
Chronic but not acute nicotine treatment reverses stress-induced impairment of LTP in anesthetized...
7
Research Report Chronic but not acute nicotine treatment reverses stress-induced impairment of LTP in anesthetized rats A.M. Aleisa a , K.H. Alzoubi b , K.A. Alkadhi a, ⁎ a Department of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, Houston, TX 77204-5515, USA b Department of Clinical Pharmacy, College of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan ARTICLE INFO ABSTRACT Article history: Accepted 17 April 2006 Available online 24 May 2006 Stress impairs long-term potentiation (LTP) and is a major cause for starting or increasing tobacco smoking. We have previously shown that chronic concurrent nicotine treatment prevents stress-induced LTP impairment. Nicotine reduces stress-induced impairment of LTP, probably, through activation of nicotinic acetylcholine receptors in the hippocampus. Herein, we investigated the effects of acute and chronic nicotine treatments on the chronic- stress-induced impairment of LTP in area CA1 of the hippocampus of urethane- anesthetized rats. Extracellular in vivo recording from the hippocampal area CA1 showed that pre-treatment with nicotine (1 mg/kg; sc twice/day for 2 weeks prior to stress) protected LTP from the inhibitory effect of subsequent chronic psychosocial stress (4 additional weeks concurrently with nicotine). In another series of experiments, 2 weeks of psychosocial stress was followed by 4 weeks of nicotine treatment concurrently with continuing stress. Nicotine treatment reversed established stress-induced impairment of LTP. However, acute nicotine treatment of rats (a single dose of 1 mg/kg; sc.) did not reverse chronic-stress-induced impairment of LTP. The results show that the impairment of LTP during chronic stress can be blocked by chronic, but not acute, nicotine treatment. © 2006 Elsevier B.V. All rights reserved. Keywords: Anesthetized rat Nicotine Chronic Acute LTP I/O curve 1. Introduction Stress is a necessary mechanism for survival, however, long-term stress alters normal brain structure and function (McEwen, 2000). The hippocampus is greatly influenced by stress hormones (McEwen and Magarinos, 1997) and the cholinergic system (Gray et al., 1996) and is implicated in cognitive function (O'Keefe, 1993; Squire, 1993; Gilbert et al., 1998; Burgess et al., 2002; Fortin et al., 2002). Chronic stress impairs hippocampus-dependent memory (Park et al., 2001; Kim and Diamond, 2002; Gerges et al., 2004b; Aleisa et al., in press-b), blocks the expression of hippocampal LTP (Foy et al., 1987; Gerges et al., 2001; Aleisa et al., in press-a, see Diamond et al., 2005) and reduces the levels of essential signaling molecules associated with memory and LTP including phosphorylated CaMKII (P-CaMKII) (Gerges et al., 2004a; Aleisa et al., in press-a) and brain-derived neuro- trophic factor (BDNF; Aleisa et al., in press-a). Stress is one of the major reasons for starting or increasing the use of tobacco products (Forgas et al., 1996; Boos and Croft, 2004). The major component of tobacco smoke, nicotine, attenuates the impairment of memory and LTP associated with aging (Fujii and Sumikawa, 2001; White and Levin, 2004) and other mental disorders including Alzheimer's disease (Sahakian et al., 1989; White and Levin, 1999; Fratiglioni and Wang, 2000). Accordingly, the BRAIN RESEARCH 1097 (2006) 78 – 84 ⁎ Corresponding author. Fax: +1 713 743 1229. E-mail address: [email protected] (K.A. Alkadhi). 0006-8993/$ – see front matter © 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.brainres.2006.04.070 available at www.sciencedirect.com www.elsevier.com/locate/brainres
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Chronic but not acute nicotine treatment reverses stress-induced impairment of LTP in anesthetized...

