![Oxidations by the system “hydrogen peroxide–[Mn 2L 2O 3][PF 6] 2 (L = 1,4,7-trimethyl-1,4,7-triazacyclononane)–oxalic acid”. Part 6. Oxidation of methane and other alkanes](https://static.fdokumen.com/doc/165x107/632307aa078ed8e56c0aabd9/oxidations-by-the-system-hydrogen-peroxidemn-2l-2o-3pf-6-2-l-147-trimethyl-147-triazacyclononaneoxalic.jpg)
Bis and tris-pyridyl amino and imino thioether Cu and Fe complexes. Thermal and microwave-assisted...
12
Journal of Molecular Catalysis A: Chemical 351 (2011) 100–111 Contents lists available at SciVerse ScienceDirect Journal of Molecular Catalysis A: Chemical jou rn al h om epa ge: www.elsevier.com/locate/molcata Bis- and tris-pyridyl amino and imino thioether Cu and Fe complexes. Thermal and microwave-assisted peroxidative oxidations of 1-phenylethanol and cyclohexane in the presence of various N-based additives Ricardo R. Fernandes a , Jamal Lasri a , M. Fátima C. Guedes da Silva a,b , José A.L. da Silva a , João J.R. Fraústo da Silva a , Armando J.L. Pombeiro a,∗ a Centro de Química Estrutural, Complexo I, Instituto Superior Técnico, TU-Lisbon, Av. Rovisco Pais, 1049-001 Lisbon, Portugal 1 b Universidade Lusófona de Humanidades e Tecnologias, ULHT Lisbon, Av. do Campo Grande, 376, 1749-024 Lisbon, Portugal a r t i c l e i n f o Article history: Received 9 July 2011 Received in revised form 15 September 2011 Accepted 25 September 2011 Available online 1 October 2011 Keywords: Copper and iron complexes N,S-ligands Oxidation N-based additives Microwave a b s t r a c t The new [Cu(OTf)(L)](OTf) 1, [Cu(L)(H 2 O)](OTf) 2 1 , [Cu(OTf)(L Me )](OTf) 2 and [Cu(L py )](OTf) 2 4, and [Fe(OTf) 2 (L Me )] 3 and [Fe(OTf)(L py )](OTf) 5 complexes, bearing bis- and tris-pyridyl amino and imino thioether ligands, have been synthesized and fully characterized by IR, ESI + -MS and elemental analyses, and also by X-ray diffraction structural analyses (for 1, 1 , 2 and 4). In the presence of various N-based additives, they show a very good catalytic activity for the solvent- and halogen-free microwave-assisted oxidation of 1-phenylethanol by t-BuOOH (model reaction). The combination of pyridazine with the Cu complex 2 provided the most efficient catalytic system, resulting in a maximum yield of acetophenone of 99% after 30 min at 80 ◦ C. The maximum turnover frequency (TOF) of 5220 h −1 (corresponding to 87% yield) was achieved just after 5 min of reaction time under the very low microwave power of 10 W. The catalytic properties of those complexes were also evaluated for the mild oxidation of cyclohexane, to cyclohexanol and cyclohexanone, in acetonitrile with H 2 O 2 , leading to yields up to 29% (based on the alkane substrate) that are comparable to the best reported Fe and Cu catalytic systems. The effects of various factors, such as nature and amount of the additives and metal centres (Cu or Fe), temperature, time and type of peroxide used, were also investigated. © 2011 Elsevier B.V. All rights reserved. 1. Introduction Selective oxidation of organic molecules is an essential tool for many chemical transformations and the permanent quest for greener and more efficient systems remains attracting much research interest to this field [1,2]. Functionalized oxygenated products are the foundation of important industrial synthetic strategies and in particular ketones are involved in multi-billion euro chemical processes, as solvents, polymer precursors and sub- strates for the syntheses of drugs and agrochemicals [3,4]. Within the vast synthetic methods for the preparation of ketones, recent efforts focus on energy- and atom-efficiency, elimination of the use of hazardous substances and reduction of the generation of toxic and heavy-metal wastes [5]. Aerobic oxidation reactions of hydrocarbons in industry often proceed at very high pressures and temperatures via radical autox- idation [1–3], whereas the more elegant bioinspired alkane [6–10] ∗ Corresponding author. Tel.: +351 218419237; fax: +351 218464455. E-mail address: [email protected] (A.J.L. Pombeiro). 1 http://cqe.ist.utl.pt/research/group5.php. or alkene [11,12] direct oxidation approaches are typically catal- ysed by synthetic model complexes of Fe [13–21], Cu [22–27] and V [28–32], using hydrogen peroxide as oxidant under very mild conditions. Only water is formed as by-product of these reactions, a feature favourable to the establishment of a green method for the preparation of ketones [5,6]. Molecular oxygen is seldom used in the biomimetic catalytic routes due to the intrinsic difficulty to activate O 2 for functionalization of the inert C–H bond at ambient temperatures. The typical non-selective and non-efficient reaction patterns, observed in C–H oxidations, impedes their wider applica- tion for the synthesis of ketones and are limited to few classes of substrates [6–10]. In addition, the classical methods for liquid-phase oxidation of alcohols to ketones are unfeasible on an industrial-scale and undesirable from the environmental viewpoint due namely to the common use of chlorinated solvents, toxic (e.g. Cr and Mn reagents) [33–35] or moisture-sensitive (e.g. N,N -dicyclohexylcarbodiimide, oxalyl chloride) oxidants [36], high catalysts loadings, presence of bases and phase-transferring agents [37–40]. Hence, aerobic [40–44] and peroxidative [45–48] oxidations of secondary alcohols are regarded as the simplest and most useful synthetic methods for the preparation of ketones, in view of the 1381-1169/$ – see front matter © 2011 Elsevier B.V. All rights reserved. doi:10.1016/j.molcata.2011.09.022
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Bis and tris-pyridyl amino and imino thioether Cu and Fe complexes. Thermal and microwave-assisted...

Bac
RJa
b
a
ARR1AA
KCNONM
1
ffrpsesteoa
pi
1d
Journal of Molecular Catalysis A: Chemical 351 (2011) 100– 111
Contents lists available at SciVerse ScienceDirect
Journal of Molecular Catalysis A: Chemical
jou rn al h om epa ge: www.elsev ier .com/ locate /molcata
is- and tris-pyridyl amino and imino thioether Cu and Fe complexes. Thermalnd microwave-assisted peroxidative oxidations of 1-phenylethanol andyclohexane in the presence of various N-based additives
icardo R. Fernandesa, Jamal Lasri a, M. Fátima C. Guedes da Silvaa,b, José A.L. da Silvaa,oão J.R. Fraústo da Silvaa, Armando J.L. Pombeiroa,∗
Centro de Química Estrutural, Complexo I, Instituto Superior Técnico, TU-Lisbon, Av. Rovisco Pais, 1049-001 Lisbon, Portugal1
Universidade Lusófona de Humanidades e Tecnologias, ULHT Lisbon, Av. do Campo Grande, 376, 1749-024 Lisbon, Portugal
r t i c l e i n f o
rticle history:eceived 9 July 2011eceived in revised form5 September 2011ccepted 25 September 2011vailable online 1 October 2011
eywords:opper and iron complexes
a b s t r a c t
The new [Cu(OTf)(L)](OTf) 1, [Cu(L)(H2O)](OTf)2 1′, [Cu(OTf)(LMe)](OTf) 2 and [Cu(Lpy)](OTf)2 4, and[Fe(OTf)2(LMe)] 3 and [Fe(OTf)(Lpy)](OTf) 5 complexes, bearing bis- and tris-pyridyl amino and iminothioether ligands, have been synthesized and fully characterized by IR, ESI+-MS and elemental analyses,and also by X-ray diffraction structural analyses (for 1, 1′, 2 and 4). In the presence of various N-basedadditives, they show a very good catalytic activity for the solvent- and halogen-free microwave-assistedoxidation of 1-phenylethanol by t-BuOOH (model reaction). The combination of pyridazine with the Cucomplex 2 provided the most efficient catalytic system, resulting in a maximum yield of acetophenoneof 99% after 30 min at 80 ◦C. The maximum turnover frequency (TOF) of 5220 h−1 (corresponding to 87%
,S-ligandsxidation-based additivesicrowave
yield) was achieved just after 5 min of reaction time under the very low microwave power of 10 W. Thecatalytic properties of those complexes were also evaluated for the mild oxidation of cyclohexane, tocyclohexanol and cyclohexanone, in acetonitrile with H2O2, leading to yields up to 29% (based on thealkane substrate) that are comparable to the best reported Fe and Cu catalytic systems. The effects ofvarious factors, such as nature and amount of the additives and metal centres (Cu or Fe), temperature,time and type of peroxide used, were also investigated.
. Introduction
Selective oxidation of organic molecules is an essential toolor many chemical transformations and the permanent questor greener and more efficient systems remains attracting muchesearch interest to this field [1,2]. Functionalized oxygenatedroducts are the foundation of important industrial synthetictrategies and in particular ketones are involved in multi-billionuro chemical processes, as solvents, polymer precursors and sub-trates for the syntheses of drugs and agrochemicals [3,4]. Withinhe vast synthetic methods for the preparation of ketones, recentfforts focus on energy- and atom-efficiency, elimination of the usef hazardous substances and reduction of the generation of toxicnd heavy-metal wastes [5].
Aerobic oxidation reactions of hydrocarbons in industry oftenroceed at very high pressures and temperatures via radical autox-
dation [1–3], whereas the more elegant bioinspired alkane [6–10]
∗ Corresponding author. Tel.: +351 218419237; fax: +351 218464455.E-mail address: [email protected] (A.J.L. Pombeiro).
1 http://cqe.ist.utl.pt/research/group5.php.
381-1169/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.molcata.2011.09.022
© 2011 Elsevier B.V. All rights reserved.
or alkene [11,12] direct oxidation approaches are typically catal-ysed by synthetic model complexes of Fe [13–21], Cu [22–27] andV [28–32], using hydrogen peroxide as oxidant under very mildconditions. Only water is formed as by-product of these reactions,a feature favourable to the establishment of a green method forthe preparation of ketones [5,6]. Molecular oxygen is seldom usedin the biomimetic catalytic routes due to the intrinsic difficulty toactivate O2 for functionalization of the inert C–H bond at ambienttemperatures. The typical non-selective and non-efficient reactionpatterns, observed in C–H oxidations, impedes their wider applica-tion for the synthesis of ketones and are limited to few classes ofsubstrates [6–10].
In addition, the classical methods for liquid-phase oxidationof alcohols to ketones are unfeasible on an industrial-scale andundesirable from the environmental viewpoint due namely to thecommon use of chlorinated solvents, toxic (e.g. Cr and Mn reagents)[33–35] or moisture-sensitive (e.g. N,N′-dicyclohexylcarbodiimide,oxalyl chloride) oxidants [36], high catalysts loadings, presence of
bases and phase-transferring agents [37–40].Hence, aerobic [40–44] and peroxidative [45–48] oxidations ofsecondary alcohols are regarded as the simplest and most usefulsynthetic methods for the preparation of ketones, in view of the

ar Cat
v(s[t[
sdaaau
etol
SeaaofaTttta
2
2
rSau(ton3eitit(ad3tobOnrtraa
R.R. Fernandes et al. / Journal of Molecul
ersatility of the metal catalysts, oxidants and alcohol substratesbenzylic and allylic compounds) used. The improvement of theseynthetic methodologies, according to green chemistry principles5,49], is a challenge to be tackled, focusing on the use of alterna-ive green solvent systems for more efficient and clean syntheses50].
