
Characterization of heme as activator of Toll-like receptor 4
9
Characterization of Heme as Activator of Toll-like Receptor 4 * Received for publication, November 20, 2006, and in revised form, April 16, 2007 Published, JBC Papers in Press, May 14, 2007, DOI 10.1074/jbc.M610737200 Rodrigo T. Figueiredo ‡ , Patricia L. Fernandez ‡§ , Diego S. Mourao-Sa ‡ , Ba ´ rbara N. Porto ‡ , Fabianno F. Dutra ‡ , Letı ´cia S. Alves ‡ , Marcus F. Oliveira ¶ , Pedro L. Oliveira ¶ , Aure ´ lio V. Grac ¸a-Souza ¶ , and Marcelo T. Bozza ‡1 From the ‡ Departamento de Imunologia, Instituto de Microbiologia, Universidade Federal do Rio de Janeiro (UFRJ) Rio de Janeiro, RJ, 21.941-590 Brasil, the § Institute of Advanced Scientific Investigations and High Technology Services, Ciudad de Panama ´, 0816-02852 Panama ´, and the ¶ Programa de Biotecnologia e Biologia Molecular, Instituto de Bioquı ´mica Me ´dica, UFRJ, Rio de Janeiro, 21.941-590 Brazil Heme is an ancient and ubiquitous molecule present in orga- nisms of all kingdoms, composed of an atom of iron linked to four ligand groups of porphyrin. A high amount of free heme, a potential amplifier of the inflammatory response, is a character- istic feature of diseases with increased hemolysis or extensive cell damage. Here we demonstrate that heme, but not its ana- logs/precursors, induced tumor necrosis factor- (TNF-) secretion by macrophages dependently on MyD88, TLR4, and CD14. The activation of TLR4 by heme is exquisitely strict, requiring its coordinated iron and the vinyl groups of the por- phyrin ring. Signaling of heme through TLR4 depended on an interaction distinct from the one established between TLR4 and lipopolysaccharide (LPS) since anti-TLR4/MD2 antibody or a lipid A antagonist inhibited LPS-induced TNF- secretion but not heme activity. Conversely, protoporphyrin IX antagonized heme without affecting LPS-induced activation. Moreover, heme induced TNF- and keratinocyte chemokine but was inef- fective to induce interleukin-6, interleukin-12, and interferon- inducible protein-10 secretion or co-stimulatory molecule expression. These findings support the concept that the broad ligand specificity of TLR4 and the different activation profiles might in part reside in its ability to recognize different ligands in different binding sites. Finally, heme induced oxidative burst, neutrophil recruitment, and heme oxygenase-1 expression independently of TLR4. Thus, our results presented here reveal a previous unrecognized role of heme as an extracellular signal- ing molecule that affects the innate immune response through a receptor-mediated mechanism. Identification of pathogens by cells of the innate immune system is performed by evolutionarily conserved receptors that recognize microorganisms and viral molecules, the so-called pathogen-associated molecular patterns (1, 2). This initial event has a critical role in the inflammatory response against infection and is a prerequisite to the subsequent activation of the adaptative immune response. In the past years, an extended concept of the functions of the innate immune system has emerged, including not only the defense against pathogens but also causing sterile inflammation and tissue remodeling after cell damage. The absence of microorganisms in several inflammatory patholo- gies indicates that endogenous molecules from damaged cells must be responsible for the triggering, amplification, or perpet- uation of the immune/inflammatory response (3, 4). Inflammation is observed in diseases of increased hemolysis or extensive cell damage, associated or not with the presence of an infectious agent. Some of the most threatening human diseases, because of the severity and/or the global distribution, have intra- or extravascular hemolysis as one of the central phenomena in the pathophysiology. The accumulation of large amounts of hemo- proteins or free heme, such as in a blood clot or after vascular deposition, overwhelms the capacity of heme scavengers (5, 6). Heme has several pro-inflammatory activities including leukocyte activation and migration, up-regulation of adhesion molecules, and induction of cytokines and acute phase proteins (7, 8). It has been assumed and widely accepted that the mechanism by which heme exerts its damaging effects relies on its amphipathic nature, allowing heme to intercalate in the cell or organelle membranes, and on its prooxidant effects (8 –11). Previous studies, however, indicated the existence of receptors in mammalian cells able to bind heme (12–15). Heme from inside the cell binds to Slo1 chan- nels and inhibits transmembrane K currents by decreasing the frequency of channel opening (13). The feline leukemia virus sub- group C receptor, a member of the major facilitator superfamily of transporter proteins, is involved in heme export across the cell membrane affecting erythroid maturation and cell survival (14). A heme importer, named heme carrier protein 1 (HCP1), was recently characterized in duodenal cells (15), and it has been shown that the mitochondrial uptake of porphyrin from the cyto- plasm requires an ATP-binding cassette transporter ABCB6 (16). A recent study demonstrated that the malaria pigment, hemozoin, and the identical synthetic molecule, -hematin, activate TLR9 2 * This work was supported by grants from Conselho de Desenvolvimento Cientı ´fico e Tecnolo ´ gico, Fundac ¸a ˜ o de Amparo a ` Pesquisa do Estado do Rio de Janeiro, Fundac ¸a ˜o Jose ´ Bonifa ´cio, Howard Hughes Medical Institute, and Programa de Nu ´ cleos de Excele ˆncia. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. 1 To whom correspondence should be addressed: Departamento de Imuno- logia, Instituto de Microbiologia, CCS Bloco I, UFRJ, Avenida Carlos Chagas Filho, 373 Cidade Universita ´ria, Rio de Janeiro, RJ, 21941-902 Brasil. Tel.: 55-21-22700990; Fax: 55-21-25608344; E-mail: [email protected]. 2 The abbreviations used are: TLR, Toll-like receptors; DC, dendritic cell; ELISA, enzyme-linked immunosorbent assay; HO-1, heme oxygenase-1; IL, interleu- kin; IP-10, IFN-inducible protein-10; IFN, interferon; LPS, lipopolysaccharide; MyD88, myeloid differentiation protein-88; PPIX, protoporphyrin IX; TNF-, tumor necrosis factor-; Pam 3 Cys, (S)-(2,3-bis(palmitoyloxy)-(2RS)-propil)- N-palmitoyl-(R)-Cys-(S)-Ser(S)-Lys(4)-OH, trihydrochloride; CM-H 2 DCFDA, 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate, acetyl ester; ROS, reactive oxygen species; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; TRIF, Toll/IL-1 receptor domain-con- taining adapter protein; KC, keratinocyte chemokine; NOD, nucleotide-bind- ing oligomerization domain-like receptors . THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 282, NO. 28, pp. 20221–20229, July 13, 2007 © 2007 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. JULY 13, 2007 • VOLUME 282 • NUMBER 28 JOURNAL OF BIOLOGICAL CHEMISTRY 20221 at CAPES/MEC - UFRJ on April 30, 2008 www.jbc.org Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Characterization of heme as activator of Toll-like receptor 4

Characterization of Heme as Activator of Toll-like Receptor 4*
Received for publication, November 20, 2006, and in revised form, April 16, 2007 Published, JBC Papers in Press, May 14, 2007, DOI 10.1074/jbc.M610737200
Rodrigo T. Figueiredo‡, Patricia L. Fernandez‡§, Diego S. Mourao-Sa‡, Barbara N. Porto‡, Fabianno F. Dutra‡,Letıcia S. Alves‡, Marcus F. Oliveira¶, Pedro L. Oliveira¶, Aurelio V. Graca-Souza¶, and Marcelo T. Bozza‡1
From the ‡Departamento de Imunologia, Instituto de Microbiologia, Universidade Federal do Rio de Janeiro (UFRJ) Rio de Janeiro,RJ, 21.941-590 Brasil, the §Institute of Advanced Scientific Investigations and High Technology Services, Ciudad de Panama,0816-02852 Panama, and the ¶Programa de Biotecnologia e Biologia Molecular, Instituto de Bioquımica Medica, UFRJ,Rio de Janeiro, 21.941-590 Brazil
Heme is an ancient and ubiquitous molecule present in orga-nisms of all kingdoms, composed of an atom of iron linked tofour ligand groups of porphyrin. A high amount of free heme, apotential amplifier of the inflammatory response, is a character-istic feature of diseases with increased hemolysis or extensivecell damage. Here we demonstrate that heme, but not its ana-logs/precursors, induced tumor necrosis factor-� (TNF-�)secretion by macrophages dependently on MyD88, TLR4, andCD14. The activation of TLR4 by heme is exquisitely strict,requiring its coordinated iron and the vinyl groups of the por-phyrin ring. Signaling of heme through TLR4 depended on aninteraction distinct from the one established betweenTLR4 andlipopolysaccharide (LPS) since anti-TLR4/MD2 antibody or alipid A antagonist inhibited LPS-induced TNF-� secretion butnot heme activity. Conversely, protoporphyrin IX antagonizedheme without affecting LPS-induced activation. Moreover,heme inducedTNF-� and keratinocyte chemokine butwas inef-fective to induce interleukin-6, interleukin-12, and interferon-inducible protein-10 secretion or co-stimulatory moleculeexpression. These findings support the concept that the broadligand specificity of TLR4 and the different activation profilesmight in part reside in its ability to recognize different ligands indifferent binding sites. Finally, heme induced oxidative burst,neutrophil recruitment, and heme oxygenase-1 expressionindependently of TLR4. Thus, our results presented here reveala previous unrecognized role of heme as an extracellular signal-ingmolecule that affects the innate immune response through areceptor-mediated mechanism.
