
Canine Distemper Virus and Measles Virus Fusion Glycoprotein Trimers: Partial Membrane-Proximal...
10
JOURNAL OF VIROLOGY, Aug. 2004, p. 7894–7903 Vol. 78, No. 15 0022-538X/04/$08.000 DOI: 10.1128/JVI.78.15.7894–7903.2004 Copyright © 2004, American Society for Microbiology. All Rights Reserved. Canine Distemper Virus and Measles Virus Fusion Glycoprotein Trimers: Partial Membrane-Proximal Ectodomain Cleavage Enhances Function Veronika von Messling, 1 † Dragana Milosevic, 1 Patricia Devaux, 1 and Roberto Cattaneo 1,2 * Molecular Medicine Program 1 and Virology and Gene Therapy Graduate Track, 2 Mayo Clinic College of Medicine, Rochester, Minnesota Received 9 February 2004/Accepted 31 March 2004 The trimeric fusion (F) glycoproteins of morbilliviruses are activated by furin cleavage of the precursor F 0 into the F 1 and F 2 subunits. Here we show that an additional membrane-proximal cleavage occurs and modulates F protein function. We initially observed that the ectodomain of approximately one in three measles virus (MV) F proteins is cleaved proximal to the membrane. Processing occurs after cleavage activation of the precursor F 0 into the F 1 and F 2 subunits, producing F 1a and F 1b fragments that are incorporated in viral particles. We also detected the F 1b fragment, including the transmembrane domain and cytoplasmic tail, in cells expressing the canine distemper virus (CDV) or mumps virus F protein. Six membrane-proximal amino acids are necessary for efficient CDV F 1a/b cleavage. These six amino acids can be exchanged with the corresponding MV F protein residues of different sequence without compromising function. Thus, structural elements of different sequence are functionally exchangeable. Finally, we showed that the alteration of a block of membrane-proximal amino acids results in diminished fusion activity in the context of a recombinant CDV. We envisage that selective loss of the membrane anchor in the external subunits of circularly arranged F protein trimers may disengage them from pulling the membrane centrifugally, thereby facilitating fusion pore formation. Fusion of cellular membranes is a fundamental biological process (21). The mechanisms of membrane fusion have been characterized in detail by using viral fusion glycoproteins (vFGps) (19). Type I vFGps include the fusion proteins of retroviruses, filoviruses, coronaviruses, orthomyxoviruses, and paramyxoviruses (7, 10, 46). Type II vFGps are exemplified by the envelope proteins of flaviviruses and alphaviruses (18, 25). Both type I and type II vFGps are arranged as trimers at fusion: rings of five to six type II homotrimers inserted in the target membrane have been visualized and modeled in atomic detail (11, 12), and a similar arrangement of type I homotri- mers has been postulated (27). Ring-like structures may facil- itate the concerted activation of vFGps and the synchronized release of their conformational energy. Type I vFGps are synthesized as monomers but trimerize after their cotranslational insertion into the membrane of the endoplasmic reticulum, glycosylation, and folding (19). Follow- ing trimerization, type I vFGps are cleaved by host proteases, an essential step in their activation (13, 23, 34). Cleavage and activation of most vFGps rely on the ubiquitous intracellular protease furin, but the activation of vFGps of certain para- and orthomyxoviruses depends on tissue-specific proteases that de- termine tropism (14, 15, 44). On proteolytic processing, vFGps are in a metastable state, essentially primed for fusion. Acti- vated type I vFGps are composed of a membrane-anchored and a membrane-distal subunit, which are named F 1 and F 2 , respectively, in paramyxoviruses. The trimer-of-heterodimer complexes formed by paramyxo- viral F proteins are remarkably similar to those formed by the other type I vFGps of orthomyxo- and retroviruses (2, 40). In particular, the membrane-anchored F 1 subunit contains two hydrophobic domains, the fusion peptide and the transmem- brane (TM) segment. These domains are adjacent to con- served heptad repeats, designated HRA and HRB, respec- tively. Distinctive differences among vFGps also exist. In the paramyxoviral F proteins, only a few linker amino acids sepa- rate the fusion peptide from HRA, and the HRB-TM linker peptide is also very short (Fig. 1). The linker regions are much longer in other vFGps (35). The mechanism of activation of paramyxoviral F proteins is similar to that of the human immunodeficiency virus envelope (Env) protein: it occurs at neutral pH and starts with binding of the viral attachment protein to its receptor (8, 49). The viral attachment protein then interacts with F, and this interaction leads to the rearrangement of HRA and HRB and the forma- tion of a stable six-helix bundle structure after insertion of the hydrophobic fusion peptide into the target membrane (2). The three-dimensional structure of a recombinantly ex- pressed ectodomain of the Newcastle disease virus F protein has been determined by X-ray crystallography at 3.3 A ˚ resolu- tion (6), and the three-dimensional structure of the proteolyti- cally processed F 1 and F 2 Sendai virus trimeric complex has been determined by electron cryomicroscopy at 16 A ˚ resolu- tion (26). These studies, together with low-resolution struc- tural analyses that identified “lollipop”- and “cone”-shaped F protein trimers in human respiratory syncytial virus (3), and * Corresponding author. Mailing address: Molecular Medicine Pro- gram, Mayo Clinic, Guggenheim 1838, 200 1st St. SW, Rochester, MN 55905. Phone: (507) 284-0171. Fax: (507) 266-2122. E-mail: cattaneo [email protected]. † Present address: INRS-Institut Armand-Frappier, Universite du Quebec, Quebec, Laval H7V 1B7, Canada. 7894
Transcript of Canine Distemper Virus and Measles Virus Fusion Glycoprotein Trimers: Partial Membrane-Proximal...

JOURNAL OF VIROLOGY, Aug. 2004, p. 7894–7903 Vol. 78, No. 150022-538X/04/$08.00�0 DOI: 10.1128/JVI.78.15.7894–7903.2004Copyright © 2004, American Society for Microbiology. All Rights Reserved.
Canine Distemper Virus and Measles Virus Fusion GlycoproteinTrimers: Partial Membrane-Proximal Ectodomain Cleavage
Enhances FunctionVeronika von Messling,1† Dragana Milosevic,1 Patricia Devaux,1 and Roberto Cattaneo1,2*
Molecular Medicine Program1 and Virology and Gene Therapy Graduate Track,2 MayoClinic College of Medicine, Rochester, Minnesota
Received 9 February 2004/Accepted 31 March 2004
The trimeric fusion (F) glycoproteins of morbilliviruses are activated by furin cleavage of the precursor F0into the F1 and F2 subunits. Here we show that an additional membrane-proximal cleavage occurs andmodulates F protein function. We initially observed that the ectodomain of approximately one in three measlesvirus (MV) F proteins is cleaved proximal to the membrane. Processing occurs after cleavage activation of theprecursor F0 into the F1 and F2 subunits, producing F1a and F1b fragments that are incorporated in viralparticles. We also detected the F1b fragment, including the transmembrane domain and cytoplasmic tail, incells expressing the canine distemper virus (CDV) or mumps virus F protein. Six membrane-proximal aminoacids are necessary for efficient CDV F1a/b cleavage. These six amino acids can be exchanged with thecorresponding MV F protein residues of different sequence without compromising function. Thus, structuralelements of different sequence are functionally exchangeable. Finally, we showed that the alteration of a blockof membrane-proximal amino acids results in diminished fusion activity in the context of a recombinant CDV.We envisage that selective loss of the membrane anchor in the external subunits of circularly arranged Fprotein trimers may disengage them from pulling the membrane centrifugally, thereby facilitating fusion poreformation.
Fusion of cellular membranes is a fundamental biologicalprocess (21). The mechanisms of membrane fusion have beencharacterized in detail by using viral fusion glycoproteins(vFGps) (19). Type I vFGps include the fusion proteins ofretroviruses, filoviruses, coronaviruses, orthomyxoviruses, andparamyxoviruses (7, 10, 46). Type II vFGps are exemplified bythe envelope proteins of flaviviruses and alphaviruses (18, 25).Both type I and type II vFGps are arranged as trimers atfusion: rings of five to six type II homotrimers inserted in thetarget membrane have been visualized and modeled in atomicdetail (11, 12), and a similar arrangement of type I homotri-mers has been postulated (27). Ring-like structures may facil-itate the concerted activation of vFGps and the synchronizedrelease of their conformational energy.
