
Articole engleza neurobiologia depresiei
53
PATHOPHYSIOLOGY OF DEPRESSION: DO WE HAVE ANY SOLID EVIDENCE OF INTEREST TO CLINICIANS? GREGOR HASLER ABSTRACT Due to the clinical and etiological heterogeneity of major depressive disorder, it has been difficult to elucidate its pathophysiology. Current neurobiological theories with the most valid empirical foundation and the highest clinical relevance are reviewed with respect to their strengths and weaknesses. The selected theories are based on studies investigating psychosocial stress and stress hormones, neurotransmitters such as serotonin, norepinephrine, dopamine, glutamate and gamma-aminobutyric acid (GABA), neurocircuitry, neurotrophic factors, and circadian rhythms. Because all theories of depression apply to only some types of depressed patients but not others, and because depressive pathophysiology may vary considerably across the course of illness, the current extant knowledge argues against a unified hypothesis of depression. As a consequence, antidepressant treatments, including psychological and biological approaches, should be tailored for individual patients and disease states. Individual depression hypotheses based on neurobiological knowledge are discussed in terms of their interest to both clinicians in daily practice and clinical researchers developing novel therapies. Keywords: Depression, pathophysiology, genetics, stress, serotonin, norepinephrine, dopamine, neuroimaging, glutamate, GABA Major depressive disorder (MDD) is a common and costly disorder which is usually associated with severe and persistent symptoms leading to important social role impairment and increased mortality 1 ,2 . It is one of the most important causes of disability worldwide 3 . The high rate of inadequate treatment of the disorder remains a serious concern 1 . This review is aimed at summarizing the solid evidence on the etiology and pathophysiology of MDD that is likely relevant for clinical psychiatry. Neurobiological findings are regarded as solid when they are consistent and convergent, i.e., they have been confirmed by several studies using the same method and fit into results from studies using different methodological approaches. GENES AND PSYCHOSOCIAL STRESS Family, twin, and adoption studies provide very solid and consistent evidence that MDD is a familial disorder and that this
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Articole engleza neurobiologia depresiei

PATHOPHYSIOLOGY OF DEPRESSION: DO WE HAVE ANY SOLID EVIDENCE OF INTEREST TO CLINICIANS?GREGOR HASLERABSTRACTDue to the clinical and etiological heterogeneity of major depressive disorder, it has been difficult to elucidate its pathophysiology. Current neurobiological theories with the most valid empirical foundation and the highest clinical relevance arereviewed with respect to their strengths and weaknesses. The selected theories are based on studies investigating psychosocialstress and stress hormones, neurotransmitters such as serotonin, norepinephrine, dopamine, glutamate and gamma-aminobutyric acid (GABA), neurocircuitry, neurotrophic factors, and circadian rhythms. Because all theories of depression apply to only some types of depressed patients but not others, and because depressive pathophysiology may vary considerably across the course of illness, the current extant knowledge argues against a unified hypothesis of depression. As a consequence, antidepressant treatments, including psychological and biologicalapproaches, should be tailored for individual patients and disease states. Individual depression hypotheses based on neurobiological knowledge are discussed in terms of their interest to both clinicians in daily practice and clinical researchers developing novel therapies.Keywords: Depression, pathophysiology, genetics, stress, serotonin, norepinephrine, dopamine, neuroimaging, glutamate, GABAMajor depressive disorder (MDD) is a common and costly disorder which is usually associated with severe and persistent symptoms leading to important social role impairment and increased mortality 1,2. It is one of the most important causes of disability worldwide 3. The high rate of inadequate treatment of the disorder remains a serious concern 1.This review is aimed at summarizing the solid evidence on the etiology and pathophysiology of MDD that is likely relevant for clinical psychiatry. Neurobiological findings are regarded as solid when they are consistent and convergent, i.e., they have been confirmed by several studies using the same method and fit into results from studies using different methodological approaches.GENES AND PSYCHOSOCIAL STRESSFamily, twin, and adoption studies provide very solid and consistent evidence that MDD is a familial disorder and that this

familiality is mostly or entirely due to genetic factors 4. This important finding suggests that parental social behavior and other familial environmental risk factors are not as important inthe pathogenesis of MDD as previously assumed and should not be the major focus of the treatment of the disorder.The above-mentioned studies consistently show that the influence of genetic factors is around 30-40% 4. Non-genetic factors, explaining the remaining 60-70% of the variance in susceptibilityto MDD, are individual-specific environmental effects (including measurement error effects and gene-environment interactions). These effects are mostly adverse events in childhood and ongoing or recent stress due to interpersonal adversities, including childhood sexual abuse, other lifetime trauma, low social support, marital problems, and divorce 5,6.These results suggest that there is a huge potential in the prevention of MDD by means of psychosocial interventions (e.g., in schools, at workplace). In addition, these results mirror the clinical practice of empirically validated psychotherapies to treat depression 7,8,9, including interpersonal, psychodynamic and cognitive behavioral psychotherapies and cognitive behavioural analysis system of psychotherapy, which all focus directly or indirectly on interpersonal difficulties and skills. This does not exclude the fact that unidentified non-genetic, non-psychosocial risk factors may also play important roles in some patients (e.g., climatic change, medical conditions).Stress sensitivity in depression is partly gender-specific. Whilemen and women are, in general, equally sensitive to the depressogenic effects of stressful life events, their responses vary depending upon the type of stressor. Specifically, men are more likely to have depressive episodes following divorce, separation, and work difficulties, whereas women are more sensitive to events in their proximal social network, such as difficulty getting along with an individual, serious illness, or death10. These findings point to the importance of gender-sensitive psychosocial approaches in the prevention and treatmentof MDD.In contrast to the very solid evidence from epidemiological studies on broad risk factor domains, there is no solid evidence for specific genes and specific gene-by-environment interactions in the pathogenesis of MDD. Genome-wide association studies have indicated that many genes with small effects are involved in complex diseases, increasing the difficulty in identifying such genes 11. While there has been progress in the search for risk

genes for several complex diseases despite this methodological problem 12, psychiatric conditions have turned out to be very resistant to robust gene identification. For example, based on a community-based prospective study, it has been proposed that a specific genetic variation in the promoter region of the serotonin transporter (a target of antidepressant drugs) interacts with stressful life events in the pathogenesis of depression13. Although there is high clinical and neurobiologicalplausibility of this interaction, a recent meta-analysis yielded no evidence that the serotonin transporter gene alone or in interaction with psychological stress was associated with the risk of depression 14.The limited success of genetic studies of depression has been related to use of current classification schemas including ICD-10and DSM-IV. These diagnostic manuals are based on clusters of symptoms and characteristics of clinical course that do not necessarily describe homogenous disorders but instead reflect common final pathways of different pathophysiolgical processes 15,16. The clinician should be aware that family history will continue to be the most solid source of information to estimate the genetic risk of MDD.STRESS HORMONES AND CYTOKINESCorticotropin-releasing hormone (CRH) is released from the hypothalamus in response to the perception of psychological stress by cortical brain regions. This hormone induces the secretion of pituitary corticotropin, which stimulates the adrenal gland to release cortisol into the plasma. The physiologic response to stress is partly gender-specific: women show generally greater stress responsiveness than men, which is consistent with the greater incidence of major depression in women 17. Moreover, men show greater cortisol responses to achievement challenges, whereas women show greater cortisol responses to social rejection challenges 18.Although MDD is considered as a stress disorder, most subjects treated for MDD have no evidence of dysfunctions of the hypothalamic-pituitary-adrenal axis (HPA) 19. However, some subjects with MDD do show abnormalities of that axis and of the extrahypothalamic CRH system 20. Altered stress hormone secretionappeared to be most prominent in depressed subjects with a history of childhood trauma 21. Elevated cortisol may act as a mediator between major depression and its physical long-term consequences such as coronary heart disease, type II diabetes, and osteoporosis22.

The importance of HPA axis dysfunction for the efficacy of antidepressants is a matter of debate 23. This axis is regulated through a dual system of mineralocorticoid (MR) and glucocorticoid (GR) receptors. Decreased limbic GR receptor function 24,25 and increased functional activity of the MR system 26 suggest an imbalance in the MR/GR ratio in stress-related conditions such as MDD. Epigenetic regulation of the glucocorticoid receptors has been associated with childhood abuse 27. Such environmental programming of gene expression may represent one possible mechanism that links early life stress to abnormal HPA axis function and increased risk of MDD in adults.While the CRH stimulation test (dex/CRH test) 28 is a sensitive measure of the HPA axis dysfunction in depression, the specificity of this test for MDD is low. However, non-suppressionin the dex/CRH test has consistently predicted increased risk fordepressive relapse during clinical remission 23. Additionally, the measurement of waking salivary cortisol concentration has been shown to be a simple and sensitive test for HPA axis hyperactivity in depression 29. Hypercortisolemia is almost exclusively found in subjects with severe and psychotic depression, in whom glucocorticoid antagonists may have some therapeutic effect 30.There is convergent evidence for CRH to play a major role in the pathogenesis of certain types of depression. Levels of CRH in thecerebrospinal fluid are elevated in some depressed subjects 31. Post-mortem studies reported an increased number of CRH secretingneurons in limbic brain regions in depression 32, likely reflecting a compensatory response to increased CRH concentrations33. In addition, CRH produces a number of physiological and behavioral alterations that resemble the symptoms of major depression, including decreased appetite, disrupted sleep, decreased libido, and psychomotor alterations 34. There is also preliminary evidence that CRH1 receptor antagonists reduce symptoms of depression and anxiety 35.“Sickness behavior” as a result of an activation of the inflammatory response system shares many symptoms with depression, including fatigue, anhedonia, psychomotor retardation, and cognitive impairment. Sickness is mediated by pro-inflammatory cytokines such as interleukin-1α, tumor necrosisfactor-α, and interleukin-6, which activate the HPA axis and impair the central serotonin system 36. The prevalence of depression as an unwanted effect of recombinant interferons is

around 30% 37. In animals, blocking pro-inflammatory cytokine-mediated signaling produces antidepressant-like effects 38. Clinical data suggest that cytokines may play a role in the pathophysiology of a subgroup of depressed subjects, particularlythose with comorbid physical conditions 36. The antidepressant enhancing effect of acetylsalicylic acid 39 points to the possible clinical relevance of psychoneuroimmunology in clinical depression research.Taken together, the laboratory tests with the highest potential to be clinically useful in the care of depressed individuals are based on abnormalities of the neuroendocrine and neuroimmune systems. Despite the large amount of basic science data suggesting that the HPA axis is importantly involved in the pathophysiology of depression, the effect of pharmacological modulation of this neuroendocrine system as antidepressant therapy has been disappointing. The link between childhood traumaand a permanently altered physiologic stress system points to theuse of specific psychotherapies in the treatment of depressed patients with a history of early life trauma 40.THE MEDIATING ROLE OF MONOAMINESMost of the serotonergic, noradrenergic and dopaminergic neurons are located in midbrain and brainstem nuclei and project to largeareas of the entire brain. This anatomy suggests that monoaminergic systems are involved in the regulation of a broad range of brain functions, including mood, attention, reward processing, sleep, appetite, and cognition. Almost every compoundthat inhibits monoamine reuptake, leading to an increased concentration of monoamines in the synaptic cleft, has been proven to be a clinically effective antidepressant 19. Inhibitingthe enzyme monoamine oxidase, which induces an increased availability of monoamines in presynaptic neurons, also has antidepressant effects. These observations led to the pharmacologically most relevant theory of depression, referred toas the monoamine-deficiency hypothesis.The monoamine-deficiency theory posits that the underlying pathophysiological basis of depression is a depletion of the neurotransmitters serotonin, norepinephrine or dopamine in the central nervous system.Serotonin is the most extensively studied neurotransmitter in depression. The most direct evidence for an abnormally reduced function of central serotonergic system comes from studies using tryptophan depletion, which reduces central serotonin synthesis. Such a reduction leads to the development of depressive symptoms

in subjects at increased risk of depression (subjects with MDD infull remission, healthy subjects with a family history of depression) 41,42, possibly mediated by increased brain metabolism in the ventromedial prefrontal cortex and subcortical brain regions42. Experimentally reduced central serotonin has been associated with mood congruent memory bias, altered reward-related behaviors, and disruption of inhibitory affective processing 16, all of which add to the clinical plausibility of the serotonin deficiency hypothesis. There is also evidence for abnormalities of serotonin receptors in depression, with the mostsolid evidence pointing to the serotonin-1A receptor, which regulates serotonin function. Decreased availability of this receptor has been found in multiple brain areas of patients with MDD 43, although this abnormality is not highly specific for MDD and has been found in patients with panic disorder 44 and temporal lobe epilepsy 45, possibly contributing to the considerable comorbidity among these conditions. However, there is no explanation for the mechanism of serotonin loss in depressed patients, and studies of serotonin metabolites in plasma, urine and cerebrospinal fluid, as well as post-mortem research on the serotonergic system in depression, have yielded inconsistent results. There is preliminary evidence that an increased availability of the brain monoamine oxidase, which metabolizes serotonin, may cause serotonin deficiency 46. In addition, loss-of-function mutations in the gene coding for the brain-specific enzyme tryptophan hydroxylase-2 may explain the loss of serotonin production as a rare risk factor for depression 47.Dysfunction of the central noradrenergic system has been hypothesized to play a role in the pathophysiology of MDD, based upon evidence of decreased norepinephrine metabolism, increased activity of tyrosine hydroxylase, and decreased density of norepinephrine transporter in the locus coeruleus in depressed patients 48. In addition, decreased neuronal counts in the locus coeruleus, increased alpha-2 adrenergic receptor density, and decreased alpha-1 adrenergic receptor density have been found in the brains of depressed suicide victims post-mortem 49. Since there is no method to selectively deplete central norepinephrine and no imaging tool to study the central norepinephrine system, solid evidence for abnormalities of this system in depression is lacking.While the classical theories of the neurobiology of depression mainly focused on serotonin and norepinephrine, there is

