
Classical and molecular cytogenetic abnormalities and outcome of childhood acute myeloid leukaemia:...
18
Classical and molecular cytogenetic abnormalities and outcome of childhood acute myeloid leukaemia: report from a referral centre in Israel Extensive cytogenetic analyses of the leukaemias have demon- strated an association of recurrent chromosomal aberrations with the clinical and biological diversity of acute myeloid leukaemia (AML) and provided insight into the genetic changes that underlie leukaemogenesis, thereby opening new horizons for profiles of microarray gene expression pathways and targeted treatment strategies (Rowley, 2000, 2001; Will- man, 2001; Haferlach et al, 2003; Yagi et al, 2003). A few of the more common specific cytogenetic/molecular abnormalities, namely, t(8;21)/AML1-ETO, t(15;17)/PML-RARA, inv(16)/ t(16;16)/CBFb-MYH11 and t(11q23)/MLL, have recently been recognized by the World Health Organization (WHO) classi- fication as distinct disease categories, associated with the French-American-British (FAB) groups (Harris et al, 1999; Vardiman et al, 2002). The strong association of diagnostic karyotype with out- come, demonstrated in some large adult studies, has rendered cytogenetics the most valuable prognostic factor for treatment selection (Bloomfield et al, 1997; Grimwade et al, 1998; Grimwade, 2001; Mro ´ zek et al, 2001). However, for some less frequent non-random aberrations, the cytogenetic prognostic classification is still inconsistent owing to the small number of patients and the variable treatment modalities used in different study groups (Grimwade et al, 1998, 2002; Slovak et al, 2000; Byrd et al, 2002). The reported childhood AML series consist of a relatively smaller number of patients and show a different distribution of cytogenetic subsets (Woods et al, 2001) and sometimes a different response to treatment from adults (Martinez-Climent et al, 1995a; Grimwade et al, 1998; Raimondi et al, 1999; Batia Stark, 1,2 Marta Jeison, 2 Leticia Glazer Gabay, 2 Jacques Mardoukh, 2 Drorit Luria, 3 Irit Bar-Am, 4 Gali Avrahami, 1 Yossef Kapeliushnik, 5 Dalia Sthoeger, 6 Gavriel Herzel, 7 David M. Steinberg, 8 Ian J. Cohen, 1 Yacov Goshen, 1 Jerry Stein, 1 Rina Zaizov 1 and Isaac Yaniv 1 1 Centre of Pediatric Hematology/Oncology, 2 Cancer Cytogenetic Laboratory and 3 Flow Cytometry Unit, Schneider Children’s Medical Centre of Israel, Petah Tiqva, and Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, 4 Applied Spectral Imaging, Ltd, Migdal Ha’emek, 5 Soroka Hospital, Beer Sheva, 6 Kaplan Hospital, Rehovot, 7 Haemek Hospital, Afula, and 8 Department of Statistics and O.R., Tel Aviv University, Tel Aviv, Israel Received 5 January 2004; accepted for publication 15 April 2004 Correspondence: B. Stark, The Center of Pediatric Hematology/Oncology and the Cancer Cytogenetic Laboratory, Schneider Children’s Medical Centre of Israel, Petah Tiqva 49202, Israel. E-mail: [email protected] Summary The incidence of cytogenetic abnormalities in childhood de novo acute myeloid leukaemia (AML) and its prognostic significance was assessed in an Israeli paediatric referral centre. Cytogenetic analysis was successful in 86 of 97 children (<20 years of age) diagnosed between 1988 and 2002 with de novo AML. Fluorescence in situ hybridization analysis detected new information in 11 of them, leading to reassignment in cytogenetic group classification. The incidence of the various cytogenetic subgroups was as follows: normal – 9%; t(11q23) – 22%; t(8;21) – 13%; t(15;17) – 8%; inv(16) – 3Æ4%; abn(3q) – 4Æ6%; 7/7q-(sole or main) – 5Æ8%; del(9q)(sole) and +21(sole) – 4Æ6% each; t(8;16) – 2Æ3%; t(6;9), t(1;22), +8(sole) – 1Æ1% each; and miscellaneous – 18%. The overall survival (OS) and event-free survival (EFS) (4 years) for 94 patients treated with the modified Berlin- Frankfu ¨rt-Mu ¨nster (BFM) AML protocols (non-irradiated) were 59Æ9% (SE ¼ 5%) and 55Æ7% (SE ¼ 5%), respectively, and for the favourable t(8;21), t(15;17) and inv(16), OS was 60% (SE ¼ 15%), 83% (SE ¼ 15%) and 100% respectively. For the normal group it was 62% (SE ¼ 17%), miscellaneous 64% (SE ¼ 12%), t(11q23) 44Æ6% (SE ¼ 11%) and of the )7/7q-, del(9q)(sole) or t(6;9), none had survived at 4 years. The incidence of cytogenetic subgroups in the Israeli childhood AML population and their outcome were similar to other recently reported paediatric series. Cytogenetic abnormalities still carry clinical relevance for treatment stratification in the context of modern chemotherapy. Keywords: cytogenetics, childhood acute myeloid leukaemia. research paper doi:10.1111/j.1365-2141.2004.05038.x ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Classical and molecular cytogenetic abnormalities and outcome of childhood acute myeloid leukaemia:...

Classical and molecular cytogenetic abnormalities and outcomeof childhood acute myeloid leukaemia: report from a referralcentre in Israel
Extensive cytogenetic analyses of the leukaemias have demon-
strated an association of recurrent chromosomal aberrations
with the clinical and biological diversity of acute myeloid
leukaemia (AML) and provided insight into the genetic
changes that underlie leukaemogenesis, thereby opening new
horizons for profiles of microarray gene expression pathways
and targeted treatment strategies (Rowley, 2000, 2001; Will-
man, 2001; Haferlach et al, 2003; Yagi et al, 2003). A few of the
more common specific cytogenetic/molecular abnormalities,
namely, t(8;21)/AML1-ETO, t(15;17)/PML-RARA, inv(16)/
t(16;16)/CBFb-MYH11 and t(11q23)/MLL, have recently been
recognized by the World Health Organization (WHO) classi-
fication as distinct disease categories, associated with the
French-American-British (FAB) groups (Harris et al, 1999;
Vardiman et al, 2002).
The strong association of diagnostic karyotype with out-
come, demonstrated in some large adult studies, has rendered
cytogenetics the most valuable prognostic factor for treatment
selection (Bloomfield et al, 1997; Grimwade et al, 1998;
Grimwade, 2001; Mrozek et al, 2001). However, for some less
frequent non-random aberrations, the cytogenetic prognostic
classification is still inconsistent owing to the small number of
patients and the variable treatment modalities used in different
study groups (Grimwade et al, 1998, 2002; Slovak et al, 2000;
Byrd et al, 2002).
The reported childhood AML series consist of a relatively
smaller number of patients and show a different distribution of
cytogenetic subsets (Woods et al, 2001) and sometimes a
different response to treatment from adults (Martinez-Climent
et al, 1995a; Grimwade et al, 1998; Raimondi et al, 1999;
Batia Stark,1,2 Marta Jeison,2
Leticia Glazer Gabay,2 Jacques
Mardoukh,2 Drorit Luria,3 Irit Bar-Am,4
Gali Avrahami,1 Yossef Kapeliushnik,5
Dalia Sthoeger,6 Gavriel Herzel,7
David M. Steinberg,8 Ian J. Cohen,1
Yacov Goshen,1 Jerry Stein,1
Rina Zaizov1 and Isaac Yaniv1
1Centre of Pediatric Hematology/Oncology,2Cancer Cytogenetic Laboratory and 3Flow
Cytometry Unit, Schneider Children’s Medical
Centre of Israel, Petah Tiqva, and Sackler Faculty
of Medicine, Tel Aviv University, Tel Aviv,4Applied Spectral Imaging, Ltd, Migdal Ha’emek,5Soroka Hospital, Beer Sheva, 6Kaplan Hospital,
Rehovot, 7Haemek Hospital, Afula, and8Department of Statistics and O.R., Tel Aviv
University, Tel Aviv, Israel
Received 5 January 2004; accepted for
publication 15 April 2004
Correspondence: B. Stark, The Center of
Pediatric Hematology/Oncology and the Cancer
Cytogenetic Laboratory, Schneider Children’s
Medical Centre of Israel, Petah Tiqva 49202,
Israel. E-mail: [email protected]
Summary
The incidence of cytogenetic abnormalities in childhood de novo acute
myeloid leukaemia (AML) and its prognostic significance was assessed in an
Israeli paediatric referral centre. Cytogenetic analysis was successful in 86 of
97 children (<20 years of age) diagnosed between 1988 and 2002 with de
novo AML. Fluorescence in situ hybridization analysis detected new
information in 11 of them, leading to reassignment in cytogenetic group
classification. The incidence of the various cytogenetic subgroups was as
follows: normal – 9%; t(11q23) – 22%; t(8;21) – 13%; t(15;17) – 8%;
inv(16) – 3Æ4%; abn(3q) – 4Æ6%; 7/7q-(sole or main) – 5Æ8%; del(9q)(sole)
and +21(sole) – 4Æ6% each; t(8;16) – 2Æ3%; t(6;9), t(1;22), +8(sole) – 1Æ1%each; and miscellaneous – 18%. The overall survival (OS) and event-free
survival (EFS) (4 years) for 94 patients treated with the modified Berlin-
Frankfurt-Munster (BFM) AML protocols (non-irradiated) were 59Æ9%(SE ¼ 5%) and 55Æ7% (SE ¼ 5%), respectively, and for the favourable
t(8;21), t(15;17) and inv(16), OS was 60% (SE ¼ 15%), 83% (SE ¼ 15%)
and 100% respectively. For the normal group it was 62% (SE ¼ 17%),
miscellaneous 64% (SE ¼ 12%), t(11q23) 44Æ6% (SE ¼ 11%) and of the
)7/7q-, del(9q)(sole) or t(6;9), none had survived at 4 years. The incidence
of cytogenetic subgroups in the Israeli childhood AML population and their
outcome were similar to other recently reported paediatric series.
Cytogenetic abnormalities still carry clinical relevance for treatment
stratification in the context of modern chemotherapy.
Keywords: cytogenetics, childhood acute myeloid leukaemia.
research paper
doi:10.1111/j.1365-2141.2004.05038.x ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337

Woods et al, 2001; Wells et al, 2002; Forestier et al, 2003;
Harrison et al, 2003). Moreover, geographic differences in the
frequency of cytogenetic subsets have been noted in adult and
childhood AML (Johansson et al, 1991; Biondi et al, 1994;
Nakase et al, 2000).
The aim of this study was to assess the demography of
cytogenetic abnormalities and their correlation with clinical
and biological features and impact on outcome in children
with de novo AML, who were diagnosed at a referral centre in
Israel and treated with the Berlin-Frankfurt-Munster (BFM)-
based AML protocols (Creutzig et al, 1999, 2001). Fluores-
cence in situ hybridization (FISH) analysis, using a panel of
commercial AML probes, was retrospectively performed on all
available, adequate-quality cytogenetic bone marrow (BM)
pellets to confirm and complement the information gained
from conventional cytogenetics.
Patients and methods
Patients
The study sample consisted of 97 newly diagnosed de novo
AML patients younger than 20 years, who were diagnosed at
the Schneider Children’s Medical Centre of Israel (88 patients),
Soroka Hospital (six patients), and Kaplan Hospital (three
patients) between July 1988 and January 2003. Four children
with Down syndrome and one with an undefined constitu-
tional non-Fanconi birth defect were included.
Treatment
Treatment for 94 patients (excluding another three patients
treated elsewhere) was based on the AML-BFM-87 (July 1988–
July 1996; 52 patients), AML-BFM-93 (August 1996–July 1998;
20 patients), and AML-BFM-98 (August 1998–January 2003;
22 patients) protocols (Creutzig et al, 1999, 2001). Modifica-
tions included mainly substitution of preventive cranial
irradiation with additional five intrathecal triple (TIT) cytara-
bine (Ara-C) methotrexate (MTX), and hydrocortisone injec-
tions during maintenance. Systemic chemotherapy was
administered without randomization, and all the patients
received the higher risk protocol. In the AML-BFM-87
protocol, patients who completed the second intensification
course were eligible for allogeneic bone marrow transplanta-
tion (BMT) from a human leucocyte antigen (HLA)-matched
sibling, if available, conditioning with cyclophosphamide and
busulphan (actually performed in three patients), or for
autologous BMT with conditioning with melphalan 180 mg/m2
(13 patients), or for continuation with maintenance chemo-
therapy for 1 year (29 patients). In the BFM-93 and -98
protocols, high-risk patients only were offered allogeneic BMT
from an HLA-matched sibling (actually performed in 11
patients). Patients in the non-high-risk group were eligible for
autologous BMT (performed in 14 patients) or maintenance
chemotherapy (nine patients). The non-high risk group was
defined similarly to the AML-BFM criteria as M3/t(15;17),
t(8;21), or M1/2 with Auer rods, inv(16) or M4 with
eosinophilia (Creutzig et al, 1999).
Diagnosis
The diagnosis and FAB subtypes of AML, with or without
evidence of dysplasia or prior myelodysplastic syndrome (MDS)
(Bennett et al, 1985a; Harris et al, 1999; Vardiman et al, 2002),
was determined by Wright-Giemsa-stained BM smears and
cytochemical staining with myeloperoxidase, Sudan-black,
a-naphthyl acetate esterase with and without sodium fluoride
inhibition, chloroacetate-esterase, periodic acid-Schiff, and acid
phosphatase. The diagnosis of M0, M6 and M7 subtypes
required confirmation of immunophenotype and/or electron
microscopywithplatelet peroxidase staining (Bennett et al, 1985b).
Immunophenotyping of the BM samples was performed by
flow cytometry using a FACScan (Becton Dickinson, San Jose,
CA, USA). The monoclonal antibodies tested were for the
panmyeloid antigens CD13 and CD33; myeloid granulocyte
marker CD15; monocytic macrophage marker CD14; erythroid
marker glycophorin A (GpA); megakaryocyte platelet markers
CD41 (GPIIb), CD42 (GPIIb), and CD61 (platelet GPIIIa);
non-lineage restricted marker CD11c; precursor markers
CD34, HLA-DR; anticytoplasmatic myeloperoxidase Cy
MPO; B-cell markers CD19 and CD10; T-cell markers CyCD3,
CD2, CD5, CD7, CD4; natural killer (NK) cell: CD56.
Cytogenetics
Cytogenetic analysis was performed on metaphase cells from
direct or short-term unstimulated BM cultures using the
trypsin-Giemsa banding technique. Chromosomal abnormal-
ities were identified and classified according to the Interna-
tional System for Human Cytogenetic Nomenclature (ISCN)
(Mitelman, 1995). Patients were classified as having a normal
karyotype after 20 normal metaphases were analysed, except for
two patients in whom only 12 and 18 metaphases were available.
Molecular-cytogenetic analysis
Since 1998, interphase FISH analysis with a panel of com-
mercially available probes has been used for the diagnostic
work-up of all new AML patients complementary to conven-
tional cytogenetics. In addition, retrospective FISH analysis
was performed on cytogenetic pellets of adequate quality from
patients treated before 1998. The AML panel included
commercially available probes for the detection of t(11q23),
t(8;21), t(15;17), inv(16), t(9;22), deletion 5/5q, deletion 7/7q,
del(20q), and extra chromosome 8. Most of the panel was used
when new or additional information was expected: if cyto-
genetic results were inadequate (i.e. non-dividing cells, non-
assessable metaphases, or normal), or non-specific chromo-
somal changes or complex karyotypes were demonstrated, or
in the presence of known additional secondary changes.
Cytogenetics, Prognosis in Childhood Acute Myeloid Leukaemia
ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337 321

