
Astrocyte-targeted expression of IL6 protects the CNSagainst a focal brain injury
19
Astrocyte-targeted expression of IL-6 protects the CNS against a focal brain injury Milena Penkowa, a,1 Mercedes Giralt, b,1 Natalia Lago, b Jordi Camats, b Javier Carrasco, b Joaquin Herna ´ndez, b Amalia Molinero, b Iain L. Campbell, c and Juan Hidalgo b, * a Department of Medical Anatomy, The Panum Institute, University of Copenhagen, DK-2200, Copenhagen, Denmark b Departamento de Biologı ´a Celular, de Fisiologı ´a y de Inmunologı ´a, Unidad de Fisiologı ´a Animal, Facultad de Ciencias, Universidad Auto ´noma de Barcelona, Bellaterra, Barcelona, Spain 08193 c Department of Neuropharmacology, The Scripps Research Institute, La Jolla, CA 92037, USA Received 23 April 2002; revised 14 November 2002; accepted 18 November 2002 Abstract The effect of CNS-targeted IL-6 gene expression has been thoroughly investigated in the otherwise nonperturbed brain but not following brain injury. Here we examined the impact of astrocyte-targeted IL-6 production in a traumatic brain injury (cryolesion) model using GFAP-IL6 transgenic mice. This study demonstrated that transgenic IL-6 production significantly increased wound healing following the cryolesion. Thus, at 20 days postlesion (dpl) the GFAP-IL6 mice showed almost complete wound healing compared to litter mate nontransgenic controls. It seems likely that a reduced inflammatory response in the long term could be responsible for this IL-6-related effect. Thus, while in the acute phase following cryolesion (1– 6 dpl) the recruitment of macrophages and T lymphocytes was higher in GFAP-IL6 mice, at 10 –20 dpl it was significantly reduced compared to controls. Reactive astrogliosis was also significantly increased up to but not including 20 dpl in the GFAP-IL6 mice. Oxidative stress as well as apoptotic cell death was significantly decreased throughout the time period studied in the GFAP-IL6 mice compared to controls. This could be linked to the altered inflammatory response as well as to the transgenic IL-6-induced increase of the antioxidant, neuroprotective proteins metallothionein-I II. These results indicate that although in the brain the chronic astrocyte-targeted expression of IL-6 spontaneously induces an inflammatory response causing significant damage, during an acute neuropathological insult such as following traumatic injury, a clear neuroprotective role is evident. © 2003 Elsevier Science (USA). All rights reserved. Keywords: Neurotrauma; Inflammation; Neuroprotection; Oxidative stress; Neurodegeneration; Apoptosis; Wound healing Introduction Injury to the CNS is followed by an inflammatory reac- tion, comprising reactive astrogliosis (Aschner 1996; Dong and Benveniste, 2001; Raivich et al., 1999), macrophage recruitment from peripheral monocytes and resident micro- glia (Popovich, 2000; Streit et al., 1999), and accumulation of lymphocytes (Hickey, 2001; Popovich et al., 1997; Raivich et al., 1999). During the CNS inflammatory response, both resident CNS cells and recruited leukocytes produce several cytokines, major histocompatibility complex (MHC), and ad- hesion/costimulatory molecules, which ultimately lead to gen- eration of reactive oxygen species (ROS) (Dong and Ben- veniste et al., 2001; Floyd, 1999a, 1999b; Hopkins and Rothwell, 1995; Martiney et al., 1998; Merrill and Benveniste, 1996; Merrill and Jonakait, 1995; Xiao and Link, 1998). ROS are highly toxic in the CNS and are key mediators of oxidative stress, which can induce neurodegeneration and cell death (Cassarino and Bennett, 1999; Floyd, 1999a, 1999b; Floyd et al., 1999; Sun and Chen, 1998). Interleukin-6 (IL-6) is a multifunctional cytokine, which is produced during the CNS inflammatory response (Ben- veniste, 1998; Gadient and Otten, 1997; Gruol and Nelson, 1997; Hirano et al., 1990; Munoz-Fernandez and Fresno, * Corresponding author. Fax: 34-93-581-23-90. E-mail address: [email protected] (J. Hidalgo). 1 Contributed equally to this article. R Available online at www.sciencedirect.com Experimental Neurology 181 (2003) 130 –148 www.elsevier.com/locate/yexnr 0014-4886/03/$ – see front matter © 2003 Elsevier Science (USA). All rights reserved. doi:10.1016/S0014-4886(02)00051-1
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Astrocyte-targeted expression of IL6 protects the CNSagainst a focal brain injury

Astrocyte-targeted expression of IL-6 protects the CNSagainst a focal brain injury
Milena Penkowa,a,1 Mercedes Giralt,b,1 Natalia Lago,b Jordi Camats,b Javier Carrasco,b
Joaquin Herna´ndez,b Amalia Molinero,b Iain L. Campbell,c and Juan Hidalgob,*a Department of Medical Anatomy, The Panum Institute, University of Copenhagen, DK-2200, Copenhagen, Denmark
b Departamento de Biologıa Celular, de Fisiologıa y de Inmunologıa, Unidad de Fisiologıa Animal, Facultad de Ciencias,Universidad Autonoma de Barcelona, Bellaterra, Barcelona, Spain 08193
c Department of Neuropharmacology, The Scripps Research Institute, La Jolla, CA 92037, USA
Received 23 April 2002; revised 14 November 2002; accepted 18 November 2002
Abstract
The effect of CNS-targeted IL-6 gene expression has been thoroughly investigated in the otherwise nonperturbed brain but not followingbrain injury. Here we examined the impact of astrocyte-targeted IL-6 production in a traumatic brain injury (cryolesion) model usingGFAP-IL6 transgenic mice. This study demonstrated that transgenic IL-6 production significantly increased wound healing following thecryolesion. Thus, at 20 days postlesion (dpl) the GFAP-IL6 mice showed almost complete wound healing compared to litter matenontransgenic controls. It seems likely that a reduced inflammatory response in the long term could be responsible for this IL-6-relatedeffect. Thus, while in the acute phase following cryolesion (1–6 dpl) the recruitment of macrophages and T lymphocytes was higher inGFAP-IL6 mice, at 10–20 dpl it was significantly reduced compared to controls. Reactive astrogliosis was also significantly increased upto but not including 20 dpl in the GFAP-IL6 mice. Oxidative stress as well as apoptotic cell death was significantly decreased throughoutthe time period studied in the GFAP-IL6 mice compared to controls. This could be linked to the altered inflammatory response as well asto the transgenic IL-6-induced increase of the antioxidant, neuroprotective proteins metallothionein-I� II. These results indicate thatalthough in the brain the chronic astrocyte-targeted expression of IL-6 spontaneously induces an inflammatory response causing significantdamage, during an acute neuropathological insult such as following traumatic injury, a clear neuroprotective role is evident.© 2003 Elsevier Science (USA). All rights reserved.
Keywords: Neurotrauma; Inflammation; Neuroprotection; Oxidative stress; Neurodegeneration; Apoptosis; Wound healing
Introduction
Injury to the CNS is followed by an inflammatory reac-tion, comprising reactive astrogliosis (Aschner 1996; Dongand Benveniste, 2001; Raivich et al., 1999), macrophagerecruitment from peripheral monocytes and resident micro-glia (Popovich, 2000; Streit et al., 1999), and accumulationof lymphocytes (Hickey, 2001; Popovich et al., 1997; Raivichet al., 1999). During the CNS inflammatory response, bothresident CNS cells and recruited leukocytes produce several
cytokines, major histocompatibility complex (MHC), and ad-hesion/costimulatory molecules, which ultimately lead to gen-eration of reactive oxygen species (ROS) (Dong and Ben-veniste et al., 2001; Floyd, 1999a, 1999b; Hopkins andRothwell, 1995; Martiney et al., 1998; Merrill and Benveniste,1996; Merrill and Jonakait, 1995; Xiao and Link, 1998). ROSare highly toxic in the CNS and are key mediators of oxidativestress, which can induce neurodegeneration and cell death(Cassarino and Bennett, 1999; Floyd, 1999a, 1999b; Floyd etal., 1999; Sun and Chen, 1998).
Interleukin-6 (IL-6) is a multifunctional cytokine, whichis produced during the CNS inflammatory response (Ben-veniste, 1998; Gadient and Otten, 1997; Gruol and Nelson,1997; Hirano et al., 1990; Munoz-Fernandez and Fresno,
* Corresponding author. Fax:�34-93-581-23-90.E-mail address: [email protected] (J. Hidalgo).1 Contributed equally to this article.
R
Available online at www.sciencedirect.com
Experimental Neurology 181 (2003) 130–148 www.elsevier.com/locate/yexnr
0014-4886/03/$ – see front matter © 2003 Elsevier Science (USA). All rights reserved.doi:10.1016/S0014-4886(02)00051-1

