
Chemisorption of HCl to the MgO(001) surface: A DFT study
9
Chemisorption of HCl to the MgO(001) surface: A DFT studyw Andreas Markmann,* a Jacob L. Gavartin b and Alexander L. Shluger b Received 20th June 2006, Accepted 14th August 2006 First published as an Advance Article on the web 22nd August 2006 DOI: 10.1039/b608719a We use plane wave and embedded cluster ab initio density functional calculations to study adsorption, dissociation and diffusion of the HCl molecule on the MgO(001) surface. The two methods yield comparable results for adsorption of an isolated HCl molecule and complement each other when considering charged species and coverage effects. We find dissociative chemisorption at a coverage smaller than 0.5 monolayer with a Cl ion electrostatically coupled to the OH ion at the surface oxygen site. The adsorption energy of the Cl (OH) complex is 1.5 eV and the activation energy of Cl diffusion away from OH is 0.6 eV. There is no significant activation energy for rotation of Cl around the adsorption site. At rising coverage, an increase in dipole–dipole repulsion between HCl molecules leads to a lowering of the adsorption energy per HCl and a change of binding towards hydrogen-bridge type as well as a lowering of the activation energy for Cl diffusion. OH formed in the surface due to HCl adsorption has a stretch frequency of 3083 cm 1 with Cl associated and 3648 cm 1 with Cl removed. 1. Introduction Understanding the nature of adsorption of polar molecules at oxide surfaces is important both fundamentally and for nu- merous technological applications. Most of the experimental and theoretical studies have been concerned with adsorption of individual water molecules and water layers. Adsorption of diatomic molecules, such as HCl and HBr, on MgO, alumina, ice and other surfaces has also been studied due to various issues pertaining to chemical processes at surfaces of atmo- spheric and interstellar dust particles and geochemistry (see, for example, ref. 1–3), mechanisms of photo-induced surface processes, 4,5 nanochemistry 6 and catalysis. 7–9 Adsorption of HCl on the MgO (100) surface is perhaps one of the simplest examples of such a system. However, even the basic features of HCl adsorption, i.e. whether it is physisorption or chemisorp- tion, whether the molecule remains intact or dissociates, how this depends on coverage, still remain controversial. Recently Wittig et al. 10,11 recorded time-of-flight (TOF) spectra of HCl scattered at the MgO(001) surface and deduced an Arrhenius activation energy for desorption of E des = 0.3 eV. On the other hand, Stark and Klabunde 6 concluded that HCl and HBr molecules chemisorb at MgO nanocluster surfaces. Interest in the adsorption behaviour of the MgO surface was motivated also by its use as an efficient nanocrystalline dehy- drohalogenation catalyst. 7–9 Large surface area and enhanced surface reactivity are responsible for a very high capacity of MgO to destructively chemisorb volatile organic compounds. It was observed that the adsorption rate of HCl varies widely with temperature, 7,8,12 so that chemisorption to special sites has been suggested to be the dominant adsorption mechanism. Adsorption of HCl to another polar surface, g-Al 2 O 3 , yields appearance of a band attributed to hydroxyl (OH ) groups at 3500 cm 1 (104.9 THz). 13,14 At the same time, no band assignable to HCl was detected. This is indicative of dissocia- tion of HCl due to adsorption. In later experimental work 15 OH groups formed at the surface were found to be mainly acidic due to OHCl interaction. A study of HCl adsorption on a-Al 2 O 3 16 yielded observa- tion of a large range of desorption temperatures (300–500 K) as the signature of adsorption at sites of different adsorption energies (e.g. defect sites vs. terrace sites) without the possibility for diffusion along the surface. A constant peak desorption temperature from the Al 2 O 3 surface at different levels of coverage was interpreted to be the signature of associative adsorption but, seemingly contradictively, the presence of OH signals was interpreted as a sign for dissociative adsorp- tion. HCl adsorption on MgO has been studied theoretically by Chacon-Taylor and McCarthy. 17 They used periodic Hartree–Fock calculations with the Colle–Salvetti correlation correction and predicted a relatively weak non-dissociative physisorption of HCl at the oxygen site. They reported an adsorption energy at one quarter monolayer coverage of about 0.48 eV. The predicted distance between H and O is 1.82 A ˚ . Another physisorbed vertical configuration with chlorine bonded to a surface magnesium ion was considered, with an adsorption energy of 0.35 eV. Both the experimental and theoretical data on HCl adsorp- tion at the MgO(001) and alumina surfaces are not conclusive. One can look for clues in other, similar systems. In particular, water adsorption at MgO surfaces is perhaps best studied. However, the character of water adsorption remains contro- versial, too, as recently reviewed in ref. 18. It was also found to a Theoretische Chemie, Technische Universita ¨t Mu ¨nchen, Lichtenbergstraße 4, 85 747 Garching, Germany. E-mail: [email protected] b Department of Physics and Astronomy and London Centre for Nanotechnology, University College London, Gower Street, London, UK WC1E 6BT w The HTML version of this article has been enhanced with colour images. This journal is c the Owner Societies 2006 Phys. Chem. Chem. Phys., 2006, 8, 4359–4367 | 4359 PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Chemisorption of HCl to the MgO(001) surface: A DFT study

Chemisorption of HCl to the MgO(001) surface: A DFT studyw
Andreas Markmann,*aJacob L. Gavartin
band Alexander L. Shluger
b
Received 20th June 2006, Accepted 14th August 2006
First published as an Advance Article on the web 22nd August 2006
DOI: 10.1039/b608719a
We use plane wave and embedded cluster ab initio density functional calculations to study
adsorption, dissociation and diffusion of the HCl molecule on the MgO(001) surface. The two
methods yield comparable results for adsorption of an isolated HCl molecule and complement
each other when considering charged species and coverage effects. We find dissociative
chemisorption at a coverage smaller than 0.5 monolayer with a Cl� ion electrostatically coupled
to the OH� ion at the surface oxygen site. The adsorption energy of the Cl�� � �(OH)� complex is
1.5 eV and the activation energy of Cl� diffusion away from OH� is 0.6 eV. There is no
significant activation energy for rotation of Cl� around the adsorption site. At rising coverage, an
increase in dipole–dipole repulsion between HCl molecules leads to a lowering of the adsorption
energy per HCl and a change of binding towards hydrogen-bridge type as well as a lowering of
the activation energy for Cl� diffusion. OH� formed in the surface due to HCl adsorption has a
stretch frequency of 3083 cm�1 with Cl� associated and 3648 cm�1 with Cl� removed.