Identification of pathogens by cells of the innate immunesystem is performed by evolutionarily conserved receptors thatrecognize microorganisms and viral molecules, the so-calledpathogen-associatedmolecular patterns (1, 2). This initial eventhas a critical role in the inflammatory response against infectionand is a prerequisite to the subsequent activation of the adaptative
immune response. In the past years, an extended concept of thefunctions of the innate immune system has emerged, includingnot only the defense against pathogens but also causing sterileinflammation and tissue remodeling after cell damage. Theabsence of microorganisms in several inflammatory patholo-gies indicates that endogenous molecules from damaged cellsmust be responsible for the triggering, amplification, or perpet-uation of the immune/inflammatory response (3, 4).Inflammation is observed in diseases of increased hemolysis or
extensive cell damage, associated or not with the presence of aninfectious agent. Some of the most threatening human diseases,because of the severity and/or the global distribution, have intra-or extravascular hemolysis as one of the central phenomena in thepathophysiology. The accumulation of large amounts of hemo-proteins or free heme, such as in a blood clot or after vasculardeposition, overwhelms the capacity of heme scavengers (5, 6).Heme has several pro-inflammatory activities including leukocyteactivation and migration, up-regulation of adhesion molecules,and induction of cytokines and acute phase proteins (7, 8). It hasbeen assumed and widely accepted that the mechanism by whichheme exerts its damaging effects relies on its amphipathic nature,allowing heme to intercalate in the cell or organelle membranes,and on its prooxidant effects (8–11). Previous studies, however,indicated the existence of receptors in mammalian cells able tobind heme (12–15). Heme from inside the cell binds to Slo1 chan-nels and inhibits transmembrane K� currents by decreasing thefrequency of channel opening (13). The feline leukemia virus sub-groupC receptor, amember of themajor facilitator superfamily oftransporter proteins, is involved in heme export across the cellmembrane affecting erythroidmaturation and cell survival (14). Aheme importer, named heme carrier protein 1 (HCP1), wasrecently characterized in duodenal cells (15), and it has beenshown that themitochondrial uptake of porphyrin from the cyto-plasm requires an ATP-binding cassette transporter ABCB6 (16).A recent studydemonstrated that themalaria pigment, hemozoin,and the identical synthetic molecule, �-hematin, activate TLR9 2
* This work was supported by grants from Conselho de DesenvolvimentoCientıfico e Tecnologico, Fundacao de Amparo a Pesquisa do Estado do Riode Janeiro, Fundacao Jose Bonifacio, Howard Hughes Medical Institute,and Programa de Nucleos de Excelencia. The costs of publication of thisarticle were defrayed in part by the payment of page charges. This articlemust therefore be hereby marked “advertisement” in accordance with 18U.S.C. Section 1734 solely to indicate this fact.
1 To whom correspondence should be addressed: Departamento de Imuno-logia, Instituto de Microbiologia, CCS Bloco I, UFRJ, Avenida Carlos ChagasFilho, 373 Cidade Universitaria, Rio de Janeiro, RJ, 21941-902 Brasil. Tel.:55-21-22700990; Fax: 55-21-25608344; E-mail: [email protected].
2 The abbreviations used are: TLR, Toll-like receptors; DC, dendritic cell; ELISA,enzyme-linked immunosorbent assay; HO-1, heme oxygenase-1; IL, interleu-kin; IP-10, IFN-inducible protein-10; IFN, interferon; LPS, lipopolysaccharide;MyD88, myeloid differentiation protein-88; PPIX, protoporphyrin IX; TNF-�,tumor necrosis factor-�; Pam3Cys, (S)-(2,3-bis(palmitoyloxy)-(2RS)-propil)-N-palmitoyl-(R)-Cys-(S)-Ser(S)-Lys(4)-OH, trihydrochloride; CM-H2DCFDA,5-(and-6)-chloromethyl-2�,7�-dichlorodihydrofluorescein diacetate, acetylester; ROS, reactive oxygen species; ERK, extracellular signal-regulated kinase;MAPK, mitogen-activated protein kinase; TRIF, Toll/IL-1 receptor domain-con-taining adapter protein; KC, keratinocyte chemokine; NOD, nucleotide-bind-ing oligomerization domain-like receptors .
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 282, NO. 28, pp. 20221–20229, July 13, 2007© 2007 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
JULY 13, 2007 • VOLUME 282 • NUMBER 28 JOURNAL OF BIOLOGICAL CHEMISTRY 20221
at CA
PE
S/M
EC
- UF
RJ on A
pril 30, 2008 w
ww
.jbc.orgD
ownloaded from

(17). These pigments consist of heme molecules crystallizedinto dimers through reciprocal iron-carboxylate bonds to oneof the propionic side chains of the porphyrin, and the dimersform chains linked by hydrogen bonds in the crystal (18). Wehypothesized whether heme would activate cells of the innateimmune system through binding to a pattern recognitionreceptor, such as a TLR. In this study, we investigated themolecular mechanism whereby heme activates macrophagesand observed that heme induces the secretion of TNF-� bymacrophages dependently of MyD88, TLR4, and CD14.
EXPERIMENTAL PROCEDURES
Mice—C57Bl/6 mice were supplied by the breeding facilitiesof Fiocruz, Rio de Janeiro, Brazil. Tlr2�/� (19), Tlr9�/� (20),Cd14�/� (21), and Myd88�/� (22) on a C57/Bl6 backgroundwere provided by Drs. Shizuo Akira (Osaka University, Osaka,Japan), Douglas Golenbock (University of Massachusetts,Worcester, MA), and Ricardo Gazzinelli (Universidade Federalde Minas Gerais, Belo Horizonte, Brazil). C3H/HePas andC3H/HeJ mice were provided by Dr. Fernando Cunha (Univer-sidade de Sao Paulo, Ribeirao Preto, Brazil). The animals werekept at a constant temperature (25 °C) with free access to chowand water in a room with a 12-h light/dark cycle. The experi-ments were approved by the Institutional Animal WelfareCommittee.Reagents—PolymixinBwas obtained fromNewBedford Lab-
oratories. Limulus amebocyte assay was obtained from Cam-brex. LPS O111:B4 was obtained from Sigma-Aldrich. FeCl3and FeSO4 were obtained from Reagen. MTS510 was a gener-ous gift from Dr. Kensuke Miyake. E5564 was a generous giftfrom Eisai Research Institute of Boston, Inc. Heme and itsanalogs were obtained from Porphyrin Products. Porphyrinswere dissolved in NaOH 0.1 N, diluted in RPMI, and filtered.Stock solutions of porphyrins were prepared in the dark toavoid free radical generation immediately before use. Hemeused contained �0.01 endotoxin units (�1 pg) in 200 �Mheme. 5-(and-6)-chloromethyl-2�,7�-dichlorodihydrofluo-rescein diacetate, acetyl ester (CM-H2DCFDA) was obtainedfromMolecular Probes.Cell Culture and Stimulation—Peritonealmacrophageswere
obtained 4 days after intraperitoneal instillation of 2 ml of thio-glycollate 3% by peritoneal washing with chilled RPMI. Cellswere seeded at 2 � 105/well in 96-well plates in RPMI. Non-adherent cells were removed by washing, and adherent cellswere stimulated in the absence of serum, and after the indicatedperiods, the supernatant was collected and frozen until cyto-kine determination. Dendritic cells (DCs) were generated asdescribed previously (23, 24), with some modifications. Briefly,bone marrow was harvested from the tibia and femur of C57/Bl6. The cells were resuspended at 106/ml in RPMI 1640(Sigma) supplemented with vitamins, amino acids, 50 �M2-mercaptoethanol, recombinant murine granulocyte-macro-phage colony-stimulating factor, and recombinantmurine IL-4at 10 ng/ml. After 5 days, fresh medium was added to culture,and with 7 days of culture, the cells were collected, and DCswere separated by Optiprep gradient (Sigma) by centrifugationat 600 � g for 30 min at 24 °C. This protocol generated morethan 75% of CD11c� cells. DCs were plated in 96-well at a
density of 2 � 105/well and incubated for 16 h with the stimuliin supplemented RPMI in the absence of serum, after which thesupernatant was recovered for the determination of cytokinesby ELISA. Human monocyte-derived macrophages fromhealthy donors were isolated by plastic adherence of peripheralblood mononuclear cells previously obtained by density gradi-ent centrifugation (Hystopaque, Sigma). In summary, 2 � 106peripheral blood mononuclear cells were plated in 48-wellplates in Dulbecco’s modified Eagle’s medium without serumfor 1 h, 5% CO2, 37 °C. Non-adherent cells were washed out,and adherent cells were cultured inDulbecco’smodified Eagle’smedium containing 10% human serum (Sigma), during 7 days,for differentiation in macrophages. Macrophage purity wasabove 90%, as checked by flow cytometry (FACScan, BD Bio-sciences) analysis using anti-CD3 and anti-CD16 antibodies(BD Biosciences).CytokineMeasurements—The concentrations ofTNF-�, KC,
