
Close linkage of the murine locus bare patches to the X-linked visual pigment gene: Implications for...
6
GENOMICS ‘7,307-312 (1990) Close Linkage of the Murine Locus Bare Patches to the X-Linked Visual Pigment Gene: Implications for Mapping Human X-Linked Dominant Chondrodysplasia Punctata GAIL E. HERMAN AND SANDY 1. WALTON Institute for Molecular Genetics and Department of Pediatrics, Baylor College of Medicine, Houston, Texas 77030 Received September 22, 1989; revised February 27, 1990 The murine X-linked dominant mutation bare patches (Bps) has a phenotype similar to and is likely homologous to human X-linked dominant chondro- dysplasia punctata (CDPXB). Classic two-point link- age analysis in the mouse with distant markers sug- gested that Bps maps near glucose-6-phosphate de- hydrogenase (G6pd). We have confirmed the regional localization using interspecific matings with Mue epretus. We have also detected a restriction fragment length polymorphism (RFLP) at the murine X-linked visual pigment (Reup) locus in inbred Bps females us- ing the restriction enzyme P&I. Cumulative data from segregation of alleles using the Pet1 RFLP and analysis of interspecific backcross progeny at the Rsvp locus suggest that Bpa is tightly linked to RSVP. Thus, the human CDPX2 gene probably maps within Xq27- Xq28 and not within Xp22.3-Xpter, where deletions associated with X-linked recessive chondrodysplasia punctata (CDPX) have been noted. This strategy should be applicable to the fine mapping of other dom- inant murine mutations. Q ieso Academic Press, IDC. INTRODUCTION Although recent advances in molecular biology have had a tremendous impact on the understanding and diagnosis of many genetic diseases, human skeletal dysplasias remain a poorly understood group of inher- ited disorders. Because many human skeletal dysplasias may prove difficult to study using a molecular approach, we have chosen to focus on a murine skeletal mutant, bare patches, for which there is presumed to be a ho- mologous skeletal dysplasia in humans. Bare patches is an X-linked dominant mutation which arose in F1 progeny of a (C3H X 101)Fi hybrid male who had received multiple doses of X-irradiation (Phillips et al., 1973). Affected heterozygous females suffer from a skeletal dysplasia with dwarfing, a short kinked tail, and occasional syndactyly or contractures. Affected male embryos apparently die shortly after im- plantation. The majority of affected females have cat- aracts, usually asymmetric or unilateral. They develop a hyperkeratotic skin eruption on Days 5-10 and they later develop bare patches (hence the mutant’s name) arranged in a linear or “blotchy” pattern over the skin. The asymmetry in the cataracts and the distribution of skin findings are presumed to be results of X-inac- tivation. Rudolf Happle has proposed that this murine dis- order is homologous to human X-linked dominant chondrodysplasia punctata (CDPXB) (Happle et al., 1983). A distinct X-linked recessive form of chondro- dysplasia punctata (CDPX) seen only in males is as- sociated with terminal deletions of Xp22.3-Xpter or X/Y translocations (Curry et aZ., 1984; Ballabio et al, 1989). All of the reported CDPX2 patients are female, and CDPXB is presumed to be lethal in hemizygous males. Human CDPX2 patients have short stature with asymmetric limb involvement and ichthyosiform skin changes evident at birth. These skin changes resolve and are replaced by linear or patchy ichthyosis and atrophoderma as well as patchy alopecia and nail ab- normalities. Over half of the affected females develop asymmetric cataracts. Forty-three of the forty-four re- ported patients have had normal intelligence, and this form of the disease is compatible with a normal life expectancy (Happle, 1979; Manzke et al., 1980; Mueller et al., 1985). The human CDPXB gene has not been mapped. It has been suggested that the locus maps within Xp22.3- Xpter deletions associated with CDPX. It has also been suggested (Davisson, 1987; Amar et al., 1988) that CDPXB maps within Xq28 on the basis of presumed homology to Bpa, which was mapped close to G6pd in 1974 by Phillips and co-workers using classic two-point linkage analysis with the distant markers tabby (Ta) and mottled (MO) (Phillips and Kaufman, 1974). Their calculation of the recombination frequencies was com- 307 o&w7543/90 $3.00 Copyright 0 1990 hy Academic Press, Inc. All rights of reproduction in any form reserved.
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Close linkage of the murine locus bare patches to the X-linked visual pigment gene: Implications for...