1. Introduction
Understanding the nature of adsorption of polar molecules at
oxide surfaces is important both fundamentally and for nu-
merous technological applications. Most of the experimental
and theoretical studies have been concerned with adsorption
of individual water molecules and water layers. Adsorption of
diatomic molecules, such as HCl and HBr, on MgO, alumina,
ice and other surfaces has also been studied due to various
issues pertaining to chemical processes at surfaces of atmo-
spheric and interstellar dust particles and geochemistry (see,
for example, ref. 1–3), mechanisms of photo-induced surface
processes,4,5 nanochemistry6 and catalysis.7–9 Adsorption of
HCl on the MgO (100) surface is perhaps one of the simplest
examples of such a system. However, even the basic features of
HCl adsorption, i.e. whether it is physisorption or chemisorp-
tion, whether the molecule remains intact or dissociates, how
this depends on coverage, still remain controversial.
Recently Wittig et al.10,11 recorded time-of-flight (TOF)
spectra of HCl scattered at the MgO(001) surface and deduced
an Arrhenius activation energy for desorption of Edes =
0.3 eV. On the other hand, Stark and Klabunde6 concluded
that HCl and HBr molecules chemisorb at MgO nanocluster
surfaces.
Interest in the adsorption behaviour of the MgO surface was
motivated also by its use as an efficient nanocrystalline dehy-
drohalogenation catalyst.7–9 Large surface area and enhanced
surface reactivity are responsible for a very high capacity of
MgO to destructively chemisorb volatile organic compounds.
It was observed that the adsorption rate of HCl varies widely
with temperature,7,8,12 so that chemisorption to special sites
has been suggested to be the dominant adsorption mechanism.
Adsorption of HCl to another polar surface, g-Al2O3, yields
appearance of a band attributed to hydroxyl (OH�) groups at
3500 cm�1 (104.9 THz).13,14 At the same time, no band
assignable to HCl was detected. This is indicative of dissocia-
tion of HCl due to adsorption. In later experimental work15
OH� groups formed at the surface were found to be mainly
acidic due to OH� � �Cl interaction.A study of HCl adsorption on a-Al2O3
16 yielded observa-
tion of a large range of desorption temperatures (300–500 K)
as the signature of adsorption at sites of different adsorption
energies (e.g. defect sites vs. terrace sites) without the possibility
for diffusion along the surface. A constant peak desorption
temperature from the Al2O3 surface at different levels of
coverage was interpreted to be the signature of associative
adsorption but, seemingly contradictively, the presence of
OH� signals was interpreted as a sign for dissociative adsorp-
tion.
HCl adsorption on MgO has been studied theoretically
by Chacon-Taylor and McCarthy.17 They used periodic
Hartree–Fock calculations with the Colle–Salvetti correlation
correction and predicted a relatively weak non-dissociative
physisorption of HCl at the oxygen site. They reported an
adsorption energy at one quarter monolayer coverage of about
0.48 eV. The predicted distance between H and O is 1.82 A.
Another physisorbed vertical configuration with chlorine
bonded to a surface magnesium ion was considered, with an
adsorption energy of 0.35 eV.
Both the experimental and theoretical data on HCl adsorp-
tion at the MgO(001) and alumina surfaces are not conclusive.
One can look for clues in other, similar systems. In particular,
water adsorption at MgO surfaces is perhaps best studied.
However, the character of water adsorption remains contro-
versial, too, as recently reviewed in ref. 18. It was also found to
a Theoretische Chemie, Technische Universitat Munchen,Lichtenbergstraße 4, 85 747 Garching, Germany.E-mail: [email protected]
bDepartment of Physics and Astronomy and London Centre forNanotechnology, University College London, Gower Street, London,UK WC1E 6BT
w The HTML version of this article has been enhanced with colourimages.
This journal is �c the Owner Societies 2006 Phys. Chem. Chem. Phys., 2006, 8, 4359–4367 | 4359
PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics

be defect related,19–23 although adsorption to a clean surface
terrace was observed at elevated vapour pressures.21,22 Theo-
retical calculations at low coverage predict weak non-disso-
ciative physisorption24–27 or even negative adsorption
energy.28,29 In contrast, both H+ and OH� ions adsorb readily
to the defect-free terraces.
Another polar molecule whose adsorption to the MgO(001)
surface has been studied theoretically is formic acid
(HCOOH). A second-order Møller–Plesset (MP2) calculation
of the interaction of formic acid with a quadratic cluster of 4 �4 MgO atoms performed by Nakatsuji et al.30 predicts a
minimum energy configuration in which the proton is disso-
ciated from the molecule and bound to a surface oxygen ion.
The adsorption energy predicted is about 2.2 eV. These cluster
calculations did not include the surface Madelung potential.
Recent, more accurate embedded cluster calculations31 predict
dissociative adsorption and formate ion formation at the
MgO(001) terrace. A plane wave density functional calcula-
tion of this system by Szymanski and Gillan32 demonstrated
that dissociative adsorption is more favorable than molecular
adsorption by about 0.3 eV. Experimentally, the dissociative
adsorption of formate on MgO(001) has been demonstrated
e.g. in ref. 33.
These results indicate that the character of adsorption can
depend on coverage and on the particular surface site of
adsorption, i.e. terrace, step, kink or a point defect, such as
a vacancy. In this paper, we study the mechanism of HCl
adsorption at the perfect MgO(001) surface terrace. Our
periodic plane wave and embedded cluster density functional
theory (DFT) simulations predict dissociative adsorption of
HCl with OH� formation due to the heterolytic splitting of
HCl at the surface.
The models used in this paper are described in section 2.
Results are presented in section 3 and discussed in section 4.
2. Models
To study the geometric and electronic structures of individual
HCl molecules and monolayers corresponding to different
surface coverage on the (001) MgO surface we employed two
methods: (i) periodic plane wave density functional theory
(PW-DFT) at the generalised gradient approximation (GGA)
level; and ii) the embedded cluster method (EC-DFT) with
atomic basis sets and the B3LYP hybrid density functional.
Both approaches have their advantages and limitations and
largely complement each other, as discussed in e.g. ref. 34.
PW-DFT calculations were performed in two rectangular
supercells containing four (001) MgO planes with surface
dimension of a0(4 � 4) and a0(6 � 6) (a0 = 2.11 A being the
nearest-neighbour Mg–O distance in the bulk crystal). With a
single HCl molecule in the supercell, this corresponds to a
coverage of 1/8 and 1/18 of a monolayer, respectively. The
vacuum gap between the slabs was set to twice the slab
thickness, which is sufficient to diminish interaction between
periodic images of the slab and leave sufficient space for a
desorbed molecule. The two layers of MgO not facing the
admolecule were fixed to the bulk structure, while the two
other layers were free to relax. We have used the VASP
package35,36 utilising PW-DFT with the PW’91 GGA density
functional by Perdew and Wang and ultrasoft pseudopoten-
tials of Vanderbilt type,37 as supplied by Kresse and Hafner.38
We used an energy cutoff of 396 eV and the G-point only in
the reduced Brillouin zone. The energy was converged to
an accuracy of 0.1 eV and the forces to an accuracy of
0.05 eV A�1.