IL-6, IP-10, and IL-12 were determined using ELISA. All themeasurements were performed in duplicate following theman-ufacturer’s instructions (Peprotec and R&D).Western Blot Analysis for MAPK Phosphorylation and I�B
Degradation—To evaluate ERK1/2 and p38 phosphorylation,elicited peritonealmacrophageswere plated in 6-well plates at adensity of 2.5 � 106 cells/well, non-adherent cells wereremoved by washing with medium, and adherent cells werestimulated with LPS or heme, as indicated in the figure legend.After 1 h, cells were lysed in a buffer consisting of Tris-HCl (50mM), NaCl (150 mM), Nonidet P-40 1%, sodium deoxycholate0.25%, EDTA (1mM), aprotinin (5 �g/ml), leupeptin (5 �g/ml),pepstatin (5 �g/ml), phenylmethylsulfonyl fluoride (1 mM),sodium orthovanadate (1 mM), and NaF (1 mM); pH 7.5. Celllysates were centrifuged, and the supernatants were boiled andsubjected to electrophoresis in SDS-polyacrylamide gel (12%)in reducing conditions. The proteins were transferred to anitrocellulose membrane at 4 °C for 2 h. After this, the mem-branes were blocked with Tris-buffered saline solution with0.05% of Tween 20 (TBS-T) and 5% of fat-free milk. The mem-branes were incubated for 2 h with anti-phospho-ERK1/2(1/2500), diluted in blocking solution, washed in TBS-T, andincubated for 1 h with horseradish peroxidase-conjugated goatanti-rabbit IgG polyclonal antibody (1/10,000), and the bandswere revealed by chemiluminescence using the ECL substrate.The normalization of MAPK phosphorylation was performedby stripping membranes during 30 min at 50 °C in strippingbuffer (100 mM �-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.7). After stripping, membranes were washed withTBS-T, blocked with 5% fat-free milk TBS-T, and incubatedwith rabbit anti-ERK1/2 (1/6000) for 2 h, and detection wasperformed as described above. The degradation of I�B proteinwas analyzed by Western blot with polyclonal rabbit anti-I�B(Sigma) diluted (1/1000) in block solution.Flow Cytometric Analysis of Costimulatory Molecule
Expression—DCs were washed with ice-cold phosphate-buff-ered saline and incubated with fluorescein isothiocyanate- orphycoeritrin-conjugated monoclonal antibody in the presenceof anti-CD16 antibody for 30 min at 4 °C. For control, non-binding isotype-matched fluorescein isothiocyanate- and phos-phatidylethanolamine-conjugated rat anti-mouse IgGs were
Heme Activates Innate Immunity
20222 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 28 • JULY 13, 2007
at CA
PE
S/M
EC
- UF
RJ on A
pril 30, 2008 w
ww
.jbc.orgD
ownloaded from
employed (anti-CD40, anti-CD80, anti-CD82). The expressionof costimulatory molecules at the surface of DCs was analyzedby flow cytometry on a FACSCalibur flow cytometer (BDBiosciences).Assay of Reactive Oxygen Species Generation—Macrophages
(106 cells/tube) were incubated with 2 �M CM-H2DCFDA inHanks’ balanced salt solution for 45min at 37 °C under 5%CO2atmosphere. Macrophages were stimulated with 30 �M hemefor 1 h. ROS productionwas analyzed by flow cytometry using aFACScalibur flow cytometer (BD Biosciences).Acute Peritonitis in Mice—Acute peritonitis was induced by
an intraperitoneal injection of heme (60 �g/cavity) in a volumeof 200 �l. Control groups received an intraperitoneal injectionof endotoxin-free saline solution or LPS (200 ng/cavity) in thesame volume. After 4 h of injection, the animals were killedunder ether anesthesia, and their peritoneal cavities wererinsed with 3 ml of cold phosphate-buffered saline. Total leu-kocytes in the peritoneal fluid were determined on Neubauerchambers after dilution in Turk’s solution. Differential count-ing of leukocytes was carried out onDiff-Quik (Baxter TravenolLaboratories)-stained slices.HO-1 Expression—Macrophages (5 � 106/well) were stimu-
lated with heme (3–30 �M) in the presence of 1% fetal calfserum for 5 h. Cells were disrupted with radioimmune precip-itation buffer (50mMTris-HCl, pH 7.4, 1%Nonidet P-40, 0.25%sodium deoxycholate, 150 mM sodium chloride, 1 mM EDTA)containing protease inhibitors (leupeptin, aprotinin, and pep-statin) and phenylmethylsulfonyl fluoride. HO-1 expressiondetection was performed byWestern blotting using a goat anti-HO-1 antibody and a peroxidase-labeled secondary antibody(Santa Cruz Biotechnology, Inc.). The detectionwas performedusing the ECL system.Statistical Analysis—Data are presented as mean � S.E.
Results were analyzed using a statistical software package(GraphPad Prism 4). Statistical differences among the experi-mental groupswere evaluated by analysis of variancewithNew-man-Keuls correction or with the Student’s t test. Values areexpressed as themean� S.E. The level of significancewas set atp � 0.05.
RESULTS
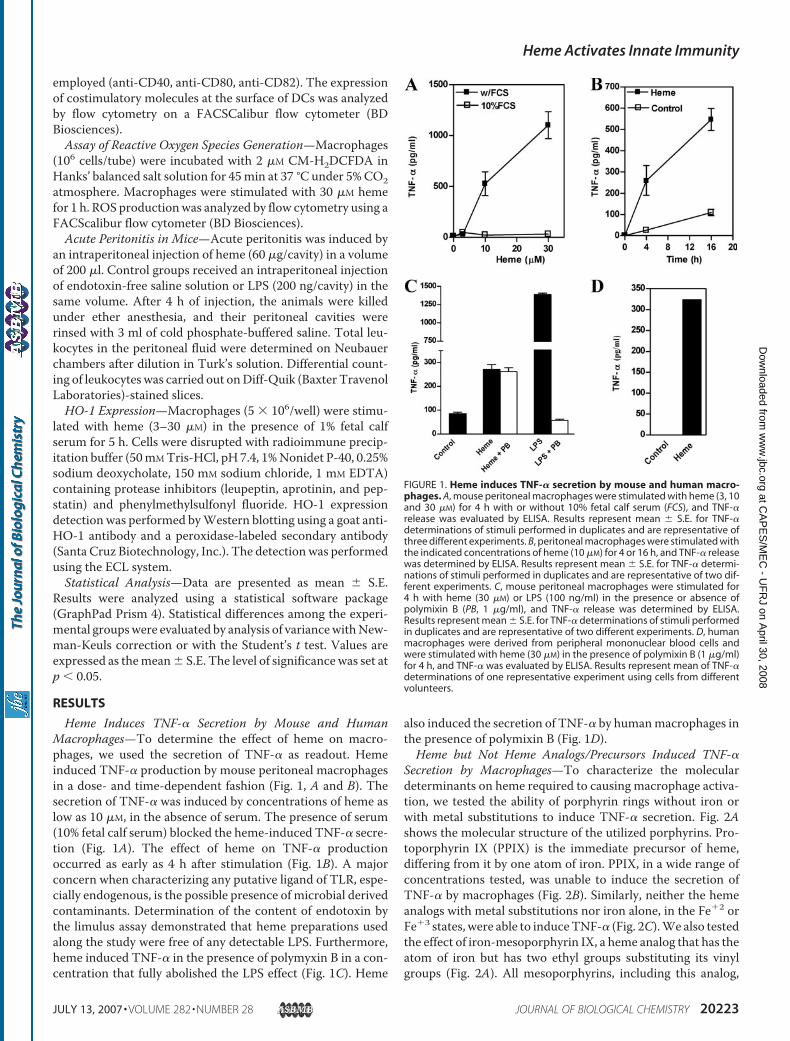
Heme Induces TNF-� Secretion by Mouse and HumanMacrophages—To determine the effect of heme on macro-phages, we used the secretion of TNF-� as readout. Hemeinduced TNF-� production by mouse peritoneal macrophagesin a dose- and time-dependent fashion (Fig. 1, A and B). Thesecretion of TNF-� was induced by concentrations of heme aslow as 10 �M, in the absence of serum. The presence of serum(10% fetal calf serum) blocked the heme-induced TNF-� secre-tion (Fig. 1A). The effect of heme on TNF-� productionoccurred as early as 4 h after stimulation (Fig. 1B). A majorconcern when characterizing any putative ligand of TLR, espe-cially endogenous, is the possible presence of microbial derivedcontaminants. Determination of the content of endotoxin bythe limulus assay demonstrated that heme preparations usedalong the study were free of any detectable LPS. Furthermore,heme induced TNF-� in the presence of polymyxin B in a con-centration that fully abolished the LPS effect (Fig. 1C). Heme
also induced the secretion of TNF-� by humanmacrophages inthe presence of polymixin B (Fig. 1D).Heme but Not Heme Analogs/Precursors Induced TNF-�
Secretion by Macrophages—To characterize the moleculardeterminants on heme required to causing macrophage activa-tion, we tested the ability of porphyrin rings without iron orwith metal substitutions to induce TNF-� secretion. Fig. 2Ashows the molecular structure of the utilized porphyrins. Pro-toporphyrin IX (PPIX) is the immediate precursor of heme,differing from it by one atom of iron. PPIX, in a wide range ofconcentrations tested, was unable to induce the secretion ofTNF-� by macrophages (Fig. 2B). Similarly, neither the hemeanalogs with metal substitutions nor iron alone, in the Fe�2 orFe�3 states, were able to induceTNF-� (Fig. 2C).We also testedthe effect of iron-mesoporphyrin IX, a heme analog that has theatom of iron but has two ethyl groups substituting its vinylgroups (Fig. 2A). All mesoporphyrins, including this analog,
FIGURE 1. Heme induces TNF-� secretion by mouse and human macro-phages. A, mouse peritoneal macrophages were stimulated with heme (3, 10and 30 �M) for 4 h with or without 10% fetal calf serum (FCS), and TNF-�release was evaluated by ELISA. Results represent mean � S.E. for TNF-�determinations of stimuli performed in duplicates and are representative ofthree different experiments. B, peritoneal macrophages were stimulated withthe indicated concentrations of heme (10 �M) for 4 or 16 h, and TNF-� releasewas determined by ELISA. Results represent mean � S.E. for TNF-� determi-nations of stimuli performed in duplicates and are representative of two dif-ferent experiments. C, mouse peritoneal macrophages were stimulated for4 h with heme (30 �M) or LPS (100 ng/ml) in the presence or absence ofpolymixin B (PB, 1 �g/ml), and TNF-� release was determined by ELISA.Results represent mean � S.E. for TNF-� determinations of stimuli performedin duplicates and are representative of two different experiments. D, humanmacrophages were derived from peripheral mononuclear blood cells andwere stimulated with heme (30 �M) in the presence of polymixin B (1 �g/ml)for 4 h, and TNF-� was evaluated by ELISA. Results represent mean of TNF-�determinations of one representative experiment using cells from differentvolunteers.
