
Advancing the collection and storage of blood micro-volumes ...
164
Advancing the collection and storage of blood micro-volumes for downstream applications A thesis submitted in fulfilment of the requirements for the degree of Master of Science Lada Staskova BBus(Business Economics)BSc(Plant Science), University of Tasmania School of Science College of Science, Engineering and Health RMIT University September, 2020
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Advancing the collection and storage of blood micro-volumes ...

Advancing the collection and storage of blood micro-volumes for
downstream applications
A thesis submitted in fulfilment of the requirements for the degree of Master of Science
Lada Staskova
BBus(Business Economics)BSc(Plant Science), University of Tasmania
School of Science College of Science, Engineering and Health
RMIT University
September, 2020

ii
Declaration
I certify that except where due acknowledgement has been made, the work is that of the
author alone; the work has not been submitted previously, in whole or in part, to qualify for
any other academic award; the content of the thesis is the result of work which has been
carried out since the official commencement date of the approved research program; any
editorial work, paid or unpaid, carried out by a third party is acknowledged; and, ethics
procedures and guidelines have been followed.
Finally, I acknowledge the support I have received for my research through the provision of
an Australian Government Research Training Program Scholarship.
Lada Staskova
Date:
4th September 2020

iii
Acknowledgment
This work would not have been possible without the help and support of many:
Firstly to all my supervisors:
To A/Prof. Jeffrey Craig, you helped me navigate my way through the research project and
gave me the opportunity to join the amazing team at the Murdoch Children’s Research
Institute. During my project your door was always open even when moving your office off
campus. You have always encouraged me when I got stuck and no question was ever too big
or too small.
To A/Prof. Robert Shellie, thank you for introducing me to the world of medical science at
Trajan Scientific and Medical. Without you I would not be where I am today. During the
project you have given me great advice and suggestions to ensure that the research is
heading in the right direction.
To Dr. Andrew Gooley, you were always very keen to see my results and encouraged me to
ask further questions. You have given me a great insight into research from an industry
point of view and I always enjoyed discussing the research with you. Thank you for giving
me the opportunity to present my work internationally.
To Professor Oliver Jones, given that my research was mainly off campus you have given me
great supervision and helped me to manoeuvre through all the university processes . You
door was always open and you have always given me excellent feedback on my research
and work ethic which has boosted my confidence.
To the epigenetics team at the Murdoch Children’s Research Institute including Dr. Jane
Loke, Dr. Pamela Leon, Ms Jennifer Snowball and Dr. Namitha Mohandas, thank you for all
your support and lunch chats, which comforted me through some of the challenging times.

iv
To the team at the Victorian Clinical Genetics Services, especially the newborn screening
laboratory, thank you for showing me the laboratory processes and assisting with sample
analysis.
To the pathology collection team at the Royal Children’s Hospital, thank you for a very
friendly atmosphere in testing the hemaPEN device. I wish you all the best and look forward
to working with you all in the future.
To my fiancé and my family, I have definitely had my ups and downs during this experience
but you were there for me every step of the way and I couldn’t have done it without you.
Last but not least, I would like to acknowledge and thank Trajan Scientific and Medical for
allowing me to conduct the research project, supporting me throughout the journey and
funding the research project. A huge thank you to Dr. Jason Hon for all your guidance and
expanding my knowledge in relevant industry topics.

v
Tableofcontent
DECLARATION .................................................................................................................................................. II
ACKNOWLEDGMENT .................................................................................................................................... III
TABLE OF CONTENT ........................................................................................................................................ V
TABLE OF FIGURES ..................................................................................................................................... VIII
LIST OF TABLES ................................................................................................................................................ X
ABBREVIATIONS ............................................................................................................................................. XI
ABSTRACT ....................................................................................................................................................... - 1 -
CHAPTER 1: LITERATURE REVIEW ....................................................................................................... - 4 -
1.1 GENERAL BACKGROUND .................................................................................................................... - 2 - 1.1.1 The blood composition ...................................................................................................................... - 2 - 1.1.2 The clinical importance of blood ...................................................................................................... - 6 - 1.1.3 Clinical and analytical method validation ......................................................................................... - 7 -
1.1.3.1 Clinical validation process ............................................................................................................................ - 7 - 1.1.3.2 Analytical uncertainty ................................................................................................................................... - 8 - 1.1.3.3 Analytical validation processes for established methods ............................................................................. - 9 - 1.1.3.4 Analytical validation processes for new methods ...................................................................................... - 10 - 1.1.3.5 The effects of preanalytical variability ....................................................................................................... - 10 -
1.1.4 Venous blood collection .................................................................................................................. - 11 - 1.1.5 Skin incision for collection of microvolumes ................................................................................. - 11 - 1.1.6 Benefits of microsampling .............................................................................................................. - 13 - 1.1.7 Collection and Storage of microvolumes ........................................................................................ - 14 -
1.2 DRIED BLOOD SPOT (DBS) ....................................................................................................................... - 14 - 1.2.1 Heel and finger prick collection ...................................................................................................... - 15 - 1.2.2 Downstream use of DBS ................................................................................................................. - 15 -
1.2.2.1 Drug development ...................................................................................................................................... - 16 - 1.2.2.2 Clinical diagnostics vs. clinical screening of DBS ..................................................................................... - 17 - 1.2.2.3 The “Omics” ............................................................................................................................................... - 18 - 1.2.2.7 Sport ........................................................................................................................................................... - 21 - 1.2.2.8 Chemical Detection ................................................................................................................................... - 22 -
1.2.3 Current challenges with DBS .......................................................................................................... - 22 - 1.2.3.1 Pre-analytical challenges in DBS ............................................................................................................... - 22 - 1.2.3.2 Analytical challenges in DBS ..................................................................................................................... - 23 -
1.3 DRIED PLASMA SPOT (DPS) ...................................................................................................................... - 25 - 1.4 CAPILLARY MICROSAMPLING (CMS) ....................................................................................................... - 26 - 1.5 EMERGING METHODS FOR MICROSAMPLING ............................................................................................. - 27 -
1.5.1 HemaSpotTM .................................................................................................................................... - 27 - 1.5.2 HemaXisTM ...................................................................................................................................... - 28 - 1.5.3 Capitainer ........................................................................................................................................ - 29 - 1.5.4 Mitra® device powered by volumetric absorptive microsampling (VAMS®) technology .............. - 30 - 1.5.5 HemaPEN® ...................................................................................................................................... - 31 -
1.6 STUDY RATIONAL ..................................................................................................................................... - 33 - 1.7 STUDY AIMS, HYPOTHESIS AND RESEARCH QUESTIONS ............................................................................ - 34 -
CHAPTER 2: GENERAL METHODS ........................................................................................................ - 36 - 2.1 ASSISTED HEMAPEN® COLLECTION ......................................................................................................... - 37 -
2.1.1 Instruction manual, video and practice collection ........................................................................... - 37 - 2.1.2 hemaPEN® collection from a donor ................................................................................................ - 38 -
2.2 ANALYTICAL TESTING OF STANDARD DBS AND HEMAPEN® AT THE VICTORIAN CLINICAL GENETICS SERVICES (VCGS) NEWBORN SCREENING LABORATORY ............................................................................. - 39 -
2.2.1 Preparation of eleven concentration levels across twenty seven analytes ...................................... - 39 - 2.2.2 Preparation of five haematocrit (HCT) levels ................................................................................. - 42 - 2.2.3 Analytical procedure at the VCGS Newborn Screening Laboratory .............................................. - 43 -

vi
2.2.4. Statistical analysis ......................................................................................................................... - 43 - 2.3 COMPARISON OF EXTRACTION KITS AND SUBSTRATES TO MAXIMISE DNA YIELD ................................... - 44 -
2.3.1 Dried blood spot preparation on different substrates (synthetic and non-synthetic) ...................... - 44 - 2.3.2 DNA extraction kits and protocol ................................................................................................... - 45 - 2.3.3 Quality control measures ................................................................................................................ - 46 - 2.3.4 Statistical analysis ........................................................................................................................... - 46 -
CHAPTER 3: USABILITY COMPARISON BETWEEN A NEW PATIENT-CENTRIC DEVICE AND THE STANDARD DBS METHOD IN NEWBORN SCREENING WORKFLOW ................................ - 47 -
3.1 INTRODUCTION ......................................................................................................................................... - 48 - 3.2. METHODS ................................................................................................................................................ - 49 - 3.3. RESULTS .................................................................................................................................................. - 50 -
3.3.1 Assisted hemaPEN® collection ....................................................................................................... - 50 - 3.3.1.1 Main feedback from phlebotomists on hemaPEN® use ............................................................................. - 51 - 3.3.1.2 Main feedback from donors on hemaPEN® use ......................................................................................... - 52 -
3.2.2 Method comparison (standard DBS and hemaPEN®) at eleven concentration levels using the newborn screening workflow ................................................................................................................... - 53 -
3.2.2.1 Overall trend between the two methods across eleven concentration levels using principle component analysis ................................................................................................................................................................... - 53 - 3.2.2.2 Method comparison for twenty-seven analytes measured .......................................................................... - 54 - 3.2.2.3 Random error in the newborn screening workflow .................................................................................... - 57 - 3.2.2.4 Systematic error (constant and proportional) ............................................................................................. - 63 - 3.2.2.5 Punch location (periphery and centre) in the standard DBS method ........................................................ - 63 - 3.2.2.6 Analyte recovery for each method across 11 different concentration levels .............................................. - 63 -
3.3 DISCUSSION .............................................................................................................................................. - 64 -
CHAPTER 4: EFFECT OF ACCURATE VOLUME CORRECTION ON THE HAEMATOCRIT BIAS IN A NEWBORN SCREENING WORKFLOW ......................................................................................... - 69 -
4.1 INTRODUCTION ......................................................................................................................................... - 70 - 4.2 METHOD ................................................................................................................................................... - 72 - 4.2 RESULTS ................................................................................................................................................... - 73 -
4.2.1 Dispersion effects associated with haematocrit .............................................................................. - 73 - 4.2.2 Comparison of standard DBS and hemaPEN® across five haematocrit levels for 20 analytes detected in the newborn screening workflow ......................................................................................................... - 73 - 4.2.3 The evaluation of standard DBS method across five haematocrit levels ........................................ - 77 - 4.2.4 The evaluation of HemaPEN® across five haematocrit levels ....................................................... - 78 - 4.2.5 Trends observed across HCTs for both methods ............................................................................ - 79 -
4.3 DISCUSSION .............................................................................................................................................. - 84 - 4.3.1 Analyte distribution in blood .......................................................................................................... - 84 - 4.3.2 HCT dependent extraction bias ....................................................................................................... - 85 -
CHAPTER 5: COMPARISON OF EXTRACTION KITS AND SUBSTRATES TO MAXIMISE DNA YIELD .............................................................................................................................................................. - 88 -
5.1 INTRODUCTION ......................................................................................................................................... - 89 - 5.1 METHODS ................................................................................................................................................. - 91 - 5.3 RESULTS ................................................................................................................................................... - 92 -
5.3.1 Blood distribution on various substrates ......................................................................................... - 92 - 5.3.2 Comparison of three DNA extraction kits: Quick-DNATM, QIAamp® MagMAX CORETM ......... - 93 - 5.2.3 Comparison of five non-synthetic substrates (GenCollectTM 2.0, FTATM Elute, FTATM Gene, GenSaverTM 2.0 and Whatman 903TM ) and one synthetic substrates (mPPM) ...................................... - 97 - 5.2.4 The combination of extraction kit and substrate for high DNA yield ............................................. - 98 -
5.3 DISCUSSION .............................................................................................................................................. - 99 -
CHAPTER 6: CONCLUSION AND FUTURE WORK ........................................................................... - 103 - 6.1 CONCLUSIONS ........................................................................................................................................ - 104 - 6.2 RECOMMENDATION FOR FUTURE WORK ................................................................................................. - 105 - 6.3 BRIDGING STUDIES ................................................................................................................................. - 107 - 6.4 THE ADOPTION OF EMERGING DEVICES ................................................................................................... - 107 -
CHAPTER 7: REFERENCES ..................................................................................................................... - 109 -
CHAPTER 8 APPENDIX ............................................................................................................................ - 128 -

vii
Appendix 8.1 Supplementary Table 1. Punch location in Standard DBS .............................................. - 129 - Appendix 8.2 Extraction kits protocol ................................................................................................... - 130 - Appendix 8.3 Ethic approval and Governance authorisation ................................................................. - 134 - Appendix 8.4 hemaPEN® instruction manual ........................................................................................ - 136 - Appendix 8.5 hemaPEN® video ............................................................................................................. - 146 - Appendix 8.6 Summary table: Feedback from donors and collectors ................................................... - 147 -

viii
TableofFigures
FIGURE 1.1 BLOOD COMPOSITION INCLUDING PLASMA AND FORMED ELEMENTS WITH FURTHER CLASSIFICATION OF DIFFERENT WHITE BLOOD CELLS. ............................................................................................................ - 4 -
FIGURE 1.2 ALTERNATIVE METHODS TO SKIN PUNCTURE: A) TAPTM, B) HEMOLINKTM INCLUDING INTEGRATABLE CARTRIDGE. ............................................................................................................................................... - 13 -
FIGURE 1.4 THE HEMASPOTTM BLOOD COLLECTION DEVICE. ............................................................................. - 28 - FIGURE 1.5 HEMAXISTM BLOOD COLLECTION AND STORAGE DEVICE. ................................................................ - 29 - FIGURE 1.6 THE CAPITAINER BLOOD COLLECTION AND STORAGE DEVICE. ........................................................ - 30 - FIGURE 1.7 MITRA® DEVICE POWERED BY VOLUMETRIC ABSORPTIVE MICROSAMPLING (VAMS®) TECHNOLOGY
FOR COLLECTION AND STORAGE OF BLOOD. ............................................................................................... - 31 - FIGURE 1.8 THE HEMAPEN® BLOOD COLLECTION AND STORAGE DEVICE. ........................................................ - 32 - FIGURE 2.1 PRACTICE HEMAPEN® COLLECTION FROM AN FLAT SURFACE OF MICRO SAMPLING TUBE. .............. - 38 - FIGURE 2.2 THE HEMAPEN® COLLECTION FROM DONOR’S FINGER. ................................................................... - 38 - FIGURE 2.3 BLOOD COLLECTION USING HEMAPEN® FROM THE LID OF A GLASS VACUTAINER. .......................... - 41 - FIGURE 2.4 STANDARD DBS PUNCH POSITION USING THE PERKINELMER PLATFORM A) ONE 3.2 MM PUNCH IN THE
CENTRE AND ONE 3.2 MM PUNCH IN THE PERIPHERY ON A 50 µL SPOT FOR AVERAGE MALE HCT B) ONE 3.2 MM PUNCH IN THE CENTRE AND THREE 3.2 MM PUNCHES IN THE PERIPHERY ON A 75 µL OF BLOOD FOR 63% HCT. .......................................................................................................................................................... - 41 -
FIGURE 3.1 MAIN FEEDBACK FROM HEMAPEN® PRACTICE AND VOLUNTEER’S ASSISTED COLLECTION FOR SIX PHLEBOTOMISTS FROM THE ROYAL CHILDREN’S HOSPITAL RANGING IN EXPERIENCE. ............................. - 52 -
FIGURE 3.2 MAIN FEEDBACK FROM FIVE VOLUNTEERS FROM HEMAPEN® COLLECTION EXPERIENCE AND THE USE OF HEMAPEN® IN COMPARISON TO STANDARD VENOUS COLLECTION ....................................................... - 53 -
FIGURE 3.3 PRINCIPLE COMPONENT ANALYSIS (PCA) PLOT FOR 27 ANALYTES BETWEEN TWO METHODS (HEMAPEN® IN TRIANGLES AND STANDARD DBS IN CIRCLES) ACROSS DIFFERENT CONCENTRATION LEVELS. EACH LEVEL AN INCREMENT OF 10 (LEVEL 1-11) I.E. LEVEL 1 (NATIVE) CONTAINED NO SPIKE AMOUNT AND LEVEL 11 (SPIKE) CONTAINED MAXIMUM SPIKE AMOUNT. FOR EACH LEVEL, EIGHT TECHNICAL REPLICATES PER METHOD WAS GRAPHED REPRESENTING 27 ANALYTES. ....................................................................... - 54 -
FIGURE 3.4 PASSING-BABLOK REGRESSION PLOTS BETWEEN TWO METHODS (STANDARD DBS – X AXIS, HEMAPEN® - Y AXIS) FOR MEASURED ANALYTES (GLY, ALA, VAL, ILE, ORN, LYS-GLN, MET, PHE, GLY-PRO, ARG, CIT, TYR, HOMOCITRULLINE, ARGINOSUCCINATE, C0, C2 , C3 , C4 , C5.1 , C5 , C6 , C8 , C10 , C12 , C14 , C16 , C18) ACROSS 11 CONCENTRATION LEVELS. LINEAR REGRESSION LINE REPRESENTED IN BLUE WITH 95% CONFIDENT BOUNDS. LINE OF IDENTITY (SLOPE = 1) INDICATING METHOD ALIGNMENT IN RED DOTTED LINE. ...................................................................................................................................... - 58 -
FIGURE 4.1 BLOOD DISPERSION OF FIVE HCT (25%, 35%, 42%, 55%, 63%) ON WHATMAN 903TM FILTER PAPER A) IN HEMAPEN® CARTRIDGE WITH THE AVERAGE SURFACE AREA COVERED BY BLOOD CALCULATED IN MM2 B) ZOOMED IN INDIVIDUAL PRE-PUNCH HEMAPEN® SAMPLES WITH 2.74µL OF BLOOD DEPOSITED VIA CAPILLARY TRANSFER C) STANDARD DBS FILTER PAPER WITH 75 µL OF BLOOD APPLIED. ....................... - 73 -
FIGURE 4.2 PRINCIPLE COMPONENT ANALYSIS PLOT REPRESENTING THE TWO DIFFERENT METHODS A) STANDARD DBS (ROUND) B) HEMAPEN® (TRIANGLE) ACROSS FIVE DIFFERENT HCTS (ORANGE - 25%, OLIVE - 35%, GREEN - 42%, BLUE - 55%, PURPLE - 63%). THE 95% CONFIDENCE ELLIPSE WAS CALCULATED FOR EACH METHOD. EACH PC SCORE ON THE PCA PLOT REPRESENTS THE VARIATION OF 20 ANALYTES IN EACH SAMPLE MEASURED. FOR EACH METHOD 160 SAMPLES (32 SAMPLES PER HCT) WERE ANALYSED. .......... - 74 -
FIGURE 4.4 BIPLOT REPRESENTING PC SCORES (SAMPLES) FOR EACH HCT (25%, 35%, 42%, 55%, 63%) AND LOADINGS OF VARIABLES (ANALYTES) FOR THE STANDARD DBS METHOD. THE LENGTH OF THE VECTOR REPRESENTS THE INFLUENCE OF THE ANALYTE AND THE DIRECTION REPRESENTS THE ANALYTE DEPENDENCY WITH RESPECT TO HCT (POSITIVE HCT DEPENDENCY – CIRCLE A, NO HCT DEPENDENCY – CIRCLE B, NEGATIVE HCT DEPENDENCY – CIRCLE C). ............................................................................................... - 77 -
FIGURE 4.5 BIPLOT REPRESENT PC SCORED (SAMPLES) FOR EACH HCT (25%, 35%, 42%, 55%, 63%) AND LOADINGS OF VARIABLES (ANALYTES) FOR HEMAPEN®. THE LENGTH OF THE VECTOR REPRESENTS THE INFLUENCE OF THE VECTORS AND THE DIRECTION REPRESENTS THE ANALYTE DEPENDENCY WITH RESPECT TO HCT (POSITIVE HCT DEPENDENCY – CIRCLE A, NO HCT DEPENDENCY – CIRCLE B, NEGATIVE HCT DEPENDENCY – CIRCLE C). ......................................................................................................................... - 78 -
FIGURE 4.6 PLOTS REPRESENTING THE PERCENTAGE CHANGE FROM THE MIDDLE HCT 42% ACROSS FIVE HCTS (25%, 35%, 42%, 55%, 63%) FOR 19 ANALYTES (VAL, ALA, GLY, ILE, MET, PHE, ARG, CIT, TYR, C0, C2, C3,C4, C5, C6, C8, C10, C16, C18). THE PLOT REPRESENTATION WAS PREVIOUSLY PUBLISHED BY (ABU-RABIE ET AL., 2015). METHODS (HEMAPEN® AND STANDARD DBS) WERE PLOTTED ONTO ONE GRAPH FOR

ix
EACH ANALYTE TO OBSERVE HCT DEPENDENT TRENDS. ERROR BAR REPRESENT 95 % CONFIDENCE INTERVAL FROM THE MEAN OF THE 32 SAMPLES MEASURED FOR EACH HCT. ........................................... - 80 -
FIGURE 5.1 DRIED BLOOD SPOTS GENERATED BY APPLYING 40 µL OF BLOOD ON SUBSTRATES (FTATM GENE, WHATMAN 903TM, FTATM ELUTE, GENCOLLECTTM2.0, GENSAVERTM 2.0) AND 12µL OF BLOOD APPLIED ONTO THE POLYMER MONOLITH (MPPM). VENOUS EDTA BLOOD WAS SOURCED FROM A FEMALE VOLUNTEER. ............................................................................................................................................... - 92 -
FIGURE 5.2 TWO-WAY ANOVA AND PAIRWISE MEAN DIFFERENCE ACROSS THREE EXTRACTION KITS (MAGMAX CORETM, QIAAMP®, QUICK-DNATM). BOX PLOTS FOR EACH KIT REPRESENTS THE DISTRIBUTION OF SIX DIFFERENT SUBSTRATE TYPES WITH 8 TECHNICAL REPLICATES (FTATM GENE, WHATMAN 903TM, FTATM ELUTE, GENCOLLECTTM 2.0, GENSAVERTM 2.0 AND MPPM) .................................................................... - 94 -
FIGURE 5.3 DNA FRAGMENTATION ON 1 % AGAROSE GEL FOR THREE EXTRACTION KITS (MAGMAX CORETM, QIAAMP® AND QUICK-DNATM) AND SIX SUBSTRATES (GENCOLLECTTM 2.0, FTATM ELUTE, FTATM GENE, MPPM, GENSAVERTM 2.0 AND WHATMAN 903TM). FOR EACH SUBSTATE AND KIT EIGHT REPLICATES WERE LOADED ON THE GEL. REFERENCE LADDER WITH FIRST 10KB BAND. ......................................................... - 96 -
FIGURE 5.4 BOX PLOT FOR EACH EXTRACTION KIT (MAGMAX CORETM, QIAAMP® AND QUICK-DNATM) AND SIX SUBSTRATES (GENCOLLECTTM 2.0, FTATM ELUTE, FTATM GENE, MPPM, GENSAVERTM 2.0 AND WHATMAN 903TM). THE TWO-WAY ANOVA STATISTICAL TEST PERFORMED FOR EACH EXTRACTION KIT. WITHIN EACH EXTRACTION KIT THE PAIRWISE MEAN DIFFERENCE WAS COMPARED AGAINST WHATMAN 903TM (NS: P> 0.05; *: P<0.05; **: P<0.001; ***: P<0.001; ***: P<0.0001) .............................................. - 98 -

x
ListofTables
TABLE 2.1 THE EXOGENOUS CONCENTRATION (µMOL/L) FOR BOTH AMINO ACIDS AND CARNITINES THAT WAS SPIKED INTO WHOLE BLOOD. ...................................................................................................................... - 40 -
TABLE 3.1 CAPILLARY TRANSFER, IN PERCENTAGE, (SUCCESSFUL BLOOD DEPOSIT FROM CAPILLARY TO THE PRE-PUNCH FILTER PAPER WITHIN THE HEMAPEN®)FOR PRACTICE HEMAPEN® DEVICES AND ONE COLLECTION FROM DONOR’S FINGER FOR ALL SIX COLLECTORS. .................................................................................... - 50 -
TABLE 3.2 THE TIME, IN SECONDS, TAKEN FOR EACH CATEGORY (ALCOHOL WIPE, LANCET FINGER PRICK, GAUZE, SAMPLE COLLECTION, CAPPING OF HEMAPEN®, FLIPPING OF HEMAPEN®, TOTAL TIME FROM WIPING OF FINGER TO FLIPPING THE HEMAPEN® AND THE TIME TAKEN TO PLACE HEMAPEN® IN A SAMPLE BAG). TIME RECORDED WAS ASSESSED FROM THE VIDEO RECORDED DURING THE DONOR’S COLLECTION. .................. - 51 -
TABLE 3.3 THE BLAND-ALTMAN MEAN DIFFERENCE (IN %), PEARSON CORRELATION, THE SLOPE AND INTERCEPT OF PASSING-BABLOK REGRESSION BETWEEN HEMAPEN® AND STANDARD DBS ACROSS 11 DIFFERENT CONCENTRATION LEVELS FOR ALL 27 ANALYTES USING THE NEWBORN SCREENING. THE PERCENTAGE DIFFERENCE BETWEEN PUNCH LOCATION (PERIPHERY MINUS CENTRE) IN THE STANDARD DBS METHOD FOR ALL 27 ANALYTES USING THE NEWBORN SCREENING WORKFLOW. ............................................................ - 56 -
TABLE 3.4 THE COEFFICIENT OF VARIATION (IN %) FOR THE ENDOGENOUS LEVEL (LEVEL 1) IN HEMAPEN® (H) AND STANDARD DBS (D). THE COEFFICIENT OF VARIATION (IN %) FOR THE TOTAL CONCENTRATION (EXOGENOUS AND ENDOGENOUS) FOR LEVEL 2,8,11 FOR HEMAPEN® AND STANDARD DBS. THE RECOVERY (IN %) FOR LEVEL 2, 8, 11 IN HEMAPEN® AND STANDARD DBS ADJUSTED FOR THE ENDOGENOUS AMOUNT (LEVEL 1) USING THE NEWBORN SCREENING WORKFLOW. ......................................................................... - 62 -
TABLE 4.1 THE ABSOLUTE PERCENTAGE CHANGE |% Δ|, THE ACTUAL CHANGE Δ (IN µMOL/L), AND P-VALUE FOR ALL 20 ANALYTES ACROSS FIVE HCTS. THE DIFFERENCE OF THE MEAN BETWEEN HAEMATOCRIT LEVELS AND ANALYTES WAS TESTING USING T-TEST. ALL P-VALUES WERE ADJUSTED FOR MULTIPLE TESTING USING BONFERRONI CORRECTION (N=105). ............................................................................................................. 76
TABLE 5.1 THE AVERAGE CONCENTRATION (NG/µL) AND YIELD (NG) FOR EIGHT TECHNICAL REPLICATES ACROSS THREE EXTRACTION KITS (MAGMAX CORETM, QIAAMP®, QUICK-DNATM) AND SIX DIFFERENT SUBSTRATES (GENCOLLECTTM, FTATM ELUTE, FTATM GENE, GENSAVERTM, WHATMAN 903TM AND METHACRYLATE MONOLITH (MPPM) THE BLOOD VOLUME USED FOR EACH EXTRACTION WAS THE SAME MAKING THE CONCENTRATIONS COMPARABLE. CONCENTRATION WAS CALCULATED USING QUBIT FLUOROMETER. .......................................................................................................................................... - 93 -
TABLE 5.2 AVERAGE NANODROP DNA PURITY (260/280 RATIO AND 260/230 RATIO) FOR ALL THREE EXTRACTION KITS (MAGMAX CORETM, QIAAMP® AND QUICK-DNATM) AND ALL SIX SUBSTRATES (GENCOLLECTTM 2.0, FTATM ELUTE, FTATM GENE, MPPM, GENSAVERTM 2.0 AND WHATMAN 903TM). ................................... - 95 -

xi
Abbreviations
CMP = Capillary microsampling
DBS = Dried blood spot
DNA = Deoxyribonucleic acid
DPS = Dried plasma spot
EDTA = Ethylenediaminetetraacetic acid
FDA = Food and Drug Administration
HCT = Haematocrit
NBS = Newborn screening
PCA = Principle component analysis
PCR = Polymerase chain reaction
PKU = Phenylketonuria
RBCs = Red blood cells
SMRT = Single molecule real time sequencing
TDM = therapeutic drug monitoring
UK = United Kingdom
USA = United State of America
VCGS = Victorian Clinical Genetic Services
WBCs = White blood cells
WHO = World Health Organisation

- 1 -
Abstract
Blood is the sample that is used as the main biological source for diagnosis and screening.
The current process for blood collection involves vein incision in the inner arm. Many find
this process invasive and painful. Furthermore, the collection process relies on a trained
health professional, which creates an extra burden for the patient who has to travel to a
local collection centre. Such collection can be particularly challenging for children and the
elderly. An alternative, less invasive method, which is used in newborn screening, called
dried blood spot (DBS) method allows the collection of a small volume of blood onto the
filter paper. The process requires a finger prick in adults or heel prick in kids and is
performed using a lancet. The blood volume collected is minimal and the benefits of storing
such a sample on filter paper reduce the cost of storage and transport. DBS is utilised in
research, however, the use is limited in a clinical setting. From an analytical perspective the
commonly discussed bias is the haematocrit (HCT), which limits the possibility for accurate
quantitative analysis. Different approaches are being developed to tackle the HCT related
issue and improve the quantitative analysis yet maintaining the benefits of DBS. Several new
blood collection and storage devices are being developed to allow easy and accurate blood
collection, together with good quality samples, one of which is tested in this thesis. The
blood collection device tested here is the hemaPEN®, which collects blood via four
borosilicate capillaries and stores samples on pre-punched filter paper within the device.
Chapter 1 reviews the literature and outlines the importance of blood, clinical use of blood,
current methods of blood collection including alternative methods and the benefits of DBS.
The chapter further outlines the use of DBS method and identifies the issues associated with
the current use of DBS. The final review identifies new emerging approaches in sample
storage including different collection and storage devices.
Chapter 2 summaries the overall methods used in the project. The chapter outlines the
processes used for hemaPEN® assisted collection, generating and analysing samples for
method comparison, HCT bias and the comparison of DNA extraction kits across different
substrates. Furthermore the chapter outlines the statistical methods.

- 2 -
Chapter 3 explains the utility of hemaPEN® from both the collection and analytical
perspective. Firstly the chapter explains the results from assisted collection at the Royal
Children’s Hospital Pathology Collection Centre, where the device was successfully used.
The results concluded that most collectors could use the device given the instruction
material but some collectors might require further practice. Furthermore, the device was
tested from an analytical perspective in the newborn screening (NBS ) laboratory. The
device was compared to strand DBS method across eleven different concentration levels at
the average male HCT. Overall linear correlation with increasing concentration levels was
observed in both methods indicating good reproducibly. However, when comparing the
method agreement, a proportional bias was observed with larger difference at higher
concentration levels. The difference in the methods was mostly due to the different volume
collected and analysed which is associated with the known volumetric inaccuracy in
standard DBS.
Chapter 4 expands on the HCT bias associated with standard DBS method. The hemaPEN®
device was used across five different haematocrit levels to test the HCT dependency when
the volume bias is corrected. The two methods were analysed in the NBS laboratory. By
collecting accurate volumes of blood followed by the whole punch analysis, few analytes
showed HCT independency in the hemaPEN®, indicating that volumetric correction can
remove the HCT related bias. However, the results also suggested that by removing the
volumetric bias associated with standard DBS, other areas of bias were uncovered outlining
the complexity of HCT related bias. The other HCT dependant areas of bias such as HCT
related extraction bias and the distribution of target analyte in whole blood must be
understood when implementing the use of any DBS related workflow.
Chapter 5 expands on the use of DBS in other fields such as genomic and epigenomics. DNA
has been successfully extracted from DBS, however, the recovery is often effected by the
spot size, sample age and extraction kit used. In this chapter, the different extraction kits
and substrates both synthetic and non-synthetic were tested to maximise the yield. The
results suggests that both extraction kit and substrate type can improve the yield.
Chemically treated cellulose based substrate WhatmanTM FTATM Elute resulted in the highest

- 3 -
DNA yield using the Quick-DNATM extraction kit. Furthermore, the chapter concludes that
the use of synthetic substrate resulted in isolating high molecular weight DNA. Further
research to optimise such substrate and tailor its properties for DNA extraction could
further improve DNA recovery.
Chapter 6 outlines the conclusions for each chapter and summaries the overarching
conclusions. The chapter highlights that to implement a novel device there are three areas
that should require further testing; the usability from a collection perspective, either
assisted or at home, use in workflow where DBS cannot be used and then move to
optimising the methods to widen the use of such a device, in this case through substrate
optimisation. The final discussion touches on the benefits of modifying the current medical
system to a more patient-centric approach that would not only benefit the people but
reduce hospital admission and medical burden.

- 4 -
Chapter1:Literaturereview

- 2 -
1.1GENERALBACKGROUND
1.1.1Thebloodcomposition
In the 1800s the anatomic concept, where the organ is the centre of disease, started to
dominate renewing the interest in examination of body fluids. In late 1900s clinical
chemistry grew with the emphasis on chemical methods for diagnosis (Rosenfeld, 2002). In
the current days modern clinical chemistry concept is a branch of medical science which
includes analysis of the biological body fluids such as blood and provides diagnostic
information on the state of the human body. In the past century the rapid change in
technological improvements have defined the method of clinical chemistry and is pushing
the boundaries even further (Kricka and Savory, 2011). Blood is one of the key fluids that
has been used in many assays and tests.
Blood is a specialised fluid circulating within the veins, arteries, and capillaries. It is a fluid
that connects the various tissues of the body. The two main components of blood are
plasma and formed elements, which refer to all cells in the blood stream (Strauss and
Mauer, 1978).
Plasma is a matrix in which formed elements - proteins, enzymes, nutrients, waste,
hormones and gases are suspended. Plasma gives blood the liquid properties and takes up
more than half (55 %) of the overall blood content (Mathew and Varacallo, 2020). There is
no specific organ creating plasma, thus, water and salts are extracted from the digestive
system. The total volume of plasma in the bloodstream can differ based on the water intake
of an individual (Mathew and Varacallo, 2020).
To isolate plasma, blood is collected into a tube that contains anticoagulant such as EDTA,
lithium heparin or sodium citrate. This is so the process of coagulation is inactivated (Bowen
and Remaley, 2014). For isolation of plasma from whole blood, centrifugation is necessary.
By centrifugation the blood elements are separated based on size and density, leaving
plasma at the top of the tube (Basu and Kulkarni, 2014). Serum is another term commonly
used when it comes to blood collection and analysis. It is a liquid part of blood after removal
of the clot elements. The substance is similar to plasma; however, fibrinogens and other
clotting factors are absent. To isolate serum from blood, the coagulation process and clot

- 3 -
formation are initiated followed by centrifugation of blood that separates serum from
formed elements based on the size and density (Leeman et al., 2018).
There are three types of formed elements in the human blood: red blood cells (RBCs), white
blood cells (WBCs) and platelets. RBCs, also known as erythrocytes, are 8 µm in diameter
and 2 µm thick and have a unique disc shape. The biconcave shape maximises the surface
area, which is key for oxygen and carbon dioxide transfer. The unique shape also increases
the elasticity to allow easier passage through small capillaries. On average, male have about
five million RBCs per cubic millimetre, and for female it is around four and a half million
(Bethesda, 2005). The RBCs are made in bone marrow, the soft tissue of bone cavities and
the lifespan is around 120 days (Diez-Silva et al., 2010). The distinct red colour of these cells
is reflected by the high amount of protein called haemoglobin (Sarode, 2018). Haemoglobin
is an iron-based protein that binds to oxygen and gives RBCs their primary function. Mature
RBCs do not contain a nucleus (DNA), mitochondria or ribosomes; this is to ensure
additional space for haemoglobin (Kuhn et al., 2017). Due to the lack of DNA, RBCs cannot
undergo mitosis and, therefore, old and damaged RBCs are usually removed by the spleen,
liver, and lymph nodes (Kuhn et al., 2017).
The second type of formed elements are WBCs. The size of WBCs ranges from 12 - 17 µm in
diameter and in contrast to RBCs, the shape varies across different WBC types. On average,
humans have between four thousand and eleven thousand WBCs in every cubic millimetre
of blood (Blumenreich, 1990). Furthermore, the WBC count can differ based on many
factors such as sex, ethnicity, age, immune response and inflammation (Chen et al., 2016,
Gwak et al., 2007). The lifespan of WBCs ranges from 13-20 days. WBCs are the only cells in
the blood stream that have nucleus and are made predominantly in bone marrow. There are
five different types of WBCs with two distinct classes: granulocytes and agranulocytes
(Figure 1.1). This classification is based on visible granules when performing blood smear
under electron microscope (Prinyakupt and Pluempitiwiriyawej, 2015).

- 4 -
.
Figure1.1Bloodcompositionincludingplasmaandformedelementswithfurtherclassificationofdifferentwhitebloodcells.There are three granulocytes subtypes: neutrophils, eosinophils and basophils each with
quite distinct functions (Prinyakupt and Pluempitiwiriyawej, 2015). Neutrophils account for
50% - 70% of all circulating WBCs and the number increases with infection, injury or other
types stress of the organisms (Sarode, 2018). The main function is immune defence, where
the key target is to attack a wide range of infectious pathogens including bacteria, fungi, and
protozoa (Mayadas et al., 2014). The invading pathogen is trapped by the cell and is then
destroyed via a process called phagocytosis. Phagocytosis allows the digestion of such
bacteria, which is taken up and creates a phagocytic vacuole within the cell. In the new
vacuole within the WBC, bacteria is destroyed by lowering the pH, and are degraded by
enzymes (Rosales, 2018).
Eosinophils take up only minor part of the WBCs and their main function is to destroy
parasites and also take part in allergenic response (Wen and Rothenberg, 2016).
Basophils are the least frequent granulocyte in human body; however, its accumulation
have been associated with allergies and allergic disease, organ rejection, autoimmunity and
cancer (Siracusa et al., 2013).
In the class of agranulocytes there are two subtypes: lymphocytes and monocytes.
Lymphocytes are further divided into two classes, the B Lymphocytes and T lymphocytes
which are bone-marrow-derived, and thymus derived, respectively. The B cell function is to
produce antibody and to restrict initial multiplication of infectious agent and therefore,
Blood
Plasma Formed elements
Red Blood Cells Platelets White blood cells
Granulocytes Agranulocytes
Neutrophils Eosinophils Basophils Lymphocytes Monocytes
Lymphocytes T Lymphocytes B

- 5 -
maintain manageable levels (Deane 2017, Macnab and Onions, 1996). The T cell function is
to act as central regulators to directly kill virus infected cells in the body (Luckheeram et al.,
2012, Suárez-Fueyo et al., 2019, LeBien and Tedder, 2008). Monocytes are the last group of
WBCs to be discuss in this chapter. Their main function is to track pathogens and remove
microorganisms, lipids and dying cells via phagocytosis (Yang et al., 2014).
Platelets are part of formed elements and are derived from thrombocytes, which comes
from the word thrombus meaning clot. Platelets are small spheroid like structures with hair
filaments made in bone marrow. These cell-like structures have no nucleus and are the
smallest cells in the blood stream. It has been debated whether platelets do classify as
“cells” and to this day some pathologist refer to platelets as cell debris (Garraud and
Cognasse, 2015). The size of platelets is around 2-4 µm. Small granules are visible using a
microscope; these contain substances that are used to trigger a clotting cascade reaction
(Garraud and Cognasse, 2015). Despite the size, there are around one hundred and fifty
thousand to four hundred thousand platelets per cubic millimetre in the blood stream
(Drachman, 2004).
The primary function of platelets is to prevent bleeding via coagulation, a process where
liquid blood is converted into a gel-like substance. Platelets and protein activators will
trigger a cascade of reactions which lead to clotting. The process of blood clotting is called
haemostasis (Smith et al., 2015). Under normal circumstances coagulation process is under
inhibitory control, which limits clot formation until this balance is interrupted by increased
coagulant factors. The coagulation pathway is complex with multiple components; however,
it is crucial to understand these systems for clinical purposes.
Evidence suggesting that platelets also play rote in homeostasis; the regulation of the
environment in which all the molecules are transported (Männel and Grau, 1997).
Homeostasis is a key component to understand as it regulates the environment through
mechanisms such as osmoregulation (fluid balance), thermoregulation (heat regulation) and
chemical regulation. The regulation is achieved by different systems in the body such as
respiratory, digestive nervous and urinary systems. The concentration of compounds
dissolved, the pH, and the temperature are maintained by feedback look mechanisms
(Modell et al., 2015) (Ribeiro et al., 2019). This balancing mechanism reduces the output
based on the triggers.