1998). IL-6 has major regulating effects upon the inflam-matory response (Benveniste, 1998; Brunello et al., 2000;Di Santo et al., 1996; Gadient and Otten, 1997; Munoz-Fernandez and Fresno, 1998; Raivich et al., 1999; VanWagoner and Benveniste, 1999). Accordingly, in IL-6-de-ficient mice (IL-6 knock-out (IL-6KO) mice) the inflamma-tory response is significantly reduced during pathologicalconditions of the brain (Eugster et al., 1998; Klein et al.,1997; Mendel et al., 1998; Penknowa et al., 1999, 2000;Raivich et al., 1999; Samoilova et al., 1998). On the otherhand, transgenic IL-6 overexpression in mice leads to neu-roinflammation and leads to significant brain damage (Brettet al., 1999; Brunello et al., 2000; Campbell, 1998a, 1998b;Campbell et al., 1993; Di Santo et al., 1996; Fattori et al.,1995). However, IL-6 belongs to the neuropoietin family ofcytokines (Hopkins and Rothwell, 1995), and has neurotro-phic effects related to the regulation of neuronal protection(Benveniste, 1998; Gruol and Nelson, 1997; Penkowa et al.,2000; Swartz et al., 2001; Van Wagoner and Benveniste,1999). Thus, IL-6 appears to exert both positive and nega-tive roles in the CNS. In order to understand the mecha-nisms underlying such paradoxical roles, we have studiedthe role of astrocyte-targeted expression of IL-6 in a modelof brain injury, the cryolesion of the cortex. To this end, wehave used the transgenic GFAP-IL6 mice, a well-knownanimal model of neurodegeneration (Brett et al., 1999;Brunello et al., 2000; Campbell, 1998a, 1998b; Campbell etal., 1993; Di Santo et al., 1996; Fattori et al., 1995). Theresults clearly demonstrated that although in the otherwiseunmanipulated brain transgenic expression of IL-6 is detri-mental, during an acute insult such as a cryolesion, IL-6may act as a neuroprotective factor.
Materials and methods
Animals
Construction and characterization of the GFAP-IL6transgenic mice was described previously (Campbell et al.,1993). Briefly, an expression vector derived from the mu-rine glial fibrillary acidic protein (GFAP) gene was used totarget expression of IL-6 to astrocytes. The GFAP-IL6 micewere originally bred and maintained for these experimentson the C57B6 � SJL hybrid genetic background. For thisparticular study, heterozygous GFAP-IL6 mice werecrossed with metallothionein-1&2 (Mt-1&2) knock-outmice, which have a 129/SvJ genetic background (Masters etal., 1994). While all the offspring are heterozygous for MT-I� II, they were genotyped for identifying the GFAP-IL6�/� and the GFAP-IL6 �/� animals as previously de-scribed (Penkowa et al., 2000). Both groups share the samegenetic background and thus were used for this study, wherewe refer to the GFAP-IL6 �/� animals as the “control”mice. Age and gender were matched for both groups.
Experimental procedure
Nine-month-old female mice were lesioned under tribro-methanol anesthesia. The skull over the right fronto-parietalcortex was exposed, and a focal cryoinjury was produced onthe surface of the brain by placing pellets of dry ice of aconsistent size onto the skull for 60 s. The animals werethen studied at different times (from 1 to 20 days postlesion(dpl)) This cryolesion method has been used extensively bythe authors for a number of years, producing very consistentand reproducible results (Hidalgo et al., 2001; Penkowa,2002).
Tissue processing
For histochemical and immunohistochemical studies, thefollowing procedure was used: the mice were deeply anes-thetized by intraperitoneal (ip) injection of 10 mg/100 gBrietal (Methohexital 10 mg/ml, Eli Lilly, DK) and wereperfused transcardially with 0.9% saline with 0.3% heparin(15000 IU/liter) for 1 min followed by Zamboni’s fixative(pH 7.4) for 8–10 min. Zamboni’s fixative consists of buff-ered 4% formaldehyde mixed (8.5/1.5, v/v) with a picricacid solution (saturated aqueous picric acid). Formaldehydewas prepared shortly before use by alkaline hydrolysis ofparaformaldehyde.
The brains were removed from the mice, dehydratedthrough graded alcohol, and embedded in paraffin, andserial 3-�m-thick sections were cut. Sections were rehy-drated, and for heat-induced antigen retrieval, boiled incitrate buffer, pH 9.1 or 6.0, in a microwave oven for 10 min(citrate buffer consists of 21,014 g citric acid and 29,410 gtrisodium citrate dihydrate dissolved in 2 liter H2O). Thesections were brought to room temperature and incubated in1.5% H2O2 in Tris-buffered saline (TBS)/Nonidet (TBS:0.05 M Tris, pH 7.4, 0.15 M NaCl; with 0.01% NonidetP-40) (Sigma–Aldrich, USA, Code N-6507) for 15 min atroom temperature to quench endogenous peroxidase. Toblock nonspecific binding, the sections were incubated with10% normal goat serum (In Vitro, DK, Code 04009-1B) or10% normal donkey serum (The Binding Site, UK, code BP005.1) in TBS/Nonidet for 30 min at room temperature.Sections prepared for incubation with monoclonal mouse-derived antibodies were in addition incubated with BlockingSolutions A � B from a HistoMouse-SP kit (Zymed Lab,Inc., USA, Code 95-9544) to quench endogenous mouseIgG.
Histochemistry
H&E staining of brain sections was performed accordingto standard procedures. Biotinylated tomato lectin fromLycopersicon esculentum (Sigma, USA, Code L9389) 1:500was used as a marker for cells of the myelomonocytic celllineages, such as microglia/macrophages, as well as amarker for blood vessels. The lectin was developed for 30
131M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

min at room temperature using streptavidin–biotin–peroxi-dase complex (StreptABComplex/HRP, Dakopatts, DK,Code K377) prepared according to the manufacturer’s rec-ommended dilutions. The reaction product was visualizedusing 0.015% H2O2 in 3,3�-diaminobenzidine/Tris-bufferedsaline (DAB/TBS), with DAB as a chromogen.
Immunohistochemistry
The sections were incubated overnight at 4°C with ratanti-mouse IL-6 1:50 (Harlan Seralab, USA, CodeMAS584); rat anti-mouse F4/80 1:15 (Serotec, UK, CodeMCA 497) (as a marker for activated microglia/macro-phages/monocytes); mouse anti-rat CD3 1:50 (Serotec, UK,Code KD MCA 772) (as a marker for all T lymphocytes);rabbit anti-bovine GFAP 1:250 (Dakopatts, DK, Code Z334) (as a marker for astrocytes); rabbit anti-rat MT-1 � 21:500 (Gasull et al., 1993; Penkowa et al., 1999a, 1999b);mouse anti-human Cu/Zn-superoxide dismutase (Cu/Zn–SOD) 1:50 (Sigma-Aldrich, USA, Code S2147); sheep anti-bovine Mn-SOD 1:50 (Biogenesis, UK, Code 8474-9550);sheep anti-human catalase 1:50 (The Binding Site, UK,Code PC136); rabbit anti-human inducible nitric-oxide syn-thase (iNOS) 1:100 (Alexis Biochemicals, USA, Code 210-503-R050) (as a marker for oxidative stress); rabbit anti-NITT 1:100 (Alpha Diagnostic Int., USA, Code NITT12-A) (a marker for peroxynitrite-induced nitration of ty-rosine residues, which monitors oxidative stress); rabbitanti-MDA 1:100 (Alpha Diagnostic Int., USA, Code M0DA11-S) (a marker for malondialdehyde (MDA) produced as abyproduct of fatty acid peroxidation, which monitors oxi-dative stress); mouse anti-calf ssDNA 1:25 (Alexis Bio-chemicals, USA, Code 804-192-R200) (as a marker forsingle stranded DNA in apoptotic cells); rabbit anti-humanactivated caspase-3 1:50 (Cell Signaling Technology Inc.,USA, Code 9661) (as a marker for apoptotic signaling).
The primary antibodies were detected using biotinylatedgoat anti-mouse IgG 1:200 (Sigma–Aldrich, USA, CodeB8774), biotinylated goat anti-mouse IgM (� chain spe-cific) 1:10 (Jackson ImmunoRes. Lab, Inc., Code 115-065-020), biotinylated goat anti-rat IgG 1:1500 (Amersham,UK, Code 1005), biotinylated mouse anti-rabbit IgG 1:400(Sigma–Aldrich, USA, Code B3275), or biotinylated don-key anti-sheep/goat IgG 1:20 (Amersham, UK Code RPN1025) for 30 min at room temperature followed by strepta-vidin–biotin–peroxidase complex (StreptABComplex/HRP,Dakopatts, DK, Code K377) prepared according to the man-ufacturer’s recommended dilutions. Sections were then in-cubated with biotinylated tyramide and streptavidin–peroxi-dase complex (NEN, Life Science Products, USA, CodeNEL700A) prepared according to the manufacturer’s rec-ommendations. Finally, the immunoreaction was visualizedusing 0.015% H2O2 in 3,3-diaminobenzidine-tetrahydro-chloride (DAB)/TBS for 10 min at room temperature.
To evaluate the extent of nonspecific binding of theprimary antibodies used in the immunohistochemical exper-
iments, the primary antibody step was omitted. A furthercontrol for some anti-CD and anti-cytokine antibodies wasto preabsorb the primary antibodies with their correspond-ing antigenic proteins before the immunohistochemical re-action was carried out. For this purpose the following block-ing peptides were used: mouse CD3 peptide (Santa CruzBiotech, Inc., USA, Code sc-1127P); human IL-1� peptide(RD Systems, UK, Code 201-LB-005); and mouse TNF-�peptide (RD Systems, UK, Code 410-MT-010). Resultswere considered only if these controls were negative.
In situ detection of DNA fragmentation
Terminal deoxynucleotidyl transferase (TdT)-mediateddeoxyuridine triphosphate (dUTP)-digoxigenin nick-end-la-beling (TUNEL) staining was performed as previously de-scribed (Penkowa et al., 1999a, 2000).
Fluorescence histochemistry
To identify cells undergoing oxidative stress, sectionswere incubated overnight at 4°C with either mouse anti-human NF 1:250 (Dakopatts, DK, Code M762) (as a neu-ronal marker) or mouse anti-porcine vimentin 1:50 (Dako-patts, DK, Code M0725) (as a marker for both reactiveastrocytes and macrophages) or mouse anti-rat CD3 (asabove) and simultaneously with either rabbit anti-humaniNOS or rabbit anti-NITT or rabbit anti-MDA (all as above).The monoclonal antibodies were detected by using goatanti-mouse IgG linked with Texas Red (TXRD) 1:50(Southern Biotechnology, USA, Code 1030-07) or goat antimouse IgM linked with TXRD 1:30 (Jackson Immuno-Research Lab, Inc., Code 115-075-020), while the poly-clonal antibodies were detected by using goat anti-rabbitIgG linked with aminomethylcoumarin (AMCA) 1:20(Jackson ImmunoResearch Lab, Inc., Code 111-155-144).
To identify cells undergoing apoptosis, triple stainingsfor TUNEL and cellular markers were performed. Hence,sections were incubated with TUNEL linked with fluores-cein (FITC) (Oncor, USA, Code S7111-KIT) according tothe manufacturer’s protocol, and afterward incubated over-night at 4°C with rabbit anti-human NSE 1:1000 (Calbio-chem, USA, Code D05059) (as a neuronal marker) andmouse anti-porcine vimentin (as above) simultaneously.Other sections were incubated with FITC-linked TUNEL(as above) and afterward incubated overnight at 4°C withmouse anti-rat CD3 (as above). The monoclonal antibodieswere detected by using goat anti-mouse IgG linked withAMCA 1:20 (Jackson ImmunoResearch Lab, Inc., Code115-155-146) or goat anti-mouse IgM linked with AMCA1:20 (Jackson ImmunoResearch Lab, Inc., Code 115-155-020). The polyclonal antibody was detected by using goatanti-rabbit IgG linked with TXRD 1:30 (Jackson Immuno-Research Lab, Inc., Code 111-075-144).
Triple labeling of TUNEL-positive cells and apoptoticsignaling factor caspase-3 and cytoplamic cytochrome c
132 M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

