
Beatriz Eugenia Ferro Ramos - Radboud Repository
193
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/162523 Please be advised that this information was generated on 2022-06-23 and may be subject to change.
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Beatriz Eugenia Ferro Ramos - Radboud Repository

PDF hosted at the Radboud Repository of the Radboud University
Nijmegen
The following full text is a publisher's version.
For additional information about this publication click this link.
http://hdl.handle.net/2066/162523
Please be advised that this information was generated on 2022-06-23 and may be subject to
change.

strategies to improve treatment of nontuberculous
mycobacterial disease
Beatriz Eugenia Ferro Ramos


Beatriz Eugenia Ferro Ramos
Strategies to improve treatment of nontuberculous mycobacterial
disease

ColofonThis research was funded by a doctoral fellowship (No. 529-2012) from the Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnología Francisco José de Caldas, COLCIENCIAS, Colombian government and the Department of Microbiology, Radboudumc, Nijmegen, the Netherlands.
Strategies to improve treatment of nontuberculous mycobacterial disease.
The research presented in this thesis was conducted in the department of Medical Microbiology, Radboudumc, associated to the Radboud Institute for Health Sciences (RIHS) and in the Center for Infectious Diseases Research and Experimental Therapeutics, Baylor Research Institute, Baylor University Medical Center, Dallas, Texas.
Thesis Radboudumc, Nijmegen.The NetherlandsISBN: 978-94-92380-15-9© by Beatriz Eugenia Ferro Ramos 2016
Layout: Matthijs PollCover design: Beatriz E. Ferro and Sandra BecerraPrinted by: Ipskamp Printing, Enschede

door
Beatriz Eugenia Ferro Ramosgeboren op 3 augustus 1973
te Palmira (Colombia)
Proefschrift
ter verkrijging van de graad van doctoraan de Radboud Universiteit Nijmegen
op gezag van de rector magnificus prof. dr. J.H.J.M. van Krieken,volgens besluit van het college van decanen
in het openbaar te verdedigen op maandag 16 januari 2017om 14.30 uur precies
Strategies to improve treatment of nontuberculous mycobacterial
disease

Promotoren:
Copromotor:
Manuscriptcommissie:
Prof. dr. J.W. MoutonProf. dr. D. van Soolingen
Dr. J. van Ingen
Prof. dr. A.J.A.M. van der VenProf. dr. A. Verbon (Erasmus MC, Rotterdam)Dr. R.E. Aarnoutse
Promotiecommissie
Paranimfen: Melanie WattenbergAnne van Stuyvenberg-Neerincx

by
Beatriz Eugenia Ferro RamosBorn on August 3, 1973in Palmira (Colombia)
Doctoral Thesis
to obtain the degree of doctorfrom Radboud University Nijmegen
on the authority of the Rector Magnificus prof. dr. J.H.J.M. van Krieken,according to the decision of the Council of Deans
to be defended in public on Monday, January 16, 2017at 14:30 hours
Strategies to improve treatment of nontuberculous mycobacterial
disease

Supervisors:
Co-supervisor:
Doctoral Thesis Committee:
Prof. dr. J.W. MoutonProf. dr. D. van Soolingen
Dr. J. van Ingen
Prof. dr. A.J.A.M. van der VenProf. dr. A. Verbon (Erasmus MC, Rotterdam)Dr. R.E. Aarnoutse
Promotion Committee
Paranymphs: Melanie WattenbergAnne van Stuyvenberg-Neerincx

In memory ofAlicia Rodríguez de Ramos †

Table of Contents
Chapter 1Introduction
Chapter 2Time-kill kinetics of antibiotics active against rapidly growing mycobacteriaJ Antimicrob Chemother. 2015;70:811-7.
Chapter 3Time-kill kinetics of slowly growing mycobacteria common in pulmonary diseaseJ Antimicrob Chemother. 2015;70:2838-43.
Chapter 4Clofazimine prevents the regrowth of Mycobacterium abscessus and Mycobacterium avium type strains exposed to amikacin and clarithromycinAntimicrob Agents Chemother. 2015;60:1097-105. Chapter 5Amikacin pharmacokinetics/pharmacodynamics in a novel hollow-fiber Mycobacterium abscessus disease model Antimicrob Agents Chemother. 2015;60:1242-8.
Chapter 6Moxifloxacin´s limited efficacy in the hollow-fiber model of Mycobacterium abscessus diseaseAntimicrob Agents Chemother. 2016;60:3779-85.
Chapter 7Tigecycline is highly efficacious against Mycobacterium abscessus pulmonary diseaseAntimicrob Agents Chemother. 2016;60:2895-900.
Chapter 8Failure of the amikacin, cefoxitin and clarithromycin combination regimen for pulmonary Mycobacterium abscessusAntimicrob Agents Chemother. 2016;60:6374-6.
Chapter 9General discussion
11
33
47
59
77
95
111
125
133

Chapter 10Summary
Chapter 11Samenvatting
Chapter 12Resumen en Español
Biography and List of Publications
Acknowledgements
PhD Portfolio
153
159
165
171
179
187


11
Chapter 1
Introduction
Partly adapted from:
Drug treatment of nontuberculous mycobacterial disease in HIV-negative patients: the evidence.
Jakko van IngenBeatriz E. FerroWouter HoefslootMartin J. BoereeDick van Soolingen
Expert Rev Anti Infect Ther. 2013;11:1065–77.

12
Chapter 1

Introduction
13
Cha
pter
1
Chapter 1
Introduction
The genus Mycobacterium consists of over 150 species, of which Mycobacterium tuberculosis complex, Mycobacterium ulcerans and Mycobacterium leprae are best known, as they are the causative agents of tuberculosis, Buruli ulcer disease and leprosy in humans. The other Mycobacterium species are collectively labeled nontuberculous mycobacteria (NTM). These NTM are increasingly recognized as causative agents of mostly opportunistic infections of humans.1,2 The epidemiology of these infections has a strong local character because the NTM species distribution differs by geographical region,3 the respective species differ in their clinical relevance,4 the prevalence of predisposing conditions differs per setting and, though this awaits experimental confirmation, strains of a single species isolated in different regions may differ in their virulence.1 Pulmonary disease caused by NTM (NTM-PD) is the most frequent disease manifestation. Although many NTM species can cause NTM-PD, its most frequent causative agents are the slow growing mycobacteria (SGM) of the Mycobacterium avium complex (MAC), Mycobacterium kansasii, Mycobacterium xenopi, Mycobacterium malmoense and the rapid growing mycobacterium (RGM) Mycobacterium abscessus. The exact composition of this top five differs by region.1,3
NTM-PD has two main radiological manifestations. The first is a fibro-cavitary disease (Figure 1A) similar to pulmonary tuberculosis, which typically affects patients with underlying lung diseases such as chronic obstructive pulmonary disease (COPD);4,6 the second is nodular-bronchiectatic disease (Figure 1B) which commonly affects an elderly population of mostly female patients without a significant history of pre-existing pulmonary disease.7 Rarely, NTM-PD presents as a solitary pulmonary lesion mimicking malignancy. NTM incidentally cause hypersensitivity pneumonitis.8
NTM can also cause a wide variety of other -extrapulmonary- diseases, which fall into three main categories. The first is lymphadenitis, typically of cervical or submandibular lymph nodes in otherwise healthy and presumably immunocompetent children. This relatively benign disease affects children under the age of 6 years, is caused mainly by M. avium, M. malmoense, M. haemophilum and M. kansasii and is usually treated by surgical resection of the affected lymph node.9,10 The second extrapulmonary NTM disease entity are the inoculation infections; these are infections of skin, soft tissues or bones in a single site, after penetrating trauma and secondary infection by NTM. The best known example is the skin disease caused by M. marinum. This so-called fish tank granuloma is acquired when damaged skin is exposed to the fish pathogen M. marinum, for example during aquarium cleaning. The result is a nodular or ulcerative skin lesion on the hand, which may spread along lymphatic vessels, similar to the Sporothrix fungus, hence called sporothrichoid spread, to more proximal on the arm.11 Other examples are catheter-related infections like peritonitis in patients using peritoneal dialysis and catheter-related bloodstream infections.12 The third category of extrapulmonary NTM diseases

14
Chapter 1
are the disseminated infections in the severely immunocompromised.13 Two distinct entities are recognized in clinical practice. The first is a disseminated skin disease, with ulcers and nodules of the skin, most prominent on the distal extremities.13 These infections are mostly seen in patients who use immunosuppressive drugs, for example after solid organ transplant. The most frequent causative agents are rapid growers like M. chelonae, M. abscessus and M. fortuitum and the slow growing M. haemophilum. The second manifestation of disseminated disease became notorious in HIV/AIDS patients and is clinically characterized by fever, night sweats, weight loss and generalized lymphadenopathy. This disease is mostly caused by M. avium, M. simiae and M. genavense.12,13
Treatment of NTM-PDThe fibro-cavitary and nodular-bronchiectatic NTM-PD types often require drug treatment, with adjunctive surgical resection of affected lobes in selected patients.1 Despite their current status as ‘emerging pathogens’, treatment of NTM-PD has a very limited evidence base. Very few trials have been performed and for many species, only small case series of patients treated with widely varying regimens of a very different duration are available. As a result, treatment of NTM-PD leans heavily on expert opinion expressed in the current treatment recommendations issued by the American Thoracic Society (ATS) and Infectious Diseases Society of America (IDSA).1
Within this chapter, we first focus on treatment and outcome as reported in clinical trials and case series available in the PubMed database (National Center for Biotechnology Information [NCBI]), to reconstruct a more evidence-based overview of possible drug treatment regimens of MAC-PD and M. abscessus-PD and their outcomes, as these species are clinically most important. Second, we describe the main non-clinical models that have been used, to provide information on the treatment of NTM-PD. At the end of the chapter, we present this thesis´ aims and objectives.
Figure 1. Main radiological manifestations of pulmonary nontuberculous mycobacterial disease. A: Fibro-cavitary disease by M. avium. B: Nodular-bronchiectatic disease by M. intracellulare
A B

Introduction
15
Cha
pter
1
Chapter 1
Clinical studies in humans
M. avium complexTreatment of pulmonary disease caused by M. avium complex (MAC-PD) has received most attention and has been subject of a small number of clinical trials. In historical perspective, three phases in the treatment of these infections can be observed. First, in the 1960s and 1970s, drug treatment often consisted of combinations of para-aminosalicylic acid (PAS), isoniazid (H) and streptomycin, identical to tuberculosis treatment. Despite the multi-drug treatment, failure and relapse were common and prolonged culture conversion could only be achieved in 30% of patients affected.14,15 Since the late 1970’s, rifampicin (R), ethambutol (E), ethionamide and streptomycin were increasingly used in the treatment of NTM pulmonary disease (PD), especially MAC-PD; these regimens could yield long-term culture conversion in up to 50% of patients.16 The third phase in NTM disease treatment started when the HIV epidemic spurred a new search for active drugs and the macrolides arose as an important new drug class to combat MAC-PD. In the 1990s, several case series revealed that macrolide monotherapy quickly led to resistance and failure.17,18 Addition of other drugs, including rifampicin (R), ethambutol (E), clofazimine and aminoglycosides could prevent macrolide resistance and these combination regimens proved highly efficacious, especially on the short term (Table 1).15-32 After the influential case series by Wallace 19 and Tanaka,20 the rifamycin-ethambutol-macrolide regimens became standard treatment regimens. Across the heterogeneous trials and case series, this regimen cured 453 of the 770 (59%) patients enrolled (Table 1).
Parallel to the inclusion of the new macrolides in RE-based MAC-PD treatment regimens, two groups have studied inclusion of macrolides in clofazimine-based regimens. Both a clofazimine-ethambutol-clarithromycin and a clofazimine-minocycline-clarithromycin regimen have been studied in retrospective non-comparative cohort studies. With culture conversion rates reaching 67% and 64%, these regimens do not seem inferior to rifamycin-ethambutol macrolide regimens (Table 1).21,25 Both these studies, however, offer limited information on their patient cohorts, thus the role of clofazimine-based regimens is now generally considered to be second-line, restricted to patients who cannot be treated with rifamycins.1
One of the subjects of ongoing debate is the role of the aminoglycoside amikacin or streptomycin as adjuncts to rifampicin ethambutol- macrolide regimens. Many of the case series describing outcomes of the macrolide-based regimens included patients who used adjunctive amikacin or streptomycin thrice weekly for the first 2–6 (mean: 3) months of treatment (Table 1).19,20,26,28,32 Yet, only a single randomized trial has assessed the efficacy of adding streptomycin. This trial, with 73 patients in each arm, revealed that sputum culture conversion rates were higher in the group that received adjunctive streptomycin and culture conversion was faster; nonetheless, relapse rates and symptom and radiological scores did not differ between the two arms.27 The only impact thus seems to be the improvement of short-term microbiological outcomes.

16
Chapter 1
Based upon the above two aspects, most experts currently recommend using a regimen that includes rifampicin, ethambutol and a macrolide, with adjunctive streptomycin or amikacin during the first 2–3 months for severe disease (Table 2).1,33
Another subject of debate is the use of intermittent -i.e. thrice weekly- therapy, which can be used successfully in mild nodular-bronchiectatic disease. 34,35
M. abscessusAmong the RGM, most clinical attention has been given to M. abscessus pulmonary disease in recent years as it is most frequent and extremely difficult to treat. This disease appears to be emerging in several settings, especially in cystic fibrosis (CF) patients.1,43 Because M. abscessus is highly resistant to most classes of antibiotics, treatment regimens are usually built on the basis of findings from in vitro drug susceptibility testing. Yet, the evidence for the relationship between in vitro susceptibility and in vivo outcome of treatment is scant.1,44
Table 1. Outcome of treatment regimens for MAC lung disease in HIV-negative patients
* Cure is defined as culture conversion without relapse during study. H, isoniazid; P, para-aminosalicylic acid (PAS); S, streptomycin; K, kanamycin; R, rifampicin/rifabutin; E, ethambutol; Cla, clarithromycin; Azm, azithromycin; Clo, clofazimine; Cip, ciprofloxacin; Mox, moxifloxacin; FQ, various fluoroquinolones. ND, no data provided.
Regimen Cure rate * Cavitary disease (%)
Mean follow-up(months)
Ref.
HPS 32% (92/285) ND ND 15
24RE 27% (10/37) 21/37 (57%) 60 23
24HRE 34% (13/38) 25/38 (66%) 60 23
Cla +/- H/R/E/Clo/FQ 71% (32/45) 26/45 (58%) 0 17
>12RECla (+/-S) 82% (32/39) 35/50 (70%) 19 19
>12RECla (+2-6K; +FQ) 84% (26/31) 18/46 (39%) 32 20
>12RECla (low-dose+3S) 58% (41/71) 39/71 (55%) ND 26
>12RECla +3S>12RECla
52% (38/73)33% (24/73)
81/146 (55%) 16 27
>12RECla (+3S) 75% (45/60) 18/60 (30%) ND 28
>12RECla 50% (7/14) 0 33 30
>12RECla/Azm 86% (154/180) 4/180 (2%) 44 22
24RECla 24% (20/83) 57/83 (69%) 60 24
24RECla 100% (14/14) 14/14 (100%) 6 29
RECla 3/wk 44% (40/91) 49/91 (54) 0 31
CloECla 67% (20/30) ND 18 21
15CloMinCla 64% (14/22) 13/29 (45%) 24 25
24RECip 23% (20/87) 57/87 (66%) 60 24
REClaMox (+S) 29% (12/41) 19/41 (46%) ND 32

Introduction
17
Cha
pter
1
Chapter 1
In recent years, several retrospective series have been published. A recent series described 69 patients treated in the USA (seven patients with CF) and revealed a culture conversion rate of just 48% for 3–5 drug regimens based on in vitro drug susceptibility test results. Patients who underwent adjunctive surgical resection of affected lung tissue more frequently achieved prolonged culture conversion (57 vs. 28%).45 Two smaller case series have assessed the outcomes of treatment with regimens based on in vitro drug susceptibility testing. In the first, 22 patients received treatment for M. abscessus pulmonary disease; after 12 months of treatment, 6 (27%) had already experienced treatment failure.46 In the second, 41 patients were retrospectively studied; 17 received a susceptibility testing. At the end of treatment 80.5% (33/41) of the patients had converted to negative cultures and treatment was considered successful. This percentage did not differ between patients treated with one or two parenteral drugs. During an average of 15 months of follow-up after treatment, four patients relapsed, thus 29 of 41 (70%) had prolonged (i.e., >6 months) conversion to negative cultures.47 Interestingly, similar results were obtained in a case series in South Korea, in which 65 non-CF patients with M. abscessus lung disease received a fixed regimen of clarithromycin, ciprofloxacin and doxycycline, combined with intravenous (iv.) amikacin and cefoxitin for the first 4 weeks. In this case series, prolonged (>12 months) culture conversion was achieved in 38 patients (58%).48 Thus, the use of drugs to which in vitro resistance is the rule (doxycycline, ciprofloxacin) can still lead to favorable clinical outcomes. All the above mentioned case series have applied a biphasic approach to treatment. The first, intensive phase is composed of multiple iv. drugs combined with oral drugs (mostly macrolides), generally selected on basis of in vitro susceptibility. This phase typically lasts 1–4 months (i.e., at least until culture conversion or clear failure) and is followed by a continuation phase of daily oral drugs or, rarely, intermittent iv. drugs or combinations thereof. 1,45–48 While most series have used drug susceptibility test results to help design continuation phase regimens,45,46 some have used drugs to which resistance in vitro is common place (e.g., ciprofloxacin and doxycycline).48 All series have used macrolides as the cornerstone for both phases of treatment;45–48 in the continuation phase, the role of the companion drugs is either to be active by themselves or prevent the emergence of macrolide resistance.1,45
In later case series, molecular tools were applied to distinguish M. abscessus subspecies abscessus from M. abscessus subspecies bolletii (particularly ‘M. massiliense’), because the former shows inducible resistance to macrolides. Across these series, it was obvious that treatment outcomes of macrolide-based regimens were better in ‘M. massiliense’ disease than in M. abscessus subspecies abscessus disease, suggesting a clear in vitro-in vivo correlation.48–50 The fixed regimen of clarithromycin, ciprofloxacin and doxycycline, combined with iv. amikacin and cefoxitin for the first 4 weeks given to 56 patients led to prolonged culture conversion in 25% (M. abscessus subspecies abscessus) and 88% (‘M. massiliense’) of patients.49 In another series from Japan, which analyzed treatment outcomes of 62 patients (42: M. abscessus subspecies abscessus; 20: ‘M. massiliense’), culture conversion rates were lowest for M. abscessus subspecies abscessus, 31% vs. 50% for ‘M. massiliense’. Moreover, relapse rates

18
Chapter 1
after initial culture conversion were higher in patients with M. abscessus subspecies abscessus disease.50 The inducible macrolide resistance is a likely cause of differences in treatment outcome between patients diseased by the two subspecies. Since these publications, separate intensive phase regimens are advised for these two subspecies of M. abscessus (Table 2).33
The taxonomy of the M. abscessus group is still subject of debate. Although ‘M. massiliense’ (partial erm gene deletion, macrolide susceptible) and ‘M. bolletii’ (erm gene intact, inducible macrolide resistance) have been described as separate species, they have most recently been lumped into a single subspecies, M. abscessus subspecies bolletii.51 This status is unfortunate, because it includes both macrolide susceptible strains and strains with inducible macrolide resistance that is likely clinically significant.49,50 Hence, measuring inducible macrolide resistance needs to become a standard laboratory test, based on either erm gene sequence analysis or phenotypic testing with prolonged incubation.49,51,52
PerspectiveThe currently available literature offers a very limited evidence base on which to select or advise treatment regimens that lead to a likely clinical cure for patients with NTM-PD. Furthermore, most currently recommended treatment regimens have grown historically by accumulation of knowledge on drug activity. These regimens were thus not rationally designed. An interesting example is the concomitant use of rifampicin and macrolides for MAC-PD despite well-known pharmacokinetic interactions leading to suboptimal macrolide concentrations.53 Much can probably be gained by designing new regimens on basis of synergistic activities, smart
Table 2. Currently recommended treatment regimens for NTM-PD, per species
AS: Amikacin or streptomycin; Clo: Clofazimine; CN: Negative cultures; E: Ethambutol; H: Isoniazid; MAC: Mycobacterium avium complex; NTM-PD: Nontuberculous mycobacterial pulmonary disease; R: Rifampicin.
Species Recommended regimen Alternative
MAC 1,34,35 RE-macrolide (+/- AS) Duration: >12 months CN
CloE-macrolide
M. kansasii 1-36-39 (rifampicin susceptible)
HRE (+/- AS) Duration: 12 months
RE-macrolide Duration: >12 months CN
M. malmoense 1,24,40 RE-macrolide Duration: >12 months CN
RE-quinolone Duration: >12 months CN
M. xenopi 1,24,41 RE-macrolide (+/- quinolone) Duration: >12 months CN
RE-quinolone Duration: >12 months CN
M. abscessus subsp. abscessusor M. abscessus subsp. bolletii(former ‘Mycobacterium bolletii’)42
Inducible macrolide resistance
3 or 4 of: amikacin, cefoxitin, imipenem, tigecycline, linezolid Duration: >12 months CN
M. abscessus subsp. bolletii(former ‘Mycobacterium massiliense’)42
No inducible macrolide resistance
Macrolide, plus 2 of: amikacin, cefoxitin, imipenem, tigecycline, linezolid Duration: >12 months CN
Amikacin, cefoxitin, macrolide, ciprofloxacin, doxycycline Duration: >12 months CN

Introduction
19
Cha
pter
1
Chapter 1
combinations of rapidly killing and slower sterilizing drugs and optimal dosing to achieve active concentrations. Another area for investigation may be the host-directed therapies. For many patients with NTM-PD, the predisposing condition is not evident. Subtle immunodeficiencies have recently been recorded in many patients with nodular-bronchiectatic NTM-PD.54,55 These, as well as the well-known predisposing conditions including COPD and CF may be improved by host-directed therapies and thereby influence the course of NTM-PD.
Preclinical models
The limitations in clinical information on NTM-PD treatment extends to the fundamental level; little is known on the dose-response relationships, pharmacokinetic and pharmacodynamic characteristics of common antibiotics used to treat NTM-PD. Pharmacokinetics describes the time course of antibiotic after the administration of a dosage regimen; providing information on the absorption, distribution, metabolism and excretion of the antibiotic in the body. Only two large studies and one small case series have investigated the pharmacokinetics of the key drugs used in NTM-PD treatment, 53,56,57 however, there is still lack of knowledge on some key antibiotics. Pharmacodynamics, on the other hand, describes the magnitude of the antibiotic effect in relation to its concentration.58 Crucial pharmacodynamic information has been gathered for Mycobacterium tuberculosis;5,60 however, that is not the case for NTM. There is still a need to know the activity, the potential to selectively amplify resistant subpopulations, and the pharmacokinetic/pharmacodynamic (PK/PD) relationships for antibiotics used in the treatment of NTM-PD.60 The determination of pharmacodynamic properties, and the establishment of PK/PD ratios may be difficult to get in humans, therefore in vitro and in vivo preclinical models have been proposed as surrogates. Diverse preclinical models can be used for this purpose as well as to guide in the selection and optimization of treatment regimens. We will shortly introduce the main models and the most important findings related to NTM-PD.
In vitro modelsDifferent classifications of in vitro pharmacodynamic models have been the matter of reviews,58,61 however, from a simple point of view we will use here the classification of static or dynamic models. In static models, there is a constant exposure of the bacteria to the antibiotic, which means that the antibiotic concentration remains static throughout the whole experiment. Dynamic models, in contrast, are meant to mimic the different exposures that usually are achieved in human fluids when treated with an antibiotic, that means that the antibiotic concentration changes during the experiment.61 In the following paragraphs we will show the main pre-clinical models that have been described for the study of NTM-PD. MIC determination and time-kill assays are the most commonly used static models, while the hollow-fiber model is the main dynamic model described so far for NTM.

20
Chapter 1
Minimum inhibitory concentration determinationMinimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) are perhaps the major pharmacodynamic parameters used to establish the in vitro activity of an antibiotic against an infectious pathogen,58 in our case NTM. According to the Clinical and Laboratory Standards Institute (CLSI) the determination of MIC by the broth microdilution method is the standard drug susceptibility procedure for NTM.62 For such determination commercially available systems (Sensititre™) or in-house assays can be used; for both, 96-well plates (with antibiotic in two-fold dilutions) are inoculated with a standardized inoculum of approximately 5×105 colony forming units (cfu) from a pure culture of NTM. The growth observed after the incubation time with the different antibiotics is compared with the growth in a drug-free control well in order to determine MICs.63 MICs for NTM are usually high for several antibiotics used in treatment regimens, compared to MICs for common Gram positive or Gram negative bacteria. The correlation between in vitro MIC and in vivo outcome of treatment remains to be determined for most antibiotics against most NTM species. What is clear so far is the relationship between clarithromycin in vitro activity and in vivo efficacy, demonstrated in patients with pulmonary MAC disease.20 To a lesser extent, this is also true for amikacin. 64 MICs of rifampicin or ethambutol are less relevant, as these antibiotics are only used as companion drugs to avoid the emergence of macrolide resistance.65
To determine the MBC, samples obtained from MICs are diluted and subcultured on antibiotic-free medium for an additional time. The lowest concentration of an antibacterial agent that results in a ≥99.9% decrease in the initial inoculum is considered the MBC. Bacteriostatic activity has been defined as a ratio of MBC/MIC >4, while bactericidal activity, on the other hand, has been defined as a ratio <4, but these definitions although very used are subject of debate. The in vitro microbiological determination of the bactericidal or bacteriostatic nature of an antibiotic may be influenced by growth conditions, bacterial density, test duration, etc, thus should be tested for each particular organism.66
Time-kill kinetic assaysAlthough MIC and MBC are considered excellent predictors of the potency of an antibiotic and measure the interaction of an antibiotic with a pathogen, they do not provide information on the time course of the antimicrobial activity.62 Time-kill assays allow the study of the pharmacodynamics, examining the rate at which different concentrations of an antibiotic kill bacteria, whether it’s killing is concentration-dependent or time-dependent. The effect of the main antibiotics against M. tuberculosis has been evaluated by time-kill assays,67,68 but only few published works are available for NTM, and even fewer are particularly related to NTM-PD.For M. avium strain 101, Bakker-Woudenberg in 2005,69 described the time-kill kinetics of five antimycobacterial agents: clarithromycin, ethambutol, rifampicin, amikacin and moxifloxacin. All antibiotics inhibited mycobacterial growth, but their bactericidal activity was different; amikacin showed the fastest killing activity and moxifloxacin the highest killing activity in the

Introduction
21
Cha
pter
1
Chapter 1
experiment over 21 days. Clarithromycin showed extremely low killing rate. No studies were found for other SGM species.
For RGM, the single and combined effect of isoniazid and the natural quinone, β-lapachone, were evaluated by time-kill curves for M. fortuitum and M. smegmatis confirming that both microorganisms are resistant to isoniazid, but the two antibiotics in combination had bactericidal effect.70 No information on M. abscessus was available.
Dynamic PK/PD modelsThe periodic administration of an antibiotic (or antibiotic combinations) to a patient, as is the case in patient treatment, implies that the concentrations of those antibiotics in human tissues will be variable or dynamic and therefore it will have a different effect on the mycobacteria. To overcome the need of a dynamic evaluation in large patient cohorts, several in vitro PK/PD models have been developed.58,61 The in vivo conditions of varying antibiotic concentrations over time, as in humans, are mimicked to allow the study of time-kill curves following the microbial kill and growth that take place under therapy. This is clearly superior to traditional approaches such as susceptibility testing or time-kill experiments performed in tubes.58 Dynamic models can adapt a variety of conditions to evaluate different microorganisms, environmental conditions, dosing schemes, single or combination therapies, etc. Although evidence suggests that there is a good correlation between certain dynamic models and in vivo findings,59,60,71 there are still limitations to be overcome; this is a field of constant evolution.
Early descriptions of the dynamic aspects of the relationship between antibiotics and NTM included the use of an in vitro dialysis model (cellophane dialyzing tube) and the membrane filtration technique applied to MAC.72-74 Clofazimine, rifabutin and amikacin were evaluated, showing a potential bactericidal action at concentrations attainable in humans; however, these investigations and the model itself were preliminary.72-74 Then the hollow-fiber system model, a robust in vitro PK/PD dynamic model was described for NTM.75,76 Hollow-fiber systems represent a two-compartment diffusion model, where the central compartment is composed of the central reservoir, the inner lumina of the hollow fiber capillaries, and connecting tubing. The other compartment, the peripheral, is the space outside the hollow-fiber capillaries that is enclosed by an impermeable plastic cartridge.78 Bacteria, in this case mycobacteria, can grow in the peripheral compartment but since the pore size of the membrane is smaller than bacteria, it selectively allows transfer of nutrients, drugs and bacterial metabolites and restrict the bacteria to the peripheral compartment. Special pumps are used to provide antibiotic(s) via a dosing port, and fresh culture medium is pumped into the central compartment while antibiotic-containing media is removed at rates programmed to simulate the drug half-life encountered in humans. By repetitive sampling of the bacterial culture exposed to the antibiotic(s) under study, the number of cfu of mycobacteria surviving the exposure can be measured by quantitative culture.78 While such models have been used more intensively to study M. tuberculosis,59,60,79 there are only

22
Chapter 1
two published studies performed for NTM, specifically modeling disseminated disease by M. avium.75,76 Yet, these studies have yielded interesting insights. The first study focused on the activity of ethambutol against MAC and concluded that in the currently recommended doses, ethambutol has little activity. The efficacy of ethambutol would be far better if doses >50 mg/kg were used, which, to avoid toxicity, should be given intermittently, for example, twice or thrice weekly.75 The second study focused on the potential role of moxifloxacin to treat MAC disease. In short, this study, too, revealed that moxifloxacin in its current dose should be considered inactive against most MAC strains, since these generally have MICs that can’t be overcome by the standard dose of moxifloxacin (400 mg/day). Moxifloxacin is only likely to have some activity if the dose would be 2–3 times higher.76 For both antibiotics, the tolerability, safety and efficacy of such dosing regimens should be subject of future clinical studies. No studies reporting the use of hollow-fiber with RGM were found.
In vivo modelsSoon after NTM were associated with human diseases, their study in laboratory animals started.80,81 Many of the initial descriptions particularly focused on pathogenicity, virulence, the type of disease or the type of immunological response associated with members of MAC, with preferential attention to the disseminated disease.82-84 Several mouse strains and routes of infection, as well as bacterial loads were tested until acute disease was produced in different organs of Beige mice, by intravenous infection of 107 viable cfu.82 Later, chronic disease was obtained with the administration of agents that promote macrophage activation, enhancing resistance to infection by M. intracellulare.84 Then the evaluation of therapy started; a landmark study investigated rifabutin alone and in combination with clofazimine. When used alone, rifabutin showed limited activity against acute or chronic M. intracellulare infection; however, complete sterilization in lungs and spleens was produced with the combination of rifabutin (10 mg/kg) and clofazimine (20 mg/kg), but only when the treatment initiated immediately after the challenge. When treatment initiated when the infection was already established, the combination was considerable less effective;85 these findings early highlighted the importance of combined therapy and that the stage of the infection is crucial for the treatment of NTM. Amikacin was also tested alone showing a remarkable activity in the treatment of early and established MAC infections The addition of clofazimine increased the activity of amikacin, but a third drug, rifabutin, did not. It was concluded then that amikacin-containing regimens are worthy of consideration for the treatment of MAC infections, as is a role for clofazimine.86 The evaluation of liposomal encapsulation of aminoglycosides in the model opened a new window to improve therapy for MAC infection; liposome-mediated delivery of amikacin and streptomycin to MAC-infected tissues in vivo enhanced the efficacy of the drugs, thus it may improve the therapy of this disease.87,88 Additional testing in C57BL/6 mice confirmed previous findings, clarithromycin and combinations with clarithromycin showed activity against M. avium infection, both in the spleen and in lungs, ethambutol was also found to be active.89,90 Some other drugs like sparfloxacin and thioacetazone, which had high in vitro activity, failed to

Introduction
23
Cha
pter
1
Chapter 1
show in vivo activity.83 A recent characterization of different mouse models for MAC infections included the comparison of BALB/c, C57BL/6, nude and beige mice. Nude mice resulted more susceptible to the infection, but the impact of treatment was clearer in BALB/c mice than in the other strains.91 Regarding treatment, the most effective combination was clarithromycin-ethambutol-rifampicin, confirming that clarithromycin-containing regimens are key in MAC treatment.91
For M. abscessus, few studies have been performed.92,93 Murine models for M. abscessus were initially used to characterize the smooth (Mab-S) and rough (Mab-R) morphotypes, finding that a mutation in the Mab-S altered the ability of this mycobacteria to persist and multiply in macrophages.92 For the evaluation of drug activity, some studies have assessed multiple mouse models. Initially, in C57BL/6 mice, antagonism was observed between moxifloxacin and macrolides for most of the M. abscessus clinical strains used, whilst indifference or synergy was observed for M. massiliense.93 In GKO and SCID mice models, the combination of clofazimine-bedaquiline was found effective in decreasing the bacterial burden in organs.94,95 Recently, Nude and GKO mice strains demonstrated to perform better for M. abscessus infections. Nude mice were chosen to evaluate the activity of single antibiotics and the combined standard treatment of amikacin, clarithromycin and cefoxitin. Cefoxitin was the most active single antibiotic, although tigecycline displayed bactericidal activity; clarithromycin and amikacin prevented death but their impact on bacillary loads was limited. The standard treatment was not more active than cefoxitin alone.96
Although animal models can provide similar growing conditions for bacteria imitating characteristics of a human infection, there is no consensus yet on the extent of this extrapolation from animals to humans, neither on which one is the best model to evaluate therapy against each NTM. Due to the growing interest in developing alternative hosts to better represent aspects of bacterial–host interactions, new models for NTM disease have been recently developed. Free living ameba (Dictyostelium discoideum), goldfish (Carassius auratus), zebrafish (Danio rerio) and fruit fly (Drosophila melanogaster) have been used to reproduce and describe the characteristics of NTM infection, some have been used for preliminary in vivo efficacy studies.97-100 However, no single model necessarily mirrors the complexity of chronic pulmonary disease seen in patients. There still a real need for a model or perhaps a combination of in vivo and in vitro models that can be used in the pre-clinical evaluation of individual and combined regimens against NTM-PD.

24
Chapter 1
Aims and outline of this thesis
There is a serious lack of evidence to support NTM-PD treatment. At the clinical level more support is needed and ideally well-designed clinical trials should be conducted to improve our knowledge on the NTM-PD therapy and patient´s clinical outcome. First, in preparation for such clinical trials, questions at the more fundamental level need to be addressed to design a more rational antibiotic treatment regimen.
Within this PhD study, we aimed to assess the pharmacodynamics of commonly used drugs to treat NTM disease by means of the time-kill kinetic assays and the development of a pharmacodynamic model on basis of the in vitro hollow-fiber system. We first studied the pharmacodynamics of antibiotics commonly used against the RGM M. abscessus and M. fortuitum, by means of time-kill kinetic assays (Chapter 2). Subsequently, we applied this same strategy to study the basic pharmacodynamics for key antibiotics used to treat M. avium and M. xenopi pulmonary disease (Chapter 3). As a next step in regimen design, we applied the time-kill kinetic assay to obtain information on the killing activity of the combinations of clofazimine/amikacin, and clofazimine/clarithromycin against the two key NTM species, M. avium and M. abscessus. To assess the extent of possible synergy, we applied two pharmacodynamic drug interaction models: the response surface model of Bliss independence and isobolographic analysis of Loewe additivity (Chapter 4). From Chapter 5 onwards, we shifted focus to M. abscessus. For this extremely drug-resistant species, we have developed a tractable model to further perform PK/PD studies and to identify the different antibiotic exposures and doses associated with optimal microbial killing and suppression of acquired drug resistance. In chapter 5 we introduce the model to study the activity -or lack thereof- of amikacin, one of the standard drugs in currently recommended regimens. In Chapter 6, we show that the commonly used fluoroquinolone, moxifloxacin, shows no activity against M. abscessus at clinically achievable doses. In Chapter 7 we describe the first antimicrobial drug that do has appreciable activity against M. abscessus in our model, tigecycline. In Chapter 8, we move away from monotherapy studies and have assessed the activity of the currently recommended combination regimen of cefoxitin, amikacin and clarithromycin against M. abscessus. This can be used as a baseline to assess activities of new combination regimens. Lastly, principal findings are summarized and discussed according to their current scientific context in the general discussion presented in Chapter 9.
These efforts should provide a basis for more rational regimen design and, in turn, clinical trials. The poor treatment outcomes in NTM-PD are unacceptable and need to be shortly improved by systematic and innovative research.

Introduction
25
Cha
pter
1
Chapter 1
1.
2.
3.
4.
5.
6.
7.
8.9.
10.
11.
12.
13.
14.15.
References
Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier J, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K, ATS Mycobacterial Diseases Subcommittee, American Thoracic Society, Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416. Jagielski T, Minias A, van Ingen J, Rastogi N, Brzostek A, Żaczek A, Dziadek J. Methodological and clinical aspects of the molecular epidemiology of Mycobacterium tuberculosis and other mycobacteria. Clin Microbiol Rev. 2016;29:239-90.Hoefsloot W, van Ingen J, Andréjak C, Angeby K, Bauriaud R, Bemer P, Beylis N, Boeree MJ, Cacho J, Chihota V, Chimara E, Churchyard G, Cias R, Daza R, Daley CL, Dekhuijzen PN, Domingo D, Drobniewski F, Esteban J, Fauville-Dufaux M, Folkvardsen DB, Gibbons N, Gómez-Mampaso E, Gonzalez R, Hoffmann H, Hsueh PR, Indra A, Jagielski T, Jamieson F, Jankovic M, Jong E, Keane J, Koh WJ, Lange B, Leao S, Macedo R, Mannsåker T, Marras TK, Maugein J, Milburn HJ, Mlinkó T, Morcillo N, Morimoto K, Papaventsis D, Palenque E, Paez-Peña M, Piersimoni C, Polanová M, Rastogi N, Richter E, Ruiz-Serrano MJ, Silva A, da Silva MP, Simsek H, van Soolingen D, Szabó N, Thomson R, Tórtola Fernandez T, Tortoli E, Totten SE, Tyrrell G, Vasankari T, Villar M, Walkiewicz R, Winthrop KL, Wagner D; Nontuberculous Mycobacteria Network European Trials Group. The geographic diversity of non-tuberculous mycobacteria isolated from pulmonary samples: an NTM-NET collaborative study. Eur Resp J. 2013;42:1604-13.van Ingen J, Bendien SA, de Lange WC, Hoefsloot W, Dekhuijzen PN, Boeree MJ, Soolingen D. Clinical relevance of nontuberculous mycobacteria isolated in the Nijmegen-Arnhem region, the Netherlands. Thorax. 2009;64:502-6.Hoefsloot W, Boeree MJ, van Ingen J, Bendien S, Magis C, de Lange W, Dekhuijzen PN, van Soolingen D. The rising incidence and clinical relevance of Mycobacterium malmoense: a review of the literature. Int J Tuberc Lung Dis. 2008;12:987-93.Andréjak C, Thomsen VO, Johansen IS, Riis A, Benfield TL, Duhaut P, Sorensen HT, Lescure FX, Thomsen RW. Nontuberculous pulmonary mycobacteriosis in Denmark: incidence and prognostic factors. Am J Respir Crit Care Med. 2010;181:514-21.Reich JM, Johnson RE. Mycobacterium avium complex pulmonary disease presenting as an isolated lingular or middle lobe pattern. The Lady Windermere syndrome. Chest. 1992;101:1605-9.Aksamit TR. Hot tub lung: infection, inflammation, or both? Semin Respir Infect. 2003;18:33-9.van Ingen J, van Soolingen D. Cervicofacial lymphadenitis caused by nontuberculous mycobacteria; host environmental or bacterial factors? Int J Pediatr Otorhinolaryngol. 2011;75:722-3.Bruijnesteijn van Coppenraet LE, de Haas PE, Lindeboom JA, Kuijper EJ, van Soolingen D. Lymphadenitis in children is caused by Mycobacterium avium hominissuis and not related to ´bird tuberculosis´. Eur J Clin Microbiol Infect Dis. 2008;27:293-9.Boyce SH. Fish tank granuloma - an unusual cause of skin infection. J Accid Emerg Med. 1997;14:400.Kasperbauer S, Huitt G. Management of extrapulmonary nontuberculous mycobacterial infections. Semin Respir Crit Care Med. 2013;34:143-50.Henkle E, Winthrop KL. Nontuberculous mycobacteria infections in immunosuppressed hosts. Clin Chest Med. 2015;36:91-9.Goldman KP. Treatment of unclassified mycobacterial infection of the lungs. Thorax. 1968;23:94-9.Field SK, Fisher D, Cowie RL. Mycobacterium avium complex pulmonary disease in patients without HIV infection. Chest. 2004;126:566-81.

26
Chapter 1
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
Dutt AK, Stead WW. Long-term results of medical treatment in Mycobacterium intracellulare infection. Am J Med. 1979;67:449-53.Dautzenberg B, Piperno D, Diot P, Truffot-Pernot C, Chauvin JP. Clarithromycin in the treatment of Mycobacterium avium lung infections in patients without AIDS. Clarithromycin Study Group of France. Chest. 1995;107:1035-40.Griffith DE, Brown-Elliott BA, Langsjoen B, Zhang Y, Pan X, Girard W, Nelson K, Caccitolo J, Alvarez J, Shepherd S, Wilson R, Graviss EA, Wallace RJ Jr. Clinical and molecular analysis of macrolide resistance in Mycobacterium avium complex lung disease. Am J Respir Crit Care Med. 2006;174:928-34.Wallace RJ Jr, Brown BA, Griffith DE, Girard WM, Murphy DT. Clarithromycin regimens for pulmonary Mycobacterium avium complex: the first 50 patients. Am J Respir Crit Care Med. 1996;153:1766-72.Tanaka E, Kimoto T, Tsuyuguchi K, Watanabe I, Matsumoto H, Niimi A, Suzuki K, Murayama T, Amitani R, Kuze F. Effect of clarithromycin regimen for Mycobacterium avium complex pulmonary disease. Am J Respir Crit Care Med. 1999;160:866-72.Field SK, Cowie RL. Treatment of Mycobacterium avium-intracellulare complex lung disease with a macrolide, ethambutol, and clofazimine. Chest. 2003;124, 1482-86.Wallace RJ Jr, Brown-Elliott BA, McNulty S, Philley JV, Killingley J, Wilson RW, York DS, Shepherd S, Griffith DE. Macrolide/Azalide therapy for nodular/bronchiectatic Mycobacterium avium complex lung disease. Chest. 2014;146:276-82.Research Committee of the British Thoracic Society. First randomised trial of treatments for pulmonary disease caused by M. avium intracellulare, M. malmoense, and M. xenopi in HIV negative patients: rifampicin, ethambutol and isoniazid versus rifampicin and ethambutol. Thorax. 2001;56:167-72.Research Committee of the British Thoracic Society. Clarithromycin vs ciprofloxacin as adjuncts to rifampicin and ethambutol in treating opportunist mycobacterial lung diseases and an assessment of Mycobacterium vaccae immunotherapy. Thorax. 2008;63:627-34.Roussel G, Igual J. Clarithromycin with minocycline and clofazimine for Mycobacterium avium intracellulare complex lung disease in patients without the acquired immune deficiency syndrome. GETIM. Groupe d’Etude et de Traitement des Infections a` Mycobactéries. Int J Tuberc Lung Dis. 1998;2:462-70.Kobashi Y, Matsushima T. The effect of combined therapy according to the guidelines for the treatment of Mycobacterium avium complex pulmonary disease. Intern Med. 2003;42:670-75.Kobashi Y, Matsushima T, Oka M. A double-blind randomized study of aminoglycoside infusion with combined therapy for pulmonary Mycobacterium avium complex disease. Respir Med. 2007;101:130-38.Kobashi Y, Abe M, Mouri K, Obase Y, Kato S, Oka M. Relationship between clinical efficacy for pulmonary MAC and drug-sensitivity test for isolated MAC in a recent 6-year period. J Infect Chemother. 2012;18:436-43.Murray MP, Laurenson IF, Hill AT. Outcomes of a standardized triple-drug regimen for the treatment of nontuberculous mycobacterial pulmonary infection. Clin Infect Dis. 2008;47:222-4.Huang JH, Kao PN, Adi V, Ruoss SJ. Mycobacterium avium-intracellulare pulmonary infection in HIV-negative patients without preexisting lung disease: diagnostic and management limitations. Chest. 1999;115:1033-40.Lam PK, Griffith DE, Aksamit TR, Ruoss SJ, Garay SM, Daley CL, Catanzaro A. Factors related to response to intermittent treatment of Mycobacterium avium complex lung disease. Am J Respir Crit Care Med. 2006;173:1283-9.

Introduction
27
Cha
pter
1
Chapter 1
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
Koh WJ, Hong G, Kim SY, Jeong BH, Park HY, Jeon K, Kwon OJ, Lee SH, Kim CK, Shin SJ. Treatment of refractory Mycobacterium avium complex lung disease with a moxifloxacin-containing regimen. Antimicrob Agents Chemother. 2013;57:2281-5.van Ingen J, Griffith DE, Aksamit TR, Wagner D. Pulmonary diseases caused by non-tuberculous mycobacteria. Eur Respir Soc Monograph (Tuberculosis). 2012;58:25-37.Griffith DE, Brown BA, Murphy DT, Girard WM, Couch L, Wallace RJ Jr. Initial (6-month) results of three-times-weekly azithromycin in treatment regimens for Mycobacterium avium complex lung disease in human immunodeficiency virus-negative patients. J Infect Dis. 1998;178:121-6.Griffith DE, Brown BA, Cegielski P, Murphy DT, Wallace RJ Jr. Early results (at 6 months) with intermittent clarithromycin-including regimens for lung disease due to Mycobacterium avium complex. Clin Infect Dis. 2000;30:288-92.Ahn CH, Lowell JR, Ahn SS, Ahn SI, Hurst GA. Short-course chemotherapy for pulmonary disease caused by Mycobacterium kansasii. Am Rev Respir Dis. 1983;128:1048-50.Santin M, Dorca J, Alcaide F, Gonzalez L, Casas S, Lopez M, Guerra MR. Long-term relapses after 12-month treatment for Mycobacterium kansasii lung disease. Eur Respir J. 2009;33:148-52.Shitrit D, Baum GL, Priess R, Lavy A, Shitrit AB, RAz M, Shlomi D, Daniela B, Kramer MR. Pulmonary Mycobacterium kansasii infection in Israel, 1999–2004: clinical features, drug susceptibility, and outcome. Chest. 2006;129:771-6.Griffith DE, Brown-Elliott BA, Wallace RJ Jr. Thrice-weekly clarithromycin-containing regimen for treatment of Mycobacterium kansasii lung disease: results of a preliminary study. Clin Infect Dis. 2003;37:1178-82.Hoefsloot W, van Ingen J, de Lange WCM, Dekhuijzen PNR, Boeree MJ, van Soolingen D. Clinical relevance of Mycobacterium malmoense isolation in the Netherlands. Eur Respir J. 2009;34:926-31.Varadi RG, Marras TK. Pulmonary Mycobacterium xenopi infection in non-HIV-infected patients: a systematic review. Int J Tuberc Lung Dis 2009;13:1210-8.van Ingen J, Griffith DE, Aksamit TR, Wagner D. Pulmonary diseases caused by nontuberculous mycobacteria. Eur Respir Monogr. 2012;58:25–37.Hill UG, Floto RA, Haworth CS. Non-tuberculous mycobacteria in cystic fibrosis. J R Soc Med. 2012;105(Suppl. 2):S14-S18.van Ingen J, Boeree MJ, van Soolingen D, Mouton JW. Resistance mechanisms and drug susceptibility testing of nontuberculous mycobacteria. Drug Resist Updat. 2012;15:149-61. Jarand J, Levin A, Zhang L, Huitt G, Mitchell JD, Daley CL. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin Infect Dis. 2011;52:565-71.Huang YC, Liu MF, Shen GH, Lin CF, Kao CC, Liu PY, Shi ZY. Clinical outcome of Mycobacterium abscessus infection and antimicrobial susceptibility testing. J Microbiol Immunol Infect. 2010;43:401-6.Lyu J, Jang HJ, Song JW, Choi CM, Oh YM, Lee SD, Kim WS, Kin DS, Shim TS. Outcomes in patients with Mycobacterium abscessus pulmonary disease treated with long-term injectable drugs. Respir Med. 2011;105:781-7.Jeon K, Kwon OJ, Lee NY, Kim BJ, Kook YH, Lee SH, Park YK, Kim CK, Koh WJ. Antibiotic treatment of Mycobacterium abscessus lung disease: a retrospective analysis of 65 patients. Am J Respir Crit Care Med. 2009;180:896-902.Koh WJ, Jeon K, Lee NY, Kim BJ, Kook YH, Lee SH, Park YK, Kim CK, Shin SJ, Huitt GA, Daley CL, Kwon OJ. Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am J Respir Crit Care Med. 2011;183:405-10.