Type I vFGps are synthesized as monomers but trimerizeafter their cotranslational insertion into the membrane of theendoplasmic reticulum, glycosylation, and folding (19). Follow-ing trimerization, type I vFGps are cleaved by host proteases,an essential step in their activation (13, 23, 34). Cleavage andactivation of most vFGps rely on the ubiquitous intracellularprotease furin, but the activation of vFGps of certain para- andorthomyxoviruses depends on tissue-specific proteases that de-termine tropism (14, 15, 44). On proteolytic processing, vFGpsare in a metastable state, essentially primed for fusion. Acti-vated type I vFGps are composed of a membrane-anchored
and a membrane-distal subunit, which are named F1 and F2,respectively, in paramyxoviruses.
The trimer-of-heterodimer complexes formed by paramyxo-viral F proteins are remarkably similar to those formed by theother type I vFGps of orthomyxo- and retroviruses (2, 40). Inparticular, the membrane-anchored F1 subunit contains twohydrophobic domains, the fusion peptide and the transmem-brane (TM) segment. These domains are adjacent to con-served heptad repeats, designated HRA and HRB, respec-tively. Distinctive differences among vFGps also exist. In theparamyxoviral F proteins, only a few linker amino acids sepa-rate the fusion peptide from HRA, and the HRB-TM linkerpeptide is also very short (Fig. 1). The linker regions are muchlonger in other vFGps (35).
The mechanism of activation of paramyxoviral F proteins issimilar to that of the human immunodeficiency virus envelope(Env) protein: it occurs at neutral pH and starts with bindingof the viral attachment protein to its receptor (8, 49). The viralattachment protein then interacts with F, and this interactionleads to the rearrangement of HRA and HRB and the forma-tion of a stable six-helix bundle structure after insertion of thehydrophobic fusion peptide into the target membrane (2).
The three-dimensional structure of a recombinantly ex-pressed ectodomain of the Newcastle disease virus F proteinhas been determined by X-ray crystallography at 3.3 A resolu-tion (6), and the three-dimensional structure of the proteolyti-cally processed F1 and F2 Sendai virus trimeric complex hasbeen determined by electron cryomicroscopy at 16 A resolu-tion (26). These studies, together with low-resolution struc-tural analyses that identified “lollipop”- and “cone”-shaped Fprotein trimers in human respiratory syncytial virus (3), and
* Corresponding author. Mailing address: Molecular Medicine Pro-gram, Mayo Clinic, Guggenheim 1838, 200 1st St. SW, Rochester, MN55905. Phone: (507) 284-0171. Fax: (507) 266-2122. E-mail: [email protected].
† Present address: INRS-Institut Armand-Frappier, Universite duQuebec, Quebec, Laval H7V 1B7, Canada.
7894

mutational studies of the HRA and HRB domains of simianvirus 5 (SV5) (41) on the one hand indicated that the F pro-teins of paramyxoviruses have similar structures and on theother identified structurally and functionally different con-formers and intermediates.
Despite a wealth of knowledge, no consensus about themechanism of fusion by type I vFGps has been reached. Nev-ertheless, one fusion model agrees with most of the experi-mental data and is widely accepted. It predicts that fusionprotein trimers are organized in rings before fusion. Uponconcerted activation, the fusion peptides are propelled over adistance of many nanometers into the target membrane. In thesecond step, the HRBs fold back on the HRAs, forming asix-helix bundle while bringing the TM segment into proximitywith the fusion peptide, pulling the two membranes togetherand forming the fusion pore. This model, however, does notexplain how bending towards the ring center is achieved by the
refolding proteins in spite of rotational symmetry of thetrimers.
Therefore, alternative models of the fusion mechanismshave been considered. One proposes that the fusion peptideinserts into the viral rather than the target membrane and thatwhen the fusion proteins are grouped in a ring around a centralpatch of lipid, a dimple is created that destabilizes the mem-brane and initiates the fusion reaction (24). Another modelconsiders that fusion peptides from the same trimer insertsimultaneously into both the target and the viral membraneand then zipper up the trimeric coiled coil to pull the twomembranes together, ultimately destabilizing the bilayer andcausing lipid exchange (47).
We show here that the ectodomain of the type I vFGps ofthree paramyxoviruses undergo partial membrane-proximalcleavage and that proteolytic processing enhances fusion func-tion. We suggest that membrane-proximal cleavage of the ex-
FIG. 1. Scheme of the CDV F protein and sequences of the membrane-proximal regions of three paramyxoviral F proteins (A) and Westernblot analysis of different paramyxoviral F proteins (B and C). (A, top) Linear drawing of the CDV F protein. This protein is synthesized with along amino-terminal precursor sequence that is cleaved posttranslationally prior to cleavage activation of the F0 precursor into the disulfide-linkedF1 and F2 subunits (53). Hydrophobic regions are indicated by hatched boxes. F protein subunits are labeled as follows: signal peptide (SP), F0precursor, F1 and F2 subunits, and the F1a and F1b fragments that result from the newly identified cleavage. The disulfide bond (SS) connectingthe F1 and F2 subunits is shown below the F protein scheme. The boxes marked HRA and HRB indicate the positions of the corresponding heptadrepeats. (Bottom) Alignment of sequences surrounding the putative cleavage regions of CDV, MV, and MuV F proteins. Identical residues areindicated by dots, hydrophobic residues in the first and fourth (a and d) positions of HRB are in bold, and predicted transmembrane domainresidues are italicized. (B and C) Characterization of the different F protein forms by Western blot analysis. Proteins were extracted from purifiedCDV or MV particles (par) or Vero cells transfected (tr) with the CDV, MV, or MuV F protein expression plasmids or infected (inf) with CDVor MV, or control uninfected cells (ctr). Lysates were separated by reducing SDS-PAGE and blotted onto polyvinylidene difluoride membranes.The F proteins were revealed with anti-cytoplasmic tail (B) or anti-F431 (C) serum. The positions of F0, F1, F1a, and F1b are indicated on the left.
VOL. 78, 2004 MEMBRANE-PROXIMAL CLEAVAGE OF TRIMERIC vFGps 7895

ternal trimer subunit may disengage it from pulling the mem-brane centrifugally, thereby enhancing fusion efficiency.
MATERIALS AND METHODS
Cells and virus purification. Vero cells (ATCC CCL-81) were used for allexperiments. The cells were maintained in Dulbecco’s modified Eagle’s medium(DMEM; Invitrogen) with 5% fetal calf serum (Invitrogen). CDV Onderstepoortstrain (CDVOS) and MV Edmonston strain (MVEdm) were propagated in Verocells.
To obtain purified viral particles, two 175-cm2 dishes were infected with eachvirus and incubated at 32°C until at least 80% of the cells were in syncytia. Thesupernatant was cleared by centrifugation at 3,000 rpm in a tabletop centrifuge(Sorvall) for 20 min at 4°C and overlaid onto a 20 to 60% sucrose gradient inTNE buffer (10 mM Tris [pH 7.8], 100 mM NaCl, 1 mM EDTA). After centrif-ugation at 28,000 rpm in a TH641 rotor (Sorvall) for 1.5 h at 4°C, the interphasewas transferred into a fresh tube, adjusted to 20% sucrose in TNE buffer and thecentrifugation was repeated at 28,000 rpm for 1.5 h at 4°C. The pellet wasresuspended in 200 �l of TNE buffer, aliquoted, and stored at �70°C. The titerof each preparation was determined, and sample volumes for Western blotanalysis were adjusted accordingly.
Construction of expression plasmids. Plasmid pCG-F (55) constituted thebasis for all CDV F protein mutants. Soluble F proteins with increasing deletionsin the F1 C terminus were generated by PCR (Expand High Fidelity PCR system;Roche Biochemicals) with the common forward primer TTTGGATCCGGTCAACCAGGTCCACCAGCCAGG, which introduces a BamHI site upstream ofthe coding region (indicated in italics), and the reverse primers TTTGCATGCTCATTACTTGTCATCGTCATCCTTGTAGTCATTAAAGGAAGAGCGCCTAACCGTCTC (pCG-sF605), TTTGCATGCTCATTACTTGTCATCGTCATCCTTGTAGTCGATCTGGTTAGAGGAGTCTATCAG (pCG-sF595), andTTTGCATGCTCATTACTTGTCATCGTCATCCTTGTAGTCAGCATCCAGTTTCTTAAGGGCG (pCG-sF585), adding the Flag peptide (underlined) between theend of the truncated F1 subunit and two stop codons, which are followed by a SphIsite (italics). Plasmid pCG-FTryps (54) was used as the template for the generationof the corresponding soluble F proteins pCG-sFtryps/605, pCG-sFtryps/595andpCG-sFtryps/585, in which the furin cleavage motif RRQRR/F was changed intothe trypsin cleavage motif RNHNR/F. The mumps virus (MuV) F protein ex-pression plasmid pCG-FMuV was obtained from P. Duprex (unpublished data).