B R A I N R E S E A R C H 1 0 9 7 ( 2 0 0 6 ) 7 8 – 8 4
ava i l ab l e a t www.sc i enced i rec t . com
www.e l sev i e r. com/ loca te /b ra in res
Research Report
Chronic but not acute nicotine treatment reversesstress-induced impairment of LTP in anesthetized rats
A.M. Aleisaa, K.H. Alzoubib, K.A. Alkadhia,⁎aDepartment of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, Houston, TX 77204-5515, USAbDepartment of Clinical Pharmacy, College of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan
A R T I C L E I N F O
⁎ Corresponding author. Fax: +1 713 743 1229.E-mail address: [email protected] (K.A. Alk
0006-8993/$ – see front matter © 2006 Elsevidoi:10.1016/j.brainres.2006.04.070
A B S T R A C T
Article history:Accepted 17 April 2006Available online 24 May 2006
Stress impairs long-term potentiation (LTP) and is a major cause for starting or increasingtobacco smoking. We have previously shown that chronic concurrent nicotine treatmentprevents stress-induced LTP impairment. Nicotine reduces stress-induced impairment ofLTP, probably, through activation of nicotinic acetylcholine receptors in the hippocampus.Herein, we investigated the effects of acute and chronic nicotine treatments on the chronic-stress-induced impairment of LTP in area CA1 of the hippocampus of urethane-anesthetized rats. Extracellular in vivo recording from the hippocampal area CA1 showedthat pre-treatment with nicotine (1mg/kg; sc twice/day for 2 weeks prior to stress) protectedLTP from the inhibitory effect of subsequent chronic psychosocial stress (4 additional weeksconcurrently with nicotine). In another series of experiments, 2 weeks of psychosocial stresswas followed by 4weeks of nicotine treatment concurrently with continuing stress. Nicotinetreatment reversed established stress-induced impairment of LTP. However, acute nicotinetreatment of rats (a single dose of 1 mg/kg; sc.) did not reverse chronic-stress-inducedimpairment of LTP. The results show that the impairment of LTP during chronic stress canbe blocked by chronic, but not acute, nicotine treatment.
© 2006 Elsevier B.V. All rights reserved.
Keywords:Anesthetized ratNicotineChronicAcuteLTPI/O curve
1. Introduction
Stress is a necessary mechanism for survival, however,long-term stress alters normal brain structure and function(McEwen, 2000). The hippocampus is greatly influenced bystress hormones (McEwen and Magarinos, 1997) and thecholinergic system (Gray et al., 1996) and is implicated incognitive function (O'Keefe, 1993; Squire, 1993; Gilbert et al.,1998; Burgess et al., 2002; Fortin et al., 2002). Chronic stressimpairs hippocampus-dependent memory (Park et al., 2001;Kim and Diamond, 2002; Gerges et al., 2004b; Aleisa et al.,in press-b), blocks the expression of hippocampal LTP (Foyet al., 1987; Gerges et al., 2001; Aleisa et al., in press-a, see
adhi).
er B.V. All rights reserved
Diamond et al., 2005) and reduces the levels of essentialsignaling molecules associated with memory and LTPincluding phosphorylated CaMKII (P-CaMKII) (Gerges et al.,2004a; Aleisa et al., in press-a) and brain-derived neuro-trophic factor (BDNF; Aleisa et al., in press-a).
Stress is one of the major reasons for starting orincreasing the use of tobacco products (Forgas et al., 1996;Boos and Croft, 2004). The major component of tobaccosmoke, nicotine, attenuates the impairment of memory andLTP associated with aging (Fujii and Sumikawa, 2001; Whiteand Levin, 2004) and other mental disorders includingAlzheimer's disease (Sahakian et al., 1989; White andLevin, 1999; Fratiglioni and Wang, 2000). Accordingly, the
.

79B R A I N R E S E A R C H 1 0 9 7 ( 2 0 0 6 ) 7 8 – 8 4
increase in tobacco smoking rate during stress is, perhaps,a form of self-medication to counter the harmful effect ofstress on cognitive function. The effects of nicotine onmemory and LTP are prevented by the non-selectivenicotinic antagonist, mecamylamine, suggesting that nico-tine induces its effects by acting on nicotinic acetylcholinereceptor (nAChR) in the hippocampus (Levin et al., 1987;Fujii et al., 1999).
We previously reported that concurrent chronic nicotinetreatment and stress prevented stress-induced impairmentof LTP (Aleisa et al., in press-a-b). In the present study, weinvestigated whether the impairment of LTP during estab-lished chronic stress could be reversed by acute or chronicnicotine treatment, and whether chronic pre-treatmentwith nicotine would protect LTP from harmful effects ofsubsequent chronic stress.
Fig. 1 – Chronic nicotine pre-treatment (chronic nic-stress)protected LTP from stress-induced impairment. LTP of areaCA1 of the hippocampus was evoked by HFS (at time zero) ofthe Schaffer collaterals pathway. LTP was measured asincreases in slope of fEPSP (A) and amplitude of pSpike (B) inurethane-anesthetized Wistar rat. Each point in the “stress”groups is significantly (P < 0.05) lower than the correspondingpoints in the other groups. All points represent themean ± SEM from 4 to 8 rats. Insets are representativepSpikes from each group before (basal) and after HFS.Calibration bars apply to all traces.