28
Chapter 1
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
Harada T, Akiyama Y, Kurashima A, Nagai H, Tsuyuguchi K, Fujii T, Yano S, Shigeto E, Kuraoka T, Kajiki A, Kobashi Y, Kokubu F, Sato A, Yoshida S, Iwamoto T, Saito H. Clinical and microbiological differences between Mycobacterium abscessus and Mycobacterium massiliense lung diseases. J Clin Microbiol. 2012;50:3556-61.Leao SC, Tortoli E, Euzéby JP, Garcia MJ. Proposal that Mycobacterium massiliense and Mycobacterium be united and reclassified as Mycobacterium abscessus subsp. bolletii comb. nov., designation of Mycobacterium abscessus subsp. abscessus subsp. nov. and emended description of Mycobacterium abscessus. Int J Syst Evol Microbiol. 2011;61:2311-3.Nash KA, Brown-Elliott BA, Wallace RJ Jr. A novel gene, erm(41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob Agents Chemother. 2009;53:1367-76.van Ingen J, Egelund EF, Levin A, Totten SE, Boeree MJ, Mouton JW, Aarnoutse RE, Heifets LB, Peloquin CA, Daley CL.The pharmacokinetics and pharmacodynamics of pulmonary Mycobacterium avium complex disease treatment. Am J Respir Crit Care Med. 2012;186:559-65.Becker KL, van Ingen J, Ten Oever J, Merkus PJ, Ferwerda G, Netea MG, Magis-Escurra C, Reijers MH, van de Veerdonk FL.Deficient interleukin-17 production in response to Mycobacterium abscessus in cystic fibrosis. Eur Respir J. 2016;47:990-3.Szymanski EP, Leung JM, Fowler CJ, Haney C, Hsu AP, Chen F, Duggal P, Oler AJ, McCormack R, Podack E, Drummond RA, Lionakis MS, Browne SK, Prevots DR, Knowles M, Cutting G, Liu X, Devine SE, Fraser CM, Tettelin H, Olivier KN, Holland SM. Pulmonary Nontuberculous Mycobacterial Infection. A Multisystem, Multigenic Disease. Am J Respir Crit Care Med. 2015;192:618-28.Koh WJ, Jeong BH, Jeon K, Lee SY, Shin SJ. Therapeutic drug monitoring in the treatment of Mycobacterium avium complex lung disease. Am J Respir Crit Care Med. 2012;186:797-802.Magis-Escurra C, Alffenaar JW, Hoefnagels I, Dekhuijzen PN, Boeree MJ, van Ingen J, Aarnoutse RE. Pharmacokinetic studies in patients with nontuberculous mycobacterial lung infections. Int J Antimicrob Agents.2013;42:256-61.Nightingale CH, Murakawa T, Ambrose, PG (ed). Antimicrobial Pharmacodynamics in Theory and Clinical Practice, 1st ed. 2001. Marcel Dekker, New York, NY. Gumbo T, Pasipanodya JG, Nuermberger E, Romero K, Hanna D. Correlations between the hollow fiber model of tuberculosis and therapeutic events in tuberculosis patients: learn and confirm. Clin Infect Dis. 2015;61 Suppl. 1:S18-24. Gumbo T, Lenaerts AJ, Hanna D, Romero K, Nuermberger E. Nonclinical models for antituberculosis drug development: a landscape analysis. J Infect Dis. 2015;211 Suppl. 3:S83-95.Gloede J, Scheerans C, Derendorf H, Kloft C. In vitro pharmacodynamic models to determine the effect of antibacterial drugs. J Antimicrob Chemother.2012;65:186-201.Clinical Laboratory Standards Institute. Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes - Approved Standard, Second Edition. 2011. Clinical Standards Institute.van Ingen J, Kuijper EJ. Drug susceptibility testing of nontuberculous mycobacteria. Future Microbiol. 2014;9:1095-110.Brown-Elliott BA, Iakhiaeva E, Griffith DE, Woods GL, Stout JE, Wolfe CR, Turenne CY, Wallace RJ Jr. In vitro activity of amikacin against isolates of Mycobacterium avium complex with proposed MIC breakpoints and finding of a 16S rRNA gene mutation in treated isolates. J Clin Microbiol. 2013;51:3389-94.Sison JP, Yao Y, Kemper CA, Hamilton JR, Brummer E, Stevens DA, Deresinski SC. Treatment of Mycobacterium avium complex infection: do the results of in vitro susceptibility tests predict therapeutic outcome in humans? J Infect Dis. 1996;173:677-683.

Introduction
29
Cha
pter
1
Chapter 1
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
Pankey GA, Sabath LD. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin Infect Dis. 2004;38:864-70.de Steenwinkel JE, de Knegt GJ, ten Kate MT, van Belkum A, Verbrugh HA, Kremer K, van Soolingen D, Bakker-Woudenberg IA. Time-kill kinetics of anti-tuberculosis drugs, and emergence of resistance, in relation to metabolic activity of Mycobacterium tuberculosis. J Antimicrob Chemother. 2010;65:2582-9.Shandil RK, Jayaram R, Kaur P, Gaonkar S, Suresh BL, Mahesh BN, Jayashree R, Nandi V, Bharath S, Balasubramanian V. Moxifloxacin, ofloxacin, sparfloxacin and ciprofloxacin against Mycobacterium tuberculosis: evaluation of in vitro and in vivo pharmacodynamic indices that best predict in vivo efficacy. Antimicrob Agents Chemother. 2007; 51:576-82.Bakker-Woudenberg IA, van Vianen W, van Soolingen D, Verbrugh HA, van Agtmael MA. Antimycobacterial agents differ with respect to their bacteriostatic versus bactericidal activities in relation to time of exposure, mycobacterial growth phase and their use in combination. Antimicrob Agents Chemother. 2005;49:2387-98.Silva JL, Mesquita AR, Ximenes EA. In vitro synergic effect of beta-lapachone and isoniazid on the growth of Mycobacterium fortuitum and Mycobacterium smegmatis. Mem Inst Oswaldo Cruz. 2009;104:580-2.Crandon JL, Schuck VJ, Banevicius MA, Beaudoin ME, Nichols WW, Tanudra MA, Nicolau DP. Comparative in vitro and in vivo efficacies of human simulated doses of ceftazidime and ceftazidime-avibactam against Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2012;56:6137-46.Gangadharam PR, Pratt PF, Damle PB, Davidson PT. Dynamic aspects of the activity of clofazimine against Mycobacterium intracellulare. Tubercle. 1981;62:201-6.Perumal VK, Gangadharam PR, Heifets LB, Iseman MD. Dynamic aspects of the in vitro chemotherapeutic activity of ansamycin (rifabutine) on Mycobacterium intracellulare. Am Rev Respir Dis. 1985;132:1278-80.Gangadharam PR, Kesavalu L, Rao PN, Perumal VK, Iseman MD. Activity of amikacin against Mycobacterium avium complex under simulated in vivo conditions. Antimicrob Agents Chemother. 1988;32:886-9.Deshpande D, Srivastava S, Meek C, Leff R, Gumbo T. Ethambutol optimal clinical dose and susceptibility breakpoint identification by use of a novel pharmacokinetic/pharmacodynamic model of disseminated intracellular Mycobacterium avium. Antimicrob Agents Chemother. 2010;54:1728-33.Deshpande D, Srivastava S, Meek C, Leff R, Hall GS, Gumbo T. Moxifloxacin pharmacokinetics/pharmacodynamics and optimal dose and susceptibility breakpoint identification for treatment of disseminated Mycobacterium avium infection. Antimicrob Agents Chemother. 2010;54:2534-9.Zinner SH, Husson M, Klatersky J. An artificial capillary in vitro kinetic model of antibiotic bactericidal activity. J Infect Dis. 1981;144:583-7.Deshpande D, Gumbo T. Pharmacokinetic/pharmacodynamic-based treatment of disseminated Mycobacterium avium. Future Microbiol. 2011;6:433-9.Gumbo T. New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob Agents Chemother. 2010; 54: 1484-91.Wolinsky E. Chemotherapy and pathology of experimental photochromogenic mycobacterial infections. Am Rev Respir Dis. 1959;80:522-34.Meissner G. The value of animal models for study of infection due to atypical mycobacteria. Rev Infect Dis. 1981;3:953-9.Gangadharam PR, Edwards CK, Murthy PS, Pratt PF. An acute infection model

30
Chapter 1
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
for Mycobacterium intracellulare disease using beige mice: preliminary results. Am Rev Respir Dis. 1983;127:648-9.Gangadharam PR. Beige mouse model for Mycobacterium avium complex disease. Antimicrob Agents Chemother. 1995;39:1647-54.Edwards CK, Hedegaard HB, Zlotnik A, Gangadharam PR, Johnston RB, Pabst MJ. Chronic infection due to Mycobacterium intracellulare in mice: association with macrophage release of prostaglandin E2 and reversal by injection of indomethacin, muramyl dipeptide, or interferon-gamma. J Immunol. 1986;136:1820-7.Gangadharam, P. R. J., V. K. Perumal, B. T. Jairam, P. N. Rao, A. K. Nguyen, D. C. Farhi, and M. D. Iseman. Activity of rifabutin alone or in combination with clofazimine or ethambutol or both against acute and chronic experimental Mycobacterium intracellulare infections. Am Rev Respir Dis. 1987;136:329-33.Gangadharam PR, Perumal VK, Podapati NR, Kesavalu L, Iseman MD. In vivo activity of amikacin alone or in combination with clofazimine or rifabutin or both against acute experimental Mycobacterium avium complex infections in beige mice. Antimicrob Agents Chemother. 1988;32:1400-3.Düzgünes N, Perumal VK, Kesavalu L, Goldstein JA, Debs RJ, Gangadharam PR. Enhanced effect of liposome-encapsulated amikacin on Mycobacterium avium-M. intracellulare complex infection in beige mice. Antimicrob Agents Chemother. 1988;32:1404-11.Düzgünes N, Ashtekar DR, Flaser DL, Ghori N, Debs RJ, Friend DS, Gangadharam PR. Treatment of Mycobacterium avium-intracellulare complex infection in beige mice with free and liposome-encapsulated streptomycin: role of liposome type and duration of treatment. J Infect Dis. 1991;164:143-51.Cohen Y, Perronne C, Lazard T, Truffot-Pernot C, Grosset J, Vilde JL, Pocidalo JJ. Use of normal C57BL/6 mice with established Mycobacterium avium infections as an alternative model for evaluation of antibiotic activity. Antimicrob Agents Chemother. 1995;39:735-8.Ji B, Lounis N, Truffot-Pernot C, Grosset J. Effectiveness of various antimicrobial agents against Mycobacterium avium complex in the beige mouse model. Antimicrob Agents Chemother. 1994;38:2521-29.Andréjak C, Almeida DV, Tyagi S, Converse PJ, Ammerman NC, Grosset JH. Characterization of mouse models of Mycobacterium avium complex infection and evaluation of drug combinations. Antimicrob Agents Chemother. 2015;59:2129-35.Byrd TF, Lyons CR. Preliminary characterization of a Mycobacterium abscessus mutant in human and murine models of infection. Infect Immun. 1999; 67:4700-7.Choi GE, Min KN, Won CJ, Jeon K, Shin SJ, Koh WJ. Activities of moxifloxacin in combination with macrolides against clinical isolates of Mycobacterium abscessus and Mycobacterium massiliense. Antimicrob Agents Chemother. 2012;56:3549-55.Ordway D, Henao-Tamayo M, Smith E, Shanley C, Harton M, Troudt J, Bai X, Basaraba RJ, Orme IM, Chan ED. Animal model of Mycobacterium abscessus lung infection. J Leukoc Biol. 2008;83:1502-11.Obregón-Henao A, Arnett KA, Henao-Tamayo M, Massoudi L, Creissen E, Andries K, Lenaerts AJ, Ordway DJ. Susceptibility of Mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob Agents Chemother. 2015;59:6904-12.Lerat I, Cambau E, Roth Dit Bettoni R, Gaillard JL, Jarlier V, Truffot C, Veziris N. In vivo evaluation of antibiotic activity against Mycobacterium abscessus. J Infect Dis. 2014;209:905-12.Talaat AM, Trucksis M, Reimschuessel R. Pathogenicity of Mycobacterium fortuitum and Mycobacterium smegmatis to goldfish, Carassius auratus. Vet Microbiol. 1999;66:151-64.

Introduction
31
Cha
pter
1
Chapter 1
98.
99.
100.
Pozos TC, Ramakrishnan L. New models for the study of Mycobacterium-host interactions. Curr Opin Immunol. 2004;16:499-505.Oh CT, Moon C. Jeong MS, Kwon SH, Jang J. Drosophila melanogaster model for Mycobacterium abscessus infection. Microbes Infect. 2013;15:788-95.Bernut A, Le Moigne V, Lesne T, Lutfalla G, Herrmann JL, Kremer L. In vivo assessment of drug efficacy against Mycobacterium abscessus using the embryonic zebrafish test system. Antimicrob Agents Chemother. 2014;58:4054-63.


33
Chapter 2
Time-kill kinetics of antibiotics active against rapidly growing mycobacteria
Beatriz E. FerroJakko van IngenMelanie WattenbergDick van SoolingenJohan W. Mouton
J Antimicrob Chemother. 2015;70:811-7.

34
Chapter 2
Summary
Objectives: This study was conducted to generate basic pharmacodynamic information on the relationship between antibiotic concentrations and the growth of rapidly growing mycobacteria (RGM), and thereby contribute to a better understanding of current and future drug regimens for diseases caused by RGM. Methods: Type strains of Mycobacterium abscessus and Mycobacterium fortuitum were used; the MICs of cefoxitin, amikacin, moxifloxacin, linezolid and clarithromycin were determined by broth microdilution. Time-kill assays were performed, exposing the bacteria to 2-fold concentrations from 0.25 to 32 times the MIC at 30 °C for 120 h. The sigmoid maximum effect (Emax) model was fitted to the time-kill curves data. Results: The highest killing of M. abscessus was observed between 24 and 72 h; amikacin had the highest Emax (0.0427 h-1), followed by clarithromycin (0.0231 h-1) and cefoxitin (0.0142 h-1). For M. fortuitum, between 3 and 24 h, amikacin also showed the highest Emax (0.1933 h-1). There were no significant differences between the Hill’s slopes determined for all the antibiotics tested against M. abscessus or M. fortuitum (P=0.2213 and P=0.2696, respectively). Conclusions: The total effect observed for all antibiotics was low and primarily determined by the Emax and not by the Hill’s slope. The limited activity detected fits well with the poor outcome of antibiotic treatment for disease caused by RGM, particularly for M. abscessus. An evaluation of drug combinations will be the next step in understanding and improving current treatment standards.

Time-kill kinetics of rapidly growing mycobacteria
35
Cha
pter
2
Chapter 2
Introduction
Infections caused by non-tuberculous mycobacteria (NTM) are emerging in many settings, particularly those where the incidence of tuberculosis is in decline.1 NTM are environmental organisms, able to produce chronic disease in patients with a local or systemic immune impairment. Pulmonary NTM disease is the most frequent manifestation, involving both slowly growing mycobacteria and rapidly growing mycobacteria (RGM), with a variation in the epidemiology by geographical region.2 The treatment of such diseases is complicated, as NTM are naturally resistant to most commonly used antibiotics and the outcome is often poor. In RGM disease, Mycobacterium abscessus disease is the most frequent but also the most difficult to treat. Currently recommended treatment regimens for M. abscessus depend on the infecting subspecies. For M. abscessus subspecies abscessus and M. abscessus subspecies bolletii strains that show inducible macrolide resistance, a combination of three or four drugs is used that includes amikacin, cefoxitin, imipenem, tigecycline or linezolid, while for M. abscessus subspecies bolletii strains that lack inducible macrolide resistance (former Mycobacterium massiliense), the recommended regimens include a macrolide in combination with two drugs from amikacin, cefoxitin, imipenem and linezolid. The choice of drugs is based on in vitro drug susceptibility testing. The treatment should continue for more than 12 months after cultures have converted to negative.3 For Mycobacterium fortuitum, the second most frequent RGM, a combination of two or three drugs is recommended and should include a fluoroquinolone, co-trimoxazole, amikacin, linezolid, imipenem or tetracycline, again based on in vitro susceptibility. However, there is limited clinical evidence to support these treatment regimens.4 Even at the more fundamental level, little is known about the pharmacodynamics of commonly used antibiotics against RGM. Time-kill assays allow the study of the pharmacodynamics of antibiotics, examining the rate at which different concentrations of an antibiotic kill bacteria; the concentration-dependent and time-dependent bactericidal activities of antimicrobial agents such as aminoglycosides, fluoroquinolones or β-lactams and macrolides can be studied using this methodology. The purpose of this study was to assess the pharmacodynamics of commonly used drugs to treat RGM disease by means of the time-kill assay and subsequent modeling of the results to assist in a more rational design of treatment regimens.
Materials and Methods
Bacterial strains We used M. abscessus subspecies abscessus CIP 104536 (Collection of Institute Pasteur, Paris, France) and M. fortuitum ATCC 6841 (ATCC, Rockville, MD, USA) for the experiments. Stock vials of each mycobacterium were preserved at -80 °C in trypticase soy broth with 40% glycerol and were thawed for each assay.

36
Chapter 2
AntibioticsMoxifloxacin, cefoxitin, amikacin and clarithromycin were obtained from Sigma-Aldrich (Zwijndrecht, the Netherlands) and linezolid was obtained from Pfizer BV (Capelle aan den Ijssel, the Netherlands) as the 2 mg/ml infusion. Water was the solvent for preparing stock solutions, except for clarithromycin, which was dissolved in methanol. Stock solutions were stored at -80 °C; prior to each experiment, one aliquot was thawed to prepare the different concentrations to be tested.
Susceptibility testing The MIC of each of the tested antibiotics was determined by broth microdilution in cation-adjusted Mueller –Hinton broth (CAMHB; BD Bioscience, Erembodegem, Belgium) at 30 °C,according to CLSI guidelines.5 For the initial evaluation, commercial panels were used (RAPMYCO Sensititre®, Trek Diagnostics/ThermoFisher, Landsmeer, the Netherlands) following the manufacturer’s recommendations. If the MIC fell outside the concentration range tested in the commercial assay, we performed manual broth microdilution with wider concentration ranges.
Time-kill assaysThe mycobacterial inoculum was prepared freshly for each experiment by growing over 24 h in CAMHB with oleic acid/bovine albumin/dextrose/catalase (OADC) growth supplement (BD Bioscience, Erembodegem, Belgium) and 0.05% Tween 80 (Sigma-Aldrich), to obtain bacteria in the early logarithmic phase of growth. Individual bottles of 20 ml of CAMHB plus OADC and 0.05% Tween 80 containing eight 2-fold increasing concentrations of each antibiotic (from 0.25 to 32 times the MIC, except for M. abscessus and cefoxitin, for which two lower concentrations were included) were cultured with the inoculum (density 105–106 cfu/ml) at 30 °C, under shaking conditions (100 rpm); ventilation through a bacterial filter (FP 30/0.2 Ca/S, Whatman GmbH, Germany) was incorporated. A growth control bottle, with inoculum but without antibiotic, as well as a sterility control (medium only) were included. At defined time intervals (3, 6, 12, 24, 36, 48, 72, 96 and 120 h), the size of the bacterial population was quantified to characterize the effect of the different antibiotics. Samples of 1 ml were taken from each bottle and serial 10-fold dilutions in 0.85% sterile saline solution were prepared. Volumes of 10 µl from undiluted samples and from each dilution were plated in triplicate on Middlebrook 7H11 (BD Bioscience, Erembodegem, Belgium) for further cfu counting after 3 –5 days of incubation at 30 °C. The theoretical detection limit was 33.3 cfu per plate, corresponding to 1.52 log10 cfu/ml.
Curve fitting and analysisThe experimental data derived from time-kill assays were analyzed using GraphPad Prism 5.03 (GraphPad Inc., San Diego, CA, USA). Log cfu values were plotted against time for each antibiotic. The kill rate was determined at different time intervals (3 –24, 3 –36, 24 –72, 24

Time-kill kinetics of rapidly growing mycobacteria
37
Cha
pter
2
Chapter 2
–96 and 24 –120 h), undertaking a linear regression to find the slope for each concentration; the logarithmic transformed concentration was then plotted against each slope and a non-linear regression analysis (dose – response) was run. The sigmoid maximum effect (Emax) model (four-parameters Hill’s equation)6,7 was fitted to the kill rate data, analyzing each assay to determine the pharmacodynamic relationship between the antibiotic concentration and bacterial growth or death. Emax, 50% effective concentration (EC50), Hill’s slope (ɣ) and R2 were calculated for each assay.
Results
SusceptibilityThe MICs determined for each antibiotic are shown in Table 1. All the MICs were higher for M. abscessus than for M. fortuitum.
Time-kill assays Figures 1 and 2 show the pattern of growth and kill by antibiotics of M. abscessus and M. fortuitum, respectively, at different concentrations of each of the tested antibiotics. The growth curves differ for each species. M. abscessus showed a lag phase of 3–12 h and its maximum growth was higher than the growth for M. fortuitum in all the experiments. The lag phase for M. fortuitum was around 3 h. In general, time-kill curves for M. abscessus showed smaller decreases in bacterial population size than those observed for M. fortuitum when exposed to antibiotics.
For M. abscessus, the cefoxitin time-kill curve was different from those of amikacin and clarithromycin. After a short lag phase, a slow decline was observed at almost all concentrations, reaching the lowest cfu counts only after 96 –120 h of incubation. In contrast, during amikacin and clarithromycin exposure, killing was observed after only 24 h and for some concentrations, in particular with clarithromycin, there appeared to be significant growth even before killing was observed. Only after 24 h did concentrations higher than 2× MIC started to effectively decrease the bacterial density, with its maximum decrease at 120 h at the two highest concentrations. Regrowth was, however, observed with 2, 4 and 8× MIC.
Table 1. Susceptibility data for M. abscessus and M. fortuitum type strains tested by broth microdilution in CAMHB, reading at 72 h.5
MIC (mg/l)
Strain Cefoxitin Amikacin Moxifloxacin Linezolid Clarithromycin
M. abscessus CIP104536 64 32 8 32 4
M. fortuitum ATCC6841 32 1 0.062 8 2

38
Chapter 2
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
Growth Control0.062 × MIC0.125 × MIC
8 × MIC16 × MIC
0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC
DetectionLimit
32 × MIC
Time (h)
Log
CFU
(a) Cefoxitin
(b) Amikacin
(c) Clarithromycin
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
Growth Control0.062 × MIC0.125 × MIC
8 × MIC16 × MIC
0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC
DetectionLimit
32 × MIC
Time (h)
Log
CFU
(a) Cefoxitin
(b) Amikacin
(c) Clarithromycin
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
Time (h)
Log
CFU
Figure 1. Time-kill curves of (A) cefoxitin, (B) amikacin and (C) clarithromycin against M. abscessus CIP 104536. Antibiotic concentrations are indicated by different symbols.
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
Growth Control0.062 × MIC0.125 × MIC
8 × MIC16 × MIC
0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC
DetectionLimit
32 × MIC
Time (h)
Log
CFU
(a) Cefoxitin
(b) Amikacin
(c) Clarithromycin
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
Time (h)
Log
CFU
Interestingly, after 48 h, part of the colonies exposed to amikacin concentrations of 2× MIC and higher converted to a rough morphology, which was observed after plating the samples for cfu counting.
Significant differences in the killing characteristics of the antibiotics were observed for M. fortuitum. Linezolid showed only slight but prolonged killing without apparent concentration dependent effects. The curves for cefoxitin, amikacin and moxifloxacin shared some characteristics and exhibited an important reduction in growth during the first 24–36 h (Figure 2); however, regrowth occurred after the initial killing and was noticeable for amikacin in particular, with the phenomenon also observed for M. abscessus. Amikacin also showed the highest kill rate and concentration-dependent activity; the main fall in cfu was observed at concentrations 4× MIC or higher. The same phenomenon of morphology changes noted previously for M. abscessus was observed for M. fortuitum colonies exposed to 2× MIC or higher of amikacin.
Time-kill modeling After a lag phase, the maximum kill rate was present over the period 24–72 h for M. abscessus and 3–24 h for M. fortuitum. Figure 3 shows the relationship between the kill rate and concentration for the two RGM species. Pharmacodynamic parameter estimates were obtained with the Emax model with a variable slope (Table 2).
The Emax model fitted well and confirmed that the kill rate for M. abscessus was relatively low. The highest killing rate was observed for amikacin, 0.0427 h-1, between 24 and 72 h. There was no significant difference in Hill’s slope estimated for the antibiotics tested (P=0.2213), indicating that the differences in effect modality are primarily determined by the maximum kill rate of each antibiotic. For M. fortuitum, the maximum kill rate was observed much earlier,
A cefoxitin
C clarithromycin
B amikacin

Time-kill kinetics of rapidly growing mycobacteria
39
Cha
pter
2
Chapter 2
between 3 and 24 h of exposure, and again amikacin showed the highest Emax, 0.1933 h-1. As with M. abscessus, no significant differences were found between the Hill’s slope calculated for each of the antibiotics tested (P=0.2696), indicating that the effect is primarily determined by the maximum effect.
Discussion
This study provides fundamental new information on the pharmacodynamic relationship between antibiotic concentrations and mycobacterial population dynamics for RGM.
Amikacin, cefoxitin and clarithromycin did not show a high killing effect on M. abscessus, although killing proceeded with time. For M. fortuitum, amikacin, cefoxitin and moxifloxacin showed highest killing rates. The higher growth level consistently reached by M. abscessus could, at least in part, explain the lesser effect observed for the antibiotics on this species. The individual analysis of each antibiotic indicated that amikacin had the highest effect on both RGM. For M. abscessus, amikacin inhibited the growth during the first 24 h, but the killing activity started after that time. For M. fortuitum, killing from amikacin started earlier. We observed morphological changes after exposure to amikacin concentrations of 2× MIC or higher. This phenomenon has been previously observed for Pseudomonas aeruginosa after 6 h of incubation in time-kill assays.8 Whether the changes we observed are related to the appearance of resistant mutants or are the result of an adaptation response should be addressed in the future.
Figure 2. Time-kill curves of (A) cefoxitin, (B) amikacin, (C) moxifloxacin and (D) linezolid against M. fortuitum ATCC 6841. Antibiotic concentrations are indicated by different symbols.
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC
32 × MIC16 × MIC8 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC
32 × MIC
0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC
32 × MIC
0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC
Time (h)
Log
CFU
(b) Amikacin
(d) Linezolid
(a) Cefoxitin
(c) Moxifloxacin
A cefoxitin
C moxifloxacin
B amikacin
D linezolid
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC
32 × MIC16 × MIC8 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC
32 × MIC
0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC
32 × MIC
0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC
Time (h)
Log
CFU
(b) Amikacin
(d) Linezolid
(a) Cefoxitin
(c) Moxifloxacin
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC
32 × MIC16 × MIC8 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC
32 × MIC
0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC
Time (h)
Log
CFU
0 12 24 36 48 60 72 84 96 108 1200123456789
1011
DetectionLimit
Growth Control0.25 × MIC
32 × MIC
0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC
Time (h)
Log
CFU
(b) Amikacin
(d) Linezolid
(a) Cefoxitin
(c) Moxifloxacin

40
Chapter 2
Recently published work with clinical strains of M. abscessus exposed to amikacin, as well as to linezolid, tigecycline and moxifloxacin, states a lack of antimicrobial bactericidal activity of these antibiotics.9 However, the assays were conducted only until 24 h, a difference from the present study, which describes the activity over 120 h assays; the observed lack of activity by the authors may therefore be largely explained by the lag phase we have also observed. Our results clearly indicate that the effects of antimicrobials on M. abscessus should be studied over at least 72 h to provide useful information.
Although it involved a different NTM, previous work on Mycobacterium avium complex (MAC) showed that amikacin appeared highly and rapidly bactericidal in the early logarithmic phase of growth.10 Moreover, with Mycobacterium tuberculosis, the time-kill kinetics of amikacin displayed a high and extremely rapid killing activity that was not time dependent and could eliminate all mycobacteria.11 Against M. abscessus and M. fortuitum, we observed a significantly weaker effect. Nonetheless, amikacin still can play an important role in the treatment of disease caused by RGM.
The effect of cefoxitin was different in the two RGM species evaluated. For M. abscessus, cefoxitin behaved like other β-lactams and the bacterial density gradually declined during exposure to it. This is in contrast to the effect observed in M. fortuitum, where cefoxitin showed a concentration-dependent effect and the second highest Emax. Differences in the effect of cefoxitin have previously been reported in MRSA. Combining cefoxitin with a variety of β-lactams enhanced their in vitro activity against community-acquired MRSA strains but not against hospital-acquired MRSA; this may result from the differential binding of cefoxitin to the target (penicillin binding protein 4, PBP4), which plays an important role only in the β-lactam resistance of community-acquired strains.12 Cefoxitin targets or mechanisms of action may not be the same in M. abscessus and M. fortuitum; this could explain the differences observed and will be an interesting subject for further evaluation.
Figure 3. The best-fit sigmoid curves obtained from the Emax model of (A) M. abscessus CIP 104536 exposed to cefoxitin, amikacin and clarithromycin, between 24 and 72 h, and (B) M. fortuitum ATCC 6841 exposed to cefoxitin, amikacin, moxifloxacin and linezolid, between 3 and 24 h. A different y-axis scale is used for (A) and (B).
0.0001 0.01 1 100 10000-0.10
-0.05
0.00
0.05ClarithromycinAmikacinCefoxitin
Concentration (mg/L)
Obs
erve
d ki
ll ra
te h
-1
(a)
(b)
0.0001 0.01 1 100 10000-0.2
-0.1
0.0
0.1
0.2
0.3MoxifloxacinAmikacinCefoxitinLinezolid
Concentration (mg/L)
Obs
erve
d ki
ll ra
te h
-1
0.0001 0.01 1 100 10000-0.10
-0.05
0.00
0.05ClarithromycinAmikacinCefoxitin
Concentration (mg/L)
Obs
erve
d ki
ll ra
te h
-1
(a)
(b)
0.0001 0.01 1 100 10000-0.2
-0.1
0.0
0.1
0.2
0.3MoxifloxacinAmikacinCefoxitinLinezolid
Concentration (mg/L)
Obs
erve
d ki
ll ra
te h
-1
0.0001 0.01 1 100 10000-0.10
-0.05
0.00
0.05ClarithromycinAmikacinCefoxitin
Concentration (mg/L)
Obs
erve
d ki
ll ra
te h
-1
(a)
(b)
0.0001 0.01 1 100 10000-0.2
-0.1
0.0
0.1
0.2
0.3MoxifloxacinAmikacinCefoxitinLinezolid
Concentration (mg/L)
Obs
erve
d ki
ll ra
te h
-1
0.0001 0.01 1 100 10000-0.10
-0.05
0.00
0.05ClarithromycinAmikacinCefoxitin
Concentration (mg/L)
Obs
erve
d ki
ll ra
te h
-1
(a)
(b)
0.0001 0.01 1 100 10000-0.2
-0.1
0.0
0.1
0.2
0.3MoxifloxacinAmikacinCefoxitinLinezolid
Concentration (mg/L)O
bser
ved
kill
rate
h-1
A B
Concentration (mg/l) Concentration (mg/l)

Time-kill kinetics of rapidly growing mycobacteria
41
Cha
pter
2
Chapter 2
Clarithromycin, long considered a cornerstone of M. abscessus treatment,1,3 showed killing capacity only at concentrations greater than 4× MIC. Similarly, previous studies of MAC observed a maximum bactericidal effect at a relatively high concentration (256 mg/l).10 Linezolid and moxifloxacin were only tested for M. fortuitum as these drugs were considered inactive against the M. abscessus type strain (Table 1); however, their killing capacity was not high for M. fortuitum. Few comparative data are available for those antibiotics.
According to the Emax model fitted to our data, Hill’s slopes were not significantly different between the antibiotics tested for each species. In this regard, the total effect observed was primarily determined by the Emax, representing the extent of kill as a function of concentration, and not by the Hill’s slope. This is different from the effect of antibiotics in other bacterial species, where a clear difference is observed for time-dependent antimicrobials such as β-lactams (lower Emax, higher Hill´s slope) and concentration-dependent drugs such as aminoglycosides (higher Emax, lower Hill´s slope).7
Our data for M. abscessus contrast with the recent findings from the nude mice model, in which cefoxitin was superior in efficacy to amikacin and clarithromycin. Only cefoxitin improved survival and reduced bacillary loads in the spleen; amikacin and clarithromycin prevented death but had little impact on bacillary loads. Interestingly, in this model the amikacin/clarithromycin/ cefoxitin combination was as active as cefoxitin alone.13
Strain Antibiotic Time (h)
Emax (h-1)
95 % CI EC50 (mg/l)
95 % CI Hill’s slope (ɣ)*
95 % CI
R2
M. abscessus CIP104536
Cefoxitin 24-72 0.0142 0.008719-0.01976
6.24 4.450-8.735
3.534 1.804-5.263
0.97
Cefoxitin 24-72 0.0427 0.03456-0.05083
51.74 41.18-65.01
0.97
Clarithromycin 24-72 0.0231 0.01430-0.03185
9.63 7.163-12.96
0.92
M. fortuitum ATCC6841
Cefoxitin 3-24 0.1247 0.1041-0.1454
18.24 12.78-26.02
1.416 1.021-1.811
0.93
Amikacin 3-24 0.1933 0.1689-0.2177
0.910 0.6848-1.209
0.99
Moxifloxacin 3-24 0.0677 0.04939-0.08590
0.021 0.01295-0.03445
0.93
Linezolid 3-24 0.0338 0.01518-0.05234
3.203 1.784-5.750
0.97
Table 2. Parameter estimates, with 95% CI, derived from inhibitory Emax.
* Pooled value for all antibiotics by species.

42
Chapter 2
Extrapolating these data to the treatment setting, the limited activity detected for amikacin, cefoxitin and clarithromycin against M. abscessus fits well with the clinical observations that treatment with regimens containing these drugs leads to poor outcomes.3 The amikacin peak serum concentration in patients with NTM pulmonary disease averages 55 mg/l,14,15 i.e. around the MIC measured for the M. abscessus type strain. Given that the activity of amikacin was best at the highest concentrations, the current dosing may not yield concentrations at the site of infection that can exhibit significant killing activity. This may in part explain the limited efficacy of amikacin against M. abscessus in the nude mice model.13 Local administration, e.g. inhaled amikacin for pulmonary disease caused by M. abscessus, may be more efficacious. The M. abscessus type strain had an MIC of clarithromycin of 4 mg/l, and the maximum effect was attained at concentrations > 2× MIC. These concentrations are above the concentration attainable in the serum of patients (which average 4 mg/l),4,14,15 but the macrolides are known to accumulate in lung tissue, epithelial lining fluid and macrophages at concentrations 2-200 times higher than serum concentrations,16 although it is not clear whether these concentrations are active.17 Hence, the high concentrations needed to achieve a significant effect in our time-kill assay may be attainable at the site of infection in M. abscessus lung disease. Unfortunately, no pharmacokinetic data are available for cefoxitin in this patient category, which needs to be investigated if cefoxitin is to continue to be included in the treatment regimens for RGM disease.
The time-kill assays performed in this study provide basic information on the individual effect of static concentrations of each antibiotic on M. abscessus and M. fortuitum. However, this setting is very different from the in vivo situation where multidrug therapy, the daily intake of drugs and their pharmacokinetics, and the localization of the causative mycobacteria create a very different scenario. Thus, the next step should be to continue these studies in dynamic pharmacokinetic/pharmacodynamic models for the evaluation of combined regimens (which are, and should be, the norm) in RGM disease. These models, despite their abstractions, will provide data that are much closer to the in vivo situation and may aid in the design of more active treatment regimens.
In conclusion, time-kill kinetic assays revealed that amikacin was more active than clarithromycin and cefoxitin against M. abscessus and that amikacin, followed by cefoxitin, moxifloxacin and linezolid, was also most active against M. fortuitum. However, we demonstrate that the activity of all the drugs tested was relatively low and the concentrations effective in vitro can hardly be reached in vivo, reminiscent of the poor outcomes of antibiotic treatment for RGM diseases.

Time-kill kinetics of rapidly growing mycobacteria
43
Cha
pter
2
Chapter 2
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
References
Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier J, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K, ATS Mycobacterial Diseases Subcommittee, American Thoracic Society, Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416. Hoefsloot W, van Ingen J, Andrejak C, Angeby K, Bauriaud R, Bemer P, Beylis N, Boeree MJ, Cacho J, Chihota V, Chimara E, Churchyard G, Cias R, Daza R, Daley CL, Dekhuijzen PN, Domingo D, Drobniewski F, Esteban J, Fauville-Dufaux M, Folkvardsen DB, Gibbons N, Gómez-Mampaso E, Gonzalez R, Hoffmann H, Hsueh PR, Indra A, Jagielski T, Jamieson F, Jankovic M, Jong E, Keane J, Koh WJ, Lange B, Leao S, Macedo R, Mannsåker T, Marras TK, Maugein J, Milburn HJ, Mlinkó T, Morcillo N, Morimoto K, Papaventsis D, Palenque E, Paez-Peña M, Piersimoni C, Polanová M, Rastogi N, Richter E, Ruiz-Serrano MJ, Silva A, da Silva MP, Simsek H, van Soolingen D, Szabó N, Thomson R, Tórtola Fernandez T, Tortoli E, Totten SE, Tyrrell G, Vasankari T, Villar M, Walkiewicz R, Winthrop KL, Wagner D; Nontuberculous Mycobacteria Network European Trials Group. The geographic diversity of non-tuberculous mycobacteria isolated from pulmonary samples: an NTM-NET collaborative study. Eur Resp J. 2013;42:1604–13.van Ingen J, Ferro BE, Hoefsloot W, Boeree MJ, van Soolingen D. Drug treatment of pulmonary antituberculous mycobacterial disease in HIV-negative patients: the evidence. Expert Rev Anti Infect Ther. 2013;11:1065–77. Magis-Escurra C, Alffenaar JW, Hoefnagels I, Dekhuijzen PN, Boeree MJ, van Ingen J, Aarnoutse RE. Pharmacokinetic studies in patients with nontuberculous mycobacterial lung infections. Int J Antimicrob Agents. 2013;42:256–61. Clinical and Laboratory Standards Institute. Susceptibility Testing of Mycobacteria, Nocardia, and Other Aerobic Actinomycetes—Second Edition: Approved Standard M24-A2. CLSI, Wayne, PA, USA, 2011. Mouton JW, Dudley MN, Cars O, Derendorf H, Drusano GL. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs. Int J Antimicrob Agents. 2002;19:355–8. Mouton JW, Vinks AA. Pharmacokinetic/pharmacodynamic modelling of antibacterials in vitro and in vivo using bacterial growth and kill kinetics: the minimum inhibitory concentration versus stationary concentration. Clin Pharmacokinet. 2005;44:201–10. Staneva M, Markova B, Atanasova I, Terziivanov D. Pharmacokinetic and pharmacodynamic approach for comparing two therapeutic regimens using amikacin. Antimicrob Agents Chemother. 1994;38:981–5.Maurer FP, Bruderer VL, Ritter C, Castelberg C, Bloemberg GV, Böttger EC. Lack of antimicrobial bactericidal activity in Mycobacterium abscessus. Antimicrob Agents Chemother. 2014;58:3828–36. Bakker-Woudenberg IA, van Vianen W, van Soolingen D, Verbrugh HA, van Agtmael MA. Antimycobacterial agents differ with respect to their bacteriostatic versus bactericidal activities in relation to time of exposure, mycobacterial growth phase, and their use in combination. Antimicrob Agents Chemother. 2005;49:2387–98. de Steenwinkel JE, de Knegt GJ, ten Kate MT, van Belkum A, Verbrugh HA, Kremer K, van Soolingen D, Bakker-Woudenberg IA. Time-kill kinetics of anti-tuberculosis drugs, and emergence of resistance, in relation to metabolic activity of Mycobacterium tuberculosis. J Antimicrob Chemother. 2010;65:2582–9.

44
Chapter 2
12.
13.
14.
15.
16.
17.
Banerjee R, Fernandez MG, Enthaler N, Graml C, Greenwood-Quaintance KE, Patel R. Combinations of cefoxitin plus other β-lactams are synergistic in vitro against community associated methicillin-resistant Staphylococcus aureus. Eur J Clin Microbiol Infect Dis. 2013;32:827–33. Lerat I, Cambau E, Roth Dit Bettoni R, Gaillard JL, Jarlier V, Truffot C, Veziris N. In vivo evaluation of antibiotic activity against Mycobacterium abscessus. J Infect Dis. 2014;209:905–12.van Ingen J, Egelund EF, Levin A, Totten SE, Boeree MJ, Mouton JW, Aarnoutse RE, Heifets LB, Peloquin CA, Daley CL. The pharmacokinetics and pharmacodynamics of pulmonary Mycobacterium avium complex disease treatment. Am J Respir Crit Care Med. 2012;186:559–65. Koh WJ, Jeong BH, Jeon K, Lee SY, Shin SJ. Therapeutic drug monitoring in the treatment of Mycobacterium avium complex lung disease. Am J Resp Crit Care Med. 2012;186:797–802. Zuckerman JM, Qamar F, Bono BR. Review of macrolides (azithromycin, clarithromycin), ketolids (telithromycin) and glycylcyclines (tigecycline). Med Clin North Am. 2011;95:761–91. Mouton JW, Theuretzbacher U, Craig WA, Tulkens PM, Derendorf H, Cars O. Tissue concentrations: do we ever learn? J Antimicrob Chemother. 2008;61:235–7.

Time-kill kinetics of rapidly growing mycobacteria
45
Cha
pter
2
Chapter 2


47
Chapter 3
Time-kill kinetics of slowly growing mycobacteria common in pulmonary disease
Beatriz E. FerroJakko van IngenMelanie WattenbergDick van SoolingenJohan W. Mouton
J Antimicrob Chemother. 2015;70:2838-43.

48
Chapter 3
Summary
Objectives: This study aimed to provide basic pharmacodynamic information for key antibiotics used to treat Mycobacterium avium and Mycobacterium xenopi pulmonary disease.Methods: M. avium subspecies hominissuis IWGMT49 and M. xenopi ATCC 19250 type strains were used; the MICs of clarithromycin, amikacin and moxifloxacin were determined by broth microdilution. Time-kill assays were performed, exposing bacteria to 2-fold concentrations from 0.062× to 32× the MIC at 37 °C for 240 h for M. avium or 42 days for M. xenopi. The sigmoid maximum effect (Emax) model was fitted to the time-kill curve data. Results: Maximum killing of M. avium by amikacin was obtained between 24 and 120 h (0.0180 h-1) and was faster and higher than with clarithromycin (0.0109 h-1); however, regrowth and amikacin-resistant mutants were observed. Killing rates for M. xenopi were higher, 0.1533 h-1 for clarithromycin and 0.1385 h-1 for moxifloxacin, yet required 42 days. There were no significant differences between the Hill´s slopes determined for all the antibiotics tested against M. avium or M. xenopi (P= 0.9663 and P= 0.0844, respectively).Conclusions: The killing effect of amikacin and clarithromycin on M. avium subspecies hominissuis was low, although amikacin activity was higher than that of clarithromycin, supporting its role in a combined therapy. Clarithromycin and moxifloxacin may have similar activity within treatment regimens for M. xenopi disease. Future studies of in vitro and in vivo pharmacokinetic/pharmacodynamic interactions are needed to improve the current regimens to treat these two important slowly growing mycobacteria in pulmonary disease.

Time-kill kinetics of slowly growing mycobacteria
49
Cha
pter
3
Introduction
Pulmonary disease is the most common clinical manifestation of non-tuberculous mycobacteria (NTM) infection.1 Several NTM can cause such disease, although their geographical distribution varies from region to region.2 Mycobacterium avium complex (MAC) is the most frequently isolated slowly growing mycobacteria (SGM) involved in pulmonary disease,2,3 but Mycobacterium xenopi has also been frequently isolated in Northern Europe as an important infectious species.2,3 Treatment of pulmonary disease caused by M. avium and M. xenopi is a challenge and the outcome is uncertain. Only macrolides, like azithromycin or clarithromycin, are active both in vitro and in vivo against M. avium, 1,4 hence treatment regimens include macrolides, but rifampicin and ethambutol are added in order to prevent acquired macrolide resistance. Amikacin is also used, during the first 2–3 months of therapy, despite clinical evidence; only the in vitro MIC supports its role.1,4 Likewise the recommended regimen for M. xenopi includes isoniazid, rifampicin, ethambutol and clarithromycin, with aminoglycosides added during the first months. Moxifloxacin has also been suggested to play a role in the treatment of such mycobacterial disease.1,4 However, there is little information on the effectiveness of those antibiotics against M. avium and M. xenopi pulmonary disease and cure rates are low.4,5 Moreover, patient cohorts of M. xenopi pulmonary disease stand out for their high mortality rates.3 The limited information on the activity of the antibiotics currently used in these regimens emphasizes the need to better understand the fundamentals of SGM treatment, in particular the potency and activity of the antimicrobials that currently play a major role. In the present study we aimed to study the basic pharmacodynamics for key antibiotics used to treat M. avium and M. xenopi pulmonary disease by performing in vitro time-kill kinetics. Because of the slow growth of SGM and the long duration of treatment, some experiments were conducted over up to 42 days of exposure.
Materials and Methods
Bacterial strains and antibioticsWe used M. avium subspecies hominissuis IWGMT49 (International Working Group on Mycobacterial Taxonomy) and M. xenopi ATCC 19250 (ATCC, Manassas, VA, USA) as test strains. Stock vials of each mycobacteria in the early logarithmic phase of growth were preserved at -80 °C in trypticase soy broth with 40% glycerol and were thawed for each assay. Moxifloxacin, amikacin and clarithromycin were obtained from Sigma-Aldrich (Zwijndrecht, The Netherlands) and dissolved in water, except for clarithromycin, which was dissolved in methanol, following CLSI recommendations.6 Stock solutions, stored at -80 °C, were thawed for each experiment to prepare the different concentrations to be tested.

50
Chapter 3
Susceptibility testing The MIC of each of the tested antibiotics was determined by broth microdilution in CAMHB (BD Bioscience, Erembodegem, Belgium) at 37 °C, according to CLSI guidelines,7 using commercial panels (SLOWMYCO Sensititre®, Trek Diagnostics/ThermoFisher-Landsmeer, The Netherlands).
Time-kill assaysIndividual bottles of 20 ml of Middlebrook 7H9 (BD Bioscience, Erembodegem, Belgium) plus OADC growth supplement (BD Bioscience) and 0.05% Tween 80 (Sigma-Aldrich), containing ten 2-fold increasing concentrations of each antibiotic (from 0.062× to 32× the MIC) were cultured with the inoculum (density 105–106 cfu/ml) at 37 °C. All bottles were shaken (100 rpm) and ventilated with air through a bacterial filter (FP 30/0.2 Ca/S, Whatman GmbH, Germany). In order to perform cfu counting of the bacterial population, samples of 200 ml were taken from each bottle and serial 10-fold dilutions in 0.85% sterile saline solution were plated on Middlebrook 7H11 (BD Bioscience) plates, at different time intervals (12, 24, 48, 72, 96, 120, 144, 168 and 240 h for M. avium and 3, 6, 9, 12, 15 18, 21, 28 and 42 days for M. xenopi).
Mutation frequency Mutants were counted at time 0 and after 240 h for M. avium exposed to both amikacin and clarithromycin, by plating the bacterial suspension and 10-fold dilutions on Middlebrook 7H11 plates containing 4× the MIC of each antibiotic. Owing to the higher mutation rate expected for amikacin, mutation frequency was determined throughout the experiment for bottles containing 1×, 4×, 8×, 16× and 32× MIC. No mutation frequency was determined for M. xenopi, since no resistant mutants were expected.
Curve fitting and analysisLog cfu values were plotted against time for each antibiotic and analyzed using Graphpad Prism 5.03 (Graphpad Inc., San Diego, CA, USA). The sigmoid maximum effect (Emax) model (four-parameter Hill’s equation)8 was fitted to the kill rate data, analyzing each assay to determine the pharmacodynamic relationship between antibiotic concentration and bacterial growth or death. Emax, 50% effective concentration (EC50), Hill’s slope (ɣ) with 95% CI and R2 were calculated for each assay.
Amikacin concentrations cfu/ml Mutation frequency One mutant in cfu/ml
Growth Control 4.00 × 109
1× MIC (8 mg/l) 2.80 × 107 7.00 × 10-3 143
4× MIC (32 mg/l) 0 - -
8× MIC (64 mg/l) 3.23 × 107 8.03 × 10-3 124
16× MIC (128 mg/l) 5.33 × 107 1.33 × 10-2 75
32× MIC (256 mg/l) 3.33 × 106 8.33 × 10-4 1200
Table 1. Mutation frequency of M. avium IWGMT49 exposed to amikacin after 240 h.