A set of CDV F protein mutants were generated by site-directed mutagenesis(Stratagene), in which the residues between E597 and L610 were changed toalanine in groups of two or three, resulting in pCG-F597/9 (A1), pCG-F600/1(A2), pCG-F602/4 (A3), pCG-F605/7 (A4), and pCG-F608/10 (A5). The com-bination of mutants A3 and A4, A4 and A5, and A3 through A5 yielded pCG-F602/7 (A3/4), pCG-F605/10 (A4/5) and pCG-F602/10 (A3-5). To interruptextended alanine stretches, the two additional constructs pCG-F602S/10 (S3-5)and pCG-F602/10S (3-5S) were generated, in which apolar residues in eitherA3/4 (S3-5) or A5 (3-5S) were changed to serine instead of alanine. Finally, thefollowing mutants were obtained: pCG-F�602/5 (�1) and pCG-F606-9 (�2),which lack the corresponding residues, and pCG-F605/10MV (M4/5) and pCG-F602/10MV (M3-5), in which the respective CDV F protein residues are re-placed by the corresponding MV F protein residues. The sequences of all con-structs were verified (ABI Prism 377 DNA Sequencer; Perkin-Elmer AppliedBiosystems).
Construction and recovery of recombinant viruses. The mutation A3-5 wasintroduced into the context of the full-length CDV genome of the vaccine strainOnderstepoort with the internal restriction sites AflII and PacI. The fragment tobe inserted was assembled by overlap extension PCR (20), combining the F genedownstream of the AflII site with the part of the untranslated region between theF and H gense that is located upstream of the PacI site. After sequence verifi-cation of the inserted fragments, recombinant viruses were recovered as de-scribed previously with an MVA-T7-based system (55). First, small syncytia weredetected around 12 days after transfection, compared to 6 days in the controltransfected with standard virus cDNA. For each virus, three syncytia werepicked, transferred to fresh Vero cells in six-well plates, and expanded into75-cm2 flasks with 10 ml of DMEM supplemented with 2% fetal calf serum.When the cytopathic effect was pronounced, the cells were scraped into themedium and subjected once to freezing and thawing. The cleared supernatantswere used for all further analysis.
Fusion assay. The quantitative fusion assay based on luciferase as the reportergene (53) was used. Briefly, Vero cells were transfected with the different Fexpression plasmids together with pCG-H/OL and pTM1-luc at a molar ratio of1:1:0.7 with Lipofectamine 2000 (Gibco-BRL). For each transfected well, asecond well of Vero cells was infected with modified vaccinia virus Ankara
expressing the T7 polymerase (MVA-T7) (31) with a multiplicity of infection of1. Twelve hours after transfection or infection, the cells were detached with 50 �lof 0.25% trypsin–EDTA (Gibco-BRL), resuspended in 1 ml of fresh DMEMwith 5% fetal calf serum, and transferred into two wells of a 24-well plate.Following visual grading of the fusion activity, luciferase activity was determinedwith the Steady-Glo luciferase assay system (Promega) and a 96-well platereading luminometer (Topcount-NXT; Packard). A fraction of each lysate wasmixed with an equal amount of 2� Laemmli sample buffer (Bio-Rad) containing100 mM dithiothreitol and subjected to Western blot analysis.
Western blot analysis. Vero cells were seeded into 12-well plates, transfectedwith the different constructs or infected with the respective viruses, and incu-bated at 37°C for 36 h or until profound cytopathic effect was observed. For theanalysis of cellular proteins, cells were washed twice with ice-cold phosphate-buffered saline (PBS; Invitrogen) before adding 100 �l of lysis buffer (150 mMNaCl, 1.0% Triton X-100, 50 mM Tris-HCl, pH 8.0) with complete proteaseinhibitor (Roche Biochemicals). After incubation for 20 min at 4°C, the lysateswere cleared by centrifugation at 5,000 � g for 15 min at 4°C, and the superna-tant was mixed with an equal amount of 2� Laemmli sample buffer (Bio-Rad)containing 100 mM dithiothreitol. For the analysis of soluble proteins, cells werewashed twice with PBS, and 500 �l of DMEM without fetal calf serum was added12 h after transfection. The supernatant was exchanged every 24 h for the next3 days, and the removed supernatant was cleared by centrifugation at 5,000 � gfor 15 min at 4°C before mixing with an equal amount of 2� Laemmli samplebuffer (Bio-Rad) containing 100 mM dithiothreitol. Samples were incubated for10 min at 95°C, followed by fractionation on a sodium dodecyl sulfate (SDS)–15% polyacrylamide gel (Bio-Rad) and blotting on polyvinylidene difluoridemembranes (Millipore). After blocking with 1% blocking reagent (Roche Bio-chemicals) overnight, the membranes were incubated with the antibody recog-nizing the epitope of interest. Following incubation with a peroxidase-conjugatedsecondary antiserum, the membranes were subjected to enhanced chemilumi-nescence (ECL) detection (Amersham Pharmacia Biotech). Band intensitieswere determined with NIH Image software version 1.6. Films from at least threeindependent experiments were used to determine the F1/F1b ratios.
Surface biotinylation. Vero cells were seeded into 12-well plates, transfectedwith the different constructs, and incubated at 37°C for 36 h. Cells were shiftedto 4°C and washed once with cold PBS before 125 mg of EZ-Link Sulfo-NHS-LC-biotin (Pierce) dissolved in 0.3 ml of cold PBS was added to each well. Afterincubation for 20 min on a rocker platform at 4°C, cells were washed with 0.5 Mglycine in PBS, followed by the addition of 1 ml of 0.5 M glycine–PBS andincubation for 20 min at 4°C to quench the excess biotin. Cells were lysed in 300�l of radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1.0% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 8.0) with completeprotease inhibitor (Roche Biochemicals), and further processed as describedabove. Instead of adding the 2� sample buffer directly, the supernatant wasmixed with 50 �l of protein A-agarose beads (Bio-Rad), and the antibody wasadded at the appropriate concentration. Following incubation at 4°C overnight,the beads were washed three times in RIPA buffer before 30 �l of 2� Laemmlisample buffer (Bio-Rad) containing 100 mM dithiothreitol was added. The sam-ples then underwent Western blot analysis as described above. The membraneswere incubated with peroxidase-conjugated streptavidin (Amersham PharmaciaBiotech) and subjected to ECL detection.
Production of antibodies. For the detection of CDV and MV F proteins, arabbit antipeptide antiserum recognizing the 14 carboxy-terminal amino acidswas used (5). A similar rabbit anti-F ectodomain peptide serum was generated.Towards this end, the peptide QVGSRRYPDAVYLHR(C), corresponding toMV F amino acids 431 to 445 with a carboxyl-terminal cysteine, was synthesizedand coupled to keyhole limpet hemocyanin. This conjugate was used to producea rabbit antiserum. Moreover, a peptide antiserum was raised against the MuVF protein. Towards this end, the peptide (C)NTISSSVDDLIRY, correspondingto the 13 carboxy-terminal residues, was coupled to keyhole limpet hemocyanin.A commercially available monoclonal antibody (M2; Sigma) was used to detectthe Flag-tagged proteins.
Membrane purification and trypsin digestion. Two wells of a six-well plateseeded with Vero cells were infected with CDVOS or transfected with plasmidsexpressing the unaltered CDV F protein, a CDV F protein in which the furinconsensus sequence was mutated to a trypsin consensus sequence (pCG-Ftryps;T), or a CDV F protein that contains an endoplasmic reticulum retention se-quence in its cytoplasmic tail (pCG-FER) (54). Thirty-six hours after transfectionor infection, the cells were washed twice with Tris-buffered saline (TBS; 50 mMTris-HCl [pH 7.5], 150 mM NaCl) and 0.45 ml of 0.1� TBS was added to eachwell. After incubation for 10 min at 4°C, cells were scraped into the supernatantand transferred into an Eppendorf tube, and 0.05 ml of 10� TBS was added. The
7896 VON MESSLING ET AL. J. VIROL.

lysate was precleared by centrifugation at 2,500 rpm for 20 min in a minicentri-fuge at 4°C. The supernatant was layered on 2.7 ml of 250 mM sucrose–TBS andcentrifuged for 30 min at 37,000 rpm in a T61 rotor (Sorvall). The pellet was usedfor all further experiments.
