
Antileishmanial activity of quinazoline derivatives: Synthesis, docking screens, molecular dynamic...
18
Original article Antileishmanial activity of quinazoline derivatives: Synthesis, docking screens, molecular dynamic simulations and electrochemical studies Cesar Mendoza-Martínez a, b , Norma Galindo-Sevilla c , Jos e Correa-Basurto d , Victor Manuel Ugalde-Saldivar e , Rosa Georgina Rodríguez-Delgado f , Jessica Hern andez-Pineda c , Cecilia Padierna-Mota g , Marcos Flores-Alamo f , Francisco Hern andez-Luis b, * a Programa de Maestría y Doctorado en Ciencias Químicas, Universidad Nacional Aut onoma de M exico, M exico, DF 04510, Mexico b Departamento de Farmacia, Universidad Nacional Aut onoma de M exico, M exico, DF 04510, Mexico c Departamento de Infectología, Instituto Nacional de Perinatología, M exico, DF 11000, Mexico d Escuela Superior de Medicina, Instituto Polit ecnico Nacional, M exico, DF 11340, Mexico e Departamento de Química Inorg anica y Nuclear, Universidad Nacional Aut onoma de M exico, M exico, DF 04510, Mexico f Facultad de Química, Universidad Nacional Aut onoma de M exico, M exico, DF 04510, Mexico g Laboratorio de Especialidades Inmunol ogicas S.A. de C.V., M exico, DF 07580, Mexico article info Article history: Received 27 August 2014 Received in revised form 23 November 2014 Accepted 28 December 2014 Available online 29 December 2014 Keywords: Leishmania mexicana Quinazoline Antiprotozoan activity abstract A series of quinazoline-2,4,6-triamine were synthesized and evaluated in vitro against Leishmania mexicana. Among them, N 6 -(ferrocenmethyl)quinazolin-2,4,6-triamine (H2) showed activity on pro- mastigotes and intracellular amastigotes, as well as low cytotoxicity in mammalian cells. Docking and electrochemical studies showed the importance of both the ferrocene and the heterocyclic nucleus to the observed activity. H2 is readily oxidized electrochemically, indicating that the mechanism of action probably involves redox reactions. © 2014 Elsevier Masson SAS. All rights reserved. 1. Introduction The search for antiparasitic molecules has recently become important because of the emergence of drug resistant strains, the toxicity of the known molecules, increased poverty and the per- centage of the affected population [1]. Parasitic tropical diseases affect hundreds of millions of people worldwide, however, it has been neglected to develop drugs against these diseases because they primarily affect people in poor regions of the world [1]. Leishmaniasis, African trypanosomiasis, and Chagas disease are vector-borne diseases caused by parasites of the kinetoplastida order [2]. Leishmaniasis is a set of devastating diseases caused by the obligate intracellular protozoa parasites of the Leishmania genus, which are transmitted by a group of 50 species and sub- species of phebotomine insects. About 1.5 million of new cases of cutaneous leishmaniasis and 500 000 new cases of visceral disease occur each year. Cutaneous leishmaniasis is endemic in more than 70 countries worldwide [3]. A major emerging problem is co- infection of Leishmania with human immunodeficiency virus, especially because there is no effective treatment for these patients [3]. Conventional chemotherapy relies on multiple parenteral ad- ministrations of pentavalent antimonials that are considerably toxic and induce resistance. Second-line drugs, such as amphoter- icin B and its lipid formulations, are either too toxic or too expen- sive for routine use in developing countries [4]. Recently, miltefosine, a phosphocholine analogue originally developed as an anticancer agent, was introduced as a drug against visceral leish- maniasis, but its effectiveness has still not been conclusively determined and there have already been reported cases of resis- tance [5,6]. Because chemotherapy for leishmaniasis is still ineffi- cient, there is an urgent need for the development of new efficient and safe drugs. Abbreviations: PTR1, Pteridine reductase 1; DHFR, Dyhydrofolate reductase; IC 50 , Half inhibitory concentration; TAQ, Quinazoline-2,4,6-triamine; PDB, Protein Data Bank. * Corresponding author. E-mail address: [email protected] (F. Hern andez-Luis). Contents lists available at ScienceDirect European Journal of Medicinal Chemistry journal homepage: http://www.elsevier.com/locate/ejmech http://dx.doi.org/10.1016/j.ejmech.2014.12.051 0223-5234/© 2014 Elsevier Masson SAS. All rights reserved. European Journal of Medicinal Chemistry 92 (2015) 314e331
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Antileishmanial activity of quinazoline derivatives: Synthesis, docking screens, molecular dynamic...

lable at ScienceDirect
European Journal of Medicinal Chemistry 92 (2015) 314e331
Contents lists avai
European Journal of Medicinal Chemistry
journal homepage: http: / /www.elsevier .com/locate/ejmech
Original article
Antileishmanial activity of quinazoline derivatives: Synthesis, dockingscreens, molecular dynamic simulations and electrochemical studies
Cesar Mendoza-Martínez a, b, Norma Galindo-Sevilla c, Jos�e Correa-Basurto d,Victor Manuel Ugalde-Saldivar e, Rosa Georgina Rodríguez-Delgado f,Jessica Hern�andez-Pineda c, Cecilia Padierna-Mota g, Marcos Flores-Alamo f,Francisco Hern�andez-Luis b, *
a Programa de Maestría y Doctorado en Ciencias Químicas, Universidad Nacional Aut�onoma de M�exico, M�exico, DF 04510, Mexicob Departamento de Farmacia, Universidad Nacional Aut�onoma de M�exico, M�exico, DF 04510, Mexicoc Departamento de Infectología, Instituto Nacional de Perinatología, M�exico, DF 11000, Mexicod Escuela Superior de Medicina, Instituto Polit�ecnico Nacional, M�exico, DF 11340, Mexicoe Departamento de Química Inorg�anica y Nuclear, Universidad Nacional Aut�onoma de M�exico, M�exico, DF 04510, Mexicof Facultad de Química, Universidad Nacional Aut�onoma de M�exico, M�exico, DF 04510, Mexicog Laboratorio de Especialidades Inmunol�ogicas S.A. de C.V., M�exico, DF 07580, Mexico
a r t i c l e i n f o
Article history:Received 27 August 2014Received in revised form23 November 2014Accepted 28 December 2014Available online 29 December 2014
Keywords:Leishmania mexicanaQuinazolineAntiprotozoan activity
Abbreviations: PTR1, Pteridine reductase 1; DHFIC50, Half inhibitory concentration; TAQ, QuinazolineData Bank.* Corresponding author.
E-mail address: [email protected] (F. Hern�andez-
http://dx.doi.org/10.1016/j.ejmech.2014.12.0510223-5234/© 2014 Elsevier Masson SAS. All rights re
a b s t r a c t
A series of quinazoline-2,4,6-triamine were synthesized and evaluated in vitro against Leishmaniamexicana. Among them, N6-(ferrocenmethyl)quinazolin-2,4,6-triamine (H2) showed activity on pro-mastigotes and intracellular amastigotes, as well as low cytotoxicity in mammalian cells. Docking andelectrochemical studies showed the importance of both the ferrocene and the heterocyclic nucleus to theobserved activity. H2 is readily oxidized electrochemically, indicating that the mechanism of actionprobably involves redox reactions.
© 2014 Elsevier Masson SAS. All rights reserved.
1. Introduction
The search for antiparasitic molecules has recently becomeimportant because of the emergence of drug resistant strains, thetoxicity of the known molecules, increased poverty and the per-centage of the affected population [1]. Parasitic tropical diseasesaffect hundreds of millions of people worldwide, however, it hasbeen neglected to develop drugs against these diseases becausethey primarily affect people in poor regions of the world [1].Leishmaniasis, African trypanosomiasis, and Chagas disease arevector-borne diseases caused by parasites of the kinetoplastidaorder [2]. Leishmaniasis is a set of devastating diseases caused bythe obligate intracellular protozoa parasites of the Leishmania
R, Dyhydrofolate reductase;-2,4,6-triamine; PDB, Protein
Luis).
served.
genus, which are transmitted by a group of 50 species and sub-species of phebotomine insects. About 1.5 million of new cases ofcutaneous leishmaniasis and 500 000 new cases of visceral diseaseoccur each year. Cutaneous leishmaniasis is endemic in more than70 countries worldwide [3]. A major emerging problem is co-infection of Leishmania with human immunodeficiency virus,especially because there is no effective treatment for these patients[3]. Conventional chemotherapy relies on multiple parenteral ad-ministrations of pentavalent antimonials that are considerablytoxic and induce resistance. Second-line drugs, such as amphoter-icin B and its lipid formulations, are either too toxic or too expen-sive for routine use in developing countries [4]. Recently,miltefosine, a phosphocholine analogue originally developed as ananticancer agent, was introduced as a drug against visceral leish-maniasis, but its effectiveness has still not been conclusivelydetermined and there have already been reported cases of resis-tance [5,6]. Because chemotherapy for leishmaniasis is still ineffi-cient, there is an urgent need for the development of new efficientand safe drugs.

Table 1Derivatives of TAQ with antiparasitic moieties.
N
NNHR
NH2
NH2
Compound R
H1
H2C NO2
H2
H3 N
NF3C
Cl
ClH2C
H4
OCH3H2C
H5OHH2C
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331 315
In order to find new drugs against leishmaniasis, the compu-tational design of new drugs against parasites has been based onthe knowledge and availability of new information from moleculartargets [7,8]. With the goal of following this strategy, we chosepteridine reductase (PTR) as the target of the study, which is aprotein that appears to be important to the resistance mechanismof Leishmania. This enzyme is NADPH-dependent with oxidereductase activity, and pterins are its natural substrate [7]. How-ever, under conditions of cellular stress or when folate metabolismis brought down by molecules that inhibit dihydrofolate reductase(DHFR), PTR may reduce folate, therefore allowing the productionof DNA through the salvage route, causing parasite resistance[9e11]. New approaches in the design of antifolates shouldconsider the importance of PTR to discover new molecules capableof inhibiting DHFR and/or PTR through rational design [7].
The isoform PTR1 of Leishmania major was co-crystallized withquinazoline-2,4,6-triamine (TAQ) [12]. By analyzing the in-teractions of TAQ with the active site of PTR1, the chemical modi-fication of TAQ at position 6 was considered because the receptionsite near this position is large and possesses both polar and non-polar regions (Fig. 1). Berman and co-workers, showed the impor-tance of the lipophilic substituents at position 6 of the quinazolinering, decreasing the ED50 against L. major. In addition, they haveshown that, by increasing the size of the substituent, the ED50increased [13]. In this case, the selection of the chemical substituentat position 6 was based on its potential to function as an anti-protozoal scaffold. With this in mind, nitrobenzene, ferrocene,benzimidazole, anisole and phenol moieties were selected as thesubstituents in H1, H2, H3, H4 and H5, respectively (Table 1).Ferrocene is present in ferroquine, an anti-malarial drug that iscurrently under clinical evaluation and has got a great potential[14]. A number of studies have shown that the introduction of theferrocene core may significantly enhance the molecule's (desirable)bioactive properties. The ferrocene unit might act as a hydrophobicspacer and/or lipophilicity/bioavailability enhancer (enabling aneasier way through cell membranes) [15]. It is also known thatferrocene Feþ2/Feþ3 redox chemistry might contribute to thebioactivity of ferrocene derivatives [16]. Benzimidazole is a privi-leged structure in antiparasitic molecules [17]; particularly, in acompound prepared in our group, 5,6-dicloro-2-(trifluoromethyl)-1H-benzimidazole, denoted in this work as G2 [18]. Nitrobenzene,anisole and phenol groups are present in several anti-parasiticdrugs [19,20].
With the aim of exploring PTR1 binding site surfaces of theprotein, we employed docking studies using TAQ as ligand and
Fig. 1. Interaction of TAQ wit
attempted to reproduce this ligand is in the previously reportedcrystal structure [12]. Next, we performed a molecular coupling ofthe proposed molecules (H1eH5) into active site of LeishmaniaPTR1 and DHFR. The affinity of all the target compounds to bothenzymes was higher than the affinity of TAQ. Subsequently, theproposed compounds were synthesized and once their structurewas elucidated by x-ray crystallographic, they were tested in vitroagainst the promastigote form of Leishmania mexicana. The mostactive was H2. For this reason, chemical modifications were carriedout to the H2 structure (Fig. 2) in order to explore changes in bio-logical responses that could cause these structural modifications.Finally, the reduction potential for this group of compounds wascarried out using DMSO as aprotic solvent. The data in aproticsolvent did not only describe the situation in that way but it alsoallowed us to obtain biological significance of interpretation.
h PTR1 (ID-PDB: 1WOC).

Fig. 2. H2 derivatives.
Scheme 1. Reagents and conditions: (a) 2-bromo-1,1-diethoxyethane, Na2CO3, DMF, 110 �C, 92%; (b) i-BBr3, CH2Cl2; ii-H2O, 99%; (c) guanidine hydrochloride, NaOH, EtOH-PrOH,reflux, 86%; (d) acetic anhydride, 110 �C, 65%; (e) H2, 10% Pd/C, MeOH, r.t. 76%; (f) MeOH, CH3COOH (a drop), 50 �C, 2 days; (g) 0 �C, NaBH4, then r.t. for 24 h, 40e90%.
