
Ambipolarity in Benzobisthiadiazole-Based Donor-Acceptor Conjugated Polymers
Upload
khangminh22Category
view
1download
0
Charge Transport in Conjugated Polymers with Pendent StableRadical GroupsYiren Zhang,† Albert M. Park,‡ Stephen R. McMillan,§ Nicholas J. Harmon,§ Michael E. Flatte,§
Gregory D. Fuchs,*,‡ and Christopher K. Ober*,†
†Materials Science and Engineering, Cornell University, Ithaca, New York 14853, United States‡Applied and Engineering Physics, Cornell University, Ithaca, New York 14853, United States§Optical Science and Technology Center and Department of Physics and Astronomy, University of Iowa, Iowa City, Iowa 52242,United States
*S Supporting Information
ABSTRACT: Synthesizing a stable radical polymer with a conjugated backbone seems like a natural way to introduceconductivity to radical polymers, which are traditionally synthesized with insulating, nonconjugated backbones. For chargestorage applications that take advantage of the redox-active nature of stable radical polymers, enhanced conductivity wouldimprove performance. To explore the interplay between stable radicals and a conjugated backbone, we prepared and studiedsoluble polythiophene with high regioregularity and various concentrations of pendent radical groups to systematically examineany change in conductivity with radical incorporation. Using electron paramagnetic resonance and electrical conductivitymeasurements, we show that there is an exponential decrease in conductivity as we increase the percentage of pendent groupsattached to repeating units, which changes the conductivity by 6 orders of magnitude between the nonradical controlpolythiophene material and the material with the highest radical content (∼80%). These findings serve as an important guide tothe future design of radical polymers on conjugated backbones with the goal of tuning conductivity as a function of stableradical content in redox-active energy storage applications.
■ INTRODUCTION
Polymers bearing pendent stable organic radicals havedemonstrated excellent properties ranging from tunableelectrochemical properties1 and rapid electron self-exchange2
to reversible redox behavior in the electrode layer2,3 and facilesolution processing.4 As a result, radical polymers have beeninvestigated as energy storage materials in batteries, and theircritical property for charge collection, conductivity, has beenstudied extensively.5−8 To date, most investigations focus onnonconjugated stable radical polymers with the existence ofelectrolytes, whose conductivity can be explained by “diffusivehopping” conduction.9 However, the charge transportmechanism in solid-state stable radical polymers remainsunclear, and the reported conductivity varies dramatically fromstudy to study even for a single model polymer, poly(2,2,6,6-tetramethylpiperidinyloxy methacrylate) (PTMA).
The stable radical group present in PTMA, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), is a common nitro-xide radical and is thought to be insulating in the molecularstate.10 It has also been reported that PTMA has a conductivitynear 10−10 S/cm.5,11−13 In a previous study, we have tested theconductivity of PTMA films made by a series of differentpolymerization methods and found the conductivity of allsamples is 10−11 S/cm over distances ranging from 2 to 10μm.14 It has been theorized that a series of redox reactions, inwhich electrons hop between redox sites with the aid ofdelocalized electrons, can result in efficient charge transport inthe pure radical solid,3,15 and some recent reports havedescribed conductivity close to 10−6 S/cm in neat PTMA films
Received: May 16, 2018Revised: June 16, 2018Published: June 18, 2018
Article
pubs.acs.org/cmCite This: Chem. Mater. 2018, 30, 4799−4807
© 2018 American Chemical Society 4799 DOI: 10.1021/acs.chemmater.8b02076Chem. Mater. 2018, 30, 4799−4807
Dow
nloa
ded
via
UN
IV O
F IO
WA
on
Aug
ust 1
9, 2
018
at 1
5:36
:35
(UT
C).
Se
e ht
tps:
//pub
s.ac
s.or
g/sh
arin
ggui
delin
es f
or o
ptio
ns o
n ho
w to
legi
timat
ely
shar
e pu
blis
hed
artic
les.

over a 100 μm path.16 In a more recent study, a relatedpolymer with TEMPO side groups and a poly(ethylene oxide)backbone, poly(4-glycidyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl), showed unexpectedly high conductivity when measuredin thin (sub-100 nm) films. It was proposed that formation ofconducting TEMPO domains explained the transport behav-ior.17
Considering its generally insulating nature, achieving long-range electron transport in a single stable radical polymerrequires a system with alternate conducting pathways besidesthe presence of organic radical sites. In stable radical polymerbatteries, the most common way to provide conductivity is byintroduction of a conducting species, like carbon black orcarbon nanotubes, at the expense of energy density.5,6,18,19
Attempts on a single-component polymer system have alsobeen made in the past two decades, mostly based onconjugated backbones as extra redox sites, such as polypyrroleand polythiophene, as potential cathode materials.20−24
Recently, for conjugated polythiophenes bearing pendentTEMPO stable radicals (PT-TEMPO), the low capacity andmodest conductivity have been explained on the basis ofinteractions between the radical unit and the conjugatedbackbone.25 Because most reported studies of conjugatedpolymers with pendent stable radical units focused onelectrochemical properties, a detailed study of electrontransport in the solid-state films remains important tounderstanding this system.Synthesis of an effective conjugated stable radical polymer
requires that enough stable radical be covalently attached tothe conjugated backbone. Meeting this criterion alone ischallenging given that the unpaired electrons on stable radicalsusually interfere with the transition metal-catalyzed couplingreactions. As a result, many reported cases are homopolymersbased on a thiophene with TEMPO radical groups, which arepolymerized via either oxidative reactions or electropolymeri-zation. However, the oxidative nature of the methodsmentioned above usually lacks control in final polymerstructures or inevitably introduces some oxidative impuritiesas dopants.In the first report of PT-TEMPO, conductivity was reported
to be 10−5 S/cm in the presence of the FeCl4− dopant
anions.20 In more recent reports of electropolymerizedundoped PT-TEMPO,21,23 conductivity values fall in therange of 10−7−10−13 S/cm, which is almost comparable withthat of nonconjugated PTMA.21,23 To date, the highestconductivity of such polymers was found to be near 10−2 S/cm in poly(3,4-ethylenedioxythiophene) (PEDOT) withpendent TEMPO radicals, but still 5 orders lower than thatof pristine PEDOT.22 Because the design of conjugatedpolymers with stable radical pendent groups can act as areplacement of two ingredients in an organic radical battery,carbon black and polymeric binders, understanding the effectof TEMPO radical functionalization on the conjugatedbackbone is important to the future fabrication of electrodematerials and organic electronics.Unlike their nonconjugated counterparts, the local organ-