Very recently, some of us reported very efficient catalyticystems based on copper(II) alkoxy-triazapentadienato [51] andiethanolaminate dicopper(II) [52] complexes for both aerobicnd peroxidative (using t-BuOOH) oxidations of different primarynd secondary alcohols, including a solvent-free microwave (MW)ssisted method which accelerates dramatically the reaction ratesing a very low microwave irradiation power.
Polydentate pyridine-type complexes have been widelymployed in biomimetic catalytic oxidations [6–8], but the collec-ion of Fe and Cu catalysts concerns almost exclusively the all-Nr N,O systems [6–8,11–16], whereas the S-derivatives remain stillittle explored [17].
In this regard, the current study aims at the syntheses of new-containing pyridine-type Cu(II) and Fe(II) complexes and theirvaluation as oxidation catalysts. The novel complexes bearing bis-nd tris-pyridyl amino or imino thioether ligands are found to acts highly efficient and selective catalysts for the solvent-free per-xidative oxidation of 1-phenylethanol to acetophenone (underocused MW irradiation) and also for the oxidation of cyclohex-ne, to cyclohexanol and cyclohexanone, at 25 ◦C in acetonitrile.he efficiency of the oxidation reactions was investigated in a sys-ematic way and the influence of various parameters, such as theype of catalyst, type and amount of various N-based additives,emperature, time and relative amounts of reagents, was evalu-ted.
. Experimental
.1. General methods
Unless noted otherwise, all reactions and operations were car-ied out at room temperature under dinitrogen by use of standardchlenk techniques. Solvents were dried and distilled before usend reagents were obtained from commercial sources (Aldrich) andsed as received. The ligand (2-pyridyl)-3-thia-5-aminopentanepyta) was obtained following a procedure previously reported inhe literature [53]. C, H and N elemental analyses were carriedut by the Microanalytical Service of the Instituto Superior Téc-ico. 1H and 13C NMR spectra were measured on Bruker Avance II00 and 400 MHz (UltraShieldTM Magnet) spectrometers at ambi-nt temperature. 1H and 13C chemical shifts (ı) are expressedn ppm relative to Si(Me)4. J values are in Hz. Infrared spec-ra (4000–400 cm−1) were recorded on a Bio-Rad FTS 3000MXnstrument in KBr pellets and the wavenumbers are in cm−1. Elec-rospray mass spectra were performed with an ion-trap instrumentVarian 500-MS LC Ion Trap Mass Spectrometer) equipped withn electrospray (ESI) ion source. For electrospray ionization, therying gas and flow rate were optimized to each sample with5 psi nebulizer pressure. Scanning was performed from m/z = 50o 1500. The compounds in solutions of CH2Cl2 or MeOH werebserved in the positive mode (capillary voltage = 80–105 V). Theatch catalytic studies in acetonitrile were performed in a Stemmni Reactio Station with capacity for 10 reactors (50 mL) witho additional concerns to remove air or moisture prior to theeactions. The catalytic investigations under microwave irradia-
ion (10 W) were performed in a focused microwave CEM Discovereactor (10 mL, 13 mm diameter), fitted with a rotational systemnd an IR detector of temperature. The catalytic reactions werenalyzed by GC (gas chromatography) using a Fisons Instrumentsalysis A: Chemical 351 (2011) 100– 111 101
model 8160 gas chromatograph equipped with a DB-WAX (col-umn length: 30 m; internal diameter: 0.32 mm; film: 0.25 �m)capillary column (helium as a carrier gas) with a FID detec-tor.
2.2. Syntheses of ligands and complexes
Preparation of L. A mixture of pyta (1.2 g, 6.5 mmol) andpyridine-2-aldehyde (0.7 g, 6.5 mmol) was refluxed in EtOH (25 mL)for 24 h. After this period the solvent was evaporated yielding theproduct as a brown oil (1.5 g, 87%). C15H17N3S (M = 271.38 g mol−1).1H NMR (CDCl3, 400 MHz) ı, ppm: 8.65 (d, J = 4.8 Hz, 1H, pyH�),8.53 (d, J = 4.8 Hz, 1H, pyH�), 8.38 (s, 1H, N CH), 7.97 (d, J = 7.8 Hz,1H, pyH�), 7.74 (t, J = 7.8 Hz, 1H, pyH�), 7.59 (t, J = 7.6 Hz, 1H,pyH�), 7.39–7.30 (m, 1H, pyH�), 7.16 (d, J = 7.8 Hz, 1H, pyH�),7.14–7.10 (m, 1H, pyH�), 3.88 (t, J = 6.8 Hz, 2H), 3.10–2.98 (m, 4H),2.90 (t, J = 6.8 Hz, 2H). 13C NMR (CDCl3, 100.6 MHz) ı, ppm: 162.9(N CH), 159.8 (pyCq), 154.1 (pyCq), 149.4 (pyC�), 149.3 (pyC�),136.6 (pyC�), 136.5 (pyC�), 125.1, 123.5, 121.5, 121.4 (pyC�), 61.5,38.8, 33.1, 32.2 (SCH2CH2N and py-CH2CH2S). ESI-MS (positivemode, CH2Cl2): m/z 294.1 {[L+Na]}+, 272.1 {[L+H]}+.
Preparation of LH. To a solution of L (1.0 g, 3.7 mmol) in EtOH(50 mL) was added NaBH4 (280 mg, 7.4 mmol) and this mixturewas stirred at room temperature for 24 h. Thereafter H2O (30 mL)was added and, by slow addition of concentrated HCl, the pHof the solution was adjusted to 1. After concentrated to dryness,H2O (50 mL) and CH2Cl2 (100 mL) were added to the residue. ThepH of the aqueous phase was brought to 13 by the addition of3 M aqueous KOH solution and the biphasic mixture was stirredfor further 1 h. The layers were separated and the aqueous phasewas further extracted with CH2Cl2 (3 × 50 mL). The CH2Cl2 frac-tions were dried over Na2SO4 and evaporated to dryness underreduced pressure to yield the product as a yellowish oil (951 mg,94%). C15H19N3S (M = 273.40 g mol−1). 1H NMR (CDCl3, 400 MHz)ı, ppm: 8.50–8.49 (m, 2H, pyH�), 7.44–7.34 (m, 2H, pyH�), 7.28(d, J = 8.0 Hz, 1H, pyH�), 6.98–6.92 (m, 3H, pyH�), 3.88 (s, 2H, py-CH2N), 3.02 (t, J = 7.6 Hz, 2H), 2.91–2.80 (m, 4H), 2.69 (t, J = 7.6 Hz,2H), 2.39 (bs, 1H, NH). 13C NMR (CDCl3, 100.6 MHz) ı, ppm: 159.3(pyCq), 158.9 (pyCq), 148.7 (pyC�), 149.6 (pyC�), 135.9 (pyC�),135.8 (pyC�), 122.6, 121.6, 121.4, 120.9 (pyC�), 54.1 (py-CH2N),47.6, 37.8, 31.8, 30.9 (py-CH2CH2S and SCH2CH2N). ESI-MS (posi-tive mode, CH2Cl2): m/z 296.1 {[LH+Na]}+, 274.1 {[LH+H]}+.
Preparation of LMe. A batch of LH (1.0 g, 3.7 mmol), 35% aque-ous solution of formaldehyde (1.3 g) and 96% formic acid (1.5 g)was heated under reflux for 24 h. After concentration to dryness,H2O (50 mL) and CH2Cl2 (100 mL) were added to the residue. ThepH of the aqueous phase was brought to 13 by the addition of a3 M aqueous KOH solution and the heterogeneous mixture stirredfor 1 h. The layers were separated and the aqueous phase was fur-ther extracted with CH2Cl2 (3 × 50 mL). The CH2Cl2 fractions weredried over K2CO3 and evaporated to dryness under reduced pres-sure to yield quantitatively the methylated product as a brown oil.C16H21N3S (M = 287.42 g mol−1). 1H NMR (CDCl3, 400 MHz) ı, ppm:8.51–8.50 (m, 2H, pyH�), 7.64–7.55 (m, 2H, pyH�), 7.42 (d, J = 7.6 Hz,1H, pyH�), 7.15–7.09 (m, 3H, pyH�), 3.66 (s, 2H, py-CH2N), 3.03 (t,J = 8.0 Hz, 2H, py-CH2CH2S), 2.91 (t, J = 8.0 Hz, 2H, py-CH2CH2S), 2.67(m, 4H, SCH2CH2N), 2.26 (b s, 3H, CH3).
13C NMR (CDCl3, 100.6 MHz) ı, ppm: 160.3 (pyCq), 159.5 (pyCq),149.7 (pyC�), 149.3 (pyC�), 136.7 (pyC�), 136.6 (pyC�), 123.4,123.3, 122.3, 121.7 (pyC�), 63.9 (py-CH2N), 57.6 (SCH2CH2N), 42.6(CH3), 38.8, 32.2, 30.2 (py-CH2CH2S and SCH2CH2N). ESI-MS (pos-itive mode, CH2Cl2): m/z 288.2 {[LMe+H]}+.
Preparation of Lpy. LH (1.6 g, 5.9 mmol), 2-picolyl chloridehydrochloride (1.0 g, 5.9 mmol) and anhydrous CH3CN (30 mL)were charged in a Schlenk tube. Na2CO3 (2.5 g) and tetrabuty-lammonium bromide, TBABr (40.0 mg), were added directly as

1 lar Cat
sNwbtthowr((p31((C3
00fsdm(C(
ot
oe
1twcawh4(C(7
o
i1rcawwaoM{c315
perature (typically 80 ◦C) was maintained using an oil bath.
02 R.R. Fernandes et al. / Journal of Molecu
olids and the suspension was vigorously stirred at reflux under2 for 18 h. After cooling to room temperature the suspensionas filtered off and the residue washed with CH2Cl2. The com-
ined filtrates were evaporated to dryness. The obtained oil wasaken in CH2Cl2 (100 mL) and H2O (100 mL) and the pH adjustedo 13 with 3 M aqueous KOH solution. After stirring for 1 h thiseterogeneous mixture, the layers were separated and the aque-us phase was extracted with CH2Cl2 (3 × 100 mL). The fractionsere dried over anhydrous Na2SO4 and the solvent removed under
educed pressure to yield a brown oil (1.96 g, 91%). C21H24N4SM = 364.51 g mol−1). 1H NMR (CDCl3, 400 MHz) ı, ppm: 8.51–8.49m, 3H, pyH�), 7.64 (dt, J = 7.6 Hz, 2H, pyH�), 7.59–7.54 (m, 3H,yH� and pyH�), 7.14–7.09 (m, 4H, pyH�), 3.83 (s, 4H, py-CH2N),.01–2.68 (m, 8H, SCH2CH2N and py-CH2CH2S). 13C NMR (CDCl3,00.6 MHz) ı, ppm: 160.3 (pyCq), 159.8 (pyCq), 149.7 (pyC�), 149.3pyC�), 136.7 (pyC�), 136.6 (pyC�), 123.5, 123.3, 122.3, 121.7pyC�), 60.5 (py-CH2–N), 54.2 (SCH2CH2N), 38.8, 32.1, 30.1 (py-H2CH2S and SCH2CH2N). ESI-MS (positive mode, CH2Cl2): m/z87.3 {[Lpy+Na]}+, 365.3 {[Lpy+H]}+.
