
Can self-assembly of copper(ii) picolinamide building blocks be controlled?
15
CrystEngComm www.rsc.org/crystengcomm Volume 15 | Number 40 | 28 October 2013 | Pages 8051–8224 COVER ARTICLE Đaković, Calhorda, Popović et al . Can self-assembly of copper(II) picolinamide building blocks be controlled?
Transcript of Can self-assembly of copper(ii) picolinamide building blocks be controlled?

Volume 15 | N
umber 40 | 2013
CrystEngC
omm
Pages 8051–8224
CrystEngCommwww.rsc.org/crystengcomm Volume 15 | Number 40 | 28 October 2013 | Pages 8051–8224
www.icces.cnRegistered charity number 207890
Themes
Bio and bioinspired systems for energy conversion
Biofuels and biomass conversion
Clean coal and fossil fuels
CO2 capture, storage and utilization
Electrochemical energy conversion and storage
Hydrogen production and storage
Materials and nanotechnology for energy systems
Photocatalysis & environmental catalysis
Solar energy conversion
Be part of the 2nd International Conference on Clean Energy Science (ICCES2) and join researchers from across the globe for a unique opportunity to discuss the latest developments in clean energy and energy storage research, and the fundamental issues surrounding the scientific challenges faced ahead.
2nd International Conference on Clean Energy Science (ICCES2)13-16 April 2014, Qingdao, China
Key Deadlines
Oral abstract submission deadline – 10 November 2013
Poster abstract submission deadline – 9 January 2014
Early bird registration deadline – 30 January 2014
Final registration – 13 March 2014
Plenary Speakers
Professor Xinhe Bao
Dalian Institute of Chemical Physics, China
Professor Guillermo Bazan UC Santa Barbara, USA
Professor James Clark
University of York, UK
Professor Shunichi Fukuzumi Osaka University, Japan
Professor Frederik Krebs
Technical University of Denmark, Denmark
Professor Ryong Ryoo
Korea Advanced Institute of Science and Technology,
South Korea
Submit your abstract now
COVER ARTICLEĐaković, Calhorda, Popović et al. Can self-assembly of copper(II) picolinamide building blocks be controlled?
CE015040_cover_PRINT.indd 1CE015040_cover_PRINT.indd 1 9/19/2013 10:57:19 AM9/19/2013 10:57:19 AM

Cite this: CrystEngComm, 2013, 15,8074
Can self-assembly of copper(II) picolinamide buildingblocks be controlled?3
Received 4th June 2013,Accepted 4th July 2013
DOI: 10.1039/c3ce41011k
www.rsc.org/crystengcomm
Marijana Ðakovic,*a Diogo Vila-Viçosa,b Nuno A. G. Bandeira,b MariaJose Calhorda,*b Bojan Kozlevcar,c Zvonko Jaglicicde and Zora Popovic*a
Four copper(II) derivatives with picolinamide (piaH), [Cu(N3)2(piaH)2] (1), [Cu(NCO)2(piaH)2] (2),
[Cu(pia)2]?2H2O (3) and [Cu(pia)(piaH)(ClO4)]2 (4) are reported. 1 and 2 were isolated as powders shortly
after precipitation, becoming crystalline (3) after prolonged standing in mother liquor. The single crystal
X-ray analysis revealed the deprotonation of the amide nitrogen (pia) in both 3 (mononuclear square
planar [Cu(pia-N,N)2] building blocks) and 4 (pia acting as a bridge between copper(II) ions within the
dimer). MAGSUS xMT versus T curves suggest only weak magnetic interactions between the copper(II)
species in 3 and 4, though 4 is dinuclear. The magnetic coupling was reproduced by DFT calculations on
the experimentally determined structures. The mechanism of formation of compounds 3 and 4 was
analyzed by DFT calculations. Models of 1 and 2 underwent transfer of two hydrogen atoms from the piaH
ligands to the two axial ligands N32 or NCO2 that were released yielding 3. In the reaction leading to 4,
the perchlorate is a very weak base that cannot deprotonate the piaH ligand and after the first hydrogen
transfer to acetate the dimer 4 is formed. This reactivity, favoring alternative pathways, may switch off an
expected self-assembly process.
Introduction
The design of supramolecular architectures by self-assembly ofsmall building blocks has become a major research area owingto its potential for practical applications.1 Although instructural engineering the metal–ligand bond plays a deter-mining role, dictating the resultant supramolecular topologyof the metal–organic frameworks (MOFs), the secondaryinteractions, such as hydrogen bonding, p–p interactions etc.also have significant influence in shaping the final architec-ture. The self-assembly of coordination frameworks is alsohighly affected by various other parameters such as the
dynamic nature of the metal–ligand bonds, structural prefer-ences of the metal centers, nature of ligating topologies of theligand, metal–ligand ratio, nature of counter ions and variousexperimental conditions such as solvents, solution pH,temperature, and preparation/crystallization methods. So, itis often difficult to predict the final outcome and, therefore,much work has to be done to improve our understanding of allthe factors influencing the final supramolecular architecture.
Pyridinecarboxamide ligands have been used as effectivetools to organize metal building blocks into extendedassemblies by combining their metal coordinating ability,robust hydrogen-bonding ability and p–p stacking betweenaromatic rings.2 We focused our research on mixed pyridine-carboxamide complexes, with the aim of examining theinterplay between different co-ligands in the first coordinationsphere on the final architecture, as well as the role of counterions in particular assembling.3
The carboxamide moiety in picolinamide complexes isnormally involved in the coordination to metal ions via theoxygen atom, and does not display its usual supramolecularbehavior. Recently, we found that the self-complementaryamide motif R2
2(8) is not the synthon of choice4 for a series oflate 3d-block metal picolinamide complexes with thiocyanate,namely Co(II), Ni(II), Cu(II) and Zn(II), but a motif involvingother accessible hydrogen-bond accepting atoms, leading toan R2
4(8) assembly, is the prevailing one.5 Moreover, the energydifference between the two motifs is rather small. Therefore, itwould be of interest to determine the ‘supramolecular
aLaboratory of General and Inorganic Chemistry, Department of Chemistry, Faculty
of Science, University of Zagreb, Horvatovac 102a, 10000 Zagreb, Croatia.
E-mail: [email protected]; [email protected]; Fax: +385-1-4606-341;
Tel: +385-1-4606-356bDepartmento de Quımica e Bioquımica, CQB, Faculdade de Ciencias, Universidade
de Lisboa, Campo Grande, Ed. C8, 1749-049 Lisbon, Portugal.
E-mail: [email protected]; Fax: +351-217500088; Tel: +351-217500196cFakulteta za kemijo in kemijsko tehnologijo, Univerza v Ljubljani, Askerceva c. 5,
1000 Ljubljana, SloveniadInstitut za matematiko, fiziko in mehaniko, Jadranska c. 19, 1000 Ljubljana,
SloveniaeFakulteta za gradbenuistvo in geodezijo, Univerza v Ljubljani, Jamova c. 2, 1000
Ljubljana, Slovenia
3 Electronic supplementary information (ESI) available: Fig. S1–S3, TG–DTAcurves for the complexes 1, 2 and 3. Fig. S4–S7, X-ray powder diffraction patternsfor the complexes 1, 2, 3 and 4. Fig. S8, reaction pathway from 1-NNN. CCDC906738 and 906739. For ESI and crystallographic data in CIF or other electronicformat see DOI: 10.1039/c3ce41011k
CrystEngComm
PAPER
8074 | CrystEngComm, 2013, 15, 8074–8087 This journal is � The Royal Society of Chemistry 2013

conditions’ in which the self-complementary amide motif is areliable and robust supramolecular synthon. As the first step,we wanted to examine other coordination possibilities of thepicolinamide (piaH) itself with intention to have some insightinto its impact on the final supramolecular assembly.
We report herein the preparation and characterization offour new compounds derived from the reactions of Cu(II)nitrate, in the presence of azide or cyanate, and Cu(II)perchlorate with picolinamide. DFT calculations6 were per-formed to understand the origin of the different productsfound and highlight the role of the base strength of theanionic ligand in promoting intramolecular proton transferreactions that prevent the formation of the self-assemblystructure found previously. Magnetic properties of polynuclearCu(II) species were studied and a different DFT approachallowed the comparison of magnetic coupling constants.
Experimental section
Materials and physical measurements
Pyridine-2-carboxamide was purchased from Sigma-Aldrichand used as received. All other chemicals were purchased fromKemika, Croatia, and used without further purification.
CHN analyses were performed with a Perkin-Elmer 2400Series II CHNS analyzer in the Analytical Services Laboratoriesof the Rud–er Boskovic Institute, Zagreb, Croatia.
The IR spectra were obtained from KBr pellets in the range4000–450 cm21 with a Perkin-Elmer Spectrum RXI FTIR-spectrometer.
Thermal measurements were performed in a Mettler-ToledoTGA/SDTA 850e instrument at heating rate of 10 uC min21 andnitrogen (purity above 99.996%) at flowing rate of 20 mLmin21 in the temperature range 25–600 uC. The samples wereplaced in a standard aluminum crucible (40 mL).