- 6 -
The assessment of platelets in whole blood can be useful indicator for diseases such as
diabetes (Tong et al., 2004), renal diseases (Ghoshal and Bhattacharyya, 2014),
tumorigenesis (Ghoshal and Bhattacharyya, 2014) and Alzheimer’s disease (Ghoshal and
Bhattacharyya, 2014, Feng et al., 2011, López and Berliner, 2017).
Due to the diverse composition and the wide distribution, the role of blood includes
transport, homeostasis, immune response and clotting response. Blood also links many
body systems such as respiratory, endocrine, digestive, urinary and immune systems.
1.1.2Theclinicalimportanceofblood
As outlined previously, blood connects many systems and organs in the body, and therefore
is a key biological sample that allows us to assess the internal environment of the body.
Blood tests are performed to evaluate the overall health of individual’s organs, to determine
future risks of a disease, to diagnose specific disease, and to indicate how well certain
treatments work (National Heart Lung and Blood Institute, 2020).
There are other less-invasive biological samples that can be collected such as urine, saliva
and cheek swabs. Using these, researchers can detect microbial (Bi et al., 2019, Lim et al.,
2017), immunologic (Pugia et al., 2007) disease specific (Jing and Gao, 2018) and molecular
biomarkers (Yoshizawa et al., 2013) as well as proteins (Lorenzo-Pouso et al., 2018),
metabolic (Kennedy et al., 2016) and hormone profiles (Gröschl, 2008). These non-invasive
biospecimens are becoming more commonly used; however, blood is still the most common
biological sample used in clinical diagnosis; up to 70% of any medical decisions by health
professionals are drawn from blood tests (Abbott, 2012). For example, at the Victorian
Clinical Genetic Services (VCGS) 10-20 % of biospecimens are cheek swabs followed by 1-5%
of dried blood spots, the remaining tests are performed on venous blood. Furthermore
overall clinical testing is performed on people of all ages, from prenatal, through newborn
testing, development delay to tests in adults.
There are two different approaches to clinical testing: screening and diagnosis. Diagnostics
is an approach that aims to gather all the information to make a clinical decision about
individual’s health based on symptoms. Many tests are routinely performed including those
to detect minerals (Harrington et al., 2014) and vitamins e.g. vitamin B12, vitamin D,

- 7 -
magnesium, iron and folate (Higgins, 1995, Zhang et al., 2018), glucose (McMillin, 1990),
cholesterol (Ranade, 1993) and other lipids, and presence of viruses such as HIV and
influenza (Baron et al., 1996).
The second approach to clinical testing is screening. This preventive testing is done on
regular basis on healthy population, without symptoms present, to allow early detection. An
example of early detection performed on people over 50, is the bowel cancer screening
(Cree, 2011, Adler et al., 2014). Furthermore, a series of antenatal screening tests is
performed on pregnant women to detect any infection, Rhesus (Rh) factor and gestational
diabetes (Dajak et al., 2014). A worldwide screening platform that is perform on all neonates
is the newborn screening (NBS). This screening aims to detect inborn errors of metabolism.
Early detection of these errors is a key for early treatment to reduce the morbidity and the
mortality rate (Kelly et al., 2016). Such screening is usually a national program and is
government-initiated and funded.
1.1.3Clinicalandanalyticalmethodvalidation
Current clinical diagnostic schemes can be referred to as “closed” where the mediator is the
healthcare professional who ensures that laboratory results are used and interpreted
correctly. The current scheme is sometimes called as “site-centric”, meaning that the
patient travels to a local clinic or hospital to see the relevant health professional
(Theodorsson and Magnusson, 2017). In this scheme, there are two separate interactions:
patient and health professional interaction, and health professional and laboratory
interaction. The two interactions are the basis for two different approaches for method
validation, clinical versus analytical. The patient and health professional interactions focus
on the clinical performance and interpretation for which the diagnostic outcome and
patient’s health is the main focus. The health professional and laboratory interaction
focuses on the analytical performance, in which the validation processes of internal and
external quality assurance is the key target (Theodorsson and Magnusson, 2017).
1.1.3.1Clinicalvalidationprocess
Clinical diagnostic performance aims to interpret results that are associated with a specific
disease or a patient’s condition. From the clinical aspect the focus is on the biological

- 8 -
variation and the reference threshold in the population. With these results, health
professionals can then diagnose a specific disease and tailor patient’s treatment accordingly
(Biswas 2016, Bossuyt, 2009). The diagnosis is usually performed based on laboratory results
and symptoms. In order to evaluate diagnosis correctly, and to determine the accuracy of a
test, validation measures such as the sensitivity and the specificity are identified (Bossuyt,
2009). These measures are population independent. Clinical sensitivity refers to the ability
of a test to correctly identify those patients with the disease (Lalkhen and McCluskey, 2008).
As an example, 95% sensitivity would identify 95% of patients with the correct disease but
5% of those with the disease would remain unidentified. Clinical specificity refers to the
ability of a test to correctly identify those patients without the disease (Parikh et al., 2008).
As an example, 95% specificity correctly identifies 95% patients without the disease and 5%
patients that do not have the disease are identified as disease positive (Lalkhen and
McCluskey, 2008).
Furthermore, the likelihood ratio measure tells us what is the likelihood that a patient with
a particular test profile has a particular disease or condition. This measure also tells us the
probability of a person with a particular result to be correctly diagnosed. This value is
calculated based on the specificity and selectivity values (Grimes and Schulz, 2005).
The next clinical evaluation is performed on population studies focused to optimise a
specific diagnostic threshold. The clinical validation process is only done when changes are
medically important. Therefore, if testing is done on a new method with known clinical
thresholds and indicators, clinical evaluation might not be necessary (Trevethan, 2017).
Clinical diagnostic performance should not be tested prior to the analytical performance.
1.1.3.2Analyticaluncertainty
From the analytical perspective, the focus is usually on the technical variation and any errors
associated with undergoing a specific test (Taverniers et al., 2004).
The following parameters are key in order to eliminate analytical uncertainty. Analytical
error can be associated with the instrument, method, reagents or the operators. The
variation in results can be due to a random error (imprecision) and a systematic error
(inaccuracy) (Flatland et al., 2014). The random error is an unpredictable error that can be
associated with a sampling error. The precision of a method shows how close measured
values are irrespective of the actual true value. Precision measures can be divided into two

- 9 -
key indicators: repeatability and reproducibility. With repeatability, the precision can be
looked at within and between sample runs in a given laboratory. The reproducibility
introduces other factors such as laboratory change or calibration change. Statistically, the
random error can be determined by coefficient of variation or standard deviation (Jennings
et al., 2009, Han et al., 2017).
The systematic error shows the similarity between two values based on expected results. In
other words, the systematic error shows how close the measured value is to the target
value. If a result is inaccurate, there is the tendency for the measured result to either
overestimate or underestimate the correct value. This bias can be divided into two types:
constant and proportional. The constant bias refers to the values being overall higher or
lower across different concentration levels. The proportional bias is increasing with
increased quantity causing greater bias at higher concentration levels. In a laboratory, these
errors are usually monitored by comparison with the quality control samples for each run
(Ludbrook, 1997, Giavarina, 2015).
1.1.3.3Analyticalvalidationprocessesforestablishedmethods
For methods that are in routine use, the standard procedure is to use internal and external
quality assurance, which are performed on a regular basis to detect any systematic and
random errors during each run (Jones et al., 2017). Internal quality control is a process
checking the between-run precision and assessing the closeness of results obtained in each
run of analysis (Kinns et al., 2013). The internal quality control measures include samples
with known concentration level of the target analyte across different concentration levels.
Such controls are included with each separate analytical run and the measured values are
compared to the actual values of the test samples. If quality controls are outside the
accepted range, results are often repeated. A calibration process might also be required to
adjust the instrument. The purpose of the calibration process is to establish the relationship
between an actual measured quantity (output) with the target quantity (input) (Danzer,
2007). This process also resets the instrument if measures are outside the values. External
quality assurance involves comparisons between laboratories to ensure that results are
comparable (Sandle, 2005).

- 10 -
1.1.3.4Analyticalvalidationprocessesfornewmethods
When establishing or comparing new methods, the following indicators need to be
identified and determined as part of validation process: analytical linearity range,
interference, recovery, carryover, limit of detection and limit of quantification. Analytical
linearity range defines the linear relationship (straight line) between the targeted
concentration and measured concentration. It is achieved by using a series of dilutions and
further determines the lower and upper levels of the specific test (Killeen et al., 2014,
Cuadros Rodríguez et al., 1996). Interference needs to be tested to determine if any
molecules or chemicals or sample type could interfere with the particular tests to ensure
the reliability of the result (Kazmierczak and Catrou, 2000). Recovery determines the
amount of analyte recovered from a sample after extraction. Recovery is identified by using
an internal standard with a known level to calculate the percentage of analyte recovered
(Thermo Scientific, 2007). Carryover is a source of potential error as sample with high
concentration can cross-contaminate neighbouring sample with low concentration.
Carryover needs to be tested in specific workflows to see if high and low values analysed
next to each other carrying over (Armbruster and Alexander, 2006). The limit of detection
tests the lowest and highest amount that can be detected in a sample and the limit of
quantification is the lowest and highest amount that can be accurately quantify for specific
analyte (Armbruster and Pry, 2008).
1.1.3.5Theeffectsofpreanalyticalvariability
The peanalytical phase refers to the process that occurs prior to the analysis of a sample.
The laboratory has no control over these processes, yet they can play key part in patient’s
diagnostics. Tournis and Makris (2018) divided these pre-analytical factors into technical
and biological. Technical factors that can cause variability include sample type, sample
collection tube, specimen storage and transport. In some cases, analytes can be measured
in different biological samples such as urine or plasma. Different sample types can have
different reference concentration numbers; therefore, it is necessary to ensure that the
right sample is used. In some analytical instruments, the type of collection tube matters, for
example, some anticoagulants can interfere with analysis.
In the biological variance, there are further two types that can influence the variability the
uncontrollable and controllable. In the uncontrollable category, factors such as age, sex,

- 11 -
ethnicity or pregnancy can affect certain tests. Semi-controllable factors include diet,
exercise or circadian rhythm (Woodworth and Pyle ,2013).
In summary, there are many procedures that need to be tested and controlled for prior to
interpretation and diagnosis by health professionals.
1.1.4Venousbloodcollection
The usual volume of blood drawn from an arm is ranging from 4 - 10 ml. The collection
involves a needle puncture into a vein in the inner elbow. If multiple collection tubes are
required (usually for multiple tests) the volume taken is much greater, sometimes up to 25 -
30 ml.
Many people find having blood collected in such a way very invasive and painful. The
collection process is even more problematic in infants, children and the elderly, whose veins
are challenging to find (Ornstein et al., 1999, Cohen et al., 2001, World Health Organization,
2010a). Moreover, individuals usually need to travel to a local pathology collection centre.
This presents a burden on individuals, especially for those needing frequent blood
collection. Collection can only be done by well-trained health professionals due to high risk
of contamination and needle injuries, making the utilisation of this method difficult in
resource-limited settings (Kralievits et al., 2015). Furthermore, the sample collection,
processing, transportation and storage is very costly (Mei, 2014). Therefore, a simpler form
of blood collection would be advantageous, not only for rapid diagnostics but also for more
frequent health checks and reducing the risk of transmission of infectious disease.
1.1.5Skinincisionforcollectionofmicrovolumes
The concept of blood micro volume collection (microsampling) involves collecting volume in
microlitres rather than millilitres. Standard microsamples ranges from 10 - 100 µL of blood
collected (Patel et al., 2019). Microsampling blood collection is generally performed by a
skin puncture rather than venepuncture. The aim for this method is to be less invasive and
quicker at collection without any basic training requirements. Such collection can be done at
home by patients and sent by post, allowing more frequent collection and reducing the
need to travel (Chapman et al., 2014).
The most common skin puncture method uses a lancet to perform a finger prick in adults or
a heel prick in infants. The lancet is used to cut or penetrate skin and create blood flow for

- 12 -
collection. It is a single-use device that varies in size, shape and sharpness to vary the blood
flow, and therefore, the volume of blood collected (Lei and Prow, 2019). There are two
types of lancets: needle and blade. Needles consist of a solid, tri-bevelled rod similar to a
medical syringe and create a puncture in the skin. Blades create an incision. The depth is
influenced by the angle of the cut and by the movement of the blade e.g. slicing or jagged
cut (Kim et al., 2012). The variation in lancets is tailored to different areas of skin, type of
capillary network and the blood volume required. Even though this collection method is less
invasive than venepuncture, some still find this method painful as some bruising is possible
if not performed correctly (Fruhstorfer et al., 1999).
There are new micro volume skin puncture methods that are currently emerging, making
microsampling cleaner, easier (with minimal training) and even less invasive (Figure 1.2). A
novel device that is being developed by Seventh Sense is called the TAPTM. A standard
needle is replaced with an array of microneedles allowing good blood flow (Blicharz et al.,
2018). Collection is performed from the forearm and once attached using an adhesive pad, a
tap (firm push of actuation button) is needed for the needles to penetrate the skin. The
main principle is the use of a vacuum for blood transfer allowing for painless collection
(Cunningham et al., 2000). The collection process takes around two minutes and leaves a
small ring of punctures similar to that of a mosquito bite. The total volume collected is
around 100 µL and blood is retained within an interior reservoir (Catala et al., 2018). The
first clinical study of this device was conducted in 2018, and involved 144 patients. The
results outlined that the pain experienced was lower than from standard venous collection.
The study also showed equivalent haemoglobin measures when compared to standard
venous blood (Blicharz et al., 2018). Further proof of concept study has shown quantitative
metabolic profile using TAPTM in comparison to standard blood showing promising use of
this device in large scale metabolic studies (Catala et al., 2018).

- 13 -
Figure1.2Alternativemethodstoskinpuncture:a)TAPTM,b)HemolinkTMincludingintegratablecartridge.Copyright: Taken from Blicharz et al. (2018) and Tasso (2020).
A similar device is being developed by Tasso called The HemoLinkTM. For this device an
integratable cartridge enables the adoption of different collection platforms such as serum
and dried blood spot (Figure 1.2) (Tasso 2020). The next stage is for the HemolinkTM to be
used in clinical trials for specific downstream applications.
1.1.6Benefitsofmicrosampling
By moving towards microsampling techniques, both the human and animal welfare can be
improved. From the human welfare perspective, quicker and less invasive collection
improves patient’s satisfaction. The ability to collect samples at home reduces the reliance
on health professionals and, therefore, lower travel requirements for a blood test, which
reduces the resources needed. In the clinical setting, when the burden for individuals is
reduced, more people are willing to be tested, allowing for earlier detection and the
possibility of wider screening (Martial et al., 2016, Zakaria et al., 2016). Early diagnosis,
better screening programs and improved monitoring of current drugs reduces hospital
admissions and thus, economic burden (Iragorri and Spackman, 2018, Lew et al.). In clinical
a
b

- 14 -
trials, these methods can reduce the cost (Amsterdam and Waldrop, 2010) and improve the
data collection by allowing additional time points to be captured (Trifonova et al., 2019).
Gathering samples from larger cohorts can improve scientific discoveries, as easier
collection expands the demographic possibilities for sample collection (Lei and Prow, 2019).
The smaller volume collected also benefits animal welfare. This is specially so in pre-clinical
settings where experiments are performed on animals. Many drugs are tested on model
animals, usually rodents. By moving towards for smaller blood volume collected the number
of timepoints that can be generated from single animal increases creating better drug
profiles. (Wickremsinhe and Perkins, 2015). The need for fewer animals also reduces
resources needed. The above benefits of microsampling have provided a great incentive for
development of new technology to allow easier, quicker and higher-quality collection of
blood (Dainty et al., 2012).
1.1.7CollectionandStorageofmicrovolumes
Different microsampling techniques for collection and storage can be used based on the
downstream requirements. There are three main approaches: the dried blood spot (DBS),
dried plasma spot (DPS) and capillary microsampling (CMS). These approaches tackle the
blood collection and storage from different perspectives, going from fresh to dry state,
separating plasma from whole blood or collecting small and accurate volumes. Based on
these three main approaches, there are also emerging techniques being developed to
improve some of the limitations of the current microsampling techniques and allow
microsampling to be commonly used in clinical setting. In the next section of this thesis I will
outline the three main approaches as well as the new emerging techniques.
1.2Driedbloodspot(DBS)
There is evidence that collecting biological fluid onto a ‘filter’ paper goes as far back as 780
AD (Hannon and Therrell, 2014). However, the use of such samples for analysis became
more successful in the 1900s (Hannon and Therrell, 2014). DBS involves depositing a small
amount (50µL) of blood onto a filter paper, drying and storing. In 1963, Robert Guthrie used
this method to develop systematic screening for phenylketonuria (PKU) in newborn babies
(Guthrie and Susi, 1962). Since then, this screening program has been applied to a large
number of inborn errors of metabolism and has been used in public health programs around

- 15 -
the world (Pitt, 2010). In many countries, these cards are stored indefinitely, representing
near-perfect national biorepositories (Cruickshank et al., 2012).
1.2.1Heelandfingerprickcollection
The source of blood for DBS is either a finger prick in adults or heel prick in infants. Earlobes
can also be used, but this is not common practice. In adults, the World Health Organization
(WHO) recommendation is to prick the index or middle finger. The size and depth of the cut
will affect the blood flow. To prepare the finger, an alcohol wipe is applied to clean the area
of the cut. Once dried, a lancet is used to create quick, continuous cut to allow good flow of
blood. The first drop of blood must be wiped off as it may contain skin tissue from the
puncture. It is recommended not to squeeze the finger as it dilutes the blood with plasma. A
single drop of blood should fall off the finger onto the filter paper. The standard NBS
collection filter paper has four circles to indicate where the sample should be collected. One
drop of blood should be applied to each circle of the collection paper. Once a sample is
collected, pressure must be applied on the puncture to stop bleeding, and a bandaid applied
to the cut (World Health Organization, 2010b).
In infants, the dept of the heel prick should not go beyond 2.4 mm. For premature neonates
an even smaller lancet is available. The depth is important for infants and children as too
much compression can cause deeper puncture that can reach the bone. For paediatric
collection, firstly the collector must physically immobilise the child. The skin preparation and
puncture procedures are same as those used in adults. To stimulate the flow of blood, the
parent or the nurse can tighten and release the child’s wrist. It is also recommended to keep
the child warm. Once a sample is collected, bleeding should be stopped by applying
pressure (World Health Organization, 2010b).
1.2.2DownstreamuseofDBS
Due to the many benefits of microsampling, DBS sampling has been introduced as a great
alternative in many fields including drug development, clinical setting, the ‘omics’
(genomics, epigenomics, metabolomics and proteomics) as well as in sport industry and
chemical detection.

- 16 -
1.2.2.1Drugdevelopment
The process of developing and marketing a drug has several phases. The first phase is
discovery and development, which involves target drug validations, compound identification
and optimisation (Horien and Yuan, 2017). After this, pre-clinical development is initiated
followed by clinical trials, which comprises of further four phases. Once enough clinical trials
are completed, the drug can then undergo the US Food and Drug Administration (FDA)
review followed by post-market monitoring (North East BioLab 2019, Mohs and Greig,
2017).
In the recent years, the DBS method has been used in pre-clinical studies as well as showing
great potential in drug discovery (Clark et al., 2010). This is because many pre-clinical
experiments are using blood from animal models. Animal ethics is placing a great emphasis
on the principle of three Rs: reduction, replacement and refinement. The key drivers are to
eliminate animal harm, the volume of blood taken and reduce the total animal number used
for experiments (Burnett 2011). As part of pre-clinical safety studies, pharmacokinetic and
toxicokinetic data are required to assess the fate of a certain substance in a living organism
and to determine the acceptable drug level. Collecting blood from animals using the DBS
method allows multiple collections from one animal rather than combining profiles from
multiple animals, thereby generating better data (Roberts et al., 2016). In addition, as fewer
animals are used, the drug dosage requirements are reduced significantly, which is
important in particular in discovery phase when the compound is limited (Wickremsinhe
and Perkins 2015). Pharmaceutical industries are implementing the use of small volume
collection and are improving not only the number of animals needed but also generating
better scientific data and reducing the cost associated with of pre-clinical testing (Burnett
2011).
After drug development and pre-clinical testing, the next step is to test the drug in clinical
trials with four phases. In the first phase, the drug is tested on small number (20-80) of
participants. If the efficiency and safety of the intervention is promising, then the
intervention is tested on a larger number (300 - 3000) of participants. In the final phase,
studies are designed to monitor efficiency and collect additional information over longer
periods of time. Developing a new drug that is prescribed by medical professionals was

- 17 -
estimated in 2016 to cost $2.6 billion (DiMasi et al., 2016), with the drug development stage
being the most expensive. Therefore, reducing costs is the main priority in drug
development. The use of the DBS method in clinical trials and therapeutic drug monitoring
can reduce both the social and the health cost as well as eliminate the dependence on
health professionals. This is even more so for children as parental supervision is required. A
study in the UK compared the cost of standard venepuncture and standard microsampling
techniques in two paediatric population requiring drug monitoring. The social cost
(calculated as combination of healthcare cost, patient related costs and costs related to loss
of productivity) dropped when implementing at home DBS collection by 43% and 61%
based on the patient health requirements (Martial et al., 2016). The cost reduction was
dependent on the number of hospital visits as well as possible implementation of home
sampling (Martial et al., 2016). The use of DBS is also favourable in longitudinal studies
which often require sampling of large number of participants. Allowing for easier and quick
collection that can, potentially, be done from home could increase demographics interest in
such studies and improve participant compliance.
1.2.2.2Clinicaldiagnosticsvs.clinicalscreeningofDBS
Despite the advantages of DBS over venepuncture as detailed above, the DBS method is not
commonly used for diagnostic purposes in developed countries. This is, however, different
in remote locations and places with limiting resources. The DBS method has been shown to
have significant impact in screening, point of care control, robust and affordable collection
where venous blood is not an option (Smith et al., 2015). The detection and diagnosis of
tropical diseases from DBS sampling has been shown to be successful with significant clinical
impact (Smith et al., 2015). For example, the detection of HIV+ patients was successful with
validated protocols using the DBS sampling method. It is critical to detect and diagnose HIV+
patients early to control the spread (Solomon et al., 2002). DBS sampling is a great tool
reducing the hazard of infection during transport, analysis and testing of a wide population
range. Researches used DBS method to detect malaria, as DNA and antibodies are present
and detectable, although the selectivity was found to be lower compared to the gold
standard (whole blood) (Al-Harthi and Jamjoom 2008). However, researches led by Ataei
and colleagues detected higher malaria specificity in DBS method compared to whole blood
(Ataei et al., 2011). Further Smit and colleagues reported that there are no commercially

- 18 -
available assays for malaria diagnosis currently available, making it a challenge for clinical
applications (Smit et al., 2014, Smith et al., 2015). The DBS method has also been used to
detect hepatitis C and B, with specificity and selectivity of > 98% (Croom et al., 2006,
Tuaillon et al., 2010); however, validation for clinical automation has been suggested as the
key next step. In summary DBS has a great potential in diagnosis; however, further research
for widespread use is needed.
The largest screening program using DBS method is the screening of newborns for inborn
errors of metabolism. There are standard first-tier tests that are standardised across
laboratories. The main focus for these tests is to detect abnormalities in metabolic pathways
and genetics. At the VCGS, the newborn screening program tests 25 different rare
conditions, for example maple syrup urine disease, cystic fibrosis, fatty acid metabolism
disorders, congenital hypothyroidism and amino acid disorders such as phenylketonuria
(Victorian Clinical Genetics Services 2020).
Each country/laboratory adds additional second-tier tests, with a constant flow of new tests
being identified. Research published in Nature used DBS method to detect spinal muscular
atrophy, which affects 1 in 6000 newborns, with the first signs observed after 3 months of
age (Kariyawasam et al., 2020). The same group also suggested that multiple second-tier
tests can be run in parallel. DBS methods can be used to detect viruses such as
cytomegalovirus, which can cause hearing problems and hearing loss (Cannon et al., 2014,
Ross et al., 2011). Moreover, Azzari et al. (2011) demonstrated detection of severe
combined immunodeficiency in DBS sampling. Therefore, in the USA, this test has recently
been added to the routine screening as this deficiency is otherwise not detectable and can
be life threatening with combination of other disease such as hepatitis or pneumonia
(Dorsey and Puck 2019). The list of tests that are performed is tailored to specific
demographics. Each test added to the screening tests add another level of complexity.
Therefore, cost benefit analysis needs to be performed to determine the efficiency and
whether it is worthwhile to add particular test to routine screening (Jansen et al., 2016).
1.2.2.3The“Omics”
The use of DBS is becoming popular and useful in the “omics” fields, for example genomics
(a field focusing on the structure, function, evolution, mapping, and editing of DNA ),

- 19 -
epigenomics (focusing on changes caused by modification of gene expression without gene
alteration), proteomics (focusing on proteins and their functions) and metabolomics
(focusing on chemical processes involving metabolites, the small molecule substrates and
products of metabolism). The ease of collection required for DBS method allows wider
population sampling and therefore, generating large data sets from longitudinal cohort (Lim
2018).
1.2.2.4Genomics
There are two main approaches to DNA sequencing that have developed as the technology
of DNA sequencing has progressed. The first DNA sequencing technology was developed in
1970s. Sanger sequencing, also known as chain termination method, allows reading of short
single sequences one at a time. The time and resources required made this method difficult
for sequencing large numbers of regions. The concept of this method was developed into
the “next gen” sequencing where multiple parallel sequences are read at one time (Koboldt
et al., 2013). High throughput is the key benefit of next generation sequencing. The
limitation of this approach is that large datasets are generated, qualified personnel and
powerful computer analysis is required for interpretation. The DNA yield requirements for
this approach are relatively low (~ 1 ng) due to an amplification step. The PCR amplification
step can, however, create potential errors in the sequencing. Furthermore, there is a lower
requirement for high molecular weight DNA as shearing process occurs prior to high
throughput sequencing (Koboldt et al., 2013).
The second approach that is being developed is part of the “third generation sequencing”.
This method allows for long reads of DNA and is sometimes referred to as long read
sequencing. This approach involves sequencing at a single molecule level (Ozsolak, 2012).
There are two methods based on different principles: the single molecule real time
sequencing (SMRT) (Ardui et al., 2018) and Oxford Nanopore technology (Lu et al., 2016).
Long reads create an advantage for specific regions of DNA where many sequences are
repeated. A high frequency of repeated sequences have been identified as a challenge for
assembly with high throughput short reads (Pollard et al., 2018). The limitation to the third
generation is a relatively high error rate; however, this technology is constantly improving.
The DNA requirements for this approach is higher compared to the next gen sequencing.

- 20 -
The DNA molecular weight required is around 40 Kbp with high DNA yield of around 1 µg
(Michigan State University, 2020).
DNA has been successfully extracted from DBS (Cruickshank et al., 2012, Wong et al., 2008,
Ghantous et al., 2014, Cruickshank et al., 2013). Researches have been working on
optimising the method to isolate high DNA yield. High quality genome wide genotyping was
performed on DBS where unamplified single nucleotide polymorphism (SNPs) were
identified, indicating great potential for genome wide association studies and large scale
genomic studies (St. Julien et al., 2013). Furthermore, whole genome sequencing was
successfully performed to detect casual mutation in a patient who inherited metabolic
diseases (Boemer et al., 2017). The use of DBS in genetic testing has great potential,
however, ethical and legal concerns around wide-spread genetic testing have been outlined
as a point to be discussed in parallel to the technical advancements.
1.2.2.5Epigenomics
Archived DBS can be useful for epigenetics studies as it allows us to access data from birth
(Filali-Ansary et al., 2016). Epigenetics is a process that refers to the modifications that
regulate DNA activity and its gene expression without the alteration of DNA sequence. Such
modification process is a biological process, which involves the addition of methyl group to
the DNA molecule. The use of DBS sampling in analysing the DNA methylation segments has
been challenging due to a stringent laboratory pre-processing steps. A successful protocol
has been published showing high quality, reproducibly and consistency in genome wide DNA
methylation analysis (Wong et al., 2008, Cruickshank et al., 2013, Ghantous et al., 2018,
Cruickshank et al., 2012). The reliability of DNA methylation measures from DBS was tested,
indicating lower reliability using the HumanMethylation450k microarray in comparison to
fresh blood, which is important when combining data from multiple large-scale studies
(Dugué et al., 2016). A good example is the DBS epigenome-wide association analysis of
monozygotic twins discordant for cerebral palsy, in which specific genes have been shown
consistently differentially methylated (Mohandas et al., 2018). These results highlight the
use of these card to accelerate life course health research post-newborn screening (Zar
Kyaw et al., 2019).

- 21 -
1.2.2.6MetabolomicsandProteomics
Over the last decade, mass spectrometry methods have been increasingly used in
metabolite and protein detection and quantification. Due to the rapidly increasing
sensitivity of these instruments, the potential mass spectrometry use in DBS is becoming
more popular.
Several studies have shown the use of DBS in protein quantification especially in
haemoglobinopathies such as sickle cell disease in both in newborns and adults (Li et al.,
2020, Martin and Cooper, 2014, Daniel et al., 2007, Boemer et al., 2008). The detection
sensitivity was not an issue as these are haemoglobin related diseases, however, moving
away from haemoglobin-related proteins, sensitivity can be an issue in DBS; therefore,
separation techniques can be required. Furthermore researchers were able to detect 97
proteins from DBS with the focus on the high throughput requirements for clinical
applications (Chambers et al., 2015).
From a metabolomics perspective, the improved sample collection methods can allow more
integrated metabolomic assessment helping with precision medicine and improve
population health studies (Drolet et al., 2017). Untargeted approaches have allowed for
cellular metabolism detection to understand metabolic pathways (Rola et al., 2019). This
approach can also help to discover novel biomarkers, and to understand disease prevention
and progression. Some studies have validated these methods in DBS. Researchers used DBS
to develop a method for detection of vitamin D (Rola et al., 2019, Hoeller et al., 2016, Eyles
et al., 2018). However, the key issue with the use of metabolomic analysis in DBS is the
associated inaccuracy in bioanalysis (Petrick et al., 2017), which will be discussed later in this
chapter.
1.2.2.7Sport
Venous blood is used in sports medicine and sports research, but is not an ideal mode of
collection. High volumes of blood taken from athletes can impact on their performance
(Thevis et al., 2020). The World Anti-Doping Agency has looked for novel methods to
increase the number of athletes tested and therefore, detect cheating (Thevis and Thomas,
2008). Researches used DBS to detect erythropoiesis-stimulating agents. These compounds
stimulate hormones that cause increased proliferation and maturation of RBC and,

- 22 -
therefore, increasing the athletes’ performance (Reverter-Branchat et al., 2018). Drug
detection is equally as important where DBS can detect cocaine and cocaine metabolites
(Ambach et al., 2019). Even though this research is showing promising results, the
importance of validation between venous and capillary blood was raised with further
research required to use DBS as a substitute for standard testing (Ellefsen et al., 2015).
1.2.2.8ChemicalDetection
Due to the collection advantages, DBS is becoming attractive for detection of blood
chemicals, especially in locations where resources are limiting. Yishai and colleagues used
DBS to determine post-exposure of warfare agents (Yishai Aviram et al., 2018). The
identification of exposed people allows for quicker and more appropriate treatment
reducing unexpected injuries and deaths. Another group detected poisonous paraquat-
based chemicals in DBS that are present in herbicides (Wen et al., 2018). The environmental
pollutant detection in human body is fast emerging filed. Researchers are suggesting the
use of DBS for detection of PFAs (Per- and Polyfluoroalkyl Substances). PFAs are present in
everyday products and in the environment and evidence has been shown that these
chemicals can be present in placenta (Ma et al., 2013).
1.2.3CurrentchallengeswithDBS
Despite the benefits of DBS, there are a few challenges, which are making this method
unattractive for some downstream applications. In this section I will summarise these from
two perspective the pre-analytical discussing the errors in sample collection and analytical
perspective associated with the errors around quantitative downstream analysis.
1.2.3.1Pre-analyticalchallengesinDBS
There are a few challenges associated with the collection procedure, which include: the
droplet depositing technique, the size of the spot, the overall collection guidelines across
different collection centres and the sample quality.
It is recommended that the droplet on the finger should fall onto the paper; however,
sometime that can be difficult when not enough blood can be generated e.g. the patient
does not bleed easily and/or is an elderly or an infant. This issue results in the need to touch
the droplet onto the filter paper. Sometimes, multiple spots are applied creating additional

- 23 -
layers of blood applied on one spot (Zakaria et al., 2016). It is estimated that one drop of
blood ranges from 15 µL – 40 µL when deposited on the filter paper (Chernonosov, 2018).
After collection, samples are usually dried on a bench in the collection centre. These drying
conditions can also impact the quality of the sample with samples need to be fully dried
prior to shipment (Denniff and Spooner, 2010). DBS card are usually not protected by any
covering allowing for possible sample contamination. Witner and colleagues tested various
newborn screening cards and found a large number of cards contaminated with urine,
faeces, or baby cream (Winter et al., 2018).
The spot size generated can vary based on the volume applied. Moat et al. (2019) tested
different diameters of spots size suggesting that spot that is too small (<8mm) or too big
(>14mm) should be rejected. The challenge of going from single site study to multi-centre/
global studies is the quality of sample collected. The collection for clinical purposes is mostly
assisted meaning that qualified health professionals are collecting those samples. However,
it has been reported that even with this control environment, the sample rejection rate can
be around 4-5 % (Zakaria and Greaves, 2019).
When moving away from assisted collection to self-collection, a precise guideline
development is crucial to ensure consistency between samples collected as the rejection
rate from at-home collection was up to 20% (Zakaria and Greaves, 2019). The ease of and
adequate collection is the key driver in developing new techniques to make DBS more user
friendly (Chernonosov, 2018, Zakaria and Greaves, 2019).
1.2.3.2AnalyticalchallengesinDBS
There are key issues with DBS limiting the use of this method for quantitative analysis.
These issues include: the unknow volume deposited, haematocrit bias, the filter paper, the
chromatography of the paper and the use of whole blood rather than plasma (the gold
standard).
During the DBS collection, the volume applied onto the filter paper is unknown, therefore,
accurate quantification using DBS has been identified as a challenge. To adjust for the
volume inaccuracy, a fixed diameter of 3-6mm (sub-punch) disk is punched out. This sub-

- 24 -
punch workflow, however, is not the ideal solution to fix the volumetric bias because other
factors, outlined below, affect the dispersion of the spot (De Kesel et al., 2013, Youhnovski
et al., 2011).
Haematocrit (HCT) is a well know factor contributing to the analytical bias in DBS. HCT is
defined as the percentage of RBCs in total volume of blood and varies across the population
based on sex, age and ethnicity (Lim et al., 2015, Mahlknecht and Kaiser, 2010). The level
can also vary depending on the time of day and can be affected by how hydrated an
individual is (Holsworth et al., 2013). For individuals with a higher HCT, the blood is more
viscous and therefore, the spot size will be smaller. For individuals with a lower HCT the
blood is less viscous, therefore, the spreading of applied spot will be greater. When using
the standard DBS workflow, punching out a fixed size spot will punch out bigger proportion
of a sample with a higher HCT (grater volume), resulting in more sample analysed and vice
versa. There have been different suggestions for adjusting the HCT including calculating the
haematocrit level from the spot by measuring the spot size (Oostendorp et al., 2016). One
study used haemoglobin measures to correct for haematocrit in detecting potassium levels
in DBS (De Kesel et al., 2013). Further reports showed single-wavelength reflectance-based
approach showing promising results in detection of HCT result which uses simplified
prediction method (Capiau et al., 2018). These methods are in some cases labour intensive,
add complexity to the analysis and create extra steps in validation and automisation.
The type of filter paper used will also affect the accuracy of DBS. The paper most commonly
used contains 100% cellulose; however, the thickness and pore size can differ between
papers. There are several manufactures that produce these cards but the two main brands
that are approved by the FDA are: Whatman 903TM and PerkinElmer 226 filter papers (Smith
et al., 2015).
The paper chromatography effect is explained by the spreading of the sample on the paper
where paper is the stationary phase and blood is the mobile phase. Depending on the paper
and the viscosity of the blood, the spreading of the analyte or blood components can be
uneven (Fan and Lee, 2012). Moreover, it has been reported that the distribution of cells
across the spot can be uneven, creating a heterogeneous spot, meaning that punch position
(centre vs periphery) can influence analyte quantification (Holub et al., 2006).