Heme Activates Innate Immunity
JULY 13, 2007 • VOLUME 282 • NUMBER 28 JOURNAL OF BIOLOGICAL CHEMISTRY 20223
at CA
PE
S/M
EC
- UF
RJ on A
pril 30, 2008 w
ww
.jbc.orgD
ownloaded from
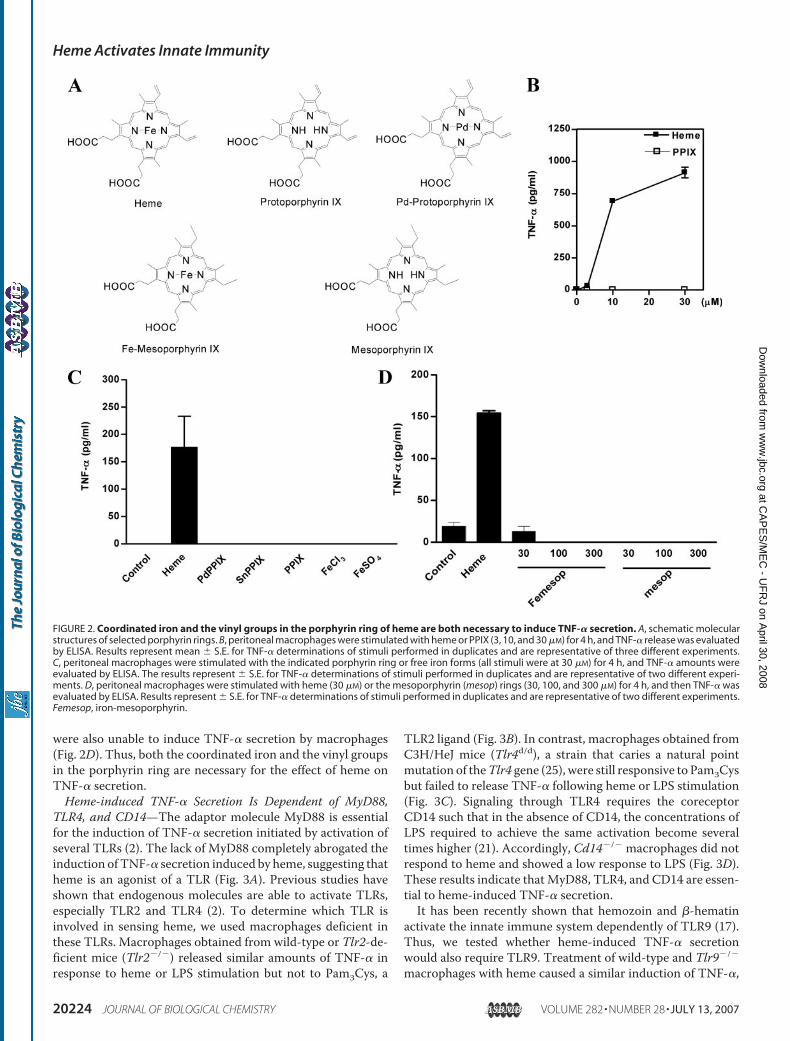
were also unable to induce TNF-� secretion by macrophages(Fig. 2D). Thus, both the coordinated iron and the vinyl groupsin the porphyrin ring are necessary for the effect of heme onTNF-� secretion.Heme-induced TNF-� Secretion Is Dependent of MyD88,
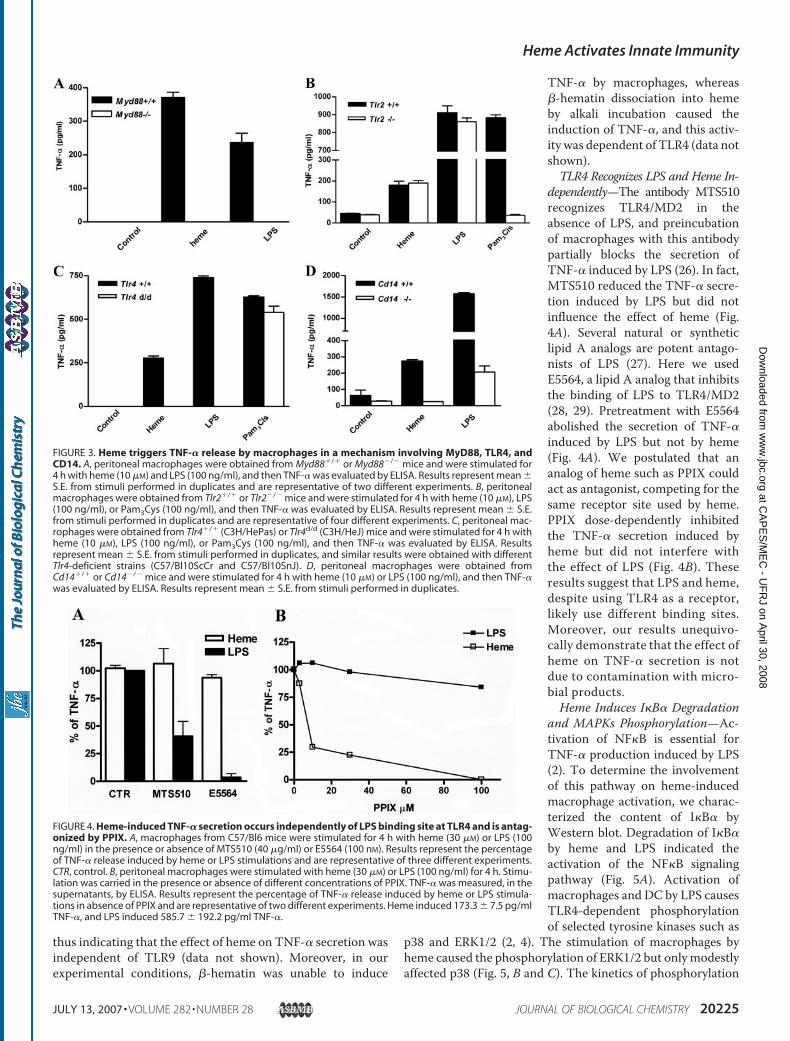
TLR4, and CD14—The adaptor molecule MyD88 is essentialfor the induction of TNF-� secretion initiated by activation ofseveral TLRs (2). The lack of MyD88 completely abrogated theinduction of TNF-� secretion induced by heme, suggesting thatheme is an agonist of a TLR (Fig. 3A). Previous studies haveshown that endogenous molecules are able to activate TLRs,especially TLR2 and TLR4 (2). To determine which TLR isinvolved in sensing heme, we used macrophages deficient inthese TLRs. Macrophages obtained from wild-type or Tlr2-de-ficient mice (Tlr2�/�) released similar amounts of TNF-� inresponse to heme or LPS stimulation but not to Pam3Cys, a
TLR2 ligand (Fig. 3B). In contrast, macrophages obtained fromC3H/HeJ mice (Tlr4d/d), a strain that caries a natural pointmutation of theTlr4 gene (25), were still responsive to Pam3Cysbut failed to release TNF-� following heme or LPS stimulation(Fig. 3C). Signaling through TLR4 requires the coreceptorCD14 such that in the absence of CD14, the concentrations ofLPS required to achieve the same activation become severaltimes higher (21). Accordingly, Cd14�/� macrophages did notrespond to heme and showed a low response to LPS (Fig. 3D).These results indicate thatMyD88, TLR4, and CD14 are essen-tial to heme-induced TNF-� secretion.It has been recently shown that hemozoin and �-hematin
activate the innate immune system dependently of TLR9 (17).Thus, we tested whether heme-induced TNF-� secretionwould also require TLR9. Treatment of wild-type and Tlr9�/�
macrophages with heme caused a similar induction of TNF-�,
FIGURE 2. Coordinated iron and the vinyl groups in the porphyrin ring of heme are both necessary to induce TNF-� secretion. A, schematic molecularstructures of selected porphyrin rings. B, peritoneal macrophages were stimulated with heme or PPIX (3, 10, and 30 �M) for 4 h, and TNF-� release was evaluatedby ELISA. Results represent mean � S.E. for TNF-� determinations of stimuli performed in duplicates and are representative of three different experiments.C, peritoneal macrophages were stimulated with the indicated porphyrin ring or free iron forms (all stimuli were at 30 �M) for 4 h, and TNF-� amounts wereevaluated by ELISA. The results represent � S.E. for TNF-� determinations of stimuli performed in duplicates and are representative of two different experi-ments. D, peritoneal macrophages were stimulated with heme (30 �M) or the mesoporphyrin (mesop) rings (30, 100, and 300 �M) for 4 h, and then TNF-� wasevaluated by ELISA. Results represent � S.E. for TNF-� determinations of stimuli performed in duplicates and are representative of two different experiments.Femesop, iron-mesoporphyrin.