Otherwise, FISH was applied with a selected probe only to
confirm a suspected specific cytogenetic AML translocation/
aberration or a specific AML FAB morphological subtype.
FISH probes. MLL-dual-colour break-apart, AML1/ETO dual-
colour dual fusion translocation, PML/RARA dual-colour
translocation, CBFb dual-colour break-apart, BCR/ABL dual-
colour extra signal, LSI EGR1 (5q31)(200 kb)/D5S721 D5S23
(5p15Æ2) (450 kb) for 5q31 deletion. LSI D7S486(7q31)
(200 kb)/CEP 7 for 7q31 deletion. D20S108 single colour for
deletion in 20q12 locus, LSI21q22Æ13-q22Æ2 (200 kb) and CEP 8.
All probes were from Vysis Inc. (Downers Grove, IL, USA).
Dual-colour FISH was performed according to the manu-
facturer’s protocol, as previously described (Stark et al, 2002).
FISH signals were visualized with an Olympus BX50 fluores-
cent microscope (Olympus Opticals, Tokyo, Japan), and
images were captured with the Cytovision System (Applied
Imaging, Newcastle-upon-Tyne, UK).
In the majority of cases, 200 interphase nuclei were screened,
but in a few cases lower number of nuclei were available. The
cut-off level for the diagnosis of deletion and trisomies, was set
at the mean percentage of false positive results + 3 SD, i.e.
9Æ5% for 7q-, 8Æ3% for 5q-, 8Æ5% for +21.
Spectral karyotype analysis
Slides were hybridized with the spectral karyotype (SKY) probe
mixture obtained from Applied Spectral Imaging (Migdal
Ha’emek, Israel) as previously described (Stark et al, 2000).
The SKY slides were analysed with the skyview software on
the SD300 Spectral Imaging system (Applied Spectral Ima-
ging).
Cytogenetic/molecular cytogenetic hierarchical classification.
Patients were categorized by the cytogenetic group that
reflected the recognized primary abnormality. In this manner,
each patient was counted only once. The cytogenetic categories
were as follows: normal karyotype, known recurrent t(8;21),
t(15;17), inv(16), t(11q23), 3q abnormality, t(6;9), t(8;16) and
variant, t(1;22), monosomy 7(sole)/del(7q), del(9q)(sole),
trisomy 8(sole), trisomy 21(sole), and miscellaneous clonal
aberrations. Monosomy 7, del(9q), +8, +21, were considered
primary when they appeared cytogenetically as the sole
aberration confirmed by FISH, ruling out any other primary
abnormality. del(7q), when appearing cytogentically in a
complex karyotytpe in all metaphases and confirmed by
FISH, was also considered primary. These abnormalities were
considered secondary when they appeared cytogenetically in
addition to other known primary translocations and/or were
detected by FISH only in a subpopulation of cells. The
cytogenetic group was usually determined by conventional
karyotyping and rarely by SKY. When FISH analysis disclosed
an AML-specific rearrangement, namely MLL splitting, PML/
RARA, or ETO/AML1 fusion, the patients were assigned to the
t(11q23), t(15;17) and t(8;21) cytogenetic groups, respectively,
even if these rearrangements were not detected by conventional
karyotyping.
Definitions of end-points and statistics. Complete remission
(CR) was defined as BM containing <5% blasts, in the
presence of regenerating normal elements and no evidence of
disease at any other site. Early death was defined as death
before or during the first 6 weeks of treatment. Patients were
considered non-responders when BM remission was not
achieved after two chemotherapy courses [after consolidation
or high dose (HD) Ara-C and mitoxantrone]. Event-free
survival (EFS) was calculated from the date of diagnosis to
the last follow-up or to the first event (i.e. failure to achieve
remission, early death, relapse, or death of any cause). Overall
survival (OS) was calculated from the date of diagnosis to the
last follow-up or to the date of death from any cause. The
last follow-up examination for all patients was performed in
April 2003.
Demographic characteristics were compared across the
groups using chi-squared test for qualitative characteristics
and t-test, Mann–Whitney test and anova were used for
quantitative characteristics. Survival curves were estimated
using the method of Kaplan and Meier (1958). Univariate
comparisons of survival curves were made by the log-rank test
(Kalbfleisch & Prentice, 1982). The procedure of Holm (1979)
was used to adjust the univariate EFS P-values for multiple
testing; unadjusted P-values are presented in the Tables. The
Cox regression model (Cox, 1972) was used for multivariate
analysis of prognostic factors for survival. P-values <0Æ05 were
considered significant.
Results
Genetic information was available for 86 of the 97 patients
(89%) with de novo AML (Table I). In eight patients, studies
were not performed, and in three, metaphases were not
obtained and FISH failed to disclose any aberration when
performed in one of these cases.
Of the 86 patients with assessable metaphases, nine had a
normal karyotype. However, molecular cytogenetics per-
formed in four of them detected an MLL split in one case,
decreasing the normal karyotype group to eight patients (9%).
In the other three patients assessed by FISH, deletion of the
ABL gene, representing possible loss of chromosome 9 or loss
of chromosome 5/5q in a subpopulation of cells, was noted in
one patient each; these were considered secondary changes.
Use of the FISH probes MLL (49 patients), ETO/AML1 (24
patients), CBFb/MYH11 (20 patients), PML/RARA (eight
patients) and BCR/ABL (37 patients) yielded new information,
not otherwise detected by conventional cytogenetics in 11
patients, resulting in a change in their cytogenetic group
classification. Specifically, MLL split was detected in nine
patients, and PML/RARA fusion in two patients. In another
four patients FISH detected del(7q) in a subpopulation of cells,
which was considered as a secondary change.
B. Stark et al
322 ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337

Table I. Abnormal G-banded karyotypes and/or SKY and/or FISH findings in 78 patients with AML.
Patient number Karyotype FISH* SKY
t(11q23) (n ¼ 19)
123 51,XY,+4,+8,t(9;11)(p22;q23),+13,+16,+19/52,
idem,+del(1)(p22)
124 46,XY,t(10;11;12)(p13;q13;q13)
131 46,XY,t(1;11)(q21;q23)/46,idem,add(19)(p13)
138 46,XX,t(6;11)(q21;q23),add(18)(q?) MLL split
140 46,XX,t(X;2)(q26;q13) MLL split
149 47,XY,+8 MLL split,7q-
153 46,XY MLL split
156 46,XY,t(6;11)(q27;q23) MLL split
161 45,XX,t(1;19)(q21;p13),add(2)(q37),)10,del(11)(q23)
MLL split
167 46,XY,i(1)(q11) MLL split 46,XY,i(1)(q11),t(9;11)(p22;q23)
180 46,XX,t(11;19)(q23;p13) MLL split
181 46,XY,t(12;17)(q13;q25)/47,XY,idem,+8 MLL split 46,XY,t(12;17)(q13;q25)/47,XY,idem,+8
184 46,XX,t(9;11)(p22;q23) MLL split
185 45,XX,t(10;11)(p13;q23),)18 MLL split
188 50,XX,+8,+9,t(9;11)(p22;q23),+19,+mar/51,
idem,+i(1)(q11)/51,idem,+del(1)(p13)
MLL split
189 47,XX,+8,del(9)(q21?)/47,idem,
der(11)t(1;11)(q23;q23)/46,XX,add (7)
(p22),)13,+22.
MLL split 47,XX,+8,del(9)(q12;q22),
der(10)t(10;11)(p13;q23)/47,idem,
der(11)t(1;11)(q23;q23)/46,XX,der(7)t(3;7)
(?;p22),t(9;10)(p22;p15?),der(11)t(10;11)(p13;q23)
202 46,XX,t(11;17)(q23;q21?) MLL split
231 46,XY,add(11)(q23) MLL split 46,XY,der(11)t(9;11)
1003 51,XY,+6,+8,add(11)(q23),+14,+21,+22 MLL split 51,XY,+6,+8,t(9;11)(p22;q23),der(12)t(12;22)
(q24;q12),+14,+19,+21
t(8;21) (n ¼ 13)
104 46,XX,t(8;21)(q22;q22)
105 46,XY,t(8;21)(q22;q22)
126 46,XX,t(8;21)(q22;q22)
130 46,XY,t(8;21)(q22;q22)
134 45,X,)Y,t(8;21)(q22;q22)136 45,X,)Y,t(8;21)(q22;q22)143 46,XX,t(8;21)(q22;q22)/46,idem,add(2)(q35)/46,
idem,add(2)(q35), del(9)(q12q22),del(11)(p13)
ABLx2
147 45,X,)Y,t(8;21)(q22;q22)158 46,XY,t(8;21)(q22;q22)/46,idem,)22,+mar
208 46,X,)Y,+8,t(8;21)(q22;q22) ETO/AML1(+),)Y233 46,XX,t(8;21)(q22;q22) ETO/AML1(+)
t(15;17) (n ¼ 7)
118 47,XX,+mar
137 46,XY,inv(12)(q12;q21),t(15;17)(q22;q21)
159 46,XY,t(15;17)(q22;q21)
198 46,XX,add(3)(p?) PML/RARA(+)
215 46,XY,t(15;17)(q22;q21)
235 46,XY,t(15;17)(q22;q21),
der(12)t(8;12)(q21;p13)
8101 N.D. PML/RARA(+)
1001 46,XX,t(15;17)(q22;q21)
inv(16) (n ¼ 3)
174 46,XY,inv(16)(p13q22) CBFB split
Cytogenetics, Prognosis in Childhood Acute Myeloid Leukaemia
ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337 323

Table I. Continued.
Patient number Karyotype FISH* SKY
183 48,XY,+9,inv(16)(p13q22),+22/47,
XY,del(7)(q11),del(9)(q12q22) inv (16)(p13q22),+22
CBFB split 7q-
216 46,XX,inv(16)(p13q22) CBFB split
3q21 (n ¼ 2)
127 46,XX,t(3;21)(q21;q22)/46,ide-
m,add(2)(q3?),add(4)(q23)/46,idem,add(1)(p36)
170 47,XY,inv(3)(q21q26),+21c
t(3;5) (n ¼ 2)
110 46,XY,t(3;5)(q25,q34)
152 46,XX,t(3;5)(q25,q34)
t(8p11) (n ¼ 2)
135 46,XX,t(8;19)(p11;q13)
213 46,XX,t(8;16)(p11;q13)
t(6;9) (n ¼ 1)
154 46,XY,t(6;9)(p23;q34)/46,idem,del(16)(q21)
t(1;22) (n ¼ 1)
191 46,XX,t(1;22)(p13;p13)/46,
idem,add(3)(q34),del(5)(q32)
46,XX,t(1;22)(p13;p13)/46,idem,t(3;5)(q34;q32)
7/7q- (n ¼ 5)
120 45,XX,)7141 45,XY,)7169 45,XY,)7 -CEP 7
232 45,XX,)7 -CEP 7
8102 50,XX,)2,add(5)(q35),add(7)(p11),del(7)(q21)x2,+19,+mar1,+mar2x2
7q-x2 50,XX,der(2)t(2;13)(q32;q21),der(5)t(2;5)(q31;q35),
der(6)t(6;10)(p23;?),der(7)t(7;10)(p21,?),del(7)(q21),
+del(7)(q21),+del(9)(q22),+del(9)(q22),der(13)
t(7;13)(q?;q21),+19
del(9q) (sole) (n ¼ 4)
108 46,XY,del(9)(q12q22)
155 46,XY,del(9)(q12q22) ABL x2
209 46,XY,del(9)(q12q22) ABL x2 46,XY,del(9)(q12q22)
228 46,XY,del(9)(q12q22) ABL x2
+21 (sole) (n ¼ 4)
164 47,XX,+21 21qx3
172 47,XY,+21/47,idem,del(20)(q12) 21qx3, 20q-,7q-
173 47,XY,+21/48,XY+8,+21 21qx3
1006 48,XY,+21,+21c
+8 (sole) (n ¼ 1)
171 47,XY,+8 CEP 8x3
One or two miscellaneous aberrations (n ¼ 7)
125 45,X,)X,add(14)(q?) CEP Xx1
163 47,XX,add(21)(p12),+21c
177 46,XY,del(6)(p21)/46,idem,inv(3)(q21q26) 7q-
182 46,XY,add(5)(q35?),del(6)(q21–22?) 46,XY,t(5;6)(q35;q21)
203 46,XX,del(16)(q22)
204 47,XY,t(16;17)(q22;q21),+21c/47,XY,
del(16)(q22),+21c/47,XY,+21c
217 45,X,)Y/45,X,)Y,del(21)(q21) )Y, 21q-Three and more miscellaneous aberrations (n ¼ 10)
128 45,XY,der(1)t(1;7)(p11?;q11?),der(1)t(1;13?)(p11;q11?),
der(11)t(1;11) (p22?;p15),add(3)(q26),add(7)(q11),
add(11)(q11),)13139 46,XX,del(6)(p23),del(9)(q34)/45,XX,del(6)(p23),)9,
der(22)t(9;22)(p11;p11)
ABL x1
165 56,XX,+del(2)(p22)?,+4,+6,+7,+8,+14,+19,+19,+20,
+mar
CEP 8x3
CEP 7x3
B. Stark et al
324 ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337

