (NBD). 1 In ClpA and ClpB, two NBDs are enclosed between N-terminal and C-terminal extensions and are separated by a 0022-2836/02/$ - see front matter q 2002 Elsevier Science Ltd. All rights reserved E-mail address of the corresponding author: [email protected] Abbreviations used: CD, circular dichroism; ClpBDN, ClpB without the N-terminal domain; DSC, differential scanning calorimetry; DTT, dithiothreitol; NBD, nucleotide binding domain. doi:10.1016/S0022-2836(02)00591-0 available online at http://www.idealibrary.com on B w J. Mol. Biol. (2002) 321, 111–120
Transcript of Conserved Amino Acid Residues within the Amino-terminal Domain of ClpB are Essential for the...

Conserved Amino Acid Residues within theAmino-terminal Domain of ClpB are Essential for theChaperone Activity
Zhonghua Liu, Vekalet Tek, Vladimir Akoev and Michal Zolkiewski*
Department of BiochemistryKansas State University, 104Willard Hall, Manhattan, KS66506, USA
ClpB from Escherichia coli is a member of a protein-disaggregating multi-chaperone system that also includes DnaK, DnaJ, and GrpE. The sequenceof ClpB contains two ATP-binding domains that are enclosed between theamino-terminal and carboxyl-terminal regions. The N-terminal sequenceregion does not contain known functional sequence motifs. Here, weperformed site-directed mutagenesis of four polar residues within theN-terminal domain of ClpB (Thr7, Ser84, Asp103 and Glu109). Theseresidues are conserved in several ClpB homologs. We found that themutations, T7A, S84A, D103A, and E109A did not significantly affect thesecondary structure and thermal stability of ClpB, nor did they inhibitthe self-association of ClpB, its basal ATPase activity, or the enhancedrate of the ATP hydrolysis by ClpB in the presence of poly-L-lysine. Weobserved, however, that three mutations, T7A, D103A, and E109A,reduced the casein-induced activation of the ClpB ATPase. The samethree mutant ClpB variants also showed low chaperone activity in theluciferase reactivation assay. We found, however, that the four ClpBmutants, as well as the wild-type, bound similar amounts of inactivatedluciferase. In summary, we have identified three essential amino acidresidues within the N-terminal region of ClpB that participate in thecoupling between a protein-binding signal and the ATP hydrolysis, andalso support the chaperone activity of ClpB.
q 2002 Elsevier Science Ltd. All rights reserved
Keywords: ClpB; molecular chaperone; site-directed mutagenesis; proteinfolding; protein aggregation*Corresponding author
Introduction
The protein family of Clp ATPases, also referredto as Hsp100, is involved in protein quality controlin prokaryotic and eukaryotic organisms.1,2 Thebest characterized Clp ATPases are bacterialproteins: ClpA, ClpB, ClpX, and ClpY (HslU).ClpA, ClpX, and ClpY form stable complexes withpeptidase subunits (ClpP, ClpQ) and are involvedin ATP-dependent protein degradation. The func-tion of Clp ATPases in such complexes is recog-nition and active unfolding of protein substrates,and their delivery to the peptidases fordegradation.3 – 5 In contrast to ClpA, ClpX, and
ClpY, ClpB does not associate with the peptidasesubunits and it is not involved in cellular proteindegradation.6 Instead, a protein disaggregationactivity has been observed for ClpB7 – 9 and itsyeast homolog, Hsp104.10 ClpB is a member of ahighly-efficient multi-chaperone system (withDnaK, DnaJ, and GrpE) that reactivates stronglyaggregated proteins. Other Clp ATPases (ClpAand ClpX) do not cooperate with the DnaK/Jsystem and do not show a ClpB-like protein-reactivation activity in vitro (M. Zolkiewski,unpublished results). Thus, Clp ATPases play twodistinct roles within the cellular protein-turnovermachinery: Whereas ClpB assists other chaperonesin rescuing misfolded and aggregated proteins,ClpA, ClpX, and ClpY participate in degradationof those proteins that cannot be rescued.
All Clp ATPases contain one or two nucleotidebinding domains (NBD).1 In ClpA and ClpB, twoNBDs are enclosed between N-terminal andC-terminal extensions and are separated by a
0022-2836/02/$ - see front matter q 2002 Elsevier Science Ltd. All rights reserved
E-mail address of the corresponding author:[email protected]
Abbreviations used: CD, circular dichroism; ClpBDN,ClpB without the N-terminal domain; DSC, differentialscanning calorimetry; DTT, dithiothreitol; NBD,nucleotide binding domain.
doi:10.1016/S0022-2836(02)00591-0 available online at http://www.idealibrary.com onBw
J. Mol. Biol. (2002) 321, 111–120

variable-length middle sequence region (see Figure1(a)). The NBDs show the highest degree ofhomology among different Clp ATPases and con-tain highly conserved Walker ATP-binding motifs.In contrast, the N-terminal, C-terminal, and middlesegments show only limited sequence similarity indifferent Clp ATPases and do not contain knownfunctional motifs.
At high protein concentration or in the presenceof ATP, Clp ATPases form ring-shaped oligomers.Whereas ClpA, ClpX, and ClpY (HslU) formhexamers,11 – 14 recently published electron-microscopy image-reconstruction analysis suggestsa heptameric structure of ClpB.15 It has been foundthat the self-association of ClpB is directlysupported by the C-terminal sequence region.16
High-resolution structure of ClpY also shows thatthe C-terminal region forms a distinct domain thatmaintains contacts with a neighboring subunitwithin the oligomeric ring of ClpY.14 We havepreviously demonstrated that the association-deficient C-terminally truncated ClpB loses itsATPase and chaperone activity.16 Thus, theC-terminal domains in Clp ATPases play animportant role in stabilizing the biologically active
oligomers. The functional role of the N-terminalregions in Clp ATPases remains, however,unknown.