was also performed in order to confirm apoptotic cell death.Sections were incubated with FITC-linked TUNEL (asabove), and afterward incubated overnight at 4°C with rab-bit anti-human activated caspase-3 (as above) and goatanti-horse cytochrome c 1:50 (Santa Cruz Biotech. Inc.,USA, Code sc-7159) (cytochrome c appears in the cyto-plasm during apoptosis). The anti-caspase-3 antibodieswere detected by using TXRD-linked donkey anti-rabbitIgG 1:30 (Jackson ImmunoResearch Lab, Inc., Code 711-
075-152), while anti-cytochrome c antibodies were detectedby using AMCA-linked donkey anti-goat IgG 1:30 (JacksonImmunoResearch Lab, Inc., Code 705-155-147).
To study apoptotic cell death in relation to oxidativestress, other sections were incubated with FITC-linkedTUNEL (as above) and afterward incubated overnight at4°C with rabbit anti-NITT (as above). The anti-NITT anti-bodies were detected by using AMCA-linked goat anti-rabbit IgG (as above).
To study apoptotic cell death in relation to MT-I � IIexpression, other sections were incubated with FITC-linkedTUNEL (as above) and afterward incubated overnight at4°C with mouse anti-horse MT-1 � 2 1:20 (Dakopatts, DK,
Fig. 1. Quantification of positively stained cells in unlesioned and lesionedcortex from control and GFAP-IL6 mice. For all specimens analyzed, cellswere counted in three sections per animal (three mice per group) frommatched areas at the border zone of the deepest part of the lesion in ablinded manner, a mean value for each animal is calculated and used foroverall quantitations. Fig. 2 shows an example of what is considered apositively stained cell. The main morphological and histopathologicalfeatures are described in detail in the following figures and under Results.Results are mean � SEM. Two-way ANOVA with strain and cryolesion asmain factors indicated that both affected significantly (P � 0.001) all thevariables analyzed, while the interaction between strain and cryolesion wassignificant only in the following variables: IL-6, P � 0.025; CD-3, lectinround and GFAP, P � 0.001; iNOS and NITT, P � 0.05; MDA, P �0.025; TUNEL, P � 0.005. A statistically significant interaction in thetwo-way ANOVA reveals that the GFAP-IL6 mice respond differentlycompared to control mice.
Fig. 2. Representative identification of positively stained cells. Fig. 1shows cell counts of positively stained cells in the border of the lesion. Weherewith provide a representative identification of GFAP-positive cells. (A)Low-power photo showing GFAP staining in control mice at 10 dpl (thesame shown in the Fig. 7I). The framed area at the border of the lesionindicates a 0.5-mm2 area where positive cells were counted from (B)High-power magnification of the framed area in (A) indicating some of thepositively stained cells (arrows) that were counted; open arrows indicatenegatively stained cells. For this particular section, the number of cellsconsidered positively stained was 63. The procedure requires considerabletraining and it is imperative that it is carried out in a blinded manner by thesame investigator. Scale bars: (A) 288 �m. (B) 56 �m.
133M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

Code 0638). Anti-MT-I � II antibodies were detected byusing TXRD-linked goat anti-mouse IgG (as above).
All the secondary fluorophore-linked antibodies wereused for 30 min at room temperature. The sections wereembedded in 20 �l fluorescent mounting (Dakopatts, DK,Code S3023) and kept in darkness at 4°C. In order toevaluate the extent of nonspecific binding of the primaryantibody in the fluorescence stainings, control sections wereincubated in the absence of primary antibody. Results wereconsidered only if these controls were negative. For thesimultaneous examination and recording of the triple stains,a Zeiss Axioplan2 light microscope equipped with a triple-band (FITC/TXRD/AMCA) filter was used.
Cell counts
For statistical evaluation of the results, positively stainedcells (defined as those cells with cytoplasmic staining or incase of TUNEL, cells with nuclear staining) were counted
from matched 0.5-mm2 areas of 3-�m-thick sections ofcortex of unlesioned mice as well as from the lesionedhemispheres (ipsilateral cortex side) for statistical evalua-tion of the results; when counting from the lesioned hemi-spheres, matched areas were chosen at the border zone ofthe deepest part of the lesion in all the mice (see Figs. 1 and2). Cell counts were performed in three mice per group in ablinded manner by a single investigator.
Statistical analysis
Results were analyzed with two-way analysis of variance(ANOVA), with strain and freeze lesion as main factors.When the interaction between both factors was significant,it was interpreted to be the consequence of a specific effectof the transgenic IL-6 expression during the lesion. Post hocanalysis has not been carried out since only two strains werecompared, and thus post hoc comparison would be justredundant of the information provided by the two-way
Fig. 3. H&E staining of brain cortex removed at various times following cryolesion. In control mice, the lesion is evident at 3 dpl (A), 6 dpl (C), and 20 dpl(E). In the GFAP-IL6 mice, the lesion is comparable to that of control mice at 3 dpl (B) and 6 dpl (D), while it is reduced dramatically at 20 dpl (F). Increasedcellularity is observed in the lesioned GFAP-IL6 mice throughout the time period studied. Also, by 20 dpl, the brain tissue of GFAP-IL6 mice is populatedwith many small and few large vessels, which were not observed to the same degree in control mice by 20 dpl. Scale bars: (A,B) 120 �m; (C,D) 90 �m;(E,F) 55 �m.
134 M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

ANOVA. Results were considered statistically significantwith a P value �0.05.
Results
As described previously (Campbell, 1998a, 1998b;Campbell et al., 1993) the GFAP-IL6 mice developedhunched posture, tremor, ataxia, and hindlimb weaknesswith aging. Also, spontaneously developing histopatholog-ical changes described for the GFAP-IL6 mice were ob-served, namely astrogliosis, microgliosis, and neurodegen-eration, which were very prominent in the cerebellum. Thus,these results will not be described in detail in this article.Following are the specific results obtained in cryolesionedcontrol, nontransgenic, and GFAP-IL6 mice in comparisonto unlesioned mice.
Morphology
H&E staining of the cortex of unlesioned mice showedno significant differences between normal and GFAP-IL6mice. However, after the cortical lesioning, the GFAP-IL6mice had increased numbers of infiltrating cells at the lesionsite at 1–6 dpl when compared to lesioned control mice(Figs. 3A–D). At 10 dpl, the GFAP-IL6 mice showed de-creased numbers of infiltrating cells and an increased brain
tissue repair relative to lesioned control mice; by 20 dpl, thetissue had largely healed in GFAP-IL6 mice, while a corti-cal lesion was still seen in lesioned control mice (Figs. 3Eand F). Throughout the time period studied, GFAP-IL6mice showed increased cellularity at the lesion site relativeto normal mice (Fig. 3). Also, by 20 dpl, the brain tissue ofGFAP-IL6 mice was populated with many small and somelarger blood vessels, which were not observed to the samedegree in normal mice (Figs. 3E and F).
IL-6
IL-6 staining was generally increased in the GFAP-IL6mice compared to normal mice throughout the brain (Fig.4). The cryolesion upregulated IL-6 expression in the areassurrounding the lesion in both control and GFAP-IL6 mice,and this upregulation was higher in the GFAP-IL6 mice(Figs. 1 and 4). IL-6 was primarily detected in macrophages,activated microglia, T lymphocytes, and reactive astrocytes.
Microglia/macrophages
As expected, unlesioned GFAP-IL6 mice showed a sig-nificant microgliosis throughout the forebrain comparedwith control mice, with microglial cells showing a bushyand activated appearance, while round or amoeboid macro-phages were hardly detected (Figs. 5A and B).
Fig. 4. IL-6 immunoreactivity in unlesioned and lesioned wild-type control and GFAP-IL6 mice. (A) In unlesioned brain of control mice, only a few cellsin the cortex show weak IL-6 staining. (B) In GFAP-IL6 mice, the number of IL-6-positive cells in the cortex is significantly increased relative to controls.(C) After cryolesion, in control mice an increased number of IL-6-expressing cells is present at the lesion site. (D) In the cryolesioned GFAP-IL6 mice, theIL-6 expression is significantly increased in astrocytes, macrophages/microglia, and T cells relative to lesioned control mice. Scale bars: (A–D) 38 �m.
135M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