2. Results
We recorded the evoked pSpike from area CA1 of adult rathippocampus before and after induction of LTP in thecontrol and experimental groups. In the “control” group,following high frequency stimulation (HFS), test pulsesshowed a marked increase in the slope of fEPSP(163 ± 13% of baseline; measured 60 min after HFS; Fig.1A) and amplitude of pSpike (277 ± 43% of baseline; Fig. 1B)in area CA1 of the hippocampus. In the “nicotine” groups(chronic, Figs. 1 and 2; acute, Fig. 3), HFS induced an LTPsimilar to that of control group. One hour after HFS, theslope of fEPSP measured from area CA1 of the nicotinegroup was 164 ± 7% of baseline (Fig. 1A) and the amplitudeof pSpike was 270 ± 28% of baseline (Fig. 1B), which werenot significantly different from those of the “control”. TheLTP magnitude in the “stress” group was markedly(P < 0.01) reduced compared to that of “control” or“nicotine”. One hour after HFS, the slope of fEPSP measuredfrom area CA1 of the stress group was 108 ± 6% of baseline(Fig. 1A) and the amplitude of pSpike was 132 ± 14% ofbaseline (Fig. 1B).
2.1. Chronic pre-treatment with nicotine protects LTP fromstress-induced impairment
Chronic nicotine treatment in the “chronic nic-stress” groupwas started 2 weeks before initiating the stress procedure.Daily stress was then initiated coupled with nicotinetreatment for 4 additional weeks. After HFS, the slope offEPSP (162 ± 9%; Fig. 1A) and amplitude of pSpike (284 ± 11%;Fig. 1B) in area CA1 were significantly (P < 0.05) higher thanthose of stress group but similar to those of “control” or“nicotine” group. This indicates that chronic pre-treatmentwith nicotine protects LTP from impairment by subsequentchronic stress.
2.2. Chronic nicotine treatment reverses LTP impairmentin established psychosocial stress
To determine whether chronic nicotine treatment reversesestablished stress-induced impairment of LTP, chronic
psychosocial stress was started 2 weeks prior to chronicnicotine treatment in the “stress-chronic nic” group. Then,nicotine was given every day for 4 weeks with continuingstress. In this treatment order, chronic nicotine reversed theimpairment of LTP induced by stress in area CA1. HFS in the“stress-chronic nic” group induced a significant (P < 0.05)increase in the slope of fEPSP (161 ± 7%; Fig. 2A) andamplitude of pSpike (280 ± 13%; Fig. 2B) compared to those ofthe stress group. No significant difference in the magnitudewas observed between this LTP and that of the “control” or“nicotine” group.
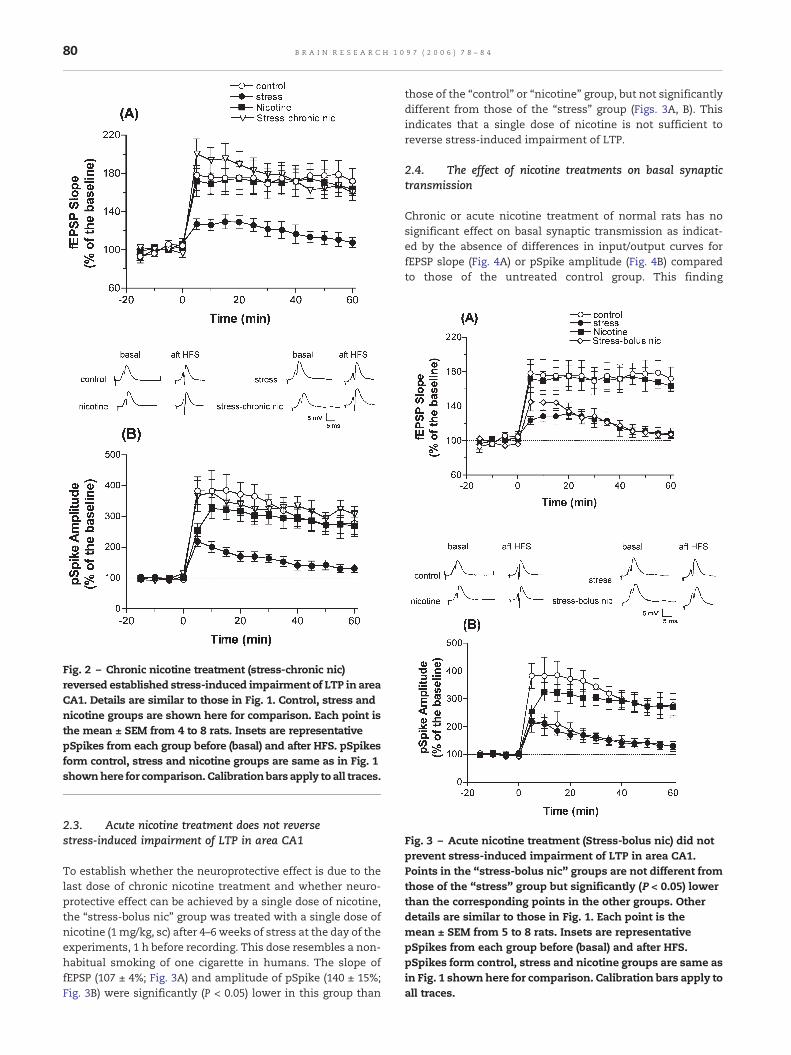
Fig. 3 – Acute nicotine treatment (Stress-bolus nic) did notprevent stress-induced impairment of LTP in area CA1.Points in the “stress-bolus nic” groups are not different fromthose of the “stress” group but significantly (P < 0.05) lowerthan the corresponding points in the other groups. Otherdetails are similar to those in Fig. 1. Each point is themean ± SEM from 5 to 8 rats. Insets are representativepSpikes from each group before (basal) and after HFS.pSpikes form control, stress and nicotine groups are same asin Fig. 1 shownhere for comparison. Calibration bars apply toall traces.