In order to estimate the adsorption energy of a single HCl
molecule (zero coverage), the energies obtained in PW-DFT
calculations were corrected for a spurious dipole–dipole inter-
action between supercells. The correction energy was calcu-
lated using effective point charges and an algorithm due to
Kantorovitch39 with the effective charges –1.426 e at the
adsorption site oxygen, 0.426 e at hydrogen as parameterised
for a free OH� molecule by Saul and Catlow,40 where e
denotes the modulus of the electron charge. Formal ionic
charges were used for Cl, Mg, and all other O ions.
PW-DFT calculations as just described have two principal
limitations:
(1) The spurious electrostatic interaction arising from the
periodic boundary conditions is significant, especially in polar
systems such as those considered here. In extreme cases this
may lead to incorrect results even with the corrections just
discussed;34,41
(2) The self-interaction error inherent in all local and semi-
local functionals (including PW’91) may lead to qualitatively
incorrect predictions in systems where electronic interaction
leads to the formation of localised states.41–44
To quantify these shortcomings of the PW-DFT approach,
we also used the embedded cluster method (EC-DFT) de-
scribed in detail elsewhere.34,45–47 The setup is illustrated in
Fig. 1a. A quantum cluster (QC) is embedded in a finite array
of point charges located near the MgO lattice sites. The part of
the ions closest to the QC (region I) is treated within the shell
model. Ions of region I interact with each other and with the
atoms in the QC via shell model potentials of Stoneham and
Sangster.48 The positions of cores and shells of classical ions in
region I are optimised self-consistently with the ionic positions
and charge density in the QC. The ions outside region I
constitute region II and are represented by non-polarisable
point charges fixed at their lattice sites and serve to provide a
correct electrostatic potential on all ions in region I. The entire
simulated system measures 19 by 19 ions along the surface and
7 ions perpendicular to the surface (i.e. about 40 � 40 � 15
A3). In order to avoid artificial polarisation of the oxygen ions
in the QC,34,47 the boundary of the quantum cluster Mg29O13
(Fig. 1b and c) is formed by effective core potential (ECP)
Mg2+ cations.49 The standard basis sets 6–311G* for O, Cl
and H and 6–31G for Mg ions were employed. These basis sets
have been validated previously in studying the electronic
properties of defects at the MgO(001) surface. The electronic
structure and geometry of the QC was calculated using the
hybrid B3LYP density functional.50,51 The self-consistent
embedding procedure as described in this section has been
implemented in the computer program GUESS,46 which in-
corporates GAUSSIAN9852 for the electronic structure calcu-
lations.
A study of defects in an oxide surface comparing PW-DFT
and cluster results has been performed previously in ref. 53 but
there, pure cluster calculations (with no embedding) were used
4360 | Phys. Chem. Chem. Phys., 2006, 8, 4359–4367 This journal is �c the Owner Societies 2006

and the PW-DFT method was parameterised according to the
cluster results. In contrast, we use EC-DFT, which models
the surface more accurately than a pure cluster and compare
the results of PW-DFT and EC-DFT without fitting one to the
other.
3. Results
We start the discussion of our results from a brief description
of surface structures obtained using both methods. As evi-
denced by experiments, the surface does not reconstruct, so
anions and cations must retain the perfect crystal structure
parallel to the surface (x and y directions) but normal to the
surface (in the z direction), the symmetry is broken, so that
anions and cations can have altered z-coordinates near the
surface. The resulting surface relaxation and rumpling can be
expressed in percentages of the lattice constant. The surface
relaxation is the change dd12 of the spacing d12 between the
surface and subsurface layers. It is given as a percentage of the
perfect lattice spacing (half of the lattice constant) d
dd12 ¼d12 � d
d100%: ð1Þ
The surface rumpling D1 is defined as the difference between
the z-coordinates of anions and cations, again as percentage of
the ideal lattice spacing
D1 ¼zanion1 � zcation1
d100%: ð2Þ
The calculated surface relaxation and rumpling parameters for
MgO are +0.81% and+3.6% in PW-DFT, and+0.05% and
+2.9% in EC-DFT. Relaxation and rumpling in the EC-DFT
model are less reliable due to the constraint caused by the
unrelaxed ions of region II and the relatively small basis set on
the oxygen ions. Additionally, the surface relaxation para-
meter is very sensitive to the choice of d (eqn (1)), which is
difficult to pinpoint in EC-DFT as this method features no
periodicity. Given this limitation, the obtained rumpling para-
meters satisfactorily reproduce earlier LDA calculations and
LEED experiments on vacuum cleaved samples (see ref. 54 for
a review).