X-ray powder diffraction experiments were performed on aPhilips PW 1850 diffractometer, Cu Ka radiation, tension 40kV, and current 40 mA. The patterns were collected in theangle region between 4u and 60u (2h) with a step size of 0.02uand 4.5 s counting per step.
X-band EPR spectra for 3 and 4 were recorded frompowdered samples at room temperature on a BrukerELEXSYS E500 spectrometer. The g values were calculated bythe equation E = hn = gbeB.
Magnetic susceptibility for 3 and 4 was investigated between2 K and 300 K in a constant magnetic field H = 1000 Oe withQuantum Design MPMS-XL-5 SQUID susceptometer. Thetemperature dependent susceptibilities x = M/H are shown.The data were corrected for sample holder contribution and atemperature independent magnetic susceptibility of innershell electrons (Larmor diamagnetism) as obtained fromPascal’s tables.7
Synthesis
[Cu(N3)2(piaH)2], (1). A warm aqueous solution of picolina-mide (0.24 g; 2.0 mmol in 50 mL of H2O) was slowly added toan aqueous solution of copper(II) nitrate trihydrate (0.24 g; 1.0mmol in 10 mL). An aqueous solution of sodium azide (0.13 g;
2.0 mmol in 10 mL) was then added to the resulting solution.Instantaneously, the light olive green microcrystalline productprecipitated. The product was filtered off, washed with waterand dried in air. Yield: 89% (0.35 g; 0.89 mmol). Microanalysis:calc. for C12H12CuN10O2 (Mr = 391.84): C, 36.78; H, 3.09; N,28.60. Found: C, 36.66; H, 3.44; N, 28.83%. FT-IR (KBr, cm21):3365 (n(NH2)as), 3288/3257/3225 (n(NH2)s), 2062 (n(NN)), 1651(n(CLO)), 1616 (d(NH2)), 1436 (n(CN)). Thermal analysis (Fig.S1, ESI3): decomposition occurs in two steps, the first onebegins at y160 uC (endothermic DTA peak at 197 uC); furtherdecomposition occurs in the temperature range 210–260 uC(two exothermic processes: DTA peaks at 222 and 240 uC).
Mechanochemical synthesis. Picolinamide (0.24 g; 2.0mmol), copper(II) nitrate trihydrate (0.24 g; 1.0 mmol) andsodium azide (0.13 g; 2.0 mmol) were ground in an agatemortar for a few minutes to obtain a light olive green powder.The powder product was washed with a small portion of waterto remove formed NaNO3. Microanalysis: calc. forC12H12CuN10O2 (Mr = 391.84): C, 36.78; H, 3.09; N, 28.60.Found: C, 36.72; H, 3.38; N, 28.87%. The powder diffractionpattern matched with that of 1 prepared by the solutionmethod (Fig. S4, ESI3).
Caution! The azide complexes are potentially explosive.Therefore, they should be prepared in small quantities andhandled with care.
[Cu(NCO)2(piaH)2], (2). A warm aqueous solution of picoli-namide (0.24 g; 2.0 mmol in 50 mL) was slowly added to anaqueous solution of copper(II) nitrate trihydrate (0.24 g; 1.0mmol in 10 mL). An aqueous solution of sodium cyanate (0.18g; 2.0 mmol in 10 mL) was then added to the resultingsolution. The light blue microcrystalline product precipitatedinstantaneously. The product was filtered off, washed withwater and dried in air. Yield: 85% (0.33 g; 0.85 mmol).Microanalysis: calc. for C14H12CuN6O4 (Mr = 391.83): C, 42.91;H, 3.09; N, 21.45. Found: C, 42.59; H, 3.16; N, 21.99%. FT-IR(KBr, cm21): 3383 (n(NH2)as), 3260 (n(NH2)s), 2225/2190(n(CN)OCN), 1656 (n(CLO)), 1616 (d(NH2)), 1439 (n(CN)).Thermal analysis (Fig. S2, ESI3): decomposition occurs in twosteps, the first one in the temperature range 160–260 uC (DTApeak at 241 uC); further decomposition occurs in the interval260–340 uC (endothermic DTA peak at 282 uC).
Mechanochemical synthesis. Picolinamide (0.24 g; 2.0mmol), copper(II) nitrate trihydrate (0.24 g; 1.0 mmol) andsodium cyanate (0.18 g; 2.0 mmol) were ground in an agatemortar for a few minutes to obtain a light blue powder. Thepowder product was washed with a small portion of water toremove formed NaNO3. Microanalysis: calc. for C14H12CuN6O4
(Mr = 391.83): C, 42.91; H, 3.09; N, 21.45. Found: C, 42.65; H,3.22; N, 22.02%. The powder diffraction pattern matched withthat of 2 prepared by the solution method (Fig. S5, ESI3).
[Cu(pia)2]?2H2O, (3). The mixture of 1 together with themother liquor was transferred into a Teflon-lined stainlesssteel vessel and heated for 5 h at 110 uC. After cooling to roomtemperature by standing overnight, the green mother liquorwas poured out and violet single crystals were found on thebottom of the vessel. The crystals were filtered off and themother liquor was left to stand for a few days. From thesolution an additional amount of single crystals were obtained
This journal is � The Royal Society of Chemistry 2013 CrystEngComm, 2013, 15, 8074–8087 | 8075
CrystEngComm Paper

in several days upon slow evaporation. Total yield: 70% (0.24 g;0.70 mmol). Anal. calc. for C12H14CuN4O4: C, 42.17; H, 4.13; N,16.39. Found: C, 42.33; H, 4.41; N, 16.58%. FT-IR (KBr, cm21):3437 (n(NH2)as), 3077 (n(NH2)s), 1631 (n(CLO)), 1604 (d(NH2)),1448 (n(CN)). Thermal analysis (Fig. S3, ESI3): dehydration inthe temperature range 60–140 uC (endothermic DTA peak at116 uC; found: 10.52%, calcd: 10.54%); the loss of one pialigand (endothermic DTA peak at 285 uC; found 37.1%,calculated 35.43%); further decomposition in the temperaturerange 300–600 uC (smooth exothermic process). EPR (295 K): g|
= 2.23, g) = 2.07. The powder diffraction pattern (bulk sample)was consistent with the pattern calculated from the singlecrystal data (Fig. S6, ESI3).
[Cu(pia)(piaH)(ClO4)]2, (4). A warm solution of picolinamide(0.24 g; 2.0 mmol) in water (50 mL) was slowly added to anaqueous solution (10 mL) of copper(II) acetate (0.18 g; 1.0mmol). An aqueous solution of sodium perchlorate (0.24 g; 2.0mmol in 10 mL) was then added to the resulting solution.Violet needle-like single crystals were formed slowly. Thecrystals were filtered off, washed with small portions of coldwater, and dried in vacuo. Yield: 76% (0.31 g; 0.38 mmol). Anal.calc. for C24H22Cu2Cl2N8O12: C, 35.48; H, 2.73; N, 13.79.Found: C, 35.61; H, 2.94; N, 13.26%. FT-IR (KBr, cm21): 3420,3354 (n(NH2)as), 3268, 3198 (n(NH2)s), 1670, 1640 (n(CLO)), 1601(d(NH2)), 1436 (n(CN)). Thermal analysis: due to highlyexothermic process at 310 uC thermal analysis was notpossible. EPR (295 K): g| = 2.17, g) = 2.07. The powderdiffraction pattern (bulk sample) was consistent with thepattern calculated from the single crystal data (Fig. S7, ESI3).
Caution! Although no problems were encountered in thiswork, perchlorate salts are potentially explosive and should behandled in small quantities.
Single-crystal X-ray structure determination
Single crystals of 3 and 4 were mounted in a randomorientation on a glass fibre. Data collection was carried outat 296 K on an Oxford Diffraction Xcalibur four-circle kappageometry single-crystal diffractometer with a Sapphire 3 CCDdetector, using a graphite monochromated MoKa (l =0.71073 Å) radiation, and applying the CrysAlis Softwaresystem.8 The crystal–detector distance was 50 mm. Datareduction, including absorption correction, was done by theCrysAlis RED program.8
The structures were solved by direct methods implementedin the SHELXS-97 program.9 The coordinates and theanisotropic displacement parameters for all non-hydrogenatoms were refined by full-matrix least-squares methods basedon F2 values using the SHELXL-97 program.9 The aromatichydrogen atoms were generated geometrically using the ridingmodel with the isotropic factor Uiso set at 1.2Ueq of the parentatom. Hydrogen atoms on the carboxamide nitrogen werelocated in the difference Fourier map at the final stage ofrefinement and were refined freely.
Graphical work was performed with the program ORTEP-3for Windows10 and Mercury 1.4.1.11 The thermal ellipsoidswere drawn at the 50% probability level. General and crystaldata and summary of intensity data collection and structurerefinement for compounds 3 and 4 are given in Table 1.
CCDC 906738 and 906739 contain the supplementarycrystallographic data for this paper.3
Computational details
All DFT6 calculations were performed using the GAUSSIAN 09software package.12 The B3LYP13–15 hybrid functional wasused in all calculations related with complexes 1, 2 and 3. Forall calculations related with complex 4, the M0616 functionalwas used, since this functional describes non covalentinteractions better.16 All optimized geometries were obtainedwith the Stuttgart/Dresden ECP (SDD) basis set17 for Cu atomsand with the 6-31G** basis set for all the others.18 All thereported energies (Gsolv) were calculated according to Gsolv =Esolv + (Ggas 2 Egas).
