
Basic Concepts in Cellular Cardiac Electrophysiology: Part II: Block of Ion Channels by...
20
Basic Concepts in Cellular Cardiac Electrophysiology: Part I: Ion Channels, Membrane Currents, and the Action Potential DAVID W. WHALLEY, DAVID J. WENDT, and AUGUSTUS O. GRANT From Duke University Medical Center, Durham, North Carolina Introduction The last two decades have seen a rapid growth in our understanding of the ionic events underly- ing normal cardiac excitability, the mechanisms of arrhythmogenesis. and the action of antiarrhyth- mic drugs. Factors contributing to this have in- cluded the ability to reliably isolate single heart cells,' the introduction of the patch voltage-clamp technique,^ and advances in molecular biology, which have enabled correlation of ion channel structure and function.^•'^ In view of the fundamental importance of cel- lular events to the understanding of clinical elec- trophysiology it seems timely to review some basic concepts of cellular cardiac electrophysiology. In the first of two parts, we review recent insights into the structure and function of transmembrane ion channels, the ionic currents underlying the ac- tion potential in ventricular and atriai cells, the mechanisms of normal and abnormal automatic- ity, and the ionic bases of excitability, conduction, and refractoriness. In Part II, we describe the mechanisms of antiarrhythmic drug—ion channel interaction, focusing on the Class I and III agents. The membrane events during the various phases of the electrical cycle of the heart can be resolved at several levels. These levels of resolu- tion are illustrated for the phase of depolarization in Figure 1. In the surface electrocardiogram, this period is registered as the QRS complex. The dura- tion of the QRS complex reflects tbe rate of depo- larization. Slowing of conduction, e.g., by admin- istration of the Na"^ channel blocking drug flecai- nide prolongs the QRS duration. The rising phase [phase 0) of the action potential recorded from a David W. VVhalley is an Overseas Research Fellow of the Na- tional Hoart Foundation of Australia. Address for reprints: Augustus O. Grant, M.D., Ph.D., Box 3504, Duke Medical Center, Durham, NC 27706. Fax: (919) 681-8978. Received May 26, 1994; accepted May 31, 1994. single cell or a tissue preparation corresponds to the period of depolarization. For many years, the rate of rise of the action potential, Vmax' was used as an indirect measure of Na * conductance. Vmax measurements have now been superseded by more direct measurements of membrane currents during depolarization. Using isolated cardiac myocytes investigators can examine the effects of a rapid change of membrane voltage [voltage clamp) and record the associated current—whole-cell or mac- roscopic current.^ Experiments using ion substitu- tion and the specific Na * channel hlocker tetrodo- toxin confirm that the initial transient inward cur- rent is carried hy Na ' ions. The application of a highly polished microelectrode to a single cell may result in the electrical isolation of a small patch of cell membrane [patch clamp). This tech- nique permits resolution of the opening and clos- ing transitions of individual Na^ channels. General Properties of Cardiac Ion Channels The passage of Na^, K ' . Ca~* and CA' ions across the cardiac plasma membrane [sarcnlemma] tbrough specialized voltage and ligand operated ion channels is the basis of normal excitability and conduction.'*'* The ion channels that mediate the cardiac action potential are large transmembrane spanning proteins, which provide pathways of rel- atively low electrical resistance across the high re- sistance, hydrophohic lipid bilayer. In contrast to active transport mechanisms such as the sarcolem- mal Na^-K^ pump, which directly couples ion fluxes to ATP hydrolysis, activated channels per- mit the passive movement of ions across the sarco- lemma down their respective electrochemical gra- dients. The resulting transmombrane ion fluxes are characteristically rapid [10' ions/s) and are be- lieved to occur tbrough aqueous pores within the channel protein. Most theoretical models suggest that ions move in single file through the channel 1556 August 1995 PACE. Vol. 18
-
Upload
dukemedschool -
Category
Documents
-
view
3 -
download
0
Transcript of Basic Concepts in Cellular Cardiac Electrophysiology: Part II: Block of Ion Channels by...

Basic Concepts in Cellular CardiacElectrophysiology: Part I: Ion Channels,Membrane Currents, and the Action Potential
DAVID W. WHALLEY, DAVID J. WENDT, and AUGUSTUS O. GRANT
From Duke University Medical Center, Durham, North Carolina
Introduction
The last two decades have seen a rapid growthin our understanding of the ionic events underly-ing normal cardiac excitability, the mechanisms ofarrhythmogenesis. and the action of antiarrhyth-mic drugs. Factors contributing to this have in-cluded the ability to reliably isolate single heartcells,' the introduction of the patch voltage-clamptechnique,^ and advances in molecular biology,which have enabled correlation of ion channelstructure and function. •'
In view of the fundamental importance of cel-lular events to the understanding of clinical elec-trophysiology it seems timely to review some basicconcepts of cellular cardiac electrophysiology. Inthe first of two parts, we review recent insightsinto the structure and function of transmembraneion channels, the ionic currents underlying the ac-tion potential in ventricular and atriai cells, themechanisms of normal and abnormal automatic-ity, and the ionic bases of excitability, conduction,and refractoriness. In Part II, we describe themechanisms of antiarrhythmic drug—ion channelinteraction, focusing on the Class I and III agents.
The membrane events during the variousphases of the electrical cycle of the heart can beresolved at several levels. These levels of resolu-tion are illustrated for the phase of depolarizationin Figure 1. In the surface electrocardiogram, thisperiod is registered as the QRS complex. The dura-tion of the QRS complex reflects tbe rate of depo-larization. Slowing of conduction, e.g., by admin-istration of the Na" channel blocking drug flecai-nide prolongs the QRS duration. The rising phase[phase 0) of the action potential recorded from a
David W. VVhalley is an Overseas Research Fellow of the Na-tional Hoart Foundation of Australia.
Address for reprints: Augustus O. Grant, M.D., Ph.D., Box 3504,Duke Medical Center, Durham, NC 27706. Fax: (919) 681-8978.
Received May 26, 1994; accepted May 31, 1994.
single cell or a tissue preparation corresponds tothe period of depolarization. For many years, therate of rise of the action potential, Vmax' was usedas an indirect measure of Na * conductance. Vmaxmeasurements have now been superseded by moredirect measurements of membrane currents duringdepolarization. Using isolated cardiac myocytesinvestigators can examine the effects of a rapidchange of membrane voltage [voltage clamp) andrecord the associated current—whole-cell or mac-roscopic current.^ Experiments using ion substitu-tion and the specific Na * channel hlocker tetrodo-toxin confirm that the initial transient inward cur-rent is carried hy Na ' ions. The application of ahighly polished microelectrode to a single cellmay result in the electrical isolation of a smallpatch of cell membrane [patch clamp). This tech-nique permits resolution of the opening and clos-ing transitions of individual Na^ channels.
General Properties of Cardiac IonChannels
The passage of Na^, K ' . Ca~* and CA' ionsacross the cardiac plasma membrane [sarcnlemma]tbrough specialized voltage and ligand operatedion channels is the basis of normal excitability andconduction.'*'* The ion channels that mediate thecardiac action potential are large transmembranespanning proteins, which provide pathways of rel-atively low electrical resistance across the high re-sistance, hydrophohic lipid bilayer. In contrast toactive transport mechanisms such as the sarcolem-mal Na^-K^ pump, which directly couples ionfluxes to ATP hydrolysis, activated channels per-mit the passive movement of ions across the sarco-lemma down their respective electrochemical gra-dients. The resulting transmombrane ion fluxes arecharacteristically rapid [10' ions/s) and are be-lieved to occur tbrough aqueous pores within thechannel protein. Most theoretical models suggestthat ions move in single file through the channel
1556 August 1995 PACE. Vol. 18

CONCEPTS IN CELLULAR CARDIAC ELECTROPHYSIOLOCY
Surface EKG
Action potential
Whole-cell Nacurrent
Sir>gle Na channelcurrent
Figure 1. Levels of resolution ofthe membrane currentduring depolarization. On the surface electrocardi-ogram (EKG). the period of rapid depolarization ismarked hy inscription of the QRS complex. This eventis marked by phase 0 of the action potential. Its rate ofrise, V,,i j(. was widely used as a measure of availableNa'^ conductance. The availability of single cardiac my-ocytes permitted step changes in membrane voltage andmeasurement ofthe associated current (voltage clamp).The application of a polished microelectrode to the cellsurface may result in the formation of a high resistanceseal and the electrical isolation of a small patch of mem-brane (patch clamp). This configuration permits the re-cording ofthe pulses of current as individual ion chan-nel molecules make their opening-closing transitions.
pore in a discontinuous fashion "hopping" be-tween discrete selective binding sites. Indeed it islikely that such preferential binding of ions, in ad-dition to the size ofthe hydrated ion, provides thebasis for selectivity of an individual channel for aparticular ion.
Gating of Cardiac Ion Channels
The process that accounts for the opening andclosing of ion channels is termed gating. Based on
their mechanisms of gating, cardiac ion channelsfall into three classes: (1) voltage gated ion chan-nels; (2) ligand gated ion channels; and (3) stretchactivated ion channels. The mechanisms of gatingare not mutually exclusive; voltage gated channelsmay be modulated by neurotransmitters and hor-mones while ligand gated ion channels may showvoltage dependence.
Voltage gated ion channels are the largestgroup of cardiac ion channels. These channelshave charges or dipoles that sense changes in themembrane field (or potential) and respond with achange in conformation. The conformationalchanges of charged regions ofthe channel generatesmall currents, termed gating currents, that pre-cede the voltage dependent conductance change.Gating currents have provided considerable in-sight into the function of ion channels.^ Voltagegated ion channels may occupy one of two conduc-tance states, open or closed. The basal closed stateis termed the resting state. Most resting channelsopen in response to depolarization. The openchannel permits ion movement across the mem-hrane. The direction of movement of a given ionthrough the open channel is determined by itselectrochemical gradient, AV. The current gener-ated by a given type of channel, I, is dependenton: (1) the fraction, p,,. of tbe total number, N. ofchannels open; (2) the magnitude of the currentthrough each open channel, i; and (3) the electro-chemical driving force for the ion:
I =
The current waveforms generated by a class ofchannels provides insight into the nature of thegating process. Figure 2 illustrates current wave-forms that may be generated by voltage gated ionchannels. The rate of channel opening or activa-tion may be maximal at the onset of the change inmembrane potential; alternatively, there may be adelay in channel opening. The number of closedstates that precede channel opening determine theactivation kinetics. The occurrence of multipleclosed states leads to a delay in activation [Fig. 2,left panel).
Some ion channels maintain their high con-ductance state for the duration of depolarization.For other channels, the change in conductance istransient, declining despite the maintenance of de-
PACE. Vol. IB August 1995 1557