In order to identify the stable adsorption configurations of
the HCl molecule on the (001) MgO terrace, we examined a
number of possible conformations including HCl bonding to a
surface oxygen site, surface Mg site, or adsorbed over a plane
interstitial site. However, our calculations predict stable ad-
sorption only at a surface oxygen site with a formation of OH�
and Cl� ions in close proximity. Several configurations (ab-
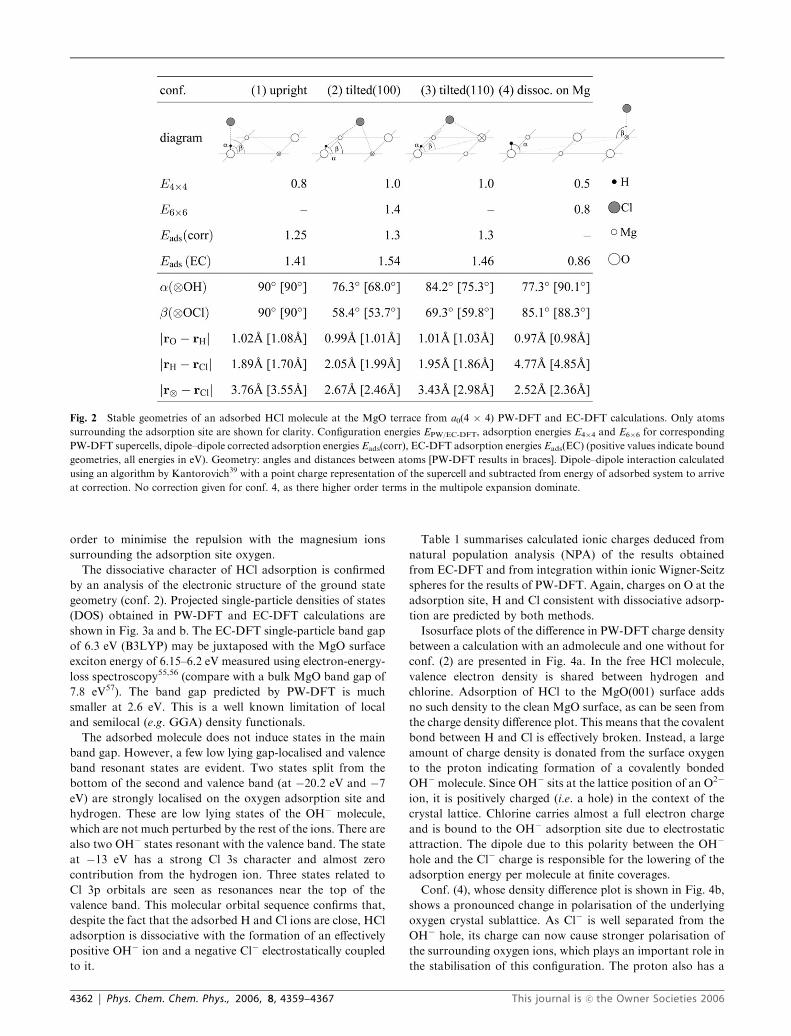
breviated ‘‘conf.’’) are found to be stable or metastable and
will be discussed in the following: The symmetric, upright HCl
molecule over the adsorption site oxygen (conf. (1)) is meta-
stable and relaxes by symmetry breaking into conf. (2),
corresponding to an H–Cl axis tilted in the (100) direction
(towards a neighbouring magnesium ion) or into conf. (3)
where the H–Cl axis is tilted in the (110) direction (towards
plane interstitial position). Conf. (4) corresponds to HCl
heterolytically dissociated at the crystal surface with the pro-
ton sitting on top of an oxygen and the chlorine on top of a
next-nearest neighbour magnesium. Calculated configura-
tional parameters and corresponding adsorption energies of
these states are summarised in Fig. 2. PW-DFT energies are
shown for the HCl coverage of 1/8 and 1/18 monolayer,
respectively, as E4�4 and E6�6. EC-DFT energies are denoted
Eads(EC). We define the adsorption energy of a molecule at a
surface as the sum of the calculated energies of a free molecule
and a clean surface minus the calculated energy of the
adsorbed molecule. Thus, a bound system has positive adsorp-
tion energy.
Most PW-DFT calculations were performed with the
a0(4 � 4) unit cell, i.e. at 1/8 monolayer coverage due to com-
putational constraints. If not specified, this unit cell is implied
for all PW-DFT results presented below. The geometries
predicted by PW-DFT and EC-DFT agree very well, i.e. the
PW-DFT unit cells are large enough to approximate an
isolated molecule and the dipole–dipole correction yields a
good extrapolation to its energy. A significant improvement is
seen in the larger a0(6� 6) unit cell only for the subtle response
of the surface ions around the adsorption site to adsorption of
HCl. The distance between the proton and chlorine in the
associated adsorption geometries (1)–(3) is, at between 1.70
and 2.05 A, considerably larger than the 1.3 A in a free
molecule. The distance between the proton and the surface
oxygen at the adsorption site is 1.0 A, i.e. similar to the 0.96 A
in a free OH� molecule. The small distances of the proton to
the surface oxygen and the large magnitude of the adsorption
energies of above 1 eV suggest dissociative chemisorption
rather than physisorption as the dominating adsorption me-
chanism. The proton always points away from the surface in
Fig. 1 Setup of HCl on MgO(001) embedded cluster calculations. An Mg29O13 cluster was used. (a) Embedding of the quantum cluster into a
lattice of shell model ions, (b) cross-section through the quantum cluster, (c) three-dimensional view of the quantum cluster. The ECP magnesium
ions surround the cluster oxygen ions at the interface.
This journal is �c the Owner Societies 2006 Phys. Chem. Chem. Phys., 2006, 8, 4359–4367 | 4361
order to minimise the repulsion with the magnesium ions
surrounding the adsorption site oxygen.
The dissociative character of HCl adsorption is confirmed
by an analysis of the electronic structure of the ground state
geometry (conf. 2). Projected single-particle densities of states
(DOS) obtained in PW-DFT and EC-DFT calculations are
shown in Fig. 3a and b. The EC-DFT single-particle band gap
of 6.3 eV (B3LYP) may be juxtaposed with the MgO surface
exciton energy of 6.15–6.2 eV measured using electron-energy-
loss spectroscopy55,56 (compare with a bulk MgO band gap of
7.8 eV57). The band gap predicted by PW-DFT is much
smaller at 2.6 eV. This is a well known limitation of local
and semilocal (e.g. GGA) density functionals.
The adsorbed molecule does not induce states in the main
band gap. However, a few low lying gap-localised and valence
band resonant states are evident. Two states split from the
bottom of the second and valence band (at �20.2 eV and �7eV) are strongly localised on the oxygen adsorption site and
hydrogen. These are low lying states of the OH� molecule,
which are not much perturbed by the rest of the ions. There are
also two OH� states resonant with the valence band. The state
at �13 eV has a strong Cl 3s character and almost zero
contribution from the hydrogen ion. Three states related to
Cl 3p orbitals are seen as resonances near the top of the
valence band. This molecular orbital sequence confirms that,
despite the fact that the adsorbed H and Cl ions are close, HCl
adsorption is dissociative with the formation of an effectively
positive OH� ion and a negative Cl� electrostatically coupled
to it.
Table 1 summarises calculated ionic charges deduced from
natural population analysis (NPA) of the results obtained
from EC-DFT and from integration within ionic Wigner-Seitz
spheres for the results of PW-DFT. Again, charges on O at the
adsorption site, H and Cl consistent with dissociative adsorp-
tion are predicted by both methods.
Isosurface plots of the difference in PW-DFT charge density
between a calculation with an admolecule and one without for
conf. (2) are presented in Fig. 4a. In the free HCl molecule,
valence electron density is shared between hydrogen and
chlorine. Adsorption of HCl to the MgO(001) surface adds
no such density to the clean MgO surface, as can be seen from
the charge density difference plot. This means that the covalent
bond between H and Cl is effectively broken. Instead, a large
amount of charge density is donated from the surface oxygen
to the proton indicating formation of a covalently bonded
OH�molecule. Since OH� sits at the lattice position of an O2�
ion, it is positively charged (i.e. a hole) in the context of the
crystal lattice. Chlorine carries almost a full electron charge
and is bound to the OH� adsorption site due to electrostatic
attraction. The dipole due to this polarity between the OH�
hole and the Cl� charge is responsible for the lowering of the
adsorption energy per molecule at finite coverages.