136 M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

After the cryolesion, all mice showed activation of mi-croglia and recruitment of macrophages (Figs. 1 and 5).Accordingly, the control mice showed round or amoeboidmacrophages at the lesion site from 1 to 20 dpl, with a peak
at 3–10 dpl (Figs. 1 and 5). However, even at 20 dpl, thelesion site was heavily infiltrated by round or amoeboidmacrophages (Figs. 5I and L). In the GFAP-IL6 mice, adifferent pattern and highly significant (P � 0.001) response
Fig. 6. CD3 immunostaining of cryolesioned cortex from wild type control and GFAP-IL6 mice. (A) Some CD3� T cells were seen at the lesion site (asterisk)of control mice at 3 dpl. (B) Numerous T cells were recruited to the lesion site (asterisk) of GFAP-IL6 mice at 3 dpl. (C) At 20 dpl in control mice, the numberof T cells recruited to the lesion site (asterisk) was increased when compared to that observed at 3 dpl. (D) The GFAP-IL6 mice showed evidence ofregenerated tissue at 20 dpl, and the number of T cells had decreased significantly relative to 3 dpl. (E) Higher magnification of (A) showing T cells in cortexof control mice. (F) Higher magnification of (B) showing T cells in cortex of GFAP-IL6 mice. Scale bars: (A,B) 150 �m; (C,D) 100 �m; (E,F) 18 �m.
Fig. 5. Lectin staining of unlesioned and lesioned wild-type control and GFAP-IL6 mice. (A) Lectin staining in the unlesioned brain of control mice. (B) Inthe brain of unlesioned GFAP-IL6 mice, the number of lectin� microglial cells is increased compared to that of control mice. (C) Following cryolesion, manyround or amoeboid macrophages are recruited to the lesion site of control mice at 3 dpl. (D) At 3 dpl, the GFAP-IL6 mice showed increased numbers oflectin�, round, or amoeboid macrophages relative to control mice. (E,F) At 6 dpl, the number of macrophages was increased in all the mice. However, thenumber of macrophages was higher in the GFAP-IL6 mice (F) when compared to that of control mice (E). (G) The control mice still showed high numbersof lectin�, round, or amoeboid macrophages at 10 dpl. (H) At 10 dpl, the GFAP-IL6 mice had significantly decreased numbers of macrophages at the lesionsite. Moreover, the lesion was decreased significantly in size and the tissue showed evidence of repair. (I) At 20 dpl, control mice showed a significant lesionassociated with the presence of several round or amoeboid macrophages. (J) In contrast, the GFAP-IL6 mice at 20 dpl showed evidence of repaired tissuewhich had only small numbers of macrophages. (K) Higher magnification of the framed area in (C) showing macrophages in control mice at 3 dpl. (L) Highermagnification of the framed area in (I) showing that round or amoeboid macrophages were still evident at 20 dpl in control mice. (M) Higher magnificationof the framed area in (D) showing macrophages in the GFAP-IL6 mice at 3 dpl. (N) Higher magnification of the framed area in (J) showing the regeneratedcortex from GFAP-IL6 mice at 20 dpl, with ramified or bushy microglial cells rather than round or amoeboid macrophages. Scale bars: (A,B) 81 �m; (C–H)182 �m; (I,J) 120 �m; (K–M) 16 �m; (N) 24 �m.
137M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

138 M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

was observed, with an increased number of macrophages atthe lesion site at 1–6 dpl (Figs. 5C–F) but a decreasednumber of these cells at 10–20 dpl (Figs. 5H and J; see alsoFig. 1). In addition, at 10–20 dpl round or amoeboid mac-rophages at the lesion site predominated in the control micewhile in GFAP-IL6 mice these cells were dramatically de-creased in number and had a more ramified appearancetypical of microglia (Figs. 5M and N). In summary, thesefindings revealed that the number of activated microglia/macrophages after the lesion was inversely related to thedegree of brain tissue repair.
Lymphocytes
Few if any, CD3� T lymphocytes were detected in theunlesioned brains (Fig. 1). After the lesion, control miceshowed many CD3� T cells infiltrating the lesion by 1–6dpl (Fig. 6A), and by 10–20 dpl the number of T cellsfurther increased at the lesion site in these mice (Figs. 1 and6C). In the lesioned GFAP-IL6 mice, the number of CD3�T cells recruited to the lesion site was significantly in-creased at 3–6 dpl relative to normal mice, while at 10–20dpl, the GFAP-IL6 mice showed fewer CD3� cells in therepairing brain tissue (Figs. 1 and 5).
Astrocytes
As expected, in the hippocampal area and less pro-nounced in the cortex of unlesioned mice, the number ofGFAP� astrocytes appeared increased in GFAP-IL6 micerelative to littermate controls (Fig. 2 and Figs. 7A and B).
After the lesion, all the mice showed reactive astrogliosisthroughout the experimental period studied (Figs. 1 and 7).However, the reactive astrogliosis, which peaked at 3 dplwas increased at all times except at 20 dpl in the GFAP-IL6mice relative to control mice (Figs. 1 and 7). At 20 dpl, thecontrol mice still showed a considerable lesion cavity,which was surrounded and infiltrated by reactive astrocytes(Fig. 7K). Because the GFAP-IL6 mice showed a fasterwound healing capacity, at 20 dpl the GFAP immunoreac-tivity looked comparable to that of cortex of unlesionedGFAP-IL6 mice (Fig. 1, 2 and Figs. 7K and L).
Expression of antioxidants
Expression of antioxidant factors was examined by usingimmunoreactivity for metallothionein-I � II (MT-I � II),Cu/Zn-SOD, Mn-SOD, and catalase. In the cortex of unle-sioned mice, the number of MT-I � II-expressing cells wasincreased in GFAP-IL6 mice relative to control mice (Fig.1). However, the expression of Cu/Zn-SOD, Mn-SOD, andcatalase was comparable in both groups of unlesioned mice(not shown).
After the lesion, MT-I � II expression increased signif-icantly in all mice at the lesion site, peaking at 3–6 dpl(Figs. 1 and 8). The number of MT-I � II-expressing cellswas significantly increased in the GFAP-IL6 mice at alltime points examined when compared to that of controlmice. Interestingly, in the GFAP-IL6 mice, the increasedMT-I � II levels were seen during the acute inflammatoryresponse as well as during the tissue repair phase. The cellsproducing MT-I � II were mainly astrocytes, with somemicroglia/macrophages also being positive. After the lesionall the mice showed increased expression of Cu/Zn-SOD,Mn-SOD, and catalase, with the levels of these antioxidantfactors being comparable in the control and GFAP-IL6 mice(Cu/Zn-SOD expression is shown in Figs. 8I and J).
Oxidative stress
Oxidative stress was assessed using immunoreactivityfor iNOS, NITT, and MDA. In the unlesioned brains of allthe mice, only a very few cells showed immunoreactivityfor iNOS, NITT, and MDA (Fig. 1). Following the lesion,oxidative stress levels increased significantly in all the mice.Thus, in control mice several cells at the lesion site showediNOS, NITT, and MDA immunostaining at 1–20 dpl, whichpeaked at 3–10 dpl (Figs. 1 and 9). As judged by doubleimmunofluorescence, the cells suffering oxidative stresswere mainly neurons and astrocytes, while some macro-phages and microglial cells also showed iNOS, NITT, andMDA immunoreactivity (Fig. 10A). Interestingly, in theGFAP-IL6 mice oxidative stress following the cryolesionwas significantly reduced relative to normal mice (Figs. 1and 9). Thus, only a few neurons and glial cells wereimmunostained for iNOS, NITT, or MDA (Fig. 10B).
Fig. 7. GFAP immunostaining of unlesioned and lesioned cortex from wild-type control and GFAP-IL6 mice. (A) GFAP� astrocytes in hippocampus ofunlesioned normal mice. (B) In the unlesioned GFAP-IL6 mice, the number of GFAP� astrocytes was increased slightly in the hippocampus. (C) Afterlesioning, control mice showed a significant reactive astrogliosis around the lesion site at 3 dpl. (D) Reactive astrogliosis was increased in the lesionedGFAP-IL6 mice at 3 dpl relative to control mice. (E) Higher magnification of the framed area in (C) showing reactive astrocytes in control mice at 3 dpl.(F) Higher magnification of the framed area in (D) showing increased astrogliosis in GFAP-IL6 mice relative to normal mice. (G) At 6 dpl, there was stillsignificant reactive astrogliosis at the lesion site in control mice. (H) The lesioned GFAP-IL6 mice also showed a significantly increased astrogliosis at 6 dplcompared to that of control mice. (I) At 10 dpl, the control mice still showed a lesion cavity surrounded by reactive astrocytes. (J) By 10 dpl, the GFAP-IL6mice still had many reactive astrocytes, but the number of cells was decreased considerably relative to 6 dpl. Furthermore, the size of the lesion had decreasedsignificantly with tissue repair evident in the GFAP-IL6 mice compared to control mice. (K) A substantial lesion is still seen in control mice at 20 dpl;however, the lesion was reduced when compared to that seen at 3–10 dpl. (L) In GFAP-IL6 mice at 20 dpl, the brain cortex had largely repaired and the tissueappeared almost similar to that of unlesioned mice. Scale bars: (A,B) 70 �m; (C,D,G–J) 224 �m; (E,F) 40 �m; (K,L) 100 �m.
139M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