Ggas and Egas were obtained from the gas phase calculationand Esolv, which is the electronic energy in solution, wasobtained using the integral equation formalism of thepolarizable continuum solvation model (IEFPCM)19 on theelectronic density (SMD).20 Water was used as solvent, as in allexperimental procedures.
Frequency calculations were performed to evaluate thenature of the optimized geometries. One imaginary frequencywas obtained for the transition states and none for theminima. All transition states were confirmed by using theintrinsic reaction coordinate (IRC) approach.21
Gaussian09 was used for discrete models (4, 39, 399, see textbelow) and the implementation of periodic boundary condi-tions22–24 (PBC) to study both spin states in the unrestrictedKohn–Sham formalism (B3LYP) for chain 3, over the X-raydetermined structures. All k-points (34 per spin) were auto-matically generated by the program24–26 and convergence wasachieved with default termination criteria. Magnetic exchangeconstants were then evaluated by the broken symmetrytechnique of Noodleman and others27,28 without spin projec-tion since this is known29–31 to exaggerate the exchangeconstant value due to lack of self-interaction error (SIE)correction in standard functionals such as B3LYP.
The simple Heisenberg–Dirac–Van Vleck Hamiltonian,32,7
which describes local spin systems can be defined as
H = 22J(SCuASCuB
) (1)
The magnetic coupling constant (J) should be taken as thedifference between the lowest energy singlet and triplet statesand should be negative (J , 0) in the anti-ferromagnetic caseand positive in the ferromagnetic case (J . 0).
Three-dimensional representations of calculated structureswere obtained with Chemcraft.33
Results and discussion
Coordination compounds 1 and 2 were prepared in astraightforward way by direct mixing of aqueous solutions ofcopper(II) nitrate, picolinamide, and sodium azide or sodiumcyanate, respectively. Also, both complexes were obtained bygrinding34,35 the mixture of starting compounds in anequimolar ratio (Scheme 1). The attempts to get single crystals
8076 | CrystEngComm, 2013, 15, 8074–8087 This journal is � The Royal Society of Chemistry 2013
Paper CrystEngComm

of 1 and 2 by a hydrothermal synthetic procedure resulted inobtaining the single crystals of complex 3. Under elevatedtemperature and pressure the deprotonation of the amide
nitrogen atom occurred, leading to N,N-chelation of picolina-mide. The resulting complex 3 is a solvatomorph of[Cu(pia)2]?4H2O36 (399) in which the deprotonation was
Table 1 Crystal data and details of the structure determination for [Cu(pia)2]?2H2O (3) and [Cu(pia)(piaH)(ClO4)]2 (4)
Compounds [Cu(pia)2]?2H2O (3) [Cu(pia)(piaH)(ClO4)]2 (4)
Empirical formula C12H14CuN4O4 C24H22Cl2Cu2N8O12
Formula weight 341.82 812.50Color and habit Violet, needle Violet, plateCrystal system, space group Triclinic, P1 Monoclinic, C2/cCrystal dimensions/mm 0.08 6 0.16 6 0.53 0.04 6 0.25 6 0.33Unit cell parameters:a/Å 5.1601(2) 24.0770(5)b/Å 7.6565(3) 6.5520(1)c/Å 9.1728(3) 19.6808(5)a/u 74.888(3) 90b/u 84.185(3) 102.133(2)c/u 71.767(2) 90V/Å3 332.23(2) 3035.3(1)Z 1 4Dcalc/g cm23 1.709 1.778m/mm21 1.666 1.654h range for the data collection/u 4.11–30.00 3.76–27.00h, k, l range 27 : 7, 210 : 10, 212 : 12 230 : 29, 28 : 8, 222 : 25Scan type v vF(000) 175 1640No. reflections collected 10 708 12 272No. independent reflections 1924 3306No. observed reflections, I ¢ 2s(I) 1730 2592No. refined parameters 106 229Ra, wRb [I ¢ 2s(I)] 0.0320, 0.0968 0.0288, 0.0742R, wR (all data) 0.0355, 0.1003 0.0397, 0.0764g1, g2 in wc 0.0793, 0.0009 0.0485, 0.000Goodness of fit on F2, Sd 1.069 0.945Max., min. electron density/e Å23 0.606/20.599 0.436/20.310Maximum D/s 0.033 ,0.001Range of transmission factors min, max 0.612, 0.874 0.563, 0.784Extinction coefficient None None
a R = g||Fo| 2 |Fc||/g|Fo|. b wR = [g(Fo2 2 Fc
2)2/gw(Fo2)2]1/2. c w = 1/[s2(Fo
2) + [(g1P)2 + g2P]] where P = (Fo2 + 2Fc
2)/3. d S = g[w(Fo2 2 Fc
2)2/(Nobs 2 Nparam)]1/2.
Scheme 1 Synthetic pathways for [Cu(pia)2]?2H2O (3).
This journal is � The Royal Society of Chemistry 2013 CrystEngComm, 2013, 15, 8074–8087 | 8077
CrystEngComm Paper

achieved by increasing the pH of the reaction mixture byaddition of Na2P2O7?10H2O (pH = 8).
Furthermore, complex 3 in the crystalline form can also beobtained at room temperature but at considerably slowerreaction rates, if 1 and 2 are not isolated as powders, but theirmother liquors left to stand. The reaction is quantitative inapproximately three weeks for 2, and in five to six months for1.
Crystal structures
The compound 3 crystallizes in the triclinic space group P1with the copper(II) ion at an inversion centre. The coordinationsphere is built up by four nitrogen atoms of two pia ligandscomprising the equatorial plane (Cu–N = 1.984(2), 1.934(2) Å),
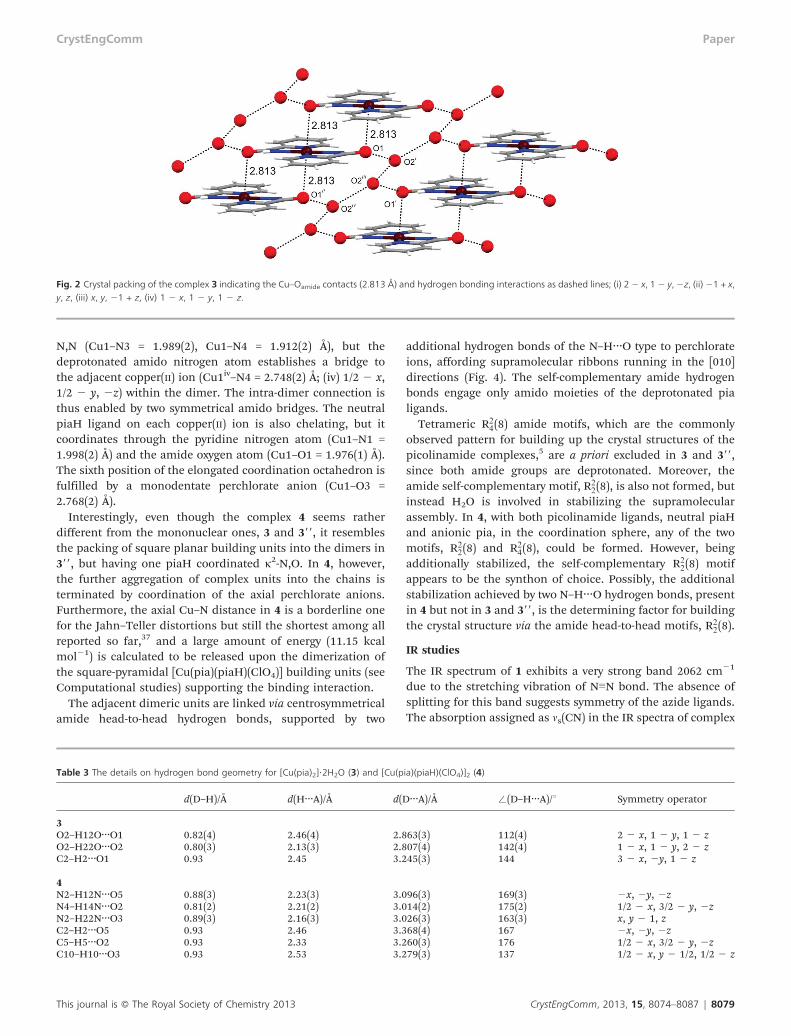
Fig. 1 and Table 2, while two amide oxygen atoms fromadjacent molecules approach the copper(II) ion at quite longdistances (Cu–O = 2.813(2) Å) for an elongated octahedralcomplex. In this way, endless chains of the complex buildingblocks running in the [100] direction, involving two interac-tions of the Cu…O type per coordination unit, are formed(Fig. 2). To stabilize the crystal structure of 3, the watermolecules, being engaged into the hydrogen bonds with theamide oxygens, act as crosslinkers between neighboringcoordination unit chains (Table 3). As a result, none of thecommonly occurring supramolecular amide motifs areobserved in 3.4
Compared with the previously reported structure by Du et al.with similarly built up square planar copper(II) coordinationunits, [Cu(pia-N,N)2]?4H2O (399),36 no difference within thesquare planar coordination units is observed, besides a slightshortening (20s) of the Cu–Npy bond distances. Distortionsfrom the regular square planar geometry for both 3 and 399 areimposed mainly by the formation of the chelate rings [the biteangles: 82.72(2)u in 3; 81.41(6)u at Cu1 and 81.81(6)u at Cu2 in399].