WHALLEY, ET AL.
Non-inactivating Inactivating
—I
* S =t(n)* Activation variable, r
• g = f(m,h)
• Activation variable, m
, Inactivation variable, h
Figure 2. Dependence of current waveforms on under-Iving kinetic scheme. The left panel shows membranecurrents from non-inactivating channels with single (n- 1) or multiple (n = 3) dosed states. When the mem-brane potential is stepped from the holding potential.VH, to the test potential Vj, there is a rise in current toa plateau level. When a single closed state. C. precedesopening. O. the rate of conductance increase is maximalat the outset. With multiple closed states, there is a delayin the rise in conductance. Current from an inactivatingchannel is shown in the right panel. The channel opensin response to depolarization however the currentreaches a maximum and declines despite the mainte-nance of depolarization. The lower part of the figureillustrates the mathematical description ofthe two typesof current waveforms. For non-inactivating currents, theconductance (g) is a function of a single activation vari-able n. Inactivating currents require both an activationvariable m. and an inactivation variable h. to fully de-scribe the conductance change.
polarization (Fig. 2, right panel). The process thatresults in this decline in current Is termed inacti-vation. The inactivated state is a second noncon-ducting state, different from the resting state sinceinactivated channels usually do not open in re-sponse to membrane depolarization. Some restingchannels bypass the open state and pass directlyinto the inactivated state. To reverse inactivationthe membrane must be repolarized to highly nega-tive values. This process is termed recovery fromor removal of inactivation. Recovery from inactiva-tion is highly voltage dependent; it is acceleratedby hyperpolarization.
Some of these properties of voltage dependention channels are illustrated with Na^ currentsrecorded from an isolated cardiac cell (Fig. 3). Na "currents elicited by 50-ms voltage-clamp stepsfrom a holding potential of -130 mV to test poten-tials of varying amplitude are illustrated in Figure3, panel A. During small depolarization steps theactivation of Na^ current is delayed. With largerdepolarization steps the current activates morerapidly and to a larger peak. In panel B, the peakamplitude ofthe current is plotted as a function ofthe test potential; the current-voltage relationship.Na" current activates at ~ - 55 mV. Peak currentincreases in amplitude, passes through a maxi-mum at - 20 mV, and decreases with further depo-larization. Panels C and D, illustrate the voltagedependence of inactivation. The membrane poten-tial is held at various conditioning levels for 1 sec-ond. The available Na^ conductance at the condi-tioning potential is determined by application oftest pulses to the fixed potential of - 30 mV. The"inactivation curve" shown in panel D relates thecurrent elicited during the test pulse to the condi-tioning potential. As the conditioning potential ismade less negative current during the test pulsedeclines hecause of increasing inactivation. Simi-lar inactivation curves can be defined for otherchannels such as the Ca ^ channel and the tran-sient K^ channel. However, the nature ofthe volt-age dependence of inactivation is characteristic foreach channel type. Factors such as pH. tempera-ture, and the concentration of divalent cations mayinfluence the voltage dependence of inactivation.
The records in Figure 3, panels A and C. re-sulted from the concerted action of all the Na^channels in the cell. In principle, this "whole-cell" or macroscopic current must be a scaled formofthe current arising from individual Na^ chan-nels in the cell. Single Na^ channel currentsrecorded from a small membrane patch in anothercell are illustrated in Figure 4. Panel A outlinesthe nomenclature used to describe single channelcurrent during a depolarizing voltage step. A chan-nel opens after a variable delay termed the latency.The channel may fail to open in response to depo-larization, resulting in a null response. Panels Band C show consecutive channel responses to de-polarizations to —60 mV and —40 mV, respec-tively. The current produced by averaging 100 de-polarizations is shown in the lower trace in each
1558 August 1995 PACE, Vol. 18

CONCEPTS IN CELLULAR CARDIAC ELEGTROPHYSIOLOGY
5 msec-90
•120
-40 10Test Potential (mV)
Conditioning Potential (mV)
Figure 3. Activation and inactivation of the cardiac Na'^ current. Panel A shows whole-cell(macroscopic) Na'^ channel current recorded from a cardiac myocyte. The holding potentialwas -100 mV and the test potential was increased in 5 mV increments. With increasing depolariza-tion, the peak of the current occurs earlier, and the amplitude increases. Peak current over awide range of membrane potentials is plotted in panel B. The current voltage relationship achievesa maximum at -20 mV. With depolarization beyond -20 mV, the current declines as the testpotential approaches the equilibrium potential for Na'^ ions. The Na ^ channel activation curveshown in the insert was obtained by adjusting the current amplitude for the change in electro-chemical gradient. Panel C shows membrane currents obtained at a fixed potential of -30 mVas the preceding conditioning potential was decreased. The peak current declines as the condi-tioning potential is made less negative. The inactivation curve in panel D is obtained by plottingthe peak values of the current during the test pulse against the conditioning potential.
panel. In panel B, channels open after a variablelatency. In the fourth trial, the channel opens re-petitively for the first time well after the peak ofthe average current. In panel C, the latency is con-sistently short, the channel opens only once, theaverage current amplitude is increased, and therising and falling phases of the current have a morerapid time course. These changes in current at thesingle channel level can account for the whole-cellcurrents observed in Figure 3, panel A.
What type of physical model may account forthe transient nature of the Na" current in responseto depolarization? Some 40 years ago, Hodgkinand Huxley" proposed a model for the Na^ chan-nel. Despite extensive studies using more complex
techniques, the basic tenets in their proposal stillstand (Fig. 5). They suggested that ion movementtiirough the Na ^ channel is controlled by activa-tion (m) and inactivation (h) gates. Resting chan-nels have the activation gate in the closed positionand the inactivation gate in the open position.Membrane depolarization moves the activationgate to the open position. With both gates in theopen position, Na^ ions move into the cell downtheir electrochemical gradient. With maintaineddepolarization, the inactivation gate moves intothe closed position and terminates inward ionmovement. With repolarization, tbe activationgate assumes the closed position almost instantly.In contrast, a significant amount of time must
PAGE, Vol. 18 August 1995 1559

WHALLEY, ET AL.
C L O S E D
O P E N
* L A T E N C Y
2 5 pA
B V40 rns
^,.t^^^t^4.^M^,.^f,^t'W|t^^
'^ jt^^^^^W^'^^^^v^'vWAJM^^
Figure 4. Single cardiac sodium channel currents. Nomenclature used to describe single sodiumchannel current is outlined in panel A. A variable period after a depolarizing step, a singlechannel opens resulting in a downward deflection in the current trace. The interval (') betweenthe change in membrane voltage and the initial opening of the channel is termed the latency.The channel may fail to open during the depolarizing pulse. Such an occurrence is termed anull. The dependence of single sodium channel current on test voltage is illustrated in panels Band C. Current in response to individual depolarizing trials is shown in the upper 5 traces. Thecurrent produced by averaging 100 such traces is shown below. The latency in response to thestep to -60 mV is quite variable, and the channel may open repetitively (trial 4). In this trial, thechannel opens for the first time when the average current is declining. The declining phase ofthe current therefore reflects the delay in opening in addition to channel closure. In response tothe step to -40 mV, latency is brief, the channel opens once and the declining phase of the averagecurrent reflects channel closure. The calibration is 10 ms and 2 and 0.75 pA for the individualand average current, respectively. The recording temperature was 22 XJ.
1560 August 1995 PACE, Vol, 18