Time-kill kinetics of slowly growing mycobacteria
51
Cha
pter
3
Results
SusceptibilityClarithromycin and amikacin MICs for M. avium were 4 and 8 mg/l. Clarithromycin and moxifloxacin MICs for M. xenopi were both 0.25 mg/l.
Time-kill assays M. aviumKill curves of M. avium exposed to clarithromycin and amikacin are shown in Figure 1. In the experiments with clarithromycin we observed a lag phase of 24 h; thereafter a slow decline in bacterial cfu/ml was noted for each concentration, except for the lowest of 0.062× MIC, which only produced inhibition of growth. Killing appeared to be maximized at relatively low multiples of the MIC and was hardly concentration dependent. Regrowth was observed after 168 h for concentrations in the lower range - 0.062×, 0.25×, 0.5× and 2× MIC. In contrast, amikacin killing was faster and higher for concentrations above 2× MIC. However, regrowth was observed earlier than with clarithromycin, after 96–120 h, except for 4× MIC.
M. xenopiKill curves of M. xenopi exposed to clarithromycin and moxifloxacin are shown in Figure 2. When clarithromycin was used, the lag phase involved 3 days and thereafter a slow decline in cfu/ml was observed at almost all concentrations; killing was maximum after 42 days. Low clarithromycin concentrations showed killing, but similar to M. avium, regrowth was observed at the lower concentrations of 0.062×, 0.125×, 0.25× and 0.5× MIC. Concentrations of 1× MIC and higher produced constant killing with no regrowth; only concentrations 4× MIC and higher resulted in undetectable cfu/ml after 42 days of exposition. When moxifloxacin was tested, a lag phase of 3 days was also observed, but only for concentrations of 0.062 to 1× MIC; concentrations 2× MIC and higher already showed a killing effect by that time. After 3 days, low concentrations of 0.062× to 0.25× MIC initially showed some killing, but regrowth appeared after 9-12 days, depending on the concentration.
Figure 1. Time-kill curves of M. avium IWGMT49 with (A) clarithromycin and (B) amikacin. Antibiotic concentrations are indicated by different symbols.
0 24 48 72 96 120 144 168 192 216 2400123456789
10
Growth Control0.062× MIC0.125× MIC0.25× MIC0.5× MIC1× MIC2× MIC4× MIC8× MIC16× MIC32× MIC
DetectionLimit
Time (h)
Log
CFU
/mL
0 24 48 72 96 120 144 168 192 216 2400123456789
10
Growth Control0.062× MIC0.125× MIC0.25× MIC0.5× MIC1× MIC2× MIC4× MIC8× MIC16× MIC32× MIC
DetectionLimit
Time (h)
Log
CFU
/mL
(a)
(a)
0 24 48 72 96 120 144 168 192 216 2400123456789
10
Growth Control0.062× MIC0.125× MIC0.25× MIC0.5× MIC1× MIC2× MIC4× MIC8× MIC16× MIC32× MIC
DetectionLimit
Time (h)
Log
CFU
/mL
0 24 48 72 96 120 144 168 192 216 2400123456789
10
Growth Control0.062× MIC0.125× MIC0.25× MIC0.5× MIC1× MIC2× MIC4× MIC8× MIC16× MIC32× MIC
DetectionLimit
Time (h)
Log
CFU
/mL
(a)
(a)B amikacinA clarithromycin

52
Chapter 3
Constant killing with no regrowth was observed with concentrations 0.5× MIC and higher, but only concentrations 1× MIC and higher resulted in undetectable cfu/ml after 42 days of exposition.
High mutation frequency in amikacin-exposed M. aviumNo amikacin-resistant or clarithromycin-resistant mutants were detected at time 0 for the M. avium inoculum used in the experiments. After 240 h of incubation of the growth control, we observed a mutation frequency of 3.24×10-7 for amikacin and a mutation frequency of 4.05×10-8 for clarithromycin. In amikacin-exposed cultures, the first M. avium resistant mutants appeared after 72-96 h of incubation. Mutation frequency after 240 h of exposure was on average 7.3×10-3, excluding 4× MIC where no mutants appeared (Table 1).
Emax model The Emax model fitted well; the highest kill rate for M. avium was observed after 24-120 h of exposure to amikacin, 0.0180 h-1 . There was no significant difference in Hill’s slope estimated for amikacin and clarithromycin (P=0.9663), indicating that the differences in effect modality are primarily determined by the maximum kill rate of each antibiotic. For M. xenopi, the maximum kill rate was observed in a different time interval, 3-42 days of exposure. The Emax values were higher than for M. avium but very similar between clarithromycin and moxifloxacin. Again, no significant differences were found between the Hill’s slope calculated for the two antibiotics tested (P=0.0844). Table 2 shows the parameter estimates obtained with the Emax model for each experiment and Figure 3 shows the best fitted sigmoid curves for the kill rate and concentration for the two SGM.
Discussion
This study presents basic pharmacodynamic information for antibiotics used to treat pulmonary disease caused by M. avium and M. xenopi. Amikacin showed a higher killing effect on M. avium than clarithromycin, although the killing was relatively low. For M. xenopi, the killing obtained with clarithromycin or moxifloxacin was similar and very slow for both agents.
The amikacin killing rate was higher than that of clarithromycin for M. avium. Its effect was faster, especially at concentrations over 2× MIC; therefore, its role as a support antibiotic in combined regimens holds promise. Strong and rapid in vitro killing by amikacin has been noted for Mycobacterium tuberculosis;9 however, no early bactericidal activity was detected in a group of patients with untreated, smear-positive, pulmonary TB in South Africa.10
Superior activity of amikacin over clarithromycin was also observed in vitro against M. avium subspecies avium11 and M. abscessus.12 Only the higher concentrations of amikacin, 2× MIC

Time-kill kinetics of slowly growing mycobacteria
53
Cha
pter
3
and above ( ≥16 mg/l), were effective against M. avium in this study, some of which can be close to the achievable levels in serum.13 Whether clinically achievable levels are good enough to attain the maximum kill is a question that should be answered in the future. The use of inhaled amikacin, currently in trials (ClinicalTrials.gov identifier NCT01315236), may overcome this pharmacodynamic barrier since higher concentrations can be obtained in lungs with potentially less nephrotoxicity and ototoxicity.14,15
Regrowth of M. avium was observed soon after the initial drop in cfu obtained with amikacin. A high mutation frequency was determined in this study after in vitro exposure of M. avium subspecies hominissuis to amikacin concentrations of 1×, 8×, 16× and 32× MIC (8, 64, 128 and 256 mg/l). The emergence of resistant mutants has been previously observed in M. tuberculosis, when exposure to an amikacin concentration window between 8 and 256 mg/l resulted in the selection of resistant bacteria, which harbored the A1401G mutation in the rrs gene.9 Previous work done with M. avium subspecies avium did not show the early and high mutation rate that we observed in this study.11
Macrolides are considered the cornerstone for the treatment of MAC disease, particularly because of the established in vitro–in vivo correlation of their activity.4,16 In this study, clarithromycin showed a slow killing for M. avium subspecies hominissuis, basically at all concentrations, which was maximum after 240 h; however, the regrowth observed after 168 h underlines that macrolide monotherapy should not be used. In addition, if there is unknown resistance to all of the companion antibiotics, clarithromycin monotherapy will favor the emergence of resistance.
When clarithromycin was tested against M. avium subspecies avium, it resulted in both concentration-dependent and time dependent effects.11 However, concentration was not the most important factor for clarithromycin effect in our study, which may be evidence of the intrinsic differences in the interaction of the same antibiotic with different M. avium subspecies.
0 3 6 9 12 15 18 21 24 27 30 33 36 39 420123456789
10
Growth Control0.062× MIC0.125× MIC0.25× MIC0.5× MIC1× MIC2× MIC4× MIC8× MIC16× MIC32× MIC
DetectionLimit
Time (days)
Log
CFU
/mL
0 3 6 9 12 15 18 21 24 27 30 33 36 39 420123456789
10
Growth Control0.062× MIC0.125× MIC0.25× MIC0.5× MIC1× MIC2× MIC4× MIC8× MIC16× MIC32× MIC
DetectionLimit
Time (days)
Log
CFU
/mL
(a)
(b)Figure 2. Time-kill curves of M. xenopi ATCC 19250 with (A) clarithromycin and (B) moxifloxacin. Antibiotic concentrations are indicated by different symbols.
0 3 6 9 12 15 18 21 24 27 30 33 36 39 420123456789
10
Growth Control0.062× MIC0.125× MIC0.25× MIC0.5× MIC1× MIC2× MIC4× MIC8× MIC16× MIC32× MIC
DetectionLimit
Time (days)
Log
CFU
/mL
0 3 6 9 12 15 18 21 24 27 30 33 36 39 420123456789
10
Growth Control0.062× MIC0.125× MIC0.25× MIC0.5× MIC1× MIC2× MIC4× MIC8× MIC16× MIC32× MIC
DetectionLimit
Time (days)
Log
CFU
/mL
(a)
(b)B moxifloxacinA clarithromycin

54
Chapter 3
In the set of experiments with M. xenopi, killing took a lot of time; it was maximal after 42 days, but we found no significant difference among the effect obtained with clarithromycin or moxifloxacin. Our observation is concordant with the characterization of the activity of anti-mycobacterial drugs against M. xenopi in the mouse model performed by Andréjak et al,5 who found no differences between clarithromycin- and moxifloxacin containing regimens. Rifampicin/ethambutol synergy in vitro is absent in M. xenopi isolates,17 which points out the role of clarithromycin during therapy. That, along with the underlying diseases in M. xenopi patients, may explain the poor results with treatment.18 The absence of correlation between the in vitro activity of clarithromycin and moxifloxacin and the in vivo outcomes of treatment with regimens based on these antimicrobials, as a result of these underlying diseases, could be hampering our understanding of M. xenopi and how to deal with it.
Hill’s slopes were not significantly different for amikacin and clarithromycin against M. avium, but the values of Emax were different. This is a confirmation of a previous observation made in a set of experiments that we conducted with rapidly growing mycobacteria,12 where the total effect observed was mainly determined by the Emax and not by the Hill’s slope. Hill’s slopes were also not significantly different for clarithromycin and moxifloxacin against M. xenopi, although neither were their Emax values. The Hill’s slope helps to infer the concentration dependency of an antibiotic. In this study both antibiotics showed similar steeper slope values for both Mycobacterium species; therefore the difference in concentration between the maximum and minimal effect was small. The overall effect is thus more of a time-dependent nature than concentration dependent.
We acknowledge several limitations of this study. Although 42 °C is the optimal growing temperature for M. xenopi, we performed our experiments at 37 °C. We preferred the physiological temperature of the human host; however, we do not know how this could have affected the results. There is a discrepancy between the in vitro MIC and the concentration
Table 2. Parameter estimates, with 95% CI, derived from inhibitory Emax
* Pooled value for the two antibiotics evaluated by species.
Strain Antibiotic Time Emax (h-1)
95 % CI EC50 (mg/l)
95 % CI Hill’s slope (ɣ)*
95 % CI R2
M. avium IWGMT49
Clarithromycin 24-120 hours
0.0109 0.0089-0.0130
0.149 0.010-0.222
1.838 1.265-2.410
0.98
Amikacin 0.0180 0.0154-0.0205
1.427 1.160-1.755
0.99
M. xenopi ATCC19250
Clarithromycin 3-42 days
0.1533 0.1356-0.1711
0.191 0.149-0.245
2.543 1.484-3.602
0.98
Moxifloxacin 0.1385 0.1232-0.1539
0.076 0.059-0.099
0.96

Time-kill kinetics of slowly growing mycobacteria
55
Cha
pter
3
that leads to 2 log kill in the time-kill curves (99% kill) at the time for MIC readout. These discrepancies result in part from technical aspects, including the fact that MICs were determined in CAMHB, while time-kill assays were conducted in Middlebrook 7H9, which has a lower pH that affects clarithromycin activity.7 Bottles used in the time-kill experiments are shaken constantly and they have a big head space and filtered vents, which allows a stable oxygen supply.
Our experiments were designed to evaluate the role of each individual antibiotic in the therapy; however, it is necessary to evaluate combined regimens as they are used in clinical practice. Dynamic models for pharmacokinetic and pharmacodynamic determinations can provide more representative data for the in vivo situation; the next step is to continue with the evaluation of current regimens with those models to help in the design of new and hopefully more effective therapies.
In conclusion, the killing effect of amikacin and clarithromycin on M. avium subspecies hominissuis is low, although amikacin activity was higher than that of clarithromycin, supporting its role in a combined therapy. Time-kill assays with M. xenopi showed no differences between clarithromycin and moxifloxacin, which did have high killing rates yet required a lot of time. Clarithromycin and moxifloxacin may have identical activity within treatment regimens for M. xenopi disease. Future studies of in vitro and in vivo pharmacokinetic/pharmacodynamic interactions are needed to be able to improve the current regimens against these two important SGM in pulmonary disease.
0.000
10.0
1 110
010
000
-0.06
-0.04
-0.02
0.00
0.02
0.04ClarithromycinAmikacin
AMK C
max
CLA C
max
Concentration (mg/L)
Obs
erve
d ki
ll r
ate
h-1
0.000
001
0.000
10.0
1 110
0-0.10
-0.05
0.00
0.05
0.10
0.15
0.20
ClarithromycinMoxifloxacin
Concentration (mg/L)
Obs
erve
d ki
ll r
ate
h-1
(a)
(b)Figure 3. Best fitted sigmoid curves obtained from the Emax model of (A) M. avium IWGMT49 exposed to clarithromycin and amikacin, between 24 and 120 h, and (B) M. xenopi ATCC 19250 exposed to clarithromycin and moxifloxacin, between 3 and 42 days. A different y-axis scale is used for (A) and (B). Serum Cmax for MAC-infected patients when treated with amikacin (AMK) or clarithromycin (CLR) is included.13
0.000
10.0
1 110
010
000
-0.06
-0.04
-0.02
0.00
0.02
0.04ClarithromycinAmikacin
AMK C
max
CLA C
max
Concentration (mg/L)
Obs
erve
d ki
ll r
ate
h-1
0.000
001
0.000
10.0
1 110
0-0.10
-0.05
0.00
0.05
0.10
0.15
0.20
ClarithromycinMoxifloxacin
Concentration (mg/L)
Obs
erve
d ki
ll r
ate
h-1
(a)
(b)A B
Concentration (mg/l) Concentration (mg/l)

56
Chapter 3
Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier J, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K, ATS Mycobacterial Diseases Subcommittee, American Thoracic Society, Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367–416.Hoefsloot W, van Ingen J, Andrejak C, Angeby K, Bauriaud R, Bemer P, Beylis N, Boeree MJ, Cacho J, Chihota V, Chimara E, Churchyard G, Cias R, Daza R, Daley CL, Dekhuijzen PN, Domingo D, Drobniewski F, Esteban J, Fauville-Dufaux M, Folkvardsen DB, Gibbons N, Gómez-Mampaso E, Gonzalez R, Hoffmann H, Hsueh PR, Indra A, Jagielski T, Jamieson F, Jankovic M, Jong E, Keane J, Koh WJ, Lange B, Leao S, Macedo R, Mannsåker T, Marras TK, Maugein J, Milburn HJ, Mlinkó T, Morcillo N, Morimoto K, Papaventsis D, Palenque E, Paez-Peña M, Piersimoni C, Polanová M, Rastogi N, Richter E, Ruiz-Serrano MJ, Silva A, da Silva MP, Simsek H, van Soolingen D, Szabó N, Thomson R, Tórtola Fernandez T, Tortoli E, Totten SE, Tyrrell G, Vasankari T, Villar M, Walkiewicz R, Winthrop KL, Wagner D; Nontuberculous Mycobacteria Network European Trials Group. The geographic diversity of non-tuberculous mycobacteria isolated from pulmonary samples: an NTM-NET collaborative study. Eur Resp J. 2013;42:1604–13.Andréjak C, Lescure FX, Pukenyte E, Douadi Y, Yazdanpanah Y, Laurans G, Schmit JL, Jounieaux V; Xenopi Group. Mycobacterium xenopi pulmonary infections: a multicentric retrospective study of 136 cases in north-east France. Thorax. 2009;64:291–6.van Ingen J, Ferro BE, Hoefsloot W, Boeree MJ, van Soolingen D. Drug treatment of pulmonary antituberculous mycobacterial disease in HIV-negative patients: the evidence. Expert Rev Anti Infect Ther. 2013;11:1065–77. Andréjak C, Almeida DV, Tyagi S, Converse PJ, Ammerman NC, Grosset JH. Improving existing tools for Mycobacterium xenopi treatment assessment of drug combinations and characterization of mouse models of infection and chemotherapy. J Antimicrob Chemother. 2013;68:659–65.Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: Twenty-first Informational Supplement M100-S21. CLSI, Wayne, PA, USA, 2011. Clinical and Laboratory Standards Institute. Susceptibility Testing of Mycobacteria, Nocardia, and Other Aerobic Actinomycetes—Second Edition: Approved Standard M24-A2. CLSI, Wayne, PA, USA, 2011. Mouton JW, Dudley MN, Cars O, Derendorf H, Drusano GL. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs. Int J Antimicrob Agents. 2002;19:355–8. de Steenwinkel JE, de Knegt GJ, ten Kate MT, van Belkum A, Verbrugh HA, Kremer K, van Soolingen D, Bakker-Woudenberg IA.. Time-kill kinetics of anti-tuberculosis drugs, and emergence of resistance, in relation to metabolic activity of Mycobacterium tuberculosis. J Antimicrob Chemother. 2010;65:2582–9. Donald PR, Sirgel FA, Venter A, Smit E, Parkin DP, Van de Wal BW, Mitchison DA. The early bactericidal activity of amikacin in pulmonary tuberculosis. Int J Tuberc Lung Dis. 2001;5:533–8.Bakker-Woudenberg IA, van Vianen W, van Soolingen D, Verbrugh HA, van Agtmael MA. Antimycobacterial agents differ with respect to their bacteriostatic versus bactericidal activities in relation to time of exposure, mycobacterial growth phase, and their use in combination. Antimicrob Agents Chemother. 2005;49:2387–98.Ferro BE, van Ingen J, Wattenberg M, van Soolingen D, Mouton JW. Time-kill kinetics of antibiotics active against rapidly growing mycobacteria. J Antimicrob Chemother. 2015;70:811–7.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
References

Time-kill kinetics of slowly growing mycobacteria
57
Cha
pter
3
van Ingen J, Egelund EF, Levin A, Totten SE, Boeree MJ, Mouton JW, Aarnoutse RE, Heifets LB, Peloquin CA, Daley CL. The pharmacokinetics and pharmacodynamics of pulmonary Mycobacterium avium complex disease treatment. Am J Respir Crit Care Med. 2012;186:559–65. Davis KK, Kao PN, Jacobs SS, Ruoss SJ. Aerosolized amikacin for treatment of pulmonary Mycobacterium avium infections: an observational case series. BMC Pulm Med. 2007;23:2.Olivier KN, Shaw PA, Glaser TS, Bhattacharyya D, Fleshner M, Brewer CC, Zalewski CK, Folio LR, Siegelman JR, Shallom S, Park IK, Sampaio EP, Zelazny AM, Holland SM, Prevots DR. Inhaled amikacin for treatment of refractory pulmonary nontuberculous mycobacterial disease. Ann Am Thorac Soc. 2014;11:30–5.van Ingen J, Boeree MJ, van Soolingen D, Mouton JW. Resistance mechanisms and drug susceptibility testing of nontuberculous mycobacteria. Drug Resist Updat. 2012;15:149–61.van Ingen J, Hoefsloot W, Mouton JW, Boeree MJ, van Soolingen D. Synergistic activity of rifampicin and ethambutol against slow-growing nontuberculous mycobacteria is currently of questionable clinical significance. Int J Antimicrob Agents. 2013;42:80–2. van Ingen J, Boeree MJ, de Lange WC, Hoefsloot W, Bendien SA, Magis-Escurra C, Dekhuijzen R, van Soolingen D. Mycobacterium xenopi clinical relevance and determinants, the Netherlands. Emerg Infec Dis. 2008;14:385–9.
13.
14.
15.
16.
17.
18.


59
Chapter 4
Clofazimine prevents the regrowth of mycobacterium abscessus and mycobacterium avium type strains exposed to amikacin and clarithromycin
Beatriz E. FerroJoseph MeletiadisMelanie WattenbergArjan de JongDick van SoolingenJohan W. MoutonJakko van Ingen
Antimicrob Agents Chemother. 2015;60:1097-105.

60
Chapter 4
Summary
Multidrug therapy is a standard practice when treating infections by nontuberculous mycobacteria (NTM), but few treatment options exist. We conducted this study to define the drug-drug interaction between clofazimine and both amikacin and clarithromycin and its contribution to NTM treatment. Mycobacterium abscessus and Mycobacterium avium type strains were used. Time-kill assays for clofazimine alone and combined with amikacin or clarithromycin were performed at concentrations of 0.25× to 2× MIC. Pharmacodynamic interactions were assessed by response surface model of Bliss independence (RSBI) and isobolographic analysis of Loewe additivity (ISLA), calculating the percentage of statistically significant Bliss interactions and interaction indices (I), respectively. Monte Carlo simulations with predicted human lung concentrations were used to calculate target attainment rates for combination and monotherapy regimens. Clofazimine alone was bacteriostatic for both NTM. Clofazimine-amikacin was synergistic against M. abscessus (I = 0.41; 95% confidence interval [CI], 0.29 to 0.55) and M. avium (I = 0.027; 95% CI, 0.007 to 0.048). Based on RSBI analysis, synergistic interactions of 28.4 to 29.0% and 23.2 to 56.7% were observed at 1× to 2× MIC and 0.25× to 2× MIC for M. abscessus and M. avium, respectively. Clofazimine-clarithromycin was also synergistic against M. abscessus (I = 0.53; 95% CI, 0.35 to 0.72) and M. avium (I = 0.16; 95% CI, 0.04 to 0.35), RSBI analysis showed 23.5% and 23.3 to 53.3% at 2× MIC and 0.25× to 0.5× MIC for M. abscessus and M. avium, respectively. Clofazimine prevented the regrowth observed with amikacin or clarithromycin alone. Target attainment rates of combination regimens were >60% higher than those of monotherapy regimens for M. abscessus and M. avium. The combination of clofazimine with amikacin or clarithromycin was synergistic in vitro. This suggests a potential role for clofazimine in treatment regimens that warrants further evaluation.

Clofazimine prevents regrowth and resistance
61
Cha
pter
4
Introduction
The treatment of diseases caused by nontuberculous mycobacteria (NTM) is a challenge, partly due to the natural resistance of NTM to most antibiotics. Treatment outcomes are generally poor, and current treatment recommendations have a very limited evidence base, since very few clinical trials have been performed.1,2
Multidrug therapy is a standard practice when treating mycobacterial infections. However, the pharmacodynamic (PD) interactions among the combined drugs are largely unknown. Understanding these interactions will help to identify synergistic combinations with increased antibacterial killing, which ultimately can result in a better treatment outcome.
One of the promising combinations is amikacin and clofazimine, given the key role of amikacin in the treatment of NTM infections2,3 and the unique characteristics of clofazimine, like its prolonged half-life, its preferential accumulation inside macrophages,4 and the recently found bactericidal activity only after 2 weeks of treatment in the mouse model of tuberculosis.5 Clarithromycin, on the other hand, has substantial in vitro and clinical activity against Mycobacterium avium complex (MAC), and it has been long considered the cornerstone for Mycobacterium abscessus treatment.3 Hence, the examination of its interaction with clofazimine is interesting.
Previous studies showed in vitro synergy between clofazimine and amikacin against both rapidly and slowly growing NTM.1,6 The combination clarithromycin-clofazimine also showed synergy against MAC strains in checkerboard evaluation.7 These checkerboard titrations offer no information on the mechanism of synergistic activity, the exact killing activity of these combinations, or its concentration dependence. We therefore investigated the pharmacodynamic interactions between clofazimine and amikacin, and clofazimine and clarithromycin, against two key NTM species, using time-kill assays analyzed with two pharmacodynamic drug interaction models: the response surface model of Bliss independence (RSBI) and isobolographic analysis of Loewe additivity (ISLA).8,9
Materials and Methods
Bacterial strains, media, and antibioticsMycobacterium abscessus subsp. abscessus CIP 104536 (Collection of Institute Pasteur) and Mycobacterium avium subsp. hominissuis IWGMT49 (International Working Group on Mycobacterial Taxonomy) type strains were used. Stocks of each strain were preserved at -80 °Cin Trypticase soy broth with 40% glycerol and were thawed for each assay. Pure powders of amikacin, clarithromycin, and clofazimine obtained from Sigma-Aldrich were dissolved in water, methanol, and dimethyl sulfoxide (DMSO), respectively.

62
Chapter 4
Susceptibility testing MICs were determined at the beginning and at the end of the experiments, following CLSI recommendations,10 by broth microdilution in cation-adjusted Mueller-Hinton broth (CAMHB); commercially available panels were used for amikacin and clarithromycin (RAPMYCO Sensititre, SLOWMYCO Sensititre; Trek Diagnostics/ Thermo Fisher), while for clofazimine, manual broth microdilution was performed in Middlebrook 7H9 broth (BD Biosciences). The unexposed M. abscessus type strain was used as a quality control during all MIC determinations.
Mutation analysisMutation analysis of rrs and rrl was performed to detect mutations associated with amikacin and clarithromycin resistance, respectively, in mycobacteria exposed to the two antibiotics in the drug-drug interaction experiments. Briefly, mycobacterial suspensions were heat inactivated at 95 °C for 30 min, and then DNA was extracted using MagNA Pure LC (Roche Life Science). Relevant fragments of rrs and rrl were amplified using previously described primers and PCR conditions.11,12 Sequence analyses were performed to detect mutations at codon 1408 in rrs and codons 2058 and 2059 in rrl.
Time-kill assays for single drugsThe inoculum consisted of mycobacteria in the early logarithmic phase of growth. Individual bottles of 20 ml of Middlebrook 7H9 with oleic acid-bovine albumin-dextrose-catalase growth supplement (BD Biosciences) and 0.05% Tween 80, containing increasing concentrations (from 0.062× to 8× MIC for M. abscessus and 0.062× to 32× MIC for M. avium) of clofazimine, were cultured with the inoculum (~105 to 106 cfu/ml) at 30 °C for M. abscessus and 37 °C for M. avium, under shaking conditions at 100 rpm (GFL); ventilation through a bacterial filter (FP 30/0.2 Ca/S; Whatman GmbH) was incorporated. A drug-free and inoculum-free bottle with medium served as the growth and sterility controls, respectively. At defined time points (3, 6, 12, 24, 36, 48, 72, 96, and 120 h for M. abscessus and 12, 24, 36, 48, 72, 96, 120, 144, 168, and 240 h for M. avium), 10 µl from undiluted samples and samples serially diluted 10-fold in normal saline were plated in triplicate on Middlebrook 7H11 agar plates (BD Biosciences). Plates were incubated for 3 to 5 days at 30 °C for M. abscessus, or 7 to 9 days at 37 °C for M. avium, and the cfu per milliliter were calculated (lower limit of detection, 33.3 cfu/ml).
Time-kill assays for drug-drug interactionFollowing the same protocol as described above, time-kill assays for M. abscessus and M. avium were performed with four concentrations (from 0.25× to 2× MIC) of each antibiotic alone and in paired combination with clofazimine. MICs for amikacin, clarithromycin, and clofazimine were checked by duplicate at the final sampling time, when colonies were present.

Clofazimine prevents regrowth and resistance
63
Cha
pter
4
Pharmacodynamic drug interaction analysisIn order to assess the nature and the magnitude of the in vitro interactions between clofazimine and amikacin or clarithromycin against NTM, the data obtained with time-kill assays were analyzed using the response surface analysis of Bliss independence (RSBI) and the isobolographic analysis of Loewe additivity (ISLA) as described previously.8,9 The RSBI was used to describe the kinetics of pharmacodynamic interactions at different time points, whereas the ISLA was used to describe the full concentration-effect relationships of the drugs alone and in combination at the end of the experiment. For that purpose, the percentage of bacterial load at each drug concentration and combination was calculated by dividing the log10 cfu per milliliter with the log10 cfu per milliliter of growth control at each sampling time throughout the experiment. Bliss synergy and antagonism was concluded if the observed bacterial load was statistically significantly (P< 0.05) lower or higher than the expected bacterial load derived from Bliss independence; otherwise, Bliss independence was deemed.8,9
In order to assess the pharmacodynamic interactions at the entire range of drug concentrations with the ISLA, concentration-effect curves were constructed using the reduction in log10 cfu per milliliter compared to growth control at the end of each experiment and analyzed with sigmoidal maximum-effect (Emax) model after nonlinear regression analysis with global fitting, i.e., shared Emax and minimum effect (Emin) (Graphpad Prism 5.03; Graphpad Inc.). The interaction index (I) was calculated for the stasis effect, i.e., 0-log10 cfu/ml reduction compared to the initial inoculum, as the ratio ECmix/ECadd, where ECmix is the effective concentration of the mixture and ECadd is the effective concentration of a theoretical additive combination, as described previously.8,9 All analyses were made using multiples of the MIC. Synergy or antagonism was concluded when the I was statistically significantly (P< 0.05) lower or higher than 1; otherwise, additivity was claimed.
Monte Carlo analysisIn order to bridge the findings to concentrations in humans, Monte Carlo analysis was performed to predict how many patients treated with standard dosing regimens will attain free lung concentrations (Cmax) associated with stasis for strains of both mycobacterial species with MICs usually observed for these species, i.e., 4 to 128 mg/l for amikacin, 0.125 to 4 mg/l for clarithromycin, and 0.03 to 4 mg/l for clofazimine. The mean ± SD target serum Cmax values were 55.3 ± 16.88 mg/l for amikacin, 2.31 ± 1.89 mg/l for clarithromycin, and 0.43 ± 0.19 mg/l for clofazimine.13 In order to determine the free lung concentrations, the epithelial lining fluid/serum concentration ratios of 0.5 for amikacin14 and 8 for clarithromycin15 were used, whereas for clofazimine, the total lung/serum concentration ratio of 30 (found in mice) was used after taking into account the 85% protein binding.5 Monte Carlo simulation analysis was performed using the normal random number generator function of Excel spreadsheet (MS Office 2007) for 10,000 patients.

64
Chapter 4
Results
SusceptibilityMICs for clofazimine, clarithromycin, and amikacin were 0.5, 4, and 32 mg/l for M. abscessus CIP 104536 and 0.06, 4, and 8 mg/l for M. avium IWGMT49.
Time-kill assays for single-drug exposureGrowth and killing curves of M. abscessus and M. avium exposed to different concentrations of clofazimine are shown in Figure 1. For M. abscessus (Figure 1A), the effect of clofazimine mainly consisted of inhibition of growth that was concentration dependent. Regrowth appeared after the initial inhibition. Because of solubility issues of clofazimine, concentrations higher than 8× MIC could not be evaluated for M. abscessus. For M. avium (Figure 1B), the clofazimine effect was in general stronger than that observed for M. abscessus. Again, the effect was a concentration-dependent inhibition of growth. The highest concentration, 32× MIC, did show some killing effect, particularly from 96 to 240 h. Single exposures of amikacin and clarithromycin were previously described for both M. abscessus16 and M. avium.17
Time-kill assays for combinationsM. abscessusThe time-kill curves of the drug combinations clofazimine-amikacin and clofazimine-clarithromycin at the different concentrations are shown in Figure 2 and 3. The effect of clofazimine was consistent with that in the single-drug assay at all concentrations (Figure 1).
Amikacin inhibited growth at concentrations 0.25× and 0.5× MIC and showed discrete killing at 1× and 2× MIC, although regrowth appeared after 96 h at both concentrations. The combination of clofazimine-amikacin showed an inhibitory effect at 0.25× and 0.5× MIC and discrete killing at 1× and 2× MIC but did not reveal the regrowth observed with amikacin alone (Figure 2A to D). The MICs of the two antibiotics for the colonies exposed
Figure 1. Time-kill curves of M. abscessus CIP 104536 (A) and M. avium IWGMT49 (B) exposed to different concentrations of clofazimine. Antibiotic concentrations are indicated by different symbols. Error bars represent SDs from the replicates.
0 12 24 36 48 60 72 84 96 108
120
0123456789
10Growth Control0.062 × MIC0.125 × MIC
8 × MIC
0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MICDetection
Limit
Time (h)
Log
CFU
/ml
0 24 48 72 96 120
144
168
192
216
240
0123456789
10Growth Control0.062 × MIC0.125 × MIC0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
DetectionLimit
Time (h)
Log
CFU
/ml
a.
b.
0 12 24 36 48 60 72 84 96 108
120
0123456789
10Growth Control0.062 × MIC0.125 × MIC
8 × MIC
0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MICDetection
Limit
Time (h)
Log
CFU
/ml
0 24 48 72 96 120
144
168
192
216
240
0123456789
10Growth Control0.062 × MIC0.125 × MIC0.25 × MIC0.5 × MIC1 × MIC2 × MIC4 × MIC8 × MIC16 × MIC32 × MIC
DetectionLimit
Time (h)
Log
CFU
/ml
a.
b.BA
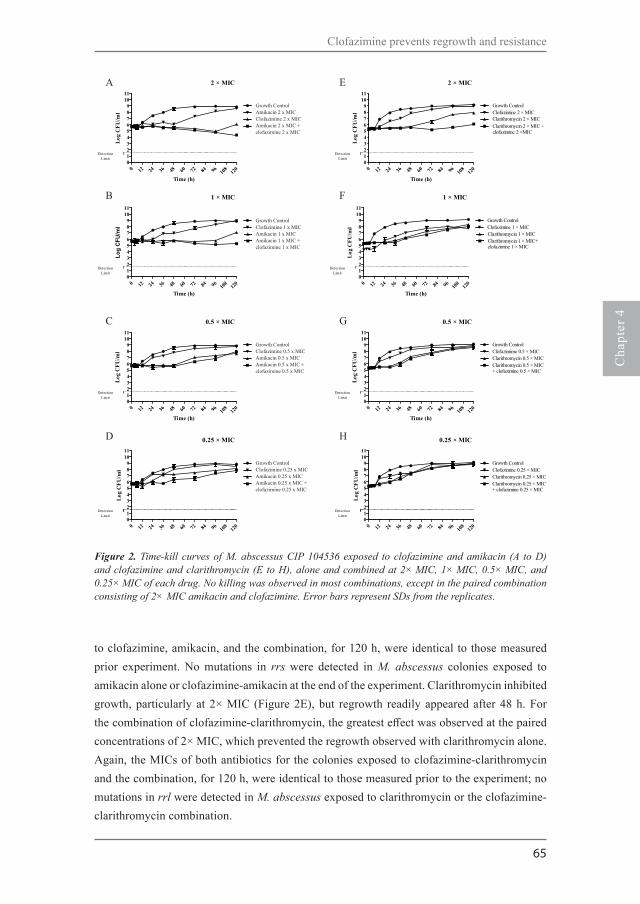
Clofazimine prevents regrowth and resistance
65
Cha
pter
4
to clofazimine, amikacin, and the combination, for 120 h, were identical to those measured prior experiment. No mutations in rrs were detected in M. abscessus colonies exposed to amikacin alone or clofazimine-amikacin at the end of the experiment. Clarithromycin inhibited growth, particularly at 2× MIC (Figure 2E), but regrowth readily appeared after 48 h. For the combination of clofazimine-clarithromycin, the greatest effect was observed at the paired concentrations of 2× MIC, which prevented the regrowth observed with clarithromycin alone. Again, the MICs of both antibiotics for the colonies exposed to clofazimine-clarithromycin and the combination, for 120 h, were identical to those measured prior to the experiment; no mutations in rrl were detected in M. abscessus exposed to clarithromycin or the clofazimine-clarithromycin combination.
2 × MIC
0 12 24 36 48 60 72 84 96 108
120
0123456789
1011
DetectionLimit
Growth Control
Clarithromycin 2 × MICClofazimine 2 × MIC
Clarithromycin 2 × MIC +clofazimine 2 ×MIC
Time (h)
Log
CFU
/ml
2 × MIC
0 12 24 36 48 60 72 84 96 108
120
0123456789
1011
DetectionLimit
Growth Control
Clarithromycin 2 × MICClofazimine 2 × MIC
Clarithromycin 2 × MIC +clofazimine 2 ×MIC
Time (h)
Log
CFU
/ml
1 × MIC
0 12 24 36 48 60 72 84 96 108
120
0123456789
1011
DetectionLimit
Growth Control
Clarithromycin 1 × MICClofazimine 1 × MIC
Clarithromycin 1 × MIC+clofazimine 1 × MIC
Time (h)
Log
CFU/
ml
1 × MIC
0 12 24 36 48 60 72 84 96 108
120
0123456789
1011
DetectionLimit
Growth Control
Clarithromycin 1 × MICClofazimine 1 × MIC
Clarithromycin 1 × MIC+clofazimine 1 × MIC
Time (h)
Log
CFU
/ml
0.5 × MIC
0 12 24 36 48 60 72 84 96 108
120
0123456789
1011
DetectionLimit
Growth ControlClofazimime 0.5 × MICClarithromycin 0.5 × MICClarithromycin 0.5 × MIC+ clofazimine 0.5 × MIC
Time (h)
Log
CFU
/ml
0.5 × MIC
0 12 24 36 48 60 72 84 96 108
120
0123456789
1011
DetectionLimit
Growth ControlClofazimime 0.5 × MICClarithromycin 0.5 × MICClarithromycin 0.5 × MIC+ clofazimine 0.5 × MIC
Time (h)
Log
CFU
/ml
0.25 × MIC
0 12 24 36 48 60 72 84 96 108
120
0123456789
1011
DetectionLimit
Growth ControlClofazimine 0.25 × MICClarithromycin 0.25 × MICClarithromycin 0.25 × MIC+ clofazimine 0.25 × MIC
Time (h)
Log
CFU
/ml
0.25 × MIC
0 12 24 36 48 60 72 84 96 108
120
0123456789
1011
DetectionLimit
Growth ControlClofazimine 0.25 × MICClarithromycin 0.25 × MICClarithromycin 0.25 × MIC+ clofazimine 0.25 × MIC
Time (h)
Log
CFU
/ml
a
b
c
d
e
f
g
h
Figure 2. Time-kill curves of M. abscessus CIP 104536 exposed to clofazimine and amikacin (A to D) and clofazimine and clarithromycin (E to H), alone and combined at 2× MIC, 1× MIC, 0.5× MIC, and 0.25× MIC of each drug. No killing was observed in most combinations, except in the paired combination consisting of 2× MIC amikacin and clofazimine. Error bars represent SDs from the replicates.
A
Growth ControlAmikacin 2 x MICClofazimine 2 x MICAmikacin 2 x MIC +clofazimine 2 x MIC
Growth ControlClofazimine 1 x MICAmikacin 1 x MICAmikacin 1 x MIC +clofazimine 1 x MIC
Growth ControlClofazimine 0.5 x MICAmikacin 0.5 x MICAmikacin 0.5 x MIC +clofazimine 0.5 x MIC
Growth ControlClofazimine 0.25 x MICAmikacin 0.25 x MICAmikacin 0.25 x MIC +clofazimine 0.25 x MIC
B
C
D
E
F
G
H

66
Chapter 4
M. aviumWhen M. avium IWGMT49 was exposed to clofazimine and amikacin, alone and in combination, in the same experiment (Figure 3A to D) the killing effect was greater than that observed for M. abscessus. Clofazimine only inhibited the growth; amikacin effect was stronger and faster, especially at 0.5×, 1×, and 2× MIC, but regrowth appeared after 96 to 120 h. The combination of clofazimine-amikacin, at all concentrations, prevented regrowth, and its effect was maximal at the end of the experiment. The MIC for the mycobacteria exposed to clofazimine after 240 h remained the same, whereas amikacin-exposed bacteria showed MICs higher than the initial MICs (>64 mg/l, versus 8 mg/l). This increase in MIC was not observed for bacteria exposed to the combination clofazimine-amikacin, in which the amikacin MIC did not change. Colonies exposed to amikacin alone, at any concentration, showed the A1408G mutation in the rrs product, but this mutation was not found in colonies exposed to any of the clofazimine-amikacin combinations.
For the set of experiments with the combination of clofazimine- clarithromycin (Figure 3E to H), clarithromycin killing effect was observed for all concentrations; however, after 144 h, regrowth was observed, especially at 0.25× and 0.5× MIC. The combination of clofazimine-clarithromycin showed sustained killing at all concentrations, and no regrowth was observed until 240 h. The MIC of clofazimine for the mycobacteria exposed to this antibiotic for 240 h remain identical to those measured prior to the experiment, but the clarithromycin MIC was higher (>64 mg/l, versus 4 mg/l) in bacteria exposed to clarithromycin alone. Again, this increase was not observed in colonies exposed to the combination of clofazimine-clarithromycin. The A2058G mutation in the rrl product was found only in colonies exposed to 0.25× and 0.5× MIC clarithromycin alone, not in colonies exposed to any of the clofazimine-clarithromycin combinations.
Interaction analysisThe RSBI analysis of the combination of clofazimine-amikacin against M. abscessus showed independent interactions within the first 12 h which converted to antagonistic interactions at 24 to 48 h and ultimately to synergistic interaction of 29.0% and 28.4% at the end of the experiment for 2× and 1× MIC, respectively (Figure 4A).
Isobolographic analysis of the full concentration-effect relationship of the same combination showed synergy at the end of the experiment at concentrations of ≥ 1× MIC with an I of 0.41 and a 95% CI of 0.29 to 0.55 (Figure 5A); stasis was achieved at 4× and 8× MIC of amikacin and clofazimine alone; for the combination, stasis was found at 1× MIC of both drugs.
The RSBI analysis of the combination clofazimine-clarithromycin against M. abscessus also showed independence in the first 12 h which converted to antagonistic interactions at high concentrations, but ultimately synergistic interactions of 23.5% were observed only at 2× MIC

Clofazimine prevents regrowth and resistance
67
Cha
pter
4
(Figure 4B). Similarly, ISLA of the same combination showed synergistic interaction reaching stasis at 2× MIC when the drugs were combined. For each drug alone, the endpoint was reached at 8× MIC (I = 0.53; 95% CI, 0.35 to 0.72) (Figure 5B). For the combination clofazimine-amikacin against M. avium, the RSBI analysis showed independence up to 48 h, then antagonism up to 120 h, and synergy at the end of the experiment, of 23.2 to 56.7% at all concentrations (Figure 4C). Strong synergy was found with the ISLA (I = 0.027; 95% CI, 0.007 to 0.048) since stasis was found at 0.5× MIC of the drugs in combination; the same endpoint was reached at 8× MIC and 32× MIC for clofazimine and amikacin alone (Figure 5C). The same profile of interactions was found
2 × MIC
0 24 48 72 96 120
144
168
192
216
240
0123456789
10
DetectionLimit
Growth Control
Clarithromycin 2 × MICClofazimine 2 × MIC
Clarithromycin 2 × MIC +clofazimine 2 ×MIC
Time (h)
Log
CFU
/ml
2 × MIC
0 24 48 72 96 120
144
168
192
216
240
0123456789
10
DetectionLimit
Growth Control
Clarithromycin 2 × MICClofazimine 2 × MIC
Clarithromycin 2 × MIC +clofazimine 2 ×MIC
Time (h)
Log
CFU
/ml
1 × MIC
0 24 48 72 96 120
144
168
192
216
240
0123456789
10
DetectionLimit
Growth Control
Clarithromycin 1 × MICClofazimine 1 × MIC
Clarithromycin 1 × MIC+clofazimine 1 × MIC
Time (h)
Log
CFU/
ml
1 × MIC
0 24 48 72 96 120
144
168
192
216
240
0123456789
10
DetectionLimit
Growth Control
Clarithromycin 1 × MICClofazimine 1 × MIC
Clarithromycin 1 × MIC+clofazimine 1 × MIC
Time (h)
Log
CFU
/ml
0.5 × MIC
0 24 48 72 96 120
144
168
192
216
240
0123456789
10
DetectionLimit
Growth ControlClofazimime 0.5 × MICClarithromycin 0.5 × MICClarithromycin 0.5 × MIC+ clofazimine 0.5 × MIC
Time (h)
Log
CFU
/ml
0.5 × MIC
0 24 48 72 96 120
144
168
192
216
240
0123456789
10
DetectionLimit
Growth ControlClofazimime 0.5 × MICClarithromycin 0.5 × MICClarithromycin 0.5 × MIC+ clofazimine 0.5 × MIC
Time (h)
Log
CFU
/ml
0.25 × MIC
0 24 48 72 96 120
144
168
192
216
240
0123456789
10
DetectionLimit
Growth ControlClofazimine 0.25 × MICClarithromycin 0.25 × MICClarithromycin 0.25 × MIC+ clofazimine 0.25 × MIC
Time (h)
Log
CFU
/ml
0.25 × MIC
0 24 48 72 96 120
144
168
192
216
240
0123456789
10
DetectionLimit
Growth ControlClofazimine 0.25 × MICClarithromycin 0.25 × MICClarithromycin 0.25 × MIC+ clofazimine 0.25 × MIC
Time (h)
Log
CFU
/ml
a
b
c
d
e
f
g
h
Figure 3. Time-kill curves of M. avium IWGMT49 exposed to clofazimine and amikacin (A to D) and clofazimine and clarithromycin (E to H), alone and combined at 2× MIC, 1× MIC, 0.5× MIC, and 0.25× MIC of each drug. Modest killing was observed with most combinations, except with the paired combination consisting of 2× MIC of amikacin and clofazimine. Error bars represent SDs from the replicates.
A
Growth ControlClofazimine 2 x MICAmikacin 2 x MICAmikacin 2 x MIC +clofazimine 2 x MIC
Growth ControlClofazimine 1 x MICAmikacin 1 x MICAmikacin 1 x MIC +clofazimine 1 x MIC
Growth ControlClofazimine 0.5 x MICAmikacin 0.5 x MICAmikacin 0.5 x MIC +clofazimine 0.5 x MIC
Growth ControlClofazimine 0.25 x MICAmikacin 0.25 x MICAmikacin 0.25 x MIC +clofazimine 0.25 x MIC
B
C
D
E
F
G
H

68
Chapter 4
for the combination clofazimine-clarithromycin; with RSBI analysis, independence was observed up to 48 h, then antagonism up to 120 h, and ultimately synergy. However, the synergistic interactions were found at concentrations of 0.25× and 0.5× MIC (53.3% and 23.3%, respectively) (Figure 4D). This was also confirmed with the ISLA (I = 0.16; 95% CI, 0.04 to 0.35), with stasis observed at combinations with < 0.25× MIC of each drug, whereas the same endpoint was reached at 0.5× MIC of clarithromycin and 8× MIC of clofazimine alone (Figure 5D). Interestingly, at combinations with > 1× MIC of each drug, the effect was similar to the effect of clarithromycin alone, indicating no interaction as the RSBI analysis showed at the end of the experiment.
Monte Carlo simulationsThe percentages of patients that will attain lung concentrations associated with stasis of both M. abscessus and M. avium are shown in Figure 6. For M. abscessus, target attainment rates with clofazimine-amikacin and clofazimine-clarithromycin combinations were higher than with monotherapy regimens, particularly for isolates with amikacin/clofazimine MICs of 16/0.25 mg/l and clarithromycin/clofazimine MICs of 2/0.25 mg/l, respectively (>60% difference between combination and monotherapy regimens). For M. avium, a >60% difference was found for the clofazimine-amikacin combination for isolates with amikacin/clofazimine MICs 32 to 128/0.25 to 1 mg/l and for the clofazimine-clarithromycin combination for isolates
M. abscessus CLO+AMK
0 12 24 36 48 60 72 84 96 108 120-30-20-10
0102030405060
2 x MIC1 x MIC0.5 x MIC0.25 x MIC
Synergy
Antagonism
Time (hours)
Bli
ss in
tera
ctio
ns
M. abscessus CLO+CLA
0 12 24 36 48 60 72 84 96 108 120-30-20-10
0102030405060
2 x MIC1 x MIC0.5 x MIC0.25 x MIC
Synergy
Antagonism
Time (hours)
Bli
ss in
tera
ctio
ns
M. avium CLO+AMK
0 24 48 72 96 120 144 168 192 216 240-30-20-10
0102030405060
2 x MIC1 x MIC0.5 x MIC0.25 x MIC
Synergy
Antagonism
Time (hours)
Bli
ss in
tera
ctio
ns
M. avium CLO+CLA
0 24 48 72 96 120 144 168 192 216 240-30-20-10
0102030405060
2 x MIC1 x MIC0.5 x MIC0.25 x MIC
Synergy
Antagonism
Time (hours)B
liss
inte
ract
ions
a.
b.
c.
d.
Figure 4. Time course of Bliss interactions of clofazimine and amikacin (CLO+AMK) and clofazimine and clarithromycin (CLO+CLA) combinations at 2×, 1×, 0.5×, and 0.25× the respective MICs against M. abscessus (A and B) and M. avium (C and D). Dotted lines represent the significance level for synergistic (+10%) and antagonistic (-10%) interactions. Note the antagonistic interaction at earlier time points followed by the synergistic interaction at later time points.
A
B
C
D