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331316

Table 2Selected bond lengths [Å] and torsion angles [�] for H2.
Fe(1)-C(1) 2.035 (2)Fe(1)-C(2) 2.044 (2)Fe(1)-C(3) 2.051 (2)Fe(1)-C(4) 2.042 (2)Fe(1)-C(5) 2.045 (2)N(1)-C(11) 1.402 (3)N(1)-C(19) 1.465 (3)C(1)-C(19) 1.498 (3)
C(19)-N(1)-C(11)-C(12) 20.8 (3)N(1)-C(11)-C(16)-C(15) �176.1 (2)C(18)-N(2)-C(17)-N(4) �179.79 (19)C(14)-N(3)-C(18)-N(2) �0.6 (3)C(14)-N(3)-C(18)-N(5) 179.9 (2)C(11)-N(1)-C(19)-C(1) 165.1 (2)C(2)-C(1)-C(19)-N(1) �106.5 (3)C(5)-C(1)-C(19)-N(1) 70.2 (3)
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331 317
2. Results and discussion
2.1. Chemistry
Compounds H4 and H5 were first synthesized and evaluated asantichagasic and antiplasmodial agents by Davoll et al. [21]. Sincethey were not evaluated against Leishmania, these molecules weresynthesized in this work in order to explore their biological activityagainst L. mexicana.
All the TAQ derivatives were synthesized according to the routeoutlined in Scheme 1. The antiprotozoal compound G2 (5,6-dicloro-2-(trifluoromethyl)-1H-benzimidazole) was prepared in our labo-ratory using known procedures [18]. As shown in Scheme 1,nucleophilic substitution of the bromine atom in 2-bromo-1,1-diethoxyethane by G2 afforded 5,6-dichloro-1-(2,2-diethoxyethyl)-2-(trifluoromethyl)-1H-benzimidazole (1). Treat-ment of 1with boron tribromide (BBr3) followed by hydrolysis gave[5,6-dichloro-2-(trifluoromethyl)-1H-benzimidazole-1-yl]acetal-dehyde (2). Preliminary attempts to obtain the aldehyde groupwithhydrochloric acid [22], I2/acetone [23] and FeCl2 [24] were unsuc-cessful. The cyclocondensation of commercially available 5-nitroanthranilonitrile with guanidine hydrochloride yielded 6-nitroquinazoline-2,4-diamine (3), which was treated with aceticanhydride followed by hydrogenation under a hydrogen atmo-sphere using palladium on charcoal to yield N,N'-(6-nitroquinazoline-2,4-diyl)diacetamide (4). Subsequent condensa-tion of 4 with the corresponding aldehyde gave the imine de-rivatives. The imine group was reduced with sodium borohydride(NaBH4) to yield the proposed molecules (H1eH5).
Of all the compounds synthesized only H2 gave crystalsadequate to carry out a single-crystal X-ray analysis. These studiesrevealed that H2 in ethanol crystallizes in the triclinic space groupP-1. A view of H2 is presented in Fig. 3, and selected bond distancesand torsion angles are listed in Table 2. The asymmetric unit of theH2 compound consists of N6-(ferrocenylmethyl)quinazolin-2,4,6-triamine and three ethanol solvate molecules in general positions.The quinazolin-2,4,6-triamine shows planarity with a rms deviationof the fitted atoms of 0.0265. The C2-C1-C19-N1 (106.47�) and C5-C1-C19-N1 (70.20�) torsion angles show a nearly perpendiculararrangement between the ferrocenyl and quinazoline groups.
In the crystal lattice, the neutral ferrocenyl complex and theethanol solvate molecules participate in multiple hydrogenbonding interactions, such as NeH$$$O, NeH$$$N and OeH$$$O(Fig. 4). The majority of the hydrogen bond distances range from1.87 to 2.25 Å (Table 3). The interaction between the nitrogen donoratom (N4) and the acceptor atom (N2) forms the R22(8) motif,whereas the N1-H1F$$$O1C hydrogen bond shows the D3
3(17)motif. All of these interactions are present in the three crystallineaxes and favor three-dimensional growth (Table 4).
Fig. 3. ORTEP diagram of H2 with displacement ellipsoids at 50% probability level fornon-H atoms. The ethanol solvent molecules were omitted for clarity.
In order to determine the part of the molecule that provides theanti-Leishmania effect when quinazoline or ferrocene are used, aseries of ferrocene derivatives (HO2, HO4, H2A, FBC) were syn-thesized via reductive aminationwith varying reagents (Scheme 2).
The structures of all the compounds synthesized were charac-terized by spectroscopic and spectrometric data. In the 1H NMRspectra of the title compounds, the doublet in the range4.5e3.9 ppm, integrating for two protons, confirmed the presenceof methylene linking the substituent to quinazoline nucleus;whereas in the 13C NMR spectra, the same signal of the secondarycarbon of the methylene was seen at d value of 46 ppm. For thesame group of compounds, the signal of proton of the NH exocyclicat position 6 was recorded as triplet at d values in the region6e5 ppm. ForH2,HO4,HO2,H2A and FBC, typical ferrocene signalswere showed in the region of 4.3e4.0 ppm in the 1H NMR spectra,and 60e90 ppm in the 13C NMR spectra. For those compoundshaving exocyclic amines groups at position 2 and 4 of quinazolinenucleus, the signal derived from two protons was observed atd values 5.6 and 7.0 ppm, respectively. Regarding 1H NMR spectra ofHO2 and HO4, the exocyclic amine group, at position 4 and 2,presented a signal to 7.73 and 5.91 ppm, respectively. Finally, in thedownfield region of the same spectrum, the signal of one proton at10.44 (HO2) and 10.78 (HO4) ppm revealed the presence of endo-cyclic NH of quinazoline moiety, suggesting the presence ofcarbonyl group at position 2 and 4, respectively. This suggestionwas supported by IR spectra of both compounds, which exhibitedstretch carbonyl characteristic band at 1624 and 1644 cm-1,respectively. Additionally, energetic stability studies confirmed thatthe most stable tautomers (Gibbs energy of 0 Kcal) for both mole-cules are those having the carbonyl group. All these data indicatethat in HO2 and HO4 the CeO bond is present as carbonyl groupand the most stable tautomeric form should be as proposed (Fig. 5).
2.2. Modeling studies
With the purpose of estimating the interactions of the synthe-sized compounds with PTR and DHFR, the affinity to both enzymeswas analyzed in a theoretical docking study. The crystal structure oftcDHFR (Trypanosoma cruzi dihydrofolate reductase ID-PBD: 2H2Q)was used to construct a homologous enzyme, L. majorwas used as amodel for L. mexicana. The DHFR sequence in L. mexicana is un-known; however, the three species (T. cruzi, L. major andL. mexicana) are evolutionarily very close. The molecular dynamicsprotocol was applied to relax the built protein, and a snapshot at10 ns was obtained for the docking of all the molecules. The rootmedia square deviation (RMSD) and the radius of gyration (RGyr)indicated that the protein reached convergence (Fig. 6). The crystal

Fig. 4. The hydrogen bonds NeH/O, NeH/N and OeH/O lead to an infinite three-dimensional in H2.
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331318
structure of L. major pteridin reductase (1 ID-PBD: 1WOC) wasobtained from the Protein Data Bank, and molecular docking wasperformed with this protein.
The free energy analysis of the molecular interaction betweenthe TAQ derivatives and the DHFR and PTR1 enzymes showed thatall of the compounds bind more strongly to the receptor site thanTAQ, and the mode of binding was very similar (Fig. 7). In the caseof the ferrocene derivatives (HO2,HO4 and H2A), the differences inthe interaction energies were a result of the substituents at posi-tions 2 and 4 (Table 5). For this study, the stability of the differenttautomers ofHO2 andHO4was analyzed (Fig. 5) with the intentionof using only the most stable.
2.3. Biological activity
Obtained compounds were tested in vitro against promastigotesform of L. mexicana. Initially, the compounds were incorporatedinto the media at 100 mM and their ability to inhibit growth of theparasite was evaluated in comparison to the control (no drug addedto the media). Glucantime was used as the leishmanicidal ofreference; pyrimethamine and trimetoprim were used as DHFRinhibitors models. Subsequently, the IC50 (50% inhibitory growthconcentration) concentration was determined. With exception ofH2 and H4, all compounds resulted with poor or null activity(Table 6). Compound H2 was the most active (IC50 ¼ 0.93 mM); thesecond place went to H4 (IC50 ¼ 14.59 mM). The components of H2(ferrocene and TAQ) had a poor antileishmanial activity individu-ally. Interestingly, H2 showed antiparasitic activity from the firsthour of being in contact with the parasites. This behavior may bedue to the ferrocene moiety contributes with its lipophilicity andredox properties. On the other hand, with exception of H4, FBCshowed slightly higher activity than the quinazoline derivatives,despite having only one phenyl group attached to ferrocene unit. InH2, the TAQ portion was necessary for leishmanicidal activitybecause when the 2- and 4-positions were modified by acetylation(H2A) or substitution of the NH2 group (HO2,HO4), the activity waslost (Table 6). In the light of these results, only H2 was selected for
the following studies in order to gain an understanding of its bio-logical activity.
There is no obvious correlation with the solubility, however, thesolubility of H2 is greater than ferrocene alone and less than tri-aminquinazoline. This, of course, could be a cause of anti-leishmanial activity shown because ferrocene is dissolved ingreater amounts. However, H2A is much more soluble than mostderivatives (except TAQ, which is extremely soluble) but this is notenough to improve the activity, so, that interaction of the amines ofTAQ are important.
2.3.1. Activity of H2 against intracellular amastigotesThe study on intracellular amastigotes revealed that H2 main-
tains strong leishmanicidal activity. This compound is able to killintracellular parasites inside the macrophages; therefore, it may beuseful for in vivo treatment of the disease. Note that although theantileishmanial activity observed is for intracellular amastigotes,H2 eliminates 97% of the parasites at 12 mM (Fig. 8B). After exposureof the infected macrophages to H2 for 72 h, removal of this com-pound and reculturing the parasites in fresh media, parasite sur-vival was observed at 3 and 6 mM, whereas concentrations of 12, 25,and 50 mM effectively killed the intracellular parasite and survivalwas not observed (Fig. 8C).
2.3.2. DHFR inhibitionAs a second approach, the inhibition of DHFR was explored, as
shown by the theoretical studies (Table 7). For experimentalstudies, the parasites were exposed to above LC50 of H2 (a con-centration that shows an effect but does not kill all of the parasites)after they had been previously exposed to folic acid, folinic acid andferulic acid. Folic acid competes for the active site of DHFR, as it isthe natural substrate. Folinic acid does not need to undergometabolism to contribute to DNA synthesis, and ferulic acid has anantioxidant effect. Although the effect anti-leishmanial of H2 wasdecreased, differences were observed among folic acid, folinic acidand ferulic acid. The greatest inhibitor of the H2 antileishmanialeffect was folic acid, which showed a 20% increase in parasite
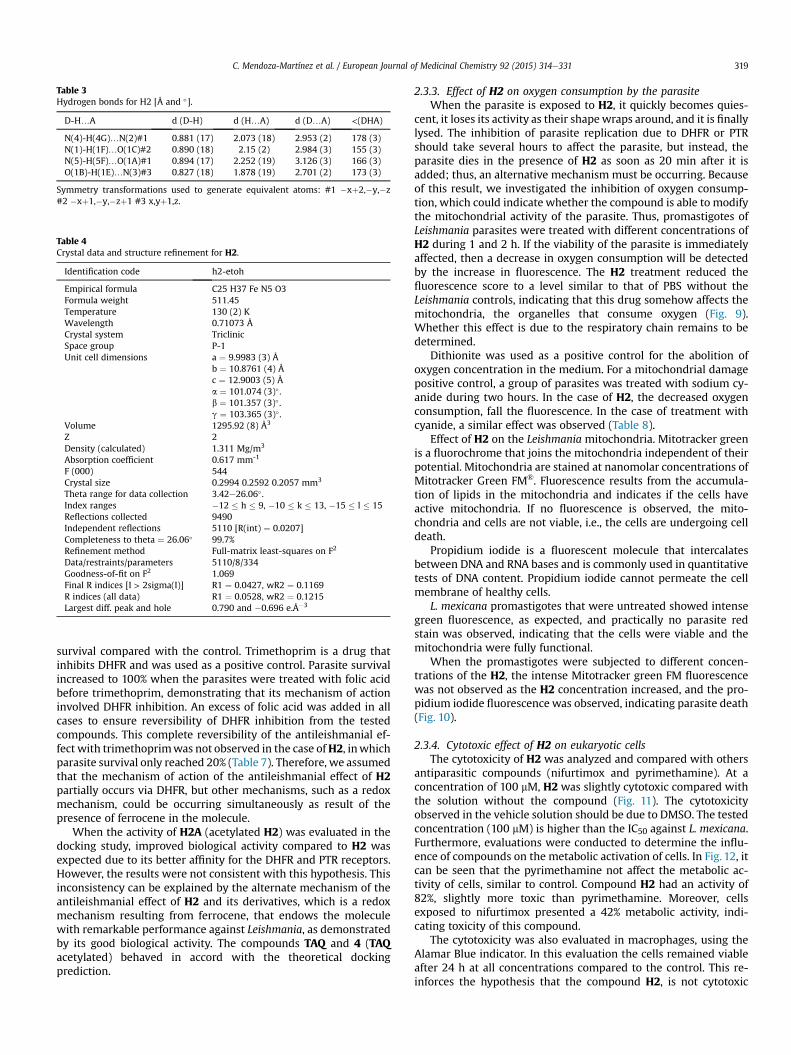
Table 3Hydrogen bonds for H2 [Å and �].