140 M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

Apoptotic cell death
Apoptotic cell death was examined by TUNEL and im-munoreactivity for ssDNA and activated caspase-3. Addi-tionally, the morphologic criteria for apoptosis was evalu-ated. In all the unlesioned mice, apoptotic cells were rarelydetected in the cortex (Fig. 1). After the cryolesion all themice showed significant increases in the number of apopto-tic cells. However, GFAP-IL6 mice showed a significantly(P � 0.001) reduced number of TUNEL� apoptotic cellswhen compared to that of controls (Figs. 1 and 11). Theseresults obtained with TUNEL were consistently confirmedby immunostaining for ssDNA and/or activated caspase-3(data not shown). To further characterize the apoptotic celldeath, triple immunofluorescence for TUNEL, cytochromec, and activated caspase-3 was performed. This showed thatthe TUNEL� cells also contained activated caspase-3 andcytoplasmic cytochrome c (Figs. 10E and F). Moreover, thecells undergoing apoptotic cell death were mainly neuronsand some astrocytes (Figs. 10C and D), while macrophagesand T cells were negative.
In addition, in order to study apoptotic cell death inrelation to oxidative stress, sections of all the mice werestained for TUNEL and NITT simultaneously. This analysisrevealed that apoptotic cells were generally in a state ofoxidative stress (Fig. 10G). Additionally, the relationshipbetween apoptosis and MT-I � II expression was studied bycostaining sections of all the mice for TUNEL and MT-I �II. The results showed that after the lesion, MT-I � II-expressing cells seem to be protected against apoptosissince in general they were TUNEL negative (Fig. 10H).
Discussion
The GFAP-IL6 mice are a well-characterized animalmodel of cytokine-induced neurodegeneration (Brett et al.,1999; Brunello et al., 2000; Campbell, 1998a, 1998b;Campbell et al., 1993; DiSanto et al., 1996; Fattori et al.,1995). Previous studies also suggest that IL-6 is a proin-flammatory cytokine in the brain that may cause significantdamage (Brett et al., 1999; Brunello et al., 2000; Campbell,1998a, 1998b; Campbell et al., 1993; DiSanto et al., 1996;Fattori et al., 1995; Gijbels et al., 1995). However, IL-6 isalso considered an important neuropoietin with neuropro-tective actions (Benveniste, 1998; Gruol and Nelson, 1997;Hirano et al., 1990; Hirota et al., 1996; Hopkins and Roth-
well, 1995; Maeda et al., 1994; Murphy et al., 1999; Pen-kowa et al., 2000; Swartz et al., 2001; Toulmond et al.,1992; Umegaki et al., 1996; Van Wagoner and Benveniste,1999). These apparent discrepancies could be related to IL-6having different roles depending on the physiological con-text. Our study here demonstrated that transgenic produc-tion of IL-6 by astrocytes during neuropathological condi-tions such as traumatic CNS injury is clearly beneficial forthe brain. Thus, wound healing and tissue repair followinga cryolesion were significantly increased in the GFAP-IL6mice relative to littermate controls. The results suggest thata significant reduction of oxidative stress and cell death inthe lesioned area could be involved in such acceleratedrecovery process. This is in agreement with studies carriedout in IL-6KO mice subjected to the same cortical freezelesion used here, which showed increased oxidative stress,decreased cell survival, and delayed wound repair (Pen-kowa et al., 1999b, 2000). Finally, other processes includingincreased revascularization of the injury site cannot be ex-cluded and may also contribute to the increased tissue repairin the GFAP-IL6 mice. In support of this, it has beenreported that GFAP-IL6 mice with a brain injury haveincreased posttraumatic revascularization of the damagedarea in comparison with control littermates (Swartz et al.,2001).
The mechanisms underlying these protective effects ofIL-6 in the cryolesion model are likely complex. We haveexamined the inflammatory response by studying macro-phage and T-cell infiltration of the damaged area. The re-sults clearly showed a different response in the GFAP-IL6mice compared to controls. Thus, in the early phase of theinjury (up to 6 dpl), the transgenic expression of IL-6 clearlypotentiated macrophage and T-cell infiltration, which likelyreflects the proinflammatory roles of IL-6 (Brett et al., 1999;Brunello et al., 2000; Campbell, 1998a, 1998b; Campbell etal., 1993; Di Santo et al., 1996; Fattori et al., 1995; Klein etal., 1997; Mendel et al., 1998; Penkowa et al., 1999b, 2000).Increased cellular infiltration after brain injury in the GFAP-IL6 mice has been reported independently (Swartz et al.,2001). Furthermore, the inflammatory response of macro-phages was clearly reduced in IL-6KO mice during differentpathological conditions of the CNS (Eugster et al., 1998;Klein et al., 1997; Mendel et al., 1998; Penkowa et al.,1999b; Raivich et al., 1999; Samoilova et al., 1998), clearlysupporting a role of IL-6 in the recruitment and activation ofmacrophages following brain injury. The number of mac-rophages and T cells returned to normal faster in the GFAP-
Fig. 8. Immunostaining for the antioxidant factors MT-I � II and Cu/Zn–SOD in lesioned cortex from control and GFAP-IL6 mice. In the control mice,expression of MT-I � II was increased in reactive astrocytes and macrophages/microglia located at the lesion site (asterisk) at 3 dpl (A), 6 dpl (C), 10 dpl(E), and 20 dpl (G). Also in the GFAP-IL6 mice, the levels of MT-I � II were significantly increased at the lesion site (asterisk) at 3 dpl (B), 6 dpl (D), 10dpl (F), and 20 dpl (H). However, the number of MT-I � II-expressing cells were, at all time points, higher in GFAP-IL6 mice than in control mice. By 3dpl, expression of the antioxidant Cu/Zn–SOD was increased at the lesion site of both control (I) and GFAP-IL6 (J) mice. However, no significant differencesin Cu/Zn–SOD levels were observed between normal and GFAP-IL6 mice. Scale bars: (A–H) 47 �m; (I,J) 35 �m.
141M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148
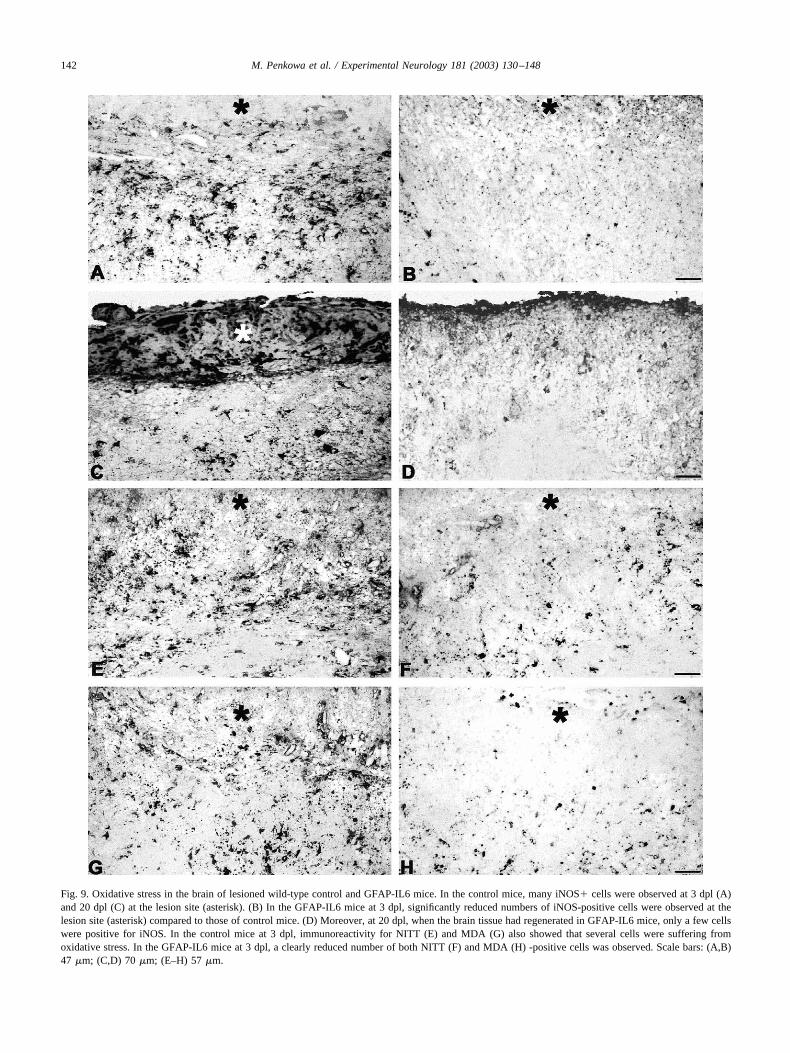
Fig. 9. Oxidative stress in the brain of lesioned wild-type control and GFAP-IL6 mice. In the control mice, many iNOS� cells were observed at 3 dpl (A)and 20 dpl (C) at the lesion site (asterisk). (B) In the GFAP-IL6 mice at 3 dpl, significantly reduced numbers of iNOS-positive cells were observed at thelesion site (asterisk) compared to those of control mice. (D) Moreover, at 20 dpl, when the brain tissue had regenerated in GFAP-IL6 mice, only a few cellswere positive for iNOS. In the control mice at 3 dpl, immunoreactivity for NITT (E) and MDA (G) also showed that several cells were suffering fromoxidative stress. In the GFAP-IL6 mice at 3 dpl, a clearly reduced number of both NITT (F) and MDA (H) -positive cells was observed. Scale bars: (A,B)47 �m; (C,D) 70 �m; (E–H) 57 �m.
142 M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

IL6 mice, in parallel with the observed accelerated tissuerepair and wound healing, suggesting a neuroprotective roleof IL-6. The latter is also supported by the dramatic reduc-tion of the number of apoptotic cells observed in the le-sioned GFAP-IL6 mice compared to littermate controls. A
positive role of IL-6 on cell survival has also been suggestedin previous reports (Bensadoun et al., 2001; Hirano et al.,1990; Hirota et al., 1996; Maeda et al., 1994; Murphy et al.,1999; Swartz et al., 2001; Toulmond et al., 1992; Umegakiet al., 1996).
Fig. 10. Cellular localization of oxidative stress markers and apoptosis in cryolesioned cortex from wild-type control and GFAP-IL6 mice. All photomi-crographs were taken in the border of the lesion. (A,B) Costaining for the oxidative stress marker NITT (blue) and NF -positive neurons (red). (A) In controlmice at 3 dpl, many NITT-positive cells were seen that included neurons (pink). (B) In the GFAP-IL6 mice at 3 dpl, the total number of NITT-positive cellswas reduced, as was the number of NITT-positive neurons. (C,D) Fluorescence staining for TUNEL-positive (green), NSE-positive neurons (red), andvimentin-positive astrocytes and macrophages (blue). (C) Many NSE-positive neurons and some vimentin-positive cells were TUNEL-positive in controlmice at 3 dpl. (D) Only a few NSE-positive neurons and vimentin-positive cells were TUNEL-positive in GFAP-IL6 mice at 3 dpl. (E,F) Triple labeling ofTUNEL (green), activated caspase-3 (red), and cytochrome c (blue) in control (E) and GFAP-IL6 (F) mice at 3 dpl. As shown, the TUNEL-positive cellsalso contained activated caspase-3 and cytoplasmic cytochrome c. (G) Dual staining analysis for TUNEL (green) and the oxidative stress marker NITT (blue)in control mice at 3 dpl, indicating that the apoptotic cells were suffering oxidative stress. (H) Dual staining analysis for TUNEL (green) and MT-I � II (red)indicated that MT-I � II-expressing cells were not apoptotic. Scale bars: (A,B,E,F) 22 �m; (C,D) 29 �m; (G,H) 32 �m.
143M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