Conf. (4), whose density difference plot is shown in Fig. 4b,
shows a pronounced change in polarisation of the underlying
oxygen crystal sublattice. As Cl� is well separated from the
OH� hole, its charge can now cause stronger polarisation of
the surrounding oxygen ions, which plays an important role in
the stabilisation of this configuration. The proton also has a
Fig. 2 Stable geometries of an adsorbed HCl molecule at the MgO terrace from a0(4 � 4) PW-DFT and EC-DFT calculations. Only atoms
surrounding the adsorption site are shown for clarity. Configuration energies EPW/EC-DFT, adsorption energies E4�4 and E6�6 for corresponding
PW-DFT supercells, dipole–dipole corrected adsorption energies Eads(corr), EC-DFT adsorption energies Eads(EC) (positive values indicate bound
geometries, all energies in eV). Geometry: angles and distances between atoms [PW-DFT results in braces]. Dipole–dipole interaction calculated
using an algorithm by Kantorovich39 with a point charge representation of the supercell and subtracted from energy of adsorbed system to arrive
at correction. No correction given for conf. 4, as there higher order terms in the multipole expansion dominate.
4362 | Phys. Chem. Chem. Phys., 2006, 8, 4359–4367 This journal is �c the Owner Societies 2006

more pronounced impact on the polarisation of its partner
oxygen. The latter is no longer polarised laterally by chlorine,
as the Cl� charge is effectively screened by the chlorine
neighbour polarisation.
Having established the structure of the stable states, we
proceed with a discussion of the activation barriers. The
barrier between configurations 2 and 3 gives an estimate for
the rotational barrier of the Cl� ion around the adsorption
site. It is very small, ato0.05 eV in both EC and PW-DFT, so
Cl� can be considered as freely rotating in a horizontal plane
around the OH bond. We also calculated the barriers for the
Cl� diffusion away from the OH� site. A direct transition
between conf. (3) and conf. (4) is not viable due to the strong
electrostatic repulsion between Cl� and the surface oxygen
O2�. The migration path via conf. (2) is found to be more
favourable energetically. A calculation with Cl� between
nearest and next-nearest neighbour Mg2+ ions (i.e. between
configurations 2 and 4) yields an energy less than 0.1 eV above
the energy of conf. (4). This corresponds to a barrier of 0.65 eV
(PW-DFT) and 0.78 eV (EC-DFT) for Cl� diffusion away
from the OH� site, and o0.1 eV for diffusion towards the
OH� site. The calculated dissociation energy of the HCl
molecule into H+ and Cl� at the MgO surface is 1.8 eV
(EC-DFT). The dissociated system comprises a proton, with
an adsorption energy of 12.3 eV and a Cl� ion with an
adsorption energy of 1.7 eV. Charged configurations were
not simulated with the PW-DFT method due to known
problems with compensation by charged backgrounds. In
contrast, the adsorption energies for H0 and Cl0 are 0.3 and
1.5 eV, respectively from both the EC and PW-DFT methods.
A diagram summarising all the processes discussed in the
previous paragraphs and their energies is shown in Fig. 5. It
can be checked that a process leading back to the starting
configuration in the diagram always results in a total transi-
tion energy of zero (reversing energies when proceeding
against the direction of an arrow). This diagram can be used
to assess processes not discussed so far. For example, removal
of chlorine as a charged ion from the surface with an asso-
ciated molecule costs 1.8 eV to dissociate OH� and Cl� at the
surface plus 1.7 eV to remove the isolated Cl� from the
surface, i.e. 3.5 eV in total. In comparison, removing Cl0 from
the adsorbed molecule as the neutral atom costs 3.5 eV alone
for dissociation at the surface plus 1.5 eV for desorption of
Cl0. This means that removal of Cl� as an anion is preferred
over the removal of Cl0 as a neutral atom and the potential
energy curves for removal of chlorine for the neutral and
anionic charge states do not cross. This is in stark contrast to
most molecular systems, for example the free HCl molecule,
where neutral separation (at 3.9 eV) is favoured over charged
separation (14.4 eV). However, both dissociation processes at
the surface are not very fast at room temperature due to their
high activation energy.
Next we discuss the effect of HCl surface coverage on the
adsorption energy. To this end, we have performed PW-DFT
calculations of up to 8 HCl molecules in the a0(4 � 4) surface
unit cell, and one molecule in the a0(6 � 6) unit cell. As the
molecule only binds to the oxygen sites, this corresponds to
coverages in the interval from 1/18th monolayer (5.6%) up to
a full monolayer of HCl molecules. Calculations predict
adsorption energies in configuration 2 (Fig. 3) to be 0.25,
0.83, 1.0 and 1.4 eV for 1/2, 1/4 , 1/8 and 1/18 monolayer,
respectively.
A coverage of 1/4 monolayer corresponds to that modelled
by Chacon-Taylor and McCarthy.17 Our value of 0.83 eV is
substantially larger than the 0.48 eV in ref. 17. However, for
1/4 monolayer coverage in conf. (2), the average OH� bond
length elongates by 0.1 A to 1.08 A and the average H–Cl
distance contracts by 0.3 A to 1.75 A. This indicates that HCl
adsorption is more like a hydrogen-bridge bond for high
coverages. Together with the tendency of Hartree–Fock to
underestimate binding energies, this explains that a different
geometry and physisorption were predicted in ref. 17.
Fig. 3 Electronic densities of state of the surface–molecule system in
conf. (2). (a) Projected densities of state (DOS) from PW-DFT. (b)
DOS from EC-DFT.
Table 1 Comparison of charges obtained from PW-DFT and EC-DFT for the tilted geometry, configuration (2). Charges are given in multiplesof the positive unit charge e. It can be seen that the spherical integration charges in the first row always deviate towards positive charge comparedto the NPA charges. This supports the NPA charges, as some (negatively charged) electron density is neglected for spherical integration
Method of calculation (population analysis) Cl H O in OH� Other O Mg
PW-DFT (spherical integration) �0.9 0.5 �1.4 �1.7 1.8EC-DFT (Mulliken analysis) �0.73 0.40 �1.10 �1.55 1.31EC-DFT (NPA) �0.85 0.47 �1.42 �1.79 1.71
This journal is �c the Owner Societies 2006 Phys. Chem. Chem. Phys., 2006, 8, 4359–4367 | 4363

This tendency can also be seen in the binding energy of the
OH� molecule, which is 2.9 eV with Hartree–Fock and 4.4 eV
with B3LYP. The B3LYP result is much closer to the value of
4.9 eV we calculated with Multi-Reference Configuration
Interaction (MRCI), the most accurate method we have used
for this molecule (all done with basis sets at the triple zeta
level).