[Cu(OTf)(L)](OTf) (1). To a stirred solution of L (100 mg,.37 mmol) in EtOH (5 mL) was added Cu(OTf)2 (134 mg,.37 mmol). The clear solution was stirred at room temperatureor 5 h and then evaporated to dryness, yielding a deep blueolid. The resulting powder was further washed with Et2O andried in vacuo (187 mg, 80%). ESI-MS (positive mode, CH3OH):/z 483.0 {[Cu(OTf)(L)]}+, 334.0 {[Cu(L)]}+. C17H17CuF6N3O6S4
M = 633.07 g mol−1): calcd. C 32.25, N 6.64, H 2.71, S 15.20; found 32.26, N 6.52, H 2.57, S 15.17. IR (KBr): 1650 (m), 1610 (m), 1485m), 1448 (m), 1249 (s), 1161 (s), 1030 (s), 779 (vs), 647 (s) cm−1.
X-ray diffraction quality single crystals of compound 1 werebtained by slow diethyl ether or pentane diffusion into a concen-rated dichloromethane solution.
[Cu(L)(H2O)](OTf)2 (1′). X-ray diffraction quality single crystalsf compound 1′ were obtained by slow diffusion of pentane into anthanol/H2O solution.
[Cu(OTf)(LMe)] (OTf) (2). A solution of Cu(OTf)2 (516 mg,.43 mmol) in THF (5 mL) was added to a vigorously stirred solu-ion of LMe (410 mg, 1.43 mmol) in THF (5 mL). The cloudy mixtureas stirred overnight at room temperature. The solution was then
oncentrated to half of the initial volume and Et2O (5 mL) wasdded, resulting in the precipitation of a blue solid. The solidas filtered off, further washed with Et2O and then dried underigh vacuum (700 mg, 75%). ESI-MS (positive mode, CH2Cl2): m/z99.0 {[Cu(OTf)(LMe)]}+, 350.1 {[Cu(LMe)]}+. C18H21CuF6N3O6S3M = 649.11 g mol−1): calcd. C 33.31, N 6.47, H 3.26, S 14.82; found
33.01, N 5.91, H 3.33, S 15.18. IR (KBr): 3500 (m), 3112 (m), 2975m), 1611 (m), 1484 (m), 1452 (m), 1274 (vs), 1156 (s), 1031 (vs),80 (m), 638 (s), 518 (m) cm−1.
X-ray diffraction quality single crystals of compound 2 werebtained by slow pentane diffusion into an acetonitrile solution.
[Fe(OTf)2(LMe)] (3). To a batch of LMe (400 mg, 1.39 mmol)n dry THF (10 mL) was added a solution of Fe(OTf)2 (493 mg,.39 mmol) in THF (5 mL). The mixture was stirred overnight atoom temperature under an N2 atmosphere. The solution was con-entrated to half of the initial volume and then dry Et2O (5 mL) wasdded, resulting in the precipitation of a beige solid. The solventas removed through a cannula and the precipitate was furtherashed with Et2O. The precipitate was then dissolved in CH2Cl2
nd the mixture stirred for 1 h. An off-white solid was finallybtained after removing the solvent in vacuo (656 mg, 69%). ESI-S (positive mode, CH2Cl2): m/z 492.2 {[Fe(OTf)(LMe)]}+, 288.2
[LMe+H]}+. C18H21F6FeN3O6S3·1/2 CH2Cl2 (M = 683.88 g mol−1):alcd. C 32.49, N 6.14, H 3.24, S 14.07; found C 32.64, N 5.80, H
.23, S 14.12. IR (KBr): 3514 (m), 3079 (w), 2928 (w), 1636 (m),609 (m), 1445 (m), 1279 (vs), 1172 (s), 1032 (m), 763 (m), 641 (s),19 (m) cm−1.alysis A: Chemical 351 (2011) 100– 111
[Cu(Lpy)](OTf)2 (4). A solution of Cu(OTf)2 (99 mg, 0.27 mmol)in anhydrous THF (3 mL) was added to a vigorously stirred solu-tion of Lpy (100 mg, 0.27 mmol) in THF (2 mL). After a few minutesthe greenish blue solution became cloudy and a blue precipitateappeared. The suspension was stirred overnight at room tem-perature, whereafter, the resulting blue solid was filtered off,washed with THF and Et2O and dried under high vacuum (178 mg,91%). ESI-MS (positive mode, CH2Cl2): m/z 576.2 {[Cu(OTf)(Lpy)]}+,427.2 {[Cu(Lpy)]}+. C23H24CuF6N4O6S3 (M = 726.19 g mol−1): calcd.C 38.04, N 7.72, H 3.33, S 13.25; found C 38.26, N 7.67, H 3.12, S 13.44.IR (KBr): 3530 (m), 3080 (w), 2973 (w), 1611 (m), 1475 (m), 1438(m), 1263 (m), 1148 (m), 1033 (vs), 765 (m), 637 (s), 518 (m) cm−1.
Suitable single crystals of 4 for X-ray diffraction were obtainedby slow diethyl ether diffusion into an acetonitrile solution.
[Fe(OTf)(Lpy)](OTf) (5). A solution of Fe(OTf)2 (97 mg,0.27 mmol) in anhydrous THF (3 mL) was slowly added to asolution of Lpy (100 mg, 0.27 mmol) in THF (5 mL) and stirredunder N2 for 20 h. Thereafter the solution was concentratedin vacuo and upon addition of Et2O a beige precipitate appeared.The solid was filtered off and dried under vacuum (140 mg, 72%).ESI-MS (positive mode, CH2Cl2): m/z 569.2 {[Fe(OTf)(Lpy)]}+.C23H24F6FeN4O6S3 (M = 718.49 g mol−1): calcd. C 38.45, N 7.80, H3.37, S 13.39; found C 38.32, N 7.59, H 3.43, S 13.41. IR (KBr): 3498(m), 2930 (w), 1606 (m), 1482 (m), 1445 (m), 1280 (vs), 1167 (m),1032 (vs), 763 (m), 640 (s), 518 (m) cm−1.
2.3. Catalytic studies
2.3.1. Peroxidative oxidations of cyclohexane and1-phenyethanol with aqueous H2O2 in acetonitrile
Typical oxidation reactions were carried out in 50 mL screwtopglass reactors (Stem Omni Reactio Station) in air as follows: cata-lysts 1–5 (10 �mol), substrate (5 mmol), CH3CN (3 mL) and additive(200 �mol). The reactors were placed in the reaction station andstirred vigorously at a controlled temperature of 25 ◦C. An aque-ous solution of hydrogen peroxide (30%) (7 mmol) was then addedand the reaction mixture stirred for 6 h. The product analysis forthe oxidation of cyclohexane was performed as follows: 90 �L ofinternal standard (cycloheptanone) and 10 mL of diethyl ether (toextract the substrate and the organic products from the reactionmixture) were added. In the case of cyclohexane oxidation, to thefinal mixture was added an excess of triphenylphosphine before theGC analysis, in order to reduce the formed cyclohexyl hydroperox-ide to the corresponding alcohol, and hydrogen peroxide to water,following a method developed by Shul’pin [54,55]. After stirringthe final reaction mixture for 10 min a sample was taken from theorganic phase and then analyzed by GC. For the oxidation of 1-phenylethanol, the GC determination of acetophenone yields wasperformed upon the addition of 2 mL of CH3CN and 300 �L of ben-zaldehyde as internal standard. Then, diethyl ether was added todilute the sample and, after mixing, a sample for GC analysis wastaken from the organic phase.
2.3.2. Peroxidative oxidation of 1-phenylethanol with aqueoust-BuOOH2.3.2.1. Conventional method. Oxidation reactions were carried outin air using 25 mL round-bottom flasks equipped with a refluxcondenser. Under typical conditions, catalyst 1–5 (10 �mol) wasplaced into the flask, followed by the addition of 1-phenylethanol(5 mmol), additive (200 �mol) and an aqueous solution of t-BuOOH(70%) (10 mmol). In all cases, the reaction solutions were vigorouslystirred for 30 min using magnetic stirrers. The desired reaction tem-
2.3.2.2. Microwave-assisted method. Typical oxidation reactionsof 1-phenylethanol were carried out in a sealed Pyrex tube

R.R. Fernandes et al. / Journal of Molecular Catalysis A: Chemical 351 (2011) 100– 111 103
Table 1Crystallographic data and structure refinement details for complexes 1, 1′ , 2 and 4.
Data 1 1′ 2 4
Empirical formula C16H17CuF3N3O3S2 C15H19CuN3OS, CF3O3S C17H21CuF3N3O3S2, CF3O3S C21H24CuN4S, 2(CF3O3S)Mr/g mol−1 483.99 502.00 649.10 726.18Crystal system Triclinic Monoclinic Orthorhombic TriclinicSpace group P-1 C 2/c P 21 21 21 P-1Temp. (K) 293(2) 150(2) 150(2) 150(2)a (Å) 9.5962(8) 25.4443(14) 9.3340(9) 9.0933(6)b (Å) 11.8804(9) 11.4041(5) 10.2920(12) 12.6712(8)c (Å) 12.5753(16) 18.1441(9) 25.993(3) 13.8153(8)˛ (◦) 113.566(5) 90.00 90.00 115.226(2)ˇ (◦) 103.206(5) 109.379(2) 90.00 91.862(3)� (◦) 99.186(4) 90.00 90.00 92.918(3)V (Å3) 1227.7(2) 4966.6(4) 2497.0(5) 1435.58(16)Z 2 8 4 2Dc (g cm−3) 1.309 1.343 1.727 1.680� (Mo K�) (mm−1) 1.100 1.093 1.211 1.064No. of collected reflns 10,616 31,791 18,718 10,974No. of unique reflns 4238 7873 5452 5182Rint 0.0930 0.0503 0.0641 0.0236Final R1a, wR2b (I ≥ 2 �) 0.1161, 0.2990 0.0541, 0.1522 0.0504, 0.1156 0.0320, 0.0873GOF on F2 1.047 1.083 1.034 1.041
a RI =∑
||F | − |F ||/∑
|F |.
u(wocw5
2Ttids
amn
2
owwwDfrSwdwArneiiec
by condensation of (2-pyridyl)-3-thia-5-aminopentane (pyta) [53]with pyridine-2-aldehyde in refluxing ethanol for 24 h (Scheme 1,reaction a). The reduction of L with sodium borohydride (NaBH4),in ethanol at r.t., afforded LH (Scheme 1, reaction b), which was
Table 2Selected bond lengths ( ´A) and angles (◦) for complexes 1, 1′ , 2 and 4.