Heme Activates Innate Immunity
20224 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 28 • JULY 13, 2007
at CA
PE
S/M
EC
- UF
RJ on A
pril 30, 2008 w
ww
.jbc.orgD
ownloaded from
thus indicating that the effect of heme on TNF-� secretion wasindependent of TLR9 (data not shown). Moreover, in ourexperimental conditions, �-hematin was unable to induce
TNF-� by macrophages, whereas�-hematin dissociation into hemeby alkali incubation caused theinduction of TNF-�, and this activ-ity was dependent of TLR4 (data notshown).TLR4 Recognizes LPS and Heme In-
dependently—The antibody MTS510recognizes TLR4/MD2 in theabsence of LPS, and preincubationof macrophages with this antibodypartially blocks the secretion ofTNF-� induced by LPS (26). In fact,MTS510 reduced the TNF-� secre-tion induced by LPS but did notinfluence the effect of heme (Fig.4A). Several natural or syntheticlipid A analogs are potent antago-nists of LPS (27). Here we usedE5564, a lipid A analog that inhibitsthe binding of LPS to TLR4/MD2(28, 29). Pretreatment with E5564abolished the secretion of TNF-�induced by LPS but not by heme(Fig. 4A). We postulated that ananalog of heme such as PPIX couldact as antagonist, competing for thesame receptor site used by heme.PPIX dose-dependently inhibitedthe TNF-� secretion induced byheme but did not interfere withthe effect of LPS (Fig. 4B). Theseresults suggest that LPS and heme,despite using TLR4 as a receptor,likely use different binding sites.Moreover, our results unequivo-cally demonstrate that the effect ofheme on TNF-� secretion is notdue to contamination with micro-bial products.Heme Induces I�B� Degradation
and MAPKs Phosphorylation—Ac-tivation of NF�B is essential forTNF-� production induced by LPS(2). To determine the involvementof this pathway on heme-inducedmacrophage activation, we charac-terized the content of I�B� byWestern blot. Degradation of I�B�by heme and LPS indicated theactivation of the NF�B signalingpathway (Fig. 5A). Activation ofmacrophages and DC by LPS causesTLR4-dependent phosphorylationof selected tyrosine kinases such as
p38 and ERK1/2 (2, 4). The stimulation of macrophages byheme caused the phosphorylation of ERK1/2 but onlymodestlyaffected p38 (Fig. 5, B and C). The kinetics of phosphorylation
FIGURE 3. Heme triggers TNF-� release by macrophages in a mechanism involving MyD88, TLR4, andCD14. A, peritoneal macrophages were obtained from Myd88�/� or Myd88�/� mice and were stimulated for4 h with heme (10 �M) and LPS (100 ng/ml), and then TNF-� was evaluated by ELISA. Results represent mean �S.E. from stimuli performed in duplicates and are representative of two different experiments. B, peritonealmacrophages were obtained from Tlr2�/� or Tlr2�/� mice and were stimulated for 4 h with heme (10 �M), LPS(100 ng/ml), or Pam3Cys (100 ng/ml), and then TNF-� was evaluated by ELISA. Results represent mean � S.E.from stimuli performed in duplicates and are representative of four different experiments. C, peritoneal mac-rophages were obtained from Tlr4�/� (C3H/HePas) or Tlr4d/d (C3H/HeJ) mice and were stimulated for 4 h withheme (10 �M), LPS (100 ng/ml), or Pam3Cys (100 ng/ml), and then TNF-� was evaluated by ELISA. Resultsrepresent mean � S.E. from stimuli performed in duplicates, and similar results were obtained with differentTlr4-deficient strains (C57/Bl10ScCr and C57/Bl10SnJ). D, peritoneal macrophages were obtained fromCd14�/� or Cd14�/� mice and were stimulated for 4 h with heme (10 �M) or LPS (100 ng/ml), and then TNF-�was evaluated by ELISA. Results represent mean � S.E. from stimuli performed in duplicates.
FIGURE 4. Heme-induced TNF-� secretion occurs independently of LPS binding site at TLR4 and is antag-onized by PPIX. A, macrophages from C57/Bl6 mice were stimulated for 4 h with heme (30 �M) or LPS (100ng/ml) in the presence or absence of MTS510 (40 �g/ml) or E5564 (100 nM). Results represent the percentageof TNF-� release induced by heme or LPS stimulations and are representative of three different experiments.CTR, control. B, peritoneal macrophages were stimulated with heme (30 �M) or LPS (100 ng/ml) for 4 h. Stimu-lation was carried in the presence or absence of different concentrations of PPIX. TNF-� was measured, in thesupernatants, by ELISA. Results represent the percentage of TNF-� release induced by heme or LPS stimula-tions in absence of PPIX and are representative of two different experiments. Heme induced 173.3 � 7.5 pg/mlTNF-�, and LPS induced 585.7 � 192.2 pg/ml TNF-�.
Heme Activates Innate Immunity
JULY 13, 2007 • VOLUME 282 • NUMBER 28 JOURNAL OF BIOLOGICAL CHEMISTRY 20225
at CA
PE
S/M
EC
- UF
RJ on A
pril 30, 2008 w
ww
.jbc.orgD
ownloaded from

of ERK1/2 by heme was different when compared with LPS.Within 20 min of stimulation, both heme and LPS caused thephosphorylation of ERK1/2; however, at 60 min, only thecells treated with heme still had a sustained ERK1/2 phos-phorylation. Similar to LPS, the phosphorylation of ERK1/2by heme was abrogated in macrophages from TLR4 mutantmice (Fig. 5D).Heme Induced TNF-� and KC but Is Unable to Induce IL-6,