ization of their nanoscale crystallized regions is usuallyimportant in determining structure−property behaviors inconjugated polymers. Besides the processing conditions,regioregularity, i.e., head−tail linkages in poly(3-hexylthio-phene) (P3HT), can influence self-organization and hencegreatly influence the resulting charge carrier transport anddevice performance.26,27 However, such systematic studies of
the structure effects have rarely been performed on conjugatedstable radical polymers. In this study, we have adopted aregioregular P3HT backbone to minimize defects in theresultant radical polymers and systematically varied theloadings of the stable radicals on each repeating unit for thefirst time. Compared with simple blends of conjugated polymerand stable radicals, the construction of a single molecularspecies will enhance mixing and also simplify study of chargetransport in these stable radical polymers.Here we report the synthesis of a series of conjugated
polythiophenes with pendent TEMPO radical groups usingGrignard metathesis polymerization (GRIM) and azide-clickreaction, affording the resultant polymer with a regioregularbackbone, controlled molecular weight, and low dispersity.Additionally, the synthesized polymers show good solubilitywithout the limitations on molecular weight reported in theliterature when polymers are made by other means.21−23 Itshould be emphasized that this study uses soluble and narrowdispersity-conjugated polymers in contrast to most of thereports in the literature. This enables detailed structuralcharacterization and physical tests from well-controlledmaterials in both solution and the solid state. We use electronparamagnetic resonance (EPR) spectroscopy to study the lineshape of these materials and to quantitatively assess theTEMPO radical content. To address the intrinsic chargetransport in all-conjugated radical polymers, we measure theconductivity of drop-cast films, in the pristine state withoutpolaron doping, for the set of synthesized polymers. We findthat as the TEMPO radical content increases linearly, theoverall conductivity decreases exponentially. This is consistentwith radical sites introducing steric distortions of theconjugated structure, which modifies both inter- and intrachainhopping conductivity. These results and the associated insightsprovide valuable guidance for the design of conjugated radical-containing polymers with controlled conductivity for energystorage applications.
■ EXPERIMENTAL SECTIONChemicals. All commercial chemicals were purchased from Sigma-
Aldrich, VWR, and A&K Scientific and used as received unlessotherwise noted. 2,5-Dibromo-3-hexylthiophene was purchased fromArk Pharm. Anhydrous tetrahydrofuran (THF) was distilled from asodium/benzophenone mixture prior to synthesis of the monomerand polymerizations. Syntheses of monomers used in the polymersynthesis are shown in Schemes S1−S3. All reactions were conductedin oven-dried glassware under a nitrogen atmosphere.
Characterization. 1H nuclear magnetic resonance (NMR) studiesof the synthesized monomers and polymers were performed in CDCl3using a Varian INOVA 400 (1H, 400 MHz) spectrometer. Fouriertransform infrared (FTIR) spectra of polymers were recorded undervacuum on a Bruker Vertex V80V Vacuum FTIR spectrometer withan attenuated total reflection (ATR) mode at a resolution of 4 cm−1.Molecular weights (Mn) and dispersities (D) of the polymers weredetermined by gel permeation chromatography (GPC). GPC wasperformed on a Waters ambient-temperature GPC instrumentequipped with a Waters 410 differential refractive index detector.GPC columns were eluted at a rate of 1.0 mL/min with THF at 40°C. Spectroscopic measurements of diluted solutions were taken on aShimadzu ultraviolet−visible−near infrared spectrophotometer. Thesamples were closed from the ambient air atmosphere, and thetemperature control was set to 25 °C. Raman spectra of polymer filmswere obtained on a Renishaw InVia confocal Raman microscope usingan excitation wavelength of 785 nm with a spectral resolution of ∼1cm−1. Polymer thin films were characterized with an Asylum MFP-3D
Chemistry of Materials Article
DOI: 10.1021/acs.chemmater.8b02076Chem. Mater. 2018, 30, 4799−4807
4800

atomic force microscope using the tapping mode (AC mode) andsilicon cantilevers (model ACCESS-NC).Polymerization and Postfunctionalization Reactions.
Grignard Metathesis (GRIM) Polymerization. Homopolymers(P3HT and P3BrHT) and copolymer P3HT-ran-P3BrHT weresynthesized following the same GRIM procedure but using variousstarting thiophene monomer ratios. Detailed synthesis of P3BrHT islisted here as an example. In a 100 mL round-bottom flask, 2,5-dibromo-3-(6-bromohexyl)thiophene (1.30 g, 4.0 mmol, 1.0 equiv)was dissolved in 4.0 mL of anhydrous THF; 4.00 mL of a t-BuMgClsolution (1.0 M in THF, 4.0 mmol, 1.0 equiv) was added, and themixture was stirred at 25 °C for 20 h. Then the mixture was dilutedwith 32.0 mL of THF, and Ni(dppp)Cl2 ([Ni]:[M] = 1:80) wasadded in one portion to start the polymerization. After 2 h, thereaction was quenched with a 1 M HCl aqueous solution, and themixture was poured into methanol to precipitate the polymer. Thecrude polythiophene was collected by filtration and purified bysequential Soxhlet extraction with methanol, hexane, and chloroform.The target polymer was recovered from the chloroform fraction anddried in vacuum at 40 °C. The 1H NMR spectra of the precursorhomopolymer and copolymers are shown in Figures S3 and S4,respectively.Azide Substitution. P3BrHT (100 mg, 0.40 mmol of Br, 1.0 equiv)
was dissolved in 4 mL of anhydrous THF. Sodium azide (210.4 mg,3.24 mmol, 8.0 equiv) and 18-crown-6 (855.42 mg, 3.24 mmol, 8.0equiv) were dissolved in 1.0 mL of N,N-dimethylformamide (DMF),and the sample was thoroughly mixed to obtain a homogeneoussolution. The DMF solution was then added to the P3BrHT solution,and the resultant mixture was stirred at 60 °C for 15 h. After thereaction had reached completion, the mixture was poured intomethanol and the precipitate was collected. The crude polymerP3HT-azide was purified by repeated Soxhlet extraction withmethanol and dried in vacuum at 40 °C.Synthesis of P3HT-TEMPO-X via Azide-Click Reaction. The azide-
click reaction was conducted under nitrogen in a Schlenck tube.P3HT-azide (71.5 mg, 341.6 μmol of azide, 1.0 equiv), alkyne-TEMPO (86.2 mg, 1.2 equiv), and copper(I) iodide (CuI) (5%equiv) were dissolved in a THF/DIPEA mixture [9:1 (v:v), 4.0 mL].Then the mixture was degassed with nitrogen for 30 min and stirredat 50 °C for 24 h. After the reaction had reached completion, themixture was diluted with THF and filtered through a short aluminacolumn to remove copper. The subsequent precipitation and filtrationgave the crude polymer P3HT-TEMPO-X, which was further purifiedby repeated Soxhlet extraction with methanol (Scheme 1).Magnetic Resonance and Conductivity Measurements.