GENOMICS ‘7,307-312 (1990)
Close Linkage of the Murine Locus Bare Patches to the X-Linked Visual Pigment Gene: Implications for Mapping Human
X-Linked Dominant Chondrodysplasia Punctata
GAIL E. HERMAN AND SANDY 1. WALTON
Institute for Molecular Genetics and Department of Pediatrics, Baylor College of Medicine, Houston, Texas 77030
Received September 22, 1989; revised February 27, 1990
The murine X-linked dominant mutation bare patches (Bps) has a phenotype similar to and is likely homologous to human X-linked dominant chondro- dysplasia punctata (CDPXB). Classic two-point link- age analysis in the mouse with distant markers sug- gested that Bps maps near glucose-6-phosphate de- hydrogenase (G6pd). We have confirmed the regional localization using interspecific matings with Mue epretus. We have also detected a restriction fragment length polymorphism (RFLP) at the murine X-linked visual pigment (Reup) locus in inbred Bps females us- ing the restriction enzyme P&I. Cumulative data from segregation of alleles using the Pet1 RFLP and analysis of interspecific backcross progeny at the Rsvp locus suggest that Bpa is tightly linked to RSVP. Thus, the human CDPX2 gene probably maps within Xq27- Xq28 and not within Xp22.3-Xpter, where deletions associated with X-linked recessive chondrodysplasia punctata (CDPX) have been noted. This strategy should be applicable to the fine mapping of other dom- inant murine mutations. Q ieso Academic Press, IDC.
INTRODUCTION
Although recent advances in molecular biology have had a tremendous impact on the understanding and diagnosis of many genetic diseases, human skeletal dysplasias remain a poorly understood group of inher- ited disorders. Because many human skeletal dysplasias may prove difficult to study using a molecular approach, we have chosen to focus on a murine skeletal mutant, bare patches, for which there is presumed to be a ho- mologous skeletal dysplasia in humans.
Bare patches is an X-linked dominant mutation which arose in F1 progeny of a (C3H X 101)Fi hybrid male who had received multiple doses of X-irradiation (Phillips et al., 1973). Affected heterozygous females suffer from a skeletal dysplasia with dwarfing, a short kinked tail, and occasional syndactyly or contractures.
Affected male embryos apparently die shortly after im- plantation. The majority of affected females have cat- aracts, usually asymmetric or unilateral. They develop a hyperkeratotic skin eruption on Days 5-10 and they later develop bare patches (hence the mutant’s name) arranged in a linear or “blotchy” pattern over the skin. The asymmetry in the cataracts and the distribution of skin findings are presumed to be results of X-inac- tivation.
Rudolf Happle has proposed that this murine dis- order is homologous to human X-linked dominant chondrodysplasia punctata (CDPXB) (Happle et al., 1983). A distinct X-linked recessive form of chondro- dysplasia punctata (CDPX) seen only in males is as- sociated with terminal deletions of Xp22.3-Xpter or X/Y translocations (Curry et aZ., 1984; Ballabio et al, 1989).
All of the reported CDPX2 patients are female, and CDPXB is presumed to be lethal in hemizygous males. Human CDPX2 patients have short stature with asymmetric limb involvement and ichthyosiform skin changes evident at birth. These skin changes resolve and are replaced by linear or patchy ichthyosis and atrophoderma as well as patchy alopecia and nail ab- normalities. Over half of the affected females develop asymmetric cataracts. Forty-three of the forty-four re- ported patients have had normal intelligence, and this form of the disease is compatible with a normal life expectancy (Happle, 1979; Manzke et al., 1980; Mueller et al., 1985).
The human CDPXB gene has not been mapped. It has been suggested that the locus maps within Xp22.3- Xpter deletions associated with CDPX. It has also been suggested (Davisson, 1987; Amar et al., 1988) that CDPXB maps within Xq28 on the basis of presumed homology to Bpa, which was mapped close to G6pd in 1974 by Phillips and co-workers using classic two-point linkage analysis with the distant markers tabby (Ta) and mottled (MO) (Phillips and Kaufman, 1974). Their calculation of the recombination frequencies was com-
307 o&w7543/90 $3.00 Copyright 0 1990 hy Academic Press, Inc.
All rights of reproduction in any form reserved.

HERMAN AND WALTON
TABLE 1
DNA Probes Used in This Study
Probe Locus RFLP
(RFLV) Wash conditions Source
pOTC (mouse cDNA) pHPT5 (mouse cDNA) Cl1 (human cDNA) F9 (human cDNA) HS7 (human cDNA) G28A, G28B (mouse genomic) St14 (human genomic) GDP25A (human cDNA) pKpn-sac (human cDNA) pXD4, pXD1 (mouse cDNA) pCAB17 (mouse genomic) R989 (human cDNA) pY2 (mouse genomic)
otc Hprt
Cf-9 RSVP
P3
G6pd Cf-8 Dltld Pgk-1
&s
TuqI Tag1 TaqI
TaqI, BanHI ToqI, PstI
‘-&I MspI
TaqI Td KpnI TuqI
A A B B B A C B B D A B D
C. T. Caskey (29) C. T. Caskey J. L. Mandel (22) J. L. Mandel(5) J. Nathans (21) P. Avner (3) ATCC M. Persico (23) Genetics Institute J. Chamberlain (6) M. McBurney (1) R. Fenwick P. Soriano (17)
Note. Washing conditions were as follows: A, sequential washes of 2X SSC, 0.1% SDS; 1X SSC, 0.1% SDS; 0.5X SSC, 0.1% SDS; and 0.1X SSC, 0.1% SDS. B, three washes with 3X SSC, 0.05% SDS. C, 2X SSC, 0.1% SDS; twice with 1X SSC, 0.1% SDS. D, protocol A omitting last wash. All washes were at 65°C for 30 min. 1X SSC is 0.15 M NaCl, 0.015 M Na citrate, pH 7.0.