WHALLEY, ET AL.
N a ®•
Resting Open Inactivated
Figure 5. The Hodgkin-Huxley model of Na* channelgating. The upper trace shows a voltage step and themiddle trace the resulting membrane current. A physi-cal model to account for the transient current is shownin the lower trace. The Na'^ channel is represented asa pore spanning the membrane. In the resting state, theactivation (m)gate is in the closed position and the inac-tivation (h)gate is in the open position. Following depo-larization, the m gate assumes the open position. Withboth gates open. Na'^ ions move into the cell. The hgate then moves into the closed position blocking ionmovement. When the membrane is returned to the rest-Ing level, the m gate moves into the closed position, aprocess termed deactivation. After a variable interval,the h gate moves into the open position, a processtermed recovery from inactivation (not shown).
elapse before the inactivation gate resumes theopen position. A similar model may hold true forother inactivating ion channels. Although thisscheme may ancount for > 95% of the behavior ofthe Na^ channels, these channels may occasion-ally show alternative modes of gating such as ap-parent failure of the inactivation process.^
Ligands such as neurotransmitters, ATP andintracellular Ca^^ ions may interact with mem-hrane bound receptors and gate ion channels. Themost widely studied example of a ligand gated ionchannel is the K^ selective channel that is openedby muscarinic (Mv) receptor activation.^" Acetyl-choline (Ach) interacts with the muscariuic recep-
tor initiating a sequence of events that result in thedissociation of the a and p-7 subunits of a guaninenucleotide regulatory G protein, G,. The a or thep-7 subnnits of Gj open a K^ channel.'" For thissystem to have a rapid response, there must bemechanisms for terminating the action of the neu-rotransmitter. These include hydrolysis of the neu-rotransmitter in the synaptic cleft and desensitiza-tion, a sequence of intracellular processes that col-lectively result in a diminution of the effect ofneurotransmitter-receptor interaction despite con-tinued neurotransmitter presence.
In contrast to the opening of an ion channelby Ach, other ligands may hind to their receptorand close an ion channel. The ATP sensitive K^channel, which is widely distributed in cardiacmuscle, is closed by normal intracellular levels ofATP. Reduction of the intracellular ATP levelbelow 0.5 mM results in opening of the channel.^^The ADP/ATP ratio and intracellular pH are im-portant modulators of the sensitivity of this chan-nel to ATP. Its activation by low levels of ATPduring myocardial ischemia may result in abbrevi-ation of the action potential.
Physical factors such as light and stretch mayalso activate ion channels; the latter is the onlyrelevant category of these channels in cardiac mus-cle. Second messenger systems are usually in-volved in the signal transduction mechanisms ofthese channels.
Structure-Function Relationships of IonChannels
For many years, the classical techniques ofprotein hiochemistry and immunology have beenused to isolate ion channels and determine theirstructure-function relationship.''''^ However, thelow surface density of most ion channels made thisprocess arduous. Over the past decade, the appli-cation of the techniques of molecular hiology hasrevolutionized the approach to the structure-func-tion studies of ion channels. The cDNAs encodingall the major voltage and ligand gated ion channelsin cardiac muscle have heen cloned and se-quenced. The amino acid sequence of the channelcan. in turn, be inferred from the respectivecDNAs. Relatively straight forward techniques canbe used to change (or mutate) specific amino acidsin the protein sequences. Mutant mRNA can besynthesized in vitro and injected into an expres-
PACE, Vol. 18 August 1995 1561

WHALLEY, ET AL.
sion system suGh as the oocyte of the frog. Theseexpression systems translate the mRNA into pro-tein, perform the necessary posttranslationalmodification and insert the channeLs into theirmembrane where they can be studied with con-ventional biophysical techniques. Progress has oc-curred at a dazzling rate. The actual three-dimen-sional structure of the channel molecules mustawait x-ray crystalographic studies, however in-formed guesses about higher order structure canbe inferred from the amino acid sequence. For ex-ample, strings of hydrophobic amino acids arelikely to occupy sites within the membrane.
The simplest structure that has been shown toform an ion channel, IminK. is responsible for theslow component of the delayed rectifier K ^ cur-rent in cardiac muscle.^'•''' It is a single polypep-tide chain of 130 amino acids that crosses themembrane once. A majority of cardiac ion chan-nels have a more complex structure. The nextorder of structural complexity is exhibited by thelarger family of voltage gated K^ channels. Thesechannels are formed by association of four sub-units. Each subunit consists of 500-1,000 amino
acids, forming six membrane spanning belices.The subunits may be tbe same, forming a bomotet-ramer or tbey may be different, forming a heterote-trameriG structure.''' The highest order of com-plexity is exhibited by tbe voltage gated Na^ andCa^^ channels. They consist of a. p,. and P2 sub-units. However, the a subunit is sufficient to formthe functional channel. The a subunit consists offour homologous domains (I-IV), with each do-main consisting of six transmembrane spanningsegments [SI-SR).''-^^ The structure of transmem-brane spanning segments is highly conserved be-tween domains; the loops connecting the segmentsshow more variability. Tbe p subunit ofthe Na^channel modulates the level of expression and ki-netics of inactivation of the a subunit."' The tenta-tive structure ofthe a subunit ofthe Na^ channelis summarized in Figure 6.
Tbe fourth transmembrane segment, S4, has apositively charged amino acid in every third posi-tion and acts as tbe voltage sensor for the activa-tion process.'^ The loop between S5 and Sfi extendsfor a considerable distance in the membrane andis believed to form the channel pore. The loop be-
Voltage sensor(m-gate)
Pore
out
Domain inactivation(h-gate)
gate Domain IV
Figure 6. Structure of the Na'*' channel. This figure illustrates the third and a portion of thefourth homologous domain ofthe Na " channel. Each domain consists of six membrane spanningsegments (Sl-SfS). The S4 segment has positive charged amino acids in every third position andacts as the voltage sensor. The loop between S5 and S6 curves into the lipid bilayer to form thechannel pore. The cytoplasmic loop between domains III and IV has a highly conserved structureand forms the inactivation gate.
1562 August 1995 PACE. Vol. 18

CONCEPTS IN CELLULAR CARDIAC ELECTROPHYSIOLOCY
tween the third and fourth homologous domainsplays a central role in normal inactivation. Sitesfor binding of the highly specific toxin tetrodo-toxin and divalent cations, have boen identi-fied.^""^" Localization of the site of antiarrhythmicdrug binding is under intense investigation.Knowledge of the structure-function relationshipsof cardiac ion channels will enable the identifica-tion of the essential current components of the ac-tion potential. Indeed, initial studies descrihingthe reconstruction of an excitable cell by co-expression of multiple channel types have alreadybeen reported.^^ These studies show that actionpotentials with pacemaker activity can be recon-structed with remarkably few current systems. Itwill be an ongoing challenge to define the preciserole of each of the many ion channel types presentin cardiac muscle.
Ionic Currents Underlying theNormal Ventricular and Atrial
Action Potentials
Resting Potential
During electrical diastole, ventricular myo-cytes maintain a stable resting membrane potential[Vr) of - 80 to - 85 mV. This primarily reflects theselective permeability of the sarcolemmal mem-brane to K*, and the transmembrane concentra-tion gradient for K * , which in turn is maintainedby the ATP dependent Na + -K^ pump. With eachcycle of the Na+-K^ pump, three intracellularNa^ions are extruded in exchange for uptake of two K 'ions. This generates an outward (hyperpolarizing)current, Ip, and maintains steep inwardly directedNa ^ and outwardly directed K " gradients.^•^ Intra-cellular K^ is constantly diffusing out of the celldown its concentration gradient. The main path-way for this K^ current is via inwardly rectifyingK^ channels. This efflux of positive charge is notbalanced by an equivalent efflux of negativecharge because the predominant intracellular an-ions are large, nondiffusable proteins. The resultis a slight excess of negative charge on the inside ofthe cell, which gives rise to the resting membranepotential. Vr is largely determined by the ratio ofextracellular to intracellular K" concentrations{[K" ],,/IK * Ii) because the sarcolemmal K * perme-ability is at least 50-fold greater than that of Na+,Cl" or Ca^^. If Vr were solely determined hy the
transmembrane K^ gradient, it would be expectedto equal the equilibrium potential for K^ (EK) pre-dicted from the Nernst equation:
where R is the universal gas constant, T is the abso-lute temperature in degrees Kelvin, z is the valenceof K+, and F is Faraday's constant. At SVC, theNernst equation for K" can be rewritten as F^ =61.5 log [K + l,y[K + li.Ifwe assume that IK^lo is 4.5mM and [K^L is 135 niM, FK is predicted to be- 91 niV. Although this is a reasonable approxima-tion of the normal resting potential, Vr is actuallyseveral millivolts positive to FK because of a smallinward "leak" current carried primarily by Na^ions that enter via channels and the Na"^-Ca^ ^ ex-changer. Figure 7 illustrates the normal intracellu-lar and extracellular concentrations of ions re-sponsible for the resting membrane potential andthe role of the Na ' -K^ pump in maintaining thetransmembrane Na^ and K' gradients. From theNernst equation it is apparent that alterations inthe extracellular to intracellular potassium con-centration ratio will significantly influence thelevel of resting membrane potential in the ventric-ular myocyte. This is clearly demonstrated duringacute myocardial ischemia when a rapid rise inextracellular potassium concentration causesmembrane depolarization; typically, the earliestelectrophysiological change of ischemia.^^^
Action Potential
The cardiac action potential is a propagatingwave of transient depolarization initiated when anexcitatory stimulus depolarizes the membrane be-yond threshold potential. It is inscribed by themovement of Na ' , K \ Ca^" , and Cl" ions throughat least nine distinct voltage or ligand operated ionchannels. Membrane transport processes includ-ing the Na ' -K^ pump and Na^-Ca^ ' exchangeralso contribute to the genesis of the action poten-tial by: (1) maintaining the transmembrane ion gra-dients, which are essential for excitability; (2) gen-erating small currents as a result of net ion move-ment; and (3) restoring the normal intracellularionic composition of the excited cell once the ac-tion potential is completed. Before detailing theionic basis of the cardiac action potential it is use-ful to review some nomenclature used to describe
PACE. Vol. 18 1995 1563