Solution EPR. Solutions for EPR were prepared in THF such thatthey contain the equivalent of a 1 mM concentration of the clickedTEMPO group. To ensure there is no other source of EPR signal,solutions of the precursor P3HT-ran-P3BrHT were also prepared inTHF as a control with the same concentration of the repeating unit.Measurements were repeated three times for each of the clickedspecies using independently prepared solutions. The solutionconcentrations of P3HT-TEMPO-X samples were prepared to targeta constant overall TEMPO concentration. The EPR data werenormalized with respect to a reference solution with the same nominalconcentration of TEMPO spins. We report the average and standarderror of the three measurements, which primarily originates fromuncertainty in the solution preparation. All measurements were madeusing a Bruker X-band EPR spectrometer at room temperature. EPR
spectra of TEMPO and the TEMPO-free precursor polymer areshown in Figures S1 and S2, respectively.
Conductivity Measurement. Microfabricated four-wire lateraltemplates with a 2.5 mm length and a 10 or 20 μm separation werepatterned using photolithography; 100 nm thick, gold contactelectrodes were prepared by sputtering and lift-off on a thermallyoxidized silicon substrate. Polymer films were prepared by drop-casting 10 mg/mL sample stock solutions on templates. The typicalfilm thickness varies from 250 nm to 2.2 μm for the drop-cast films.The P3HT-TEMPO-X samples were dissolved in chlorobenzene,except for P3HT-TEMPO-100, which was dissolved in 1,4-dioxanebecause of its limited solubility in chlorobenzene. To obtain theconductivity without a contribution from parasitic contact resistance,all four electrodes were used for measurement, applying current to theouter electrodes with a Keithley 6610 precision current sourcemeterand reading voltages from the inner electrodes with a Keithley 6514electrometer. The film thickness was measured using a TencorAlphastep profilometer. As a control sample, P3HT without TEMPOgroups was prepared in toluene and measured in a similar fashionusing the four-wire template. All measurements were taken in a dark,vacuum-sealed, and continuously pumped chamber.
■ RESULTS AND DISCUSSION
Because the commonly used and effective methods for directpolymerization of the radical-bearing thiophene monomer,such as oxidative polymerization or electropolymerization,usually produce materials with poor control of molecularweight and backbone regioregularity, it is necessary to employalternative synthetic strategies to provide a sample with a goodsolubility and a low polydispersity index. Our approach relieson Grignard metathesis (GRIM) polymerization, which affordshighly regioregular polythiophene backbones based on the Ni-catalyzed transfer reaction.28−30 To avoid the deactivation ofthe TEMPO radical by the Grignard reagent,31 we initiallystarted with direct polymerization of a novel thiophenemonomer bearing 2,2,6,6-tetramethylpiperidine, a TEMPOprecursor moiety used in our previous work.14 Unfortunately,this amine precursor of TEMPO can deprotonate thethiophene ring in the presence of a Grignard reagent.32
Instead, alkoxyamine-containing thiophene was synthesizedbecause of its stability toward the Grignard reagent.33
However, the subsequent polymerization after Grignardmetathesis produced only oligomers, which may be ascribedto interference of the large alkoxyamine structure with thepolymerization process.As an alternative, a postfunctionalization method was
developed for this study. Poly[3-(6-bromohexyl)thiophene](P3BrHT) was chosen as the precursor polymer because of itshigh regioregularity, similar to that of P3HT, while thebromine at the side chain terminus acts as a handle forchemical modifications. To covalently attach the TEMPOradical, etherification in THF using the 4-hydroxy-TEMPO/sodium hydride combination (40 equiv with respect to Br) wasattempted. For higher conversion, the reaction mixture washeated at 72 °C for 72 h. However, solution EPR showed a low
Scheme 1. Synthesis Route of P3HT-TEMPO-X
Chemistry of Materials Article
DOI: 10.1021/acs.chemmater.8b02076Chem. Mater. 2018, 30, 4799−4807
4801
radical content in the product, which indicates the deactivationof the TEMPO radical under extended strong basic conditions.To achieve a high level of TEMPO attachment, the azide-
click reaction was used for the preparation of the targetpolymer because of its compatibility with the TEMPO stableradical.34 P3HT-azide was prepared by reacting P3BrHT withsodium azide, and complete substitution was demonstrated bythe shift of the proton peak (Figure S3). The subsequent clickreaction with alkyne-TEMPO was performed at 50 °C for 24 hin a THF/DIPEA mixture [9:1 (v:v)] with CuI as the catalyst.After the reaction had reached completion, a red fiberlikeprecipitate formed, which can be easily filtered and purified bysubsequent Soxhlet extraction. However, as in previous reports,the resultant P3HT-TEMPO-100 (target molecular weight of22 kDa) was insoluble in many solvents, including THF,chloroform, toluene, chlorobenzene, dimethyl sulfoxide, anddichlorobenzene, even when heated. Successful modification ofthe azide group is indicated by the absence of a strong azidepeak at 2083 cm−1, while the removal of excess alkyne-TEMPO is suggested by the absence of an alkyne peak at 3230cm−1 (Figure 1).