The ClpB gene contains an internal translationinitiation site that corresponds to Val149 at the Nterminus of NBD1 (see Figure 1(a)). Expression ofthe ClpB gene results in production of the full-length 95 kDa ClpB as well as the N-terminallytruncated 80 kDa ClpBDN.6,17 In vivo, both formsof ClpB contribute to the survival of bacteriaduring heat shock.18
In ClpA, deletion of the N-terminal region doesnot produce significant defects in activity.19,20 TheN-terminally truncated ClpA remains active as acomponent of the ATP-dependent protease. In con-trast, the N-terminal truncation of ClpB stronglyinhibits its chaperone activity in vitro.16 Also,casein-induced activation of the ClpB ATPase isreduced after removal of the N-terminal region.16,21
Recently, we have shown that the N-terminalregion of ClpB folds into a stable domain thatdoes not form strong tertiary contacts with theremaining domains of ClpB.22 The full-lengthClpB as well as ClpBDN bind casein and inacti-vated luciferase with similar affinity. Thus, the
middle C-terminalN-terminal
Q769 Q857V149M1
NBD1 NBD2
* Hsp101 1 --MNPE-KFTHKTNETIATAHELAVNAGHAQFTPLHLAGALISD-PTGIFP--QAISSAG TbClp 1 MAHSDR-QCTNAAQTALSDAVESARKHNNGFVDPAHLALVLFKN-EDGLAS--RVLRKLN ClpB 1 --MRLD-RLTNKFQLALADAQSLALGHDNQFIEPLHLMSALLNQ-EGGSVS--PLLTSAG Hsp104 1 --MNDQTQFTERALTILTLAQKLASDHQHPQLQPIHILAAFIETPEDGSVPYLQNLIEKG * * * Hsp101 55 GENAAQSAERVINQALKKLPSQSPPPDDIPASSSLIKVIRRAQAAQKSRGDTHLAVDQLI TbClp 57 ----AGTVLEPLAARVGALPEQRPRPRSITFSSDGGCAQHRRAEANR-VGDSLIAVDHLL ClpB 55 --INAGQLRTDINQALNRLPQVEGTGGDVQPSQDLVRVLNLCDKLAQKRGDNFISSELFV Hsp104 59 -RYDYDLFKKVVNRNLVRIPQQQPAPAEITPSYALGKVLQDAAKIQKQQKDSFIAQDHIL Hsp101 115 MGLLEDS-QIRDLLNEVGVATARVKSEVEKLRGKEGKK TbClp 112 IGLFECK-EVEAIMKAAHASKKAVEGALLELR--KGKK ClpB 113 LAALESRGTLADILKAAGATTANITQAIEQMR--GGES Hsp104 118 FALFNDS-SIQQIFKEAQVDIEAIKQQALELR--GNTR
(a)
(b)
Figure 1. Postulated domain structure of ClpB and positions of mutations within the N-terminal domain. (a) Thediagram shows two nucleotide binding domains in ClpB (NBD1, NBD2) enclosed between the N-terminal andC-terminal regions and separated with the middle domain. In ClpB, the N terminus of NBD1 (Val149) corresponds tothe alternative translation initiation site.21 (b) Alignment of the N-terminal sequences from four Clp ATPases: Hsp101from A. thaliana (Hsp101), ClpB analog from T. brucei (TbClpB), ClpB from E. coli (ClpB), and Hsp104 from S. cerevisiae(Hsp104). The N-terminal sequence region corresponds to amino acid residues 1–148 in ClpB (see (a)). Sequence align-ment has been obtained using the program ClustalW (http://www.ch.embnet.org/software/ClustalW.html). Asterisks( p ) show the positions of residues that were changed to alanine in the present work.
112 Mutagenesis of the N-terminal Domain of ClpB

N-terminal domain is not required for binding ofprotein substrates to ClpB.22
Here, we have focused on the role of conservedamino acid residues within the N-terminal regionof ClpB. Figure 1(b) shows the alignment of theN-terminal sequences of ClpB from Escherichia coliand three ClpB homologs from eukaryoticorganisms: Hsp101 from Arabidopsis thaliana,TbClpB from Trypanosoma brucei, and Hsp104 fromSaccharomyces cerevisiae. According to a recentclassification, ClpB, Hsp101, TbClpB, and Hsp104belong to subgroup B of Clp ATPases.1 Hsp101and Hsp104 are molecular chaperones that playcrucial role in thermotolerance of plants and yeast,respectively,23,24 which mirrors the function ofClpB in bacteria.17,25
As shown in Figure 1(b), sequence similaritybetween different Clp ATPases from the B-sub-family is low within the N-terminal regions. Here,we performed site-directed mutagenesis of fourpolar residues (Thr7, Ser84, Asp103, Glu109),which are conserved among ClpB, Hsp101,TbClpB, and Hsp104. We found that replacementof each of these residues with an alanine does notaffect the structure of ClpB. However, in three outof the four mutants, T7A, D103A, and E109A, weobserved an inhibition of the chaperone activitywithout a decrease in substrate binding capability.This result demonstrates an essential role of
specific sequence motifs within the N-terminalregion in supporting the function of ClpB andpossibly other related Clp ATPases.
Results
First, we tested whether point mutations withinthe N-terminal region of ClpB affect the structureof ClpB. Circular dichroism (CD) spectra of wild-type and the four mutant ClpB variants (Figure 2)show a negative band with a double minimum at208 and 222 nm, which indicates that ClpB con-tains a-helical structures. More importantly, noapparent differences have been found between theCD spectra of wild-type ClpB and the mutants.Thus, substitutions of the four amino acid residueschosen in this study do not affect the overallsecondary structure of ClpB.