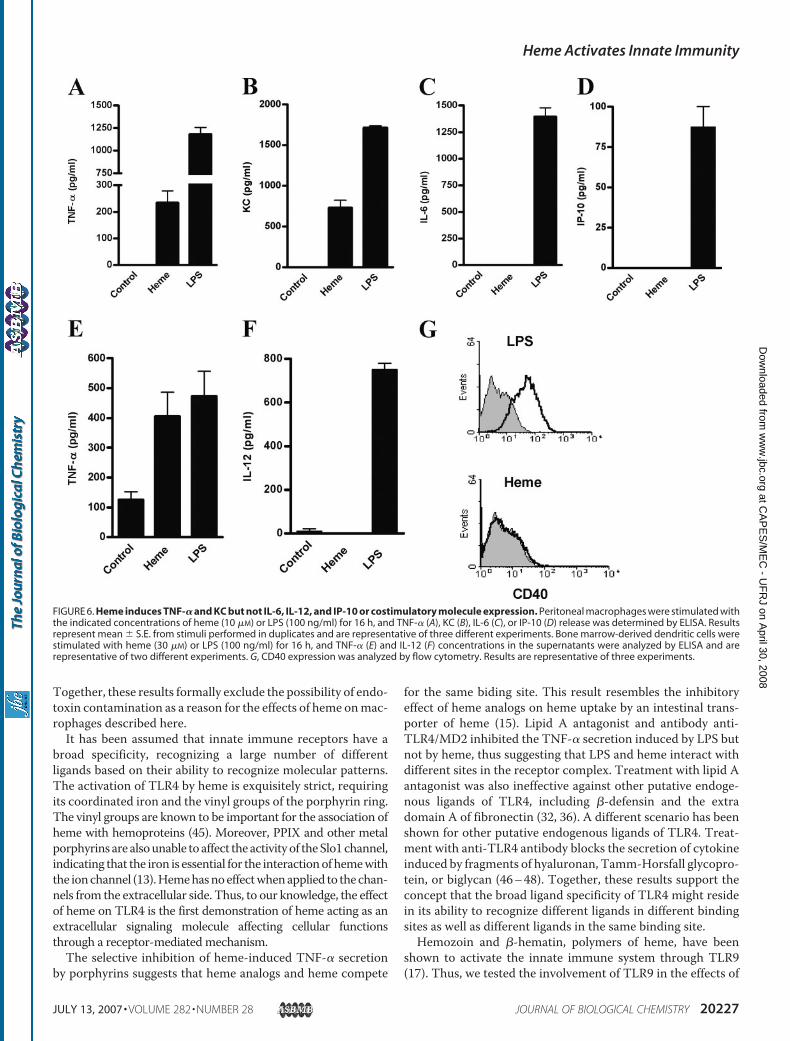
IL-12, and IP-10 or Costimulatory Molecule Expression—Acti-vation of TLR4 by LPS triggers the MyD88-dependent and theTRIF-dependent pathways (2). The MyD88 pathway leads tothe activation of NF�B and MAPKs and the induction of cyto-kines such as TNF-�, KC, IL-6, and IL-12. The TRIF pathwaysignals through IRF-3 and is required for the induction ofIFN-�, IP-10, and co-stimulatory molecule expression. Thesimultaneous analysis of TNF-�, KC, IL-6, and IP-10 concen-trations in the supernatants of macrophages stimulated withheme or LPS demonstrated that heme induced TNF-� and KCbut was unable to induce IL-6 or IP-10 secretion (Fig. 6, A–D).The effect of heme on KC also depended on a functional TLR4(data not shown). Heme, although inducing TNF-� on bonemarrow-derivedDC, did not induce IL-12 secretion or increasethe expression of co-stimulatory molecules, including CD40,CD80, and CD82, or presented adjuvant properties uponimmunization of mice with ovalbumin (Fig. 6, E–G; data notshown). Similarly, heme induced the expression of TNF-�mRNA but not of IL-6 or IFN-� by macrophages (data notshown). The intraperitoneal or intravenous injection ofheme (0, 5–10, 0 mg/mouse) did not induce TNF-� in theplasma or in the peritoneal cavity (data not shown), thussuggesting that serum molecules block interaction of hemeand TLR4 in vivo.Heme Induces ROS Generation, HO-1 Expression, and Neu-
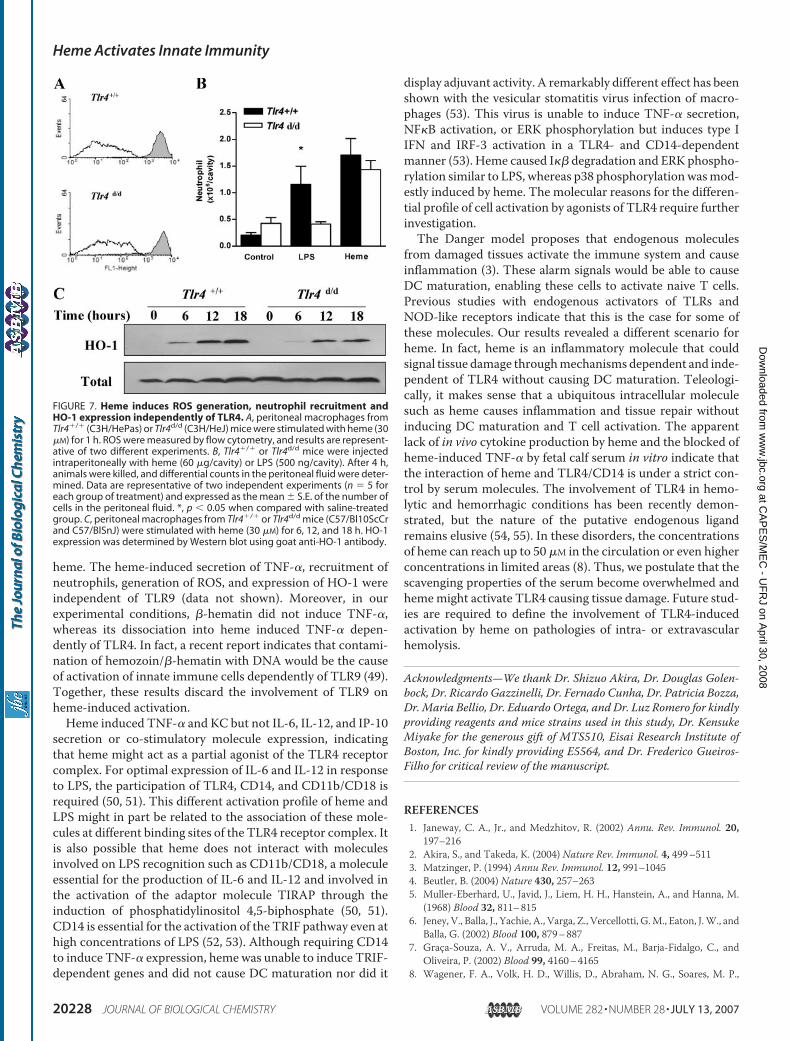
trophil Recruitment Independent of TLR4—LPS causes a num-ber of biological functions besides the secretion of cytokines.However, these biological effects of LPS also require a func-tional TLR4. Considering that heme also induces ROS genera-tion, neutrophil recruitment, and HO-1 expression, we tested
the dependence of TLR4 on heme-induced change on theseparameters. Treatment ofmacrophages fromTlr4d/dmice withheme still caused ROS generation, albeit slightly reduced whencompared with Tlr4�/� (Fig. 7A). Moreover, the intraperito-neal injection of heme on Tlr4�/� and Tlr4d/d mice caused asimilar increase of neutrophils, whereas LPS caused neutrophilrecruitment only on Tlr4�/� (Fig. 7B). The induction of HO-1by heme was independent of a functional TLR4 (Fig. 7C). Asmall reduction on HO-1 expression was observed on macro-phages from Tlr4d/d when compared with Tlr4�/�. No differ-ences on ROS generation, HO-1 expression, or neutrophilrecruitment were observed comparing the effect of heme onwild-type and Tlr9�/� mice (data not shown). We recentlyobserved that the ability of heme to induce neutrophil recruit-ment and activation is dependent on a G� inhibitory protein.3These results indicate that heme is also able to activate immunecells independently of TLR4.
DISCUSSION
Several studies have shown that endogenous moleculeswould be able to activate innate immune cells dependently ofreceptors such as TLRs or NOD-like receptors (2, 30–39). Thepossibility of bacterial product contaminants was not formallyexcluded for all these molecules. In fact, at least for heat shockproteins, the effect on TNF-� secretion was later attributed toLPS contamination (40–44). For this reason, our major con-cern was to exclude that the effect of heme on TNF-� secretionwas due to endotoxin contamination. Highly purified hemeused was free of endotoxin contamination, and polymixin B,anti-TLR4/MD2, and lipid A antagonist all inhibited the effectsof LPS but did not interfere with the induction of TNF-� byheme. Serum and PPIX blocked the effects of heme on TNF-�secretion but did not inhibit the activity of LPS. Moreover,heme was ineffective to induce several cytokines or co-stimu-latory molecules expression typical of TLR4 activation by LPS.
3 B. N. Porto, L. S. Alves, P. L. Fernandez, T. P. Dutra, R. T. Figueiredo, A. V.Graca-Souza, and M. T. Bozza, manuscript in preparation.
FIGURE 5. Heme induces I�B degradation and MAPKs phosphorylation. Elicited peritoneal macrophages were either non-stimulated or stimulated withheme (30 �M) or LPS (100 ng/ml) during the indicated periods of time. Cell extracts were prepared and submitted to electrophoresis. Detection of non-phos-phorylated ERK1/2 was performed to normalize the amount of protein run on the lanes. A–C, I�B degradation and p38 and ERK1/2 phosphorylation (p-p38 andp-ERK1/2) were detected by immunoblotting using anti-I�B, anti-phopho-p38, or anti-phopho-ERK1/2 polyclonal antibodies, respectively. D, heme inducesERK1/2 phosphorylation through TLR4 signaling. Elicited peritoneal macrophages obtained from wild-type or TLR4d/d mice were either non-stimulated orstimulated with heme (30 �M) or LPS (100 ng/ml) for 20 min. Cell extracts were prepared and submitted to electrophoresis. ERK1/2 phosphorylation wasdetected by immunoblotting. The figures are representative of two different experiments with similar results.