The main difference between 3 and 399, apart from thenumber of co-crystallized water molecules, is in the arrange-ment of square planar building blocks in the crystal structures.While the same separation and the same slippage betweeneach two neighboring building units are observed in thechains of 3, those in 399 are formed by alternation of twosymmetrically independent square planar blocks, one withCu(II) ions at inversion centers (Cu2) and the other one withCu(II) ions at general positions (Cu1). Moreover, in 399 therepeating unit comprises three complex units, two with Cu(II)at general positions, mutually related by the center ofinversion, and one unit with Cu(II) at the inversion center.Consequently, two intra-chain separations are revealed, thesmaller one within the two symmetrically dependent mole-cules and the larger one between two symmetrically indepen-dent molecules. Furthermore, in 3, the slippage between theadjacent molecules is such that it allows each amide oxygen tocomplete the coordination sphere of the neighboring Cu(II),while in 399 it enables the deprotonated amide nitrogen atomsto approach the neighboring copper(II) ions.
According to our survey of the Cambridge StructuralDatabase (CSD),37 confined to picolinamide complexes, theN,N-coordination mode of pyridine-2-carboxamide is quiterare. There are only 8 out of 58 listed reports in which piabinds to metal ions through both nitrogen atoms:[Cu(pia)2]?4H2O,36 [Ni(pia)2]?2H2O,38 [AuCl2(pia)],39 [N(C4H9)4][Pd2{C4(COOMe)4}2(pia)],40 [Ru(pia)(pTol-COCH2CO-pTol)2]?CH3CN,41 [Ru(pia)(2,29-bipy)2][BF4],40 [PtClPPh3(pia)]42 and[Rh(C8H12)(pia)],43 and only two of them are bis(pia) com-plexes. In the others, piaH binds to metal ions in its usualpiaH–N,O chelation mode, and no other than only these twocoordination modes, k2-N,O and k2-N,N, have been observedso far.
The crystal structure of compound 4, prepared by the roomtemperature synthetic procedure, revealed centrosymmetricalbinuclear complex units (Fig. 3). Each copper(II) ion iscoordinated by one deprotonated pia ligand. However, incontrast to 3, the anionic pia does not coordinate only as k2-
Fig. 1 Molecular structure of the square planar [Cu(pia)2]?2H2O (3) with theatom numbering scheme.
Table 2 Selected bond distances (Å) and angles (u) for [Cu(pia)2]?2H2O (3) and[Cu(pia)(piaH)(ClO4)]2 (4)a
Bond distances Bond angles
3Cu1–N1 1.984(2) N1–Cu1–N2 82.72(6)Cu1–N2 1.934(2) O1i–Cu1–N1 91.76(5)Cu1–O1i 2.813(2) N1–Cu1–N1ii 180
N1–Cu1–N2ii 97.28(6)O1iii–Cu1–N1 88.24(5)O1–Cu1–N2ii 93.72(6)O1iii–Cu1–N2 86.28(6)
4Cu–O1 1.976(1) O1–Cu1–O3 88.72(6)Cu–O3 2.768(2) O1–Cu1–N1 81.80(6)Cu–N1 1.998(2) O1–Cu1–N3 94.71(6)Cu–N3 1.989(2) O1–Cu1–N4 176.50(7)Cu–N4 1.912(2) O1–Cu1–N4iv 84.88(6)Cuiv–N4 2.748(2) O3–Cu1–N1 86.18(6)
O3–Cu1–N3 85.71(6)O3–Cu1–N4 93.26(7)O3–Cu1–N4iv 173.32(6)N1–Cu1–N3 171.24(7)N1–Cu1–N4 101.20(7)N1–Cu1–N4iv 94.77(6)N3–Cu1–N4 82.57(7)N3–Cu1–N4iv 92.91(6)N4–Cu1–N4iv 93.04(7)
a (i) x 2 1, y, z; (ii) 1 2 x, 1 2 y, 1 2 z; (iii) 2 2 x, 1 2 y, 1 2 z; (iv)1/2 2 x, 1/2 2 y, 2z.
8078 | CrystEngComm, 2013, 15, 8074–8087 This journal is � The Royal Society of Chemistry 2013
Paper CrystEngComm
N,N (Cu1–N3 = 1.989(2), Cu1–N4 = 1.912(2) Å), but thedeprotonated amido nitrogen atom establishes a bridge tothe adjacent copper(II) ion (Cu1iv–N4 = 2.748(2) Å; (iv) 1/2 2 x,1/2 2 y, 2z) within the dimer. The intra-dimer connection isthus enabled by two symmetrical amido bridges. The neutralpiaH ligand on each copper(II) ion is also chelating, but itcoordinates through the pyridine nitrogen atom (Cu1–N1 =1.998(2) Å) and the amide oxygen atom (Cu1–O1 = 1.976(1) Å).The sixth position of the elongated coordination octahedron isfulfilled by a monodentate perchlorate anion (Cu1–O3 =2.768(2) Å).
Interestingly, even though the complex 4 seems ratherdifferent from the mononuclear ones, 3 and 399, it resemblesthe packing of square planar building units into the dimers in399, but having one piaH coordinated k2-N,O. In 4, however,the further aggregation of complex units into the chains isterminated by coordination of the axial perchlorate anions.Furthermore, the axial Cu–N distance in 4 is a borderline onefor the Jahn–Teller distortions but still the shortest among allreported so far,37 and a large amount of energy (11.15 kcalmol21) is calculated to be released upon the dimerization ofthe square-pyramidal [Cu(pia)(piaH)(ClO4)] building units (seeComputational studies) supporting the binding interaction.
The adjacent dimeric units are linked via centrosymmetricalamide head-to-head hydrogen bonds, supported by two
additional hydrogen bonds of the N–H…O type to perchlorateions, affording supramolecular ribbons running in the [010]directions (Fig. 4). The self-complementary amide hydrogenbonds engage only amido moieties of the deprotonated pialigands.
Tetrameric R24(8) amide motifs, which are the commonly
observed pattern for building up the crystal structures of thepicolinamide complexes,5 are a priori excluded in 3 and 399,since both amide groups are deprotonated. Moreover, theamide self-complementary motif, R2
2(8), is also not formed, butinstead H2O is involved in stabilizing the supramolecularassembly. In 4, with both picolinamide ligands, neutral piaHand anionic pia, in the coordination sphere, any of the twomotifs, R2
2(8) and R24(8), could be formed. However, being
additionally stabilized, the self-complementary R22(8) motif
appears to be the synthon of choice. Possibly, the additionalstabilization achieved by two N–H…O hydrogen bonds, presentin 4 but not in 3 and 399, is the determining factor for buildingthe crystal structure via the amide head-to-head motifs, R2
2(8).
IR studies
The IR spectrum of 1 exhibits a very strong band 2062 cm21
due to the stretching vibration of NMN bond. The absence ofsplitting for this band suggests symmetry of the azide ligands.The absorption assigned as ns(CN) in the IR spectra of complex
Fig. 2 Crystal packing of the complex 3 indicating the Cu–Oamide contacts (2.813 Å) and hydrogen bonding interactions as dashed lines; (i) 2 2 x, 1 2 y, 2z, (ii) 21 + x,y, z, (iii) x, y, 21 + z, (iv) 1 2 x, 1 2 y, 1 2 z.
Table 3 The details on hydrogen bond geometry for [Cu(pia)2]?2H2O (3) and [Cu(pia)(piaH)(ClO4)]2 (4)
d(D–H)/Å d(H…A)/Å d(D…A)/Å /(D–H…A)/u Symmetry operator
3O2–H12O…O1 0.82(4) 2.46(4) 2.863(3) 112(4) 2 2 x, 1 2 y, 1 2 zO2–H22O…O2 0.80(3) 2.13(3) 2.807(4) 142(4) 1 2 x, 1 2 y, 2 2 zC2–H2…O1 0.93 2.45 3.245(3) 144 3 2 x, 2y, 1 2 z
4N2–H12N…O5 0.88(3) 2.23(3) 3.096(3) 169(3) 2x, 2y, 2zN4–H14N…O2 0.81(2) 2.21(2) 3.014(2) 175(2) 1/2 2 x, 3/2 2 y, 2zN2–H22N…O3 0.89(3) 2.16(3) 3.026(3) 163(3) x, y 2 1, zC2–H2…O5 0.93 2.46 3.368(4) 167 2x, 2y, 2zC5–H5…O2 0.93 2.33 3.260(3) 176 1/2 2 x, 3/2 2 y, 2zC10–H10…O3 0.93 2.53 3.279(3) 137 1/2 2 x, y 2 1/2, 1/2 2 z
This journal is � The Royal Society of Chemistry 2013 CrystEngComm, 2013, 15, 8074–8087 | 8079
CrystEngComm Paper

2 (2225/2190 cm21) is shifted towards higher wave numberscompared with that of free cyanate (2170 cm21),44 which is inagreement with the coordination of cyanate ligand to themetal cation.