Next, we investigated the thermal stability ofwild-type ClpB and the mutants using differentialscanning calorimetry (DSC). The DSC thermogramof wild-type full-length ClpB (Figure 3, data set a)shows two prominent unfolding transitions at53.5 8C and 63.8 8C. The area of the high-tempera-ture transition is significantly decreased inClpBDN (Figure 3, data set f) and in a doublytruncated ClpBDNC, which does not contain theN-terminal and the C-terminal domain (data not
Figure 2. CD spectra of wild-type ClpB and the mutants. CD spectra have been measured at 25 8C in a 0.2 mm cellfor the buffer: 50 mM Hepes/KOH (pH 7.5), 0.2 M KCl, 20 mM MgCl2, 1 mM EDTA, 2 mM b-mercaptoethanol (thindotted line) and for 2 mg/ml wt ClpB (thick continuous line), T7A (thin continuous line), S84A (thick broken line),D103A (thin broken line), E109A (thick dotted line).
Mutagenesis of the N-terminal Domain of ClpB 113

shown). Thus, the thermal unfolding of theN-terminal domain of ClpB occurs within thehigh-temperature DSC endotherm. As shown inFigure 3 (data sets b–e), the four ClpB mutants pro-duced in this study show the double-endothermunfolding patterns, which mirror that found inwild-type ClpB. Whereas the position of the low-temperature endotherm is similar in wild-typeClpB and the mutants, the temperature of thesecond endotherm is lower in the mutants than inthe wild-type. The S84A mutant shows the lowesttemperature of the second endotherm at 59.8 8C.We conclude that the mutations shown in Figure1(b) decrease the thermodynamic stability of theN-terminal domain. However, the DSC resultsindicate that the N-terminal domain and otherdomains of ClpB are folded in the four mutants aswell as in the wild-type at the temperature ofbiochemical assays (25–37 8C) performed here.
As has been documented before, wild-type ClpBdisplays a protein concentration-dependent self-
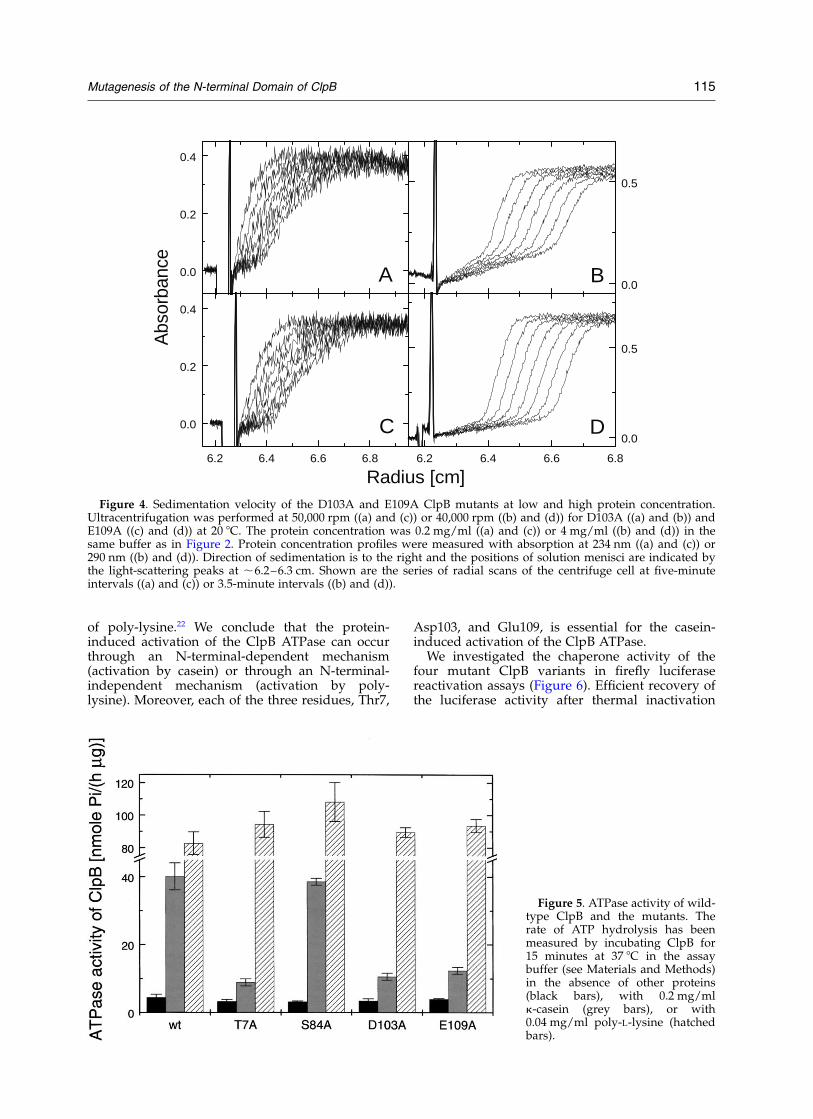
association.16,26 Using analytical ultracentrifuga-tion, we investigated whether the four mutatedClpB variants also form oligomers at high proteinconcentration. In a sedimentation velocity experi-ment, the rate of movement of a protein-concen-tration boundary is related to the sedimentationcoefficient of molecular species in solution. Asshown in Figure 4 for two of the ClpB mutants,D103A and E109A, the predominant proteinspecies sedimented faster at 4 mg/ml (Figure 4(b)and (d)) than at 0.2 mg/ml (Figure 4(a) and (c)).The results shown in Figure 4 are similar to thoseobtained earlier for wild-type ClpB.16
Table 1 lists the values of the sedimentationcoefficient for wild-type ClpB and the mutants atlow and high protein concentration. The sedimen-tation velocity results indicate that the fourmutants, T7A, S84A, D103A, and E109A, self-associate at 4 mg/ml and that the size of theoligomers is the same as that for the wild-type. Asshown below (see Figure 5), the basal ATPaseactivity of ClpB is not affected by the N-terminalmutations either. Taken together, the above resultsindicate that the structure and folding stability ofthe four mutant forms of ClpB produced here arevery similar to those of the wild-type.