D-H…A d (D-H) d (H…A) d (D…A) <(DHA)
N(4)-H(4G)…N(2)#1 0.881 (17) 2.073 (18) 2.953 (2) 178 (3)N(1)-H(1F)…O(1C)#2 0.890 (18) 2.15 (2) 2.984 (3) 155 (3)N(5)-H(5F)…O(1A)#1 0.894 (17) 2.252 (19) 3.126 (3) 166 (3)O(1B)-H(1E)…N(3)#3 0.827 (18) 1.878 (19) 2.701 (2) 173 (3)
Symmetry transformations used to generate equivalent atoms: #1 �xþ2,�y,�z#2 �xþ1,�y,�zþ1 #3 x,yþ1,z.
Table 4Crystal data and structure refinement for H2.
Identification code h2-etoh
Empirical formula C25 H37 Fe N5 O3Formula weight 511.45Temperature 130 (2) KWavelength 0.71073 ÅCrystal system TriclinicSpace group P-1Unit cell dimensions a ¼ 9.9983 (3) Å
b ¼ 10.8761 (4) Åc ¼ 12.9003 (5) Åa ¼ 101.074 (3)� .b ¼ 101.357 (3)� .g ¼ 103.365 (3)� .
Volume 1295.92 (8) Å3
Z 2Density (calculated) 1.311 Mg/m3
Absorption coefficient 0.617 mm-1
F (000) 544Crystal size 0.2994 0.2592 0.2057 mm3
Theta range for data collection 3.42e26.06� .Index ranges �12 � h � 9, �10 � k � 13, �15 � l � 15Reflections collected 9490Independent reflections 5110 [R(int) ¼ 0.0207]Completeness to theta ¼ 26.06� 99.7%Refinement method Full-matrix least-squares on F2
Data/restraints/parameters 5110/8/334Goodness-of-fit on F2 1.069Final R indices [I > 2sigma(I)] R1 ¼ 0.0427, wR2 ¼ 0.1169R indices (all data) R1 ¼ 0.0528, wR2 ¼ 0.1215Largest diff. peak and hole 0.790 and �0.696 e.Å�3
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331 319
survival compared with the control. Trimethoprim is a drug thatinhibits DHFR and was used as a positive control. Parasite survivalincreased to 100% when the parasites were treated with folic acidbefore trimethoprim, demonstrating that its mechanism of actioninvolved DHFR inhibition. An excess of folic acid was added in allcases to ensure reversibility of DHFR inhibition from the testedcompounds. This complete reversibility of the antileishmanial ef-fect with trimethoprimwas not observed in the case ofH2, inwhichparasite survival only reached 20% (Table 7). Therefore, we assumedthat the mechanism of action of the antileishmanial effect of H2partially occurs via DHFR, but other mechanisms, such as a redoxmechanism, could be occurring simultaneously as result of thepresence of ferrocene in the molecule.
When the activity of H2A (acetylated H2) was evaluated in thedocking study, improved biological activity compared to H2 wasexpected due to its better affinity for the DHFR and PTR receptors.However, the results were not consistent with this hypothesis. Thisinconsistency can be explained by the alternate mechanism of theantileishmanial effect of H2 and its derivatives, which is a redoxmechanism resulting from ferrocene, that endows the moleculewith remarkable performance against Leishmania, as demonstratedby its good biological activity. The compounds TAQ and 4 (TAQacetylated) behaved in accord with the theoretical dockingprediction.
2.3.3. Effect of H2 on oxygen consumption by the parasiteWhen the parasite is exposed to H2, it quickly becomes quies-
cent, it loses its activity as their shape wraps around, and it is finallylysed. The inhibition of parasite replication due to DHFR or PTRshould take several hours to affect the parasite, but instead, theparasite dies in the presence of H2 as soon as 20 min after it isadded; thus, an alternative mechanism must be occurring. Becauseof this result, we investigated the inhibition of oxygen consump-tion, which could indicate whether the compound is able to modifythe mitochondrial activity of the parasite. Thus, promastigotes ofLeishmania parasites were treated with different concentrations ofH2 during 1 and 2 h. If the viability of the parasite is immediatelyaffected, then a decrease in oxygen consumption will be detectedby the increase in fluorescence. The H2 treatment reduced thefluorescence score to a level similar to that of PBS without theLeishmania controls, indicating that this drug somehow affects themitochondria, the organelles that consume oxygen (Fig. 9).Whether this effect is due to the respiratory chain remains to bedetermined.
Dithionite was used as a positive control for the abolition ofoxygen concentration in the medium. For a mitochondrial damagepositive control, a group of parasites was treated with sodium cy-anide during two hours. In the case of H2, the decreased oxygenconsumption, fall the fluorescence. In the case of treatment withcyanide, a similar effect was observed (Table 8).
Effect of H2 on the Leishmania mitochondria. Mitotracker greenis a fluorochrome that joins the mitochondria independent of theirpotential. Mitochondria are stained at nanomolar concentrations ofMitotracker Green FM®. Fluorescence results from the accumula-tion of lipids in the mitochondria and indicates if the cells haveactive mitochondria. If no fluorescence is observed, the mito-chondria and cells are not viable, i.e., the cells are undergoing celldeath.
Propidium iodide is a fluorescent molecule that intercalatesbetween DNA and RNA bases and is commonly used in quantitativetests of DNA content. Propidium iodide cannot permeate the cellmembrane of healthy cells.
L. mexicana promastigotes that were untreated showed intensegreen fluorescence, as expected, and practically no parasite redstain was observed, indicating that the cells were viable and themitochondria were fully functional.
When the promastigotes were subjected to different concen-trations of the H2, the intense Mitotracker green FM fluorescencewas not observed as the H2 concentration increased, and the pro-pidium iodide fluorescence was observed, indicating parasite death(Fig. 10).
2.3.4. Cytotoxic effect of H2 on eukaryotic cellsThe cytotoxicity of H2 was analyzed and compared with others
antiparasitic compounds (nifurtimox and pyrimethamine). At aconcentration of 100 mM, H2 was slightly cytotoxic compared withthe solution without the compound (Fig. 11). The cytotoxicityobserved in the vehicle solution should be due to DMSO. The testedconcentration (100 mM) is higher than the IC50 against L. mexicana.Furthermore, evaluations were conducted to determine the influ-ence of compounds on the metabolic activation of cells. In Fig. 12, itcan be seen that the pyrimethamine not affect the metabolic ac-tivity of cells, similar to control. Compound H2 had an activity of82%, slightly more toxic than pyrimethamine. Moreover, cellsexposed to nifurtimox presented a 42% metabolic activity, indi-cating toxicity of this compound.
The cytotoxicity was also evaluated in macrophages, using theAlamar Blue indicator. In this evaluation the cells remained viableafter 24 h at all concentrations compared to the control. This re-inforces the hypothesis that the compound H2, is not cytotoxic
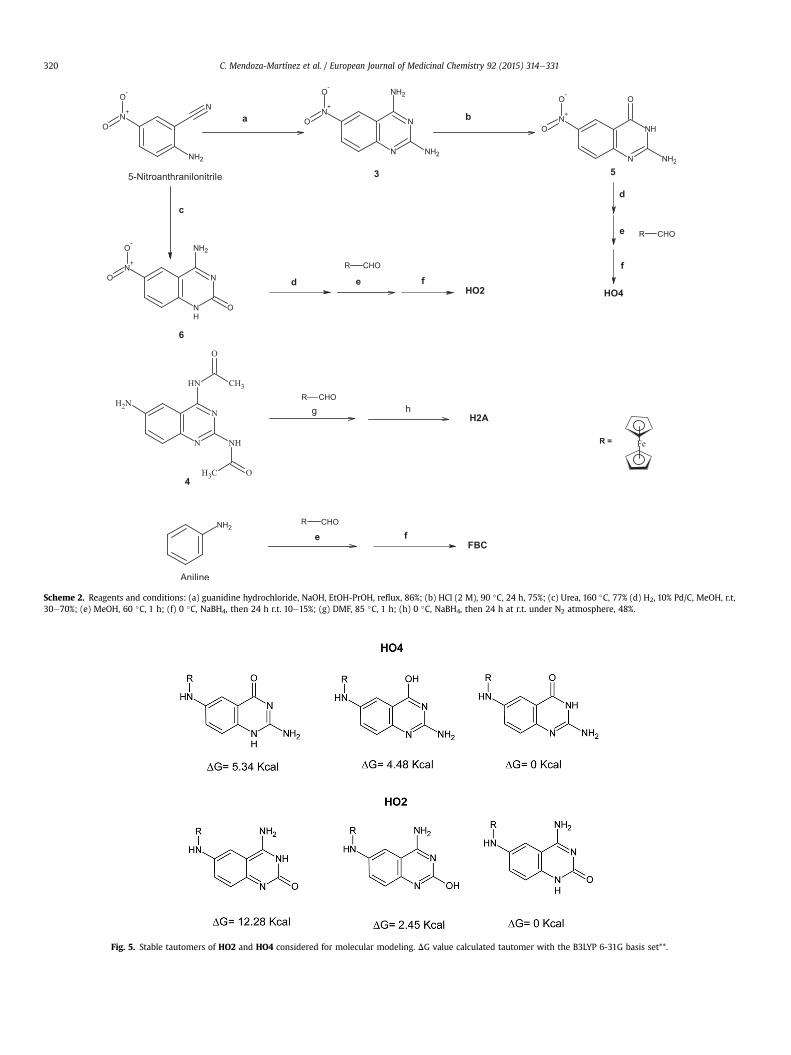
Scheme 2. Reagents and conditions: (a) guanidine hydrochloride, NaOH, EtOH-PrOH, reflux, 86%; (b) HCl (2 M), 90 �C, 24 h, 75%; (c) Urea, 160 �C, 77% (d) H2, 10% Pd/C, MeOH, r.t,30e70%; (e) MeOH, 60 �C, 1 h; (f) 0 �C, NaBH4, then 24 h r.t. 10e15%; (g) DMF, 85 �C, 1 h; (h) 0 �C, NaBH4, then 24 h at r.t. under N2 atmosphere, 48%.
Fig. 5. Stable tautomers of HO2 and HO4 considered for molecular modeling. DG value calculated tautomer with the B3LYP 6-31G basis set**.
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331320
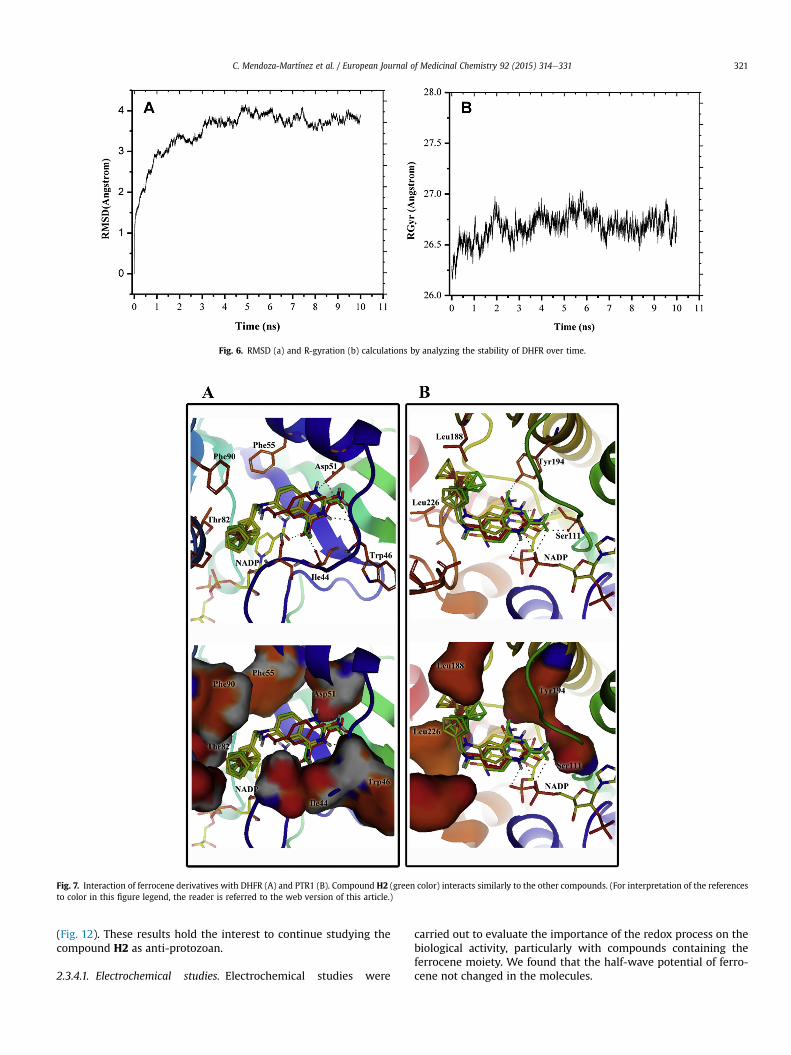
Fig. 6. RMSD (a) and R-gyration (b) calculations by analyzing the stability of DHFR over time.