Concerning the CLO stretching frequencies of the freeligand (piaH) and its metal complexes 1–4, a negative shift isobserved only in complexes 1, 2 and 4. In complex 3, the amideoxygen atom is not involved into the coordination to the metalion, but is involved into two hydrogen bonds instead. Incomplexes 1 and 2, a small negative shift of the CLO stretchingfrequency is expected, owing to the usual chelating-N,Ocoordination of the picolinamide ligands. In complex 4,splitting of the CLO band was found, suggesting the presenceof coordinated as well as free carboxamide oxygen atom in thestructure.
EPR and dc magnetic susceptibility
The presence of polymeric chains (3) and dimers (4) of Cu(II)prompted us to analyze their magnetic properties. The roomtemperature EPR spectrum of 3 shows two S = 1/2 signals withg)(x,y) = 2.07 and g|(z) = 2.23 and without hyperfine splitting.Both signals are in accordance with four short coordinationbonds Cu–N in one plane around copper(II) ion seen in thesquare planar coordination sphere. Similar, though lessrefined, splitting of the axial symmetry signals, g)(x,y) (2.07)and g|(z) (2.17), is seen for dinuclear 4. These data are inaccordance with the structural data, where no obvious pathwayis present to maintain the magnetic interaction between the3.264(2) Å separated copper(II) ions within the dinuclearcoordination unit of 4.
The susceptibility of both compounds 3 and 4 increases withdecreasing temperature and approximately follows the Curielaw (Fig. 5). The Curie constants and the corresponding
Fig. 4 Supramolecular ribbons in the crystal structure of complex 4 running in the [010] direction; hydrogen bonding interactions are indicated as dashed lines. (i) 1/22 x, 3/2 2 y, 2z; (ii) 1/2 2 x, 1/2 2 y, 2z; (iii) x, 1 + y, z.
Fig. 5 Magnetic susceptibility xM and product xMT (inset) as a function oftemperature for compounds 3 and 4. Full lines in the inset are the best fits withfunctions as described in text.Fig. 3 Molecular structure of [Cu(pia)(piaH)(ClO4)]2 (4) with the atom number-
ing scheme.
8080 | CrystEngComm, 2013, 15, 8074–8087 This journal is � The Royal Society of Chemistry 2013
Paper CrystEngComm

effective magnetic moments (both calculated per one Cu2+ ion)at room temperature for compounds 3 and 4 of C = 0.392 cm3
K mol21 (meff = 1.75 BM) and C = 0.405 cm3 K mol21 (meff = 1.80BM), respectively, are in agreement with a d9 Cu(II) ion withone unpaired electron.45
Only a detailed inspection of the product xMT versustemperature (graph in the inset in Fig. 5) shows a smalldeviation from the Curie law at low temperatures. The productxMT of 3 slightly decreases below 30 K indicating a weakantiferromagnetic coupling between magnetic Cu2+ ions.According to the structure, one might suggest the semi-coordination magnetic path from the amido oxygen atom fromone [Cu(pia)2] coordination unit to the copper(II) ion axialposition of adjacent [Cu(pia)2] unit (Fig. 2; Cu–O = 2.813(2) Å).The weakly coordinated Cu{dx22y2}1–N…Cu{dz2}2 moiety mayenable a certain degree of magnetic interaction, thoughobviously only of a weak magnitude. Nevertheless, we triedto fit the experimental data with the Bonner–Fisher model46
for a chain of S = 1/2 spins. The result is shown as a full greenline in the inset in Fig. 5 and reproduces the measuredsusceptibility excellently. The intra-chain interaction betweenthe Cu2+ ions as a results of the fitting procedure is indeedweak (J = 20.3 cm21).
Compound 4 deviates even less from the ideal paramagneticbehavior, as the product xMT is almost constant in the wholeinvestigated temperature range. Only below 15 K does theproduct first slightly increase and, after achieving a maximumat 6 K, decrease when the temperature approaches 2 K. A smallincrease of the product xMT suggests a weak ferromagneticinteraction, which was additionally confirmed after applying aCurie–Weiss law xM = C/(T 2 H) to the measured data above 50K. Indeed, the fit shown as a full line in the inset in Fig. 5exhibits a positive Weiss constant h # 1.1 K. Using a meanfield approximation,7 this value yields an estimation of J/kB y1.5 cm21 if the number of nearest neighbours around a givenmagnetic molecule z is assumed to be 1. Owing to the natureof the dimer, the obvious magnetic interaction path occursthrough the Cu–N–Cu bridges. Its magnitude is clearlydiminished by unfavorable bonding Cu{dx22y2}1–N…Cu{dz2}2
(Cu1–N4 = 1.912(2) Å; Cu1iv–N4 = 2.748(2) Å; (iv) 1/2 2 x, 1/2 2
y, 2z). Another potential magnetic interaction pathway takesplace through the self-complementary amide hydrogen bondsbetween neighboring dinuclear molecules which extends thispathway into a chain. Contrary to the inter-dimer path, thisone takes place via both equatorial positions of eachcoordination sphere, and these bridges extend over five atomsCu{dx22y2}1–N–C–O…H–N–Cu9{dx22y2}1. Despite convenient Cud orbitals involved in the inter-dimer path, the five atombridges diminish the potential of this exchange pathway.Therefore, the magnetic interactions in 3 and 4 can be only ofa weak magnitude.
Computational studies – magnetism
Calculations (see Computational details) were performed inorder to determine the magnetic exchange coupling constantsof compounds 3 and 4, as well as in the solvatomorph[Cu(pia)2]?4H2O36 (399) which can be seen as an assembly ofdimeric complexes each interspaced by one square planarmonomer (Fig. 6). The Cu–N axial bond lengths (2.926 Å) in
the dimer are slightly larger than the ones in 4 and themonomeric spacer is over 3.1 Å apart on either side. The chain3 and the dimer 4 were taken as in the X-ray structure. Twomodels were built for the solvatomorph chain (Fig. 6).36 In 39,the dimer with Cu–N distance 2.926 Å was taken; in 399, atrimer was built by addition of a monomer at 3.117 Å.
The calculated J values are listed in Table 4. The complexes4, 39, and 399 with Cu–N axial bonds exhibit slightlyferromagnetic properties whereas chain 3 with Cu–O axialbonds is nearly paramagnetic. The available experimentalvalues compare well with the calculated ones with Jexp (4). Thevalues of Jcalc (399) show that the magnetic coupling in 39 isbarely affected by inserting the ‘loose’ monomer.
In chain 3 the magnetic pathway is quite long both throughthe superexchange path (coupling via bridging ligands) orthrough space metal–metal contacts. The ligand bridge ismade up of three atoms (Cu–N–CLO–Cu) and the Cu…Cudistance is 5.160 Å. It is difficult to account for theantiferromagnetic contribution in J (3) since there is someband dispersion between the highest occupied bands (HOB).At the origin of the Brillouin zone (k = 0) the singly occupiedcrystal orbital gap is 70 meV whereas at the zone edge (k = p/a)the gap is zero and there is a steady decrease in the gap alongthe whole Brillouin zone. The average gap over the number of kpoints is 45 meV.
The difference in J values between 4 and 39 can berationalized by taking into account the Hay–Thibeault–Hoffmann47 expression for one electron–hole magnetic dimers.
J~2Kab{(e1{e2)2
U(2)
Fig. 6 Part of the crystal structure obtained by Du et al.,36 where 399
corresponds to the dimer with short Cu–N distance (2.926 Å) and 399 to a trimerincluding also the most distant unit (.3.1 Å).
Table 4 Magnetic exchange constants evaluated with DFT/B3LYP (Jcalc) andcorresponding experimental values (Jexp)
Compound Jcalc/cm21 Jexp/cm21
[Cu2(pia)2(piaH)2(ClO4)2] (4) +2.0 y+1.5[Cu(pia)2]n?2H2O (3) 20.2 20.3[Cu(pia)2]2 (39) +1.1 —[Cu(pia)2]3 (399) +1.0/20.1 —
This journal is � The Royal Society of Chemistry 2013 CrystEngComm, 2013, 15, 8074–8087 | 8081
CrystEngComm Paper

Kab is the exchange integral between magnetic fragments a andb, U is the (positive) energy gap between the neutral and ionicforms (Cu3+…Cu0 « Cu0…Cu3+) and e1, e2 the energies of thetriplet state SOMOs (or down spin LUMOs). The first termcorresponds thus to the ferromagnetic part of J (JF) and thesecond (always negative) to the antiferromagnetic part JAF.