plicated by the presence of another locus causing X0 offspring, but Bpa was eventually separated from it. The data place Bpa 9-36 CM from MO and 2-15 CM from Tu at 95% confidence limits. No direct mapping of Bpa with other loci has been performed.
The clinical similarities between the phenotype of Bpa mice and that of human CDPXB patients are striking and very likely represent mutations in ho- mologous genes. As a first step toward cloning this in- teresting locus, we have confirmed the map position of Bpa on the mouse X chromosome. The strategy em- ployed should be applicable to mapping other dominant murine mutations.
MATERIALS AND METHODS
Mouse Strains and Crosses
Heterozygous Bpa female mice (B6CBA-AWeJ/A- Bps), Mus spretus (Spain) males, and (C57BL/6JAwWJ X CBA)F1 hybrid males were purchased from The Jackson Laboratory (Bar Harbor, ME). For routine breeding, Bpa females were mated to (C57BL/6JAW” X CBA) males. Affected females were identified at 5- 7 days after birth by a hyperkeratotic skin eruption and later development of linear bare patches. For in- terspecific crosses, Bpa females were mated with M. spretus males, and F1 affected females were backcrossed to (C57BL/6JA”J X CBA)Fr males. Since these hybrid males are always derived from a C57BL/6JAW” female mated with a CBA male, the X chromosome is always derived from the former strain. The hybrid males pro- duced many more progeny than did the inbred C57BL/ 6JAwJ males. The number of affected females identified
was approximately 50% of that expected, and likely reflects in utero or early postnatal loss. The sex of progeny was confirmed by analysis of DNA using the male-specific Y chromosome probe pY2 (see Table 1).
Southern Blotting
Genomic DNA was prepared from mouse tails or or- gans by using SDS/proteinase K lysis and phenol/ chloroform extraction. Restriction enzymes were pur- chased from Boehringer-Mannheim, and digests were performed as recommended by the manufacturer. Gel electrophoresis in 0.7-0.8% agarose and Southern transfer to GeneScreen Plus were performed as rec- ommended by DuPont. DNA probes were prepared us- ing random oligohexamer labeling (Feinberg and Vo- gelstein, 1984) with [a-32P]dCTP (Amersham). Pre- hybridization and hybridization were performed at 42°C in 40% formamide, 1 M NaCl, 10% dextran sul- fate (Pharmacia), 1% SDS, and 200 wg/ml denatured herring sperm DNA. The DNA probes used and the specific washing conditions are listed in Table 1.
RESULTS
Analysis of Interspecific Backcross Progeny
To date, 22 normal male and affected female back- cross progeny from the interspecific matings have been analyzed with eight DNA probes spanning the X chro- mosome (Fig. 1). Because of the possibility of incom- plete penetrance, normal-appearing females were ex- cluded from the analysis. Several single recombinant offspring suggest that Bpa maps between Cf-9 and Pgk-1 .