Earlier biochemical studies have shown thatClpB and Hsp104 are protein-activated ATPases.6,27
In ClpB, deletion of the whole N-terminal domainreduces the ATPase activation by casein16,21 with-out inhibiting the binding of casein to ClpB.22 Wemeasured the rate of ATP hydrolysis by wild-typeClpB and the four mutants in the absence of otherproteins (basal activity) and in the presence of,100-fold molar excess of k-casein and poly-L-lysine. As shown in Figure 5, the basal activity ofthe four ClpB mutants is similar to that of thewild-type. However, whereas the ATPase activitiesof wild-type ClpB and S84A are activated , tenfoldby casein, the activities of T7A, D103A, and E109Aare activated only , two- to threefold. In contrast,the ATPase activation by poly-lysine is similar forwild-type ClpB and the four mutants. Similarly,deletion of the whole N-terminal region of ClpBdoes not inhibit the ATPase activity in the presence
Figure 3. DSC of wild-type ClpB, the mutants, and theN-terminally truncated ClpBDN. Shown are the DSCthermograms obtained with a 1 K/minute scanning ratefor 0.73 mg/ml wild-type ClpB (a), 0.65 mg/ml T7A(b), 0.77 mg/ml S84A (c), 0.73 mg/ml D103A (d),0.84 mg/ml E109A (e), and 0.56 mg/ml ClpBDN (f).Baselines obtained with the dialyzate buffer (50 mMHepes/KOH, pH 7.5) without the proteins have beensubtracted from the protein scans. The data have beennormalized for protein concentration and variable offsetshave been applied to the data sets for clarity ofpresentation.
Table 1. Apparent sedimentation coefficients of wild-type ClpB and the mutants
Protein samples20,w [S]a
0.2 mg/ml 4.0 mg/ml
wt ClpB 4.7b 16.4b
T7A n.d. 16.05S84A n.d. 16.1D103A 4.3 16.1E109A 4.6 15.9
a Observed sedimentation coefficients were obtained from thetime-derivative analysis of the sedimentation velocity data (seeFigure 4 and Materials and Methods), corrected to the valuescorresponding to the density and viscosity of water, andreported as s20,w in Svedberg units (S).
b The values for wild-type ClpB are from Barnett et al.16
114 Mutagenesis of the N-terminal Domain of ClpB
of poly-lysine.22 We conclude that the protein-induced activation of the ClpB ATPase can occurthrough an N-terminal-dependent mechanism(activation by casein) or through an N-terminal-independent mechanism (activation by poly-lysine). Moreover, each of the three residues, Thr7,
Asp103, and Glu109, is essential for the casein-induced activation of the ClpB ATPase.
We investigated the chaperone activity of thefour mutant ClpB variants in firefly luciferasereactivation assays (Figure 6). Efficient recovery ofthe luciferase activity after thermal inactivation
0.0
0.2
0.4
A
0.0
0.5
Abs
orba
nce
B
6.2 6.4 6.6 6.8
0.0
0.2
0.4
C
6.2 6.4 6.6 6.8
0.0
0.5
Radius [cm]
D
Figure 4. Sedimentation velocity of the D103A and E109A ClpB mutants at low and high protein concentration.Ultracentrifugation was performed at 50,000 rpm ((a) and (c)) or 40,000 rpm ((b) and (d)) for D103A ((a) and (b)) andE109A ((c) and (d)) at 20 8C. The protein concentration was 0.2 mg/ml ((a) and (c)) or 4 mg/ml ((b) and (d)) in thesame buffer as in Figure 2. Protein concentration profiles were measured with absorption at 234 nm ((a) and (c)) or290 nm ((b) and (d)). Direction of sedimentation is to the right and the positions of solution menisci are indicated bythe light-scattering peaks at ,6.2–6.3 cm. Shown are the series of radial scans of the centrifuge cell at five-minuteintervals ((a) and (c)) or 3.5-minute intervals ((b) and (d)).
Figure 5. ATPase activity of wild-type ClpB and the mutants. Therate of ATP hydrolysis has beenmeasured by incubating ClpB for15 minutes at 37 8C in the assaybuffer (see Materials and Methods)in the absence of other proteins(black bars), with 0.2 mg/mlk-casein (grey bars), or with0.04 mg/ml poly-L-lysine (hatchedbars).
Mutagenesis of the N-terminal Domain of ClpB 115

(see Figure 6(a)) requires ClpB and three otherE. coli chaperones: DnaK, DnaJ, and GrpE. Wefound that, among the four ClpB mutants investi-gated here, three mutants, T7A, D103A, andE109A, showed an inhibition of the chaperoneactivity and the S84A mutant was more effectivein the luciferase reactivation than wild-type ClpB.To verify these results, we have also performedthe assay using chemically denatured luciferase(Figure 6(b)). As has been shown before, underconditions of this assay, strong aggregation ofluciferase during refolding prevents its spon-taneous reactivation.7 Strong inhibition of thechaperone activity of T7A, D103A, and E109A isevident under the conditions of Figure 6(b). Thedefect in chaperone activity of T7A, D103A, andE109A, mirrors their deficiency in the casein-
induced activation of the ATPase activity (compareFigures 5 and 6).
Since the concentration of ClpB is higher thanthe concentration of luciferase under the experi-mental conditions of Figure 6, we investigatedwhether the apparent inhibition of the chaperoneactivity observed for three ClpB mutants may bedue to a modulation of their binding affinity forthe substrate, luciferase. As shown in Figure 7,thermally inactivated luciferase binds to wild-typeClpB, as well as to the four mutants in the presenceof ATP (see Materials and Methods). When bindingassays were performed in the absence of ATP, nosignificant difference in the ELISA signals wasobserved between the ClpB-covered and the con-trol plates (not shown). Moreover, the amount ofbound luciferase is similar for wild-type ClpB andthe mutants. We conclude that the loss of ,70–80% of the chaperone activity in T7A, D103A, andE109A, as compared to the wild-type (see Figure6(a)), is not due to the inhibition of interactions ofinactivated luciferase with these mutant forms ofClpB.