Our results show that the coverage dependence of the
adsorption energy is dominated by the dipole–dipole repulsion
between the molecules which almost completely can be ac-
counted for by Ewald summation over the molecule effective
charges in the lateral directions. This is demonstrated in Fig. 6,
where we have plotted the difference between the adsorption
energy calculated with EC-DFT (1.54 eV, conf. 2) and
PW-DFT adsorption energies per molecule at different cov-
erages. This line is matched (taking into account error bars for
the crude point charge model) by the dipole–dipole repulsion
energy.
Coverage of every surface oxygen with an HCl molecule
(full monolayer) results in an unstable system, i.e. upon
relaxation the molecules migrate into the vacuum. However,
an additional HCl molecule can be adsorbed on top of a half
monolayer of HCl with an adsorption energy of 0.3 eV. The
additional HCl molecule is oriented with its positive (i.e.
hydrogen) end towards the underlying Cl� ion. As the lower-
most proton is bonded chemically to a surface oxygen and the
first and second Cl� layer are chemically equivalent, the
Fig. 4 Isosurface plots of the difference in charge density between
calculations of a surface with an admolecule and without. Surfaces
shown match a charge density difference value of rcutoff = 0.56 e A�3.
(a) Adsorption of an associated molecule, configuration (2). The
charge density change on the far oxygen ion is due to the PBC. (b)
Adsorption of a dissociated molecule, configuration (4) [Cl sits over
the magnesium ion (210) from the proton adsorption site]. The
apparent cutoff of the density on the leftmost O ion is due to a
limitation of the plot area. More oxygen ions are polarised in (b) which
partially accounts for the discrepancy between the differences in H–Cl
electrostatic energy and PW-DFT energy.
Fig. 5 Adsorption and dissociation energies of HCl. Negative energies stand for energy gained and positive ones for energy to spend. Left side:
dissociation into charged species. Right side: dissociation into neutral species.
Fig. 6 This comparison shows that the dipole–dipole interaction
energy is the dominant contribution in the coverage dependence of
the adsorption energy per admolecule. Conf. (2), dipole–dipole en-
ergies evaluated with a rigid ion model of the unit cell (hence the large
error bars). The energy difference is Eads(EC-DFT) � Eads(PW-DFT),
where Eads(EC-DFT) = 1.5 eV.
4364 | Phys. Chem. Chem. Phys., 2006, 8, 4359–4367 This journal is �c the Owner Societies 2006

proton in the middle mediates a hydrogen bond between the
first and second admolecule layer.
We have used the EC-DFT method to calculate vibrational
frequencies of some localised vibrational modes. Table 2
shows an overview of mode frequencies found for OH� in
conf. (2) and in the protonated surface, i.e. without the
associated chlorine ion. It can be seen that the non-transla-
tional modes have frequencies which are well-separated from
the top of the MgO surface slab vibrational band (22 THz in
shell model lattice dynamics58). This means they are unlikely
to couple strongly to the normal modes of the MgO substrate.
The electrostatic attraction of the chlorine ion softens the
translational and stretch modes of OH�, while it hardens the
librational modes with respect to the proton without asso-
ciated Cl�. The changes in the intramolecular OH� frequen-
cies are due mostly to the slight elongation of the OH� bond
which makes OH� reside in the sloping part of its (approxi-
mately Morse-type) potential.
In experimental adsorption of HCl to MgO nanocrystals6 at
high pressure, broad bands from 3000 to 3700 cm�1 were
measured. The broad bandwidth measured indicates a wide
range of OH� intramolecular distances present in the sample
due to dipole–dipole repulsion and chlorine diffusion.
Chlorine readily diffuses away from the adsorption site
(most likely to surface defects) at high coverage: a Cl� ion
can rotate freely around its associated OH� but it can also
rotate freely around an OH� ion at the neighbouring oxygen
lattice position as one is likely to be available at the high
coverages in these experiments, which can remove chlorine
from its adsorption site. Hence, the activation energy for Cl�
diffusion goes from 0.6 eV for an isolated molecule to below
0.05 eV for high coverage.
A band at 3750 cm�1 was present prior to HCl adsorption
due to surface contamination with OH�.6 Note that these
OH� species are not embedded in the surface like the one
formed due to HCl adsorption at the terrace. Bands at 1600
and 1700 cm�1 appeared on HCl adsorption which are in the
region of the water bending mode (1400–1600 cm�1, depend-
ing on isotopes).59 These are likely due to hydrochloric acid
protonating the contaminating OH� species already present at
the surface.
4. Discussion
The results presented above clearly demonstrate that at low
coverage the HCl molecule adsorbs dissociatively on the MgO
(001) terrace. Our DFT calculations predict adsorption of HCl
to the MgO(001) surface at low coverage to be caused pri-
marily by a chemical bond between a surface oxygen and the
molecular proton. The adsorption energy in the most stable
configurations, (2) and (3), is 1.5 eV. In unison with the
breaking of the covalent bond between H and Cl, their
distance increases to between 1.7 and 2.0 A, compared to 1.3
A in the free molecule. Chlorine is held at the adsorption site
by its electrostatic attraction to the OH� molecule, which
represents a hole in the context of the crystal. Hence HCl
at the surface is chemically dissociated but electrostatically
associated.
EC-DFT simulations of HCl adsorbed to the MgO(001)
surface confirmed and extended our PW-DFT results. The fact
that both methods using different basis sets and density
functionals predict the dissociative character of the adsorption
further supports our conclusions.
In comparison to molecular hydrogen, which adsorbs to
MgO dissociatively only at low-coordinated special sites or
vacancies60–63 and to water, which does so at steps, vacancies
or corners of nanocrystals,19–23 HCl adsorbs dissociatively at
the MgO(001) surface terrace. In this respect, HCl represents a
limiting case. Out of the HX compounds, where X is a halogen
ion, dissociative adsorption to the MgO(001) terrace may be
hindered only for HF, which has a dissociation energy of
almost 6 eV. For all other hydrohalogenic acids, dissociative
adsorption is to be expected.
The adsorption energy of HCl may be viewed as consisting
of the HCl vacuum dissociation energy, the protonation
energy and the adsorption energy of Cl� to the proton
adsorption site (Fig. 5). The accuracy of the surface protona-
tion component determines the accuracy of the total adsorp-
tion energy. It has been determined previously by embedded
cluster Hartree–Fock (HF) calculations64 at 10.4 eV and by
flame-ion mass spectrometry experiments65 at around 10 eV.