1 1′ 2 4
Cu1–N1 2.023(7) 2.045(2) 1.997(4) 1.9654(19)Cu1–N2 1.971(8) 1.961(2) 2.032(4) 2.0180(19)Cu1–N3 1.955(7) 1.963(2) 2.004(4) 2.014(2)Cu1–N4 – – – 2.111(2)Cu1–S 2.350(3) 2.3700(7) 2.3508(15) 2.5073(7)Cu1–O 2.227(7) 2.176(2) 2.245(4) –
Bite angles5-Membered rings
N–Cu1–N 80.1(3) 80.80(9) 83.58(18) 95.85(8)
o c o
b wR2 = [∑
[w(F2o − F2
c )2]/
∑[w(F2
o )2]]
1/2.
nder focused microwave irradiation as follows: 1-phenylethanol5 mmol), catalysts 1–5 (2.5–10 �mol) and additive (10–200 �mol)ere introduced into the reactor. Thereafter, an aqueous solution
f t-BuOOH (70%) (5–15 mmol) was added and finally the cylindri-al Pyrex reactor was placed in the microwave reactor. The systemas left under stirring and irradiation (10 W) for 5–240 min at
0–100 ◦C temperature range.
.3.2.3. Extraction and GC analysis for 1-phenylethanol oxidations.he reaction mixtures were allowed to cool to room tempera-ure and, upon product extraction with 5 mL of CH3CN, 300 �L ofnternal standard (benzaldehyde) was added and centrifuged. Then,iethyl ether was added to dilute the sample and, after mixing, aample for GC analysis was taken from the organic phase.
Blank tests indicate that only traces of acetophenone are gener-ted in the absence of the copper or iron catalyst in the conventionalethod, while under MW-irradiation the formation of acetophe-
one does not exceed 6% in the metal-free system.
.4. X-ray structure determination
X-ray diffraction quality single crystals of 1, 1′, 2 and 4 werebtained as indicated above. They were mounted in an inert oilithin the cold N2 stream of the diffractometer. Intensity dataere collected using a Bruker AXS-KAPPA APEX II diffractometerith graphite monochromated Mo K� radiation (� = 0.71073 A).ata were collected at 293 and 150 K using omega scans of 0.5◦ per
rame and a full sphere of data was obtained. Cell parameters wereetrieved using Bruker SMART software and refined using BrukerAINT [56] on all the observed reflections. Absorption correctionsere applied using SADABS [56]. The structures were solved byirect methods by using the SHELXS-97 package [57]. Calculationsere performed using the WinGX System-Version 1.80.03 [58].ll hydrogens were inserted in calculated positions. Least-squaresefinements with anisotropic thermal motion parameters for all theon-hydrogen atoms and isotropic for the remaining atoms weremployed. In the structures of 1 and 1′ there are disordered groups
n voids. The number of electrons in the voids were determined,n each case, by means of PLATON/Squeeze Program [59] and fitxactly for one disordered trifluoromethane sulfonate anion in eachase. Tables 1 and 2 contain the crystallographic parameters for thedescribed crystals. CCDC 823159–823162 contains the supplemen-tary crystallographic data for this paper. These data can be obtainedfree of charge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data request/cif.
3. Results and discussion
3.1. Synthesis and characterization of the copper(II) and iron(II)complexes
The first part of this work focused on the preparation of novelcopper(II) and iron(II) complexes based on mixed-N,S tetraden-tate and pentadentate imino and amino thioether ligands, that canbe regarded as biomimetic models of certain oxidation enzymes(e.g. multicopper oxidases and cytochromes P-450), in view oftheir nitrogen and sulfur rich coordination spheres [6–8]. The sec-ond part concerned the investigation of the functional propertiesof these complexes in the two model oxidation reactions of 1-phenylethanol and cyclohexane.
The work started with the synthesis of the organic potentialligands L, LH, LMe and Lpy. The imine product L was produced
– – – 81.58(8)N–Cu1–S 85.1(2) 84.32(7) 87.68(13) 87.22(6)
6-Membered ringsN–Cu1–S 90.8(2) 91.33(8) 87.87(13) 89.17(6)

104 R.R. Fernandes et al. / Journal of Molecular Catalysis A: Chemical 351 (2011) 100– 111
pyridy
fguthtoN
Scheme 1. Syntheses of bis- and tris-
urther functionalized in two different ways. Firstly, the aminoroup (NH) in LH was methylated by an Eschweiler–Clarke reactionsing an excess of HCHO/HCOOH to yield LMe (Scheme 1, reac-ion c). Secondly, LH was treated with 2-(chloromethyl)pyridine
ydrochloride in the presence of K2CO3 to furnish ultimately theris-pyridyl amino-thioether ligand Lpy (Scheme 1, reaction d). Therganic species L, LH, LMe and Lpy were characterized by 1H and 13CMR spectroscopies and ESI+-MS (see Section 2).Scheme 2. Syntheses of Cu(II) (1, 2 and
l amino and imino thioether ligands.
A simple treatment of L, LMe or Lpy with copper triflate in EtOHor THF gives the corresponding Cu(II) complex 1, 2 or 4, respec-tively (Scheme 2, reactions a–c), whereas the reaction of LMe or Lpy
with iron triflate in anhydrous THF under a dinitrogen atmosphere
affords the Fe(II) complex 3 or 5, correspondingly (Scheme 2, reac-tions d and e). All the complexes have been fully characterized byIR, ESI+-MS and elemental analyses, and, for 1, 2 and 4, also by X-raydiffraction structural analyses (Figs. 1–3 and Section 2).4) and Fe(II) (3 and 5) complexes.

R.R. Fernandes et al. / Journal of Molecular Catalysis A: Chemical 351 (2011) 100– 111 105
F tion ol rity.
3
t1adT6cto
Nbrtta
Fsa
Fsa
ig. 1. Ortep diagrams of (a) [Cu(OTf)(L)]+ (cation of 1) and (b) [Cu(L)(H2O)]2+ (dicaevel and the hydrogen atoms and counter-ion (in 1′) are omitted for the sake of cla
.2. X-ray crystal structures
The coordination environments of copper in 1, 1′ and 2 includehe tetradentate N,N,N,S-ligand and the O atom of an OTf anion (in
and 2) or a water molecule (in 1′). In these cases the geometryround the pentacoordinated Cu atom is distorted square pyrami-al with the O-containing moieties in the apical sites (Figs. 1 and 2).he binding of the bis-pyridyl amino thioether ligand involves one-membered chelate ring, Cu–N–C–C–C–S, and two 5-memberedhelate rings, Cu–N–C–C–N and Cu–N–C–C–S, the bite angles ofhese (see Table 2) being lower than that of the former, as a resultf ring constriction.
In the structure of 4 the ligand acts as a pentadentate4S-chelator and its geometry is better described as trigonalipyramidal with the sulfur and two N-pyridyl atoms in the equato-
ial plane and the remaining N-pyridyl in the axial site together withhe N-amine atom (Fig. 3). In this case the metal atom is involved inhree 5-membered rings, one Cu–N–C–C–S and two Cu–N–C–C–N,nd in one 6-membered chelate ring, Cu–N–C–C–C–S. Addison et al.ig. 2. Ortep diagram of [Cu(OTf)(LMe)]+ (cation of 2) showing the atom labellingcheme. The ellipsoids are drawn at the 50% probability level and the hydrogentoms and counter-ion omitted for the sake of clarity.
ig. 3. Ortep diagram of [Cu(Lpy)]2+ (dication of 4) showing the atom labellingcheme. The ellipsoids are drawn at the 50% probability level and the hydrogentoms and counter-ions omitted for the sake of clarity.
f 1′) with atom labelling schemes. The ellipsoids are drawn at the 50% probability
[60] introduced a parameter (�5) to describe the geometry of afive-coordinate metal system which is determined by the equa-tion �5 = ( − ˛)/60 where and are the largest angles involvingthe metal. By means of this simple criterium, perfect square pyra-mid or trigonal bipyramid geometries will have �5 values of 0 or1, respectively. For our complexes such parameter assume valuesof 0.05 (1), 0.12 (1′), 0.059 (2) or 0.71 (4), thus showing that theformer structures are clearly square pyramidal while the latter istrigonal bipyamidal.
In the structure of 1, the neighbouring copper(II) units are posi-tioned relatively far from each other, resulting in Cu· · ·Cu shortestseparations of 8.694 A, and their association via weak H-bonds(Fig. S1) lead to 1D chains along the crystallographic c axis. In 1′, thecomplex cations are even closer (Cu· · ·Cu separation of 8.119 A) asa result of an effective medium intense �· · ·� interaction involvingthe pyridyl rings (Fig. S2). In 2 and 4, the complex cations are fur-ther apart (Cu· · ·Cu separations of 8.719 and 8.885 A, respectively)what may result from steric hindrance due to the O-containing lig-and (in 2) or to the bulkiness of the thioether ligand (in 4). Resultingfrom the conformation adopted by the tioether ligand around cop-per, one of their methylene groups tilts towards the metal atom,the distances from Cu to the inner hydrogen H10A (in 1), H10D (in1′), H11A (in 2) or H7A (in 4) being ca. 3.03 A, 2.68 A, 2.66 or 2.68 Arespectively, with C–H· · ·Cu angles of ca. 100◦ in all cases. Sincethose distances are small compared to the van der Waals contacts,they may evidence the formation of intramolecular weak agosticbonds. According to the criteria of Braga et al. [61] who analyzedX· · ·M and H· · ·M distances and X–H· · ·M angles simultaneously,our values appear to indicate a pseudo-agostic type interaction, asituation commonly observed with electron deficient metal atoms.In our cases such interactions are intramolecular. Infrared spec-troscopy of the complexes in KBr did not provide evidence foragostic interactions.
3.3. MW-assisted solvent-free oxidation of 1-phenylethanol
The investigation of the catalytic properties of the new cop-per(II) 1, 2 and 4, and iron(II) 3 and 5 complexes was undertakenfollowing our recently developed procedure [51,52] for the oxi-dation of secondary alcohols, namely by reacting, at 80 ◦C in asolvent-free medium and under microwave (MW) irradiation, 1-phenylethanol with t-BuOOH (aq. 70%) in the presence of catalyst1, 2, 3, 4 or 5 (Scheme 3).
The copper(II) complexes 1, 2 and 4 catalyse the peroxidativeoxidation of 1-phenylethanol under a low MW irradiation power(10 W), leading, respectively, to 31, 33 and 46% yields of acetophe-
none after 30 min at 80 ◦C of reaction (Table 3, entries 2, 4 and 6).However, the reaction catalysed by 1 for an extended period of240 min results in an acetophenone yield boost from 31% to 45%(Table 3, entry 2 vs. entry 3).
