
A Saccharomyces cerevisiae Genome-Wide Mutant Screen for Altered Sensitivity to K1 Killer Toxin
20
Copyright 2003 by the Genetics Society of America A Saccharomyces cerevisiae Genome-Wide Mutant Screen for Altered Sensitivity to K1 Killer Toxin Nicolas Page ´, 1 Manon Ge ´rard-Vincent, Patrice Me ´nard, Maude Beaulieu, Masayuki Azuma, 2 Gerrit J. P. Dijkgraaf, Huijuan Li, Jose ´ Marcoux, Thuy Nguyen, Tim Dowse, Anne-Marie Sdicu and Howard Bussey 3 Biology Department, McGill University, Montreal, Quebec H3A 1B1, Canada Manuscript received November 5, 2002 Accepted for publication December 12, 2002 ABSTRACT Using the set of Saccharomyces cerevisiae mutants individually deleted for 5718 yeast genes, we screened for altered sensitivity to the antifungal protein, K1 killer toxin, that binds to a cell wall -glucan receptor and subsequently forms lethal pores in the plasma membrane. Mutations in 268 genes, including 42 in genes of unknown function, had a phenotype, often mild, with 186 showing resistance and 82 hypersensitiv- ity compared to wild type. Only 15 of these genes were previously known to cause a toxin phenotype when mutated. Mutants for 144 genes were analyzed for alkali-soluble -glucan levels; 63 showed alterations. Further, mutants for 118 genes with altered toxin sensitivity were screened for SDS, hygromycin B, and calcofluor white sensitivity as indicators of cell surface defects; 88 showed some additional defect. There is a markedly nonrandom functional distribution of the mutants. Many genes affect specific areas of cellular activity, including cell wall glucan and mannoprotein synthesis, secretory pathway trafficking, lipid and sterol biosynthesis, and cell surface signal transduction, and offer new insights into these processes and their integration. T HE sequenced and analyzed Saccharomyces cerevisiae are involved in cell wall synthesis and regulation (Shahi- nian and Bussey 2000). Here we describe the results genome has enabled a program of precise targeted of global screens of haploids and homozygous and het- gene disruption, resulting in a collection of mutant strains erozygous diploid mutants for altered K1 toxin sensi- deficient in each gene (Winzeler et al. 1999; Giaver tivity. et al. 2002; see also: http://sequence-www.stanford.edu/ group/yeast/yeast_deletion_project/deletions3.html). Such a collection promotes the discovery of cellular MATERIALS AND METHODS roles for genes by facilitating the characterization of mutant phenotypes and allows a comprehensive exami- Strains and media: Wild-type strains were BY4742 (MAT) nation of the genetic complexity of a phenotype. We and BY4743 (MATa/MAT; Brachmann et al. 1998), except have used the S. cerevisiae gene disruption set to screen where noted in Figure 1B, which also presents some results from strain SEY6210 (Robinson et al. 1988). Deletant strains for K1 killer toxin phenotypes. Toxin resistance has were from the Saccharomyces Genome Deletion Consortium been extensively studied by classical genetics, and many (Giaver et al. 2002) and are available at Research Genetics genes have been identified. This toxin is encoded on (http://www.resgen.com/products/YEASTD.php3; see Table the M1 satellite virion of the L dsRNA virus of S. cerevisiae 1 for complete genotype descriptions). Haploid big1 and pkc1 mutants were obtained by dissection of the heterozygous dip- (Wickner 1996). Toxin sensitivity results from binding loid strains on media supplemented with 0.6 m and 1.0 m of the protein to the cell surface and its subsequent sorbitol, respectively. To improve spore viability of pkc1 tetrads, action at the plasma membrane promoting a lethal loss 1.0 m sorbitol was added during the zymolyase treatment of of cellular ions (reviewed in Bussey 1991; Breinig et asci. Yeast were grown in standard YPD medium (Sherman al. 2002). Defects in the genes involved in these pro- 1991), unless otherwise stated. YPD/G418 medium, used to pregrow the mutants for 18 hr on 2% agar plates, is made of cesses may change cellular sensitivity to this toxin, and YPD supplemented with 200 mg/liter geneticin (GIBCO-BRL, known resistant mutants define genes whose products Grand Island, NY). To test for drug sensitivity, YPD plates contained 25 or 50 g/ml of calcofluor white, 30 or 80 g/ ml of hygromycin B, or 0.05% SDS. K1 killer toxin assay: K1 toxin sensitivity was measured as 1 Present address: Institute of Biochemistry, Swiss Federal Institute of follows (for details see Brown et al. 1994). Yeast mutant strains Technology, Zurich CH-8093, Switzerland. (haploid MAT as well as the homozygous and heterozygous 2 Present address: Department of Bioapplied Chemistry, Osaka City diploids) were pregrown for 18 hr at 30 on YPD/G418 in University, 3-3-138 Sugimoto, Sumiyoshi-ku Osaka, 558-8585, Japan. parallel with corresponding wild types pregrown on YPD. To 3 Corresponding author: Department of Biology, McGill University, control for variation in toxin activity between experiments, 1205 Ave. Docteur Penfield, Montreal, Quebec H3A 1B1, Canada. E-mail: [email protected] three wild-type controls were incorporated into every batch Genetics 163: 875–894 ( March 2003)