increasing interest in the role of dopamine 50. Dopamine reuptakeinhibitors (e.g., nomifensine) and dopamine receptor agonists (e.g., pramipexole) had antidepressant effects in placebo-controlled studies of MDD 51. In the cerebrospinal fluid and jugular vein plasma, levels of dopamine metabolites were consistently reduced in depression, suggesting decreased dopamineturnover 52. Striatal dopamine transporter binding and dopamine uptake were reduced in MDD, consistent with a reduction in dopamine neurotransmission 53. Degeneration of dopamine projections to the striatum in Parkinson’s disease was associatedwith a major depressive syndrome in about one half of cases, which usually preceded the appearance of motor signs 54. Experimentally reduced dopaminergic transmission into the accumbens has been associated with anhedonic symptoms and performance deficits on a reward processing task in subjects at increased risk of depression 55,56. These findings are consistentwith the clinical observation that depressed patients have a blunted reaction to positive reinforcers and an abnormal responseto negative feedback 57.Almost all established antidepressants target the monoamine systems 58. However, full and partial resistance to these drugs and their delayed onset of action suggest that dysfunctions of monoaminergic neurotransmitter systems found in MDD represent thedownstream effects of other, more primary abnormalities. Despite this limitation, the monoamine-deficiency hypothesis has proved to be the most clinically relevant neurobiological theory of depression. New findings on the role of dopamine in depression emphasize the scientific potential of this theory, and promising reports of antidepressant effects of drugs that modulate the dopaminergic system (e.g., pramipexole, modafinil) in difficult-to-treat depression underline its clinical relevance 51,59.THE NEUROIMAGING OF DEPRESSIONAlthough many historical attempts to localize mental functions have failed, they have considerably contributed to a modern neuroscientific understanding of mental disorders 60. The development of neuroimaging techniques has opened up the potential to investigate structural and functional abnormalities in living depressed patients. Unfortunately, the diversity of imaging techniques used, the relatively small and heterogeneous study samples studied, and the limited overlap of results across imaging paradigms 61 make it difficult to reliably identify neuronal regions or networks with consistently abnormal structureor function in MDD.

Functional imaging studies have provided the most limited overlapof findings. This may be due to methodological limitations and/orthe complexity of neurocircuitry involved in MDD. A recent meta-analytic study found the best evidence for abnormal brain activity in MDD in lateral frontal and temporal cortices, insula,and cerebellum. In these brain regions activity was decreased at rest, they showed a relative lack of activation during induction of negative emotions, and an increase in activity following treatment with serotonin reuptake inhibitors. Opposite changes may exist in ventromedial frontal areas, striatum and possibly other subcortical brain regions 61.More solid evidence has been provided by structural imaging and post-mortem studies. A recent meta-analytic study on brain volumeabnormalities in MDD revealed relatively large volume reductions in the ventromedial prefrontal cortex, particularly in the left anterior cingulate and in the orbitofrontal cortex. Moderate volume reductions were found in the lateral prefrontal cortex, hippocampus and striatum 62. Post-mortem studies consistently identified a reduction in glia cell density in dorsal, orbital and subgenual prefrontal cortices, as well as in the amygdala 63,64.Overall, functional, structural and post-mortem studies suggest that structural and functional abnormalities in the left subgenual cingulate cortex are the most solid neuroanatomical finding in MDD. Volume reduction in this region was found early in illness and in young adults at high familial risk for MDD 65, suggesting a primary neurobiological abnormality associated with the etiology of the illness. Humans with lesions that include thesubgenual prefrontal cortex showed abnormal autonomic responses to social stimuli 66, and rats with left-sided lesions in this region had increased sympathetic arousal and corticosterone responses to restraint stress 67. Most importantly, chronic deep brain stimulation to reduce the potentially elevated activity in the subgenual cingulated cortex produced clinical benefits in patients with treatment-resistant depression 68.In summary, despite the considerable heterogeneity of findings from neuroimaging studies, there is convergent evidence for the presence of abnormalities in the subgenual prefrontal cortex in some patients with MDD. Neuroanatomical research in depression isof great clinical interest, since novel antidepressant treatmentssuch as deep brain stimulation can target specific brain regions.In addition, there are promising leads for neuroimaging findings to predict the likelihood of responses to specific treatments 69.

THE NEUROTROPHIC HYPOTHESIS OF DEPRESSIONRisk factors for depressive episodes change during the course of the illness. The first depressive episode is usually “reactive”, i.e., triggered by important psychosocial stressors, while subsequent episodes become increasingly “endogenous”, i.e., triggered by minor stressors or occurring spontaneously 70. Thereis consistent evidence that the volume loss of the hippocampus and other brain regions is related to the duration of depression 71, suggesting that untreated depression leads to hippocampal volume loss, possibly resulting in increased stress sensitivity 72 and increased risk of recurrence 73.Glucocorticoid neurotoxicity, glutamatergic toxicity, decreased neurotrophic factors, and decreased neurogenesis have been proposed as possible mechanisms explaining brain volume loss in depression. There is no solid evidence on any of these mechanisms, since there are no imaging tools to directly examine neurotoxic and neurotrophic processes in vivo. Brain derived neurotrophic factor (BDNF) has attracted considerable interest. Specifically, preclinical studies have shown correlations betweenstress-induced depressive-like behaviors and decreases in hippocampal BDNF levels, as well as enhanced expression of BDNF following antidepressant treatment 74. The clinician should be aware of the potentially brain-damaging effect of depression and treat depressed patients as early and effectively as possible.ALTERED GLUTAMATERGIC AND GABAERGIC NEUROTRANSMISSIONA series of magnetic resonance spectroscopy studies consistently showed reductions in total gamma-aminobutyric acid (GABA) concentrations in the prefrontal and occipital cortex in acute depression 75. This may reflect acute stress effects, since psychological stress seems to induce presynaptic down-regulation of prefrontal GABAergic neurotransmission 76. Alternatively, low total GABA concentration may reflect reduction in the density andsize of GABAergic interneurons 77. In addition, chronic stress may reduce GABA-A receptor function, possibly through changes in neuroactive steroid synthesis 78. Contradictory evidence of the GABA hypothesis of depression includes the lack of effects of GABAergic drugs on core depressive symptoms 79 and normal prefrontal GABA concentration in subjects with remitted MDD 80.Several lines of evidence suggest a dysfunction of the glutamate neurotransmitter system in MDD: a single dose of the glutamate N-methyl-D-aspartate (NMDA) receptor antagonist ketamine produced rapid and large antidepressant effects in patients with treatment-resistant MDD 81; inhibitors of glutamate release

(e.g., lamotrigine, riluzole) demonstrated antidepressant properties82; abnormal glutamate levels were found in depressed subjects as determined by magnetic resonance spectroscopy 75; andthere is evidence for abnormal NMDA signaling in post-mortem tissue preparations 83. Since glutamate is the major excitatory neurotransmitter involved in almost every brain activity, the characterization of the specific role of glutamate in depression deserves further investigation (e.g., there are promising leads that the metabotropic glutamate receptor 5 is specifically involved in MDD 84).CIRCADIAN RHYTHMSSleep disturbances and daytime fatigue are diagnostic criteria for MDD, suggesting impaired sleep-wake regulation in depressed patients. In addition, some depressive symptoms may show diurnal variations (mood, psychomotor activity, accessibility of memoriesof positive and negative experiences), and a subgroup of patientswith MDD may have a circadian rhythm disorder 85. In healthy young subjects, moderate changes in the timing of the sleep-wake cycle had specific effects on subsequent mood 86. In depressed patients, manipulations of circadian rhythms (light therapy, sleep deprivation, phase advance treatment) can have antidepressant efficacy.Based on these findings, circadian abnormalities have been hypothesized to be etiologically associated with MDD 16. The association between phase advance of the sleep-wake cycle and phase advances in nocturnal cortisol secretion; shortened REM latency in some subjects with MDD; and the effect of antidepressants on circadian rhythms of behavior, physiology, andendocrinology contribute to the biological foundation of this hypothesis 85,87,88. Despite of the many promising findings, the molecular and genetic underpinnings of this hypothesis are largely unknown. It remains to be determined whether antidepressant effects of new therapeutics such as agomelatine directly relate to normalization of circadian rhythms 87.Based onthese findings, circadian abnormalities have been hypothesized tobe etiologically associated with MDD 16. The association between phase advance of the sleep-wake cycle and phase advances in nocturnal cortisol secretion; shortened REM latency in some subjects with MDD; and the effect of antidepressants on circadianrhythms of behavior, physiology, and endocrinology contribute to the biological foundation of this hypothesis 85,87,88. Despite ofthe many promising findings, the molecular and genetic underpinnings of this hypothesis are largely unknown. It remains

to be determined whether antidepressant effects of new therapeutics such as agomelatine directly relate to normalizationof circadian rhythms 87.CONCLUSIONSThe main strengths and weaknesses of the various neurobiological hypotheses of depression are summarized in Table 1. The many theories of depression and the relatively low response rate of all available antidepressant treatments clearly argue against a “unified hypothesis of depression” and suggest that depression isa clinically and etiologically heterogeneous disorder.This encourages research on predictors of the response to therapeutic interventions using biomarkers such as neuroimaging and neuroendocrine tests in combination with genotyping for inter-individual variability with respect to stress sensitivity and antidepressant drug action.The identification of reliable predictors of therapeutic outcomeswill allow for the development of personalized medicine that has the potential to individually tailor interventions and to open upnew pathways in the evaluation of novel therapeutic approaches.
ARTICLE INFORMATIONWorld Psychiatry. 2010 October; 9(3): 155–161.GREGOR HASLER1………………………..International Journal of Clinical PracticeWiley-Blackwell, John Wiley & SonsNeurobiology of depression: an integrated view of key findingsV Maletic, M Robinson, [...], and J RussellAdditional article informationAbstractAimsThe objectives of the present review were to summarise the key findings from the clinical literature regarding the neurobiology of major depressive disorder (MDD) and their implications for maximising treatment outcomes. Several neuroanatomical structuresin the prefrontal and limbic areas of the brain are involved in affective regulation. In patients with MDD, alterations in the dynamic patterns of activity among these structures have profoundimplications for the pathogenesis of this illness.DiscussionThe present work reviews the evidence for the progressive nature of MDD along with associated changes in neuroanatomical structureand function, especially for the hippocampus. The role of

glucocorticoids, inflammatory cytokines and brain-derived growth factors are discussed as mediators of these pathological alterations. From this integrated model, the role of antidepressant therapy in restoring normative processes is examined along with additional treatment guidelines.ConclusionMajor depressive disorder is an illness with significant neurobiological consequences involving structural, functional andmolecular alterations in several areas of the brain. Antidepressant pharmacotherapy is associated with restoration of the underlying physiology. Clinicians are advised to intervene with MDD using an early, comprehensive treatment approach that has remission as the goal.Review CriteriaThe search strategies used for this review involved literature searches of the MEDLINE and Psychinfo electronic databases. The main heading terms included major depressive disorder, neurobiology, antidepressant, hippocampus, brain-derived neurotrophic growth factor, glucocorticoids and monoamines. As part of the research strategy, each article's bibliography was reviewed for additional potential research findings relevant to these terms.Message for the ClinicMajor depressive disorder (MDD) is an illness with significant neurobiological consequences involving structural, functional andmolecular alterations in several areas of the brain. Antidepressant pharmacotherapy is associated with restoration of the underlying physiology. Clinicians are advised to intervene with MDD using an early, comprehensive treatment approach that has remission as the goal.IntroductionMajor depressive disorder (MDD) remains one of the most frequently seen psychiatric illnesses in primary care settings (1). Although family and primary care physicians have greatly increased their recognition and treatment of this illness, MDD remains an unresolved treatment challenge for many physicians andpatients (2). Increasing evidence has accrued in recent years regarding the impact of MDD on the structural and functional processes occurring in the brain. From the initial views that depression was caused by ‘chemical imbalance’ in the brain, this body of research has developed into a complex theory involving neuronal networks and plasticity (3). The network model has also led to a greater understanding of the mechanisms of effective

treatment interventions and their role in mitigating the deleterious effects of MDD (4).The objectives of the present review were to summarise the key findings from the clinical literature regarding the neurobiology of MDD and their implications for maximising treatment outcomes. First, the evidence that MDD is not only a chronic and recurrent illness, but also a progressive illness will be presented. Second, the impact of MDD on the primary neuroanatomical sites associated with mood regulation will be described at the structural and functional level. Third, the molecular processes that have been implicated for mediating these structural and functional changes will be explored. Fourth, the role of multipleneurotransmitter systems will be reviewed for their involvement in restoration and recovery from MDD. The last section will discuss the treatment guidelines for obtaining remission in the context of this neurobiological model.Major depressive disorder as a progressive illnessEpidemiological studies have consistently shown that MDD is one of the most prevalent lifetime psychiatric disorders. In the National Comorbidity Replication Survey, based on DSM-IV criteriafor MDD, the lifetime prevalence rate was 16.2%, with a 12-month estimate of 6.6% (5). The presentation of MDD is heterogeneous with respect to both core and associated symptoms (6). In the Diagnostic and Statistical Manual of Mental Disorders Fourth Edition, Text Revision (7), the diagnosis of MDD requires the experience of major depressive episodes that are defined by at least five of the following symptoms for at least 2 weeks duration: loss of interest, depressed mood, appetite/weight disturbance, sleep disturbance, psychomotor change, loss of energy, worthlessness/guilt, concentration difficulties/indecisiveness and thoughts of death/suicide. Depressed mood or loss of interest must be one of the symptoms, but with the inclusion of compound criteria (e.g. worthlessness or guilt), a diagnosis of MDD can be met by various permutations,and episodes may then be further qualified by other associated features (e.g. postpartum, seasonal pattern, with melancholy or psychotic symptoms).Even though MDD is characterised as an episodic illness, prospective studies have found that recurrence is the norm ratherthan the exception. For example, in a naturalistic, 15-year follow-up of a sample of 380 patients experiencing an index MDD episode, 73% experienced a recurrent episode (8), with each subsequent episode increasing the probability of further episodes