Fig. 7. Interaction of ferrocene derivatives with DHFR (A) and PTR1 (B). Compound H2 (green color) interacts similarly to the other compounds. (For interpretation of the referencesto color in this figure legend, the reader is referred to the web version of this article.)
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331 321
(Fig. 12). These results hold the interest to continue studying thecompound H2 as anti-protozoan.
2.3.4.1. Electrochemical studies. Electrochemical studies were
carried out to evaluate the importance of the redox process on thebiological activity, particularly with compounds containing theferrocene moiety. We found that the half-wave potential of ferro-cene not changed in the molecules.

Table 5Docking results by DHFR and PTR from L. major.
Compound DHFR PTR
Kd (mM) DG (Kcal) Kd (mM) DG (Kcal)
H1 0.241 �9.03 1.430 �7.98H2 0.379 �8.76 1.460 �7.96H3 0.752 �8.35 0.093 �9.59H4 1.580 �7.91 2.850 �7.57H5 1.020 �8.17 2.400 �7.67HO2 (most stable tautomer) 3.650 �7.42 1.350 �8.01HO4 (most stable tautomer) 1.700 �7.87 0.223 �9.08H2A 0.087 �9.63 0.008 �13.45TAQ 83.98 �5.56 58.75 �5.773 15.81 �6.55 0.2615 �8.98
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331322
It is important mentioning that of all of the compounds, H2 isoxidizedmost easily.H2 has an oxidation potential of�0.086 V (IA),and the magnitude of the current intensity observed in this processis three times higher than those of TAQ and the other compounds.That could be very important for the biological activity (Fig. 13). Wepropose that the nitrogen in position 1 of quinazoline nucleus isoxidized because this is the location of the highest electron density(Fig. 14).
The presence of the ferrocene group is important for the bio-logical activity, which is demonstrated by the parasite growth in-hibition caused by ferrocene and FBC. However, for ferrocenederivatives, the oxidation potential of the Fc/Fcþ couple does notsubstantially change (Table 9); therefore, the difference in activitybetween them results from the modification of the quinazolinenucleus.
3. Conclusions
Leishmaniasis has become a health issue due to its high inci-dence, especially in underdeveloped countries. There is an urgentneed to develop molecules that can better treat the disease becauseexisting molecules have become inefficient. In this study, we havetwo new antileishmanial agents (H2 and H4). The first presentedthe most pronounced potency and activity against promastigotesand amastigotes stages of parasite. The results demonstrated thatleishmanicidal activity of H2 is due to multiple mechanisms ofaction, which are the most significant advantage of this molecule,because they could reduce the likelihood of the parasite developingresistance. It is interesting to note that this antiparasitic effect islargely due to the presence of the ferrocene group. This moietyaffords distinct characteristics to the molecule, primarily as resultof the readily oxidized iron atom. Previous studies have shown thatthe ferrocene group can produce ROS, principally hydroxyl radicals,to exert its effect. Additionally, the quinazoline-2,4-diamine scaf-fold is required for biological activity, as demonstrated by the poorbiological activities of HO2, HO4, H2A and FBC. In this sense, theexocyclic amino groups must be retained because they are tocontribute to receptor interaction. It is worth mentioning thatH2 iselectrochemically oxidized more easily with respect other quina-zoline derivatives, indicating that the mechanism of action involvesa type of redox reaction. Regarding the inhibition of DHFR or PTR isstill involved, but it is not the primary mechanism of action of thiscompound because the consequences of their enzymatic inhibitionwould require several hours for their display; H2 initiates its bio-logical action from the first hour after being applied to parasites.
Other possibility is that the mitochondria are actively affectedduring the action of H2. The decrease in oxygen consumption(similar to that observed with cyanide) clearly indicates the rela-tionship of the mechanism to inhibition of the respiratory chain,
which results in apoptosis of the parasite. However, more studiesare required to identify the extent of this influence on mitochon-drial function. Furthermore, the fact that the promastigotes becomepermeable to propidium iodide is an indicator of the cell membranedamage caused by H2. We are currently clarifying these hypothe-ses. A feature last, in cytotoxicity studies, H2 did not showed severetoxicity in eukaryotic cells.
In regard to H4, which also presented an interesting activity, wecould indicate that likely it has a good interaction with its receptorowing to the substituent lipoid that constitutes. Its mechanism ofaction may be different from H2 because it does not present in itsstructure the ferrocene entity, and its antiparasitic effect is man-ifested after 24 h of exposure. We will focus on studying in greaterdetail the mechanism of activity and optimization of this com-pounds in the future.
Finally, we emphasize that there are new opportunities todevelop novel drugs that are more active against intracellularparasites, such as Leishmania, and that present only low toxicity tothe host. The results presented in this study advance the fightagainst orphan parasitic diseases, which often cause death.
4. Experimental section
4.1. Chemistry
Melting points were determined in open capillary tubes with aBüchi B-540 melting point apparatus and are uncorrected. Re-actions were monitored by TLC on 0.2 mm precoated silica gel 60F254 plates (E. Merck) and were visualized by irradiation with a UVlamp. Silica gel 60 (70e230 mesh) was used for column chroma-tography. IR spectra were recorded with a FT Perkin Elmer 16 PCspectrometer on KBr disks. 1H NMR spectra were measured with aVarian EM-390 (300 MHz) spectrometer. Chemical shifts are givenin ppm relative to tetramethylsilane (Me4Si, d ¼ 0), and J values aregiven in Hz. Splitting patterns have been designated as follows: s,singlet; d, doublet; q, quartet; dd, doublet of doublet; t, triplet; m,multiplet; br. s, broad singlet. MS data were recorded on a JEOLJMS-SX102A spectrometer by FAB [(FAB(þ)]. Elemental analyses (C,H, N) for the new compounds were performed on a Fisons EA 1108instrument and were within ±0.4% of the theoretical values of theproposed structures. The catalytic hydrogenation reactions werecarried out in a Parr shaker hydrogenation apparatus. The startingmaterials 5-nitroanthranilonitrile, guanidine hydrochloride, andaniline are commercially available (Sigma Aldrich).
The percent purity for compounds H1, H2, H3, H4, H5, H2A,HO2, HO4 and FBC was calculated by chromatographic analysis.Standard solutions of all of the compounds were dissolved inmethanol grade HPLC, and the volume of injection was 20 mL. Themethanol and acetonitrile used for the mobile phase were ofchromatographic grade (J.T. Baker, Phillipsburg, NJ, U.S.A). Waterwas deionized and osmosed using a Milli-Q® system (Millipore,Bedford, MA, U.S.A). The phosphates were of analytical grade. TheHPLC system consisted of a Waters Alliance e2695 separationmodule autosampler and a 2489 dual l absorbance UV/Visibledetector coupled with Empower™ software (Waters, Milford, MA,U.S.A). The analytical column was an Acentis® RP-Amide column(150 mm� 4.6 mm ID, particle size 5 mm) (Supelco, SigmaeAldrich,U.S.A) protected by a compatible guard column. The mobile phaseconsisted of 10 mM phosphate buffer at pH 3.3 and acetonitrile(60:40 v/v). The assay run time was 8 min with a flow rate of1.00 mL/min, and it was carried out at 30 �C. Absorbance wasmeasured at 240 and 272 nm.
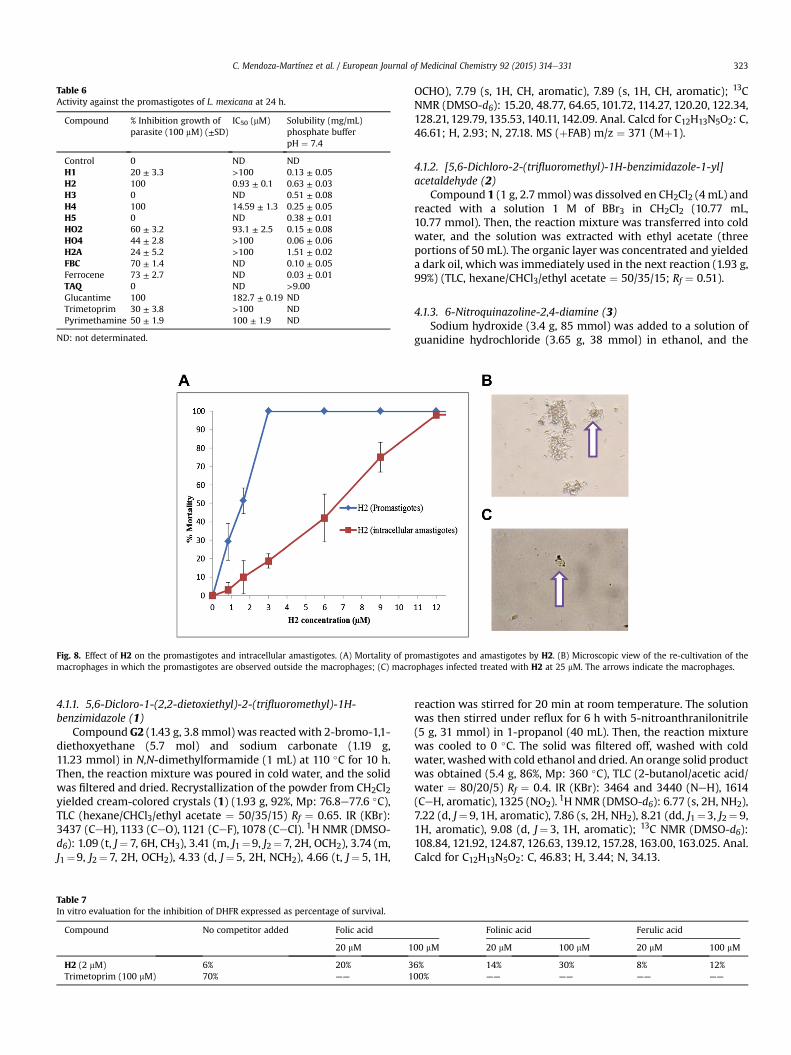
Table 6Activity against the promastigotes of L. mexicana at 24 h.
Compound % Inhibition growth ofparasite (100 mM) (±SD)
IC50 (mM) Solubility (mg/mL)phosphate bufferpH ¼ 7.4
Control 0 ND NDH1 20 ± 3.3 >100 0.13 ± 0.05H2 100 0.93 ± 0.1 0.63 ± 0.03H3 0 ND 0.51 ± 0.08H4 100 14.59 ± 1.3 0.25 ± 0.05H5 0 ND 0.38 ± 0.01HO2 60 ± 3.2 93.1 ± 2.5 0.15 ± 0.08HO4 44 ± 2.8 >100 0.06 ± 0.06H2A 24 ± 5.2 >100 1.51 ± 0.02FBC 70 ± 1.4 ND 0.10 ± 0.05Ferrocene 73 ± 2.7 ND 0.03 ± 0.01TAQ 0 ND >9.00Glucantime 100 182.7 ± 0.19 NDTrimetoprim 30 ± 3.8 >100 NDPyrimethamine 50 ± 1.9 100 ± 1.9 ND
ND: not determinated.
Fig. 8. Effect of H2 on the promastigotes and intracellular amastigotes. (A) Mortality of promastigotes and amastigotes by H2. (B) Microscopic view of the re-cultivation of themacrophages in which the promastigotes are observed outside the macrophages; (C) macrophages infected treated with H2 at 25 mM. The arrows indicate the macrophages.
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331 323
4.1.1. 5,6-Dicloro-1-(2,2-dietoxiethyl)-2-(trifluoromethyl)-1H-benzimidazole (1)
Compound G2 (1.43 g, 3.8 mmol) was reactedwith 2-bromo-1,1-diethoxyethane (5.7 mol) and sodium carbonate (1.19 g,11.23 mmol) in N,N-dimethylformamide (1 mL) at 110 �C for 10 h.Then, the reaction mixture was poured in cold water, and the solidwas filtered and dried. Recrystallization of the powder from CH2Cl2yielded cream-colored crystals (1) (1.93 g, 92%, Mp: 76.8e77.6 �C),TLC (hexane/CHCl3/ethyl acetate ¼ 50/35/15) Rf ¼ 0.65. IR (KBr):3437 (CeH), 1133 (CeO), 1121 (CeF), 1078 (CeCl). 1H NMR (DMSO-d6): 1.09 (t, J¼ 7, 6H, CH3), 3.41 (m, J1¼9, J2¼ 7, 2H, OCH2), 3.74 (m,J1¼9, J2¼ 7, 2H, OCH2), 4.33 (d, J¼ 5, 2H, NCH2), 4.66 (t, J¼ 5, 1H,
Table 7In vitro evaluation for the inhibition of DHFR expressed as percentage of survival.
Compound No competitor added Folic acid
20 mM 1
H2 (2 mM) 6% 20% 3Trimetoprim (100 mM) 70% —— 1
OCHO), 7.79 (s, 1H, CH, aromatic), 7.89 (s, 1H, CH, aromatic); 13CNMR (DMSO-d6): 15.20, 48.77, 64.65, 101.72, 114.27, 120.20, 122.34,128.21, 129.79, 135.53, 140.11, 142.09. Anal. Calcd for C12H13N5O2: C,46.61; H, 2.93; N, 27.18. MS (þFAB) m/z ¼ 371 (Mþ1).