WHALLEY, ET AL.
- 140 mM
small "leak"
[Na+li~iOmM
[K+]. -135 mM
I*—M (proteinsn
-85 mV [Q32+] ^ 200 nM - 20 mM
mM
Figure 7. Ionic determinants of the normal resting potential (V,.) in the ventricular cell. Typicalintracellular and extracellular concentrations of Na'^. K^ , Ca^*. and CI' are shown. The steepoutwardly directed concentration gradient for K *" and inward gradient for Na " are maintainedby the ATP dependent Na'^-K'^ pump. The high permeability of the membrane to K* relative toother ions and the transmemhrane concentration gradient for K^ are the principle determinantsof Vf. A small inward leak of Na^ ions keeps V^ slightly positive to £'K. The Na*-K* pumpgenerates a small net outward current. The Na*-Ca^* exchanger generates a small net inwardcurrent. The predominant intracellular anions ([A]) are large, impermeant proteins.
the underlying membrane currents. By conven-tion, inward currents are defined as being equiva-lent to the movement of positive charge into themyocyte. Inward currents are, therefore, depolar-izing currents. The Na ^ current, IM ,, which is re-sponsible for the action potential upstroke inatriaL His-Purkinje and ventricular cells and theCa^^ current (Ica), which generates the upstrokein sinoatrial nodal and atrioventricular (AV) nodalcells are the major physiological inward currents.Outward currents are equivalent to the efflnx ofpositive charge from the cell. Outward currents re-polarize the cell after the action potential upstrokeand plateau and may cause hyperpolarization ofVp. K " ions are the major charge carrier of outwardcurrent in the heart. Influx of CI" ions also contrib-utes outward, repolarizing current during the car-diac action potential.
Rectification describes the dependence of re-sistance to ion flow on membrane voltage and isillustrated in Figure 8. Ohmic or nonrectifying cur-
rent obeys Ohm's law, which states that currentis linearly dependent on membrane voltage, andresistance is voltage independent (I = V/R]. Out-ward rectification occurs when the membrane con-ducts an outward flow of current at voltages posi-tive to the reversal potential of the ion (Erev) moreeasily than it conducts inward current at voltagesnegative to Er,,v. Examples of currents displayingoutward rectification include the delayed rectifierK^ current [IK) and the transient outward current(Ii,J. Inward rectification occurs when the mem-brane passes inward current at voltages negativeto Eppv more readily than outward current at poten-tials positive to E^,,^. Perhaps a simpler way of con-ceptualizing this property is that currents showingoutward rectification increase with progressivemembrane depolarization whereas the conduc-tance of inwardly rectifying currents decreaseswith progressive depolarization.
We will initially describe the ionic currentsunderlying the ventricular action potential (Fig. 9,
1564 August 1995 PACE, Vol. 18

CONCEPTS IN CELLULAR CARDIAC ELECTROPHYSIOLOGY
Current-Voltage (I-V) Relationship
IOutward {+ve) Outward Rectification
Linear (ohmic)
Inward Rectification
~^ V
Figure 8. Current-voltage relationships ofK^ channelsillustrating the property of rectification. Ionic current(ordinate) is shown as a function of membrane voltage(abscissa). Net K^ current is zero at the reversal poten-tial, which equals the value of £'K derived from theNernst equation (-90 mV). K^ current is outwardly di-rected at potentials positive to Ef- and inwardly directedat potentials negative to Ef^. An ohmic current changeslinearly as a function of voltage since resistance is con-stant at all potentials. Channels displaying outward rec-tification pass outward current at potentials positive to£K more readily than inward currents at potentials nega-tive to EK_. Inwardly rectifying K^ channels pass inwardcurrent at potentials negative to .EK more readily thanoutward current at potentials positive to Em-
A) and then highlight the important differ-ences seen in the atrial action potential (Fig. 9,panel B). The action potential upstroke (phase 0)is generated by a very large but brief (< 1-2 ms)increase in membrane Na^ conductance (I^J.^^'^^As discussed earlier, depolarization increases theprobability of Na^ channel activation. Thresholdpotential (V,h) represents the membrane voltage atwhich enough inward Na^ current is activated toovercome the repolarizing influence of the out-ward K^ conductances. Once V,h is reached, theinflux of Na^ ions produces a regenerative depo-larization, which drives the membrane potential(Vm) to +40 mV thus inscribing the action poten-tial upstroke. V^ at the peak of the action potential,therefore, approaches but does not achieve theNa* equilibrium potential (approximately +70
mV). The maximum rate of rise of the upstroke,(Vmax). reflects, albeit nonlinearly, the magnitudeof IN.T^ ' ' In this regard, it is of interest to notethat Purkinje cells, which have the fastest Vmax(500-700 V/s), also have a significantly higherdensity of Na^ channels than atrial or ventricularmyocytes in which Vmnx ranges from 200-400 V/s. As will be discussed subsequently, Vmax and theaction potential amplitude are important determi-nants of conduction velocity throughout the myo-cardium.
The peak of the action potential upstroke isfollowed by a phase of early repolarization (phase1) that returns V^ into the region of +10 ± 10mV. Rapid repolarization results from voltage de-pendent inactivation of 1 ^ together with activa-tion of the transient outward current (Ito). Ito in the
Ventricular
Vm
{mV)
-80
40
Vm
(mV)
'Na
BO 1—
'K1
Atrlal
iK(Ach)
Figure 9. Membrane currents underlying the normalventricular (panel A) and atrial (panel B] action poten-tials.I^^.^ — Na* carrent;Ifa - transient outward current;
= Ca^'^ current; - Na*- exchanger;= plateau K'*^ current; I^ ^ delayed (outwardly rectify-ing) K^ current; 1^1 — inwardly rectifying K*^ current;/pump — Na*-K^ pump current;/K(Ach) - acetylcholineactivated K*^ current. Note the absence of a well-definedplateau phase and faster repolarization in the atrial cell.
PACE, Vol. 18 August 1995 1565

WHALLEY, ET AL.
mammalian heart appears to consist of at least twocomponents.^"-^^ Itot is a voltage dependent, rap-idly activating K" current that displays outwardrectification. It undergoes voltage and time depen-dent inactivation and its channels are characteris-tically slow to recover from inactivation.^" Thislatter behavior may diminish its importance inearly repolarization as heart rate is increased andthe diastolic recovery interval shortens. I,,, is he-lieved to be a Ca^ ^ dependent Cl" current [inwardflow of Cl" ions).^^ Its activation correlates withCa^^ release from the sarcoplasmic reticulumwhich in turn, is triggered by influx of Ca^ ^through L-type channels (ICH.L)- There is a markeddegree of regional and interspecies variation in theprominence of It . This heterogeneity is particu-larly evident when comparing the action potentialof epicardial ventricular cells that have a promi-nent I,,, and phase 1 repolarization ("spike anddome" configuration) with endocardial cells inwhich I,,, is weakly expressed and phase 1 is negli-gible.''^ In addition to Ito2. Cl ions may contributeto early ropolarization during adrenergic stimula-tion via a rapidly activating, noninactivating Cl"current.^^
The plateau phase (phase 2) reflects a delicatebalance between inward and ontward currents. In-activation of INO together with ihe decrease in con-ductance of IKI imposed by its inward rectificationincrease membrane resistance (R,n)- As a result,small changes in net ionic current may signifi-cantly alter V,,i. The inward currents during phase2 include: (1) Ca^ * currents through channels thatwere activated by the action potential upstroke; (2)the Na"' -Ca^^ exchange current; and (3) a slowlyinactivating or "late" Na^ current.^ I a is carriedthrough two distinct channels.^"^ Current throughT type channels (Ica.T) i activated rapidly but tran-siently at potentials positive to - 70 niV. It is ofminor importance in ventricular cells but maycontribute to the pacemaker potential of cellswithin the SA node and His-Purkinje system. Ica.L-the long-lasting form of Ca current is activatedat potentials positive to —40 mV. It inactivatesslowly (~ 100 ms) and its primary role is to triggerthe release of Ca " from the sarcoplasmic reticn-lum, which activates the contractile myofilaments.Inactivation of Ica.L is a function of voltage, time,and the intracellular Ca^ ^ concentration. TheNa -Ca^ " exchanger, by virtue of its 3 Na ^ :1 Ca' ^
transport ratio generates a small net membranecurrent [Fig. 7).' ^ The direction of exchanger cur-rent during the action potential is determined bythe relationship of V^ to the combined equilib-rium potential (ENaCa)- At rest ENaCa is approxi-mately - 3 0 to - 4 0 mV, and since Vp is negativeto this value, inward current is generated. At thepeak of the action potential upstroke and duringearly repolarization, V,,, is positive to ENa ca. whichmay result in a brief phase of outward current dueto reversal of the exchanger (i.e., Na^ efflux/Ca^^influx).'''' During the plateau phase as intracellularcalcium increases ENaCi becomes more positiveand the exchanger reverts to its Na^ influx/ Ca^^efflux mode thereby generating net inward cur-rent. In ventricular and Purkinje cells a small, per-sistent inward Na^ current continues to flowthroughout the action potential plateau. It is notpresently known whether this current is due todelayed or failed inactivation of the Na^ channelsresponsible for the action potential upstroke (per-sistent I^a), or represents a distinct subpopulationofNa^ channels that open with long latencies (i.e.,"late" Na channels).^ Inhibition of this persistentcomponent of IjMa is thought to be responsible forthe shortening of ventricular action potential dura-tion seen with lidocaine and other Class IB antiar-rhythmic drugs.•'^ I^i. the dominant K^ conduc-tance at resting potentials contributes little out-ward current during the plateau phase due to itsproperty of strong inward rectification. The de-layed, outward rectifier K^ current (IK), is gener-ally considered to be the dominant outward con-ductance during the plateau phase, although arecently discovered rapidly activating, nonin-activating K ^ current [iKp) may also contribute sig-nificantly.•''' IK is activated slowly during the pla-teau phase. In several species it consists of twocomponents with markedly different kinetics andconductance properties.^''•^^ IKP is the rapidly acti-vating component of IK. It shows strong inwardrectification and is selectively blocked by thenewer Class III antiarrhythmic agents E-4031 anddofetilide.•'"•'*" IKS is the larger, slowly activatingcomponent. Its conductance is linear [nonrectify-ing) and sensitive to p-adrenergic stimulation. IKSis not blocked by E-4031. There are considerableinterspecies differences in the relative abundanceof Ijcs and I|Cr- As yet the relative importance of theindividual components of I^ to repolarization in
1566 August 1995 PACE, Vol. 18