106 R.R. Fernandes et al. / Journal of Molecular Catalysis A: Chemical 351 (2011) 100– 111
with t
aa
(Cct[tatuaHaw
aa1
3
mhtFch2
(tehv8i
TM
ai
p
presence of 200 �mol of pyridine (py) (n(py)/n(catalyst 1) = 20).Broadening the type of additives to other N-based compoundswith basic character, such as pyrazine, piperazine, pyridazine,
Table 4Effects of various additives in the MW-assisted solvent-free peroxidative oxidationof 1-phenylethanol catalysed by 1.a
Entry Additive Yieldb (%) TONc TOF (h−1)d
1 HNO3 4.5 22.5 45.0
2 5.0 25.0 50.0
3 8.6 43.0 86.0
4 75.6 378 756
5 52.4 262 524
6 57.4 287 574
Scheme 3. Microwave-assisted oxidation of 1-phenylethanol
In the case of the reactions catalysed by the iron complexes 3nd 5, significantly different yields of 17 and 64%, respectively, werettained after 30 min of reaction (Table 3, entries 5 and 7).
For both Fe(II) and Cu(II) catalysts, those with Lpy are more activein the absence of additives) than those with LMe (Fe 5 > 3 andu 4 > 2). The presence, in the former, of an extra pyridyl group,an eventually promote the catalytic activity, e.g. by assisting H+-ransfer steps (see below) [10,29,54,62,63]. In previous studies6,8,13], it was postulated that peroxidative oxidations catalysed byhe so-called FeII(N4Py) systems (N4Py is a all-N pentadentate lig-nd analogous to Lpy), with just one labile site as in 5, could occur viahe formation of Fe(III)–OOR peroxo intermediates that are likely tondergo rapid O–O homolytic cleavages generating the active HO•
nd RO• radical oxidizing species. Moreover, addition of t-BuOOH or2O2 to more concentrated solutions of 5 (in acetonitrile) leads to
very strong catalase-type activity (fast peroxide decomposition),hich was never detected in the presence of complex 3.
Control experiments (blank tests) were carried out in thebsence of any metal catalyst and only 6.5% of acetophenone waschieved under the same experimental conditions (Table 3, entry).
.4. Effect of different additives in the reactions catalysed by 1
In view of the moderate activity of 1 (31–45%) in solvent-freeicrowave-assisted peroxidative oxidation of 1-phenylethanol, we
ave investigated the influence of the presence of various addi-ives (co-catalysts) on the acetophenone product yield (Table 4 andig. 4). Therefore, we have chosen the Cu complex 1 as a benchmarkatalyst and initiated the tests by using some mineral (HNO3) andeteroaromatic N-based acids, as 2-pyridinecarboxylic (Hpic) and-pyrazinecarboxylic acids (Hpca), as additives.
The presence of 200 �mol of HNO3 as an additiven(HNO3)/n(catalyst 1) = 20) decreases dramatically the ace-ophenone yield from 31% (Table 3, entry 2) to 4.5% (Table 4,ntry 1 and Fig. 4). Alternatively, in the presence of the N-basedeteroaromatic acids Hpic and Hpca (n(additive)/n(catalyst 1) = 20)
ery poor yields of acetophenone were also achieved (5.0% and.6%, respectively) (Table 4, entries 2 and 3 and Fig. 4). Thisnhibitory behaviour contrasts with the usual beneficial effect
able 3W-assisted solvent-free oxidation of 1-phenylethanol catalysed by 1–5.a
Entry Catalyst Yieldb (%) TONc
1 – 6.5 32.52 [Cu(OTf)(L)](OTf) (1) 30.9 1553d [Cu(OTf)(L)](OTf) (1) 45.1 2264 [Cu(OTf)(LMe)](OTf) (2) 32.9 1655 [Fe(OTf)2(LMe)] (3) 16.7 83.56 [Cu(Lpy)](OTf)2(4) 46.0 2307 [Fe(OTf)(Lpy)](OTf) (5) 64.2 321
a Reaction conditions (unless stated otherwise): 1-phenylethanol (5 mmol), cat-lyst (10 �mol, 0.2 mol%), t-BuOOH (10 mmol, aq. 70%), reaction time (30 min), MWrradiation power (10 W), temperature (80 ◦C).
b Molar yield (%) based on 1-phenylethanol, i.e. number of moles of acetophenoneer 100 mol of 1-phenylethanol, determined by GC.c Turnover number (moles of acetophenone per mol of metal catalyst).d Reaction carried out for 240 min.
-BuOOH catalysed by Cu(II) (1, 2, 4) or Fe(II) (3, 5) complexes.
observed for both Cu [22–24] and Fe [15,19–21] oxidation systemsin slight acidic medium, and prompted us to search for other typesof co-catalysts.
The recognized affinity of certain heterocyclic amines for coppercentres [40] and their vital role as coenzymes in copper-catalysedbiological oxidations [64,65] hint at the possible positive co-catalytic influence of N-based heteroaromatic bases as additives.Indeed, a significant increase from 31% (Table 3, entry 2) to 76%(Table 4, entry 4) on the acetophenone yield was observed in the
7 80.8 404 808
8 80.8 404 808
9 55.6 278 556
10 Et3N 81.4 407 81411 NaOtBu 65.1 326 65212 K2CO3 42.2 211 42213e – 30.9 155 310
a Reaction conditions (unless stated otherwise): 1-phenylethanol (5 mmol), cat-alyst 1 (10 �mol, 0.2 mol%), additives (200 �mol), t-BuOOH (10 mmol, aq. 70%),reaction time (30 min), MW irradiation power (10 W), temperature (80 ◦C).
b Molar yield (%) based on 1-phenylethanol, i.e. number of moles of acetophenoneper 100 mol of 1-phenylethanol, determined by GC.
c Turnover number (moles of acetophenone per mol of metal catalyst).d Turnover frequency (TON per hour).e For comparative purposes (entry 2, Table 3).
R.R. Fernandes et al. / Journal of Molecular Cat
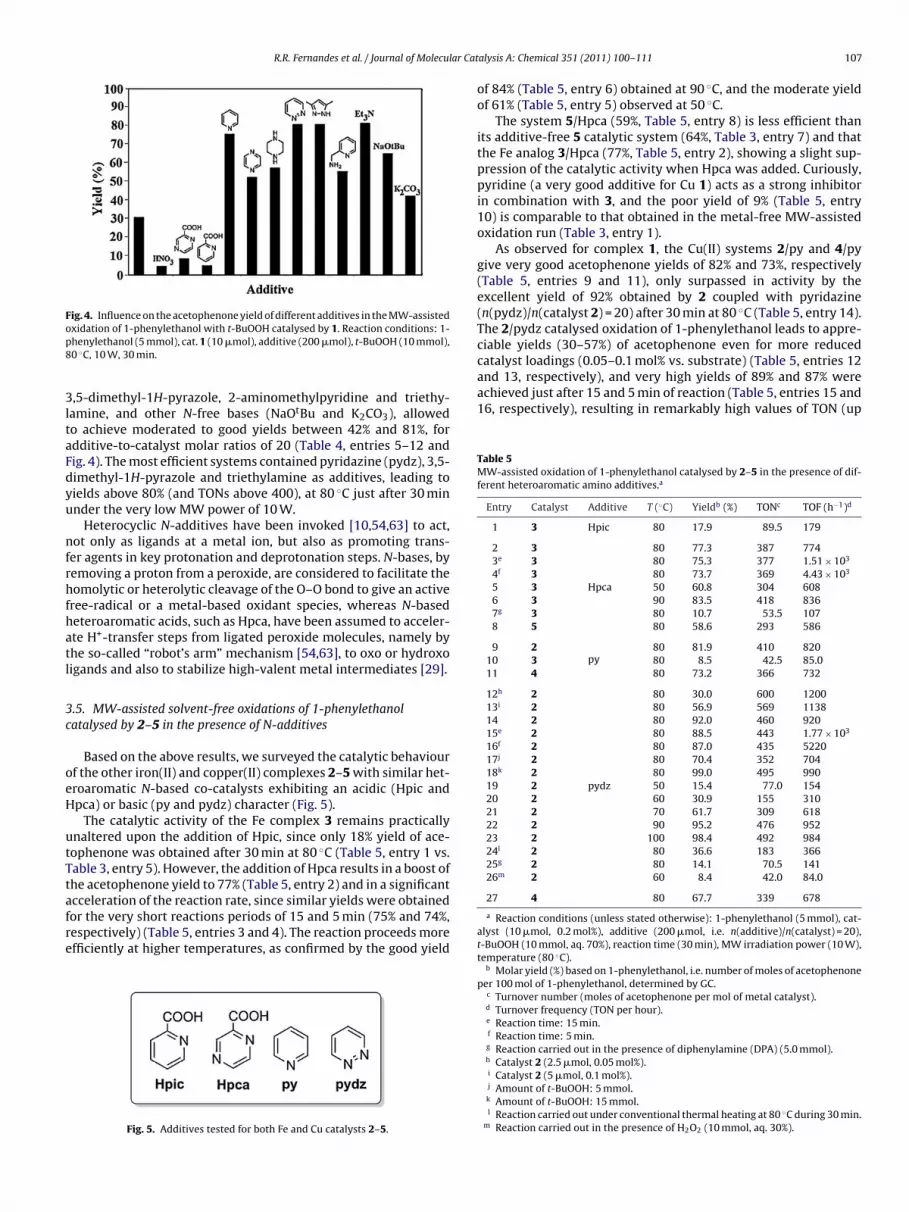
Fig. 4. Influence on the acetophenone yield of different additives in the MW-assistedoxidation of 1-phenylethanol with t-BuOOH catalysed by 1. Reaction conditions: 1-p8
3ltaFdyu
nfrhfhatl
3c
oeH
utTtafre
and 13, respectively), and very high yields of 89% and 87% wereachieved just after 15 and 5 min of reaction (Table 5, entries 15 and16, respectively), resulting in remarkably high values of TON (up
Table 5MW-assisted oxidation of 1-phenylethanol catalysed by 2–5 in the presence of dif-ferent heteroaromatic amino additives.a
Entry Catalyst Additive T (◦C) Yieldb (%) TONc TOF (h−1)d
1 3 Hpic 80 17.9 89.5 179
2 3
Hpca
80 77.3 387 7743e 3 80 75.3 377 1.51 × 103
4f 3 80 73.7 369 4.43 × 103
5 3 50 60.8 304 6086 3 90 83.5 418 8367g 3 80 10.7 53.5 1078 5 80 58.6 293 586
9 2py
80 81.9 410 82010 3 80 8.5 42.5 85.011 4 80 73.2 366 732
12h 2
pydz
80 30.0 600 120013i 2 80 56.9 569 113814 2 80 92.0 460 92015e 2 80 88.5 443 1.77 × 103
16f 2 80 87.0 435 522017j 2 80 70.4 352 70418k 2 80 99.0 495 99019 2 50 15.4 77.0 15420 2 60 30.9 155 31021 2 70 61.7 309 61822 2 90 95.2 476 95223 2 100 98.4 492 98424l 2 80 36.6 183 36625g 2 80 14.1 70.5 14126m 2 60 8.4 42.0 84.0
27 4 80 67.7 339 678
henylethanol (5 mmol), cat. 1 (10 �mol), additive (200 �mol), t-BuOOH (10 mmol),0 ◦C, 10 W, 30 min.