Cytogenetic changes, incidence, and biologicalcharacteristics
The distribution of the major AML-specific cytogenetic
groups, as identified by cytogenetics and molecular cytogenetic
analysis, and their associated clinical and biological character-
istics is shown in Tables I and II.
11q23 and/or MLL gene split. These were the most frequent
changes, encountered in 19 patients (22%). In nine cases, they
were identified only by MLL splitting on FISH (Table I). The
partner chromosomes could be identified only by SKY in four
of these cases; 9p (three patients) and 10p (one patient) (Stark
et al, 2000), and in one patient, even SKY did not disclose the
translocation with 11q23. In 12 patients, translocation 11q23
was accompanied by additional structural and/or numerical
aberrations, such as extra chromosome 8 in five patients, and
extra 1q in three patients (Table I).
The clinical features of the children with t(11q23) were
unique (Table II), with extramedullary involvement in eight
patients; central nervous system (CNS) and skin involvement
in three patients each; ocular, subcutaneous masses and pleural
effusion in two patients each; and pericardial effusion and
testicular involvement in one patient each. Leucopenia
preceded the leukaemia by 6 months, and was accompanied
by severe fasciitis in one patient and extramedullary involve-
ment and leucopenia of 1 month’s duration in another. Most
of the children had an immunophenotype that was compatible
with monocytic leukaemia: positive for CD33, CD11b, CD11c,
and CD4 antigens, and occasionally CD14.
t(8;21)(q22;q22). This was the second most frequent
aberration, detected in 11 patients (13%) (Table I). It was
accompanied by loss of chromosome Y in four patients,
including one patient with an extra chromosome 8. Two
patients presented with orbital myelosarcoma, one of whom
transformed to overt leukaemia 6 weeks later. In two other
patients, refractory anaemia with excess of blasts (RAEB) or
with transformation (RAEB-t) preceded the overt leukaemia
by 1–2 months. Two additional patients showed bilineage
myelodysplastic morphological changes, with approximately
30% BM blasts at presentation. On immunophenotyping,
blasts were positive for HLA-DR in 92%, CD34 in 92%, CD13
in 75%, and CD33 in 50%. CD15 expression was documented
in five of the eight examined patients, and CD56 in four of the
seven examined patients. CD14 was negative in all patients.
t(15;17)(q22;q11–12) and/or PML/RARA rearrangement. This
aberration was observed in six patients, in four by karyotype
with FISH confirmation, and in two by FISH only (Table I). In
one of the latter, conventional cytogenetics showed add(3p);
cytogenetics was not performed in the other case. Another
(seventh) patient with an extra marker, in whom t(15;17)
could not be detected because FISH was not performed, was
nevertheless added to this group because he had FAB M3
confirmed by electron microscopy and immunophenotyping.
Immunophenotypically, all patients were negative for HLA-DR
antigen, had a low expression of CD34 antigen, and were
positive for CD13 and CD33 (Hrusak et al, 2002).
inv(16)(p13q22), CBFb/MYH11. This aberration was detected
cytogenetically and confirmed by FISH in three patients
(3Æ6%), all displaying break-apart of the CBFb gene probe
located on 16q22. FISH for inv(16) was performed in 20
additional patients [three no mitosis, three normal, three
deleted 9q, two trisomy 8, one t(6;9) and eight miscellaneous],
none of whom was positive. In one of the three positive
patients, additional numerical and structural aberrations of
del(7q), del(9q) and +22 were observed in the sideline and
confirmed by FISH (Table I). All three patients were classified
as FAB M4 with eosinophilia. Immunophenotypically, all were
strongly positive for CD34, CD13, CD33, and CD15, and only
one patient was positive for CD11b and CD4.
Monosomy 7(sole)/del(7q). This group consisted of four
patients with a simple karyotype of monosomy 7 as the sole
aberration and one patient with a complex karyotype, including
Table I. Continued.
Patient number Karyotype FISH* SKY
176 46,XY,del(2)(q31),der(12),)18,+mar 46,XY,
del(2)(q31),i(12)(q11),der(18)t(4;18)(q13;q23)
194 47,X,)X, add(11)(p13),+13,+mar/
48,XX,add(11)(p13),+13,+mar
TEL x3
CEP X x3
47,X,del(X)(p?;or q?),der(6)t(X;6) (?;q21),der(11)
t(X;11)(?;p13),+13/48,idem,+12
196 51,XX,add(2)(q35),)3,add(6)(q25),+15,+19,+22,+3mar
BCR x3
234 49,XY,+4,+6,del(7)(p11;?),+10,add(19)(q11?) 7/7q x2,
4x3, 10x3
CEP 4x3
CEP 10x3
49,XY,+4,+6,der(7)t(7;19)(p11;?),+10,der(19)
t(17;19)(q?;q11?)
1002 47,XY,+2,del(6)(q21),t(15;19)(q15;p13)
1005 46,XX,del(1)(p13)/50,XX,+del(1)(p13),+8,+8,+16 CEP 8x4 46,XX,der(1)t(1;19)(p13;p13)/50,XX,idem
+8,+8,+16
*FISH – regular font indicates the karyotype was confirmed by FISH; bold font indicates new information.
SKY, spectral karyotype ; FISH, fluorescence in situ hybridization; AML, acute myeloid leukaemia.
Cytogenetics, Prognosis in Childhood Acute Myeloid Leukaemia
ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337 325

Table
II.Clinico-biologicalcharacteristicsof97
AMLchildrenincludingthe86
analysed
patientswithcytogenetic
inform
ation.
Cytogenetic
subgroup
Patients
number
(%)
Gender
(M/F)
Origin
(J/non-J)
Age
(years)
median(range)
WBC(109/l)
median(range)
Platelets(109/l)
median(range)
Extra
medl
FAB
PriorMDS
FAB
dysp
02
34
57
Totalcytogenetics
86(100)
48/38
68/18
6Æ5(0Æ0–18Æ5)
16(1–280)
62(3–408)
166
167
1825
1415
10
Norm
al8(9)
4/4
7/1
11Æ6
(6–17Æ6)
24(1–62)
80(24–298)
0–
1–
24
10
1
11q23/M
LL
19(22)
10/9
15/4
2Æ4(0–15)
53(6–280)
84(42–234)
8–
1–
215
12
0
t(8;21)
11(13)
7/4
8/3
8Æ3(2Æ9–16Æ8)
20(6–124)
40(15–120)
3–
10–
1–
–3
4
t(15;17)–
7(8)
4/3
7/0
14Æ9
(6Æ4–17Æ1)
2(2–140)
27(14–80)
0–
–7
––
–0
0
inv(16)
3(3Æ4)
2/1
3/0
12Æ2
(3Æ4–18Æ5)
92(6–115)
24(23–28)
1–
––
3–
–0
0
inv(3)/t(3;21)
2(2Æ3)
1/1
1/1
0Æ6
–(14–59)
–(15–180)
01
––
––
1§1
0
t(3;5)
2(2Æ3)
1/1
1/1
–(4Æ3–9Æ7)
–(8–10)
–(9–27)
0–
1–
––
1�1
1
t(8;16)
2(2Æ3)
0/2
2/0
–(15–15Æ4)
–(3–4)
–(22–26)
0–
––
2–
–0
0
t(6;9)
1(1Æ1)
1/0
1/0
13Æ0
1864
0–
1–
––
–0
0
t(1;22)
1(1Æ1)
0/1
1/0
0Æ3
2568
0–
––
––
10
0
)7/7q-(sole,m
ain)
5(5Æ8)
2/3
5/0
9Æ0(0Æ5–14Æ7)
22(1–30)
23(4–54)
02*
––
11
11
1
9q-(sole)
4(4Æ6)
4/0
4/0
9Æ9(2Æ5–16Æ1)
48(14–84)
120(46–255)
0–
2–
2–
–1
0
+21
(sole)
4(4Æ6)
3/1
2/2
2Æ3(1Æ6–4Æ5)
10(6–18)
14(3–38)
0–
––
–1
3§3
0
+8(sole)
1(1Æ1)
1/0
1/0
12Æ0
284
0–
––
––
10
0
Miscellaneous<3abnorm
alities
7(8)
4/3
4/3
2Æ8(1Æ5–16Æ9)
10(2–180)
61(14–99)
12�
––
21
2�§
22
Miscellaneous‡3
abnorm
alities
9(10)
4/5
6/3
5Æ5(0Æ2–18Æ4)
13(3–73)
104(27–408)
31�
––
23
21
1
Nomitoses
32/1
1/2
7Æ4(1Æ5–15Æ5)
13(8–82)
67(30–444)
1–
––
2–
10
0
Notdone
83/5
3/5
5Æ0(0Æ4–20)
23(5–320)
43(6–380)
12�
2–
21
10
0
TotalAML
9753/44
72/25
6Æ9(0–20)
18(1–320)
61(3–408)
188
187
2226
1615
10
*FABM1,
onepatient.
�FABM6,
onepatient.
�Basophylic
leukaem
iabyelectronmicroscope,fourpatientspreviouslyreported
(Shvidelet
al,2003).
§Downsyndrome,fourpatients(twoofthem
inthemiscellaneousgroup).
–OnepatientwithFABM3andmarkerchromosomewas
added
tothis
group.J/non-J,Jewish/non-Jew
ish;Extra
medl,extram
edullaryinvolvem
ent;FABdysp,FABdysplasia;
AML,acute
myeloid
leukaem
ia;FAB,French-A
merican-British;MDS,
myelodysplastic
syndrome.
B. Stark et al
326 ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337

del(7q) · 2, confirmed by FISH and SKY (Table I). In another
five patients, loss of 7q was considered a secondary change: one
patient (no. 128) with incomplete complex miscellaneous
karyotype with add(7)(q11) in whom FISH failed; one (no. 183)
with the primary aberration inv(16) (15 metaphases) and
del(7q11) identified cytogenetically in six of the 15 metaphases
and by FISH in a subpopulation (32% of cells); and three
patients with the loss of submicroscopic 7q31 detected only by
FISH in a subpopulation (20–45%) of interphase nuclei (50–
100), including one patient with MLL split (no. 149), one with
del(6p) (no. 177), and one with trisomy 21 (no. 172). FISH for
7/7q was normal in the other 37 patients [including 13 with
miscellaneous aberrations, six with 11q23 abnormalities, and
three with del(9q), normal, or no mitosis]. FAB subtypes varied
and refractory anaemia preceded the leukaemic transformation
by 3 years in one patient (Table II).
del(9)(q12;q22). Interstitial deletion of 9q12q22 as the sole
karyotypic abnormality was found in four patients. FISH
analysis was performed in three patients, and the BCR/ABL
probes disclosed the remainder of the two ABL genes located on
9q34, in accordance with the interstitial deletion. FISH with all
AML panel probes showed a normal pattern. Another five
patients had a visible deletion of (9q) in addition to other
primary structural aberrations (Table I): in a subclone of t(8;21),
inv(16), t(10;11); in a complex karyotype with del(7q); and in a
subclone of del(6)(p23). The deletion in the first four patients
seemed to be interstitial, with preservation of the two ABL genes
(by FISH), and was more distal with a loss of ABL gene locus in
the last patient. There was no difference inmedian age and white
blood cell count (WBC) between the solitary del(9q) group and
the whole series. However, three patients in the solitary del(9q)
group had a higher platelet count (over 100 · 109/l). Except for
some lymph node enlargement, no extramedullary involvement
was noted. FAB M2 and M4 leukaemias were found in two
patients each, with strong myeloperoxidase staining, and Auer
rods in all. One patient presented with RAEB, transforming
within 3 weeks to leukaemia with 30% BM blasts. All four
patients were positive for HLA-DR, CD34, and CD33, and three
of four, for CD13 and CD7.
Trisomy 21. Trisomy 21 as the sole acquired aberration was
noted in four patients, three of them with M7 leukaemia, one of
these with Down syndrome, and the other two were identical
twins, described in an earlier report (Stark et al, 2002). FISH
analysis, performed in three patients, confirmed the cytogenetic
findings and ruled out an underlying cryptic MLL split, del(7q)
and del(5q). Extra chromosome 21 as a secondary change was
observed in one patient in a sideline of monosomy 7.
Trisomy 8. One patient had trisomy 8 as the sole abnormality,
confirmed by FISH using the whole AML probe panel.
Secondary extra chromosome 8 was seen in another nine
patients; six had the primary 11q23 aberrations, one t(8;21),
one trisomy 21, one miscellaneous (Table I).
Rare recurrent chromosomal abnormalities. The
t(8;16)(p11;p13) and its variant t(8;19)(p11;q13) (Stark et al,
1995) were detected as the sole aberration in one patient each
with FAB M4-5 morphology with erythrophagocytosis.
Abnormal (3q) was detected in four patients.
t(3;5)(q25;q34) was the sole aberration detected in two
patients; one had erythroleukaemia (M6) and the other had
RAEB, which transformed after a few weeks to leukaemia;
t(3;5)(q25;q34) also appeared in a sideline to the primary
t(1;22) (Table I). In the other two patients, both infants,
t(3;21)(q21;q22) and inv(3)(q21q26) were detected in one
each. The latter infant, with Down syndrome, had M7
leukaemia following a short transient myeloproliferative dis-
order. In addition, inv(3)(q21;q26) was detected in a subclone
of del(6p).
Other rare recurrent abnormalities were t(6;9)(p23;q34) in
one patient with FAB M2 leukaemia, and t(1;22)(p13;q13) in
one infant with M7.
Miscellaneous. Miscellaneous clonal abnormalities were initially
found by cytogenetics in 26 patients. With the application of
FISH, MLL split was detected in seven of the 22 patients tested,
and PML/RARA rearrangements in one of the four patients
tested. One additional patient, with an extra marker
chromosome in whom FISH failed, was transferred to the
t(15;17) group because he had proven FAB M3 morphology.
This left only 16 patients with unclassified miscellaneous
aberrations (Table I): seven patients with one or two structural
or numerical aberrations and nine patients with three or more
structural changes. FISH analysis showed a 7q deletion in one of
the 14 patients examined, no 5 or 5q deletion in 13 patients and
no ABL/BCR rearrangement in 12 patients examined. The
clinical and biological characteristics of the miscellaneous group
are detailed in Table II. Two young children had Down
syndrome and M7 leukaemia. There was a relatively high
proportion of M0 (three), and M7 (five) subtypes. In three
patients (two with M0, one M7) basophilic leukaemia was
detected by electron microscopy, two of them (nos 203, 204)
have been previously reported (Shvidel et al, 2003).
There were four children with Down syndrome (Table I, nos
170, 1006, 163, 204) with chromosome abnormalities:
inv(3)(q21;q26), acquired +21, add(21)(p12), t(16;17)(q22;
q21) respectively. Their ages ranged between 0Æ6 and 1Æ6 years,
all with M7 leukaemia.
Outcome by cytogenetic subgroups and clinicobiologicalparameters
Overall, outcome analysis of the 94 patients with newly
diagnosed AML treated with BFM-based protocols yielded, at a
median follow-up of 69 months (range 4–177), a remission
rate of 93%, with an estimated probability of 4-year EFS of
55Æ7 ± 5% and OS of 59Æ9 ± 5% (Table III). Among the 83
patients who were evaluated cytogenetically, EFS was
52Æ1 ± 6% and OS 57Æ1 ± 6%.
Cytogenetics, Prognosis in Childhood Acute Myeloid Leukaemia
ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337 327