- 25 -
The current gold standard for diagnosis is plasma isolated from venous blood. When
switching from venous to DBS collection and storage of whole blood, a few additional
factors need to be considered. Since DBS include cells, which are usually removed in plasma
samples, the integration of DBS can be challenging. Furthermore, additional components
may need to be considered and investigated when dealing with dried whole blood such as
the compound affinity and kinetics to the cellular components of blood, the overall
adjustment of blood to plasma ratio and the bioavailability of the drug /analyte in whole
blood rather than plasma (Rowland and Emmons, 2010). Therefore, when switching from
fresh plasma to DBS in current, additional bridging experiments may be necessary. One
research group outlined a possible statistical analysis that can characterize the relationship
and concordance between DBS and fresh plasma (Wickremsinhe et al., 2013). However,
such bridging experiments are compound-specific. Therefore, it is sensible to introduce the
use of DBS at the method development level, which would eliminate the need for bridging
studies (Amsterdam and Waldrop, 2010).
Given these challenges different methods have been developed to address some issues
based on the downstream application.
1.3Driedplasmaspot(DPS)
Microsampling storage of dried plasma spots (DPS) is a method that involves depositing
isolated plasma onto filter paper. For this method there is no need for comparison between
whole blood and plasma as the gold standard for many applications is plasma. The absence
of RBCs eliminates the potential interference with the quantitative analysis due to the
haematocrit effect. There are two methods of DPS collection. The first involves standard
blood collection (finger or heel) into a collection tube containing anticoagulant. This is then
spun, and plasma is applied onto a filter paper either using a pipet or glass capillary. Since
plasma is usually close to translucent, coloured filter papers are used to indicate plasma
application. Different research groups compared DBS vs. DPS to suggest the potential in
detection of HIV-1 viral load in infected African patients (Rodriguez-Auad et al., 2015,
Andreotti et al., 2010). Furthermore, multiple studies have used DPS to show the potential
in pharmacokinetics (Barfield and Wheller, 2011, Nijenhuis et al., 2016, Meesters, 2016).
Use of this method was also suggested in clinical setting. The detection of specific marker

- 26 -
has been used to identify glomerular filtration rate in kidney, which is particularly important
in resource limiting places as the DPS method is reasonably reliable in comparison to DBS
due to inaccuracy in low HCT (Hagan et al., 2013). DPS have potential in therapeutic drug
monitoring where DPS is used for monitoring antiviral drugs (Conti et al., 2018). DPS have
been tested in the potential detection of tuberculosis (Aabye et al., 2013). However, the
collection process of DPS methods is not ideal as the process involves additional steps
including spinning and depositing plasma on filter paper (Clarke and Dasgupta, 2016). The
use of this method will only be useful if standard DBS method is shown to be inadequate
due to RBC interference (Emmons and Rowland, 2010).
The second method of collecting DPS is using a filter device that separates the RBCs and
plasma. A blood drop is applied onto the filtration device as per usual DBS protocol and the
sample is dried. During drying, RBCs are trapped in the separation membrane and plasma
travels through to the collection membrane (Li et al., 2012). Once dried plasma is separated,
RBCs are removed by pealing of the filtration membrane. The polymeric membrane
separates the blood though absorption and filtration processes (Li et al., 2012). It is
important to note that plasma in the standard venous protocol sample is centrifuged rather
than filtered, therefore, it is important to validate that the plasma-like matrix on the
collection membrane is equivalent to plasma generated by centrifugation. Limited number
of studies have used such membranes for drug detection (Li et al., 2012). These new
filtration membranes are of great potential as they remove the need for centrifugation after
sample is collected, however, further studies are required to validate this method.
1.4Capillarymicrosampling(CMS)
Microsamples can also be collected using glass capillaries. The idea behind this method is to
collect and store a defined liquefied blood volume in a capillary, therefore, removing the
issues associated when going from fresh to dry state. Once blood is collected using the
capillary, one end of the capillary is wax sealed. Collected capillaries can then be centrifuged
allowing plasma and RBC separation. Plasma from spun capillaries can then be extracted
either by capillary snapping and plasma elution into a micro-container or by end to end
capillary transfer to smaller capillary (Coleman, 2020). Drummond sampling capillary is

- 27 -
engineered to make capillary collection and processing easier (Bowen et al., 2013). Firstly, a
glass capillary is sealed with a plastic wrapping for easier use and minimal breakage.
Secondly, the capillary is coated with anticoagulant to ensure adequate mixing and
comparison with laboratory internal standards and quality controls. Other improvements
include a sealable plug (Bowen et al., 2013). When the plug comes in contact with blood for
more than 30 seconds, it swells up, sealing the capillary. This removes issues such as
overfilling, leakage and contamination. A thixotropic gel present in the capillary moves with
blood density during centrifugation separation the red blood cell and plasma. The capillary
collection end can also be trimmed to fit into butterfly needle to allow easy collection and
constant flow to prevent bubbles (Bowen et al., 2013).
Despite some of the advantages, capillary microsampling methods are not currently
implemented in clinical settings due to the highly involved collection process. Furthermore,
the standard capillary microsampling method is used predominantly in pharmacokinetics
(Korfmacher et al., 2015) and toxicokinetic (Verhaeghe et al., 2017) in rats or mice as the
collection environment can be controlled by scientists. The use of liquid matrix removes the
benefit of low transport cost and storage that is associated with DBS.
1.5Emergingmethodsformicrosampling
Despite the advantages of the above methods, microvolumes are still not a standard
protocol especially in clinical setting. New devices have been developed to address the
disadvantages and allow easier user-friendly collection. Each device addressed this issue in a
different way.
1.5.1HemaSpotTM
HemaSpotTM is a device that collects blood from a finger prick into a moisture-tight cartridge
that has absorbent paper embedded within. The device comprises of application surface,
desiccant, absorbent paper in a moisture-tight cartridge with a tamper-resistant latch
(Figure 1.4). The recommended collection method is similar to the standard DBS, with the
first droplet being wiped off. For each collection a maximum of three drops should be
applied onto the applications surface (Spot On Sciences, 2020).

- 28 -
Figure1.4TheHemaSpotTMbloodcollectiondevice.Copyright: Taken from Spot on Science (2020).
The HemaSpotTM collection device claims to allow easier collection and better sample
quality. The cartridge contains an absorbent paper with a desiccant for quicker drying time.
Once three drops of blood are applied, the cartridge is closed and locked in for transport,
limiting any sample contamination. The cartridge is equipped with a barcode for sample
identification. A study to detect an HIV-1 drug was conducted using the HemaSpotTM
showing high improved viral load and good resistance mutation when compared to plasma
(Brooks et al., 2016). From the collection perspective, this device may improve some
challenges associated with standard DBS, but not the analytical challenges that accompany
the haematocrit effect and volumetric inaccuracy.
1.5.2HemaXisTM
HemaXisTM is a plastic foldout device that uses the DBS format and allows for accurate
amount (10 µL per spot) of blood to be collected (Figure 1.5). The collection involves the
standard DBS collection protocol. Blood drops generated on a finger have to come in
contact with the capillary inlet where blood is wicked into the capillary channel. On each
card, four microfluidic capillary inlets are available to collect for four spots. Once samples
are collected, the Whatman 903TM paper that is integrated in a cover is clicked on and
pressure applied for ten seconds. A microfluidic capillary channel comes in contact with the
paper and blood transfer occurs, the sample stays within the plastic cover. The DBS card can
be taken out and inserted in an automated workflow (HemaXis, 2018).

- 29 -
Figure1.5HemaXisTMbloodcollectionandstoragedevice.Copyright: Taken from HemaXis, (2018).
A clinical trial exploring drug metabolism enzymes is currently being conducted using
HemaXisTM (Lloret-Linares et al., 2017). This study aims to recruit 205 patients with multiple
data points collected using this device (Lloret-Linares et al., 2017). Further studies have used
HemaXisTM to detect the dosage of immunosuppressant tacrolimus and mycophenolic acid
that are monitored in patients after renal transplant. The use of this device is showing
promising results comparable to those using whole blood (Zwart et al., 2018). The study also
emphasises that patient training and good guidance for sample collection is essential (Zwart
et al., 2018). The outcome of this study shows a great potential in tests where the golds
standard is whole blood rather than plasma. Given the accurate volume collection and
whole spot analysis, this device can eliminate the haematocrit volumetric related bias.
1.5.3Capitainer
Capitainer is another device that allows accurate collection of blood volumes. One drop of
blood is applied onto an inlet as per standard protocol for DBS collection (Figure 1.6). The
contact of blood with the surface activates the metering channel, which then transfers an
13.5 µL of blood onto the specimen collection membrane. Once blood is transferred via the
channel, blood is absorbed onto a paper pad. The pad with dried blood is protected by a
paper film that can be peeled off for analysis. On one card there are two channels to allow
collection of two different spots (Capitainer, 2020).

- 30 -
Figure1.6TheCapitainerbloodcollectionandstoragedevice.Copyright: Taken from Capitainer (2020).
Few studies have been done as proof of concept to validate this device. The precise
dispensing was tested showing on average volume precision (Velghe and Stove, 2018). The
recovery across different haematocrit levels (25-65%) of labelled analyte was tested
showing promising haematocrit independence. Patients’ samples were also used to test
caffeine and paraxanthine with promising results showing quantification accuracy (Velghe
and Stove, 2018). An alcohol biomarker was also compared to whole blood indicating close
agreement (Beck et al., 2018). All studies performed have been, however, small proof-of-
concept studies, in which samples were generated using a lab pipette. Therefore, larger
scale validation and clinical trial testing is required.
1.5.4Mitra®devicepoweredbyvolumetricabsorptivemicrosampling(VAMS®)
technology
Volumetric absorptive microsampling (VAMS®) technology has been derived from the DBS
method but does not maintain the traditional filter paper format (Figure 1.7). The
technology uses an absorbent polymeric tip. The Mitra® device collects an accurate volume.
The tip of the device should touch the droplet of blood and 10 µL, 20 µL or 30 µL of blood is
collected (Neoteryx, 2019).

- 31 -
Figure1.7Mitra®devicepoweredbyvolumetricabsorptivemicrosampling(VAMS®)technologyforcollectionandstorageofblood.Copyright: Taken from Neoteryx (2019).
One study looked at the accuracy during sample collection and recommended careful
handling of the device, as any contamination, such as touching the tip, could alter the
volume precision (Denniff and Spooner, 2014). Haematocrit dependency was also tested
indicating volume precision at various haematocrit levels; however, several studies
indicated the possible issue with analyte recovery with increased haematocrit (De Kesel et
al., 2015, Mano et al., 2015). Evidence suggests that optimisation of extraction protocol
might be necessary in order to move from standard filter paper substrate to the polymeric
substrate in some workflows. Additional steps, such as sonication, increasing temperature
or use of different reagents can improve recovery (Velghe et al., 2019). From an analytical
perspective, this device has been tested by many research groups (Volani et al., 2017, Fang
et al., 2018, Ye and Gao, 2017); however, further studies are required to show applications
including clinical trials.
1.5.5HemaPEN®
HemaPEN® is a hand-held pen-like device designed for accurate blood collection, storage
and transport of DBS. The hemaPEN® enables an individual to collect a pre-defined volume
of blood. Blood is wicked into device’s tip along four capillaries onto four secure, pre-
punched FDA approved Whatman 903TM or PerkinElmer 226 paper discs 3.5 mm in
diameter. The capacity of each capillary is 2.74 μl. Therefore, the total volume of blood

- 32 -
collected by each pen is 10.92 μl. At this stage of the developmental process, a hemaPEN®
prototype is available for a trained person (e.g., phlebotomist, nurse, or the donor) who will
collect a drop of fresh finger prick blood. Figure 1.8 shows the principle of collection using
the hemaPEN®(Trajan Scientific and Medical, 2020).
Figure1.8ThehemaPEN®bloodcollectionandstoragedevice.Copyright: Taken from Trajan Scientific and Medical (2020).
The hemaPEN® eliminates some of the challenges in DBS. Firstly, blood is collected via
borosilicate capillaries allowing accurate volume collection. Blood is further deposited onto
a filter paper where the entire spot is then analysed eliminating the non-homogeneity bias
and the sub-punch bias. Its intuitive design allows easier and quicker collection. The blood
volume required for one hemaPEN® to be collected successfully (equivalent to 4 x 3mm
punches) is around 25 µL (pea size drop) with minimal biological sample wastage.
This device also eliminates the possible inconsistency in collection if sample is correctly
collected (the capillary is filled) blood is then deposited onto the filter paper allowing
uniform, high quality samples. Dried samples stay within the device preventing any
contamination and desiccant present in the hemaPEN® allows faster drying time. This device
is in developmental stage and validation studies are required to test it. In this thesis, the aim
is to test the hemaPEN® from both the collection and analytical perspective, to demonstrate
its usability and accuracy of such blood collection device.

- 33 -
1.6Studyrational
Blood is a key biological sample for clinical diagnosis and research, however, current
methods are often invasive, creating a burden for patients, the cost of collection and sample
transport is extensive as cold chain logistics are required. Alternative method, DBS has been
proven to eliminate some of the disadvantage of standard blood collection, however, by
switching from fresh blood to dry storage some issues were introduced. Such issues are
mainly associated with the analytical performance where the inaccuracy in the volume
collected, together with naturally occurring phenomenon haematocrit and the quality of
sample collected create challenges in quantification for downstream applications. The
current use of DBS in clinical setting is in newborn screening where semi quantification is
acceptable as errors in metabolism result in a large spike. To allow for quantitative analysis
and therefore, expand the use of DBS in clinical setting good quality sample with accurate
volume collected is required. The hemaPEN® device has been developed to eliminate the
volume inaccuracy around DBS and to allow easier and better sample collection. The
hemaPEN® is a novel device with a need for validation and clinical testing.
The use of DBS have recently expended from metabolic detection to genomics, including
epigenetics. Such studies are showing great potential in both research and clinical
applications as DNA can be extracted from DBS. However, the DNA recovery has been
outlined as a challenge with the need for protocol optimisation. The current use of filter
paper in newborn screening is FTA approved Whatman 903TM There are other filter paper
designed for DNA extraction. Such papers claim to increase the yield extracted from filter
papers, however, few studies compared these papers to understand if selecting different
filter paper can increase the DNA yield.

- 34 -
1.7Studyaims,hypothesisandresearchquestions
In this thesis a new blood collection device – hemaPEN® - that uses the DBS concept was
tested. This device aims to improve the collection and storage of blood samples to ensure
better sample quality and further accurate quantitative analysis. In this thesis the device will
be tested from different perspectives and for different downstream applications.
A. Collecting perspective
Aim 1: To test the collection usability of the new device in the Pathology centre at the Royal
Children’s Hospital.
Hypothesis 1: Written and video training material will be sufficient for the collectors at the
Royal Children’s Hospital to use the hemaPEN® and no one-on one training will be required.
Research Questions:
1. Can phlebotomists at the Royal Children’s Hospital use the device without training
(given video and written instructions)?
2. Are the video and instructions sufficient mode of training?
3. Would the phlebotomists use the device?
4. What is the general feedback form volunteers and phlebotomists?
B. Analytical perspective
Aim 2: To compare the standard dried blood spot workflow with the new device in well-
established newborn screening at the Victorian Clinical Genetics Services.
Hypothesis 2: No difference between the two methods (hemaPEN® and standard DBS) will
be observed at standard male haematocrit level across different concentration levels in the
newborn screening workflow.
Research Questions:
1. Can we use hemaPEN® in the standard NBS workflow?
2. Is there difference between the standard dried blood spot method in comparison to
the new device?
3. Does the hemaPEN® affect the NBS workflow? If yes, How?
4. Is the hemaPEN® fit for analytical purpose?
5. Does capillary collection affect the recovery?

- 35 -
Aim: 3: Explore the haematocrit bias in the dried blood spot using the new device in a
newborn screening workflow.
Hypothesis 3: There will be difference between the methods (hemaPEN® and standard DBS)
across haematocrit levels with hemaPEN® showing haematocrit independency across all
analytes in the newborn screening workflow.
Research Questions:
1. Is there a volume difference between the methods across haematocrit levels?
2. Can collecting accurate volume remove the haematocrit bias?
Aim 4: Test the DNA extraction from dried blood spots using different extraction methods
and different substrates (synthetic vs non-synthetic) to achieve high DNA yield.
Hypothesis 4: The synthetic substrate tested will outperform the non-synthetic cellulose
substrates for an optimized DNA extraction protocol.
Research Questions:
1. Can we optimise the DNA extraction protocol for DBS?
2. Is there a difference between different extraction kits used?
3. Can we improve DNA yield by changing the substrate used?

- 36 -
Chapter2:GeneralMethods

- 37 -
Chapter 2: General Methods This study was approved by the Health Research Ethics Committee, Royal Children’s
Hospital, Melbourne Australia HREC – LNR/17/RCHM/411. Protocol development and
ethical submission was done as part of the project timeline (see appendix 8.3 for approval
letter).
2.1AssistedhemaPEN®collection
The total of six consented phlebotomists from the Royal Children’s Hospital Pathology
Collection Centre ranging from less to very experienced (2.5-26 years) were selected. Each
phlebotomist received an instruction manual (see appendix 8.4) and video prior to
collection (see appendix 8.5). On the day, each collector watched the video and then
practiced hemaPEN® based blood collection. After practice, one collection was performed
from a donor’s finger. Both practice and donor collection were performed in a standard
pathology collection room. After the hemaPEN® collection, participants (donors and
collectors) were asked to fill out a questionnaire to provide feedback. Collection was also
recorded. No in-person training or feedback were provided. Two researchers observed the
collection (masters student and Trajan Scientific representative) even if mistakes were
made. The role of the two researcher was purely to record and observe the collection.
2.1.1Instructionmanual,videoandpracticecollection
Each collector received written instructions and a video prior to collection. The instruction
manual described all six steps recommended for hemaPEN® use. These steps include
opening the package, preparing for use, collecting blood sample (both from a finger and an
micro sampling tube), capping the hemaPEN®, flipping the hemaPEN® and repackaging (see
appendix 8.4). The instruction manual did not include any recommendations on the
technique of finger pricking. All collectors were advised to follow the standard method used
at the pathology collection centre for finger pricking including the lancet type.
The instruction video demonstrated all six steps in a visual manner (see appendix 8.5). The
video also included some tips on what NOT to do during the collection. The key tips were
collection angle - no collection at 45°, no collection for less than 10 seconds, and no tapping
to provoke capillary transfer.

- 38 -
Practice collection involved collection from a flat surface of a micro sampling tube to get the
feel of hemaPEN® use, especially the capillary collection and capping. Researcher pipetted
25 µL of EDTA blood onto the lid of a micro sampling tube. The tube was then given to the
collector who collected the blood droplet using hemaPEN®. In total each phlebotomist
practiced collection 3 times (Figure 2.1).
Figure2.1PracticehemaPEN®collectionfromanflatsurfaceofmicrosamplingtube.Copyright 2020 by Trajan Scientific and Medical. Printed with permission.
2.1.2hemaPEN®collectionfromadonor
After practice collection each collector had to collect one hemaPEN® from a donor’s finger.
The donor was seated and the collector initiated the collection as per standard practice
(Figure. 2.2). The standard DBS lancet that was used at the Royal Children’s Hospital
Pathology Collection Centre was the Safety-lancet super - purple, volume 100-500 µL
(Sarstedt, Numbrecht, Germany) .Total of five collectors participated in this study. One
consented donor did not show up on the day.
Figure2.2ThehemaPEN®collectionfromdonor’sfinger.Copyright 2020 by Trajan Scientific and Medical. Printed with permission.

- 39 -
2.2AnalyticaltestingofstandardDBSandhemaPEN®attheVictorian
ClinicalGeneticsServices(VCGS)NewbornScreeningLaboratory
2.2.1Preparationofelevenconcentrationlevelsacrosstwentysevenanalytes
Two six millilitre lithium heparin tubes (BD Vacutainer®, Oakville, Canada) were collected
from a consented healthy male volunteer. Venous blood was pooled and inverted 10 x
before separating again into two equal six millilitre aliquots (native and spiked). One aliquot
was spiked with 250µL mixture containing amino acids and carnitines, which was obtained
from the VCGS newborn screening laboratory. The total concentration of exogenous
amount that was added into the blood is outlined in Table 2.1. Spiked aliquot contained
both endogenous and exogenous amount of analytes. Samples were mixed for 20 minutes
on a rotator using the slowest speed. After the mixing step the native and spiked aliquots
were used to generate 11 different aliquot levels. Samples were stored in a glass vacutainer.
Each level was an increment of 10 (Level 1-11) i.e. Level 1 (native) contained no spike
amount, Level 11 (spike) the highest spiked concentration level. Once all levels were
aliquoted samples were placed on a rotator for 20 minutes on the slowest speed.

- 40 -
Table2.1Theexogenousconcentration(µmol/l)forbothaminoacidsandcarnitinesthatwasspikedintowholeblood.
Analyte Spiked amount in µmol/l
Analyte Spiked amount in µmol/l
Gly 695.0 C0 86.9 Ala 1739.3 C2 87.0 Val 696.2 C3 17.4 Ile 1740.2 C4 8.7 Orn 351.9 C5:1 8.7 Lys-Gln 695.1 C5 8.7 Met 347.37 C6 8.7 Phe 1739.2 C8 8.7 Gly-Pro 43.4 C 10 8.7 Arg 174.2 C12 8.7 Cit 173.7 C14 8.7 Tyr 869.6 C16 17.4 Homocitrulline 43.2 C18 8.7 Arginosuccinate 43.7
Gly, glycine; Ala, alanine; Val, valine; Ile, l-isoleucine; Orn, ornithine; Lys-Gln, Lysyl-Glutamine;
Met, methionine; Phe, phenylalanine; Gly-Pro, Glycyl-L-proline; Arg, arginine; Cit, citrulline;
Tyr, tyrosine; homocitrulline, N(6)-carbamoyl-L-lysine; Arginosuccinate, L-Argininosuccinic
acid; C0, free carnitine; C2, acetylcarnitine; C3, propionylcarnitine; C4, butyryl-
/isobutyrylcarnitine; C5.1, tiglylcarnitine; C6, hexanoylcarnitine; C8, octanoylcarnitine; C10,
decanoylcarnitine; C 12, dodecanoylcarnitine; C14, tetradecanoylcarnitine; C16,
palmitoylcarnitine; C18, stearylcarnitine.
The lid from the glass vacutainer was used as a surface to collect blood using the hemaPEN®
(Figure 2.3). The hemaPEN®’s four capillaries were brought into contact with the blood and
collection was initiated. Following filling of the capillaries ( ~ 10 sec) the hemaPEN® was
clicked into the base as per hemaPEN® collection instructions (appendix 8.4 step 4), which
brought the paper cartridge into contact with the outlet of each capillary, initiating transfer
of the blood onto the paper disc (Whatman 903TM). Each capillary collects 2.74 µL of blood,
therefore, for each hemaPEN®, four 2.74 µL of blood was transferred onto individually pre-
punched paper of 3.5 mm in diameter was collected. Two hemaPEN® devices were
generated for each level and left to dry overnight. The next day, all hemaPEN® devices were
manually opened and samples were transferred from hemaPEN® cartridge onto standard 96
well microplates.

- 41 -
Figure2.3BloodcollectionusinghemaPEN®fromthelidofaglassvacutainer.Copyright 2020 by Trajan Scientific and Medical. Printed with permission.
For the standard DBS method, eight spot for each level were generated by pipetting 50 µL of
blood onto Whatman 903TM. Standard DBS were dried overnight. The next day, a 3.2 mm
punch was punched out using DBS punch platform (PerkinElmer, Massachusetts, United
States). For each standard DBS spot one punch was punch in the middle and one in the
periphery (Figure 2.4 a). All samples were punched out directly onto the microplate and
sorted to ensure minimal carryover.
Figure2.4StandardDBSpunchpositionusingthePerkinElmerplatforma)one3.2mmpunchinthecentreandone3.2mmpunchintheperipheryona50µLspotforaveragemaleHCTb)one3.2mmpunchinthecentreandthree3.2mmpunchesintheperipheryona75µLofbloodfor63%HCT.Copyright 2020 by Lada Staskova . Printed with permission.
a
b

- 42 -
2.2.2Preparationoffivehaematocrit(HCT)levels
Two six millilitre lithium heparin tubes (BD Vacutainer®, Oakville, Canada) were obtained
from a consented volunteer. The HCT was measured using a HemoCue Hb 201+ (HemoCue,
Angelholm, Sweden) from the venous blood collected. Five different HCTs were prepared
manually by centrifugation and adjustment of the plasma levels to prepare 25%, 35%, 42%,
55%, 63% HCT (measured by HemoCue Hb 201+). Prior to the preparation of each HCT,
Octanoyl-L-Carnitine was added to plasma to prepare a working solution and an identical
volume was added to each HCT to achieve a final concentration of 5 mmol/L.
A total of 25 µL per HCT sample was dispensed onto the cap of a micro sampling tube and
the device’s four capillaries were brought into contact with the droplet (see appendix 8.4
step 3 – Blood collection from tube). Eight hemaPEN® devices were generated for each HCT.
The hemaPEN® devices were dried overnight and then manually disassembled. The four 3.5
mm pre-punched spots were manually transferred from hemaPEN® cartridge to standard 96
well microplate.
A total of 75 µL per HCT was dispensed by a pipette onto the four spots of the newborn
screening (Whatman 903TM) filter paper. Samples were dried overnight in a fume hood. For
each standard DBS spot four 3.2 mm semi-automated punches (one in the centre and three
in the periphery, Figure 2.4b) were prepared using the DBS punch platform (PerkinElmer,
Massachusetts, United States) and dispensed into the corresponding microplate.
Samples were randomised, however, DBS were grouped by HCT and organised from lowest
HCT to the highest HCT to eliminate any carryover. A total of 20 analytes were further
analysed across the five different HCT (Val, Ala, Gly, Ile, Orn, Met, Phe, Arg, Cit, Tyr, C0, C2,
C3, C4, C5, C6, C8 , C10, C16, C18)
The blood area covered on the filter paper for the hemaPEN® was calculated using image
processing and analysis Java program called ImageJ version 1.8.0. (public domain: LOCI,
University of Wisconsin) in which a photo of each sample was taken with a scale reference.
The boundaries of each spot were outlined by the program and the area of the blood spot
was expressed in mm2.

- 43 -
2.2.3AnalyticalprocedureattheVCGSNewbornScreeningLaboratory
The following process was conducted at the VCGS and all consumables were used from
routine stock supply at the newborn screening laboratory. Samples in the microplates were
extracted by adding, 150 µL of methanol containing internal standards. Deuterated internal
standard contained labelled amino acids and carnitines. Samples were incubated in the
methanol solution for 1 hour on a shaker at room temperature. The total volume was then
transferred into a new microplate well. After transfer, samples were air dried for 20 minutes
on a dry block heater set to 65°C. Once dried, 50 µL of butanolic HCl was added followed by
incubation for 30 minutes at 65°C. Samples were sealed to prevent evaporation. After this
derivatization process, samples were air dried and reconstituted in 200 µL acetonitrile
(ACN), the mobile phase and placed on shaker for 5 minutes at room temperature. A total of
10 µL was injected into a triple quadrupole mass spectrometer by flow injection analysis
(FIA-MS/MS) (Waters Acquity TQ detector, Waters Acquity UPLC FTN Sample manager,
Waters Acquity UPLC Binary solvent manager). Amino acids, carnitines and equivalent
internal standards were detected in positive ionization multiple reaction monitoring (MRM).
MS/MS data were generated using the peak ratio of known concentration of internal
standard of a particular analyte to calculate the concentration of each analyte. For each run,
five point calibration curve for each analyte was used to determine the final concentration
in μmol/L.
2.2.4.Statisticalanalysis
Statistical analysis was performed using R Studio version 1.3.1056 (R Studio Team 2020,
Integrated Development Environment for R., PBC, Boston, MA).
Principle component analysis (PCA) was computed by scaled “prcomp” function as part of
the R ‘stats' package (R Core Team 2020). The scores for PC 1 and PC 2 were plotted using
the “ggplot2 “ package (Wickham, 2016).
The Passing Bablock regression analysis was calculated across 11 concentration levels and
plotted using the “mcreg” function within the “mcr” package (Manuilova and
Schuetzenmeister, 2014). The mean difference between the methods (reference method -
standard DBS, new method – hemaPEN®) across 11 concentration levels was calculated with

- 44 -
the Bland-Altman framework using a website with the R programming framework (Bahar,
2020).
The biplot across five HCTs with 20 analytes was calculated and plotted using scaled
“fviz_pca_biplot” function within the “Factoextra” package (Kassambara and Mundt, 2020).
A simple linear regression model (one way variance analysis) using the “lm” function from
the R ‘stats' package (R Core Team 2020) was performed for each analyte and haematocrit.
All P-values were then adjusted using the p.adjust function with the Bonferroni method (n =
105) (Armstrong, 2014) from the R ‘stats' package (R Core Team 2020).
Haematocrit dependency for each method was calculated by adjusting all values as
percentage change from the average value at HCT 42% for each analyte as previously done
by (Abu-Rabie et al., 2015) . The mean and standard error of the percentage change data set
were calculated using “summarySE” function withing the “Rmisc" package (Hope, 2013) and
plotted using “ggplo2t” package.
The exogenous recovery of each analytes across 11 concentration levels was calculated
using Microsoft Excel (Version 16.35) and reported for level 2,8 and 11 to capture the
overall trend. The endogenous level was calculated based on the level 1 values for each
method and analyte and then the mean for each method and analyte was used to calculate
the recovery of the exogenous (spike level) for all levels.
2.3ComparisonofextractionkitsandsubstratestomaximiseDNA
yield
2.3.1Driedbloodspotpreparationondifferentsubstrates(syntheticandnon-
synthetic)
One ten millilitre EDTA tube (BD Vacutainer®, Oakville, Canada) was collected from a female
healthy volunteer. Venous blood was processed within one hour of collection. To generate
DBS, 40 µL was pipetted onto paper-based substrates and 12 µL of blood onto the synthetic
substrate. For the non-synthetic cellulose-based the following substrates were used:
WhatmanTM FTATM Gene (GE Healthcare, United States), WhatmanTM FTATM Elute (GE
Healthcare, United States), Whatman 903TM (GE Healthcare, United States), GenSaverTM 2.0
(Ahlstrom-Munksjö, Finland) and GenCollectTM 2.0 (Ahlstrom-Munksjö, Finland). A specially

- 45 -
designed synthetic substrate, methacrylate porous polymer monolith (mPPM) was
developed by Trajan Scientific and Medical (Australia) for storage blood microvolumes
(Ferreira Neto et al., 2019).
All samples were dried in a fume hood overnight and stored in a polystyrene box each
covered in a paper bag to avoid contamination. Samples were stored without a desiccant.
For paper-based substrates, four punches were taken (one in the middle and three in the
periphery). For mPPM one monolith was used per extraction. All samples were extracted
within the first month of collection. For each substrate and extraction kit, eight technical
replicates were extracted.
The reasoning behind the volume pipetted onto the monolith was to ensure similar blood
volumes used for extraction. From previous research it has been reported that 1 x 3.00 mm
punch from DBS contained around 3µL of blood (Di Caprio et al., 2015). Therefore, one
extraction with four punches is roughly equal to 12µL of blood.
2.3.2DNAextractionkitsandprotocol
Three different extraction kits were tested in this study: Quick-DNA™ Miniprep Plus Kit
(Zymo Research, United States), QIAamp® DNA Blood Mini (Qiagen, Germany) and
MagMAX™ CORE Nucleic Acid Purification Kit (Life Technologies Thermo Fisher Scientific,
United States). The extraction protocol was developed based on previous research done by
Mohandas et al. (2018), Cruickshank et al. (2013) and manufacturer’s instructions (for
detailed protocols see appendix 8.2). The protocol included the following steps: pre-
treatment (incubation and mechanical beads), lysis & protein digestion, purification (column
vs magnetic beads) and elution.
Beads and the lysis solution were ordered separately. For Quick-DNATM protocol, 2 mm
Bashing Beads (Zymo Research, United States) and the lysis solution (Zymo Research, United
States) were used. For QIAamp® protocol, one 3 mm Tungsten bead (Qiagen, Germany) and
Buffer ATL Tissue Lysis (Qiagen, Germany) was used. For MagMAX™ CORE, PK Buffer (Life
Technologies Thermo Fisher Scientific, United States) and one 3 mm Tungsten bead as per
QIAamp DNA kit was used.
All cellulose substrates were incubated overnight in 360 µL of PBS, 40 µL of Proteinase K (20
mg/mL) (Sigma-Aldrich, Germany) and beads at 37°C. Next day 400 µL of lysis solution was
added, and samples were homogenized in a Tissue lyser (Qiagen, Germany). Next steps

- 46 -
were as per the manufacture instructions. Supernatant was then passed through a column
or magnetic beads were added. As the final step elution buffer was applied to recover DNA.
Column purification was performed in Quick-DNA™ and QIAamp®. Magnetic beads
purification step was performed in MagMAX™ CORE, where manual magnetic stand was
used.
The synthetic substrate, mPPM was not soaked overnight; instead, the lysis solution and
beads were added followed by tissue lyser homogenization to break up the solid matter and
further extraction described above. Due to the difference in elution volumes across
extraction kits all samples were reconstituted in 40 µL of 1xTE using Savant SpeedVac
(Thermo Fisher Scientific, United States).
2.3.3Qualitycontrolmeasures
To assess the quantity of the DNA the concentration was recorded using a Qubit
fluorometer V 2.0 (Thermo Fisher Scientific, United States). To look at the quality of DNA the
260/280 ratio and 230/260 was measured using Nanodrop (Thermo Fisher Scientific, United
States). Further, 1% agarose gel that was made manually in the laboratory based on
previously published protocol by Lee et al., (2012) was performed to look at the
fragmentation of extracted gDNA.
2.3.4Statisticalanalysis
The difference between the groups mean was calculated and plotted using the “ggboxplot”
function within the “ggpubr” package (Kassambara, 2020). The two-way ANOVA test was
calculated using “stat_compare_means” function within the R ‘stats' package (R Core Team
2020) to determine the overall p-value.A pair wise comparison was calculated using the
“stat_compare_means” function withing the R ‘stats' package (R Core Team 2020) with the
t-test method. Values recorded were adjusted for multiple testing.

- 47 -
Chapter3:USABILITYCOMPARISONBETWEENANEWPATIENT-CENTRIC
DEVICEANDTHESTANDARDDBSMETHODINNEWBORNSCREENINGWORKFLOW

- 48 -
Chapter 3: Usability comparison between a new
patient-centric device and the standard DBS method
in newborn screening workflow 3.1Introduction
Blood is a biospecimen that can be used to identify changes in metabolic profiles for early
disease detection or for diagnosis of a specific disease (Stringer et al., 2015, Fiehn and Kind,
2007). The blood collection is often invasive, unfavourable to many, and the collection
requirements are complex including commuting burden for the patient. Collecting blood
from an arm is a skill that requires training and can only be done by a health professional
(Ialongo and Bernardini, 2016). Due to the reliance on health professionals and current
invasive methods of collecting blood from an arm, venous blood collection can be a
challenge, especially, where multiple timepoints are required or collection is required from
the elderlies or infants whose veins are often difficult to find (Lim, 2018).
Dried blood spot (DBS) sampling eliminates some of these challenges as the collection
process requires less blood without the need for vein incision. Current DBS collection
involves a finger prick in adults or heel prick in children using a lancet, after which a drop of
blood is collected onto a filter paper (Mei, 2014). A world-wide screening scheme has been
utilising DBS as the sampling method for clinical detection of metabolic inborn errors. This
so-called newborn screening was introduced in 1960 by Guthrie to identify single metabolic
error and to reduce the morbidity and mortality in children (Guthrie and Susi, 1963). Initially
this program identified newborns with phenylketonuria (PKU), which causes decreased
metabolism of phenylalanine. Untreated PKU can cause seizures, behavioural problems and
mental disorders (Bickel et al., 1953). Treatment of this inborn error consists of a strict diet
and regular monitoring of phenylalanine levels (Williams et al., 2008). The newborn
screening program is now implemented around the world with a wide range of metabolites
detected including essential amino acid and carnitines for various different inborn errors
(Bhattacharya et al., 2014). Samples are collected within 48 hours of birth and any inborn
error is detected as a multi-fold increase in targeted analyte concentration (McHugh et al.,

- 49 -
2011). Apart from the use of DBS in newborn screening and some serology tests such as
measles, mumps and viruses, this sampling method is still not commonly used for clinical
diagnosis (Gupta and Mahajan, 2018).
A poor quality DBS sample can occur when a sample is collected from a patient who is
lacking a stable blood flow, which can happen especially in infants or elderlies (George and
Moat, 2016, Panchal et al., 2016). It has been reported that a 4-5% rejection rate has been
identified for DBS collected by phlebotomists (Zakaria and Greaves, 2019). The rejection
rate is even higher for at-home DBS sample collection (Veenhof et al., 2017, Al-Uzri et al.,
2017). The contamination of samples has also been reported to have substantial impact on
the final result in newborn screening (Winter et al., 2018). Furthermore, the unknown
volume collected on the card creates additional problems especially for quantitative
downstream metabolic analysis (Spooner, 2010).
In this chapter, the blood collection using the hemaPEN® is described and compared to the
standard DBS method. The hemaPEN® allows for easier and cleaner blood collection due to
its intuitive pen-like design (see section 1.5.5 for further details).The hemaPEN® was tested
in assisted collection where qualified phlebotomists used the device to collect blood from a
single donor. Further, the chapter tests the analytical performance of hemaPEN® in the
newborn screening workflow. Eleven concentration levels with twenty-seven analytes
routinely screened for at the VCGS were detected and compered between the standard DBS
and hemaPEN® (see specific section of chapter 2 outlined below).
3.2.Methods
See the following sections from chapter 2: GENERAL METHODS
2.1Assisted hemaPEN® collection
2.1.1 Instruction manual, video and practice collection
2.1.2 hemaPEN® collection from a donor
2.2 Analytical testing of standard DBS and hemaPEN® at the Victorian Clinical Genetics
Services (VCGS) Newborn Screening Laboratory
2.2.1 Preparation of eleven concentration levels for twenty seven analytes
2.2.3 Analytical procedure at the VCGS newborn screening laboratory
2.2.4. Statistical analysis

- 50 -
3.3.Results
The total of six qualified phlebotomists from the Royal Children’s Hospital tested the
hemaPEN®. Each collector received the instruction manual, video and three practice
hemaPEN®. After practice, one hemaPEN® device was collected from a donor’s finger.
Feedback from all participant was reported.
3.3.1AssistedhemaPEN®collection
Four collectors improved the capillary transfer from the first practice to the final donor’s
collection from a finger (Table 3.1 ). Collector Two and Three successfully transferred all four
capillaries from the donor’s finger. Collectors One and Four managed to transfer three out
of four capillaries. Collector Five and Six worsened in capillary transfer during the donor’s
collection compared to the practice. Collector number Five who failed collection panicked
during the collection due to a high bleeding volume. Collector Six successfully transferred
only one out of four capillaries due to the failure during capping.
Table3.1Capillarytransfer,inpercentage,(successfulblooddepositfromcapillarytothepre-punchfilterpaperwithinthehemaPEN®) forpracticehemaPEN®devicesandonecollectionfromdonor’sfingerforallsixcollectors.
The time length, in seconds, of collection was assessed from the video captured during the
donor’s collection. The time length was divided into 7 categories, from alcohol wipe through
capping and repacking (Table 3.2). The average time from alcohol wipe to flipping the
hemaPEN® was only 26 seconds. The repacking required after collection was the most time
consuming part of the process with the mean time of 47 seconds. With the exception of one
collector where the device was not placed back into the plastic base cover which resulted in
short repacking of 6 seconds.
Collector Practice 1 Practice 2 Practice 3 Donors
1 0 50 75 75
2 50 75 25 100
3 100 75 100 100
4 0 75 75 75
5 100 100 75 0
6 75 25 100 25

- 51 -
Table3.2Thetime,inseconds,takenforeachcategory(alcoholwipe,lancetfingerprick,gauze,samplecollection,cappingofhemaPEN®, flippingofhemaPEN®,totaltimefromwipingoffingertoflippingthehemaPEN®andthetimetakentoplacehemaPEN® inasamplebag).Timerecordedwasassessedfromthevideorecordedduringthedonor’scollection.
Collector Alcohol
wipe
Lancet
finger prick Gauze Collect Cap flip
Wipe to
flip
repacking
1 11 3 2 7 4 NA 16 6
2 9 6 2 7 2 4 21 50
3 11 10 3 12 5 2 32 60
4 13 3 2 10 4 2 21 60
5 8 8 4 13 4 3 32 60
6 14 5 2 17 7 3 34 43
average 11 6 3 11 4 3 26 47
3.3.1.1MainfeedbackfromphlebotomistsonhemaPEN®use
The feedback showed that five collectors found the practice very useful (Figure 3. 1).
Furthermore, the video was reported to be more beneficial in comparison to the written
instructions. When asking what form of learning was the most preferred, video scored the
highest followed by the practice hemaPEN® session. This feedback indicated that practicing
hemaPEN® use prior to collection as well as video aids can improve the usability of the
device.
Written feedback outlined that visual aids are preferred for those whose first language is
not English. A split feedback was observed where three of the six phlebotomists would
consider the use of hemaPEN® in the paediatric setting and three would not. Those that
would not use hemaPEN® in a paediatric setting has been in the field for 4.5 - 26 years. The
main reason for not using the hemaPEN® was that collectors were happy with the standard
DBS method and did not see why hemaPEN® was needed; to them the device was
“sophisticated” and “fancy”. Those who would use the hemaPEN® in the paediatric setting
ranged from 2.5 - 5 years of experience. The reason given was that it is quick to collect, not
messy and the blood requirement is minimal. When using hemaPEN® in the paediatric

- 52 -
setting, a comment was raised that holding the device on infant’s foot might be a challenge.
Despite unsuccessful collection, Collector Five liked the device and explained that with
practice, better collection was possible (for full summary of the feedback see appendix 8.6).
Figure3.1MainfeedbackfromhemaPEN®practiceandvolunteer’sassistedcollectionforsixphlebotomistsfromtheRoyalChildren’sHospitalranginginexperience.
3.3.1.2MainfeedbackfromdonorsonhemaPEN®use
One out of the five donors reported to feel anxious when having blood collected from an
arm (Figure 3.2). None of the donors reported that the finger prick was painful during
collection. All five donors indicated that they could prick the finger themselves with further
four out of the five donors being able to collect the sample using the hemaPEN® device.
Three out of the five would prefer the hemaPEN® over standard collection from the arm,
however, when the concept of frequent collection/at home (given the appropriate training)
was introduced, all five participants would prefer to use hemaPEN®(Figure 3.2).
Feedback from donors revealed that all donors liked the device due to the “intuitive design
with easy use” and “non-hazardous properties” (for the summary of the entire feedback see
appendix 8.6).