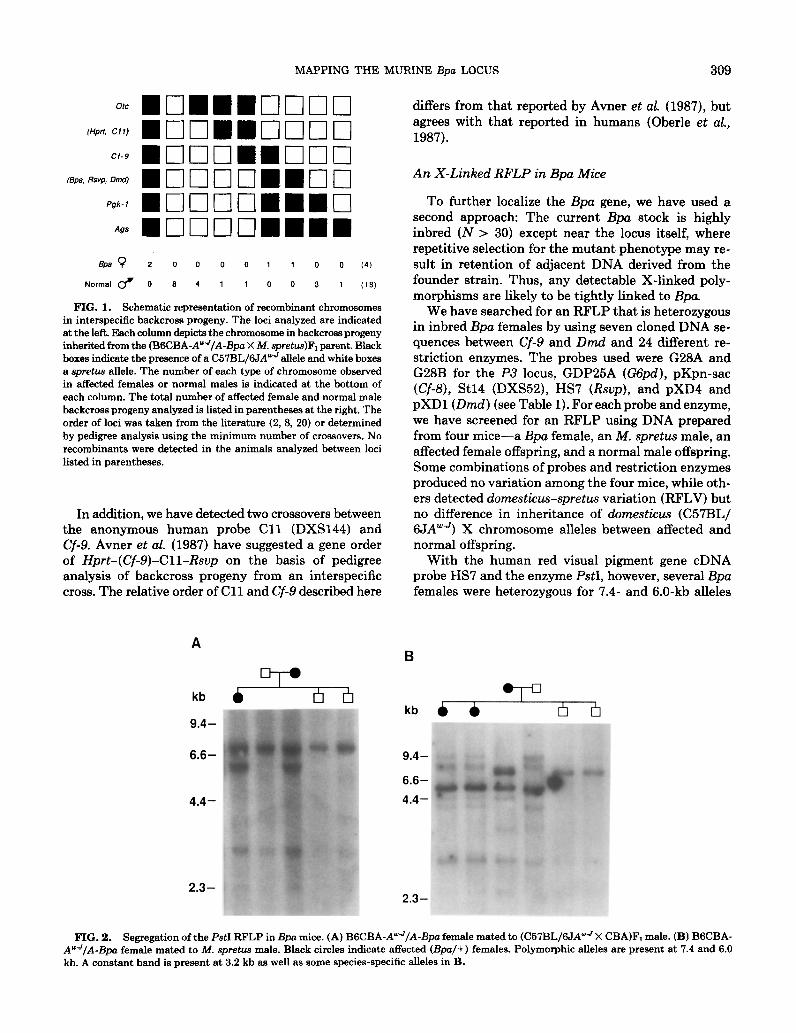
MAPPING THE MURINE &a LOCUS 309
@Pa9 2 0 0 0 0110 0 (4)
Normal # 0 8 4 1 1 0 0 3 1 (18)
FIG. 1. Schematic representation of recombinant chromosomes in interspecific backcross progeny. The loci analyzed are indicated at the left. Each column depicts the chromosome in backcross progeny inherited from the (BSCBA-AwJ/A-Bpo X M. spretu.s)Fi parent. Black boxes indicate the presence of a C57BL/6JA”’ allele and white boxes a spretua allele. The number of each type of chromosome observed in affected females or normal males is indicated at the bottom of each column. The total number of affected female and normal male backcross progeny analyzed is listed in parentheses at the right. The order of loci was taken from the literature (2, 8, 20) or determined by pedigree analysis using the minimum number of crossovers. No recombinants were detected in the animals analyzed between loci listed in parentheses.
In addition, we have detected two crossovers between the anonymous human probe Cl1 (DXS144) and Cf-9. Avner et al. (1987) have suggested a gene order of Hprt-(Cf-9)-Cll-Rsup on the basis of pedigree analysis of backcross progeny from an interspecific cross. The relative order of Cl 1 and Cf-9 described here
2.3-
differs from that reported by Avner et al. (1987), but agrees with that reported in humans (Oberle et aZ., 1987).
An X-Linked RFLP in Bpa Mice
To further localize the Bpa gene, we have used a second approach: The current Bpa stock is highly inbred (N > 30) except near the locus itself, where repetitive selection for the mutant phenotype may re- sult in retention of adjacent DNA derived from the founder strain. Thus, any detectable X-linked poly- morphisms are likely to be tightly linked to Bpa.
We have searched for an RFLP that is heterozygous in inbred Bpa females by using seven cloned DNA se- quences between Cf-9 and Dmd and 24 different re- striction enzymes. The probes used were G28A and G28B for the P3 locus, GDP25A (GGpd), pKpn-sac (Cf-8), St14 (DXS52), HS7 (Rsup), and pXD4 and pXD1 (Dmd) (see Table 1). For each probe and enzyme, we have screened for an RFLP using DNA prepared from four mice-a Bpu female, an M. spretus male, an affected female offspring, and a normal male offspring. Some combinations of probes and restriction enzymes produced no variation among the four mice, while oth- ers detected domesticus-spretus variation (RFLV) but no difference in inheritance of domesticus (C57BL/ 6JAweJ) X chromosome alleles between affected and normal offspring.
With the human red visual pigment gene cDNA probe HS7 and the enzyme PstI, however, several Bpa females were heterozygous for 7.4- and 6.0-kb alleles
kb & ()
2.3-
FIG. 2. Segregation of the PstI RFLP in Bpa mice. (A) BSCBA-AWJ/A-Bpo female mated to (C57BL/6JAwJ X CBA)FI male. (B) BGCBA- AYJ/A-Bpa female mated to M. spretus male. Black circles indicate affected @pa/+) females. Polymorphic alleles are present at 7.4 and 6.0 kh. A constant band is present at 3.2 kb as well as some species-specific alleles in B.