Because of the insoluble nature of the model polymerP3HT-TEMPO-100 with a target molecular weight of 22 kDa,we attempted to increase solubility by decreasing the degree ofpolymerization of the polythiophene backbone. Finally, P3HT-TEMPO-100 with molecular weight ∼10 kDa was synthesized,which can be easily dissolved in hot THF or 1,4-dioxane, butnot in hot toluene or chlorobenzene. After dissolution in hotTHF and aging for several days at room temperature, a redP3HT-TEMPO-100 precipitate started to form. Upon
reduction of the TEMPO radical by addition of hydrazinehydrate, the precipitate dissolved immediately, which indicatesthat the poor solubility may originate from the interactionsbetween pendent TEMPO radicals. To further improve thesolubility, 2,5-dibromo-3-hexylthiophene was introduced as aco-monomer in the precursor polymer synthesis, and a series ofstatistical polymers of P3HT-ran-P3BrHT were made (25, 50,and 75% Br). Because of the similar structure and reactivity,copolymerization of two thiophene monomers produced acopolymer with a similar Mn and a low dispersity, but withoutlarge deviations in composition from the starting monomerratio (Table 1). Finally, P3HT-TEMPO-X (X = 25, 50, and75% TEMPO) was prepared using the same azide-clickreaction and is soluble in many solvents, including tolueneand chlorobenzene.We characterized the radical yield of TEMPO clicked on the
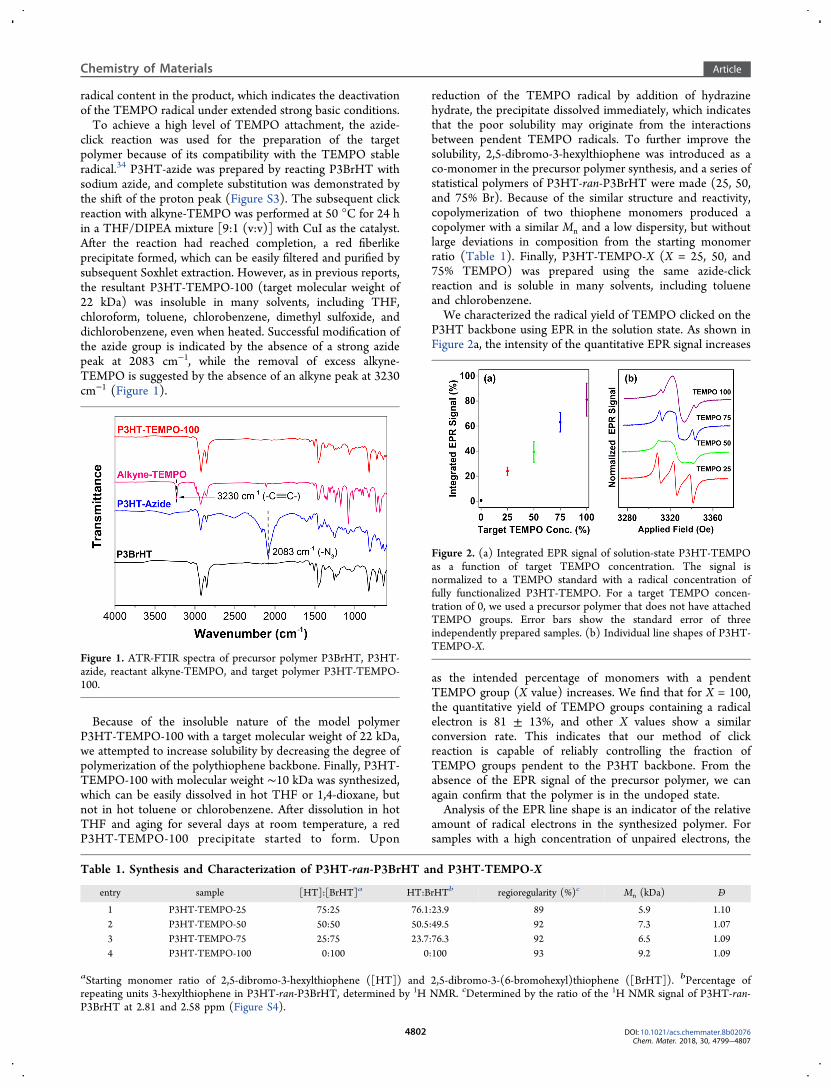
P3HT backbone using EPR in the solution state. As shown inFigure 2a, the intensity of the quantitative EPR signal increases
as the intended percentage of monomers with a pendentTEMPO group (X value) increases. We find that for X = 100,the quantitative yield of TEMPO groups containing a radicalelectron is 81 ± 13%, and other X values show a similarconversion rate. This indicates that our method of clickreaction is capable of reliably controlling the fraction ofTEMPO groups pendent to the P3HT backbone. From theabsence of the EPR signal of the precursor polymer, we canagain confirm that the polymer is in the undoped state.Analysis of the EPR line shape is an indicator of the relative
amount of radical electrons in the synthesized polymer. Forsamples with a high concentration of unpaired electrons, the
Figure 1. ATR-FTIR spectra of precursor polymer P3BrHT, P3HT-azide, reactant alkyne-TEMPO, and target polymer P3HT-TEMPO-100.
Table 1. Synthesis and Characterization of P3HT-ran-P3BrHT and P3HT-TEMPO-X
entry sample [HT]:[BrHT]a HT:BrHTb regioregularity (%)c Mn (kDa) D
1 P3HT-TEMPO-25 75:25 76.1:23.9 89 5.9 1.102 P3HT-TEMPO-50 50:50 50.5:49.5 92 7.3 1.073 P3HT-TEMPO-75 25:75 23.7:76.3 92 6.5 1.094 P3HT-TEMPO-100 0:100 0:100 93 9.2 1.09
aStarting monomer ratio of 2,5-dibromo-3-hexylthiophene ([HT]) and 2,5-dibromo-3-(6-bromohexyl)thiophene ([BrHT]). bPercentage ofrepeating units 3-hexylthiophene in P3HT-ran-P3BrHT, determined by 1H NMR. cDetermined by the ratio of the 1H NMR signal of P3HT-ran-P3BrHT at 2.81 and 2.58 ppm (Figure S4).
Figure 2. (a) Integrated EPR signal of solution-state P3HT-TEMPOas a function of target TEMPO concentration. The signal isnormalized to a TEMPO standard with a radical concentration offully functionalized P3HT-TEMPO. For a target TEMPO concen-tration of 0, we used a precursor polymer that does not have attachedTEMPO groups. Error bars show the standard error of threeindependently prepared samples. (b) Individual line shapes of P3HT-TEMPO-X.