It is essential to realize that oxidative stress was signif-icantly decreased in the GFAP-IL6 mice relative to controls,since this could be causatively linked to the findings ob-served for tissue repair as well as for cellular survival(Benveniste, 1998; Cassarino and Bennett, 1999; Floyd,1999a, 1999b; Floyd et al., 1999; Maedu et al., 1994; Mar-
tin, 2001; Sun and Chen, 1998). Thus, for understanding theneuroprotective roles of IL-6 during brain injury it seemsimportant to know all the proteins upregulated by this cy-tokine, particularly the antioxidant proteins. From a numberof antioxidant proteins examined in this study, only MT-I �II differed in the GFAP-IL6 mice compared to littermate
Fig. 11. Analysis of TUNEL-positive apoptotic cells in unlesioned and lesioned cortex from wild-type control and GFAP-IL6 mice. Control mice shownumerous TUNEL-positive apoptotic cells at the lesion site at 3 dpl (A), 10 dpl (C), and 20 dpl (E). In contrast, the GFAP-IL6 mice showed significantlyreduced numbers of TUNEL-positive cells at the lesion site at 3 dpl (B), 10 dpl (D); and 20 dpl (F). (G) Higher magnification of (C) showing numerousTUNEL-positive cells inside of the lesion of control mice. (H) Higher magnification of (D) showing a reduced number of TUNEL-positive cells of GFAP-IL6mice relative to control mice. (I) Few TUNEL-positive cells are seen in the brains of unlesioned control mice. Scale bars: (A) 158 �m; (B) 150 �m; (C) 147�m; (D) 140 �m; (E,F) 147 �m; (G–I) 47 �m.
144 M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

controls. We have previously shown that the GFAP-IL6mice have a dramatic upregulation of MT-I � II that par-allels the inflammatory response displayed by these mice(Hidalgo et al., 1997), which is fully confirmed in this study.Interestingly, MT-I � II expression following the cryole-sion was significantly potentiated in the GFAP-IL6 mice,which is consistent with studies in IL-6KO mice (Penkowaet al., 1999b, 2000). MT-I � II are considered major anti-oxidant proteins (Cai et al., 2000; Ghoshal et al., 1999;Kling and Olsson, 2000; Lazo et al., 1995, 1998; Penkowa,2002; Rana and Kumar, 2000; Sato and Bremner, 1993;Schwarz et al., 1994, 1995; Suzuki et al., 2000; Tamai et al.,1993; Thornalley and Vasak, 1985), also in the CNS(Carrasco et al., 2000; Giralt et al., 2002; Hidalgo et al.,2001; Penkowa, 2002; Penkowa et al., 1999a, 2000, 2001),and thus it is tempting to speculate that the higher upregu-lation of these proteins observed in the GFAP-IL6 micefollowing the cryolesion contributes significantly to the di-minished oxidative stress and thus to their increased tissuerepair and wound healing capacity. Interestingly, IL-6 per sehas apparently no effects upon nitric oxide synthesis, amajor factor contributing to oxidative stress (De Laurentiiset al., 2000).
MT-I � II might also be related to the decreasedapoptosis rate observed in the GFAP-IL6 mice since theyare considered significant antiapoptotic proteins (Carrascoet al., 2000; Giralt et al., 2002; Hidalgo et al., 2001; Pen-kowa, 2002; Penkowa et al., 1999a, 2000, 2001). The pre-cise mechanisms of action underlying this antiapoptotic roleof MT-I � II is unclear but presumably could affect thecritical steps in the signal transduction cascade leading toapoptosis. Both regulation and expression of proto-onco-genes and tumor suppressor protein p53 are critical in thesteps affecting apoptotic cell death (Kerr et al., 1994; Mar-tin, 2001; Sun and Chen, 1998). The levels of p53 wereincreased in Mt-1&2 knock-out mice relative to normalmice (Kondo et al., 1997; Zheng et al., 1996), and antisensedownregulation of MT-I � II was followed by decreasedlevels of proto-oncogenes bcl-2 and c-myc and increasedlevels of p53 while the opposite was observed in MT-II-overexpressing cells (Absel-Mageed and Agrawal, 1997).One of the mechanisms for this action could be the seques-tration of Zn by MT-I � II in the cell nucleus, as MT-I �II bind and regulate Zn (Aschner, 1996, 1997; Hidalgo etal., 1997, 2001), whereby the Zn-dependent gene expressionand signaling pathways could be significantly altered (An-drews, 2000; Ghoshal and Jacob, 2000; Zeng et al., 1991a,1991b). Also, Zn release by glutamatergic neurons has beenshown to be deleterious to postsynaptic neurons followingseizures or ischemia, at least in part through increased ROSproduction (see Choi and Koh, 1998, and Sensi et al., 1999,and references therein), and thus, by binding Zn, MT-I � IIcould exert significant antiapoptotic effects. In addition,Mt-1&2 knock-out mice display increased immunoreactiv-ity for caspase-1 and activated caspase-3 (Carrasco et al.,2000; Penkowa et al., 2001), while MT-II treatment of rats
during CNS inflammation could significantly lead to re-duced levels of caspase-1, caspase-3, and cytochrome c(Penkowa, 2002), which are also involved in the criticalsteps leading to apoptosis (Cassarino and Bennett, 1999).Furthermore, the levels of nuclear transcription factor kappaB (NF� B), which is critical in the signal transductioncascade leading to apoptosis (Sun and Chen, 1998), weresignificantly increased during CNS inflammation whenMT-I � II levels were decreased (Penkowa, 2002). Thenotion of MT-I � II having influence upon apoptotic signaltransduction has been supported, in that cells from Mt-1&2knock-out mice display increased NF�B activity relative tocontrols (Crowthers et al., 2000). Also, MT-I � II mayinteract with NF�B (Abdel-Mageed and Agrawal, 1998)and reduce its activity (Sakurai et al., 1999). Taken together,these data support the idea that MT-I � II most likely havea significant effect upon the critical steps in the signaltransduction cascade leading to apoptotic cell death. How-ever, it remains to be elucidated whether MT-I � II influ-ence apoptotic signal transduction per se or the above-mentioned changes are epiphenomena, which may becaused by MT-I � II-associated alterations in the levels ofoxidative stress.
Finally, it is noteworthy to mention that Mt-1&2 knock-out mice show a drastically decreased wound healing ca-pacity in the cryolesion injury model (Penkowa et al.,1999a, 2000), which could be linked to their deficient ex-pression of angiogenic and regenerative growth factors(Hopkins and Rothwell, 1995) such as basic fibroblastgrowth factor (bFGF), transforming growth factor-� (TGF-�), neurotrophin-3 (NT-3) and 4(NT-4), and vascular endo-thelial growth factor (VEGF) (Penkowa et al., 2000). Con-versely, transgenic mice overexpressing MT-I show anincreased wound healing capacity in the same injury model(Giralt et al., 2002). Thus, it seems feasible that the higherMT-I � II upregulation observed in the GFAP-IL6 micecould contribute significantly to the increased tissue repairby means of their proangiogenic and other trophic effects.
In summary, the present study demonstrates that al-though IL-6 may cause spontaneous brain damage in atransgenic mouse model of astrocyte-targeted IL-6 expres-sion, it shows clearcut neuroprotective roles following trau-matic brain injury and thus demonstrate a dual role of thiscytokine. Regardless of the exact mechanism, it seemslikely that the antioxidant and antiapoptotic functions ofMT-I � II could be major underlying mechanisms of actionbehind the increased tissue repair and wound healing ob-served in the cryolesioned cortex of the GFAP-IL6 micerelative to wild-type controls. Work is in progress to verifythis possibility.
Acknowledgments
The authors are grateful to H. Hadberg, M. Søberg, P.S.Thomsen, J. Sørensen, G. Hahn, K. Stub, and B. Risto for
145M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