CONCEPTS IN CELLULAR CARDIAC ELECTROPHYSIOLOGY
human ventricular myocytes has not been clearlydefined.
Progressive decay of Ica combined with an in-creasing outward K^ conductance terminates theplateau phase and initiates the late rapid phaseof repolarization (phase 3). Pbase 3 repolarizationreflects the combined influence of outward currentthrough IK and IKI together with a small contribu-tion from the Na^-K* pump current. Early phase3 repolarization is predominantly due to IK. AS re-polarization proceeds, IKI channels, which wereclosed during the plateau phase, are progressivelyreactivated thereby causing a regenerative repolar-ization of the membrane back to its resting poten-tial.
The major differences between atrial and ven-tricular action potentials lie in the distribution andmagnitude of the ionic currents responsible for V,.and action potential repolarization (Fig. 9. panelB).'*^'^^IKI is the dominant resting K^ conductancein the atrial cell, although it is considerablysmaller than in the ventricular cell.'*'' Another K ^channel, activated by Ach via muscarinic (M2) re-ceptors (IK.ACH). shares with IKI the role of stabiliz-ing the atrial resting potential and is responsiblefor the hyperpolarization of Vr observed duringvagal stimulation. The atrial action potential up-stroke is followed by a pronounced phase of rapidrepolarization with only a brief, ill-defined plateauphase. This configuration reflects the predomi-nance of a large I,,, current over a relatively smallIcii-*'' "* The slow terminal phase of repolarizationis mediated by IK and iK.Ach with the latter currentprobably playing the major role. Activation ofiK.Ach by vagal stimulation shortens the atrial ac-tion potential duration and refractory period.
Mechanisms of Automaticity
Normal Automaticity
In tbe normal heart, the property of sponta-neous impulse generation or automaticity residesin the cells of the sinus node and the specializedconducting system. The sinus node has the highestintrinsic rate and acts as the dominant pacemaker.However, cells in the AV node or His-Purkinje sys-tem direct the cardiac rhythm when the sinus nodefails or when their intrinsic automatic rates are en-hanced. We shall review the ionic mechanisms ofsinus node automaticity and highlight some of the
200
t t out
in
Figure 10. Membrane currents during normal automa-ticity. Spontaneous action potentials recorded from asinus node cell are shown in the upper panel. The lowerpanel outlines the membrane currents that underlie thephase of spontaneous depolarization (phase 4). Threecurrents contribute to the membrane potential duringphase 4: a decreasing outward current. ly^; a time inde-pendent inward current, Ii,; and a hyperpolarization ac-tivated inward current. If.
differences that may exist between the sinus nodeand other cardiac pacemakers.
The transmembrane potential recorded froma sinus node cell is shown in Figure 10. The dia-stolic membrane potential is not stable. From amaximum diastolic potential of about - 60 mV,the cell undergoes slow diastolic (phase 4) depo-larization; this phase merges smoothly into the up-stroke (phase 0) of the action potential. This slowdiastolic depolarization is characteristic of auto-matic cells. Despite intense study, controversyabout the role of specific ionic currents duringphase 4 remains."*^ Since the cell membrane is de-polarizing, the net membrane current must be in-ward. Three current mechanisms play an impor-tant role: the delayed rectifier K current, I^, acti-vated during the preceding action potential, thetime independent background current, I|,, carriedpredominantly by Na ' ions, and a hyperpolariza-tion activated current. I|. Repolarization results inthe closure or deactivation of K^ channels openedduring the preceding action potential, This resultsin a decline in K^ conductance during phase 4 ofthe action potential. The nature of l[, is uncertain.It may arise from a different channel than that re-sponsible for the transient inward current, IM . The
PACE. Vol. 18 August 1995 1567

WHALLEY, ET AL.
decline of the outward K^ current in the face of afixed inward background current can account forthe diastolic depolarization. The inward current,If, also plays a role. If is carried by Na" and K"ions, is activated at about - 4 5 mV and has a rever-sal potential of - 2 0 mV. In the normal potentialrange for the sinus node pacemaker. If is inward.The kinetics of If are slow (activation time ~ 1 s).Therefore, there is a phase lag between the degreeof activation of If and its steady-state value at agiven potential. Because of this lag. If actually in-creases during the pacemaker potential. In Pur-kinje cells where the normal diastolic potential isbetween - 70 and - 90 mV, If is the major currentresponsible for diastolic depolarization. In thesinus node, Ca''^ channels account for the phaseof rapid depolarization, while in Purkinje cells,Na" channels fulfill this role. Membrane trans-porters such as the Na -Ca^ " exchanger cause netion movement across the cell membrane and maycontribute to cellular automaticity.'*^ Because ofthe nature of the currents underlying pacemakeractivity, the intrinsic rate of automatic cells maybe influenced by modulators of K , Ca^ , and Na currents.
The autonomic nervous system modulatesheart rate by an action on membrane currents insinus node cells; vagal stimulation induces a brad-ycardia whereas sympathetic stimulation inducesa tachycardia. The conventional view of vagal ac-tion on the sinus node cell is that Ach interactingwith the M2 receptor effects three changes; (1) acti-vation of an inwardly rectifying K^ current, iKAchI(2) reduction of I,:,,; and [3) reduction of If.'' ''*IKAHII hyperpolarizes the membrane potential andabbreviates the action potential duration. This ac-tion, together with a decrease in If slows the rateof diastolic depolarization. Inhibition of I g de-creases the rate of phase 0 depolarization. The ac-tion of direct vagal stimulation in intact tissue ishowever different.*'' The slope of phase 4 depolari-zation is decreased without a change in action po-tential configuration. This suggest that vagal stim-ulation may activate a different receptor-effectorsequence of events than those observed when Achis applied to the entire cell.
Catecholamines accelerate the sinus rate bymoving the potential for If activation to more posi-tive potentials, and by increasing Ica and IK. Eachof these factors may increase the slope of the pace-
maker potential. The adrenergic receptors acti-vated by catecholamine application and sympa-thetic nerve stimulation may also differ."*
Abnormal Automaticity
There are at least three mechanisms that mayresult in repetitive activity in normally quiescentatrial and ventricular cells: (1) depolarizing cur-rent; (2) early afterdepolarizations (EADs); and (3)delayed afterdepoiarizations (DADs).
For more than a century, it has been appreci-ated that application of constant depolarizing cur-rent to the myocardium may result in automatic-ity.^^ The applied current, in the face of decliningK^ current may result in diastolic depolarizationto the threshold for Na^ and Ca^^ current activa-tion. This mechanism has been postulated to occurduring myocardial ischemia.'*' Currents of injurybetween the normal and the ischemic zones mayresult in repetitive depolarization.
Action potentials illustrating mechanisms band c are shown in Figure 11. They are examplesof triggered activity in which repetitive depolari-zation is dependent on the preceding action poten-
0
0 —
-90—I
\ \
-90—"
60/min
Figure 11. Abnormal automaticity in cardiac cells.Early afterdepoiarizations are illustrated in panels A-C;delayed afterdepoiarizations in panels D and E. In panelA, action potential prolongation precedes the develop-ment of single (panel B) or multiple (panel C) early after-depoiarizations. Panel D illustrates delayed afterdepo-iarizations. In panel E, they reach threshold and resultin repetitive activity. (Reproduced with permission.^^)
1568 August 1995 PACE, Vol. 18