The difference between our EC-DFT result of 12.3 eV and HF
can be attributed to the tendency of Hartree–Fock to under-
estimate binding energies and of GGA DFT to overestimate
them slightly. The mixed B3LYP functional, however, is fairly
reliable, as demonstrated for the OH� molecule in section 3.
The same tendencies of the methods, together with lowering
of the adsorption energy due to coverage effects is responsible
for the previous prediction of physisorption of HCl on
MgO(001).17
The adsorption energy of 1.5 eV predicted by our results is
significantly higher than that estimated in molecular scattering
experiments. Wittig et al.10 derived an activation energy of
0.3 eV by applying the Arrhenius equation to the measured
desorption rate. As we have demonstrated that the adsorption
energy per HCl molecule decreases with coverage, it may be
supposed that the discrepancy to our predicted adsorption
energy was due to coverage. However, the experiment gives
Table 2 Overview of OH� mode frequencies from EC-DFT calculations. The electrostatic attraction to the associated chlorine ion softens theOH� translational and stretch modes and hardens the librational mode
OH� in HCl on MgO terrace OH� in protonated terrace
Mode Wavenumbers/cm�1 Mode Wavenumbers/cm�1
Translational modes 312–394 Translational modes 235–290Libration 1007 Libration 835Rotation 1021 Libration 835Stretch 3083 Stretch 3648
This journal is �c the Owner Societies 2006 Phys. Chem. Chem. Phys., 2006, 8, 4359–4367 | 4365

repeatable results for many molecular pulses,66 which indicates
that the surface coverage remains low throughout the experi-
ment. Due to the ratio of HCl in the molecular beam (1–2%)
the surface coverage is estimated to be of the order of 10�3
monolayers HCl.66 Coverage can therefore be excluded as a
reason for the discrepancy.
Instead we propose a dynamical mechanism: OH� at the
surface has vibrational and librational frequencies well sepa-
rated from the bulk phonon density of states of MgO. We
argue that such spectrally and spatially localised modes are
characterised by a low dissipation rate, and thus can retain
vibrational excitations for a considerably long time. The
smallest incident kinetic energy in the experiments of Wittig
et al. corresponds to a velocity of 763 m s�1. At an incidence
angle of 151, this corresponds to an approach velocity per-
pendicular to the surface of 737 m s�1. Assuming an interac-
tion region of 5 A above the surface (a generous estimate, as
the Madelung potential decays exponentially with distance),
the molecule has about 1.35 ps to reorient, dissipate its excess
energy and get trapped. If this succeeds, a chemical bond
between the proton and a surface oxygen can be formed. The
dissipated energy (adsorption energy + translational kinetic
energy of the molecule) is likely to be initially transformed into
a vibrational excitation of the OH� stretching mode. The
vibrational frequencies of OH� libration and vibration
(30–92 THz) are well above the maximum crystal frequency
(omax(MgO) = 22 THz). Therefore, excitation of these modes
may survive for many pico- or even nanoseconds due to the
spectral localisation of these modes. However, the activation
energy for desorption of a ‘hot’ molecule is the difference
between the vacuum level and the stored vibrational energy.
This can significantly reduce the measured desorption energy
in scattering experiments, assuming that mostly desorption of
‘hot’ molecules is observed. Accounting for the zero vibration
energy of the OH� stretch mode lowers the desorption barrier
by approximately 0.2 eV ( 1.4 eV � (0.38 eV/2)E 1.2 eV). The
desorption energy involving OH� in its first vibrational excited
state would lower this energy by a further 0.3 eV, and so on.
Thermalised rotational distributions at all incident energies
were previously interpreted as a sign of trapping–desorption.10
However, molecular dynamics67 predict that the collision of
randomly oriented diatomic molecules with a surface should
lead to such a rotational distribution naturally. The depen-
dence of the characteristic rotational temperature on the
temperature of the crystal can be ascribed to events where
molecules collide with oncoming surface atoms. Overall, this
demonstrates that the experimental observations in ref. 10, 11,
13–15 are not contradicting the theoretical predictions pre-
sented in this article. At special adsorption sites, such as steps
and kinks, the frequency of the stretch mode of OH� should
be increased due to a stronger chemical bond. This means that
the OH� stretch mode is spectrally separated further from the
MgO band, maintaining weak coupling. Hence, the qualitative
features of the dynamics of HCl adsorbed on a step or kink
oxygen should be similar to adsorption at the terrace.
Low-kinetic energy, low coverage trapping–desorption ex-
periments involving H35Cl and D37Cl may illuminate whether
HCl molecules dissociate and recombine (in mixed combina-
tions) at the surface prior to desorption. This would yield an
unambiguous answer to the question whether molecules dis-
sociate and whether chlorine can diffuse at the surface at long
time scales.
Acknowledgements
The authors are indebted to P. V. Sushko for his support
running GUESS and to L. Kantorovich for aiding the calcula-
tion of dipole–dipole interaction energies. Both colleagues
have provided useful further advice throughout the project.
W. Eisfeld has lent support for the MRCI calculation of the
OH� molecule. A. Markmann would like to thank the Physics
and Astronomy Department of University College London for
a studentship and the Delbrucksche Familienstiftung for
further financial support. J. L. Gavartin would like to thank
the Leverhulme trust for funding.
References
1 A. D. Pradhan, K. P. Kirby and A. Dalgarno, J. Chem. Phys.,1991, 95, 9009.
2 C. Toubin, S. Picaud and C. Girardet, Chem. Phys., 1999, 244, 227.3 J. B. Fegley, in Treatise on Geochemistry, ed. Andrew M. Davisand Heinrich D. Holland, Elsevier, Amsterdam, 2003, vol. 1,pp. 487–507.
4 T. Seideman and H. Guo, J. Chem. Phys., 1995, 103, 2745.5 M. Hintenender, F. Rebentrost, R. B. Gerber and R. Kosloff,J. Chem. Phys., 1995, 102, 578.
6 J. V. Stark and K. J. Klabunde, Chem. Mater., 1996, 8, 1913.7 U. Rappold and G. Luft, Chem. Eng. Technol., 1999, 22, 843.8 M. Li and H. Shaw, Ind. Eng. Chem. Res., 2000, 39, 1898.9 I. V. Mishakov, A. F. Bedilo, R. M. Richards, V. V. Chesnokov,A. M. Volodin, V. I. Zaikovskii, R. A. Buyanov and K. J.Klabunde, J. Catal., 2002, 206, 40.