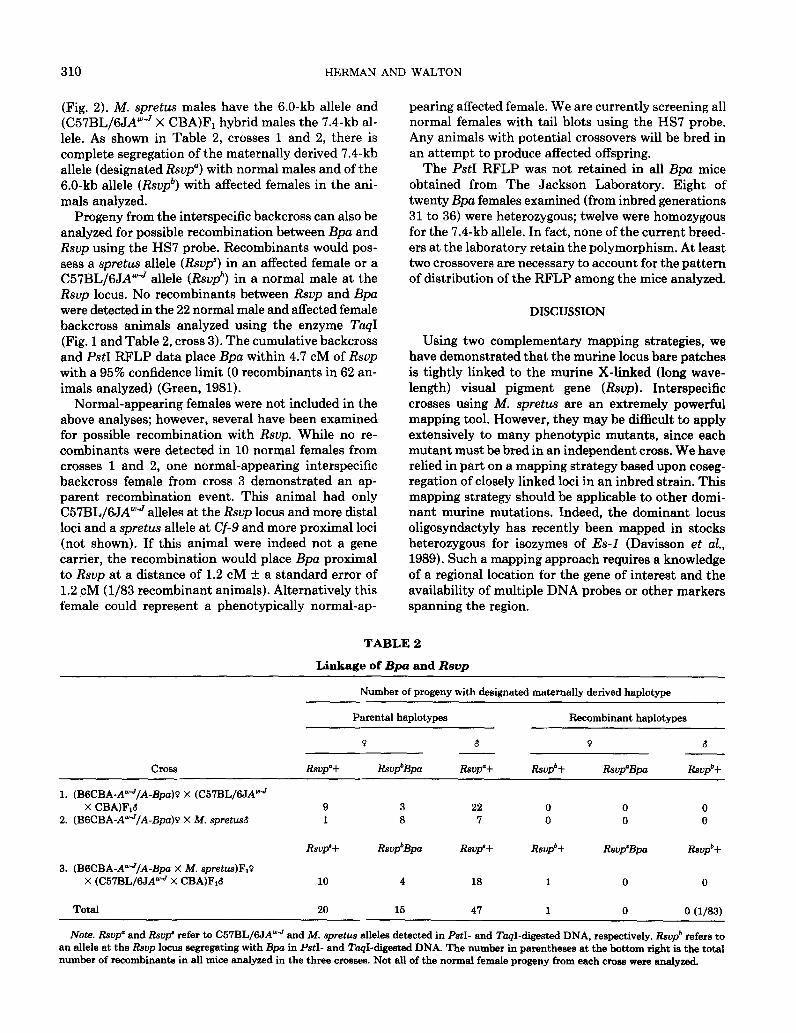
310 HERMAN AND WALTON
(Fig. 2). M. spretus males have the 6.0-kb allele and (C57BL/6JAweJ X CBA)Fi hybrid males the 7.4-kb al- lele. As shown in Table 2, crosses 1 and 2, there is complete segregation of the maternally derived 7.4-kb allele (designated Rsvp”) with normal males and of the 6.0-kb allele (R&‘) with affected females in the ani- mals analyzed.
Progeny from the interspecific backcross can also be analyzed for possible recombination between Bpa and Rsvp using the HS7 probe. Recombinants would pos- sess a spretus allele (Rsup’) in an affected female or a C57BL/6JA”-$ allele (Rsvp*) in a normal male at the Rsvp locus. No recombinants between Rsvp and Bpa were detected in the 22 normal male and affected female backcross animals analyzed using the enzyme TaqI (Fig. 1 and Table 2, cross 3). The cumulative backcross and PstI RFLP data place Bpa within 4.7 CM of Rsvp with a 95% confidence limit (0 recombinants in 62 an- imals analyzed) (Green, 1981).
Normal-appearing females were not included in the above analyses; however, several have been examined for possible recombination with RSVP. While no re- combinants were detected in 10 normal females from crosses 1 and 2, one normal-appearing interspecific backcross female from cross 3 demonstrated an ap- parent recombination event. This animal had only C57BL/6JA”‘J alleles at the Rsup locus and more distal loci and a spretus allele at Cf-9 and more proximal loci (not shown). If this animal were indeed not a gene carrier, the recombination would place Bpa proximal to Rsvp at a distance of 1.2 CM + a standard error of 1.2 cM (l/83 recombinant animals). Alternatively this female could represent a phenotypically normal-ap-
pearing affected female. We are currently screening all normal females with tail blots using the HS7 probe. Any animals with potential crossovers will be bred in an attempt to produce affected offspring.
The PstI RFLP was not retained in all Bpa mice obtained from The Jackson Laboratory. Eight of twenty Bpa females examined (from inbred generations 31 to 36) were heterozygous; twelve were homozygous for the 7.4-kb allele. In fact, none of the current breed- ers at the laboratory retain the polymorphism. At least two crossovers are necessary to account for the pattern of distribution of the RFLP among the mice analyzed.