CONCEPTS IN CELLULAR CARDIAC ELECTROPHYSIOLOGY
tial. Each mechanism may account for certain clin-ical arrhythmias.
Early afterdepolarizations are secondary de-polarizations arising from the incompletely repo-larized memhrane. They are nsnally associatedwith prolongation ofthe action potential durationand tho QT interval on the ECG. Slowing of theheart rate results in further action potential pro-longation and enhances the occurrence of EADswhile acceleration of heart rate suppresses the oc-currence of EADs. They may occur at any potentialhetween the plateau and the point of complete re-polarization. In the normal action potential, thenet membrane current is outward in this range ofmemhrane potential. Any factor that causes the netmemhrane current to shift transiently in the in-ward direction may result in EADs. There are twomechanisms that may result in such a shift of thenet membrane current in the inward direction:blockade of the outward current(s), or enhance-ment of the inward current. The former may beparticularly important in the genesis of arrhyth-mias. As a part of their therapeutic action, manydrugs such as quinidine, disopyramide, sotalol,and other Class III drugs block the outward K ^current. Other drugs, e.g., the phenothiazines andterfenadine block K" channels as a secondary ef-fect. All these agents may cause marked prolonga-tion ofthe action potential and QT interval, settingthe stage for EADs, The ionic current responsiblefor tbe rapid upstroke of EADs depends on therange of potentials at which they occur. In the pla-teau range of potential, the Ca^ channel is respon-sible for tbe upstroke while at more negative mem-brane potentials the Na^ channel also contributes.
Available evidence strongly suggests thatEADs are the cellular basis of torsades de pointes.Drugs such as quinidine, which cause torsades depointes also cause EADs in vitro.^^'^^ EADs maybe recorded in tbe in situ heart under conditionsassociated with torsades de pointes.
Delayed afterdepolarizations are tbe thirdmechanism of abnormal automaticity. These aresecondary depolarizations that arise from tbe fullyrepolarized membrane. Acceleration of beart rateincreases the amplitude of DADs. Eventually, theymay depolarize the membrane to threshold poten-tial and result in repetitive activity. DADs mayarise under conditions that result in intracellularCa^^ overload such as ischemia and digitalis in-
toxication. Ca^ overload may result in the oscilla-tory release of Ca^^ from intracellular storage sitessuch as the sarcoplasmic reticulum. The high in-tracellular Ca^ ^ levels may then activate a nonspe-cific cation channel resulting in tbe DAD. An addi-tional or alternative mechanism for the generationof the transient inward current is an electrogeniccurrent generated by the Na^-Ca^^ exchangemechanism.''^ Drugs tbat block Ca^^ movementinto cells, e.g., Ca^^ antagonists, are effectiveagainst arrhythmias arising from DADs.
Cellular Determinants ofExcitability, Conduction Velocity,
and Refractoriness
Excitability
The term excitability describes the ease withwhich a regenerative action potential is initiatedin response to a depolarizing stimulus and propa-gated to neighboring cells.^^ Excitability is depen-dent upon tbe complex interaction of membraneproperties, which have been broadly classified byArnsdorP'' into active (generator) properties andpassive (resistive) properties. Active properties in-clude: (1) Tbe amplitude and kinetics of ionic cur-rents responsible for tbe action potential upstroke(iNa in the atria, ventricles and His-Purkinje sys-tem; IQ, in the SA and AV nodes); (2) tbe thresholdpotential, Vii,, for activation of I a or Ica; (3) thedifference between the resting potential or maxi-mum diastolic potential and V,h, which deter-mines tbe minimum transfer of positive chargeneeded to evoke an action potential. Passive prop-erties refer to the impedance elements or "cable"properties of tbe myocyte and include the mem-brane resistance (R,,,). and the resistance to longi-tudinal flow of ionic current witbin tbe cell (Rj),and through the extracellular space [R,J and capac-itance Ri comprises the myoplasmic resistance andthe resistance of gap junctional channels that inter-connect adjoining myocytes. ^•^*'
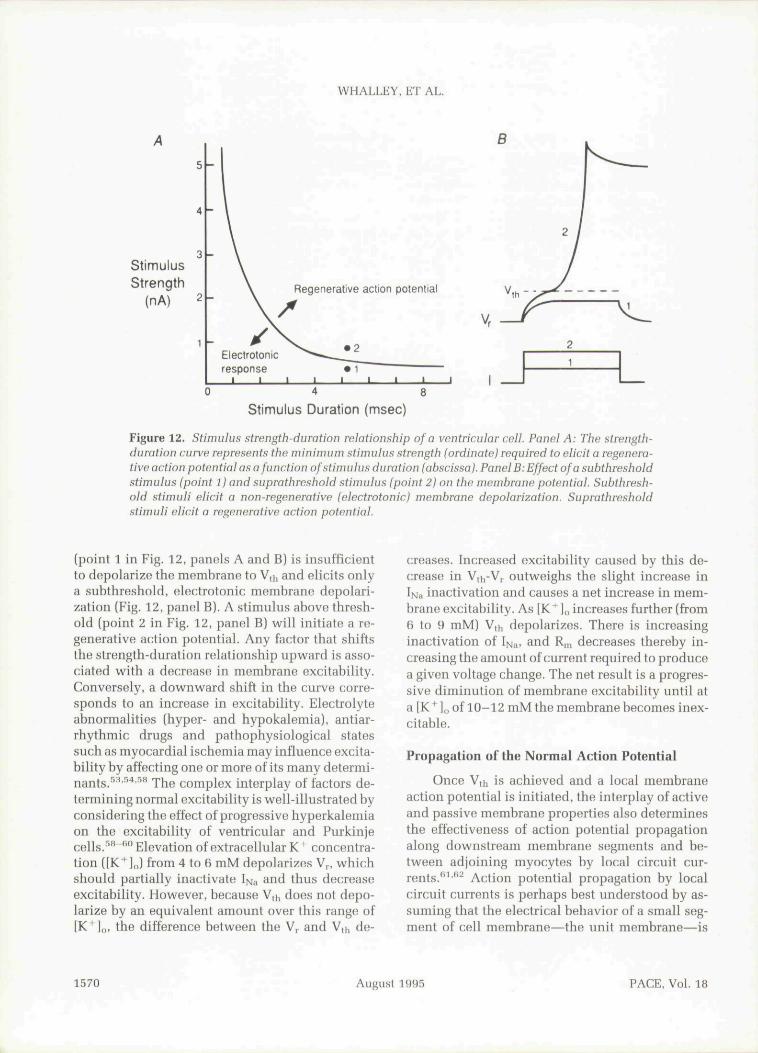
Experimentally, the concept of excitability isbest illustrated by tbe strength-duration relation-ship of an isolated cell as illustrated schematicallyin Eigure 12.^' Tbe strength-duration curve (Fig.12, panel A) represents the minimum depolarizingcurrent required to elicit a regenerative action po-tential at each pulse duration. At any given dura-tion, a current stimulus below tbis threshold
PACE, Vol. 18 August 1995 1569
WHALLEY, ET AL.
f 1
StimulusStrength
(nA)
5
4
3
2
1
-
Electrotonicresponse
Regenerative
H
• 11 1
action potential
1 1 1
0
LStimulus Duration (msec)
Figure 12. Stimulus strength-duration relationship of a ventricular cell. Panel A: The strength-duration curve represents the minimum stimulus strength (ordinate) required to elicit a regenera-tive action potential as a function of stimulus duration (abscissa). Panel B: Effect of a subthresholdstimulus (point 1) and suprathreshold stimulus (point 2) on the niembrane potential. Subthresh-old stimuli elicit a non-regenerative (electrotonic) membrane depolarization. Suprathresholdstimuli elicit a regenerative action potential.
(point 1 in Fig. 12, panels A and B) is insufficientto depolarize the membrane to Vth and elicits onlya subthreshold. electrotonic membrane depolari-zation (Fig. 12, panel B). A stimulus above thresh-old (point 2 in Fig. 12, panel B) will initiate a re-generative action potential. Any factor that shiftsthe strength-duration relationship upward is asso-ciated with a decrease in membrane excitability.Conversely, a downward shift in the curve corre-sponds to an increase in excitability. Electrolyteabnormalities (hyper- and hypokalemia). antiar-rhythmic drugs and pathophysiological statessuch as myocardial ischemia may influence excita-bility by affecting one or more of its many determi-nants/' ' '•'•' " The complex interplay of factors de-termining normal excitability is well-illustrated byconsidering the effect of progressive hyperkalemiaon the excitability of ventricular and Purkinjecells. '*"*'" Elevation of extracellular K" concentra-tion ([K" ],J from 4 to 6 mM depolarizes V , whichshould partially inactivate I>j;, and thns decreaseexcitability. However, because V,h does not depo-larize by an equivalent amount over this range of[K^lo, the difference between the Vp and Vth de-
creases. Increased excitability caused by this de-crease in V,i -V,. outweighs the slight increase inIiMa inactivation and causes a net increase in mem-brane excitability. As [K^ ]„ increases further (from6 to 9 mM) V,h depolarizes. There is increasinginactivation of If^^, and R,n decreases thereby in-creasing tbe amount of current required to producea given voltage change. The net result is a progres-sive diminution of membrane excitability until ata IK^],, of 10-12 mMtbe membrane becomes inex-citable.
Propagation of the Normal Action Potential
Once V,h is achieved and a local membraneaction potential is initiated, the interplay of activeand passive membrane properties also determinesthe effectiveness of action potential propagationalong downstream membrane segments and be-tween adjoining myocytes by local circuit cur-rents.''^ " Action potential propagation by localcircuit currents is perhaps best understood by as-suming that tbe electrical behavior of a small seg-ment of cell membrane—the unit membrane—is
1570 August 1995 PACE. Vol. 18