(9). Similarly, in the STAR*D Project (Sequenced Treatment Alternatives to Relieve Depression) that includes 1500 patients with MDD, 74% of patients had experienced more than one episode (10). Recurrence of MDD appears to be driven in part by neurobiological vulnerabilities. In the STAR*D Project, patients who experienced multiple episodes were more likely to have a positive family history of depressive illness and an earlier age of onset of their index depressive episode compared with patientswho were in their first episode (10).Consistent evidence has also supported a ‘kindling hypothesis’ inwhich depressive episodes become more easily triggered over time (11). As the number of depressive episodes increase, future episodes are predicted more by the number of prior episodes rather than by life stress (12) (Figure 1). Kindling can be described as a process which occurs by a lowering of the threshold for the impact of stressful life events (i.e. sensitisation to minor events) or by an increase in spontaneous dysregulation, both of which could indicate progressive effects of MDD (13). An analysis of the risk of recurrence in a large study of twins also suggests genetic contributions as patients with a high genetic risk were ‘prekindled’; that is, they had a lower association between stressful life events and the onset of depressive episodes compared with patients having a low genetic risk (14).
Figure 1Major depression as a progressive illness. As the number of majordepressive episodes increase, the risk for subsequent episodes ispredicted more from the number of prior episodes and less from the occurrence of a recent life stress. Figure adapted from ...Early adverse experiences may also contribute to long-term neurobiological alterations associated with depression. In preclinical studies, maternal deprivation of rat pups during critical development periods resulted in subsequent hyper-reactivity to stress and marked behavioural changes in adult rats(15). In children who had a history of early maltreatment, the risk for depressive symptoms was associated with an interaction between genotypes [e.g. serotonin (5-HT) transporter] and historyof maltreatment (16). Considering these findings, some researchers have suggested that greater neurobiological changes occur in patients with depression who have early adverse

experiences compared with patients who are depressed but do not have such a history, indicating that these patients may representan especially vulnerable subtype of depressive illness (17).Chronicity also suggests long-term neurobiological consequences associated with the MDD illness. In the STAR*D Project, 25% of the patients (with single or recurrent MDD) were identified as having a chronic episode of more than 2 years duration (10). In another large multicentre treatment study (n= 681), patients’ depression was classified using DSM-IV modifiers into four categories: chronic MDD (episodes > 2 years), MDD with incompleterecovery (partial response), MDD superimposed on dysthymia (double depression) and chronic MDD superimposed on dysthymia (depressive symptoms > 4 years). Despite multiple comparisons across a broad range of clinical and psychological variables, fewdifferences were found among the four groups, resulting in the conclusion that various manifestations of chronic depression represent the same illness (18).As the duration of depressive episodes increases, the probabilityof recovery substantially decreases over time. In a 5-year prospective study of outpatients with depression, approximately half recovered within the first 6 months, but afterwards the rateof recovery diminished substantially. For example, patients who had experienced depressive episodes of 1-year duration had a recovery rate of 16% compared with a 1% recovery rate for patients whose episodes persisted > 5 years (19). Similarly, in aprospective study of new onset depressive episodes, a longer duration (> 12 weeks) of previous episodes reduced the likelihoodof recovery from the new onset episode by 37% (20).Even if patients no longer meet full criteria for an MDD episode,studies have found that a substantial subset of patients continueto experience residual symptoms and diminished functioning. In a 3-year longitudinal epidemiological study, 165 patients were assessed before and after an MDD episode. Although mean values onfunctional measures returned to premorbid levels, 15–40% of patients experienced a worsening in psychosocial functioning thatpersisted after the episode, and the overall functioning of the entire sample continued to be lower than that of a healthy cohort(21). In a 10-year, naturalistic longitudinal study, patients whoexperienced subthreshold depressive symptoms following an MDD episode were at significantly greater risk for a recurrence, and they also had a much faster onset of their next episode compared with patients whose episode had fully remitted, suggesting that

residual symptoms represent vulnerability because of an active disease state (22).The recurrence and chronicity of MDD along with possible kindlingeffects have shifted the perspective of the appropriate treatmentgoal. The gold standard for treatment outcome has been raised from response (reduction in symptoms) to remission (absence of symptoms) or recovery (extended period of remission) (23). However, obtaining recovery implies not only the remission of symptoms but also a restoration of the underlying physiology associated with the illness. Therefore, further understanding of the neurobiological changes associated with MDD is necessary for identifying true recovery processes.Functional and structural changes in MDDAlthough much information still needs to be attained, imaging andother methods have begun to elucidate the neurobiological abnormalities associated with MDD. In particular, several prefrontal and limbic structures and their interconnected circuits have been implicated in affective regulation (Figure 2).These neuroanatomical areas include the ventromedial prefrontal cortex (VMPFC), lateral orbital prefrontal cortex (LOPFC), dorsolateral prefrontal cortex (DLPFC), anterior cingulated cortex (ACC), ventral striatum (including nucleus accumbens), amygdala and the hippocampus. Abnormalities in these areas have been shown in patients with MDD compared with healthy controls and thus suggest a foundation for the symptomatic expression of MDD (24, 25). However, these deviations may be obscured or not present at the individual patient level and thus, these findings cannot necessarily be considered pathognomic.
Figure 2Major depressive disorder affects the dynamic connectivity among neuroanatomical structures involved in regulation of mood and stress response. Limbic structures (amygdala, hippocampus and nucleus accumbens) have reciprocal connections with ‘para-limbic’ ...As an integrated circuit, the prefrontal cortex, cingulate, amygdala, and hippocampus serves not only mood regulation, but also learning and contextual memory processes. Within the prefrontal cortex, the VMPFC mediates pain, aggression, sexual functioning and eating behaviours whereas the LOPFC assesses riskand modulates maladaptive and perseverative affective states

(behaviours). These two areas have a reciprocal pattern of activity with the DLFPC, which maintains executive function, effortful sustained attention, and working memory processes (26).Subdivisions within the ACC assume diverse roles, with the dorsalACC being part of the cognitive/executive functioning network andthe ventral ACC being involved in assessing emotional and motivational information. The ACC also monitors outcomes of behaviour and cognition and makes adjustments based on changing contingencies (27, 28).In patients with MDD, regional blood flow studies suggest hyperactivity in the VMPFC and LOPFC and hypoactivity in the DLFPC compared with controls (24). Given the functions of these regions, as previously described, this abnormal activity pattern may be responsible for the manifestations of symptoms associated with MDD. Hyperactivity of the VMPFC is associated with enhanced sensitivity to pain, anxiety, depressive ruminations and tension whereas hypoactivity of the DLFPC may produce psychomotor retardation, apathy, and deficits in attention and working memory. Using fMRI paradigms, connectivity studies have also suggested a decrement in the ‘communication’ between amygdala andACC regions (29). A consequence of this loss of connectivity could be a failure of the ACC to serve its inhibitory role in emotional regulation (30), resulting in further motivational and affective disruption (31).At the intersection of limbic, cognitive/executive and neuroendocrine regulatory circuits, including the hypothalamic-pituitary-adrenal axis (HPA), the hippocampus may be particularlyvulnerable in depression. Imaging studies of hippocampal volume have been of particular interest. In a meta-analysis of 12 studies, hippocampal volume was found to be consistently and significantly reduced in patients with MDD compared with controls, and these reductions occurred bilaterally with a slightly greater decrement in right hippocampal volume (32). Other studies have shown that the degree of hippocampal reductionis directly proportional to the number and the duration of untreated depressive episodes (33). Among depressed inpatients, while controlling for the effect of age, hippocampal volume was significantly correlated with duration of illness prior to hospitalisation (34). Even after remission of an episode, patients with recurrent MDD have continued to show significantly smaller hippocampal volume compared with healthy controls (35).Differences in hippocampal volume between patients with depression and healthy controls may not be fully attributable to

the disease state. Heritability studies of hippocampal volume suggest both environmental and genetic contributions with heritability estimates of 54% in nonhuman primates and 40% in adult male twins (36, 37). Several genomic imaging studies, comparing patients with MDD and healthy controls, have shown associations between hippocampal volume and specific genes that are implicated in mood disorders (38, 39). In a 1-year prospective study of 30 patients with MDD, hippocampal volume didnot significantly change during the study period, but patients whose depression failed to remit had a significantly smaller hippocampus at baseline and at 1 year than did patients who did remit (40). Combining the evidence from these genetic, cross-sectional, and clinical treatment studies suggests that morphological differences in the hippocampus may be a predisposing factor in MDD, but changes can also accumulate in the course of the disease and thereby create an obstacle to full recovery.Molecular processes mediating neurobiological changesThe alteration in the hippocampus signifies a potential outcome of injurious feedback that occurs via neuroendocrine dysregulation. A consistent finding in patients with MDD is a high level of the stress hormone cortisol, which may cause impairment in neuroplasticity and cellular resistance (41). An imbalance between glucocorticoid and mineral corticoid receptors in MDD along with high-density glucocorticoid receptors (GRs) mayalso contribute to the hippocampus’ susceptibility to neuronal damage (42). Subsequent hippocampal atrophy could result in further neuroendocrine dysfunction and hence a potential ‘run-away’ system (43). Postmortem comparisons of brain tissue in patients with MDD and age-matched healthy controls have shown hippocampal shrinkage in depressed subjects that was caused by increased density of neuronal cells and a significant reduction in neuropil (i.e. decreased dendridic branching and spine complexities) (44).A corollary of elevated glucocorticoids and compromised hippocampal functioning may also be the down-regulation of the GRsensitivity. Under conditions of chronic stress, decrease in GR sensitivity can have negative consequences as GR signalling becomes insufficient to ‘turn off’ the initial responses to stress as part of a negative feedback process (45, 46) (Figure 3). Subsequently, HPA hypothalamic overactivity, in conjunction with amygdala activation, leads to increased sympathetic tone, which promotes the release of cytokines from macrophages.

Increase in pro-inflammatory cytokines has been associated with loss of insulin and GR sensitivity, which further perpetuates metabolic and neuroendocrine disruption (47). Symptomatically, disruptions as a result of proinflammatory cytokines may be experienced as fatigue, loss of appetite and libido as well as hypersensitivity to pain (48).
Figure 3Molecular processes are impacted by stress and depression. Stressresults in release of glucocorticoids and corticotrophin releasing hormones (CRH) and pro-inflammatory cytokines (TNF, IL-1, IL-6). In depression, disruption of serotonin (5-HT), norepinephrine...Proinflammatory cytokines may also diminish neurotrophic support and monoamine neurotransmission that can lead to neuronal apoptosis and glial damage. Alterations in glia–neuron relationships have been recently emphasised in the aetiology of neuropathic pain and MDD (47, 49). Glia cells are involved in an intricate interaction with neurons in which astroglia and microglia maintain homeostasis of the neuronal environment by modulating electrolytes, neurotransmitters, cytokines and neurotrophic factors (50). Neurons reciprocate support of glial function via neurotrophin signalling. Stress, depression and ensuing peripheral immune dysregulation lead to activation of microglia that then contribute to the existing immune disruption by additional release of inflammatory cytokines (51).An integral part of maintaining the health of these glial–neuron interactions may be mediated by brain-derived neurotrophic factor(BDNF) (52). Involved in neurogenesis, BDNF is the primary neurotrophin of the hippocampus. As a dimeric protein involved incell maintenance, plasticity, growth and death (apoptosis), BDNF is structurally related to nerve growth factor and is distributedwidely throughout the brain (53). When BDNF interacts with tyrosine receptor kinase receptors (TRkB), it promotes cellular resilience and long-term potentiation. However, the precursor form of BDNF (pro-BDNF) can also precipitate reduction in dendritic spines and cell death when it binds with the p75 receptor. Thus, depending upon its expression, BDNF can prune neural networks in an activity dependent manner that is regulated