Clofazimine prevents regrowth and resistance
69
Cha
pter
4
with clarithromycin/clofazimine MICs of 32 to 124/0.5 to 2 mg/l. High (>95%) and low (<5%) target attainment rates were found for all monotherapy and combination regimens for M. abscessus isolates with amikacin/clofazimine/clarithromycin MICs of ≤4/0.06/0.5 mg/l and ≥64/1/8 mg/l and for M. avium isolates with amikacin/clofazimine/clarithromycin MICs of ≤8/0.06/8 mg/l and ≥128/1/256 mg/l, respectively (except amikacin monotherapy against M. avium, for which target was not attained even for isolates with very low MICs).
Discussion
This study evaluated the activity of clofazimine and the drug-drug interaction in the combinations clofazimine-amikacin and clofazimine-clarithromycin against M. abscessus and M. avium. Although the effect of clofazimine alone was poor, we found synergy, at specific concentrations, for both combinations against both species. The combined therapy prevented the regrowth observed when amikacin or clarithromycin was used alone.
Clofazimine is the prototype riminophenazine antibiotic with in vitro activity against most mycobacteria.1,4 Its main clinical use so far is in the multidrug treatment of leprosy and multidrug-resistant tuberculosis.18 For the treatment of MAC pulmonary disease, a clofazimine-
-4
-3
-2
-1
0
1
2
3
4
AmikacinClofazimineCombination
I (CI 95%) for stasis =0.41 (0.29-0.55)
clofazimine-amikacin combinationagainst M. abscessus
210.50.2
50.1
20.06 4 8 16 32
0.030
Multiples of MIC
120h
log 1
0 CFU
/ml r
educ
tion
-4
-3
-2
-1
0
1
2
3
4
ClarithromycinClofazimineCombination
I (CI 95%) for stasis =0.53 (0.35-0.72)
clofazimine-clarithromycincombination against M. abscessus
210.50.2
50.1
20.06 4 8 16 32
0.030
Multiples of MIC
120h
log 1
0 CFU
/ml r
educ
tion
-5-4-3-2-1012345
AmikacinClofazimineCombination
I (CI 95%) for stasis =0.027 (0.007-0.048)
clofazimine-amikacin combinationagainst M. avium
210.50.2
50.1
20.06 4 8 16 320
Multiples of MIC
240h
log 1
0 CFU
/ml r
educ
tion
-5-4-3-2-1012345
ClarithromycinClofazimineCombination
I (CI 95%) for stasis =0.16 (0.04-0.35)
clofazimine-clarithromycincombination against M. avium
210.50.2
50.1
20.06 4 8 16 320
Multiples of MIC
240h
log 1
0 CFU
/ml r
educ
tion
a.
b.
c.
d.
Figure 5. Concentration-effect curves of clofazimine and amikacin (left graphs) and clofazimine and clarithromycin (right graphs) combinations against M. abscessus CIP 104536 after 120 h (A and B) and M. avium IWGMT49 after 240 h (C and D). I, interaction index.
A
B
C
D

70
Chapter 4
ethambutol-macrolide regimen was suggested to be as effective as a rifampin-ethambutol-macrolide regimen.2,19 Our in vitro results support the role of clofazimine for the treatment of M. avium disease, as we found synergistic effects between clofazimine and amikacin or clarithromycin. Its role still needs to be determined but may be to replace rifamycins in patients who do not tolerate those drugs, or as an add-on in the early phase of treatment of severe fibro-cavitary MAC pulmonary disease, when amikacin is also added to the rifampin-ethambutol-macrolide regimen.2,3 The regrowth observed in experiments with M. avium exposed to amikacin and clarithromycin was associated with an increase in MIC and the appearance of mutations in rrs (A1408G) and rrl (A2058G).11,12 Clofazimine prevented this mutational resistance from emerging.
For the treatment of M. abscessus, clofazimine is used, but there is less supportive clinical evidence of its efficacy.20 Amikacin and clarithromycin are cornerstones of treatment, but outcomes remain poor.2,3 The evaluation of new multidrug regimens is urgently needed, and the inclusion of clofazimine should be considered. In contrast to M. avium, M. abscessus showed regrowth when exposed to amikacin and clarithromycin alone, but without increases in MICs for these compounds; no mutation was found in the target genes and codons evaluated. This suggests the presence of other mechanisms to decrease susceptibility; adaptive resistance through the expression of efflux pumps has been hypothesized to be linked to reversible low
AMK+CLO againt M. abscessus
64/1
32/0.
5
16/0.
25
8/0.12
54/0
.062/0
.030
20
40
60
80
100AMK aloneCLO aloneAMK+CLO
AMK/CLO MICs (mg/l)
% p
atie
nts a
ttan
ing
seru
m C
max
asso
ciat
ed w
ith
stas
is
CLA+CLO against M. abscessus
8/1 4/0.5
2/0.25
1/0.12
5
0.5/0.
06
0.25/0
.030
20
40
60
80
100CLA aloneCLO aloneCLA+CLO
CLA/CLO MICs (mg/l)
% p
atie
nts a
ttan
ing
seru
m C
max
asso
ciat
ed w
ith
stas
is
AMK+CLO against M. avium
128/1
64/0.
5
32/0.
25
16/0.
125
8/0.06
4/0.03
0
20
40
60
80
100AMK aloneCLO aloneAMK+CLO
AMK/CLO MICs (mg/l)
% p
atie
nts a
ttan
ing
seru
m C
max
asso
ciat
ed w
ith
stas
is
CLA+CLO against M. avium
256/4
124/2 64
/132
/0.5
16/0.
25
8/0.12
50
20
40
60
80
100CLA alone
CLA+CLOCLO alone
CLA/CLO MICs (mg/l)
% p
atie
nts a
ttan
ing
seru
m C
max
asso
ciat
ed w
ith
stas
is
Figure 6. Percentages of patients achieving serum concentrations associated with stasis for M. abscessus and M. avium isolates with increasing MICs for mean daily doses of 15.05 ± 3.62 mg/kg of amikacin (AMK), 14.33 ± 5.09 mg/kg of clarithromycin (CLA), and 1.62 ± 0.28 mg of clofazimine (CLO) alone and in combination corresponding to Cmax of 55.30 ± 16.88 mg/l, 2.31± 1.89 mg/l, and 0.43 ± 0.19 mg/l, respectively).13

Clofazimine prevents regrowth and resistance
71
Cha
pter
4
levels of resistance in other mycobacteria.21,22 Clofazimine may prevent the activation of these mechanisms.
The current 100-mg-once-daily dose of clofazimine19,20 has a very limited evidence base. In MAC-infected patients, it yields a peak plasma drug concentration of 0.43 mg/l,13 close to the MIC we found in this study for M. abscessus CIP 104536 and 8× MIC of M. avium IWGMT49. Clofazimine levels are much higher in tissues than in blood, and the drug accumulates particularly in macrophages, similar to the macrolides.18 Whether the current 100-mg-once-daily dose leads to active concentrations at the site of infection in patients with M. abscessus or M. avium pulmonary disease is not known. A pharmacokinetic-pharmacodynamic (PK-PD) study of clofazimine in the mouse model suggested that lower doses could be effectively used for treatment of tuberculosis,5 but this should be investigated for NTM.
Monte Carlo analysis showed that the number of patients attaining concentrations associated with stasis in vitro was higher in combination than in monotherapy regimens for a wide range of MICs for both mycobacterial species, particularly for M. avium because of the stronger synergy found for this species. The lowest target attainment rates were found for amikacin, which may be explained by the lower amikacin concentrations in tissue than in serum. The currently evaluated inhaled liposomal amikacin could overcome this by delivering larger amounts of the antibiotic directly to the lung.23
The use of Bliss independence and Loewe additivity analyses8,9 allowed us to describe the kinetics of pharmacodynamic interactions and the full concentration-effect relationship. The use of a global response surface model to describe the entire time concentration-response surface is challenging,24 because of the complexity of pharmacodynamic interactions ranging from antagonistic to synergistic interactions at different time points. We found that after a short period of independent interactions, first antagonism was observed and later synergy. The initial antagonistic interaction, where the effect of combination is similar to the effect of the most active drug without the effect of the least active drug, might be explained by slow intracellular accumulation of the least active drug or a differential effect on mycobacterial subpopulations. Antagonism has been found previously for amikacin combinations against mycobacterial species as well as other antimycobacterial drugs targeting both RNA and DNA synthesis against M. avium.25,26 The exact mechanisms of these interactions need further investigation.
Our study had some limitations. First, only combinations at fixed-ratio concentrations of 1×:1× MIC were analyzed, prohibiting the investigation of complex concentration-dependent pharmacodynamic interactions. Second, for comparative purposes, all analyses were intentionally performed using multiples of the MICs determined with broth microdilution formats that most labs are using as reference standards. Because MICs may be different in different test systems and media, extrapolation of the present findings should be done using

72
Chapter 4
MICs determined as in the present study, i.e., RAPMYCO Sensititre panels in CAMHB for amikacin and clarithromycin and broth microdilution method in Middlebrook 7H9 broth for clofazimine. Third, although time kill assays allow a more dynamic evaluation than MIC, they do not provide actual dynamic concentrations of the antibiotics, as it occurs during the course of therapy in humans. Nevertheless, we found synergistic combinations that can be further tested in dynamic PK-PD models. Fourth, the emergence of resistance was investigated partially by detecting MIC increases after exposure and resistance-associated mutations in selected colonies. An alternative approach could have been plating of samples on plates containing several magnitudes of the MIC of the drugs under study.
In conclusion, clofazimine is not very active itself but prevents regrowth of M. avium and M. abscessus exposed to amikacin and clarithromycin. This synergistic activity supports a role for clofazimine as part of a combined therapy with amikacin or clarithromycin for M. avium and, to some extent, for M. abscessus. Its role should be further evaluated with dynamic PK-PD models and hopefully later in clinical trials.

Clofazimine prevents regrowth and resistance
73
Cha
pter
4
van Ingen J, Totten SE, Helstrom NK, Heifets LB, Boere MJ, Daley CL. In vitro synergy between clofazimine and amikacin in treatment of nontuberculous mycobacterial disease. Antimicrob Agents Chemother. 2012;56:6324–27. van Ingen J, Ferro BE, Hoefsloot W, Boeree MJ, van Soolingen D. Drug treatment of pulmonary antituberculous mycobacterial disease in HIV-negative patients: the evidence. Expert Rev Anti Infect Ther. 2013;11:1065–77. Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier J, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K, ATS Mycobacterial Diseases Subcommittee, American Thoracic Society, Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367–416.Reddy VM, O’Sullivan JF, Gangadharam PR. Antimycobacterial activities of riminophenazines. J Antimicrob Chemother. 1999;43:615–23.Swanson RV, Adamson J, Moodley C, Ngcobo B, Ammerman NC, Dorasamy A, Moodley S, Mgaga Z, Tapley A, Bester LA, Singh S, Grosset JH, Almeida DV. Pharmacokinetics and pharmacodynamics of clofazimine in the mouse model of tuberculosis. Antimicrob Agents Chemother. 2015;59:3042–3305.Shen GH, Wu BD, Hu ST, Lin CF, Wu KM, Chen JH. High efficacy of clofazimine and its synergistic effect with amikacin against rapidly growing mycobacteria. Int J Antimicrob Agents. 2010;35:400–4.Fattorini L, Piersimoni C, Tortoli E, Xiao Y, Santoro C, Ricci ML, Orefici G. In vitro and ex vivo activities of antimicrobial agents used in combination with clarithromycin, with or without amikacin, against Mycobacterium avium. Antimicrob Agents Chemother. 1995;39:680–5.Meletiadis J, Petraitis V, Petraitiene R, Lin P, Stergiopoulou T, Kelaher AM, Sein T, Schaufele RL, Bacher J, Walsh TJ. Triazole-polyene antagonism in experimental invasive pulmonary aspergillosis: in vitro and in vivo correlation. J Infect Dis. 2006; 194:1008–18. Meletiadis J, te Dorsthorst DTA, Verweij PE. The concentration dependent nature of the in vitro amphotericin B-itraconazole interaction against Aspergillus fumigatus: isobolographic and response surface analysis of complex pharmacodynamic interactions. Int J Antimicrob Agents. 2006;28:439–49.Clinical and Laboratory Standards Institute. Susceptibility Testing of Mycobacteria, Nocardia, and Other Aerobic Actynomycetes—Second Edition: Approved Standard M24-A2. CLSI, Wayne, PA, USA, 2011. Nessar R, Reyrat JM, Murray A, Gicquel B. Genetic analysis of new 16S rRNA mutations conferring aminoglycoside resistance in Mycobacterium abscessus. J Antimicrob Chemother. 2011; 66:1719–24.Bastian S, Veziris N, Roux AL, Brossier F, Gaillard JL, Jarlier V, Cambau E. Assessment of clarithromycin susceptibility in strains belonging to the Mycobacterium abscessus group by erm(41) and rrl sequencing. Antimicrob Agents Chemother. 2011;55:775–81.van Ingen J, Egelund EF, Levin A, Totten SE, Boeree MJ, Mouton JW, Aarnoutse RE, Heifets LB, Peloquin CA, Daley CL. The pharmacokinetics and pharmacodynamics of pulmonary Mycobacterium avium complex disease treatment. Am J Respir Crit Care Med. 2012;186:559–65. Tayman C, El-Attug MN, Adams E, van Schepdael A, Debeer A, Allegaert K, Smits A. Quantification of amikacin in bronchial epithelial lining fluid in neonates. Antimicrob Agents Chemother. 2011;55:3990–3.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
References

74
Chapter 4
Ikawa K, Kikuchi E, Kikuchi J, Nishimura M, Derendorf H, Morikawa N. Pharmacokinetic modelling of serum and bronchial concentrations for clarithromycin and telithromycin, and site-specific pharmacodynamic simulation for their dosages. J Clin Pharm Ther. 2014;39:411–7.Ferro BE, van Ingen J, Wattenberg M, van Soolingen D, Mouton JW. Time-kill kinetics of antibiotics active against rapidly growing mycobacteria. J Antimicrob Chemother. 2015;70:811–7. Ferro BE, van Ingen J, Wattenberg M, van Soolingen D, Mouton JW. Time-kill kinetics of slowly growing mycobacteria common in pulmonary disease. J Antimicrob Chemother. 2015;70:2838-43. Cholo MC, Steel HC, Fourie PB, Germishuizen WA, Anderson R. Clofazimine: current status and future prospects. J Antimicrob Chemother. 2012; 67:290–8.Field SK, Cowie RL. Treatment of Mycobacterium avium-intracellulare complex lung disease with a macrolide, ethambutol and clofazimine. Chest. 2003;124:1482–6. Jarand J, Levin A, Zhang L, Huitt G, Mitchell JD, Daley CL. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin Infect Dis. 2011;52:565–71.Burian J, Ramón-García S, Howes CG, Thompson CJ. WhiB7, a transcriptional activator that coordinates physiology with intrinsic drug resistance in Mycobacterium tuberculosis. Expert Rev Anti Infect Ther. 2012;10:1037–47.Schmalstieg AM, Srivastava S, Belkaya S, Deshpande D, Meek C, Leff R, van Oers NS, Gumbo T. The antibiotic resistance arrow of time: efflux pump induction is a first step in the evolution of mycobacterial drug resistance. Antimicrob Agents Chemother. 2012;56:4806–15. Olivier KN, Gupta R, Daley CL, Winthrop KL, Ruoss S, Addrizzo- Harris DJ, Flume P, Dorgan D, Salathe MA, Brown-Elliott BA, Wallace RJ, Griffith DE. A randomized, double-blind, placebo-controlled study of liposomal amikacin for inhalation (Arikace®) in patients with recalcitrant nontuberculous mycobacterial lung disease. Am J Respir Crit Care Med. 2014;189:A4126.Drusano GL, Neely M, van Guilder M, Schumitzky A, Brown D, Fikes S, Peloquin C, Louie A. Analysis of combination drug therapy to develop regimens with shortened duration of treatment for tuberculosis. PLoS One. 2014; 9:e101311. Chiodini RJ. Antimicrobial activity of rifabutin in combination with two and three other antimicrobial agents against strains of Mycobacterium paratuberculosis. J Antimicrob Chemother. 1991;27:171–6. Kent RJ, Bakhtiar M, Shanson DC. The in-vitro bactericidal activities of combinations of antimicrobial agents against clinical isolates of Mycobacterium avium-intracellulare. J Antimicrob Chemother. 1992;30:643–50.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.

Clofazimine prevents regrowth and resistance
75
Cha
pter
4


77
Beatriz E. FerroShashikant SrivastavaDevyani DeshpandeCarleton M. ShermanJotam G. PasipanodyaDick van SoolingenJohan W. Mouton Jakko van IngenTawanda Gumbo
Chapter 5
Amikacin pharmacokinetics / pharmacodynamics in a novel hollow-fiber mycobacterium abscessus disease model
Antimicrob Agents Chemother. 2015;60:1242-8.

78
Chapter 5
Summary
The treatment of pulmonary Mycobacterium abscessus disease is associated with very high failure rates and easily acquired drug resistance. Amikacin is the key drug in treatment regimens, but the optimal doses are unknown. No good preclinical model exists to perform formal pharmacokinetics/pharmacodynamics experiments to determine these optimal doses. We developed a hollow-fiber system model of M. abscessus disease and studied amikacin exposure effects and dose scheduling. We mimicked amikacin human pulmonary pharmacokinetics. Both amikacin microbial kill and acquired drug resistance were linked to the peak concentration-to-MIC ratios; the peak/MIC ratio associated with 80% of maximal kill (EC80) was 3.20. However, on the day of the most extensive microbial kill, the bacillary burden did not fall below the starting inoculum. We performed Monte Carlo simulations of 10,000 patients with pulmonary M. abscessus infection and examined the probability that patients treated with one of 6 doses from 750 mg to 4,000 mg would achieve or exceed the EC80. We also examined these doses for the ability to achieve a cumulative area under the concentration-time curve of 82,232 mg·h/l × days, which is associated with ototoxicity. The standard amikacin doses of 750 to 1,500 mg a day achieved the EC80 in <21% of the patients, while a dose of 4 g/day achieved this in 70% of the patients but at the cost of high rates of ototoxicity within a month or two. The susceptibility breakpoint was an MIC of 8 to 16 mg/l. Thus, amikacin, as currently dosed, has limited efficacy against M. abscessus. It is urgent that different antibiotics be tested using our preclinical model and new regimens developed.

79
Cha
pter
5
Amikacin in M. abscessus hollow-fiber model
Introduction
Mycobacterium abscessus is a rapidly growing mycobacterium (RGM) responsible for about 80% of all pulmonary infections caused by RGM.1 It is one of the most drug-resistant microorganisms encountered in the clinic, far worse than extensively and totally drug-resistant Mycobacterium tuberculosis; this is a reason why it is considered the “new antibiotic nightmare”.2 The current treatment for M. abscessus diseases varies according to the infecting subspecies;3 in general, it involves a backbone of amikacin in combination with clarithromycin and either cefoxitin or imipenem early during therapy, followed by subsequent use of oral antibiotics, which is analogous to the initial and continuation phases of tuberculosis treatment.1 Unfortunately, at least half of the patients either fail this therapy, relapse, or die; there is no reliable antibiotic regimen that cures M. abscessus lung disease.1,4 Several other regimens have been tried and found wanting. In such regimens and the standard regimen, the doses were chosen based on what has worked well in mundane Gram negative bacilli. No formal antimicrobial pharmacokinetic/pharmacodynamic (PK/PD) analyses have been performed with M. abscessus. Our time-kill assays in the past demonstrated that amikacin was the most active antibiotic compared to cefoxitin and clarithromycin.5 Here, we performed formal PK/PD studies of amikacin, which is considered the most active parenteral antibiotic against M. abscessus, according to treatment guidelines.1 Recent attempts to evaluate antibiotics and regimens in preclinical M. abscessus disease models have included the use of Drosophila melanogaster and zebra fish.6,7 However, a tractable model in which PK/PD experiments can be performed still needs to be developed. Here, we adapted the hollow-fiber system models (HFS) of slower-growing mycobacteria8-11 and developed a novel PK/PD system for M. abscessus pulmonary disease in order to identify the amikacin exposures and dose schedules associated with optimal microbial kill and suppression of acquired drug resistance (ADR).
Materials and Methods
Bacterial and drugsM. abscessus ATCC 19977 (American Type Culture Collection) was used as the test strain. Stock cultures of the mycobacteria were preserved at -80 °C in Middlebrook 7H9 broth supplemented with 10% oleic acid-albumin-dextrose-catalase (OADC) and 15% glycerol. For each assay, one vial was thawed and incubated for 24 to 48 h at 30 °C to achieve logarithmic-growth phase. Amikacin sulfate powder for intravenous administration was purchased from the Baylor University Medical Center pharmacy (Dallas, TX) and then diluted in sterile water to desired concentrations for the assays. Apramycin was purchased from Sigma.

80
Chapter 5
MIC and mutation frequencyBroth macrodilution in Middlebrook 7H9 broth (here termed “broth”) was used to identify the MIC).12 In addition to the turbidity test, the cfu per milliliter were enumerated for each concentration evaluated in the broth macrodilution test, and the MIC was defined as the lowest concentration associated with ≥99% inhibition of growth. The Etest (bioMérieux) on Middlebrook 7H10 agar (here termed “agar”) was also used as a screening test when an MIC evaluation was needed for HFS liquid cultures. Mutation frequency was determined for the inoculum by culturing 0.2 ml each on 20 agar plates supplemented with 3 times the amikacin MIC. This concentration was chosen since 3 times the MIC is the lowest fold change associated with efflux pump-related resistance in other mycobacteria.13
Concentration-effect studies in test tubesM. abscessus was grown in broth in log phase to the equivalent of a 0.5 McFarland standard turbidity based on optical density measurements at 600 nm, and it was then back diluted to reach a bacterial density of approximately 106 log10 cfu/ml. M. abscessus was exposed to amikacin concentrations of 0, 1/8, 1/4, 1/2,1, 2, 4, and 8 times the MIC at 30 °C in test tubes. After 72 h of incubation, the contents of each tube were washed with saline and then serially diluted and cultured on agar at 30 °C in order to enumerate the cfu per milliliter. The inhibitory sigmoid maximum effect (Emax) model was used to identify the relationship between amikacin concentration and M. abscessus cfu per milliliter.
Hollow-fiber model for M. abscessusThe HFS model has two physical compartments: the central compartment, in which drug circulates, and the peripheral compartment, which houses the mycobacteria.8 The peripheral compartment is separated from the central compartment by the semi permeable hollow fibers with a pore size of 42 Da, through which fresh medium (broth with 10% dextrose) circulates. Thus, the concentrations of chemicals in the peripheral compartment are in equilibrium with the central compartment. Twenty milliliters of 106 log10 cfu/ml log phase M. abscessus cells were inoculated into the peripheral compartment of each cellulosic hollow-fiber cartridge (FiberCell Systems). M. abscessus cells are much larger than the pores and thus stay confined to the peripheral compartment, where they are continuously bathed by dynamic concentrations of chemicals and drugs. All systems were permanently kept in a 30 °C incubator.
Pharmacokinetic/pharmacodynamic studyTwelve HFS were inoculated with M. abscessus, as described above. For the dose-effect experiments, amikacin doses that mimic the serum area under the concentration-time curve from 0 to 24 h (AUC0–24) and peak concentrations (Cmax) achieved in humans exposed to human-equivalent doses of 66.7, 125, 250, 500, 1,000, 2,000, 4,000, and 6,400 mg were administered once daily to the central compartment via computerized syringe pumps. Dose-scheduling studies were simultaneously performed; we tested human-like doses of 500, 2,000, and 4,000

81
Cha
pter
5
Amikacin in M. abscessus hollow-fiber model
mg, chosen based on the pilot concentration-effect study in test tubes, to be administered thrice daily in order to match some of the Cmax/MIC and percent time the concentration remains above the MIC (%T MIC) in the once-a-day regimens. The dilution rates were set to achieve an amikacin half-life of 3 h, as encountered in serum and bronchial secretions.14,15 The amikacin concentrations achieved in all the HFS were validated by sampling each central compartment during the last 2 days at 0, 0.5, 2.7, 5.4, 18.7, 23.5, 24.5, 26.7, 29.4, 42.7, and 47.5 h after the drug administration; the sampling times were chosen based on optimal sampling theory for the pharmacokinetics of amikacin.16-18 In order to quantify the M. abscessus cfu per milliliter, 1 ml of the peripheral compartment contents was removed from each system on days 0, 1, 2, 3, 5, 7, 10, and 14. The samples were washed with saline to avoid antibiotic carryover, after which samples were serially diluted and cultured on agar. To quantify the amikacin-resistant M. abscessus cfu per milliliter, the same samples were also inoculated on agar supplemented with 3 times the amikacin MIC.
Drug assaysApramycin was used as the internal standard. Calibrator, controls, and apramycin were included in each analytical run for quantitation. Stock solutions of amikacin and apramycin were prepared in 80:20 methanol-water at a concentration of 1 mg/ml and stored at -20 °C. An eight-point calibration curve was prepared by diluting the amikacin stock solution in drug-free broth (1, 2, 10, 20, 50, 100, 200, and 400 µg/ml). Quality control samples were prepared by spiking media with stock standards for two levels of controls. Samples were prepared in 96-well microtiter plates, 10 µl of calibrator, quality controls, or sample was added to 190 µl of 0.1% formic acid in water containing 10 µg/ml apramycin, and the samples were vortexed. Chromatographic separation was achieved on an Acquity ultrahigh-performance liquid chromatography (UPLC) high strength silica (HSS) T3, 1.8 µm, 50 by 2.1-mm analytical column (Waters) maintained at 30 °C at a flow rate of 0.2 ml/min, with a binary gradient and a total run time of 6 min. The observed ion (m/z) values of the fragment ions were amikacin (m/z 586 →425) and apramycin (m/z 540→378). Sample injection and separation were performed using an Acquity UPLC interfaced with a XevoTQ mass spectrometer (Waters). All data were collected using MassLynx version 4.1 SCN810. The limit of quantitation for this assay was 0.5 µg/ml.
Pharmacokinetic and PK/PD modelingThe drug concentrations from each HFS were comodeled as a one-compartment pharmacokinetic model, a two-compartment model, and a three-compartment model using the ADAPT 5 program.19 The best model was chosen using the Akaike information criterion (AIC) and parsimony.20,21 The pharmacokinetic parameter estimates were then used to calculate the observed AUC0–24, the AUC0–24/MIC ratio, and %TMIC. The observed Cmax in each HFS was used to calculate the Cmax/MIC. Dose response was modeled using the inhibitory sigmoid Emax model with the total bacterial burden used as the response parameter, while drug exposure was expressed as either the AUC0–24/MIC ratio, %TMIC, or Cmax/MIC ratio. However, the
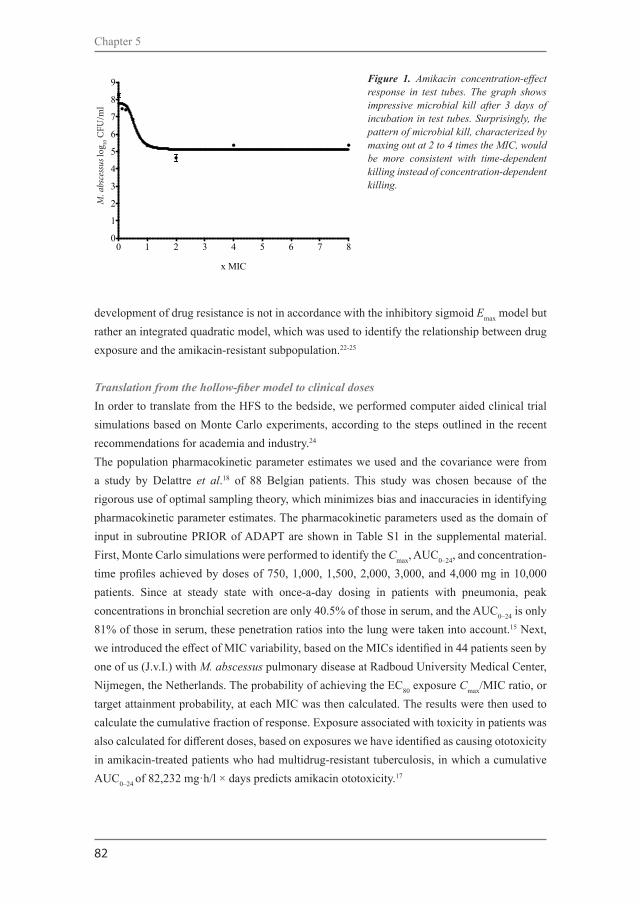
82
Chapter 5
development of drug resistance is not in accordance with the inhibitory sigmoid Emax model but rather an integrated quadratic model, which was used to identify the relationship between drug exposure and the amikacin-resistant subpopulation.22-25
Translation from the hollow-fiber model to clinical dosesIn order to translate from the HFS to the bedside, we performed computer aided clinical trial simulations based on Monte Carlo experiments, according to the steps outlined in the recent recommendations for academia and industry.24 The population pharmacokinetic parameter estimates we used and the covariance were from a study by Delattre et al.18 of 88 Belgian patients. This study was chosen because of the rigorous use of optimal sampling theory, which minimizes bias and inaccuracies in identifying pharmacokinetic parameter estimates. The pharmacokinetic parameters used as the domain of input in subroutine PRIOR of ADAPT are shown in Table S1 in the supplemental material. First, Monte Carlo simulations were performed to identify the Cmax, AUC0–24, and concentration-time profiles achieved by doses of 750, 1,000, 1,500, 2,000, 3,000, and 4,000 mg in 10,000 patients. Since at steady state with once-a-day dosing in patients with pneumonia, peak concentrations in bronchial secretion are only 40.5% of those in serum, and the AUC0–24 is only 81% of those in serum, these penetration ratios into the lung were taken into account.15 Next, we introduced the effect of MIC variability, based on the MICs identified in 44 patients seen by one of us (J.v.I.) with M. abscessus pulmonary disease at Radboud University Medical Center, Nijmegen, the Netherlands. The probability of achieving the EC80 exposure Cmax/MIC ratio, or target attainment probability, at each MIC was then calculated. The results were then used to calculate the cumulative fraction of response. Exposure associated with toxicity in patients was also calculated for different doses, based on exposures we have identified as causing ototoxicity in amikacin-treated patients who had multidrug-resistant tuberculosis, in which a cumulative AUC0–24 of 82,232 mg·h/l × days predicts amikacin ototoxicity.17
Figure 1. Amikacin concentration-effect response in test tubes. The graph shows impressive microbial kill after 3 days of incubation in test tubes. Surprisingly, the pattern of microbial kill, characterized by maxing out at 2 to 4 times the MIC, would be more consistent with time-dependent killing instead of concentration-dependent killing.
0 1 2 3 4 5 6 7 80
1
2
3
4
5
6
7
8
9
x MIC
M. a
bsce
ssus
log 1
0 CFU
/mL
M. a
bsce
ssus
log 10
CFU
/ml

83
Cha
pter
5
Amikacin in M. abscessus hollow-fiber model
Results
The amikacin MIC for the M. abscessus laboratory strain was 32 mg/l by all methods, on repeat analysis. The minimum bactericidal concentration (MBC), which is the concentration associated with ≥99.9% kill, was 64 mg/l, so that the MBC/MIC ratio was 2, making amikacin a bactericidal agent by standard definitions.26 The mutation frequency to 3 times the amikacin MIC was 2.73 ± 0.31 × 10-6 in repeat experiments.
The relationship between amikacin concentration as a multiple of the MIC and microbial burden after 3 days of incubation in test tubes is shown in Figure 1. This experimental design, which uses static concentrations of amikacin, was associated with a maximum effect (Emax) of 2.65 log10 cfu/ml (95% confidence interval [95% CI], 2.26 to 3.04 log10 cfu/ml), a Hill´s slope (H) of 3.88 (95% CI, 1.13 to 6.63), and a 50% effective concentration (EC50) of 0.57 times the MIC (95% CI, 0.46 to 0.69 times the MIC) (r2 =0.93). Figure 1 shows that the bacterial burden at the Emax was 5.12 log10 cfu/ml, which is >1.0 log10 cfu/ml less than the bacterial burden of 6.13 log10 cfu/ml at the beginning of incubation, with just 3 days of incubation with static amikacin concentrations. This magnitude of microbial kill is consistent with the notion that amikacin is very active against M. abscessus.
Next, we performed HFS studies that used dynamic concentrations of amikacin, and we co-modeled the 121 amikacin concentrations that were measured in all the HFS. The concentrations were best described by a two-compartment pharmacokinetic model, which was characterized by a total clearance of 1.5 l/h, volume of central compartment of 7.98 l, intercompartmental clearance of 22.3 l/h, and a peripheral volume of 0.04 l, which are virtually similar to those parameters in patients treated with amikacin.17
The day 0 bacterial burden in the HFS (i.e., just prior to start of therapy) was 6.69 log10 cfu/ml. The changes in total bacterial burden and the amikacin-resistant subpopulation for each drug exposure are shown in Figure 2. The figure shows that the amikacin efficacy (i.e., Emax) was minimal. By day 3, the bacterial burden at the Emax was only 0.50-log10 cfu/ml lower than starting bacterial burden, which is considerably lower than that with static concentrations. Figure 2 also shows that amikacin microbial kill was effectively terminated by day 5, and starting on day 7, the overall bacterial burden had begun to reflect the replacement of the total population by the amikacin-resistant subpopulation.
We performed an Etest at the beginning and end of the experiments, and as shown in Figure 2C and D, there was a change in the MIC in the two highest concentration treatments, from 32 to >256 mg/l on both the once-a-day and three-times-a-day treatment schedules. Inhibitory sigmoid Emax models for microbial kill up to 7 days, using either observed Cmax/MIC, AUC/MIC, or %TMIC, revealed the Akaike information criterion (AIC) scores shown in Table 1. The lowest AIC score was for Cmax/MIC for all days up to day 5. If one simply used the r2 for

84
Chapter 5
model fit, the r2 values were highest for Cmax/MIC up to day 5. It should be noted that while the day 7 results seemed to indicate the lowest AIC score for %TMIC, the result was judged to be spurious, since the EC50 exceeded the possible extrema of the %TMIC function; in this case, the maximum possible %TMIC of 100% was exceeded by the EC50. Taken together, amikacin efficacy was primarily driven by Cmax/MIC ratios, which had the lowest AIC scores for up to 5 days, beyond which the total population was replaced by drug-resistant subpopulation in some systems.
The relationship between the Cmax/ MIC ratio and bacterial burden on day 5 is shown in Figure 3. The relationship was characterized by Emax of 2.40 ± 0.22 log10 cfu/ml, a Hill´s slope of 3.79 ± 2.44, and an EC50 that was a Cmax/MIC of 2.22 ± 0.44 (r2=0.97). Thus, the EC50 is considerably higher than that in the static amikacin concentration experiments (Figure 1), while the H is virtually identical. The EC80, which is considered the optimal exposure, was a Cmax/MIC ratio of 3.20. Table 2 shows the AIC for Cmax/MIC, %TMIC, and AUC/MIC versus the amikacin-resistant subpopulation modeled on the quadratic function for drug resistance. While the quadratic function for resistance emergence 22-25 is integrated over time, we utilized piecewise functions for each day of sampling, since we have found that PK/PD indices “wobble” with the
Figure 2. Changes in bacterial burden and amikacin-resistant subpopulation with time. (A to D) The total M. abscessus population (solid lines) and amikacin resistant subpopulation (dashed lines) over the course of 14 days of exposure to different Cmax/MIC exposures are shown. There was a higher amikacin-resistant subpopulation of M. abscessus as the Cmax/MIC increased, so that at higher Cmax/MIC ratios (C and D), the total population had been completely replaced by the amikacin-resistant subpopulation by day 14.
0 2 4 6 8 10 12 140
1
2
3
4
5
6
7
8
9
10
11
12
Cmax/MIC: 0.11Cmax/MIC: 0.06Control
Time (days)
M. a
bsce
ssus
log 1
0 CFU
/mL
0 2 4 6 8 10 12 140
1
2
3
4
5
6
7
8
9
10
11
12 Cmax/MIC: 0.20Cmax/MIC: 0.38Cmax/MIC: 0.71
Time (days)
M. a
bsce
ssus
log 1
0 CFU
/mL
0 2 4 6 8 10 12 140
1
2
3
4
5
6
7
8
9
10
11
12 Cmax/MIC: 1.74Cmax/MIC: 8.93Cmax/MIC: 12.12
Time (days)
M. a
bsce
ssus
log 1
0 CFU
/mL
0 2 4 6 8 10 12 140
1
2
3
4
5
6
7
8
9
10
11
12
Cmax/MIC: 4.73Cmax/MIC: 9.38
Cmax/MIC: 1.70
Time (days)
M. a
bsce
ssus
log 1
0 CFU
/mL
A.
B.
C.
D.
A
B
C
D
M. a
bsce
ssus
log 10
CFU
/ml
M. a
bsce
ssus
log 10
CFU
/ml
M. a
bsce
ssus
log 10
CFU
/ml
M. a
bsce
ssus
log 10
CFU
/ml

85
Cha
pter
5
Amikacin in M. abscessus hollow-fiber model
duration of therapy.27 Table 2 shows the same wobble phenomenon, and that the PK/PD index associated with the size of the amikacin-resistant subpopulation during the first 3 days was %TMIC; by day 14, this had changed to Cmax/MIC. Figure 4 shows a system of inverted “U” curves during the first 2 days, with a switch to a straight line on day 3, followed by “U” curves from day 3 onwards. The day 14 relationship between Cmax/MIC and the amikacin-resistant subpopulation was described by log10 cfu/ml = 4.333 + 0.364 × (Cmax/MIC) + 0.006 × (Cmax/MIC)2; r2=0.906. This means that by day 14, there was no amikacin concentration that could reduce the size of the drug-resistant subpopulation to the 3.8 log10 cfu/ml encountered in non treated controls.
To translate our HFS findings to the clinic, we performed Monte Carlo simulations to identify the probability that different amikacin doses could achieve or exceed the EC80 exposure Cmax/MIC ratio of 3.2. The MIC distribution in 44 clinical isolates used in the simulations is shown in Figure 5; the median MIC was 16 mg/l (range, 4 to 128 mg/l). Figure 5A and B show the proportions of patients treated with doses of 750, 1,000, and 1,500 mg who achieved this target exposure, which give cumulative response percentages of 3.94%, 8.41%, and 20.77% of patients, respectively. Figure 5B and C also show target attainment by the higher doses of 2,000, 3,000, and 4,000 mg, for which the cumulative response fractions were 39.21%, 61.43%, and 70.05%, respectively. This means that none of the doses tested would achieve or exceed the EC80 in >90% of the patients, which is considered an adequate proportion of patients in other mycobacterial infections. Figure 5A and B also illustrate the MIC below which there is
Table 1. Akaike information criteria scores for pharmacokinetic/pharmacodynamic (PK/PD) indices linked to microbial kill.
PK/PD parameter Day 2 Day 3 Day 5 Day 7
% time above MIC 3.272 No convergence -0.7837 -4.925
AUC/MIC -5.173 -1.303 3.955 -0.9094
Cmax/MIC -5.173 -2.244 -14.56 -3.525
Figure 3. Amikacin exposure effect in the hollow-fiber system. The results are shown up to day 7, at which point the drug-resistant subpopulation had begun to replace the total population in some systems. This is reflected by the increase in Emax each day until day 5, after which it began to decline. At the maximal microbial kill, the bacterial burden barely fell below the stasis line, suggesting that in fact amikacin is not bactericidal in the hollow-fiber system.
0 1 2 3 4 5 6 7 8 9 10 11 12 130123456789
Day 2Day 3Day 5Day 7Stasis line
Cmax/MIC
M. a
bsce
ssus
log 1
0 CFU
/mL
M. a
bsce
ssus
log 10
CFU
/ml
Cmax/MIC

86
Chapter 5
reduced microbial kill in at least 10% of patients with the standard dose, which is 8 mg/l, while Figure 5B and C show that with higher doses of ≥2,000 mg, this is 16 mg/l. Thus, the resistance breakpoint drug concentration in Middlebrook broth would be set at 16 mg/l at the most, and it should be considered that beyond this MIC, patients will not have good responses unless the dose is increased. However, such dose increases would increase the chances of toxicity. As an example, the duration of therapy and a cumulative AUC0–24 of 82,232 mg·h/l × days predict amikacin ototoxicity, and Figure 5D shows the probability of achieving that threshold concentration associated with an increased risk of ototoxicity for the six different daily doses. There is a rapid increase in odds of deafness at doses of 3,000 to 4,000 mg a day.
Discussion
Amikacin has long been considered the most active parenteral antibiotic against M. abscessus.1 We found that while models of static concentrations suggested that it is a bactericidal drug with time-driven efficacy, they overestimated amikacin efficacy compared to that in the HFS model. The HFS model’s poor amikacin efficacy correlates better with clinical observations of
Table 2. Akaike information criteria scores for pharmacokinetic/pharmacodynamic (PK/PD) indices linked to acquired resistance.
PK/PD parameter Day 1 Day 2 Day 3 Day 5 Day 7 Day 10 Day 14
% time above MIC -8.984 -19.28 -0.7686 11.37 10.44 19.89 11.27
AUC/MIC -6.262 -16.93 0.5867 5.903 1.006 1.087 7.316
Cmax/MIC -7.126 -18.72 0.1416 9.673 6.695 7.812 0.2809
Figure 4. Amikacin-resistant subpopulation evolution with duration of treatment. The curves for each day are piecewise for the U-shaped relationship between exposure versus size of the drug-resistant subpopulation. With increasing time, the baseline amikacin-resistant subpopulation in the absence of drug exposure grows so that curves start at a different y intercept on each day. For days 1 and 2, the curve shape is an inverted U curve; at day 3, it is a straight line, and then it switches to an upright “U.” However, by day 14, the right side
0 2 4 6 8 10 12 140
2
4
6
8
10
12Day 1Day 2Day 3Day 5Day 7Day 10Day 14
Cmax/MIC
Res
istan
tM. a
bsce
ssus
log 1
0 CFU
/mL
of the curve has straightened out, which means that there was no amikacin Cmax/MIC ratio high enough to reduce the size of the drug-resistant subpopulation, and the curve is no longer a “U.”
Resi
stan
t M. a
bsce
ssus
log 10
CFU
/ml
Cmax/MIC

87
Cha
pter
5
Amikacin in M. abscessus hollow-fiber model
amikacin containing regimens in the antibiotic treatment of pulmonary M. abscessus, which entail high failure rates, “the talking M. abscessus blues”.28,29 We found that both optimal amikacin microbial kill and ADR were associated with the Cmax/MIC ratio; for ADR, however, the PK/PD response parameter was %TMIC in the first 3 days but changed to Cmax/MIC beyond day 3. The optimal Cmax/MIC ratio for microbial kill was 3.2, which is considerably lower than the Cmax/MIC of 8 to 12 identified for mundane bacteria.30,31 However, we show that even for that relatively lower Cmax/MIC ratio, the standard amikacin doses failed to achieve the target in more than three-quarters of patients, and much higher intravenous doses would need to be administered to even achieve the target in >70% of patients. In any case, this EC80 target would merely hold bacterial burden constant at best, which is just “cuddling” the bacteria, while with the higher doses, the patients would pay with a high risk of deafness. Alternative delivery methods, such as the use of inhaled liposomal amikacin, could overcome this: the efficacy, safety, and tolerability of daily dosing of 590 mg of liposomal amikacin for inhalation versus placebo in patients with recalcitrant nontuberculous mycobacterial lung disease are currently
Figure 5. Probability target attainment of different amikacin doses in 10,000 patients. (A) At doses of 750 mg and 1,000 mg, which are standard doses, there was very poor target attainment at the most commonly encountered MICs. The target attainment fell below 90% at the 8 mg/l MIC. (B) At doses of 1,500 mg to 2,000 mg, 90% of the patients achieved the target Cmax/MIC after the MIC of 8 mg/l. At a dose of 15 mg/kg of body weight, patients weighing up 100 kg would have 1,500 mg as normal dose, while the 2,000-mg dose is higher than currently administered. (C) Doses of 3,000 mg and 4,000 mg would better attain target concentrations, but even at these high doses, target attainment falls below 90% at an MIC of 32 mg/l. (D) Probability of ototoxicity in 10,000 patients exposed to different amikacin concentrations for different durations of therapy, indicating a rapid increase in the odds of attaining toxic cumulative AUCs (cAUC) with just 2 months of therapy for doses of ≥2,000 mg.
1 2 4 8 1 6 3 2 6 4 1 2 8 2 5 60 .0
0 .1
0 .2
0 .3
0 .4
0 .5
0 .6
0 .7
0 .8
0 .9
1 .0
A m ik a c in M IC (m g /L )
Pro
ba
bili
ty t
arg
et
att
ain
me
nt
M IC
7 5 0 m g
1 ,0 0 0 m g9 0 %
A .
1 2 4 8 1 6 3 2 6 4 1 2 8 2 5 60 .0
0 .1
0 .2
0 .3
0 .4
0 .5
0 .6
0 .7
0 .8
0 .9
1 .0
A m ik a c in M IC (m g /L )
Pro
ba
bili
ty t
arg
et
att
ain
me
nt
M IC
1 ,5 0 0 m g
2 ,0 0 0 m g
9 0 %
B .
1 2 4 8 1 6 3 2 6 4 1 2 8 2 5 60 .0
0 .1
0 .2
0 .3
0 .4
0 .5
0 .6
0 .7
0 .8
0 .9
1 .0
A m ik a c in M IC (m g /L )
Pro
ba
bili
ty t
arg
et
att
ain
me
nt
M IC
3 ,0 0 0 m g
4 ,0 0 0 m g
9 0 %
C .
0 1 2 3 4 5 60 .0
0 .1
0 .2
0 .3
0 .4
0 .5
0 .6
0 .7
0 .8
0 .9
1 .0
T im e in m o n th s
Pro
ba
bili
ty o
f cA
UC
ass
cosi
ate
d w
ith t
oxi
city
7 5 0 m g1 0 0 0 m g1 5 0 0 m g2 0 0 0 m g3 0 0 0 m g4 0 0 0 m g
D .
A
C
B
D
A m i k a c i n M I C ( m g / l ) A m i k a c i n M I C ( m g / l )
A m i k a c i n M I C ( m g / l )

88
Chapter 5
being assessed (ClinicalTrials.gov identifier NCT01315236). However, by definition, even such delivery methods would not have a higher Emax, so that effect would still be compromised. Regardless, our findings mean that the role of intravenous amikacin in the current regimens used for pulmonary M. abscessus should be revisited and perhaps be replaced by more active antibiotics.
Current M. abscessus regimens are partly guided by in vitro results, but the clinical correlation is lacking. The existing susceptibility breakpoint for amikacin, tested in cation-adjusted Mueller-Hinton medium, is ≥64 mg/l.12 However, this breakpoint has failed to discriminate patients who would respond to therapy. Based on HFS findings and Monte Carlo simulations, we propose a breakpoint of 16 mg/l in Middlebrook broth. The poor correlation observed between in vitro tests and patient outcomes might be explained by this discrepancy, since most isolates would be considered resistant based on this proposed breakpoint. While these are merely simulations of a single agent used in combination therapy in real life, experience with antituberculosis drugs has demonstrated that such HFS- and Monte Carlo simulation- derived breakpoints for monotherapy are highly accurate in predicting the breakpoints above which therapy fails that are actually observed in the clinic in combination therapy studies.32-36 Thus, our results will likely be borne out in the clinic setting in the future.Finally, our new HFS model of M. abscessus disease is a tractable model that can be used to study the roles of other antibiotics, both as monotherapy and as combination therapy. The model allows repetitive sequential sampling in the same system of bacterial cultures and drug concentrations, which let us follow the evolution of the bacteria when exposed to different treatment regimens. This approach, which is not possible with animal models, for which harvesting of lungs is a terminal procedure, or with fruit flies, permits more powerful statistical computations, such as time-to-event analyses, repeated-measures analyses, and construction of systems equations for phenomena, such as resistance emergence.
Our study has some limitations. First, we examined only the effect of amikacin alone. Amikacin is used in combination with other antibiotics in patients. However, for optimal combination regimen design, it was necessary to start with the optimization of each component of the regimen as monotherapy for those combinations in which the drugs are additive. In the case of a synergistic combination, it may be possible to achieve more microbial kill without optimization of the combination. Second, we show the results of an HFS model for extracellular infection. However, M. abscessus, like all mycobacteria, is both an extracellular and intracellular pathogen. Nevertheless, amikacin does not kill intracellular bacteria due to only 5% penetration into macrophages,37 so amikacin efficacy might be worse than what we observed. We have intracellular HFS models of M. abscessus, which we will use to study those agents that are active intracellularly. Third, we report results with one M. abscessus strain. A larger number of isolates would give a more robust EC80 target;38 this, however, is limited by costs associated with performing each HFS study.