- 53 -
Figure3.2MainfeedbackfromfivevolunteersfromhemaPEN®collectionexperienceandtheuseofhemaPEN®incomparisontostandardvenouscollection
3.2.2Methodcomparison(standardDBSandhemaPEN®)atelevenconcentration
levelsusingthenewbornscreeningworkflow
3.2.2.1Overalltrendbetweenthetwomethodsacrosselevenconcentrationlevels
usingprinciplecomponentanalysis
The two methods (hemaPEN® and standard DBS) were compared in the newborns screening
workflow across 11 different analyte concentration levels. Samples were analysed at the
VCGS, using routine NBS clinical processes, where the metabolite concentration levels were
detected for 27 analytes. A PCA plot for the overall data was performed to examine a trend
between the methods (Figure 3.3). PC 1 represented 91.22% of the total variance and PC 2
represents 3.78%, therefore, the first two principal components represent 95% of the total
variance. PC 1 separates the data set according to the metabolite concentration levels,
lower levels in the negative PC 1 scores and higher levels in the positive PC 1 scores. The
lower metabolite concentration levels (level 1-5) showed overlap indicating no difference
between the two methods. At higher metabolite concentration levels (level 5-11), however,
the difference between methods was observed where this difference was separated by PC
2.

- 54 -
Figure3.3Principlecomponentanalysis(PCA)plotfor27analytesbetweentwomethods(hemaPEN® in triangles and standard DBS in circles) across different concentrationlevels.Eachlevelanincrementof10(Level1-11)i.e.Level1(native)containednospikeamount andLevel11 (spike) containedmaximumspikeamount. For each level, eighttechnicalreplicatespermethodwasgraphedrepresenting27analytes.3.2.2.2Methodcomparisonfortwenty-sevenanalytesmeasured
A positive percentage mean difference was observed for all analytes (except
Arginosuccinate) indicating that greater analyte concentration was recorded from
hemaPEN® in comparison to the standard DBS method (Table 3.3). The percentage mean
difference between the methods across 11 concentration levels was larger in some analytes
more than others. Twelve analytes (Gly, Ala, Val, Met, Phe, Tyr, Homocitrulline, C0, C12,
C14, C16, C18) indicated a between-method difference lower than 15%. Ornithine showed
mean difference of 28%, higher in hemaPEN®. Arginine appeared to have the greatest mean
difference between the two methods with, 49% higher in hemaPEN®. Similarly, most
−2.5
0.0
2.5
−10 −5 0 5 10PC1
PC2
HTC1
2
3
4
5
6
7
8
9
10
11
deviceg
h
method
Level
PC2
3.78
%
PC1 91.22%
Standard DBS hemaPEN®

- 55 -
carnitines showed a mean between-methods difference of around 20%. Lysyl-Glutamine
exhibited difference of 46%; however, the linear correlation with increased concentration
levels was 0.19 indicating low reproducibility for both methods. The only opposite trend was
observed for Arginosuccinate, where the mean difference was - 43% indicating a lower level
of recovery for hemaPEN® with linear correlation of 0.88.

- 56 -
Table3.3TheBland-Altmanmeandifference(in%),Pearsoncorrelation,theslopeandinterceptofPassing-BablokregressionbetweenhemaPEN®andstandardDBSacross11different concentration levels for all 27 analytes using the newborn screening. Thepercentagedifferencebetweenpunchlocation(peripheryminuscentre)inthestandardDBSmethodforall27analytesusingthenewbornscreeningworkflow.
*highlighted analytes with less than 0.9 correlation ** highlighted analytes with slope and 95% CI grater then 1 *** highlighted analytes with intercept and 95% CI not equal 0
Analyte
Mean difference between method (in %)
Pearson correlation* Slope [95%CI]** Intercept [95% CI]***
Punch location
mean difference
Gly 11 0.95 1.13 [1.05, 1.2] -0.5 [-39.6, 31.9] 7%
Ala 15 0.97 1.11 [1.06, 1.18] 22.6 [-20.4, 57.5] 5%
Val 14 0.97 1.08 [1.03, 1.15] 19.4 [-21.4, 60.3] 5%
Ile 16 0.98 1.09 [1.05, 1.14] 26.8 [2, 56.8] 5%
Orn 28 0.86 1.7 [1.5, 2] -118.2 [-182.8, -58.4] 14%
Lys-Gln 46 0.19 0.73 [0.49, 1.10] 46.1 [16.45, 65.67] 28%
Met 13 0.96 1.14 [1.08, 1.22] -0.5 [-7.3, 5.7] 5%
Phe 9 0.98 1.06 [1.03, 1.1] 3.8 [-11.6, 30.0] 6%
Gly-Pro 5 0.84 0.99 [0.9, 1.10] -0.4 [-2.2, 1.8] 29%
Arg 49 0.95 1.65 [1.54, 1.76] -3.7 [-6.5, -0.3] 13%
Cit 8 0.64 0.92 [0.75, 1.13] 5.7 [-19.3, 28.5] 28%
Tyr 6 0.99 1.04 [1.01, 1.07] 4.5 [-1.8, 14.9] 7%
HomoCitrulline 6 0.96 1.03 [0.98, 1.08] 0.0 [-0.6, 0.9] 14%
Arginosuccinate -43 0.88 0.54 [0.46, 0.61] -0.4 [-1.4, 0.9] 13%
C0 12 0.98 1.14 [1.1, 1.18] 1.0 [-4.5, 2.4] 6%
C2 32 0.96 1.22 [1.16, 1.29] 1.1 [0.1, 1.9] 5%
C3 26 0.98 1.26 [1.21, 1.31] 0.0 [-0.2, 0.1] 6%
C4 20 0.99 1.21 [1.18, 1.25] 0.01 [-0.04,.02] 6%
C5.1. 20 0.99 1.23 [1.2, 1.26] 0.0 [-0.06, 0.07] 10%
C5 19 0.99 1.20 [1.17, 1.24] 0.0 [-0.06, 0.04] 8%
C6 17 0.99 1.15 [1.12, 1.18] 0.01 [-0.02, 0.05] 7%
C8 17 0.99 1.14 [1.11, 1.16] 0.04 [0.01, 0.07] 13%
C10 19 0.99 1.19 [1.12, 1.23] 0.00 [-0.04, 0.05] 16%
C12 14 0.99 1.18 [1.14, 1.21] -0.02 [-0.06, 0.06] 17%
C14 13 0.98 1.14 [1.101, 173] -0.01 [-0.05, 0.1] 14%
C16 9 0.98 1.11 [1.08, 1.15] -0.10 [-0.24, 0.01] 11%
C18 4 0.97 1.07 [1.03, 1.12] -0.04 [-0.09, 0.01] 9%

- 57 -
3.2.2.3Randomerrorinthenewbornscreeningworkflow
The workflow precision was assessed by summarising the results from the Pearson
correlation where linear correlation with increasing concentration level was measured
between the two methods hemaPEN® and standard DBS (Table 3.3). Five analytes (Orn, Lys-
Gln, Gly-Pro, Cit, Arginosuccinate) showed a Pearson correlation less than 0.9 indicating
some imprecision in the newborn screening workflow (Figure 3.4). Lysyl-Glutamine showed
the lowest correlation of 0.19, indicating a very poor linear relationship with increasing
concentration levels for both methods. Second lowest Pearson correlation was Citrulline
with 0.64 showing moderate imprecision in the workflow. For Ornithine, Glycyl-Proline and
Arginosuccinate the correlation was raging from 0.83 - 0.87 indicating lower yet still
acceptable correlation with increasing concentration levels.
The coefficient of variation was also examined for both hemaPEN® and standard DBS (Table
3.4) . For level 1 (the endogenous level) the coefficient of variation was higher in
comparison to level 11 in most analytes for both hemaPEN® and standard DBS indication
that low endogenous levels were at the border of detection. Analytes with lower Pearson
correlation exhibited on average higher coefficient of variation confirming the imprecision in
the extraction and recovery of the analytes in the newborn screening workflow.

- 58 -
Figure3.4Passing-Bablokregressionplotsbetweentwomethods(standardDBS–xaxis,hemaPEN®-yaxis)formeasuredanalytes(Gly,Ala,Val,Ile,Orn,Lys-Gln,Met,Phe,Gly-Pro,Arg,Cit,Tyr,HomoCitrulline,Arginosuccinate,C0,C2,C3,C4,C5.1,C5,C6,C8,C10,C12,C14,C16,C18)across11concentrationlevels.Linearregressionlinerepresentedin blue with 95% confident bounds. Line of identity (slope = 1) indicating methodalignmentinreddottedline.
15
3.4 Supplementary data
300 400 500 600 700 800
200
400
600
800
1000
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.955
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.53 + 1.13 * guthrie
identity
Glycine
300 400 500 600 700 800
200
400
600
800
1000
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.955
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.53 + 1.13 * guthrie
identity
200 400 600 800 1000 1200
200
400
600
800
1000
1200
1400
Passing Bablok Regression Fit
guthriehe
ma
Pearson's r = 0.971
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
22.59 + 1.11 * guthrie
identity
Alanine
200 400 600 800 1000 1200
200
400
600
800
1000
1200
1400
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.971
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
22.59 + 1.11 * guthrie
identity
200 400 600 800 1000 1200
200
400
600
800
1000
1200
1400
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.97
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
19.43 + 1.09 * guthrie
identity
Valine
200 400 600 800 1000 1200
200
400
600
800
1000
1200
1400
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.97
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
19.43 + 1.09 * guthrie
identity
200 400 600 800 1000 1200 1400
500
1000
1500
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.983
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
26.82 + 1.09 * guthrie
identity
200 400 600 800 1000 1200 1400
500
1000
1500
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.983
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
26.82 + 1.09 * guthrie
identity
I-isoleucine
150 200 250 300 350 400 450 500
100
200
300
400
500
600
700
800
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.861
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−118.19 + 1.72 * guthrie
identity
Ornithine
150 200 250 300 350 400 450 500
100
200
300
400
500
600
700
800
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.861
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−118.19 + 1.72 * guthrie
identity
50 100 150 200 250
050
100
150
200
250
300
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.961
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.52 + 1.14 * guthrie
identity
Methionine
50 100 150 200 250
050
100
150
200
250
300
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.961
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.52 + 1.14 * guthrie
identity
0 200 400 600 800 1000 1200 1400
050
010
0015
00
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.985
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
3.8 + 1.06 * guthrie
identity
Phenylalanine
0 200 400 600 800 1000 1200 1400
050
010
0015
00
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.985
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
3.8 + 1.06 * guthrie
identity
50 100 150
5010
015
020
025
0
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.191
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
46.12 + 0.73 * guthrie
identity
Lysyl Glutamine
50 100 150
5010
015
020
025
0
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.191
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
46.12 + 0.73 * guthrie
identity

- 59 -
16
0 10 20 30 40 50 60 70
020
4060
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.838
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.43 + 0.99 * guthrie
identity
0 10 20 30 40 50 60 70
020
4060
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.838
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.43 + 0.99 * guthrie
identity
Glycyl Proline
10 20 30 40 50 60
2040
6080
100
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.954
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−3.71 + 1.65 * guthrie
identity
Arginine
10 20 30 40 50 60
2040
6080
100
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.954
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−3.71 + 1.65 * guthrie
identity
50 100 150 200 250
050
100
150
200
250
300
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.641
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
5.69 + 0.92 * guthrie
identity
Citrulline
50 100 150 200 250
050
100
150
200
250
300
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.641
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
5.69 + 0.92 * guthrie
identity
100 200 300 400 500 600
100
200
300
400
500
600
700
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.99
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
4.51 + 1.04 * guthrie
identity
Tyrosine
100 200 300 400 500 600
100
200
300
400
500
600
700
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.99
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
4.51 + 1.04 * guthrie
identity
0 10 20 30 40 50
010
2030
4050
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.96
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0 + 1.03 * guthrie
identity
0 10 20 30 40 50
010
2030
4050
Passing Bablok Regression Fit
guthrie
hema
Pearson's r = 0.96
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0 + 1.03 * guthrie
identity
Homocitrulline
0 10 20 30 40 50 60
010
2030
40
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.878
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.39 + 0.54 * guthrie
identity
Arginosuccinate
40 60 80 100 120 140
4060
8010
012
014
016
0
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.984
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−1 + 1.14 * guthrie
identity
Free Carnitine
40 60 80 100 120 140
4060
8010
012
014
016
0
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.984
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−1 + 1.14 * guthrie
identity
10 15 20
1015
2025
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.956
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
1.05 + 1.22 * guthrie
identity
C2 Carnitine
10 15 20
1015
2025
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.956
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
1.05 + 1.22 * guthrie
identity
C2 carnitine

- 60 -
17
1 2 3 4 5
12
34
56
7
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.981
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.03 + 1.26 * guthrie
identity
C3 Carnitine
1 2 3 4 5
12
34
56
7
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.981
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.03 + 1.26 * guthrie
identity
0.5 1.0 1.5 2.0
0.5
1.0
1.5
2.0
2.5
3.0
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.99
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.01 + 1.21 * guthrie
identity
C4 Carnitine
0.5 1.0 1.5 2.00.
51.
01.
52.
02.
53.
0
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.99
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.01 + 1.21 * guthrie
identity
0 1 2 3 4 5 6
02
46
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.992
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0 + 1.23 * guthrie
identity
0 1 2 3 4 5 6
02
46
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.992
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0 + 1.23 * guthrie
identity
C5.1 Carnitine
0 1 2 3 4
01
23
45
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.992
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0 + 1.2 * guthrie
identity
0 1 2 3 4
01
23
45
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.992
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0 + 1.2 * guthrie
identity
C5 Carnitine
0.0 0.5 1.0 1.5 2.0 2.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.993
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0.04 + 1.14 * guthrie
identity
C8 Carnitine
0.0 0.5 1.0 1.5 2.0 2.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.993
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0.04 + 1.14 * guthrie
identity
0.5 1.0 1.5 2.0 2.5 3.0
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.991
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0 + 1.19 * guthrie
identity
C10 Carnitine
0.5 1.0 1.5 2.0 2.5 3.0
0.00.5
1.01.5
2.02.5
3.03.5
Passing Bablok Regression Fit
guthrie
hema
Pearson's r = 0.991
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0 + 1.19 * guthrie
identity
0.0 0.5 1.0 1.5 2.0 2.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.993
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0.01 + 1.15 * guthrie
identity
C6 Carnitine
0.0 0.5 1.0 1.5 2.0 2.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.993
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
0.01 + 1.15 * guthrie
identity
0 1 2 3 4 5
01
23
45
6
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.985
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.02 + 1.18 * guthrie
identity
C12 Carnitine
0 1 2 3 4 5
01
23
45
6
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.985
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.02 + 1.18 * guthrie
identity

- 61 -
18
0 1 2 3 4
01
23
45
6
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.981
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.01 + 1.14 * guthrie
identity
0 1 2 3 4
01
23
45
6
Passing Bablok Regression Fit
guthrie
he
ma
Pearson's r = 0.981
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.01 + 1.14 * guthrie
identity
C14 Carnitine
2 4 6 8
02
46
810
12
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.979
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.1 + 1.11 * guthrie
identity
2 4 6 8
02
46
810
12
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.979
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.1 + 1.11 * guthrie
identity
C 16 Carnitine
0.5 1.0 1.5 2.0 2.5 3.0 3.5
12
34
Passing Bablok Regression Fit
guthrie
hem
a
Pearson's r = 0.972
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.04 + 1.07 * guthrie
identity
0.5 1.0 1.5 2.0 2.5 3.0 3.5
12
34
Passing Bablok Regression Fit
guthrie
he
ma
Pearson's r = 0.972
The 0.95−confidence bounds are calculated with the bootstrap(quantile) method.
Passing Bablok RegressionFit (n=88)
−0.04 + 1.07 * guthrie
identity
C 18 Carnitine

- 62 -
Table 3.4 The coefficient of variation (in %) for the endogenous level (Level 1) inhemaPEN® (H)andstandardDBS (D).Thecoefficientofvariation (in%) for the totalconcentration(exogenousandendogenous)forlevel2,8,11forhemaPEN®andstandardDBS.Therecovery(in%)forlevel2,8,11inhemaPEN®andstandardDBSadjustedfortheendogenousamount(Level1)usingthenewbornscreeningworkflow.
analyte
Level 1 Level 2 Level 8 Level 11
CV (in %)
CV (in %)
Recovery (in%)
CV (in %)
Recovery (in%)
CV (in %)
Recovery (in%)
H D H D H D H D H D H D H D
Gly 13 10 14 5 100 98 4 8 91 85 5 6 94 84
Ala 15 23 9 7 89 79 3 7 65 60 4 9 63 53
Val 26 31 11 8 132 116 8 10 145 136 8 7 151 135
Ile 22 31 8 6 90 80 3 9 75 70 3 4 76 67
Orn 12 15 20 10 113 96 7 12 98 79 14 10 118 79
Lys-Gln 28 60 13 59 53 58 20 26 20 16 16 35 17 15
Met 17 19 18 10 88 82 4 12 76 65 8 8 73 64
Phe 15 23 9 9 89 81 4 8 78 77 5 6 80 75
Gly-Pro 55 20 26 41 126 123 9 8 108 110 1 17 105 116
Arg 11 12 19 10 67 47 6 11 42 29 13 8 49 30
Cit 44 34 24 43 89 95 11 9 77 82 19 17 76 88
Tyr 9 10 9 6 88 83 3 7 72 70 5 3 73 70
HomoCitrulline 33 47 15 13 120 100 10 8 105 101 13 11 104 103
Arginosuccinate 40 40 35 20 56 102 24 12 52 97 9 19 57 101
Free 6 7 6 7 108 106 2 6 117 110 3 5 120 108
C2 5 7 3 8 63 58 5 8 30 25 5 7 28 22
C3 5 7 4 8 62 56 3 8 37 30 3 6 35 28
C4 13 11 5 8 45 39 3 6 31 26 3 6 30 25
C5.1. 56 48 8 8 90 72 3 7 78 64 2 5 78 62
C5 13 17 6 7 64 54 3 6 53 45 2 5 52 43
C6 17 35 7 8 41 36 3 6 34 30 2 4 34 29
C8 10 20 5 9 46 38 2 8 34 30 2 5 34 30
C10 11 22 8 7 49 43 2 9 36 32 3 5 36 30
C12 38 39 7 9 73 64 5 10 64 55 7 5 63 53
C14 15 31 8 7 67 62 7 10 58 52 8 4 59 50
C16 8 13 6 8 70 68 13 12 57 50 8 3 55 49
C18 11 7 7 12 57 60 12 11 40 39 8 4 39 35

- 63 -
3.2.2.4Systematicerror(constantandproportional)
The method agreement was represented by two measures the intercept (constant bias) and
slope (proportional bias). The 95% confidence intervals for the intercept were assessed. Six
(Ile, Orn, Lys-Gln, Arg, C2 and C8) of the 27 analytes showed between-method difference
where the confidence intervals values did not include zero (Table 3.3). The two carnitines,
C2 and C8 were slight above zero with the confidence intervals [0.1, 1.9] and [0.01, 0.07],
respectively. These results suggest that for these six analytes, a constant systematic error
was present across the two methods.
The slope, together with the corresponding 95% confidence intervals was evaluated for all
analytes (Table 3.3). Between-method Difference were present for 23 of 27 analytes with
the confidence interval not including the slope of 1 (the identity line), suggesting that
methods are not in an agreement. This significant difference suggests proportional
difference between the method where the difference is greater at higher concentration
levels. The proportional bias (increase with increased quantity) was larger in the higher
concentration levels, which was also seen in the overall trend for the PCA analysis.
3.2.2.5Punchlocation(peripheryandcentre)inthestandardDBSmethod
The effect of punch location on analyte concentration was compared between hemaPEN®
and DBS. The percentage mean difference (periphery minus centre location) are shown in
Table 3.3. The analyte concentration levels indicated that more analyte was recovered on
the periphery in comparison to the centre punch. This difference was more prominent in
some analytes more than others. However, when performing Student’s T-test with
Bonferroni correction (n=27) for multiple testing no significant patterns were observed
between the punch location (see appendix 8.1 for supplementary data).
3.2.2.6Analyterecoveryforeachmethodacross11differentconcentrationlevels
The analyte recovery for levels 2, 8 and 11 was calculated based on the level 1 (endogenous
amount) for each analyte and method (Table 3.4). An overall trend was seen, in which
better extraction recovery was observed at higher concentration levels for both methods.
The overall recovery for hemaPEN® was also slightly higher than in standard DBS for all
levels. Four analytes (Val, Glycyl-Proline, Homocitrulline, C0) exhibited recovery over 100%.
Interestingly, Arginosuccinate is the only analyte that showed different recovery between

- 64 -
the methods where hemaPEN® was showing half the recovery in comparison to standard
DBS.
3.3Discussion
In this chapter, the hemaPEN® used in assisted collection was compared against the
standard DBS to measure the concentration of the 27 analytes measures in the VCGS
newborn screening laboratory. Firstly, the potential utility of this device by qualified
phlebotomist was demonstrated. From a patient’s point of view this device received positive
feedback where donors would use this device in the need for frequent monitoring given
relevant training. From the collectors’ point of view, some resistance towards this device
was observed. The donor capillary transfer success rate varied with only two collectors
succeeded 100% transfer meaning that all four samples (3.2mm disks) were collected at
once. For the newborn screening only one out of the four samples is required, therefore,
only one out of the 6 hemaPEN® devices would not give result in newborns screening
setting. Furthermore, collectors were not given any one-on one training and were not
corrected during the practice if mistakes were made, therefore, higher failure rate can be
expected. Furthermore, the current recommendation for the use of hemaPEN® is to receive
training prior to clinical use of hemaPEN. Further research and development of the device is
being conducted to maximise the success rate of capillary transfer.
The two main issues observed during the donor’s collection were the estimation of the 10
second collection interval (the length of having capillary in contact with the blood droplet)
recommended followed by inadequate clicking of the device into the base and intense
bleeding during collection. The The high-volume bleeding may be controlled by the lancet
type. Lancet in this study was tailored for high blood volume collection. There are many
lancets on the market varying in the incision (needle vs blade), size, motion and how deep
they cut. The lancet criteria affect the pain as well as the bleeding volume (Grady et al.,
2014, Goto et al., 2020). Currently, there are no recommendations on what lancet type to
use for hemaPEN®, therefore, the device is compatible with different collection centres and
health professionals. It might be beneficial to look at how the lancet size affects bleeding,
pain and give some recommendation on the size of lancet with the hemaPEN® use.

- 65 -
This collection study is the first study to be conducted with the hemaPEN® providing first
insights in the collection workflow of this device.
The main limitations of this study is the small number of phlebotomists and donors tested,
therefore, the results and feedback reported are preliminary. The donor participation in this
small study were recruited at the Royal Children’s Hospital, therefore, possible bias in
usability where people have medical education or experience must be outlined. Further
studies will need to be done to validate assisted collection across multiple collection centres
with varying donor population demographics.
As with the analytical performance, a positive correlation between the methods across
linearly increasing concentration levels was demonstrated indicating good workflow
reproducibility. The between-method difference was larger in some analytes more than
others, with consistently higher concentration recovered in hemaPEN®. Twelve analytes
indicated a between-method difference lower than 15%, which has been reported as
acceptable by the international regulatory agencies (Shah et al., 2000, Denniff and Spooner,
2010). The between-method difference can be explained by the volume collected in both
methods. The hemaPEN® collects known volume of 2.74 µL per punch, however, the volume
used in the DBS can differ. The punch volume in standard DBS can differ substantially based
on the patient’s HCT, the amount of red blood cells in total volume of blood (Denniff and
Spooner, 2010). The HCT level for average male, which was used as the reference point for
this study, ranges between 40 - 54 % (Walker et al., 1990). In the newborn screening
workflow, it has been reported that 3.0 mm punch spot collected on Whatman 903TM paper
contains ~ 3µL of blood (Li and Tse, 2010). Hewawasam et al. (2018) further reported that at
50.1% HCT, a 3.0 mm punch contains 3.1 µL of blood and a 53% HCT contains 3.2 µL volume
of sample (Hewawasam et al., 2018). The same group also reported the volume of 2.2 µL
from a 3.0 m punch based on average HCT range (Hewawasam et al., 2018). For 3.2 mm
punch the volume reported was around 2.6 - 2.8 µL in the HCT range of 40 - 45% (Hall et al.,
2015). Therefore, the difference observed in this study can be predominantly due to the
volume difference between the methods where more blood was collected in the hemaPEN®
in comparison to the standard DBS method. Therefore, these results could indicate that the
3.2 mm punch contains less than 2.74 µL of blood. The limitation of this study is that the

- 66 -
HCT was not measured at the time of collected, therefore, it is hard to estimate the volume
collected In the DBS method.
The volume of blood used to create the overall spot can further contribute to the punch
volume. Hall et al. (2015) showed that by reducing the spot volume from75 µL to 50 µL, a
4% punch volume reduction was observed (Hall et al., 2015). Furthermore multiple spotting
of blood on the same spot can also result in higher volume per punch (Hall et al., 2015).
When two times 20 µL was applied, the 3.0 mm spot volume changed from 1.4 µL to 1.6 µL
(Hewawasam et al., 2018); however, this was compared using different filter paper.
The next factor that can affect the volume and that is the filter paper used to generate the
sample. Even though in this study, same type of paper was used (Whatman 903TM paper), it
has been reported that in many newborn screening laboratories the thickness can differ
based on the storage conditions and, therefore, further impact the volumetric accuracy (Mei
et al., 2010).
The literature suggests that punch location can affect the concentration recovered (O'Mara
et al., 2011). With the hemaPEN®, the whole spot that is collected is further analysed
removing sample heterogeneity. In the standard DBS workflow, the distribution of blood on
the filter paper is heterogenous (Uribe et al., 2019). Therefore, punching a spot in the
middle vs in the periphery can alter the final concentration. In this study, the mean
difference between punch locations was observed, however, the difference was not
statistically significant when corrected for multiple testing. Heterogeneity has been reported
to be more prominent in samples with lower HCT (20 - 35 %) (Holub et al., 2006). A UK study
showed that the concentration recovered from a punch taken in the periphery was greater
by up to 35 % with the effect being noticeable when larger volumes (50 – 70 µL) are applied
onto the filter paper (Lawson et al., 2016). In summary, evidence suggests that total spot
volume, punch size, punch position and filter paper properties can affect the volume of the
spot.
The extraction recovery in this study showed some variability across analytes with both
methods. Extraction bias has been discussed as a component of DBS bias because the form
of the sample storage involves moving from a fresh state (venous as the gold standard) to

- 67 -
dry a state. In the newborn screening workflow, the same extraction process is used to
detect all metabolites (De Jesús et al., 2010). Therefore, the recovery of some analytes will
be better than for others. In some cases, more elaborate extraction protocols with
increasing temperatures, altering solvents or addition of sonication steps, for example, can
help to make the extraction process more efficient (De Jesús et al., 2010, Odoardi et al.,
2014, la Marca et al., 2012). For newborn screening, metabolite levels are multi-fold higher
in those with an inborn error in metabolism compared to healthy individual. Therefore, even
if there is a statistically significant variability across samples from healthy individual, this
doesn’t necessarily have an impact on clinical decisions i.e. not clinically significant. For
example, the healthy range of phenylalanine is around 50 µmol/L with the cut off for PKU
ranging around 97-135 µmol/L (McHugh et al., 2011). Therefore, higher noise i.e. higher
variability in the assay is acceptable. Furthermore, multiple metabolites of the same
pathway are detected. A good example is maple syrup urine disease (a rare genetic disorder
characterized by deficiency of enzymes that break down specific branch amino acid -
leucine, isoleucine, and valine) where I-isoleucine and valine are measured to detect this
disease (McHugh et al., 2011). Despite some imprecision in the workflow it is very crucial to
eliminate any false negatives as later detection can result in great stress to the child
development (Kwon and Farrell, 2000).
In conclusion, the newborn screening allowed to assess hemaPEN® performance testing in a
clinical setting because the current workflow already uses the DBS method. Between-
method difference in metabolite concentrations at the average haematocrit between the
two methods was observed, which was larger at the higher metabolite levels. As a limitation
of this study the haematocrit level was not measured in this chapter, however, the large
sample size with eleven concentration level measured across 27 analytes in the clinically
used, world- wide newborn screening workflow provides high data confidence. The
difference observed can be mainly due to the different volume collected within each
collection method, as there are many factors that can affect the volume in the standard DBS
which has been discussed in this chapter. A strong correlation was observed across
increasing analyte concentration levels for both methods, indicating good method
reproducibility. As discussed, newborn screening is designed to detect multi-fold changes in
concentration and therefore, some noise in data for healthy individuals is acceptable with

- 68 -
has no impact on the clinical decisions. Moreover, to test the analytical accuracy and
precision in hemaPEN®, further testing is recommended especially in workflows with low
data noise requirements.

- 69 -
CHAPTER4:EFFECTOFACCURATEVOLUMECORRECTIONONTHE
HAEMATOCRITBIASINANEWBORNSCREENING
WORKFLOW

- 70 -
Chapter 4 Effect of accurate volume correction on the
haematocrit bias in a newborn screening workflow 4.1Introduction
In recent years the dried blood spot (DBS) method, which involves depositing a small (~ 50
µL) amount of blood onto the filter paper, has gained prominence in research and clinical
settings, suggesting its use as an alternative to venous collection (Su et al., 2018). Despite
benefits such as lower costs, lower reliance on health professionals and the overall
collection being less invasive, it is still not widely used in clinical settings (Lim, 2018). The
main limitation of the DBS method is the semi quantitative bioanalysis it provides, which
makes subsequent clinical diagnosis challenging (Denniff and Spooner, 2010).
The main component contributing to the inaccuracy in bioanalysis via DBS is a natural
occurring phenomenon called the haematocrit (HCT) (Timmerman et al., 2011). Haematocrit
refers to the volume of red blood cells in a set volume of blood expressed as a percentage. It
has been estimated that the HCT range in adult females is between 36 – 44 %, in adults
males it is 41-50 %, and in children from 1 month to 2 years, 28 – 55 % (Denniff and
Spooner, 2010). The HCT can further differ across the population based on age, gender and
ethnicity (Lim et al., 2015, Mahlknecht and Kaiser, 2010).
Furthermore, the viscosity of blood positively correlates with the HCT (Pasquini et al., 1983),
which affects the dispersion of blood on the filter paper when DBS method is used. In the
standard analytical process a punch ranging from 3 to 6 mm in diameter is taken often from
the centre of the blood spot. This so called sub-punch undergoes liquid extraction to isolate
the target analyte before downstream analysis (Dénes et al., 2012). Due to the HCT effect,
when a sub-punch is taken from a sample with a high HCT, a larger proportion (higher
volume) of sample is taken for downstream analysis, creating issues in analyte
quantification (Hall et al., 2015). Volume estimation for a 3 mm punch is approximately 3.1
μL at 50.1% HCT and 3.2 μL at 53% HCT (Holub et al., 2006). Furthermore, the distribution of
cells and analytes on DBS paper is not uniform across the whole blood spot. It has been

- 71 -
demonstrated that some accumulate on the periphery of the spot (Hall et al., 2015)
therefore, the punch location matters.
There are two approaches to nullify the HCT bias 1) post-analysis - by determining the HCT
and further adjusting the results accordingly and 2) pre-analysis - depositing an accurate
volume of blood onto the filter paper followed by whole punch analysis. For the post
analysis approach Oostendrop et al. (2016) outlined the promising use of near-infrared
spectroscopy to calculate the area of the spot and further estimate the HCT. Furthermore,
De Kesel et al. (2013) used Potassium [K+] levels in the spot to determine the HCT. The post-
analytical methods create an additional steps during analysis with data adjustments and
doesn’t solve the punch heterogeneity issue associated with the sub punch analysis. The
pre-analytical approach requires an accurate volume of blood to be collected onto the filter
paper followed by whole spot analysis. This approach is widely used in pharmacokinetic
studies where animal models are required and the accurate collection is often done using
capillary tube coated with anticoagulant which is controlled by experienced scientists
(Aabye et al., 2013, Stokes et al., 2011). Collecting an accurate volume onto the filter paper
creates extra step in the collection making it unattractive in clinical setting (Abu-Rabie et al.,
2015). The accurate volume collection combined with the whole spot analysis eliminates the
volume and heterogeneity issue making it an approach worth investigating.
A new device is being developed combining the use of capillary for collection and the dry
blood spot method for storage. The device called hemaPEN® is a single-use device
introducing easy yet precise collection with the potential for clinical use. This device
requires less blood (~ 11 µL) in comparisons to standard DBS (~ 50 µL). Samples are stored
within the device, preventing any contamination (Trajan Scientific and Medical, 2020).
In this chapter, the new hemaPEN® device with standard DBS sub-punch process across five
different HCTs is compared. Samples were tested in the newborn screening laboratory at
the VCGS where the concentrations of 20 analytes were detected that are otherwise
detected in routine testing for errors in metabolism in newly born babies (Victorian Clinical
Genetics Services, 2020). The effects of accurate volume collection and whole spot analysis
using the hemaPEN® in comparison to the standard DBS process was explored.
Furtheremore the hemaPEN® was tested to assess if the known HCT bias associated with

- 72 -
volume and heterogeneity for the targeted analyte can be eliminated (see specific section of
chapter 2 outlined below).
4.2Method
See the following sections from chapter 2: GENERAL METHODS
2.2 Analytical testing of standard DBS and hemaPEN® at the Victorian Clinical
Genetics Services (VCGS) Newborn Screening Laboratory
2.2.2 Preparation of five haematocrit (HCT) levels
2.2.3 Analytical procedure at the VCGS newborn screening laboratory
2.2.4. Statistical analysis

- 73 -
4.2Results
4.2.1Dispersioneffectsassociatedwithhaematocrit
A blood dispersion effect was seen when collecting 2.74 µL of blood onto a pre-punched 3.5
mm spot (Figure 4. 1). The size of the area covered by blood was negatively associated with
the haematocrit. At low HCT the full pre-punch spot was covered with blood (Figure 4.1 b)
The surface area covered by blood for four spots was on average 9.34 mm2 at HCT 25%.
Furthermore the surface area from four spots was on average around 6.99 mm2 at HCT 63%.
The area of the standard DBS spot also changed with increased HCT. Even though the spot
size was not measured in mm2 as this has been previously reported by Hall et al., (2015) the
spot size being smaller at higher HCT for standard DBS was also observed (Figure 4.1 c).
Figure4.1BlooddispersionoffiveHCT(25%,35%,42%,55%,63%)onWhatman903TMfilterpapera) inhemaPEN®cartridgewiththeaveragesurfaceareacoveredbybloodcalculatedinmm2b)zoomedinindividualpre-punchhemaPEN®sampleswith2.74µLofblooddepositedviacapillarytransferc)standardDBSfilterpaperwith75µLofbloodapplied.4.2.2ComparisonofstandardDBSandhemaPEN®acrossfivehaematocritlevels
for20analytesdetectedinthenewbornscreeningworkflow
A PCA plot for the overall dataset was used to visualise the two largest components of
sample variation in the data (Figure 4.2). PC 1 and PC 2 represented 76.42 % and 22.06% of
sample variation across the dataset, respectively. In total the first two principal components
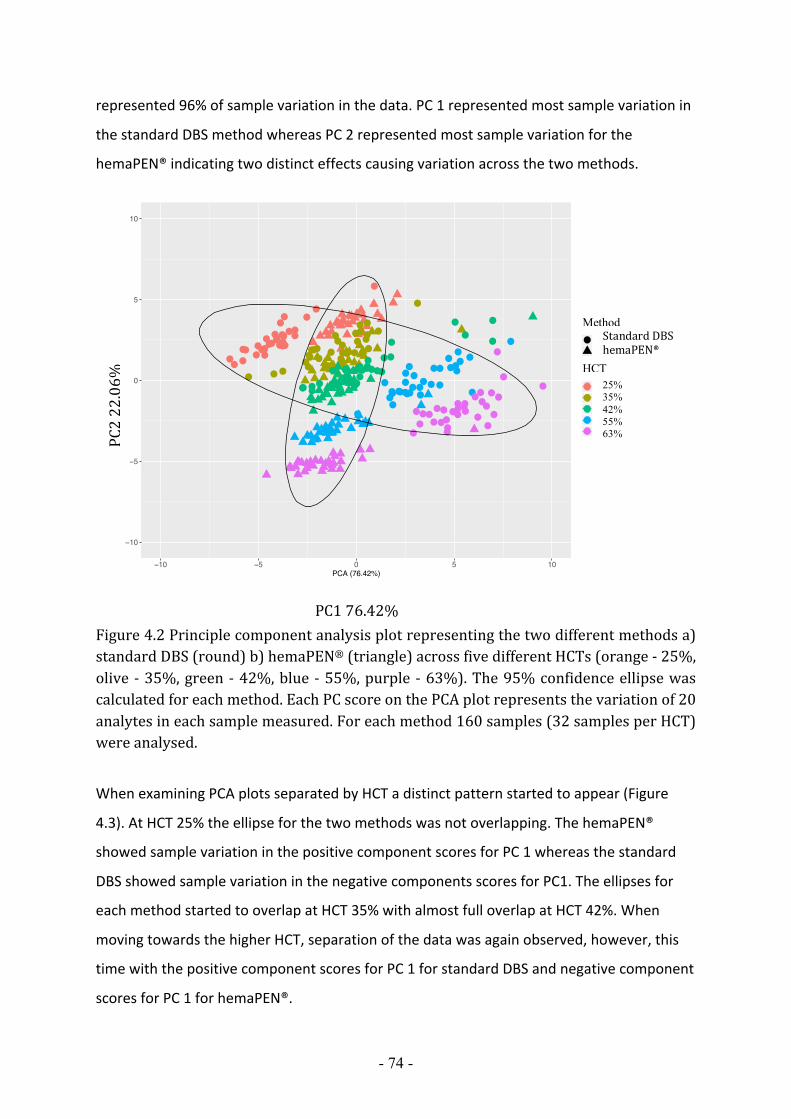
- 74 -
represented 96% of sample variation in the data. PC 1 represented most sample variation in
the standard DBS method whereas PC 2 represented most sample variation for the
hemaPEN® indicating two distinct effects causing variation across the two methods.
Figure4.2Principlecomponentanalysisplotrepresentingthetwodifferentmethodsa)standardDBS(round)b)hemaPEN®(triangle)acrossfivedifferentHCTs(orange-25%,olive -35%,green-42%,blue-55%,purple -63%).The95%confidenceellipsewascalculatedforeachmethod.EachPCscoreonthePCAplotrepresentsthevariationof20analytesineachsamplemeasured.Foreachmethod160samples(32samplesperHCT)wereanalysed.
When examining PCA plots separated by HCT a distinct pattern started to appear (Figure
4.3). At HCT 25% the ellipse for the two methods was not overlapping. The hemaPEN®
showed sample variation in the positive component scores for PC 1 whereas the standard
DBS showed sample variation in the negative components scores for PC1. The ellipses for
each method started to overlap at HCT 35% with almost full overlap at HCT 42%. When
moving towards the higher HCT, separation of the data was again observed, however, this
time with the positive component scores for PC 1 for standard DBS and negative component
scores for PC 1 for hemaPEN®.
−10
−5
0
5
10
−10 −5 0 5 10PCA (76.42%)
PCA
(22.
06%
)
deviceG
H
HTC1
2
3
4
5
PCA analysis for hemaPEN and Guthrie card across all hematocrit levels
StandardDBShemaPEN®
Method
PC222.06%
PC176.42%
25% 35% 42% 55% 63%
HCT

- 75 -
Figure 4.3 Individual principle component analysis plots separated by HCT (25%, 35%, 42%,
55%, 63%). Each plot represents data separation between two methods tested - standard DBS
(red) and hemaPEN® (green). The 95% confidence ellipse was calculated for each method.
Each PC score (dot) on the PCA plot represent the variation of 20 analytes in each sample
measured. For each method and haematocrit 32 samples (8 samples with 4 replicates) were
analysed.
The mean difference between the methods was also tested statistically for each analyte and
haematocrit (Table 4.1). For HCT 25%, 14/20 analytes were showing significant (P <0.05)
mean difference between the methods. The positive difference at HCT 25% indicated that
greater amount was recovered for all analytes in hemaPEN® compared to standard DBS with
an average mean difference around 12%. For HCT 42%, all analytes except Ornithine,
showed no significant difference in the mean between the methods with the mean
difference on average around 4%. At HCT 63%, 18/20 showed significant difference, this
time with negative mean difference indicating that greater amount was recovered for
analytes using the standard DBS compared to hemaPEN®. On average, the difference was
around 23%.