by various neurotransmitters [glutamate, GABA, 5-HT, norepinephrine (NE), acetylcholine, dopamine and hormones] (54).Preclinical and clinical studies have suggested dysregulation in BDNF occurs under conditions of chronic stress and depression. Inanimal models, acute and chronic immobilisation stress resulted in decreased BDNF expression using mRNA assays. Similar results were also observed following administration of acute and chronic pain stimuli (55). Within humans, levels of serum BDNF has been found to be significantly lower in untreated patients with MDD compared with treated patients or healthy controls (56). Similarly, postmortem analyses of brains of persons who committedsuicide showed that BDNF and another neurotrophin (NT-3) were significantly reduced compared with non-suicide controls (57).From the above observations, the neurotrophic hypothesis has emerged as a major theory for the pathogenesis of major depression. In this model, stress and genetic vulnerability elevate glucocorticoid steroids and alter cellular plasticity viadownregulation of growth factors and receptor sensitivity (4). The reduction in growth factors, such as BDNF, impacts negativelyon the structural and functional processes within the limbic system, especially for the hippocampus. Chronic and recurrent MDDmay result in subsequent atrophy and further disruptions in neurocircuitry. From this hypothesis, recovery and remission of MDD would be dependent upon a reversal of these processes, such as an increase in BDNF levels.Complementing the neurotrophic hypothesis of MDD is the monoaminetheory, which postulates that depression is associated with low levels of monoamines, particularly, 5-HT and NE. A recent imagingstudy of patients with untreated depression found a high global receptor density for the monoamine oxidase A (MAO-A), which nonspecifically metabolises these neurotransmitters. In this updated theory, long-term monoamine loss because of this global MAO-A activity interacts with regional specific transporter densities (i.e. 5-HT, NE), resulting in the expression of the depressive illness (58). Both 5-HT and NE ascending fibres originate from brainstem nuclei and innervate the limbic system, prefrontal cortex and associated structures involved in the regulation of mood. Descending pathways project through the dorsolateral spinal column and are instrumental in the regulationof pain (59, 60). Therefore, depending upon the specific transporter densities within these regions, various symptoms of depression (mood, cognition and pain) will be manifested within

the context of the overall global reduction in monoamine levels (58).Role of neurotransmitters in recovery from MDDTherapeutically, selective serotonergic reuptake inhibitors (SSRIs) and NE reuptake inhibitors (NRIs) are known to increase their respective monoamine levels in the brain. Chronic treatmentwith monoamine reuptake inhibitors increases activation of cyclicadenosine 3-5 monophosphatase (cAMP), which in turn stimulates protein kinase A. Activation of this protein enzyme regulates target genes leading to an increase in BDNF synthesis (52). The antidepressant-induced cAMP activity can also enhance GR sensitivity and inhibit cytokine signalling, further assisting inthe restoration of the neurocircuitry feedback loops (61).The effect of increasing monoamine levels (dopamine, 5-HT and NE)on BDNF and growth factors may be one mechanism that produces theantidepressant response. Preclinical study of rat brain cells hasdemonstrated that monoamenergic activity (NE, 5-HT) upregulates BDNF synthesis in astrocytes (62). Clinically, successful treatment with antidepressants results in normalisation of serum BDNF level, which is considered an indirect measure of cortical BDNF activity. Support for the relationship between serum and cortical BDNF levels has been derived from correlations in animalstudies as well as findings that serum BDNF passes the blood–brain barrier and reflects stored and circulating BDNF in humans (63, 64). In a study of 10 patients who were treated for 12 weekswith a dual reuptake inhibitor, improvement in depressive symptoms was correlated with increases in BDNF levels, and the BDNF levels of remitted patients had normalised to the same levelobserved in healthy controls (65). Response to various SSRI and 5-HT noradrenalin reuptake inhibitors (SNRI) treatments has been similarly associated with restoration of normative BDNF values (66) (Figure 4). Postmortem analysis of brain tissue has shown that subjects who had been treated with an antidepressant at timeof death had greater hippocampal BDNF expression as measured by immunoreactivity than did untreated subjects with mood disorders (67).
Figure 4

Antidepressant therapy is associated with restoring normative processes. Treatment with various selective serotonin antidepressant treatments and serotonergic noradrenergic reuptakeinhibitors resulted in increases in serum brain-derived neurotrophic ...Antidepressant therapeutic response is also associated with re-establishment of normative cortical activity. A study of 17 inpatients with MDD examined regional activity changes following 1 week and 6 week fluoxetine treatment. At 1 week, all patients showed increases in hippocampal activity and decreases in posterior cingulate and prefrontal cortex activity. After 6 weeksof treatment, patients who had responded to treatment showed a reversal of this pattern with decreased limbic activity and increased prefrontal cortical activity whereas non-responders continued to show the 1-week pattern (68). Normalisation in the amygdala and ACC has also been associated with positive response to treatment. Using a masking paradigm for subconscious activation, patients with MDD showed a baseline hyper-reactivity of the left amygdala that attenuated following 8-week treatment with sertraline (69).Other lines of evidence also support the restorative nature of antidepressant therapy. Structural and functional MRI assessmentsof patients with MDD who were treated with fluoxetine indicated the importance of ACC grey matter volume for treatment response as there was a positive association among grey matter volume, normalisation of ACC activity, and response to treatment (70). Conversely, in patients with MDD who failed to respond to antidepressant treatment, plasma levels of proinflammatory cytokines were elevated compared with healthy controls or euthymic patients with MDD (71).Symptomatically, improvements in specific MDD symptoms have been associated with regional improvements in brain metabolic activity. In 39 outpatients with MDD, improvement in cognitive symptoms was correlated with increases in DLPFC and improvements in fatigue/psychomotor retardation was associated with decreases in VMPFC activity. Interestingly, these changes were seen in responders regardless of whether treatment was pharmacological orpsychological (72). Restoration of the neurobiological regulationin MDD via neurotrophic factors and neurogenesis appears to be a common factor across various effective treatments for MDD, including pharmacological, psychological and somatic treatments, such as diet and exercise (73).Treatment implications of the neurobiological model

The neurobiological sequelae and repercussions of chronic or recurrent MDD indicate that interventions for MDD should be focused on achieving optimal treatment early. Longitudinal studies have shown that one of the best predictors of remission status at 2 years was response to acute treatment, i.e. initial 6weeks (74). In addition, the adequacy of treatment may also have prognostic implications. For patients with late-life depression, exposure to previous inadequate trials of antidepressants resulted in a reduced response rate to pharmacological intervention augmented by psychotherapy compared with treatment of naive patients, even after controlling for baseline severity (75). Similarly, in a large observational study of 996 patients with MDD, non-response or incomplete response to initial antidepressant treatment was a significant predictor of eventual treatment resistance (76). On the positive side, an early response to antidepressants has been shown to predict greater treatment adherence (77).One way of maximising early response is to apply a comprehensive treatment that increases activity of multiple monoaminergic systems. In a double-blind, randomised treatment study, 39 inpatients with MDD received either fluoxetine (a serotonergic intervention), desipramine (a noradrenergic intervention) or their combination. After 6 weeks of treatment, patients who had been given the combination treatment were more likely to achieve remission (53.8%) than either intervention alone (0 % and 7.1%) (78). Similarly, a recent large meta-analysis encompassing 93 trials and 17,036 patients compared efficacy outcomes of SSRI with SNRI treatments for MDD that showed a modest but significantadvantage in efficacy with SNRI treatments (79). An earlier meta-analysis did not find a difference in efficacy between SSRIs and dual acting agents (mostly tricyclic antidepressants), with the exception of the inpatient populations, where dual acting tricyclic antidepressants had an advantage (80). Thus, although current treatment algorithms for MDD usually are initiated with SSRIs, the role of combination treatment or dual reuptake inhibitors are increasingly being considered as a preferred option (81).Another advantage of targeting both of 5-HT and NE systems is improvement not only in the core features of MDD, but also in associated physical symptoms. Painful physical symptoms are prevalent in patients with MDD, and these symptoms increase the illness burden and impair the ability to attain remission (82, 83). In a study of primary care patients with MDD who were

treated with SSRIs for 9 months, mood symptoms continued to improve over time while painful physical symptoms persisted (84).The occurrence of painful physical symptoms and MDD reflects the shared underlying pathophysiology between mood and pain regulation. Importantly, there may be also a synergistic interaction between the 5-HT and NE systems to obtain analgesia. In an animal model of pain, treatment with dual reuptake inhibitors or combination treatment (5-HT/NE) appeared to enhancethe effectiveness of pain alleviation (85). Clinically, patients with MDD who experienced a 50% or greater reduction in pain were more likely to achieve remission than patients whose pain reduction was < 50% (86).With remission and recovery as the goal, the treatment guidelinesderived from the neurobiological model emphasise the need for notonly early and comprehensive intervention, but also vigorous attention to residual symptoms. In a 2-year study of outpatients with MDD, patients who obtained only a partial remission of symptoms were more likely to relapse (67.5%) than patients who had attained full remission (15.2%) (87). Specific recommendations for the treatment of residual symptoms have not been determined empirically, but likely require additional augmentation with other pharmacological and psychological treatments; in addition to reducing the risk of relapse, the treatment of residual symptoms may enhance compliance and long-term outcomes (88).ConclusionsAs the underlying neurobiological model of depression is increasingly understood, treatment providers are directed to recognise that the factors that may initiate a MDD episode and those that maintain the illness are likely to be very different. Genetic and stress vulnerabilities interplay to initiate a cascade of neurobiological alterations that disrupt a dynamic system. Progressive effects of recurrent and chronic MDD may thenbe potentiated by further structural and functional abnormalities.Given these long-term consequences, an essential objective of treatment must be to restore normative functioning and prevent further neurobiological structural alterations. Increasing 5-HT and NE neurotransmission is likely to initiate true recovery withthe restoration of neurotrophic support, glucocorticoid signalling and neuroendocrine regulation. The use of dual reuptake inhibitors enhances the probability of remission as it addresses the complex interplay of the emotional and physical

symptoms of MDD. Painful physical symptoms are increasingly recognised as having a significant impact on functioning and recovery; thus, affirming the need for antidepressant treatments that can effectively reduce these symptoms as well.From the neurobiological model, the treatment guidelines of early, comprehensive and progressive treatment require a change in perspective for both patients and providers. A residual symptom may be interpreted as a proxy of an active disease state,with ensuing structural alterations and systemic consequences. With remission and recovery as the goal, patients will need to beeducated about the benefits of long-term treatment rather than episodic or incomplete intervention. A biopsychosocial treatment model that incorporates cognitive-behavioural or interpersonal therapy along with pharmacological interventions serves to address both the initiation and maintenance factors and can reduce the risk of relapse (89). Once remission is attained, maintenance of effect may become the more appropriate term, rather than relapse prevention, to emphasise the necessity for anongoing collaboration between patient and physician in order to maintain neurobiological homeostasis.Article informationInt J Clin Pract. 2007 December; 61(12): 2030–2040.doi: 10.1111/j.1742-1241.2007.01602.xPMCID: PMC2228409V Maletic,1 M Robinson,2 T Oakes,2 S Iyengar,2 S G Ball,2,3 and JRussell2……………………………………………Stress, Serotonin Receptors, and the Neurobiology of Depressionhttp://www.cyberounds.comJuan F. López, M.D.Department of Psychiatry and Mental Health Research InstituteUniversity of Michigan, Ann ArborEducational ObjectivesUpon completion of this Cyberounds®, the participant should be able to:1) List some of the signs and symptoms of Major Depressive Disorder.2) Describe research findings showing hyperactivity of the Hypothalamic-Pituitary Adrenal Axis in Major Depressive Disorder3) Describe some of the serotonin receptor changes found in the brains of suicide victims.4) Discuss some of the ways in which the Hypothalamic-Pituitary-Adrenal Axis and the Serotonin system interact.

5) Discuss the evidence linking stress with the onset of depressive symptoms.IntroductionMajor Depressive Disorder (MDD), also known as Major Depression, is a psychiatric syndrome characterized by pervasive disturbancesin mood, sleep, appetite, energy, motivation, hedonic capacity, and thinking. According to the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), a depressive episode must be diagnosed if a patient has had depressed mood, or has lost interest or pleasure in most activities, for a duration of at least two weeks. However, depressive episodes often last months, sometimes years, and they carry a significant impairment in social and occupational functioning. Depressive episodes also tend to be recurrent, and if left untreated, most patients will have multiple episodes during their lifetime. The episodes tend to become more frequent, and/or more severe as the disease progresses (Post 1992). This is known as the "kindling" or "sensitization" hypothesis of mood disorders.Table I. Diagnostic criteria for a Major Depressive Episode (Adapted from DSM-IV)At least 5 of the following symptoms have been present during thesame 2-week period and represent a change from previous functioning
1 Depressed mood most of the day, nearly every day2. Markedly diminished interest or pleasure in all, or almost all, activities3. Significant weight loss when not dieting or weight gain, or decrease or increase in appetite nearly every day4. Insomnia or hypersomnia nearly every day5. Psychomotor agitation or retardation nearly every day6. Fatigue or loss of energy nearly every day7. Feelings of worthlessness or excessive or inappropriate guilt8. Diminished ability to think or concentrate, or indecisiveness9. Recurrent thoughts of death, recurrent suicidal ideation, or asuicide attempt or a specific plan for committing suicideMDD is a common medical disorder, with a lifetime prevalence of 17.1%, and a 12 month prevalence of 10.3%, in the general population (Kessler et al., 1994). However, the prevalence of MDDin medical populations is even higher, with rates ranging from 30% to 50%, depending on the specific medical condition. This psychiatric disorder also carries a significant morbidity and