Treatment protocols, type of BMT, and outcome by
cytogenetic subgroup are demonstrated in Table III and
Fig 1. The 4-year EFS in patients with a normal karyotype
and in patients with t(8;21), most of whom were treated with
the earlier protocol, was 62% and 60% respectively. Of the
seven patients with t(15;17), one died early in induction from
disseminated intravascular coagulopathy and one relapsed
4 years after diagnosis, leading to a drop in EFS from 85Æ7% to
57Æ1 ± 25%; however, since the latter patient was salvaged, OS
at 4 years was 85Æ7 ± 13%. All three patients with inv(16) are
alive in first remission. Of the 19 patients with 11q23
abnormalities, all but one who lived longer than 6 months
underwent allogeneic transplantation (four patients) or autol-
ogous transplantation (nine patients), according to HLA-
matched sibling availability. The 4-year EFS in this subgroup
was 33Æ8 ± 11% and OS 44Æ6 ± 11%. Similar results were
noted for the subgroup of six patients with t(9;11) (EFS
22 ± 19%) and the other t(11q;23) (EFS 38 ± 13%)
(P ¼ 0Æ24). Of the five patients with monosomy 7, two did
not achieve remission, and two relapsed within 1 year. Only
one, transplanted early, is alive in remission after 9 months. All
three patients with solitary del(9q) relapsed within 18 months
and died. Patients with miscellaneous abnormalities had an
EFS of 60Æ2 ± 12%. No difference was found between those
with more or less than three abnormalities (EFS 66Æ6 ± 15%
vs. 57Æ1 ± 18% respectively, P ¼ 0Æ9). The appearance of
del(9q), del(7q), and trisomy 8 as additional secondary
changes did not add any prognostic significance to the primary
aberrations.
A complex karyotype of three or more unrelated cytogenetic
abnormalities, excluding the t(8;21), inv(16), t(15;17), and
normal karyotype groups, had a trend-level effect on 4-year
EFS: 51 ± 8% for the 37 patients with a simple karyotype vs.
29 ± 12% for the 17 patients with a complex one (P ¼ 0Æ08).Within the complex karyotype group, the 1-year EFS for the 12
patients with three to four abnormalities was 33 ± 15%, and
for the five patients with five or more abnormalities, it was
20 ± 18% (P ¼ 0Æ43). A complex karyotype with three or
more abnormalities had a poor prognostic impact within the
t(11q23) group, with a 4-year EFS of 0 vs. 50Æ3 ± 14% for
those with simple t(11q23) (P ¼ 0Æ019).This retrospective series covered two main treatment
periods: the early one, in which treatment was based on the
AML-BFM-87 protocol (Creutzig et al, 1999), and the later
one, when treatment was based on the BFM-93, -98
protocols (Creutzig et al, 2001). The protocols differed
mainly in the postinduction timing of HD Ara-C: in the
BFM-87, it followed the 6 weeks of consolidation, whereas
in the BFM-93/-98, it was administered earlier, immediately
after induction. In addition, in the second period, autolo-
gous and allogeneic BMT were performed in a higher
percentage of patients: 34% and 27% vs. 25% and 6% in the
Table III. Treatment and outcome of 97 acute myeloid leukaemia (AML) children by cytogenetic subgroups.
Cytogenetic subgroup
Number
of patients
Treatment Outcome
BFM
BMT
(auto/allo)87 93/98 Other* No CR Relp
4-year EFS�(% ± SE)
4-year OS�(% ± SE)
Total cytogenetics 86 44 39 3 23/13 5 31 52Æ1 ± 6 57Æ1 ± 6
Normal 8 7 1 0 3/0 0 3 62Æ5 ± 17 62Æ5 ± 17
11q23/MLL 19 9 10 0 9/4 1 11 33Æ8 ± 11 44Æ6 ± 11
t(8;21) 11 9 2 0 1/2 0 4§ 60Æ0 ± 15 60Æ0 ± 15
t(15;17) 7 2 5 0 0/0 1� 1 57Æ1 ± 25 85Æ7 ± 13
inv(16) 3 0 3 0 2/0 0 0 100 100
t(8;16) 2 1 1 0 2/0 0 0
)7/7q-(sole,main) 5 3 2 0 0/1 3 2 0 0
9q- (sole) 4 2 1 1 0/0 0 3 0 0
+21 (sole) 4 1 2 1 1/2 0 0 100 100
Miscellaneous <3 abnormalities 7 2 5 0 2/0 0 3 57Æ1 ± 18 57Æ1 ± 18
Miscellaneous ‡3 abnormalities 9 3 6 0 1/4 2 1 66Æ6 ± 15 36Æ4 ± 27
No mitoses 3 2 1 0 1/0 0 1 66Æ6 ± 27 66Æ6 ± 27
Not done 8 6 2 0 3/1 0 1 87Æ5 ± 11 87Æ5 ± 11
Total AML 97 52 42 3 27/14 5 33 55Æ7 ± 5 59Æ9 ± 5
*Another protocol was used for three patients, who were treated elsewhere and lost to follow-up. They were not included in the assessment of EFS and
OS.
�EFS (event-free survival) and OS (overall survival) were calculated for the 94 newly diagnosed patients treated with the modified AML-BFM-87, -
93/-98 protocols.
�Early death within 9 d.
§One patient died in first remission from chronic GVHD.
B. Stark et al
328 ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337