89
Cha
pter
5
Amikacin in M. abscessus hollow-fiber model
In summary, we developed a preclinical model for M. abscessus disease in which we identified the low efficacy of intravenous amikacin. The model will be useful for understanding the efficacy and ADR during the treatment of M. abscessus infection, dose-ranging and dose-scheduling studies, and the design of new and more effective combination regimens.

90
Chapter 5
Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier J, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K, ATS Mycobacterial Diseases Subcommittee, American Thoracic Society, Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367–416.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother. 2012; 67:810–18.van Ingen J, Ferro BE, Hoefsloot W, Boeree MJ, van Soolingen D. Drug treatment of pulmonary antituberculous mycobacterial disease in HIV-negative patients: the evidence. Expert Rev Anti Infect Ther. 2013;11:1065–77.Jarand J, Levin A, Zhang L, Huitt G, Mitchell JD, Daley CL. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin Infect Dis. 2011;52:565–71.Ferro BE, van Ingen J, Wattenberg M, van Soolingen D, Mouton JW. Time-kill kinetics of antibiotics active against rapidly growing mycobacteria. J Antimicrob Chemother. 2015;70:811–7.Bernut A, Le Moigne V, Lesne T, Lutfalla G, Herrmann JL, Kremer L. In vivo assessment of drug efficacy against Mycobacterium abscessus using the embryonic zebrafish test system. Antimicrob Agents Chemother. 2014;58:4054–63.Oh CT, Moon C, Jeong MS, Kwon SH, Jang J. Drosophila melanogaster model for Mycobacterium abscessus infection. Microbes Infect. 2013;15:788–95.Srivastava S, Gumbo T. In vitro and in vivo modeling of tuberculosis drugs and its impact on optimization of doses and regimens. Curr Pharm Des. 2011;17:2881–8.Deshpande D, Srivastava S, Meek C, Leff R, Gumbo T. Ethambutol optimal clinical dose and susceptibility breakpoint identification by use of a novel pharmacokinetic-pharmacodynamic model of disseminated intracellular Mycobacterium avium. Antimicrob Agents Chemother. 2010;54:1728–33.Gumbo T, Louie A, Deziel MR, Parsons LM, Salfinger M, Drusano GL. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis. 2004;190:1642–51.Pasipanodya JG, Nuermberger E, Romero K, Hanna D, Gumbo T. Systematic analysis of hollow fiber model of tuberculosis experiments. Clin Infect Dis. 2015 61(Suppl 1):S10 –S17.Clinical and Laboratory Standards Institute. Susceptibility Testing of Mycobacteria, Nocardia, and Other Aerobic Actynomycetes—Second Edition: Approved Standard M24-A2. CLSI, Wayne, PA, USA, 2011.Schmalstieg AM, Srivastava S, Belkaya S, Deshpande D, Meek C, Leff R, van Oers NS, Gumbo T. The antibiotic resistance arrow of time: efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob Agents Chemother. 2012;56:4806–15.Dull WL, Alexander MR, Kasik JE. Bronchial secretion levels of amikacin. Antimicrob Agents Chemother. 1979;16:767–771.Santré C, Georges H, Jacquier JM, Leroy O, Beuscart C, Buguin D, Beaucaire G. Amikacin levels in bronchial secretions of 10 pneumonia patients with respiratory support treated once daily versus twice daily. Antimicrob Agents Chemother. 1995;39:264–7.Tam VH, Preston SL, Drusano GL. Optimal sampling schedule design for populations of patients. Antimicrob Agents Chemother. 2003;47:2888–91.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
References

91
Cha
pter
5
Amikacin in M. abscessus hollow-fiber model
Modongo C, Pasipanodya JG, Zetola NM, Williams SM, Sirugo G, Gumbo T. Amikacin concentrations predictive of ototoxicity in multidrug-resistant tuberculosis patients. Antimicrob Agents Chemother. 2015;59:6337–43.Delattre IK, Musuamba FT, Nyberg J, Taccone FS, Laterre PF, Verbeeck RK, Jacobs F, Wallemacq PE. Population pharmacokinetic modeling and optimal sampling strategy for Bayesian estimation of amikacin exposure in critically ill septic patients. Ther Drug Monit. 2010;32:749–56.D’Argenio DZ, Schumitzky A, Wang X. 2009. ADAPT 5 user’s guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource, University of Southern California, Los Angeles, CA. Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr. 1974;19:716–23.Ludden TM, Beal SL, Sheiner LB. Comparison of the Akaike information criterion, the Schwarz criterion and the F test as guides to model selection. J Pharmacokinet Biopharm. 1944;22:431–45.Gumbo T, Louie A, Liu W, Brown D, Ambrose PG, Bhavnani SM, Drusano GL. Isoniazid bactericidal activity and resistance emergence: integrating pharmacodynamics and pharmacogenomics to predict efficacy in different ethnic populations. Antimicrob Agents Chemother. 2007;51:2329–36.Gumbo T, Dona CS, Meek C, Leff R. Pharmacokinetics/pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother. 2009;53:3197–204.Gumbo T, Angulo-Barturen I, Ferrer-Bazaga S. Pharmacokineticpharmacodynamic and dose-response relationships of antituberculosis drugs: recommendations and standards for industry and academia. J Infect Dis. 2015;211(Suppl 3):S96 –S106.Gumbo T. Integrating pharmacokinetics, pharmacodynamics and pharmacogenomics to predict outcomes in antibacterial therapy. Curr Opin Drug Discov Devel. 2008;11:32–42.Pankey GA, Sabath LD. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin Infect Dis. 2004;38:864–70.Musuka S, Srivastava S, Siyambalapitiyage Dona CW, Meek C, Leff R, Pasipanodya J, Gumbo T. Thioridazine pharmacokineticpharmacodynamic parameters “wobble” during treatment of tuberculosis: a theoretical basis for shorter-duration curative monotherapy with congeners. Antimicrob Agents Chemother. 2013;57:5870–7.Griffith DE. The talking Mycobacterium abscessus blues. Clin Infect Dis. 2011;52:572–4.van Ingen J, Egelund EF, Levin A, Totten SE, Boeree MJ, Mouton JW, Aarnoutse RE, Heifets LB, Peloquin CA, Daley CL. The pharmacokinetics and pharmacodynamics of pulmonary Mycobacterium avium complex disease treatment. AmJ Respir Crit Care Med. 2012;186:559–65.Ambrose PG, Bhavnani SM, Rubino CM, Louie A, Gumbo T, Forrest A, Drusano GL. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it’s not just for mice anymore. Clin Infect Dis. 2007;44:79–86.Craig WA, Redington J, Ebert SC. Pharmacodynamics of amikacin in vitro and in mouse thigh and lung infections. J Antimicrob Chemother. 1991;27(Suppl C):29–40.Gumbo T. New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob Agents Chemother. 2010;54:1484–91.Gumbo T, Chigutsa E, Pasipanodya J, Visser M, van Helden PD, Sirgel FA, McIlleron H. The pyrazinamide susceptibility breakpoint above which combination therapy fails. J Antimicrob Chemother. 2014;69:2420–5.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.

92
Chapter 5
Gumbo T, Pasipanodya JG, Wash P, Burger A, McIlleron H. Redefining multidrug-resistant tuberculosis based on clinical response to combination therapy. Antimicrob Agents Chemother. 2014;58:6111–15.Ocheretina O, Escuyer VE, Mabou MM, Royal-Mardi G, Collins S, Vilbrun SC, Pape JW, Fitzgerald DW. Correlation between genotypic and phenotypic testing for resistance to rifampin in Mycobacterium tuberculosis clinical isolates in Haiti: investigation of cases with discrepant susceptibility results. PLoS One. 2014; 9:e90569.Gumbo T, Pasipanodya JG, Romero K, Hanna D, Nuermberger E. Forecasting accuracy of the hollow fiber model of tuberculosis for clinical therapeutic outcomes. Clin Infect Dis. 2015; 61(Suppl 1):S25–S31.Rey-Jurado E, Tudo G, Soy D, Gonzalez-Martin J. Activity and interactions of levofloxacin, linezolid, ethambutol and amikacin in three drug combinations against Mycobacterium tuberculosis isolates in a human macrophage model. Int J Antimicrob Agents. 2013;42:524–30.MacGowan AP, Reynolds R, Noel AR, Bowker KE. Bacterial strain-to-strain variation in pharmacodynamic index magnitude, a hitherto unconsidered factor in establishing antibiotic clinical breakpoints. Antimicrob Agents Chemother. 2009;53:5181–4.
34.
35.
36.
37.
38.

93
Cha
pter
5
Amikacin in M. abscessus hollow-fiber model


95
Chapter 6
Moxifloxacin´s limited efficacy in the hollow-fiber model of Mycobacterium abscessus disease
Antimicrob Agents Chemother. 2016;60:3779-85.
Beatriz E. FerroShashikant SrivastavaDevyani DeshpandeJotam G. PasipanodyaDick van SoolingenJohan W. MoutonJakko van IngenTawanda Gumbo

96
Chapter 6
Summary
Current regimens used to treat pulmonary Mycobacterium abscessus disease have limited efficacy. There is an urgent need for new drugs and optimized combinations and doses. We performed hollow fiber system studies in which M. abscessus was exposed to moxifloxacin lung concentration-time profiles similar to human doses between 0 and 800 mg/day. The minimum bactericidal concentration and MIC were 8 and 2 mg/l in our M. abscessus strain, suggesting bactericidal activity. Measurement of moxifloxacin concentrations in each hollow fiber system revealed an elimination rate (kel) of 0.11 ± 0.05 h-1 (half-life of 9.8 h). Inhibitory sigmoid maximal effect (Emax) modeling revealed that the highest Emax was 3.15 ± 1.84 log10 cfu/ml on day 3, and the exposure mediating 50% of Emax (EC50) was a 0-24 h area under the concentration time curve (AUC0-24) to MIC ratio of 41.99 ± 31.78 (r2=0.99). The EC80 was an AUC0-24/MIC of 102.11. However no moxifloxacin concentration killed the bacteria to burdens below the starting inoculum. There was re-growth beyond day 3 in all doses, with replacement by resistant subpopulation that had an MIC >32 mg/l by the end of experiment. A quadratic function best described the relationship between AUC0-24/MIC and moxifloxacin-resistant subpopulation. Monte Carlo simulations of 10,000 patients revealed that the 400-800 mg/day doses would achieve or exceed the EC80 in ≤12.5% of patients. The moxifloxacin susceptibility breakpoint was 0.25 mg/l, which means almost all M. abscessus clinical strains were moxifloxacin-resistant by these criteria. While moxifloxacin´s efficacy against M. abscessus was poor, formal combination therapy studies with moxifloxacin are still recommended.

97
Chapter 6
Cha
pter
6
Moxifloxacin in M. abscessus hollow-fiber model
Introduction
Mycobacterium abscessus is a rapidly growing mycobacterium that is notorious because of resistance to most antibiotics.1 Pulmonary disease due to M. abscessus infection is chronic and relentless. Current regimens used to treat M. abscessus consist of a combination of amikacin, a macrolide, and either cefoxitin or imipenem; however the regimens fail in most patients.2 Based on static in vitro models, amikacin is considered the key antibiotic in the treatment regimens.3-6 We recently demonstrated that the efficacy of amikacin based on concentration-time profiles achievable in human lungs as recapitulated in the hollow-fiber system model of M. abscessus (HFS-Mab) was poor, and failed to kill the bacteria below stasis.7 This, as well as clinical experience of high failure rates of amikacin-based regimens, means that there is an urgent need to find new antibiotics and optimize their doses. Here we approached this by conducting recommended formal pharmacokinetic/pharmacodynamic (PK/PD) evaluation of alternative antibiotics with potential bactericidal activity.8
The 8-methoxy fluoroquinolone, moxifloxacin, has been shown to have excellent efficacy against M. tuberculosis, M. avium and M. kansasii.9-12 In addition, continuation regimens currently used to treat pulmonary M. abscessus include moxifloxacin in the combination, starting from the second month of therapy. Given these prior successes of moxifloxacin, as well as its current role in the treatment of M. abscessus, we also tested it against this species. We performed formal moxifloxacin PK/PD work in the hollow fiber system model of pulmonary M. abscessus. We exposed M. abscessus to moxifloxacin concentration-time profiles as encountered in lungs of patients with pneumonia.13 Repetitive day-to-day sampling of hollow fiber systems is a major advantage in identifying the evolution of bacteria in response to the periodic fluctuation of antibiotic concentrations, vital given M. abscessus’ propensity to develop acquired drug resistance.8,14-17
Materials and Methods
Bacteria, antibiotic and growth conditionsStock cultures of M. abscessus ATCC 19977 (American Type Culture Collection, Manassas, VA) were stored at -80 °C in Middlebrook 7H9 broth supplemented with 10% OADC (Remel, Lenexa, KS) and 15% glycerol, were used for all the experiments. One vial was thawed before each assay and incubated for 24-48 h at 30 °C to achieve logarithmic growth phase (log-phase). Moxifloxacin hydrochloride was purchased from Baylor University Medical Center pharmacy. On the day of use, the moxifloxacin was diluted in sterile water to desired concentrations for the assays.

98
Chapter 6
Antimicrobial susceptibility testing and mutation frequencyThe MIC was identified using broth macrodilution in Middlebrook 7H9 broth (here termed “broth”) as well as by use of the E-test (BioMerieux, Durham, NC). In addition to the turbidity test, colony-forming units (cfu) per ml were enumerated for each concentration evaluated in the broth macrodilution test. In this test, the MIC was defined as the lowest concentration associated with ≥99% decrease in cfu/ml compared to untreated control and MBC as the concentration corresponding to >99.9% of kill. Mutation frequency was determined for the inoculum by culturing 0.2 ml on Middlebrook 7H10 agar plates (here termed “agar”) supplemented with 3 times the moxifloxacin MIC. Cultures were incubated for 5 days.
Exposure-response studies in the Hollow-fiber systemThe hollow fiber system model of pulmonary M. abscessus (HFS-Mab) has been previously used to perform PK/PD evaluation of amikacin.7 Twenty milliliters of 6 log10 cfu/ml M. abscessus in log-phase were inoculated into the peripheral compartment of each of seven hollow fiber cartridges (FiberCell Systems, Frederick, MD). Doses that mimicked the non-protein bound plasma 0-24h area under the concentration time curve (AUC0-24), peak concentrations, and time to maximum concentration, achieved in humans treated with moxifloxacin doses of 0, 25, 50, 100, 200, 400 and 800 mg were administered to the central compartment once daily via computerized syringe pumps.18
These exposures were chosen because they represent the range of clinically tolerated doses of moxifloxacin. Treatment was for 21 days of daily therapy. A plasma-to-lung epithelial lining fluid penetration ratio of 1 was assumed, based on the literature.13,19,20 As an example, the standard 400 mg a day dose was expected to achieve a peak concentration of 4.2 mg/l, and a half-life of 12 h, translating to an AUC0-24/MIC ratio of 28.3. Actual moxifloxacin concentrations achieved in all the systems were validated by repetitive sampling of 1 ml from the central compartment of each HFS-Mab during the first three days at 0, 1, 6, 9, 12, 18, 23.5, 25, 30, 33, 36, 42, and 47.5 h post dose. In order to quantify the M. abscessus burden, 1 ml of the peripheral compartment culture contents was removed from each system on days 0, 1, 2, 3, 5, 7, 10,14 and 21. The samples were washed with saline to avoid antibiotic carryover, after which samples were serially diluted and cultured on agar. To quantify the moxifloxacin-resistant M. abscessus cfu/ml, the same samples were also inoculated on agar supplemented with 3 times the moxifloxacin MIC.
Drug assayMoxifloxacin concentrations in the samples collected from the central compartment of HFS-Mab were analyzed by LC/MS/MS. Moxifloxacin and moxifloxacin-13Cd3 (internal standard) were purchased from SIGMA (St. Louis, MO) and Santa Cruz Biotech (Santa Cruz, CA), respectively. Calibrator, controls and internal standard were included in each analytical run for quantitation. Stock solutions of moxifloxacin and internal standard were prepared in 80:20

99
Chapter 6
Cha
pter
6
Moxifloxacin in M. abscessus hollow-fiber model
methanol:water at a concentration of 1mg/ml and stored at -20 ºC. A 7-point calibration curve was prepared by diluting moxifloxacin stock solution in drug-free media (0.1, 0.2, 1, 2, 5, 10, 20 mg/l). Quality control samples were prepared by spiking media with stock standards for two levels of controls. Samples were prepared in 96-well microtiter plates by the addition of 10 µl of calibrator, quality controls or sample to 190 µl 0.1% formic acid in water containing 10 mg/l internal standard followed by vortex. Chromatographic separation was achieved on an Acquity UPLC HSS T3 1.8 μm 50 x 2.1 mm analytical column (Waters) maintained at 30 °C at a flow of 0.2 ml/min with a binary gradient with a total run time of 6 min. The observed ions (m/z) values of the fragment ions were moxifloxacin (m/z 402.2→384.2) and internal standard, Moxifloxacin-13CD (m/z 406.2→388.3). Sample injection and separation was performed by an Acquity UPLC interfaced with a Xevo TQ mass spectrometer (Waters). All data were collected using MassLynx version 4.1 SCN810. The limit of quantitation for this assay was 0.1 mg/l. The inter- and intra-day variation was 1.5% and 9.4%, respectively.
Pharmacokinetic and pharmacodynamic modelingAll drug concentrations from each of the HFS-Mab units at all time points were co-modeled using the ADAPT 5 software (Biomedical Simulations Resource, University of Southern California, CA). Steps used in the pharmacokinetic parameter analysis were as described in detail in prior studies.10,21 The pharmacokinetic parameter estimates identified were used to calculate the observed AUC0-24 and AUC0-24/MIC ratios. Exposure-response was modeled using the inhibitory sigmoid Emax model, a standard model recommended for examination of effect of antibiotics whether they are static or cidal, and can be seen in both clinical and laboratory data.22-24 Total bacterial burden was used as the response parameter, while drug exposure was expressed as AUC0-24/MIC ratio. For moxifloxacin-resistance emergence, we used the quadratic model we have identified in the past, with drug exposure examined versus size of moxifloxacin-resistant sub-population each sampling day.8,25
Monte Carlo simulationsIn order to put our findings into clinical context, we performed a 10,000 patient Monte Carlo simulation to identify two important clinical aspects. First, we wanted to identify the clinical dose best able to achieve or exceed the EC80, which is the exposure mediating 80% of maximal kill (Emax). This exposure is considered “optimal” since Emax is on an asymptote, and is independent of whether a drug is cidal or static. Second, we wanted to identify the susceptibility breakpoint, which we considered as the MIC below which >10% of patients achieve the EC80 with standard dosing or with the highest dose that can be administered. Steps in the Monte Carlo simulations were as detailed in recommendations and in our prior work.8,26 We examined moxifloxacin doses of 400 mg a day, 600 mg a day, and 800 mg a day. We entered the two compartment model pharmacokinetic parameter estimates and covariance identified by Kees et al. into subroutine PRIOR of ADAPT 5, as shown in Table 1.27 We assumed a 1:1 AUC0-24 ratio in the lung versus plasma.13An MIC distribution from Korea 28

100
Chapter 6
was used; as comparators data on MIC distribution from Nijmegen, the Netherlands and the Dallas-Fort Worth metroplex, USA was included for comparison of the impact of the new susceptibility breakpoint.
Results
In this study the moxifloxacin MIC for the M. abscessus laboratory strain was 2 mg/l. In addition, the concentration associated with >99.9% of kill, or MBC, was 8 mg/l. Thus the MBC/MIC ratio was 4, which means that moxifloxacin would be considered bactericidal according to standard convention.29 The mutation frequency to 3 times the moxifloxacin MIC was 2.11 ± 0.16 x 10-5 in repeat experiments.
In the HFS-Mab dose-effect studies, the measured moxifloxacin concentrations in each HFS-Mab unit revealed an elimination rate constant (kel) of 0.11 ± 0.05 h-1, which translates to a half-life of 9.8 h, consistent with moxifloxacin pharmacokinetics in a recent 241 patient clinical study.18 As an example, the r2 for intended drug concentrations, exposures and half-life of the 400 mg standard dose versus observed concentrations was 0.98, which means intended pharmacokinetics were recapitulated well. Figure 1 shows the time-kill curves for each of the moxifloxacin exposures in the HFS-Mab. None of the evaluated exposures achieved a considerable killing effect of the M. abscessus.
The relationship between the AUC0-24/MIC and M. abscessus burden in the HFS-Mab is shown in Figure 2; the highest Emax was encountered on day 3. The day 3 parameters were an Emax
of 3.15 ± 1.84 log10 cfu/ml, a Hill´s slope of 1.56 ± 0.63, and an EC50 that was an AUC0-24/MIC ratio of 41.99 ± 31.78 (r2=0.987). Based on the inhibitory sigmoid Emax relationship for day 3, the EC80 was calculated as an AUC0-24/MIC ratio of non-protein bound moxifloxacin of 102.11. The Emax appears high mainly because in non-treated controls the M. abscessus grew
Pharmacokinetic parameter Observed in patients (entered into subroutine PRIOR)
Simulated in 10,000 patients
Parameter estimate
IIV as % CV Parameter estimate
IIV as % CV
Total clearance (l/h) 11.3 23.7 11.3 14.58Intercompartmental clearance (l/h) 47.7 * 47.7 19.19Absorption rate constant (h-1) 1.09 135 1.09 143Central volume (l) 55.6 * 55.6 18.40Peripheral volume (l) 59.6 15.3 59.6 14.99
Table 1. Pharmacokinetic parameter estimates utilized in Monte Carlo simulations.
* Fixed in the pharmacokinetic model by Kees et al.27

101
Chapter 6
Cha
pter
6
Moxifloxacin in M. abscessus hollow-fiber model
to large bacterial burdens of about 10 log10 cfu/ml, starting from 6.0 log10 cfu/ml at the start of the experiment. Indeed, Figure 2 shows that even on day 3, moxifloxacin never reduced the bacterial burden below the starting inoculum (stasis).
Figure 3 shows that acquired drug resistance emerged after 10 days of exposure. The figure further shows that the moxifloxacin resistant sub-population had replaced the total population in the systems exposed to the 3 highest doses by day 21.The relationship between size of resistant subpopulation and moxifloxacin exposure was described by a system of changing “U” shaped curves, in effect a quadratic function. However, even at the highest AUC0-24/MIC ratios tested, moxifloxacin was not able to suppress acquired drug resistance. Indeed, the MIC of cultures from the systems exposed to the 3 highest doses had changed from 2 mg/l to >32 mg/l by day 21.
Figure 4 shows the result of Monte Carlo simulations of 10,000 simulated subjects, based on the largest published moxifloxacin MIC distribution of M. abscessus, which is from Korea.25 Overall, the cumulative fraction of response or proportion of patients that achieved or exceeded EC80, given the MIC distribution, was poor for all doses. For the standard dose of 400 mg daily, only 4.7% of patients would achieve the EC80, for 600 mg a day 9.7%, and for 800 mg a day 12.5%. If the penetration ratio of moxifloxacin AUC achieved in the lung versus plasma were 2, as suggested by two outlier pharmacokinetic studies,19,20 the cumulative fraction of response would become 12.5% for 400 mg a day, 21.5% for 600 mg a day, and 26.6% for 800 mg a day. Either way, standard and high doses of moxifloxacin performed poorly.
Figure 1. Moxifloxacin exposure-effect against M. abscessus in the HFS. None of the moxifloxacin exposures evaluated attained killing below the starting inoculum of 6.0 log10 cfu/ml at any point during the study. Regrowth was observed after day 3 in all the systems.
Figure 2. Moxifloxacin exposure versus M. abscessus burden. Inhibitory sigmoid Emax curves are shown for the first 3 days, prior to replacement of microbial population by drug-resistant subpopulation. The highest Emax was encountered on day 3, but not even that produced kill below the stasis level.
0 3 6 9 12 15 18 210
1
2
3
4
5
6
7
8
9
10
11
AUC/MIC: 0 (control)
AUC/MIC: 53.93
AUC/MIC: 12.66AUC/MIC: 32.85
AUC/MIC: 1.89
AUC/MIC: 6.79AUC/MIC: 3.87
Day
M. a
bsce
ssus
Log 1
0 CFU
/mL
0 10 20 30 40 50 600
1
2
3
4
5
6
7
8
9
10
Day 1Day 2Day 3Stasis
Moxifloxacin AUC0-24/MIC
M. a
bsce
ssus
log 1
0 CFU
/mL
M. a
bsce
ssus
log 10
CFU
/ml
M. a
bsce
ssus
log 10
CFU
/ml

102
Chapter 6
Figure 4 also shows the target attainment at different moxifloxacin MICs. The Figure shows that at the standard dose, and at 600 mg a day, the target attainment to achieve EC80 fell to below 0.9 (i.e., 90%) after the MIC of 0.25 mg/l. This is the PK/PD derived moxifloxacin susceptibility breakpoint for M. abscessus, since above this MIC most patients will not achieve optimal microbial effect. It should be borne in mind that even this “optimal kill” would still not kill below bacterial burden at the start of treatment. If dose were increased to 800 mg a day, the susceptibility breakpoint only changes by one tube dilution. In contrast, the epidemiologic cut-off point for the Korean MIC distribution would be 16 mg/l. In Figure 5, we show the MIC distributions of M. abscessus from our two clinical practice locations in the Netherlands and the Dallas-Fort Worth metroplex in Texas. Given the MIC distributions in Figure 5, and the proposed susceptibility breakpoint of 0.25 mg/l, then almost all M. abscessus isolates in North Texas and the Netherlands as of 2015 were intrinsically resistant to moxifloxacin. Accordingly, the cumulative fraction of responses in these two locales would fall to 0% even for a dose of 800 mg a day.
0 3 6 9 12 15 18 210123456789
101112
AUC/MIC: 1.89AUC/MIC: 3.87AUC/MIC: 6.79
ControlA.
Time (days)
M. a
bsce
ssus
Log
10 C
FU/m
L
0 3 6 9 12 15 18 210123456789
101112
AUC/MIC: 32.85AUC/MIC: 12.66
AUC/MIC: 53.93
B.
Time (days)
M. a
bsce
ssus
Log
10 C
FU/m
L
0 5 10 15 20 25 30 35 40 45 50 550
2
4
6
8
10
12
14 Day 1Day 2Day 3Day 7Day 10Day 14Day 21
C.
AUC/MIC
M. a
bsce
ssus
Log
10 C
FU/m
L
0 3 6 9 12 15 18 210123456789
101112
AUC/MIC: 1.89AUC/MIC: 3.87AUC/MIC: 6.79
ControlA.
Time (days)
M. a
bsce
ssus
Log
10 C
FU/m
L
0 3 6 9 12 15 18 210123456789
101112
AUC/MIC: 32.85AUC/MIC: 12.66
AUC/MIC: 53.93
B.
Time (days)
M. a
bsce
ssus
Log
10 C
FU/m
L
0 5 10 15 20 25 30 35 40 45 50 550
2
4
6
8
10
12
14 Day 1Day 2Day 3Day 7Day 10Day 14Day 21
C.
AUC/MIC
M. a
bsce
ssus
Log
10 C
FU/m
L
Figure 3. Changes in total and moxifloxacin-resistant subpopulations with time. Total M. abscessus population (solid line) and moxifloxacin-resistant subpopulation (hatched line) over the course of 21 days of treatment with different moxifloxacin AUC0-24/MIC exposures. A: Control and three lower exposures. While the drug-resistant subpopulation increased with time, especially beyond day 10, it had not completely replaced the total population in these lower doses, with the highest dose among these lower doses associated with the largest moxifloxacin-resistant subpopulation. B: At the three highest exposures, the total population had been completely replaced by moxifloxacin-resistant subpopulation by day 14. C: The relationship between moxifloxacin AUC/MIC and size of sub-resistant subpopulation is described by systems of inverted “U” curves. At day 1 it was a straight line, then it switches to an inverted “U” curve that was more pronounced by day 21.
A B
C
M. a
bsce
ssus
log 10
CFU
/ml
M. a
bsce
ssus
log 10
CFU
/ml
M. a
bsce
ssus
log 10
CFU
/ml

103
Chapter 6
Cha
pter
6
Moxifloxacin in M. abscessus hollow-fiber model
Discussion
Moxifloxacin, and quinolones in general, are considered ideal antibiotics given that they are bactericidal, are active against a broad spectrum of bacteria, are available both in oral and parenteral formulations, have a broad distribution in organs and tissues and can achieve therapeutic concentrations both in the intra and extracellular environment.21,30 Indeed, these agents have now found broad use in treatment of many slow growing and rapidly growing mycobacteria. The MBC/MIC ratio calculated in this study would seem to indicate that the drug would work well against M. abscessus. However, in our formal PK/PD study we found that even at high exposures, moxifloxacin does not reduce bacterial burden below starting inoculum and resistance arises fairly quickly during monotherapy. Therefore, the meaning of standard MBC/MIC assays used to determine bactericidal effect is questionable.
Moxifloxacin has been used for M. abscessus pulmonary disease at 400 mg/daily, after the discontinuation of parenteral antibiotics that are administered during the first 1-2 months. In that role, it is used as part of combination therapy. However, we show that its performance in the HFS-Mab does not inspire continued confidence even for that role. Inhibitory sigmoid Emax based target exposures such as EC80-EC90 have been used as target exposures to identify susceptibility breakpoints of several bacteria, including mycobacteria.8,10-12,31-37 We chose the EC80 exposure as optimal kill, which we have found to best translate to the clinic for other mycobacteria such as M. tuberculosis and M. avium, and indeed to other Gram-negative and Gram-positive organisms, even with drugs that do not kill below stasis.31-35 Indeed, indices of microbial such as 3.0 log10 cfu/ml kill that are commonly used in work with Gram-positive cocci
Figure 5. Moxifloxacin MIC distribution in three different practice locations. The MIC distribution from Korea was used for the Monte Carlo simulations, the other two settings are used as comparators. Notably, MIC distribution from the Netherlands and from Texas, USA, is showing most isolates as resistant to moxifloxacin.
0.125 0.25 0.5 1 2 4 8 16 32 640.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
400 mg600 mg800 mgMIC proportion
Minimum inhibitory concentration (mg/L)
Prop
ortio
n of
pat
ient
s (%
)
Figure 4. Probability target attainment of three different moxifloxacin doses in 10,000 patients.Target attainment probability at each MIC fell after the 0.25 mg/l to 0% for a dose of 400 mg, and just above 60% for 600 mg. That for doses of 800 mg a day plummeted after 0.5 mg/l. Since the proportion of clinical isolates with an MIC >0.25 mg/l is large (and 0% of these achieve the EC80), the fraction of all patients who will achieve the EC80 is very small.
0.13 0.25 0.50 1.00 2.00 4.00 8.00 16.00 32.000
5
10
15
20
25
30
35
40
Seoul, Korea (n= 64)
Dallas-Fort Worth, Texas (n=23)Nijmegen, Netherlands (n=47)
Minimum inhibitory concentration (mg/L)
M. a
bsce
ssus
isol
ate
coun
t
Minimum inhibitory concentration (mg/l) Minimum inhibitory concentration (mg/l)
0.13 0.25 0.50 1.00 2.00 4.00 8.00 16.00 32.000
5
10
15
20
25
30
35
40
Seoul, Korea (n= 64)
Dallas-Fort Worth, Texas (n=23)Nijmegen, Netherlands (n=47)
Minimum inhibitory concentration (mg/L)
M. a
bsce
ssus
isol
ate
coun
t

104
Chapter 6
and Gram-negative bacilli are rarely achieved in work with mycobacteria with a single agent. Nevertheless, a similar approach to ours has been used to identify susceptibility breakpoints even at exposures that are merely associated with stasis for several drugs, with tigecyline and Enterobacteriaceae.35-37 Work done with M. tuberculosis in the hollow fiber system to identify the EC80 in inhibitory sigmoid Emax model for drugs that kill below stasis as well as those that do, in tandem with Monte Carlo simulations, has shown to predict optimal clinical exposures, doses, and susceptibility breakpoints to within 94% of the value later on identified in the clinic, based on FDA and EMA submissions.8,38-42 This hollow fiber work, which identified these exposures and breakpoints later confirmed in the clinic, was done with single strains of the M. tuberculosis, with the MIC distribution taken into account only at the stage of Monte carlo simulations. We report here on moxifloxacin dosing and optimal exposures using the same hollow fiber system and the same inhibitory sigmoid Emax models and EC80 exposures in tandem with Monte Carlo simulations, demonstrating limited activity at doses up to 800 mg a day, and expect the same degree of accuracy as in tuberculosis.
In the treatment of M. abscessus, moxifloxacin is used as part of combination therapy. Thus, it could be that it would show efficacy in combination therapy. Formal combination therapy studies with moxifloxacin in the treatment of M. abscessus will need to be performed in the HFS-Mab, in order to explore potential synergy with other agents before definitive recommendations can be made to remove it from current regimens. So far, we have examined moxifloxacin high dose in combination with tigecyline and clarithromycin, and found that it did not add to the effect of these drugs (manuscript in preparation). Similarly, we found that amikacin which also failed to kill M. abscessus appreciably as monotherapy, did not add to the effect of clarithromycin and cefoxitin, which suggest that findings in monotherapy studies reflect well the efficacy of drugs in combination regimens used to treat this infection (manuscript in preparation). Thus, findings of moxifloxacin monotherapy are likely to be borne out in combination therapy.
Acquired moxifloxacin resistance has been described in mycobacteria, particularly for M. tuberculosis.11 In the absence of other companion antibiotics, moxifloxacin monotherapy rapidly leads to resistance as was the case here for M. abscessus. Unfortunately, in this study no moxifloxacin exposure was associated with resistance suppression. The relationship between acquired moxifloxacin resistance and moxifloxacin AUC/MIC was best described by a quadratic function, similar to our findings with M. tuberculosis.25 However, the underlying mechanism for moxifloxacin resistance in M. abscessus deserves further exploration; DNA gyrase is still an interesting bacterial target against which new inhibitors are being developed with evaluation of their potential against M. abscessus and other nontuberculous mycobacteria.43
Finally, according to guidelines, the current breakpoint to determine moxifloxacin resistance to rapidly growing mycobacteria such as M. abscessus is ≥4 mg/l in cation-adjusted Mueller Hinton broth44, which in a wide MIC distribution28 will barely allow the use of moxifloxacin

105
Chapter 6
Cha
pter
6
Moxifloxacin in M. abscessus hollow-fiber model
in 50% of the cases. A 1.0 mg/l susceptibility breakpoint has also been used by the CLSI.44 In this study, although using a different type of broth (Middlebrook 7H9), we found a PK/PD derived breakpoint of 0.25 mg/l. Examination of MIC distributions in our clinical practices demonstrated that at this proposed breakpoint, most isolates would be a priori resistant to moxifloxacin. This comports well with failure of the moxifloxacin-containing regimens in clinical practice. Our findings suggest that the poor outcome observed when treating M. abscessus is no more than a reflection of the amount of intrinsically resistant isolates that are circulating and infecting patients. The approach that utilizes the hollow fiber system model and the EC80 or EC90, followed by Monte Carlo simulations, has been found to be highly accurate in predicting MICs above which combination therapy fails in clinical studies, even for drugs that do not kill below the stasis line, in the case of tuberculosis and other more mundane bacteria such as Gram-negative rods.25,45-53 Thus our findings are likely accurate, but this will require clinical validation in the future. Such confirmation will require multivariable pharmacometric analyses of clinical data, which does not currently exist for moxifloxacin and M. abscessus pulmonary disease.54 Indeed, no such data exists as far as we know for any of the drugs used in treatment of pulmonary M. abscessus, since there is lack of clinical trials and large prospective clinical cohort studies for this disease. Thus, the PK/PD derived breakpoint is a proposed breakpoint to be used for clinical decision making until a large enough data set could better identify further breakpoints.
There are some limitations in this study. We performed our evaluation with one laboratory strain. Although the strain used was the type strain, ideally this work should be extended to clinical isolates with different susceptibility profiles. Indeed, the type strain was not from a patient with pneumonia, but an abscess after trauma. It is not clear if strains that cause lung disease are intrinsically different from those that cause extrapulmonary disease. Nevertheless, even when single strains have been used in PK/PD studies in the hollow fiber with other bacteria, results have been found to be fairly accurate and to allow generalizations.11,25,38-42,45-53 Moreover, here we only report the effect of moxifloxacin alone and there still a need to evaluate its performance as part of combination therapy. However, studies evaluating different moxifloxacin-based combinations to explore the potential of synergistic combinations as well to explore the intracellular model for pulmonary disease have been performed, and are being prepared for publication.
In summary, we used the recently developed pre-clinical model for M. abscessus disease to evaluate and then identify the efficacy of moxifloxacin, which demonstrated rapid appearance of resistance. Efficacy was poor, and moxifloxacin even at high doses is not expected to work in the clinic. We used the PK/PD studies to identify and propose a new moxifloxacin susceptibility breakpoint of 0.25 mg/l.

106
Chapter 6
Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother. 2012;67:810-8.Jarand J, Levin A, Zhang L, Huitt G, Mitchell JD, Daley CL. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin Infect Dis. 2011;52:565–571.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace RJ, Jr, Winthrop K. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175: 367–416.Ferro BE, van Ingen J, Wattenberg M, van Soolingen D, Mouton JW. Time-kill kinetics of antibiotics active against rapidly growing mycobacteria. J Antimicrob Chemother. 2015;70:811–817.van Ingen J, Ferro BE, Hoefsloot W, Boeree MJ, van Soolingen D. Drug treatment of pulmonary nontuberculous mycobacterial disease in HIV-negative patients: the evidence. Expert Rev Anti Infect Ther. 2013;11:1065– 1077.Lee MR, Sheng WH, Hung CC, Yu CJ, Lee LN, Hsueh PR. Mycobacterium abscessus complex infections in humans. Emerg Infect Dis. 2015;21:1638–1646.Ferro, BE, Srivastava S, van Ingen J, Deshpande D, Sherman C, Pasipanodya JG, van Soolingen D, Mouton, JW, Gumbo T. Amikacin pharmacokinetics/pharmacodynamics in a novel hollow-fiber Mycobacterium abscessus disease model. Antimicrob Agents Chemother. 2016; 60:1242– 1248. Gumbo T, Angulo-Barturen I, Ferrer-Bazaga S. Pharmacokineticpharmacodynamic and dose-response relationships of antituberculosis drugs: recommendations and standards for industry and academia. J Infect Dis. 2015; 211(Suppl 3):S96 –S106. Sindelar G, Zhao X, Liew A, Dong Y, Lu T, Zhou J, Domagala J, Drlica K. Mutant prevention concentration as a measure of fluoroquinolone potency against mycobacteria. Antimicrob Agents Chemother. 2000;44: 3337–3343. Deshpande D, Srivastava S, Meek C, Leff R, Hall GS, Gumbo T. Moxifloxacin pharmacokinetics/pharmacodynamics and optimal dose and susceptibility breakpoint identification for treatment of disseminated Mycobacterium avium infection. Antimicrob Agents Chemother. 2010;54:2534– 2539. Gumbo T, Louie A, Deziel MR, Parsons LM, Salfinger M, Drusano GL. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis. 2004;190:1642–1651. Srivastava S, Pasipanodya J, Sherman CM, Meek C, Leff R, Gumbo T. Rapid drug tolerance and dramatic sterilizing effect of moxifloxacin monotherapy in a novel hollow-fiber model of intracellular Mycobacterium kansasii disease. Antimicrob Agents Chemother. 2015;59:2273–2279. Simon N, Sampol E, Albanese J, Martin C, Arvis P, Urien S, Lacarelle B, Bruguerolle B. Population pharmacokinetics of moxifloxacin in plasma and bronchial secretions in patients with severe bronchopneumonia. Clin Pharmacol Ther. 2003;74:353–363. Pasipanodya JG, Nuermberger E, Romero K, Hanna D, Gumbo T. Systematic analysis of hollow fiber model of tuberculosis experiments. Clin Infect Dis. 2015;61(Suppl 1):S10 –S17. Srivastava S, Gumbo T. In vitro and in vivo modeling of tuberculosis drugs and its impact on optimization of doses and regimens. Curr Pharm Des. 2011;17:2881–2888. Gumbo T, Lenaerts AJ, Hanna D, Romero K, Nuermberger E. Nonclinical models for antituberculosis drug development: a landscape analysis. J Infect Dis. 2015;211(Suppl 3):S83–S95.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
References

107
Chapter 6
Cha
pter
6
Moxifloxacin in M. abscessus hollow-fiber model
Schmalstieg AM, Srivastava S, Belkaya S, Deshpande D, Meek C, Leff R, van Oers NS, Gumbo T. The antibiotic resistance arrow of time: efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob Agents Chemother. 2012;56:4806–4815. Zvada SP, Denti P, Sirgel FA, Chigutsa E, Hatherill M, Charalambous S, Mungofa S, Wiesner L, Simonsson US, Jindani A, Harrison T, McIlleron HM. Moxifloxacin population pharmacokinetics and model-based comparison of efficacy between moxifloxacin and ofloxacin in African patients. Antimicrob Agents Chemother. 2014;58:503–510. Capitano B, Mattoes HM, Shore E, O’Brien A, Braman S, Sutherland C, Nicolau DP. Steady-state intrapulmonary concentrations of moxifloxacin, levofloxacin, and azithromycin in older adults. Chest. 2004;125:965–973. Soman A, Honeybourne D, Andrews J, Jevons G, Wise R. Concentrations of moxifloxacin in serum and pulmonary compartments following a single 400 mg oral dose in patients undergoing fibre-optic bronchoscopy. J Antimicrob Chemother. 1999;44:835–838. Musuka S, Srivastava S, Siyambalapitiyage Dona CW, Meek C, Leff R, Pasipanodya J, Gumbo T. Thioridazine pharmacokineticpharmacodynamic parameters “wobble” during treatment of tuberculosis: a theoretical basis for shorter-duration curative monotherapy with congeners. Antimicrob Agents Chemother. 2013;57:5870–5877. Gumbo T. 2011. General principles of antimicrobial therapy, p 1365– 1382. In Brunton LL, Chabner BA, Knollmann BC (ed), Goodman & Gilman’s the pharmacological basis of therapeutics, 12th ed. McGraw Hill Medical, New York, NY.Mouton JW, Punt N, Vinks AA. Concentration-effect relationship of ceftazidime explains why the time above the MIC is 40 percent for a static effect in vivo. Antimicrob Agents Chemother. 2007;51:3449–3451. Muller AE, Punt N, Mouton JW. Optimal exposures of ceftazidime predict the probability of microbiological and clinical outcome in the treatment of nosocomial pneumonia. J Antimicrob Chemother. 2013; 68:900– 906.Gumbo T, Dona CS, Meek C, Leff R. Pharmacokineticspharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother. 2009; 53:3197–3204. Pasipanodya J, Gumbo T. An oracle: antituberculosis pharmacokinetics- pharmacodynamics, clinical correlation, and clinical trial simulations to predict the future. Antimicrob Agents Chemother. 2011;55:24–34. Kees MG, Schaeftlein A, Haeberle HA, Kees F, Kloft C, Heininger A. Population pharmacokinetics and pharmacodynamic evaluation of intravenous and enteral moxifloxacin in surgical intensive care unit patients. J Antimicrob Chemother. 2013;68:1331–1337. Koh WJ, Jeon K, Lee NY, Kim BJ, Kook YH, Lee SH, Park YK, Kim CK, Shin SJ, Huitt GA, Daley CL, Kwon OJ. Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am J Respir Crit Care Med. 2011;183:405–410. Pankey GA, Sabath LD. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin Infect Dis. 2004;38:864–870. Owens RC, Jr, Ambrose PG. 2001. Pharmacodynamics of quinolones, 155–176. In Nightingale CH, Murakawa T, Ambrose PG (ed), Antimicrobial pharmacodynamics in theory and clinical practice, 1st ed. Marcel Dekker, New York, NY.Drusano GL, Preston SL, Hardalo C, Hare R, Banfield C, Andes D, Vesga O, Craig WA. Use of preclinical data for selection of a phase II/III dose for evernimicin and identification of a preclinical MIC breakpoint. Antimicrob Agents Chemother. 2001;45:13–22.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.

108
Chapter 6
Deshpande D, Srivastava S, Meek C, Leff R, Gumbo T. Ethambutol optimal clinical dose and susceptibility breakpoint identification by use of a novel pharmacokinetic-pharmacodynamic model of disseminated intracellular Mycobacterium avium. Antimicrob Agents Chemother. 2010;54:1728–1733.Srivastava S, Musuka S, Sherman C, Meek C, Leff R, Gumbo T. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J Infect Dis. 2010;201:1225–1231. Deshpande D, Pasipanodya JG, Gumbo T. Azithromycin dose to maximize efficacy and suppress acquired drug resistance in pulmonary Mycobacterium avium disease. Antimicrob Agents Chemother. 2016;60:2157– 2163. Labreche MJ, Graber CJ, Nguyen HM. Recent updates on the role of pharmacokinetics-pharmacodynamics in antimicrobial susceptibility testing as applied to clinical practice. Clin Infect Dis. 2015;61:1446–1452. Ambrose PG, Meagher AK, Passarell JA, Van Wart SA, Cirincione BB, Rubino CM, Korth-Bradley JM, Babinchak T, Ellis-Grosse E. Use of a clinically derived exposure-response relationship to evaluate potential tigecycline-Enterobacteriaceae susceptibility breakpoints. Diagn Microbiol Infect Dis. 2009;63:38–42. Gumbo T, Pasipanodya JG, Romero K, Hanna D, Nuermberger E. Forecasting accuracy of the hollow fiber model of tuberculosis for clinical therapeutic outcomes. Clin Infect Dis. 2015;61(Suppl 1):S25–S31. Gumbo T, Pasipanodya JG, Nuermberger E, Romero K, Hanna D. Correlations between the hollow fiber model of tuberculosis and therapeutic events in tuberculosis patients: learn and confirm. Clin Infect Dis. 2015;61(Suppl 1):S18 –S24. Cavaleri M, Manolis E. Hollow fiber system model for tuberculosis: the European Medicines Agency experience. Clin Infect Dis. 2015;61(Suppl 1): S1–S4. Romero K, Clay R, Hanna D. Strategic regulatory evaluation and endorsement of the hollow fiber tuberculosis system as a novel drug development tool. Clin Infect Dis. 2015;61(Suppl 1):S5–S9. Chilukuri D, McMaster O, Bergman K, Colangelo P, Snow K, Toerner JG. The hollow fiber system model in the nonclinical evaluation of antituberculosis drug regimens. Clin Infect Dis. 2015;61(Suppl 1):S32–S33. Locher CP, Jones SM, Hanzelka BL, Perola E, Shoen CM, Cynamon MH, Ngwane AH, Wiid IJ, van Helden PD, Betoudji F, Nuermberger EL, Thomson JA. A novel inhibitor of gyrase B is a potent drug candidate for treatment of tuberculosis and nontuberculosis mycobacterial infections. Antimicrob Agents Chemother. 2015;59:1455–1465. Clinical Laboratory Standards Institute. 2011. Susceptibility testing of Mycobacteria, Nocardiae and other aerobic Actinomycetes—approved standard M24-A2, 2nd ed. CLSI, Wayne, PA. Booker BM, Smith PF, Forrest A, Bullock J, Kelchlin P, Bhavnani SM, Jones RN, Ambrose PG. Application of an in vitro infection model and simulation for reevaluation of fluoroquinolone breakpoints for Salmonella enterica serotype Typhi. Antimicrob Agents Chemother. 2005;49:1775– 1781. Defife R, Scheetz MH, Feinglass JM, Postelnick MJ, Scarsi KK. Effect of differences in MIC values on clinical outcomes in patients with bloodstream infections caused by gram-negative organisms treated with levofloxacin. Antimicrob Agents Chemother. 2009;53:1074–1079. Gumbo T. New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob Agents Chemother. 2010;54:1484–1491. Chigutsa E, Pasipanodya JG, Visser ME, van Helden PD, Smith PJ, Sirgel FA, Gumbo T, McIlleron H. Impact of nonlinear interactions of pharmacokinetics and MICs on sputum bacillary kill rates as a marker of sterilizing effect in tuberculosis. Antimicrob Agents Chemother. 2015;59:38–45.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.