Discussion
Here, we have produced four variants of ClpB,each with an alanine replacing a conserved polarresidue within the N-terminal region (see Figure1). We found that the mutations did not apparentlyaffect the folding and structural integrity of ClpB(see Figures 2–4 and Table 1). Thus, observedeffects of the mutations T7A, S84A, D103A, andE109A can be directly attributed to the function ofthe modified amino acid residues.
Our previous work on the N-terminally trun-cated ClpB showed a strong inhibition of thechaperone activity after deletion of the wholeN-terminal region (amino acid residues 1–148).16
This result has been corroborated in the presentstudy. We have now identified three amino acidresidues within the N-terminal domain, Thr7,Asp103, and Glu109, which apparently play animportant role in the chaperone function of ClpB(see Figure 6). Moreover, the same three aminoacid residues support the casein-inducedactivation of the ClpB ATPase (see Figure 5). Ourresults demonstrate that specific amino acid resi-dues within the N-terminal domain of ClpB areinvolved in responding to a protein-binding signal,which results in acceleration of the ATP hydrolysisrate. Whereas casein-induced activation of theClpB ATPase has been used in the past as a simplebiochemical assay of ClpB,6 we now show that theClpB response to casein mirrors its activity in aprotein reactivation assay. Since k-casein is a highlyheterogeneous, aggregated protein in solution,28,29
it is most likely recognized by ClpB as a substratefor disaggregation. In contrast, strong activation ofthe ClpB ATPase in the presence of poly-lysineapparently occurs through a different mechanism,
0 20 40 60 80 1000
1
2
3 B
Time [min]
Lum
ines
cenc
e [a
rb. u
.]
0 20 40 60 80 100 1200
2
4
6
A
Figure 6. Chaperone activity of wild-type ClpB andthe mutants in the multi-chaperone system includingDnaK, DnaJ, and GrpE. (a) Reactivation of thermallyinactivated luciferase at 25 8C. (b) Reactivation of urea-denatured luciferase at 25 8C. The activity of firefly luci-ferase during its chaperone-assisted reactivation hasbeen monitored using luminescence (see Materials andMethods). Protein concentrations were: 33 nM (a) or25 nM (b) luciferase, 0.3 mM ClpB (oligomer), 1.0 mMDnaK, 1.0 mM DnaJ, 1.0 mM GrpE. The data sets corre-spond to the luminescence measured in the presence ofDnaK, DnaJ, GrpE, and wild-type ClpB (filled circles),T7A (filled triangles), S84A (open triangles), D103A(filled squares), or E109A (open squares). Due to a non-linear signal response of the scintillation counter, relativeerrors of the data points obtained with high luciferaseactivity (wt ClpB and S84A at .50 minutes) are , twoto threefold greater than those of the data pointsobtained with low luciferase activity.
116 Mutagenesis of the N-terminal Domain of ClpB

which does not directly involve the three residues,Thr7, Asp103 and Glu109 (see Figure 5).
A study by Lo et al.19 identified two apparentsequence repeats within the N-terminal region ofa group of Clp ATPases. The two sequence repeatsin ClpB correspond to residues 1–77 and 78–145,respectively. Interestingly, the position of Thr7 inthe first repeat corresponds to the position ofSer84 in the second repeat. We found that themutation of Thr7, but not that of Ser84, inhibitschaperone activity of ClpB. The other two essentialresidues, Asp103 and Glu109 belong to the secondsequence repeat and do not have correspondingpartner residues within the first repeat.19 Thus,the two apparent sequence repeats within theN-terminal domain of ClpB are not functionallyequivalent, in spite of some similarity between thetwo parts of the sequence.
So far, it has not been clear how the N-terminaldomains of Clp ATPases are involved in thefunction of these proteins as either molecularchaperones (ClpB and Hsp104) or ATP-dependentproteases (ClpA). No high-resolution structuralinformation is currently available for theN-terminal regions of Clp ATPases. Previousstudies suggest that the N-terminal regions ofClpA and ClpB form distinct folding domains thatare only weakly associated with the remainingparts of a protein.19,20,22 In the oligomeric ClpAand ClpB, the N-terminal domains may form ringsof exposed structures that are conformationallyflexible and are potentially accessible to proteinsubstrates.20 The N-terminal domain of ClpA isnot required for its activity, but it does appear tomodulate substrate recognition by ClpA.19 TheN-terminal domain of ClpB is required for reacti-vation of luciferase, but not for luciferase bindingto ClpB.16,22 Consistently, our studies on the bind-ing of inactivated luciferase to the N-terminal
mutants of ClpB (see Figure 7) show that thedecrease of chaperone activity in three of themutants cannot be directly attributed to defects insubstrate binding. Thus, specific amino acid resi-dues within the N-terminal domain (Thr7, Asp103and Glu109) are involved in supporting confor-mational rearrangements in ClpB and/or in a pro-tein substrate that occur during substratereactivation.
Here, we have identified three residues withinthe N-terminal domain, Thr7, Asp103, andGlu109, that directly support the chaperoneactivity of ClpB in the luciferase reactivationassay. In vivo, the ClpB/DnaK/DnaJ multi-chaper-one system assists hundreds of misfolded andaggregated proteins during heat shock.25 Both thefull-length ClpB and the N-terminally truncatedClpBDN contribute to the thermotolerance ofSynechococcus.18 Thus, some of the many proteinsubstrates may not require the N-terminal domainof ClpB for their refolding and/or reactivation. Itis not known if the distinction between N-terminaldomain-dependent and independent substrates ofClpB is related to their different extent of misfold-ing/aggregation or to some specific structuralcharacteristics. Our discovery of essential func-tion-supporting residues within the N-terminaldomain may contribute to delineating the substratediscrimination rules for ClpB and, ultimately, tounderstanding the mechanism of the ClpB-assistedprotein reactivation.