Analyte 25 35 42 55 63
|% Δ| Δ p-value |% Δ| Δ p-value |% Δ| Δ p-value |% Δ| Δ p-value |% Δ| Δ p-value
Val 12% 14.3 2.1x10-14 2% 2.6 NS 3% -3.8 NS 11% -11.2 2.4x10-13 18% -18.6 2.1x10-14
Ala 12% 20.5 2.1x10-14 0% 0.8 NS 3% -4.3 NS 11% -17.5 2.3x10-13 18% -28.1 2.1x10-14
Gly 16% 46.3 2.1x10-14 3% 10.1 NS 2% 6.8 NS 8% -23.8 5.8x10-07 10% -31.3 1.6x10-05
Ile 12% 10.3 2.1x10-14 1% 1.2 NS 3% -2.5 NS 12% -10.4 NS 19% -16.2 2.1x10-14
Orn 6% -6.7 NS 18% -18.3 4.8x10-11 22% -20.8 4.1x10-05 29% -28.0 2.1x10-14 37% -31.2 NS
Met 10% 1.3 5.1x10-07 9% 1.1 1.1x10-03 2% -0.2 NS 6% -0.7 NS 13% -1.4 3.5x10-04
Phe 10% 3.6 2.1x10-14 1% -0.4 NS 5% -1.7 NS 16% -5.6 4.6x10-13 19% -6.6 3.4x10-13
Arg 10% -0.5 2.3x10-02 4% -0.2 NS 5% -0.2 NS 20% -0.8 5.0x10-06 21% -0.8 NS
Cit 7% 1.9 NS 1% 0.3 NS 5% -1.2 NS 19% -4.3 1.6x10-11 25% -5.0 2.1x10-14
Tyr 7% 3.2 NS 5% -2.1 3.7x10-02 6% -2.5 NS 16% -6.4 2.1x10-14 21% -8.2 2.3x10-14
C0 9% 2.3 2.1x10-14 4% -1.0 3.6x10-02 2% -0.4 NS 13% -2.5 4.01x10-14 18% -3.5 2.1x10-14
C2 29% 1.5 2.1x10-14 18% 1.2 NS 1% 0.06 NS 2% -0.2 NS 18% -1.7 6.5x10-14
C3 19% 0.1 NS 16% 0.2 5.4x10-13 3% 0.03 NS 1% 0.01 NS 11% -0.2 1.5x10-06
C4 2% 0.00 NS 0% 0.00 NS 5% -0.01 NS 27% -0.04 2.53x10-13 47% -0.06 2.1x10-14
C5 15% 0.01 5.7x10-11 5% 0.00 NS 1% 0.00 NS 7% -0.01 4.8x10-01 15% -0.01 7.8x10-08
C6 14% 0.02 6.5x10-13 1% 0.00 NS 3% 0.00 NS 19% -0.02 3.1x10-10 42% -0.03 2.8x10-10
C8 15% 0.6 2.1x10-14 1% 0.03 NS 5% -0.20 NS 18% -0.7 2.1x10-14 36% -1.3 2.1x10-14
C10 17% 0.2 2.1x10-14 2% 0.02 NS 3% -0.02 NS 14% -0.09 7.9x10-10 37% -0.18 2.1x10-14
C16 8% 0.02 3.1x10-05 2% -0.01 NS 3% -0.01 NS 2% -0.01 NS 16% -0.08 1.0x10-08
C18 6% 0.01 NS 0% 0.00 NS 3% -0.01 NS 3% -0.01 NS 14% -0.05 1.5x10-09
Table4.1Theabsolutepercentagechange|%Δ|,TheactualchangeΔ(inµmol/L),andp-valueforall20analytesacrossfiveHCTs.ThedifferenceofthemeanbetweenhaematocritlevelsandanalyteswastestingusingT-test.Allp-valueswereadjustedformultipletestingusingBonferronicorrection(n=105).

4.2.3TheevaluationofstandardDBSmethodacrossfivehaematocritlevels
A biplot for the standard DBS method across five HCT levels was visualised (Figure 4.4).The
biplot allowed the visualisation of both samples (dots) and the influence of variables
(arrows). There were eleven analytes (Phe, Ala, Val, Gly, Ile, C2, C3, C5, C8, C16, C18) driven
by high values at high HCT (i.e. positive HCT dependency), represented by vectors pointing
towards the higher HCT (circle a). The opposite was observed where three analytes (C10, C6,
Arg) were driven by high values in the low HCT (i.e. negative HCT dependency) (circle c).
There were six analytes (Orn, C0, Cit, Met, Tyr, C4) showing independency to HCT (circle b).
Overall the biplot showed the influence spectrum for each analyte across HCTs for the
standard DBS, which reflect HCT dependency for each analyte. The results showed that
some analytes are more susceptible to HCT bias compared to other analytes. These results
indicated that there are other factors apart from the volumetric inaccuracy affecting HCT
bias.
Figure4.4BiplotrepresentingPCscores(samples)foreachHCT(25%,35%,42%,55%,63%)andloadingsofvariables(analytes)forthestandardDBSmethod.ThelengthofthevectorrepresentstheinfluenceoftheanalyteandthedirectionrepresentstheanalytedependencywithrespecttoHCT(positiveHCTdependency–circlea,noHCTdependency–circleb,negativeHCTdependency–circlec).
GlyAlaVal
Ile
Orn
MetPhe
Arg Cit TyrC0
C2C3
C4
C5
C6
C8
C10
C16C18
−10
−5
0
5
10
−4 0 4Dim1 (57.8%)
Dim
2 (1
8%)
Groups25
35
42
55
63
PCA − Biplot
PC1 57.8%
PC2
18%
HCT
a b c
Standard DBS

- 78 -
4.2.4TheevaluationofHemaPEN®acrossfivehaematocritlevels
A biplot for the hemaPEN® across five HCTs was visualised (Figure 4.5). Interestingly, few
analytes (C2, C16, C18 and C3) were driven by high HCT (circle a), represented by vectors
pointing towards the higher HCT, indicating HCT dependency despite accurate volume
collection. The opposite was observed for wide range of analytes where high values were
driven by the low HCT (circle c) i.e. negative dependency with increasing HCT. Four analytes
(Phe, Gly, Ala, Ile) were showing HCT independency (circle b) suggesting that HCT related
volumetric difference can be corrected but in this case can only be applied to four out of the
20 analytes detected in the study. The biplot showed the spectrum of HCT dependency
across analytes in hemaPEN®.
Figure4.5BiplotrepresentPCscored(samples)foreachHCT(25%,35%,42%, 55%,63%) and loadings of variables (analytes) for hemaPEN®. The length of the vectorrepresents the influence of the vectors and the direction represents the analytedependencywithrespecttoHCT(positiveHCTdependency–circlea,noHCTdependency–circleb,negativeHCTdependency–circlec).
No
Gly
Ala
Val
Ile
Orn
MetPhe
ArgCit
Tyr
C0
C2C3
C4
C5
C6
C8
C10
C16C18
0
5
10
−10 −5 0 5 10Dim1 (58%)
Dim
2 (1
8.6%
)
Groups25
35
42
55
63
PCA − Biplot
PC1 58%
PC2
18.6
%
HCT
a
b
c
hemaPEN®

- 79 -
4.2.5TrendsobservedacrossHCTsforbothmethods
The percentage mean difference from the middle HCT 42% was plotted for each analyte to
observe HCT dependent tendency between the two methods. (Figure 4.6). Ornithine was
not plotted as that is the only analyte that showed significant difference between the
methods at HCT 42%. In the standard DBS, a volumetric bias was found thought a
concentration increase with increased HCT. There were four analytes (Gly, Ala ,Phe, Ile) that
showed HCT dependency in standard DBS but showed no HCT dependency in the hemaPEN®
indicating that by collecting an accurate volume the HCT bias was eliminated.
However, no or negative HCT dependency was also observed in standard DBS indicating that
volumetric bias is not the only bias associated with haematocrit in standard DBS. For most
analytes measured using the hemaPEN®, some degree of negative HCT dependency was
observed indicating that by removing the volumetric bias other areas of bias were
uncovered that will need to be investigated.

- 80 -
Figure4.6PlotsrepresentingthepercentagechangefromthemiddleHCT42%acrossfiveHCTs(25%,35%,42%,55%,63%)for19analytes(Val,Ala,Gly,Ile,Met,Phe,Arg,Cit,Tyr,C0,C2,C3,C4,C5,C6,C8,C10,C16,C18).Theplotrepresentationwaspreviouslypublished by (Abu-Rabie et al., 2015).Methods (hemaPEN® and StandardDBS)wereplotted onto one graph for each analyte to observeHCTdependent trends. Error barrepresent95%confidenceintervalfromthemeanofthe32samplesmeasuredforeachHCT.
−20.0%
0.0%
20.0%
25 35 42 55 63HTC
GLY
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
Glycine
−20.0%
0.0%
20.0%
25 35 42 55 63HTC
ALA Method
Sub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
Alanine
−20.0%
0.0%
20.0%
25 35 42 55 63HTC
MET
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
Methionine
−20.0%
0.0%
20.0%
25 35 42 55 63HTC
VAL Method
Sub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
Valine
−20.0%
0.0%
20.0%
25 35 42 55 63HTC
PHE Method
Sub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
Phenylalanine
−20.0%
0.0%
20.0%
25 35 42 55 63HTC
Ile_Leu Method
Sub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
Leu-Leu-Pro-OHl-isoleucine

- 81 -
−20.0%
0.0%
20.0%
25 35 42 55 63HTC
ARG Method
Sub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
Arginine
−20.0%
0.0%
20.0%
25 35 42 55 63HTC
CITR
ULLINE
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata (
%)
Hematocrit level
Standard DBShemaPEN
Citrulline
−20.0%
0.0%
20.0%
25 35 42 55 63HTC
TYR Method
Sub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
Tyrosine
−50%
0%
50%
25 35 42 55 63HTC
C2.CAR
N
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
C2 Carnitine
−50%
0%
50%
25 35 42 55 63HTC
C3.CAR
N
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
C3 Carnitine
−20.0%
0.0%
20.0%
25 35 42 55 63HTC
FREE
.CAR
N
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
Free Carnitine

- 82 -
−50%
0%
50%
25 35 42 55 63HTC
C4.CAR
N
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
C4 Carnitine
−50%
0%
50%
25 35 42 55 63HTC
C5.CAR
N
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
C5 Carnitine
−50%
0%
50%
25 35 42 55 63HTC
C6.CAR
N
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
C6 Carnitine
−50%
0%
50%
25 35 42 55 63HTC
C8.CAR
N
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
C8 Carnitine
−50%
0%
50%
25 35 42 55 63HTC
C10.CAR
N
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
C10 Carnitine
−50%
0%
50%
25 35 42 55 63HTC
C16.CAR
N
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
C16 Carnitine

- 83 -
−50%
0%
50%
25 35 42 55 63HTC
C18.CAR
N
MethodSub−punchPre−punch
Bias
com
pare
d to
42%
hem
atoc
rit d
ata
(%)
Hematocrit level
Standard DBShemaPEN
C18 Carnitine

- 84 -
4.3DiscussionIn this chapter, the difference between the hemaPEN® and standard DBS across five
different HCTs in the newborn screening workflow was evaluated. The main aim was to
assess how the volumetric accuracy affects the HCT bias. In the middle range of HCTs, 42%,
no significant mean difference between the two methods was observed. At higher and
lower HCT, however, there was significant difference between the methods. At the higher
HCT (55%, 63%) higher analyte concentration was recovered for the standard DBS in
comparison to hemaPEN®. The reverse was observed at the lower HCT; greater analyte
concentration in hemaPEN® compared to the standard DBS. The standard DBS trend seen in
most analytes, mainly a positive increase with increased HCT, was reported in 2010 by
Denniff and Spooner (2010). The so called HCT effect was then identified as a “single most
important parameter influencing the spread of blood on DBS cards, which could impact the
validity of the results” (Fan and Lee, 2012, Timmerman et al., 2011). The HCT bias has been
responsible for the lack of quantitative analysis limiting the use of DBS in therapeutic drug
monitoring and bioanalysis (Denniff and Spooner, 2010, De Kesel et al., 2014). By collecting
accurate volumes using the hemaPEN®, these results demonstrated that volumetric bias is
one of the main factors contributing to the HCT bias and that different areas of bias such as
analyte distribution in blood and extraction bias need to be considered when dealing with
HCT bias.
4.3.1Analytedistributioninblood
In several analytes tested, a weaker yet significant positive association with increased HCT
was seen in the hemaPEN®. The interpretation of this effect can be explained by
investigating the analyte distribution in blood (Wilhelm et al., 2014). At higher HCT the
proportion of RBC is greater, thus, if a drug or analyte is associated with RBCs, more analyte
will be collected with higher HCT. By removing the volumetric bias, potential bias has been
uncovered, indicating the importance of understanding the analyte distribution in blood for
accurate quantification. Volumetric and blood bias are positively correlated with HCT,
causing even stronger HCT dependency in the standard DBS. In this chapter, the majority of
carnitines tested were showing a RBC bias in the hemaPEN®. Research has shown the
presence of carnitines and acetyl carnitines in RBC (Cooper et al., 1988) with the evidence
for function cell maintenance and cell efficiency of RBC (Toptas et al., 2006).

- 85 -
In most diagnostic tests, plasma isolated from whole blood is used rather than whole blood,
eliminating the RBC bias altogether (Sepetiene et al., 2018). Therefore, when switching from
using plasma to whole blood for quantitative analysis, additional tests may need to be
performed to understand i) analyte composition in whole blood versus plasma, ii) analyte-
specific interactions in blood and iii) additional equipment calibrations processes might be
required (Wickremsinhe et al., 2013).
Recent research has been trying to develop methods to collect dried plasma spots without
the need of centrifugation. A RBC filtration membrane has been introduced to separate
whole blood from plasma via separation science (Sturm et al., 2015). This membrane
consists of two layers the filtration membrane and collection unit. The filtration layer
comprises of at least one filtration membrane, which filters particles from 1-10 microns. The
filtration membrane has two distinct surfaces, which can vary in thickness to allow the
capture of desired particles such as RBCs (Henion et al., 2017). Once blood is applied and
sample is dried, the top layer – the filtration membrane can be peeled off to access the
collection unit where filtered dried plasma can be used for analysis. This membrane is
consistent with the workflow of standard DBS, therefore, from workflow compatibility point
of view it is promising, however, initial studies tested the haematocrit dependency showing
inconsistency when varying volumes of blood applied. Despite the volume roadblock this
approach is promising for future use in clinical settings (Li et al., 2012).
4.3.2HCTdependentextractionbias
In several analytes, the decrease in concentration with increased HCT with both methods
and in some cases, an even steeper decrease within the hemaPEN® method was observed. A
similar trend was observed by Youhnovski group, where a pre-cut, accurate volume
approach on filter paper was tested with the suggestion that extraction efficiency can differ
across HCTs (Youhnovski et al., 2011). Different extraction processes and cellulose-based
papers were tested with the recommendation that sample extraction recovery must be
tested at various HCT levels to ensure the accuracy and precision of such methods. The
same group also reported that analytes with recovery > 90% across all HCTs showed no
significant association with haematocrit-related extraction bias (Youhnovski et al., 2011).
Extraction bias associated with HCT can, therefore, be one of the contributing factors to the
HCT bias.

- 86 -
The overall analyte extraction process is labour intensive, as soaking, elution and chemical
derivatisation is required. Because blood is deposited onto the filter paper during collection,
an internal standard must be added during the extraction process, making it more
challenging to adjust for the extraction bias (Li et al., 2020). Since the method of extraction
used in this chapter is designed for screening purposes only and is uniform for all analytes, it
is reasonable to assume that this extraction is more efficient for some analytes than others.
Therefore, extraction bias can negatively associate with increased haematocrit and in some
cases counteracts the positive volumetric bias in standard DBS, nullifying the haematocrit
bias altogether. Furthermore, in the hemaPEN® the two effect (accurate volume and
haematocrit- related extraction bias) enhance the negative association with increased HCT
and further worsen the accuracy of the method.
Different approaches have been used to attempt and nullify the extraction bias (Abu-Rabie
et al., 2015). The most practical approach suggested has been to spray the internal standard
onto the dried blood sample before extraction using a spray module. (Velghe et al., 2019). A
different approach to improve extraction across HCT was identified by Hempen et al. in
which a temperature-enhanced desorption method was applied to assess the recovery of
four immunosuppressants (Hempen et al., 2015). This method improved the recovery which
was largely independent to HCT. For both approaches, the spray module and the heated
flow desorption are available commercially.
This chapter has shown promising results and outlined the key factors associated with HCT
bias to allow quantitative analysis. One of the limitations of this study is that only one
volunteer was used to investigate the HCT bias. Further studies including the use of multiple
volunteers to understand the effects of biological variation are required. Moreover,
validation of this device not only from the analytical perspective but also the collection on
infants on whom the newborns screening would provide further insight for clinical
applications. Furthermore, the workflow is designed to screen infants with metabolic inborn
errors, which results in multiple-fold increase in analyte concentrations. Since samples were
generated from a healthy volunteer, the concentration level of analytes are at the level of
detection that creates extra noise in the data set. Despite some of these limitations, the
large sample size and distinct significant HCT patterns suggest strong evidence that other

- 87 -
areas of bias associated with HCT need to be considered when using DBS for quantitative
analysis.
In Conclusion, it is clear that volume is a factor contributing to the HCT bias. By collecting
accurate volumes followed by whole spot analysis, HCT independency can be achieved for
some analytes. Creating an HCT independent workflow allows for quantitative analysis and,
therefore, opens up new possibilities of use of DBS metabolic and drug quantitative analysis.
However, this study also highlights other areas of bias potentially associated with HCT,
which need to be considered when using dried blood spot methods or other DBS related
methods for quantitative analysis. When using DBS methods for detection of a single or
multiple analytes further tests need to be done to understand the analyte distribution in
blood as well as recovery of such analyte dependent on HCT level.

- 88 -
CHAPTER5:COMPARISONOFEXTRACTIONKITSAND
SUBSTRATESTOMAXIMISEDNAYIELD

- 89 -
Chapter 5: Comparison of extraction kits and
substrates to maximise DNA yield 5.1IntroductionThe possibility of extracting genomic DNA from DBS has been successfully reported and has
led to DBS downstream applications being possible in various fields including genomics and
epigenomics (Wong et al., 2008, Cruickshank et al., 2012). However, these applications are
limited by the fact that the yield extracted depends on how much sample is used, how old
the sample is and what extraction kit is used (Ghantous et al., 2014). According to the
literature, the recovery of genomic DNA from DBS varies and therefore, there is a need to
optimise the method not only through DNA extraction but also optimising the substrate
used to collect the sample (Ghantous et al., 2014). The current substrate used for DBS is a
hydrophilic, 90-99 % cellulose-based filter paper (Smit et al., 2014). The paper contains set
of cavity pores with capillary dimensions that allow liquid absorption. The two advantages
of using such filter paper are the compatible hydrophilic properties allowing blood collection
and minimal cost (Smit et al., 2014).
The most commonly used FDA approved filter papers is Whatman 903TM. This ‘protein saver’
card is used for newborn screening and is often the reference substrate (Mei et al., 2010).
It’s wide use in the screening program creates great opportunity for large scale genetic and
epidemiology studies (Sjöholm et al., 2007, Wong et al., 2008). In Sweden, the newborn
screening registry now comprises of 3 million individuals, almost the entire population less
than 30 years old (Hannelius et al., 2005). However, in the United States, security concerns
have been raised around the use of residual newborn screening samples with a request for
clear definition of potential sample accessibility, law enforcement and inappropriate
government control with such samples. Further ethical concerns have been raised by
parents around the privacy of medical information and the proprietary rights (Institute of
Medicine (US), 2010). The key challenge is to accommodate the safety and ethical concerns
with the rapid technological development and expansion of NBS programs to allow
adequate use of such repositories (Green et al., 2006).

- 90 -
There are various cellulose-based filter papers that are chemically treated to better preserve
DNA, prevent bacteria growth and increase yield during extraction. The WhatmanTM FTATM
Elute is a substrate that contains chaotropic salt that allows lysis of cells upon contact
allowing the release of DNA (Lipic et al., 2018). The Ahlstrom-Munksjö GenSaverTM 2.0 card
allows for long term stabilisation and prevents degradation for up to 20 years (GenTegra,
2017). The limitation of the cellulose paper is that modifications to its chemical properties
are difficult. Furthermore, studies have demonstrated that DNA tends to stick to cellulose
making DNA extraction harder and sufficient beading or bashing is needed, which can result
in DNA fragmentation (Boese and Breaker, 2007). Cruickshank et al. (2013) introduced the
idea of mechanical beads in combination with soaking to increase the DNA yield extracted
from filter paper. This addition to the protocol, however, adds an extra step during
extraction making it less attractive for clinical adoption.
The steps for DNA extraction involve 1) cell lysis and release of DNA 2) enzymatic digestion
of cellular proteins 3) DNA precipitation 4) DNA binding (purification) and 5) DNA elution
(Ghantous et al., 2014). There are different protocols and extraction kits commercially
available; however, the protocol is always aligns with these five steps. For the DNA binding
there are two different methods available, the dispersed solid approach (magnetic beads) or
the microfluidic solid phase approach (column). The magnetic bead approach has recently
become popular in other fields, especially as it allows easy automisation (Bordelon et al.,
2013). The magnetic beads are coated to target the biomarker - in this case DNA. During the
purification step beads capture DNA from a modified/lysed biological sample. Beads are
magnetically transferred into the elution buffer where DNA is released and the beads are
removed (He et al., 2017).
The microfluidic solid phase extraction method has also been used for DNA extraction (Fan
et al., 2013). The purification step involves selective adsorption of DNA from a
modified/lysed biological sample that passes through a solid stationary phase, the column.
After desorption of unwanted compounds, elution medium is used to release DNA from the
stationary solid support (Günal et al., 2017). For the microfluidic solid phase, silica-based
monolithic columns are used due to the integral porous body with a mesh-like skeleton and
specific properties including high surface area (Wen et al., 2006, Liu et al., 2009), high filter

- 91 -
performance and selective adsorption (Galarneau et al., 2016). These columns are now
widely used for the purification step during DNA extraction, making it a multi-step
extraction process and difficult for automisation (Katevatis et al., 2017).
In early 2000s, a diverse range of monolith substrates (single structure consisting of thin-
walled narrow channels) have been identified including methacrylates (Vlakh and
Tennikova, 2007, Groarke and Brabazon, 2016). This porous polymer monolith can be
grafted with different chemical properties. The advantage of these methacrylate monoliths
is that they are easy to make, they can be formed into almost any form, created with a high
degree of macro porosity and the chemical properties can be tailored to specific
applications (Groarke and Brabazon, 2016). A study has demonstrated the possibility of
grafting such monoliths with specific properties that can be tailor for genomic applications
(Wen et al., 2006). By introducing these monoliths the aim is to improve the DNA quality
with the removal of all contaminants and reduce the protocol, to a single step (Smrekar et
al., 2013).
In this chapter, methacrylate monolith is used as blood storage substrate rather than the
base for solid phase extraction (Ferreira Neto et al., 2019). This methacrylate porous
polymer monolith (mPPM), have been grafted with hydrophilic properties with meso (2-50
nm) and macro (>50 nm) pores to allow blood absorption. The aim was to compare the DNA
yield extracted from different non-synthetic cellulose-based substrates and compare those
with synthetic methacrylate polymer monolith. The different extraction kits were tested
with modified protocol as per the manufacture’s recommendations and literature
recommendations (Ghantous et al., 2014, Ghantous et al., 2018, Mohandas et al., 2018) (for
methods used in this chapter see specific section of chapter 2 outlined below).
5.1MethodsSee the following sections from chapter 2: GENERAL METHODS
2.3 Comparison of extraction kits and substrates to maximise DNA yield
2.3.1 Dried blood spot preparation on different substrates
2.3.2 DNA extraction kits and protocol
2.3.3 Quality control measures
2.3.4 Statistical analysis

- 92 -
5.3Results5.3.1Blooddistributiononvarioussubstrates
Applying a fixed volume (cellulose-based substrates - 40 µL , mPPM – 12 µL) onto different
substrates resulted in visible difference in blood distribution on the filter papers (Figure 5.1).
FTATM Elute exhibited a distinct pattern compared to the other substrates with a light red
band on the outer edge of a dark red centre. The GenSaverTM resulted in red centre with a
distinct edge around the spot. The remaining three cellulose-based substrates had similar
distributions, with slightly darker and more defined edges. The mPPM did not show any
blood distribution patterns and the surface was either fully or partially covered by blood.
Figure5.1Driedbloodspotsgeneratedbyapplying40µLofbloodonsubstrates(FTATMGene,Whatman903TM,FTATMElute,GenCollectTM2.0,GenSaverTM2.0)and12µLofbloodappliedonto thepolymermonolith (mPPM).VenousEDTAbloodwas sourced fromafemalevolunteer.
FTATM Gene Whatman 903TM FTATM Elute
GenCollectTM 2.0 GenSaverTM 2.0 mPPM

- 93 -
5.3.2ComparisonofthreeDNAextractionkits:Quick-DNATM,QIAamp®MagMAX
CORETM
DNA concentration and yield were extracted and the average for eight technical replicates
reported for each substrate and extraction kit (Table 5.1). The best performing extraction kit
was Quick-DNATM with the average yield and concentration of 205.3 ng and 5.1 ng/µL. The
second-best kit was QIAamp® with the yield and concentration of 129.7 ng and 3.2 ng/µL.
The lowest yield and concentration extracted was using the MagMAX CORETM with 68.7 ng
and 1.7 ng/µL. The coefficient of variation across all samples within each kit was higher in
Quick-DNATM and QIAamp® with 29% and 30%, respectively. In comparison, the MagMAX
CORETM had a coefficient of variation of only 19%. The high coefficient of variation observed
in Quick-DNATM and QIAamp® was due to the impact of different substrate used.
Table5.1Theaverageconcentration(ng/µL)andyield(ng)foreighttechnicalreplicatesacrossthreeextractionkits(MagMAXCORETM,QIAamp®,Quick-DNATM)andsixdifferentsubstrates (GenCollectTM, FTATM Elute, FTATM Gene, GenSaverTM,Whatman 903TM andmethacrylatemonolith(mPPM)Thebloodvolumeusedforeachextractionwasthesamemaking the concentrations comparable. Concentration was calculated using QubitFluorometer.
Extraction Kit
Substrate
MagMAX CORETM QIAamp® Quick-DNATM
yield (ng)
concentration ng/µL
yield (ng)
concentration ng/µL
yield (ng)
concentration ng/µL
GenCollectTM2.0 57.1 1.4 77.7 1.9 161.6 4.0
FTATM Elute 66.0 1.7 160.8 4.0 284.3 7.1
FTATM Gene 68.6 1.7 144.7 3.6 162.7 4.1
mPPM 55.2 1.4 179.0 4.5 270.9 6.8
GenSaverTM2.0 91.9 2.3 93.2 2.3 205.8 5.1
Whatman 903TM 73.6 1.8 122.7 3.1 146.9 3.7
Mean 68.7 1.7 129.7 3.2 205.3 5.1
Coefficient of variation
19% 30% 29%

- 94 -
The mean difference between extraction kits was tested using a two-way ANOVA. A
significant difference between the kits was observed with the P value < 2.2 x 10-16 (Figure
5.2). A box plot was utilised to assess the distribution across samples within each extraction
kit. There was greater variation in the distribution for Quick-DNATM and QIAamp® in
comparison to MagMAX CORETM. This is due to the DNA yield variability across different
substrates.
The extraction kit mean difference was significant for all pairwise combination with the
greatest significance between Quick-DNATM and MagMAX CORETM with the P value of 1.5 x
10-15 (Figure 5.2).
Figure5.2Two-wayANOVAandpairwisemeandifferenceacrossthreeextractionkits(MagMAX CORETM, QIAamp®, Quick-DNATM). Box plots for each kit represents thedistribution of six different substrate types with 8 technical replicates (FTATM Gene,Whatman903TM,FTATMElute,GenCollectTM2.0,GenSaverTM2.0andmPPM)
For the QIAamp® and Quick-DNATM the average 260/280 ratios were 2.38 and 1.94,
respectively (Table 5.2). The reference value for 260/280 ratio is ~ 1.8 (Thermo Fisher
Scientific, 2009). Therefore, for Quick-DNATM samples were close to the reference value
3.3e−09
1.5e−15
1.7e−07
Anova, p < 2.2e−16
100
200
300
400
500
MagMax Core QIAamp DNA Quick−DNA MiniExtraction kit
yiel
d (n
g)
Kit.1 magmax qiagen zymo

- 95 -
indicating low proteins contamination. For the 260/230 ratio average values measured were
around 0.13 for QIAamp® and 0.4 Quick-DNATM, which was much lower than the accepted
purity value 2.0, indicating the presence of salts and other contaminants absorption at the
230 nm wavelength. The NanoDrop readings for the MagMAX CORETM were concerning for
both the average 260/280 and 260/230 ratios, indicating high contamination from both
protein, salts and other contaminates across all samples.
Table5.2AveragenanodropDNApurity(260/280ratioand260/230ratio)forallthreeextraction kits (MagMAX CORETM, QIAamp® and Quick-DNATM) and all six substrates(GenCollectTM 2.0, FTATM Elute, FTATM Gene, mPPM, GenSaverTM 2.0 and Whatman903TM).
Extraction Kit
Substrate MagMAX CORETM QIAamp® Quick-DNATM
260/280 260/230 260/280 260/230 260/280 260/230
GenCollectTM2.0 6.73 -2.0 2.48 0.10 2.05 0.35
FTATM Elute 6.68 -5.22 1.94 0.13 1.96 0.38
FTATM Gene 3.88 8.01 1.76 0.21 1.51 0.28
mPPM 5.93 1.17 2.41 0.13 2.06 0.41
GenSaverTM2.0 5.28 -1.60 2.81 0.10 1.95 0.33
Whatman 903TM 4.19 13.55 2.87 0.10 2.08 0.30
Mean 5.45 2.32 2.38 0.13 1.94 0.4
DNA quality was assessed using 1% agarose gel electrophoresis (Figure 5.3). The Quick-
DNATM kit exhibited higher fragmentation (molecular weight <10kb) compared to the
QIAamp®. This could be due to the different beading system used where multiple small
beads were used in comparison to one tungsten bead in the QIAamp® kit. The DNA ladder
used presented first band at 10kb. For both QIAamp® and Quick-DNATM the first band was
above the first DNA ladder band suggesting molecular weight higher than 10kb. Minimal
DNA was detecting for the MagMAX CORETM across all substrates. The three different
extraction kits were run on a separate gel.

Figure 5.3 DNA fragmentation on 1% agarose gel for three extraction kits (MagMAX CORETM, QIAamp® and Quick-DNATM) and sixsubstrates (GenCollectTM2.0,FTATMElute,FTATMGene,mPPM,GenSaverTM2.0andWhatman903TM).Foreachsubstateandkiteightreplicateswereloadedonthegel.Referenceladderwithfirst10kbband.
FTATM Gene Whatman 903TM
GenSaverTM 2.0 GenCollectTM 2.0
FTATM Elute mPPM Q
IAam
p®
Qui
ck-D
NATM
M
agM
AX
CO
RETM
10kb
10kb
10kb
3kb
1 kb
4kb
3kb

5.2.3Comparisonoffivenon-syntheticsubstrates(GenCollectTM2.0,FTATMElute,
FTATMGene,GenSaverTM2.0andWhatman903TM)andonesyntheticsubstrates
(mPPM)
The highest yield and concentration extracted was using the FTATM Elute with 284.3 ng and
7.1 µL. The second highest yield and concentrations extracted was using the mPPM with
179 ng and 4.5 µL (Table 5.1). The mean difference across substrates was statistically tested
using Two-way ANOVA with P values of 5.8 x 10-6 for QIAamp® and 1.1 x 10-9 for Quick-
DNATM indicating statistical differences between substrates (Figure 5.4).
The Two way ANOVA for MagMAX CORETM was not significant with p value of 0.2. The purity
ratio for both 260/280 and 260/230 did not vary across substrates for Quick-DNATM and
QIAamp® (Table 5.2). The purity ratios varied for the MagMAX CORETM significantly,
however, all values were not near to the reference point suggesting the contamination was
kit specific rather than substrate-specific. The fragmentation differed slightly across
different substrates with the mPPM showing higher molecular weight (molecular weight
>10kb) extracted (Figure 5.3). For the mPPM using the Quick-DNATM a batch effect was
observed where first batch (first four replicates) were showing slightly less fragmented DNA.
The commonly used substrate Whatman 903TM performed worse off in comparison to the
FTATM Elute and mPPM.