first period respectively (P ¼ 0Æ005). There was no signifi-
cant difference in the distribution of the patient character-
istics (gender, origin, WBC, platelet count and cytogenetic
risk groups) between the two periods, except for a
marginal difference in FAB subtypes with some higher
incidence of M5, M7 of 33% and 19%, respectively in the
second period, compared with 23% and 11% in the first
period (P ¼ 0Æ08).With respect to outcome, remission was achieved in 95% of
the patients treated by the later protocols compared with 92%
of those treated by the early one (P ¼ 0Æ69); death in remission
was 0% vs. 2Æ3% (P ¼ 0Æ4), cumulative incidence of relapse
was 31Æ6 ± 8% vs. 44Æ7 ± 7% (P ¼ 0Æ35). The 4-year EFS was
65Æ1 ± 8% in the second period, with a median follow-up of
49 months (range 4–74), and 49Æ8 ± 7% in the first period,
with a median follow-up of 125 months (range 26–177), with
no significant difference (P ¼ 0Æ24) at the time of analysis, and
the same for OS, 66Æ6 ± 8% vs. 55Æ8 ± 7% respectively
(P ¼ 0Æ35).Because of the relatively small number of children and the
non-significant differences in their presenting features and
outcome, the results of both treatment periods were combined
for univariate analysis of prognosis (Table IV). Male gender,
FAB dysplasia, WBC 50 · 109/l to 100 · 109/l, and platelet
count over 50 · 109/l were associated with lower EFS. FAB M5
leukaemia was associated with a trend of worse 4-year EFS
compared with the rest of the group (42% vs. 61%, P ¼ 0Æ12).The small subgroup of FAB M7 had a better, but statistically
insignificant, 4-year EFS (79% vs. 51%, P ¼ 0Æ12). Three (nos170, 163, 204) of the four patients with Down syndrome were
treated with modified BFM protocols, one of them underwent
an autotransplant. In one patient (no. 204) the disease
recurred and the child died.
Multivariate analysis was performed with the variables
found to be significant on univariate analysis: cytogenetic risk
group (unfavourable versus others), gender, platelet count
(>50 · 109/l), FAB dysplasia, and WBC (<50, 50–100, >100).
Of these, unfavourable cytogenetic risk group [P < 0Æ001,
relative risk (RR) 4Æ08, 95% confidence interval (CI): 1Æ92–8Æ68]; FAB dysplasia (P ¼ 0Æ007, RR 4Æ00, 95% CI: 1Æ62–9Æ88);and male gender (P ¼ 0Æ029, RR 2Æ12, 95% CI: 1Æ06–4Æ26),remained significant adverse prognostic factors.
Outcome by cytogenic risk groups
On the basis of outcome in terms of EFS probability, and the
previously published cytogenetic risk classification in adults
Fig 1. Overall survival by cytogenetic abnormalities.
Table IV. Prognostic factors of the 94 newly diagnosed children
treated with the modified acute myeloid leukaemia Berlin-Frankfurt-
Munster (AML-BFM) protocols.
Variable
Patients/events
(n)
4-year EFS
(% ± SE)
P-value
(log-rank)
Total patients 94/39 55Æ7 ± 5 –
Gender
Female 44/12 70Æ8 ± 7 0Æ006Male 50/27 42Æ3 ± 7
Origin
Non-Jew 24/7 70Æ8 ± 9 0Æ15Jew 70/32 50Æ0 ± 6
Age (years)
<2 24/10 58Æ0 ± 10 0Æ58<10 36/17 49Æ8 ± 8
<15 22/9 55Æ1 ± 11
>15 12/3 67Æ3 ± 16
WBC (·109/l)<50 71/27 59Æ5 ± 6 0Æ02<100 12/8 27Æ8 ± 13
>100 11/4 60Æ6 ± 15
Platelets (·109/l)<50 44/12 72Æ3 ± 7 0Æ011‡50 48/26 41Æ2 ± 7
FAB type
0, 1 8/3 58Æ3 ± 18 0Æ482 18/8 55Æ6 ± 12
3 7/2 57Æ1 ± 25
4 21/9 53Æ2 ± 11
5 26/14 42Æ0 ± 11
6, 7 14/3 78Æ6 ± 11
FAB-dysplasia
Negative 84/32 59Æ4 ± 6 0Æ010Positive 10/7 22Æ5 ± 14
Prior MDS
Negative 80/29 61Æ3 ± 6 0Æ007Positive 14/10 23Æ8 ± 12
Cytog. risk groups* (83 patients)
Favourable 21/6 66Æ8 ± 11 0Æ005Intermediate 21/6 71Æ1 ± 10
Unfavourable 41/25 33Æ6 ± 8
*Cytogenetic risk groups: favourable: t(8;21), inv(16), t(15;17), FAB
M3.
Intermediate: normal, t(8;16), t(1;22), +21(sole), miscellaneous <3
abnormalities.
Unfavourable: t(11q23), del(9q)(sole), abn(3q), t(6;9), )7/7q), mis-
cellaneous ‡3 abnormalities.
Cytogenetics, Prognosis in Childhood Acute Myeloid Leukaemia
ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337 329
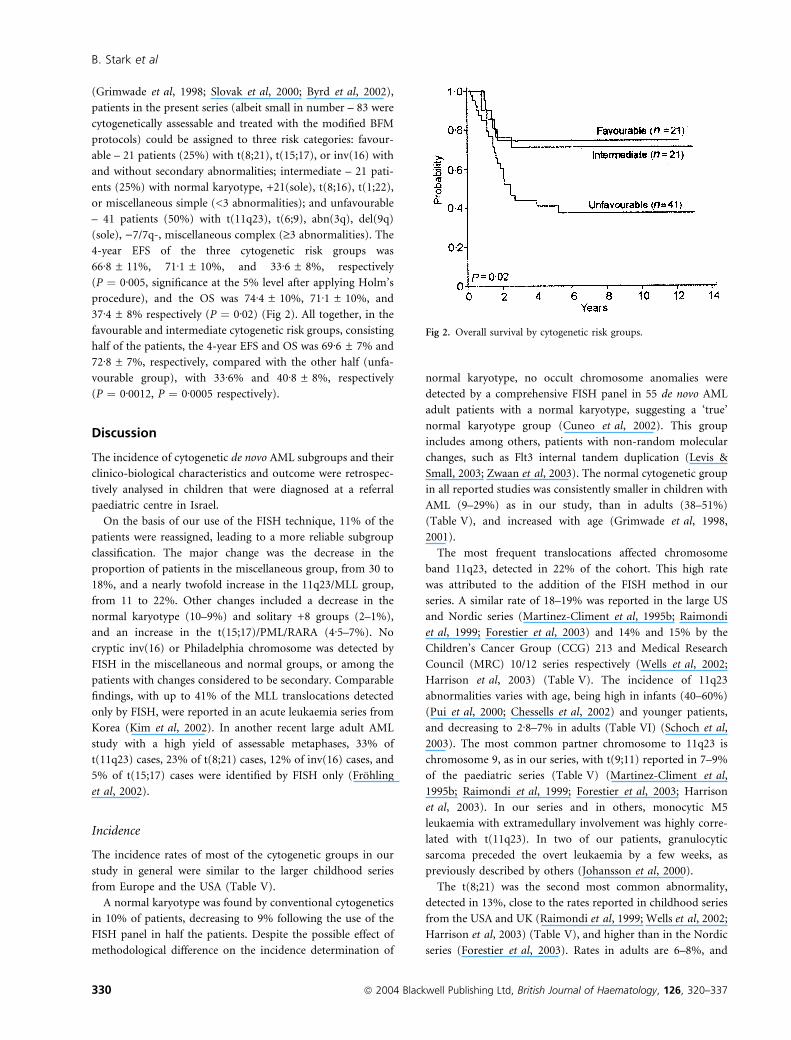
(Grimwade et al, 1998; Slovak et al, 2000; Byrd et al, 2002),
patients in the present series (albeit small in number – 83 were
cytogenetically assessable and treated with the modified BFM
protocols) could be assigned to three risk categories: favour-
able – 21 patients (25%) with t(8;21), t(15;17), or inv(16) with
and without secondary abnormalities; intermediate – 21 pati-
ents (25%) with normal karyotype, +21(sole), t(8;16), t(1;22),
or miscellaneous simple (<3 abnormalities); and unfavourable
– 41 patients (50%) with t(11q23), t(6;9), abn(3q), del(9q)
(sole), )7/7q-, miscellaneous complex (‡3 abnormalities). The
4-year EFS of the three cytogenetic risk groups was
66Æ8 ± 11%, 71Æ1 ± 10%, and 33Æ6 ± 8%, respectively
(P ¼ 0Æ005, significance at the 5% level after applying Holm’s
procedure), and the OS was 74Æ4 ± 10%, 71Æ1 ± 10%, and
37Æ4 ± 8% respectively (P ¼ 0Æ02) (Fig 2). All together, in the
favourable and intermediate cytogenetic risk groups, consisting
half of the patients, the 4-year EFS and OS was 69Æ6 ± 7% and
72Æ8 ± 7%, respectively, compared with the other half (unfa-
vourable group), with 33Æ6% and 40Æ8 ± 8%, respectively
(P ¼ 0Æ0012, P ¼ 0Æ0005 respectively).
Discussion
The incidence of cytogenetic de novo AML subgroups and their
clinico-biological characteristics and outcome were retrospec-
tively analysed in children that were diagnosed at a referral
paediatric centre in Israel.
On the basis of our use of the FISH technique, 11% of the
patients were reassigned, leading to a more reliable subgroup
classification. The major change was the decrease in the
proportion of patients in the miscellaneous group, from 30 to
18%, and a nearly twofold increase in the 11q23/MLL group,
from 11 to 22%. Other changes included a decrease in the
normal karyotype (10–9%) and solitary +8 groups (2–1%),
and an increase in the t(15;17)/PML/RARA (4Æ5–7%). No
cryptic inv(16) or Philadelphia chromosome was detected by
FISH in the miscellaneous and normal groups, or among the
patients with changes considered to be secondary. Comparable
findings, with up to 41% of the MLL translocations detected
only by FISH, were reported in an acute leukaemia series from
Korea (Kim et al, 2002). In another recent large adult AML
study with a high yield of assessable metaphases, 33% of
t(11q23) cases, 23% of t(8;21) cases, 12% of inv(16) cases, and
5% of t(15;17) cases were identified by FISH only (Frohling
et al, 2002).
Incidence
The incidence rates of most of the cytogenetic groups in our
study in general were similar to the larger childhood series
from Europe and the USA (Table V).
A normal karyotype was found by conventional cytogenetics
in 10% of patients, decreasing to 9% following the use of the
FISH panel in half the patients. Despite the possible effect of
methodological difference on the incidence determination of
normal karyotype, no occult chromosome anomalies were
detected by a comprehensive FISH panel in 55 de novo AML
adult patients with a normal karyotype, suggesting a ‘true’
normal karyotype group (Cuneo et al, 2002). This group
includes among others, patients with non-random molecular
changes, such as Flt3 internal tandem duplication (Levis &
Small, 2003; Zwaan et al, 2003). The normal cytogenetic group
in all reported studies was consistently smaller in children with
AML (9–29%) as in our study, than in adults (38–51%)
(Table V), and increased with age (Grimwade et al, 1998,
2001).
The most frequent translocations affected chromosome
band 11q23, detected in 22% of the cohort. This high rate
was attributed to the addition of the FISH method in our
series. A similar rate of 18–19% was reported in the large US
and Nordic series (Martinez-Climent et al, 1995b; Raimondi
et al, 1999; Forestier et al, 2003) and 14% and 15% by the
Children’s Cancer Group (CCG) 213 and Medical Research
Council (MRC) 10/12 series respectively (Wells et al, 2002;
Harrison et al, 2003) (Table V). The incidence of 11q23
abnormalities varies with age, being high in infants (40–60%)
(Pui et al, 2000; Chessells et al, 2002) and younger patients,
and decreasing to 2Æ8–7% in adults (Table VI) (Schoch et al,
2003). The most common partner chromosome to 11q23 is
chromosome 9, as in our series, with t(9;11) reported in 7–9%
of the paediatric series (Table V) (Martinez-Climent et al,
1995b; Raimondi et al, 1999; Forestier et al, 2003; Harrison
et al, 2003). In our series and in others, monocytic M5
leukaemia with extramedullary involvement was highly corre-
lated with t(11q23). In two of our patients, granulocytic
sarcoma preceded the overt leukaemia by a few weeks, as
previously described by others (Johansson et al, 2000).
The t(8;21) was the second most common abnormality,
detected in 13%, close to the rates reported in childhood series
from the USA and UK (Raimondi et al, 1999; Wells et al, 2002;
Harrison et al, 2003) (Table V), and higher than in the Nordic
series (Forestier et al, 2003). Rates in adults are 6–8%, and
Fig 2. Overall survival by cytogenetic risk groups.
B. Stark et al
330 ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337

Table
V.Frequency
ofcytogenetic
abnorm
alitiescompared
withrecentlargechildhoodandadult
de
nov
oAMLseries.
Children
Adults
Present
study
No.(%
)
Harrison
etal
(2003)
MRC10/12
No.(%
)
Forestier
etal
(2003)
NOPHO
93
No.(%
)
Wells
etal
(2002)
CCG
213
No.(%
)
Raimondi
etal
(1999)
POG
8821
No.(%
)
Martinez-C
liment
etal
(1995a)
No.(%
)
Grimwade
etal
(1998,
2001)
MRC10/11
No.(%
)
Mrozek
etal
(2001),
Byrd
etal
(2002)
CALGB8461
No.(%
)
Slovak
etal
(2000)
SWOG/ECOG
No.(%
)
Totalnumber
ofpatients
97255
498
610
120
1795
808
Age
(years)
<20
<15
<18
<21
<21
<20
15–55
15–86
16–55
Totalanalysed
86(88)
808
239(93)
269(54)
478(78)
115(96)
1244
1213
(77)
609(80)
Norm
al8(9)
179(22)
67(28)
77(29)
109(23)
17(15)
560(45)
582(48)
t(8;21)
11(13)
94(12)
18(7)
35(13)
56(12)
9(8)
–(7)
81(7)
50(8)
t(15;17)
7(8)
67(8)
8(3)
NI
55(11)
12(10)
–(13)
88(7)
27(4)
inv(16)/t(16;16)
3(3)
46(6)
13(5)
17(5)
28(6)
9(8)
–(4)
96(8)
53(9)
t(11q23)*
19(22)
119(15)
44(18)
38(14)
88(18)
21(19)
–(3)
54(5)
42(7)
t(9;11)
6(7)
51(6)
21(9)
35(7)
10(9)
–(<1)
27(3)
t(8;16)
2(2)
–(1)
3(1)
–(<1)
)7/del(7q)
5(6)
59(7)
8(3)
11(4)
9(2)
6(5)
–(6)
95(8)
del(9q)(sole)
4(4)
14(4)
1–(1)
11(1)
17(3)
+21(sole)
4(4)
1(0)
7(1)
5(<1)
Miscellaneous
16(18)
89(19)
NI,notincluded;AML,acute
myeloid
leukaem
ia.
*t(11q23)withallpartner
chromosomes
includingchromosome9.
Cytogenetics, Prognosis in Childhood Acute Myeloid Leukaemia
ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337 331

drop to 1–2% at older ages (Slovak et al, 2000; Grimwade et al,
2001; Schoch et al, 2001a). In adults, racial differences were
noted, with an increased incidence of t(8;21) in African-
Americans compared with Caucasians (Sekeres et al, 2002) and
in Asian-Japanese compared with Australians (Nakase et al,
2000). The t(8;21) associated with FAB M2 morphology, in
rare cases, may occur with MDS (Kojima et al, 1998; Felice
et al, 2000), or MDS may precede the leukaemia (Taj et al,
1995) or leukaemia may be associated with dysplastic changes
(Haferlach et al, 1996; Nakamura et al, 1997). This was noted
in a few of our patients as well. Granulocytic sarcoma in the
orbit, the epidural and malar regions, as documented in two of
our patients, were described in higher proportions in children
from Argentina (Felice et al, 2000) and black children from
South Africa (Schwyzer et al, 1998).
The frequency of t(15;17) in our series was in the range of
the childhood series from the USA and UK, namely, 8–11%
(Table V). A lower (3%) frequency was reported in the Nordic
series (Forestier et al, 2003), and a higher one (17%) in the
paediatric Italian series (Biondi et al, 1994). Geographic
variations and a higher prevalence was also observed in adult
Latino and African-American series (Douer et al, 1996; Sekeres
et al, 2002). The absence of non-Jewish representation in this
subgroup is of note (Table II) and may suggest an ethnic
variation.
The chromosome 16 aberration inv(16)(p13;q22) occurred
in only 3% of our cohort. Testing by FISH in another 19
patients with normal or miscellaneous aberrations was negat-
ive. In the USA, UK and Nordic childhood series, rates ranged
from 5 to 8% (Table V). This difference warrants a future
study of a larger cohort for possible geographic heterogeneity,
because of the ethnic genetic make-up of specific gene
polymorphisms. For example, the C609T polymorphism in
the NQO1 (quinone oxyreductase 1) gene, which lowers the
activity of the enzyme, thereby leading to a decrease in quinone
detoxification, was found to be associated with an increased
risk of inv(16) in adult AML (Smith et al, 2001).
The recurrent balanced translocations t(8;16), t(6;9), t(3;5)
and t(1;22) occurred rarely, together accounting for 6% of the
cases, in line with the findings of Raimondi et al (1999). As in
other childhood series, we did not detect the rare Philadelphia
chromosome, even by FISH (performed on 41 patients).
The incidence of the numerical changes of monosomy 7 and
7q- and extra chromosome 21 as sole aberrations was also
within the range of previous childhood studies (Table V).
Trisomy 8 as the sole aberration was detected in only one
patient. A similar rate was noted in the series of Raimondi et al
(1999), lower than in other paediatric series (Forestier et al,
2003) and adult series (Byrd et al, 1998; Wolman et al, 2002).
In another of our patients, an apparent solitary +8 was later
determined to be a MLL split, by FISH.
In the present series, solitary int del(9q) was observed in 4%
of patients. This group was mentioned briefly in only one
paediatric series (Harrison et al, 2003), and the reported rate
in adult series is <1% (Grimwade et al, 1998, 2002; Byrd et al,
2002). Like others (Ferrara et al, 1996; Wan et al, 2003), we
noted an association of del(9q) with FAB M2 and M4, or
preceding MDS (RAEB-t), presence of Auer rods and CD7 and
CD34 expression.
The rare miscellaneous abnormalities accounted for 18% of
our cases, as reported also by Raimondi et al (1999) and the
Nordic group (Forestier et al, 2003). This group included
some aberrations described anecdotally by Raimondi et al
(1999), such as del(16)(q23) and t(16;17)(q24;q11), in addi-
tion to t(X;6) (Dastugue et al, 1997), t(1;19) (Tchinda et al,
2002), or -Y(sole), which accounted for 1Æ5% of the adult series
of the Cancer and Leukemia Group B (CALGB) (Byrd et al,
2002).
Treatment, prognosis and cytogenetic risk groups
The children in the present series were treated with the AML-
BFM-87, -93/-98 protocols, with extended TIT injections
replacing cranial irradiation. Overall, CR was achieved in 95%;
the 4-year EFS and OS were 55Æ7 ± 5%, and 59Æ9 ± 5%
respectively. These results are in line with the improved EFS
reported by multicentre paediatric clinical studies (Arceci,
2002): BFM-93, 51% at 5 years (Creutzig et al, 2001); MRC 10
and 12, 48% and 52% at 5 years (Webb et al, 2001; Gibson
et al, 2002); Nordic Society of Pediatric Oncology and
Hematology (NOPHO) 93, 49% at 7 years (Lie et al, 2003);
and CCG 2891 intensive timing, 49% survival at 8 years
(Woods et al, 2001).
The cytogenetic subgroups had different impacts on prog-
nosis. Patients with t(8;21), treated with the modified BFM-87
protocol had a relatively favourable prognosis, with a 4-year
EFS and OS of 60% each, close to the rates achieved in other
series based on the AML-BFM-87 (Felice et al, 2000), the
NOPHO 93 (EFS 56%) and the Pediatric Oncology Group
(POG) 8821 (EFS 45%) (Raimondi et al, 1999). An OS of 81%
was recently reported in the MRC 10/12 (Harrison et al, 2003).
All these protocols included HD Ara-C. The benefit of
repetitive HD Ara-C in adult t(8;21) AML was previously
proven by the CALGB group (with an OS of 76% compared
with 44% in patients receiving lower doses) (Bloomfield et al,
1998; Byrd et al, 1999, 2002), and confirmed by the UK series
(OS 69%) (Grimwade et al, 1998). Some studies claimed that
within the t(8;21) group, additional secondary del(9q) or loss
of sex chromosome may have an adverse impact on outcome
(Haferlach et al, 1996; Schoch et al, 1996; Slovak et al, 2000),
although this has not been supported by others (Grimwade
et al, 1998; Raimondi et al, 1999; Byrd et al, 2002; Harrison
et al, 2003) or our series. In the present series, relapse occurred
in three of four patients with myelodysplastic changes, whereas
dysplastic features had no prognostic significance in the adult
t(8;21) series of Haferlach et al (1996).
Patients with the other core-binding factor type leukaemia
with inv(16), are considered a favourable prognostic group. In
our series too, this group had a very good outcome, and all
three patients are alive in first CR. Small paediatric series with
B. Stark et al
332 ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337