mortality, with a negative impact not only for the patient itself, but also for the family, and for society in general. About two thirds of severely depressed patients exhibit suicidal ideation, and 10% to 15% commit suicide (Kaplan and Sadock, 1991).The specific pathogenetic cause of depression is unknown, but it is widely accepted that its etiology, course, and long-term prognosis are influenced by genetic, environmental and neurobiological factors. A greater understanding of the neurobiology of this disorder is leading us to better treatment strategies that can prevent or decrease the impact of this disease. Although many neurotransmitters and neurohormones have been linked to the pathophysiolgy of depression, (e.g. norepinephrine, dopamine, thyroid hormones), research studies have implicated disturbances in the serotonin (5-HT) system and the Hypothalamic-Pituitary-Adrenal (HPA) axis as two of the neurobiological alterations most consistently associated with affective illness (Gold et al., 1988; Meltzer, 1988; Kathol et al., 1989; Nemeroff, 1998). Therefore, this presentation will focus on the regulation of these two systems, as they relate to depression.Historically, the role of each of these two biological systems inmood disorders have been studied independently, but more recently, their interaction in the brain, as it relates to the pathophysiology of depression, has received increased attention. In this presentation, we will review recent animal and human findings, from the perspective of the 5-HT and HPA axis, which shed new light into the neurobiology of MDD. We will discuss someof the ways in which adrenal glucocorticoids and 5-HT receptors interact during conditions of chronic stress, or severe "allostatic load". We will also discuss what is the potential significance of these findings for the pathophysiology and treatment of mood disorders.That disturbances in the Hypothalamic-Pituitary-Adrenal axis and in serotonin may share a common pathophysiological mechanism is not surprising, since we know from animal studies that they interact extensively and that they are related in a variety of ways (McEwen, 1987; Chalmers et al., 1993; López et al., 1999). The hippocampus, and the paraventricular nucleus of the hypothalamus (PVN), in particular, are anatomical regions in which components of the Hypothalamic-Pituitary-Adrenal axis and the 5-HT systems have a rich representation (see Figure 1 for an illustration of how serotonin neurons from the raphe nucleus

project to the PVN and hippocampus). These regions are also part of the limbic system, an area implicated in the regulation of several vegetative functions (arousal, sleep, appetite and hedonic capacity) as well as in the control of mood. The hippocampus has also been implicated in memory and cognitive function. The recognition that the hippocampus is an integral component of the Hypothalamic-Pituitary-Adrenal axis has led someinvestigators (including our laboratory) to refer to this neuroendocrine system as the "Limbic"-Hypothalamic-Pituitary-Adrenal (LHPA) axis. We will therefore, for the purpose of this presentation, use the term "LHPA", instead of the more commonly used term "HPA", when referring to this system.The Limbic-Hypothalamic-Pituitary-Adrenal Axis and StressThe LHPA is the classic neuroendocrine system that responds to stress. Perception of stress by an organism results in a series of events, the final result of which is the secretion of glucocorticoids (cortisol in humans, corticosterone in rats) fromthe adrenal cortex (Dallman et al., 1987). Activation and termination of the adrenocortical stress response is critical foradaptation and survival. Inhibition of stress responsiveness is partly achieved by the binding of circulating glucocorticoids to specific cytoplasmic receptors in hypothalamus, where they inhibit corticotropin releasing hormone (CRH) and consequently pituitary adrenocorticotropin (ACTH) secretion. Additional modulation of the system is apparently achieved in limbic structures, especially the hippocampus, a structure that is linked to the hypothalamus through neuronal connections that converge on the paraventricular nucleus of the hypothalamus (PVN), where the stress responsive CRH and vasopressin (AVP) neurons reside (López et al., 1991).There are several lines of evidence that highlight the importanceof the hippocampus for LHPA feedback mechanisms (McEwen, 1991). Pioneer work by McEwen demonstrated that the hippocampus containsa high abundance of two types of glucocorticoid receptors which are thought to control negative feedback: Type I (also known as Mineralocorticoid Receptor, or "MR") and Type II (also known as Glucocorticoid receptors or "GR"). Type I (or MR) resembles the kidney mineralocorticoid receptor and has stringent specificity, binding selectively corticosterone, the main glucocorticoid of the rat. In the rodent brain, MR is most densely localized in hippocampal and septal neurons. Type II or GR receptors are widely distributed in rat brain, including hippocampus, hypothalamus, and prefrontal cortex. However, they bind

corticosterone with a lower affinity compared to the Type I (or MR) receptor. Their highest affinity is for potent synthetic glucocorticoids, such as dexamethasone (McEwen, 1991). These receptor characteristics complement each other and put the MR andGR in a position to modulate LHPA responses (see DeKloet et al., 1998, for a recent excellent review on the neurobiology of GR andMR). The MR receptors appear to be operative at low corticosterone concentrations and may offer tonic inhibition to the axis during the nadir of the circadian rhythm (De Kloet et al., 1988; McEwen 1991). When high concentrations are present, MRreceptors saturate, and the GR receptors ensure the return of homeostasis. As stated above, it is also well established that the hippocampus is a central component of limbic circuitry and isfundamental in controlling aspects of cognitive and behavioral functions, putting these receptors in a position to modulate simultaneously the neuroendocrine and cognitive response of the organism to stress.
The Limbic-Hypothalamic-Pituitary-Adrenal Axis in DepressionHyperactivity of the LHPA axis is a well documented phenomena in MDD. This dysregulation is manifested, among other things, by cortisol hypersecretion, failure to suppress cortisol secretion after dexamethasone administration, exaggerated adrenal responsesto endocrine challenges, and blunted ACTH response to CRH

administration (Carroll et al., 1976; Kalin et al., 1987; López et al., 1987; Gold et al., 1988; Kathol et al., 1989b). This lastobservation has been interpreted as evidence of pituitary downregulation of CRH receptors secondary to an increase in secretion of CRH. There is indeed good evidence of increased central drive, based on increased activity at the nadir of the circadian rhythm (Young et al., 1995) as well as more direct findings of elevated CRH in the CSF of depressed patients (Nemeroff et al., 1984), and increased CRH immunoreactivity and mRNA levels in the PVN (Raadsheer et al., 1994, 1995). Interestingly, post-mortem studies have also found evidence of chronic LHPA activation in suicide victims, such as adrenal hyperplasia (Dorovini-Zis and Zis, 1987), downregulation of CRH receptors (Nemeroff et al., 1988), and increases in proopiomelanocortin mRNA, the precursor for ACTH, in the pituitary (López et al., 1992). It is not known whether these LHPA changes in suicide are due to the fact that a significant subset of suicide victims are patients with depressive disorders,to the stress surrounding the suicide itself, or to a neurobiological "abnormality" common to all suicides irrespectiveof diagnosis.We have found, in a group of suicide victims with a history of depression, decreases in hippocampal MR mRNA levels (López et al., 1998), and more recently, a decrease in GR mRNA levels in prefrontal cortex (unpublished observation) an area of the brain which is associated with higher cognitive, and executive function. We don�t know whether these changes represent a genetic, or developmental, vulnerability "marker" for suicide, orfor depression. Nevertheless, these findings are consistent with a history of exposure to chronic stress and/or to high peripheralglucocorticoid levels (Herman and Watson, 1994; Herman et al., 1995).Historically, the presence of LHPA overactivity in patients with depression was believed by many to be a "secondary" phenomena of the illness, reflecting either a central monoaminergic disturbance, the stress of the illness, or both. However, over the past few years, it has become clearer that the LHPA abnormalities in MDD are intimately linked to the pathohysiology of the disease. This change in perspective was stimulated in partby the increased awareness that glucocorticoids, the final products of the LHPA axis have been shown to have profound effects on mood and behavior (McEwen 1987). For example, a high incidence of depression is linked to pathologies involving

elevated corticosteroid levels, such as Cushing's syndrome. This corticosteroid-induced depression usually disappears when corticosteroid levels return to normal (Kathol 1985; Murphy 1991). In fact, it has become increasingly clear, from both animal and clinical studies, that circulating glucocorticoid levels provide important hormonal control of affect, which may bemediated by steroid-induced modulation of central limbic circuitry (McEwen 1987). The precise mechanism by which corticosteroids exert this influence on affect is not well understood. However, this mechanism is likely to involve interactions with brain neurotransmitters, since we know that central control of affect is intimately associated with the actions of the monoamines serotonin, norepinephrine and dopamine.Serotonin Receptors and DepressionThe serotonin system has been widely investigated as a key element in the pathophysiology of Depression (Meltzer, 1989), andof suicide (Mann et al., 1989), and as a mediator of the therapeutic action of antidepressants (Berendsen, 1995). Althoughthe 5-HT system has many components, the three 5-HT molecules believed to be most closely associated with the neurobiology of mood are the serotonin transporter (5-HTt), the serotonin 1a receptor (5-HT1a), and the serotonin 2a receptor (5-HT2a, formerly 5-HT2). Most new (as well as older) antidepressants inhibit the re-uptake of serotonin from the synapse, and alter 5-HTt protein and mRNA levels (López et al., 1994; Owens and Nemeroff, 1998). Animal studies have also demonstrated that chronic antidepressant administration affects the function and number of the 5-HT1a and 2a receptors. Many electrophysiological studies have shown that antidepressants "upregulate" or "sensitize" 5-HT1a function in the hippocampus, while at the sametime they "down-regulate" or "desensitize" 5-HT1a function in theraphe, where it acts as an inhibitory somatodendritic receptor (Blier and de Montigny, 1994). Some studies have also reported a modest increase in 5-HT1a receptor number in hippocampus following antidepressant administration to rodents (Welner et al., 1989; Klimek et al., 1994). We have found that suicide victims with a history of depression have decreases in 5-HT1a gene expression in hippocampus (López et al., 1998). These changes are in the opposite direction of what is observed with antidepressant treatment in rat hippocampus.The 5-HT2a receptor has also been shown to be affected by chronicantidepressant treatment. Most, but not all, studies have reported decreases in 5-HT2a binding in the prefrontal cortex

after chronic antidepressant administration (Peroutka and Snyder,1980; Bourin and Baker, 1996). The opposite changes (5-HT2a upregulation) are found in the prefrontal cortex of suicide victims (Mann et al., 1986; Arango et al., 1990; Hrdina et al., 1993), although these findings are not universal (Stockmeier, 1997). In addition, subjects with a history of MDD dying of natural causes have increases in 5-HT2a binding in the prefrontalcortex (Yates et al., 1990).These findings have led several investigators to propose that postsynaptic 5-HT1a and 5-HT2a receptors have functionally opposing effects (Schreiber and De Vry, 1993), that a disturbed balance of these receptors may be contributing to the pathophysiology of Depression (Berendsen, 1995), and that restoring this balance is necessary for antidepressant action (Borsini, 1994).Interaction Between the LHPA axis and SerotoninAnother level of complexity is added by the fact that 5-HT and the LHPA axis interact at multiple levels. For example, it is known that some of the 5-HT neurons arising from the nucleus raphe dorsalis and nucleus raphe magnus project to the PVN and synapse onto CRH neurons (Fuller, 1992). 5-HT neurons also project to other brain areas, such as the amygdala and the suprachiasmatic nucleus, which are thought to modulate the function of the PVN (Törk, 1990). Pharmacological stimulation with 5-HT agents can activate ACTH and cortisol release (Fuller, 1992). Many brain areas that express 5-HT receptors also have abundant concentrations of corticosteroid receptors. In the limbic system in particular, the hippocampus has high concentrations of 5-HT1a in the same neurons that contain abundant GR and MR receptor levels, and the prefrontal cortex is rich in 5-HT2a receptors (Pazos and Palacios, 1987), as well as GR receptors.Interestingly, unlike rodents, human prefrontal cortex also has significant amounts of MR mRNA. This constitutes anatomical evidence suggesting that MR may have a more expanded role in modulating brain function in humans than in rodents. Furthermore,the co-localization of MR and GR in prefrontal cortex suggest that both of these receptors are capable of modulating higher brain functions, such as mood, social behavior, and cognitive processing, perhaps by interacting with 5-HT receptors.Serotonin and corticosteroid receptors not only interact anatomically, but also functionally. It has been reported that administration of serotonin can up-regulate GR in the

hippocampus, and that conversely, pharmacological destruction of serotonergic projections decreases GR and MR mRNA levels in the hippocampus (Betitto et al., 1990; Seckl et al., 1990). Regulation in the other direction, i.e. glucocorticoid regulationof 5-HT receptors, has also been reported. Animal studies have demonstrated that adrenalectomy and corticosteroid administrationpowerfully regulate 5-HT1a receptor number and mRNA in the hippocampus (Chalmers et al., 1992, 1994; Kuroda et al., 1994). This effect of glucocorticoids seems to be specific for the postsynaptic 5-HT1a receptor, since the somatodendritic 5-HT1a isnot affected by these manipulations (Chalmers et al., 1992).The 5-HT2a receptor is also sensitive to changes in peripheral glucocorticoid levels. Upregulation of 5-HT2a in rat neocortex cortex has been reported following ten days of exogenous administration of ACTH (Kuroda et al., 1992). This effect is abolished by adrenalectomy and mimicked by corticosterone administration for ten days. Dexamethasone treatment for the sameamount of time also causes a dose dependent increase in 5-HT2a binding in cortex (Kuroda et al., 1993), suggesting that this effect is mediated by GR, since dexamethasone is a potent GR agonist. Interestingly, the effect of corticosteroids on 5-HT2a receptor number seems to be specific for the cortex. Dexamethasone did not alter 5-HT2a binding in rat hippocampus. Given the relatively equal abundance of MR and GR in the hippocampus, and the preponderance of GR in rat frontal cortex, it is plausible that 5-HT receptor regulation in the cortex is mediated through different mechanisms than in the hippocampus.Stress, Serotonin and the LHPA axisIn reviewing the clinical, psychological and biological literature on depressive illness, one factor that emerges as being closely associated with depression is stress. Stress and depression have been linked in a variety of ways: For example, both physical and psychological stressors have been shown to be temporally (and perhaps causally) related to the onset of depressive episodes (Post 1992). Some studies have suggested that, at least for recurrent depression, stressful life events are more common in "non-endogenous depression" (Frank et al., 1994). Other studies have found that stressful life events are significantly correlated even with the first episode of psychotic/endogenous depression (Brown et al., 1994). This is notto say that stress "causes" depression in people. Rather, stress is very likely interacting with an endogenous genetic predisposition, such that in some vulnerable individuals, a