- 98 -
Figure 5.4 Box plot for each extraction kit (MagMAX CORETM, QIAamp® and Quick-DNATM) and six substrates (GenCollectTM 2.0, FTATM Elute, FTATM Gene, mPPM,GenSaverTM2.0andWhatman903TM).TheTwo-wayANOVAstatisticaltestperformedforeach extraction kit. Within each extraction kit the pairwise mean difference wascomparedagainstWhatman903TM(ns:p>0.05;*:p<0.05;**:p<0.001;***:p<0.001;***:p<0.0001)
5.2.4ThecombinationofextractionkitandsubstrateforhighDNAyield
Within each extraction category multiple pairwise tests were performed against a reference
group, Whatman 903TM (Figure 5.4). For the Quick-DNATM extraction kit significant
difference was seen for mPPM and FTATM Elute (P<0.0001), further significant difference
was seen for GenSaverTM (P<0.001) and no significant difference using GenCollectTM and
FTATM Gene (NS). For QIAamp® extraction kit significant difference was seen using
GenCollectTM (P<0.01). The DNA yield, however, was less in GenCollectTM in comparison to
Whatman 903TM. Further significant difference was observed using FTATM Elute and mPPM
(P<0.05) in comparison to Whatman 903TM. The overall best combination was using Quick-
DNATM extraction kit on FTATM Elute. When using QIAamp® extraction kit the best
performing substrate was mPPM. For the reference substrate Whatman 903TM the highest
yield was extracted using Quick-DNATM.
p = 0.2
nsnsns nsns nsnsns nsns
p = 5.8e−06
**** *ns **** *ns
p = 1.1e−09
********ns ***ns ********ns ***ns
Kit.1: magmax Kit.1: qiagen Kit.1: zymo
Collect
elute
gene
mili Sav
er
Whatman
Collect
elute
gene
mili Sav
er
Whatman
Collect
elute
gene
mili Sav
er
Whatman
100
200
300
Substrates
yiel
d (n
g)
paper.1Collectelute
genemili
SaverWhatman
GenColle
ctFTA
Elute
FTA Gen
e mPPM
GenSa
ver
Whatm
an 90
3Gen
Collect
FTA El
ute FTA
Gene
mPPMGen
Save
rW
hatman
903
GenColle
ctFTA
Elute
FTA Gen
e mPPM
GenSa
ver
Whatm
an 90
3
Mag Max Core QIAamp DNA Blood Mini Quick DNA Mini

- 99 -
5.3DiscussionFrom the three extraction kits tested, the Quick-DNATM extraction kit showed the highest
DNA yield; however, the fragmentation was higher in comparison to the QIAamp®
extraction kit. The substrate with the highest DNA yield was the cellulose-based FTATM Elute
followed by the synthetic mPPM monolith. The synthetic substrate mPPM also showed
promising results by isolating higher molecular weight DNA in comparison to the FTATM
Elute. In this chapter, the mechanical bead system (one tungsten vs multiple silicon) that
breaks up the cellulose substrates to increase DNA elution into the solution matrix was
utilised. Based on the data mechanical beads don’t necessary affect the integrity of DNA and
that high molecular weight DNA can be extracted using tungsten beads also increasing the
yield.
In the literature, different protocols have been tested to optimise DNA extraction from DBS.
Studies have demonstrated the use of QIAamp® extraction kit to isolate DNA from DBS for
both viral (Fischer et al., 2004) and genomic applications (Hue et al., 2011). The Quick-
DNATM kit is similar to the now discontinued ZR DNA Card Extraction Kit (Zymo Research,
USA) used by Mohandas et al. (2018), which showed sufficient yield for bisulfite conversion
to allow methylation analysis. For the MagMAX CORETM the use of manual magnetic stand
together with current protocol is not sufficient for high quality and quantity DNA extraction
from DBS. The high contamination could be due to the different purification methods in
which a manual magnetic stand was used with limited magnetic transfer to the new tube.
Using automated processes could allow for better sample purity. However, further protocol
optimisation and validation are recommended.
Different substrates both cellulose-based and synthetic were also tested. Few studies have
looked at the cellulose-based substrate effect on DNA extraction from DBS. Halsall and
colleagues compared FTATM Elute with Whatman 903TM indicating that FTATM Elute resulted
in increased DNA yield (Halsall et al., 2008). However, not many studies compared more
than the two substrates. The current use of the methacrylate polymer monoliths has been
used in separation science as part of DNA purification step during extraction. Such methods
were tested predominantly for plasmid DNA purification (Groarke and Brabazon, 2016)
rather than genomic DNA purification. Researchers have tested the polymer-based

- 100 -
substrate as a stationary phase for a purification of DNA using lysed/modified blood (Günal
et al., 2017). The polymer was compared with silica-based monoliths and exhibited better
purification properties and isolating large amounts of high molecular DNA. The use of
polymeric monoliths as a substrate rather than microfluidic solid phase column have not
been tested before. The same mPPM that was used in this chapter has been used for fatty
acid analysis using GS-MS, highlighting the potential for mPPM as an alternative substrate
resulting in lower contamination rate in comparison to standard Whatman 903TM. This
substrate has been patented in December 2019 (Ferreira Neto et al., 2019).
In this chapter, the highest DNA yield extracted from four 3.2 mm punches using Whatman
903TM was 147 ng. Many studies have extracted DNA from the same substrate but in many
cases, larger punch size or number of punches was used (Choi et al., 2014) (Hannelius et al.,
2005, Sjöholm et al., 2007, Strøm et al., 2014, Saavedra-Matiz et al., 2013). Few attempts
were successfully reported to extract DNA from older samples some being from 18-27 years
old (Wong et al., 2008, Sjöholm et al., 2007). However, the recovery of total DNA is
estimated to be approximately 15-25% which highlights the need for protocol optimisation
from both the substrate and extraction perspective (Choi et al., 2014). For low DNA yield
samples an amplification step can be used to increase the DNA concentration and therefore,
allow further genetic application. The key advantage is that even very small amounts (1ng)
of DNA can be used for amplification process of short sequences. There are, however, some
disadvantages to using amplification. Like all enzymes, errors in reads can occur causing
possible misinterpretation of data (Ben-Ezra, 1995). An amplification step generates large
libraries and can cause further data loss (Staunstrup et al., 2016). For whole exome
sequencing (WES) and whole genome sequencing (WGS) generating libraries is necessary to
align short read sequences. The interpretation of the data is complex, relying on complex
programs and expert health professionals (Punetha and Hoffman, 2013, Whiteford et al.,
2005).
A recent study published in Nature performed whole exome sequencing form DBS using the
linked reads sequence approach that may improve the contextual information of short
reads (Mortensen et al., 2019). DNA extracted from DBS was also used for genome wide
screening arrays, which shows great potential to improve genome-wide association study

- 101 -
and aim towards personalised medicine (Hollegaard et al., 2011, Suwinski et al., 2019). A
further pilot study outlined the possible identification of multiple polymorphisms from DBS
(Boemer et al., 2017). Bassaganyas and his colleagues have demonstrated WES and WGS
without amplification using DBS (Bassaganyas et al., 2018). DNA profiling for DNA
methylation studies from DBS have also been tested with studies now supporting the use of
archived DBS for epigenetic especially in large scale studies (Cruickshank et al., 2012, Wong
et al., 2008) (Walker et al., 2019)(Ghantous et al., 2018, Dugué et al., 2016). Such studies
can ‘go back in time’ and access samples that were collected at birth, some even decades
ago (Joo et al., 2013).
The big shift in DNA sequencing is towards third generation sequencing, which uses longer
sequences (Mantere et al., 2019, Mortensen et al., 2019). This technology include label-free,
ultra-long reads (104–106 bases) with direct real time sequencing (Loose et al., 2016). The
current DNA requirements for single molecule nanopore technology are between 1.5-2 µg
of DNA with the purity of being strictly near the reference values and the fragmentation not
less than 40kb (Michigan State University). The third generation technology is rapidly
emerging and has created a greater need for longer reads and therefore, high molecular
weight requirements.
In this chapter, when extracting DNA using four 3.2 mm punches, the yield isolated from any
substrate using any kit is not sufficient for downstream applications without amplification or
for long read third generation sequencing. The high molecular weight DNA extracted using
the mPPM, however, showed promising results for third generation sequencing. Further
study collecting larger volume of blood on the mPPM for DNA extraction could investigate
how much blood would be required for the use of mPPM for third generation sequencing.
The results of this study outlined the importance of substrate optimisation in order to
maximise DNA yield. The mPPM used in this chapter has been design for blood storage. Due
to the nature of these methacrylate monoliths there are possibilities to tailor such monolith
further to allow for cell lysis treatment and ion exchange properties that could further
maximise the yield extracted.
The samples from different extraction kit were ran on separate gel and the length of time
that the gels were run varied, therefore, the molecular bands on the ladder for MagMAX

- 102 -
CORETM are not as separated compared to the QIAamp® and Quick-DNATM. However, the
DNA concentration extracted using MagMAX CORETM across all substrates was
comparatively lower with high contamination. The first band visible on the ladder is 10kb,
therefore, it is difficult to identify the size of the larger genomic DNA recovered.
Furthermore, the amount of DNA loaded on the gel varied across different samples as
different DNA concentration was recovered creating the limitation of quantitative
interpretation based on the gel results. Thus, further studies would be required to load
same DNA concentration using technologies such as TapeStation to determine exact
molecular weight and fragment size of genomic DNA.
It is the combination of optimising the i) extraction protocol ii) substrate to extract good
quality and high quantity of DNA as well as the iii) advancing sequencing technology that
will allow to quick, good quality sequencing from DBS in the future. The introduction of
genomic testing especially as part of NBS is an opportunity to identify more infants with
potential severe illnesses and prevent death. However, key challenges have been identified
and remain to be discussed. These challenges tap into the ethical and legal issues when
genomic data are generated and the related professional responsibility of outcomes based
on genetic testing, especially so in paediatric setting (Boemer et al., 2017, Friedman et al.,
2017). It is the combination of ethical, legal policies and technical advances that need to be
addressed simultaneously.

- 103 -
CHAPTER6:CONCLUSIONANDFUTUREWORK

- 104 -
Chapter 6 Conclusions and future work 6.1ConclusionsIn this project the hemaPEN® was assessed from both the collection and analytical
perspective as the first steppingstone in the use of this device in clinical setting.
In Aim 1, the usability of hemaPEN® in an assisted setting was demonstrated where
phlebotomists at the Royal Children’s Hospital Pathology Collection Centre collected
hemaPEN® samples. For future usability in clinics some collectors would require more
practice than others given the current instructions and video material. A slight resistance
from phlebotomists was observed in adopting this device in a clinical setting but general
feedback received was positive. Further studies are required to outline the usability
potential and beneficial value of this device for diagnosis.
In the Aim 2, when comparing the standard DBS and hemaPEN® at an average HCT, a
difference was observed especially at higher concentration level. The difference observed
was due to the blood volume collected where more analyte was recovered in hemaPEN®.
Both methods showed a strong correlation with linearly increasing concentration levels in
the NBS analytical workflow indicating that recovery is not affected by the capillary action in
the device. From a workflow point of view, the transfer of samples from the device to the
micro plate is different in comparison to the standard DBS punch platform and therefore,
further automisation is required in order to use such device in NBS laboratory.
In Aim 3, when testing the device and standard DBS to understand the HCT related bias, a
standard DBS semi-quantitative bias was confirmed in most analytes. When correcting for
the volume inaccuracy across HCTs, some analytes showed HCT independency; however, for
most analytes, other areas of bias were uncovered such as HCT related extraction bias and
analyte blood distribution outlining the complexity of HCT bias.
For newborn screening workflow, qualified nurses should be able to use hemaPEN® even
though further and practice would be required especially on newborn babies. As for the
analytical workflow, hemaPEN® could be used in the newborn screening laboratory;
however, the clinical significance between the method due to the volume difference and

- 105 -
extraction bias associated with high HCT requires further validation to ensure that accurate
volume collection together with higher haematocrit level (which is often in newborn babies)
would not affect the clinical outcome. Furthermore, the sample transfer from hemaPEN® to
the micro plate during analysis requires further automisation as manual opening of
hemaPEN® devices would not be viable for routine NBS testing.
In Aim 4, DNA extracted from four 3.2 mm non-synthetic cellulose substrates and a
synthetic methacrylate monolith were not sufficient for downstream applications without
amplification. However, the results suggest that both substate and extraction kit contribute
to maximising DNA recovery. The synthetic monolithic substrate was the second best
substrate using the best performing extraction kit. The DNA extracted exhibited higher
molecular weight showing promising results especially for third generation long read
sequencing. Chemically treated FTATM Elute exhibited the highest DNA yield extracted
suggesting that treatment and lysis of cells when blood is deposited on filter paper can
increase the DNA yield.
6.2RecommendationforfutureworkIn order to expand the usability of such hemaPEN®, three research concepts should to be
considered: 1) mode of collection: assisted versus self-collection 2) the use of hemaPEN® in
current workflows that cannot use DBS due to volume inaccuracy 3) optimisation of
workflows including substrate development to allow wider range of downstream
applications.
From the collection perspective, the device should be tested in larger population studies
and clinical trials in both assisted collection and self-collection.
There could be different types of assisted collection. Firstly, where nurses and qualified
health professional collects blood from the patient in the clinic or hospital. Secondly,
parental collection of such samples from own children at home. Furthermore, exploring
testing of hemaPEN® in a self-collection setting, where patients collects blood sample by
themselves at home.

- 106 -
It has been demonstrated that by switching from assisted collection in clinics and pathology
centres, the cost can be reduced not only form the health care perspective but also form the
patients’ perspective (DiMasi et al., 2016). Therefore, commencing such studies with
selected population where patient undergo frequent blood collection can provide an
incentive for use of hemaPEN® and reduce the current burden of travel and cost associated
with frequent blood collection (DiMasi et al., 2016). It is important to state that it has been
outlined in few studies testing novel blood collection devices that instruction manuals and
training to ensure sample integrity is crucial (Zwart et al., 2018, Panchal et al., 2016).
Secondly, identify workflows where the advantages of DBS including the lower burden on
participant, lower cost and more frequent monitoring are very attractive but not used due
to the current impression related to HCT. For example, accuracy and precision is crucial for
therapeutic drug monitoring to monitor patient health and disease progression and to
provide optimal treatment for patients (Kang and Lee, 2009). A good example is a study by
Zwart et al, (2018) where a novel DBS method was utilised to detect tacrolimus as a
immunosuppressive drug for patents post-transplant allowing at home collection with
better patient satisfaction. Furthermore, use in research could increase participant
compliance and recruitment rate as easier bio sample collection can reducing the burden on
participants.
Lastly, optimising workflows to allow wider use of hemaPEN® in various downstream
applications including genomics, metabolomics, lipidomics and proteomics. The need for
workflow optimisation is not only through good quality sample collection but through to
extraction and substrate optimisation. Based on the DNA study using synthetic methacrylate
monolith (mPPM) tailoring such substrates to specific downstream application could open
additional opportunities to hemaPEN® use. Due to the initiative design with the ability to
collect four samples simultaneously different substrates could be introduced in one
cartridge i.e. one hemaPEN® device.

- 107 -
6.3BridgingstudiesIt is important to note that when switching or introducing the DBS concept there are few
things that need to be taken into consideration, as the current gold standard is the use of
plasma. When using DBS, the analysis is moving from a wet sample to dry and therefore, the
extraction protocol optimisation is crucial. When using DBS the analysis is moving from
plasma to whole blood, therefore, the interaction and distribution of target analyte need to
be investigated. Lastly, when using DBS most often capillary blood is used which can differ
from venous blood; therefore, reference points and threshold need to be tested to ensure
validity between methods. So-called bridging studies are often required in order to switch
from the gold standard to DBS (Amsterdam and Waldrop, 2010, Wickremsinhe et al., 2013).
6.4TheadoptionofemergingdevicesWider use of hemaPEN® or any emerging novel blood collection devices require
implementing changes to the current system, which can be difficult. This comes down to the
fact that promoting behavioural change in medical setting is a complex intervention
(Johnson and May, 2015). Wilkes et al., (2018) have outlined the issue of current one-way
teaching practice of medicine and pointed out the need of continuous learning and the need
for, interactive, integrational and interdisciplinary learning. The transformation requires so
called “tomorrow’s doctors” to anticipate, embrace and manage changes (Wilkes et al.,
2018).
Passive sensing technology, which captures data about a person without extra effort on
their part is fast emerging. Smartphones are key as they can capture behavioural change in
situ. Smartphones can track data that can assess our physical activity, sleep but also
depression or loneliness. Therefore, this patient specific, accurate data may result in
smarter and more personalised healthcare (Cornet and Holden, 2018). The need to change
towards patient-centric scheme is crucial to solve the key challenges in the current form of
healthcare. The population demographics is changing. It is predicted that by 2030, the
number of 65-year-old will double (National Institute on Aging, 2018). The burden on
healthcare service will be bigger than ever and the growing cost of the health care system is
unsustainable.

- 108 -
Patient-centred care is defined as an approach to “providing care that is respectful of and
responsive to individual patient preferences, needs, and values and ensuring that patient
values guide all clinical decisions” (Wilkerson et al., 2010). It is up to the patient to make the
final decision regarding their lifestyle choices and in some degree the medical intervention
they decide to undergo (Jo Delaney, 2018). This patient centric scheme can improve the
knowledge of individuals’ health leading to better choices made, prevention, positive life
change and hence improve overall population health. A great example is the type 2 diabetes
cohort where a patient centric scheme improved the knowledge, better self- care, glycaemic
levels and higher quality of life (Brown et al., 2013). From an industry point of view, patient-
centric schemes reduce the overall cost of healthcare and decrease readmission to hospital.
This scheme also increases competitive advantage, so more hospitals would have to
compete for patients based on cost as well as quality of care (Rivers and Glover, 2008).
In the US where private insurance dominates, arguments have been raised where insurance
companies could give consumers incentive to reduce their premiums by making better
health choices. Or is government interventions required to create better screening and
monitoring programs to incentive the population to make better health choices?
In conclusion, this thesis outlines the importance of blood as a biological sample. It further
outlines methods of blood collection including those commonly used in clinical setting,
alternative less invasive approaches and emerging devices. In this thesis I have outlined
some of the required tests for novel device implementation and also possible challenges.
Future publications:
No results have been published yet due to industry collaboration but several chapter/
section from this thesis are planned to be publish after submission.

- 109 -
CHAPTER7:REFERENCES

- 110 -
Chapter 7 Reference 1. AABYE,M.G.,LATORRE,I.,DIAZ,J.,MALDONADO,J.,MIALDEA,I.,EUGEN-
OLSEN,J.,RAVN,P.,DOMINGUEZ,J.&RUHWALD,M.2013.Driedplasmaspotsinthediagnosisoftuberculosis:IP-10releaseassayonfilterpaper.EurRespirJ,42,495.
1. ABBOTT.2012.Thesciencebehindwhybloodtestsmatter[Online]Available:https://www.lifetothefullest.abbott/en/articles/science-behind-why-blood-tests-matter.html[Accessed6June2020].
2. ABU-RABIE,P.,DENNIFF,P.,SPOONER,N.,CHOWDHRY,B.Z.&PULLEN,F.S.2015.InvestigationofdifferentapproachestoincorporatinginternalstandardinDBSquantitativebioanalyticalworkflowsandtheireffectonnullifyinghematocrit-basedassaybias.AnalChem,87,4996-5003.
3. ADLER,A.,GEIGER,S.,KEIL,A.,BIAS,H.,SCHATZ,P.,DEVOS,T.,DHEIN,J.,ZIMMERMANN,M.,TAUBER,R.&WIEDENMANN,B.2014.ImprovingcompliancetocolorectalcancerscreeningusingbloodandstoolbasedtestsinpatientsrefusingscreeningcolonoscopyinGermany.BMCGastroenterol,14,183.
4. AL-HARTHI,S.A.&JAMJOOM,M.B.2008.PCRassayinmalariadiagnosisusingfilterpapersamplesfromJazanregion,SaudiArabia.JEgyptSocParasitol,38,693-706.
5. AL-UZRI,A.,FREEMAN,K.A.,WADE,J.,CLARK,K.,BLEYLE,L.A.,MUNAR,M.&KOOP,D.R.2017.Longitudinalstudyontheuseofdriedbloodspotsforhomemonitoringinchildrenafterkidneytransplantation.PediatrTransplan,21.
6. AMBACH,L.,MENZIES,E.,PARKIN,M.C.,KICMAN,A.,ARCHER,J.R.H.,WOOD,D.M., DARGAN, P. I. & STOVE, C. 2019. Quantification of cocaine and cocainemetabolites indriedblood spots froma controlledadministration studyusingliquidchromatography–tandemmassspectrometry.DrugTestAnal,11,709-720.
7. AMSTERDAM,P.V.&WALDROP,C.2010.Theapplicationofdriedbloodspotsamplinginglobalclinicaltrials.Bioanalysis,2,1783-1786.
8. ANDREOTTI,M.,PIRILLO,M.,GUIDOTTI,G.,CEFFA,S.,PATURZO,G.,GERMANO,P., LUHANGA, R., CHIMWAZA, D., MANCINI, M. G., MARAZZI, M. C., VELLA, S.,PALOMBI, L. & GIULIANO, M. 2010. Correlation between HIV-1 viral loadquantification in plasma, dried blood spots, and dried plasma spots using theRocheCOBASTaqmanassay.JClinVirol,47,4-7.
9. ARDUI,S.,AMEUR,A.,VERMEESCH,J.R.&HESTAND,M.S.2018.Singlemoleculereal-time(SMRT)sequencingcomesofage:applicationsandutilitiesformedicaldiagnostics.NucleicAcidsRes,46,2159-2168.
10. ARMBRUSTER,D.A.&ALEXANDER,D.B.2006.Sampletosamplecarryover:asourceofanalyticallaboratoryerroranditsrelevancetointegratedclinicalchemistry/immunoassaysystems.ClinChimActa,373,37-43.
11. ARMBRUSTER,D.A.&PRY,T.2008.Limitofblank,limitofdetectionandlimitofquantitation.ClinBiochemRev,29,49-52.
12. ARMSTRONG,R.A.2014.WhentousetheBonferronicorrection.OphthalmicPhysiolOpt2014;34,502–508.
13. ATAEI,S.,NATEGHPOUR,M.,HAJJARAN,H.,EDRISSIAN,G.H.&FOROUSHANI,A.R.2011.Highspecificityofsemi-nestedmultiplexPCRusingdriedbloodspotsonDNABankingCardincomparisonwithfrozenliquidbloodfordetectionofPlasmodiumfalciparumandPlasmodiumvivax.JClinLabAnal,25,185-190.

- 111 -
14. BAHAR,B.2020.MethodComparisonandBiasEstimationUsingRandShiny[Online].Available:https://bahar.shinyapps.io/method_compare/[Accessed19May2020].
15. BARFIELD,M.&WHELLER,R.2011.Useofdriedplasmaspotsinthedeterminationofpharmacokineticsinclinicalstudies:validationofaquantitativebioanalyticalmethod.AnalChem,83,118-124.
16. BARON,S.,FONS,M.&ALBRECHT,T.1996.ViralPathogenesis.4ed.TheUniversityofTexasMedicalGalveston.
17. BASU, D. & KULKARNI, R. 2014. Overview of blood components and theirpreparation.IndianJAnaesth,58,529-537.
18. BECK,O.,KENANMODEN,N.,SEFERAJ,S.,LENK,G.&HELANDER,A.2018.Studyofmeasurementofthealcoholbiomarkerphosphatidylethanol(PEth)indriedbloodspot(DBS)samplesandapplicationofavolumetricDBSdevice.ClinChimActa,479,38-42.
19. BETHESDA,D.L.2005.BloodGroupsandRedAntigens,NationalCenterforBiotechnologyInformation(US).
20. BHATTACHARYA,K.,WOTTON,T.&WILEY,V.2014.Theevolutionofblood-spotnewbornscreening.TranslPediatrs,3,63-70.
21. BI,H.,TIAN,Y.,SONG,C.,LI,J.,LIU,T.,CHEN,Z.,CHEN,C.,HUANG,Y.&ZHANG,Y.2019. Urinary microbiota – a potential biomarker and therapeutic target forbladdercancer.JMedMicrobiol,68,1471-1478.
22. BICKEL,H.,GERRARD,J.&HICKMANS,E.M.1953.Influenceofphenylalanineintakeonphenylketonuria.Lancet,265,812-3.
23. BISWAS,B.2016.ClinicalPerformanceEvaluationofMolecularDiagnosticTests.JMolDiagn,18,803-812.
24. BLICHARZ,T.M.,GONG,P.,BUNNER,B.M.,CHU,L.L.,LEONARD,K.M.,WAKEFIELD,J.A.,WILLIAMS,R.E.,DADGAR,M.,TAGLIABUE,C.A.,ELKHAJA,R.,MARLIN,S.L.,HAGHGOOIE,R.,DAVIS,S.P.,CHICKERING,D.E.&BERNSTEIN,H.2018.Microneedle-baseddevicefortheone-steppainlesscollectionofcapillarybloodsamples.NatBiomedEng,2,151-157.
25. BLUMENREICH,M.S.1990.TheWhiteBloodCellandDifferentialCount.3ed.ButterworthsBoston.
26. BOEMER,F.,FASQUELLE,C.,D’OTREPPE,S.,JOSSE,C.,DIDEBERG,V.,SEGERS,K.,GUISSARD,V.,CAPRARO,V.,DEBRAY,F.G.&BOURS,V.2017.Anext-generationnewbornscreeningpilotstudy:NGSondriedbloodspotsdetectscausalmutationsinpatientswithinheritedmetabolicdiseases.SciRep,7,17641.
27. BOEMER,F.,KETELSLEGERS,O.,MINON,J.M.,BOURS,V.&SCHOOS,R.2008.Newbornscreeningforsicklecelldiseaseusingtandemmassspectrometry.ClinChem,54,2036-2041.
28. BOSSUYT,X.2009.Clinicalperformancecharacteristicsofalaboratorytest.Apracticalapproachintheautoimmunelaboratory.AutoimmunRev,8,543-548.
29. BOWEN,C.L.,LICEA-PEREZ,H.,KARLINSEY,M.Z.,JURUSIK,K.,PIERRE,E.,SIPLE,J.,KENNEY,J.,STOKES,A.,SPOONER,N.&EVANS,C.A.2013.Anovelapproachtocapillaryplasmamicrosamplingforquantitativebioanalysis.Bioanalysis,5,1131-1135.
30. BOWEN,R.A.R.&REMALEY,A.T.2014.Interferencesfrombloodcollectiontubecomponentsonclinicalchemistryassays.Biochemmed,24,31-44.
31. BROOKS,K.,DELONG,A.,BALAMANE,M.,SCHREIER,L.,ORIDO,M.,CHEPKENJA,M.,KEMBOI,E.,D'ANTUONO,M.,CHAN,P.A.,EMONYI,W.,DIERO,L.,COETZER,

- 112 -
M.&KANTOR,R.2016.HemaSpot,aNovelBloodStorageDeviceforHIV-1DrugResistanceTesting.JClinMicrobiol,54,223-225.
32. BURNETT,J.E.2011.Driedbloodspotsampling:practicalconsiderationsandrecommendationforusewithpreclinicalstudies.Bioanalysis,3,1099-10107.
33. CANNON,M.J.,GRIFFITHS,P.D.,ASTON,V.&RAWLINSON,W.D.2014.UniversalnewbornscreeningforcongenitalCMVinfection:whatistheevidenceofpotentialbenefit?RevMedVirol,24,291-307.
34. CAPIAU,S.,WILK,L.S.,DEKESEL,P.M.M.,AALDERS,M.C.G.&STOVE,C.P.2018.CorrectionfortheHematocritBiasinDriedBloodSpotAnalysisUsingaNondestructive,Single-WavelengthReflectance-BasedHematocritPredictionMethod.AnalChem,90,1795-1804.
35. CAPITAINER.2020.Thenewbloodtesttechinque[Online].Available:https://capitainer.se/technology/[Accessed30May2020].
36. CATALA,A.,CULP-HILL,R.,NEMKOV,T.&D’ALESSANDRO,A.2018.QuantitativemetabolomicscomparisonoftraditionalblooddrawsandTAPcapillarybloodcollection.Metabolomics,14,100.
37. CHAMBERS,A.G.,PERCY,A.J.,YANG,J.&BORCHERS,C.H.2015.MultipleReactionMonitoringEnablesPreciseQuantificationof97ProteinsinDriedBloodSpots.MolCellProteomics,14,3094-104.
38. CHAPMAN,K.,BURNETT,J.,CORVARO,M.,MITCHELL,D.,ROBINSON,S.,SANGSTER,T.,SPARROW,S.,SPOONER,N.&WILSON,A.2014.Reducingpre-clinicalbloodvolumesfortoxicokinetics:toxicologists,pathologistsandbioanalystsunite.Bioanalysis,6,2965-2968.
39. CHEN,Y.,ZHANG,Y.,ZHAO,G.,CHEN,C.,YANG,P.,YE,S.&TAN,X.2016.DifferenceinLeukocyteCompositionbetweenWomenbeforeandafterMenopausalAge,andDistinctSexualDimorphism.PLOSONE,11.
40. CHERNONOSOV,A.2018.TheUseofDriedBloodSpotsfortheQuantificationofAntihypertensiveDrugs.IntJAnalChem,2018.
41. CLARK,G.T.,HAYNES,J.J.,BAYLISS,M.A.&BURROWS,L.2010.UtilizationofDBSwithindrugdiscovery:developmentofaserialmicrosamplingpharmacokineticstudyinmice.Bioanalysis,2,1477-1488.
42. CLARKE,W.&DASGUPTA,A.2016.ClinicalChallengesinTherapeuticDrugMonitoringpecialPopulations,PhysiologicalConditions,andPharmacogenomics.ElsevierInc.
43. COHEN,L.L.,BLOUNT,R.L.,COHEN,R.J.,BALL,C.M.,MCCLELLAN,C.B.&BERNARD,R.S.2001.Children'sexpectationsandmemoriesofacutedistress:short-andlong-termefficacyofpainmanagementinterventions.JPediatrPsychol,26,367-374.
44. COLEMAN,D.2020.Practicalcapillarymicrosampling–avoidingthethepitfalls.ENVIGO,NC3Rs.
45. CONTI,M.,MATULLICAVEDAGNA,T.,RAMAZZOTTI,E.,MANCINI,R.,CALZA,L.,RINALDI,M.,BADIA,L.,GUARDIGNI,V.,VIALE,P.&VERUCCHI,G.2018.Multiplexedtherapeuticdrugmonitoring(TDM)ofantiviraldrugsbyLC–MS/MS.ClinMassSpectrom,7,6-17.
46. COOPER,M.B.,FORTE,C.A.&JONES,D.A.1988.Carnitineandacetylcarnitineinredbloodcells.BiochimBiophysActa,959,100-105.
47. CREE,I.A.2011.Improvedbloodtestsforcancerscreening:generalorspecific?BMCCancer,11,499.
48. CROOM,H.A.,RICHARDS,K.M.,BEST,S.J.,FRANCIS,B.H.,JOHNSON,E.I.,DAX,

- 113 -
E.M.&WILSON,K.M.2006.CommercialenzymeimmunoassayadaptedforthedetectionofantibodiestohepatitisCvirusindriedbloodspots.JClinVirol,36,68-71.
49. CRUICKSHANK,M.N.,OSHLACK,A.,THEDA,C.,DAVIS,P.G.,MARTINO,D.,SHEEHAN,P.,DAI,Y.,SAFFERY,R.,DOYLE,L.W.&CRAIG,J.M.2013.Analysisofepigeneticchangesinsurvivorsofpretermbirthrevealstheeffectofgestationalageandevidenceforalongtermlegacy.GenomeMed,5,96.
50. CRUICKSHANK,M.N.,PITT,J.&CRAIG,J.M.2012.GoingbacktothefuturewithGuthrie-poweredepigenome-wideassociationstudies.GenomeMed,4,83.
51. CUADROSRODRÍGUEZ,L.,GARCÍACAMPAÑA,A.M.&BOSQUESENDRA,J.M.1996.StatisticalEstimationofLinearCalibrationRange.AnalLett,29,1231-1239.
52. CUNNINGHAM,D.D.,HENNING,T.P.,SHAIN,E.B.,YOUNG,D.F.,ELSTROM,T.A.,TAYLOR,E.J.,SCHRODER,S.M.,GATCOMB,P.M.&TAMBORLANE,W.V.2000.Vacuum-assistedlancingoftheforearm:aneffectiveandlesspainfulapproachtobloodglucosemonitoring.DiabetesTechnolTher,2,541-548.
53. DAINTY,T.C.,RICHMOND,E.S.,DAVIES,I.&BLACKWELL,M.P.2012.Driedbloodspotbioanalysis:anevaluationoftechniquesandopportunitiesforreductionandrefinementinmouseandjuvenilerattoxicokineticstudies.IntJToxicol,31,4-13.
54. DAJAK,S.,ROJE,D.,HAŠPL,Ž.H.&MAGLIĆ,P.E.2014.TheimportanceofantenatalpreventionofRhDimmunisationinthefirstpregnancy.BloodTransfus,12,410-415.
55. DANIEL,Y.A.,TURNER,C.,HAYNES,R.M.,HUNT,B.J.&DALTON,R.N.2007.QuantificationofHemoglobinA2byTandemMassSpectrometry.ClinChem,53,1448-1454.
56. DANZER,K.2007.CalibrationinAnalyticalChemistry.1ed.SpringerBerlin.57. DEJESÚS,V.R.,CHACE,D.H.,LIM,T.H.,MEI,J.V.&HANNON,W.H.2010.
Comparisonofaminoacidsandacylcarnitinesassaymethodsusedinnewbornscreeningassaysbytandemmassspectrometry.ClinicaChimicaActa,411,684-689.
58. DEKESEL,P.M.M.,LAMBERT,W.E.&STOVE,C.P.2015.Doesvolumetricabsorptivemicrosamplingeliminatethehematocritbiasforcaffeineandparaxanthineindriedbloodsamples?Acomparativestudy.AnalyticaChimicaActa,881,65-73.
59. DEKESEL,P.M.M.,SADONES,N.,CAPIAU,S.,LAMBERT,W.E.&STOVE,C.P.2013.Hemato-criticalissuesinquantitativeanalysisofdriedbloodspots:challengesandsolutions.Bioanalysis,5,2023-2041.
60. DEKESEL,P.M.,CAPIAU,S.,LAMBERT,W.E.&STOVE,C.P.2014.Currentstrategiesforcopingwiththehematocritproblemindriedbloodspotanalysis.Bioanalysis,6,1871-4.
61. DEANE,P.2017.BCells:TheAntibodyFactoriesoftheImmuneSystem[Online].Available:https://www.lifespan.io/news/b-cells/[Accessed19May2020].
62. DÉNES,J.,SZABÓ,E.,ROBINETTE,S.L.,SZATMÁRI,I.,SZŐNYI,L.,KREUDER,J.G.,RAUTERBERG,E.W.&TAKÁTS,Z.2012.MetabonomicsofNewbornScreeningDriedBloodSpotSamples:ANovelApproachintheScreeningandDiagnosticsofInbornErrorsofMetabolism.AnalChem,84,10113-10120.
63. DENNIFF,P.&SPOONER,N.2010.Effectofstorageconditionsontheweightandappearanceofdriedbloodspotsamplesonvariouscellulose-basedsubstrates.

- 114 -
Bioanalysis,2,1817-1822.64. DENNIFF,P.&SPOONER,N.2010.Theeffectofhematocritonassaybiaswhen
usingDBSsamplesforthequantitativebioanalysisofdrugs.Bioanalysis,2,1385-95.
65. DENNIFF,P.&SPOONER,N.2014.Volumetricabsorptivemicrosampling:adriedsamplecollectiontechniqueforquantitativebioanalysis.AnalChem,86,8489-95.
66. DICAPRIO,G.,STOKES,C.,HIGGINS,J.M.&SCHONBRUN,E.2015.Single-cellmeasurementofredbloodcelloxygenaffinity.ProcNatlAcadSci,112,9984-9999.
67. DIEZ-SILVA,M.,DAO,M.,HAN,J.,LIM,C.T.&SURESH,S.2010.ShapeandBiomechanicalCharacteristicsofHumanRedBloodCellsinHealthandDisease.MRSBull,35,382-388.
68. DIMASI,J.A.,GRABOWSKI,H.G.&HANSEN,R.W.2016.Innovationinthepharmaceuticalindustry:NewestimatesofR&Dcosts.JHealthEcon,47,20-33.
69. DORSEY,M.J.&PUCK,J.M.2019.NewbornScreeningforSevereCombinedImmunodeficiencyintheUnitedStates:LessonsLearned.ImmunolAllergyClinNorthAm,39,1-11.
70. DRACHMAN,J.G.2004.Inheritedthrombocytopenia:whenalowplateletcountdoesnotmeanITP.Blood,103,390-398.
71. DROLET,J.,TOLSTIKOV,V.,WILLIAMS,B.A.,GREENWOOD,B.P.,HILL,C.,VISHNUDAS,V.K.,SARANGARAJAN,R.,NARAIN,N.R.&KIEBISH,M.A.2017.IntegratedMetabolomicsAssessmentofHumanDriedBloodSpotsandUrineStrips.Metabolites,7.
72. DUGUÉ,P.-A.,ENGLISH,D.R.,MACINNIS,R.J.,JUNG,C.-H.,BASSETT,J.K.,FITZGERALD,L.M.,WONG,E.M.,JOO,J.E.,HOPPER,J.L.,SOUTHEY,M.C.,GILES,G.G.&MILNE,R.L.2016.ReliabilityofDNAmethylationmeasuresfromdriedbloodspotsandmononuclearcellsusingtheHumanMethylation450kBeadArray.SciRep,6,30317.
73. ELLEFSEN,K.N.,DACOSTA,J.L.,CONCHEIRO,M.,ANIZAN,S.,BARNES,A.J.,PIRARD,S.,GORELICK,D.A.&HUESTIS,M.A.2015.CocaineandmetaboliteconcentrationsinDBSandvenousbloodaftercontrolledintravenouscocaineadministration.Bioanalysis,7,2041-2056.
74. EMMONS,G.&ROWLAND,M.2010.Pharmacokineticconsiderationsastowhentousedriedbloodspotsampling.Bioanalysis,2,1791-1796.
75. EYLES,D.W.,TRZASKOWSKI,M.,VINKHUYZEN,A.A.E.,MATTHEISEN,M.,MEIER,S.,GOOCH,H.,ANGGONO,V.,CUI,X.,TAN,M.C.,BURNE,T.H.J.,JANG,S.E.,KVASKOFF,D.,HOUGAARD,D.M.,NØRGAARD-PEDERSEN,B.,COHEN,A.,AGERBO,E.,PEDERSEN,C.B.,BØRGLUM,A.D.,MORS,O.,SAH,P.,WRAY,N.R.,MORTENSEN,P.B.&MCGRATH,J.J.2018.TheassociationbetweenneonatalvitaminDstatusandriskofschizophrenia.SciRep,8,17692-17692.
76. FAN,L.&LEE,J.A.2012.ManagingtheeffectofhematocritonDBSanalysisinaregulatedenvironment.Bioanalysis,4,345-347.
77. FANG,K.,BOWEN,C.L.,KELLIE,J.F.,KARLINSEY,M.Z.&EVANS,C.A.2018.Drugmonitoringbyvolumetricabsorptivemicrosampling:methoddevelopmentconsiderationstomitigatehematocriteffects.Bioanalysis,10,241-255.
78. FENG,Y.,LI,L.&SUN,X.-H.2011.MonocytesandAlzheimer'sdisease.NeurosciBull,27,115-122.
79. FERREIRANETO,R.J.,HILDER,E.F.,GOOLEY,A.A.&HON,W.B.2019.Device

- 115 -
andmethodsforcollectingandstoringfluidsamplesforanalysis,PCT/AU2019/000069.
80. FIEHN,O.&KIND,T.2007.Metaboliteprofilinginbloodplasma.MethodsMolBiol,358,3-17.
81. FILALI-ANSARY,A.,LUNVEN,C.,TURPAULT,S.,BEYER,Y.-J.,OʼBRIEN,A.,DELFOLIE,A.,BOYANOVA,N.,SANDERINK,G.-J.&BALDINETTI,F.2016.DriedBloodSpotMethodologyinCombinationWithLiquidChromatography/TandemMassSpectrometryFacilitatestheMonitoringofTeriflunomide.TherDrugMonit,38,471-482.
82. FLATLAND,B.,FRIEDRICHS,K.R.&KLENNER,S.2014.Differentiatingbetweenanalyticalanddiagnosticperformanceevaluationwithafocusonthemethodcomparisonstudyandidentificationofbias.VetClinPathol,43,475-486.
83. FRUHSTORFER,H.,SCHMELZEISEN-REDEKER,G.&WEISS,T.1999.Capillarybloodsampling:relationbetweenlancetdiameter,lancingpainandbloodvolume.EurJPain,3,283-286.
84. FUHRMAN,J.2018.TheHiddenDangersofFastandProcessedFood.AmJLifestyleMed,12,375-381.
85. GARRAUD,O.&COGNASSE,F.2015.ArePlateletsCells?AndifYes,areTheyImmuneCells?FrontImmuno,6,70-70.
86. GEORGE,R.S.&MOAT,S.J.2016.EffectofDriedBloodSpotQualityonNewbornScreeningAnalyteConcentrationsandRecommendationsforMinimumAcceptanceCriteriaforSampleAnalysis.ClinChem,62,466-75.
87. GHANTOUS,A.,HERNANDEZ-VARGAS,H.&HERCEG,Z.2018.DNAMethylationAnalysisfromBloodSpots:IncreasingYieldandQualityforGenome-WideandLocus-SpecificMethylationAnalysis.MethodsMolBiol,1708,605-619.
88. GHANTOUS,A.,SAFFERY,R.,CROS,M.-P.,PONSONBY,A.-L.,HIRSCHFELD,S.,KASTEN,C.,DWYER,T.,HERCEG,Z.&HERNANDEZ-VARGAS,H.2014.OptimizedDNAextractionfromneonataldriedbloodspots:applicationinmethylomeprofiling.BMCBiotechnology,14,60.
89. GHOSHAL,K.&BHATTACHARYYA,M.2014.Overviewofplateletphysiology:itshemostaticandnonhemostaticroleindiseasepathogenesis.ScientificWorldJournal,2014,781857.
90. GIAVARINA,D.2015.UnderstandingBlandAltmananalysis.Biochemmed,25,141-151.
91. GOTO,T.,INOUE,T.,KAMIYA,C.,KAWABE,H.,HIGUCHI,M.,SUYAMA,M.,GOTO,T.,KOIDE,W.,MAKI,K.,USHIJIMA,K.,BAN,K.&YAMADA,Y.2020.Neonatalpainresponsetoautomaticlancetversusneedleheel-prickbloodsampling:Aprospectiverandomizedcontrolledclinicaltrial.PediatrInt,62,357-362.
92. GRADY,M.,PINEAU,M.,PYNES,M.K.,KATZ,L.B.&GINSBERG,B.2014.Aclinicalevaluationofroutinebloodsamplingpracticesinpatientswithdiabetes:impactonfingerstickbloodvolumeandpain.JDiabetesSciTechnol,8,691-698.
93. GRIMES,D.A.&SCHULZ,K.F.2005.Refiningclinicaldiagnosiswithlikelihoodratios.Lancet,365,1500-1505.
94. GRÖSCHL,M.2008.CurrentStatusofSalivaryHormoneAnalysis.ClinChem,54,1759.
95. GUPTA,K.&MAHAJAN,R.2018.ApplicationsandDiagnosticPotentialofDriedBloodSpots.IntJApplBasicMedRes,8,1-2.
96. GUTHRIE,R.&SUSI,A.1963.AsimplePhenylalaninemethodfordetectingphenylketonuriainlargepopulationsofnewborninfants.Pediatrics,32,338.