For proteolysis with trypsin, the pellet was resuspended in 80 �l of TBS with2 mM tetracaine and 2 mM CaCl2 and divided into two equal aliquots. Trypsinwas either omitted or added to a final concentration of 25 �g/ml. After incuba-tion for 1 h on ice, 30 �g of aprotinin per ml was added to terminate theproteolysis. Following an additional incubation for 20 min on ice, all sampleswere adjusted to 0.5% NP-40 and solubilized for 20 min on ice. The samples werecentrifuged for 15 min at 14,000 rpm in a minicentrifuge at 4°C, and the super-natant was mixed with an equal amount of 2� Laemmli buffer (Bio-Rad) with100 mM dithiothreitol and subjected to Western blot analysis as described above.
RESULTS
Small F protein carboxy-terminal fragment is detected intransfected and infected cells and in purified particles of threeparamyxoviruses. With an F protein cytoplasmic tail-specificantiserum, we occasionally observed a small band in Westernblots of lysates of cells expressing the F proteins of twoparamyxoviruses, the morbilliviruses MV and CDV. To bettercharacterize this fragment, we analyzed lysates of cells trans-fected with F protein expression plasmids or cells infected withMV or CDV or purified viral particles. With the same anti-serum, we detected an approximately 10-kDa band in lysates oftransfected and infected cells as well as purified viral particles(Fig. 1B). Some variability in band size or a double band wasdetected in some samples. We suspect this may be due tofurther proteolytic degradation of the small fragment. Detec-tion of this F protein carboxy-terminal fragment in all samplesindicates that it is produced in both transfected and infectedcells and that it is incorporated into viral particles. We suspectthat this small fragment has been previously overlooked due toits size and its lack of reactivity against antibodies raisedagainst the F protein ectodomain.
We then asked whether the complementary fragment couldalso be detected. For this we raised a serum against a peptidecorresponding to a hydrophilic sequence of the MV F1 ectodo-main. With this antiserum, we detected an approximately 30-kDa band in purified viral particles (Fig. 1C, band F1a; thisantiserum does not react with the CDV F protein). The factthat a strong F1 band was maintained in all lysates (Fig. 1B andC) indicates that only a fraction of all F proteins undergo theadditional cleavage. We refer to the newly identified fragmentsas F1a and F1b. Since the CDV F protein is more stable thanthe MV F protein, further analyses focused on it.
We then asked whether an F1b-like fragment is produced bya representative of another Paramyxoviridae subfamily (30),the rubulavirus MuV. Indeed, with an antiserum raised againstthe 13 carboxy-terminal residues of the MuV F protein, a smallfragment was detected in a cell lysate (Fig. 1B, right panel). Wedetermined the ratio of cleaved (F1b) to uncleaved (F1) pro-teins by densitometry. In purified virions we measured an av-erage 1:2 ratio for CDV (Fig. 1B, left panel, and data notshown) and an apparently lower ratio for MV, in the range of1:3 to 1:4 (Fig. 1B, center panel). However, the MV F1a to F1
ratio also ranged around 1:2 (Fig. 1C), suggesting that approx-imately one-third of morbillivirus F proteins incorporated inviral particles have undergone the additional cleavage.
We then compared the amino acid sequences in the regionspreceding the predicted transmembrane domain of CDV, MV,
and MuV (30). The hydrophobic residues in positions 1 and 4(Fig. 1A, bottom) of the HRB domain allow alignment of thethree sequences, but otherwise little conservation exists.Downstream of the HRB amphipathic helix, five to sevenamino acids precede the hydrophobic region predicted to crossthe membrane. A single residue (serine) was conserved amongthe three viruses.
F1a/b cleavage requires endoplasmic reticulum transit andF1/2 cleavage. To determine the sequence of events occurringduring F protein processing and maturation, we relied on twomutants, Ftryps, in which the furin cleavage sequence ischanged to a trypsin cleavage sequence, resulting in a proteinthat is efficiently processed and transported to the cell surfacein its uncleaved form; and FER, in which an endoplasmic re-ticulum retention signal has been introduced (54). Both FER
and Ftryps can be cleaved by trypsin after cell lysis. Membranepreparations were used for these experiments because theywere less prone to nonspecific proteolytic cleavage (data notshown).
In membrane protein extracts of CDV-infected cells or cellstransfected with the plasmid expressing the unaltered F pro-tein, trypsin digestion led to the disappearance of F0, nomarked increase in F1, and slightly reduced F1b (Fig. 2, lanesCDV� and F�), suggesting that trypsin may not cleave at theF1a/b junction. In the case of Ftryps, F0 was the dominant formdetected in the undigested sample (Fig. 2, lane Ftryps�). Tryp-sin digestion of this protein resulted in increases of not only F1
but also F1b, suggesting the possibility that F1/2 cleavage is aprerequisite for F1a/b processing (Fig. 2, lane Ftryps�). WhileFER was also predominantly detected in its uncleaved F0 formin the undigested sample (Fig. 2, lane FER�), mainly F1 wasdetected in the trypsin-digested aliquot (Fig. 2, lane FER�). Insummary, these findings indicate that F1a/b cleavage is a lateevent in F protein maturation that requires endoplasmic retic-ulum transit and F1/2 cleavage.
FIG. 2. Trypsin digestion of CDV F protein mutants. Western blotanalysis of crude membrane preparations of Vero cells infected withCDV or transfected with different F protein expression plasmids wasperformed. Cells underwent hypotonic lysis prior to pelleting through250 mM sucrose in TBS. The pellet was resuspended and divided intotwo equal aliquots, one of which was subjected to trypsin digestion (�),while the other served as a control (�). Lysates were separated byreducing SDS-PAGE (15% acrylamide) and blotted onto polyvinyli-dene difluoride membranes. The F proteins were revealed with ananti-cytoplasmic tail serum. The positions of F0, F1, and F1b are indi-cated on the left. Note that trypsin digestion reduced the amount of allF protein forms detected regardless of the mutant analyzed. C, controlnontransfected cells; F, Ftryps, and FER, cells transfected with pCG-F,pCG-Ftryps, and pCG-FER, respectively.
VOL. 78, 2004 MEMBRANE-PROXIMAL CLEAVAGE OF TRIMERIC vFGps 7897

F1a/b cleavage is conformation dependent and occurs be-tween residues 595 and 608. To localize the F1a/b cleavage site,we reasoned that we could monitor the loss of a terminal tagsequence added at different positions towards the end of theHRB sequence or further downstream (see scheme in Fig. 3A).We generated F proteins with a Flag tag inserted after aminoacid 585 (deleting the last two heptad repeats; Fig. 3B), afterresidue 595 (deleting the last heptad repeat), or after residue608 (leaving the whole ectodomain intact). Since the Flag tagwas followed by a stop codon, secretion of these proteins wasexpected. We reasoned that F1a/b cleavage may result in loss ofprotein detection, thus allowing us to map the cleavage siteposition.
With the anti-Flag antibody, we detected the F1 ectodomain
in the supernatant of cells transfected with sF585 and sF595 butnot in the supernatant of sF608-transfected cells (Fig. 3C),indicating that the cleavage site is located upstream of residue608 but downstream of amino acid 595. In control furin cleav-age-resistant F proteins, F0 ectodomains were secreted (Fig.3C, lanes sFtryps/585, sFtryps/595, and sFtryps/608), confirming thatF1a/b processing depends on F1/2 cleavage. However, thesFtryps/608 signal was reduced compared to the intensity of thecorresponding bands of sFtryps/585 and sFtryps/595, which mayreflect inefficient cleavage independent of F1/2 cleavage orlower stability of this protein form. Thus, the F1a/b cleavage siteis located between residues I595 and S608 in close proximity tothe transmembrane region. In contrast to the membrane-bound F protein, the soluble form is completely cleaved mem-brane proximally, suggesting that membrane anchor loss makesthe cleavage site more accessible.