4.1.2. [5,6-Dichloro-2-(trifluoromethyl)-1H-benzimidazole-1-yl]acetaldehyde (2)
Compound 1 (1 g, 2.7 mmol) was dissolved en CH2Cl2 (4 mL) andreacted with a solution 1 M of BBr3 in CH2Cl2 (10.77 mL,10.77 mmol). Then, the reaction mixture was transferred into coldwater, and the solution was extracted with ethyl acetate (threeportions of 50 mL). The organic layer was concentrated and yieldeda dark oil, which was immediately used in the next reaction (1.93 g,99%) (TLC, hexane/CHCl3/ethyl acetate ¼ 50/35/15; Rf ¼ 0.51).
4.1.3. 6-Nitroquinazoline-2,4-diamine (3)Sodium hydroxide (3.4 g, 85 mmol) was added to a solution of
guanidine hydrochloride (3.65 g, 38 mmol) in ethanol, and the
reaction was stirred for 20 min at room temperature. The solutionwas then stirred under reflux for 6 h with 5-nitroanthranilonitrile(5 g, 31 mmol) in 1-propanol (40 mL). Then, the reaction mixturewas cooled to 0 �C. The solid was filtered off, washed with coldwater, washedwith cold ethanol and dried. An orange solid productwas obtained (5.4 g, 86%, Mp: 360 �C), TLC (2-butanol/acetic acid/water ¼ 80/20/5) Rf ¼ 0.4. IR (KBr): 3464 and 3440 (NeH), 1614(CeH, aromatic), 1325 (NO2). 1H NMR (DMSO-d6): 6.77 (s, 2H, NH2),7.22 (d, J¼ 9, 1H, aromatic), 7.86 (s, 2H, NH2), 8.21 (dd, J1¼3, J2¼ 9,1H, aromatic), 9.08 (d, J¼ 3, 1H, aromatic); 13C NMR (DMSO-d6):108.84, 121.92, 124.87, 126.63, 139.12, 157.28, 163.00, 163.025. Anal.Calcd for C12H13N5O2: C, 46.83; H, 3.44; N, 34.13.
Folinic acid Ferulic acid
00 mM 20 mM 100 mM 20 mM 100 mM
6% 14% 30% 8% 12%00% —— —— —— ——

Fig. 9. L. mexicana treated with different concentrations of H2. L. mexicana (5 � 106) treated with different concentrations of H2 (0e100 mM) during 1 h (A) and 2 h (B) before thedeterminations. The data represent the percentage of fluorescence; the control was considered 100%. At least four independent experiments were performed. * p < 0.05 vs controlwithout the drug. (Tukey Test). A. U.
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331324
4.1.4. N-[2-(acetylamino)-6-aminoquinazolin-4-yl]acetamide (4)Compound 3 (1 g, 4.87 mmol) and acetic anhydride (1 mL) were
heated at 100 �C for 12 h. Then, the reaction mixture was pouredinto cold water, and the solid was filtered off, washed with coldwater at pH 7 and dried. A yellow solid (3a) was obtained (0.89 g,65%, Mp: 278 �C), TLC (CHCl3/MeOH ¼ 60/40) Rf ¼ 0.8. Anal. Calcdfor C12H11N5O4: C, 49.83; H, 3.83; N, 24.21. Found: C, 49.99; H, 3.11;N, 23.81.
The catalytic reduction of 3a (0.5 g, 1.73 mmol) with hydrogenand Pd/C (10%) (0.05 g) was carried out on a Parr assembly at 60 psiat room temperature for 1 h. The catalyst was then removed byfiltration, and the filtrate was concentrated on a rotavapor underreduced pressure to yield 4 (0.34 g, 76%, Mp ¼ 238 �C), TLC (CHCl3/MeOH ¼ 60/40) Rf ¼ 0.67. IR (KBr): 3353 and 3230 (NeH). 1H NMR(DMSO-d6): 2.2 (s, 3H, CH3), 2.34 (s, 3H, CH3), 5.61 (br. s, 2H, NH2),7.04 (d, J¼ 3, 1H, aromatic), 7.28 (dd, J1 ¼8, J2 ¼ 3, 1H, aromatic), 7.5(d, J ¼ 8, 1H, aromatic), 10.46 (br. s, 1H, NH amide), 10.22 (br. s, 1H,NH amide); 13C NMR (DMSO-d6): 24.51, 102.37, 116.82, 126.02,127.48, 145.16, 146.55, 150.08, 156.08, 169.18, 170.73. Anal. Calcd forC12H13N5O2: C, 55.59; H, 5.05; N, 27.01. Found:C, 57.0; H, 4.52; N,25.20.
4.1.5. 2-amino-6-nitroquinazolin-4(3H)-one (5)Compound 3 (2 g, 9.7 mmol) was refluxed in 75 mL of HCl (6 N)
for 2 weeks. The solution was then cooled and adjusted to pH 7with a saturated solution of Na2CO3. The solid was filtered andwashed repeatedly with water. A yellow solid was obtained (1.5 g,75%, Mp: 351.5e352.5 �C), TLC (CHCl3/MeOH ¼ 80/20) Rf ¼ 0.53. IR(KBr): 3572 and 3437 (NeH), 1704 (C]O), 1330 (NO2). 1H NMR(DMSO-d6): 7.06 (br. s, 2H, NH2), 7.28 (d, J ¼ 9, 1H, aromatic), 8.30(dd, J1 ¼ 3, J2 ¼ 9, 1H, aromatic), 8.61 (d, J ¼ 3, 1H, aromatic), 11.74(br. s, 1H, NH); 13C NMR (DMSO-d6): 116.71, 123.13, 125.23, 127.76,141.05, 154.87, 156.84, 162.04. Anal. Calcd for C12H13N5O2: C, 46.61;H, 2.93; N, 27.18. Found:C, 47.0; H, 2.71; N, 28.12.
4.1.6. 4-amino-6-nitroquinazolin-2(1H)-one (6)Urea and 5-nitroantranilonitrile (2 g) were heated at 160 �C for
2 h with agitation. The mixture was then transferred into coldwater. The solid was filtered and washed repeatedly with water. Ayellow compound was obtained (0.34 g, 77%, Mp: 351.7e354.4 �C),TLC (CHCl3/MeOH ¼ 80/20) Rf ¼ 0.82. IR (KBr): 3444 and 3333(NeH), 1703 (C]O), 1317 (NO2). 1H NMR (DMSO-d6): 7.24 (d, J ¼ 9,1H, aromatic), 8.09 (br. s, 2H, NH2), 8.37 (dd, J1 ¼ 2, J2 ¼ 9, 1H, ar-omatic), 9.07 (d, J ¼ 2, 1H, aromatic), 11.29 (br. s, 1H, NHCO). 13CNMR (DMSO-d6): 108.22, 116.29, 122.19, 128.96, 131.89, 141.24,
147.67, 156.09, 163.73. Anal. Calcd for C12H13N5O2: C, 46.61; H, 2.93;N, 27.18. Found: C, 45.64; H, 2.74; N, 28.01.
4.1.7. Synthesis of compounds H1, H2, H3, H4 and H5A solution of the appropriate aldehyde (1.6 mmol) and com-
pound 4 (0.4144 g, 1.6 mmol) in methanol (80 mL) was stirred at60 �C for 2 days. Then, the reaction mixture was cooled to 0 �C, andNaBH4 (0.09 g, 2.37 mmol) was added. Subsequently, the reactionmixture was stirred at room temperature for 12 h. Next, the reac-tion mixture was concentrated on a rotavapor apparatus underreduced pressure, and cold water was added. The solid was filteredoff, washed with cold water and dried.
4.1.8. N6-(4-nitrobenzyl)quinazoline-2,4,6-triamine (H1)Red powder (0.41 g, 82%, Mp: 215e218 �C). TLC (2-butanol/
acetic acid/water ¼ 80/20/5) Rf ¼ 0.54. IR (KBr): 3369 (NeH), 1517(eNO2); 1H NMR (DMSO-d6): 4.45 (d, J ¼ 6, 2H, CH2), 5.71 (br. s, 2H,NH2), 6.25 (t, J ¼ 6, 1H, NH), 7.03 (m, 5H, NH2, CeH quinazoline),7.67 (d, J ¼ 8, 2H, p-nitrobenzyl), 8.21 (d, J ¼ 8, 2H, p-nitrobenzyl);13C NMR (DMSO-d6): 46.16 (CH2), 100.66 (CH, quinazoline), 110.93(C, quinazoline), 123.17 (CH, quinazoline), 123.32 (CH, p-nitro-benzyl), 128.44 (CH, p-nitrobenzyl), 142.35 (CeNH, quinazoline),144.44 (C, quinazoline), 146.36 (C, p-nitrobenzyl), 148.91 (CeNO2),158.18 (CeNH2, quinazoline), 161.435 (CeNH2, quinazoline); Anal.Calcd for C15H14N6O2: C, 58.06; H, 4.55; N, 27.08. Found: C, 57.0; H,4.52; N, 25.20. HPLC purity of 99.9% (retention time ¼ 3.841 min).
4.1.9. N6-(ferrocenmethyl)quinazolin-2,4,6-triamine (H2)Dark yellow powder (62%, Mp: 210.6e211 �C). TLC (2-butanol/
acetic acid/water ¼ 80/20/5) Rf ¼ 0.53. IR (KBr): 3393 (NeH), 1613(CeN), 823 (CeH ferrocene). 1H NMR (DMSO-d6): 3.98 (t, J¼ 6, 2H,CH2), 4.12 (s, 2H, ferrocene), 4.20 (s, 5H, ferrocene), 4.32 (s, 2H,ferrocene), 5.68 (t, J ¼ 6, 2H, NH2), 6.23 (s, 2H, NH2), 7.03 (m, 3H,aromatic quinazoline), 11.24 (br. s, 1H, NH). 13C NMR (DMSO-d6):43.01 (CH2), 67.33 (CH, ferrocene), 68.40 (CH, ferrocene), 86.10 (C,ferrocene), 100.25 (CH, quinazoline), 110.73 (C, quinazoline), 123.52(CH, quinazoline), 124.99 (CH, quinazoline), 142.84 (CeNH, quina-zoline), 145.11 (CeNH, quinazoline), 158.34 (CeNH2, quinazoline),161.48 (CeNH2, quinazoline). Anal. Calcd for C19H19FeN5: C, 61.14;H, 5.13; N, 18.76. Found: C, 61.14; H, 4.92; N, 18.03. HPLC purity of99.9% (retention time ¼ 3.947 min). The X-ray structure wasdetermined.

Table 8Percent oxygen consumption for the treatment of the promastigotes before 2 h.
Treatment % Oxygen consumption
Leishmania 100PBS 20.4% ± 1.2L. mexicana treated with sodium cyanide (0.02 mM) 20.5% ± 1L. mexicana treated with H2 (100 mM) 22.5% ± 1Sodium dithionite (5.74 mM) 175.9% ± 5.8
Fig. 10. The promastigotes of L. mexicana exposed to H2. (A) 10 mM, (B) 20 mM, (C) 40 mM, and (D) 100 mM. Although an intense fluorescence with the parasites continued to beobserved, some of them had little movement; note that some of the promastigotes were completely immobile. The promastigotes that were less fluorescent and also tested positivefor propidium iodide (E).
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331 325
4.1.10. N6-{2-[5,6-dichloro-2-(trifluoromethyl)-1H-benzimidazol-1-yl]ethyl}quinazoline-2,4,6-triamine (H3)
Brown powder (0.62 g, 85%, Mp: 133e136 �C), TLC (2-butanol/acetic acid/water ¼ 80/20/5) Rf ¼ 0.59. IR (KBr): 3350 (NeH), 1273(CeN), 742 (CeF); 1H NMR (DMSO-d6): 3.53 (br. s, 2H, CH2), 4.55 (br.s, 2H, CH2), 5.61 (s, 1H, NH), 5.65 (s, 2H, NH2). 6.84 (s, 1H, quina-zoline), 7.07 (m, 4H, NH2, CH quinazoline), 7.92 (br. s, 1H, benz-imidazole), 8.13 (s, 1H, benzimidazole); 13C NMR (DMSO-d6): 42.89(CH2), 44.21 (CH2),100.21 (CH, quinazoline),110.67 (C, quinazoline),114.29 (CH, benzimidazole), 117.29 (C, benzimidazole), 121.91 (CeH,benzimidazole), 123.27 (CH, quinazoline), 124.89 (CH, quinazoline),126.31 (CeCl, benzimidazole), 127.88 (CeCl, benzimidazole), 135.31(C, benzimidazole), 139.76 (C-CF3), 140.00 (CF3), 141.80 (CeNH,quinazoline), 144.45 (C, quinazoline), 156.66 (CeNH2, quinazoline),161.98 (CeNH2, quinazoline). MS (þFAB) m/z ¼ 456 (Mþ1); HRMS(þFAB) calcd for C18H15Cl2F3N7: 456.07. Found: 456.07. HPLC purityof 99.9% (retention time ¼ 3.188 min).