Heme Activates Innate Immunity
20226 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 28 • JULY 13, 2007
at CA
PE
S/M
EC
- UF
RJ on A
pril 30, 2008 w
ww
.jbc.orgD
ownloaded from
Together, these results formally exclude the possibility of endo-toxin contamination as a reason for the effects of heme onmac-rophages described here.It has been assumed that innate immune receptors have a
broad specificity, recognizing a large number of differentligands based on their ability to recognize molecular patterns.The activation of TLR4 by heme is exquisitely strict, requiringits coordinated iron and the vinyl groups of the porphyrin ring.The vinyl groups are known to be important for the association ofheme with hemoproteins (45). Moreover, PPIX and other metalporphyrins are alsounable toaffect theactivityof theSlo1channel,indicating that the iron is essential for the interactionofhemewiththe ionchannel (13).Hemehasnoeffectwhenapplied to thechan-nels from the extracellular side. Thus, to our knowledge, the effectof heme on TLR4 is the first demonstration of heme acting as anextracellular signaling molecule affecting cellular functionsthrough a receptor-mediatedmechanism.The selective inhibition of heme-induced TNF-� secretion
by porphyrins suggests that heme analogs and heme compete
for the same biding site. This result resembles the inhibitoryeffect of heme analogs on heme uptake by an intestinal trans-porter of heme (15). Lipid A antagonist and antibody anti-TLR4/MD2 inhibited the TNF-� secretion induced by LPS butnot by heme, thus suggesting that LPS and heme interact withdifferent sites in the receptor complex. Treatment with lipid Aantagonist was also ineffective against other putative endoge-nous ligands of TLR4, including �-defensin and the extradomain A of fibronectin (32, 36). A different scenario has beenshown for other putative endogenous ligands of TLR4. Treat-ment with anti-TLR4 antibody blocks the secretion of cytokineinduced by fragments of hyaluronan, Tamm-Horsfall glycopro-tein, or biglycan (46–48). Together, these results support theconcept that the broad ligand specificity of TLR4 might residein its ability to recognize different ligands in different bindingsites as well as different ligands in the same binding site.Hemozoin and �-hematin, polymers of heme, have been
shown to activate the innate immune system through TLR9(17). Thus, we tested the involvement of TLR9 in the effects of
FIGURE 6. Heme induces TNF-� and KC but not IL-6, IL-12, and IP-10 or costimulatory molecule expression. Peritoneal macrophages were stimulated withthe indicated concentrations of heme (10 �M) or LPS (100 ng/ml) for 16 h, and TNF-� (A), KC (B), IL-6 (C), or IP-10 (D) release was determined by ELISA. Resultsrepresent mean � S.E. from stimuli performed in duplicates and are representative of three different experiments. Bone marrow-derived dendritic cells werestimulated with heme (30 �M) or LPS (100 ng/ml) for 16 h, and TNF-� (E) and IL-12 (F) concentrations in the supernatants were analyzed by ELISA and arerepresentative of two different experiments. G, CD40 expression was analyzed by flow cytometry. Results are representative of three experiments.
Heme Activates Innate Immunity
JULY 13, 2007 • VOLUME 282 • NUMBER 28 JOURNAL OF BIOLOGICAL CHEMISTRY 20227
at CA
PE
S/M
EC
- UF
RJ on A
pril 30, 2008 w
ww
.jbc.orgD
ownloaded from
heme. The heme-induced secretion of TNF-�, recruitment ofneutrophils, generation of ROS, and expression of HO-1 wereindependent of TLR9 (data not shown). Moreover, in ourexperimental conditions, �-hematin did not induce TNF-�,whereas its dissociation into heme induced TNF-� depen-dently of TLR4. In fact, a recent report indicates that contami-nation of hemozoin/�-hematin with DNA would be the causeof activation of innate immune cells dependently of TLR9 (49).Together, these results discard the involvement of TLR9 onheme-induced activation.Heme induced TNF-� and KC but not IL-6, IL-12, and IP-10
secretion or co-stimulatory molecule expression, indicatingthat heme might act as a partial agonist of the TLR4 receptorcomplex. For optimal expression of IL-6 and IL-12 in responseto LPS, the participation of TLR4, CD14, and CD11b/CD18 isrequired (50, 51). This different activation profile of heme andLPS might in part be related to the association of these mole-cules at different binding sites of the TLR4 receptor complex. Itis also possible that heme does not interact with moleculesinvolved on LPS recognition such as CD11b/CD18, a moleculeessential for the production of IL-6 and IL-12 and involved inthe activation of the adaptor molecule TIRAP through theinduction of phosphatidylinositol 4,5-biphosphate (50, 51).CD14 is essential for the activation of the TRIF pathway even athigh concentrations of LPS (52, 53). Although requiring CD14to induce TNF-� expression, heme was unable to induce TRIF-dependent genes and did not cause DC maturation nor did it
display adjuvant activity. A remarkably different effect has beenshown with the vesicular stomatitis virus infection of macro-phages (53). This virus is unable to induce TNF-� secretion,NF�B activation, or ERK phosphorylation but induces type IIFN and IRF-3 activation in a TLR4- and CD14-dependentmanner (53). Heme caused I�� degradation and ERK phospho-rylation similar to LPS, whereas p38 phosphorylationwasmod-estly induced by heme. The molecular reasons for the differen-tial profile of cell activation by agonists of TLR4 require furtherinvestigation.The Danger model proposes that endogenous molecules
from damaged tissues activate the immune system and causeinflammation (3). These alarm signals would be able to causeDC maturation, enabling these cells to activate naive T cells.Previous studies with endogenous activators of TLRs andNOD-like receptors indicate that this is the case for some ofthese molecules. Our results revealed a different scenario forheme. In fact, heme is an inflammatory molecule that couldsignal tissue damage throughmechanisms dependent and inde-pendent of TLR4 without causing DC maturation. Teleologi-cally, it makes sense that a ubiquitous intracellular moleculesuch as heme causes inflammation and tissue repair withoutinducing DC maturation and T cell activation. The apparentlack of in vivo cytokine production by heme and the blocked ofheme-induced TNF-� by fetal calf serum in vitro indicate thatthe interaction of heme and TLR4/CD14 is under a strict con-trol by serum molecules. The involvement of TLR4 in hemo-lytic and hemorrhagic conditions has been recently demon-strated, but the nature of the putative endogenous ligandremains elusive (54, 55). In these disorders, the concentrationsof heme can reach up to 50 �M in the circulation or even higherconcentrations in limited areas (8). Thus, we postulate that thescavenging properties of the serum become overwhelmed andhememight activate TLR4 causing tissue damage. Future stud-ies are required to define the involvement of TLR4-inducedactivation by heme on pathologies of intra- or extravascularhemolysis.
Acknowledgments—We thank Dr. Shizuo Akira, Dr. Douglas Golen-bock, Dr. Ricardo Gazzinelli, Dr. Fernado Cunha, Dr. Patricia Bozza,Dr. Maria Bellio, Dr. Eduardo Ortega, and Dr. Luz Romero for kindlyproviding reagents and mice strains used in this study, Dr. KensukeMiyake for the generous gift of MTS510, Eisai Research Institute ofBoston, Inc. for kindly providing E5564, and Dr. Frederico Gueiros-Filho for critical review of the manuscript.
REFERENCES1. Janeway, C. A., Jr., and Medzhitov, R. (2002) Annu. Rev. Immunol. 20,
197–2162. Akira, S., and Takeda, K. (2004) Nature Rev. Immunol. 4, 499–5113. Matzinger, P. (1994) Annu Rev. Immunol. 12, 991–10454. Beutler, B. (2004) Nature 430, 257–2635. Muller-Eberhard, U., Javid, J., Liem, H. H., Hanstein, A., and Hanna, M.
(1968) Blood 32, 811–8156. Jeney, V., Balla, J., Yachie, A., Varga, Z., Vercellotti, G.M., Eaton, J.W., and
Balla, G. (2002) Blood 100, 879–8877. Graca-Souza, A. V., Arruda, M. A., Freitas, M., Barja-Fidalgo, C., and
Oliveira, P. (2002) Blood 99, 4160–41658. Wagener, F. A., Volk, H. D., Willis, D., Abraham, N. G., Soares, M. P.,
FIGURE 7. Heme induces ROS generation, neutrophil recruitment andHO-1 expression independently of TLR4. A, peritoneal macrophages fromTlr4�/� (C3H/HePas) or Tlr4d/d (C3H/HeJ) mice were stimulated with heme (30�M) for 1 h. ROS were measured by flow cytometry, and results are represent-ative of two different experiments. B, Tlr4�/� or Tlr4d/d mice were injectedintraperitoneally with heme (60 �g/cavity) or LPS (500 ng/cavity). After 4 h,animals were killed, and differential counts in the peritoneal fluid were deter-mined. Data are representative of two independent experiments (n � 5 foreach group of treatment) and expressed as the mean � S.E. of the number ofcells in the peritoneal fluid. *, p � 0.05 when compared with saline-treatedgroup. C, peritoneal macrophages from Tlr4�/� or Tlr4d/d mice (C57/Bl10ScCrand C57/BlSnJ) were stimulated with heme (30 �M) for 6, 12, and 18 h. HO-1expression was determined by Western blot using goat anti-HO-1 antibody.
Heme Activates Innate Immunity
20228 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 28 • JULY 13, 2007
at CA
PE
S/M
EC
- UF
RJ on A
pril 30, 2008 w
ww
.jbc.orgD
ownloaded from

Adema, G. J., and Figdor, C. G. (2003) Pharmacol. Rev. 55, 551–5719. Chou, A. C., and Fitch, C. D. (1980) J. Clin. Investig. 66, 856–85810. Schmitt, T. H., Frezzatti, W. A., Jr., and Schreier, S. Arch. Biochem. Bio-
phys. (1993) 307, 96–10311. Balla, J., Balla, G., Jeney, V., Kakuk, G., Jacob, H., and Vercellotti, G. (2000)
Blood 95, 3442–345012. Galbraith, R. A. (1990) J. Hepatol. 10, 305–31013. Tang, X., Xu, R., Reynolds, M., Garcia, M., Heinemann, S., and Hoshi, T.