109
Chapter 6
Cha
pter
6
Moxifloxacin in M. abscessus hollow-fiber model
48.
49.
50.
51.
52.
53.
Gumbo T, Pasipanodya JG, Wash P, Burger A, McIlleron H. Redefining multidrug-resistant tuberculosis based on clinical response to combination therapy. Antimicrob Agents Chemother. 2014;58:6111–6115. Gumbo T, Chigutsa E, Pasipanodya J, Visser M, van Helden PD, Sirgel FA, McIlleron H. The pyrazinamide susceptibility breakpoint above which combination therapy fails. J Antimicrob Chemother. 2014;69: 2420–2425. Williamson DA, Roberts SA, Bower JE, Vaughan R, Newton S, Lowe O, Lewis CA, Freeman JT. Clinical failures associated with rpoB mutations in phenotypically occult multidrug-resistant Mycobacterium tuberculosis. Int J Tuberc Lung Dis. 2012;16:216–220. van Ingen J, Aarnoutse R, de Vries G, Boeree MJ, van Soolingen D. Low-level rifampicin-resistant Mycobacterium tuberculosis strains raise a new therapeutic challenge. Int J Tuberc Lung Dis. 2011;15:990–992. Christianson S, Voth D, Wolfe J, Sharma MK. Re-evaluation of the critical concentration for ethambutol antimicrobial sensitivity testing on theMGIT960. PLoS One. 2014;9(9):e108911. Ambrose PM. 2016. Rational susceptibility test interpretive criteria. Perspectives from the USCAST. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM374226.pdf.


111
Antimicrob Agents Chemother. 2016;60:2895-900.
Chapter 7
Tigecycline is highly efficacious in Mycobacterium abscessus pulmonary disease
Beatriz E. FerroShashikant SrivastavaDevyani DeshpandeJotam G. PasipanodyaDick van SoolingenJohan W. MoutonJakko van IngenTawanda Gumbo

112
Chapter 7
Summary
Mycobacterium abscessus causes chronic pulmonary infections that are extremely difficult to cure. The currently recommended combination therapy is associated with high failure rates and relapse. Tigecycline has been explored in salvage regimens, with a response rate of 43% in those who received at least a month of therapy. We performed a dose-response study in a hollow-fiber system model of pulmonary M. abscessus infection in which we recapitulated tigecycline human pulmonary concentration-time profiles of 8 different doses for 21 days. We identified the maximal kill or efficacy in cfu per milliliter and the ratio of the 0- to 24-h area under the concentration-time curve to MIC (AUC/MIC) associated with 80% efficacy (EC80). The tigecycline efficacy was 5.38 ± 2.35 log10 cfu/ml, and the drug achieved the unprecedented feat of a bacterial level of 1.0 log10 cfu/ml below the pretreatment inoculum (1-log kill) of M. abscessus in the hollow-fiber system. The EC80 AUC/MIC ratio was 36.65, while that for a 1-log kill was 44.6. Monte Carlo experiments with 10,000 patients were used to identify the clinical dose best able to achieve the EC80 or 1-log kill. The standard dose of 100 mg/day had cumulative fractions of response of 51% for the EC80 and 46% for 1-log kill. For both the EC80 target and 1-log kill, the optimal tigecycline clinical dose was identified as 200 mg/day. The susceptibility breakpoint was <0.5 mg/l. Tigecycline is the most active single agent evaluated to date, and we propose that 200 mg/day be examined as the backbone of new combination therapy regimens to replace current treatment.

113
Cha
pter
7
Tigecycline efficacy against M. abscessus
Introduction
Mycobacterium abscessus is, for all the correct reasons, considered an “antibiotic nightmare”.1 This pathogen primarily causes chronic pulmonary infections that are extremely difficult to cure owing to the extensive drug resistance intrinsic to this organism.2 Personalized treatment regimens based on in vitro drug susceptibility testing and aggressive surgical resection yielded “cure” rates of only 57% in one case series.3 Even then, over the years there is relapse and death among those who have been “cured.” Thus, there has been a continued search for new drugs, with the hope to craft a new effective regimen.
Tigecycline, the first developed glycylcycline, was designed to overcome mechanisms of resistance that are known for older tetracyclines, such as the active efflux and the ribosome protection mechanisms. It is considered a bacteriostatic antibiotic, sometimes bactericidal, with demonstrated broad in vitro effects on several Gram-positive and Gram-negative bacteria, including some rapidly growing mycobacteria.4-7 This broad antimicrobial spectrum, its large volume of distribution (≥7 to 10 l/kg), and the known intracellular accumulation represent a sound rationale for the clinical use of tigecycline.4,5
Although tigecycline is FDA approved to be used for the treatment of complicated skin/soft tissue infections, complicated intra-abdominal infections, and community-acquired bacterial pneumonia, in a recent case series, tigecycline salvage regimens in 30 patients were associated with improvement in 43% of patients who received >1 month of therapy.8 These patients received doses of tigecycline designed for Gram-negative and Gram-positive bacterial infections. In patients treated with tigecycline for pneumonia caused by these other bacteria, the main determinant of a clinical response has been identified as a ratio of the free-drug area under the concentration-time curve from 0 to 24 h (fAUC0–24) to the MIC of >12.8 in plasma.9,10 This suggests a concentration-dependent effect of tigecycline and that doses could be optimized based on the free-drug AUC0–24/MIC ratios.
Here, we wanted to identify the efficacy of tigecycline and utilize it to identify optimal doses for treatment of pulmonary disease caused by M. abscessus.
Materials and Methods
MaterialsThe test strain was M. abscessus subsp. abscessus ATCC 19977 (American Type Culture Collection, Manassas, VA). Stocks of the mycobacteria were kept at -80 °C in Middlebrook 7H9 broth (Remel, Thermo Fisher Scientific, Lenexa, KS) supplemented with 10% oleic acid-albumin-dextrose-catalase (OADC) and 15% glycerol. Twenty-four hours before each experiment, one vial was thawed and incubated at 30 °C to achieve logarithmic growth phase

114
Chapter 7
(log phase). Tigecycline powder was purchased from Baylor University Medical Center pharmacy. For each experiment, the antibiotic was reconstituted, sterile filtered, and then diluted in sterile water to the desired concentrations. Hollow-fiber cartridges were purchased from FiberCell Systems (Frederick, MD).
Drug susceptibility testingThe MIC was determined two times by broth macrodilution in Middlebrook 7H9 broth (here referred to as “broth”) read at 72 h.11 In addition, cfu per ml were enumerated from each concentration evaluated in the broth macrodilution test to determine the lowest concentration associated with >99% inhibition of growth, corresponding to the MIC.
Hollow-fiber studyThe hollow-fiber system (HFS) model of pulmonary M. abscessus infection was previously developed and used to perform pharmacokinetic/pharmacodynamic (PK/PD) evaluation of amikacin and moxifloxacin.12,13 The peripheral compartments of 8 HFSs were each inoculated with 20 ml of 6 log10 cfu/ml M. abscessus. Treatment was administered daily for 21 days at doses that mimicked the non-protein bound or free AUC0–24, peak concentrations, and times to maximum concentration achieved in the lungs of humans treated with tigecycline doses of 0, 12.5, 25, 50, 100, 200, 400, and 800 mg, assuming linear increases in AUC0–24 with dose since clearance is constant over a range of doses.14-16 The doses were administered to the central compartment of each HFS once daily via computerized syringe pumps. Tigecycline concentrations achieved in all the systems were validated by repetitive sampling from the central compartment of each HFS at 0, 1, 6, 9, 12, 18, 23.5, 25, 30, 33, 36, 42, and 47.5 h after the first dose. The M. abscessus burden was quantified by taking 1-ml samples from the peripheral compartment culture contents of each system on days 0, 1, 2, 3, 5, 7, 10, 14, and 21 of treatment. After washing by centrifugation with saline to avoid antibiotic carryover, samples were serially 10-fold diluted and cultured on Middlebrook 7H10 agar. The tigecycline MIC was identified at the end of the experiment in each HFS by broth macrodilution.
Drug assayTigecycline concentrations in the samples collected from the central compartment of each HFS were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Tigecycline and tigecycline-D9 (internal standard) were purchased from Toronto Research Chemicals (Toronto, Canada). A calibrator, controls, and the internal standard were included in each analytical run for quantitation. Stock solutions of tigecycline and internal standard were prepared in 80:20 methanol-water at a concentration of 1 mg/ml and stored at -20 °C. A seven point calibration curve was prepared by diluting tigecycline stock solution in drug-free medium (0.1, 0.2, 1, 2, 5, 10, and 20 mg/l). Quality control samples were prepared by spiking the medium with stock standards for two levels of controls. Samples were prepared in 96-well microtiter plates by the addition of 10 µl of calibrator, quality controls, or sample to 190 µl 0.1% formic

115
Cha
pter
7
Tigecycline efficacy against M. abscessus
acid in water containing 10 mg/l internal standard followed by vortex mixing. Chromatographic separation was achieved on an Acquity ultraperformance liquid chromatography (UPLC) HSS T3 analytical column (1.8 µm, 50 by 2.1 mm; Waters) maintained at 30 °C at a flow rate of 0.2 ml/min with a binary gradient and a total run time of 6 min. The observed m/z values of the fragment ions were 586.33 to 569.3 for tigecycline and the internal standard and 595.4 to 578.4 for tigecycline 13CD. Sample injection and separation was performed with an Acquity UPLC interfaced with a Xevo TQ mass spectrometer (Waters). All data were collected using Mass Lynx version 4.1 SCN810. The limit of quantitation for this assay was 0.1 mg/l. The inter- and intraday variations were 12.1% and 11.8%, respectively.
Pharmacokinetic analysis and PK/PD modelingTigecycline concentrations from each of the HFS units at all time points were co-modeled using the ADAPT 5 software (Biomedical Simulations Resource, University of Southern California) following steps for the pharmacokinetic parameter analysis described in detail in prior studies.17-19 The observed AUC0–24 and AUC0–24/MIC ratios were calculated from the pharmacokinetic parameter estimates identified. The dose response was modeled using the inhibitory sigmoid Emax model with the total bacterial burden used as the response parameter and the steady-state AUC0–24/MIC ratio as drug exposure, with maximal kill (Emax) or efficacy and effect in log10 cfu/ml. This relationship was used to calculate the EC80, which is the exposure mediating 80% of Emax, since 100% Emax lies on an asymptote of the inhibitory sigmoid Emax model. In addition, we also calculated the AUC0–24/MIC ratio at which the bacterial burden is similar to that at time zero (“stasis”), as well as that associated with a 1-log10 cfu/ml decrease compared to time zero (“1-log kill”).
Monte Carlo simulationsMonte Carlo simulations were performed in order to translate the HFS-derived AUC/MIC exposures to an optimal clinical dose. Steps for the Monte Carlo experiments are detailed in recommendations and in our prior work19,20 and were followed to identify the clinical dose best able to achieve or exceed specific target exposures in lungs of 10,000 patients with pulmonary M. abscessus. Three target exposures were examined: (i) EC80, (ii) AUC0–24/MIC ratio associated with stasis, and (iii) AUC0–24/MIC ratio associated with 1-log kill. The same exercise was also used to identify the PK/PD-derived susceptibility breakpoint, which we consider to be the MIC below which >10% of patients achieve the EC80 with standard dosing or, alternatively, with the optimal dose that can be tolerated by patients, based on prior work.19-23 We examined doses of 50 mg, 100 mg, 150 mg, and 200 mg per day. The population pharmacokinetic parameters and covariance used as prior data were those from Rubino et al.14 In addition, we took into account the fact that the tigecycline AUC in epithelial lining fluid (ELF) in 1.32-fold higher than that in plasma.15 Drug concentrations in ELF are considered concentration surrogates at the site of effect for pneumonias, including those caused by mycobacteria.14,15,17-26 Since the concentration of proteins in ELF in patients is low,25-27 we considered ELF protein binding negligible. An

116
Chapter 7
alternate view could be that a large portion of M. abscessus in lungs is intracellular (this is by no means resolved), in which case it would be the intracellular tigecycline concentration that should be simulated. Since tigecycline’s concentration in alveolar macrophages is >58-fold than that in ELF, our use of ELF exposures would constitute a worst-case scenario.15 We also examined the probability that these doses would be likely to achieve a plasma AUC of >6.87 mg · h/l, which was identified by Rubino et al. as being associated with greater incidence of nausea and vomiting.10 The tigecycline MIC distribution was from isolates of patients treated in northern Texas, as reported by Wallace and colleagues.7 The cumulative fraction of response (CFR) is a summation calculated as:
where PTA is the probability of target attainment at each MIC and F is the proportion of isolates at each MIC.19,28
Results
MIC and pharmacokinetic analysisThe tigecycline MIC for the M. abscessus strain was 3.12 mg/l. Tigecycline pharmacokinetics in the HFS model of pulmonary M. abscessus infection were best described by a two-compartment model, based on Akaike information criterion and Bayesian information criterion scores, as intended in the study design. The pharmacokinetic parameters achieved in the HFS model were a total clearance of 20.6 ± 29.6 l/h, a volume of the central compartment of 459 ± 13.1 l, an intercompartmental clearance of 3.41 l/h, and a peripheral volume of 0.80 ± 0.45 l.
Time-kill curve in the HFS modelFigure 1 shows time-kill curves for the HFS model of pulmonary M. abscessus infection, which
0 3 6 9 12 15 18 210123456789
1011
AUC/MIC=0AUC/MIC=0.71AUC/MIC=0.89AUC/MIC=3.12AUC/MIC=7.85AUC/MIC=21.31
AUC/MIC=44.60AUC/MIC=79.85
Day
Myc
obac
teri
um a
bsce
ssus
log 1
0 CFU
/ml
Figure 1. Tigecycline time-kill curves in the hollow-fiber system model of M. abscessus infection. The two higher exposures lead to considerable killing, which was maximum at 7 days. Regrowth started at day 10 and after day 14 was observed in all systems.
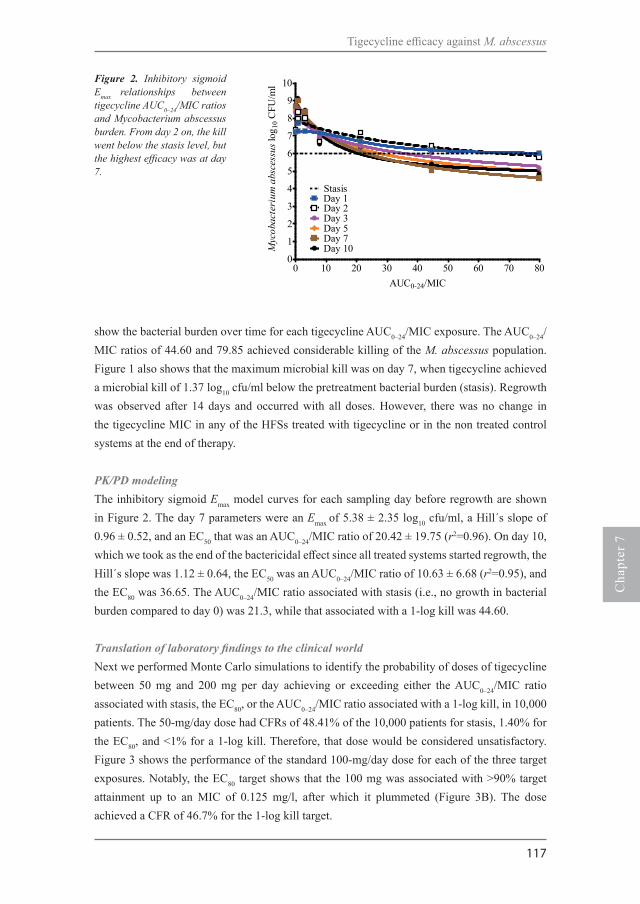
117
Cha
pter
7
Tigecycline efficacy against M. abscessus
show the bacterial burden over time for each tigecycline AUC0–24/MIC exposure. The AUC0–24/MIC ratios of 44.60 and 79.85 achieved considerable killing of the M. abscessus population. Figure 1 also shows that the maximum microbial kill was on day 7, when tigecycline achieved a microbial kill of 1.37 log10 cfu/ml below the pretreatment bacterial burden (stasis). Regrowth was observed after 14 days and occurred with all doses. However, there was no change in the tigecycline MIC in any of the HFSs treated with tigecycline or in the non treated control systems at the end of therapy.
PK/PD modelingThe inhibitory sigmoid Emax model curves for each sampling day before regrowth are shown in Figure 2. The day 7 parameters were an Emax of 5.38 ± 2.35 log10 cfu/ml, a Hill´s slope of 0.96 ± 0.52, and an EC50 that was an AUC0–24/MIC ratio of 20.42 ± 19.75 (r2=0.96). On day 10, which we took as the end of the bactericidal effect since all treated systems started regrowth, the Hill´s slope was 1.12 ± 0.64, the EC50 was an AUC0–24/MIC ratio of 10.63 ± 6.68 (r2=0.95), and the EC80 was 36.65. The AUC0–24/MIC ratio associated with stasis (i.e., no growth in bacterial burden compared to day 0) was 21.3, while that associated with a 1-log kill was 44.60.
Translation of laboratory findings to the clinical worldNext we performed Monte Carlo simulations to identify the probability of doses of tigecycline between 50 mg and 200 mg per day achieving or exceeding either the AUC0–24/MIC ratio associated with stasis, the EC80, or the AUC0–24/MIC ratio associated with a 1-log kill, in 10,000 patients. The 50-mg/day dose had CFRs of 48.41% of the 10,000 patients for stasis, 1.40% for the EC80, and <1% for a 1-log kill. Therefore, that dose would be considered unsatisfactory. Figure 3 shows the performance of the standard 100-mg/day dose for each of the three target exposures. Notably, the EC80 target shows that the 100 mg was associated with >90% target attainment up to an MIC of 0.125 mg/l, after which it plummeted (Figure 3B). The dose achieved a CFR of 46.7% for the 1-log kill target.
Figure 2. Inhibitory sigmoid Emax relationships between tigecycline AUC0–24/MIC ratios and Mycobacterium abscessus burden. From day 2 on, the kill went below the stasis level, but the highest efficacy was at day 7.
0 10 20 30 40 50 60 70 800
1
2
3
4
5
6
7
8
9
10
Day 1Day 2Day 3
Day 7Day 10
Stasis
Day 5
AUC0-24/MICM
ycob
acte
rium
abs
cess
us lo
g 10 C
FU/m
l

118
Chapter 7
The performance of the 150-mg/day dose is shown in Figure 4. More than 90% of patients would achieve stasis at this dose (Figure 4A). In regard to the EC80, the MIC above which >10% would fail to achieve that exposure was 0.25 mg/l; at 0.5 mg/l, the proportion fell to 0%. Figure 4C shows that the proportion that achieved an AUC0–24/MIC ratio associated with a 1-log kill at 150 mg/day was approximately 63%.
The 200-mg/day dose achieved target exposures as shown in Figure 5. Both stasis (Figure 5A) and EC80 exposures (Figure 5B) were achieved in >90% of patients. Figure 5B shows target attainment of >98% at an MIC of 0.25 mg/l, which then fell to 2.80% at an MIC of 0.5 mg/l. If this dose was adopted for pulmonary M. abscessus infection, the pharmacokinetic/pharmacodynamic based susceptibility breakpoint would be 0.5 mg/l, as shown in Figure 5B. The AUC0–24/MIC ratio associated with a 1-log kill was achieved or exceeded in 87.34% of patients, which though short of 90% is close, suggesting that this is the optimal dose for rapid microbial kill in pulmonary M. abscessus disease. Since the tigecycline AUC0–24 threshold that predicts nausea and vomiting is a plasma AUC of >6.87 mg·h/l, we also examined the probability of achieving that plasma concentration (as opposed to lung concentrations as described above) at the different doses. Figure 6 shows a steep increase in the proportion of the patients achieving this at above 150 mg/day, achieving a probability of close to 1 at 200 mg/day.
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
CFR=88.69%
A.
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
MIC distribution90% achievement
B.
CFR=51.06%
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
CFR=46.70%
C.
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
CFR=88.69%
A.
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
MIC distribution90% achievement
B.
CFR=51.06%
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
CFR=46.70%
C.
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
Figure 3. Probability of target attainment in patients treated with 100 mg/day.Proportions of 10,000 patients achieving the AUC/MIC ratio associated withstasis (A) EC80 target (B), and the target AUC/MIC ratio associated with a1-log kill (C) are shown. The cumulative fraction of response (CFR) is low forthe EC80 and 1-log kill targets. Moreover, in panel A, the standard dose will notachieve even stasis if the MIC is 0.5 mg/l.
A
B
C
Tigecycline MIC (mg/l) Tigecycline MIC (mg/l)
Tigecycline MIC (mg/l)

119
Cha
pter
7
Tigecycline efficacy against M. abscessus
Discussion
Tigecycline is efficacious against the notoriously resistant M. abscessus in the hollow-fiber model system for pulmonary disease. For the first time in the HFS model of pulmonary M. abscessus infection, a drug exhibited a considerable microbial kill below the stasis line, exceeding what has been previously observed for amikacin or moxifloxacin.12,13 This magnitude of the efficacy means that tigecycline could enter a regimen as the first truly bactericidal drug for M. abscessus. This alone should qualify this drug to be considered as a first-line agent early during therapy and the backbone for new combination regimens. Perhaps aggressive regimens, including combination The tigecycline EC80 exposure for M. abscessus microbial kill was calculated as an AUC0–24/MIC ratio of 36.65. In patients with community-acquired pneumonia due to a variety of Gram-positive and Gram-negative pathogens, the ratio associated with faster time to fever resolution for tigecycline was an fAUC0–24/MIC ratio of ≥12.8.10 Thus, the exposure associated with optimal efficacy that we identified is much higher than that for other pathogens. Nevertheless, the microbial kill indices (either EC80, stasis, or 1-log kill) that translate from the HFS model of pulmonary M. abscessus infection to patients are still unclear. However, in our simulations, it is interesting that the CFR of 46.70% based on the 1-log kill target was the one closet to clinical response rates of 43% in tigecycline-containing regimens in pulmonary M. abscessus disease studies by Wallace et al.8 This is consistent with the notion that other antibiotics currently used exert very limited antimicrobial effects, as seen in our HFS model and with clinical experience.1-3,8,12,13 The 200-mg/day dose achieved this 1-log kill target exposure in close to 90% of patients, suggesting that it is the optimal dose for a bactericidal
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
CFR=92.19%
A.
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
MIC distribution
90% achievement
B.
CFR=82.58%
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
CFR=62.76%
C.
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
CFR=92.19%
A.
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
MIC distribution
90% achievement
B.
CFR=82.58%
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
CFR=62.76%
C.
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
ntsFigure 4. Probability of target attainment in
patients treated with 150 mg/day forthe targets AUC/MIC ratio associated with stasis (A), EC80 (B), and AUC/MICratio associated with a 1-log kill (C). The CFRs for the EC80 and 1-log kill targets are below 90%, and thus the dose should be considered suboptimal.
A B
C
Tigecycline MIC (mg/l) Tigecycline MIC (mg/l)
Tigecycline MIC (mg/l)
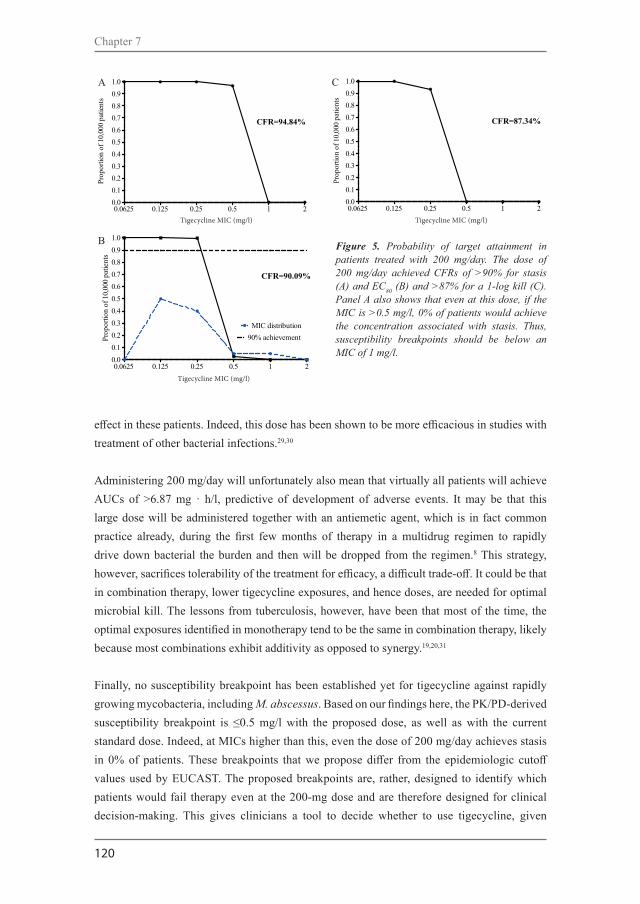
120
Chapter 7
effect in these patients. Indeed, this dose has been shown to be more efficacious in studies with treatment of other bacterial infections.29,30
Administering 200 mg/day will unfortunately also mean that virtually all patients will achieve AUCs of >6.87 mg · h/l, predictive of development of adverse events. It may be that this large dose will be administered together with an antiemetic agent, which is in fact common practice already, during the first few months of therapy in a multidrug regimen to rapidly drive down bacterial the burden and then will be dropped from the regimen.8 This strategy, however, sacrifices tolerability of the treatment for efficacy, a difficult trade-off. It could be that in combination therapy, lower tigecycline exposures, and hence doses, are needed for optimal microbial kill. The lessons from tuberculosis, however, have been that most of the time, the optimal exposures identified in monotherapy tend to be the same in combination therapy, likely because most combinations exhibit additivity as opposed to synergy.19,20,31
Finally, no susceptibility breakpoint has been established yet for tigecycline against rapidly growing mycobacteria, including M. abscessus. Based on our findings here, the PK/PD-derived susceptibility breakpoint is ≤0.5 mg/l with the proposed dose, as well as with the current standard dose. Indeed, at MICs higher than this, even the dose of 200 mg/day achieves stasis in 0% of patients. These breakpoints that we propose differ from the epidemiologic cutoff values used by EUCAST. The proposed breakpoints are, rather, designed to identify which patients would fail therapy even at the 200-mg dose and are therefore designed for clinical decision-making. This gives clinicians a tool to decide whether to use tigecycline, given
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0A.
CFR=94.84%
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
MIC distribution90% achievement
B.
CFR=90.09%
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0C.
CFR=87.34%
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
Figure 5. Probability of target attainment in patients treated with 200 mg/day. The dose of 200 mg/day achieved CFRs of >90% for stasis (A) and EC80 (B) and >87% for a 1-log kill (C). Panel A also shows that even at this dose, if the MIC is >0.5 mg/l, 0% of patients would achieve the concentration associated with stasis. Thus, susceptibility breakpoints should be below an MIC of 1 mg/l.
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0A.
CFR=94.84%
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0
MIC distribution90% achievement
B.
CFR=90.09%
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
0.0625 0.125 0.25 0.5 1 20.00.10.20.30.40.50.60.70.80.91.0C.
CFR=87.34%
Tigecycline MIC (mg/liter)
Prop
ortio
n of
10,
000
patie
nts
A
B
C
Tigecycline MIC (mg/l) Tigecycline MIC (mg/l)
Tigecycline MIC (mg/l)

121
Cha
pter
7
Tigecycline efficacy against M. abscessus
higher probabilities of failure even to achieve stasis, with an agent that would be associated with high rates of adverse events.
This study has some limitations. First, the assay to monitor and detect the size of the resistant subpopulation was not optimized. Thus, we could determine neither the mutation frequency nor the proportion of the resistant subpopulation throughout the study. Second, only the M. abscessus type strain was examined. Ideally several clinical isolates should be evaluated. Nevertheless, taking into account a wider MIC distribution in our simulations should in part compensate for that. Third, these are results from an HFS model for extracellular pulmonary M. abscessus infection. It is unknown what proportion of the pathogen is extracellular or intracellular in lungs of patients. However, given tigecycline’s high ratios of penetration into alveolar macrophages, reflected by an alveolar cell-to-serum AUC ratio of 77.5 times15, even the standard dose may achieve either the EC80 or 1-log kill in patients. Thus, our dose recommendations may be too conservative. Nevertheless, in other mycobacterial pneumonias, the ELF has been accurate as a surrogate in prediction in patient clinical studies.31-33
In summary, tigecycline has the potential to improve treatment outcomes in M. abscessus disease. To date, it is one of the most active drugs evaluated in the HFS model of pulmonary M. abscessus infection. We recommend 200 mg; however, the caveat is that this is based on the assumption of a predominantly extracellular bacterial location. The role of tigecycline in regimens to treat pulmonary M. abscessus infection deserves further exploration, including development of combination therapy regimens.
Figure 6. Proportion of 10,000 patients who achieve the tigecycline AUC threshold predictive of nausea and vomiting.
0 50 100 150 2000
10
20
30
40
50
60
70
80
90
100
Tigecycline dose (mg/day)%
of p
atie
nts a
chie
ving
AU
C0-
24>6
.87
mg*
h/lit
er%
of p
atie
nts a
chie
ving
AU
C0-
24 >
6.87
mg*
h/l

122
Chapter 7
Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother. 2012;67:810-8.Griffith DE. The talking Mycobacterium abscessus blues. Clin Infect Dis. 2011;52:572-4. Jarand J, Levin A, Zhang L, Huitt G, Mitchell JD, Daley CL. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin Infect Dis. 2011;52:565-71.Stein GE, Craig WA. Tigecycline: a critical analysis. Clin Infect Dis. 2006;15:518-24.Estes KS, Derendorf H. Comparison of the pharmacokinetic properties of vancomycin, linezolid, tigecyclin and daptomycin. Eur J Med Res. 2010;15:533-43.Giamarellou H, Poulakou G. Pharmacokinetic and pharmacodynamic evaluation of tigecycline. Expert Opin Drug Metab Toxicol. 2011;7:1459-70.Wallace RJ, Jr, Brown-Elliott BA, Crist CJ, Mann L, Wilson RW. Comparison of the in vitro activity of the glycylcycline tigecycline (formerly GAR-936) with those of tetracycline, minocycline, and doxycycline against isolates of nontuberculous mycobacteria. Antimicrob Agents Chemother. 2002; 46:3164-7.Wallace RJ, Jr, Dukart G, Brown-Elliott BA, Griffith DE, Scerpella EG, Marshall B. Clinical experience in 52 patients with tigecyclinecontaining regimens for salvage treatment of Mycobacterium abscessus and Mycobacterium chelonae infections. J Antimicrob Chemother. 2014;69:1945-53.Bhavnani SM, Rubino CM, Hammel JP, Forrest A, Dartois N, Cooper CA, Korth-Bradley J, Ambrose PG. Pharmacological and patient specific response determinants in patients with hospital-acquired pneumonia treated with tigecycline. Antimicrob Agents Chemother. 2012;56:1065-72. Rubino CM, Bhavnani SM, Forrest A, Dukart G, Dartois N, Cooper A, Korth-Bradley J, Ambrose PG. Pharmacokinetics pharmacodynamics of tigecycline in patients with community-acquired pneumonia. Antimicrob Agents Chemother. 2012;56:130-6. Clinical and Laboratory Standards Institute. Susceptibility Testing of Mycobacteria, Nocardia, and Other Aerobic Actinomycetes - Second Edition: Approved Standard M24-A2. CLSI, Wayne, PA, USA, 2011. Ferro BE, Srivastava S, Deshpande D, Sherman CM, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. Amikacin pharmacokinetics/pharmacodynamics in a novel hollow fiber Mycobacterium abscessus disease model. Antimicrob Agents Chemother. 2015;60:1242-48.Ferro BE, Srivastava S, Deshpande D, Sherman CM, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2015. Moxifloxacin efficacy in a hollow-fiber model of Mycobacterium abscessus disease, abstr A-014c. Program Abstr 55th Intersci Conf Antimicrob Agents Chemother-28th Int Congr Chemother Meet, San Diego, CA.Rubino CM, Forrest A, Bhavnani SM, Dukart G, Cooper A, Korth- Bradley J, Ambrose PG. Tigecycline population pharmacokinetics in patients with community- or hospital-acquired pneumonia. Antimicrob Agents Chemother. 2010;54:5180-6. Conte JE, Jr, Golden JA, Kelly MG, Zurlinden E. Steady-state serum and intrapulmonary pharmacokinetics and pharmacodynamics of tigecycline. Int J Antimicrob Agents. 205;25:523-9.Barbour A, Schmidt S, Ma B, Schiefelbein L, Rand KH, Burkhardt O, Derendorf H. Clinical pharmacokinetics and pharmacodynamics of tigecycline. Clin Pharmacokinet. 2009;48:575-84.Deshpande D, Srivastava S, Meek C, Leff R, Hall GS, Gumbo T. Moxifloxacin pharmacokinetics/pharmacodynamics and optimal dose and susceptibility breakpoint identification for treatment of disseminated Mycobacterium avium infection. Antimicrob Agents Chemother. 2010;54:2534-9.
1.
2.3.
4.5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
References

123
Cha
pter
7
Tigecycline efficacy against M. abscessus
Musuka S, Srivastava S, Siyambalapitiyage Dona CW, Meek C, Leff R, Pasipanodya J, Gumbo T. Thioridazine pharmacokinetic pharmacodynamic parameters “wobble” during treatment of tuberculosis: a theoretical basis for shorter-duration curative monotherapy with congeners. Antimicrob Agents Chemother. 2013;57:5870-7. Gumbo T, Angulo-Barturen I, Ferrer-Bazaga S. Pharmacokinetic/pharmacodynamic and dose-response relationships of antituberculosis drugs: recommendations and standards for industry and academia. J Infect Dis. 2015;211(Suppl 3):S96-S106.Pasipanodya J, Gumbo T. An oracle: antituberculosis pharmacokinetics- pharmacodynamics, clinical correlation, and clinical trial simulations to predict the future. Antimicrob Agents Chemother. 2011;55:24-34.Gumbo T, Pasipanodya JG, Wash P, Burger A, McIlleron H. Redefining multidrug-resistant tuberculosis based on clinical response to combination therapy. Antimicrob Agents Chemother. 2014;58:6111-5.Gumbo T, Chigutsa E, Pasipanodya J, Visser M, van Helden PD, Sirgel FA, McIlleron H. The pyrazinamide susceptibility breakpoint above which combination therapy fails. J Antimicrob Chemother. 2014;69:2420-5.Gumbo T. New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob Agents Chemother. 2012;54:1484-91.Craig WA. 2007. Pharmacodynamics of antimicrobials: general concepts and applications, p 1-19. In Nightingale CH, Ambrose PG, Drusano GL, Murakawa T (ed), Antimicrobial pharmacodynamics in theory and practice 2nd ed, vol 44. Informa Healthcare USA, Inc., New York, NY. Craig WA, Redington J, Ebert SC. Pharmacodynamics of amikacin in vitro and in mouse thigh and lung infections. J Antimicrob Chemother. 1991;27(Suppl C):29-40. Kiem S, Schentag JJ. Interpretation of antibiotic concentration ratios measured in epithelial lining fluid. Antimicrob Agents Chemother. 2008;52:24-36. Gumbo T, Siyambalapitiyage Dona CS, Meek C, Leff R. Pharmacokinetics-pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother. 2009;53:3197-204.Gumbo T. Integrating pharmacokinetics, pharmacodynamics and pharmacogenomics to predict outcomes in antibacterial therapy. Curr Opin Drug Discov Dev. 2008;11:32-42. Falagas ME, Vardakas KZ, Tsiveriotis KP, Triarides NA, Tansarli GS. Effectiveness and safety of high-dose tigecycline-containing regimens for the treatment of severe bacterial infections. Int J Antimicrob Agents. 2014;44:1-7.De Pascale G, Montini L, Pennisi M, Bernini V, Maviglia R, Bello G, Spanu T, Tumbarello M, Antonelli M. High dose tigecycline in critically ill patients with severe infections due to multidrug-resistant bacteria. Crit Care. 2014;18:R90.Gumbo T, Pasipanodya JG, Romero K, Hanna D, Nuermberger E. Forecasting accuracy of the hollow fiber model of tuberculosis for clinical therapeutic outcomes. Clin Infect Dis. 2015;61(Suppl 1):S25-S31.Chigutsa E, Pasipanodya JG, Visser ME, van Helden PD, Smith PJ, Sirgel FA, Gumbo T, McIlleron H. Impact of nonlinear interactions of pharmacokinetics and MICs on sputum bacillary kill rates as a marker of sterilizing effect in tuberculosis. Antimicrob Agents Chemother. 2015;59:38-45. Pasipanodya JG, McIlleron H, Burger A, Wash PA, Smith P, Gumbo T. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J Infect Dis. 2013;208:1464-73.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.


125
Antimicrob Agents Chemother. 2016;60:6374-6.
Chapter 8
Beatriz E. FerroShashikant SrivastavaDevyani DeshpandeJotam G. PasipanodyaDick van SoolingenJohan W. Mouton Jakko van IngenTawanda Gumbo
Failure of the amikacin, cefoxitin and clarithromycin combination regimen for pulmonary mycobacterium abscessus

126
Chapter 8
Summary
In a hollow-fiber model, we mimicked the drug exposures achieved in the lungs of humans treated with standard amikacin, clarithromycin, and cefoxitin combination therapy for Mycobacterium abscessus infection. At optimal dosing, a kill rate of -0.09 (95% confidence interval [CI], -0.04 to 0.03) log10 cfu per ml/day was achieved over the first 14 days, after which there was regrowth due to acquired drug resistance. Thus, the standard regimen quickly failed. A new regimen is needed.

127
Cha
pter
8
Failure of standard regimen to treat M. abscessus
Clinicians treat Mycobacterium abscessus pulmonary disease with a combination of amikacin, clarithromycin, and cefoxitin for 1 to 2 months, followed by an oral maintenance regimen, usually with a fluoroquinolone, based on the recommendations of experts.1 However, the use of this approach “cures” only 50% even with adjunctive surgery, most of whom relapse or die.2 We have developed a hollow-fiber model of M. abscessus (HFS-M. abscessus) and evaluated the effect of monotherapy of amikacin and moxifloxacin in the system. Each drug failed dramatically, and acquired drug resistance (ADR) developed.3,4 Since these drugs are given as combination therapy in patients, they could still work in combination. Here, we evaluated the performance of the standard combination regimen of amikacin, cefoxitin, and clarithromycin in the HFS-M. abscessus to determine potential synergy, and even more importantly, whether the combination therapy could prevent ADR.
Antibiotics were purchased from the Baylor University Medical Center Pharmacy (Dallas, TX) and from Sigma-Aldrich (St. Louis, MO). Antibiotics were dissolved in water-methanol (clarithromycin), sterile filtered, and diluted to the desired concentrations in Middlebrook 7H9 broth (here broth; Remel, Lenexa, KS). Stock solutions of M. abscessus ATCC 19977 (ATCC, Manassas, VA) were grown to logarithmic-growth phase in the broth. MICs were measured using broth microdilution.5 The amikacin, cefoxitin, and clarithromycin MICs were 32, 16, and 8 mg/l, respectively.
The peripheral compartments of six HFS-M. abscessus systems were inoculated with 20 ml of 6.0 log10 cfu/ml M. abscessus, as previously described.3 Three systems were immediately treated with a combination of amikacin, cefoxitin, and clarithromycin, while three replicate systems were non-treated controls. Amikacin, cefoxitin, and clarithromycin were administered via syringe pumps for 28 days, as in patients. We mimicked free-drug area under the concentration-time curve from 0 to 24 h (fAUC0–24), peak concentration (fCmax), and time to maximum concentration achieved in the lungs of humans treated with amikacin and clarithromycin administered once daily, at 1,000 mg, and cefoxitin administered at 2 g four times a day.6,7 Different half-lives in each HFS-M. abscessus system were achieved, as described by us in the past.8 Antibiotic concentrations achieved in all the systems were validated by sampling from the central compartment of each system at seven time points over 24 h post dose, after which concentrations were assayed in a validated multiplexed assay using liquid chromatography-tandem mass spectrometry in our laboratory at the Baylor University Medical Center. We achieved an AUC0–24/MIC of 5.3 mg·h/l for clarithromycin, a percentage of time above the MIC of 100% for cefoxitin, and an fCmax/MIC of 4.0 of amikacin, so that each drug was at optimal free-drug exposure.3 These exposures are actually better than those achieved in most patients with standard dosing, based on prior work.3,4,7,9
We sampled each system’s peripheral compartment on days 0, 1, 2, 3, 5, 7, 10, 14, 21, and 28 of treatment, washed the cultures in saline to avoid antibiotic carryover, serially diluted the

128
Chapter 8
cultures, and cultured them on antibiotic-free Middlebrook 7H10 agar. The size of the resistant subpopulation was quantified by culturing on Middlebrook agar plates containing 3 times the MIC of each antibiotic. The day-to-day bacterial burdens with standard therapy and in the controls are shown in Figure 1A. The combination therapy killed at the low rate of -0.09 (95% confidence interval [CI], -0.04 to 0.03) log10 cfu/ml/day, to achieve 1.22 log10 cfu/ml kill on day 14 compared to the day 0 bacterial burden. After day 14, regrowth was observed. Thus, the combination regimen failed quickly, despite optimal exposures. The proportions of amikacin- and cefoxitin-resistant subpopulations to total population remained at a low level in the untreated control throughout the experiment, as shown in Figure 1B. However, in the systems exposed to combination therapy, the cefoxitin-resistant subpopulation increased considerably after day 14, while the amikacin resistant subpopulation proportion did not change (Figure 1C). Clarithromycin resistance could not be reliably assessed because of inducible macrolide resistance in M. abscessus.10
The standard regimen tested failed to effectively kill M. abscessus in the 28-day evaluation in the HFS-M. abscessus model that mimicked lung disease. Cefoxitin seemed to be the driving
0 2 4 6 8 10 12 14 16 18 20 22 24 26 280123456789
10111213
Standard regimenUntreated control
Stasis
days
M. a
bsce
ssus
log 1
0 CFU
/mL
0 2 4 6 8 10 12 14 16 18 20 22 24 26 280123456789
10111213
Cefoxitin-resistant
Untreated controlAmikacin-resistant
days
M. a
bsce
ssus
log 1
0 CFU
/mL
0 2 4 6 8 10 12 14 16 18 20 22 24 26 280123456789
10111213
Cefoxitin-resistant
Standard therapyAmikacin-resistant
days
M. a
bsce
ssus
log 1
0 CFU
/mL
a.
b.
c.
0 2 4 6 8 10 12 14 16 18 20 22 24 26 280123456789
10111213
Standard regimenUntreated control
Stasis
days
M. a
bsce
ssus
log 1
0 CFU
/mL
0 2 4 6 8 10 12 14 16 18 20 22 24 26 280123456789
10111213
Cefoxitin-resistant
Untreated controlAmikacin-resistant
days
M. a
bsce
ssus
log 1
0 CFU
/mL
0 2 4 6 8 10 12 14 16 18 20 22 24 26 280123456789
10111213
Cefoxitin-resistant
Standard therapyAmikacin-resistant
days
M. a
bsce
ssus
log 1
0 CFU
/mL
a.
b.
c.Figure 1. Efficacy of standard combination regimen against M. abscessus and resistance emergence during 1 month of treatment .(A) The initial very slow decline, slower than the doubling time of the bacteria, was reversed after 14 days and all treated systems started to grow.(B) Changes in bacterial burden and the emergence of acquired drug resistance with time during repetitive sampling in non-treated controls. Total M. abscessus population in untreated control (solid line) and amikacin- or cefoxitin-resistant subpopulation (dashed lines) over the course of 28 days. The amikacin- and cefoxitin-resistant proportion was stable in the untreated control. (C) Changes in bacterial burden and the emergence of acquired drug resistance with time during repetitive sampling in systems treated with standard therapy. Starting with no cfu per milliliter resistant to 3 times the cefoxitin MIC, there developed a higher cefoxitin-resistant subpopulation of M. abscessus in the systems exposed to standard therapy, coincident with cessation of effect of the triple regimen.
A B
M. a
bsce
ssus
log 10
CFU
/ml
M. a
bsce
ssus
log 10
CFU
/ml
M. a
bsce
ssus
log 10
CFU
/ml
C

129
Cha
pter
8
Failure of standard regimen to treat M. abscessus
force of the regimen, as the HFS-M. abscessus shows that while there was initial kill, this was abrogated by ADR to cefoxitin. In the nude mouse M. abscessus disseminated disease model, cefoxitin has played a key role as well, as the activity of the triple regimen was equal to that of cefoxitin alone.11 Mouse studies, while useful, however, do not allow for repetitive sampling that would allow for an assessment of the evolution of ADR with time in the same mouse. Even though the proportion of bacteria resistant to 3 times the MIC of amikacin did not change, the biphasic nature of response on the current amikacin-containing regimen suggests resistance as well, albeit with <3-fold increases in MIC. This type of low-level resistance, which rapidly emerges within days, has been shown to be due to evolutionarily conserved efflux pumps in mycobacteria that are part of the “antibiotic resistance arrow of time”.12,13
Even at above-optimal amikacin and cefoxitin exposures, the regimen still failed. The situation observed here resembles the poor outcome often faced when patients are treated with the standard combined regimen; if they tolerate it, they may initially respond, but in the end, many simply fail to respond at all, and some respond and then relapse. In other words, it is going nowhere fast. The propensity to develop multiple drug resistances quickly is in part why M. abscessus has been termed the “antibiotic resistance nightmare”.14 Since dosing to achieve greater-than-optimal exposure would not result in any greater effectiveness, the solution is a de novo building of new regimens that are more bactericidal and can suppress the emergence of ADR. One of the alternatives could be to use tigecycline as an anchor drug in such a regimen, which has been shown to be effective.15,16 However, tigecycline will need to be combined with other effective antibiotics, and the search for such antibiotics is ongoing.