- 116 -
97. GWAK,M.S.,CHOI,S.J.,KIM,J.A.,KO,J.S.,KIM,T.H.,LEE,S.M.,PARK,J.A.&KIM,M.H. 2007. Effects of gender onwhite blood cell populations and neutrophil-lymphocyteratiofollowinggastrectomyinpatientswithstomachcancer.JKoreanMedSci,22,104-108.
98. HAGAN,A.S.,JONES,D.R.&AGARWAL,R.2013.UseofDriedPlasmaSpotsfortheQuantificationofIothalamateinClinicalStudies.ClinJAmSocNephrol,8,909.
99. HALL,E.M.,FLORES,S.R.&DEJESÚS,V.R.2015.InfluenceofHematocritandTotal-SpotVolumeonPerformanceCharacteristicsofDriedBloodSpotsforNewbornScreening.IntJNeonatalScreen,1,69-78.
100. HAN,A.,ANDRE,M.P.,ERDMAN,J.W.,LOOMBA,R.,SIRLIN,C.B.&O'BRIEN,W.D.2017.RepeatabilityandReproducibilityofaClinicallyBasedQUSPhantomStudyandMethodologies.IEEETransUltrasonFerroelectrFreqControl,64,218-231.
101. HANNON,W.H.&THERRELL,B.L.2014.OverviewoftheHistoryandApplicationsofDriedBloodSamples.JohnWiley&Sons.
102. HARRINGTON,J.M.,YOUNG,D.J.,ESSADER,A.S.,SUMNER,S.J.&LEVINE,K.E.2014.AnalysisofhumanserumandwholebloodformineralcontentbyICP-MSandICP-OES:developmentofamineralomicsmethod.BiolTraceElemRes,160,132-42.
103. HEMAXIS.2018.HemaXisDB10WholeBloodCollectionDevice[Online].Available:https://hemaxis.com/products/hemaxis-db10/[Accessed30May2020].
104. HEMPEN,C.M.,MAARTENKOSTER,E.H.&OOMS,J.A.2015.Hematocrit-independentrecoveryofimmunosuppressantsfromDBSusingheatedflow-throughdesorption.Bioanalysis,7,2019-29.
105. HENION,J.D.,RYONA,L.&BOWERS,L.D.2017.Multilayerdeviceforseparatingbloodcomponentsandusethereof,PCT/US2017/019405.
106. HEWAWASAM,E.,LIU,G.,JEFFERY,D.W.,GIBSON,R.A.&MUHLHAUSLER,B.S.2018.EstimationoftheVolumeofBloodinaSmallDiscPunchedFromaDriedBloodSpotCard.Eur.J.LipidSci.Technol,120,1700362.
107. HIGGINS,C.1995.Deficiencytestingforiron,vitaminB12andfolate.NursTimes,91,38-39.
108. HOELLER,U.,BAUR,M.,ROOS,F.F.,BRENNAN,L.,DANIEL,H.,FALLAIZE,R.,FORSTER,H.,GIBNEY,E.R.,GIBNEY,M.,GODLEWSKA,M.,HARTWIG,K.,KOLOSSA,S.,LAMBRINOU,C.P.,LIVINGSTONE,K.M.,LOVEGROVE,J.A.,MACREADY,A.L.,MANIOS,Y.,MARSAUX,C.F.,MARTINEZ,J.A.,CELIS-MORALES,C.,MOSCHONIS,G.,NAVAS-CARRETERO,S.,O'DONOVAN,C.B.,SAN-CRISTOBAL,R.,SARIS,W.H.,SURWIŁŁO,A.,TRACZYK,I.,TSIRIGOTI,L.,WALSH,M.C.,WOOLHEAD,C.,MATHERS,J.C.&WEBER,P.2016.ApplicationofdriedbloodspotstodeterminevitaminDstatusinalargenutritionalstudywithunsupervisedsampling:theFood4Meproject.BrJNutr,115,202-211.
109. HOLSWORTH,R.E.,JR.,CHO,Y.I.&WEIDMAN,J.2013.Effectofhydrationonwholebloodviscosityinfirefighters.AlternTherHealthMed,19,44-49.
110. HOLUB,M.,TUSCHL,K.,RATSCHMANN,R.,STRNADOVA,K.A.,MUHL,A.,HEINZE,G.,SPERL,W.&BODAMER,O.A.2006.Influenceofhematocritandlocalisationofpunchindriedbloodspotsonlevelsofaminoacidsandacylcarnitinesmeasuredbytandemmassspectrometry.ClinChimActa,373,27-31.

- 117 -
111. HOPE,R.M.2013.Rmisc.[Online].Available:https://CRAN.R-project.org/package=Rmisc[Accessed20May2020].
112. HORIEN,C.&YUAN,P.2017.DrugDevelopment.YaleJ.Biol.Med,90,1-3.113. IALONGO,C.&BERNARDINI,S.2016.Phlebotomy,abridgebetween
laboratoryandpatient.Biochemmed,26,17-33.114. IRAGORRI,N.&SPACKMAN,E.2018.Assessingthevalueofscreeningtools:
reviewingthechallengesandopportunitiesofcost-effectivenessanalysis.PublicHealthRev,39,17.
115. JAGER,W.2003.Breaking‘badhabits’:adynamicalperspectiveonhabitformationandchange.UniversityofGroningen
116. JANSEN,M.E.,METTERNICK-JONES,S.C.&LISTER,K.J.2016.Internationaldifferencesintheevaluationofconditionsfornewbornbloodspotscreening:areviewofscientificliteratureandpolicydocuments.EurJHumGenet,25,10-16.
117. JENNINGS, L., DEERLIN, V. M. V. & GULLEY, M. L. 2009. RecommendedPrinciplesandPracticesforValidatingClinicalMolecularPathologyTests.ArchPatholLabMed,133,743-755.
118. JING,J.&GAO,Y.2018.Urinebiomarkersintheearlystagesofdiseases:currentstatusandperspective.DiscovMed,25,57-65.
119. JONES,G.R.D.,ALBAREDE,S.,KESSELER,D.,MACKENZIE,F.,MAMMEN,J.,PEDERSEN,M.,STAVELIN,A.,THELEN,M.,THOMAS,A.,TWOMEY,P.J.,VENTURA,E.&PANTEGHINI,M.2017.Analyticalperformancespecificationsforexternalqualityassessment-definitionsanddescriptions.ClinChemLabMed,55,949-955.
120. KANG,J.S.&LEE,M.H.2009.Overviewoftherapeuticdrugmonitoring.KoreanJInternMed,24,1-10.
121. KARIYAWASAM, D. S. T., RUSSELL, J. S.,WILEY, V., ALEXANDER, I. E. &FARRAR, M. A. 2020. The implementation of newborn screening for spinalmuscularatrophy:theAustralianexperience.GenetMed,22,557-565.
122. KASSAMBARA,A.&MUNDT,F.2020.factoextra:ExtractandVisualizetheResultsofMultivariateDataAnalyses[Online].Available:https://CRAN.R-project.org/package=factoextra[Accessed20May2020].
123. KASSAMBARA,A.2020.ggpubr:'ggplot2'BasedPublicationReadyPlots.[Online].Available:https://CRAN.R-project.org/package=ggpubr[Accessed19May2020]
124. KAZMIERCZAK,S.C.&CATROU,P.G.2000.Analyticalinterference.Morethanjustalaboratoryproblem.AmJClinPathol,113,9-11.
125. KELLY,N.,MAKAREM,D.C.&WASSERSTEIN,M.P.2016.ScreeningofNewbornsforDisorderswithHighBenefit-RiskRatiosShouldBeMandatory.JLawMedEthics,44,231-240.
126. KENNEDY, A. D., MILLER, M. J., BEEBE, K., WULFF, J. E., EVANS, A. M.,MILLER,L.A.D.,SUTTON,V.R.,SUN,Q.&ELSEA,S.H.2016.MetabolomicProfilingofHumanUrineasaScreenforMultipleInbornErrorsofMetabolism.GenetTestMolBiomarkers,20,485-495.
127. KILLEEN,A.A.,LONG,T.,SOUERS,R.,STYER,P.,VENTURA,C.B.&KLEE,G.G.2014.Verifyingperformancecharacteristicsofquantitativeanalyticalsystems:calibrationverification,linearity,andanalyticalmeasurementrange.ArchPatholLabMed,138,1173-81.
128. KIM,Y.-C.,PARK,J.-H.&PRAUSNITZ,M.R.2012.Microneedlesfordrug

- 118 -
andvaccinedelivery.AdvDrugDelivRev,64,1547-1568.129. KINNS,H.,PITKIN,S.,HOUSLEY,D.&FREEDMAN,D.B.2013.Internal
qualitycontrol:bestpractice.JClinPathol,66,1027-32.130. KOBOLDT,D.C.,STEINBERG,K.M.,LARSON,D.E.,WILSON,R.K.&
MARDIS,E.R.2013.Thenext-generationsequencingrevolutionanditsimpactongenomics.Cell,155,27-38.
131. KORFMACHER,W.,FITZGERALD,M.,LUO,Y.,HO,S.,WANG,J.,WU,Z.,SNOW,G.&O'SHEA,T.2015a.CapillarymicrosamplingofwholebloodformousePKstudies:aneasyroutetoserialbloodsampling.Bioanalysis,7,449-61.
132. KORFMACHER,W.,LUO,Y.,HO,S.,SUN,W.,SHEN,L.,WANG,J.,WU,Z.,GUO,Y.,SNOW,G.&O'SHEA,T.2015b.Utilityofcapillarymicrosamplingforratpharmacokineticstudies:Comparisonoftail-veinbleedtojugularveincannulasampling.JPharmacolToxicolMethods,76,7-14.
133. KRALIEVITS,K.E.,RAYKAR,N.P.,GREENBERG,S.L.&MEARA,J.G.2015.Theglobalbloodsupply:aliteraturereview.Lancet,385Suppl2,S28.
134. KRICKA,L.J.&SAVORY,J.2011.InternationalYearofChemistry2011:AGuidetotheHistoryofClinicalChemistry.ClinChem,57,1118-1126.
135. KUHN,V.,DIEDERICH,L.,KELLER,T.C.S.T.,KRAMER,C.M.,LUCKSTADT,W.,PANKNIN,C.,SUVORAVA,T.,ISAKSON,B.E.,KELM,M.&CORTESE-KROTT,M.M.2017.RedBloodCellFunctionandDysfunction:RedoxRegulation,NitricOxideMetabolism,Anemia.AntioxidRedoxSignal,26,718-742.
136. KWASNICKA,D.,DOMBROWSKI,S.U.,WHITE,M.&SNIEHOTTA,F.2016.Theoreticalexplanationsformaintenanceofbehaviourchange:asystematicreviewofbehaviourtheories.HealthPsycholRev,10,277-296.
137. KWON,C.&FARRELL,P.M.2000.TheMagnitudeandChallengeofFalse-PositiveNewbornScreeningTestResults.ArchPediatrAdolescMed, 154, 714-718.
138. LAMARCA,G.,GIOCALIERE,E.,VILLANELLI,F.,MALVAGIA,S.,FUNGHINI,S.,OMBRONE,D.,FILIPPI,L.,DEGAUDIO,M.,DEMARTINO,M.&GALLI,L.2012.DevelopmentofanUPLC-MS/MSmethodforthedeterminationofantibioticertapenemondriedbloodspots.JPharmBiomedAnal,61,108-13.
139. LALKHEN,A.G.&MCCLUSKEY,A.2008.Clinicaltests:sensitivityandspecificity.BJAEducation,8,221-223.
140. LAWSON,A.J.,BERNSTONE,L.&HALL,S.K.2016.NewbornscreeningbloodspotanalysisintheUK:influenceofspotsize,punchlocationandhaematocrit.JMedScreen,23,7-16.
141. LEBIEN,T.W.&TEDDER,T.F.2008.Blymphocytes:howtheydevelopandfunction.Blood,112,1570-1580.
142. LEE, P. Y., COSTUMBRADO, J., HSU, C.-Y. & KIM, Y. H. 2012. Agarose gel electrophoresis for the separation of DNA fragments. Journal of visualized experiments. JoVE, 3923.
143. LEEMAN,M.,CHOI,J.,HANSSON,S.,STORM,M.U.&NILSSON,L.2018.Proteinsandantibodiesinserum,plasma,andwholeblood-sizecharacterizationusingasymmetricalflowfield-flowfractionation(AF4).AnalBioanalChem,410,4867-4873.
144. LEI,B.U.W.&PROW,T.W.2019.Areviewofmicrosamplingtechniquesandtheirsocialimpact.Biomedmicrodevices,21,81.
145. LEW,J.-B.,FELETTO,E.,WADE,S.,CARUANA,M.,KANG,Y.-J.,NICKSON,C.,SIMMS,K.,PROCOPIO,P.,TAYLOR,N.,WORTHINGTON,J.,SMITH,D.&CANFELL,

- 119 -
K.2019.Benefits,harmsandcost-effectivenessofcancerscreeninginAustralia:anoverviewofmodellingestimates.PublicHealthResPract,29.
146. LI,W.&TSE,F.L.2010.DriedbloodspotsamplingincombinationwithLC-MS/MSforquantitativeanalysisofsmallmolecules.BiomedChromatogr,24,49-65.
147. LI,W.,JIAN,W.&FU,Y.2020.BasicSamplePreparationTechniquesinLC-MSBioanalysis.JohnWiley&Sons,Inc.
148. LI,Y.,HENION,J.,ABBOTT,R.&WANG,P.2012.Theuseofamembranefiltrationdevicetoformdriedplasmaspotsforthequantitativedeterminationofguanfacineinwholeblood.RapidCommun.MassSpectrom,26,1208-1212.
149. LIM,E.,MIYAMURA,J.&CHEN,J.J.2015.Racial/Ethnic-SpecificReferenceIntervalsforCommonLaboratoryTests:AComparisonamongAsians,Blacks,Hispanics,andWhite.HawaiiJMedPublicHealth,74,302-310.
150. LIM,M.D.2018.DriedBloodSpotsforGlobalHealthDiagnosticsandSurveillance:OpportunitiesandChallenges.AmJTropMedHyg,99,256-265.
151. LIM,Y.,TOTSIKA,M.,MORRISON,M.&PUNYADEERA,C.2017.OralMicrobiome:ANewBiomarkerReservoirforOralandOropharyngealCancers.Theranostics,7,4313-4321.
152. LLORET-LINARES,C.,DAALI,Y.,CHEVRET,S.,NIETO,I.,MOLIÈRE,F.,COURTET,P.,GALTIER,F.,RICHIERI,R.-M.,MORANGE,S.,LLORCA,P.-M.,EL-HAGE,W.,DESMIDT,T.,HAESEBAERT,F.,VIGNAUD,P.,HOLTZMANN,J.,CRACOWSKI,J.-L.,LEBOYER,M.,YRONDI,A.,CALVAS,F.,YON,L.,LECORVOISIER,P.,DOUMY,O.,HERON,K.,MONTANGE,D.,DAVANI,S.,DÉGLON,J.,BESSON,M.,DESMEULES,J.,HAFFEN,E.&BELLIVIER,F.2017.Exploringvenlafaxinepharmacokineticvariabilitywithaphenotypingapproach,amulticentricfrench-swissstudy(MARVELstudy).BMCpharmacolToxico,18,70-70.
153. LÓPEZ,J.A.&BERLINER,N.2017.Introductiontoaseriesofreviewsonclinicalplateletdisorders.Blood,129,2821-2822.
154. LORENZO-POUSO,A.I.,PEREZ-SAYANES,M.,BRAVO,S.B.,LOPEZ-JORNET,P.,GARCIA-VENCE,M.,ALONSO-SAMPEDRO,M.,CARBALLO,J.,GARCIA-GARCIA,A.2018.Protein-BasedSalivaryProfilesasNovelBiomarkersforOralDiseases.DiseaseMarkers,2018,22.
155. LU, H., GIORDANO, F. & NING, Z. 2016. Oxford Nanopore MinIONSequencingandGenomeAssembly.GenomicsProteomicsBioinformatics,14,265-279.
156. LUCKHEERAM,R.V.,ZHOU,R.,VERMA,A.D.&XIA,B.2012.CD4(+)Tcells:differentiationandfunctions.ClinDevImmunol,2012,925135.
157. LUDBROOK,J.1997.Comparingmethodsofmeasurements.ClinExpPharmacolPhysiol,24,193-203.
158. MA,W.,KANNAN,K.,WU,Q.,BELL,E.M.,DRUSCHEL,C.M.,CAGGANA,M.&ALDOUS,K.M.2013.AnalysisofpolyfluoroalkylsubstancesandbisphenolAindriedbloodspotsbyliquidchromatographytandemmassspectrometry.AnalBioanalChem,405,4127-38.
159. MACNAB,J.C.M.&ONIONS,D.1996.MedicalMicrobiology.4edTheUniversityofTexasMedicalBranchGalveston.
160. MAHLKNECHT,U.&KAISER,S.2010.Age-relatedchangesinperipheralbloodcountsinhumans.Exp.Ther.Med,1,1019-1025.
161. MÄNNEL,D.N.&GRAU,G.E.1997.Roleofplateletadhesionin

- 120 -
homeostasisandimmunopathology.MolPathol.,50,175-185.162. MANO,Y.,KITA,K.&KUSANO,K.2015.Hematocrit-independentrecovery
isakeyforbioanalysisusingvolumetricabsorptivemicrosamplingdevices,Mitra™.Bioanalysis,7,1821-9.
163. MANUILOVA,E.&SCHUETZENMEISTER,A.2014.MethodComparisonRegression[Online].Available:https://CRAN.R-project.org/package=mcr[Accessed19April2020].
164. MARTIAL,L.C.,AARNOUTSE,R.E.,SCHREUDER,M.F.,HENRIET,S.S.,BRÜGGEMANN,R.J.M.&JOORE,M.A.2016.CostEvaluationofDriedBloodSpotHomeSamplingasComparedtoConventionalSamplingforTherapeuticDrugMonitoringinChildren.PloSone,11.
165. MARTIN,N.J.&COOPER,H.J.2014.Challengesandopportunitiesinmassspectrometricanalysisofproteinsfromdriedbloodspots.ExpertRevProteom,11,685-695.
166. MATHEW,J.&VARACALLO,M.2020.Physiology,BloodPlasma.StatPearlsPublishing.
167. MAYADAS,T.N.,CULLERE,X.&LOWELL,C.A.2014.Themultifacetedfunctionsofneutrophils.AnnuRevPathol,9,181-218.
168. MCHUGH,D.,CAMERON,C.A.,ABDENUR,J.E.,ABDULRAHMAN,M.,ADAIR,O.,ALNUAIMI,S.A.,AHLMAN,H.,ALLEN,J.J.,ANTONOZZI,I.,ARCHER,S.,AU,S.,AURAY-BLAIS,C.,BAKER,M.,BAMFORTH,F.,BECKMANN,K.,PINO,G.B.,BERBERICH,S.L.,BINARD,R.,BOEMER,F.,BONHAM,J.,BREEN,N.N.,BRYANT,S.C.,CAGGANA,M.,CALDWELL,S.G.,CAMILOT,M.,CAMPBELL,C.,CARDUCCI,C.,BRYANT,S.C.,CAGGANA,M.,CALDWELL,S.G.,CAMILOT,M.,CAMPBELL,C.,CARDUCCI,C.,CARIAPPA,R.,CARLISLE,C.,CARUSO,U.,CASSANELLO,M.,CASTILLA,A.M.,RAMOS,D.E.,CHAKRABORTY,P.,CHANDRASEKAR,R.,RAMOS,A.C.,CHEILLAN,D.,CHIEN,Y.H.,CHILDS,T.A.,CHRASTINA,P.,SICA,Y.C.,DEJUAN,J.A.,COLANDRE,M.E.,ESPINOZA,V.C.,CORSO,G.,CURRIER,R.,CYR,D.,CZUCZY,N.,D'APOLITO,O.,DAVIS,T.,DESAIN-VANDERVELDEN,M.G.,DELGADOPECELLIN,C.,DIGANGI,I.M.,DISTEFANO,C.M.,DOTSIKAS,Y.,DOWNING,M.,DOWNS,S.M.,DY,B.,DYMERSKI,M.,RUEDA,I.,ELVERS,B.,EATON,R.,ECKERD,B.M.,ELMOUGY,F.,EROH,S.,ESPADA,M.,EVANS,C.,FAWBUSH,S.,FIJOLEK,K.F.,FISHER,L.,FRANZSON,L.,FRAZIER,D.M.,GARCIA,L.R.,BERMEJO,M.S.,GAVRILOV,D.,GERACE,R.,GIORDANO,G.,IRAZABAL,Y.G.,GREED,L.C.,GRIER,R.,GRYCKI,E.,GU,X.,GULAMALI-MAJID,F.,HAGAR,A.F.,HAN,L.,HANNON,W.H.,HASLIP,C.,HASSAN,F.A.,HE,M.,HIETALA,A.,HIMSTEDT,L.,HOFFMAN,G.L.,HOFFMAN,W.,HOGGATT,P.,etal.2011.Clinicalvalidationofcutofftargetrangesinnewbornscreeningofmetabolicdisordersbytandemmassspectrometry:aworldwidecollaborativeproject.GenetMed,13,230-54.
169. MCMILLIN,J.1990.BloodGlucose.3ed.ButterworthsBoston.170. MCPHEE,J.S.,FRENCH,D.P.,JACKSON,D.,NAZROO,J.,PENDLETON,N.&
DEGENS,H.2016.Physicalactivityinolderage:perspectivesforhealthyageingandfrailty.Biogerontology,17,567-580.
171. MEESTERS,R.2016.BioanalyticalevaluationofdriedplasmaspotmicrosamplingmethodologiesinpharmacokineticstudiesapplyingAcetaminophenasmodeldrug.JApplBioanal,2,25-37.
172. MEI,J.2014.DriedBloodSpotSampleCollection,Storage,andTransportation.JohnWiley&Sons.

- 121 -
173. MEI,J.V.,ZOBEL,S.D.,HALL,E.M.,DEJESUS,V.R.,ADAM,B.W.&HANNON,W.H.2010.Performancepropertiesoffilterpaperdevicesforwholebloodcollection.Bioanalysis,2,1397-403.
174. MICHIGANSTATEUNIVERSITY.2020.OxfordNanoporesamplerequirements[Online].Available:https://rtsf.natsci.msu.edu/genomics/sample-requirements/oxford-nanopore-sample-requirements/[Accessed16May2020].
175. MOHANDAS,N.,BASS-STRINGER,S.,MAKSIMOVIC,J.,CROMPTON,K.,LOKE,Y.J.,WALSTAB,J.,REID,S.M.,AMOR,D.J.,REDDIHOUGH,D.&CRAIG,J.M.2018.Epigenome-wideanalysisinnewbornbloodspotsfrommonozygotictwinsdiscordantforcerebralpalsyrevealsconsistentregionaldifferencesinDNAmethylation.ClinEpigenetics,10,25.
176. MOHS,R.C.&GREIG,N.H.2017.Drugdiscoveryanddevelopment:Roleofbasicbiologicalresearch.AlzheimersDement(NY),3,651-657.
177. NATIONALHEARTLUNGANDBLOODINSTITUTE.2020.BloodTests[Online].Available:https://www.nhlbi.nih.gov/health-topics/blood-tests[Accessed19May2020].
178. NATIONALINSTITUTEONAGING.2018.World’solderpopulationgrowsdramatically[Online].Available:https://www.nih.gov/news-events/news-releases/worlds-older-population-grows-dramatically[Accessed19April2020].
179. NEOTERYX.2019.Theuser-friendlysolutionforaccurateremotebloodcollection[Online].Available:https://www.neoteryx.com/[Accessed5June2020].
180. NIJENHUIS,C.M.,HUITEMA,A.D.,MARCHETTI,S.,BLANK,C.,HAANEN,J.B.,VANTHIENEN,J.V.,ROSING,H.,SCHELLENS,J.H.&BEIJNEN,J.H.2016.TheUseofDriedBloodSpotsforPharmacokineticMonitoringofVemurafenibTreatmentinMelanomaPatients.JClinPharmacol,56,1307-12.
181. NORTHEASTBIOLAB,N.2019.AllEssentialStepsInvolvedInDrugDiscoveryandDevelopmentProcess[Online].Available:https://www.nebiolab.com/drug-discovery-and-development-process/[Accessed22May2020].
182. O'MARA,M.,HUDSON-CURTIS,B.,OLSON,K.,YUEH,Y.,DUNN,J.&SPOONER,N.2011.Theeffectofhematocritandpunchlocationonassaybiasduringquantitativebioanalysisofdriedbloodspotsamples.Bioanalysis,3,2335-47.
183. ODOARDI,S.,ANZILLOTTI,L.&STRANO-ROSSI,S.2014.Simplifyingsamplepretreatment:applicationofdriedbloodspot(DBS)methodtobloodsamples,includingpostmortem,forUHPLC-MS/MSanalysisofdrugsofabuse.ForensicSciInt,243,61-7.
184. OOSTENDORP,M.,ELAMRANI,M.,DIEMEL,E.C.,HEKMAN,D.&VANMAARSEVEEN,E.M.2016.MeasurementofHematocritinDriedBloodSpotsUsingNear-InfraredSpectroscopy:Robust,Fast,andNondestructive.ClinChem,62,1534-1536.
185. ORNSTEIN,P.A.,MANNING,E.L.&PELPHREY,K.A.1999.Children'smemoryforpain.JDevBehavPediatr,20,262-77.
186. OZSOLAK,F.2012.Third-generationsequencingtechniquesandapplicationstodrugdiscovery.Expertopinionondrugdiscovery,7,231-243.
187. PADRÃO,P.,LUNET,N.,SANTOS,A.C.&BARROS,H.2007.Smoking,

- 122 -
alcohol,anddietarychoices:evidencefromthePortugueseNationalHealthSurvey.BMCpublichealth,7,138-138.
188. PANCHAL,T.,SPOONER,N.&BARFIELD,M.2016.Ensuringthecollectionofhigh-qualitydriedbloodspotsamplesacrossmultisiteclinicalstudies.Bioanalysis,9,209-213.
189. PARIKH,R.,MATHAI,A.,PARIKH,S.,CHANDRASEKHAR,G.&THOMAS,R.2008.Understandingandusingsensitivity,specificityandpredictivevalues.Indianjophthalmol,56,45-50.
190. PASQUINI,G.,ALBANESE,B.,MANESCALCHI,P.G.&MORINI,R.1983.Relationofbloodviscosity,plasmaviscosityandhematocrit.RicClinLab,13,327-331.
191. PATEL,S.R.,BRYAN,P.,SPOONER,N.,TIMMERMAN,P.&WICKREMSINHE,E.2019.Microsamplingforquantitativebioanalysis,anindustryupdate:outputfromanAAPS/EBFsurvey.Bioanalysis,11,619-628.
192. PETRICK,L.,EDMANDS,W.,SCHIFFMAN,C.,GRIGORYAN,H.,PERTTULA,K.,YANO,Y.,DUDOIT,S.,WHITEHEAD,T.,METAYER,C.&RAPPAPORT,S.2017.Anuntargetedmetabolomicsmethodforarchivednewborndriedbloodspotsinepidemiologicstudies.Metabolomics,13,27.
193. POLLARD,M.O.,GURDASANI,D.,MENTZER,A.J.,PORTER,T.&SANDHU,M.S.2018.Longreads:theirpurposeandplace.HumnMolGenet,27,234-241.
194. PRINYAKUPT,J.&PLUEMPITIWIRIYAWEJ,C.2015.Segmentationofwhiteblood cells and comparison of cell morphology by linear and naïve Bayesclassifiers.BiomedEngOnline,14,63-63.
195. PUGIA,M.J.,JORTANI,S.A.,BASU,M.,SOMMER,R.,KUO,H.H.,MURPHY,S.,WILLIAMSON,D.,VRANISH,J.,BOYLE,P.J.,BUDZINSKI,D.,VALDES,R.,JR.&BASU,S.C.2007.Immunologicalevaluationofurinarytrypsininhibitorsinbloodandurine:roleofN-&O-linkedglycoproteins.GlycoconjJ,24,5-15.
196. RANADE,V.1993.Cholesteroldetection,diagnosisandevaluation.IntJClinPharmacolTherToxicol,31,313-21.
197. REVERTER-BRANCHAT,G.,VENTURA,R.,EZZELDIN,M.,MATEUS,J.,PEDRO,C.&SEGURA,J.2018.Detectionoferythropoiesis-stimulatingagentsinasingledriedbloodspot.DrugTestAnal,10,1496-1507.
198. RIBEIRO,L.S.,MIGLIARIBRANCO,L.&FRANKLIN,B.S.2019.RegulationofInnateImmuneResponsesbyPlatelets.FrontImmunol,10.
199. RIVERS,P.A.&GLOVER,S.H.2008.Healthcarecompetition,strategicmission,andpatientsatisfaction:researchmodelandpropositions.JHealthOrganManag,22,627-641.
200. ROBERTS,J.,WILSON,I.,HENLEY,A.T.,RICHARDS,M.,THAI,C.&RAYNAUD,F.I.2016.CapillaryMicrosamplingofMouseBloodinEarlyPre-ClinicalStudies:APreferredAlternativetoDriedBloodSpotSampling.JBioanalBiomed,8,1-6.
201. RODRIGUEZ-AUAD,J.P.,ROJAS-MONTES,O.,MALDONADO-RODRIGUEZ,A.,ALVAREZ-MUÑOZ,M.T.,MUÑOZ,O.,TORRES-IBARRA,R.,VAZQUEZ-ROSALES,G.&LIRA,R.2015.UseofDriedPlasmaSpotsforHIV-1ViralLoadDeterminationandDrugResistanceGenotypinginMexicanPatients.BiomedResInt,2015,240407.
202. ROLA,R.,KOWALSKI,K.,BIEŃKOWSKI,T.,KOŁODYŃSKA-GOWOREK,A.&STUDZIŃSKA,S.2019.DevelopmentofamethodformultiplevitaminDmetabolitemeasurementsbyliquidchromatographycoupledwithtandemmass

- 123 -
spectrometryindriedbloodspots.Analyst,144,299-309.203. ROSALES,C.2018.Neutrophil:ACellwithManyRolesinInflammationor
SeveralCellTypes?Frontinphysiol,9,113-113.204. ROSENFELD,L.2002.ClinicalChemistrySince1800:Growthand
Development.ClinChem,48,186-197.205. ROSS,S.A.,NOVAK,Z.,PATI,S.&BOPPANA,S.B.2011.Overviewof the
diagnosisofcytomegalovirusinfection.InfectDisordDrugTargets,11,466-474.206. ROWLAND,M.&EMMONS,G.T.2010.Useofdriedbloodspotsindrug
development:pharmacokineticconsiderations.TheAAPSjournal,12,290-293.207. SANDLE,L.N.2005.Themanagementofexternalqualityassurance.JClin
Pathol,58,141-144.208. SARODE,R.2018.FormationofBloodCells[Online].Available:
https://www.msdmanuals.com/en-nz/home/blood-disorders/biology-of-blood/formation-of-blood-cells[Accessed19May2020].
209. SEPETIENE,R.,SIDLAUSKIENE,R.&PATAMSYTE,V.2018.PlasmaforLaboratoryDiagnostics,PlasmaMedicine-ConceptsandClinicalApplications,IntechOpen.
210. SHAH,V.P.,MIDHA,K.K.,FINDLAY,J.W.A.,HILL,H.M.,HULSE,J.D.,MCGILVERAY,I.J.,MCKAY,G.,MILLER,K.J.,PATNAIK,R.N.,POWELL,M.L.,TONELLI,A.,VISWANATHAN,C.T.&YACOBI,A.2000.BioanalyticalMethodValidation—ARevisitwithaDecadeofProgress.PharmRes,17,1551-1557.
211. SIRACUSA,M.C.,KIM,B.S.,SPERGEL,J.M.&ARTIS,D.2013.Basophilsandallergicinflammation.JAllergyClinImmunol,132,789-801.
212. SMIT,P.W.,ELLIOTT,I.,PEELING,R.W.,MABEY,D.&NEWTON,P.N.2014.Anoverviewoftheclinicaluseoffilterpaperinthediagnosisoftropicaldiseases.AmJTropMedHyg,90,195-210.
213. SMITH,S.A.,TRAVERS,R.J.&MORRISSEY,J.H.2015.Howitallstarts:Initiationoftheclottingcascade.CritRevBiochemMolBiol,50,326-36.
214. SOLOMON,S.S.,SOLOMON,S.,RODRIGUEZ,II,MCGARVEY,S.T.,GANESH,A.K.,THYAGARAJAN,S.P.,MAHAJAN,A.P.&MAYER,K.H.2002.Driedbloodspots(DBS):avaluabletoolforHIVsurveillanceindeveloping/tropicalcountries.IntJSTDAIDS,13,25-8.
215. SPOONER,N.2010.Driedbloodspotsamplingforquantitativebioanalysis:timeforarevolution?Bioanalysis,2,1781.
216. SPOTONSCIENCES.2020.HemaSpot[Online].Available:https://www.spotonsciences.com/hemaspot-hf[Accessed30May2020].
217. ST.JULIEN,K.R.,JELLIFFE-PAWLOWSKI,L.L.,SHAW,G.M.,STEVENSON,D.K.,O’BRODOVICH,H.M.&KRASNOW,M.A.2013.HighQualityGenome-WideGenotypingfromArchivedDriedBloodSpotswithoutDNAAmplification.PLOSONE,8,64710.
218. STOKES,A.H.,MOOSE,T.A.,PARRY,S.P.,BARFIELD,M.,LOVATT,C.A.,DOPSON,W.J.,MELICH,D.,OVERVOLD,C.R.,GADE,S.D.&SPOONER,N.2011.Determinationofdrugconcentrationsusingdriedbloodspots:investigationofbloodsamplingandcollectiontechniquesinCrl:CD(SD)rats.LabAnim,45,109-13.
219. STRAUSS,R.G.&MAUER,A.M.1978.FormedElementsofHumanBloodSpringerBoston.
220. STRINGER,K.A.,YOUNGER,J.G.,MCHUGH,C.,YEOMANS,L.,FINKEL,M.A.,PUSKARICH,M.A.,JONES,A.E.,TREXEL,J.&KARNOVSKY,A.2015.Whole

- 124 -
BloodRevealsMoreMetabolicDetailoftheHumanMetabolomethanSerumasMeasuredby1H-NMRSpectroscopy:ImplicationsforSepsisMetabolomics.Shock,44,200-208.
221. STURM,R.,HENION,J.,ABBOTT,R.&WANG,P.2015.Novelmembranedevicesandtheirpotentialutilityinbloodsamplecollectionpriortoanalysisofdriedplasmaspots.Bioanalysis,7,1987-2002.
222. SU,X., CARLSON,B. F.,WANG,X., LI,X., ZHANG,Y.,MONTGOMERY, J. P.,DING,Y.,WAGNER,A.L.,GILLESPIE,B.&BOULTON,M.L.2018.Driedbloodspots:Anevaluationofutilityinthefield.JInfectPublicHealth,11,373-376.
223. SUÁREZ-FUEYO,A.,CRISPÍN,J.C.&TSOKOS,G.C.2019.10-TCells.In:WALLACE,D.J.&HAHN,B.H.(eds.)Dubois'LupusErythematosusandRelatedSyndromes9ed.ElsevierLondon
224. TASSO.2020.ExpandVirtualCare[Online].Available:https://www.tassoinc.com/[Accessed20June2020].
225. TAVERNIERS,I.,DELOOSE,M.&VANBOCKSTAELE,E.2004.Trendsinqualityintheanalyticallaboratory.II.Analyticalmethodvalidationandqualityassurance.TrACTrendAnallChem,23,535-552.
226. THEODORSSON,E.&MAGNUSSON,B.2017.Fullmethodvalidationinclinicalchemistry.AccredQualAssur,22,235-246.
227. THERMOSCIENTIFIC.2007.Spike-and-recoveryandlinearity-of-dilutionassessment[Online].Available:http://tools.thermofisher.com/content/sfs/brochures/TR0058-Spike-and-Recovery.pdf[Accessed22May2020].
228. THEVIS,M.&THOMAS,A.2008.DriedBloodSpots(DBS)inSportsDrugTesting:ComplementaryMatrixProvidingUniqueFeaturesand.utilitiestomodernanti-dopingfights.TheWorldAnti-DopingAgency.Available:https://www.wada-ama.org/en/resources/research/dried-blood-spots-dbs-in-sports-drug-testing-complementary-matrix-providing[Accessed8June2020].
229. THEVIS,M.,KUURANNE,T.,DIB,J.,THOMAS,A.&GEYER,H.2020.Dodriedbloodspots(DBS)havethepotentialtosupportresultmanagementprocessesinroutinesportsdrugtesting?DrugTestAnal,12,704-710.
230. TIMMERMAN,P.,WHITE,S.,GLOBIG,S.,LÜDTKE,S.,BRUNET,L.&SMERAGLIA,J.2011.EBFrecommendationonthevalidationofbioanalyticalmethodsfordriedbloodspots.Bioanalysis,3,1567-1575.
231. TONG,P.C.,LEE,K.-F.,SO,W.-Y.,NG,M.H.,CHAN,W.-B.,LO,M.K.,CHAN,N.N.&CHAN,J.C.2004.WhiteBloodCellCountIsAssociatedWithMacro-andMicrovascularComplicationsinChinesePatientsWithType2Diabetes.DiabetesCare,27,216.
232. TOPTAS,B.,BAYKAL,A.,YESILIPEK,A.,ISBIR,M.,KUPESIZ,A.,YALCIN,O.&BASKURT,O.K.2006.L-carnitinedeficiencyandredbloodcellmechanicalimpairmentinbeta-thalassemiamajor.ClinHemorheolMicrocirc,35,349-57.
233. TOURNIS,S.&MAKRIS,K.2018.ClinicalUseofBoneTurnoverMarkersinOsteoporosis.Elsevier.
234. TRAJANSCIENTIFICANDMEDICAL.2020.Advancedprecisionmicrosampling[Online].Available:https://www.trajanscimed.com/pages/hemapen[Accessed5thJune2020].
235. TREVETHAN,R.2017.Sensitivity,Specificity,andPredictiveValues:Foundations,Pliabilities,andPitfallsinResearchandPractice.Frontiersinpublichealth,5,307-307.