Inhibition of F1a/b cleavage leads to cell-cell fusion reduc-tion. To identify residues important for cleavage within thepreviously defined 14-amino-acid segment, we then systemati-cally mutated or exchanged blocks of two to three neighboringresidues (Fig. 4A). The resulting mutants were analyzed in aquantitative cell-cell fusion assay, with luciferase as the re-porter gene. While mutants A1 (residues 597 to 599) and A2(residues 600 and 601) were efficiently cleaved membraneproximally (Fig. 4C, lanes A1 and A2, F1b signals; Fig. 4A, %processing column) and exhibited wild-type fusion activity(Fig. 4A, % fusion column), a 40 to 50% reduction of fusionefficiency was observed for mutants A3 and A4 (residues 602 to604 and residues 605 to 607, respectively), which coincidedwith detection of less F1b (Fig. 4B, lanes A3 and A4). MutantA5 (residues 608 to 610) cleavage analysis results were morevariable than for the other mutants, as reflected by the stan-dard deviation in Fig. 4A.
Throughout these experiments, we observed some variabilityin the amount of F1b detected for the different mutants (Fig.4A, % processing column). Nevertheless, the standard F pro-tein and the five mutants that retained 95 to 110% fusionefficiency (A1, A2, M4/5, �1, and �2; see below) all had 37 to42% membrane-proximal processing, whereas those combina-tion mutants that had low fusion efficiency (A3/4, A3-5, and3-5S) had 5 to 7% membrane-proximal processing (Fig. 4A,middle column). Figure 4E shows that the fusion efficiencies ofmost mutants correlated well with the levels of their F1a/b
processing.To assess whether the effects of A3, A4, and A5 are cumu-
lative, we generated plasmids with different combinations ofthese mutations. The combination of A3 and A4 reduced fu-sion activity to near the background (Fig. 4A, A3/4, % fusioncolumn), and little F1b was detected (Fig. 4B, lane A3/4). Incontrast, fusogenicity and F1b production of the A4/5 combi-nation mutant remained in the range of A4 (Fig. 4A, % fusionand % processing columns). For the 3-5 triple combinationmutant, in addition to A3-5 with eight consecutive alanines,two constructs with serines interrupting the alanine stretcheswere generated (3-5S and S3-5, Fig. 4A). Fusion activity andmembrane-proximal processing of mutants A3-5 and 3-5S weresimilar to those of A3/4, while those of S3-5 were in the rangeof A4 and A4/5 (Fig. 4A, % fusion and % processing columns).Since surface expression of all mutants reached at least wild-type levels (Fig. 4D) and was thus not a limiting factor, these
FIG. 3. Scheme of the F protein (A), sequence of its membrane-proximal region (B), and initial mapping of the membrane-proximalcleavage site (C). (A) Scheme of the membrane-bound F protein (left)and of the truncated F proteins sF608, sF595, and sF585. The disulfidebond connecting the F1 and F2 subunits is symbolized by a line. Thecell membrane is indicated by a gray rectangle, the Flag tag by a whitebox, and the cleavage site by an arrow. (B) The position of the lastresidue of different proteins is indicated with an ordinal number and abent arrow. The hydrophobic a and d residues of HRB are in bold, andpredicted transmembrane domain residues are italicized. (C) Westernblot analysis of soluble F proteins. Supernatants of Vero cells trans-fected with the indicated plasmids were subjected to reducing SDS-PAGE (10% acrylamide) and blotted onto polyvinylidene difluoridemembranes. Soluble F proteins were detected with a monoclonal an-tibody directed against the Flag peptide that was added at the trunca-tion site. The positions of F0 and F1 are indicated on the left.
7898 VON MESSLING ET AL. J. VIROL.

FIG. 4. Sequence, processing, fusion activity, and surface expression of CDV F protein cleavage mutants. (A) Sequence of the region of interest ofthe original and mutant F proteins, efficacy of their membrane-proximal processing, and fusion activity. The names of the mutant F proteins are indicatedon the left. Mutated residues are bold. The end of HRB and the beginning of the transmembrane region (TM) are marked by bent arrows. Forquantitative processing assays (means and standard deviation are indicated in the % processing column), Western blots from three independentexperiments similar to those shown in panels B and C were evaluated. For quantitative fusion assays, Vero cell monolayers were either infected withMVA-T7 (multiplicity of infection of 1) or transfected with the different F constructs, a plasmid coding for the H protein, and a plasmid containing theluciferase reporter gene under control of the T7 promoter. Twelve hours after transfection, both cell populations were mixed and seeded into fresh plates.After 36 h at 37°C, fusion was quantified by measuring luciferase activity. For each experiment, the value measured for the parental F protein was setto 100%. The means and standard deviations of four independent experiments done in duplicate are indicated in the % fusion column. (B and C) Westernblot analysis of CDV F protein mutants. Lysates were separated by reducing SDS-PAGE and blotted onto polyvinylidene difluoride membranes. The Fproteins were revealed with an anti-cytoplasmic tail serum. The positions of F0, F1, and F1b are indicated on the left. (D) Surface biotinylation. A duplicatewell of the cells used for Western blot analysis was shifted to 4°C, biotin labeled, lysed, and immunoprecipitated overnight with the anti-Fcyt rabbitantipeptide antibody. Samples were separated by reducing SDS-PAGE, blotted onto polyvinylidene difluoride membranes, and probed with peroxidase-coupled streptavidin. (E) Correlation of fusion activity (y axis) with processing efficiency (x axis) for the 15 proteins characterized.
7899
results indicate that the residues most relevant for cleavage aresituated between S602 and G607.
Different primary amino acid sequences can be efficientlycleaved. Having noted that the primary sequences of CDV andMV differ in the cleavage region, we asked whether MV se-quences could nevertheless functionally substitute the corre-sponding CDV segment. Towards this end, we replaced resi-dues 602 to 607 or residues 602 to 610 with the correspondingMV F protein amino acids (Fig. 4A, M4/5 and M3-5, respec-tively). In contrast to the alanine mutant A3/4, M4/5 displayedfusion activity similar to that of standard F (Fig. 4A, % fusion)and was efficiently cleaved (Fig. 4C, lane M4/5); the fusoge-nicity of M3-5 was only slightly reduced (Fig. 4A and C, lanesM3-5). The fact that two divergent primary sequences can becleaved with similar efficiency suggests that an exposed struc-ture may be the primary determinant of proteolytic cleavage.
To complete the characterization of the requirements forefficient cleavage of CDV F at this site, we generated twomutants in which four amino acids, either residues 602 to 605(�1) or 606 to 609 (�2), were deleted (Fig. 4A, bottom twolines). Both mutants were efficiently cleaved and displayedwild-type fusion activity (Fig. 4A, % fusion columns). Theseresults concur with the MV F segment substitution analysis insuggesting that the main cleavage requirements are close prox-imity to the transmembrane region and a tertiary structurecompatible with protease accessibility and cleavage.