stressor can precipitate a mood disorder (i.e. vulnerability + stress = depression). In fact, studies in twins by Kendler et al have demonstrated a clear interaction between genetic substrate and a recent stressful life event in the precipitation of a depressive episode: the more the genetic "loading" for depression, the more likely than a stressful event will trigger an episode. There are of course cases in which the genetic "loading" or predisposition is so high, that an episode of depression can occur in the absence of any apparent precipitatingfactors.Another important link between depression and stress is the fact that both the LHPA and 5-HT systems, in addition to been involvedin the pathophysiology of depression, are also, as we have discussed above, critical contributors to the neurobiology of stress (McEwen 1987) . Therefore, studying the neurobiology of stress by focusing on these two systems has given us important clues into the pathophysiology of affective illness, shed light on the actions of antidepressants, and begin to reveal how stressand mood disorders are related.We have used chronic unpredictable stress, a postulated animal model of depression (Katz and Sibell, 1982; Armario et al., 1988)to investigate the parallel changes in 5-HT and LHPA related molecules, and to study how chronic stress and circulating glucocorticoids influence the 5-HT receptor system (López et al.,1997, 1998,1999b). In this paradigm, rats are exposed to different mild to moderate stressors everyday, therefore making the stress "unpredictable" from day to day. Rats that undergo this treatment show LHPA overactivity, and increases in peripheral glucocorticoids, very similar to those found in MDD (Chapel et al., 1996; Armario et al., 1988). We found that rats subjected to this paradigm show a significant decrease in 5-HT1a mRNA and binding in the hippocampus, as well as decreases in MR mRNA levels in this same region (López et al., 1998). Chronic unpredictable stress also causes a significant increase in 5-HT2areceptor and mRNA levels in the prefrontal cortex (López et al, submitted). As stated above, these "opposite" effects (hippocampal 5-HT1a downregulation, cortical 5-HT2a upregulation)are also found in suicide victims with a history of depression (Mann et al., 1989; López et al., 1997). It is not clear if these5-HT receptor changes are due to a direct effect of corticosteroids on the receptor themselves, or if they are secondary to corticosteroid induced changes in serotonin synthesis and turnover. However, the effects of chronic stress on

these 5-HT receptors are prevented if the stress-related glucocorticoid increase is abolished. No changes are found in thepresynaptic 5-HT transporter sites after chronic stress, suggesting that the effects are mainly in the postsynaptic receptors (López et al., 1997, 1998).Antidepressant Medications, the LHPA Axis and 5-HT ReceptorsThe LHPA overactivity observed with chronic unpredictable stress can also be prevented by the chronic administration of either imipramine or desipramine, two tricyclic antidepressants (López et al., 1998). Both desipramine and imipramine also reverse the stress induced downregulation of 5-HT1a in hippocampus, and the 5-HT2a upregulation in cortex. On the other hand, zimelidine and fluoxetine, two specific serotonin reuptake inhibitors, are unable to prevent the stress-induced elevation in corticosterone levels. This failure to prevent the LHPA overactivity is associated with a failure to restore the 5-HT receptor changes tobaseline levels. This suggests that failure of an antidepressant to reverse the LHPA hypersecretion is associated with a failure to prevent the 5-HT receptor "dysregulation" secondary to chronicstress. We have proposed that this may be one of the neurobiological mechanisms underlying "treatment resistance" in patients with severe depression (López et al., 1997).There is some clinical evidence that these mechanisms may be operating in depressive disorders. Persistence of hypercortisolemia after antidepressant administration in depressed patients has been associated with relapse and poorer treatment outcome (Greden et al., 1983; Ribeiro et al., 1995). Inaddition, some clinical studies have found that tricyclics are more effective than SSRIs in the treatment of melancholia (DanishUniversity Antidepressant Group, 1986, 1990; Roose et al., 1994),Venlafaxine, an antidepressant with both norepinephrine and 5-HT reuptake activity, was reported to be more effective than fluoxetine in treating melancholic depression (Clerc et al., 1994), and in comorbid depression and anxiety (Silverstone et al., 1999). Since melancholia and severity of depression are associated with a higher incidence of hypercortisolemia (Kathol et al., 1989; Meador-Woodruff et al., 1990), it is possible that the presence of a severely disturbed LHPA axis in this populationmay be contributing to the relative resistance to SSRI treatment.Interestingly, many augmentation strategies in treatment resistant patients are in effect attempts to broaden the biochemical profile of the pharmacological treatment, which may

be more effective in reversing LHPA overactivity than a treatmentwhose main impact is in a single neurotransmitter system.Interplay Between Stress, the LHPA Axis, Serotonin, and AntidepressantsBased on the animal and human studies reviewed here, we can generate a working model of the interplay between stress, the LHPA axis, 5-HT receptors, and their potential interactions in suicide and depression. Figure 2 illustrates this model (solid arrows represent known effects, dashed arrows represent possible effects).While it is possible that some monoamine receptor changes are dueto antecedent changes in their endogenous ligands, we propose that many of the receptor changes observed may be a result of theLHPA overactivity present in at least some suicide victims, in particular those with a history of affective disorders. The rationale for this hypothesis derives from the above evidence, which can be summarized as follows:1) Depressed patients, as well as suicide victims, show evidence of overactivity of the LHPA axis. 2) Chronic stress and/or high steroid levels in rats result in an alteration of specific 5-HT receptors (e.g. increases in cortical 5-HT2a, decreases in hippocampal 5-HT1a). 3) Many human studies show the same receptorchanges in the brain of suicide victims (increases in cortical 5-HT2a, decreases in hippocampal 5-HT1a) as found in hypercorticoidstates. 4) Chronic antidepressant administration causes opposite 5-HT receptor changes to those seen with chronic stress 5) Antidepressant administration reverses the overactivityof the LHPA axis.If indeed the 5-HT1a and 5-HT2a receptors have at least a partialrole in controlling affective states (either directly or secondarily through other systems), then their modulation by corticosteroids provides a potential mechanism by which these hormones may regulate mood. This of course does not exclude the possibility that stress can be simultaneously acting through other systems, such as the CRH receptors, thereby synergisticallyaffecting mood and behavior. Antidepressants can counteract this phenomenon by affecting 5-HT receptor function directly and by simultaneously regulating stress induced corticosteroid secretion.
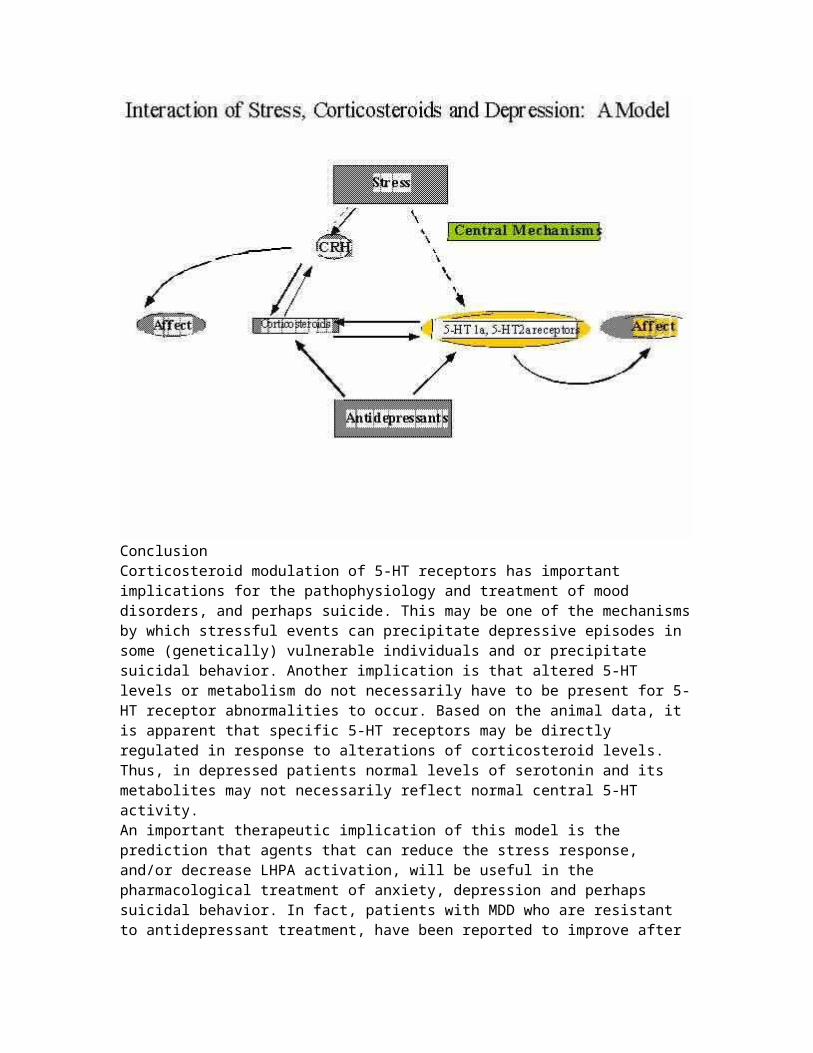
ConclusionCorticosteroid modulation of 5-HT receptors has important implications for the pathophysiology and treatment of mood disorders, and perhaps suicide. This may be one of the mechanismsby which stressful events can precipitate depressive episodes in some (genetically) vulnerable individuals and or precipitate suicidal behavior. Another implication is that altered 5-HT levels or metabolism do not necessarily have to be present for 5-HT receptor abnormalities to occur. Based on the animal data, it is apparent that specific 5-HT receptors may be directly regulated in response to alterations of corticosteroid levels. Thus, in depressed patients normal levels of serotonin and its metabolites may not necessarily reflect normal central 5-HT activity.An important therapeutic implication of this model is the prediction that agents that can reduce the stress response, and/or decrease LHPA activation, will be useful in the pharmacological treatment of anxiety, depression and perhaps suicidal behavior. In fact, patients with MDD who are resistant to antidepressant treatment, have been reported to improve after

receiving steroid suppression agents, like ketoconazole (Murphy et al 1991; Wolkowitz et al 1993). However, these agents have many side effects, and are often difficult for patients to tolerate. In this respect, CRH receptor antagonists, which are currently under development, may provide us with a new therapeutic weapon to treat these patients (De Souza 1995, Chalmers et al., 1996). These compounds could be used in conjunction with antidepressants, as adjuvants or augmenting agents, and may decrease treatment resistance. This agents may also be useful in monotherapy, since preventing hypercortisolemiamay be translated into an improvement of monoaminergic receptor function. The use of modern biochemical and pharmacological tools, coupled with our increased understanding of the neurobiology of depression, should allow us to test these hypotheses, first in animal models and then directly in patients with affective illness.
http://www-personal.umich.edu/~jflopez/Depression.html…………………………………………..CMAJ February 3, 2009 vol. 180 no. 3 doi: 10.1503/cmaj.080697ReviewNeurobiological mechanisms in major depressive disorderMarije aan het Rot PhD, Sanjay J. Mathew MD, Dennis S. Charney MDNearly 1 in 5 people will experience a major depressive episode at some point in their lives.1 In this review, we discuss data describing how genes, psychosocial adversity in childhood, and ongoing or recent psychosocial stress may impact multiple neurobiological systems relevant to major depressive disorder. Major depressive disorder may be caused by the cumulative effectsof these 3 factors on the brain.2A major depressive episode is characterized by a low mood or an inability to experience pleasure (anhedonia), or both, for more than 2 weeks, combined with several cognitive and vegetative symptoms and the occurrence of distress or impairment.3 A diagnosis of major depressive disorder can be made if a person suffers at least 1 such episode (without ever experiencing mania). However, most people with major depressive disorder have multiple episodes. Importantly, several medical illnesses such asdiabetes, heart disease, autoimmune disorders and pain are commoncomorbid diagnoses.4,5 The relation between major depressive disorder and these chronic and disabling conditions appears to be

bidirectional because one may influence the prognosis of the other.Investigations into the neurobiology of major depressive disorderhave traditionally focused on the monoamine neurotransmitters serotonin and norepinephrine. The monoamine hypothesis initially posited that depressed individuals are likely to have low levels of these neurotransmitters because various antidepressant drugs acutely increase their levels.6 However, even though monoaminergic antidepressants are generally used for first-line treatment, they do not exert their clinical benefit immediately and for some people they do not provide any benefit at all. We review the neurobiological research that may help explain this. A revised monoamine hypothesisStudies on the pathophysiology of major depressive disorder tend to focus on people who are currently depressed. Although informative, data derived from such studies often do not allow for a distinction to be made between cause and effect. These studies also do not allow researchers to distinguish between coremechanisms responsible for the disease and epiphenomena. For example, the finding that the rate of serotonin synthesis may be low in depressed patients can be explained in multiple ways.7 A reduction in serotonin synthesis may result in depression, depression may result in a reduction in serotonin synthesis, or athird factor may be responsible for both lowering serotonin synthesis rates and triggering depression.Experimental studies involving patients whose depression is currently in remission may help clarify the role of serotonin in major depressive disorder. Indeed, one of the few areas of investigation with studies including such patients is concerned with the effects of experimental serotonin manipulations on mood.In most of these studies, patients ingest a tryptophan-deficient amino acid mixture that transiently decreases serotonin levels inthe brain, because serotonin is derived from tryptophan (Figure 1).8 Patients taking medications at the time of study may experience a brief relapse, especially if they are taking drugs that affect the serotonin system. Patients not taking any medications are also likely to experience a brief relapse of symptoms during tryptophan depletion, especially if their depression has been in remission for only a few months. This suggests that a lowering of serotonin levels may result in depression.