DISCUSSION
Using two complementary mapping strategies, we have demonstrated that the murine locus bare patches is tightly linked to the murine X-linked (long wave- length) visual pigment gene (Rsup). Interspecific crosses using M. spretus are an extremely powerful mapping tool. However, they may be difficult to apply extensively to many phenotypic mutants, since each mutant must be bred in an independent cross. We have relied in part on a mapping strategy based upon coseg- regation of closely linked loci in an inbred strain. This mapping strategy should be applicable to other domi- nant murine mutations. Indeed, the dominant locus oligosyndactyly has recently been mapped in stocks heterozygous for isozymes of Es-l (Davisson et al., 1989). Such a mapping approach requires a knowledge of a regional location for the gene of interest and the availability of multiple DNA probes or other markers spanning the region.
TABLE 2
Linkage of Bps and Rsvp
Number of progeny with designated maternally derived haplotype
Parental haplotypes Recombinant haplotypes
Q 6 0 6
Cross RSVP’+ Rsvp*Bpa RSVP’+ Rwp*+ Rsvp’Bpa RSVP*+
1. (BsCBA-AY’JIA-Bpa)P X (C57BL/6JAWJ X CBAlFi.3 9 3 22 0 0 0
2. (B6CBA-AWJ/A-Bpa)g X hf. spretusd 1 8 7 0 0 0
RSVP’+ Rsvp*Bpa Rsvp’+ RSVP*+ Rsvp’Bpa RSVP*+
3. (BSCBA-AwJ/A-&a X M. spretus)F,?+ X (C57BL/6JA“‘-’ X CBA)FiG 10 4 18 1 0 0
Total 20 15 47 1 0 0 (l/83)
Note. RSVP” and Rsu@ refer to C67BL/6JA”J and M. spretus alleles detectad in PstI- and TaqI-digested DNA, respectively. RSVP* refers to an allele at the Rsvp locus segregating with Bpa in PstI- and TaqI-digested DNA. The number in parentheses at the bottom right is the total number of recombinanta in all mice analyzed in the three crosses. Not all of the normal female progeny from each cross were analyzed.

MAPPING THE MURINE Bps LOCUS 311
The backcross animals analyzed also suggest a gene order for Cl1 and Cf-9 that is an alternative to that previously reported by Avner et al. (1987). Their anal- ysis also utilized an interspecific cross with M. spretw (BGCBARI X SPE/Pas). However, the Cf-9 probe pro- duced weak signals and only MspI produced detectable RFLVs. With BumHI, the Cf-9 probe employed here detects an easily scored C57BL/6JA”” allele of ap- proximately 18 kb and a spretus allele of 9.5 kb (not shown). It is possible that a small inversion involving Cl1 and Cf-9 exists in the strains used by Avner et al.; however, it would require the same gene order in both strains or recombination would be suppressed.
Bare patches is very likely homologous to CDPXB. Nomenclature for this group of human chondrodys- plasias is extremely confusing. At least four different inherited types of chondrodysplasia punctata (CDP) have been described, including a severe recessive dis- ease (rhizomelic CDP) associated with peroxisomal abnormalities (Heymans et al., 1985; Poulos et al., 1988; Hoefler et al., 1988) and one or more milder autosomal dominant forms (Sheffield et al., 1989).
There are two distinct X-linked forms of CDP. The X-linked recessive form (CDPX) is seen only in males and is associated with terminal deletions of Xp22.3- Xpter or X/Y translocations (Curry et al., 1984; Bal- labio et al., 1989). Ichthyosis in these patients is as- sociated with deletions involving steroid sulfatase (STS). Recently, combined cytogenetics and molecular analyses of multiple males affected with this “contig- uous gene syndrome” suggest that the CDPX locus maps distal to STS (Ballabio et al., 1989). The only abnormality in female carriers of Xp22.3-Xpter dele- tions compared to noncarriers is short stature (Curry et al., 1984).
In contrast, the X-linked dominant form of chon- drodysplasia punctata (CDPXB) has been reported ex- clusively in females. On the basis of presumed homol- ogy to Bpa, we propose that CDPXB maps within Xq27-Xq28. There have been at least five major chro- mosomal rearrangements between murine and human X chromosomes (Davison, 1987; Amar et aZ., 1988). It could be argued that CDPX2 maps near the human DMD locus since Rsup and Dmd are linked in the mouse (Fig. 3). We believe that this is unlikely because human males with deletions extending several million base pairs on either side of the DMD locus have been described (Francke et al., 1985; Towbin et al., 1989). If CDPXB maps in this region, these males should not survive. Further, the one mouse demonstrating a re- combination described here places Bpa proximal to Rsvp in a region of conserved linkage between the mouse and human Xq27-Xq28.