(2003) Nature 425, 531–53514. Quigley, J. G., Yang, Z., Worthington, M. T., Phillips, J. D., Sabo, K. M.,
Sabath, D. E., Berg, C. L., Sassa, S., Wood, B. L., and Abkowitz, J. L. (2004)Cell 118, 757–766
15. Shayeghi, M., Latunde-Dada, G. O., Oakhill, J. S., Laftah, A. H., Takeuchi,K., Halliday, N., Khan, Y., Warley, A., McCann, F. E., Hider, R. C., Frazer,D.M., Anderson,G. J., Vulpe, C.D., Simpson, R. J., andMcKie, A. T. (2005)Cell 122, 789–801
16. Krishnamurthy, P. C., Du, G., Fukuda, Y., Sun, D., Sampath, J., Mercer,K. E., Junfeng Wang, J., Sosa-Pineda, B., Murti, K. G., and Schuetz, J. D.(2006) Nature 443, 586–589
17. Coban, C., Ishii, K. J., Kawai, T., Hemmi, H., Sato, S., Uematsu, S.,Yamamoto, M., Takeuchi, O., Itagaki, S., Kumar, N., Horii, T., and Akira,S. (2005) J. Exp. Med. 201, 19–25
18. Pagola, S., Stephens, P. W., Bohle, D. S., Andrew, D., Kosar, A. D., andMadsen, S. K. (2000) Nature. 404, 307–310
19. Takeuchi, O., Hoshino, K., Kawai, T., Sanjo, H., Takada, H., Ogawa, T.,Takeda, K., and Akira, S. (1999) Immunity 11, 443–451
20. Hemmi, H., Takeuchi, O., Kawai, T., Kaisho, T., Sato, S., Sanjo, H., Mat-sumoto, M., Hoshino, K., Wagner, H., Takeda, K., and Akira, S. (2000)Nature 408, 740–745
21. Haziot, A., Ferrero, E., Kontgen, F., Hijiya, N., Yamamoto, S., Silver, J.,Stewart, C. L., and Goyert, S. M. (1996) Immunity 4, 407–414
22. Adachi, O., Kawai, T., Takeda, K., Matsumoto, M., Tsutsui, H., Sakagami,M., Nakanishi, K., and Akira, S. (1999) Immunity 9, 143–150
23. Inaba, K., Inaba, M., Romani, N., Aya, H., Deguchi, M., Ikehara, S., Mura-matsu, S., and Steinman, R. M. (1992) J. Exp. Med. 176, 1693–1702
24. Bittencourt, V. C., Figueiredo, R. T., da Silva, R. B., Mourao-Sa, D. S.,Fernandez, P. L., Sassaki, G. L., Mulloy, B., Bozza, M. T., and Barreto-Bergter, E. (2006). J. Biol. Chem. 281, 22614–22623
25. Poltorak, A., He, X., Smirnova, I., Liu, M. Y., Van Huffel, C., Du, X., Bird-well, D., Alejos, E., Silva, M., Galanos, C., Freudenberg, M., Ricciardi-Castagnoli, P., Layton, B., and Beutler, B. (1998) Science 282, 2085–2088
26. Akashi, S., Shimazu, R., Ogata, H., Nagai, Y., Takeda, K., Kimoto, M., andMiyake, K. (2000) J. Immunol. 164, 3471–3475
27. Rossignol, D. P., and Lynn, M. (2005) Curr. Opin. Investig. Drugs 6,496–502
28. Mullarkev, M., Rose, J. R., Bristol, J., Kawata, T., Kimura, A., Kobayashi, S.,Przetak, M., Chow, J., Gusovsky, F., Christ, W. J., and Rossignol, D. P.(2003) J. Pharmacol. Exp. Ther. 304, 1093–1102
29. Visintin, A., Halmen, K. A., Latz, E., Monks, B. G., and Golenbock, D. T.(2005) J. Immunol. 175, 6465–6472
30. Ohashi, K., Burkart, V., Flohe, S., and Kolb, H. (2000) J. Immunol. 164,558–561
31. Vabulas, R. M., Ahmad-Nejad, P., da Costa, C., Miethke, T., Kirschning,C. J., Hacker, H., andWagner, H. (2001). J. Biol. Chem. 276, 31332–31339
32. Okamura, Y., Watari, M., Jerud, E. S., Young, D. W., Ishizaka, S. T., Rose,
J., Chow, J. C., and Strauss, J. F., 3rd. (2001). J. Biol. Chem. 276,10229–10233
33. Asea, A., Rehli, M., Kabingu, E., Boch, J. A., Bare, O., Auron, P. E., Steven-son, M. A., Smiley, S. T., King, J. A., and Hancock, W.W. (2001) J. Immu-nol. 167, 2887–2894
34. Guillot, L., Balloy, V., McCormack, F. X., Golenbock, D. T., Chignard, M.,and Si-Tahar, M. (2002) J. Immunol. 168, 5989–5992
35. Calderwood, S. K. (2002) J. Biol. Chem. 277, 15028–1503436. Biragyn, A., Ruffini, P. A., Leifer, C. A., Klyushnenkova, E., Shakhov, A.,
Chertov, O., Shirakawa, A. K., Farber, J. M., Segal, D. M., Oppenheim, J. J.,and Kwak, L. W. (2002). Science 298, 1025–1029
37. Vabulas, R. M., Braedel, S., Hilf, N., Singh-Jasuja, H., Herter, S., Ahmad-Nejad, P., Kirschning, C. J., Da Costa, C., Rammensee, H. G., Wagner, H.,and Schild, H. (2002) J. Biol. Chem. 277, 20847–20853
38. Park, J. S., Svetkauskaite, D., He, Q., Kim, J. Y., Strassheim, D., Ishizaka, A.,and Abraham, E. (2004) J. Biol. Chem. 279, 7370–7377
39. Martinon, F., Petrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006)Nature 440, 237–241
40. Bausinger, H., Lipsker, D., Ziylan, U., Manie, S., Briand, J. P., Cazenave,J.-P., Muller, S., Haeuw, J.-F., Davanat, C., de la Salle, H., and Hanau, D.(2002). Eur. J. Immunol. 32, 3708–3713
41. Gao, B., and Tsan, M. F. (2003) J. Biol. Chem. 278, 174–17942. Gao, B., and Tsan, M. F. (2003) J. Biol. Chem. 278, 22523–2252943. Reed, R. C., Berwin, B., Baker, J. P., and Nicchitta, C. V. (2003) J. Biol.
Chem. 278, 31853–3186044. Tsan, M. F., and Gao, B. (2004) J. Leukocyte Biol. 76, 514–51945. Daltrop, O., Allen, J., Willis, A., and Ferguson, S. (2002) Proc. Natl. Acad.
Sci. U. S. A. 99, 7872–787646. Termeer, C., Benedix, F., Sleeman, J., Fieber, C., Voith, U., Ahrens, T.,
Miyake, K., Freudenberg, M., Galanos, C., and Simon, J. C. (2002) J. Exp.Med. 195, 99–111
47. Saemann, M. D., Weichhart, T., Zeyda, M., Staffler, G., Schunn, M., Stu-hlmeier, K. M., Sobanov, Y., Stulnig, T. M., Akira, S., von Gabain, A., vonAhsen, U., Horl, W. H., and Zlabinger, G. J. (2005). J. Clin. Investig. 115,468–475
48. Schaefer, L., Babelova, A., Kiss, E., Hausser, H. J., Baliova, M., Kr-zyzankova, M., Marsche, G., Young, M. F., Mihalik, D., Gotte, M., Malle,E., Schaefer, R.M., andGrone,H. J. (2005) J. Clin. Investig. 115, 2223–2233
49. Parroche, P., Lauw, F. N., Goutagny, N., Latz, E.,Monks, B. G., Visintin, A.,Halmen, K. A., Lamphier, M., Olivier, M., Bartholomeu, D. C., Gazzinelli,R. T., and Golenbock, D. T. (2007) Proc. Natl. Acad. Sci. U. S. A. 104,1919–1924
50. Perera, P. Y., Mayadas, T. N., Takeuchi, O., Akira, S., Zaks-Zilberman,M.,Goyert, S. M., and Vogel, S. N. (2001) J. Immunol. 166, 574–581
51. Kagan, J. C., and Medzhitov, R. (2006) Cell 125, 943–95552. Perera, P. Y., Vogel, S. N., Detore, G. R., Haziot, A., and Goyert, S. M.
(1997) J. Immunol. 158, 4422–442953. Jiang, Z., Georgel, P., Du, X., Shamel, L., Sovath, S., Mudd, S., Huber, M.,
Kalis, C., Keck, S., Galanos, C., Freudenberg, M., and Beutler, B. (2005)Nat. Immunol. 6, 565–670
54. Barsness, K. A., Arcaroli, J., Harken, A. H., Abraham, E., Banerjee, A.,Reznikov, L., and McIntyre, R. C. (2004) Am. J. Physiol. 287, R592–R599
55. Oyama, J., Blais, C., Jr., Liu, X., Pu,M., Kobzik, L., Kelly, R. A., andBourcier,T. (2004). Circulation 109, 784–789
Heme Activates Innate Immunity
JULY 13, 2007 • VOLUME 282 • NUMBER 28 JOURNAL OF BIOLOGICAL CHEMISTRY 20229
at CA
PE
S/M
EC
- UF
RJ on A
pril 30, 2008 w
ww
.jbc.orgD
ownloaded from




