inv(16) AML and a larger adult series demonstrated an EFS of
58–76% (Raimondi et al, 1999; Razzouk et al, 2001; Wells
et al, 2002; Lie et al, 2003) and an OS of 57–81% respectively
(Grimwade et al, 1998; Byrd et al, 2002; Delaunay et al, 2003;
Harrison et al, 2003). All protocols but one (Delaunay et al,
2003) used HD Ara-C.
Our third favourable group was t(15;17), treated with all-
trans-retinoic acid (ATRA) combined with BFM-based che-
motherapy; EFS was 57% and OS was 83%. Similar results
were achieved with the NOPHO 93 (Lie et al, 2003) and the
MRC 10/12 (Harrison et al, 2003), and adult protocols based
on concurrent ATRA and chemotherapy in induction and
maintenance (Tallman et al, 2002).
The normal karyotype group is considered to be of
intermediate risk by all studies, with an EFS of 37–43% in
paediatric series (Raimondi et al, 1999; Wells et al, 2002;
Harrison et al, 2003; Lie et al, 2003) and an OS of 24–45% in
adult series (Grimwade et al, 1998, 2002; Byrd et al, 2002). In
our normal group, EFS and OS were both 62%.
The outcome of the largest group of t(11q23)/MLL split was
poor, with a 4-year EFS of 34% and OS of 45%. There was no
difference between the patients with t(9;11) and the rest of the
t(11q23) group. While the CCG 213 (Wells et al, 2002), and
POG 8821 series (Raimondi et al, 1999) noted similar results, a
few other paediatric centres showed a better outcome for the
t(9;11) group, with an EFS of 56–85% (Martinez-Climent et al,
1995b; Rubnitz et al, 2002; Lie et al, 2003). In the adult
CALGB 8461 series, t(9;11) also carried a better prognosis [OS
41% intermediate risk vs. 0% high risk for the rest of t(11q23)
group] (Mrozek et al, 2001; Byrd et al, 2002). In the adult
MRC 10/12 trial, OS was 42% for the whole abn(11q23) group,
with no significant difference from t(9;11) (5-year OS, 56%)
and the other 11q23 partners (Grimwade et al, 2002; Harrison
et al, 2003). These authors classified the whole group of
abn(11q23) as intermediate risk. However, the Southwest
Oncology Group (SWOG) study classified abn(11q23) to be an
unfavourable risk (Slovak et al, 2000). In one in vitro study
using the cell kill methyl-thiazoltetrazolium (MTT) assay,
t(9;11) blasts showed higher sensitivity to Ara-C, etoposide,
anthracyclines and 2-chlorodeoxyadenosine than the other
11q23 translocations and the other AMLs (Zwaan et al, 2002).
In the present study, within the t(11q23) group, the presence
of a complex karyotype with three or more secondary
aberrations conferred a fatal outcome, as opposed to 50%
survival for those with fewer or no additional chromosomal
changes. However, in the CALGB 8461 study, neither complex
karyotype nor secondary aberrations affected the outcome of
patients with t(9;11) (Byrd et al, 2002).
The small group with monosomy 7 fared extremely poorly.
Only one of four patients is currently in remission following
early BMT, which is in line with the unfavourable classification
in other paediatric and adult series (Grimwade et al, 1998,
2002; Byrd et al, 2002; Wells et al, 2002).
The small del(9q) (sole) group was categorized in the adult
MRC 10/12 series and in the CALGB trial as intermediate risk,
with an OS of 36–55% (Grimwade et al, 1998, 2002; Byrd et al,
2002), and by the SWOG study (Slovak et al, 2000) as
unfavourable risk. In one recent paediatric series, survival was
not different from the rest of the group (Harrison et al, 2003).
In the present series, all three patients relapsed within
5–20 months; none underwent transplantation.
Trisomy 21 was considered an intermediate risk group in the
adult CALGB and MRC series. However, a high remission rate
was achieved in the paediatric POG series (Raimondi et al,
1999). In the present series, all three children with trisomy 21
are in long-term remission following the BFM protocol and
BMT (Stark et al, 2002).
Abn(3q), especially inv(3)(q21;q26) or t(3;3), is categorized
in all adult series as an extremely poor prognostic group
(Grimwade et al, 1998, 2002; Byrd et al, 2002). Patients with
t(3;5)(q25;q34) in the POG series fared poorly (Raimondi
et al, 1999), but in a recent adult series, OS was 45%
(Grimwade et al, 2002). In the present study, two of four
children with abn(3q) are in remission.
The child with t(6;9) in this series relapsed. Accordingly, all
earlier paediatric and adult series classified t(6;9) as a worst
prognostic group (Raimondi et al, 1999; Byrd et al, 2002;
Grimwade et al, 2002).
The recurrent t(8;16) remains unclassified in the large series.
However, in the few affected patients in the POG series
(Raimondi et al, 1999), and on review of other reports on a
limited number of patients (Stark et al, 1995), outcome
seemed poor. By contrast, in the present series, both patients
were cured.
The infant with t(1;22) in our sample was cured with the
BFM-98 protocol and autologous BMT. In an earlier review
(Bernstein et al, 2000), and in the POG series (Raimondi et al,
1999), the t(1;22) group had an adverse outcome, whereas in a
recent French report of childhood megakaryocytic leukaemia,
t(1;22) carried a 3-year EFS of 50% (Dastugue et al, 2002).
The del(16)(q22) was detected in one patient who relapsed.
In some classifications, del(16)(q22) was combined with the
favourable inv(16), whereas in others, it was excluded from the
inv(16) group because it often differed in both characteristics
and outcome, with an OS of only 10% (Marlton et al, 1995;
Byrd et al, 2002).
Complex karyotype, in all but the favourable risk group, was
considered in all studies as a predictor of adverse outcome.
Complex karyotype was defined by the MRC as ‡5 unrelated
abnormalities (Grimwade et al, 1998), and by the CALGB,
SWOG and German groups as ‡3 abnormalities (Slovak et al,
2000; Schoch et al, 2001b; Byrd et al, 2002). In our series, EFS
was 52% for <3 abnormalities, 33% for ‡3 abnormalities, and
20% for ‡5 abnormalities.
On the basis of outcome in the present series and the
published criteria (Bloomfield et al, 1997; Grimwade et al,
1998; Slovak et al, 2000; Byrd et al, 2002), we identified three
cytogenetic risk groups. The well-defined favourable risk group
consisted of t(8;21), t(15,17), and inv(16), even when accom-
panied by secondary changes or complex karyotype (Byrd et al,
Cytogenetics, Prognosis in Childhood Acute Myeloid Leukaemia
ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337 333

2002), excluding del(16q). This group accounted for about
25% of our sample. Four-year EFS and OS were 67% and 74%,
respectively, comparable with the OS of 65% in the adult MRC
10 series (Grimwade et al, 1998) and the 55% in the CALGB
study (Byrd et al, 2002).
The definition of the intermediate risk group is more
variable. All series included a normal karyotype. The CALGB
and MRC studies also included trisomy 21(sole) and
del(9q)(sole). Like in the SWOG study, we moved the
del(9q) from the intermediate to the unfavourable risk group,
and added t(8;16), t(1;22) (previously unclassified) and
miscellaneous with <3 abnormal aberrations. The MRC also
included the miscellaneous abnormalities in the intermediate
group. Our intermediate risk group accounted for 25% of the
cohort. Outcome was similar to the favourable risk group,
with both the 4-year EFS and OS being 71%. In a recent
paediatric MRC 12 series, survival from the time of CR was
79% (Gibson et al, 2002). In adults, the intermediate risk
group is much larger (46–61%) because of the larger normal-
karyotype subgroup and the inclusion of either t(9;11) or all
abn(11q23) by the CALGB and MRC series, respectively,
which report a 5-year OS of 24–41% (Grimwade et al, 1998;
Byrd et al, 2002).
Our unfavourable risk group comprised t(11q23), as in the
SWOG study (Slovak et al, 2000), in addition to t(6;9),
abn(3q), )7/7q-, del(9q)(sole) and miscellaneous complex
karyotype (‡3 unrelated abnormalities) – similar, for the most
part, to the adult series (Slovak et al, 2000; Grimwade et al,
2001; Byrd et al, 2002). It accounted for 50% of the sample,
and EFS and OS were 33% and 37% respectively. The poor
prognosis was confirmed also by multivariate analysis. In
adults, the unfavourable risk group consists of 10–30% of
patients, with a very poor OS of 5–14% (Slovak et al, 2000;
Grimwade et al, 2001; Byrd et al, 2002).
All together, two main cytogenetic risk categories of similar
size could be expected; one consisting of patients in the
favourable and intermediate groups, in whom EFS was 70%,
compared with the remaining patients with an unfavourable
prognosis and EFS of 33% (P ¼ 0Æ0012).In conclusion, the cytogenetic analysis of childhood AML
using FISH yielded a higher number of patients with
cytogenetic information and a more accurate risk classification,
with optimal clinical relevance. Although modern therapeutic
protocols have led to a better outcome in AML, cytogenetic
aberrations still show a significant impact on prognosis, as
demonstrated also in our relatively small series. Within the
favourable/intermediate risk groups, additional cytogenetic or
molecular genetic markers may improve prediction of relapse
and treatment stratification. In the higher risk group, earlier
BMT and new modalities directed at overcoming drug
resistance are needed.
The recently acquired knowledge on gene expression profiles
in relation to cytogenetic subsets will further refine risk
assessment and optimize targeted therapy.
Acknowledgments
The authors wish to thank Dina Kugel for her expert data
management; Meira Zoldan for the morphological and cyto-
chemistry staining; and Yona Kodman for the immunophen-
otypic assessment. Authors also thank Ilana Gelernter and Hila
Shemer of the Statistical Laboratory of Tel Aviv University for
their excellent assistance with the statistical analysis (I.G.) and
database management (H.S.), and Gloria Ginzach and Marian
Propp for their editorial and secretarial assistance.
References
Arceci, R.J. (2002) Progress and controversies in the treatment of
pediatric acute myelogenous leukemia. Current Opinion in Hema-
tology, 9, 353–360.
Bennett, J.M., Catovsky, D., Daniel, M.T., Flandrin, G., Galton, D.A.G.,
Gralnick, H.R. & Sultan, C. (1985a) Proposed revised criteria for the
classification of acute myeloid leukemia. A report of the French-
American-British Cooperative Group. Annals of Internal Medicine,
103, 620–629.
Bennett, J.M., Catovsky, D., Daniel, M.T., Flandrin, G., Galton,
D.A.G., Gralnick, H.R. & Sultan, C. (1985b) Criteria for the diag-
nosis of acute leukemia of megakaryocyte lineage (M7). A report of
the French-American-British Cooperative Group. Annals of Internal
Medicine, 103, 460–462.
Bernstein, J., Dastugue, N., Haas, O.A., Harbott, J., Heerema,
N.A., Huret, J.L., Landman-Parker, J., LeBeau, M.M., Leonard, C.,
Mann, G., Pages, M.P., Perot, C., Pirc-Danoewinata, H., Roitzheim,
B., Rubin, C.M., Slociak, M. & Viguie, F. (2000) Nineteen cases of
the t(1;22)(p13;q13) acute megakaryoblastic leukaemia of infants/
children and a review of 39 cases: report from a t(1;22) study group.
Leukemia, 14, 216–218.
Biondi, A., Rovelli, A., Cantu-Rajnoldi, A., Fenu, S., Basso, G.,
Luciano, A., Rondelli, R., Mandelli, F., Masera, G. & Testi, A.M. for
the Associazione Italiana di Ematologia e Oncologia Pediatrica
(AIEOP) (1994) Acute promyelocytic leukemia in children: experi-
ence of the Italian Pediatric Hematology and Oncology Group
(AIEOP). Leukemia, 8, 1264–1268.
Bloomfield, C.D., Shuma, C., Regal, L., Philip, P.P., Hossfeld,
D.K., Hageneijer, A.M., Garson, O.M., Peterson, B.A., Sakurai, M.,
Alimena, G., Berger, R., Rowley, J.D., Ruutu, T., Mitelman, F.,
Dewald, G.W. & Swansbury, J. (1997) Long-term survival of patients
with acute myeloid leukemia. A third follow-up of the Fourth
International Workshop on Chromosomes in Leukemia. Cancer, 80,
2191–2198.
Bloomfield, C.D., Lawrence, D., Byrd, J.C., Carroll, A., Pettenati, M.J.,
Tantravahi, R., Patil, S.R., Davey, F.R., Berg, D.T., Schiffer, C.A.,
Arthur, D.C. & Mayer, R.J. (1998) Frequency of prolonged remis-
sion duration after high-dose cytarabine intensification in acute
myeloid leukemia varies by cytogenetic subtype. Cancer Research, 58,
4173–4179.
Byrd, J.C., Lawrence, D., Arthur, D.C., Pettenati, M.J., Tantravahi, R.,
Qumsiyeh, M., Stamberg, J., Davey, F.R., Schiffer, C.A. & Bloom-
field, C.D. (1998) Patients with isolated trisomy 8 in acute myeloid
leukemia are not cured with cytarabine-based chemotherapy: results
from Cancer and Leukemia Group B 8461. Clinical Cancer Research,
4, 1235–1241.
B. Stark et al
334 ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337