10 M. Korolik, D. W. Arnold, M. J. Johnson, M. M. Suchan, H.Reisler and C. Wittig, Chem. Phys. Lett., 1998, 284, 164.
11 M. Korolik, M. M. Suchan, M. J. Johnson, D. W. Arnold, H.Reisler and C. Wittig, Chem. Phys. Lett., 2000, 326, 11.
12 Y. Kajiwara, N. Yoshida and S. Kishimoto, J. Colloid InterfaceSci., 1987, 118, 297.
13 J. B. Peri, J. Phys. Chem., 1965, 70, 1482.14 M. Tanaka and S. Ogasawara, J. Catal., 1970, 16, 157.15 A. Kytokivi and M. Lindblad, J. Chem. Soc., Faraday Trans.,
1995, 91, 941.16 J. W. Elam, C. E. Nelson, M. A. Tolbert and S. M. George, Surf.
Sci., 2000, 450, 64.17 M. R. Chacon-Taylor and M. I. McCarthy, J. Phys. Chem., 1996,
100, 7610.18 M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1.19 W. Langel and M. Parrinello, Phys. Rev. Lett., 1994, 73, 504.20 J. L. Anchell and A. C. Hess, J. Phys. Chem., 1996, 100, 18317.21 P. Liu, T. Kendelewicz, J. Gordon, E. Brown and G. A. Parks,
Surf. Sci., 1998, 412/413, 287.22 P. Liu, T. Kendelewicz and J. Gordon E. Brown, Surf. Sci., 1998,
412/413, 315.23 F. Finocchi and J. Goniakowski, Phys. Rev. B, 2001, 64, 125426.24 D. Ferry, C. Girardet and J. Suzanne, Surf. Sci., 1998, 409, 101.25 S. Joyce and B. Kay, J. Chem. Phys., 1996, 105, 1295.26 M. A. Johnson, E. V. Stefanovich, T. N. Truong, G. Gunster and
D. W. Goodman, J. Phys. Chem. B, 1999, 103, 3391.27 M. I. McCarthy, G. K. Schenter, C. A. Scamehorn and J. B.
Nicholas, J. Phys. Chem., 1996, 100, 16989.28 A. L. Shluger, J. D. Gale and C. R. A. Catlow, J. Phys. Chem.,
1992, 96, 10389.29 J. Goniakowski, S. Bouette-Russo and C. Noguera, Surf. Sci.,
1993, 284, 315.30 H. Nakatsuji, M. Yoshimoto, M. Hada, K. Domen and C. Hirose,
Surf. Sci., 1995, 336, 232.31 M. L. Sushko, A. Y. Gal, M. Watkins and A. L. Shluger,
Nanotechnology, 2006, 17, 2062.
4366 | Phys. Chem. Chem. Phys., 2006, 8, 4359–4367 This journal is �c the Owner Societies 2006

32 M. Szymanski and M. Gillan, Surf. Sci., 1996, 367, 135.33 X. D. Peng and M. A. Barteau, Catal. Lett., 1991, 7, 395.34 A. Shluger, A. Foster, J. Gavartin and P. Sushko, inNano and Giga
Challenges in Microelectronics, ed. J. Greer, A. Korkin and J.Labanowski, Elsevier, Amsterdam, 2003.
35 G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558.36 G. Kresse and J. Furthmuller, Phys. Rev. B, 1996, 54, 11169.37 D. Vanderbilt, Phys. Rev. B, 1990, 41, 7892.38 G. Kresse and J. Hafner, J. Phys.: Condens. Matter, 1994, 6, 8245.39 L. N. Kantorovich, Phys. Rev. B, 1999, 60, 15476.40 P. Saul and C. R. A. Catlow, Philos. Mag. B, 1985, 51, 107.41 J. L. Gavartin, P. V. Sushko and A. L. Shluger, Phys. Rev. B, 2003,
67, 035108.42 G. Pacchioni, F. Frigoli, D. Ricci and J. A. Weil, Phys. Rev. B,
2001, 63, 054102.43 J. Lægsgaard and K. Stokbro, Phys. Rev. Lett., 2001, 86, 2834.44 J. L. Gavartin and A. L. Shluger, Phys. Rev. B, 2001, 64, 245111.45 P. V. Sushko and A. L. Shluger, Surf. Sci., 1999, 421(3), L157.46 P. V. Sushko, A. L. Shluger and C. R. A. Catlow, Surf. Sci., 2000,
450, 153.47 P. V. Sushko, J. L. Gavartin and A. L. Shluger, J. Phys. Chem. B,
2002, 106, 2269.48 A.M. Stoneham andM. J. L. Sangster, Philos. Mag. B, 1985, 52, 717.49 W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284.50 A. D. Becke, J. Chem. Phys., 1993, 98, 5648.51 C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785.52 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A.
Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr,R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D.Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V.Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo,S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K.
Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B.Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov,G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R.L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A.Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B.Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M.Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98(Revision a.7), Gaussian, Inc., Pittsburgh, PA, 1998.
53 R. Coquet and D. J. Willock, Phys. Chem. Chem. Phys., 2005, 7,3819.
54 A. R. Gerson and T. Bredow, Phys. Chem. Chem. Phys., 1999, 1,4889.
55 V. E. Henrich, G. Dresselhaus and H. J. Zeiger, Phys. Rev. B, 1980,22, 4764.
56 P. A. Cox and A. A. Williams, Surf. Sci., 1986, 175, L782.57 D. M. Roessler and W. C. Walker, Phys. Rev., 1967, 159, 733.58 A. Markmann, J. L. Gavartin and A. L. Shluger, unpublished
work.59 P. F. Bernath, Phys. Chem. Chem. Phys., 2002, 4, 1501.60 K. Sawabe, N. Koga, K. Morokuma and Y. Iwasawa, J. Chem.
Phys., 1994, 101, 4819.61 H. Kobayashi, D. Salahub and T. Ito, J. Phys. Chem., 1994, 98,
5487.62 H. Kobayashi and T. Ito, Nippon Kagaku Kaishi, 1996, 9, 765.63 E. N. Gribov, S. Bertarione, D. Scarano, C. Lamberti, G. Spoto
and A. Zecchina, J. Phys. Chem. B, 2004, 108, 16174.64 J. Oviedo, C. J. Calzado and J. F. Sanz, J. Chem. Phys., 1997, 108,
4219.65 Q. Chen and J. M. Goodings, Int. J. Mass Spectrom., 1998, 181,
181.66 M. Suchan, private communication.67 J. Blomer and A. E. Beylich, Surf. Sci., 1999, 423, 127.
This journal is �c the Owner Societies 2006 Phys. Chem. Chem. Phys., 2006, 8, 4359–4367 | 4367
















![Simultaneous HCl and Hg Control-Final [Compatibility Mode]](https://static.fdokumen.com/doc/165x107/6337d8c99e5ed58fcb099e56/simultaneous-hcl-and-hg-control-final-compatibility-mode.jpg)