Assuming that U does not vary significantly with distanceand that its value is nearly identical in 39 and 4, and that theenergy gap (e1 2 e2) between the triplet SOMOs, obtained fromcalculations, is the same in 39 and 4 (54 meV), the discrepancyin the values of J (39) and J (4) should arise from the JF term.The shortest interplanar distance in 4 is 2.749 Å whereas in 39
it is 2.926 Å. The Cu–Cu distances in 4 are 3.264 Å and in 39
they are 3.465 Å. So the magnetic pathway, whether we takeinto account the bridging ligands or the metal–metaldistances, is always longer in 39. Thus the value of JF = 2Kab
will be smaller in 39 than in 4. Therefore the DFT calculationsof the magnetic interactions for 3 and 4 are in accordance withthe corresponding experimental data.
Computational studies – the formation of 3 and 4
DFT calculations were performed in order to understand theformation of the final products 3 and 4, as well as tocharacterize the two complexes 1 and 2, based on infraredspectroscopic data. We assumed that the two complexes 1 and2 were octahedral, [Cu(N3)2(piaH)2] (1) and [Cu(NCO)2(piaH)2](2), when starting the optimization. Given the square planargeometry of complex 3, with two deprotonated pia ligands, theazido and cyanato ligands in 1 and 2 were considered axial,with two piaH ligands in the equatorial plane. In the cyanatocomplex trans-[Cu(NCO)2(piaH)2] (2), there are six possiblearrangements. The bidentate piaH ligands can coordinate ask2-N,N or k2-N,O and ambidentate NCO through N or O. Foreach of these coordination modes of piaH, it is possible tohave two NCO, two NCO, or NCO and NCO bound ligands. Thesix optimized structures (see Computational details) are shownin Fig. 7.
All the isomers exhibit two intramolecular hydrogen bonds.Only one complex (2-NOO), with two N–H…N hydrogen bonds,can be considered octahedral with the expected Jahn–Tellertetragonal distortion. The Cu–O(piaH) distance (2.444 Å) ismuch longer than the Cu–OCN bond (2.016 Å). 2-NO(N,O) is atetragonal bipyramid, with two different axial Cu…O bonds of2.475 and 3.004 Å, much longer than the other Cu–O/N bonds.The geometry of the other four isomers can be described assquare planar, with very long distances (above 2.7 Å) betweenthe metal and the two axial ligands. The piaH ligand actsessentially as a monodentate ligand. This explains why 2-NNNand 2-NON have almost the same energy: both have four Cu–Nbonds, and only differ by the hydrogen bond, N–H…N or N–H…O, respectively. The potential fifth and sixth coordinatingatoms are too far away (y3 Å).
The most stable isomer (2-NNN) has all the ligandscoordinated by a nitrogen atom and the piaH and NCOligands are associated through strong intramolecular N–H…Nhydrogen bonds. When the bidentate ligand becomes k2-N,O,the energy only rises y2 kcal mol21. The hydrogen bondbecomes N–H…O, which is normally weaker. Changing the
coordination of the cyanate from NCO to OCN has a dramaticeffect. It is less pronounced (energy increase of 21 kcal mol21)in 2-NOO, probably because there are six Cu–O/N bonds andtwo intramolecular N–H…N hydrogen bonds. In 2-NNO,however, the hydrogen bonds are weaker (N–H…O), and theycoexist with only two Cu–N and two Cu–O bonds, leading to anenergy 31 kcal mol21 higher. The two isomers with one NCOand one NCO ligand, 2-NN(N,O) and 2-NO(N,O), have inter-mediate energies.
In the azide complex [Cu(N3)2(piaH)2] (1) there are only twoisomers, 1-NNN and 1-NON, since N3 always binds throughnitrogen. They are shown in Fig. 8 with their relative energies.
These two models have very similar energies because thereare four strong Cu–N bonds in both, ranging between 1.973and 2.106 Å in 1-NNN, and 1.991 and 2.119 Å in 1-NON. Thetwo remaining bonds are too long for an octahedral complex,even taking into account the Jahn–Teller distortion. Therefore,it is not very important that the two long distances are Cu–N orCu–O, since they are weak anyway. Two intramolecular N–H…N hydrogen bonds are present in both isomers, involvingthe unbound NH2 group of the piaH ligand and the bound (1-NNN) or terminal (1-NON) nitrogen atom. The shorter H…Ndistances observed in 1-NNN (1.951, 1.971 Å) probablycontribute to make this isomer slightly more stable than 1-NON, where the same distance rises to 2.298, 3.284 Å.
The Cu–N(azide) distances in N3 complexes with similarenvironments described in the literature37 fall into two groups,from 1.94 to 2.05 Å and from and 2.46 to 2.67 Å. The first set ofvalues compares well with the Cu–N calculated distances
Fig. 7 Optimized structures of six isomers of [Cu(NCO)2(piaH)2] (2), with relativeenergies (kcal mol21) and some relevant distances (Å).
8082 | CrystEngComm, 2013, 15, 8074–8087 This journal is � The Royal Society of Chemistry 2013
Paper CrystEngComm

(about 2 Å, Fig. 8). The second set refers to long bonds due toJahn–Teller distortion, which we did not observe for the azideligand. Structures with coordinate isocyanate are less com-mon, and Cu–N distances lie around 1.93–1.97 Å, in goodagreement with those calculated (Fig. 7).37
The calculated n(CN) infrared frequencies for the boundNCO ligands are 2302 (s) and 2289 (as) cm21 for the lowestenergy isomer 2-NNN, very close to the experimental ones,2225 and 2190 cm21, supporting the assignment. Thecalculated frequencies for the other isomer (2-NON) with Nbound NCO are very similar, while in isomers with NCOcoordination frequencies are very different from the experi-mental ones.
The calculated infrared frequencies for the bound N3
ligands in 1-NNN are 2194 (as) and 2198 (s) cm21, and 2164(as) and 2167 (s) cm21 in 1-NON, both very close to theexperimental one 2062 cm21, suggesting that they appear as abroader band. The very similar energies indicate that there isno structural preference.
In all isomers of the two complexes, the presence of N–H…Nor N–H…O hydrogen bonds opens the way for the hydrogentransfer to the azide ligand in 1 and cyanate ligand in 2, withloss of the protonated axial ligands and formation of thesquare planar complex 3. We started to analyze the protontransfer reaction in the [Cu(NCO)2(piaH)2] (2) complex. It wasnot possible to find a pathway starting from the 2-NON, 2-NNO, 2-NN(N,O) and 2-NO(N,O) complexes. The other twoisomers, where an N–H…N hydrogen bond exists, allowed todetermine a full reaction path for the double hydrogentransfer, leading from 2 to 3. Considering that the reactionbarriers do not differ significantly, it is more likely that thereaction takes place from 2-NNN, than from the higher energy(21 kcal mol21) isomer 2-NOO. Also, the path from 2-NOOleads to the wrong isomer of the square planar complex[Cu(pia)2] (3), with a k2-N,O ligand, instead of k2-N,N.
Since there are two successive proton transfers, we checkedthe possibility to have a concerted and a stepwise reaction, andthe last option proved to be the more favorable one. Thereaction path for the double hydrogen transfer from the NH2
group of the coordinated bidentate piaH ligand to the axialcyanate ligands is shown in Fig. 9.
The reaction starts from the 2-NNN complex. The NH protonof the unbound NH2 group is involved in a hydrogen bondwith the coordinated nitrogen of the cyanate ligand (H…Ndistances 1.897 and 1.958 Å). In the first transition state (TS1),the hydrogen atom involved in the stronger hydrogen bond(shortest distance) is being transferred. The N–H bond haselongated from 1.026 to 1.225 Å, while the N…H distance ofthe bond being formed has shortened to 1.335 Å, and there is anew Cu–N bond (2.165 Å) with the N atom of the halfdeprotonated NH2 group. The coordination environment ofthe metal is square pyramidal and the other side of themolecule remains unchanged. In the Int1* intermediate, thetransfer is complete, with the HNCO molecule formed andhydrogen bonded via a N…N–H to the coordinated nitrogenatom of the deprotonated pia ligand (N…H 2.073, N–H1.024 Å). The Cu–N bond became shorter (1.939 Å). Theactivation barrier for this transfer was y14 kcal mol21 and thereaction is endergonic. The intermediate Int1* loses thehydrogen bonded HNCO, forming Int1. This intermediatehas a very regular square planar geometry (N–Cu–N angles ofalmost 90 or 180u), and the NH2 group of the second piaHligand (unbound, Cu…N distance 3.00 Å) is involved in aNH…H hydrogen bond with the remaining NCO ligand.
The second step occurs in a similar way, with the transfer ofthe hydrogen from the intramolecular hydrogen bond of Int1.At TS2 it is at 1.230 Å from its initial position and 1.327 Å fromthe cyanate. The coordination around Cu is distorted squarepyramidal. The Cu–N(cyanate) bond is lengthening (2.502 Å),while the new Cu–N bond to the half deprotonated NH2 groupis already formed (2.175 Å). Loss of HNCO proceeds throughintermediate 3* (N…H 1.952, N–H 1.030 Å). The activationbarrier for this second transfer reaction is y16 kcal mol21,and the energies of the intermediate Int1 and the final product3 are very close, 3 being more stable by only 1.5 kcal mol21.