Materials and Methods
Proteins
Site-directed mutagenesis of ClpB has been performedusing the QuickChange method (Stratagene). Plasmid
Figure 7. Binding of heat-inacti-vated luciferase to wild-type ClpBand the mutants. Wells in anELISA plate have been coveredwith wild-type ClpB (filled circles),T7A (filled triangles), S84A (opentriangles), D103A (filled squares),or E109A (open squares), blocked,and subsequently incubated withsolutions of inactivated luciferaseat the indicated concentrations. Asa control, a group of wells have notbeen covered with ClpB (opencircles). After washing, the amountsof luciferase remaining on the platehave been probed with anti-lucifer-ase antibodies. The data pointsrepresent average readings fromsix independent determinations.Standard deviations of the averagesare shown for the control data set,wild-type ClpB, and T7A and arerepresentative of the accuracy of alldata sets.
Mutagenesis of the N-terminal Domain of ClpB 117

pET-20b containing the ClpB coding sequence16 was usedas the template. The following single-base mutationshave been introduced into the ClpB DNA sequence: pos-ition 19, A ! G (T7A); position 250, T ! G (S84A); pos-ition 308, A ! C (D103A); position 326, A ! C (E109A).Identity of the mutations has been confirmed by theDNA Sequencing Facility at Iowa State University,Ames, Iowa.
Wild-type full-length ClpB and the mutants have beenoverexpressed in E. coli and purified as describedbefore.16 After purification, the protein samples were dia-lyzed against 50 mM Tris–HCl (pH 7.5), 0.2 M KCl,20 mM MgCl2, 10% (v/v) glycerol, 1 mM EDTA, 1 mMDTT and stored at 220 8C. Protein concentration wasmeasured using the absorption coefficient A280 ¼0:38 cm2=mg: Rabbit polyclonal anti-ClpB antibodieswere produced by Cocalico Biologicals (Reamstown,Pennsylvania) using purified wild-type ClpB as animmunogen.
Poly-L-lysine and k-casein were obtained from Sigma.Firefly luciferase was purchased from Promega and theanti-luciferase antibody was from Sigma. Other E. colichaperones, DnaK, DnaJ, and GrpE were obtained fromStressGen Biotechnologies (Victoria, BC, Canada).
Circular dichroism
CD spectra were measured using a Jasco J-720 spectro-polarimeter and a 0.02 cm cylindrical cuvette.
Differential scanning calorimetry
DSC experiments were performed using a VP-DSCcalorimeter (MicroCal Inc., Northampton, MA) at ascan-rate of 1 K/minute. For each protein sample, theinstrument baseline was obtained first by measuringthe thermogram of the dialysis buffer. Subsequently, thebuffer in the sample cell was replaced with a ClpB sol-ution and the protein thermogram was measured. Thethermal unfolding of ClpB was irreversible, as shownby the lack of endotherms in the repetitive scans of eachprotein sample.
Analytical ultracentrifugation
Sedimentation velocity experiments were performedwith a Beckman Optima XL-I analytical ultracentrifugeusing double-sector 1.2 cm cells with aluminum center-pieces, as described.16 The sedimentation velocity datawere analyzed with a time-derivative method30 usingsoftware supplied with the ultracentrifuge. The time-derivative method gives apparent sedimentation velocitydistributions for the molecular species in solution andthe values of the observed sedimentation coefficients.The sedimentation coefficients corresponding to thedensity and viscosity of water were calculated as s20;w ¼1:022sobs; using previously measured values of thedensity and viscosity of the buffer.31
ClpB ATPase activity assays
Wild-type ClpB and mutant samples (80 mg/ml) wereincubated for 15 minutes at 37 8C in the assay buffer(100 mM Tris–HCl (pH 8.0), 10 mM MgCl2, 5 mM ATP,1 mM EDTA, 1 mM DTT) without other proteins, with0.2 mg/ml k-casein, or with 0.04 mg/ml poly-L-lysine.Concentration of inorganic phosphate produced from
ATP was determined using the malachite greencolorimetric assay.32,33
ClpB chaperone activity assays
In the assay shown in Figure 6(a), firefly luciferase(61 kDa, 12.6 mg/ml) was diluted 300-fold into PBSbuffer containing 1 mg/ml BSA and incubated at 45 8Cfor 12 minutes. After the heat treatment, the luciferasesample was cooled to room temperature and diluted 20-fold into the renaturation buffer (30 mM Hepes–KOH(pH 7.6), 120 mM KCl, 10 mM MgCl2, 5 mM ATP, 1 mMEDTA, 1 mM DTT, 0.1 mg/ml bovine serum albumin(BSA)) containing wild-type ClpB or the mutants andDnaK, DnaJ, GrpE. The activity of luciferase in therefolding solution was measured using a Beckman LS3801 scintillation counter as described before.7
In the assay shown in Figure 6(b), luciferase (15 mg/ml) was diluted 100-fold into the denaturation buffer(30 mM Hepes–KOH (pH 7.6), 60 mM KCl, 10 mMMgCl2, 1 mM EDTA, 10 mM DTT, 7 M urea) andincubated for one hour at room temperature. Next, thedenatured luciferase was diluted 100-fold into therenaturation buffer (see above) and the luciferase activitywas measured.
ClpB–luciferase interaction assay
Interactions between ClpB and inactivated luciferasewere probed using an ELISA procedure. ClpB or themutants (100 ml of 0.1 mg/ml protein in buffer A(100 mM Tris–HCl (pH 8.0), 10 mM MgCl2, 2 mM ATP,1 mM EDTA, 1 mM DTT)) was incubated in the wells ofMaxiSorp 96-well plates (Nalge Nunc International,Denmark) for three hours at room temperature.