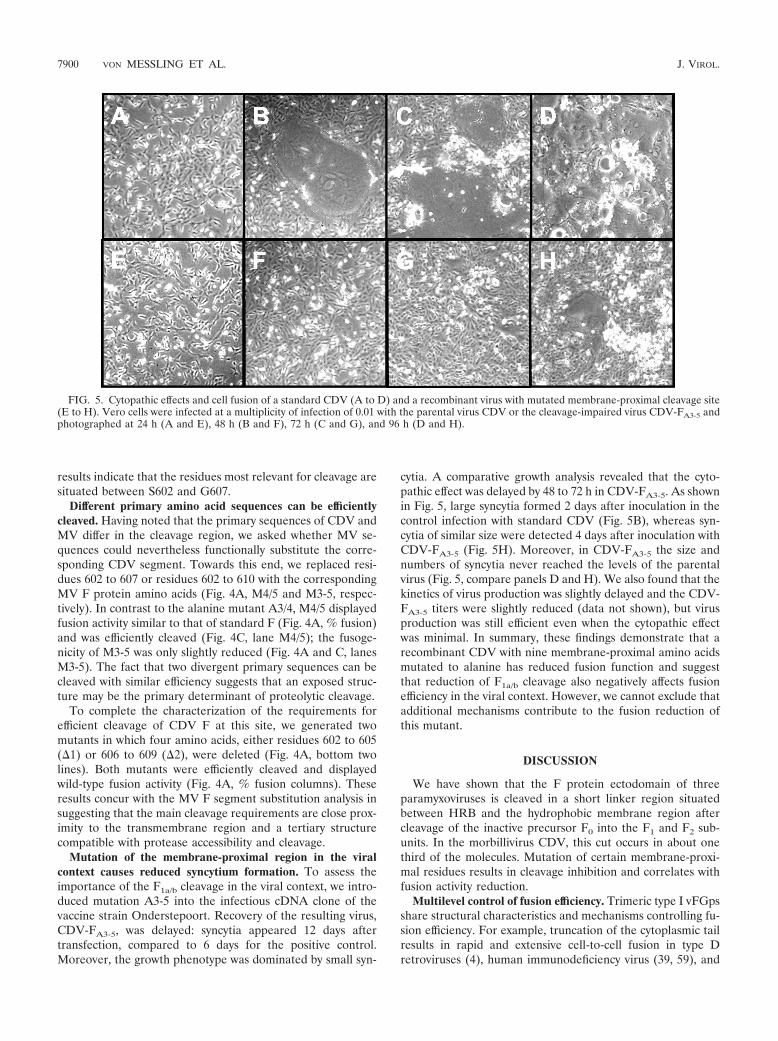
Mutation of the membrane-proximal region in the viralcontext causes reduced syncytium formation. To assess theimportance of the F1a/b cleavage in the viral context, we intro-duced mutation A3-5 into the infectious cDNA clone of thevaccine strain Onderstepoort. Recovery of the resulting virus,CDV-FA3-5, was delayed: syncytia appeared 12 days aftertransfection, compared to 6 days for the positive control.Moreover, the growth phenotype was dominated by small syn-
cytia. A comparative growth analysis revealed that the cyto-pathic effect was delayed by 48 to 72 h in CDV-FA3-5. As shownin Fig. 5, large syncytia formed 2 days after inoculation in thecontrol infection with standard CDV (Fig. 5B), whereas syn-cytia of similar size were detected 4 days after inoculation withCDV-FA3-5 (Fig. 5H). Moreover, in CDV-FA3-5 the size andnumbers of syncytia never reached the levels of the parentalvirus (Fig. 5, compare panels D and H). We also found that thekinetics of virus production was slightly delayed and the CDV-FA3-5 titers were slightly reduced (data not shown), but virusproduction was still efficient even when the cytopathic effectwas minimal. In summary, these findings demonstrate that arecombinant CDV with nine membrane-proximal amino acidsmutated to alanine has reduced fusion function and suggestthat reduction of F1a/b cleavage also negatively affects fusionefficiency in the viral context. However, we cannot exclude thatadditional mechanisms contribute to the fusion reduction ofthis mutant.
DISCUSSION
We have shown that the F protein ectodomain of threeparamyxoviruses is cleaved in a short linker region situatedbetween HRB and the hydrophobic membrane region aftercleavage of the inactive precursor F0 into the F1 and F2 sub-units. In the morbillivirus CDV, this cut occurs in about onethird of the molecules. Mutation of certain membrane-proxi-mal residues results in cleavage inhibition and correlates withfusion activity reduction.
Multilevel control of fusion efficiency. Trimeric type I vFGpsshare structural characteristics and mechanisms controlling fu-sion efficiency. For example, truncation of the cytoplasmic tailresults in rapid and extensive cell-to-cell fusion in type Dretroviruses (4), human immunodeficiency virus (39, 59), and
FIG. 5. Cytopathic effects and cell fusion of a standard CDV (A to D) and a recombinant virus with mutated membrane-proximal cleavage site(E to H). Vero cells were infected at a multiplicity of infection of 0.01 with the parental virus CDV or the cleavage-impaired virus CDV-FA3-5 andphotographed at 24 h (A and E), 48 h (B and F), 72 h (C and G), and 96 h (D and H).
7900 VON MESSLING ET AL. J. VIROL.

paramyxoviruses (5, 29). Moreover, in paramyxoviruses thelateral interactions of the vFGps and the attachment proteinmodulate the efficiency with which the signal elicited by recep-tor binding is transmitted (8, 16, 38, 49). In addition, N-linkedglycans modulate fusion efficiency in paramyxo- and ortho-myxoviral vFGps (33, 54).
Membrane-proximal F1 protein cleavage is another level atwhich viral fusion can be controlled. Cleavage and activation ofthe vFGps of certain para- and orthomyxoviruses depends ontissue-specific proteases and determines tropism by restrictingfusion to selected tissues (15, 23, 44). Analogously, F1a/b cleav-age may modulate fusion efficiency in different tissues. How-ever, we do not know which protease cleaves in the membrane-proximal region. The primary sequence does not give hints,because it is highly variable even among pairs of closely relatedparamyxoviruses such as the morbilliviruses CDV and MV(Fig. 1) or the rubulaviruses MuV and simian virus 5 (data notshown). Thus, it seems likely that structural elements, andpossibly different proteases, may be involved in membrane-proximal cleavage. Analogously, it has been proposed thatstructural elements as well as recognition sequences contributeto the cleavage susceptibility of cell surface proteins whoseectodomains are cleaved membrane proximally (1, 17).
Two common characteristics emerge from the mutationalanalysis of the vFGps membrane-proximal region of the threeparamyxoviruses simian virus 5 (58), human parainfluenza vi-rus 2 (52), and now CDV: (i) insertion of additional aminoacids is not tolerated, while up to four residues can be deletedwithout negatively affecting fusion activity; (ii) a majority ofresidues are polar, and an increase in apolar amino acids re-sults in reduction of fusion activity. In contrast, an unusuallyhigh concentration of aromatic amino acids exists in the mem-brane-proximal regions of the vFGps of retroviruses, filovi-ruses, orthomyxoviruses, rhabdoviruses, alphaviruses, and fla-viviruses (48). It is known that in the lentiviral vFGps, certainmembrane-proximal tryptophan residues are essential for fu-sion (42) and may have a membrane-partitioning function.Since there are no membrane proximal tryptophan residues inMV, CDV, and MuV, paramyxoviral F proteins cannot rely onthose to distort membranes at fusion.
Mechanism of fusion pore formation. Fusion induced bymost paramyxoviruses relies on two viral proteins, the attach-ment (H or HN for hemagglutinin and hemagglutinin-neura-minidase, respectively) protein that contacts the cellular recep-tor(s), and the F protein that coalesces the viral with thecellular membrane (40). In this respect, paramyxoviral fusion isdifferent from fusion induced by the influenza virus hemagglu-tinin or the retroviral Env; both hemagglutinin and Env can actalone to fuse membranes. Figure 6A shows the cellular recep-FIG. 6. Model of the morbillivirus fusion mechanism. (A) Viral
and cellular proteins involved in fusion; (B) target membrane invasion;and (C) pore formation. (A, bottom) Viral membrane (gray) with theattachment (H) protein tetrameric complex (green) and the F proteintrimer. (Top) Cellular membrane (gray) with the receptor (dark blue).The H protein has a six-blade propeller structure (9, 56) and consistsof two noncovalently linked dimers of covalently linked dimers (36,37). The main receptor protein for both MV and CDV is the signalinglymphocytic activation molecule (SLAM, also called CD150) of theimmunoglobulin superfamily (50, 51). The contact surface areas bothon SLAM (32) and the attachment protein (45, 56) have been defined.The F protein consist of a fusion peptides (blue horizontal cylindricalstructure), a short linker (light gray cylinder), HRA (yellow cylindersnumbered 1 to 3), the body of the F2 and F1 subunits (large elongated red
object), HRB (orange cylinders numbered 4 to 6), another short linker(a gray continuous cylinder in two monomers; an interrupted gray andblue cylinder in the cleaved monomer), and the TM segment with thecytoplasmic tail (blue thin cylindrical structure). (B) Six circularlyarranged F protein trimers in the target membrane invasion confor-mation. (C) Trimers in their most stable six-helix bundle conformationarranged circularly around a fusion pore. Three of the six trimers havebeen removed for visual clarity. The inset is a coronal section of thesix-helix bundle. For details, see the text.
VOL. 78, 2004 MEMBRANE-PROXIMAL CLEAVAGE OF TRIMERIC vFGps 7901

tor and viral proteins involved in morbillivirus fusion: one ofthe F trimer subunits is cleaved membrane proximally (Fig. 6A,black arrow). Figures 6B and C show how membrane-proximalcleavage of the F protein ectodomain may facilitate the fusionprocess.
The model of morbillivirus fusion is based on the assump-tion (21, 57) that type I vFGp trimers are arranged into aring-like structure at the site where the fusion pore will form.It postulates that the fusion process begins with the contact ofa cellular receptor (SLAM; Fig. 6A, top) with the H proteintetrameric complex. This contact may elicit an H protein con-formational change, transmitting the signal laterally to the Fprotein trimer (38, 49). The F protein trimer, in a metastableconformation after cleavage and activation, may then propelthe fusion peptide over a distance of several nanometers to-wards the tip of the molecule and into the target membrane(Fig. 6B, top). In the resulting intermediate conformation, theextended F protein is connected with both membranes and theHRAs form a trimeric coiled-coil (Fig. 6B, top). In a secondstep, the HRBs fold back and dock on the trimeric coiled coil,repositioning the TM segments near the fusion peptides whileforming the fusion pore (Fig. 6C).