Fig. 2 – Chronic nicotine treatment (stress-chronic nic)reversed established stress-induced impairment of LTP in areaCA1. Details are similar to those in Fig. 1. Control, stress andnicotine groups are shown here for comparison. Each point isthe mean ± SEM from 4 to 8 rats. Insets are representativepSpikes from each group before (basal) and after HFS. pSpikesform control, stress and nicotine groups are same as in Fig. 1shownhere for comparison. Calibrationbars apply to all traces.
80 B R A I N R E S E A R C H 1 0 9 7 ( 2 0 0 6 ) 7 8 – 8 4
2.3. Acute nicotine treatment does not reversestress-induced impairment of LTP in area CA1
To establish whether the neuroprotective effect is due to thelast dose of chronic nicotine treatment and whether neuro-protective effect can be achieved by a single dose of nicotine,the “stress-bolus nic” group was treated with a single dose ofnicotine (1 mg/kg, sc) after 4–6 weeks of stress at the day of theexperiments, 1 h before recording. This dose resembles a non-habitual smoking of one cigarette in humans. The slope offEPSP (107 ± 4%; Fig. 3A) and amplitude of pSpike (140 ± 15%;Fig. 3B) were significantly (P < 0.05) lower in this group than
those of the “control” or “nicotine” group, but not significantlydifferent from those of the “stress” group (Figs. 3A, B). Thisindicates that a single dose of nicotine is not sufficient toreverse stress-induced impairment of LTP.
2.4. The effect of nicotine treatments on basal synaptictransmission
Chronic or acute nicotine treatment of normal rats has nosignificant effect on basal synaptic transmission as indicat-ed by the absence of differences in input/output curves forfEPSP slope (Fig. 4A) or pSpike amplitude (Fig. 4B) comparedto those of the untreated control group. This finding

Fig. 4 – Acute or chronic nicotine treatment does not enhancebasal synaptic transmission. Input/output (I/O) curvesrepresenting synaptic responses (A) fEPSP and (B) pSpikeresulting from progressively increasing stimulus intensities.The abscissa numbers for stimulus intensity are arbitraryunits where 1 and 8 are the intensity that gaveminimum andmaximum responses, respectively. Each point in each groupis the mean ± SEM from 8 to 10 animals.
81B R A I N R E S E A R C H 1 0 9 7 ( 2 0 0 6 ) 7 8 – 8 4
suggests that nicotine selectively restores impaired synapticplastic response without interfering with the basal synaptictransmission.
3. Discussion
Individually, stress and nicotine have widely different effectson LTP. Whereas stress impairs LTP in the hippocampus(Gerges et al., 2001; see Diamond and Rose, 1994), nicotineprevents the stress-induced impairment of LTP when nico-tine treatment is initiated simultaneously with stress (Aleisaet al., in press-b; Aleisa et al., 2006). In this work, we haveshown that chronic psychosocial stress markedly decreasesLTP in area CA1 of hippocampus, which confirms ourprevious findings (Gerges et al., 2001; Aleisa et al., in press-b). We have, also, determined the effect of different orders ofnicotine treatment and application of stress on the expres-sion of LTP.
The stress-induced impairment of the cellular correlate oflearning and memory, LTP, may be the result of a reduction inthe levels of associated essential signaling molecules includ-ing P-CaMKII and BDNF (Gerges et al., 2004a; Aleisa et al., inpress-a). Moreover, others have demonstrated that stressinduces down-regulation of nAChR in the brain (Pauly andCollins, 1993; Takita and Muramatsu, 1995; Takita et al., 1999),similar to that observed in the brains of Alzheimer's diseasepatients (Whitehouse and Au, 1986; Aubert et al., 1992;Warpman and Nordberg, 1995), suggesting a relationshipbetween the reduction in nAChR and cognitive impairment.