130
Chapter 8
Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K. 2007. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 175:367-416. Jarand J, Levin A, Zhang L, Huitt G, Mitchell JD, Daley CL. 2011.Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin Infect Dis. 52:565–571. Ferro BE, Srivastava S, Deshpande D, Sherman CM, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2015. Amikacin pharmacokinetics/pharmacodynamics in a novel hollow fiber Mycobacterium abscessus disease model. Antimicrob Agents Chemother. 60:1242–48. Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2016. Moxifloxacin efficacy in a hollow-fiber model of Mycobacterium abscessus disease. Antimicrob Agents Chemother. 60:3779-85.Clinical Laboratory Standards Institute. 2011. Susceptibility testing of Mycobacteria, Nocardiae and other aerobic Actinomycetes- Second Edition: approved standard M24-A2.CLSI, Wayne PA. Dull WL, Alexander MR, Kasik JE. 1979. Bronchial secretion levels of amikacin. Antimicrob Agents Chemother. 16:767-771. Santre C, Georges H, Jacquier JM, Leroy O, Beuscart C, Buguin D, Beaucaire G. 1995. Amikacin levels in bronchial secretions of 10 pneumonia patients with respiratory support treated once daily versus twice daily. Antimicrob Agents Chemother. 39:264-267. Srivastava S, Pasipanodya JG, Meek C, Leff R, Gumbo T. 2011. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis. 204:1951-9. van Ingen J, Egelund EF, Levin A, Totten SE, Boeree MJ, Mouton JW, Aarnoutse RE, Heifets LB, Peloquin CA, Daley CL. 2012. The pharmacokinetics and pharmacodynamics of pulmonary Mycobacterium avium complex disease treatment. Am J Respir Crit Care Med. 186:559-65.Bastian S, Veziris N, Roux AL, Brossier F, Gaillard JL, Jarlier V, Cambau E. 2011. Assessment of clarithromycin susceptibility in strains belonging to the Mycobacterium abscessus group by erm (41) and rrl sequencing. Antimicrob Agents Chemother. 55:775-781.Lerat I, Cambau E, Roth Dit BR, Gaillard JL, Jarlier V, Truffot C, Veziris N. 2014. In vivo evaluation of antibiotic activity against Mycobacterium abscessus. J Infect Dis. 209:905-912.Schmalstieg AM, Srivastava S, Belkaya S, Deshpande D, Meek C, Leff T, van Oers NS, Gumbo T. 2012. The antibiotic resistance arrow of time: efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob Agents Chemother. 56:4806-15.Gumbo T. 2013. Biological variability and the emergence of multidrug-resistant tuberculosis. Nat Genet. 45:720-1.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. 2012. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother. 67:810-8.Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2016. Tigecycline is highly efficacious in Mycobacterium abscessus pulmonary disease. Antimicrob Agents Chemother. 60:2895-900.Wallace RJ Jr, Dukart G, Brown-Elliott BA, Griffith DE, Scerpella EG, Marshall B. 2014. Clinical experience in 52 patients with tigecycline-containing regimens for salvage treatment of Mycobacterium abscessus and Mycobacterium chelonae infections. J Antimicrob Chemother. 69:1945-53.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
References

131
Cha
pter
8
Failure of standard regimen to treat M. abscessus


133
Chapter 9
Discussion, conclusions and future steps

134
Chapter 9

Discussion, conclusions and future steps
135
Chapter 9
Cha
pter
9
„the drugs don’t work, they just make you worse…“(The Verve / Richard Ashcroft; Urban Hymns, 1997)
This thesis summarizes the results of studies performed in a preclinical development track, to understand the current treatment and to find new and more efficacious treatment regimens for nontuberculous mycobacterial pulmonary disease (NTM-PD), with focus on Mycobacterium abscessus. The outcome of NTM-PD treatment, particularly when caused by M. abscessus, is often poor.1-3 Many factors determine this outcome, including the immune status of the patient, age, co-morbidities, extent of disease, tolerance of the antibiotics and, of course, the efficacy of the antibiotics administered. The latter aspect is the focus of the research described in this thesis.
Four main issues can be inferred from our experimental work. First, the efficacy of most currently recommended antibiotics to treat NTM-PD is low. Second, clofazimine has a potential role as part of combination therapy against M. avium and M. abscessus. Third, tigecycline is bactericidal against M. abscessus and should be part of a new combination regimen to be tested. Fourth, β-lactam antibiotics are likely to be the driving force in the currently recommended treatment regimen for M. abscessus disease. With this new knowledge we hope to contribute to the understanding and improvement of NTM disease treatment, including the design of more effective treatment regimens for NTM-PD.
-New insights in antimicrobial susceptibilities of slowly growing NTM (SGM)We standardized and performed static time-kill assays for several antibiotics, where we observed that all the key antibiotics tested had low activity (Chapter 3). Despite the low activity observed, the individual evaluation of antibiotics demonstrated that amikacin had the strongest killing effect against M. avium in the time-kill assays (Chapter 3), although regrowth was observed soon after the initial killing.
Previous in vitro experiments showed complete sterilization when amikacin was used against M. avium.4 In vivo evaluations in the mouse model also showed remarkable activity, particularly when amikacin was combined with either rifabutin or clofazimine.5 The role of adjunctive streptomycin was the topic of one of the few randomized clinical trials in NTM-PD. The addition of streptomycin to a regimen of rifampicin, ethambutol, and clarithromycin for M. avium complex pulmonary disease (MAC-PD) led to a faster conversion to negative cultures, but little to no long-term benefits.6 Based hereupon, current guidelines only advocate the use of amikacin or streptomycin, early during therapy in patients with severe, mostly fibro-cavitary, MAC-PD.7 Our findings support this limited role and use.
Clarithromycin was also tested in our time-kill experiments against M. avium and M. xenopi; its effect was also low, particularly for M. avium. Killing took place even at relatively low multiples

136
Chapter 9
of the MIC (0.125× MIC) and was hardly concentration dependent. When clarithromycin was previously evaluated against M. avium strain 101, in time-kill assays, both concentration and time dependent effect were described, and the effect observed was maximal at relatively high concentrations (256 mg/l).8 In vivo evidence from the Balb/c and nude mice model, also infected with M. avium 101 and treated with clarithromycin-containing regimens, led to the conclusion that clarithromycin was the most active antibiotic.9 Macrolides, i.e. clarithromycin and azithromycin, are considered the cornerstone for M. avium complex treatment, due to the activity observed in the early case series.10,11 However, much more remains to be deciphered about macrolides. We clearly observed regrowth after the initial killing by clarithromycin in time-kill assays, as well as for amikacin, and that occurred with both M. avium and M. xenopi. Therefore, we underline the important concept that macrolides should never be used alone in any treatment regimen for NTM; combination therapy should be the rule (Chapter 4).7
Fluoroquinolones have been suggested to play a role in the treatment of M. xenopi as a substitute for one of the core antibiotics, particularly the macrolide;7 a clinical trial comparing clarithromycin and moxifloxacin as adjuncts to rifampicin and ethambutol is currently under way (ClinicalTrials.gov Identifier NCT01298336). We also evaluated moxifloxacin in time-kill assays against M. xenopi; as with clarithromycin low moxifloxacin activity was observed and killing took a lot of time, but no significant difference was found on the effect obtained with the two antibiotics. This observation is in line with the in vivo characterization of drugs against M. xenopi in the mouse model, where no differences between clarithromycin- and moxifloxacin-containing regimens were observed.12 Whether there is a benefit in including both macrolides and moxifloxacin in M. xenopi treatment regimens remains to be investigated.
The potential role of clofazimine in M. avium treatmentPrevious in vitro evaluations by checkerboard titrations detected synergy between clofazimine and both amikacin and clarithromycin activity against several NTM,13-15 thus we moved one step further in time-kill experiments to evaluate the possible contribution of such combinations in the treatment of M. avium disease. Although the effect of clofazimine alone was mainly a concentration-dependent inhibition of growth, clofazimine-amikacin and clofazimine-clarithromycin combinations prevented the regrowth observed when amikacin or clarithromycin were used alone against M. avium (Chapter 4). Synergy was found at specific concentrations and the regrowth observed in the systems exposed to single antibiotics was associated with mutational resistance to amikacin or clarithromycin in M. avium, evidenced by a change in the MIC of the corresponding antibiotic as well as by the occurrence of established resistance-conferring mutations in rrs and rrl genes.In a recent study of 107 patients treated for M. avium complex pulmonary disease (MAC-PD) that were followed for a long term, a significantly higher proportion of patients treated with clofazimine-containing regimens converted to negative cultures compared to patients treated with rifampicin-containing regimens; in addition clofazimine was well tolerated by the majority

Discussion, conclusions and future steps
137
Chapter 9
Cha
pter
9
of patients.16 Another series of cases reported that regimens that include clofazimine were easier and less demanding to monitor.17 Treatment outcomes in the few available case series proved equal for clofazimine-ethambutol-macrolide and rifampicin-ethambutol-macrolide regimens (Chapter 1); a randomized controlled clinical trial comparing these two in MAC-PD is currently underway in our center (Eudra CT Register 2015-003786-28). However, much more remains to be unraveled about SGM disease treatment. Particularly for M. avium, for which a dynamic pharmacokinetic/pharmacodynamic hollow-fiber model is already available,18 there is a need to move forward into more research on currently recommended combination regimens as well as new regimens.
-New insights in antimicrobial susceptibilities of rapid growing NTM (RGM)Static models demonstrated the limited activity of most commonly used antibiotics against M. abscessus and M. fortuitum (Chapter 2); this fits well with the generally poor outcomes of treatment for NTM, particularly for M. abscessus disease. Here too, amikacin showed the strongest killing effect when evaluated against M. abscessus and M. fortuitum, but for these species, too, regrowth appeared soon. Time-kill assays performed by other groups, although only for 24 hours, evidenced lack of amikacin activity for M. abscessus.19 Based on in vitro assays with M. abscessus, others have proposed that the role of amikacin is to help to limit the spread of the mycobacteria from infected to uninfected macrophages.20 Treatment evaluation in a mouse model for M. abscessus disseminated disease, showed little activity of amikacin on the reduction of bacterial load in lungs, therefore the long term use of amikacin in the treatment of M. abscessus was questioned.21,22 The current treatment guidelines by the American Thoracic Society and Infectious Diseases Society of America (ATS/IDSA) state that amikacin, in combination with cefoxitin or imipenem, and a third agent like clarithromycin or other (according to the susceptibility profile), may help in controlling the symptoms and progression of M. abscessus pulmonary disease, but that is still considered a chronic incurable disease for the majority of patients.7
Cefoxitin also showed low activity in our experiments against M. abscessus and M. fortuitum, and its effect was very different for both RGM species evaluated (Chapter 2). For M. fortuitum, the effect was faster and stronger than for M. abscessus and it was concentration-dependent, while for M. abscessus cefoxitin activity was slow and concordant with the expected time-dependent effect for a β-lactam antibiotic. So, genetically related NTM may differ widely in their response to antibiotic treatment; this observation is in line with clinical experiences. In opposition to the cefoxitin effect that we (Chapter 2) and others19 have observed in time-kill assays, an in vitro and intra-macrophage activity evaluation of cefoxitin and imipenem indicated that both are active against M. abscessus, although imipenem´s activity might be superior to cefoxitin.23 In addition, an in vivo study in the nude mice model for M. abscessus disease found that cefoxitin was the most active drug in the currently recommended regimen to treat M. abscessus. The pivotal role of cefoxitin (or other β- lactams) in the treatment of M.

138
Chapter 9
abscessus was also evident from our hollow-fiber model experiments, which will be presented later in this chapter.
The effect of clarithromycin was also low against M. abscessus, perhaps because of erm gene induction in M. abscessus 24 at lower concentrations; killing was only observed at higher concentrations (> 4× MIC), which is different to the clarithromycin-mediated kill observed for SGM; this may represent another example that findings from one NTM are not necessarily pertinent to all NTM species. Regrowth was also observed. In vitro information on the evaluation of M. abscessus treatment is scarce, but in vivo experiments in the nude mice model showed clarithromycin´s limited effect on M. abscessus: it prevented the death of the mice, but its impact on bacillary loads was limited.21
Despite erm gene associated inducible resistance in M. abscessus subspecies abscessus and some strains of M. abscessus subspecies bolletii, macrolides are still used in M. abscessus treatment; medical reasons support its use as well as some in vitro findings like those we will discuss below about the potentiality to stop the switch on of the erm gene by using other companion drugs like clofazimine.
A combination of two or three antibiotics, including a fluoroquinolone is recommended for M. fortuitum treatment, based on the in vitro susceptibility evaluation;7 therefore, we choose to evaluate moxifloxacin in our time-kill assays, finding moxifloxacin activity against M. fortuitum, particularly at concentration 2× MIC or higher. Because of high MICs (8 mg/l), we did not perform time-kill analyses of moxifloxacin against M. abscessus.
The potential role of clofazimine in M. abscessus treatmentWe also tested clofazimine-containing combinations to treat M. abscessus; here too, clofazimine-amikacin and clofazimine-clarithromycin, prevented the regrowth observed when amikacin or clarithromycin were used alone. However, opposite to what we observed for M. avium, neither changes in the MIC nor known mutations in rrs and rrl genes were found for M. abscessus when regrowth appeared in the systems exposed to either amikacin or clarithromycin. Adaptive resistance through modifications in cell wall permeability, inducible resistance mechanisms like transcriptional activators or the methylases blocking access to ribosomal targets, and the expression of efflux pumps has been associated in other mycobacteria with decrease susceptibility to antimicrobials;25,26 this may be also the case for M. abscessus, for which an erm gene has been identified, associated with inducible macrolide resistance.24 Clofazimine seems to prevent the activation of such mechanisms. Here, it was evidenced that M. abscessus does not necessarily share the same mechanism associated with resistance to amikacin and clarithromycin in other NTM, e.g. M. avium. Information coming from RNA and DNA sequencing will help to clarify this theory, at least for amikacin. Although more evidence is needed, particularly for M. abscessus, ours and other researchers´ findings call for the future inclusion and evaluation of clofazimine in combination regimens

Discussion, conclusions and future steps
139
Chapter 9
Cha
pter
9
to treat both M. avium and M. abscessus disease. Treatment of M. abscessus infected mice with a combination of clofazimine and bedaquiline was effective in decreasing the burden of mycobacteria in the organs of the animals, including the lung.22 The exact mechanism of action of clofazimine is not entirely understood yet, but what is known is that its role in the electron transfer chain at the plasma membrane is related to that of bedaquiline, and both antibiotics have synergistic in vivo effect.22,27 In the search for better antibiotic alternatives for NTM treatment these two drugs may play an important role in the future.
-What does the hollow-fiber system model tell us about treatment of M. abscessus The hollow-fiber system model (HFS) is an in vitro dynamic pharmacokinetic/pharmacodynamic model used to mimic the in vivo conditions of varied antibiotic concentrations in human compartments over time and its effect against an infecting microorganism. The original HFS for mycobacteria was developed for M. tuberculosis and has shown high accuracy (94.4%) in forecasting optimal drug exposures, doses, and dosing schedules for clinical use when compared to clinical studies.28,29 Since M. abscessus is the most notorious causative agent of NTM-PD, this species was selected for the evaluation in the HFS. We standardized and used the HFS to evaluate single antibiotic exposures, first for amikacin, the key representative of M. abscessus treatment, then for moxifloxacin and tigecycline. Finally, we evaluated the guideline-recommended combination therapy of amikacin, cefoxitin and clarithromycin against M. abscessus.
Individual exposuresAmikacin showed minimal efficacy in the HFS (Chapter 5), which was considerably lower than the efficacy observed in static time-kill assays (Chapter 2). Regrowth was also observed, associated with the replacement of the total population by an amikacin-resistant subpopulation when exposed to higher doses (Chapter 5). These findings highlight a concern on the discrepant results from static vs. dynamic models for NTM, particularly for M. abscessus. While models of static concentrations suggested amikacin bactericidal activity, although maximal at 2-4x MIC, they overestimated its efficacy compared to the HFS model.Despite the toxicity associated with the use of aminoglycosides, amikacin is considered a key antibiotic in NTM therapy.7 However, findings from the nude and IFN-γ knockout (GKO) mice models for M. abscessus have shown limited activity of amikacin in lungs, consistent with our in vitro findings.21,22 We consider that the role of amikacin in M. abscessus pulmonary disease, in the current doses and by intravenous administration, needs to be reconsidered. Other investigators have proposed to minimize amikacin toxicity by alternating inhaled amikacin with parenteral amikacin, but the efficacy of this combination and dosing strategy remains to be determined.30 The analysis of the results of a recently completed clinical trial on the use of nebulized liposomal amikacin as an alternative for NTM treatment will provide useful information on the future use of this drug, although preliminary information have only shown promising results for MAC.31,32

140
Chapter 9
Moxifloxacin´s efficacy against some slowly growing mycobacteria, particularly M. tuberculosis and M. kansasii has been established in the past.33-35 Given this and the current role of moxifloxacin in the so called ´continuation phase´ of the treatment of M. abscessus, after the initial months of parenteral antibiotics, we also tested it in the HFS (Chapter 6). Surprisingly, even at high exposures in the HFS, moxifloxacin did not significantly reduce the M. abscessus population and resistance was found soon during monotherapy. Therefore, moxifloxacin´s performance does not encourage its continued use in the treatment of M. abscessus, unless a synergistic role with other agents is found in combination therapy.
Based on the findings from HFS and Monte Carlo simulations new breakpoints for amikacin and moxifloxacin have been proposed (Chapter 5 and 6). The currently used breakpoints need to be reformulated to better correlate with antibiotic efficacy in dynamic models or with in vivo outcomes; many are now based on wild type MIC distributions only.36 One possible explanation for the poor correlation between in vitro tests and treatment outcomes in clinical practice 2 might be that most isolates may have been considered susceptible when they are actually resistant to the given doses of amikacin and moxifloxacin.
The search for new therapeutic options against NTM, led us to use our HFS to test more options against M. abscessus. Tigecycline has previously demonstrated in vitro effect on RGM and its use in vivo was associated with improvement in 43% of patients who were treated for more than a month.37,38 When tigecycline was evaluated in the HFS model of pulmonary M. abscessus infection, we observed considerable microbial kill below the stasis line for the first time. Its killing exceeded what we previously found for amikacin or moxifloxacin. Tigecycline was highly efficacious and the magnitude of this efficacy means that it can be the first truly bactericidal antibiotic to be included in a regimen against M. abscessus (Chapter 7). A dose of 200 mg/day achieved 1 log kill in close to 90% of patients in the Monte Carlo simulations, therefore we consider this the optimal dose. There is limited information on the effectiveness and safety of high doses of tigecycline; 200 mg/ day has been used and found to be more efficacious in combination regimens against other bacterial pathogens, although mainly in patients intensive care units, who have increased volumes of distribution.39Adverse effects associated with the use of tigecycline at such doses should be carefully considered. The use of anti-emetics is a common practice when administering tigecycline that can help with the adverse effects, 40 but candidates and strategies for treatment with tigecycline particularly at high-doses should be defined.Regrowth was also observed after 10 days of tigecycline monotherapy in the HFS, but in the absence of changes in the MIC, we may have faced adaptive changes in M. abscessus. Anyhow, this is an area of particular attention for future research, since resistance to tigecycline is uncommon and it will be very interesting to decipher how M. abscessus can grow in the presence of tigecycline. Thinking on a better future for NTM treatment, aggressive combination regimens including tigecycline should be evaluated in M. abscessus pulmonary disease.

Discussion, conclusions and future steps
141
Chapter 9
Cha
pter
9
Combination regimenWe used the HFS model to evaluate the performance and benchmark the standard regimen composed by amikacin, clarithromycin and cefoxitin (Chapter 8). Initial killing was observed and lasted 14 days, after which regrowth appeared. The standard combination regimen fails to kill M. abscessus after 28 days, even under optimal exposures for all the antibiotics. This situation resembles the poor outcome commonly observed when treating patients with the standard combination regimen: patients seem to respond at the beginning, but then fail or relapse. Even with customized treatment regimens, based on in vitro drug susceptibility testing, and adjunctive surgery, cure rates were only 57% in a well-documented series of 69 patients.2
Interestingly, the regrowth occurred at the same time an increase of cefoxitin-resistant subpopulation occurred. Bringing back the previous discussion, we consider that cefoxitin, or β-lactam antibiotics in general, are key drivers of the efficacy of M. abscessus treatment. This is in agreement with the in vivo evaluation of the standard combination regimen in the nude mice model, where cefoxitin was the only active agent.21 Whether cefoxitin or other β-lactam antibiotics will feature in new regimens remains to be determined. Different β-lactams and combinations with β-lactamase inhibitors should be tested in the HFS and in vivo first. For now, we advise to include them.
-Designing a new regimen against M. abscessusAccording to recent reports, bedaquiline may have some potential against M. abscessus and other NTM.22,41 In two different mouse models for M. abscessus disease, bedaquiline treatment during eight days reached significant reduction of the bacterial load in several organs, including lung. More importantly, when clofazimine was added to bedaquiline, the combination achieved more significant reduction in bacterial burden and resulted in less inflammation and cellular aggregates in major organs. The evaluation against M. avium showed bacteriostatic activity of bedaquiline in the mouse model; provided that bedaquiline’s safety profile was acceptable, it has been proposed as a good companion drug of clarithromycin in the combination regimen.42 Bedaquiline-containing treatment regimens showed only a modest and temporary clinical response when it was included for at least 6 months as part of salvage therapy to treat 10 patients with advanced and refractory lung disease by MAC or M. abscessus. Further studies of optimal combination regimens are required to determine the place and use of bedaquiline in the management of NTM-PD.41 However, the possible complications of this combination, such as cardiovascular safety and cross-resistance, will require further studies.
There is a growing interest on different β- lactams to treat mycobacteria23,43-47 and important information is being released. The question is: what is the optimal β-lactam antibiotic to treat M. abscessus disease and what is the role of β-lactamase inhibitors? The production of a broad spectrum β-lactamase (BlaMab) by M. abscessus was recently described, explaining why the in vivo activity of certain β-lactams can be limited when they are susceptible to the hydrolysis;44 luckily that is not the case for cefoxitin, whose hydrolysis is extremely slow.44 β-lactamase

142
Chapter 9
inhibitors have been also evaluated with the goal to reduce the effect of BlaMab.44 The new non-
β-lactam β-lactamase inhibitor avibactam proved most promising, in vitro, when combined with imipenem as well as the 5th generation cephalosporin ceftaroline.45,46 Nevertheless, the search and evaluation of active β-lactams and their combination with β-lactamase inhibitors continues, to provide better options for a new regimen. In fact, we have performed preliminary work with ceftaroline-avibactam in the HFS (manuscript in preparation), and it showed bactericidal effect against M. abscessus, but this needs further evaluation.
Considering all this information, if we would now have to propose a new regimen for M. abscessus, it would be the combination of tigecycline, clofazimine, liposomal amikacin, a macrolide and any active β-lactam, like ceftaroline with avibactam, cefoxitin, or imipenem; perhaps bedaquiline will also have a role in this combination.
-Knowledge gaps in regimen design Before we advance into regimen design, critical knowledge gaps need to be filled: completion of individual drug assessment, exploration of synergistic combinations, understanding of resistance mechanisms, etc. One of the important questions that need an answer is for example, is there tigecycline-clofazimine synergy? A preliminary report on the combined activity of clofazimine and tigecycline showed synergy in 42% of the 19 strains tested, and indifference in 10 strains;48 however, more studies and a better characterization of such potential synergism should be performed. On the other hand, other report on the combination of tigecycline with amikacin showed antagonism in >18% of each RGM species evaluated, including M. abscessus,49 that should be considered if amikacin will continue being part of the treatment as it is now (Figure 1). On the contrary, the combination of tigecycline and clarithromycin was synergistic in >90% of M. abscessus isolates,49 this also may indicate that macrolides could still have a role in the treatment of M. abscessus, even in the presence of a functional erm gene, as discussed above. Our in vitro findings with the combination of clofazimine and clarithromycin also support this.
-Other observations from the preclinical modelsDuring the various experiments, several observations apart from the primary study endpoints were made. Some of these warrant mention as they may be interesting subjects of further study and may learn us more about NTM and their stress responses to antibiotic exposure. First, we observed morphological changes in colonies, associated with NTM exposure to amikacin in M. abscessus and M. fortuitum (Chapter 2). Perhaps the same phenomenon also took place in the series of experiments with M. avium, but the varied morphology common to this species prevented us to clearly determine it. Interestingly, changes in M. abscessus included the switch from smooth to rough phenotype in the colonies exposed to amikacin at concentrations 2× MIC or higher in time-kill assays, or to doses greater than the standard daily dose in the HFS (data not shown). The switch in morphology type in M. abscessus has been considered a selective

Discussion, conclusions and future steps
143
Chapter 9
Cha
pter
9
advantage that is linked in vitro to loss of glycopeptidolipid in the cell wall and increased pathogenicity, while in patients it occurs with the change from acute to chronic infection, linked to clinical deterioration.50,51 Other studies have detected that sub-inhibitory concentrations of amikacin produce changes in M. abscessus colony morphology, associated with increased adherence, aggregation and resistance to phagocytosis, besides an increased stimulation of macrophages with the accompanying severity of lung inflammation.52 Whether the changes in morphology we observed were part of adaptive response of M. abscessus in the presence of amikacin, decoded into the expression of this virulence factor, is a question that needs to be answered in the future. Regrowth after initial amikacin killing was observed with all the NTM species, but known mutations in rrs gene,53 associated with resistance to aminoglycosides in mycobacteria were only demonstrated in M. avium (Chapter 4). In the absence of known mutations in the rrs gene, we hypothesize that adaptive changes, such as over expression of aminoglycoside modifying enzymes,19,54 are behind the regrowth observed in M. abscessus experiments. To further understand the phenomenon of amikacin resistance and the mechanism responsible for this phenomenon in M. abscessus, RNA and DNA samples from the HFS experiments are being sequenced.
Second, the lowest antibiotic effect was found for M. abscessus within RGM and for M. avium within SGM; interestingly those two species reached higher growth levels in the untreated controls (up to 1010 - 1011 cfu/ml) than the others. The consistently high growth levels may, to a degree, explain why the observed effect of the antibiotics was lower for M. abscessus and M. avium. If the number of logs cfu kill over time is fixed per drug and concentration, the killing will appear less prominent in species that can grow to high bacterial loads. As additional
Figure 1. Synergy and antagonism against M. abscessus in vitro. Green arrows correspond to synergy; red arrows correspond to antagonism observed; Question marks reflects that interactions are not well studied yet.
CLOFAZIMINE
AMIKACIN(inhaled liposomal) BEDAQUILINE
Beta-LACTAM/AVIBACTAM TIGECYCLINE

144
Chapter 9
evidence, also high growth for M. abscessus (up to 1012 cfu/ml) was observed in the assays performed in the HFS (Chapter 5), despite the differences in the culture conditions compared to time-kill assays. Other reports on time-kill assays have also shown high growth, like we observed, for M. abscessus or M. avium.8,55 Whether the ability of NTM to replicate in vitro and reach high loads is related to their ability to replicate in the host lungs,56 translated into virulence and antibiotic resistance, is an issue that will require further study.
Third, when standardizing HFS experiments with M. abscessus, a green/blue pigmentation and bacterial aggregates were observed in polysulphone hollow-fiber cartridges, permanently bathed in cation-adjusted Mueller Hinton broth. Subculture of bacteria taken from the model still produced this pigment, visible as a black discoloration of Columbia sheep blood agar. The chemical structure of this pigment is currently under investigation. Are both conditions, the pigment and the aggregates (associated with biofilms)57 induced in M. abscessus a response that resembles the pathogenicity needed to invade and persist in human lungs? This is another question that surely deserves future research in the field of host-pathogen interaction, with possible effect for M. abscessus treatment.
Finally, we observed that clumping of M. xenopi was related, in our experiments, to the bacterial load. When we started the growth curves and time-kill kinetic experiments with a low bacterial load, despite incubation under shaking conditions, clumping was observed during the first sampling days, also in the plates that were used to enumerate the cfu/ml of bacteria. However, when cultures reached higher loads, clumps were less evident and suspensions turned more even, as well as the distribution of cfu on plates. Is this due to the activation of quorum sensing in M. xenopi? Since quorum sensing is a mode of cell to-cell communication amongst bacteria, partly related with virulence factors, some researchers have questioned if it can become an important target for controlling the spread of antibiotic resistant bacteria.58 Whether what we observed is quorum sensing, possibly related to M. xenopi virulence, will be also be a subject for future research.
-Limitations of the preclinical modelsWe have detected several limitations in the work we conducted within this thesis. First, the experiments were performed only with the NTM type strains; mainly because of the costs associated with the extensive evaluations performed, we could not include other strains. Ideally clinical isolates should be part of further evaluations and validation of findings. Second, our experiments only represent an evaluation of the extracellular infection. Although mycobacteria are both extracellular and intracellular pathogens, we prioritized the extracellular evaluation, with the idea of a posterior validation of the main and most relevant findings in the intracellular environment. In addition, for M. abscessus for example, there is scarce information on the proportion of bacteria in the extra or intracellular location in lungs, neither on the different oxygen conditions or metabolic states. More fundamental studies into these aspects will benefit

Discussion, conclusions and future steps
145
Chapter 9
Cha
pter
9
therapy simulations. Third, none of our models took into account the role of the immune system in the treatment of NTM and here too, future experimental conditions considering these variables will provide useful information for the treatment. Fourth, a detailed study of resistance mechanisms to amikacin, moxifloxacin and tigecycline was out of the reach of our work. However, we took samples from the HFS for future analysis. Hopefully, whole genome sequencing and RNA sequencing will provide interesting information on this matter. Fifth, there is limited information on the pharmacokinetics of some antibiotics used in the treatment of NTM infected patients, particularly on pharmacokinetics at the site of infection. Such data are needed to improve infection and treatment simulations in dynamic models.
-Bridging this thesis to other worlds...In The Netherlands and Europe in general, as well as in the USA and other developed countries, there is more awareness of the big challenge NTM-PD and their treatment represents. Where I come from, Colombia, and in many other developing countries the situation can be different. It was previously conceived that NTM were not really a problem in countries where tuberculosis (TB) transmission was an issue, and that NTM and TB were in opposite proportions in several settings. However, how much of the patients diagnosed with TB actually have NTM-PD? What is the incidence of NTM in our countries? Are developing countries prepared to diagnose and treat NTM disease? Those are questions with not answered yet.In a group of 106 patients with pulmonary TB from Cali, Colombia, recruited in 2005-2006 to participate in a clinical trial, one patient was initially diagnosed as having TB by clinical symptoms and direct smear, but the disease was actually NTM-produced.59 M. kansasii was detected, but it happened after months of TB treatment, when solid cultures were positive and we exhausted the available tools to identify the strain. Later we found in the same city two more patients that were part of the local TB programme, and were facing the same fate. After years of being treated for TB and recurrent TB, finally a diagnosis of MAC-PD in one patient and M. simiae in the other was made.60 Unfortunately, to decipher that the infection and disease is not TB but NTM-produced is not necessarily good news for the patients, but somehow changed their lives. These are only examples of what can happen when the protocols for pulmonary TB diagnosis have only little room and resources for other mycobacteria producing pulmonary disease. There is scarce data on NTM-PD in Colombia, the available information comes from TB surveillance performed by the Colombian National Institute of Health (INS), and for NTM in general is mainly associated to few outbreaks due to RGM after cosmetic surgery.61,62 As recently suggested the efforts in developing new treatments and diagnostics for TB can be harnessed in the fight against NTM-associated diseases.63 We need to face the growing significance of NTM infections and the great challenge that their treatment represents. A global picture of the epidemiology of this new treat is needed as much as we need to gather work and information from different settings in the world. NTM are for sure present in every scenery, if we forget of them, they will not forgive us...

146
Chapter 9
Griffith D. Mycobacterium abscessus subsp. abscessus lung disease: ‘trouble ahead, trouble behind…’ Prime Reports. 2014;6:107.Jarand J, Levin A, Zhang L, Huitt G, Mitchell JD, Daley CL. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin Infect Dis. 2011;52:565-71.Jeon K, Kwon OJ, Lee NY, Kim BJ, Kook YH, Lee SH, Park YK, Kim CK, Koh WJ. Antibiotic treatment of Mycobacterium abscessus lung disease: a retrospective analysis of 65 patients. Am J Respir Crit Care Med. 2009;180:896-902.Gangadharam PR, Kesavalu L, Rao PN, Perumal VK, Iseman MD. Activity of amikacin against Mycobacterium avium complex under simulated in vivo conditions. Antimicrob Agents Chemother. 1998;32:886-9.Gangadharam PR, Perumal VK, Podapati NR; Kesavalu L, Iseman MD. In vivo activity of amikacin alone or in combination with clofazimine or rifabutin or both against acute experimental Mycobacterium avium complex infections in beige mice. Antimicrob Agents Chemother. 1998;32:1400–3.Kobashi Y, Matsushima T, Oka M. A double-blind randomized study of aminoglycoside infusion with combined therapy for pulmonary Mycobacterium avium complex disease. Respir Med. 2007;101:130-38.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier J, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K, ATS Mycobacterial Diseases Subcommittee, American Thoracic Society, Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.Bakker-Woudenberg IAJM, van Vianen W, van Soolingen D, Verbrugh HA, van Agtmael MA. Antimycobacterial agents differ with respect to their bacteriostatic versus bactericidal activities in relation to time of exposure, mycobacterial growth phase, and their use in combination. Antimicrob Agents Chemother. 2005; 49:2387-98.Andréjak C, Almeida DV, Tyagi S, Converse PJ, Ammerman NC, Grosset JH. Characterization of Mouse Models of Mycobacterium avium complex Infection and Evaluation of Drug Combinations. Antimicrob Agents Chemother. 2015;59:2129-35.Wallace RJ Jr, Brown BA, Griffith DE, Girard WM, Murphy DT. Clarithromycin regimens for pulmonary Mycobacterium avium complex: the first 50 patients. Am J Respir Crit Care Med. 1996;153:1766-72.Tanaka E, Kimoto T, Tsuyuguchi K, Watanabe I, Matsumoto H, Niimi A, Suzuki K, Murayama T, Amitani R, Kuze F. Effect of clarithromycin regimen for Mycobacterium avium complex pulmonary disease. Am J Respir Crit Care Med. 1999;160:866-72.Andréjak C, Almeida DV, Tyagi S, Converse PJ, Ammerman NC, Grosset JH. Improving existing tools for Mycobacterium xenopi treatment: assessment of drug combinations and characterization of mouse models of infection and chemotherapy. J Antimicrob Chemother. 2013;68:659-65. van Ingen J, Totten SE, Helstrom NK, Heifets LB, Boeree MJ, Daley CL. In vitro synergy between clofazimine and amikacin in treatment of nontuberculous mycobacterial disease. Antimicrob Agents Chemother. 2012;56:6324-7. Shen GH, Wu BD, Hu ST, Lin CF, Wu KM, Chen JH. High efficacy of clofazimine and its synergistic effect with amikacin against rapidly growing mycobacteria. Int J Antimicrob Agents. 2010;35:400-4.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
References

Discussion, conclusions and future steps
147
Chapter 9
Cha
pter
9
Fattorini L, Piersimoni C, Tortoli E, Xiao Y, Santoro C, Ricci ML, Orefici G. In vitro and ex vivo activities of antimicrobial agents used in combination with clarithromycin, with or without amikacin, against Mycobacterium avium. Antimicrob Agents Chemother. 1995;39:680-5.Jarand J, Davis JP, Cowie RL, Field SK, Fisher DA. Long Term Follow Up of Mycobacterium avium Complex Lung Disease In Patients Treated With Regimens Including Clofazimine and/or Rifampin. Chest. 2016;149:1285-93.Field SK, Cowie RL. Treatment of Mycobacterium avium-intracellulare complex lung disease with a macrolide, ethambutol, and clofazimine. Chest. 2003;124:1482-6.Deshpande D, Srivastava S, Meek C, Leff R, Gumbo T. Ethambutol optimal clinical dose and susceptibility breakpoint identification by use of a novel pharmacokinetic pharmacodynamic model of disseminated intracellular Mycobacterium avium. Antimicrob Agents Chemother. 2010;54:1728-33.Maurer FP, Bruderer VL, Ritter C, Castelberg C, Bloemberg GV, Böttger EC. Lack of antimicrobial bactericidal activity in Mycobacterium abscessus. Antimicrob Agents Chemother. 2014;58:3828-36. Greendyke R, Byrd TF. Differential antibiotic susceptibility of Mycobacterium abscessus variants in biofilms and macrophages compared to that of planktonic bacteria. Antimicrob Agents Chemother. 2008;52:2019-26.Lerat I, Cambau E, Roth DitBettoni R, Gaillard JL, Jarlier V, Truffot C, Veziris N. In vivo evaluation of antibiotic activity against Mycobacterium abscessus. J Infect Dis. 2014;209:905-12.Obregón-Henao A, Arnett KA, Henao-Tamayo M, Massoudi L, Creissen E, Andries K, Lenaerts AJ, Ordway DJ. Susceptibility of Mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob Agents Chemother. 2015;59:6904-12.Lefebvre AL, Dubée V, Cortes M, Dorchêne D, Arthur M, Mainardi JL. Bactericidal and intracellular activity of β-lactams against Mycobacterium abscessus. J Antimicrob Chemother. 2016; 29. pii: dkw022. [Epub ahead of print]Nash KA, Brown-Elliott BA, Wallace RJ Jr. A novel gene, erm (41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob Agents Chemother. 2009;53:1367-76.Schmalstieg AM, Srivastava S, Belkaya S, Deshpande D, Meek C, Leff R, van Oers NS, Gumbo T. The antibiotic resistance arrow of time: efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob Agents Chemother. 2012;56:4806-15.Burian J, Ramón-García S, Howes CG, Thompson CJ. WhiB7, a transcriptional activator that coordinates physiology with intrinsic drug resistance in Mycobacterium tuberculosis. Expert Rev Anti Infect Ther. 2012;10:1037-47.Lechartier B, Cole ST. Mode of action of clofazimine and combination therapy with benzothiazinones against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2015;59:4457-63.Gumbo T, Pasipanodya JG, Romero K, Hanna D, Nuermberger E. Forecasting Accuracy of the Hollow Fiber Model of Tuberculosis for Clinical Therapeutic Outcomes. Clin Infect Dis. 2015;15;61 Suppl 1:S25-31.Gumbo T, Pasipanodya JG, Nuermberger E, Romero K, Hanna D. Correlations Between the Hollow Fiber Model of Tuberculosis and Therapeutic Events in Tuberculosis Patients: Learn and Confirm. Clin Infect Dis. 2015;15;61 Suppl 1:S18-24.Benwill JL, Wallace RJ Jr. Mycobacterium abscessus: challenges in diagnosis and treatment. Curr Opin Infect Dis. 2014;27:506-10.Winthrop KL, Eagle G, McGinnis JP, Micioni L, Daley CL, Ruoss SJ, Addrizzo-Harris DJ, Flume PA, Dorgan DJ, Salathe M, Brown-Elliott BA, Wallace RJ, Griffith DE, Olivier KN. Subgroup Analyses of Baseline Demographics and Efficacy in Patients with Refractory Nontuberculous
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.

148
Chapter 9
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
Mycobacteria (NTM) Lung Infection Treated with Liposomal Amikacin for Inhalation (LAI). Meeting abstract, Am J Respir Crit Care Med. 191;2015:A6294.Biller JA, Eagle G, McGinnis JP, Micioni L, Daley CL, Winthrop KL, Ruoss SJ, Addrizzo-Harris DJ, Flume PA, Dorgan DJ, Salathe M, Brown-Elliott BA, Wallace RJ, Griffith DE, Olivier KN. Efficacy of Liposomal Amikacin for Inhalation (LAI) in Achieving Nontuberculous Mycobacteria (NTM) Culture Negativity in Patients Whose Lung Infection is Refractory to Guideline-Based Therapy. Meeting abstract, Am J Respir Crit Care Med. 191;2015:A6295.Sindelar G, Zhao X, Liew A, Dong Y, Lu T, Zhou J, Domagala J, Drlica K. Mutant prevention concentration as a measure of fluoroquinolone potency against mycobacteria. Antimicrob Agents Chemother. 2000;44:3337-43.Srivastava S, Pasipanodya J, Sherman CM, Meek C, Leff R, Gumbo T. Rapid drug tolerance and dramatic sterilizing effect of moxifloxacin monotherapy in a novel hollow-fiber model of intracellular Mycobacterium kansasii disease. Antimicrob Agents Chemother. 2015;59:2273-79.Gumbo T, Louie A, Deziel M, Parsons LM, Salfinger M, Drusano GL. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis. 2004;190:1642-51. Clinical Laboratory Standards Institute. 2011. Susceptibility testing of Mycobacteria, Nocardiae and other aerobic Actinomycetes—approved standard M24-A2, 2nd ed. CLSI, Wayne, PA.Wallace RJ Jr, Brown-Elliott BA, Crist CJ, Mann L, Wilson RW. Comparison of the in vitro activity of the glycylcycline tigecycline (formerly GAR-936) with those of tetracycline, minocycline, and doxycycline against isolates of nontuberculous mycobacteria. Antimicrob Agents Chemother. 2002;46:3164-7.Wallace RJ Jr, Dukart G, Brown-Elliott BA, Griffith DE, Scerpella EG, Marshall B. Clinical experience in 52 patients with tigecycline containing regimens for salvage treatment of Mycobacterium abscessus and Mycobacterium chelonae infections. J Antimicrob Chemother. 2014;69:1945-53.Falagas ME, Vardakas KZ, Tsiveriotis KP, Triarides NA, TansarliGS. Effectiveness and safety of high-dose tigecycline-containing regimens for the treatment of severe bacterial infections. Int J Antimicrob Agents. 2014;44:1-7.Floto RA, Olivier KN, Saiman L, Daley CL, Herrmann JL, Nick JA, Noone PG, Bilton D, Corris P, Gibson RL, Hempstead SE, Koetz K, Sabadosa KA, Sermet-Gaudelus I, Smyth AR, van Ingen J, Wallace RJ, Winthrop KL, Marshall BC, Haworth CS; US Cystic Fibrosis Foundation and European Cystic Fibrosis Society. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. Thorax. 2016 Jan;71 Suppl 1:i1-22Philley JV, Wallace RJ Jr, Benwill JL, Taskar V, Brown-Elliott BA, Thakkar F, Aksamit TR, Griffith DE. Preliminary Results of Bedaquiline as Salvage Therapy for Patients With Nontuberculous Mycobacterial Lung Disease. Chest. 2015;148:499-506.Lounis N, Gevers T, Van den Berg J, Vranckx L, Andries K.ATP synthase inhibition of Mycobacterium avium is not bactericidal. Antimicrob Agents Chemother. 2009; 53: 4927-4929.Lavollay M, Fourgeaud M, Herrmann JL, Dubost L. Marie A, Gutmann L, Arthur M, Mainardi JL. The peptidoglycan of Mycobacterium abscessus is predominantly cross-linked by L,D-transpeptidases. J Bacteriol. 2011;173:778-82.Soroka D, Dubée V, Soulier-Escrihuela O, Cuinet G, Hugonnet JE, Gutmann L, Mainardi JL, Arthur M. Characterization of broad-spectrum Mycobacterium abscessus class A β-lactamase. J Antimicrob Chemother. 2014;69:691-6.

Discussion, conclusions and future steps
149
Chapter 9
Cha
pter
9
Dubée V, Soroka D, Cortes M, Lefebvre AL, Gutmann L, Hugonnet JE, Arthur M, Mainardi JL. Impact of β-lactamase inhibition on the activity of ceftaroline against Mycobacterium tuberculosis and Mycobacterium abscessus. Antimicrob Agents Chemother. 2015;59:2938-41.Dubée V, Bernut A, Cortes M, Lesne T, Dorchene D, Lefebvre AL, Hugonnet JE, Gutmann L, Mainardi JL, Herrmann JL, Gaillard JL, Kremer L, Arthur M. β-Lactamase inhibition by avibactam in Mycobacterium abscessus. J Antimicrob Chemother. 2015;70:1051-8.Lavollay M, Dubée V, Heym B, Herrmann JL, Gaillard JL, Gutmann L, Arthur M, Mainardi JL. In vitro activity of cefoxitin and imipenem against Mycobacterium abscessus complex. Clin Microbiol Infect. 2014;20:O297-300.Singh S, Bouzinbi N, Chaturvedi V, Godreuil S, Kremer L. In vitro evaluation of a new drug combination against clinical isolates belonging to the Mycobacterium abscessus complex. Clin Microbiol Infect. 2014;20:O1124-7.Huang CW, Chen JH, Hu ST, Huang WC, Lee YC, Huang CC, Shen GH. Synergistic activities of tigecycline with clarithromycin or amikacin against rapidly growing mycobacteria in Taiwan. Int J Antimicrob Agents. 2013;41:218-23.Howard ST, Rhoades E, Recht J, Pang X, Alsup A, Kolter R, Lyons CR, Byrd TF. Spontaneous reversion of Mycobacterium abscessus from a smooth to a rough morphotype is associated with reduced expression of glycopeptidolipid and reacquisition of an invasive phenotype. Microbiology. 2006;152:1581-90.Park IK, Hsu AP, Tettelin H, Shallom SJ, Drake SK, Ding L, Wu UI, Adamo N, Prevots DR, Olivier KN, Holland SM, Sampaio EP, Zelazny AM. Clonal Diversification and Changes in Lipid Traits and Colony Morphology in Mycobacterium abscessus Clinical Isolates. J Clin Microbiol. 2015;53:3438-47.Tsai SH, Lai HC, Hu ST. Subinhibitory doses of aminoglycoside antibiotics induce changes in the phenotype of Mycobacterium abscessus. Antimicrob Agents Chemother. 2015;59:6161-9.Brown-Elliott BA, Iakhiaeva E, Griffith DE, Woods GL, Stout JE, Wolfe CR, Turenne CY, Wallace RJ Jr. In vitro activity of amikacin against isolates of Mycobacterium avium complex with proposed MIC breakpoints and finding of a 16S rRNA gene mutation in treated isolates. J Clin Microbiol. 2013;51:3389-94.Maurer FP, Bruderer VL, Castelberg C, Ritter C, Scherbakov D, Bloemberg GV, Böttger EC. Aminoglycoside-modifying enzymes determine the innate susceptibility to aminoglycoside antibiotics in rapidly growing mycobacteria. J Antimicrob Chemother. 2015;70:1412-9.Kaushik A,MakkarN,Pandey P, Parrish N, Singh U, Lamichhane G. Carbapenems and Rifampin Exhibit Synergy against Mycobacterium tuberculosis and Mycobacterium abscessus. Antimicrob Agents Chemother. 2015;59:6561-67.Sohn H, Kim HJ, Kim JM, Jung Kwon O, Koh WJ, Shin SJ. High virulent clinical isolates of Mycobacterium abscessus from patients with the upper lobe fibrocavitary form of pulmonary disease. Microb Pathog. 2009;47:321-8.Qvist T, Eickhardt S, Kragh KN, Andersen CB, Iversen M, Høiby N, Bjarnsholt T. Chronic pulmonary disease with Mycobacterium abscessus complex is a biofilm infection. Eur Respir J. 2015;46:1823-6Polkade AV, Mantri SS, Patwekar UJ, Jangid K. Quorum Sensing: An Under-Explored Phenomenon in the Phylum Actinobacteria. Front Microbiol. 2016;7:131.Rojas CM, Villegas SL, Piñeros HM, Chamorro EM, Durán CE, Hernández EL, Pacheco R, Ferro BE. Clinical, epidemiological and microbiological characteristics of a cohort of pulmonary tuberculosis patients in Cali, Colombia. Biomedica. 2010;30:482-91
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.

150
Chapter 9
60.
61.
62.
63.
Villegas SL, García P, Rozo JC, Nieto LM, Ferro BE. Infección por Micobacterias atípicas y enfermedad pulmonar crónica en pacientes inmunocompetentes: dos casos clínicos. Meeting abstract. Biomedica. 2009;29 Suppl. 1:199-200.Llerena C, Valbuena A, Gómez T. Micobacterias no tuberculosas causantes de micobacteriosis en Colombia. Meeting abstract. Biomedica. 2015;35 Suppl. 216.Correa NE, Cataño JC, Mejía GI, Realpe T, Orozco B, Estrada S, Vélez A, Vélez L, Barón P, Guzmán A, Robledo J. Outbreak of mesotherapy-associated cutaneous infections caused by Mycobacterium chelonae in Colombia. Jpn J Infect Dis. 2010;63:143-5.Raju RM, Raju SM, Zhao Y, Rubin EJ. Leveraging advances in tuberculosis diagnosis and treatment to address nontuberculous mycobacterial disease. Emerg Infect Dis. 2016;22:365-9.