The general model of type I vFGps fusion does not explainhow bending of the trimers toward the pore center is achievedin spite of the rotational symmetry of their structures. Themodel shown in Fig. 6B and C illustrates how cleavage of theexternal subunits in a ring of trimers facilitates fusion poreformation by breaking symmetry. The trimers are poised toreach the most stable six-helix bundle conformation: in thisprocess HRB 5 will dock between HRA 1 and HRA 2, HRB 6between or HRA 2 and HRA 3, and HRB 4 between HRA 1and HRA 3 (scheme in the inset of Fig. 6C). In the six-helix-bundle formation process HRB 4 and HRB 6 will drag thelower membrane towards the upper membrane that is concom-itantly pulled down by the trimeric coiled coil. If still connectedwith its TM segment, the subunit with HRB 5 would counteractthe action of the two others. However, membrane-proximalcleavage disengages the HRB 5 subunit from pulling the mem-brane centrifugally; the protein body flips over and connects tothe trimeric coiled coil from the top (HRB 5 in Fig. 6C, centerand inset), whereas its TM segment and cytoplasmic tail areleft behind in the membrane (Fig. 6C, bottom).
Why does membrane-proximal cleavage remain partial? Ourdata suggest that a major determinant for cleavage is an ex-posed structure. We propose that cleavage of a single subunitmay induce a conformational change of the trimer, interferingwith efficient cleavage of the two other subunits. Self-limitingcleavage proximal to the membrane may also favor the recon-figuration of the F protein trimers in a ring.
Can membrane-proximal cleavage modulate the function ofother type I vFGps? We have shown that the MuV F proteinis cleaved proximal to the membrane, and it is therefore con-ceivable that the F protein of other rubulaviruses, includingsimian virus 5, undergoes similar processing. Viruses whoseglycoproteins undergo elaborate proteolytic trimming, such asthe filoviruses Ebola virus and Marburg virus (22, 43), areother candidates for partial membrane-proximal vFGps cleav-age.
How do type I vFGps that are not processed membraneproximally efficiently execute fusion? Structural data have re-
cently suggested that in type II vFGps, a long interdomainlinker permits independent domain rotation and allows spon-taneous symmetry breaking (28). It is possible that certain typeI vFGps have adopted a similar mechanism.
ACKNOWLEDGMENTS
We thank Paul Duprex for providing the MuV F protein expressionplasmid, Cristina Ghenoiu for help with the virus purification, Som-pong Vongpunsawad for excellent technical support, David Smyrk forthe graphics, and Bruce Horazdowsky, Dick Pagano, Robert A. Lamb,and Richard Kuhn for comments on the manuscript.
This work was supported by grants from the Mayo and Siebensfoundations, NIH grant CA90636 to R.C., and an Emmy Noetheraward from the German Research Foundation to V.V.M.
REFERENCES
1. Althoff, K., J. Mullberg, D. Aasland, N. Voltz, K. Kallen, J. Grotzinger, andS. Rose-John. 2001. Recognition sequences and structural elements contrib-ute to shedding susceptibility of membrane proteins. Biochem. J. 353:663–672.
2. Baker, K. A., R. E. Dutch, R. A. Lamb, and T. S. Jardetzky. 1999. Structuralbasis for paramyxovirus-mediated membrane fusion. Mol. Cell 3:309–319.
3. Begona Ruiz-Arguello, M., L. Gonzalez-Reyes, L. J. Calder, C. Palomo, D.Martin, M. J. Saiz, B. Garcia-Barreno, J. J. Skehel, and J. A. Melero. 2002.Effect of proteolytic processing at two distinct sites on shape and aggregationof an anchorless fusion protein of human respiratory syncytial virus and fateof the intervening segment. Virology 298:317–326.
4. Brody, B. A., S. S. Rhee, M. A. Sommerfelt, and E. Hunter. 1992. A viralprotease-mediated cleavage of the transmembrane glycoprotein of Mason-Pfizer monkey virus can be suppressed by mutations within the matrix pro-tein. Proc. Natl. Acad. Sci. USA 89:3443–3447.
5. Cathomen, T., H. Y. Naim, and R. Cattaneo. 1998. Measles viruses withaltered envelope protein cytoplasmic tails gain cell fusion competence. J. Vi-rol. 72:1224–1234.
6. Chen, L., J. J. Gorman, J. McKimm-Breschkin, L. J. Lawrence, P. A. Tul-loch, B. J. Smith, P. M. Colman, and M. C. Lawrence. 2001. The structure ofthe fusion glycoprotein of Newcastle disease virus suggests a novel paradigmfor the molecular mechanism of membrane fusion. Structure (Cambridge)9:255–266.
7. Colman, P. M., and M. C. Lawrence. 2003. The structural biology of type Iviral membrane fusion. Nat. Rev. Mol. Cell. Biol. 4:309–319.
8. Corey, E. A., A. M. Mirza, E. Levandowsky, and R. M. Iorio. 2003. Fusiondeficiency induced by mutations at the dimer interface in the Newcastledisease virus hemagglutinin-neuraminidase is due to a temperature-depen-dent defect in receptor binding. J. Virol. 77:6913–6922.
9. Crennell, S., T. Takimoto, A. Portner, and G. Taylor. 2000. Crystal structureof the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat.Struct. Biol. 7:1068–1074.
10. Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusionand its inhibition. Annu. Rev. Biochem. 70:777–810.
11. Gibbons, D. L., I. Erk, B. Reilly, J. Navaza, M. Kielian, F. A. Rey, and J.Lepault. 2003. Visualization of the target-membrane-inserted fusion proteinof Semliki Forest virus by combined electron microscopy and crystallogra-phy. Cell 114:573–583.
12. Gibbons, D. L., M. C. Vaney, A. Roussel, A. Vigouroux, B. Reilly, J. Lepault,M. Kielian, and F. A. Rey. 2004. Conformational change and protein-proteininteractions of the fusion protein of Semliki Forest virus. Nature 427:320–325.
13. Gonzalez-Reyes, L., M. B. Ruiz-Arguello, B. Garcia-Barreno, L. Calder, J. A.Lopez, J. P. Albar, J. J. Skehel, D. C. Wiley, and J. A. Melero. 2001. Cleavageof the human respiratory syncytial virus fusion protein at two distinct sites isrequired for activation of membrane fusion. Proc. Natl. Acad. Sci. USA98:9859–9864.
14. Goto, H., and Y. Kawaoka. 1998. A novel mechanism for the acquisition ofvirulence by a human influenza A virus. Proc. Natl. Acad. Sci. USA 95:10224–10228.
15. Gotoh, B., T. Ogasawara, T. Toyoda, N. M. Inocencio, M. Hamaguchi, andY. Nagai. 1990. An endoprotease homologous to the blood clotting factor Xas a determinant of viral tropism in chick embryo. EMBO J. 9:4189–4195.
16. Gravel, K. A., and T. G. Morrison. 2003. Interacting domains of the HN andF proteins of newcastle disease virus. J. Virol. 77:11040–11049.
17. Hahn, D., A. Pischitzis, S. Roesmann, M. K. Hansen, B. Leuenberger, U.Luginbuehl, and E. E. Sterchi. 2003. Phorbol 12-myristate 13-acetate-in-duced ectodomain shedding and phosphorylation of the human meprinbetametalloprotease. J. Biol. Chem. 278:42829–42839.
18. Heinz, F. X., and S. L. Allison. 2000. Structures and mechanisms in flavivirusfusion. Adv. Virus Res. 55:231–269.
19. Hernandez, L. D., L. R. Hoffman, T. G. Wolfsberg, and J. M. White. 1996.Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol. 12:627–661.
7902 VON MESSLING ET AL. J. VIROL.

20. Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, and L. R. Pease. 1989.Site-directed mutagenesis by overlap extension using the polymerase chainreaction. Gene 77:51–59.
21. Jahn, R., T. Lang, and T. C. Sudhof. 2003. Membrane fusion. Cell 112:519–533.
22. Jeffers, S. A., D. A. Sanders, and A. Sanchez. 2002. Covalent modifications ofthe ebola virus glycoprotein. J. Virol. 76:12463–12472.