Byrd, J.C., Dodge, R.K., Carroll, A., Baer, M.R., Edwards, C., Stamberg,
J., Qumsiyeh, M., Moore, J.O., Mayer, R.J., Davey, F., Schiffer, C.A.
& Bloomfield, C.D. (1999) Patients with t(8;21)(q22;q22) and acute
myeloid leukemia have superior failure-free and overall survival
when repetitive cycles of high-dose cytarabine are administered.
Journal of Clinical Oncology, 17, 3767–3775.
Byrd, J.C., Mrozek, K., Dodge, R.K., Carroll, A.J., Edwards, C.G.,
Arthur, D.C., Pettenati, M.J., Patil, S.R., Rao, K.W., Watson, M.S.,
Koduru, P.R.K., Moore, J.O., Stone, R.M., Mayer, R.J., Feldman,
E.J., Davey, F.R., Schiffer, C.A., Larson, R.A. & Bloomfield, C.D.
(2002) Pretreatment cytogenetic abnormalities are predictive of
induction success, cumulative incidence of relapse, and overall
survival in adult patients with de novo acute myeloid leukemia:
results from Cancer and Leukemia Group B (CALGB 8461). Blood,
100, 4325–4336.
Chessells, J.M., Harrison, C.J., Kempski, H., Webb, D.K.H., Wheatley,
K., Hann, I.M., Stevens, R.F., Harrison, G. & Gibson, B.E. (2002)
Clinical features, cytogenetics and outcome in acute lymphoblastic
and myeloid leukaemia of infancy: report from the MRC Childhood
Leukaemia working party. Leukemia, 16, 776–784.
Cox, D.R. (1972) Regression models and life-tables (with discussion).
Journal of the Royal Statistical Society, Series B, 34, 187–220.
Creutzig, U., Zimmermann, M., Ritter, J., Henze, G., Graf, N., Loffler,
H. & Schellong, G. (1999) Definition of a standard-risk group in
children with AML. British Journal of Haematology, 104, 630–639.
Creutzig, U., Ritter, J., Zimmermann, M., Reinhardt, D., Jermann, J.,
Berthold, F., Henze, G., Jurgens, H., Kabisch, H., Havers, W., Reiter,
A., Kluba, U., Niggli, F. & Gadner, H. for the Acute Myeloid
Leukemia-Berlin-Frankfurt-Munster Study Group (2001) Improved
treatment results in high-risk pediatric acute myeloid leukemia
patients after intensification with high-dose cytarabine and mitox-
antrone: results of study Acute Myeloid Leukemia-Berlin-Frankfurt-
Munster 93. Journal of Clinical Oncology, 19, 2705–2713.
Cuneo, A., Bigoni, R., Cavazzini, F., Bardi, A., Roberti, M.G., Agostini,
P., Tammiso, E., Ciccone, N., Mancini, M., Nanni, M., De Cuia, R.,
Divona, M., La Starza, R., Crescenzi, B., Testoni, N., Rege Cambrin,
G., Mecucci, C., Lo Coco, F., Saglio, G. & Castoldi, G. (2002)
Incidence and significance of crytpic chromosome aberrations
detected by fluorescence in situ hybridization in acute myeloid
leukemia with normal karyotype. Leukemia, 16, 1745–1751.
Dastugue, N., Duchayne, E., Kuhlein, E., Rubie, H., Demur, C., Aurich,
J., Robert, A. & Sie, P. (1997) Acute basophilic leukemia and
translocation t(X;6)(p11;q23). British Journal of Haematology, 98,
170–176.
Dastugue, N., Lafage-Pochitaloff, M., Pages, M.-P., Radford, I.,
Bastard, C., Talmant, P., Mozziconacci, M.J., Leonard, C., Bilhou-
Nabera, C., Cabrol, C., Capodano, A.-M., Cornillet-Lefebvre, P.,
Lessard, M., Mugneret, F., Perot, C., Taviaux, S., Fenneteaux, O.,
Duchayne, E. & Berger, R. (2002) Cytogenetic profile of childhood
and adult megakaryoblastic leukemia (M7): a study of the Groupe
Francais de Cytogenetique Hematologique (GFCH). Blood, 100,
618–626.
Delaunay, J., Vey, N., Leblanc, T., Fenaux, P., Rigal-Huguet, F., Witz,
F., Lamy, T., Auvrignon, A., Blaise, D., Pigneux, A., Mugneret,
F., Bastard, C., Dastugue, N., Van den Akker, J., Fiere, D., Reiffers, J.,
Castaigne, S., Leverger, G., Harousseau, J.-L. & Dombret, H. for the
French Acute Myeloid Leukemia (AML) Intergroup (2003) Prog-
nosis of inv(16)/t(16;16) acute myeloid leukemia (AML): a survey of
110 cases from the French AML Intergroup. Blood, 102, 462–469.
Douer, D., Preston-Martin, S., Chang, E., Nichols, P.W., Watkins, K.J.
& Levine, A.M. (1996) High frequency of acute promyelocytic
leukemia among latinos with acute myeloid leukemia. Blood, 87,
308–313.
Felice, M.S., Zubizarreta, P.A., Alfaro, E.M., Gallego, M.S., Cygler,
A.M., Rosso, D.A., Rossi, J.G. & Sackmann-Muriel, F. (2000) Good
outcome of children with acute myeloid leukemia and
t(8;21)(q22;q22), even when associated with granulocytic sarcoma.
A report from a single institution in Argentina. Cancer, 88, 1939–
1944.
Ferrara, F., Scognamiglio, M., Di Noto, R., Schiavone, E.M., Poggi, V.,
Fiorillo, A., Libertini, R., Vicari, L., Del Vecchio, L. & Sebastio, L.
(1996) Interstitial 9q deletion is associated with CD7+ acute leuke-
mia of myeloid and T lymphoid lineage. Leukemia, 10, 1990–1992.
Forestier, E., Heim, S., Blennow, E., Borgstrom, G., Holmgren, G.,
Heinonen, K., Johannsson, J., Kerndrup, G., Klarskov Anderson, M.,
Lundin, C., Nordgren, A., Rosenquist, R., Swolin, B. & Johansson, B.
on behalf of the Nordic Society of Paediatric Haematology and
Oncology (NOPHO), the Swedish Cytogenetic Leukaemia Study
Group (SCLSG) and the NOPHO Leukaemia Cytogenetic Study
Group (NLCSG) (2003) Cytogenetic abnormalities in childhood
acute myeloid leukaemia: a Nordic series comprising all children
enrolled in the NOPHO-93-AML trial between 1993 and 2001.
British Journal of Haematology, 121, 566–577.
Frohling, S., Skelin, S., Liebisch, C., Scholl, C., Schlenk, R.F., Dohner,
H. & Dohner, K. for the Acute Myeloid Leukemia Study Group Ulm
(2002) Comparison of cytogenetic and molecular cytogenetic
detection of chromosome abnormalities in 240 consecutive adult
patients with acute myeloid leukemia. Journal of Clinical Oncology,
20, 2480–2485.
Gibson, B., Webb, D., de-Graaf, S. & Wheatley, K. (2002) Improved
outcome in MRC AML 12 paediatrics. Has the limit of conventional
chemotherapy been reached? Blood, 100, 35a.
Grimwade, D. (2001) The clinical significance of cytogenetic
abnormalities in acute myeloid leukaemia. Best Practice and Research
Clinical Haematology, 14, 497–529.
Grimwade, D., Walker, H., Oliver, F., Wheatley, K., Harrison, C.,
Harrison, G., Rees, J., Hann, I., Stevens, R., Burnett, A. & Godstone,
A. on behalf of the Medical Research Council Adult and Children’s
Leukaemia Working Parties (1998) The importance of diagnostic
cytogenetics on outcome in AML: analysis of 1,612 patients entered
into the MRC AML 10 trial. Blood, 92, 2322–2333.
Grimwade, D., Walker, H., Harrison, G., Oliver, F., Chatters, S.,
Harrison, C.J., Wheatley, K., Burnett, A.K. & Goldstone, A.H. on
behalf of the Medical Research Council Adult Leukaemia Working
Party (2001) The predictive value of hierarchical cytogenetic clas-
sification in older adults with acute myeloid leukemia (AML):
analysis of 1065 patients entered into the United Kingdom Medical
Research Council AML11 trial. Blood, 98, 1312–1320.
Grimwade, D., Moorman, A.V., Holt, M.P., Walker, H., Chatters, S.,
Harrison, C.J., Hann, I., Rees, J., Stevens, R., Webb, D.K.H., Gibson,
B.E., Goldstone, A.H., Burnett, A.K. & Wheatley, K. (2002)
Refinement of cytogenetic classification in AML: determination of
prognostic significance of rare recurring chromosomal abnormalit-
ies amongst patients entered into the UK MRC AML10 and 12 trials.
Blood, 100, 195a.
Haferlach, T., Bennett, J.M., Loffler, H., Gassmann, W., Andersen,
J.W., Tuzuner, N., Casslleth, P.A., Fonatsch, C., Schoch, C., Schle-
gelberger, B., Becher, R., Thiel, E., Ludwig, W.-D., Sauerland, M.C.,
Cytogenetics, Prognosis in Childhood Acute Myeloid Leukaemia
ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337 335