Analysis of the I/O relationships indicates that nicotineadministered chronically or as a single dose does not seem toaffect basal synaptic transmission in normal or chronicallystressed rats (Aleisa et al., in press-a). The lack of effect ofnicotine, at the dose level we use, on synaptic plasticity innormal animals is not due to the phenomenon of saturation.This is indicated by a similar absence of effect of nicotine onresponses evoked by trains of subthreshold stimuli in botharea CA1 and dentate gyrus of the hippocampus (Aleisa et al.,in press-b; Aleisa et al., 2006). Chronic nicotine treatmentupregulates α4β2 and α7 nAChR subtypes in most brainregions including the hippocampus (Ridley et al., 2001;Mugnaini et al., 2002; Parker et al., 2004). The activation bynicotine of presynaptic α4β2 and α7 nAChR in the glutama-tergic pyramidal neurons enhances glutamate release and,consequently, increases pyramidal neurons excitability.Therefore, it is reasonable to expect that nicotine mayenhance basal synaptic transmission, facilitate expressionand enhance themagnitude of LTP. However, in this as well asprevious studies, we observe that chronic nicotine treatment,although clearly prevents impairment of LTP induced bychronic stress (Aleisa et al., in press-a-b), it does not affectbasal synaptic transmission or the magnitude of LTP innormal animals. This finding is at variance with that of Fujiiet al. (1999) who report that nicotine facilitates the inductionof LTP in CA1 region in hippocampal slice preparation.Although the slice preparation has some advantages, the invivo physiological conditions may not have been sufficientlysimulated. Therefore, by using in vivo recording, we eliminat-ed the disadvantages of slice preparation such as loss ofneurotransmitters, disruption of excitatory and inhibitorypathways, and temperature variation that may affect theresults. The absence of significant effects of the present doseof nicotine in normal (control) animals has been consistentlyseen not only with electrophysiological parameters doneunder urethane anesthesia (Aleisa et al., in press-a; Aleisa etal., 2006), but also in behavioral (Aleisa et al., in press-b) andmolecular studies (Aleisa et al., 2006) from this laboratory.
Treating stressed rats chronically with nicotine reversesthe established stress-induced impairment of LTP. Unlikechronic treatment, a single dose of nicotine does not reversethe stress-induced impairment of LTP. The absence ofneuroprotective effect of acute nicotine treatment on stress-induced impairment of LTP is, perhaps, related to the effect ofthese treatments on the levels of key signaling moleculesassociated with memory and LTP. We have shown that thebasal levels of BDNF in area CA1 are increased after chronicnicotine treatment of normal rats (Aleisa et al., in press-a).However, acute nicotine treatment decreases the expression

82 B R A I N R E S E A R C H 1 0 9 7 ( 2 0 0 6 ) 7 8 – 8 4
of BDNF in rat hippocampus (Kenny et al., 2000). In addition,chronic nicotine treatment normalizes stress-induced reduc-tion in the levels of BDNF (Aleisa et al., in press-a), which mayexplain the expression of normal LTP in stressed rats treatedchronically with nicotine.
Nicotine in cigarette smoke has a calming effect, while atthe same time it can activate some of the same physiologicalresponses seen with stress. Even with this calming effect,nicotine activates several stress reactive systems. Nicotinecan activate the hypothalamic–pituitary–adrenal axis leadingto higher plasma levels of the stress hormones and stimulatethe release of catecholamines from nerve terminals (Haassand Kubler, 1997). Chronic nicotine administration does notalter basal plasma corticosterone levels in normal animals(Serova et al., 1999; Cheng et al., 2005), but decreases stress-induced increase of these levels (Cheng et al., 2005). Theseresults indirectly support present and previous findings fromthis laboratory (Aleisa et al., in press-a-b); Aleisa et al., 2006).
The different effect of acute and chronic nicotine treat-ments on stress-induced impairment of LTP could be alsoexplained by the fact that only chronic nicotine treatmentinduces upregulation of α4β2 and α7 nAChR subtypes in thehippocampus (Ridley et al., 2001; Mugnaini et al., 2002; Parkeret al., 2004). As a result, activation of a higher number ofnAChR may lead to enhanced activation of downstreamprotein kinases and genes and initiation of synthesis ofproteins associated with synaptic plasticity and neuronalregeneration. Moreover, chronic nicotine treatment has beenreported to prevent stress-induced down-regulation of nAChR(Takita et al., 1999), which may explain the expression ofnormal LTP in stressed rats treated chronically with nicotine.Thus, changing the dynamics of nAChR activation anddistribution influences the release of neurotransmitters andaffects activity-dependent synaptic plasticity.
In conclusion, we investigated various orders of applicationof nicotine and stress and observed that chronic, but notacute, nicotine treatment reversed established impairment ofLTP in stressed rats and that chronic treatment of normal ratswith nicotine prevented the inhibitory effect of subsequentstress on LTP.