,5-dimethyl-1H-pyrazole, 2-aminomethylpyridine and triethy-amine, and other N-free bases (NaOtBu and K2CO3), allowedo achieve moderated to good yields between 42% and 81%, fordditive-to-catalyst molar ratios of 20 (Table 4, entries 5–12 andig. 4). The most efficient systems contained pyridazine (pydz), 3,5-imethyl-1H-pyrazole and triethylamine as additives, leading toields above 80% (and TONs above 400), at 80 ◦C just after 30 minnder the very low MW power of 10 W.
Heterocyclic N-additives have been invoked [10,54,63] to act,ot only as ligands at a metal ion, but also as promoting trans-
er agents in key protonation and deprotonation steps. N-bases, byemoving a proton from a peroxide, are considered to facilitate theomolytic or heterolytic cleavage of the O–O bond to give an active
ree-radical or a metal-based oxidant species, whereas N-basedeteroaromatic acids, such as Hpca, have been assumed to acceler-te H+-transfer steps from ligated peroxide molecules, namely byhe so-called “robot’s arm” mechanism [54,63], to oxo or hydroxoigands and also to stabilize high-valent metal intermediates [29].
.5. MW-assisted solvent-free oxidations of 1-phenylethanolatalysed by 2–5 in the presence of N-additives
Based on the above results, we surveyed the catalytic behaviourf the other iron(II) and copper(II) complexes 2–5 with similar het-roaromatic N-based co-catalysts exhibiting an acidic (Hpic andpca) or basic (py and pydz) character (Fig. 5).
The catalytic activity of the Fe complex 3 remains practicallynaltered upon the addition of Hpic, since only 18% yield of ace-ophenone was obtained after 30 min at 80 ◦C (Table 5, entry 1 vs.able 3, entry 5). However, the addition of Hpca results in a boost ofhe acetophenone yield to 77% (Table 5, entry 2) and in a significantcceleration of the reaction rate, since similar yields were obtained
or the very short reactions periods of 15 and 5 min (75% and 74%,espectively) (Table 5, entries 3 and 4). The reaction proceeds morefficiently at higher temperatures, as confirmed by the good yieldFig. 5. Additives tested for both Fe and Cu catalysts 2–5.
alysis A: Chemical 351 (2011) 100– 111 107
of 84% (Table 5, entry 6) obtained at 90 ◦C, and the moderate yieldof 61% (Table 5, entry 5) observed at 50 ◦C.
The system 5/Hpca (59%, Table 5, entry 8) is less efficient thanits additive-free 5 catalytic system (64%, Table 3, entry 7) and thatthe Fe analog 3/Hpca (77%, Table 5, entry 2), showing a slight sup-pression of the catalytic activity when Hpca was added. Curiously,pyridine (a very good additive for Cu 1) acts as a strong inhibitorin combination with 3, and the poor yield of 9% (Table 5, entry10) is comparable to that obtained in the metal-free MW-assistedoxidation run (Table 3, entry 1).
As observed for complex 1, the Cu(II) systems 2/py and 4/pygive very good acetophenone yields of 82% and 73%, respectively(Table 5, entries 9 and 11), only surpassed in activity by theexcellent yield of 92% obtained by 2 coupled with pyridazine(n(pydz)/n(catalyst 2) = 20) after 30 min at 80 ◦C (Table 5, entry 14).The 2/pydz catalysed oxidation of 1-phenylethanol leads to appre-ciable yields (30–57%) of acetophenone even for more reducedcatalyst loadings (0.05–0.1 mol% vs. substrate) (Table 5, entries 12
a Reaction conditions (unless stated otherwise): 1-phenylethanol (5 mmol), cat-alyst (10 �mol, 0.2 mol%), additive (200 �mol, i.e. n(additive)/n(catalyst) = 20),t-BuOOH (10 mmol, aq. 70%), reaction time (30 min), MW irradiation power (10 W),temperature (80 ◦C).
b Molar yield (%) based on 1-phenylethanol, i.e. number of moles of acetophenoneper 100 mol of 1-phenylethanol, determined by GC.
c Turnover number (moles of acetophenone per mol of metal catalyst).d Turnover frequency (TON per hour).e Reaction time: 15 min.f Reaction time: 5 min.g Reaction carried out in the presence of diphenylamine (DPA) (5.0 mmol).h Catalyst 2 (2.5 �mol, 0.05 mol%).i Catalyst 2 (5 �mol, 0.1 mol%).j Amount of t-BuOOH: 5 mmol.k Amount of t-BuOOH: 15 mmol.l Reaction carried out under conventional thermal heating at 80 ◦C during 30 min.
m Reaction carried out in the presence of H2O2 (10 mmol, aq. 30%).

108 R.R. Fernandes et al. / Journal of Molecular Catalysis A: Chemical 351 (2011) 100– 111
Fig. 6. Effect of the temperature variation on the acetophenone yield, in the MW-a2(
twIto
att
oate
ctlma
ot2e
pHea
o(s4s
ftl2Dyt1a1
Fig. 7. Dependence of the acetophenone yield on the amount of additive present in
ssisted solvent-free mild peroxidative oxidation of 1-phenylethanol catalysed by/pydz. Reaction conditions: 1-phenylethanol (5 mmol), cat. 2 (10 �mol), pydz200 �mol), t-BuOOH (10 mmol), 10 W, 30 min.
o 435) and TOF (up to 5220 h−1). The TOF value obtained in thisork is within the best values reported so far in the field [51,52].
t should also be noted that very high selectivities (>98% to ace-ophenone) are typically observed in the MW-assisted oxidationsf 1-phenylethanol, as confirmed by mass balances.
By simply varying the relative amount of t-BuOOH used we wereble to raise the acetophenone yield from 70% (1 equiv. of t-BuOOH)o 92% (2 equiv.) and maximize it at 99% (3 equiv., corresponding tohe high TON of 495) (Table 5, entries 17, 14 and 18, respectively).
The reaction temperature has an important effect on the MWxidation of 1-phenylethanol by the 2/pydz/t-BuOOH system, asttested by a gradual increase of the acetophenone yield from 15%o 98% on rising the temperature in the 50–100 ◦C range (Table 5,ntries 19–23 and Fig. 6).
Our system efficiently operates (99% yield) with a relatively lowatalyst loading (0.2 mol% vs. substrate), under the reaction condi-ions of entry 18 of Table 5. Such a catalyst loading is significantlyower than those commonly applied (up to 5 mol% vs. substrate) in
ost of the state-of-the-art systems for the oxidation of secondarylcohols [40].
For comparison, the conventional oxidation (heating in anil bath) of 1-phenylethanol has been also performed. Hence,he thermal oxidation (80 ◦C, 30 min) of 1-phenylethanol by the/pydz/t-BuOOH system affords only 37% of acetophenone (Table 5,ntry 24)
It is worthwhile to mention that the oxidation of 1-henylethanol is not efficient upon replacement of t-BuOOH by2O2 (aq. 30%), resulting in only 8% yield of acetophenone (Table 5,ntry 26). Further augmentation of the temperature was notttempted for safety reasons.
The divergent activity, under the same experimental conditions,f the copper(II) systems 2/pydz (92%, Table 5, entry 14) and 4/pydz68%, Table 5, entry 27) is likely to be associated with the higheraturation and steric effects around the Cu(II) centre in complex, which contrasts with the possible availability of two trans labileites in 2.
Although the detailed mechanistic study of the described trans-ormations was out of the scope of the present work, we haveested the influence of a radical scavenger [51,52], i.e. dipheny-amine (DPA), on the MW oxidation of 1-phenylethanol by the/pydz/t-BuOOH or 3/Hpca/t-BuOOH system. Thus, the addition ofPA results in a very significant suppression of the acetophenoneield that drops from 92% down to 14%, when performed under
he reaction conditions of entry 25 (Table 5), or from 77% down to1% (Table 5, entry 7), respectively. These observations substanti-te the involvement of a free radical mechanism in the oxidation of-phenylethanol [51,52].the (a) Cu 2 – py (�) or 2 – pydz (�) and (b) Fe 3 – Hpic (�) or 3 – Hpca (�) cat-alytic systems. Reaction conditions: 1-phenylethanol (5 mmol), catalyst (10 �mol),t-BuOOH (10 mmol), 80 ◦C, 10 W, 30 min.
3.6. Effect of the additive amount in the reactions catalysed by 2and 3
The results discussed above highlight the disparate behaviour ofthe related Cu 2 and Fe 3 complexes, bearing the same tetradentateligand LMe, in the presence of the heteroaromatic N-based Hpca(acidic character) or pydz (basic character). The optimization of theacetophenone yields as a function of the co-catalyst amount (Hpic,Hpca, py and pydz) (Fig. 5) was performed for the systems 2/base(py and pydz) and 3/acid (Hpic and Hpca) and is depicted in Fig. 7aand b (see Supplementary data, Table S1). All experiments werecarried out under low MW irradiation power (10 W) at 80 ◦C for30 min by using 2 equiv. (10 mmol) of t-BuOOH.
A similar reaction profile was observed for the 2/py and 2/pydzcopper oxidation systems (Fig. 7a) and the optimal conditions wereattained for 10–20 additive-to-catalyst 2 molar ratios leading tomaximum acetophenone yields of 85% and 92%, respectively. Thereaction efficiency does not improve with a further growth of thepy and pydz concentrations, and in both cases a slight yield dropoccurs (to 76% and 79% yield, respectively) at the much higher molarratio of 100 which corresponds to 1000 �mol of additive.
Among the two iron(II) complexes (3 and 5) evaluated as cata-lysts, we verified that Hpca is a very good additive in combinationwith complex 3, and consequently we have decided to study for thissystem the influence of the relative amounts of the heteroaromaticacids Hpic and Hpca on the acetophenone yield.
As we can see in Fig. 7b, the addition of 10 �mol of Hpca, which
corresponds to an additive-to-catalyst molar ratio of 1, results ina yield boost from 17% (in the absence of Hpca) to 47%. The grad-ual increase of the acetophenone yield from 47% to 86% resultedfrom the augmentation of the Hpca amount in the 10–1000 �mol
R.R. Fernandes et al. / Journal of Molecular Catalysis A: Chemical 351 (2011) 100– 111 109
I) (1, 2
rawct(
sapat2(nea
31
llf
mswuc
d
TE
a
o
Scheme 4. Oxidation of cyclohexane with H2O2 catalysed by Cu(I
ange (molar ratios between 1 and 100, respectively). Surprisingly, quite different behaviour was found when the amount of Hpicas increased from 10 to 1000 �mol, resulting in a very signifi-
ant suppression of acetophenone yield that drops from 61% downo 14% (Fig. 7b). The latter yield is even lower than that observed17%) in the absence of Hpic.