The reaction converting 2-NOO in [Cu(pia)2] (3) proceeds ina very similar way, with activation barriers of y5 and y9 kcalmol21 for the first and the second hydrogen transfer,respectively. Although this seems very promising, the startingcomplex has a very high energy (21 kcal mol21 above 2-NNN)and the reaction leads to an isomer of 3, which has not beenobserved, and requires an isomerization to take the bidentateligands from k2-N,O to k2-N,N coordination.
Fig. 8 Optimized structures of two isomers of [Cu(N3)2(piaH)2] (1), with relative energies (kcal mol21) and some relevant distances (Å).
This journal is � The Royal Society of Chemistry 2013 CrystEngComm, 2013, 15, 8074–8087 | 8083
CrystEngComm Paper

In the final complex 3, the pia ligand is k2-N,N coordinated.This arrangement is 6 kcal mol21 more stable than the k2-N,Oalternative, in agreement with the crystal structures available,where it is always observed. On the other hand, the protonatedpiaH is always k2-N,O bound, in known crystal structures.37
The calculated species cannot be easily compared, as otherfactors are involved.
The reaction leading from the azide complex 1-NNN followsthe same stepwise path as described for 2-NNN, with verysimilar energies for the first (y13 kcal mol21) and the secondtransfer (y16 kcal mol21) and, after loss of the second HN3
molecule, the final complex 3 is reached (Fig. S8, ESI3). It wasnot possible to find a transition state for the first protontransfer when starting from 1-NON. This leads us to proposethat the 1-NNN isomer is the most likely to form.
The dimeric complex [Cu(pia)(piaH)(ClO4)]2 (4) resultedfrom the reaction between piaH and another Cu(II) precursor(acetate), with sodium perchlorate added later. Although atfirst sight it looks very different from 3, the removal of theaxial perchlorate ligands leaves two monomeric units eachbearing one pia and one piaH ligands, pia being k2-N,N asbefore, and piaH k2-N,O. It therefore seems that only the firsthydrogen transfer from coordinated piaH has taken place andthen the reaction proceeded in a different way.
The first question addressed was the geometry of the dimer,which required a different computational approach (see
Computational details), namely the M06 functional, in orderto reproduce the Cu…Cu and Cu…ligand distances. Thecoordination pattern described above for 4 was found to havea lower energy than the alternative with two k2-N,N ligands (4*,Fig. 10). This preference suggests that if a reaction similar tothe one described above is to occur, only a single hydrogentransfer takes place. This example clearly exemplifies thepreference for k2-N,N pia and k2-N,O piaH.
This result indicates that the proposed pathway to 3 from 1or 2 can also be followed in this system, but with some
Fig. 9 Reaction pathway for the conversion of the 2-NNN isomer of [Cu(NCO)2(piaH)2] (2), into the square planar complex [Cu(pia)2] (3) (DG in kcal mol21).
Fig. 10 Optimized structures of two isomers of [Cu(pia)(piaH)(ClO4)]2 (4), withrelative energies (kcal mol21).
8084 | CrystEngComm, 2013, 15, 8074–8087 This journal is � The Royal Society of Chemistry 2013
Paper CrystEngComm

modifications. If we take as initial complex an analogue of 1 or2 with bound ClO4
2, there is a problem because this ion is tooweak a base. Therefore, as the starting reagent wasCu(CH3COO)2, and NaClO4 was added later, we began thereaction mechanism study from the octahedral[Cu(piaH)2(CH3COO)2] complex, with axial acetate ligands (4-NNO). The optimized geometry reveals an elongated octahe-dron, with piaH bound through the pyridine nitrogen atomstrans to each other, the remaining positions being occupied bytwo k2-O,O-acetate (Cu–O distances of 1.970 and 2.450 Å or1.927 and 2.842 Å, reflecting to the Jahn–Teller distortion).Two N–H…O hydrogen bonds between the unbound amineand one carboxylate oxygen complete the description of thestructure and prepare the molecule for the hydrogen transfer(Fig. 11). This movement is apparent in the transition stateTS5, where the acetic acid is almost formed (O–H distance1.073 Å), remaining coordinated with one Cu–O bond(2.111 Å). This transition state relaxes to an intermediate(Int5*) where the acetic acid is weakly bound (Cu–O 2.424 Å)and is easily lost to afford another pentacoordinate inter-mediate (Int5).
At this stage, several pathways can be envisaged. Followingthe mechanisms described above, one might expect a secondhydrogen transfer to the acetate, leading to the same squareplanar complex 3. It has not been detected and the activationbarrier is indeed high (TS6, 17.8 kcal mol21). An alternativecan be approximately described as an isomerization. The N–
H…O hydrogen bond is broken by the rotation of the pialigand, which approaches the metal and binds through theamine nitrogen. The barrier for this step is only 2.9 kcal mol21
(TS7), so that this path is well preferred compared to the firstoption.
In order to form the dimeric complex 4, the acetate in Int7should be replaced by perchlorate (added as excess reagent) toform the pentacoordinate species [Cu(pia)(piaH)(ClO4)], aprocess that we did not study. It should be added that theweak perchlorate base is unable to deprotonate the piaHligand, as was confirmed by several attempts at calculatingsuch an alternative pathway. The initial reaction must takeplace from the acetate derivative. Finally, two monomers gettogether, releasing 11.2 kcal mol21 and forming the dimer 4.
Despite the different products obtained, 3 and 4, thereaction pathway is determined by the base strength of thecoordinated monodentate ligand, azide or cyanate, or acetate,as perchlorate is too weak. The different behavior of acetateprevents the second deprotonation from occurring, ending inthe dimer formation.
These intramolecular hydrogen transfer reactions did nottake place when the ligand was thiocyanate.5 Two factors areresponsible for that behavior, namely the lower electronega-tivity of sulfur compared with oxygen, giving rise to weak N–H…S hydrogen bonds (more stable isomer) or N–H…Nhydrogen bonds leading to high energy products.
Fig. 11 Reaction pathway for the conversion of the 4-NNO isomer of [Cu(piaH)2(CH3COO)2], (4), into the square planar complex [Cu(pia)2] (3) or the square pyramidal[Cu(pia)(piaH)(CH3COO)] (Int7) (DG in kcal mol21).
This journal is � The Royal Society of Chemistry 2013 CrystEngComm, 2013, 15, 8074–8087 | 8085
CrystEngComm Paper

Besides the models discussed, the deprotonation of thepicolinamide ligand could also take place in a large variety ofalternative pathways, namely by an external base, but suchconsiderations are probably outside the scope of the currentwork.
Conclusions
Four copper(II) coordination compounds with picolinamide,namely [Cu(N3)2(piaH)2] (1), [Cu(NCO)2(piaH)2] (2),[Cu(pia)2]?2H2O (3) and [Cu(pia)(piaH)(ClO4)]2 (4) were synthe-sized and characterized. The compounds 1 and 2 were isolatedas pure powders only shortly after their precipitation, since theprolonged standing of powder products in their motherliquors leads to the formation of 3, suggesting that kineticsfavors 1 and 2, while 3 is thermodynamically more stable, asDFT calculations showed. Assuming the structure of 1 and 2 tobe octahedral with axial L ligands (L = N3
2 or NCO2), inagreement with infrared spectra, their transformation into 3consists of a stepwise double protonation of L by the piaHligand, with low reaction barriers. The single crystal X-raystructure determination of 3 revealed that the amido groups ofthe starting piaH ligands are deprotonated (pia), chelating viaboth nitrogen atoms, as in the few crystal structures available.The mononuclear square planar [Cu(pia)2] coordinationbuilding blocks in 3 are thus neutral, and arranged in chains,involving two interactions of the Cu…O type per coordinationunit.
On the other hand, the molecular structure of 4 reveals adinuclear coordination units, with one neutral k2-N,O and oneanionic k2-N,N ligand bound to each Cu(II) ion. Besidesbinding as a chelate, the deprotonated amido nitrogen atomenables also an additional semi-coordination bond to theadjacent copper(II) ion within the coordination dimer. Theformation of 4 starts also with an intramolecular protontransfer from piaH, according to the DFT calculations.However, the perchlorate anion is too weak a base to undergothe same reaction, but the acetate, also present, can accept oneproton from piaH in the octahedral species[Cu(piaH)2(CH3COO)2]. Substitution of acetate by excess ofperchlorate and dimerization afford the dimer 4.
The experimentally weak magnetic interactions detectedbetween the copper(II) species in the chains of 3 and dinuclearunits of 4 were reproduced by DFT calculations.
The calculations clearly demonstrated the thermodynamicstability of the pia k2-N,N coordination mode seen in 3 and 4relative to the kinetically favorable piaH k2-N,O in 1 and 2, aswell as the importance of an intramolecular N–H…N hydrogenbond for proton transfer being therefore the driving force forthe formation of 3. Furthermore, the chains in 3 and thedimers in 4, assembled via water mediated H-bonds and theself-complementary amide synthons, respectively, gave aninsight into importance of the additional stabilization forthe building up the supramolecular structure of picolinamidecomplexes through the amide self-complementary synthons.
We can thus conclude that, besides the template effect ofthe metal and the complementarity of supramolecular inter-actions, the metal center structural preferences (S, N or O), theJahn–Teller distortion and the nature of the intramolecularhydrogen bond network may, as we showed, reverse theexpectations and make the control of the self-assembly processdifficult.