4. Experimental procedures
Adult male Wistar rats (200–250 g Harlan, Indianapolis, IN)were housed 6 per cage. Animals had free access to food andwater and were subjected to a 12 h light/dark cycle at 25 °C. Allprocedures involving animals were carried out in accordancewith the National Research Council's Guide for the Care and Useof Laboratory Animals and on approval of the University ofHouston Institutional Animal Care and Use Committee.
4.1. Treatments
There were six treatment groups in this study; control, stress,chronic nicotine (nicotine), stress followed by bolus nicotine(stress-bolus nic), stress followed by chronic nicotine (stress-chronic nic), and chronic nicotine followed by stress (chronicnic-stress) groups. The “control” and “stress” groups receivedsaline (0.9% w/v NaCl, sc twice/day) for 4–6 weeks. In addition,
the stress groups were subjected to the “intruder” model ofstress for 4–6 weeks. The “nicotine” group was treated withnicotine (Sigma, St. Louis, MO; 1 mg/kg sc twice/day) for 4–6 weeks. Electrophysiological procedures were carried out 1 hafter the last dose of nicotine. In the “stress-bolus nic” group,rats were stressed for 4–6 weeks then a single dose of nicotine(1 mg/kg sc) was given 1 h before the electrophysiologicalprocedures. In the “stress-chronic nic” group, chronic stresswas started 2 weeks prior to nicotine treatment, then, nicotinewas given for 4 weeks with continuing stress. The “chronicnic-stress” group was treated chronically with nicotine2 weeks before the stress procedure, and then rats werestressed daily and concurrently treated with nicotine for 4additional weeks.
4.2. Induction of chronic psychosocial stress
Stress groups were subjected to a form of “intruder” psycho-social stress as described (Gerges et al., 2001; Alkadhi et al.,2005; Aleisa et al., in press-a-b). After allowing the rats toestablish social hierarchy within a group (kept with the samecage mates for at least 1 week), stress was then generated bydaily random exchanging of two animals between the cagesfor a period of 4–6 weeks. This model of stress, termed“intruder psychosocial stress,” is known to produce stress bydisrupting established social hierarchy such that rats mustcontinuously adjust to new situations. It models the kind ofstress that humans experience daily in social settings. Stressby this procedure was indicated by a 50% increase in serumcorticosterone levels (Gerges et al., 2001) and a markedelevation of blood pressure (Alkadhi et al., 2005).
4.3. Electrophysiological procedures
Standard procedures for in vivo recording from CA1 regionof anesthetized rats were performed between 8 AM and 6 PMas described (e.g. Gerges et al., 2001; Aleisa et al., in press-a).At the end of treatment, rats were anesthetized withurethane (1.2 g/kg ip; Sigma, USA) and placed in astereotaxic frame (nose bar at 0.0). Then, the skull wasexposed and burr holes were drilled. A concentric bipolarstimulating electrode was placed through a burr hole in areaCA3 of the left hippocampus at an angle of 5° toward themidline to stimulate the Schaffer collaterals/commissuralpathway (AP, −3; L, 3.5; D, 2.8). A recording glass microelec-trode (tip resistance; 1–5 mΩ; filled with 1% Fast Green dyein 2 M NaCl) was placed through another burr hole in thestratum pyramidale of area CA1 of the right hippocampus(AP, −3; L, 1.8; D, 2.0). A train of 8 pulses (400 Hz) was appliedevery 10 s for a period of 70 s to the Schaffer collaterals/commissural pathway of the contralateral (left) CA3 regionto evoke LTP in area CA1. An input/output curve for synaptictransmission in area CA1 was constructed for each rat. Astimulus intensity of approximately 30–50% of the maxi-mum response was chosen to evoke test responses. Thisintensity was found to cause maximum enhancement offield excitatory postsynaptic potential (fEPSP) slope after HFS(Gerges et al., 2001). Evoked population spikes (pSpike) wereamplified (Axoclamp 2A amplifier, Axon Instruments, Inc.,Foster City, CA, USA) and recorded from area CA1 of the

83B R A I N R E S E A R C H 1 0 9 7 ( 2 0 0 6 ) 7 8 – 8 4
right hippocampus. The slope of fEPSP, which represents theintensity of synaptic activity, and amplitude of populationspike (pSpike), which reflects the number of neuronsreaching threshold as a result of synaptic activity, weremeasured as described (Gerges et al., 2001). Computer-basedstimulation and recording were accomplished by the use ofpCLAMP 8.2 software and DigiData 1322A (Axon Instru-ments, Inc.).
4.4. Statistical analysis
Comparison among groups was carried out by a computer-based (Prism, GraphPad) one-way ANOVA followed by Tukey'sposttest. Data were expressed as means ± SEM, and P < 0.05was considered statistically significant.
R E F E R E N C E S
Aleisa, A.M., Alzoubi, K.H., Alkadhi, K.A., 2006. Nicotine preventsstress-induced enhancement of LTD: electrophysiological andmolecular studies. J. Neurosci. Res. 83 (2), 309–317.