4.1.11. N6-(4-methoxybenzyl)quinazoline-2,4,6-triamine (H4)Yellow powder (0.19 g, 40%, Mp: 204.1e205.2 �C). TLC (CHCl3/
MeOH ¼ 80/20) Rf ¼ 0.73. IR (KBr): 3450 and 3328 (NeH), 1639(CeN), 1469 and 1563 (C]C Ar), 1243 (CeOeC); 1H NMR (DMSO-d6) ppm: 3.72 (s, 3H, CH3), 4.20 (d., J ¼ 6, 2H, CH2), 5.53 (br. s, 2H,NH2), 5.81 (t, J ¼ 6, 1H, NH), 6.89 (d, J ¼ 9, 2H, p-methoxybenzyl),6.96 (br. s, 3H, NH2, CH quinazoline), 7.03 (m, 2H, CH quinazoline),7.35 (d, J ¼ 9, 2H, p-methoxybenzyl); 13C NMR (DMSO-d6): 46.69(CH2), 55.45 (CH3eO), 100.89 (CH, quinazoline), 111.15 (C, quina-zoline), 114.02 (CH, p-methoxybenzyl), 123.91 (CH, quinazoline),125.35 (CH, quinazoline), 129.42 (CH, p-methoxybenzyl), 132.31 (C,p-methoxybenzyl), 143.26 (CeNH, quinazoline), 145.37 (C,
quinazoline), 158.57 (CeO, p-methoxybenzyl), 158.70 (CeNH2,quinazoline), 161.85 (CeNH2, quinazoline). MS (þFAB) m/z ¼ 296(Mþ1); HRMS (þFAB) calcd for C16H18N5O: 296.15. Found: 296.15.HPLC purity of 99.9% (retention time ¼ 3.597 min).
4.1.12. 4-{[(2,4-Diaminoquinazolin-6-yl)amino]methyl}phenol(H5)
Yellow powder (0.36 g, 79%, Mp: 192.1e193.7 �C). TLC (CHCl3/
MeOH ¼ 70/30) Rf ¼ 0.65. IR (KBr): 3355 (OH), 3206 (NH), 1626(CN), 1466 and 1568 (C]C Ar); 1H NMR (DMSO-d6): 4.14 (d, J ¼ 3,2H, CH2), 5.51 (br. s, 2H, NH2), 5.70 (t, J ¼ 3, 1H, NH), 6.73 (d, J ¼ 8,2H, p-hydroxybenzyl), 6.96 (br. s, 3H, NH2, CH quinazoline), 7.03 (m,2H, quinazoline), 7.22 (d, J¼ 8, 2H, p-hydroxybenzyl), 9.23 (br. s, 1H,OH); 13C NMR (DMSO-d6): 47.01 (CH2), 101.79 (CH, quinazoline),110.78 (C, quinazoline), 115.45 (CH, p-hydroxybenzyl), 124.31 (CH,quinazoline), 126.11 (CH, quinazoline), 129.48 (CH, p-hydrox-ybenzyl), 130.10 (C, p-hydroxybenzyl), 144.21 (CeNH, quinazoline),156.79 (CeO, p-hydroxybenzyl), 159.71 (CeNH2), 162.49 (CeNH2,quinazoline); MS (þFAB) m/z ¼ 282 (Mþ1); HRMS (þFAB) calcd forC15H16N5O: 282.13. Found: 282.13. HPLC purity of 92% (retentiontime ¼ 3.081 min).
4.1.13. N-{2-(acetylamino)-6-[(ferrocenmethyl)amin]quinazolin-4-yl}acetamide (H2A)
A solution of ferrocencarboxaldehyde (0.5317 g, 2.48 mmol),compound 4 (0.5850 g, 2.25 mmol), and acetic acid (one drop) inN,N-dimethylformamide (1 mL) was stirred at 85 �C for 1 h. Then,the reaction mixture was cooled to 0 �C, and NaBH4 (0.09 g,2.43 mmol) was added. Subsequently, the reaction mixture wasstirred at room temperature for 12 h. Then, the reaction mixturewas concentrated on a rotavapor apparatus under reduced pres-sure, and cold water was added. The solid was filtered off, washedwith cold water and dried. Yellow powder (0.495 g, 48%, Mp:218.3e220.9 �C). TLC (CHCl3/MeOH ¼ 80/20) Rf ¼ 0.76. IR (KBr):3369 and 3244 (NeH), 1693 y 1668 (C]O), 825 (CeH ferroceno). 1HNMR (DMSO-d6): 2.2 (s, 3H, CH3), 2.49 (s, 3H, CH3), 4.1 (d, J ¼ 6, 2H,CH2), 4.13 (t, J ¼ 2, 2H, ferrocene), 4.22 (s, 5H, ferrocene), 4.34 (t,J ¼ 2, 2H, ferrocene), 6.17 (t, J ¼ 6, 1H, NH), 7.10 (d, J ¼ 2, 1H,

Fig. 11. Fibroblast exposed to the various compounds tested at 24 h of exposure.
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331326
quinazoline), 7.4 (dd, J ¼ 9, J ¼ 2.4, 1H, quinazoline), 7.47 (d, J ¼ 9,1H, quinazoline), 10.52 (br. s, 1H, NH amide), 10.2 (br. s, 1H, NHamide). 13C NMR (DMSO-d6): 24.38 and 25.31 (CH3,amide), 42.40(CH2), 67.43 (CH, ferrocene), 68.43 (CH, ferrocene), 85.55 (C,ferrocene), 97.99 (CH, quinazoline), 115.45 (C, quinazoline), 125.86(CH, quinazoline), 127.13 (CH, quinazoline), 145.11 (CeNH, quina-zoline), 146.13 (C, quinazoline), 149.87 (C-amide, quinazoline),155.58 (CeNHCO, quinazoline), 169 (C]O, amide), 171.75 (C]O,amide). EM-[FAB(þ)] m/z ¼ 457. HRMS (þFAB) calcd forC23H24FeN5O2: 457.1196. Found: 457.1170. HPLC purity of 99.9%(retention time ¼ 3.435 min).
4.1.14. 2-amino-6-[(4-ferrocenylmethyl)amino]quinazolin-4(3H)-one (HO4)
The catalytic reduction of 4 (1 g, 4.85 mmol) with hydrogen andPd/C (10%) (0.1 g) was performed on a Parr assembly at 60 psi atroom temperature for 1 h. The catalyst was filtered off, and thefiltrate was concentrated on a rotavapor apparatus under reducedpressure. A light brown compound (0.6 g) was obtained (4a,yield ¼ 70%), and it rapidly darkened; therefore, it was usedimmediately. A solution of ferrocencarboxaldehyde (0.21 g,0.9813 mmol) and the compound obtained from the above reduc-tion (4a) (0.15 g, 0.8514 mmol) in methanol (80 mL) was stirred at60 �C for 2 days. Then, the reaction mixture was cooled to 0 �C, andNaBH4 (0.064 g, 1.69 mmol) was added. Subsequently, the reactionmixture was stirred at room temperature for 12 h. Then, the reac-tion mixture was concentrated on a rotavapor apparatus underreduced pressure, and cold water was added. The solid was filtered
off, washed with cold water and dried. Brown powder (0.030 g, 10%,Mp: 171.2e173.1),TLC (CHCl3/MeOH ¼ 80/20) Rf ¼ 0.6. IR (KBr):3410 (NeH), 1644 (C]O), 825 (CeH ferrocene). 1H NMR (DMSO-d6): 3.99 (d, J¼ 6, 2H, CH2), 4.12 (br. s, 2H, ferrocene), 4.18 (s, 5H,ferrocene), 4.26 (br. s., 2H, ferrocene), 5.64 (t, J ¼ 6, 1H, NH), 5.9 (br.s, 2H, NH2), 7.02 (m, 3H, CH quinazoline), 10.78 (br. s, 1H, NHCO). 13CNMR (DMSO-d6): 42.77 (CH2), 67.26 (CH, ferrocene), 68.26 (CH,ferrocene), 68.14 (CH, ferrocene), 86.28 (C, ferrocene), 104.28 (CH,quinazoline), 117.62 (CH, quinazoline), 122.33 (CH, quinazoline),133.40 (C, quinazoline), 143.99 (C, quinazoline), 151.62 (CeNH2),166.48 (C]O). EM-[FAB(þ)] m/z ¼ 375. HRMS (þFAB) calcd forC19H19FeN4O: 375.0903. Found: 375.0865. HPLC purity of 98%(retention time ¼ 4.364 min).
4.1.15. 4-Amino-6-[(4-ferrocenylmethyl)amino]quinazolin-2(1H)-one (HO2)
The catalytic reduction of 5 (1 g, 4.85 mmol) with hydrogen andPd/C (10%) (0.1 g) was performed on a Parr assembly at 60 psi atroom temperature for 1 h. The catalyst was then filtered, and thefiltrate was concentrated on a rotavapor apparatus under reducedpressure. A light brown compound (0.3 g) was obtained (5a, 35%),and it rapidly darkened; therefore, it was used immediately. A so-lution of the appropriate aldehyde (0.21 g, 0.9813 mmol) and thecompound obtained from the above reduction (5a) (0.15 g,0.8514 mmol) in methanol (80 mL) was stirred at 60 �C for 2 days.Then, the reaction mixture was cooled to 0 �C, and NaBH4 (0.09 g,1.5 eq.) was added. Subsequently, the reaction mixture was stirredat room temperature for 12 h. Then, the reaction mixture was
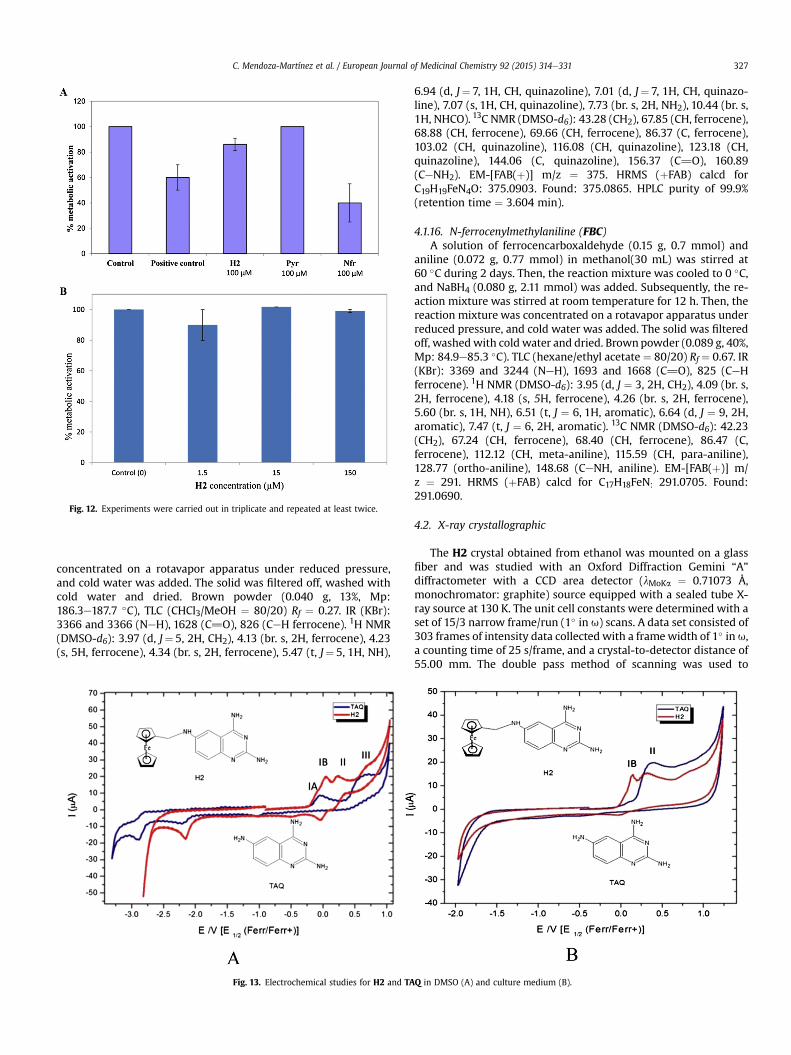
Fig. 12. Experiments were carried out in triplicate and repeated at least twice.
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331 327
concentrated on a rotavapor apparatus under reduced pressure,and cold water was added. The solid was filtered off, washed withcold water and dried. Brown powder (0.040 g, 13%, Mp:186.3e187.7 �C), TLC (CHCl3/MeOH ¼ 80/20) Rf ¼ 0.27. IR (KBr):3366 and 3366 (NeH), 1628 (C]O), 826 (CeH ferrocene). 1H NMR(DMSO-d6): 3.97 (d, J¼ 5, 2H, CH2), 4.13 (br. s, 2H, ferrocene), 4.23(s, 5H, ferrocene), 4.34 (br. s, 2H, ferrocene), 5.47 (t, J¼ 5, 1H, NH),
Fig. 13. Electrochemical studies for H2 and TA
6.94 (d, J¼ 7, 1H, CH, quinazoline), 7.01 (d, J¼ 7, 1H, CH, quinazo-line), 7.07 (s, 1H, CH, quinazoline), 7.73 (br. s, 2H, NH2), 10.44 (br. s,1H, NHCO). 13C NMR (DMSO-d6): 43.28 (CH2), 67.85 (CH, ferrocene),68.88 (CH, ferrocene), 69.66 (CH, ferrocene), 86.37 (C, ferrocene),103.02 (CH, quinazoline), 116.08 (CH, quinazoline), 123.18 (CH,quinazoline), 144.06 (C, quinazoline), 156.37 (C]O), 160.89(CeNH2). EM-[FAB(þ)] m/z ¼ 375. HRMS (þFAB) calcd forC19H19FeN4O: 375.0903. Found: 375.0865. HPLC purity of 99.9%(retention time ¼ 3.604 min).