Recycling in the Cu 2/pydz [n(pydz)/n(catalyst 2) = 20] catalyticystem was also attempted. Thus, after the first oxidation run,ffording acetophenone in 87% yield (Table 5, entry 16), the organicroducts were extracted with diethyl ether, the remaining cat-lyst suspension was dried in vacuo and 1-phenylethanol and-BuOOH were again added, in the absence or presence of additional00 �mol of pyridazine. These reactions, under MW irradiation10 W, 80 ◦C, 5 min), afford in both cases much lower acetophe-one yields (only 9 or 26%, respectively). Such results indicate anxtensive decomposition of our catalytic system to inactive or lessctive species, under MW irradiation, during the first oxidation run.
.7. Effect of various additives in the cyclohexane and-phenylethanol oxidations in acetonitrile
The application of polydentate pyridine-type Fe and Cu cata-ysts covers a broad range of oxidative transformations and theast decades witnessed a particular research interest on the mildunctionalization of alkanes [6–8,11–18].
Hence, we extended our investigations to the oxidation of theore inert C(sp3)–H bond, choosing cyclohexane as a model sub-
trate (easy product identification/increased reactivity comparingith light alkanes) under mild conditions (25 ◦C) in acetonitrile
sing as oxidant H2O2 (aq. 30%), to yield cyclohexanol (CyOH) andyclohexanone (Cy’O) (Scheme 4).All the complexes 1–5 were screened as catalysts in the oxi-ation of cyclohexane in the presence of diverse acid additives
able 6ffect of different additives in the oxidation of cyclohexane and 1-phenylethanol catalyse
Entry Substrate Additive Yieldb,c (%)
1
1 – 9.0
2 Hpca 0.2
3 HNO3 3.6
4 HOTf 2.9
5 TFA 3.1
6 pydz 16.7 (1.2 d)
7 Hpca –
8 pydz
a Reaction conditions (unless stated otherwise): CH3CN (3 mL), substrate (5 mmol), cataq. 30%), reaction time (6 h), temperature (25 ◦C).b Molar yield (%) based on substrate, i.e. number of moles of product per 100 mol of s
xidation).c Cyclohexanol and cyclohexanone (in the case of cyclohexane oxidation) or acetophend Reaction carried out in the presence of diphenylamine (DPA) (5.0 mmol).e Cyclohexane amount: 1 mmol.f t-BuOOH (7 mmol, aq. 70%) was used as oxidant.
, 4) or Fe(II) (3, 5) complexes in the presence of several additives.
[21–24,54,55], such as 2-pyrazinecarboxylic acid (Hpca), nitric acid(HNO3), triflic acid (HOTf) and trifluoroacetic acid (TFA), and alsoin the presence of pyridazine (pydz) (basic character). In Table 6(see also Supplementary data, Table S2) are compiled the resultsobtained, and the overall yield dependence on the types of catalystand additive is represented.
The catalysts 1–5 performance, in the presence of the acid addi-tives, exhibits a parallel behaviour to that found in the MW-assistedoxidation of 1-phenylethanol. Therefore, the Cu(II) oxidation sys-tems 1, 2 and 4 are inhibited by the addition of any acid and theiractivity drops, in the case of 1 and 2, from overall yields of 9% and12% (corresponding to TON of 45 and 62, respectively) (Table 6,entry 1) to less than ca. 4%. The tris-pyridyl complex 4 is much lessactive affording only 3% of oxygenated products in the absence ofany acid and <1% upon the addition of an acid additive.
The Fe-catalysed (3 and 5) oxidation reactions of cyclohexaneproceed more efficiently in a slight acidic medium and the overallyields achieved are typically in the range of 20–25% (Table 6, entries2–5), which correspond to TON values of ca. 100–125. Complex 5 ismuch more efficient than 3 in the absence of any acid additive (10%vs. 1%, Table 6, entry 1), and the activities are comparable whenHNO3, HOTf or TFA, were added. The maximum yield of 25% andTON up to 127 (Table 6, entry 5) were attained with the system3/TFA after 6 h at 25 ◦C.
The additions of pydz to the Cu(II) (1, 2 and 4) and Fe(II) (3 and5) systems lead to dramatic different effects on the reactivities. TheCu-catalysed oxidations resulted in overall yields of ca. 16%, whilefor the Fe systems no improvement was observed in the oxygenatedproduct yields (Table 6, entry 6).
The peroxidative oxidation of cyclohexane catalysed by Fe andCu systems leads frequently to the formation, via the involvementof radical species, of cyclohexyl hydroperoxide (CyOOH) as pri-mary product [21,54,55]. The marked difference on the CyOH and
d by 1–5.a
2 3 4 5
12.3 1.0 3.1 9.50.2 20.5 (2.5d) 0.4 14.13.3 23.4 0.4 21.83.7 24.2 0.4 22.92.9 25.3 0.4 22.0
16.9 (29.0e) 0.9 15.3 8.5
– 18.7 (25.4f) –8.3 (10.4f)
lyst (10 �mol), additive (200 �mol, i.e. n(additive)/n(catalyst) = 20), H2O2 (7 mmol,
ubstrate (determined by GC after treatment with PPh3, in the case of cyclohexane
one (in the case of 1-phenylethanol oxidation).

1 lar Cat
Car3trrt6
tsbdsa
3
attahw2
ab[SctotrridaD12sitcvup
4
aipsaptv
a
[
10 R.R. Fernandes et al. / Journal of Molecu
y’O concentrations in the chromatograms obtained before andfter treatment of the final reaction mixtures with PPh3, in theeactions catalysed by complexes 2/pydz and 3/Hpca (but also for/HNO3, 3/HOTf and 3/TFA) reveals [54,55] an extensive forma-ion of CyOOH, thus indicating [21,29,54,55,62] the involvement ofadical species in the oxidation reactions. This fact was further cor-oborated by the dramatic yield drop observed when DPA, a radicalrapping agent, was added to these reactions (Table 6, entries 2 and).
For comparison with our MW-assisted method, we performedhe oxidation of 1-phenylethanol using the most active Cu and Feystems 2/pydz and 3/Hpca in acetonitrile at 25 ◦C for 6 h usingoth H2O2 and t-BuOOH as oxidants. The results evidence a strongependence of the catalytic activity on the reaction temperature,ince rather modest yields (<25%) were obtained after a consider-ble long reaction time of 6 h (Table 6, entries 7 and 8).
.8. ESI+-MS studies of the 2/pydz catalytic system
The mechanism of the transformation of 1-phenylethanol tocetophenone is still not clear, but the lower yields obtained inhe presence of a radical trap (Table 5, entries 7 and 25) are indica-ive of a radical pattern. In order to get a further insight on thective species and on the role of the various N-based additives, weave studied firstly the ESI+-MS spectrum of [Cu(OTf)(LMe)](OTf) 2ith pyridazine (pydz), and secondly the ESI+-MS spectrum of the
/pydz catalytic system after the oxidation reaction.Thus, complex 2 was treated with 20 equiv. of pydz, in
cetonitrile at room temperature for 10 min, to give possi-ly the reduced (under ESI+-MS conditions) [66,67] complexCu(LMe)(pydz)(CH3CN)]+ as suggested by ESI+-MS m/z 472.1 (seeupplementary data, Fig. S3). The latter complex was formed viaoordination of 2 with pydz and one solvent molecule. We inves-igated also by ESI+-MS the MW-assisted peroxidative oxidationf 1-phenylethanol to acetophenone catalysed by the 2/pydz sys-em. After the oxidation reaction, the acetophenone product wasemoved by simple extraction with diethyl ether, and the catalyticesidue was directly analyzed by ESI+-MS. The reduced [66,67]ntermediate [Cu(LMe)(pydz)(CH3CN)]+ is still present after the oxi-ation reaction together with its analog [Cu(LMe)(pydz)]+ (withoutcetonitrile ligand), also detected by ESI+-MS at m/z 430.0 (Fig. S3).ifferent uncharacterized products have been observed in the m/z00–300 range due to the decomposition of complex 2 and/or the/pydz system under focused MW irradiation. Presumably, the firsttep involves the displacement of OTf− by pydz in complex 2 lead-ng to [Cu(LMe)(pydz)](OTf)2, which will then react with t-BuOOHo generate the oxidizing radical active species. The role of pydzan be associated with a rapid and reversible coordination to theacant Cu sites, precluding the deactivation of the catalytic centrepon coordination of the alcohol substrate as it was proposed inrevious Cu-catalysed oxidation studies [40].
. Conclusion
Bis- and tris-pyridyl amino and imino thioethers display a goodnd flexible coordination ability towards Cu(II) and Fe(II), lead-ng to the formation of complexes of these metals with vacant (orotentially so) coordination positions, which, in the presence ofimple and inexpensive N-based additives, show a very good cat-lytic activity for the solvent- and halogen-free microwave-assistederoxidative oxidation of 1-phenylethanol to acetophenone. Reac-
ions are fast, selective, require a small catalyst load (0.2–0.05 mol%s. substrate) and proceed at mild temperatures (50–80 ◦C).For instance, the Fe(II) complex 3 in the presence of Hpca as andditive shows a very good catalytic activity for the solvent-free
[[
[
alysis A: Chemical 351 (2011) 100– 111
MW-assisted oxidation of 1-phenylethanol by t-BuOOH, in a veryshort reaction time (5 min) at 80 ◦C, and the acetophenone prod-uct was obtained in 74% yield with TON of 369 and TOF of4.43 × 103 h−1. Furthermore, the Cu(II) 2 complex in combinationwith pydz gives an excellent yield of acetophenone (up to 99%)at 80 ◦C for 30 min, and 87% after 5 min at the same temperature,which corresponds to TON of 435 and TOF of 5220 h−1.
Apart from the high efficiency in terms of yields and TOF,important features of the present peroxidative oxidation of 1-phenylethanol, with respect to some prior studies on relatedMW-assisted alcohol transformations [68,69], consist also in theuse of solvent-free systems and a very low MW irradiation power(10 W).
The complexes 1–5 are also active in the mild oxidation of cyclo-hexane with H2O2 as oxidant and the reaction efficiency, yieldsup to 29% and maximum TON of ca. 125, is particularly base- oracid-sensitive, depending if a Cu or Fe centre catalyses the reaction.
In general, the addition of an acid-type additive (e.g. Hpca orHNO3) promotes the reactions catalysed by the Fe systems, whereasan inhibiting effect was disclosed for the Cu-catalysed peroxidativeoxidations of both 1-phenylethanol and cyclohexane. In contrast,the use of heteroaromatic additives with a basic character (e.g. pyor pydz) was shown to drastically enhance the efficiency of the Cusystems and suppress the activity of the analogous Fe catalysts.
Favourable features, a priori, of these complexes include theirbiologically inspired nature, easy synthesis, cheapness, environ-mental tolerance and very good solubility in water and in thealcohol substrate, thus allowing to avoid the use of other solventsand phase-transferring agents. The search for new catalytic systemsand the establishment of detailed reaction mechanisms is underconsideration in our laboratory.