Chemistry of Materials Article
DOI: 10.1021/acs.chemmater.8b02076Chem. Mater. 2018, 30, 4799−4807
4802

exchange interaction between closely spaced radicals quenchesthe hyperfine-induced triplet line shape of small-moleculeTEMPO in solution (Figure S1), and we observe instead broadLorentzian singlets. In lower-concentration samples, however,many of the radical electron spins are comparably isolated, andthus, they have narrow triplet line shapes.35 From the lineshapes of P3HT-TEMPO-X (Figure 2b), we see thatincreasing the concentration of radical electrons follows thistrend. P3HT-TEMPO-25 is dominated by a triplet line shape,whereas P3HT-TEMPO-100 has little triplet contribution andis dominated by a broad singlet. We also confirm there is nodistinguishable EPR signal from the P3HT precursor polymerthat does not have attached TEMPO (Figure S2).Next, we measured the conductivity of the polymer series in
the pristine state to study the correlation between TEMPOconcentration and electrical conductivity. Details of theelectrical measurement procedure are given in ref 16. Figure3 shows that the film conductivity decreases monotonically
from 7 × 10−5 to 3.8 × 10−11 S/cm as the normalized TEMPOcontent per monomer increases from 0 for the control sampleto 0.81 for TEMPO-100. The high conductivity at a low radicalconcentration and the decaying trend of conductivity with anincreasing radical content suggest that the main mechanism ofconductivity in this material is charge transport along theconjugated backbone rather than variable-range hopping(VRH) between radical pendent sites.
In the P3HT-TEMPO system, when the concentration ofclicked TEMPO is low, the conjugated backbone can beorganized to form crystalline regions, which leads to higherconductivity. However, as more TEMPO groups areintroduced, the ordered packing of the polythiophenebackbone decreases because of the steric hindrance from theTEMPO side chains. This mechanism also agrees withprevious results that showed that differences in crystallineordering of domains in conjugated polymers can lead todifferences in conductivity of orders of magnitude.36
Considering the bulky methyl substituents of a TEMPOradical, we used ultraviolet−visible (UV−vis) spectroscopy toassess the extent of conjugation and its effect on conductivitygiven the possible steric interactions upon introduction ofTEMPO groups. UV−vis absorption spectra were recorded forall four polymers studied in solution (THF) and as-cast filmson glass substrates. In a THF solution, all the polymers showedan absorption maximum around 444 nm (Figure 4a), which isred-shifted by around 20 nm as compared to previous reportsof regiorandom analogues.23 This result suggests extendedconjugation caused by a regioregular polythiophene backbonein the solution state. No further absorption features can beobserved upon variation of the TEMPO content, indicatingthat the attachment of the bulky TEMPO structure to thepolythiophene backbone did not change its electronic structuresignificantly in the solution state. This result is also consistentwith an earlier report that the TEMPO radical has no dopingeffect on neutral polythiophenes. The results in ref 25 shouldbe distinguished from our findings because, while we studyconductivity in the solid state where radicals are all in theneutral state, ref 25 included electrochemical measurementsthat involve oxidation of the radical, redox-active sites leadingto doping and charged species.25
In the as-cast films, the absorption maxima of four TEMPO-containing polymers were red-shifted with respect to thesolution absorption maxima, with P3HT-TEMPO-25 showingthe largest red-shift and P3HT-TEMPO-100 showing thesmallest. Such phenomena can be ascribed to planarization ofthe conjugated segments or enhanced interchain interactionsbetween adjacent polythiophene backbones in solid films(Figure 4b). As the level of TEMPO radical loading increases,the vibronic features usually observed in P3HT filmsdisappeared, suggesting the degree of interchain packing ofthe conjugated backbones has been reduced. This can be
Figure 3. Semilog plot of the conductivity of P3HT-TEMPO-X as afunction of calibrated TEMPO content. The dashed line is a fit to thesimple model described by eq 4.
Figure 4. (a) Ultraviolet−visible (UV−vis) absorption of P3HT-TEMPO-X in a THF solution. (b) UV−vis absorption of a drop-casted P3HT-TEMPO-X film on a glass substrate. (c) Raman spectra of drop-cast P3HT and P3HT-TEMPO-X films on a Si substrate.
Chemistry of Materials Article
DOI: 10.1021/acs.chemmater.8b02076Chem. Mater. 2018, 30, 4799−4807
4803

explained if the bulky TEMPO radical groups create sterichindrance.The conjugation length distribution can be reflected not
only in the UV−vis spectra but also in the Raman spectra.With a more planar structure, the conjugated polymers have anextended conjugation length relative to a more twistedstructure, which can provide further information about theeffect of the TEMPO radical on backbone planarization. InFigure 4c, the Raman spectra (λexc = 785 nm) of regioregularP3HT and P3HT-TEMPO-X are presented. The Ramanresults show that when the level of TEMPO radical loading is<50%, the intensity of the band corresponding to symmetricCα−Cβ stretching deformations slowly increases from 1446.5to 1447.8 cm−1.37 This indicates that the introduction ofTEMPO radicals at a low level mainly affects the interchainpacking rather the chain planarization. However, as the level ofTEMPO loading exceeds 50%, the band shifts to a higherwavenumber, which is 1461.8 cm−1 for P3HT-TEMPO-75 and1465.1 cm−1 for P3HT-TEMPO-100, signifying a decrease inthe planarity or degree of conjugation along the backbone.38
As a result, intrachain electron transport is impeded and theoverall electron conductivity further decreases. This observa-tion is consistent with the disappearance of vibronic featuresfor a high TEMPO content, and it is also consistent with theprevious reports.39 Thus, we conclude that the main cause ofthe dramatic decrease in the electrical conductivity with anincreasing TEMPO percentage is a decrease in the P3HTcrystallinity caused by steric hindrance.It should be noted that in a prior study it was found that loss
of conductivity in another TEMPO-containing conjugatedpolymer was due to dedoping of the polythiophene back-bone.25 To avoid confusion, we remind the reader that thepolymer in the study presented here was examined in the solidstate with no electrochemical bias while the prior study ofelectropolymerized TEMPO-containing polythiophene wasperformed in an electrochemical cell under bias, and as aresult, the two cases are difficult to compare.To further elucidate the aggregation of P3HT-TEMPO-X in
the solid state, we investigated solvent-annealed thin films ofthese polymers by atomic force microscopy (AFM). P3HT is asemicrystalline polymer, and fibers composed of well-packedconjugated segments can be obtained by annealing the film inthe presence of organic solvent vapor or by thermal treatment.We first prepared a series of thin films of P3HT and P3HT-TEMPO-X by spin-coating, and then the films were annealedin THF vapor overnight prior to the AFM measurements. InFigure 5a, we observe that short fibers of P3HT formed aftersolvent vapor annealing, indicating interchain packing betweenthe conjugated polymer backbones. As the level of TEMPOloading increases to 25%, we note that domains with a highercrystallinity can be observed in the P3HT-TEMPO-25 film,but the domains are disordered with negligible or only smallgrain size (Figure 5b). Upon introduction of more TEMPOradical substituents, fiberlike aggregation disappears andpolymer chains start to form large amorphous aggregates(Figure 5c), which become smaller at a higher level of TEMPOloading (Figure 5d). This indicates that the π−π interactionsbetween the conjugated backbones have been impeded by thelarge steric hindrance of TEMPO groups. As the result, theefficient electron transport pathway cannot be achieved in theconjugated homopolymers with high TEMPO contentsbecause of the twisted backbone and amorphous nature of