In a control experiment, the amounts of different ClpBvariants in the wells were compared with direct ELISAusing anti-ClpB antibodies. After decanting the liquid,the wells were washed three times with 200 ml of PBST(PBS þ 0.05% Tween-20) and three times with 200 ml of0.2% (w/v) BSA in PBS. To block the wells, 200 ml of0.2% BSA in PBS were incubated in wells for two hoursat room temperature. After washing four times withPBST, 100 ml of anti-ClpB serum diluted in PBS wasadded to the wells and incubated overnight at 4 8C.After incubation with the antibody, the wells werewashed six times with 200 ml of PBST and then incubatedwith 100 ml of goat anti-rabbit IgM þ IgG conjugated tohorseradish peroxidase (Southern Biotechnology Associ-ates, Birmingham, AL) for 45 minutes at room tempera-ture. The wells were then washed ten times with 200 mlof PBST and the amount of secondary antibody wasmeasured using the TMB peroxidase substrate kit(Pierce, Rockford, IL). The absorbance at 450 nm wasmeasured with a UV micro-plate reader (MolecularDevices). We found that similar amounts of wild-typeClpB and the four mutants bound to the ELISA plate(,6% variation between different ClpB samples).
To investigate the binding of inactivated luciferase toClpB and the mutants, the wells covered with ClpB andsubsequently blocked with BSA (see the procedureabove) were washed twice with 200 ml of buffer B(25 mM Hepes–KOH (pH 7.6), 150 mM KCl, 25 mMNaCl, 1 mM DTT, 0.1 mM EDTA, 10 mM MgCl2, 2 mMATP, 2.5% glycerol, 0.05% Triton-X-100, 0.2% BSA). Fire-fly luciferase was inactivated by incubation at 45 8C for12 minutes at the concentration of 0.04 mg/ml in PBS.After diluting the heat-treated luciferase to a desired
118 Mutagenesis of the N-terminal Domain of ClpB

concentration with buffer B, the luciferase solutions wereadded to the wells covered with wild-type ClpB, themutants, or only blocked with BSA (control). Wells werewashed twice with 200 ml of buffer B, three times with200 ml of 0.2% BSA in PBS, and once with 200 ml of bufferA. Following this, 100 ml of anti-luciferase serum (in PBS)was added and the plates were incubated for two hoursat room temperature. After washing three times with200 ml of 0.2% BSA in PBST and twice with 0.2% BSA inPBS, 100 ml of the secondary antibody was added to thewells and the ELISA signal was measured as describedabove.
Acknowledgments
We thank Micheal E. Barnett for assistance in purifyingClpB and performing circular dichroism and sedimen-tation experiments as well as for helpful discussions.This work was supported by the American Heart Associ-ation, Heartland Affiliate (grant 0060445Z) and by theNational Institutes of Health (grant GM58626). This iscontribution 02-172-J from the Kansas AgriculturalExperiment Station.
References
1. Schirmer, E. C., Glover, J. R., Singer, M. A. &Lindquist, S. (1996). HSP100/Clp proteins: a com-mon mechanism explains diverse functions. TrendsBiochem. Sci. 21, 289–296.
2. Wickner, S., Maurizi, M. R. & Gottesman, S. (1999).Posttranslational quality control: folding, refolding,and degrading proteins. Science, 286, 1888–1893.
3. Weber-Ban, E. U., Reid, B. G., Miranker, A. D. &Horwich, A. L. (1999). Global unfolding of a sub-strate protein by the Hsp100 chaperone ClpA.Nature, 401, 90–93.
4. Singh, S. K., Grimaud, R., Hoskins, J. R., Wickner, S.& Maurizi, M. R. (2000). Unfolding and internaliz-ation of proteins by the ATP-dependent proteasesClpXP and ClpAP. Proc. Natl Acad. Sci. USA, 97,8898–8903.
5. Kim, Y. I., Burton, R. E., Burton, B. M., Sauer, R. T. &Baker, T. A. (2000). Dynamics of substrate denatura-tion and translocation by the ClpXP degradationmachine. Mol. Cell, 5, 639–648.
6. Woo, K. M., Kim, K. I., Goldberg, A. L., Ha, D. B. &Chung, C. H. (1992). The heat-shock protein ClpB inEscherichia coli is a protein-activated ATPase. J. Biol.Chem. 267, 20429–20434.
7. Zolkiewski, M. (1999). ClpB cooperates with DnaK,DnaJ, and GrpE in suppressing protein aggregation.A novel multi-chaperone system from Escherichiacoli. J. Biol. Chem. 274, 28083–28086.
8. Motohashi, K., Watanabe, Y., Yohda, M. & Yoshida,M. (1999). Heat-inactivated proteins are rescued bythe DnaK.J-GrpE set and ClpB chaperones. Proc.Natl Acad. Sci. USA, 96, 7184–7189.
9. Goloubinoff, P., Mogk, A., Zvi, A. P., Tomoyasu, T. &Bukau, B. (1999). Sequential mechanism of solu-bilization and refolding of stable protein aggregatesby a bichaperone network. Proc. Natl Acad. Sci. USA,96, 13732–13737.
10. Glover, J. R. & Lindquist, S. (1998). Hsp104, Hsp70,and Hsp40: a novel chaperone system that rescuespreviously aggregated proteins. Cell, 94, 73–82.
11. Kessel, M., Maurizi, M. R., Kim, B., Kocsis, E., Trus,B. L., Singh, S. K. & Steven, A. C. (1995). Homologyin structural organization between E. coli ClpAP pro-tease and the eukaryotic 26 S proteasome. J. Mol. Biol.250, 587–594.