The existence of a murine model for a human skeletal dysplasia provides a unique opportunity to apply mo- lecular techniques to disorders that have been difficult
FIG. 3. Comparison of human and murine X chromosomes. Gene locations were taken from Refs. (2,8,20). GLA and Ags indicate the human and murine a-galactosidase loci, respectively. GCP/RCP are human green and red visual pigment loci corresponding to the murine Rsvp locus.
to understand. We are currently trying to demonstrate linkage in human CDPXB pedigrees to Xq28 markers. We are also continuing to refine our mapping studies of Bpa as more backcross animals are generated and more DNA probes between Cf-9 and Rsvp become available. Pulsed-field gel analysis of the region is being used to search for a submicroscopic deletion encom- passing the Bpa locus. The eventual isolation of the Bpa locus and its human homolog should provide new information concerning cartilage and/or bone bio- chemistry and biogenesis.
ACKNOWLEDGMENTS
The authors thank Dr. J. Fielding Hejtmancik, Baylor College of Medicine, and the individuals listed in Table 1 for providing DNA probes and Dr. Eva Either and Ms. Belinda Harris, Jackson Labo- ratory, for many helpful discussions. G.E.H. is a Lucille P. Markey Scholar and this research was supported by a grant from the Lucille P. Markey Charitable Trust and USPHS Grant RR-05425.
REFERENCES
1. ADRA, C. N., BOER, P. H., AND MCBURNEY, M. W. (1987). Cloning and expression of the mouse pgk-I gene and the nu- cleotide sequence of its promoter. Gene 60: 65-74.
2. AMAR, L. C., DANDOLO, L., HANALJER, A., COOK, A. R., ARNA~D, D., MANDEL, J-L., AND AVNER, P. (1988). Conservation and reorganization of loci on the mammalian X chromosome: A

312 HERMAN AND WALTON
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
molecular framework for the identification of homologous sub- chromosomal regions in man and mouse. Genomics 2: 220-230.
AVNER, P., AMAR, L., ARNAUD, D., HANAUER, A., AND CAMBROU, J. (1987). Detailed ordering of markers localizing to the Xq26- Xqter region of the human X chromosome by the use of an interspecific Mus spretus mouse cross. Proc. Natl. Acad. Sci.
USA 84: 1629-1633.
BALLABIO, A., BARDONI, B., CARROZZO, R., ANDRIA, G., BICK, D., CAMPBELL, L., HAMEL, B., FERGUSON-SMITH, M. A., GI-
MELLI, G., FRACCARO, M., MARASCHIO, P., ZUFFARDI, O., GUIOLI, S., AND CAMERINO, G. (1989). Contiguous gene syn- dromes due to deletions in the distal short arm of the human X chromosome. Proc. Natl. Acad. Sci. USA 86: lO,OOl-10,005.
CAMERINO, G., MATTEI, M. G., MAITEI, J. F., JAYE, M., AND MANDEL, J-L. (1983). Close linkage of fragile X-mental retar- dation syndrome to hemophilia B and transmission through a normal male. Nature U.ondon) 306: 701-704.
CHAMBERLAIN, J. S., PEARLMAN, J. A., MUZNY, D. M., GIBBS, R. A., RANIER, J. E., REEVES, A. A., AND CASKEY, C. T. (1988). Expression of the murine Duchenne muscular dystrophy gene in muscle and brain. Science 239: 1416-1418.
CURRY, C. J. R., MAGENIS, R. E., BROWN, M., LANMAN, J. T., JR., TSAI, J., O’LAGUE, P., GOODFELLOW, P., MOHANDAS, T., BERGNER, E. A., AND SHAPIRO, L. J. (1984). Inherited chon- drodysplasia punctata due to a deletion of the terminal short arm of an X chromosome. N. Engl. J. Med. 311: 1010-1015.
DAVISSON, M. T. (1987). Review: X-linked genetic homologies between mouse and man. Gerwmics 1: 213-227.
DAVISSON, M. T., Fox, R. R., RODERICK, T. H., SWEET, H. O.,
AND SPENCER, C. (1989). E&erase-l tightly linked to OS. Mouse NewsLett. 84:90. FEINBERG, A. P., AND VOGELSTEIN, B. (1984). A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity: Addendum. Anal. Biochem. 137: 266-277. FRANCKE, U., OCHS, H. D., DE MARTINVILLE, B., GIACALONE, J., LINDGREN, V., DISTECHE, C., PAGON, R. A., HOFKER, M. H., VAN OMMEN, G. B., PEARSON, P. L., AND WEDGWOOD, R. J. (1985). Minor Xp21 chromosome deletion in a male as- sociated with expression of Duchenne muscular dystrophy, chronic granulomatous disease, retinitis pigmentosa, and McLeod syndrome. Amer. J. Hum. Genet. 37: 250-267.
GREEN, E. L. (1981). “Genetics and Probability in Animal Breeding Experiments,” p. 27, Oxford Univ. Press, New York.