- 125 -
236. TRIFONOVA,O.P.,MASLOV,D.L.,BALASHOVA,E.E.&LOKHOV,P.G.2019.EvaluationofDriedBloodSpotSamplingforClinicalMetabolomics:EffectsofDifferentPapersandSampleStorageStability.Metabolites,9,277.
237. TUAILLON,E.,MONDAIN,A.M.,MEROUEH,F.,OTTOMANI,L.,PICOT,M.C.,NAGOT,N.,VANDEPERRE,P.&DUCOS,J.2010.DriedbloodspotforhepatitisCvirusserologyandmoleculartesting.Hepatology,51,752-8.
238. URIBE,B.,GONZÁLEZ,O.,BLANCO,M.E.,ALBÓNIGA,O.E.,ALONSO,M.L.&ALONSO,R.M.2019.AnalysisoftheHeterogeneousDistributionofAmilorideandPropranololinDriedBloodSpotbyUHPLC-FLDandMALDI-IMS.Molecules,24,4320.
239. VEENHOF,H.,KOSTER,R.A.,ALFFENAAR,J.-W.C.,BERGER,S.P.,BAKKER,S.J.L.&TOUW,D.J.2017.ClinicalValidationofSimultaneousAnalysisofTacrolimus,CyclosporineA,andCreatinineinDriedBloodSpotsinKidneyTransplantPatients.Transplantation,101,1727-1733.
240. VELGHE,S.&STOVE,C.P.2018.EvaluationoftheCapitainer-BMicrofluidicDeviceasaNewHematocrit-IndependentAlternativeforDriedBloodSpotCollection.AnalChem,90,12893-12899.
241. VELGHE,S.,DELAHAYE,L.&STOVE,C.P.2019.Isthehematocritstillanissueinquantitativedriedbloodspotanalysis?JPharmBiomedAnal,163,188-196.
242. VERHAEGHE,T.,DILLEN,L.,STIELTJES,H.,ZWART,L.,FEYEN,B.,DIELS,L.,VROMAN,A.&TIMMERMAN,P.2017.TheapplicationofcapillarymicrosamplinginGLPtoxicologystudies.Bioanalysis,9,531-540.
243. VICTORIANCLINICALGENETICSSERVICES.2020.NewbornScreening[Online].Available:https://www.vcgs.org.au/tests/newborn-bloodspot-screening[Accessed22May2020].
244. VOLANI,C.,CAPRIOLI,G.,CALDERISI,G.,SIGURDSSON,B.B.,RAINER,J.,GENTILINI,I.,HICKS,A.A.,PRAMSTALLER,P.P.,WEISS,G.,SMARASON,S.V.&PAGLIA,G.2017.Pre-analyticevaluationofvolumetricabsorptivemicrosamplingandintegrationinamassspectrometry-basedmetabolomicsworkflow.Analyticalandbioanalyticalchemistry,409,6263-6276.
245. Walker,H.K.,Hall,W.D.,Hurst,J.W.1990.ClinicalMethods:TheHistory,Physical,andLaboratoryExaminations.3ed.ButterworthBoston.
246. WEN,D.,YANG,Y.,XIANG,P.,YU,F.,ZHENG,F.,LIU,T.,SHI,Y.,ZHANG,X.,DONG,M.,CONG,B.&MA,C.2018.Anovelapproachfordeterminationofparaquatbasedondriedbloodspot(DBS)extractionandUHPLC-HRMSanalysis.JournalofPharmaceuticalandBiomedicalAnalysis,159,11-17.
247. WEN,T.&ROTHENBERG,M.E.2016.TheRegulatoryFunctionofEosinophils.MicrobiolSpectr,4.
248. WICKHAM,H.2016.ggplot2:ElegantGraphicsforDataAnalysis.2ed.SpringerInternationalPublishing
249. WICKREMSINHE,E.R.&PERKINS,E.J.2015.Usingdriedbloodspotsamplingtoimprovedataqualityandreduceanimaluseinmousepharmacokineticstudies.JAmAssocLabAnimSci,54,139-44.
250. WICKREMSINHE,E.R.,HUANG,N.H.,ABDUL,B.G.,KNOTTS,K.,RUTERBORIES,K.J.&MANRO,J.R.2013.Preclinicalbridgingstudies:understandingdriedbloodspotandplasmaexposureprofiles.Bioanalysis,5,159-70.
251. WILHELM,A.J.,DENBURGER,J.C.G.&SWART,E.L.2014.Therapeutic

- 126 -
drugmonitoringbydriedbloodspot:progresstodateandfuturedirections.Clinicalpharmacokinetics,53,961-973.
252. WILLIAMS,R.A.,MAMOTTE,C.D.S.&BURNETT,J.R.2008.Phenylketonuria:aninbornerrorofphenylalaninemetabolism.TheClinicalbiochemist.Reviews,29,31-41.
253. WINTER,T.,LANGE,A.,HANNEMANN,A.,NAUCK,M.&MULLER,C.2018.Contaminationofdriedbloodspots-anunderestimatedriskinnewbornscreening.ClinChemLabMed,56,278-284.
254. WONG,N.C.,MORLEY,R.,SAFFERY,R.&CRAIG,J.M.2008.ArchivedGuthriebloodspotsasanovelsourceforquantitativeDNAmethylationanalysis.BioTechniques,45,423-430.
255. WOODWORTH,A.&PYLE,A.L.2013.SampleProcessingandSpecimenMisidentificationIssues.2ed.ElsevierSanDiego
256. WORLDHEALTHORGANIZATION2010a.Bestpracticeinphlebotomyandbloodcollection,Geneva:WorldHealthOrganization.
257. WORLDHEALTHORGANIZATION2010b.Capillarysampling,Geneva:WorldHealthOrganization.
258. YANG,J.,ZHANG,L.,YU,C.,YANG,X.-F.&WANG,H.2014.Monocyteandmacrophagedifferentiation:circulationinflammatorymonocyteasbiomarkerforinflammatorydiseases.BiomarkerResearch,2,1.
259. YE,Z.&GAO,H.2017.Evaluationofsampleextractionmethodsforminimizinghematocriteffectonwholebloodanalysiswithvolumetricabsorptivemicrosampling.Bioanalysis,9,349-357.
260. YISHAIAVIRAM,L.,MAGEN,M.,CHAPMAN,S.,NEUFELDCOHEN,A.,LAZAR,S.&DAGAN,S.2018.DryBloodSpotsamplecollectionforpost-exposuremonitoringofchemicalwarfareagents-InvivodeterminationofphosphonicacidsusingLC-MS/MS.JChromatogrBAnalytTechnolBiomedLifeSci,1093-1094,60-65.
261. YOSHIZAWA,J.M.,SCHAFER,C.A.,SCHAFER,J.J.,FARRELL,J.J.,PASTER,B.J.&WONG,D.T.W.2013.Salivarybiomarkers:towardfutureclinicalanddiagnosticutilities.Clinicalmicrobiologyreviews,26,781-791.
262. YOUHNOVSKI,N.,BERGERON,A.,FURTADO,M.&GAROFOLO,F.2011.Pre-cutdriedbloodspot(PCDBS):analternativetodriedbloodspot(DBS)techniquetoovercomehematocritimpact.RapidCommunMassSpectrom,25,2951-8.
263. ZAKARIA,R.&GREAVES,R.F.2019.There-emergenceofdriedbloodspotsampling–areweready?.ClinChemLabMed,57,1805.
264. ZAKARIA,R.,ALLEN,K.J.,KOPLIN,J.J.,ROCHE,P.&GREAVES,R.F.2016.AdvantagesandChallengesofDriedBloodSpotAnalysisbyMassSpectrometryAcrosstheTotalTestingProcess.EJIFCC,27,288-317.
265. ZARKYAW,T.,YAMAGUCHI,S.,IMAI,C.,UEMATSU,M.&SATO,N.2019.Theutilityofpost-testnewbornbloodspotscreeningcardsforepigeneticassociationanalyses:associationbetweenHIF3Amethylationandbirthweight-for-gestationalage.JournalofHumanGenetics,64,795-801.
266. ZHANG,Y.,ZHOU,W.-E.,YAN,J.-Q.,LIU,M.,ZHOU,Y.,SHEN,X.,MA,Y.-L.,FENG,X.-S.,YANG,J.&LI,G.-H.2018.AReviewoftheExtractionandDeterminationMethodsofThirteenEssentialVitaminstotheHumanBody:AnUpdatefrom2010.Molecules,23,1484.
267. ZWART,T.C.,GOKOEL,S.R.M.,VANDERBOOG,P.J.M.,DEFIJTER,J.W.,

- 127 -
KWEEKEL,D.M.,SWEN,J.J.,GUCHELAAR,H.-J.&MOES,D.J.A.R.2018.Therapeuticdrugmonitoringoftacrolimusandmycophenolicacidinoutpatientrenaltransplantrecipientsusingavolumetricdriedbloodspotsamplingdevice.Britishjournalofclinicalpharmacology,84,2889-2902.

- 128 -
Chapter8Appendix
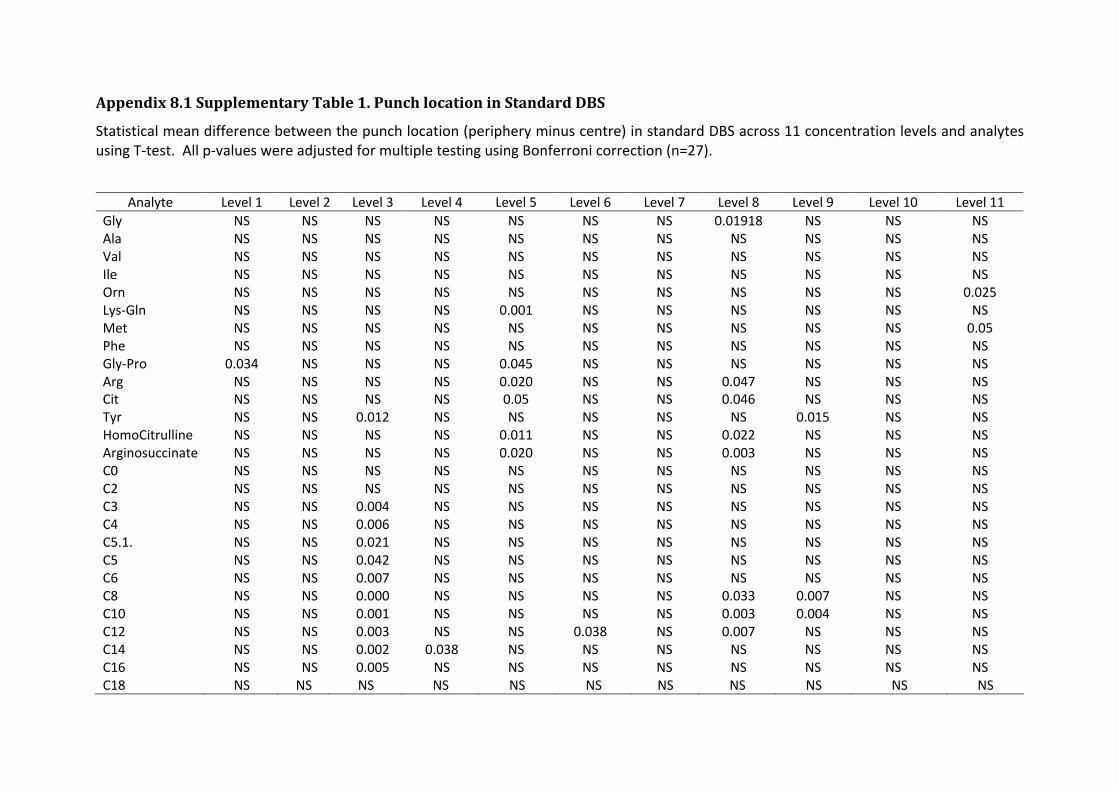
Appendix8.1SupplementaryTable1.PunchlocationinStandardDBS
Statistical mean difference between the punch location (periphery minus centre) in standard DBS across 11 concentration levels and analytes using T-test. All p-values were adjusted for multiple testing using Bonferroni correction (n=27).
Analyte Level 1 Level 2 Level 3 Level 4 Level 5 Level 6 Level 7 Level 8 Level 9 Level 10 Level 11 Gly NS NS NS NS NS NS NS 0.01918 NS NS NS Ala NS NS NS NS NS NS NS NS NS NS NS Val NS NS NS NS NS NS NS NS NS NS NS Ile NS NS NS NS NS NS NS NS NS NS NS Orn NS NS NS NS NS NS NS NS NS NS 0.025 Lys-Gln NS NS NS NS 0.001 NS NS NS NS NS NS Met NS NS NS NS NS NS NS NS NS NS 0.05 Phe NS NS NS NS NS NS NS NS NS NS NS Gly-Pro 0.034 NS NS NS 0.045 NS NS NS NS NS NS Arg NS NS NS NS 0.020 NS NS 0.047 NS NS NS Cit NS NS NS NS 0.05 NS NS 0.046 NS NS NS Tyr NS NS 0.012 NS NS NS NS NS 0.015 NS NS HomoCitrulline NS NS NS NS 0.011 NS NS 0.022 NS NS NS Arginosuccinate NS NS NS NS 0.020 NS NS 0.003 NS NS NS C0 NS NS NS NS NS NS NS NS NS NS NS C2 NS NS NS NS NS NS NS NS NS NS NS C3 NS NS 0.004 NS NS NS NS NS NS NS NS C4 NS NS 0.006 NS NS NS NS NS NS NS NS C5.1. NS NS 0.021 NS NS NS NS NS NS NS NS C5 NS NS 0.042 NS NS NS NS NS NS NS NS C6 NS NS 0.007 NS NS NS NS NS NS NS NS C8 NS NS 0.000 NS NS NS NS 0.033 0.007 NS NS C10 NS NS 0.001 NS NS NS NS 0.003 0.004 NS NS C12 NS NS 0.003 NS NS 0.038 NS 0.007 NS NS NS C14 NS NS 0.002 0.038 NS NS NS NS NS NS NS C16 NS NS 0.005 NS NS NS NS NS NS NS NS C18 NS NS NS NS NS NS NS NS NS NS NS

Appendix8.2Extractionkitsprotocol
Quick-DNA™ Miniprep Plus Kit
Extra order: 1. 2 mm Bashing Bead (Zymo Research, United States) 2. lysis solution (Zymo Research, United States)
Day 1 Paper based substrates:
1. Punch out 4 x 3 mm spots 2. Add 2 mm Bashing Beads 3. Soak over-night in 360 µL PBS and 40 µL proteinase K (20 mg/mL ) 4. Centrifuge briefly and incubate at 37°C overnight
Day 2 1. mPPM and paper-based substrates: Add 400µL of lysis solution (and
beads for mPPM) and homogenise with beads in Tissue Lyser for 20 sec 30Hz flip it over and repeat
2. Pre-warm DNA elution buffer at 55-60°C 3. Centrifuge the sample tube for 1 minute no more than 10 000 rpm 4. To the sample add:
a. 390µL of digestion buffer b. 10µL Proteinase K (20 mg/mL )
5. Mix and incubate tube at 55°C for 30 minutes 6. Add 1.3 ml of DNA isolation buffer into new flacon tube 7. Let sample to cool at room temperature for 3 - 4 minutes 8. Centrifuge sample for 1 minute for no more than 8 000 rpm 9. Transfer 650 µL of supernatant to the flacon tube that contains 1.3 ml
of isolation buffer (the ratio should be 1:2 supernatant : isolation buffer)
10. Transfer 650 µL ( max 800 µL) of mixture into the spin column in a collection tube
11. Spin for 1 min at 14 000 rpm, discard flow through and repeat until the entire volume of the mixture in the flacon tube has passed through the column
12. Add 200 µL of DNA wash buffer to the spin column (check if ethanol was added)
13. Centrifuge for 1 minute at 14 000 rpm 14. Repeat step 12 15. Transfer the spin column into a clean 1.5ml microcentrifuge tube.
Allow the spin columns to dry by leaving the caps open for 4 minutes in room temperature
16. Add 20 µL of pre-warmed DNA elution buffer into the column carefully (aiming for the middle). Incubate for 15 minutes at room temperature
17. Centrifuge it for 2 min at 14 000rpm 18. Add another 20 µL pre-warmed DNA elution buffer into a spin column
and leave for another 15 minute 19. Centrifuge it for 2 min at 14000 rpm

- 131 -
20. Speed vac samples and reconstitute in 40µL TE QIAamp® DNA Blood Mini (Qiagen, Germany) Extra order:
1. QIAamp DNA, 3 mm Tungsten bead (Qiagen, Germany) 2. Buffer ATL Tissue Lysis (Qiagen, Germany)
Day 1 1. Paper based substrates: 2. Punch out 4x 3 mm spots 3. Add 1x Tungsten bead 4. Soak over-night in 360 µL PBS and 40 µL proteinase K (20 mg/mL ) 5. Centrifuge briefly and incubate at 37°C overnight
Day 2 1. mPPM and paper based substrate: Add 400 µ l of ATL Tissue Lysis buffer
(and 1x tungsten bead to mPPM) 2. Homogenise samples with beads in Tissue Lyser 5 x 20 seconds 30Hz 3. Spin down and transfer supernatant to 2 ml tube that contains proteinase
K (10 mg/mL ) 4. Add 200µL Buffer ATL into substrate homogenate and incubate for 10
minute at 55°C 5. Spin down and pool supernatant 6. Incubate mixture that contains proteinase K for 1 hours at 56°C 7. Add 400 µL Buffer AL and incubate at 70°C for 10 minute 8. Add 400 µL of 100% ethanol vortex and spin 9. Apply ~ 620 µL of mixture to spin column and centrifuge at 8000 rpm for 3
min. Place column in new tube and continue until all sample passed through column
10. Place column in fresh collection tube. Carefully open the spin column and add 500 µL buffer AW1 (wash 1)without wetting the rim. Close the cap and centrifuge at 8000 rpm for 3 minute
11. Replace the spin column in a clean 2 ml tube and discard the tube containing filtrate. Carefully open the spin column and add 500 µL buffer AW2 (wash 2). Close the cap and centrifuge at full spread for 3 minute.
12. Place the spin column in a new 1.5 ml tube and discard the tube with filtrate. Centrifuge at full spread for 1 minute
13. Place the spin column in a new tube and discard the tube with filtrate 14. Carefully open the spin column and add 100 µL of AE incubate at room
temperature for 10 minute 15. Centrifuge at 8000 rpm for 1 minute 16. Repeat Carefully open the spin column and add 100 µL of AE (elution)
incubate at room temperature for 10 minute 17. Centrifuge at 8000 rpm for 1 minute 18. Carefully open the spin column and add 50 µL of AE (elution) incubate at
room temperature for 10 minute 19. Centrifuge at 8000 rpm for 1 minute 20. Speed vac samples and reconstitute in 40µL TE

- 132 -
MagMAX™ CORE Nucleic Acid Purification Kit Extra order:
1. QIAamp DNA, 3 mm Tungsten bead (Qiagen, Germany) 2. PK Buffer (Life Technologies Thermo Fisher Scientific, United States)
Day 1 1. Paper based substrates: 2. Punch out 4x 3 mm spots 3. Add 1x Tungsten bead 4. Soak over-night in 360 µL PBS and 40 µL proteinase K (20 mg/mL ) 5. Centrifuge briefly and incubate at 37°C overnight
Day 2 1. mPPM and paper based substrates: Add 400µL of multi DNA lysis buffer
(and 1x tungsten bead to mPPM) 2. homogenise with beads in Tissue Lyser 5 x 20sec 30 Hz 3. Prepare enzyme mix (16µL proteinase K, 184µL PK digestion buffer per
reaction) 4. Add 200 µL of enzyme mix into samples containing DBS 5. Incubate for 20 min at 60°C 6. Quick spin, transfer liquid into new tube leaving card material behind 7. Shake tube on vortex adaptor for 3 min 8. Prepare DNA binding bead mix (350 µL binding solution, 20µL Binding
beads, 30µ water) 9. Add 400 µL of binding bead mix into a sample and shake for anther 3
minutes 10. Add 480 µL of 100 % isopropanol to the sample and shake for another 3
minutes 11. Place the tube on the magnetic stand for 5 minutes or until solution clears 12. While the tube is on the magnet discard supernatant, being careful not to
disturb beads 13. Wash sample with 300 µL was solution 1 (remove the tube from magnetic
stand add 300 µL of wash solution then vortex the tube in pulses for 10 seconds being careful to prevent beads from sticking to the side of the tube)
14. Place tube on the magnetic stand for 1 minute until all beads are against the magnet
15. While the tube is on the magnet discard supernatant 16. Repeat step 14 with wash solution 2 17. Air dry uncapped sample on magnetic stand for 3 minutes 18. Add 50 µL of elution buffer to sample incubate sample on heat block for 30
min at 70°C 19. Vortex sample until resuspended, 20. Place the tube on magnetic stand for 5 minutes or until solution clears 21. Keeping the plate on the magnet transfer eluate which now contains DNA
to new 2 mm tube 22. Speed vac samples and reconstitute in 40µL TE

- 133 -

- 134 -
Appendix8.3EthicapprovalandGovernanceauthorisation
Page 1 of 2
ETHICS APPROVAL & GOVERNANCE AUTHORISATION 9 February 2018 Associate Professor J Craig Environmental and Genetic Epidemiology Research Murdoch Children’s Research Institute Dear A/Prof. Craig, Project Title: Advancing the collection and storage of blood micro-volumes for clinical and research applications RCH HREC Reference Number: 37299B I am pleased to advise that the below amendment has received ethical approval from The Royal Children’s Hospital Melbourne Human Research Ethics Committee (HREC). The HREC confirms that your proposal meets the requirements of the National Statement on Ethical Conduct in Human Research (2007). This HREC is organised and operates in accordance with the National Health and Medical Research Council’s (NHRMC) National Statement on Ethical Conduct in Human Research (2007), and all subsequent updates, and in accordance with the Note for Guidance on Good Clinical Practice (CPMP/ICH/135/95), the Health Privacy Principles described in the Health Records Act 2001 (Vic) and Section 95A of the Privacy Act 1988 (and subsequent Guidelines). The amendment has also received governance authorisation at the Melbourne Children’s Campus (incorporating The Royal Children’s Hospital, Murdoch Children’s Research Institute and the University of Melbourne Department of Paediatrics). HREC Approval Date: 9 February 2018 Participating Sites: Ethical approval for this project applies at the following sites:
Site Name
x Melbourne Children’s Campus (incorporating The Royal Children’s Hospital, Murdoch Children’s Research Institute and the University of Melbourne Department of Paediatrics).
Approved Documents: The following documents have been reviewed and approved:
Document Version Date Protocol 3.0 6 February 2018 PICF - Phlebotomists 3.0 31 January 2018 PICF - Participant 3.0 31 January 2018 PICF - Venous donors 3.0 31 January 2018 hemaPEN Instructions for use 1709-014v3 6 February 2018 hemaPEN disassembly instructions for use 1709-019v1 22 May 2017 Video on hemaPEN use VI-0010-G Rev C 2 February 2018 Video on hemaPEN disassembly VI-0013-G-Rev A 22 May 2017

- 135 -
Page 2 of 2
Conditions of Ethics Approval: x You are required to submit to the HREC:
x An Annual Progress Report (that covers all sites listed on approval) for the duration of the project. This report is due on the anniversary of HREC approval. Continuation of ethics approval is contingent on submission of an annual report, due within one month of the approval anniversary. Failure to comply with this requirement may result in suspension of the project by the HREC.
x A comprehensive Final Report upon completion of the project. x Submit to the reviewing HREC for approval any proposed amendments to the project including any
proposed changes to the Protocol, Participant Information and Consent Form/s and the Investigator Brochure.
x Notify the reviewing HREC of any adverse events that have a material impact on the conduct of the research in accordance with the NHMRC Position Statement: Monitoring and reporting of safety for clinical trials involving therapeutic products May 2009.
x Notify the reviewing HREC of your inability to continue as Coordinating Principal Investigator. x Notify the reviewing HREC of the failure to commence the study within 12 months of the HREC
approval date or if a decision is taken to end the study at any of the sites prior to the expected date of completion.
x Notify the reviewing HREC of any matters which may impact the conduct of the project. x If your project involves radiation, you are legally obliged to conduct your research in accordance
with the Australian Radiation Protection and Nuclear Safety Agency Code of Practice ‘Exposure of Humans to Ionizing Radiation for Research Purposes’ Radiation Protection series Publication No.8 (May 2005)(ARPANSA Code).
x The HREC, authorising institution and/or their delegate/s may conduct an audit of the project at any time.
RCH HREC do NOT require existing consented participants to be re-consented on this new approved version of the PICF. This version should be used for future participants (if applicable). Yours sincerely,
Deeptika Chauhan Research Ethics and Governance Officer Research Ethics and Governance The Royal Children’s Hospital Melbourne Phone : (03) 9345 5044 Email : [email protected] Web : www.rch.org.au

- 136 -
Appendix8.4hemaPEN®instructionmanual
1709-014 rev3 ©Trajan Scientific and Medical 02/2018 Page 1 of 10
hemaPEN® Instructions for Use
hemaPEN® Instructions for Use
PLEASE READ THESE INSTRUCTIONS FOR USE BEFORE OPERATING THE HEMAPEN DEVICE
The hemaPEN device is in DEVELOPMENT and FOR RESEARCH or INVESTIGATIONAL PURPOSES ONLY.
This device is NOT FOR USE IN HUMAN APPLICATIONS OR DIAGNOSTIC PROCEDURES
Manufacturer: Trajan Scientific Australia Pty Ltd, 7 Argent Place Ringwood VIC 3134 Australia.
Tel: +61 (0) 2 9609 1755 Web: www.trajanscimed.com
Description:
The hemaPEN has been designed and assembled by Trajan Scientific and Medical for use as a micro sampling device specifically for the collection and storage of blood in four discrete dried blood spots (DBS). The device is supplied as non-sterile and is single use only.
Figure 1: hemaPEN Components
Data Matrix Label
Base (Cap)
Main Body
Top
Capillaries
Adhesive Tape
Green Tab

- 137 -
1709-014 rev3 ©Trajan Scientific and Medical 02/2018 Page 2 of 10
hemaPEN® Instructions for Use
Instructions:
These instructions for use may be used in conjunction with a device training video.
The instructions are intended for the user to collect blood from the participant. It is always recommended that you first wash your hands with soap and warm water, rinse and dry completely before using the hemaPEN. You may choose to wear gloves when taking or handling blood from the participant. Follow your institutions’ biohazard policy and wear the appropriate personal protective equipment.
Before unpacking and using the device ensure you have:
x A clean flat work surface
x A permanent marker to write on a label
x Sufficient time to complete the procedure uninterrupted
Lay out the contents of the hemaPEN kit on the work surface.
Verify the kit contents:
1 x instructions for use
1x silver opaque Zip Lock heat sealed bag which contains the hemaPEN device
Inspect the silver Zip Lock bag and check that the heat seal is intact and that the bag is not damaged.
PRECAUTION: IF NOT USING IMMEDIATELY STORE THE UNOPENED hemaPEN PACKAGE IN A DRY ENVIRONMENT AT ROOM TEMPERATURE AND NOT EXPOSED TO DIRECT SUNLIGHT.

- 138 -
1709-014 rev3 ©Trajan Scientific and Medical 02/2018 Page 3 of 10
hemaPEN® Instructions for Use
Step 1 – Opening the Package
PRECAUTION: USE THE hemaPEN WITHIN 30 MINUTES ONCE REMOVED FROM THE SILVER ZIP LOCK BAG.
WARNING: THE hemaPEN CONTAINS GLASS COMPONENTS. CARE IS REQUIRED WHEN OPENING THE PACKAGE IN THE RARE EVENT THAT CAPILLARIES ARE BROKEN DURING TRANSPORT.
Tear open the silver Zip Lock bag using the notch as a guide to remove the heat sealed section.
The bag is intended for reuse for storage and transport of the hemaPEN after the sampling procedure.
Do not tear or damage the bag below the Zip Lock section.
Do not remove the desiccant sachet from inside the Zip Lock bag and do not discard the bag.
Step 2 – Preparation of hemaPEN for Use
Describes the procedure to remove the hemaPEN from its packaging and prepare the device for sample collection
Remove the hemaPEN located in its protective plastic tray from inside the bag and place on the work surface and do not ‘tip’ it out of the bag.

- 139 -
1709-014 rev3 ©Trajan Scientific and Medical 02/2018 Page 4 of 10
hemaPEN® Instructions for Use
Reseal the Zip Lock bag and set aside for later use. The bag is used for transport, so do not discard.
Pick up the plastic tray, containing the hemaPEN, and cradle in your hand with the open side of the protective tray facing upwards as illustrated.
To remove the hemaPEN from the tray insert a finger into the bottom opening of the clear plastic base and gently lift the device from the protective plastic as illustrated in the cartoon.
A piece of adhesive tape with a green tab holds together the white Main Body of the device and the clear plastic base. Hold the white Main Body of the device and place the hemaPEN upright on the work surface, resting on the clear plastic base.
PRECAUTION: DO NOT APPLY PRESSURE OR PUSH THE WHITE MAIN BODY INTO THE BASE. THIS WOULD RENDER THE DEVICE UNUSABLE. THE DEVICE IS RESTING LOOSLY ON ITS BASE. DO NOT KNOCK THE hemaPEN AS THE DEVICE COULD FALL AND DAMAGE THE CAPILLARIES.
Approach the hemaPEN side on with your hand and hold the Main Body and the base together being careful not to apply downward pressure onto the base. Take hold of the green tab and peel off the tape by pulling upwards and discard the tape.
The hemaPEN is now ready for use and will be resting loosely on its base.

- 140 -
1709-014 rev3 ©Trajan Scientific and Medical 02/2018 Page 5 of 10
hemaPEN® Instructions for Use
PRECAUTION: TO PREVENT CONTAMINATION OR BREAKAGE DO NOT TOUCH THE CAPILLARIES AT THE TIP OF THE hemaPEN PRIOR TO BLOOD COLLECTION.
PRECAUTION: AS SOON AS THE SAMPLE COLLECTION IS INITIATED ALL STEPS MUST BE CARRIED OUT IN A TIMELY MANNER TO ENSURE OPTIMUM DEVICE PERFORMANCE. THE PROCEDURE SHOULD TAKE LESS THAN ONE (1) MINUTE.
COLLECT – CAP – FLIP
Step 3 – Collect Blood Sample
Describes how the hemaPEN is used to ensure the optimal collection of blood sample from a variety of sources, eg tubes, fingers etc
Finger capillary blood
Finger Capillary Blood may be collected in authorised sites and facilities. The hemaPEN is not supplied with a lancet and institutional protocols must be followed if this will be the source of blood.
Where a lancet is available and the hemaPEN has been approved for clinical use in approved studies, institutional protocols for the use of a lancet and collection of capillary blood must be followed.
We recommend following the WHO guidelines for capillary blood sampling. The user and/or individual whose blood will be collected (Study Participant) should wash their hands with soap and warm water, rinse and dry them. Blood flow should be encouraged by massaging the hand of the Study Participant in a downward motion towards the finger tips.
Wipe the proposed puncture site with an alcohol swab and allow to dry. Hold the middle or ring finger firmly and puncture the side of the finger using a sterile lancet. Encourage blood flow and wipe away the first droplet of blood with a piece of sterile gauze or tissue.

- 141 -
1709-014 rev3 ©Trajan Scientific and Medical 02/2018 Page 6 of 10
hemaPEN® Instructions for Use
Allow the blood to bead into a small droplet.
Remove the hemaPEN from the base using your dominant hand as if using a pen to write with.
Hold the hemaPEN horizontal to the sample and gently touch the tip onto the surface of the blood. The hemaPEN should be held steady in the blood for approximately 10 seconds. Blood can be observed entering and moving up the capillaries.
PRECAUTION: DO NOT SUBMERGE THE CAPILLARIES INTO THE BLOOD DROPLET SUCH THAT THEY CONTACT THE SURFACE UNDERNEATH. IF CONTACT OCCURS THIS MAY BLOCK THE CAPILLARY TIP AND PREVENT OR SLOW BLOOD COLLECTION
Replace the body of the hemaPEN onto the clear plastic base in a vertical (upright) position.
Proceed immediately to Step 4 – Cap the hemaPEN
Blood Collection from a Tube
In cases where the blood source contains anti-coagulant a blood droplet may be deposited onto a clean surface such as a centrifuge tube cap. The blood can be collected using the hemaPEN as illustrated below.

- 142 -
1709-014 rev3 ©Trajan Scientific and Medical 02/2018 Page 7 of 10
hemaPEN® Instructions for Use
This procedure may be used for training in the hemaPEN use.
Follow the blood collection instructions as described above and proceed to Step 4.
Step 4 – Cap the hemaPEN
Describes the procedure to ensure the hemaPEN is correctly engaged with the base to seal the device and prevent reuse. Performing the correct capping procedure initiates the sample transfer to the DBS paper and maintains the sample integrity. This should be performed within 30 seconds of blood collection.

- 143 -
1709-014 rev3 ©Trajan Scientific and Medical 02/2018 Page 8 of 10
hemaPEN® Instructions for Use
Firmly hold the clear base (cap) of the hemaPEN with your non-dominant hand.
Using your dominant hand push down the top of the hemaPEN until it engages with the base (cap) with a “CLICK”.
Step 5 – Flip hemaPEN
Describes the confirmation that the sample transfer has either initiated or completed by inverting the hemaPEN

- 144 -
1709-014 rev3 ©Trajan Scientific and Medical 02/2018 Page 9 of 10
hemaPEN® Instructions for Use
Turn the hemaPEN upside down (Flip). Hold the inverted hemaPEN up at eye level and inspect the glass capillaries to check that the blood transfer is underway or completed.
The blood in the glass capillaries will now empty (transfer) onto the paper pads inside the body of the hemaPEN. The transfer may take 60 seconds to complete.
You do not have to wait until the transfer is complete before going to Step 6.
PRECAUTION: DO NOT ATTEMPT TO FORCEABLY REMOVE THE ENGAGED hemaPEN FROM THE CLEAR PLASTIC BASE. THIS WILL DAMAGE THE DEVICE AND COMPROMISE THE INTEGRITY OF THE COLLECTED SAMPLE.
Step 6 – Repackaging
Describes the additional labelling requirements, placement of the used hemaPEN into the Zip Lock bag for drying the samples as dried blood spots and storage under dry conditions to maintain sample integrity.
Open the original Zip Lock bag and check that the desiccant sachet is in the bag.
Return the hemaPEN to the bag with the desiccant and reseal the bag.
The hemaPEN should remain in the bag (for at least 60 minutes) so the samples dry completely before attempting to perform any analysis on the DBS.

- 145 -
1709-014 rev3 ©Trajan Scientific and Medical 02/2018 Page 10 of 10
hemaPEN® Instructions for Use
The hemaPEN may be shipped to a separate location for testing using standard mail or courier services however it is subject to aviation security and clearing procedures as identified in the aviation security and dangerous goods declaration.
The Zip Lock bag must identify the content as a “dried blood spot not restricted by International Air Transport Association (IATA)” and display a biohazard symbol.
The bag can then be placed and shipped in a standard postal bag or envelope with the delivery and return address clearly marked.
PRECAUTION: IF THE SAMPLE IS KNOWN TO CONTAIN INFECTIOUS MATERIAL CONSULT A MEDICAL PRACTITIONER TO ENSURE THE SAMPLE IS SAFE TO TRANSPORT VIA THE MAIL SERVICE. SPECIAL PRECAUTIONS MAY BE REQUIRED. ALWAYS FOLLOW INTERNATIONAL, NATIONAL AND LOCAL REGULATIONS WITH RESPECT TO PACKAGING AND LABELLING.
Dispose of any waste materials and biological samples in accordance with local waste management policies and procedures.
References
GHTF/SG1/N70:2011 Label and Instructions for Use for Medical Devices
Australian Regulatory Guidelines for Medical Devices: Essential Principle 13
http://www.cdc.gov/labstandards/pdf/nsqap/Bloodspot_Transportation_Guidelines.pdf
World Health Organization Guidelines on drawing blood: best practices in phlebotomy (2010) ISBN 978 924 159922 1 Chapter 7: Capillary Sampling

- 146 -
Appendix8.5hemaPEN®video
To view the hemaPEN video used for assisted collection please click on the below link.
https://www.youtube.com/watch?v=M92pTN2WKiM&feature=youtu.be
Copyright 2020 by Trajan Scientific and Medical. Printed with permission

- 147 -
Appendix8.6Summarytable:Feedbackfromdonorsandcollectors
Summary table of feedback from donors on hemaPEN® use 1 age 55,60,55,40,34 2 sex 5 x female 3 Occupation Purchasing, receptionist,
admin assistant, research assistant, team coordinator
4 Did you like the design of hemaPEN 5 x yes 5 What did you like about the hemaPEN “Painless”, “easy to use”,
“great design”, “it’s not threatening”, “quick and easy collection”
6 What didn’t you like about the hemaPEN 5 x nothing 7 Is there anything you would change and what? 5 x nothing 8 Do you feel anxious having blood collected from a
vain in your arm 4 x no 1 x yes
9 After having your blood collected by the phlebotomist, would you prefer hemaPEN collection over collection from a vein in your arm?
3 x yes 2 x no
10 How painful was the finger prick 5 x not painful 0 x somewhat painful 0 x painful
11 finger prick yourself 5 x yes 0 x no
12 Collect blood sample using hemaPEN yourself 4 x yes 1 x no – didn’t work
13 If you needed your blood tested frequently, do you think you would be able to collect it yourself using the hemaPEN?
5 x yes 0 x no
14 If you needed your blood tested frequently, do you think you would prefer to use hemaPEN over blood collected from your arm (given appropriate training)?
5 x yes 0 x no

- 148 -
Summary table of feedback from phlebotomists on hemaPEN® use
1 In our opinion, do you consider yourself:
0 x less experienced
3 x experienced
3 x very experienced
2
How long have you been qualified to
collect blood for?
Please state the word "year" or "month"
4 years 6 months
9 years
2.5 years
26 years
5.5 years
4 years
3
In our opinion, how experienced would
you consider yourself in lancet finger
pricking?
0 x less experienced
1 x experienced
5 x very experienced
4 What lancet do you use:
Sarstedt lancet
Video tutorial question
5 Did you find the video tutorial useful? 6 x yes
0 x no
6 Did you find the video easy to follow?
6x yes
0 x no
7
How beneficial was the video prior to
collection?
Not beneficial b) somewhat beneficial c)
very beneficial
0 x not beneficial
2x Somewhat beneficial
4x very beneficial
8 Other comments regarding the video “It's been really helpful to have visual
teaching”.
Written instructions
10 Did you find the instruction useful?
6x yes
0 x no
11 How beneficial were the written
instructions prior to collection?
0 x not beneficial
3 x somewhat beneficial

- 149 -
3 x Very beneficial
12 Were the instructions:
5 x Clear and easy to follow
2 x detailed-all relevant information
provided
1 x too long
13 Other comments regarding the
instructions
“It's better with pictures so people with
NESB will be easy to follow too”
Practice use of hemaPEN prior to volunteer’s collection
14 Testing the hemaPEN prior collection was
0 x not useful
1 x Somewhat useful
5 x very useful
15 What form of learning did you prefer?
4 x video
1 x instruction
3 x practice collection
16 What form of learning gave you the best
idea on how to use the hemaPEN?
4x video
1 x instruction
2 x practice collection
Use of hemaPEN when collecting samples
17 How easy was collection using hemaPEN?
4 x easy
2 x somewhat easy
0 x not easy
18 Did you have any issues using hemaPEN
6 x no
0 x yes
19 Was there something that surprised you 2 x yes
4 x no
If Yes:
“The blood didn't fully draw from micro
tubes”.
“It was harder than I thought”
20
Would you consider using the hemaPEN
instead of the current method for
newborn screening?
3 x yes
3 x no

- 150 -
If Yes:
“If it's fully approved”
“With practice makes for a better collection”
“Only requiring one decent drop of blood”
“Quicker”
22
Would you consider using the hemaPEN in
the paediatric setting?
3x yes
2x no
If Yes:
“Quick and easy”
“Will be interesting device to use”
23 Do you like the design of the hemaPEN
6 x yes
0 x no
24 What did you like about the hemaPEN
“Easy to use”, “Ease of use”, ”quick and
not messy”, “It’s Fancy”, “It’s ok”, “It’s
function overall”
25 What didn't you like about the hemaPEN
“Having to hold it steady for 10 seconds”
“Some of the device didn't drawn or
click properly and affected the blood
drawing”
26 Is there anything you would change 5 x No
1 x yes
If Yes: Quicker collection




