4.1.16. N-ferrocenylmethylaniline (FBC)A solution of ferrocencarboxaldehyde (0.15 g, 0.7 mmol) and
aniline (0.072 g, 0.77 mmol) in methanol(30 mL) was stirred at60 �C during 2 days. Then, the reaction mixture was cooled to 0 �C,and NaBH4 (0.080 g, 2.11 mmol) was added. Subsequently, the re-action mixture was stirred at room temperature for 12 h. Then, thereaction mixture was concentrated on a rotavapor apparatus underreduced pressure, and cold water was added. The solid was filteredoff, washedwith cold water and dried. Brownpowder (0.089 g, 40%,Mp: 84.9e85.3 �C). TLC (hexane/ethyl acetate ¼ 80/20) Rf ¼ 0.67. IR(KBr): 3369 and 3244 (NeH), 1693 and 1668 (C]O), 825 (CeHferrocene). 1H NMR (DMSO-d6): 3.95 (d, J ¼ 3, 2H, CH2), 4.09 (br. s,2H, ferrocene), 4.18 (s, 5H, ferrocene), 4.26 (br. s, 2H, ferrocene),5.60 (br. s, 1H, NH), 6.51 (t, J ¼ 6, 1H, aromatic), 6.64 (d, J ¼ 9, 2H,aromatic), 7.47 (t, J ¼ 6, 2H, aromatic). 13C NMR (DMSO-d6): 42.23(CH2), 67.24 (CH, ferrocene), 68.40 (CH, ferrocene), 86.47 (C,ferrocene), 112.12 (CH, meta-aniline), 115.59 (CH, para-aniline),128.77 (ortho-aniline), 148.68 (CeNH, aniline). EM-[FAB(þ)] m/z ¼ 291. HRMS (þFAB) calcd for C17H18FeN: 291.0705. Found:291.0690.
4.2. X-ray crystallographic
The H2 crystal obtained from ethanol was mounted on a glassfiber and was studied with an Oxford Diffraction Gemini “A”diffractometer with a CCD area detector (lMoKa ¼ 0.71073 Å,monochromator: graphite) source equipped with a sealed tube X-ray source at 130 K. The unit cell constants were determined with aset of 15/3 narrow frame/run (1� in u) scans. A data set consisted of303 frames of intensity data collected with a framewidth of 1� in u,a counting time of 25 s/frame, and a crystal-to-detector distance of55.00 mm. The double pass method of scanning was used to
Q in DMSO (A) and culture medium (B).

Fig. 14. Electrostatic potential mapped on the electronic density from H2, a level B3LYP 6-31G**.
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331328
exclude any noise. The collected frames were integrated using anorientation matrix determined from the narrow frame scans. TheCrysAlisPro and CrysAlis RED software packages were used for datacollection and data integration [25]. Analysis of the integrated datadid not show any decay. The final cell constants were determinedby a global refinement of 6196 reflections (q < 26.06�). Thecollected data were corrected for absorbance using an analyticalnumerical absorption correction, which employs a multifacetedcrystal model based on expressions of the Laue symmetry usingequivalent reflections [26]. Structure solution and refinement wereperformed with the following programs: the SHELXL97 programfor molecular graphics, the ORTEP-3 program for Windows and theWinGX software for the preparation of materials for publication[27e29].
The full-matrix least-squares refinement was conducted byminimizing (Fo2 � Fc2)2. All of the non-hydrogen atoms wererefined anisotropically. The H atoms of the methanol solvent (HeO)and the amine group (HeN) were located in a difference map andrefined isotropically with a Uiso(H) value of 1.5 and 1.2 Ueq for (O)and (N), respectively. The H atoms attached to C atoms were placedin geometrically idealized positions and refined as riding on theirparent atoms, with CeH ¼ 0.95e0.99 Å with Uiso (H)¼ 1.2Ueq(C) formethylene and aromatic groups, and Uiso (H) ¼ 1.5 Ueq(C) for themethyl group. The crystal data and experimental details of thestructure determinations are listed in Table 5.
4.3. Modeling studies
To construct DHFR Leishmania major, the Trypanosoma cruzienzyme (ID-PDB: 2H2Q Key) was used as the template. First, theamino acids were corrected with the Modeler program [30]. Clas-sical MD simulations were performed using the NAMD2.6 programusing the CHARMM27 force field, and water molecules were addedusing the VMD program [31,32]. The structure was neutralized with4 sodium ions after being immersed in a TIP3P water box con-taining 9249 water molecules. The equilibration protocol beganwith 1500 minimization steps followed by 30 ps of MD at 310 Kwith fixed protein atoms. Then, the entire system was minimizedfor 1500 steps (at 0 K) and then heated gradually from 10 to 310 Kby temperature reassignment during the first 60 ps of 100 psequilibration dynamics without restraints. The final step was a30 ps NTP dynamics using the Nose-Hoover Langevin piston pres-sure control 28 at 310 K and 1.01325 bar for density (volume)fitting. From this point, the simulation was continued in the NTVensemble during 5 ns. The periodic boundary conditions and theparticle-mesh Ewald method were applied for a complete electro-statics calculation. The dielectric water constant was used, and thetemperature was maintained at 310 K using Langevin dynamics.
The structure was allowed to converge, which was reached at 10 ns.A snapshot was obtained at this time for the computationaldocking.
The docking was also performed for PTR from the structure forthis enzyme obtained after crystallization with triaminoquinazo-line (ID-PDB: 1WOC), which also validated the study. The programAutoDock4 was used [33]. For the docking of both proteins, thewater molecules were removed, and the active site was defined asall of the residues within a grid of 60 A� � 60 A� � 60 A� centered inthe active site, with an initial population of 100 randomly placedindividuals and amaximumnumber of 1.0�107energy evaluations.The compounds for docking were drawn in Gauss view 3.0. Beforedocking, the compounds were subjected to energy minimizationusing the hybrid functional B3LYP with a 6, 31 G(d,p) basis set. Thecrystallographic structure was taken as the starting point for con-struction of the compounds. These constructs were minimizedusing the Gaussian 03 program. The most stable tautomers werechosen. We assumed that ferrocene has aromatic character. Thevalues of Kd and DG were taken from the conformation with theminimum energy, and a large Kd value has been reported else-where. This study was validated by comparing the conformation oftriaminoquinazoline from the crystalline structure and the dockingresult, which achieved a satisfactory result. The graphics wereprepared with OriginLab, and the figures were prepared with ACD/ChemSketch for the structures (ACD/Structure Elucidator, version12.01, Advanced Chemistry Development, Inc., Toronto, ON, Canada,www.acdlabs.com, 2013) and PyMOL for the proteins and ligands(The PyMOL Molecular Graphics System, Version 1.2r3pre,Schr€odinger, LLC).
4.4. Biological studies
4.4.1. Antiprotozal activityThe antileishmanial activity was tested in an in vitro culture of
promastigotes the L. mexicana strain MHOM/BZ/61/M379 growingin Dulbecco's modified Eagle's medium (DMEM) containing L-glutamine and glucose (4500 mg per liter), without sodium bicar-bonate (GIBCO, Grand Island, NY), supplemented with 10% fetal calfserum (GIBCO). The experiments were performed in 24-well platesfor tissue culture by adding 0.5 mL of one of the serial dilutions ofthe compound solubilized in a specified solvent (in a ratio of 1:10 ofcompound:media) to eachwell alongwith 0.5 mL 2�106 parasites/mL. The plates were stored at 26 �C for 72 h. The parasite densitywas counted with a hemacytometer in triplicate.
The effect of the compounds was tested on intracellular mac-rophages as previously described [34]. Briefly, the intracellularparasites were prepared by the addition of 107 parasites, withgrowth at 32 �C to a monocyte monolayer in a 24-well plate of

Table 9Oxidation potential of the ferrocene derivatives.
Compound N-Oxidation Ferrocene N-Oxidation
DMSO DMSO Culture medium DMSO Culture medium
IA Ea IB II III II
H2 �0.086 0.037 0.134 0.21 0.681 0.31HO2 0.020 0.105 0.46 0.21HO4 0.021 0.126 0.267 0.86 0.39H2A 0.047 0.127 0.619 0.33TAQ �0.026 0.668 0.3833 0.665 0.652FBC 0.011 0.136 0.614 0.68
a E1/2Ferr/Ferrþ
C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331 329
peritoneal resident macrophages harvested from BALB/c male adultmice. The culture plates were incubated at 32 �C under an atmo-sphere of 5% CO2 for 48 h to allow internalization of the parasites bythe macrophages. The cultures were washed in triplicate withprewarmed sterile phosphate-buffered saline (PBS) to removeextracellular parasites, and 1 mL of media containing the variousconcentrations of the compoundwas added to eachwell. The plateswere returned to 32 �C under an atmosphere of 5% CO2 for anadditional 24, 48 or 72 h incubation. Then, the compound wasremoved, and fresh medium was added. Subsequently, the plateswere cooled to 26 �C to promote the transformation of the livingparasites to the motile form and to cause their release from themacrophages. The parasites were counted with a hematocytometer24 h after compound removal.
The results for promastigotes and intracellular amastigotes wereexpressed as percentage of growth inhibition relative to the control.It is also possible to calculate the percent survival subtracting from100 the percentage of growth inhibition.
4.4.2. DHFR inhibitionThis experimental study was carried out on the in vitro growth
assay for promastigotes. The concentrations evaluated of H2 andtrimetoprim on the leishmania parasites were of 2 mM and 100 mM,respectively. To test DHFR inhibition by folinic acid, ferulic acid andfolic acid, the compounds were incubated with 20 or 100 mM with106 leishmania at the logarithmic phase of growth, washed andresuspended with PBS and incubated for 1 h at room temperature[35]. Then, the parasites were centrifuged, the media was elimi-nated, and the parasites were resuspended in the culturemedia anddistributed in a 24-well plate. H2, or trimetoprim, were then addedat the desired concentration, and the plates were incubated for48 h. The percentage of living parasites was calculated using theformula: % AP ¼ 100 � (Tc� Tp)/Tc, where % AP is the percentage ofgrowth inhibition for each period, and each compound concen-tration, Tc, is the number of parasites/mL in the control wells, andTp is the average number of moving parasites/mL.
4.4.3. Measurement of oxygen consumption of the Leishmaniaparasites
The effects of the H2 treatment on leishmania oxygen con-sumption were determined using a BDtm Oxygen Biosensor Systemthat incorporates an oxygen-sensitive fluorophore into the wells ofan automation-friendly BD Falcon microplate (BD Biosciences,Bedford, MA, USA). The leishmania promastigotes were depositedon the plate with the indicated concentration of H2 or the controlfor 1 or 2 h, and oxygen consumption was measured in a Synergy 2Microplate Reader (Bio Tek Instruments, Inc, Winooski, VT, USA).Sodium cyanide was used a positive control for mitochondrialdamage, and dithionite alone was considered a positive control forthe abolition of oxygen content in the medium. The experiments
were performed in triplicate. The promastigotes of L. mexicanaweretreated with different concentrations ofH2 for 1 and 2 h. Dithionitewas used as a positive control for the abolition of oxygen concen-tration in the medium; for the mitochondrial damage positivecontrol, a group of parasites were treated with sodium cyanide.
4.4.4. Mitochondrial functionality of Leishmania promastigotes inthe presence of H2
Themitochondrial function of the L. mexicana strain M379 in thepresence of H2 was tested using a MitoTracker probe (Invitrogen,Eugene, OR, USA) according to the manufacturer's instructions.Briefly, the parasites were washed three times with PBS - 0.1%glucose to prevent cell lysis due to lack of nutrients. This stepmaintains the parasites under optimal conditions before treatmentwith H2. The parasites were counted with a hematocytometerchamber and adjusted to 5 � 106 parasites/mL. Serial concentra-tions of H2were added to each well, and after 2 h of treatment, theparasites were stained with 50 mg/mL propidium iodide and200 nM MitoTracker® Green FM. The preparations were observedusing an epifluorescencemicroscope. In the control withoutH2, theparasites showed high fluorescence, which indicates the functionalrespiratory process. The parasites without H2, H2 without Leish-mania, sodium cyanide with Leishmania and H2 alone wereobserved for fluorescence, and no fluorescence was observed (datanot shown).
4.4.5. Cytotoxicity studies4.4.5.1. Method 1. The agar diffusion method (USP <87> cytotox-icity test) was used to this evaluation [36]. The cell line used fortesting was L929, fibroblast cell from subcutaneous, adipose andareolar mouse tissues. The L929 cells were derived from ATCCcatalog number CCL-1. Cells were grown in MEM medium supple-mented with 10% FBS and incubated at 37 �C under 5% CO2, asshown in the data sheet ATCC, and until a monolayer, with greater80%, confluence, was obtained. The agar layer was tin enough topermit diffusion of test compound solution. Then compounds wereplaced on the agar surface on the cells. Cells cultures were incubatefor 24 h at 37 �C. Each culture was examined under microscopic.Positive control was USP High density polyethylene RS. After 48 hincubation proceeded to remove the test compound and the agarlayer with the purpose of determining the metabolic ability of cells.This capacity was quantified by the metabolic dye Alamar Blue ®.The cells were incubated for 6 h with this dye at 37 �C and 5% CO2and were then quantified in a plate reader by measuring fluores-cence emission of 535 nm and excitation of 595 nm. All evaluationswere in triplicate.