films, which explains why in earlier attempts with such apolymer no obvious increase in conductivity was achieved.To understand this effect more quantitatively, we consider a
charge hopping model. As charge moves through pristineP3HT, there are relatively low potential barriers between sitesalong the same polymer chain (intrachain hopping) comparedto the potential barriers between chains (interchain hopping),due to the nature of the conducting backbone. This results in apreference for backbone conduction along the length of thepolymer until it terminates. After traveling along the length ofthe polymer, the charge hops to another nearby chain. Becausethe interchain hops are unavoidable and correspond to thelargest barriers, they dominate the conductance. In the case ofpure, regioregular P3HT, the chains can stack into ordered,crystalline domains that facilitate interchain hopping. Asdiscussed above, the addition of pendent TEMPO side chainsdistorts the stacking and thus adds to the degree of disorder inchain−chain orientation, which increases the interchainhopping barrier.In this treatment, we model charge conduction in P3HT as
tunneling through interchain barriers, which are arranged in arandom three-dimensional network.40 The average number ofsites per volume, N, can be written as the product of density ofstates g(μ) and bandwidth ε0
μ ε=N g2 ( ) 0 (1)
where the factor of 2 accounts for sites in the range μ ± ε0.The addition of TEMPO side chains acts to reduce thenumber of available sites by pushing them farther apart inposition or energy. This suggests the volumetric density ofstates is reduced by an amount that is proportional to theTEMPO percentage, f. In the simplest model, each TEMPOhas a probability p for pushing one site out of reach of thetransport states. This allows one to write the density of statesas a function of f
μπ ε
=−
gpf
l( )
3(1 )8 3
0 (2)
Figure 5. Atomic force microscopy phase images of (a) P3HT, (b)P3HT-TEMPO-25, (c) P3HT-TEMPO-50, and (d) P3HT-TEMPO-75 after spin-coating from solutions in chlorobenzene and annealingin THF.
Chemistry of Materials Article
DOI: 10.1021/acs.chemmater.8b02076Chem. Mater. 2018, 30, 4799−4807
4804

where l is the average intersite distance. Applying eq 2 to theexpression for the resistance of a unit segment of a Miller−Abrahams type network41 yields an expression for theconductivity
σ σ= −−
Ä
Ç
ÅÅÅÅÅÅÅÅÅÅ
É
Ö
ÑÑÑÑÑÑÑÑÑÑl
a pfexp
2(1 )0 1/3
(3)
where a represents the carrier wave function localization and σ0is a material- and temperature-dependent term that describesthe conductivity caused by carrier−phonon interactions. Torecover an exponential dependence of conductivity onTEMPO concentration, we consider the perturbative limitwhere fp is ≪1. This permits the expansion of eq 3 about fp =0
σ σ≈− +
Ä
Ç
ÅÅÅÅÅÅÅÅÅÅÅÅÅÅÅ
É
Ö
ÑÑÑÑÑÑÑÑÑÑÑÑÑÑÑ
( )l
aexp
2 1 pf
03
(4)
If we take p = 1, the slope of the fit line in Figure 3 near f = 0gives an estimate for l/a ∼ 30. Although this simple modelcannot simultaneously account for the low-f and high-fbehavior of σ, it qualitatively captures the trend in the data.For larger values of f, we expect that one would need toexplicitly account for the dependence of l on f, and there couldbe contributions from disorder that further decrease the levelof intrachain hopping transport, which would also decrease theconductivity exponentially. We do not make these extensionsbecause five data points cannot constrain a more complexmodel. Taking a ∼ 0.7 Å, corresponding roughly to thecovalent radius of carbon,42 we estimate that the averageinterchain hopping distance is around 2 nm, which iscomparable to but smaller than the average chain length.This result suggests that the crystalline domains have a sizableaverage separation and that increasing the TEMPO percentagereduces the average number of accessible crystalline regionsavailable for conductivity, thus exponentially suppressing theconductivity.Because TEMPO is a radical spin, there is a localized site,
typically filled, at some energy below the chemical potentialand a localized site, typically empty, above the chemicalpotential. We see no evidence of transport through suchTEMPO-associated sites for carrier hopping and thus suggestthat these sites are not within the range of energies accessiblefor transport within the materials as we have fabricated them.
■ CONCLUSIONSIn conclusion, we have synthesized and studied regioregularpolythiophene polymers with pendent TEMPO groups in asystematic range of 0−80% of the repeating units. Thismaterial remains soluble even for the highest TEMPO fraction,has a controlled molecular weight, and has a low dispersity. Wefound that as we increase the percentage of pendent TEMPO,the conductivity of the film decreases exponentially, which weunderstand in the context of a disordered network hoppingtheory. This result based on both experiment and a simple butappropriate model is also consistent with UV−vis absorptionmeasurements of the solid films that show decreasingcrystallinity with an increasing TEMPO percentage. In lightof energy storage applications of stable radical polymers inwhich one would ideally combine the high conductivity fromthe polythiophene backbone with the redox activity of a stable
radical group, our results demonstrate that for this directapproach to increasing radical polymer conductivity, acompromise will be necessary. We point out that for a verylarge TEMPO fraction, the conductivity is similar to theconductivity that we recently reported in nonconjugatedPTMA.14,43 However, we have shown that by reducing thefraction of radical groups linearly, one can increaseconductivity exponentially, by orders of magnitude. Therefore,the insights from this work can guide the engineering of futureenergy storage materials, and they can motivate theinvestigation of new copolymers for combining stable radicalgroups and conjugated conducting units in a single polymer.
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.chemma-ter.8b02076.
Monomer synthesis, NMR spectra, and details of
precursor polymer characterization (PDF)
■ AUTHOR INFORMATIONCorresponding Authors*E-mail: [email protected].*E-mail: [email protected] K. Ober: 0000-0002-3805-3314NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work was supported the U.S. Department of EnergyOffice of Basic Energy Science (Grant DE-SC0014336). Theauthors acknowledge use of the facilities of the CornellNanoScale Facility, a member of the National NanotechnologyCoordinated Infrastructure (NNCI), supported by the Na-tional Science Foundation (NSF) (Grant ECCS-1542081).The authors also acknowledge the use of shared facilities of theCornell Center for Materials Research, which is supportedthrough the NSF MRSEC program (Grant DMR-1719875),and the shared facilities of the National Biomedical Center forAdvanced Electron Spin Resonance Technology, which issupported by the National Institute of General MedicalSciences, a part of the National Institutes of Health (GrantP41GM103521).