View larger version:
Figure 1: The serotonin synapse. Serotonin is synthesized from tryptophan by the enzyme tryptophan hydroxylase. Serotonin is then packaged into vesicles for release into the synaptic cleft, which occurs when there is sufficient stimulation of the neuron. Serotonin released from the serotonin neuron into the synaptic cleft has multiple actions. (1) Serotonin binds to its receptors on other neurons. Activation of postsynaptic receptors results intransduction of the signal that initially stimulated the serotonin neuron. (2) Serotonin also binds to presynaptic serotonin receptors on the neuron from which it was released, which provides feedback and regulates plasticity of the neuron. (3) Serotonin is taken up back into the presynaptic serotonin neuron by the serotonin transporter. Serotonin is then recycled for future release or broken down by monoamine oxidase and excreted in urine. Image by: Lianne Friesen and Nicholas WoolridgeHowever, individuals without a personal or family history of major depressive disorder tend to not show any mood changes following tryptophan depletion,8despite the fact that tryptophan depletion alters the activity of mood-regulating regions of the brain, such as the amygdala, in these individuals as it does in patients with major depressive disorder.9 Thus, lowering serotonin levels does not induce depression in all people. It is possible that a depressive episode alters the serotonin system such that a person becomes more vulnerable to the effects of future changes in serotonin levels. However, even in the absence of a personal history of major depressive disorder, people with afamily history may report mood-worsening following tryptophan depletion.8Clearly, there are several possible factors that contribute to a person's vulnerability to the effects of altered levels of serotonin on mood.10

Genes influencing serotonin metabolism moderate the impact of stressScientists have not identified a gene or a series of genes that cause depression. Rather, certain variations in genes, called polymorphisms, may increase risk for depression. Genes can predispose individuals to major depressive disorder in many ways.For example, genes help control the metabolism of neurotransmitters and their receptors, the numbers of particular types of neurons and their synaptic connections, the intracellular transduction of neuronal signals, and the speed with which all of these can change in response to environmental stressors.11 The serotonin transporter gene is the most studied in major depressive disorder (Figure 1).12 This gene is of interest because it contains a polymorphism that gives rise to 2 different alleles (long and short). People usually have 2 copies of each gene in their DNA; therefore, a person can be homozygous for the long allele, homozygous for the short allele or heterozygous (1 long and 1 short allele). The short allele slows down the synthesis of the serotonin transporter. This is thought to reduce the speed with which serotonin neurons can adapt to changes in their stimulation.13 Given that an acute stressor increases serotonin release, the polymorphism may influence a person's sensitivity to stress. Indeed, healthy people with the short allele display exaggerated amygdala activation when exposedto stress-evoking stimuli.14 These people may also have a greaterlikelihood of mood-worsening following tryptophan depletion.15Stress is a common precipitating factor for depression.16,17 However, not everyone exposed to stress becomes depressed. Rather, stress interacts with a person's genetic makeup to influence his or her risk for developing major depressive disorder (Figure 2). A gene–environment interaction suggesting that carriers of the short allele of the serotonin transporter may be especially vulnerable to depression when understress was first suggested in 2003 in a prospective study involving a birth cohort. In a group of 26-year-old men and women, having had a major depressive episode in the past year wasbest predicted by the combination of having the short allele of the serotonin transporter and having had multiple stressful life events in the past 5 years.18The interactive effects of the serotonin transporter polymorphism and stress on a person's risk for major depressive disorder have been reported by various groups.19–22 Nevertheless, controversy remains. Not all studies

have observed this gene–environment interaction. In a study including patients in remission from a major depressive episode, tryptophan depletion had less pronounced effects on mood in carriers of the short allele than in people homozygous for the long allele, not more as might have been expected.23 In any case,the impact of individual genes on the risk of major depressive disorder is thought to be small. The interactive effects of multiple genes and psychosocial stress on the risk of depression are only starting to be explored.24
View larger version:
Figure 2: Schematic representation of gene–environment interaction. Genetic polymorphisms may influence a person's risk for major depressive disorder in certain psychosocial environments. Genes may contribute to a person's risk for major depressive disorder through polymorphisms or variations in their DNA sequence that may influence the expression or activity of thegene product. A person may have 2 identical alleles of a gene (homozygous) or 2 different alleles (heterozygous). Although carrying a certain allele may in itself not affect a person's risk for developing major depressive disorder, several genetic polymorphisms have been shown to interact with the environment interms of their effect on disease vulnerability. Please note that this figure is a schematic representation. Individual polymorphisms and individual studies may differ in terms of how aperson's risk for major depressive disorder is affected by carrying 1 or 2 vulnerability alleles. Limitations of studies to date include the method used to assess disease vulnerability (e.g., some studies used a person's score on a depression questionnaire rather than a formal diagnosis of major depressive disorder as the outcome variable) and the retrospective nature ofmost studies' assessment of psychosocial stress exposure.5–7,9–13
Brain-derived neurotrophic factorOne polymorphism that may moderate the interactive effect of the serotonin transporter polymorphism and psychosocial stress is located in the gene that codes for brain-derived neurotrophic factor. This growth factor plays a major role in the birth,

survival and maturation of brain cells during development. Brain-derived growth factor is important for cell growth and for allowing changes in the synapses between neurons (synaptic plasticity) throughout life. Brain-derived neurotrophic factor contributes to these processes primarily by activating DNA-binding factors that stimulate gene transcription. For example, in the raphe nuclei located in the brain stem, brain-derived neurotrophic factor stimulates transcription of genes involved inserotonin function, such as the serotonin transporter and tryptophan hydrolase (the serotonin synthesizing enzyme). In turn, activation of serotonin receptors by serotonin released from the raphe nuclei stimulates expression of the brain-derived neurotrophic factor gene. During brain development, this cyclic process promotes outgrowth, synapse formation and survival of serotonin neurons, and the eventual innervation of multiple brainregions. The ability of the serotonin system to adapt and change in response to various stimuli continues to be influenced by brain-derived neurotrophic factor throughout life.25A common polymorphism in the gene that codes for brain-derived neurotrophic factor produces alleles called “Val” and “Met.” Thispolymorphism affects the intracellular transport and secretion ofbrain-derived neurotrophic factor.26 People with the Met allele have been found to have a relatively small hippocampus at birth and to display hippocampal hypoactivity in a resting state, hippocampal hyperactivation during learning, and relatively poor hippocampus-dependent memory function.27,28 This may contribute to a hippocampal hypersensitivity to stress. It could also explain why studies have found that having the Met allele, in addition to having the short allele of the serotonin transporter and psychosocial stress, increases vulnerability to depression more than having the short allele of the serotonin transporter and psychosocial stress alone.29 These 3 factors combined may increase depression vulnerability even if the stress took place in childhood.30,31Further evidence for a role of brain-derived neurotrophic factor in the pathophysiology of major depressive disorder comes from postmortem studies, which have found low levels of brain-derived neurotrophic factor in the hippocampus and prefrontal cortex of symptomatic depressed patients.27,28 In addition, a recent reviewreported that serum levels of brain-derived neurotrophic factor in patients with major depressive disorder are abnormally low.32 In healthy people, serum levels of brain-derived neurotrophic factor correlate negatively with sensitivity to

stress and positively with brain levels of N-acetyl-aspartate, a putative marker of neuronal integrity that can be measured by neuroimaging.33
Psychosocial adversity in childhood and the hypothalamic–pituitary–adrenal axisStressful events often do not occur at random. This may be especially true of childhood stressors. A child's environment is influenced by his or her own behaviour as well as by the behaviour of his or her parents. The same genes influence the behaviour of both generations (provided the child grows up with his or her biological parents). In studies of the impact of psychosocial adversity during childhood on the risk of adult depression, it is often difficult to separate the effects of genes from those of the environment.2,11Experimental studies involving nonhuman primates and other mammals can be more informative. For example, monkeys temporarilyreared by peers rather than by their mothers develop exaggerated stress responses. These responses have been associated with abnormalities in serotonin activity as well as in the hypothalamic–pituitary–adrenal axis (Figure 3).34 Data from studies in rats also suggest that childhood experiences can alterthe reactivity of the hypothalamic–pituitary–adrenal axis and show that these alterations are at least partially mediated by modifications in genes that do not involve any actual changes in the underlying DNA (epigenetic changes).35 Specifically, the hippocampi of adult rats deprived of maternal care during infancydisplay these epigenetic modifications in the gene for the glucocorticoid receptor, which helps mediate the effects of cortisol released from the adrenal glands in response to stress (Figure 3). The process that leads to epigenetic changes, which affect gene transcription, is known to be influenced by serotonin.36 The resulting changes in glucocorticoid receptor expression in the hippocampus increase the reactivity of the hypothalamic–pituitary–adrenal axis.37 This epigenetic process may even occur in utero38 and is thought to be extremely long lasting. This might help explain why people with major depressivedisorder often show abnormalities in this neuroendocrine system.39

View larger version:
Figure 3: The hypothalamic–pituitary–adrenal axis. This system isactivated by stress directly at the level of the hypothalamus or indirectly at the level of the amygdala. The hypothalamus produces and releases corticotropin-releasing factor. Local stimulation of corticotropin-releasing factor type 1 receptors results in additional release of corticotropin-releasing factor. This creates a feed-forward loop, which facilitates a rapid response to the stressor. In the pituitary, stimulation of corticotropin-releasing factor type 1 receptors results in release of corticotropin (also known as adrenocorticotropic hormone). The adrenal glands are stimulated by corticotropin to produce the stress hormone cortisol, which affects many organs, including the brain. The hippocampus is an important target of cortisol. Local activation of glucocorticoid receptors helps the hippocampus control the hypothalamic–pituitary–adrenal axis. Glucocorticoid receptors are also found in the hypothalamus and pituitary. Chronic stress increases the level of corticotropin-releasing factor and cortisol and decreases expression of corticotropin-releasing factor type 1 receptors and glucocorticoid receptors. Similar changes have been found in somepatients with major depressive disorder. Stress-and depression-associated changes at the level of the hippocampus in particular are thought to underlie the structural changes seen in this brainregion, which in turn may contribute to chronic disinhibition of the hypothalamic–pituitary–adrenal axis.
The role of corticotropin-releasing factor in determining sensitivity to stressStress may activate the hypothalamus and therefore may activate the hypothalamic–pituitary–adrenal axis directly, by stimulating

local synthesis and release of corticotropin-releasing factor (Figure 3).37 Stress may also activate this axis indirectly by releasing corticotropin-releasing factor from neurons in other regions of the brain, including the amygdala.40 These neurons mayalso contribute to activation of the serotonin and norepinephrinesystems.41,42 Reciprocal connections between the norepinephrine system and the hypothalamus create a feed-forward cascade in which stress progressively activates corticotropin-releasing factor and norepinephrine signalling. Activation of this system is thought to increase vigilance and fear.43,44The combined dysregulation of the hypothalamic and extrahypothalamic corticotropin-releasing factor systems may helpexplain why patients with major depressive disorder often have inappropriately high levels of corticotropin-releasing factor andelevated levels of norepinephrine in their blood plasma and cerebrospinal fluid, and why they display faulty processing of environmental threats and exaggerated stress reactions.39 Childhood adversity might also contribute to the abnormalities seen in these systems.43A recent study suggests that the impact of childhood abuse on a person's vulnerability for depression may be moderated by polymorphisms in the corticotropin-releasing factor type 1 receptor gene.45 Evidence for gene–stress interactions affecting the risk of major depressive disorder can be found across neurotransmitter systems (Figure 2).
Stress-induced changes in the dopamine systemPersistent changes in the body's stress response during infancy and childhood are thought to increase responses to even relatively low-threat negative events experienced later in life. Sensitization to negative events may help explain why recurrent depressive episodes are more likely than the first episode to occur independently of stress.46 A complex relation between serotonin dysfunction, hypersensitivity to stress, and vulnerability to developing major depressive disorder has been suggested.47In addition, dopamine is increasingly thought to play an important role in the pathophysiology of major depressive disorder. Environmental threats perceived by the amygdala increase the levels of dopamine in the prefrontal cortex and the ventral striatum.48 Local inhibitory feedback ensures a return tohomeostasis. However, a severe stressor may disrupt this feedback