Acknowledgements
M. D. and Z. P. acknowledge the financial support of thisresearch by Ministry of Science, Education and Sport of theRepublic of Croatia, Zagreb (Grant No. 119-1193079-1332). M.J. C. and D. V. V. thank FCT, POCI and FEDER (PEst-OE/QUI/UI0612/2011) for financial support and D. V. V. for a grant(SFRH/BD/81017/2011). M. J. C. and D. V. V. also thank P. J.Costa for the useful discussions regarding the reactionmechanisms. B. K. thanks the ARRS (Javna agencija zaraziskovalno dejavnost Republike Slovenije; P1-0175). TheEPR experiments were conducted at the Jozef StefanInstitute, Laboratory of Biophysics, Ljubljana, Slovenia.
References
1 (a) J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29,1304–1319; (b) G. R. Desiraju, Angew. Chem., Int. Ed. Engl.,1995, 34, 2311–2327; (c) M. M. Conn and J. Rebek Jr, Chem.Rev., 1997, 97, 1647–1668.
2 (a) A. Angeloni and A. G. Orpen, Chem. Commun., 2001,343–344; (b) C. B. Aakeroy, A. M. Beatty, J. Desper,M. O’Shea and J. Valdes-Martinez, Dalton Trans., 2003,3956–3962; (c) C. B. Aakeroy, B. M. Scott, M. M. Smith, J.F. Urbina and J. Desper, Inorg. Chem., 2009, 48, 4052–4061.
3 (a) M. Ðakovic and Z. Popovic, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 2007, 63, m507–m509; (b)M. Ðakovic and Z. Popovic, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 2007, 63, m557–m559; (c)M. Ðakovic, Z. Popovic, G. Giester and M. Rajic-Linaric,Polyhedron, 2008, 27, 210–222; (d) M. Ðakovic andZ. Popovic, Acta Crystallogr., Sect. E: Struct. Rep. Online,2008, 64, m311–m312; (e) M. Ðakovic, B.-M. Kukovec andZ. Popovic, Transition Met. Chem., 2010, 36, 65–71; (f)M. Ðakovic, M. Dosen and Z. Popovic, J. Chem. Crystallogr.,2010, 41, 180–185; (g) M. Ðakovic, M. Benko andZ. Popovic, J. Chem. Crystallogr., 2010, 41, 343–348; (h)M. Ðakovic, M. Vinkovic, S. Roca, Z. Popovic, I. Vickovic,D. Vikic-Zopic, J. Lukac, N. Ðakovic and Z. Kusic, J. Coord.Chem., 2012, 65, 1017–1032.
4 (a) M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126; (b)J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang,Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573.
5 M. Ðakovic, D. Vila-Viçosa, M. J. Calhorda and Z. Popovic,CrystEngComm, 2011, 13, 5863–5871.
6 R. G. Parr and W. Yang, in Density Functional Theory ofAtoms and Molecules, Oxford University Press, New York,1989.
7 O. Kahn, in Molecular Magnetism, VCH Publishers, NewYork, 1993.
8086 | CrystEngComm, 2013, 15, 8074–8087 This journal is � The Royal Society of Chemistry 2013
Paper CrystEngComm

8 Oxford Diffraction, Xcalibur CCD System, CrysAlis SoftwareSystem, Version 171.31, Oxford Diffraction Ltd., 2004.
9 G. M. Sheldrick, Acta Crystallogr., Sect. A: Found.Crystallogr., 2008, A64, 112–122.
10 L. J. Farrugia, ORTEP-3, J. Appl. Crystallogr., 1997, 30,565–566.
11 I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C.F. Macrae, P. McCabe, J. Pearson and R. Taylor, ActaCrystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397.
12 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M.A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone,B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato,X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda,J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao,H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta,F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin,V. N. Staroverov, R. Kobayashi, J . Normand,K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar,J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J.E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo,R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin,R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin,K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J.B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox,Gaussian 09, Gaussian Inc., Wallingford C. T., 2009.
13 A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652.14 P. J. Stephens, J. F. Devlin, C. F. Chabalowski and M.
J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627.15 C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens.
Matter Mater. Phys., 1988, 37, 785–789.16 Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120,
215–241.17 (a) U. Haeusermann, M. Dolg, H. Stoll and H. Preuss, Mol.
Phys., 1993, 78, 1211–1224; (b) W. Kuechle, M. Dolg,H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100,7535–7542; (c) T. Leininger, A. Nicklass, H. Stoll, M. Dolgand P. Schwerdtfeger, J. Chem. Phys., 1996, 105, 1052–1059.
18 (a) A. D. McClean and G. S. Chandler, J. Chem. Phys., 1980,72, 5639–5648; (b) R. Krishnan, J. S. Binkley, R. Seeger andJ. A. Pople, J. Chem. Phys., 1980, 72, 650–654; (c) A.H. Wachters, Chem. Phys., 1970, 52, 1033–1036; (d) P.J. Hay, J. Chem. Phys., 1977, 66, 4377–4384; (e)K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989,91, 1062–1065; (f) R. C. Binning and L. A. Curtiss, J.Comput. Chem., 1995, 103, 6104–6113; (g) M. P. McGrathand L. Radom, J. Chem. Phys., 1991, 94, 511–516.
19 G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132,114110–114115.
20 A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys.Chem. B, 2009, 113, 6378–6396.
21 K. Fukui, Acc. Chem. Res., 1981, 14, 363–638.
22 V. R. Saunders, Faraday Symp. Chem. Soc., 1984, 19, 79–84.23 M. D. Towler, A. Zupan and M. Causa, Comput. Phys.
Commun., 1996, 98, 181–205.24 K. Kudin and G. E. Scuseria, Chem. Phys. Lett., 1998, 289,
611–616.25 K. N. Kudin and G. E. Scuseria, Phys. Rev. B: Condens.
Matter Mater. Phys., 2000, 61, 16440–16553.26 O. Yazyev, K. N. Kudin and G. E. Scuseria, Phys. Rev. B:
Condens. Matter Mater. Phys., 2002, 65, 205117.27 L. Noodleman and J. G. Norman, J. Chem. Phys., 1979, 70,
4903–4906.28 L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743.29 R. Caballol, O. Castell, F. Illas, I. de P. R. Moreira and J.
P. Malrieu, J. Phys. Chem. A, 1997, 101, 7860–7866.30 E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput.
Chem., 1999, 20, 1391–1400.31 E. Ruiz, S. Alvarez, J. Cano and V. Polo, J. Chem. Phys., 2005,
123, 164110-1–154110-7.32 O. Kahn and B. Briat, J. Chem. Soc., Faraday Trans. 2, 1976,
72, 268–281.33 http://www.chemcrattprog.com/index.html.34 T. Friscic, Chem. Soc. Rev., 2012, 41, 3493–3510.35 D. Cincic and B. Kaitner, CrystEngComm, 2011, 13,
4351–4357.36 Q.-Y. Du, L.-Y. Xin, Y.-P. Li and D.-S. Cao, Chin. J. Struct.
Chem., 2006, 25, 295–299.37 H. F. Allen and W. D. S. Motherwell, Acta Crystallogr., Sect.
B: Struct. Sci., 2002, 58, 380–388.38 (a) Y. Nawata, H. Iwasaki and Y. Saito, Bull. Chem. Soc. Jpn.,
1967, 40, 515–521; (b) S. C. Chang, D. Y. Park and N. C. Li,Inorg. Chem., 1968, 7, 2144–2150.
39 (a) D. Fan, C.-T. Yang, J. D. Ranford and J. J. Vittal, DaltonTrans., 2003, 4749–4753; (b) D. T. Hill, K. Burns, D.D. Titus, G. R. Girard, W. M. Reiff and L. M. Mascavage,Inorg. Chim. Acta, 2003, 346, 1–6.
40 G. Sanchez, J. Vives, G. Lopez, J. L. Serrano, L. Garcia andJ. Perez, Eur. J. Inorg. Chem., 2005, 2360–2367.
41 P. Wang, J. E. Miller, L. M. Henling, C. L. Stern, N. L. Frank,A. L. Eckermann and T. J. Meade, Inorg. Chem., 2007, 46,9853–9862.
42 A. Albinati, F. Lianza, B. Muller and P. S. Pregosin, Inorg.Chim. Acta, 1993, 208, 119–121.
43 H. Brunner, B. Nuber and M. Prommesberger, J.Organomet. Chem., 1996, 523, 179–185.
44 M. A. Goher, A. Escuer, A. M. Abu-Youssef and F.A. Mautner, Polyhedron, 1998, 17, 4265.
45 N. W. Ashcroft and N. D. Mermin, in Solid State Physics,Saunders College Publishing, USA, 1976.
46 J. C. Bonner and M. E. Fisher, Phys. Rev. A, 1964, 135,A640–A658.
47 P. J. Hay, J. C. Thibeault and R. Hoffmann, J. Am. Chem.Soc., 1975, 97, 4884–4899.
This journal is � The Royal Society of Chemistry 2013 CrystEngComm, 2013, 15, 8074–8087 | 8087
CrystEngComm Paper




