■ REFERENCES(1) Janoschka, T.; Hager, M. D.; Schubert, U. S. Powering up theFuture: Radical Polymers for Battery Applications. Adv. Mater. 2012,24, 6397−6409.(2) Oyaizu, K.; Nishide, H. Radical Polymers for Organic ElectronicDevices: A Radical Departure from Conjugated Polymers? Adv. Mater.2009, 21, 2339−2344.(3) Nakahara, K.; Oyaizu, K.; Nishide, H. Organic Radical BatteryApproaching Practical Use. Chem. Lett. 2011, 40, 222−227.(4) Zhang, K.; Hu, Y.; Wang, L.; Fan, J.; Monteiro, M. J.; Jia, Z. TheImpact of the Molecular Weight on the Electrochemical Properties ofpoly(TEMPO Methacrylate). Polym. Chem. 2017, 8, 1815−1823.(5) Nakahara, K.; Iwasa, S.; Satoh, M.; Morioka, Y.; Iriyama, J.;Suguro, M.; Hasegawa, E. Rechargeable Batteries with OrganicRadical Cathodes. Chem. Phys. Lett. 2002, 359, 351−354.
Chemistry of Materials Article
DOI: 10.1021/acs.chemmater.8b02076Chem. Mater. 2018, 30, 4799−4807
4805

(6) Nishide, H.; Iwasa, S.; Pu, Y. J.; Suga, T.; Nakahara, K.; Satoh,M. Organic Radical Battery: Nitroxide Polymers as a Cathode-ActiveMaterial. Electrochim. Electrochim. Acta 2004, 50, 827−831.(7) Yonekuta, Y.; Susuki, K.; Oyaizu, K.; Honda, K. Battery-Inspired,Nonvolatile, and Rewritable Memory Architecture: A RadicalPolymer-Based Organic Device. J. Am. Chem. Soc. 2007, 129,14128−14129.(8) Suga, T.; Ohshiro, H.; Sugita, S.; Oyaizu, K.; Nishide, H.Emerging N-Type Redox-Active Radical Polymer for a TotallyOrganic Polymer-Based Rechargeable Battery. Adv. Mater. 2009, 21,1627−1630.(9) Sato, K.; Ichinoi, R.; Mizukami, R.; Serikawa, T.; Sasaki, Y.;Lutkenhaus, J.; Nishide, H.; Oyaizu, K. Diffusion-Cooperative Modelfor Charge Transport by Redox-Active Nonconjugated Polymers. J.Am. Chem. Soc. 2018, 140, 1049−1056.(10) Yu, X.; Mailman, A.; Lekin, K.; Assoud, A.; Robertson, C. M.;Noll, B. C.; Campana, C. F.; Howard, J. A. K.; Dube, P. A.; Oakley, R.T. Semiquinone-Bridged Bisdithiazolyl Radicals as Neutral RadicalConductors. J. Am. Chem. Soc. 2012, 134, 2264−2275.(11) Kim, J. K.; Cheruvally, G.; Choi, J. W.; Ahn, J. H.; Lee, S. H.;Choi, D. S.; Song, C. E. Effect of Radical Polymer Cathode Thicknesson the Electrochemical Performance of Organic Radical Battery. SolidState Ionics 2007, 178, 1546−1551.(12) Kim, J.-K.; Kim, Y.; Park, S.; Ko, H.; Kim, Y. Encapsulation ofOrganic Active Materials in Carbon Nanotubes for Application toHigh-Electrochemical-Performance Sodium Batteries. Energy Environ.Sci. 2016, 9, 1264−1269.(13) Kim, J.-K.; Scheers, J.; Ahn, J.-H.; Johansson, P.; Matic, A.;Jacobsson, P. Nano-Fibrous Polymer Films for Organic RechargeableBatteries. J. Mater. Chem. A 2013, 1, 2426−2430.(14) Zhang, Y.; Park, A.; Cintora, A.; McMillan, S. R.; Harmon, N.J.; Moehle, A.; Flatte, M. E.; Fuchs, G. D.; Ober, C. K. Impact of theSynthesis Method on the Solid-State Charge Transport of RadicalPolymers. J. Mater. Chem. C 2018, 6, 111−118.(15) Oyaizu, K.; Ando, Y.; Konishi, H.; Nishide, H. NernstianAdsorbate-like Bulk Layer of Organic Radical Polymers for High-Density Charge Storage Purposes. J. Am. Chem. Soc. 2008, 130,14459−14461.(16) Rostro, L.; Baradwaj, A. G.; Boudouris, B. W. ControlledRadical Polymerization and Quantification of Solid State ElectricalConductivities of Macromolecules Bearing pendent Stable RadicalGroups. ACS Appl. Mater. Interfaces 2013, 5, 9896−9901.(17) Joo, Y.; Agarkar, V.; Sung, S. H.; Savoie, B. M.; Boudouris, B.W. A Nonconjugated Radical Polymer Glass with High ElectricalConductivity. Science (Washington, DC, U. S.) 2018, 359, 1391−1395.(18) Nakahara, K.; Iriyama, J.; Iwasa, S.; Suguro, M.; Satoh, M.;Cairns, E. J. High-Rate Capable Organic Radical Cathodes forLithium Rechargeable Batteries. J. Power Sources 2007, 165, 870−873.(19) Suguro, M.; Iwasa, S.; Nakahara, K. Fabrication of a Practicaland Polymer-Rich Organic Radical Polymer Electrode and Its RateDependence. Macromol. Rapid Commun. 2008, 29, 1635−1639.(20) Iragi, A.; Crayston, J. A.; Walton, J. C.; Harrison, A. AminoxylFunctionalized Polythiophene: Synthesis and Electrochemical Appli-cations. J. Mater. Chem. 1995, 5, 1291−1295.(21) Aydın, M.; Esat, B.; Kılıc, C.; Kose, M. E.; Ata, A.; Yılmaz, F. APolythiophene Derivative Bearing TEMPO as a Cathode Material forRechargeable Batteries. Eur. Polym. J. 2011, 47, 2283−2294.(22) Casado, N.; Hernandez, G.; Veloso, A.; Devaraj, S.; Mecerreyes,D.; Armand, M. PEDOT Radical Polymer with Synergetic Redox andElectrical Properties. ACS Macro Lett. 2016, 5, 59−64.(23) Li, F.; Zhang, Y.; Kwon, S. R.; Lutkenhaus, J. L. Electro-polymerized Polythiophenes Bearing pendent Nitroxide Radicals. ACSMacro Lett. 2016, 5, 337−341.(24) Xu, L.; Yang, F.; Su, C.; Ji, L.; Zhang, C. Synthesis andProperties of Novel TEMPO-Contained Polypyrrole Derivatives asthe Cathode Material of Organic Radical Battery. Electrochim. Acta2014, 130, 148−155.