Aleisa, A.M., Alzoubi, K.H., Gerges, N.Z., Alkadhi, K.A., in press-a.Chronic psychosocial stress-induced LTP impairment ofhippocampal LTP: possible role of BDNF. Neurobiology ofDisease.
Aleisa, A.M., Alzoubi, K.H., Gerges, N.Z., Alkadhi, K.A., in press-b.Nicotine blocks stress-induced impairment of spatial memoryand LTP of the hippocampal CA1 region. Intl. J.Neuropsychopharmacol. (Epub).
Alkadhi, K.A., Alzoubi, K.H., Aleisa, A.M., Tanner, F.L., Nimer, A.S.,2005. Psychosocial stress-induced hypertension results from invivo expression of long term potentiation in sympatheticganglia. Neurobiol. Dis. 20, 849–857.
Aubert, I., Araujo, D.M., Cecyre, D., Robitaille, Y., Gauthier, S.,Quirion, R., 1992. Comparative alterations of nicotinic andmuscarinic binding sites in Alzheimer's and Parkinson'sdiseases. J. Neurochem. 58, 529–541.
Boos, C.J., Croft, A.M., 2004. Smoking rates in the staff of a militaryfield hospital before and after wartime deployment. J. R. Soc.Med. 97, 20–22.
Burgess, N., Maguire, E.A., O'Keefe, J., 2002. The humanhippocampus and spatial and episodic memory. Neuron 35,625–641.
Cheng, S.Y., Glazkova, D., Serova, L., Sabban, E.L., 2005. Effect ofprolonged nicotine infusion on response of rat catecholaminebiosynthetic enzymes to restraint and cold stress. Pharmacol.Biochem. Behav. 82 (3), 559–568.
Diamond, D.M., Rose, G.M., 1994. Stress impairs LTP andhippocampal-dependent memory. Ann. N. Y. Acad. Sci. 746,411–414 (Nov. 30).
Diamond, D.M., Park, C.R., Campbell, A.M., Woodson, J.C., 2005.Competitive interactions between endogenous LTD and LTP inthe hippocampus underlie the storage of emotional memoriesand stress-induced amnesia. Hippocampus 15 (8), 1006–1025.
Forgas, L.B., Meyer, D.M., Cohen, M.E., 1996. Tobacco use habitsof naval personnel during Desert Storm. Mil. Med. 161,165–168.
Fortin, N.J., Agster, K.L., Eichenbaum, H.B., 2002. Critical role of thehippocampus in memory for sequences of events. Nat.Neurosci. 5, 458–462.
Foy, M.R., Stanton, M.E., Levine, S., Thompson, R.F., 1987.Behavioral stress impairs long-term potentiation in rodenthippocampus. Behav. Neural Biol. 48, 138–149.
Fratiglioni, L., Wang, H.X., 2000. Smoking and Parkinson's andAlzheimer's disease: review of the epidemiological studies.Behav. Brain Res. 113, 117–120.
Fujii, S., Sumikawa, K., 2001. Acute and chronic nicotineexposure reverse age-related declines in the induction oflong-term potentiation in the rat hippocampus. Brain Res.894, 347–353.
Fujii, S., Ji, Z., Morita, N., Sumikawa, K., 1999. Acute and chronicnicotine exposure differentially facilitate the induction of LTP.Brain Res. 846, 137–143.
Gerges, N.Z., Stringer, J.L., Alkadhi, K.A., 2001. Combination ofhypothyroidism and stress abolishes early LTP in the CA1 butnot dentate gyrus of hippocampus of adult rats. Brain Res. 922,250–260.
Gerges, N.Z., Aleisa, A.M., Schwarz, L.A., Alkadhi, K.A., 2004a.Reduced basal CaMKII levels in hippocampal CA1 region:possible cause of stress-induced impairment of LTP inchronically stressed rats. Hippocampus 14, 402–410.
Gerges, N.Z., Alzoubi, K.H., Park, C.R., Diamond, D.M., Alkadhi,K.A., 2004b.Adverse effect of the combination of hypothyroidismand chronic psychosocial stress on hippocampus-dependentmemory in rats. Behav. Brain Res. 155, 77–84.
Gilbert, P.E., Kesner, R.P., DeCoteau, W.E., 1998. Memory for spatiallocation: role of the hippocampus in mediating spatial patternseparation. J. Neurosci. 18, 804–810.
Gray, R., Rajan, A.S., Radcliffe, K.A., Yakehiro, M., Dani, J.A., 1996.Hippocampal synaptic transmission enhanced by lowconcentrations of nicotine. Nature 383, 713–716.