4.4.5.2. Method 2. H2 toxicity on peritoneal macrophages fromBALB/c mice. Peritoneal macrophages were obtained from healthyBALB/c mice, washed twice with PBS, suspended in DMEM at 105/mL, dispensed in 96 wells plate at volume of 100 mL. H2, solvent, ormeglumine antimoniate were added to the wells, as was previouslydescribed for efficacy experiment. Macrophages were incubated by24 h with the test compounds and then washed. Fresh media wasused for additional 48 h. Alamar blue was added to measure cellviability. The change in fluorescence were measured using fluo-rescence spectrophotometry. In this case, the reduction of the dyecomes from macrophages. Experiments were carried out in tripli-cate and repeated at least twice.
4.5. Solubility studies
The solubility was calculated according to Lipinsky et al. [37].Compound was dissolved in DMSO at a concentration of 10 mg/mL.One microliter of this solution was added at a time to a non-

C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331330
chloride solution containing pH 7 phosphate buffer, at room tem-perature. The additions of solutions are spaced a minute apart. Atotal of 14 additions were made. This correspond to solubility in-crements of 5 pg/mL to a top value of 65 mg/mL when the buffervolume is 2.5 mL (as in a UV cuvette). If it is clear that precipitationwas occurring early in the addition sequence, the addition wasstopped, so that we had two consecutive readings after the pre-cipitate was detected. Precipitation can be detected by an absor-bance increase due to light scattering by precipitated particulatematerial in a UV spectrophotometer. In its simplest implementa-tion, the precipitation point was calculated from a bilinear curve fitto the absorbance versus concentration plot and is reported in mg/mL [37]. Experiments were carried out in triplicate and repeated atleast twice.
4.6. Electrochemical studies
Electrochemical measurements were performed on apotentiostat-galvanostat Autolab model PAR263A device with athree-electrode system in a 0.1 M solution of tetrabutylammoniumhexafluorophosphate (Bu4NPF6) in DMSO as the supporting elec-trolyte. A carbon glass disc (0.071 cm2) was used as the workingelectrode, a Pt wire was used as the auxiliary electrode, and 0.1 M(Bu4N)Br/AgBr(s)/Ag was used as the reference electrode. Theworking electrode (C) was polished with alumina to ensure theabsence of residues on the surface. All of the voltammograms wereinitiated from the open circuit potential (Ei ¼ 0), and the scan wasinitiated in both the positive and negative potential directions. Toreport the potentials used according to the IUPAC convention, thevoltammogramswere obtained for approximately 10�3 M solutionsof ferrocene (Fc) in a supporting electrolyte. For the working con-ditions, the electroactive domain was between �2.70 and 1.60 V/Fcþ-Fc. The halfwave potentials were estimated from E1/2¼ (Eap þ Ecp)/2, where Eap and Ecp are the anodic and cathodicpeak potentials, respectively.
Acknowledgments
This study was supported by a fellowship and infrastructurepurchased with funds from PAPIIT-UNAM IN213914 and researchfunds from CONACYT-6248. We are grateful to Rosa Isela del Villar,Georgina Duarte, Margarita Guzm�an, Nayeli L�opez and MariselaGuti�errez from Facultad de Química, USAI, UNAM for the determi-nation of all of the spectra. For the theoretical studies, we aregrateful to DGSCA-UNAM for allowing the use of the KanBalamsupercomputer.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2014.12.051.
References
[1] K.T. Andrews, G. Fisher, T.S. Skinner-Adams, Drug repurposing and humanparasitic protozoan diseases, Int. J. Parasitol. Drugs Drug Resist 4 (2014)95e111.
[2] World Health Organization. Control of the Leishmaniases. WHO TechnicalReport Series. http://www.who.int/leishmaniasis/resources/en.
[3] T.S. Tiumana, A.O. Santos, T. Ueda-Nakamuraa, B.P. Dias Filhoa,C.V. Nakamuraa, Recent advances in leishmaniasis treatment, Int. J. Infect. Dis.15 (2011) e525ee532.
[4] R.M. Reguera, E. Calvo-�Alvarez, R. �Alvarez-Velilla, R. Bala~na-Fouce, Target-based vs. phenotypic screenings in Leishmania drug discovery: a marriage ofconvenience or a dialogue of the deaf? Int. J. Parasitol. Drugs Drug Resist.(2014). http://dx.doi.org/10.1016/j.ijpddr.2014.05.001.
[5] N. Singh, M. Kumar, R.K. Singh, Leishmaniasis: current status of availabledrugs and new potential drug targets, Asian Pac. J. Trop. Med. 5 (2012)
485e497.[6] J. Mishra, S. Singh, Miltefosine resistance in Leishmania donovani involves
suppression of oxidative stress-induced programmed cell death, Exp. Para-sitol. 135 (2013) 397e406.
[7] I.V. Ogungbe, W.R. Erwin, W.N. Setzer, Antileishmanial phytochemical phe-nolics: molecular docking to potential protein targets, J. Mol. Graph. Model. 48(2014) 105e117.
[8] P. Gahtori, S.K. Ghosh, P. Parida, A. Prakash, K. Gogoi, H.R. Bhat, U.P. Singh,Antimalarial evaluation and docking studies of hybrid phenylthiazolyl-1,3,5-triazine derivatives: a novel and potential antifolate lead for pf-DHFR-TS in-hibition, Exp. Parasitol. 130 (2012) 292e299.
[9] I.M. Kompis, K. Islam, L.R. Then, DNA and RNA synthesis: antifolates, Chem.Rev. 105 (2005) 593e620.
[10] B. Nare, L.W. Hardy, S.M. Beverley, The roles of pteridine reductase 1 anddihydrofolate reductase-thymidylate synthase in pteridine metabolism in theProtozoan parasite Leishmania major, J. Biol. Chem. 272 (1997) 13883e13891.
[11] H.B. Ong, N. Sienkiewicz, S. Wyllie, A.H. Fairlamb, Dissecting the metabolicroles of pteridine reductase 1 in Trypanosoma brucei and Leishmania major,J. Biol. Chem. 286 (2011) 10429e10438.
[12] K. McLuskey, F. Gibellini, P. Carvalho, M.A. Avery, W.N. Hunter, Inhibition ofLeishmania major pteridine reductase by 2,4,6-triaminoquinazoline: structureof the NADPH ternary complex, Acta Cryst. D60 (2004) 1780e1785.
[13] J.D. Berman, M. King, N. Edwards, Antileishmanial activities of 2,4-diaminoquinazoline putative dihydrofolate reductase inhibitors, Antimicrob.Agents Chemother. 33 (1989) 1860e1863.
[14] D. Dive, C. Biot, Ferrocene conjugates of chloroquine and other antimalarials:the development of ferroquine, a new antimalarial, ChemMedChem 3 (2008)383e391.
[15] A. Pejovi�c, M.S. Deni�c, D. Stevanovi�c, I. Damljanovi�c, M. Vukicevi�c, K. Kostova,M. Tavlinova-Kirilov, P. Randjelovi�c, N.M. Stojanovi�c, G.A. Bogdanovi�c,P. Blagojevi�c, M. D'hooghe, N.S. Radulovi�c, R.D. Vuki�cevi�c, Discovery of anxi-olytic 2-ferrocenyl-1,3-thiazolidin-4-ones exerting GABAA receptor interac-tion via the benzodiazepine-binding site, Eur. J. Med. Chem. 83 (2014) 57e73.
[16] I.S. Damljanovi�c, M.D. Vuki�cevi�c, N.S. Radulovi�c, R.M. Pali�c, E. Ellmerer,Z.R. Ratkovi�c, M.D. Joksovi�c, R.D. Vuki�cevi�c, Synthesis and antimicrobial ac-tivity of some new pyrazole derivatives containing a ferrocene unit, Bioorg.Med. Chem. Lett. 19 (2009) 1093e1096.
[17] S. Gurvinder, K. Maninderjit, C. Mohan, Benzimidazoles: the latest informationon biological activities, Int. Res. J. Pharm. 4 (2013) 82e87.
[18] G. Navarrete-V�azquez, R. Cedillo, A. Hern�andez-Campos, L. Y�epez,F. Hern�andez-Luis, J. Valdez, R. Morales, R. Cort�es, M. Hern�andez, R. Castillo,Synthesis and antiparasitic activity of 2-(trifluoromethyl)-benzimidazole de-rivatives, Bioorg. Med. Chem. Lett. 11 (2001) 187e190.
[19] F. Palomares-Alonso, H. Jung-Cook, J. P�erez-Villanueva, J.C. Piliado,S. Rodríguez-Morales, G. Palencia-Hern�andez, N. L�opez-Balbiaux,A. Hern�andez-Campos, R. Castillo, F. Hern�andez-Luis, Synthesis and in vitrocysticidal activity of new benzimidazole derivatives, Eur. J. Med. Chem. 44(2009) 1794e1800.
[20] L. Kedzierski, J.M. Curtis, M. Kaminska, J. Jodynis-Liebert, M. Murias, In vitroantileishmanial activity of resveratrol and its hydroxylated analogues againstLeishmania major promastigotes and amastigotes, Parasitol. Res. 102 (2007)91e97.
[21] J. Davoll, A.M. Johnson, H.J. Davies, O.D. Bird, J. Clarke, E.F. Elslager, 2,4-Dia-mino-6-{[aralkyl and (heterocyclic)methyl]amino}quinazolines, a novel classof antimetabolites of interest in drug-resistant malaria and Chagas' disease,J. Med. Chem. 15 (1972) 812e826.
[22] C.E. Ballou, H.O.L. Fischer, The synthesis of dihydroxyacetone phosphate,J. Am. Chem. Soc. 78 (1956) 1659e1661.
[23] J. Sun, Y. Dong, L. Cao, X. Wang, S. Wang, Y. Hu, Highly efficient chemo-selective deprotection of O,O-acetals and O,O-ketals catalyzed by moleculariodine in acetone, J. Org. Chem. 69 (2004) 8932e8934.
[24] S.E. Sen, S.L. Roach, J.K. Boggs, G.J. Ewing, J. Magrath, Ferric chloride hexa-hydrate: a mild hydrolytic agent for the deprotection of acetals, J. Org. Chem.62 (1997) 6684e6686.
[25] Oxford Diffraction, CrysAlis CCD, and CrysAlis RED, Oxford Diffraction Ltd,Abingdon, UK, 2009.
[26] R.C. Clark, J.S. Reid, The analytical calculation of absorption in multifacetedcrystals, Acta Crystallogr. A51 (1995) 887e897.
[27] G.M. Sheldrick, A short history of SHELX, Acta Crystallogr. A64 (2008)112e122.
[28] L.J. Farrugia, ORTEP-3 for Windows e a version of ORTEP-III with a GraphicalUser Interface (GUI), Appl. Crystallogr. 30 (1997) 565.
[29] L.J. Farrugia, WinGX suite for small-molecule single-crystal crystallography,Appl. Crystallogr. 32 (1999) 837e838.
[30] A. Sali, T.L. Blundell, Comparative protein modelling by satisfaction of spatialrestraints, J. Mol. Biol. 234 (1993) 779e815.
[31] J.C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot,R.D. Skeel, L. Kale, K. Schulten, Scalable molecular dynamics with NAMD,J. Comput. Chem. 26 (2005) 1781e1802.
[32] W. Humphrey, A. Dalke, K. Schulten, VMD e visual molecular dynamics,J. Molec. Graph. 14 (1996) 33e38.
[33] G.M. Morris, R. Huey, W. Lindstrom, M.F. Sanner, R.K. Belew, D.S. Goodsell,A.J. Olson, Autodock4 and AutoDockTools4: automated docking with selectivereceptor flexibility, J. Comput. Chem. 16 (2009) 2785e2791.
[34] H. Martinez-Rojano, J. Mancilla-Ramirez, L. Qui~nonez, N. Galindo-Sevilla,

C. Mendoza-Martínez et al. / European Journal of Medicinal Chemistry 92 (2015) 314e331 331
Activity of hydroxyurea against Leishmania mexicana, Antimicrob. AgentsChemother. 52 (2008) 3642e3647.
[35] L. Qui~nonez-Díaz, J. Mancilla, M. Avila-García, J. Ortíz-Avalos, A. Berron,S. Gonzalez, Y. Paredes, N. Galindo-Sevilla, Effect of ambient temperature onthe clinical manifestation of experimental diffuse cutaneous leishmaniasis in arodent model, Vector Borne Zoonotic Dis. 12 (2012) 851e860.
[36] US Pharmacopeia (USP29). http://www.pharmacopeia.cn/v29240/usp29nf24s0_c87.html (accessed 01.30.13).
[37] C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney, Experimental andcomputational approaches to estimate solubility and permeability in drugdiscovery and development settings, Adv. Drug Deliv. Rev. 23 (1997) 3e25.



