CONCEPTS IN CELLULAR CARDIAC ELECTROPHYSIOLOCY
equivalent to a resistance (R,,,) and capacitance(C,,i) arranged in parallel.^^'^^ The resistive proper-ties arise from the presence of ion channels. Thelipid bilayer with its bydrophobic core and polarcytoplasmic and extracellular surfaces exhibits theproperty of capacitance, i.e., the ability to separateand store charge. The entire cell has been modeledas an electrical cable, consisting of many unitmembranes each coupled to adjacent units by theresistances of the cytoplasm (Rj) and the extracel-lular fluid (Ro).
Local circuit current is initiated by the ioniccurrent responsible for the action potential up-stroke in the particular cell (1^., or Ic.J. The result-ing influx of positive charge moves longitudinallythrough the cytoplasmic resistance R, and dis-charges the capacitance of the adjacent nonexcitedmembrane segment thereby bringing it to thresh-old for activation of excitatory inward current. Thelocal circuit is completed when positive chargesreleased to the extracellular space from the C^ ofthe downstream membrane segment return to theinitiating active membrane segment via RQ. A1-tbougb the action potential is initiated by I ., orIca- local circuit currents are predominantly car-ried intracellularly by K^ ions since these are byfar tbe most abundant intracellular cations (Fig.7).
Propagation between adjacent cells occurs byspecialized low resistance gap junctional channelslocated in greatest density at tbe ends of ventricu-lar and His-Purkinje cells to facilitate preferentialspread of excitation in a longitudinal direc-tion/''^"'^'' Tbe velocity of impulse propagationthrough the myocardium is determined by severalfactors. Active membrane properties include: (l)the amplitude and kinetics of IN., or I ;, (reflectedby V,n,,x and action potential amplitude); and (2)the difference between the resting and thresholdpotentials. Passive properties include: (l) Themembrane capacitance, C,,,; (2) membrane resis-tance, Rn^; and (3) the internal and external longitu-dinal resistances (R; and RQ).
Conduction velocity may be increased by fac-tors that enhance the magnitude of inward cur-rents (e.g., hyperpolarization of Vm), increase R,,,(e.g., hypokalemia), or decrease R, (catechola-mines).^'''^^ Conduction slowing may result froma decrease of inward current (membrane depolari-zation, ischemia); a decrease in Rm (hyperka-
iemia), or an increase in longitudinal resistancevia Rj (ischemia, acidosis. elevated intracellularCa^^ or Mg^^ concentration) or R (ischemia).During early ischemia (< 10 min) ventricular con-duction slowing is mainly due to a decrease in in-ward Na * current resulting from membrane depo-larization.''^**" Late conduction stowing (after15-20 min) mainly reflects an increase in Rj,whicb may eventually lead to cellular uncoup-ling/'^''^'^" This rise in R, is attributable to decreas-ing conductance of gap junctions caused by a pro-gressive rise in intracellular Ca and the develop-ment of intracellular
Cellular Determinants of MyocardialRefractoriness
Refractoriness denotes the inability to reexcitea cell or tissue immediately following the initia-tion of an action potential. The state of refractori-ness of a ventricular cell depends upon both theprevailing Na * channel availability (i.e., the num-ber of Na^ channels that have recovered from inac-tivation) and the ability of the membrane to depo-larize to threshold for activation of these availablechannels. As discussed in an earlier section, theprocess of recovery of Na^ (and Ca^^) channelsfrom inactivation is both time and voltage depen-dent and it is these properties that largely deter-mine the duration of refractoriness. In the ventric-ular cell during the action potential plateau andearly phase 3 repolarization almost all Na^ chan-nels are in the inactive state. A stimulus deliveredduring this time, regardless of its magnitude, willnot initiate a regenerative membrane depolariza-tion. This period, which begins with the actionpotential upstroke and lasts until V,,, has repolar-
ized to 50 mV, is the absolute refractory period.As Vm repolarizes from - 50 mV to more negativepotentials an increasing proportion of Na ^ chan-nels recover from inactivation as a function of timeand voltage. " - ^ Regenerative action potentialsmay he elicited during this phase of repolariza-tion—tbe relative refractory period—however,this requires a stimulus larger than the normalthreshold. In addition, action potentials initiatedduring the relative refractory period will have re-duced V,iiax and amplitude, and will be conductedmore slowly than normal because a proportion ofNa" channels remain inactivated.
PACE, Vol. 18 August 1995 1571

WHALLEY, ET AL.
In atrial, ventricular and His-Purkinje cells,the time course of recovery of excitability closelyfollows that of membrane repolarization. When V,,,is fully repolarized, Na ' channel availahility hasalmost returned to its baseline value prior to theaction potential. Total recovery of membrane ex-citability usually outlasts full membrane repolari-zation by < 50 ms. This reflects the rapid timecourse of recovery of Na^ channels from inactiva-tion. In SA and AV nodal cells tbe slower timecourse of recovery of l^^ from inactivation meansthat refractoriness may outlast full repolarizationby > 100 ms. Tbis prolonged postrepolarizationrefractoriness is a characteristic feature of nodalcells and is an important determinant of their dec-remental conduction properties. Prolonged postre-polarization refractoriness, while not a feature ofthe normal ventricular cell, may occur duringearly ischemia when membrane depolarizationand other factors slow the recovery of Na^ chan-nels from inactivation.*'^
Recovery of membrane excitability in the ven-tricular cell has traditionally been equated withthe availability of Na^ channels. In contrast, theimportant role of deactivation of K^ channels inmodulating excitability is less appreciated. Thisconcept has heen reemphasized recently by Del-
who has shown that dynamic changes in IKIand IK. by altering R,n, play important roles in de-termining tbe ease with wbicb the membrane de-polarizes to threshold. During tbe action potentialIK activates slowly and is believed to be responsi-ble for initiating membrane repolarization. As re-polarization progresses, IK deactivates with a timeconrse, which outlasts full repolarization and typi-cally extends into the diastolic interval followingthe action potential. If a previously supratbresholdexcitatory stimulus is delivered before IK is fullydeactivated, outward current through tbe resid-ually activated IK cbannels will oppose tbe inwardstimulating current and may result in a subthresb-old response despite full recovery of Na " channelsfrom inactivation.'^ To summarize, both tbe timecourses of recovery of IN;, from inactivation and ofIK deactivation are important determinants of thetime course of recovery of excitability and bencethe refractory period. The relationship betweenthe kinetics of the Na" and K * cbannels, the ac-tion potential duration, and membrane excitabilityand refractoriness provide the background for un-derstanding the antiarrhythmic properties of drugsthat block the Na^ cbannel and prolong tbe car-diac action potential. Tbe ionic bases of antiar-rbythmic drug action will he considered further inPart II.
References
1. Powell T, Twist VW. A rapid technique for the iso-lation and purification of adult cardiac musclecells having respiratory control and a tolerance tocalcium. Biochem Biophys RRS Comm 1976; 72:327-333.
2. Hamill OP, Marty A. Neher E, et al. Improvedpatch-clamp techniques for high-resolution cur-rent recording from cell and cell-free membranepatches. Pflugers Arch 1981; 391:85-100.
3. Catterall WA. Structure and function of voltage-sensitive ion channels. Science 1988; 242:50-61.
4. Catterail WA. Cellular and molecular biology ofvoltage-gated sodium channels. Physiol Rev 1992;72:515-548.
5. Brown AM, Lee KS. Powell T. Voltage clamp andinternal perfusion of single rat heart muscle cells.J Physiol (Lond) 1981; 318:455-477.
6. Hille B. Ionic channels of excitable membranes.Sunderland Sinauer Assoc, Inc., 1984, pp. 1-19.
7. Armstrong CM. Sodium channels and gating cur-rents. Physiol Rev 1981; 61:644-683.
8. Hodgkin AL, Huxley AF. A quantitative descrip-tion of membrane current and its application Io
conduction and excitation in nerve. J Physiol(Lond) 1952; 117:500-544.
9. Patlak JB, Ortiz M, Slow currents through singlesodium channels of adult rat heart. } Gen Physiol1985; 86:89-104.
10. Brown AM. Ion channels as G protein effectors.NIPS 1991; 6:158-161.
11. Nichols CG, Lederer WJ. Adenosine triphosphate-sensitive potassium channels in the cardiovascu-lar system. Am J Physiol 1991; 261:H1675-H1686.
12. Cohen SA, Barchi RL. Voltage-dependent sodiumchannels. Internat Rev Cytol 1993; 137C:55-103.
13. PhilipsonLH, Miller RJ. A small K" channel loomslarge. TIPS 1992; 13:8-11.
14. Freeman LC, Kass RS. Expression of a minimal K"channel protein in mammalian cells and immuno-localization in guinea pig heart. Circ Res 1993; 73:968-973.
15. Po S, Roberds S, Snyders DJ, et al. Heteromultim-eric assembly of human potassium channels. Mo-lecular basis of a transient outward current? CircRes 1993; 72:1326-1336.
16. Isom LL, De Jongh KS, Patton DE, et al. Primary
1572 August 1995 PACE, Vol. 18