system by altering striatal levels of brain-derived neurotrophic factor.Abnormal feedback in the striatal dopamine system may help explain why depressed patients often attribute inappropriate salience to even mildly negative stimuli. Further, changes in thestriatal dopamine system are thought to underlie the anhedonia reported by many patients.48,49 Evidence for dopamine abnormalities in major depressive disorder is mixed but does exist.50 A polymorphism in the dopamine type 2 receptor gene was recently found to influence the effect of past stressful life events on current mood.51The genetic makeup of the dopamine system may help influence vulnerability to major depressive disorder via interaction with the environment (Figure 2).
Structural changes in the depressed brainMajor depressive disorder has traditionally been viewed as an illness in which depressive episodes are followed by periods of euthymic mood. Nonetheless, patients showing clinical signs of remission may present with persistent neurobiological abnormalities. These abnormalities may worsen over time, and somepatients may become chronically depressed. Similar to multiple sclerosis, there may be variability in the course of the disease over time. In multiple sclerosis, there are 2 common patterns of disease progression: relapsing–remitting and primary–progressive.The existing data for major depressive disorder suggest that although many patients may have a relapsing–remitting variant of the disease, some may have a primary–progressive variant.Most relevant to this idea are structural neuroimaging studies which indicate that individuals with recurrent major depressive episodes may have relatively small hippocampi even during periodsof clinical remission. Recurring or enduring illness and lack of antidepressant treatment are thought to contribute to progressivevolume reductions of the hippocampus, which in turn may explain the memory problems of some patients, as well as several other symptoms of the disorder.52–54 Patients may also present with volumetric abnormalities in other subcortical brain regions, including the amygdala and ventral striatum, and in cortical regions, including the anterior cingulate cortex, orbitofrontal cortex and prefrontal cortex (Figure 4).53–55These too may persist during remission and might help explain why patients in remission continue to overreact to threatening stimuli.56 This cognitive reactivity contributes to the risk of future relapse.57
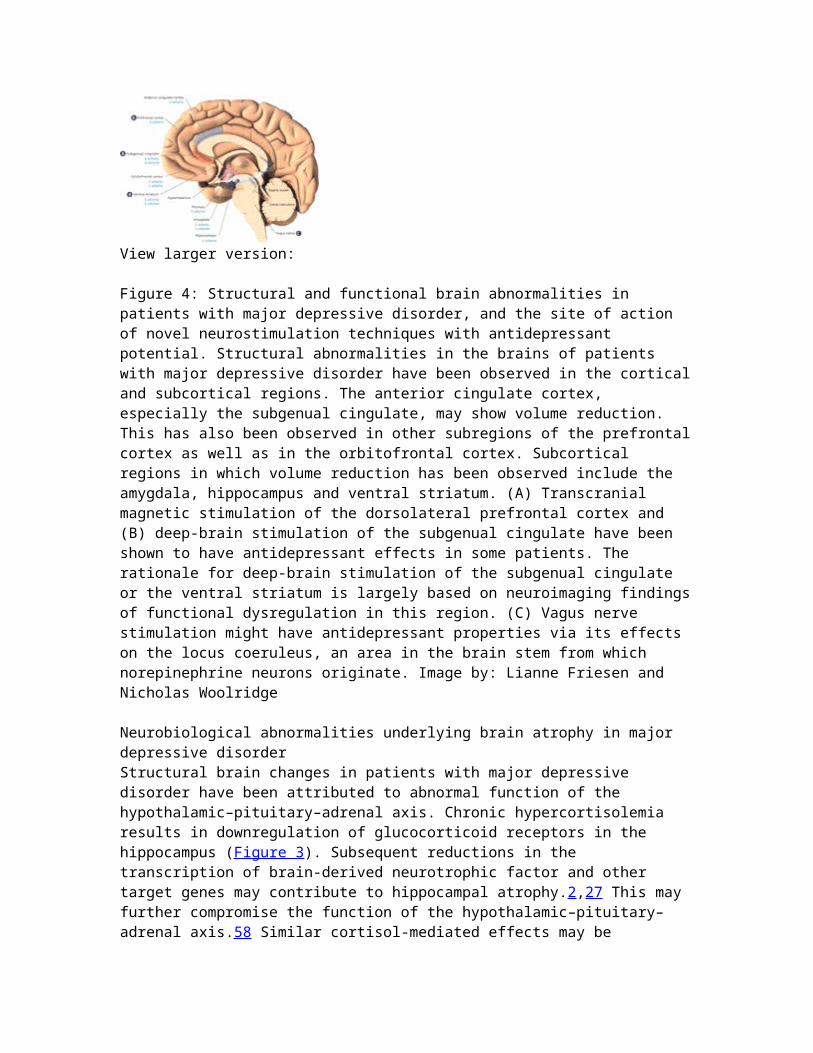
View larger version:
Figure 4: Structural and functional brain abnormalities in patients with major depressive disorder, and the site of action of novel neurostimulation techniques with antidepressant potential. Structural abnormalities in the brains of patients with major depressive disorder have been observed in the corticaland subcortical regions. The anterior cingulate cortex, especially the subgenual cingulate, may show volume reduction. This has also been observed in other subregions of the prefrontalcortex as well as in the orbitofrontal cortex. Subcortical regions in which volume reduction has been observed include the amygdala, hippocampus and ventral striatum. (A) Transcranial magnetic stimulation of the dorsolateral prefrontal cortex and (B) deep-brain stimulation of the subgenual cingulate have been shown to have antidepressant effects in some patients. The rationale for deep-brain stimulation of the subgenual cingulate or the ventral striatum is largely based on neuroimaging findingsof functional dysregulation in this region. (C) Vagus nerve stimulation might have antidepressant properties via its effects on the locus coeruleus, an area in the brain stem from which norepinephrine neurons originate. Image by: Lianne Friesen and Nicholas Woolridge
Neurobiological abnormalities underlying brain atrophy in major depressive disorderStructural brain changes in patients with major depressive disorder have been attributed to abnormal function of the hypothalamic–pituitary–adrenal axis. Chronic hypercortisolemia results in downregulation of glucocorticoid receptors in the hippocampus (Figure 3). Subsequent reductions in the transcription of brain-derived neurotrophic factor and other target genes may contribute to hippocampal atrophy.2,27 This may further compromise the function of the hypothalamic–pituitary–adrenal axis.58 Similar cortisol-mediated effects may be

responsible for structural changes in the amygdala, prefrontal cortex and other brain regions. All or some of the structural changes may contribute to the risk for relapse following a periodof apparent homeostasis.59,60The growth and survival of brain cells also involves multiple actions of glutamate, an amino acid neurotransmitter.2 Normal glutamate signalling includes activation of multiple types of receptors.61 Acute stress moderately increases synaptic glutamateneurotransmission in, for example, the hippocampus and amygdala, thereby increasing the levels of brain-derived neurotrophic factor and stimulating neuroplasticity. Termination of the stressor usually results in a return to the steady state. In times of chronic stress, however, excessive glutamate levels may lead to activation of N-methyl-D-aspartic acid type glutamate receptors outside the synapse. Hyperactivation of these receptors, located on both neurons and glial cells, increases intracellular calcium to a level that decreases rather than increases levels of brain-derived neurotrophic factor, induces atrophy and causes cell death.61 The degeneration of glial cells,which normally help avoid the toxic buildup of glutamate releasedby neurons, may accelerate this process. Continued disruptions inthe glutamate system may ultimately result in hypoactivity of cortical brain regions. Patients with depression may have reducedlevels of glutamate and associated proteins in the prefrontal cortex, as indicated by neuroimaging studies.61,62 This provides evidence that glutamate toxicity plays a role in the pathophysiology of major depressive disorder.
Abnormal brain activation patterns may help explain mood labilityStructural brain abnormalities in patients with major depressive disorder have been linked to changes in metabolic activity (Figure 4).2 These functional abnormalities have been directly associated with disease severity.63 Overall, neuroimaging studiesindicate that major depressive episodes may be characterized by multiple abnormalities in the interconnectivity of various subcortical (especially limbic) and cortical brain regions.64 A relative lack of cortical regulation of the limbic system in the face of psychosocial adversity may help explain the stress sensitivity, emotional lability, irritability and suicidality often seen in people with depression.42,65 Various brain regions continue to show functional abnormalities upon clinical remission.63 Inducing a brief depressive relapse by lowering serotonin levels (via tryptophan depletion) or by lowering

norepinephrine and dopamine levels (via administration of the drug α-methyl-para-tyrosine) activates both overlapping and distinct brain regions.66,67 Thus, different neurotransmitter systems may contribute in different ways to the cortical regulation of mood.
Treatment strategiesAntidepressant medicationsIt is clear that even a revised monoamine hypothesis does not do justice to the multitude of interconnected systems involved in the pathophysiology of major depressive disorder. This explains why treatment with existing antidepressant drugs, most of which target monoamines, frequently does not lead to clinical remission.68 The time lag between the start of and response to antidepressants in patients whose symptoms improve can be explained by the effects of these drugs on brain-derived neurotrophic factor and other growth-regulating systems.25,69 Theeffects of these drugs on neuroplasticity are particularly relevant given that treatment may reverse or even prevent structural brain abnormalities. Among the new drugs currently under investigation are those that target the corticotropin-releasing hormone, dopamine and glutamate systems.70 However, a recent study found no significant antidepressive effect of a drugwith selective action on the corticotropin-releasing factor type 1 receptor.71 In contrast, in a study that used the anesthetic ketamine, which has actions on both dopamine and glutamate, a single intravenous administration of a subanesthetic dose was found to have robust and rapid antidepressant effects even in patients whose depression is considered resistant to conventionaldrug treatment strategies.72 However, most patients experienced arelapse within 1 week. Other glutamate-modulating agents currently under investigation for treatment of major depressive disorder include memantine and riluzole, and there are others in development.61,73,74 Finally, there has been a recent interest indrugs targeting other neurotransmitters such as gamma-aminobutyric acid, melatonin and substance P.70Studies are underway to advance personalized medicine based on pharmacogenetics. A recent study found no evidence for an effect of the polymorphism in the serotonin transporter gene on the clinical response to citalopram (a selective serotonin reuptake inhibitor). Yet, patients with the short allele reported more adverse outcomes.75 A polymorphism in the gene for the serotonin

type 2A receptor, however, has been found to influence the outcome of treatment with citalopram.76Other polymorphisms with possible effects on patients' responses to antidepressant drugs include those resulting from allelic variations in other genes with roles in the serotonin system. Genes involved in the function of the hypothalamic–pituitary–adrenal axis and the corticotropin-releasing factor and norepinephrine systems have also been implicated.77 In addition, polymorphisms in genes for certain liver enzymes involved in drugmetabolism may influence plasma drug levels and may therefore impact efficacy.77 A screening kit to identify how a person will process certain medicines based on his or her genetic makeup and thus help guide doctors to prescribe effective and tolerable medications has recently been developed but is not currently commercially available in Canada.Nonpharmacologic approachesThe most traditional approach for treating major depressive disorder by nonpharmacologic means is psychotherapy. This approach may be especially helpful for patients with a history ofchildhood adversity or recent stress.78Another approach that has been available for decades is electroshock or electroconvulsive therapy; however, its use is limited by its invasive nature, which includes the requirement of general anesthesia and the riskof retrograde amnesia, which may be irreversible in some patients.79A small number of other neurostimulation techniques have recentlybeen developed based on current insights into the neurobiology ofmajor depressive disorder. These include electrical stimulation of the vagus nerve, transcranial magnetic stimulation of the prefrontal cortex, and electrical stimulation of the subgenual cingulate or the ventral striatum (Figure 4).68 The development of transcranial magnetic stimulation has been guided by findings of low activity in the prefrontal cortex of patients with depression (Figure 4). The rationale for deep-brain stimulation procedure is based on findings of functional impairments in the subgenual cingulate and ventral striatum and abnormalities in corticolimbic interconnectivity.64Deep brain stimulation is still under experimental investigation.Transcranial magnetic stimulation and vagus nerve stimulation have been approved in Canada for treatment of major depressive disorder. However, because of their relatively invasive nature, techniques such as electroconvulsive therapy are currently only

indicated for patients whose depression is resistant to conventional treatments.
Future directionsMajor depressive disorder is thought to result from the complex interplay of multiple inherited genetic factors and subsequent exposure to a wide range of environmental variables throughout life. The exact roles of the monoamine and other neurotransmittersystems as well as their extracellular, intracellular, local and regional targets continue to be defined. The reciprocal connections between various brain regions and the neurochemical messengers involved in these connections are being increasingly studied from a systems perspective. Furthermore, researchers are increasingly considering the impact of an individual's psychosocial environment on brain chemistry, activity and anatomy, and vice versa. The impact of early childhood events in particular may have long-lasting effects,80 especially if epigenetic changes are involved.28,35 Clearly, researchers as well as clinicians must pay careful attention to the possible contribution of childhood adversity to adulthood psychopathology,for example, by incorporating family history and childhood background into diagnostic interviews with patients.This review focused mainly on psychosocial adversity as an environmental risk factor, which largely reflects the current interest of this field. Much remains to be explored in terms of how genes interact with other environmental variables to influence the risk of major depressive disorder. For example, we have yet to discover why some people become depressed when they develop inflammatory disease or spend a winter at higher latitude, while others do not. The genetic factors influencing this differential susceptibility are unknown. Major depressive disorder is likely to have a large number of causes, both geneticand environmental. We have attempted to provide a framework for acomplex disease that requires a multifaceted approach in research, diagnosis and treatment.Key pointsMajor depressive disorder is caused by the cumulative impact of genetics, adverse events in childhood and ongoing or recent stress.Gene–environment interactions seem to predict a person's risk formajor depressive disorder better than genes or environment alone.Structural and functional brain abnormalities in patients with major depressive disorder may be associated with low levels of

brain-derived neurotrophic factor, abnormal function of the hypothalamic–pituitary–adrenal axis and glutamate-mediated toxicity.These abnormalities are thought to contribute to recurrent episodes of major depressive disorder and chronic illness.Existing options for antidepressant treatment are limited by their delayed onset of action, lack of efficacy and adverse outcomes.Future developments include the advancement of personalized medicine by means of genotyping for interindividual variability in drug action and metabolism.………………………………..Alte articole (urmati link-ul)A revised monoamine hypothesisGenes influencing serotonin metabolism moderate the impact of stressBrain-derived neurotrophic factorPsychosocial adversity in childhood and the hypothalamic–pituitary–adrenal axisThe role of corticotropin-releasing factor in determining sensitivity to stressStress-induced changes in the dopamine systemStructural changes in the depressed brain…………………………………….