Heinecke, A. & Buchner, T. for the AML Cooperative Group and
ECOG (1996) Acute myeloid leukemia with translocation (8;21).
Cytomorphology, dysplasia and prognostic factors in 41 cases.
Leukemia and Lymphoma, 23, 227–234.
Haferlach, T., Kohlmann, A., Kern, W., Hiddemann, W., Schnittger, S.
& Schoch, C. (2003) Gene expression profiling as a tool for the
diagnosis of acute leukemias. Seminars in Hematology, 40, 281–295.
Harris, N.L., Jaffe, E.S., Diebold, J., Flandrin, G., Muller-Hermelink,
H.K., Vardiman, J., Lister, T.A. & Bloomfield, C.D. (1999) World
Health Organization classification of neoplastic diseases of the
hematopoietic and lymphoid tissues: Report of the Clinical Advisory
Committee meeting – Airlie House, Virginia, November 1997.
Journal of Clinical Oncology, 17, 3835–3849.
Harrison, C.J., Moorman, A.V., Hills, R.K., Hann, I.M., Grimwade, D.,
Webb, D.K.H., Stevens, R., Wheatley, K. & Gibson, B.E. on behalf of
the UK NCRI Childhood Leukaemia Working Party (2003) Cyto-
genetics of childhood AML from UK Medical Research Council
Treatment trials. Blood, 102, 98a.
Holm, S. (1979) A simple sequentially rejective multiple test proce-
dure. Scandinavian Journal of Statistics, 6, 65–70.
Hrusak, O. & Porwit-MacDonald, A. (2002) Antigen expression patterns
reflecting genotype of acute leukemias. Leukemia, 16, 1233–1258.
Johansson, B., Mertens, F. & Mitelman, F. (1991) Geographic het-
erogeneity of neoplasia-associated chromosome aberrations. Genes
Chromosomes and Cancer, 3, 1–7.
Johansson, B., Fioretos, T., Kullendorff, C.-M., Wiebe, T., Bekassy,
A.N., Garwicz, S., Forestier, E., Roos, G., Akerman, M., Mitelman, F.
& Billstrom, R. (2000) Granulocytic sarcomas in body cavities in
childhood acute myeloid leukemias with 11q23/MLL rearrange-
ments. Genes, Chromosomes and Cancer, 27, 136–142.
Kalbfleisch, J.D. & Prentice, R.L. (1982) The Statistical Analysis of
Failure Time Data. John Wiley & Sons, New York.
Kaplan, E.L. & Meier, P. (1958) Non-parametric estimation from
incomplete observations. Journal of the American Statistical Asso-
ciation, 53, 457–481.
Kim, H.J., Cho, H.I., Kim, E.C., Ko, E.K., See, C.J., Park, S.Y. & Lee,
D.S. (2002) A study of 289 consecutive Korean patients with acute
leukaemias revealed fluorescence in situ hybridization detects the
MLL translocation without cytogenetic evidence both initially and
during follow-up. British Journal of Haematology, 119, 930–939.
Kojima, K., Omoto, E., Hara, M., Sasaki, K., Katayama, Y., Nawa, Y.,
Kimura, Y., Azuma, T., Takimoto, H. & Harada, M. (1998) Mye-
lodysplastic syndrome with translocation (8;21): a distinct myelod-
ysplastic syndrome entity or M2-acute myeloid leukemia with
extensive myeloid maturation? Annals of Hematology, 76, 279–282.
Levis, M. & Small, D. (2003) FLT3: it does matter in leukemia.
Leukemia, 17, 1738–1752.
Lie, S.O., Abrahamsson, J., Clausen, N., Forestier, E., Hasle, H., Hovi,
L., Jonmundsson, G., Mellander, L. & Gustafsson, G. (2003)
Treatment stratification based on initial in vivo response in acute
myeloid leukaemia in children without Down’s syndrome: results of
NOPHO-AML trials. British Journal of Haematology, 122, 217–225.
Marlton, P., Keating,M., Kantarjian,H., Pierce, S., O’Brien, S., Freireich,
E.J. & Estey, E. (1995) Cytogenetic and clinical correlates in AML
patients with abnormalities of chromosome 16. Leukemia, 9, 965–971.
Martinez-Climent, J.A., Lane, N.J., Rubin, C.M., Morgan, E., John-
stone, H.S., Mick, R., Murphy, S.B., Vardiman, J.W., Larson, R.A.,
Le Beau, M.M. & Rowley, J.D. (1995a) Clinical and prognostic
significance of chromosomal abnormalities in childhood acute
myeloid leukemia de novo. Leukemia, 9, 95–101.
Martinez-Climent, J.A., Espinosa, R. III, Thirman, M.J., Le Beau, M.M.
& Rowley, J.D. (1995b) Abnormalities of chromosome band 11q23
and the MLL gene in pediatric myelomonocytic and monoblastic
leukemias. Identification of the t(9;11) as an indicator of long sur-
vival. Journal of Pediatric Hematology/Oncology, 17, 277–283.
Mitelman, F. (1995) ISCN. An International System for Human Cyto-
genetic Nomenclature (ed. by F. Mitelman), S. Karger, Basel.
Mrozek, K., Heinonen, K. & Bloomfield, C.D. (2001) Clinical
importance of cytogenetics in acute myeloid leukaemia. Best Practice
and Research Clinical Haematology, 14, 19–47.
Nakamura, H., Kuriyama, K., Sadamori, N., Mine, M., Itoyama, T.,
Sasagawa, I., Matsumoto, K., Tsuji, Y., Asou, N., Kageyama, S.-I.,
Sakamaki, H., Emi, N., Ohno, R. & Tomonaga, M. (1997) Mor-
phological subtyping of acute myeloid leukemia with maturation
(AML-M2): homogeneous pink-colored cytoplasm of mature neu-
trophils is most characteristic of AML-M2 with t(8;21). Leukemia,
11, 651–655.
Nakase, K., Bradstock, K., Sartor, M., Gottlieb, D., Byth, K., Kita, K.,
Shiku, H., Kamada, N. and the Japanese Cooperative Group of
Leukemia/Lymphoma (2000) Geographic heterogeneity of cellular
characteristics of acute myeloid leukemia: a comparative study of
Australian and Japanese adult cases. Leukemia, 14, 163–168.
Pui, C.-H., Raimondi, S.C., Srivastava, D.K., Tong, X., Behm, F.G.,
Razzouk, B., Rubnitz, J.E., Sandlund, J.T., Evens, W.E. & Ribeiro, R.
(2000) Prognostic factors in infants with acute myeloid leukemia.
Leukemia, 14, 684–687.
Raimondi, S.C., Chang, M.N., Ravindranath, Y., Behm, F.G., Gresik,
M.V., Steuber, C.P., Weinstein, J.H. & Carroll, A.J. for the Pediatric
Oncology Group (1999) Chromosomal abnormalities in 478 chil-
dren with acute myeloid leukemia: clinical characteristics and
treatment outcome in a Cooperative Pediatric Oncology Group
Study – POG 8821. Blood, 94, 3707–3716.
Razzouk, B.I., Raimondi, S.C., Srivastava, D.K., Pritchard, M., Behm,
F.G., Tong, X., Sandlund, J.T., Rubnitz, J.E. & Pui, C.-H. (2001)
Impact of treatment of the outcome of acute myeloid leukemia with
in version 16: a single institution’s experience. Leukemia, 15, 1326–
1330.
Rowley, J.D. (2000) Cytogenetic analysis in leukemia and lymphoma:
an introduction. Seminars in Hematology, 37, 315–319.
Rowley, J.D. (2001) Chromosome translocations: dangerous liaisons
revisited. Nature Reviews, 1, 245–250.
Rubnitz, J.E., Raimondi, S.C., Tong, X., Srivastava, D.K., Razzouk, B.I.,
Shurtleff, S.A., Downing, J.R., Pui, C.-H., Ribeiro, P.C. & Behm,
F.G. (2002) Favorable impact of the t(9;11) in childhood acute
myeloid leukemia. Journal of Clinical Oncology, 20, 2302–2309.
Schoch, C., Haase, D., Haferlach, T., Gudat, H., Buchner, T., Freund,
M., Link, H., Lengfelder, E., Wandt, H., Sauerland, M.C., Loffler, H.
& Fonatsch, C. (1996) Fifty-one patients with acute myeloid leu-
kemia and translocation t(8;21)(q22;q22): an additional deletion in
9q is an adverse prognostic factor. Leukemia, 10, 1288–1295.
Schoch, C., Haferlach, T., Schnittger, S., Kern, W., Dugas, M. &
Hiddemann, W. (2001a) AML with recurring chromosome abnor-
malities as defined in the new WHO classification: incidence of
subgroups, additional genetic abnormalities, FAB subtype and age
distribution in an unselected series of 1897 cytogenetically and
molecular genetically analysed AML. Blood, 98, 457a.
B. Stark et al
336 ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337

Schoch, C., Haferlach, T., Haase, D., Fonatsch, C., Loffler, H.,
Schlegelberger, B., Staib, P., Sauerland, M.C., Heinecke, A., Buchner,
T. & Hiddemann, W. for the German AML Cooperative Study
Group (2001b) Patients with de novo acute myeloid leukaemia and
complex karyotype aberrations show a poor prognosis despite
intensive treatment: a study of 90 patients. British Journal of
Haematology, 112, 118–126.
Schoch, C., Schnittger, S., Klaus, M., Kern, W., Hiddemann, W. &
Haferlach, T. (2003) AML with 11q23/MLL abnormalities as defined
by the WHO classification: incidence, partner chromosomes, FAB
subtype, age distribution, and prognostic impact in an unselected
series of 1897 cytogenically analyzed AML cases. Blood, 120, 2395–
2402.
Schwyzer, R., Sherman, G.G., Cohn, R.J., Poole, J.E. & Willem, P.
(1998) Granulocytic sarcoma in children with acute myeloblastic
leukemia and t(8;21). Medical and Pediatric Oncology, 31, 144–149.
Sekeres, M.A., Dodge, R.K., Bloomfield, C.D., Larson, R.A. & Stone,
R.M. (2002) Racial differences in prognostic factors and outcome in
acute myeloid leukemia (AML): a Cancer and Leukemia Group B
(CALGB) Study. Blood, 100, 88a.
Shvidel, L., Shaft, D., Stark, B., Shtalrid, M., Berrebi, A. & Resnitzky, P.
(2003) Acute basophilic leukaemia: eight unsuspected new cases
diagnosed by electron microscopy. British Journal of Haematology,
120, 774–781.
Slovak, M.L., Kopecky, K.J., Cassileth, P.A., Harrington, D.H., Theil,
K.S., Mohamed, A., Paietta, E., Willman, C.L., Head, D.R., Rowe,
J.M., Forman, S.J. & Appelbaum, F.R. (2000) Karyotypic analysis
predicts outcome of preremission and postremission therapy in
adult acute myeloid leukemia: a Southwest Oncology Group/Eastern
Cooperative Oncology Group Study. Blood, 96, 4075–4083.
Smith, M.T., Wang, Y., Kane, E., Rollinson, S., Wiemels, J.L., Roman,
E., Roddam, P., Cartwright, R. & Morgan, G. (2001) Low
NAD(P)H:quinone oxidoreductase 1 activity is associated with
increased risk of acute leukemia in adults. Blood, 97, 1422–1426.
Stark, B., Resnitzky, P., Jeison, M., Luria, D., Blau, O., Avigad, S., Shaft,
D., Kodman, Y., Gobuzov, R., Ash, S., Stein, J., Yaniv, I., Barak, Y. &
Zaizov, R. (1995) A distinct subtype of M4/M5 acute myeloblastic
leukemia (AML) associated with t(8:16)(p11:p13), in a patient with
the variant t(8:19)(p11:q13) – case report and review of the litera-
ture. Leukemia Research, 19, 367–379.
Stark, B., Jeison, M., Gobuzov, R., Finkelshtein, S., Ash, S., Avrahami,
G., Cohen, I.J., Stein, J., Yaniv, I., Zaizov, R. & Bar-Am, I. (2000)
Apparently unrelated clones shown by spectral karyotyping to
represent clonal evolution of cryptic t(10;11)(p13;q23) in a patient
with acute monoblastic leukemia. Cancer, Genetics and Cytogenetics,
120, 105–110.
Stark, B., Jeison, M., Preudhomme, C., Fenaux, P., Ash, S., Korek, Y.,
Stein, J., Zaizov, R. & Yaniv, I. (2002) Acquired trisomy 21 and
distinct clonal evolution in acute megakaryoblastic leukaemia in
young monozygotic twins. British Journal of Haematology, 118,
1082–1086.
Taj, A.S., Ross, F.M., Vickers, M., Choudhury, D.N., Harvey, J.F.,
Barber, J.C.K., Barton, C. & Smith, A.G. (1995) t(8;21) mye-
lodysplasia, an early presentation of M2 AML. British Journal of
Haematology, 89, 890–892.
Tallman, M.S., Andersen, J.W., Schiffer, C.A., Appelbaum, F.R.,
Feusner, J.H., Woods, W.G., Ogden, A., Weinstein, H., Shepherd, L.,
Willman, C., Bloomfield, C.D., Rowe, J.M. & Wiernik, P.H. (2002)
All-trans retinoic acid in acute promeylocytic leukemia: long-term
outcome and prognostic factor analysis from the North American
Intergroup protocol. Blood, 100, 4298–4302.
Tchinda, J., Volpert, S., Neumann, T., Kennerknecht, I., Ritter, J.,
Buchner, T., Berdel, W.E. & Horst, J. (2002) Novel der(1)t(1;19) in
two patients with myeloid neoplasias. Cancer Genetics and Cyto-
genetics, 133, 61–65.
Vardiman, J.W., Harris, N.L. & Brunning, R.D. (2002) The World
Health Organization (WHO) classification of the myeloid neo-
plasms. Blood, 100, 2292–2302.
Wan, T.S.K., Ma, E.S.K., Lam, C.C.K., Chan, L.C., Lee, K.K. & Au,
W.Y. (2003) Deletion 9q as the sole karyotypic abnormality in
myelocytic disorders: a new case of myelodysplastic syndrome and
its prognostic implications in acute myelocytic leukemia. Cancer
Genetics and Cytogenetics, 145, 184–186.
Webb, D.K.H., Harrison, G., Stevens, R.F., Gibson, B.G., Hann, I.M. &
Wheatley, K. (2001) Relationships between age at diagnosis, clinical
features, and outcome of therapy in children treatment in the
Medical Research Council AML 10 and 12 trials for acute myeloid
leukemia. Blood, 98, 1714–1720.
Wells, R.J., Arthur, D.C., Srivastava, A., Heerema, N.A., Le Beau, M.,
Alonzo, T.A., Buxton, A.B., Woods, W.G., Howells, W.B., Benjamin,
D.R., Betcher, D.L., Buckley, J.D., Feig, S.A., Kim, T., Odom, L.F.,
Ruymann, F.B., Smithson, W.A., Tannous, R., Whitt, J.K., Wolff, L.,
Tjoa, T. & Lampkin, B.C. (2002) Prognostic variables in newly
diagnosed children and adolescents with acute myeloid leukemia:
Children’s Cancer Group Study 213. Leukemia, 16, 601–607.
Willman, C.L. (2001) Targeted AML therapy: new biologic paradigms
and therapeutic opportunities. Leukemia, 15, 690–694.
Wolman, S.R., Gundacker, H., Appelbaum, F.R. & Slovak, M.L. for the
Southwest Oncology Group (2002) Impact of trisomy 8(+8) on
clinical presentation, treatment response, and survival in acute
myeloid leukemia: a Southwest Oncology Group Study. Blood, 100,
29–35.
Woods, G.W., Neudorf, S., Gold, S., Sanders, J., Buckley, J.D., Barnard,
D.R., Dusenbery, K., DeSwarte, J., Arthur, D.C., Lange, B.J. &
Kobrinsky, N.L. (2001) A comparison of allogeneic bone marrow
transplantation, autologous bone marrow transplantation, and
aggressive chemotherapy in children with acute myeloid leukemia in
remission; a report from the Children’s Cancer Group. Blood, 97,
56–62.
Yagi, T., Morimoto, A., Eguchi, M., Hibi, S., Sako, M., Ishii, E.,
Mizutani, S., Imashuku, S., Ohki, M. & Ichikawa, H. (2003) Iden-
tification of a gene expression signature associated with pediatric
AML prognosis. Blood, 102, 1849–1856.
Zwaan, C.M., Kaspers, G.J.L., Pieters, R., Hahlen, K., Huismans, D.R.,
Zimmermann, M., Harbott, J., Slater, R.M., Creutzig, U. &
Veerman, A.J.P. (2002) Cellular drug resistance in childhood acute
myeloid leukemia is related to chromosomal abnormalities. Blood,
100, 3352–3360.
Zwaan, C.M., Meshinchi, S., Radich, J.P., Veerman, A.J.P., Huismans,
D.R., Munske, L., Podleschny, M., Hahlen, K., Pieters, R.,
Zimmermann, M., Reinhardt, D., Harbott, J., Creutzig, U., Kaspers,
G.J.L. & Griesinger, F. (2003) FLT3 internal tandem duplication in
234 children with acute myeloid leukemia: prognostic significance
and relation to cellular drug resistance. Blood, 102, 2387–2394.
Cytogenetics, Prognosis in Childhood Acute Myeloid Leukaemia
ª 2004 Blackwell Publishing Ltd, British Journal of Haematology, 126, 320–337 337




