Acknowledgements
This work has been partially supported by the Fundac ão paraa Ciência e a Tecnologia (FCT), Portugal, and its PPCDT program(FEDER funded). J.L. expresses gratitude to the FCT and the Insti-tuto Superior Técnico (IST) for his research contracts (CIÊNCIA2007 program). R.R.F. expresses gratitude to FCT for a fellowship(grant SFRH/BD/31150/2006). The authors gratefully acknowledgethe Portuguese NMR Network (IST-UTL Center) for providing accessto the NMR facility. Thanks are also due to Dr. M.C. Vaz (microana-lytic service) and Dr. C. Oliveira (ESI-MS).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.molcata.2011.09.022.
References
[1] A.E. Shilov, G.B. Shul’pin, Activation and Catalytic Reactions of Saturated Hydro-carbons in the Presence of Metal Complexes, Kluwer Academic Publishers,Dordrecht, 2000.
[2] J.-E. Bäckvall, Modern Oxidation Methods, Wiley-VCH, Weinheim, 2004.[3] Ullmann’s Encyclopedia of Industrial Chemistry, 6th ed., Wiley-VCH, Wein-
heim, 2002.[4] R.A. Smiley, H.L. Jackson, Chemistry and the Chemical Industry, CRC Press, 2002.[5] R.A. Sheldon, I. Arends, U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH,
2007.[6] M. Costas, K. Chen, L. Que Jr., Coord. Chem. Rev. 200–202 (2000) 517.[7] M. Costas, M.P. Mehn, M.P. Jensen, L. Que Jr., Chem. Rev. 104 (2004) 939.[8] L. Que, W.B. Tolman, Nature 455 (2008) 333.[9] G.B. Shul’pin, Org. Biomol. Chem. 8 (2010) 4217.10] G.B. Shul’pin, Mini-Rev. Org. Chem. 6 (2009) 95.
11] M.C. White, A.G. Doyle, E.N. Jacobsen, J. Am. Chem. Soc. 123 (2001) 7194.12] R. Mas-Balleste, M. Costas, T. van den Berg, L. Que Jr., Chem. Eur. J. 12 (2006)7489.13] G. Roelfes, M. Lubben, R. Hage, L. Que Jr., B.L. Feringa, Chem. Eur. J. 6 (2000)
2152.

ar Cat
[[
[
[
[
[
[[
[
[
[
[[
[
[[
[
[
[
[[[
[
[
[
[
[
[[[[
[
[
[[[[[
[
[
[[[[[[[
[
[[
[
[
[
R.R. Fernandes et al. / Journal of Molecul
14] R. Mas-Balleste, L. Que Jr., J. Am. Chem. Soc. 129 (2007) 15964.15] L. Gomez, I. Garcia-Bosch, A. Company, J. Benet-Buchholz, A. Polo, X. Sala, X.
Ribas, M. Costas, Angew. Chem. Int. Ed. 48 (2009) 5720.16] J. England, C.R. Davies, M. Banaru, A.J.P. White, G.J.P. Britovsek, Adv. Synth. Catal.
350 (2008) 883.17] J. England, R. Gondhia, L. Bigorra-Lopez, A.R. Petersen, A.J.P. White, G.J.P.
Britovsek, Dalton Trans. (2009) 5319.18] C.J. Whiteoak, R.T.M. de Rosales, A.J.P. White, G.J.P. Britovsek, Inorg. Chem. 49
(2010) 11106.19] S. Tanase, C. Foltz, R. de Gelder, R. Hage, E. Bouwman, J. Reedijk, J. Mol. Catal. A
225 (2005) 161.20] J.K. Tang, P. Gamez, J. Reedijk, Dalton Trans. (2007) 4644.21] R.R. Fernandes, M.V. Kirillova, J.A.L. da Silva, J.J.R. Fraústo da Silva, A.J.L.
Pombeiro, Appl. Catal. A: Gen. 353 (2009) 107.22] A.M. Kirillov, M.N. Kopylovich, M.V. Kirillova, M. Haukka, M.F.C. Guedes da Silva,
A.J.L. Pombeiro, Angew. Chem. Int. Ed. 44 (2005) 4345.23] A.M. Kirillov, M.N. Kopylovich, M.V. Kirillova, E.Y. Karabach, M. Haukka, M.F.C.
Guedes da Silva, A.J.L. Pombeiro, Adv. Synth. Catal. 348 (2006) 159.24] M.V. Kirillova, Y.N. Kozlov, L.S. Shul’pina, O.Y. Lyakin, A.M. Kirillov, E.P. Talsi,
A.J.L. Pombeiro, G.B. Shul’pin, J. Catal. 268 (2009) 26.25] T. Punniyamurthy, L. Rout, Coord. Chem. Rev. 252 (2008) 134.26] C. Di Nicola, F. Garau, Y.Y. Karabach, L. Martins, M. Monari, L. Pandolfo, C.
Pettinari, A.J.L. Pombeiro, Eur. J. Inorg. Chem. (2009) 666.27] T.F.S. Silva, G.S. Mishra, M.F.G. da Silva, R. Wanke, L. Martins, A.J.L. Pombeiro,
Dalton Trans. (2009) 9207.28] P.M. Reis, J.A.L. Silva, J. da Silva, A.J.L. Pombeiro, Chem. Commun. (2000) 1845.29] M.V. Kirillova, M.L. Kuznetsov, V.B. Romakh, L.S. Shul’pina, J.J.R. Fraústo da Silva,
A.J.L. Pombeiro, G.B. Shul’pin, J. Catal. 267 (2009) 140.30] T.F.S. Silva, E. Alegria, L. Martins, A.J.L. Pombeiro, Adv. Synth. Catal. 350 (2008)
706.31] G.S. Mishra, T.F.S. Silva, L. Martins, A.J.L. Pombeiro, Pure Appl. Chem. 81 (2009)
1217.32] T.F.S. Silva, K.V. Luzyanin, M.V. Kirillova, M.F.C. Guedes da Silva, L.M.D.R.S. Mar-
tins, A.J.L. Pombeiro, Adv. Synth. Catal. 352 (2010) 171.33] W.G. Dauben, M. Lorber, D.S. Fullerton, J. Org. Chem. 24 (1969) 3587.34] G. Rothenberg, H. Wiener, Y. Sasson, J. Mol. Catal. A: Chem. 136 (1998) 253.35] J. Singh, M. Sharma, M. Chhibber, J. Kaur, G.L. Kad, Synth. Commun. 30 (2000)
3941.36] M. Hudlicky, Oxidations in Organic Chemistry, ACS Monograph 186, Washing-
ton, DC, 1990.37] I.E. Markó, P.R. Giles, M. Tsukazaki, S.M. Brown, C.J. Urch, Science 274 (1996)
2044.38] G. Rothenberg, L. Feldberg, H. Wiener, Y. Sasson, J. Chem. Soc. Perkin Trans. 2
(1998) 2429.39] R. Chakrabarty, P. Sarmah, B. Saha, S. Chakravorty, B.K. Das, Inorg. Chem. 48
(2009) 6371.
[
[
[
alysis A: Chemical 351 (2011) 100– 111 111
40] I.E. Markó, A. Gautier, R. Dumeunier, K. Doda, F. Philippart, S.M. Brown, C.J. Urch,Angew. Chem. Int. Ed. 43 (2004) 1588.
41] S.S. Stahl, Angew. Chem. Int. Ed. 43 (2004) 3400.42] G.-J. ten Brink, I.W.C.E. Arends, R.A. Sheldon, Science 287 (2000) 1636.43] Y. Uozumi, R. Nakao, Angew. Chem. Int. Ed. 42 (2003) 194.44] D.S. Bailie, G.M.A. Clendenning, L. McNamee, M.J. Muldoon, Chem. Commun.
46 (2010) 7238.45] D. Sloboda-Rozner, P.L. Alsters, R. Neumann, J. Am. Chem. Soc. 125 (2003)
5280.46] V.R. Choudary, D.K. Dumbre, B.S. Uphade, V.S. Narkhede, J. Mol. Catal. A: Chem.
215 (2004) 129.47] J. Boudreau, M. Doucette, A.N. Ajjou, Tetrahedron Lett. 47 (2006) 1695.48] W. Zhao, Y. Zhang, B. Ma, Y. Ding, W. Qiu, Catal. Commun. 11 (2010) 527.49] P. Anastas, N. Eghbali, Chem. Soc. Rev. 39 (2010) 301.50] S. Liu, J. Xiao, J. Mol. Catal. A: Chem. 270 (2007) 1.51] P.J. Figiel, M.N. Kopylovich, J. Lasri, M.F.C. Guedes da Silva, J.J.R. Fraústo da Silva,
A.J.L. Pombeiro, Chem. Commun. 46 (2010) 2766.52] P.J. Figiel, A.M. Kirillov, M.F.C. Guedes da Silva, J. Lasri, A.J.L. Pombeiro, Dalton
Trans. 39 (2010) 9879.53] V.E. Kaasjager, L. Puglisi, E. Bouwman, W.L. Driessen, J. Reedijk, Inorg. Chim.
Acta 310 (2000) 183.54] G.B. Shul’pin, J. Mol. Catal. A 189 (2002) 39.55] G.B. Shul’pin, C. R. Chim. 6 (2003) 163.56] Bruker, APEX2 & SAINT, Bruker, AXS Inc., Madison, WI, USA, 2004.57] G.M. Sheldrick, Acta Crystallogr. Sect. A 64 (2008) 112.58] L.J. Farrugia, J. Appl. Crystallogr. 32 (1999) 837.59] A.L. Speck, Acta Crystallogr. Sect. A 46 (1990) C34.60] A.W. Addison, T.N. Rao, J. Reedijk, J. van Rijn, G.C. Verschoor, J. Chem. Soc. Dalton
Trans. (1984) 1349.61] D. Braga, F. Grepioni, E. Tedesco, K. Biradha, G.R. Desiraju, Organometallics 16
(1997) 1846.62] M.L. Kuznetsov, A.J.L. Pombeiro, Inorg. Chem. 48 (2009) 307.63] G.B. Shul’pin, Y.N. Kozlov, G.V. Nizova, G. Suss-Frank, S. Stanislas, A. Kitaygorod-
skiy, V.S. Kulikova, J. Chem. Soc. Perkin Trans. 2 (2001) 1351.64] J.J.R. Fraústo da Silva, R.J.P. Williams, The Biological Chemistry of the Elements
– The Inorganic Chemistry of Life, 2nd ed., Oxford University Press, Oxford,2001.
65] H.-B. Kraatz, N. Metzler-Nolte, Concepts and Models in Bioinorganic Chemistry,Wiley-VCH, Weinheim, 2006.
66] W. Henderson, J.S. McIndoe, Mass Spectrometry of Inorganic, Coordination andOrganometallic Compounds, Wiley, Chichester, 2005.
67] L. Gianelli, V. Amendola, L. Fabbrizzi, P. Pallavicini, G.G. Mellerio, Rapid Com-mun. Mass Spectrom. 15 (2001) 2347.
68] A.M. Balu, J.M. Hidalgo, J.M. Campelo, D. Luna, R. Luque, J.M. Marinas, A.A.Romero, J. Mol. Catal. A: Chem. 293 (2008) 17.
69] V. Polshettiwar, R.S. Varma, Acc. Chem. Res. 41 (2008) 629.




