Discussion, conclusions and future steps
151
Chapter 9
Cha
pter
9


153
Chapter 10
Summary

154
Chapter 10

Summary
155
The focus of this thesis was to investigate the pharmacodynamic foundation of current treatment regimens for nontuberculous mycobacterial pulmonary disease (NTM-PD); next, we searched for new and more efficacious treatment regimens, with focus on Mycobacterium abscessus.
In Chapter 1 we present a general introduction that provides the background for the study. We briefly introduce the reader to the NTM-PD, explaining the main clinical presentations and radiological findings. Then, we focused on the treatment of NTM-PD, showing the evidence that supports the currently recommended treatment regimens for Mycobacterium avium complex and Mycobacterium abscessus disease. The second part of the chapter shifts to the evidence that has been provided by preclinical studies, both in vitro and in vivo, through different assays and models. Finally, we present the aim of the thesis that is to assess the pharmacodynamics of commonly used drugs to treat NTM disease by means of the time–kill kinetic assays and the development of a pharmacodynamic model on basis of the in vitro hollow fiber system.
In Chapter 2, the results of time-kill kinetics of antibiotics against rapidly growing mycobacteria (RGM) are presented. We tested amikacin and cefoxitin against M. abscessus and M. fortuitum, clarithromycin only against M. abscessus, and moxifloxacin and linezolid only against M. fortuitum. Although amikacin was the most active antibiotic, in general, all antibiotics showed low activity, reminiscent of the poor outcome observed in patients with RGM-pulmonary disease, particularly when produced by M. abscessus. Regrowth of the mycobacteria after several days of exposure was prominent for all tested antibiotics, suggestive of the emergence of resistance or tolerance.
In Chapter 3, the outcome of the evaluation of antibiotics against slowly growing mycobacteria (SGM) common in pulmonary disease is exposed. As we had observed for RGM, the key antibiotics tested against M. avium and M. xenopi also had low activity in time-kill assays. Amikacin had the strongest killing capacity against M. avium, supporting its role in a combined therapy. For M. xenopi, clarithromycin and moxifloxacin proved equally efficacious, indicating that both antibiotics may have identical activity within treatment regimens and that perhaps both should be part of treatment regimens.
Both chapter 2 and 3 pointed out that -given the low activity of single drugs- the evaluation of antibiotic combinations or combined regimens should be the next step in our study, therefore within Chapter 4, we assessed the activity of clofazimine and the activity of the combinations of clofazimine-amikacin and clofazimine-clarithromycin against M. abscessus and M. avium, by time-kill assays. For both species, clofazimine alone was inactive, but it prevented the regrowth observed when amikacin or clarithromycin were used alone, suggesting a potential adjunctive role for clofazimine in treatment regimens.
Sum
mar
y

156
Chapter 10
Moving forward into the direction of performing dynamic pharmacokinetic/pharmacodynamic (PK/PD) evaluations, which are more representative of the in vivo situation, Chapter 5 details on the standardization and development of the hollow-fiber system model (HFS) for M. abscessus pulmonary disease. Amikacin, the most active antibiotic in static models (Chapter 2 and 3), was chosen for this initial PK/PD evaluation; however, minimal efficacy was demonstrated in the HFS. Both microbial kill and the acquisition of drug resistance were linked to the peak concentration-to-MIC ratio (Cmax/MIC). The peak/MIC ratio associated with 80% of the maximal kill (EC80) was 3.20; EC80 is considered the optimal exposure. In Monte Carlo simulations, the highest of the standard amikacin dose, 1,500 mg a day, achieved the EC80 in less than 21% of the patients, therefore the use of intravenous amikacin in the current dose needs to be revisited.
Given the urgent need to find better treatment options for M. abscessus disease, further experiments were performed in the HFS. In Chapter 6, the evaluation of moxifloxacin is presented. The EC80 was an area under the concentration-time curve (AUC) AUC0–24/MIC ratio of 102.11, however, no moxifloxacin concentration killed M. abscessus to bacterial loads below the starting inoculum; besides, regrowth was observed after day 3 with all doses. Monte Carlo simulations revealed that doses of 400 to 800 mg/day would achieve or exceed the EC80 in <12.5% of patients. A moxifloxacin PK/PD susceptibility breakpoint of 0.25 mg/l is proposed, which means that almost all M. abscessus clinical isolates should be considered resistant. Despite moxifloxacin´s poor efficacy as monotherapy, formal combination therapy studies with moxifloxacin still need to be performed in the HFS-M. abscessus to explore potential synergy with other agents, before definite recommendations can be made to remove it from current regimens. In Chapter 7, we aimed to perform a dose-response study with tigecycline to identify its efficacy and determine optimal doses for the treatment of M. abscessus pulmonary disease. For the first time in the HFS-M. abscessus model, a drug exhibited a considerable microbial kill below the stasis line, exceeding what has been previously observed for amikacin or moxifloxacin. The AUC/MIC ratio required to attain the EC80 was 36.65; Monte Carlo simulation experiments with 10,000 patients identified that the optimal tigecycline clinical dose for adults is 200 mg/day. Since tigecycline proved bactericidal against M. abscessus, it should be considered as a first-line agent early during therapy and as a backbone for new combination regimens. However, tigecycline is known to cause severe adverse events and its clinical use should be cautious, until its safety and efficacy have been established in clinical trials.
Clinicians treat M. abscessus pulmonary disease with a combination of amikacin, clarithromycin and either cefoxitin or imipenem, that is the guideline-recommended combination regimen for the initial phase of treatment. In Chapter 8 we evaluate the performance of this combination regimen in the HFS-M. abscessus, to determine whether there is synergy or prevention of the drug resistance observed with single drug exposures. At optimal doses, there was initial kill during the first 14 days, but then there was regrowth due to acquired resistance; the standard

Summary
157
combination regimen failed after 28 days of evaluation. Cefoxitin seemed to be the driving force of this regimen since acquired resistance to cefoxitin abrogated the initial kill. The situation observed here resembles the poor long-term outcomes often faced when patients are treated with the standard combination regimen.
Finally, Chapter 9 presents the discussion of the main findings, conclusions and future steps. Four main issues can be inferred from our experimental work. First, the efficacy of most currently recommended antibiotics to treat NTM-PD is low. Second, clofazimine has a potential role as part of combination therapy against M. avium and M. abscessus. Third, tigecycline is bactericidal against M. abscessus and should be part of a new combination regimen to be tested. Fourth, β-lactam antibiotics are likely to be the driving force in the currently recommended treatment regimen for M. abscessus disease. Based on our and others’ in vitro findings as well as recent clinical information, we propose a combination of tigecycline, clofazimine, liposomal amikacin, a macrolide and an active β-lactam antibiotic, like ceftaroline with avibactam, cefoxitin, or imipenem as a new regimen to treat M. abscessus disease.With this new knowledge we hope to contribute to the understanding and improvement of NTM disease treatment, including the design of more effective treatment regimens for NTM-PD. Critical gaps still need to be filled and these include completion of individual drug assessment, exploration of synergistic combinations and understanding of -and influencing- resistance mechanisms for all drugs with established or supposed potential in NTM treatment.
Sum
mar
y


159
Nederlandstalige samenvatting
Chapter 11

160
Chapter 11

Nederlandstalige samenvatting
161
Het doel van dit proefschrift was om de farmacodynamiek van belangrijke antibiotica in de huidige behandeling van niet-tuberculeuze mycobacteriële longinfecties (NTM-PD) te onderzoeken. Tegelijkertijd evalueerden we nieuwe en mogelijk effectievere behandelingen, met name voor Mycobacterium abscessus infecties.
In hoofdstuk 1 geven we een algemene inleiding die de achtergrond voor onze studies beschrijft. We introduceren eerst de klinische en radiologische manifestaties van NTM-PD. Daarna worden de huidige behandelingen voor NTM-PD en de gegevens waarop deze zijn gebaseerd besproken, met de focus op ziekte die veroorzaakt wordt door Mycobacterium avium complex en Mycobacterium abscessus. In het tweede deel van het hoofdstuk beschrijven we de resultaten van preklinische studies, zowel in vitro als in vivo uitgevoerd, met de verschillende testplatforms en modellen. Ten slotte stellen we het doel van deze thesis vast: het evalueren van de farmacodynamiek van belangrijke antibiotica die gebruikt worden in de behandeling van NTM-PD, door het uitvoeren van time-kill kinetiek testen en het opzetten van een Hollow Fiber farmacodynamisch model (hollow fiber model; HFM) om therapie van M. abscessus longinfecties te simuleren.In hoofdstuk 2 presenteren wij de resultaten van de time-kill kinetiek testen van de belangrijkste antibiotica gebruikt voor infecties door snel groeiende mycobacteriën (rapid growing mycobacteria; RGM). We testten de werkzaamheid van amikacine en cefoxitine tegen M. abscessus en M. fortuitum, claritromycine tegen M. abscessus en moxifloxacine en linezolid tegen M. fortuitum. Amikacine was het meest effectieve antibioticum, maar alle geteste antibiotica vertoonden lage activiteit, passend bij de slechte uitkomst van therapie bij patiënten met RGM-longziekte, met name veroorzaakt door M. abscessus. Na enkele dagen blootstelling groeiden de mycobacteriën doorgaans weer uit, passend bij het opkomen van resistentie of tolerantie.
In hoofdstuk 3 evalueerden we de effectiviteit van de meest gebruikte antibiotica tegen langzaam groeiende mycobacteriën (slow growing mycobacteria; SGM). Zoals wij al hadden vastgesteld voor RGM, hadden de belangrijkste antibiotica waarvan de werkzaamheid getest werd tegen M. avium en M. xenopi, wederom een lage activiteit in time-kill testen. Amikacine had het sterkste effect tegen M. avium en dit middel heeft dus mogelijk een plaats in combinatietherapieën voor M. avium infecties. Claritromycine en moxifloxacine waren even effectief tegen M. xenopi; beide antibiotica kunnen een rol spelen in behandelregimes, eventueel in combinatie.Hoofdstuk 2 en 3 toonden -met de beperkte activiteit van individuele antibiotica- het belang van therapie met meerdere antibiotica aan en dus ook de noodzakelijkheid van in vitro evaluatie van combinaties van antibiotica. De volgende stap in ons onderzoek was derhalve het onderzoeken, in hoofdstuk 4, van de individuele activiteit van clofazimine, maar ook van de combinaties clofazimine-amikacine en clofazimine-claritromycine tegen M. abscessus en M. avium, wederom middels time-kill testen. Clofazimine was niet erg actief tegen deze beide species, maar het verhinderde wel de uitgroei die werd waargenomen wanneer amikacine of Sam
enva
tting

162
Chapter 11
claritromycine afzonderlijk werden getest. Dit suggereert een mogelijke potentiërende rol voor clofazimine in de antibiotische behandeling van M. avium en M. abscessus infecties.
De volgende stap was het uitvoeren van farmacokinetiek/farmacodynamiek (PK/PD) evaluaties, die representatiever zijn voor de in vivo situatie. Hoofdstuk 5 beschrijft de ontwikkeling en standaardisatie van een Hollow Fiber model (HFM) voor M. abscessus longziekte. Amikacine, dat als het effectiefste antibioticum uit de statische modellen naar voren kwam, (Hoofdstuk 2 en 3) werd gekozen voor deze eerste PK/PD evaluatie. In het HFM met M. abscessus werd echter een minimale werkzaamheid aangetoond in klinisch haalbare doseringen. Zowel het bacteriedodend effect als het verwerven van resistentie bleken gerelateerd te zijn aan de verhouding van de piekconcentratie en minimaal remmende concentratie (MIC)(Cmax/MIC). De verhouding die 80% van het maximale bacteriedodend effect (EC80) opleverde was een Cmax/MIC ratio van 3,20. Met Monte Carlo simulaties toonden wij aan dat de standaard amikacine doses van 750 tot 1500 mg eenmaal daags de EC80 werd bereikt in minder dan 21% van de patiënten. Gezien dit beperkte effect dient de plaats en het gebruik van intraveneuze amikacine in behandelregimes voor M. abscessus infecties te worden herzien.
De resultaten van antibiotische behandeling van M. abscessus longinfecties zijn bedroevend te noemen. Om dit falen te begrijpen en om nieuwe behandelopties te verkennen, voerden wij verdere experimenten in het HFM met M. abscessus uit. In hoofdstuk 6 beschrijven wij de evaluatie van moxifloxacine, een middel dat veel wordt gebruikt in de M. abscessus behandeling. De EC80 was een oppervlak onder de tijd-concentratie curve: MIC (AUC0-24/MIC) verhouding van 102,11. Geen enkele van de geteste moxifloxacine doseringen kon het aantal M. abscessus bacteriën terugbrengen tot onder het initiële inoculum. Bovendien werd bij alle doseringen uitgroei waargenomen na dag 3, passend bij het opkomen van resistentie of tolerantie. Monte Carlo simulaties lieten zien dat doses van 400-800 mg/dag bij minder dan 12,5% van de patiënten zou leiden tot het halen of overstijgen van de EC80. De rol van moxifloxacine lijkt dus zeer beperkt. Op basis van deze PK/PD analyse stelden wij een MIC breekpunt voor resistentie voor moxifloxacine vast van 0,25 mg/l. Gezien de normale MIC verdeling moeten vrijwel alle klinische M. abscessus stammen als resistent beschouwd worden. Om eventueel synergistisch potentieel van moxifloxacine te onderzoeken dient nog wel de combinatietherapie onderzocht te worden, uitgevoerd in het HFM met M. abscessus. In hoofdstuk 7 hebben we de werkzaamheid en optimale dosering van tigecycline voor de behandeling van M. abscessus longinfecties onderzocht. Tigecycline bleek het eerste middel dat in het HFM met M. abscessus bactericide was en het aantal M. abscessus bacteriën kon reduceren tot onder het initiële inoculum, de zogenaamde stasis lijn. De EC80 werd bereikt bij een AUC/MIC ratio van 36,65; Monte Carlo experimenten met 10.000 patiënten toonden aan dat de optimale tigecycline dosis 200 mg/dag is; het dubbele van de huidige dosis. Aangezien tigecycline bactericide bleek tegen M. abscessus, lijkt het een plek te verdienen in behandelregimes, zeker in de initiële fase van behandeling. Gezien de aanzienlijke

Nederlandstalige samenvatting
163
bijwerkingen van tigecycline dient dit echter met voorzichtigheid te gebeuren en bij voorkeur pas na bevestiging van veiligheid en effectiviteit in klinische studies.
Richtlijnen voor de behandeling van M. abscessus longinfecties adviseren momenteel een combinatieregime van amikacine, claritromycine en cefoxitine of imipenem. In Hoofdstuk 8 evalueren wij van de effectiviteit van die standaard combinatiebehandeling van amikacine, claritromycine en cefoxitine, om te bepalen of deze combinatie synergistisch is en of deze resistentievorming, zoals steeds geobserveerd in de monotherapie experimenten, voorkomt. Bij optimale dosering, was er een duidelijke bactericide werking tijdens de eerste 14 dagen, maar in de daaropvolgende 14 dagen trad weer uitgroei op, als gevolg van verworven resistentie. Cefoxitine leek de drijvende kracht achter dit combinatieregime, aangezien de uitgroei pas optrad bij het opkomen van verworven resistentie tegen cefoxitine. Deze observatie is goed te rijmen met klinische observaties; als patiënten de intensieve therapie tolereren, laten zij vaak initieel een microbiologische respons zien. Helaas wordt in latere stadia van behandeling toch vaak terugkeer van ziekte gezien. Een nieuw regime is dus hard nodig.
Hoofdstuk 9 is de afsluitende bespreking van de belangrijkste bevindingen, conclusies en toekomstige stappen. Vier belangrijke bevindingen kunnen worden afgeleid uit ons experimentele werk. Ten eerste, de effectiviteitvan de meeste antibiotica die gebruikt worden in de behandeling van NTM-PD is laag. Ten tweede, clofazimine heeft een mogelijke potentiërende rol binnen combinatieregimes tegen M. avium en M. abscessus. Ten derde, tigecycline is bactericide tegen M. abscessus en zou deel uit kunnen maken van nieuw te ontwerpen combinatieregimes. Ten vierde, β-lactam antibiotica (cefoxitine, imipenem) zijn waarschijnlijk de drijvende kracht achter het momenteel aanbevolen behandelregime voor M. abscessus infecties. Op basis van onze in vitro bevindingen en die van anderen, maar ook van recente klinische studies, stellen wij een nieuw regime voor van tigecycline, clofazimine, liposomale amikacine, een macrolide en een β-lactam antibioticum, zoals ceftaroline met avibactam, cefoxitine of imipenem, voor behandeling van M. abscessus infecties. Met deze nieuwe kennis hopen we bij te dragen aan het begrip en het verbeteren van de behandeling van NTM infecties, met name het ontwerpen van effectievere behandelingen voor NTM longinfecties. Er zijn echter nog vele hiaten in onze kennis op te vullen, te beginnen bij de voltooiing van de evaluatie van alle individuele antibiotica die een bijdrage kunnen leveren in de behandeling van NTM infecties, het verder verkennen van synergistische combinaties, en het vergroten van onze kennis van resistentie mechanismen in NTM en de beïnvloeding hiervan.
Sam
enva
tting


165
Resumen en Español
Chapter 12

166
Chapter 12

Resumen en Español
167
El enfoque de esta tesis fue el de investigar los fundamentos farmacodinámicos del tratamiento actual de la enfermedad pulmonar por micobacterias no tuberculosas (NTM-PD); posteriormente también buscamos y evaluamos nuevos y más eficaces regímenes de tratamiento, con particular interés en el tratamiento de Mycobacterium abscessus.
En el capítulo 1 se presenta una introducción general que proporciona las bases del estudio. Se hace una breve introducción al lector sobre NTM-PD, en la cual se incluye información sobre las principales presentaciones clínicas y los hallazgos radiológicos. Posteriormente, el capítulo se centra en el tratamiento de la NTM-PD, y se presenta la evidencia que soporta los actuales regímenes de tratamiento para la enfermedad producida por Mycobacterium avium y Mycobacterium abscessus. En la segunda parte, se vira hacia la evidencia que los estudios pre-clínicos, tanto in vitro como in vivo, han proporcionado a través de diferentes ensayos y modelos. Por último, se presenta el objetivo general de la tesis, que es evaluar la farmacodinámica de los antibióticos comúnmente utilizados para tratar la NTM-PD, por medio de ensayos cinéticos de tiempo-muerte, y el desarrollo de un modelo farmacodinámico in vitro, basado en el sistema de fibra hueca.
En el capítulo 2, se exponen los resultados de los ensayos de cinética de tiempo-muerte de los antibióticos contra las micobacterias de crecimiento rápido (RGM). Se evaluó amikacina y cefoxitina contra dos especies: M. abscessus y M. fortuitum, también se evaluó claritromicina contra M. abscessus, y moxifloxacina y linezolid contra M. fortuitum. Aunque amikacina fue el antibiótico más activo, en general los antibióticos mostraron una baja actividad, lo que concuerda con la mala respuesta clínica observada en los pacientes con enfermedad pulmonar producida por RGM, especialmente por M. abscessus. Con todos los antibióticos evaluados se observó re-crecimiento de las micobacterias, luego de varios días de exposición, lo cual sugiere la aparición de resistencia o tolerancia.
En el capítulo 3, se presenta el resultado de la evaluación de los antibióticos contra micobacterias de crecimiento lento (SGM) comunes en la enfermedad pulmonar. Como se mencionó anteriormente para RGM, los antibióticos clave evaluados contra M. avium y M. xenopi también mostraron una baja actividad en ensayos cinéticos de tiempo-muerte. Amikacina mostró actividad bactericida contra M. avium, lo cual soporta su rol en una terapia combinada. Para M. xenopi, claritromicina y moxifloxacina no mostraron diferencia en el efecto, lo que indica que ambos antibióticos podrían tener una actividad equivalente dentro de los regímenes de tratamiento; tal vez ambos antibióticos deben ser parte del régimen de tratamiento.
En los capítulos 2 y 3 se señala que la evaluación de las combinaciones de antibióticos o regímenes combinados debería ser el siguiente paso en nuestro estudio, dada la baja actividad observada en la evaluación individual de los antibióticos; por lo tanto, en el capítulo 4, se evaluó la actividad de la clofazimina y la actividad de las combinaciones de clofazimina- Res
umen

168
amikacina y clofazimina-claritromicina frente a M. avium y M. abscessus, mediante ensayos cinéticos de tiempo-muerte. Para ambas especies, clofazimina no fue muy activo, pero evitó el re-crecimiento observado cuando amikacina o claritromicina se utilizaron solos, lo que sugiere un posible papel de clofazimina como coadyuvante en los regímenes de tratamiento.
Avanzando en la dirección de realizar evaluaciones más representativas de la situación in vivo, el capítulo 5 muestra la estandarización y el desarrollo de un modelo dinámico farmacocinético/farmacodinámico (PK/PD): el sistema de fibra hueca (HFS) para la enfermedad pulmonar producida por M. abscessus. Amikacina, el antibiótico más activo en los modelos estáticos (capítulo 2 y 3), fue elegido para esta evaluación inicial de PK/PD; sin embargo, demostró mínima eficacia en el HFS. Tanto la muerte microbiana como la adquisición de resistencia a los medicamentos se asociaron con la relación entre el pico o concentración máxima (Cmax) y el MIC: Cmax/MIC. La relación Cmax/MIC asociada con el 80% del efecto máximo (EC80) fue 3.20; Se considera que el EC80 es la exposición ideal. En simulaciones de Monte Carlo, la dosis estándar más alta de amikacina, 1500 mg/día, logró el EC80 en menos del 21% de los pacientes, por lo tanto, el uso intravenoso de amikacin a la dosis utilizada actualmente, debe ser revisado.
Dada la urgente necesidad de probar diferentes antibióticos y encontrar mejores opciones contra M. abscessus, más experimentos se llevaron a cabo en el HFS. En el capítulo 6 se presenta la evaluación de moxifloxacina. El EC80 fue una relación del área bajo la curva (AUC0-24)con MIC: AUC0-24/MIC de 102.11; sin embargo, moxifloxacina a ninguna concentración logró matar a M. abscessus a niveles por debajo del inoculo de partida; adicionalmente, se observó re-crecimiento después del tercer día. Las simulaciones de Monte Carlo revelaron que dosis de moxifloxacina de 400 a 800 mg/día lograrían superar el EC80 solo en menos del 12.5% de los pacientes. Se propuso un punto de corte de susceptibilidad para moxifloxacina de 0.25 mg/l, lo que se traduce en que casi la totalidad de las cepas clínicas de M. abscessus deberían ser consideradas resistentes. A pesar de que en nuestros experimentos se observó poca eficacia de moxifloxacina, antes de hacer recomendaciones definitivas acerca de la exclusión de este antibiótico de los regímenes actuales, su potencial de sinergia con otros agentes debe ser evaluada en estudios de terapia combinada contra M. abscessus. El capítulo 7 tuvo como objetivo identificar la eficacia de tigeciclina y evaluar en un estudio de dosis-respuesta las dosis óptimas para el tratamiento de la enfermedad pulmonar producida por M. abscessus. Por primera vez en el modelo HFS, un antibiótico mostró producir una considerable muerte microbiana, por debajo de la línea de estasis, lo cual es superior a lo observado previamente con amikacina y moxifloxacina. La relación AUC/MIC necesaria para obtener el EC80 fue 36.65. Las simulaciones de Monte Carlo con 10,000 pacientes identificaron que la dosis clínica óptima de tigeciclina es de 200 mg/día. Dado que la tigeciclina mostró ser bactericida contra M. abscessus, se debe considerar como un agente de primera línea al comienzo del tratamiento, jugando un papel fundamental en los nuevos regímenes combinados. Sin embargo, es bien sabido que la tigeciclina puede producir reacciones adversas severas, por lo
Chapter 12

Resumen en Español
169
que su uso clínico debe ser cauteloso, hasta tanto su seguridad y eficacia haya sido establecida en ensayos clínicos.
La enfermedad pulmonar producida por M. abscessus se trata generalmente con una combinación de amikacina, claritromicina y, cefoxitina o imipenem, lo cual constituye el régimen combinado estándar, recomendado para la fase inicial de tratamiento en las guías de expertos. En el capítulo 8 se muestra la evaluación de dicho régimen, de amikacina, claritromicina y cefoxitina, con el fin de determinar si estos antibióticos exhiben potencial de sinergia y previenen la resistencia a los medicamentos que se observa con las exposiciones individuales. A dosis óptimas, durante los primeros 14 días hubo destrucción inicial de las micobacterias, pero luego se observó re-crecimiento debido a resistencia adquirida; el régimen de combinación estándar falló después de 28 días de evaluación. La cefoxitina parece ser la fuerza impulsadora de este régimen, ya que la resistencia adquirida a cefoxitina abrogó la matanza inicial. Esta situación se asemeja a la mala evolución que a menudo se observa cuando los pacientes son tratados con el régimen combinado estándar: si lo toleran, pueden responder inicialmente, pero al final, muchos pacientes simplemente no responden, y algunos responden pero luego recaen.
Por último, el capítulo 9 presenta la discusión de los principales hallazgos de este trabajo, las conclusiones y los pasos futuros. Cuatro argumentos principales se pueden inferir de este trabajo experimental. En primer lugar, la eficacia de los antibióticos más recomendados en la actualidad para el tratamiento de la NTM-PD es pobre. En segundo lugar, la clofazimina tiene un papel potencial como parte de una terapia combinada contra M. avium y M. abscessus. En tercer lugar, la tigeciclina es bactericida contra M. abscessus y debería ser parte de un nuevo régimen combinado a evaluar. En cuarto lugar, los antibióticos β-lactámicos son probablemente la fuerza impulsora en el régimen de tratamiento actualmente recomendado para la enfermedad pulmonar por M. abscessus.
Con base en estos hallazgos, así como en información clínica reciente, proponemos un nuevo régimen para tratar la enfermedad pulmonar producida por M. abscessus, basado en la combinación de tigeciclina, clofazimina, amikacina liposomal, un macrólido y un β-lactámico activo, tal como ceftarolina con avibactam, cefoxitina o imipenem.
Con el nuevo conocimiento generado con este trabajo esperamos contribuir a la comprensión y a la mejora del tratamiento de la enfermedad pulmonar producida por NTM, incluido el diseño de regímenes de tratamiento más eficaces. Aún persisten vacíos de conocimiento que son críticos y que deben ser llenados; es necesario, por ejemplo, completar la evaluación individual de medicamentos, explorar combinaciones sinérgicas y profundizar en el entendimiento de los diversos mecanismos de resistencia para todos los medicamentos que tienen potencial, conocido o supuesto, para el tratamiento de NTM-PD.
Res
umen


Biography andList of Publications

172
Biography and List of Publications

Biography and List of Publications
173
Biography
Beatriz Eugenia Ferro Ramos was born on August 3rd, 1973 in Palmira, Colombia, as the youngest of the two children from the family formed by Joaquin Ferro García † and Yolanda Ramos Rodríguez. She grew up and attended to a Catholic school, Sagrado Corazón de Jesús - Bethlemitas, in her home town, where she did primary and secondary studies. In the final year of the secondary school, Beatriz got the highest score from her class in the national evaluation exam for Colombian students, ICFES, then she was admitted and started to study a Bachelor in Bacteriology and Clinical Laboratory Science at Universidad del Valle, Cali (the third largest city in Colombia), where she received scholarships for high academic performance in several semesters. She finished universitary studies in 1994 and did the social service year working at the Instituto de Inmunología- Universidad del Valle, as a research assistant in several projects on malaria. After this year, she started a Master of Science, also at the Universidad del Valle, where she conducted her thesis project “Performance of OptiMAL® in the diagnosis of Plasmodium vivax and Plasmodium falciparum infections in a malaria referral center in Colombia”. During that time, Beatriz also worked as a teaching assistant and specialized professional at the Microbiology Department of the Universidad del Valle.
Just before she finished her master, she joined CIDEIM- International Center for Medical Research and training, Cali, where she worked for 13 years in different research projects, first on malaria, then on other infectious diseases, including tuberculosis. Beatriz was first research assistant, then laboratory coordinator and researcher, having in charge the conduction of several projects, both in research and services. She also worked ad honorem as teacher at Universidad ICESI, Cali, as part of the collaboration between CIDEIM and the university, designing the courses and teaching Microbiology and Medical Microbiology for undergraduate students.
In 2012, Beatriz participated in a Colombian government call, where she got one of the approximately 250 scholarships that were awarded that year to support PhD studies abroad, in all scientific areas. Based on previous collaborations with Prof. Dr. Dick van Soolingen and because of the prestige of Radboud University, Beatriz decided to start the process to be admitted at the Medical Microbiology Department of Radboud University Medical Center, where she joined to the multi-disciplinary research group lead by Prof. Dr. Johan W. Mouton, Prof. Dr. Dick van Soolingen and Dr. Jakko van Ingen, supervisors of her work. In line with the group´s goal to provide fundamental information on antibiotics efficacy for the treatment of non-tuberculous mycobacteria, Beatriz performed her thesis work. She also participated in the inter-institutional collaboration held with Dr. Tawanda Gumbo, director of the center for infectious diseases research and experimental therapeutics from Baylor Institute of Immunology research at Dallas, USA, where she did a 7 months internship with significant advances in the establishment of a new pharmacokinetic/pharmacodynamic model, the hollow-fiber system for M. abscessus.

174
This PhD work has led to 7 published papers in international peer-reviewed journals, as well as 6 presentations at conferences in USA and Europe.
Biography and List of Publications

Biography and List of Publications
175
Ferro BE, González I, de Carvajal F, Palma G, Saravia N. Performance of OptiMAL® in the diagnosis of Plasmodium vivax and Plasmodium falciparum infections in a malaria referral center in Colombia. Mem Inst Oswaldo Cruz. 2002;97:731-5.
Osorio L, Ferro BE, Castillo C. Effects of chloroquine and sulfadoxine/pyrimethamine on gametocytes in patients with uncomplicated P. falciparum malaria. Mem Inst Oswaldo Cruz. 2002; 97:1221-1223.
Hazbon MH, Guarin N, Ferro BE, Rodriguez AL, Labrada LA, Tovar R, Riska PF, Jacobs WR Jr. Photographic and luminometric detection of luciferase reporter phages for drug susceptibility testing of clinical Mycobacterium tuberculosis isolates. J Clin Microbiol. 2003;41:4865-9.
Gonzalez IJ, Varela RE, Murillo C, Ferro BE, Salas J, Giraldo LE, Zalis MG, Saravia NG. Polymorphisms in cg2 and pfcrt genes and resistance to chloroquine and other antimalarials in itro in Plasmodium falciparum isolates from Colombia. Trans R Soc Trop Med Hyg. 2003;97:318-24.
Moreira CA, Hernandez HL, Arias NL, Castaño MC, Ferro BE, Jaramillo E. [Initial drug resistance as a threat for tuberculosis control: the case of Buenaventura, Colombia]. Biomedica. 2004;24 Supp 1:73-9.
Rodríguez AL, Ferro BE, Varona MX, Santafe M. [Exposure to Leptospira in stray dogs in the city of Cali]. Biomedica. 2004;24:291-5.
Ferro BE, Rodriguez AL, Perez M, Travi BL. [Seroprevalence of Leptospira infection in inhabitants of peripheral neighborhoods in Cali, Colombia]. Biomedica. 2006;26:250-7.
Rojas CM, Villegas SL, Piñeros HM, Chamorro EM, Duran CE, Hernández EL, Pacheco R, Ferro BE. Características clínicas, epidemiológicas y microbiológicas de una cohorte de pacientes con tuberculosis pulmonar en Cali, Colombia. Biomédica. 2010:30:482-91.
Lienhardt C; Cook SV; Burgos M; Yorke-Edwards V; Rigouts L; Anyo G; Kim SJ: Jindani A; Enarson DA; Nunn AJ; Study C Trial Group.Efficacy and safety of a 4-drug fixed-dose combination regimen compared withseparate drugs for treatment of pulmonary tuberculosis: the Study C randomized controlled trial. JAMA. 2011;305:1415-1423.
List of Publications
1.
2.
3.
4.
5.
6.
7.
8.
9.

176
Ferro BE, Nieto LM, Rozo JC, Forero L, van Soolingen D. Multidrug-Resistance Mycobacterium tuberculosis, Southwestern Colombia. Emerg Infect Dis.2011;17:1259-62.
Nieto LM, Ferro BE, Villegas SL, Mehaffy C, Forero L, Moreira C, Rastogi N, van Soolingen D. Characterization of extensively drug resistant tuberculosis (XDR-TB) cases from Valle del Cauca, Colombia. J Clin Microbiol.2012;50:4185-7.
Villegas SL, Ferro BE, Perez-Velez CM, Moreira CA, Forero L, Martínez E, Rastogi N, Caminero JA. High initial multidrug-resistant tuberculosis rate in Buenaventura, Colombia: a public-private initiative. Eur Respir J. 2012;40:1569-72.
Ferro BE, García PK, Nieto LM, van Soolingen D. Predictive value of molecular drug resistance testing of Mycobacterium tuberculosis isolates in Valle del Cauca, Colombia. J Clin Microbiol. 2013;51:2220-4.
van Ingen J, Ferro BE, Hoefsloot W, Boeree MJ, van Soolingen D. Drug treatment of pulmonar nontuberculous mycobacterial disease in HIV-negative patients: the evidence. Expert Rev Anti Infect Ther. 2013;11:1065-77.
Realpe T, Correa N, Rozo JC, Ferro BE, Gomez V, Zapata E, Ribon W, Puerto G, Castro C, Nieto LM, Diaz ML, Rivera O, Couvin D, Rastogi N, Arbelaez MP, Robledo J. Population structure among Mycobacterium tuberculosis isolates from pulmonary tuberculosis patients in Colombia. PLoS One. 2014 Apr 18;9(4):e93848. doi: 10.1371/journal.pone.0093848.
de Beer JL, Ködmön C, van Ingen J, Supply P, van Soolingen D; Global Network for Molecular Surveillance of Tuberculosis 2010. Second world wide proficiency study on variable number of tandem repeats typing of Mycobacterium tuberculosis complex. Int J Tuberc Lung Dis. 2014;18:594-600.
Villegas SL, Ferro BE, Rojas CM, Perez-Velez CM. Assessment of children exposed to adult pulmonary tuberculosis in Cali, Colombia. Paediatr Int Child Health. 2014;34:170-7.
Ferro BE, van Ingen J, Wattenberg M, van Soolingen D, Mouton JW. Time Kill Kinetics of antibiotics active against rapidly growing mycobacteria. J Antimicrob Chemother. 2015;70:811-7.
Biography and List of Publications
10.
11.
12.
13.
14.
15.
16.
17.
18.

Biography and List of Publications
177
Ferro BE, van Ingen J, Wattenberg M, van Soolingen D, Mouton JW. Time Kill Kinetics of slowly growing mycobacteria common in pulmonary disease. J Antimicrob Chemother.2015;70:2828-43.
Rueda J, Realpe T, Mejia GI, Zapata E, Rozo JC, Ferro BE, Robledo J. Genotypic analysis of genes associated with independent and cross-resistance to Isoniazid and Ethionamide in M. tuberculosis clinical isolates. Antimicrob Agents Chemother. 2015;59:7805-10.
Ferro BE, Meletiadis J, Wattenberg M, de Jong A, van Soolingen D, Mouton JW, van Ingen J. Clofazimine prevents the regrowth of Mycobacterium abscessus and Mycobacterium avium type strains exposed to amikacin and clarithromycin. Antimicrob Agents Chemother. 2016;60:1097-105.
Ferro BE, Srivastava S, Deshpande D, Sherman CM, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. Cuddling Nightmare Bacteria: Amikacin Pharmacokinetics/Pharmacodynamics in a Novel Hollow Fiber Mycobacterium abscessus Disease Model. Antimicrob Agents Chemother. 2016;60:1242–48.
Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. Tigecycline is highly efficacious in Mycobacterium abscessus pulmonary disease. Antimicrob Agents Chemother. 2016;60:2895-900.
Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. Moxifloxacin’s Limited Efficacy in the Hollow-Fiber Model of Mycobacterium abscessus Disease. Antimicrob Agents Chemother. 2016;60:3779-85.
Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. Failure of the amikacin, cefoxitin and clarithromycin combination regimen for pulmonary Mycobacterium abscessus. Antimicrob Agents Chemother. 2016;60:6374-6.
19.
20.
21.
22.
23.
24.
25.


Acknowledgements / Dankwoord

180
Acknowledgements

181
“What matters in life is not what happens to you but what you remember and how you remember it.”
Gabriel García Márquez
I will always remember all of you with greatest gratitude, for being you, for all what you have done, for all your kind help in several ways, for being part of this PhD adventure in a wonderful country!
Thank you very much for the financial and administrative support to conduct this PhD to Colciencias, Colombian govenment and Colfuturo, who provided and managed the PhD scholarship, to RadboudUMC: Medical Microbiology Department, RIHS, international office, Radboudhotel, to Radboud University and all the parties involved in my work in the Netherlands. Also special thanks to the Center for Infectious Diseases Research and Experimental Therapeutics, Baylor Research Institute, Dallas, USA, where I conducted an important part of this work.
I would like to express my sincere gratitude to my supervisors: Prof. Dr. Johan W. Mouton and Prof. Dr. Dick van Soolingen, you believed in me and I felt your support along the way! Thank you very much and forever for your patience, motivation, and immense knowledge.
Thank you very much to my co-supervisor, Dr. Jakko van Ingen, for all your teaching and support in laboratory and extra-laboratory issues. Thank you for your time every week to discuss my findings and making me believe that all my observations were important! You, Hilde, Tom and Koen made me feel the warm of a friendly family, there are no words to thank for everything! You all are in my hearth!
Besides my supervisors and co-supervisor in the Netherlands, I would like to thank to Dr. Tawanda Gumbo, from Dallas, USA, for all your guidance, for the time you prioritized to have our discussions, for the great support to my experiments! Also special thanks to your wife, Mrs. Sipho Gumbo, who had time even in her busy days to show me a different and important part of women´s life in Texas.
My mentor, my friend, my Dutch sister, Melanie, thank you for all!!! Since the very first day you offered me your friendship, your support, everything! There was nothing I didn´t receive from you to help me... you were always there for me and you will be forever in my heart!!! Also special thanks to Vera and Joris! very sweet, smart, tender and blessed persons... Also thanks to Jos, Hetty, Jessica and family, Piet, Raymond and family, I shared with all of you very nice moments in Millingen aan the Rijn and Cuijk, that made me experience Dutch celebrations in family!
Acknowledgements

182
My sincere thank also goes to Nicole, Yvonne and Dirk! Without your precious help in the TB laboratory it would not be possible to conduct this research. Nicole, we spent more time together, you always had the patience to show me everything and helped me to solve different situations. I enjoyed the Carnival time spent with Fabian, Yanniek and Mirthe in Lent. Yvonne, you also made my time special there! I loved every day I saw you, you always helped me to get all what I needed to run my experiments, for that and for all thank you! Also thanks to Bas, Bart and Anne, you are a lovely family! It was a pleasure to see all the family business, the beautiful landscape of Groesbeek and our trip to the zoo. Dirk, our days at BSL3 will be always the funniest! Thank you for the music, for all your advice and for being such a gentleman! Thank you all for changing your Dutch, perhaps relaxing conversations, into English, even when you were tired...just to made me feel better! Thank you very much also to the other ladies in the TB lab, Hanke and Myriam, we shared less time, although it was nice and special! I was there in a time of changes, but our conversations were always interesting and inspiring!
I would like to thank to my fellow permanent office mates John and Nel, you two were simple great! Thank you also to Marian that joined us later and was very kind. John I am very grateful for the time you spent helping me to get all the stuff to build the hollow-fiber model at the UMC, thank you for your connection with Johan, who kindly provided us lots of help with the model. Also thanks to Bea and Tis, we had a lovely time in your house. Dear Nel, how great it was to observe and learn about your decency and patience, then to discover the beautiful pieces of art you create and receive your tips to have a better experience in the Netherlands, thank you for all! Also special thank to Saskia, it was really nice to receive your warm greeting “Hola, cómo estás?” everytime I saw you! Thank you for your interest in our mycobacterial work and for loving Latin American culture!
I would like to express my gratitude to the whole MMB department! To the secretaries: Monica, Diana and Marian, I am deeply grateful for your kind help, you always had a smile for me, despite how busy you were! Also special thanks to the ladies in the media laboratory: Angela, Schawn, Marian, you showed me that there is no need of language when the heart is big enough to help! To all the staff in the laboratories, thank you very much for being nice and welcome me. I only interacted with few of you, but with every conversation in the cantine I had the opportunity to see that you are a great team. Special thanks to those who I bother the most with my questions and requests: Karla, Rob, Judith, Hein, Arjan and other staff from molecular biology laboratory. Thank you also to the students I had the privilege to work with: Eran, always nice and clever, Esther, Michelle and Josien, Med students Paul, Daan, Koen, Mirthe, Michiel; Kim and Danique.
Dear PK/PD group, thank you for all what we shared together, for the meetings, for the time out: Ria, Anita, Roger, Claudia, Mark, Amir, Hanneke, Fiona, Anne and all the students that did internships there. Dear Ria, you were always available for questions and ready to help,
Acknowledgements

183
thank you also for being like a mother trying to have all of us in track!. Thank you Anita, Claudia and Mark for your kindness and conversations about life and everything. Also thank you Amir, the other foreign in the group, every time you were there you changed the meetings, it was also a relief to have somebody to speak in English and to receive your advice. Thank you Hanneke for being always kind, I enjoyed a lot the time spent with your family: Peter, Boudewijn and Irene! Thank you Fiona for all the hard work and fun in the lab, building the in house hollow-fiber model, etc. It was also great to be there to see you and Tommie with little Vivianne newborn! Thank you very much for your hospitality in San Diego, when we assisted to ICAAC, it was really nice from you to help me with that! Dear Anne, step by step we built a friendship that relief me in many ways... it was always a pleasure to talk to you, to meet your parents and to see you and Fudge in the horse competition! but most importantly it was great to be there to see all the success you have had during your PhD and the beautiful human being you are!
I would like to thank also Dr. Martin Boeree, Dr. Cecile Magis-Escurra and all the staff at Dekkerswald, particularly Wouter and Sami. It was always a pleasure to be there for the TB sandwich meeting, to present every year my results and receive your feedback and comments. Dear Martin, your greeting and smile was always light for those meetings. Dear Cecile, it was a pleasure to talk to you always, mostly in Spanish, also to be in your house and to share time with your nice family! thank you for the invitations and lovely moments! A special thank also to Rob Aarnoutse from the pharmacy department, who always showed interest and collaborate with our work! A very special thank to Prof. Reinout van Crevel and the staff of his laboratory, particularly Arjan, Ekta and Lidya. We had few, but interesting conversations about mycobacteria that were really nice. Ekta, it was really nice to talk to you, to exchange experiences and to hear from your expertise about all PhD issues. Dear Lidya, you literally changed my life when introduced me to Mien Janssen, where I lived the most beautiful part of my stay in Nijmegen! Thank you for that and for all the time we spend together as roomies! I know you are close to the end, good luck finishing your PhD! Thank you very much also Mien, your house was like a heaven: a warm place with a beautiful garden, two cats and your smile that made me feel really special! Thank you forever for your concern in our welfare, for the Sinterklaas candies, for everything!!! Thank you also to the people I met at Radboudhotel: Hetty, Fabiola, Robel, Nora, to the teachers and girls I met in the Radboud University courses: Annette, Donna, Lidya, Annelie, Giuliana, to the instructors at the Yoga and Zumba classes, you made those hours and days better! My sincere gratitude also goes to Matthijs, who helped me a lot editing this book!
From my time in Dallas, more people were part of this voyage, for whom I am deeply grateful. As already mentioned, Dr. Gumbo and his wife, also Shashikant, Devyani, Vijay, Jotam thank you very much for your help and for the time we spent together working at the laboratory and revising data from experiments. Shashi, thank you for being always there, but also for the
Acknowledgements

184
tricky moments and hard questioning, that incented me to grow and perform the research to the best that I could. Thank you to the administrative staff from Baylor Research Institute: Cindy, Angela, Pam, you all were very nice and opened with me, thank you for the support and help! A special and big thank to my lab mates: Kayle, Carleton, Wasana, Stephen, Brian, Lena, Theresa! We had a really intense and nice time together, thank you for all what you did for me!!!! Dear Kayle, you were always the sun there! I loved our conversations, our fun in the middle of hard times, our lunch time, your recipes, your stories, everything! You are a wonderful woman! It was also nice to have met Chris! I wish we will see each other again one day... here or there. Thank you also to Carleton for all the kind help and support in many ways; thank you to sweet Wasana for you kindness and help, you and your husband, even far from home, keep the charm of Sri Lankan people and spread it! I am also sincerely grateful to other people that were very special with me in the USA: Dawn and Mark Thames, you hosted me in your house and I loved our conversations, your advice, everything! I hope you will continue receiving the blessings you also give to all that meet you! Maria Adelaida, what a pleasure to share part of the time in Dallas with you, biking to the laboratory, cooking, talking. Thank you for your support and for introducing me to Ximena! Xime, your kindness and help really changed my stay there... thank you for all the time we spent in leisure activities... to keep me out of the insanity! Thank you for offering your help for everything and for being there for me! I hope to see you soon! Thank you also to Luz Stella another wonderful Colombian in Dallas, it was always nice to talk to you! An a special thank to the Colombian team in Houston. Dear Chris, you have always been one of the most special guys I have ever met, I really enjoyed the 4th of July trip to Houston, the fireworks, our conversations, etc. Thank you for being so special with me. Dear Diana, I loved to see you and your family in Houston, thanks for bringing me all the good old memories, that made me happy! Also special thanks to Carlos Mario, even living in Arizona, you were always kind with me and ready to help! Thank you for your concern when I first moved to the Netherlands, I still have the book you sent me!
This brings to my mind all the other Latin American people I have met or found in this journey, which are loved friends. Dear Hanna, you were the first contact in the Netherlands, thank you for your welcome, for showing me Amsterdam, for the first Dutch lessons, for opening my mind! I loved the time we shared together, also later to meet Claudia and little Emilia. Anita and Alejandro, I enjoyed the weekend together in Utrecht, during the first King´s day, thank you for your time, for the bike lessons and all the fun! Iveth, Ricardo, Leticia and Ernesto, thank you for your welcome to the Colombian headquarters in Switzerland, I really enjoyed the time in Laussane! Daniel, Sonia, Matt, thank you for the celebration of my first birthday in the Netherlands, it will remain in my memory forever! And the second birthday was also great in Berlin! lots of fun! Also thank you Sonyi for being always there for me, for your continuous calls and messages! You and Cesar, made unforgettable the time in the TB reunion in Athens, we have to do another meeting soon!!! It was also nice to meet Gladys, Freddy
Acknowledgements

185
and Maria Jose in Amsterdam, thank you for the invitation to sightseeing and for your love! Thank you Clara for your visit to Nijmegen, it was a nice time to share and talk!. Sandra, it was a pleasure to meet you in Nijmegen and to have done all what we did in the three months together... I hope I will visit you soon in Paraguay! Then, I unexpectedly found one Colombian in a supermarket in Nijmegen! Paola, to hear your voice with Colombian accent in the middle of a Dutch crowd was miraculously for me! Thank for offering your help, for all what we did together and with Laury, for the time we spent watching movies, preparing Colombian food, for the conversations and advice, for being so open to us. Life has changed and I only hope you are happy now in your new position, you will always be in my heart. Dear Laury, it was great to meet you in Nijmegen! We have lived a lot in these two years, but these experiences are saved in our hearts! Thank you for your kindness and company, for being so lively, for the light you brought, and for share it with others, thank you for showing me that one can always talk about the problems. I am pretty sure you will successfully finish your PhD, Colombia is waiting for you! As Colombia is waiting for Doris, I wish you the best in your PhD!
And last but not least, I want to thank to all my family for their love, the calls, the messages and things sent from Colombia; also thank you Flor and Marita for the visit to the Netherlands. I left you all one day of March in 2013 with many tears in our eyes, but also with promise: I will honor your tears and the suffering by finishing my PhD! Now I can say I did it! and this is also for you all! To my mother, Yolanda, who had always showed me the value of working hard, in a way this is a dream coming true for you, thank you for all the support. To my brother, Juan Carlos and my niece and nephew, Manuela and Alessandro, who are in the biggest part of my heart, thank you for all you love, for coming to my defense, for always being the brother, the friend, the accomplice... Special thanks my aunts: Maria Nelssy, Mirian and Florencia, who have always given me the extra love I needed to succeed, to my uncles Hector Fabio, Alberto and Ricardo, who in a way or another have been examples of how to be brave and good professionals. Thank you to my cousins, Claudia and Andres, who are like a sister and a brother to me, Martha, Adriana, Maria Isabel, Paola, Evelyn, Mario, Marcela, Tayna, Shary, Valeria and Daniel, and the little cousins that have brighten our days: Sebastian, Ariel Alejandro, Mariana, Natalia, Maria Adelaida, Thiago, Martina, Emiliano and Isabella. Special thanks to the extended family: Marquitos, his daughter Vicky and Carlos, Ariel, Huguito †, Carmen Eugenia, Adry Gastaldi, Javier, Carlos and my friend Alexandra, you are all family in my heart. Thank you for the support of my father´s family, aunt Carmen Julia, Mario, and the whole family. Dear Robin, there is no word big enough to express my gratitude, you were always there for me, there was no day without receiving your support, your help or your joy, all of what was the extra strength I needed to survive! Thank you very much for all your love, for your visits, for everything! Also thank you to your parents Isaac and Mercedes, to Sara and the whole family, your good wishes and blessings always reached me and made me feel protected!
Acknowledgements

186
None of this would have been possible without God guidance! I have always felt protected by the person who most influenced who I am today, my grandmother and now personal angel: Alicia! The highest price I paid to do this PhD was not being there when the time came to say the final goodbye to you... You are always is my heart and in my mind, your love have made all the miracles in my life. I hope you and my grandfather Aulio are now together in peace; please let him know that I finally deserve how he named me “Doctora Titis”!
Acknowledgements

PHD PORTFOLIOName PhD candidate: B.E. Ferro RamosDepartment: Medical MicrobiologyGraduate School: Radboud Institute for Health Sciences
PhD period: 18-03-2013 – 16-01-2017Promotor(s): Prof. dr. JW Mouton, Prof. dr. D. van SoolingenCo-promotor(s): Dr. J. van Ingen
Year(s) ECTSTRAINING ACTIVITIES
a) Courses & Workshops- Introduction day Radboudumc - PK/PD weekly meeting (technical forums)- Academic writing course- Presentation skills course- Advanced conversation course- PK/PD full day course International Congress. 54th Interscience
Conference on Antimicrobial Agents and Chemotherapy, ICAAC - Biosafety and spill drill training training- Scientific Integrity course - Basic laboratory and BSL2 laboratory training at Baylor Research
Institute- University pedagogy diploma course by e-learning, Universidad
San Buenaventura, Cali, Colombia
20132013-20152014201420142014
201420152015
2016
0.5631.51.51.0
0.51.00.5
5
b) Seminars & lectures- PK/PD weekly meeting (oral presentation)- Broodje mycobacterie, monthly meeting- Broodje mycobacterie lecture (oral presentation)- Medical Microbiology department lectures- Baylor Research Institute Lectures
2013-20152013-20152013-201520152015
220.750.60.4
c) Symposia & congresses- National symposia . Mini-symposium “The nature of the bug”,
Amsterdam- International Congress. 54th Interscience Conference on
Antimicrobial Agents and Chemotherapy, ICAAC (poster presentation)
- National symposia. Mini-symposium “Whole genome sequencing for Mycobacterium tuberculosis”, Nijmegen
- International Congress. 25th European Congress of Clinical Microbiology and Infectious Diseases, Copenhagen, Denmark (oral presentation).
- International Congress. 55th Interscience Conference on Antimicrobial Agents and Chemotherapy (3 poster presentation)
- National symposia. Genetic Screening , Amsterdam- National symposia. M. tuberculosis Beijing strains genome.
Bogotá, Colombia- International tuberculosis research working meeting. Cali,
Colombia
2013
2014
2014
2015
2015
20152016
2016
0.25
1.5
0.25
1.5
2.5
0.250.5
0.75
TEACHING ACTIVITIES
d) Supervision of internships / other- Supervision Bachelor student internships- Review of manuscripts for journals
2013-20152013-2016
30.5
TOTAL 37.25