CONCEPTS IN CELLULAR CARDIAC ELECTROPHYSIOLOCY
structure and functional expression of the pi sub-unit of the rat hrain sodium channel. Science 1992;256:839-842.
17. Stuehmer W, Conti F, Suzuki H, et al. Structuralparts involved in activation and inactivation of thesodium channel. Nature 1993; 339:597-603.
18. Satin J, Kyle JW, Chen M, et al, A mutant of TTX-resistant cardiac sodium channels with TTX-sensi-tive properties. Science 1992; 256:1202-1205,
19. Backx PH, Yue DT, Lawrence |H, et al. Molecularlocalization of an ion-binding site within the poreof mammalian sodium channels. Science 1992;257:248-251.
20. Noda M. Suzuki S, Numa S, et al, A single pointmutation confers tetrodotoxin and saxitoxin insen-sitivity on the sodium channel II. FEBS Lett 1989;259:213-216,
21. Hsu H, Huang X-cY, Karschin A, et al. Slow andincomplete inactivations of voltage-gated chan-nels dominate encoding in synthetic neurons. Bio-phys I 1993; 65:1196-1206.
22. Gadshy DC, Kimura ], Noma A. Voltage-depen-dence of Na/K pump current in isolated heart cells.Nature [Lond) 1985; 315:63-65.
23. Hill JL, Gettes LS. Effect of acute coronary arteryocclusion on local myocardial extracellular K * ac-tivity in swine. Circulation 1980; 61:768-778.
24. Cettes LS, Reuter H, Slow recovery from inactiva-tion of inward currents in mammalian myocardialfibres, J Physiol (Lond] 1974; 240:703-724.
25. Weidmann S. The effect of the cardiac membranepotential on the rapid availahility of the sodium-carrying system, J Physiol (Lond) 1955; 127:213-224.
26. Sheets MF, Hanck DA, Nonlinear relation betweenVmax and INH in canine cardiac Purkinje cells. CircRes 1988; 63:386-398.
27. Cohen C|, Bean BP. Tsien RW, Maximum upstrokevelocity as an index of available sodium conduc-tance. Circ Res 1984; 54:636-651,
28. Corahoeuf E, Carmeliet E, Existence of two tran-sient outward currents in sheep cardiac Purkinjefihers. Pflugers Arch 1982; 392:352-359,
29. Tseng G-N, Hoffman BF, Two components of tran-sient outward current in canine ventricular myo-cytes. Circ Res 1989; 64:633-647.
30. Fermini B, Wang Z, Duan D, et al. Differences inrate dependence of transient outward current inrabbit and human atrium. Am J Physiol 1992; 263:H1747-H1754.
31. Zygmunt AC, Gibbons WR. Calcium-activatedchloride current in rabbit ventricular myocytes.Circ Res 1991; 68:424-437.
32. Litovsky SH, Antzelevitch C. Transient outwardcurrent prominent in canine ventricular epicar-dium but not endocardium. Circ Res 1988; 62:116-126.
33. Harvey RD, Clark CO, Hume JR. A chloride currentin mammalian cardiac myocytes: Novel mecha-nism for autonomic regulation of action potential
duration and resting membrane potential. J GenPhysiol 1990; 95:1077-1102.
34. Bean BP. Two kinds of calcium channels in canineatriai cells: Differences in kinetics, selectivity andpharmacology. J Gen Physiol 1985; 86:1-31.
35. Eisner DA, Lederer WT. Na-Ca exchange: Stoichi-ometry and electrogenicity. Am } Physiol 1985;248:C189-C2O2.
36. Wasserstrom }A, Salata ]]. Basis for tetrodotoxinand lidocaine effects on action potentials in dogventricular myocytes. Am } Physiol 1988; 254:H1157-H1166.
37. Yue DT, Marban E, A novel cardiac potassiumchannel that is active and conductive at depolar-ized potentials. Pflugers Arch 1988; 413:127-133.
38. Colatsky TJ, Follmer CH, Starmer CF. Channelspecificity in antiarrhythmic drug action. Mecha-nism of potassium channel block and its role insuppressing and aggravating cardiac arrhythmias.Circulation 1990; 82:2235-2242.
39. Sanguinetti MC, Jurkiewicz NK. Two componentsof cardiac delayed rectifier K ^ current, ] Gen Phys-iol 1990; 96:195-215.
40. Carmeliet E, Voltage- and time-dependent block ofthe delayed K * current in cardiac myocytes hy do-fetilide. J Pharmacol Exp Ther 1992;"262:809-817.
41. Giles WR. Imaizumi Y. Comparison of potassiumcurrents in rabbit atriai and ventricular cells. JPhysiol (Lond) 1988; 405:123-145,
42. Hume JR, Uehara A. Ionic basis of the different ac-tion potential configurations of single guinea-pigatriai and ventricular myocytes, J Physiol (Lond)1985; 368:525-544.
43. Heidbuchel H, Vereecke J, Carmeliet E. Three dif-ferent potassium channels in human atrium, CircRes 1990; 66:1277-1286.
44. Escande D, Coulombe A, Faivre J-F, Deroubaix E,Coraboeuf E, Two types of transient outward cur-rents in adult human atriai cells. Am ] Physiol1987; 252:H843-H850.
45. Irisawa H, Brown HF, Giles W. Cardiac pacemak-ing in the sinoatrial node. Physiol Rev 1993; 73:197-227.
46. Campbell DL, Rasmusson RL, Strauss HC. Ioniccurrent mechanisms generating vertebrate primarycardiac pacemaker activity at the single cell level:An integrative view. Ann Rev Physiol 1992; 54:279-302.
47. Hirst GDS, Edwards FR, Bramich NJ, et al. Neuralcontrol of cardiac pacemaker potentials. NIPS1991; 6:185-190.
48. Foster M, Dew-Smith AG, The effects of constantcurrent on the heart, J Anat Physiol 1876; 10:735-771,
49. Katzung B, Electrically induced automaticity inventricular myocardium. Lifn Sciences 1974; 14:1133-1140,
50. Roden DM, Hoffman B. Action potential prolonga-tion and induction of abnormal automaticity bylow quinidine concentrations in canine Purkinjefibers. Circ Res 1985; 56:857-867.
PACE, Vol. 18 August 1995 1573

WHALLEY, ET AL.
51. Starmer CF. Grant AO, Phasic ion cbannel block- 62.ado: A kinetic and parameter estimation proce-dure. Mol Pharmacol 1985; 28:348-356.
52. Wit AL. Rosen MR. Afterdepolarizations and trig-gered activity: Distinction from automaticity as an 63.arrhythmogenit: mechanism. In HA Fozzard. RBJennings. E Haber, et al. (eds.): The Heart and Car-diovascular System, New York, NY, Raven Press, 64.1992, pp, 2113-2163.
53. Arnsdorf MF, Arnsdorfs paradox. J CardiovascElectrophysiol 1990: 1:42-52. 65.
54. Arnsdorf MF, Basic: understanding of electrophysi-ological actions of antiarrhythmic drugs: Soiirces,sinks and matrices of information, Med Clin North 66,Am 1984; 68:1247-1280.
55. Kleber AG. Riegger CB, Electrical constants of arte-rially perfused rahbit papillary muscle. J Physiol(Lond) 1987; 385:307-324.
56. Weidmann S. Passive properties of cardiac fibers. 67.In MR Rosen, MJ Janse, AL Wit (eds.): Cardiac Elec-Irophysiology: A Textbook, Mount Kisco, NY, Fu-tura Publishing Co., Inc. 1990. pp, 29-35.
57. Fozzard HA, Schoenberg M, Strength-durationcurves in cardiac Purkinje fibres: Effects of liminal 68.length and charge distribution. J Physiol (Lond)1972; 266:593-618.
58. Dominguez G, Fozzard HA. Influence of extracellu-lar K + concentration on cable properties and nxcit- 69.ability of sheep cardiac Purkinje fihers. Circ Res1970;'26:565-574,
59. Fnzzard HA. The roles of membrane potential andinward Na" and Ca " currents in determiningconduction. In MR Rosen. MJ Janse, AL Wit (eds.): 70,Cardiac Electrophysiology: A Textbook. MountKisco, NY. Futura Publishing Co., Inc.. 1990, pp.415-425.
60. Peon J. Ferrier GR. Moe GK. The relationship ofexcitability to conduction velocity in canine Pur- 71.kinje tissue. Circ Res 1978; 43(l):125-135.
61. Fozzard HA. Conduction of the action potential. InRM Berne (ed,): Handbook of Physiology Section 72.2. The Cardiovascular System, V ol 1. Bethesda,MD, Am Physiol Soc, 1979. pp, 335-356.
Fozzard HA, Arnsdorf MF. Cardiac electrophysiol-ogy. In HA Fozzard, RB Jennings. E Haber. et al.(eds,): The Heart and Cardiovascular System. NewYork, NY, Raven Press, 1992, pp. 63-98.Spray DC, Bennett MVL, Physiology and Pharma-cology of gap junctions. Ann Rev Physiol 1985; 47:281-303,Burt JM, Block of intercellular communication: In-teraction of intracellular H " and Ca" *. Am J Phys-iol (Coll Physiol 22) 1987; 253:C607-C612.Veonstra RD, Physiological modulation of cardiacgap junction channels, J Cardiovasc Electrophysiol1991; 2:168-189,Gettes LS. Effects of ionic changes on impulsepropagation. In MR Rosen. MJ Janse, AL Wit (eds.):Cardiac Electrophysiology: A Texthook, MountKisco. NY. Futura Publishing Co,. Inc, 1990, pp,459-479.Gettes LS, Gascio WE. Effect of acute ischemia oncardiac electrophysiology. In HA Fozzard, EHaber, RB Jennings, et al, (eds,): The Heart andCardiovascular System. New York, NY, RavenPress, 1992, pp. 2021-2054.Kleber AG. Riegger CB. Janse MJ. Extracellular K +and H" shifts in early ischemia: Mechanisms andrelation to changes in impulse propagation, J MolCell Cardiol 1987; 19(Suppl, 5):35-44,Kleber AC. Riegger GB. Janse MJ. Electrical un-coupling and increase of extracellular resistanceafter induction of ischemia in isolated, arteriailyperfused rabbit papillary muscle. Circ Res 1987;61:271-279.Cascio WE, Yan G-X, Kleber AC. Passive electricalproperties, mechanical activity and extracellularpotassium in arteriaily perfused and ischemicrabbit ventricular muscle. Circ Res 1990; 66:1461-1473,Delmar M. Role of potassium currents on cell excit-iibility in cardiac ventricular myocytes. J Cardiov-asc Electrophysiol 1992; 3:474-486.Wit AL, Rosen MR. Cellular electrophysiology ofcardiac arrhythmias. Mod Concepts CardiovascDis 1981; 50:1-6.
1574 August 1995 PACE, Vol. 18





