valuable help. These studies were supported by The DanishMedical Research Council, Novo Nordisk Fonden, Univer-sity of Copenhagen/SVF Fond; The Danish Medical Asso-ciation Research Fund, Schrøders Fond, Holger Rabitz Min-delegat, Dir. Ib Henriksens Fond, Dir. Jacob Madsen’sFond, Fonden af 17.12.1981, Kong Christian X’s Fond,Fonden til Lægevidenskabens Fremme, Warwara LarsensFond, Scleroseforeningen, Gerda og Aage Haensch’s Fond,Øjenfonden, Toyota Fonden, Dagmar Marshalls Fond,Bernhard og Marie Kleins Legat, Lily Benthine LundsFond, Eva og Henry Frænkels Mindefond, Øster-JørgensensFond, Karen A. Tolstrups Fond, Eva og Robert Voss Han-sens Fond, Dansk Parkinsonforening, Ragnhild Ibsens Le-gat for Medicinsk Forskning, Kong Christian IX og Dron-ning Louises Jubilæumslegat, Øjenfonden, Læge Eilif Trier-Hansen og Hustru Ane Trier-Hansens Legat, Fru GudrunElboth, født Døbelins, Mindelegat (to M.P.); and bySAF2002-01268, PSPGC PM98-0170, Direccio General deRecerca 2001SGR 00203, and Fundacion “La Caixa” 97/102-00 (J.H.).
References
Abdel-Mageed, A., Agrawal, K.C., 1997. Antisense down-regulation ofmetallothionein induces growth arrest and apoptosis in human breastcarcinoma cells. Cancer Gene Ther. 4, 199–207.
Abdel-Mageed, A.B., Agrawal, A.C., 1998. Activation of nuclear factorkappaB: potential role in metallothionein-mediated mitogenic re-sponse. Cancer Res. 58, 2335–2338.
Andrews, G.K., 2000. Regulation of metallothionein gene expression byoxidative stress and metal ions. Biochem Pharmacol. 59, 95–104.
Aschner, M., 1996. The functional significance of brain metallothioneins.FASEB J. 10, 1129–1136.
Aschner, M., 1997. Astrocyte metallothioneins (MTs) and their neuropro-tective role. Ann. N.Y. Acad. Sci. 825, 334–347.
Aschner, M., 1998. Astrocytes as mediators of immune and inflammatoryresponses in the CNS. Neurotoxicology 19, 269–281.
Bensadoun, J.C., Almeida, L.P., Dreano, M., Aebischer, P., Degion, N.J.,2001. Neuroprotective effect of interleukin-6 and IL6/IL6R cimera inthe quinolinic acid rat model of Huntington’s syndrome. Eur. J. Neu-rosci. 14, 1753–1761.
Benveniste, E.N., 1998. Cytokine actions in the central nervous system.Cytokine Growth Factor Rev. 9, 259–275.
Brett, F.M., Mizisin, A.P., Powell, H.C., Campbell, I.L., 1995. Evolution ofneuropathologic abnormalities associated with blood-brain barrierbreakdown in transgenic mice expressing interleukin-6 in astrocytes.J. Neuropathol. Exp. Neurol. 54, 766–775.
Brunello, A.G., Weissenberger, J., Kappeler, A., Vallan, C., Peters, M.,Rose-John, S., Weis, J., 2000. Astrocytic alterations in interleukin-6/Soluble interleukin-6 receptor alpha double-transgenic mice. Am JPathol. 157, 1485–1593.
Cai, L., Klein, J.B., Kang, Y.J., 2000. Metallothionein inhibits peroxyni-trite-induced DNA and lipoprotein damage. J. Biol. Chem. 275, 38957–38960.
Campbell, I.L., 1998a. Structural and functional impact of the transgenicexpression of cytokines in the CNS. Ann. N.Y. Acad. Sci. 840, 83–96.
Campbell, I.L., 1998b. Transgenic mice and cytokine actions in the brain:bridging the gap between structural and functional neuropathology.Brain Res. Rev. 26, 327–336.
Campbell, I.L., Abraham, C.R., Masliah, E., Kemper, P., Inglis, J.D.,Oldstone, M.B., Mucke, L., 1993. Neurologic disease induced in trans-
genic mice by cerebral overexpression of interleukin 6. Proc. Natl.Acad. Sci. USA 90, 10061–10065.
Carrasco, J., Penkowa, M., Hadberg, H., Molinero, A., Hidalgo, J., 2000.Enhanced seizures and hippocampal neurodegeneration followingkainic acid induced seizures in metallothionein-I � II deficient mice.Eur. J. Neurosci. 12, 2311–2322.
Cassarino, D.S., Bennett Jr., J.P., 1999. An evaluation of the role ofmitochondria in neurodegenerative diseases: mitochondrial mutationsand oxidative pathology, protective nuclear responses, and cell death inneurodegeneration. Brain Res. Rev. 29, 1–25.
Choi, D.W., Koh, J.Y., 1998. Zinc and brain injury. Annu. Rev. Neurosci.21, 347–375.
Crowthers, K.C., Kline, V., Giardina, C., Lynes, M.A., 2000. Augmentedhumoral immune function in metallothionein-null mice. Toxicol. Appl.Pharmacol. 166, 161–172.
De Laurentiis, A., Pisera, D., Lasaga, M., Diaz, M., Theas, S., Duvilanski,B., Seilicovich, A., 2000. Effect of interleukin-6 and tumor necrosisfactor-alpha on GABA release from mediobasal hypothalamus andposterior pituitary. Neuroimmunomodulation 7, 77–83.
Di Santo, E., Alonzi, T., Fattori, E., Poli, V., Ciliberto, G., Sironi, M.,Gnocchi, P., Ricciardi-Castagnoli, P., Ghezzi, P., 1996. Overexpressionof interleukin-6 in the central nervous system of transgenic miceincreases central but not systemic proinflammatory cytokine produc-tion. Brain Res. 740, 239–244.
Dong, Y., Benveniste, E.N., 2001. Immune function of astrocytes. Glia 36,180–190.
Eugster, H.P., Frei, K., Kopf, M., Lassmann, H., Fontana, A., 1998.IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induceautoimmune encephalomyelitis. Eur. J. Immunol. 28, 2178–2187.
Fattori, E., Lazzaro, D., Musiani, P., Modesti, A., Alonzi, T., Ciliberto, G.,1995. IL-6 expression in neurons of transgenic mice causes reactiveastrocytosis and increase in ramified microglial cells but no neuronaldamage. Eur. J. Neurosci. 7, 2441–2449.
Floyd, R.A., 1999a. Neuroinflammatory processes are important in neuro-degenerative diseases: an hypothesis to explain the increased formationof reactive oxygen and nitrogen species as major factors involved inneurodegenerative disease development. Free Radic. Biol. Med. 26,1346–1355.
Floyd, R.A., 1999b. Antioxidants, oxidative stress, and degenerative neu-rological disorders. Proc. Soc. Exp. Biol. Med. 222, 236–245.
Floyd, R.A., Hensley, K., Jaffery, F., Maidt, L., Robinson, K., Pye, Q.,Stewart, C., 1999. Increased oxidative stress brought on by pro-inflam-matory cytokines in neurodegenerative processes and the protectiverole of nitrone-based free radical traps. Life Sci. 65, 1893–1899.
Gadient, R.A., Otten, U.H., 1997. Interleukin-6 (IL-6)—a molecule withboth beneficial and destructive potentials. Prog. Neurobiol. 52, 379–390.
Gasull, T., Rebollo, D.V., Romero, B., Hidalgo, J., 1993. Development ofa competitive double antibody radioimmunoassay for rat metallothio-nein. J. Immunoassay 14, 209–225.
Ghoshal, K., Jacob, S.T., 2000. Regulation of metallothionein gene ex-pression. Prog. Nucleic Acid Res. Mol. Biol. 66, 357–384.
Ghoshal, K., Majumder, S., Li, Z., Bray, T.M., Jacob, S.T., 1999. Tran-scriptional induction of metallothionein-I and -II genes in the livers ofCu,Zn-superoxide dismutase knockout mice. Biochem. Biophys. Res.Commun. 264, 735–742.
Gijbels, K., Brocke, S., Abrams, J.S., Steinman, L., 1995. Administrationof neutralizing antibodies to interleukin-6 (IL-6) reduces experimentalautoimmune encephalomyelitis and is associated with elevated levelsof IL-6 bioactivity in central nervous system and circulation. Mol. Med.1, 795–805.
Giralt, M., Penkowa, M., Lago, N., Camats, J., Hernandez, J., Molinero,A., Hidalgo, J., 2002. Metallothionein-1 � 2 protect the CNS after afocal brain injury. Exp. Neurol. 173, 114–128.
Gruol, D.L., Nelson, T.E., 1997. Physiological and pathological roles ofinterleukin-6 in the central nervous system. Mol. Neurobiol. 15, 307–339.
146 M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