23. Klenk, H. D., and W. Garten. 1994. Host cell proteases controlling viruspathogenicity. Trends Microbiol. 2:39–43.
24. Kozlov, M. M., and L. V. Chernomordik. 1998. A mechanism of protein-mediated fusion: coupling between refolding of the influenza hemagglutininand lipid rearrangements. Biophys. J. 75:1384–1396.
25. Lescar, J., A. Roussel, M. W. Wien, J. Navaza, S. D. Fuller, G. Wengler, andF. A. Rey. 2001. The Fusion glycoprotein shell of Semliki Forest virus: anicosahedral assembly primed for fusogenic activation at endosomal pH. Cell105:137–148.
26. Ludwig, K., B. Baljinnyam, A. Herrmann, and C. Bottcher. 2003. The 3Dstructure of the fusion primed Sendai F-protein determined by electroncryomicroscopy. EMBO J. 22:3761–3771.
27. Markovic, I., E. Leikina, M. Zhukovsky, J. Zimmerberg, and L. V. Cherno-mordik. 2001. Synchronized activation and refolding of influenza hemagglu-tinin in multimeric fusion machines. J. Cell Biol. 155:833–844.
28. Modis, Y., S. Ogata, D. Clements, and S. C. Harrison. 2004. Structure of thedengue virus envelope protein after membrane fusion. Nature 427:313–319.
29. Moll, M., H. D. Klenk, and A. Maisner. 2002. Importance of the cytoplasmictails of the measles virus glycoproteins for fusogenic activity and the gener-ation of recombinant measles viruses. J. Virol. 76:7174–7186.
30. Morrison, T., and A. Portner. 1991. Structure, function, and intracellularprocessing of the glycoproteins of paramyxoviruses, p. 347–382. In D. W.Kingsbury (ed.), The paramyxoviruses. Plenum Press, New York, N.Y.
31. Moss, B., O. Elroy-Stein, T. Mizukami, W. A. Alexander, and T. R. Fuerst.1990. New mammalian expression vectors. Nature 348:91–92.
32. Ohno, S., F. Seki, N. Ono, and Y. Yanagi. 2003. Histidine at position 61 andits adjacent amino acid residues are critical for the ability of SLAM (CD150)to act as a cellular receptor for measles virus. J. Gen. Virol. 84:2381–2388.
33. Ohuchi, R., M. Ohuchi, W. Garten, and H. D. Klenk. 1997. Oligosaccharidesin the stem region maintain the influenza virus hemagglutinin in the meta-stable form required for fusion activity. J. Virol. 71:3719–3725.
34. Ortmann, D., M. Ohuchi, H. Angliker, E. Shaw, W. Garten, and H. D. Klenk.1994. Proteolytic cleavage of wild type and mutants of the F protein ofhuman parainfluenza virus type 3 by two subtilisin-like endoproteases, furinand Kex2. J. Virol. 68:2772–2776.
35. Park, H. E., J. A. Gruenke, and J. M. White. 2003. Leash in the groovemechanism of membrane fusion. Nat. Struct. Biol. 10:1048–1053.
36. Parks, G. D., and R. A. Lamb. 1990. Defective assembly and intracellulartransport of mutant paramyxovirus hemagglutinin-neuraminidase proteinscontaining altered cytoplasmic domains. J. Virol. 64:3605–3616.
37. Plemper, R. K., A. L. Hammond, and R. Cattaneo. 2000. Characterization ofa region of the measles virus hemagglutinin sufficient for its dimerization.J. Virol. 74:6485–6493.
38. Plemper, R. K., A. L. Hammond, D. Gerlier, A. K. Fielding, and R. Cattaneo.2002. Strength of envelope protein interaction modulates cytopathicity ofmeasles virus. J. Virol. 76:5051–5061.
39. Ritter, G. D., Jr., M. J. Mulligan, S. L. Lydy, and R. W. Compans. 1993. Cellfusion activity of the simian immunodeficiency virus envelope protein ismodulated by the intracytoplasmic domain. Virology 197:255–264.
40. Russell, C. J., T. S. Jardetzky, and R. A. Lamb. 2001. Membrane fusionmachines of paramyxoviruses: capture of intermediates of fusion. EMBO J.20:4024–4034.
41. Russell, C. J., K. L. Kantor, T. S. Jardetzky, and R. A. Lamb. 2003. A
dual-functional paramyxovirus F protein regulatory switch segment: activa-tion and membrane fusion. J. Cell Biol. 163:363–374.
42. Salzwedel, K., J. T. West, and E. Hunter. 1999. A conserved tryptophan-richmotif in the membrane-proximal region of the human immunodeficiencyvirus type 1 gp41 ectodomain is important for Env-mediated fusion and virusinfectivity. J. Virol. 73:2469–2480.
43. Sanger, C., E. Muhlberger, B. Lotfering, H. D. Klenk, and S. Becker. 2002.The Marburg virus surface protein GP is phosphorylated at its ectodomain.Virology 295:20–29.
44. Scheid, A., and P. W. Choppin. 1974. Identification of biological activities ofparamyxovirus glycoproteins. Activation of cell fusion, hemolysis, and infec-tivity of proteolytic cleavage of an inactive precursor protein of Sendai virus.Virology 57:475–490.
45. Seki, F., N. Ono, R. Yamaguchi, and Y. Yanagi. 2003. Efficient isolation ofwild strains of canine distemper virus in Vero cells expressing canine SLAM(CD150) and their adaptability to marmoset B95a cells. J. Virol. 77:9943–9950.
46. Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusionin virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569.
47. Stegmann, T., R. W. Doms, and A. Helenius. 1989. Protein-mediated mem-brane fusion. Annu. Rev. Biophys. Biophys. Chem. 18:187–211.
48. Suarez, T., W. R. Gallaher, A. Agirre, F. M. Goni, and J. L. Nieva. 2000.Membrane interface-interacting sequences within the ectodomain of thehuman immunodeficiency virus type 1 envelope glycoprotein: putative roleduring viral fusion. J. Virol. 74:8038–8047.
49. Takimoto, T., G. L. Taylor, H. C. Connaris, S. J. Crennell, and A. Portner.2002. Role of the hemagglutinin-neuraminidase protein in the mechanism ofparamyxovirus-cell membrane fusion. J. Virol. 76:13028–13033.
50. Tatsuo, H., N. Ono, K. Tanaka, and Y. Yanagi. 2000. SLAM (CDw150) is acellular receptor for measles virus. Nature 406:893–897.
51. Tatsuo, H., N. Ono, and Y. Yanagi. 2001. Morbilliviruses use signaling lym-phocyte activation molecules (CD150) as cellular receptors. J. Virol. 75:5842–5850.
52. Tong, S., F. Yi, A. Martin, Q. Yao, M. Li, and R. W. Compans. 2001. Threemembrane-proximal amino acids in the human parainfluenza type 2 (HPIV2) F protein are critical for fusogenic activity. Virology 280:52–61.
53. von Messling, V., and R. Cattaneo. 2002. Amino-terminal precursor se-quence modulates canine distemper virus fusion protein function. J. Virol.76:4172–4180.
54. von Messling, V., and R. Cattaneo. 2003. N-linked glycans with similarlocation in the fusion protein head modulate paramyxovirus fusion. J. Virol.77:10202–10212.
55. von Messling, V., G. Zimmer, G. Herrler, L. Haas, and R. Cattaneo. 2001.The hemagglutinin of canine distemper virus determines tropism and cyto-pathogenicity. J. Virol. 75:6418–6427.
56. Vongpunsawad, S., N. Oezgun, W. Braun, and R. Cattaneo. 2004. Selectivelyreceptor-blind measles viruses: identification of residues necessary forSLAM- or CD46-induced fusion and their localization on a new hemagglu-tinin Structural model. J. Virol. 78:302–313.
57. Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wiley.1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430.
58. Zhou, J., R. E. Dutch, and R. A. Lamb. 1997. Proper spacing between heptadrepeat B and the transmembrane domain boundary of the paramyxovirusSV5 F protein is critical for biological activity. Virology 239:327–339.
59. Zingler, K., and D. R. Littman. 1993. Truncation of the cytoplasmic domainof the simian immunodeficiency virus envelope glycoprotein increases envincorporation into particles and fusogenicity and infectivity. J. Virol. 67:2824–2831.
VOL. 78, 2004 MEMBRANE-PROXIMAL CLEAVAGE OF TRIMERIC vFGps 7903




