(25) Li, F.; Gore, D. N.; Wang, S.; Lutkenhaus, J. L. UnusualInternal Electron Transfer in Conjugated Radical Polymers. Angew.Chem., Int. Ed. 2017, 56, 9856−9859.(26) Kim, Y.; Cook, S.; Tuladhar, S. M.; Choulis, S. A.; Nelson, J.;Durrant, J. R.; Bradley, D. D. C.; Giles, M.; McCulloch, I.; Ha, C.-S.;et al. A Strong Regioregularity Effect in Self-Organizing ConjugatedPolymer Films and High-Efficiency Polythiophene:fullerene SolarCells. Nat. Mater. 2006, 5, 197−203.(27) Shen, X.; Hu, W.; Russell, T. P. Measuring the Degree ofCrystallinity in Semicrystalline Regioregular poly(3-Hexylthiophene).Macromolecules 2016, 49, 4501−4509.(28) Yokoyama, A.; Miyakoshi, R.; Yokozawa, T. Chain-GrowthPolymerization for Poly(3-Hexylthiophene) with a Defined MolecularWeight and a Low Polydispersity. Macromolecules 2004, 37, 1169−1171.(29) Loewe, R. S.; Ewbank, P. C.; Liu, J.; Zhai, L.; McCullough, R.D. Regioregular, Head-to-Tail Coupled poly(3-Alkylthiophenes)Made Easy by the GRIM Method: Investigation of the Reactionand the Origin of Regioselectivity. Macromolecules 2001, 34, 4324−4333.(30) Tkachov, R.; Senkovskyy, V.; Komber, H.; Sommer, J. U.; Kiriy,A. Random Catalyst Walking along Polymerized poly(3-Hexylth-iophene) Chains in Kumada Catalyst-Transfer Polycondensation. J.Am. Chem. Soc. 2010, 132, 7803−7810.(31) Maji, M. S.; Studer, A. Transition-Metal-Free OxidativeHomocoupling of Aryl, Alkenyl, and Alkynyl Grignard Reagentswith TEMPO. Synthesis 2009, 2009, 2467−2470.(32) Zhang, M. X.; Eaton, P. E. BuMgNiPr2: A New Base forStoichiometric, Position-Selective Deprotonation of CyclopropaneCarboxamides and Other Weak CH. Angew. Chem., Int. Ed. 2002, 41,2169−2171.(33) Kaul, E.; Senkovskyy, V.; Tkachov, R.; Bocharova, V.; Komber,H.; Stamm, M.; Kiriy, A. Synthesis of a Bifunctional Initiator forControlled Kumada Catalyst-Transfer Polycondensation/nitroxide-Mediated Polymerization and Preparation of poly(3-Hexylthio-phene)-Polystyrene Block Copolymer Therefrom. Macromolecules2010, 43, 77−81.(34) Kunz, T. K.; Wolf, M. O. Electrodeposition and Properties ofTEMPO Functionalized Polythiophene Thin Films. Polym. Chem.2011, 2, 640−644.(35) Bobela, D. C.; Hughes, B. K.; Braunecker, W. A.; Kemper, T.W.; Larsen, R. E.; Gennett, T. Close Packing of Nitroxide Radicals inStable Organic Radical Polymeric Materials. J. Phys. Chem. Lett. 2015,6, 1414−1419.(36) Sirringhaus, H.; Brown, P. J.; Friend, R. H.; Nielsen, M. M.;Bechgaard, K.; Langeveld-Voss, B. M. W.; Spiering, A. J. H.; Janssen,R. A. J.; Meijer, E. W.; Herwig, P.; et al. Two-Dimensional ChargeTransport in Self-Organized, High-Mobility Conjugated Polymers.Nature 1999, 401, 685−688.(37) Louarn, G.; Trznadel, M.; Buisson, J. P.; Laska, J.; Pron, A.;Lapkowski, M.; Lefrant, S. Raman Spectroscopic Studies ofRegioregular poly(3-Alkylthiophenes). J. Phys. Chem. 1996, 100,12532−12539.(38) Kim, J.; Baeg, K. J.; Khim, D.; James, D. T.; Kim, J. S.; Lim, B.;Yun, J. M.; Jeong, H. G.; Amegadze, P. S. K.; Noh, Y. Y.; et al.Optimal Ambipolar Charge Transport of Thienylenevinylene-BasedPolymer Semiconductors by Changes in Conformation for High-Performance Organic Thin Film Transistors and Inverters. Chem.Mater. 2013, 25, 1572−1583.(39) Burkhart, B.; Khlyabich, P. P.; Thompson, B. C. Influence ofthe Ethylhexyl Side-Chain Content on the Open-Circuit Voltage inRr-poly(3-Hexylthiophene- Co −3-(2-Ethylhexyl)thiophene) Copoly-mers. Macromolecules 2012, 45, 3740−3748.(40) Shklovskii, B. I.; Efros, A. L. Electronic Properties of DopedSemiconductors; Springer: Heidelberg, Germany, 1984.(41) Miller, A.; Abrahams, E. Inpurity Conduction at LowConcentrations. Phys. Rev. 1960, 120, 745.(42) Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry PartA: Structure and Mechanisms; Springer: Boston, 2007.
Chemistry of Materials Article
DOI: 10.1021/acs.chemmater.8b02076Chem. Mater. 2018, 30, 4799−4807
4806

(43) Suga, T.; Takeuchi, S.; Ozaki, T.; Sakata, M.; Oyaizu, K.;Nishide, H. Synthesis of Poly(oxoammonium Salt)s and TheirElectrical Properties in the Organic Thin Film Device. Chem. Lett.2009, 38, 1160−1161.
Chemistry of Materials Article
DOI: 10.1021/acs.chemmater.8b02076Chem. Mater. 2018, 30, 4799−4807
4807
Copyright © 2022 FDOKUMEN




