HAPPLE, R. (1979). X-linked dominant chondrodysplasia punctata. Hum. Genet. 53: 65-73.
HAPPLE, R., PHILLIPS, R. J. S., ROESSNER, A., AND JUNEMANN, G. (1983). Homologous genes for X-linked chondrodysplasia punctata in man and mouse. Hum. Genet. 63: 24-27.
HEYMANS, H. S. A., OORTHUYS, J. W. E., NELCK, G., WANDERS, R. J. A., AND SCHUTGENS, R. B. H. (1985). Rhizomelic chon- drodysplasia punctata: Another peroxisomal disorder. N. Engl. J. Med. 313: 187-188.
HOEFLER, G., HOEFLER, S., WATKINS, P. A., CHEN, W. W., MOSER, A., BALDWIN, V., MCGILLIVARY, B., CHARROW, J.,
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
FRIEDMAN, J. M., RUTLEDGE, L., HASHIMOTO, T., AND MOSER, H. W. (1988). Biochemical abnormalities in rhizomelic chon- drodysplasia punctata. J. Pediatr. 112: 726-733. LAMAR, E. E., AND PALMER, E. (1984). Y-encoded species-spe- cific DNA in mice: Evidence that the Y chromosome exists in two polymorphic forms in inbred strains. Cell 37: 171-177.
MANZKE, H., CHRISTOPHERS, E., AND WIEDEMANN, H-R. (1980). Dominant sex-linked inherited chondrodysplasia punctata: a distinct type of chondrodysplasia punctata. C&n. Genet. 17:
97-107.
MUELLER, R. F., CROWLE, P. M., JONES, R. A. K., AND DAVISON, B. C. C. (1985). X-linked dominant chondrodysplasia punctati a case report and family studies. Amer. J. Med. Genet. 20: 137- 144.
MULLINS, L. J., GRANT, S. G., STEPHENSON, D. A., AND CHAP- MAN, V. M. (1988). Multilocus molecular mapping of the mouse X chromosome. Genomics 3: 187-194.
NATHANS, J., THOMAS, D., AND HOGNESS, D. S. (1986). MO- lecular genetics of human color vision: The genes encoding blue,
green, and red pigments. Science 232: 193-202.
OBERLE, I., CAMERINO, G., WROGEMANN, K., ARVEILER, B., HANAUER, A., RAIMONDI, E., AND MANDEL, J. L. (1987). Mul- tipoint genetic mapping of the Xq26-q28 region in families with fragile X mental retardation and in normal families reveals tight linkage of markers in q26-q27. Hum. Genet. 77: 60-65.
PERSICO, M. G., VIGLIETTO, G., MARTINI, G., TONIOLO, D., PAONESSA, G., MOSCATELLI, G., DONO, R., VULLIAMY, T., Luz- ZATTO, L., AND D’URSO, M. (1986). Isolation of human glucose- 6-phosphate dehydrogenase (GGPD) cDNA clones: Primary structure of the protein and unusual 5’ non-coding region. Nu- cleic Acids Res. 14: 2511-2522.
PHILLIPS, R. J. S., HAWKER, S. G., AND MOSELEY, H. J. (1973). Bare-patches, a new sex-linked gene in the mouse, associated with a high production of X0 females. Genet. Res. 22: 91-99.
PHILLIPS, R. J. S., AND KAUFMAN, M. H. (1974). Bare-patches, a new sex-linked gene in the mouse, associated with a high production of X0 females. Genet. Res. 24: 27-41.
POULOS, A., SHEFFIELD, L., SHARP, P., SHERWOOD, G., JOHN- SON, D., BECKMAN, K., FELLENBERG, A. J., WRAITH, J. E., CHOW, C. W., USHER, S., AND SINGH, H. (1988). Rhizomelic chondrodysplasia punctata: Clinical, pathologic, and biochem- ical findings in two patients. J. Pediatr. 113: 685-690. SHEFFIELD, L. J., HALLIDAY, J. L., DANKS, D. M., ROGERS, J. G., POULOS, A., AND MORRISON, N. (1989). Clinical, radio- logical and biochemical classification of chondrodysplasia punctata. Amer. J. Hum. Genet. 45: A64.
TOWBIN, J. A., WV, D., CHAMBERLAIN, J., LARSEN, P. D., SELTZER, W. K., AND MCCABE, E. R. B. (1989). Characterization of patients with glycerol kinase deficiency utilizing cDNA probes for the Duchenne muscular dystrophy locus. Hum. Genet. 83: 122-126. VERES, G., GIBBS, R. A., SCHERER, S. E., AND CASKEY, C. T. (1987). The molecular basis of the sparse fur mouse mutation. Science 237:415-417.




