Haass, M., Kubler, W., 1997. Nicotine and sympatheticneurotransmission. Cardiovasc. Drugs Ther. 10, 657–665.
Kenny, P.J., File, S.E., Rattray, M., 2000. Acute nicotine decreases,and chronic nicotine increases the expression of brain-derivedneurotrophic factor mRNA in rat hippocampus. Brain Res. Mol.Brain Res. 85, 234–238.
Kim, J.J., Diamond, D.M., 2002. The stressed hippocampus,synaptic plasticity and lost memories. Nat. Rev., Neurosci. 3 (6),453–462 (Jun).
Levin, E.D., Castonguay, M., Ellison, G.D., 1987. Effects of thenicotinic receptor blocker mecamylamine on radial-armmaze performance in rats. Behav. Neural Biol. 48,206–212.
McEwen, B.S., 2000. Effects of adverse experiences for brainstructure and function. Biol. Psychiatry 48, 721–731.
McEwen, B.S., Magarinos, A.M., 1997. Stress effects on morphologyand function of the hippocampus. Ann. N. Y. Acad. Sci. 821,271–284.
Mugnaini, M., Tessari, M., Tarter, G., Merlo Pich, E., Chiamulera, C.,Bunnemann, B., 2002. Upregulation of [3H]methyllycaconitinebinding sites following continuous infusion of nicotine,without changes of alpha7 or alpha6 subunit mRNA: anautoradiography and in situ hybridization study in rat brain.Eur. J. Neurosci. 16, 1633–1646.
O'Keefe, J., 1993. Hippocampus, theta, and spatial memory. Curr.Opin. Neurobiol. 3, 917–924.
Park, C.R., Campbell, A.M., Diamond, D.M., 2001. Chronicpsychosocial stress impairs learning and memory andincreases sensitivity to yohimbine in adult rats. Biol. Psychiatry50, 994–1004.
Parker, S.L., Fu, Y., McAllen, K., Luo, J., McIntosh, J.M., Lindstrom,J.M., Sharp, B.M., 2004. Up-regulation of brain nicotinicacetylcholine receptors in the rat during long-term self-administration of nicotine: disproportionate increase of thealpha6 subunit. Mol. Pharmacol. 65, 611–622.
Pauly, J.R., Collins, A.C., 1993. An autoradiographic analysis ofalterations in nicotinic cholinergic receptors following 1 weekof corticosterone supplementation. Neuroendocrinology 57,262–271.
Ridley, D.L., Rogers, A., Wonnacott, S., 2001. Differential effects ofchronic drug treatment on alpha3* and alpha7 nicotinic

84 B R A I N R E S E A R C H 1 0 9 7 ( 2 0 0 6 ) 7 8 – 8 4
receptor binding sites, in hippocampal neurones and SH-SY5Ycells. Br. J. Pharmacol. 133, 1286–1295.
Sahakian, B., Jones, G., Levy, R., Gray, J., Warburton, D., 1989. Theeffects of nicotine on attention, information processing, andshort-term memory in patients with dementia of theAlzheimer type. Br. J. Psychiatry 154, 797–800.
Serova, L., Danailov, E., Chamas, F., Sabban, E.L., 1999. Nicotineinfusion modulates immobilization stress-triggered inductionof gene expression of rat catecholamine biosynthetic enzymes.J. Pharmacol. Exp. Ther. 291, 884–892.
Squire, L.R., 1993. The hippocampus and spatial memory. TrendsNeurosci. 16, 56–57.
Takita, M., Muramatsu, I., 1995. Alteration of brain nicotinicreceptors induced by immobilization stress and nicotine inrats. Brain Res. 681, 190–192.
Takita, M., Taniguchi, T., Zhu, J., Piao, H.L., Tsai, T.Y., Muramatsu,I., 1999. Effects of chronic treatment with (+)-nicotine on the
stress-induced hypertension and downregulation of centralnicotinic receptors in rats: comparative study with (−)-nicotine.Gen. Pharmacol. 33, 29–33.
Warpman, U., Nordberg, A., 1995. Epibatidine and ABT 418 revealselective losses of alpha 4 beta 2 nicotinic receptors inAlzheimer brains. NeuroReport 6, 2419–2423.
White, H.K., Levin, E.D., 1999. Four-week nicotine skin patchtreatment effects on cognitive performance inAlzheimer's disease. Psychopharmacology (Berlin) 143,158–165.
White, H.K., Levin, E.D., 2004. Chronic transdermal nicotine patchtreatment effects on cognitive performance in age-associatedmemory impairment. Psychopharmacology (Berlin) 171,465–471.
Whitehouse, P.J., Au, K.S., 1986. Cholinergic receptors in aging andAlzheimer's disease. Prog. Neuro-psychopharmacol. Biol.Psychiatry 10, 665–676.




