Hickey, W.F., 2001. Basic principles of immunological surveillance of thenormal central nervous system. Glia. 36, 118–124.
Hidalgo, J., Aschner, M., Zatta, P., Vasak, M., 2001. Roles of the metal-lothionein family of proteins in the central nervous system. Brain Res.Bull. 55, 133–145.
Hidalgo, J., Castellano, B., Campbell, I.L., 1997. Regulations of brainmetallothioneins. Curr. Topics Neurochem. 1, 1–26.
Hirano, T., Akira, S., Taga, T., Kishimoto, T., 1990. Biological and clinicalaspects of interleukin 6. Immunol. Today 11, 443–449.
Hirota, H., Kiyama, H., Kishimoto, T., Taga, T., 1996. Accelerated nerveregeneration in mice by upregulated expression of interleukin (IL) 6and IL-6 receptor after trauma. J. Exp. Med. 183, 2627–2634.
Hopkins, S.J., Rothwell, N.J., 1995. Cytokines and the nervous system: I.Expression and recognition. Trends Neurosci. 18, 83–88.
Kerr, J.F., Winterford, C.M., Harmon, B.V., 1994. Apoptosis. Its signifi-cance in cancer and cancer therapy. Cancer 73, 2013–2026.
Klein, M.A., Moller, J.C., Jones, L.L., Bluethmann, H., Kreutzberg, G.W.,Raivich, G., 1997. Impaired neuroglial activation in interleukin-6 de-ficient mice. Glia 19, 227–233.
Kling, P.G., Olsson, P., 2000. Involvement of differential metallothioneinexpression in free radical sensitivity of RTG-2 and CHSE-214 cells.Free Radic. Biol. Med. 28, 1628–1637.
Kondo, Y., Rusnak, J.M., Hoyt, D.G., Settineri, C.E., Pitt, B.R., Lazo, J.S.,1997. Enhanced apoptosis in metallothionein null cells. Mol. Pharma-col. 52, 195–201.
Lazo, J.S., Kondo, Y., Dellapiazza, D., Michalska, A.E., Choo, K.H., Pitt,B.R., 1995. Enhanced sensitivity to oxidative stress in cultured embry-onic cells from transgenic mice deficient in metallothionein I and IIgenes. J. Biol. Chem. 270, 5506–5510.
Lazo, J.S., Kuo, S.M. Woo, E.S. Pitt, B.R., 1998. The protein thiol metal-lothionein as an antioxidant and protectant against antineoplastic drugs.Chem. Biol. Interact. 111–112, 255–262.
Maeda, Y., Matsumoto, M., Hori, O., Kuwabara, K., Ogawa, S., Yan, S.D.,Ohtsuki, T., Kinoshita, T., Kamada, T., Stern, D.M., 1994. Hypoxia/reoxygenation-mediated induction of astrocyte interleukin 6: a para-crine mechanism potentially enhancing neuron survival. J. Exp. Med.180, 2297–2308.
Martin, L.J., 2001. Neuronal cell death in nervous system development,disease, and injury. Int. J. Mol. Med. 7, 455–478.
Martiney, J.A., Cuff, C., Litwak, M., Berman, J., Brosnan, C.F., 1998.Cytokine-induced inflammation in the central nervous system revisited.Neurochem. Res. 23, 349–359.
Masters, B.A., Kelly, E.J., Quaife, C.J., Brinster, R.L., Palmiter, R.D.,1994. Targeted disruption of metallothionein I and II genes increasessensitivity to cadmium. Proc. Natl. Acad. Sci. USA 91, 584–588.
Mendel, I., Katz, A., Kozak, N., Ben-Nun, A., Revel, M., 1998. Interleu-kin-6 functions in autoimmune encephalomyelitis: a study in gene-targeted mice. Eur. J. Immunol. 28, 1727–1737.
Merrill, J.E., Benveniste, E.N., 1996. Cytokines in inflammatory brainlesions: helpful and harmful. Trends Neurosci. 19, 331–338.
Merrill, J.E., Jonakait, G.M., 1995. Interactions of the nervous and immunesystems in development, normal brain homeostasis, and disease.FASEB J. 9, 611–618.
Munoz-Fernandez, M.A., Fresno, M., 1998. The role of tumour necrosisfactor, interleukin 6, interferon-gamma and inducible nitric oxide syn-thase in the development and pathology of the nervous system. Prog.Neurobiol. 56, 307–340.
Murphy, P.G., Borthwick, L.S., Johnston, R.S., Kuchel, G., Richardson,P.M., 1999. Nature of the retrograde signal from injured nerves thatinduces interleukin-6 mRNA in neurons. J. Neurosci. 19, 3791–3800.
Penkowa, M., 2002. Metallothionein expression and roles in the centralnervous system . Biomedical Rev. 13, 1–11.
Penkowa, M., Carrasco, J., Giralt, M., Molinero, A., Hernandez, J., Camp-bell, I.L., Hidalgo, J., 2000. Altered central nervous system cytokine-growth factor expression profiles and angiogenesis in metallothionein-I� II deficient mice. J. Cereb. Blood Flow Metab. 20, 1174–1189.
Penkowa, M., Carrasco, J., Giralt, M., Moos, T., Hidalgo, J., 1999a. CNSwound healing is severely depressed in metallothionein I- and II-deficient mice. J. Neurosci. 19, 2535–2545.
Penkowa, M., Espejo, C., Martinez-Caceres, E.M., Poulsen, C.B., Mont-alban, X., Hidalgo, J., 2001. Altered inflammatory response and in-creased neurodegeneration in metallothionein I � II deficient miceduring experimental autoimmune encephalomyelitis. J. Neuroimmunol.119, 248–260.
Penkowa, M., Giralt, M., Camats, J., Hidalgo, J., 2002. Metallothionein 1� 2 protect the CNS during neuroglial degeneration induced by 6-ami-nonicotinamide. J. Comp. Neurol. 444, 174–189.
Penkowa, M., Giralt, M., Carrasco, J., Hadberg, H., Hidalgo, J., 2000.Impaired inflammatory response and increased oxidative stress andneurodegeneration after brain injury in interleukin-6-deficient mice.Glia. 32, 271–285.
Penkowa, M., Moos, T., Carrasco, J., Hadberg, H., Molinero, A., Blueth-mann, H., Hidalgo, J., 1999b. Strongly compromised inflammatoryresponse to brain injury in interleukin-6 deficient mice. Glia 25, 343–357.
Popovich, P.G., 2000. Immunological regulation of neuronal degenerationand regeneration in the injured spinal cord. Prog. Brain Res. 128,43–58.
Popovich, P.G., Wei, P., Stokes, B.T., 1997. Cellular inflammatory re-sponse after spinal cord injury in Sprague–Dawley and lewis rats.J. Comp. Neurol. 377, 443–464.
Raivich, G., Bohatschek, M., Kloss, C.U., Werner, A., Jones, L.L.,Kreutzberg, G.W., 1999. Neuroglial activation repertoire in the injuredbrain: graded response, molecular mechanisms and cues to physiolog-ical function. Brain Res. Rev. 30, 77–105.
Rana, S.V., Kumar, A., 2000. Metallothionein induced by cadmium or zincinhibits lipid peroxidation in rats exposed to dimethylnitrosamine. Arh.Hig. Rada Toksikol. 51, 279–286.
Sakurai, A., Hara, S., Okano, N., Kondo, Y., Inoue, J., Imura, N., 1999.Regulatory role of metallothionein in NF-kappaB activation. FEBSLett. 455, 55–58.
Samoilova, E.B., Horton, J.L., Hilliard, B., Liu, T.S., Chen, Y., 1998.IL-6-deficient mice are resistant to experimental autoimmune enceph-alomyelitis: roles of IL-6 in the activation and differentiation of auto-reactive T cells. J Immunol. 161, 6480–6486.
Sato, M., Bremner, I., 1993. Oxygen free radicals and metallothionein.Free Radic. Biol. Med. 14, 325–337.
Schwarz, M.A., Lazo, J.S., Yalowich, J.C., Allen, W.P., Whitmore, M.,Bergonia, H.A., Tzeng, E., Billiar, T.R., Robbins, P.D., Lancaster, J.R.,Pitt, B.R., 1995. Metallothionein protects against the cytotoxic andDNA-damaging effects of nitric oxide. Proc. Natl. Acad. Sci. USA 92,4452–4456.
Schwarz, M.A., Lazo, J.S., Yalowich, J.C., Reynolds, I., Kagan, V.E.,Tyurin, V., Kim, Y.M., Watkins, S.C., Pitt, B.R., 1994. Cytoplasmicmetallothionein overexpression protects NIH 3T3 cells from tert-butylhydroperoxide toxicity. J. Biol. Chem. 269, 15238–15243.
Sensi, S.L., Yin, H.Z., Weiss, J.H., 1999. Glutamate triggers preferentialZn2� flux through Ca2� permeable AMPA channels and consequentROS production. NeuroReport 10, 1723–1727.
Streit, W.J., Walter, S.A., Pennell, N.A., 1999. Reactive microgliosis. Prog.Neurobiol. 57, 563–581.
Sun, A.Y., Chen, Y.M., 1998. Oxidative stress and neurodegenerativedisorders. J. Biomed. Sci. 5, 401–414.
Suzuki, Y., Apostolova, M.D., Cherian, M.G., 2000. Astrocyte culturesfrom transgenic mice to study the role of metallothionein in cytotox-icity of tert-butyl hydroperoxide. Toxicology 145, 51–62.
Swartz, K.R., Liu, F., Sewell, D., Schochet, T., Campbell, I., Sandor, M.,Fabry, Z., 2001. Interleukin-6 promotes post-traumatic healing in thecentral nervous system. Brain Res. 896, 86–95.
Tamai, K.T., Gralla, E.B., Ellerby, L.M., Valentine, J.S., Thiele, D.J.,
147M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148

1993. Yeast and mammalian metallothioneins functionally substitutefor yeast copper–zinc superoxide dismutase. Proc. Natl. Acad. Sci.USA 90, 8013–8017.
Thornalley, P.J., Vasak, M., 1985. Possible role for metallothionein inprotection against radiation-induced oxidative stress. Kinetics andmechanism of its reaction with superoxide and hydroxyl radicals.Biochim. Biophys. Acta 827, 36–44.
Toulmond, S., Vige, X., Fage, D., Benavides, J., 1992. Local infusion ofinterleukin-6 attenuates the neurotoxic effects of NMDA on rat striatalcholinergic neurons. Neurosci. Lett. 144, 49–52.
Umegaki, H., Yamada, K., Naito, M., Kameyama, T., Iguchi, A., Na-beshiama, T., 1996. Protective effect of interleukin-6 against the deathof PC 12 cells caused by serum deprivation or by the addition of acalcium ionophore. Biochem. Pharmacol. 52, 911–916.
Van Wagoner, N.J., Benveniste, E.N., 1999. Interleukin-6 expression andregulation in astrocytes. J. Neuroimmunol. 100, 124–139.
Xiao, B.G., Link, H., 1998. Immune regulation within the central nervoussystem. J. Neurol. Sci. 157, 1–12.
Zeng, J., Heuchel, R., Schaffner, W., Kagi, J.H., 1991a. Thionein (apo-metallothionein) can modulate DNA binding and transcription activa-tion by zinc finger containing factor Sp1. FEBS Lett. 279, 310–312.
Zeng, J., Vallee, B.L., Kagi, J.H., 1991b. Zinc transfer from transcriptionfactor IIIA fingers to thionein clusters. Proc. Natl. Acad. Sci. USA 88,9984–9988.
Zheng, H., Liu, J., Choo, K.H., Michalska, A.E., Klaassen, C.D., 1996.Metallothionein-I and -II knock-out mice are sensitive to cadmium-induced liver mRNA expression of c-jun and p53. Toxicol. Appl.Pharmacol. 136, 229–235.
148 M. Penkowa et al. / Experimental Neurology 181 (2003) 130–148




