12. Grimaud, R., Kessel, M., Beuron, F., Steven, A. C. &Maurizi, M. R. (1998). Enzymatic and structural simi-larities between the Escherichia coli ATP-dependentproteases, ClpXP and ClpAP. J. Biol. Chem. 273,12476–12481.
13. Kessel, M., Wu, W., Gottesman, S., Kocsis, E., Steven,A. C. & Maurizi, M. R. (1996). Six-fold rotationalsymmetry of ClpQ, the E. coli homolog of the 20 Sproteasome, and its ATP-dependent activator, ClpY.FEBS Letters, 398, 274–278.
14. Bochtler, M., Hartmann, C., Song, H. K., Bourenkov,G. P., Bartunik, H. D. & Huber, R. (2000). The struc-tures of HsIU and the ATP-dependent proteaseHsIU–HsIV. Nature, 403, 800–805.
15. Kim, K. I., Cheong, G. W., Park, S. C., Ha, J. S., Woo,K. M., Choi, S. J. & Chung, C. H. (2000). Heptamericring structure of the heat-shock protein ClpB, aprotein-activated ATPase in Escherichia coli. J. Mol.Biol. 303, 655–666.
16. Barnett, M. E., Zolkiewska, A. & Zolkiewski, M.(2000). Structure and activity of ClpB from Escherichiacoli. Role of the amino- and -carboxyl-terminaldomains. J. Biol. Chem. 275, 37565–37571.
17. Squires, C. L., Pedersen, S., Ross, B. M. & Squires, C.(1991). ClpB is the Escherichia coli heat shock proteinF84.1. J. Bacteriol. 173, 4254–4262.
18. Clarke, A. K. & Eriksson, M. J. (2000). The truncatedform of the bacterial heat shock protein ClpB/HSP100 contributes to development of thermo-tolerance in the cyanobacterium Synechococcus sp.strain PCC 7942. J. Bacteriol. 182, 7092–7096.
19. Lo, J. H., Baker, T. A. & Sauer, R. T. (2001). Character-ization of the N-terminal repeat domain ofEscherichia coli ClpA-A class I Clp/HSP100 ATPase.Protein Sci. 10, 551–559.
20. Singh, S. K., Rozycki, J., Ortega, J., Ishikawa, T., Lo, J.,Steven, A. C. & Maurizi, M. R. (2001). Functionaldomains of the ClpA and ClpX molecular chaper-ones identified by limited proteolysis and deletionanalysis. J. Biol. Chem. 276, 29420–29429.
21. Park, S. K., Kim, K. I., Woo, K. M., Seol, J. H., Tanaka,K., Ichihara, A. et al. (1993). Site-directed muta-genesis of the dual translational initiation sites ofthe clpB gene of Escherichia coli and characterizationof its gene products. J. Biol. Chem. 268, 20170–20174.
22. Tek, V. & Zolkiewski, M. (2002). Stability and inter-actions of the amino-terminal domain of ClpB fromEscherichia coli. Protein Sci. 11, 1192–1198.
23. Queitsch, C., Hong, S. W., Vierling, E. & Lindquist, S.(2000). Heat shock protein 101 plays a crucial role inthermotolerance in Arabidopsis. Plant Cell, 12,479–492.
24. Sanchez, Y. & Lindquist, S. L. (1990). HSP104required for induced thermotolerance. Science, 248,1112–1115.
25. Tomoyasu, T., Mogk, A., Langen, H., Goloubinoff, P.& Bukau, B. (2001). Genetic dissection of the roles ofchaperones and proteases in protein folding anddegradation in the Escherichia coli cytosol. Mol.Microbiol. 40, 397–413.
Mutagenesis of the N-terminal Domain of ClpB 119

26. Zolkiewski, M., Kessel, M., Ginsburg, A. & Maurizi,M. R. (1999). Nucleotide-dependent oligomerizationof ClpB from Escherichia coli. Protein Sci. 8,1899–1903.
27. Schirmer, E. C. & Lindquist, S. (1998). Purificationand properties of Hsp104 from yeast. MethodsEnzymol. 290, 430–444.
28. Groves, M. L., Wickham, E. D. & Farrell, H. M., Jr(1998). Environmental effects on disulfide bondingpatterns of bovine kappa-casein. J. Protein Chem. 17,73–84.
29. Farrell, H. M., Jr, Wickham, E. D., Dower, H. J.,Piotrowski, E. G., Hoagland, P. D., Cooke, P. H. &Groves, M. L. (1999). Characterization of the particlesof purified kappa-casein: trypsin as a probe of sur-face-accessible residues. J. Protein Chem. 18, 637–652.
30. Stafford, W. F., III (1992). Boundary analysis in sedi-mentation transport experiments: a procedure forobtaining sedimentation coefficient distributionsusing the time derivative of the concentration profile.Anal. Biochem. 203, 295–301.
31. Maurizi, M. R., Singh, S. K., Thompson, M. W.,Kessel, M. & Ginsburg, A. (1998). Molecular proper-ties of ClpAP protease of Escherichia coli: ATP-depen-dent association of ClpA and ClpP. Biochemistry, 37,7778–7786.
32. Hess, H. H. & Derr, J. E. (1975). Assay of inorganicand organic phosphorus in the 0.1–5 nanomolerange. Anal. Biochem. 63, 607–613.
33. Lanzetta, P. A., Alvarez, L. J., Reinach, P. S. & Candia,O. A. (1979). An improved assay for nanomoleamounts of inorganic phosphate. Anal. Biochem. 100,95–97.
Edited by P. Wright
(Received 12 April 2002; received in revised form 11 June 2002; accepted 11 June 2002)
120 Mutagenesis of the N-terminal Domain of ClpB




![The Drowsy Chaperone Program [2012] - USM Digital ...](https://static.fdokumen.com/doc/165x107/6322912f887d24588e044a66/the-drowsy-chaperone-program-2012-usm-digital-.jpg)