Transcript of A Saccharomyces cerevisiae Genome-Wide Mutant Screen for Altered Sensitivity to K1 Killer Toxin

Copyright 2003 by the Genetics Society of America
A Saccharomyces cerevisiae Genome-Wide Mutant Screen for AlteredSensitivity to K1 Killer Toxin
Nicolas Page,1 Manon Gerard-Vincent, Patrice Menard, Maude Beaulieu, Masayuki Azuma,2
Gerrit J. P. Dijkgraaf, Huijuan Li, Jose Marcoux, Thuy Nguyen, Tim Dowse,Anne-Marie Sdicu and Howard Bussey3
Biology Department, McGill University, Montreal, Quebec H3A 1B1, Canada
Manuscript received November 5, 2002Accepted for publication December 12, 2002
ABSTRACTUsing the set of Saccharomyces cerevisiae mutants individually deleted for 5718 yeast genes, we screened
for altered sensitivity to the antifungal protein, K1 killer toxin, that binds to a cell wall �-glucan receptorand subsequently forms lethal pores in the plasma membrane. Mutations in 268 genes, including 42 ingenes of unknown function, had a phenotype, often mild, with 186 showing resistance and 82 hypersensitiv-ity compared to wild type. Only 15 of these genes were previously known to cause a toxin phenotype whenmutated. Mutants for 144 genes were analyzed for alkali-soluble �-glucan levels; 63 showed alterations.Further, mutants for 118 genes with altered toxin sensitivity were screened for SDS, hygromycin B, andcalcofluor white sensitivity as indicators of cell surface defects; 88 showed some additional defect. Thereis a markedly nonrandom functional distribution of the mutants. Many genes affect specific areas ofcellular activity, including cell wall glucan and mannoprotein synthesis, secretory pathway trafficking, lipidand sterol biosynthesis, and cell surface signal transduction, and offer new insights into these processesand their integration.
THE sequenced and analyzed Saccharomyces cerevisiae are involved in cell wall synthesis and regulation (Shahi-nian and Bussey 2000). Here we describe the resultsgenome has enabled a program of precise targetedof global screens of haploids and homozygous and het-gene disruption, resulting in a collection of mutant strainserozygous diploid mutants for altered K1 toxin sensi-deficient in each gene (Winzeler et al. 1999; Giavertivity.et al. 2002; see also: http://sequence-www.stanford.edu/
group/yeast/yeast_deletion_project/deletions3.html).Such a collection promotes the discovery of cellular
MATERIALS AND METHODSroles for genes by facilitating the characterization ofmutant phenotypes and allows a comprehensive exami- Strains and media: Wild-type strains were BY4742 (MAT�)nation of the genetic complexity of a phenotype. We and BY4743 (MATa/MAT�; Brachmann et al. 1998), excepthave used the S. cerevisiae gene disruption set to screen where noted in Figure 1B, which also presents some results
from strain SEY6210 (Robinson et al. 1988). Deletant strainsfor K1 killer toxin phenotypes. Toxin resistance haswere from the Saccharomyces Genome Deletion Consortiumbeen extensively studied by classical genetics, and many(Giaver et al. 2002) and are available at Research Genetics
genes have been identified. This toxin is encoded on (http://www.resgen.com/products/YEASTD.php3; see Tablethe M1 satellite virion of the L dsRNA virus of S. cerevisiae 1 for complete genotype descriptions). Haploid big1 and pkc1
mutants were obtained by dissection of the heterozygous dip-(Wickner 1996). Toxin sensitivity results from bindingloid strains on media supplemented with 0.6 m and 1.0 mof the protein to the cell surface and its subsequentsorbitol, respectively. To improve spore viability of pkc1 tetrads,action at the plasma membrane promoting a lethal loss1.0 m sorbitol was added during the zymolyase treatment of
of cellular ions (reviewed in Bussey 1991; Breinig et asci. Yeast were grown in standard YPD medium (Shermanal. 2002). Defects in the genes involved in these pro- 1991), unless otherwise stated. YPD/G418 medium, used to
pregrow the mutants for 18 hr on 2% agar plates, is made ofcesses may change cellular sensitivity to this toxin, andYPD supplemented with 200 mg/liter geneticin (GIBCO-BRL,known resistant mutants define genes whose productsGrand Island, NY). To test for drug sensitivity, YPD platescontained 25 or 50 �g/ml of calcofluor white, 30 or 80 �g/ml of hygromycin B, or 0.05% SDS.
K1 killer toxin assay: K1 toxin sensitivity was measured as1Present address: Institute of Biochemistry, Swiss Federal Institute offollows (for details see Brown et al. 1994). Yeast mutant strainsTechnology, Zurich CH-8093, Switzerland.(haploid MAT� as well as the homozygous and heterozygous2Present address: Department of Bioapplied Chemistry, Osaka Citydiploids) were pregrown for 18 hr at 30� on YPD/G418 inUniversity, 3-3-138 Sugimoto, Sumiyoshi-ku Osaka, 558-8585, Japan.parallel with corresponding wild types pregrown on YPD. To3Corresponding author: Department of Biology, McGill University,control for variation in toxin activity between experiments,1205 Ave. Docteur Penfield, Montreal, Quebec H3A 1B1, Canada.
E-mail: [email protected] three wild-type controls were incorporated into every batch
Genetics 163: 875–894 ( March 2003)

876 N. Page et al.
TABLE 1
Yeast strains
Strain Genotype Source
BY4742 MAT� his3�1 leu2�0 lys2�0 ura3�0 Brachmann et al. (1998)BY4743 MATa/MAT� his3�1/his3�1 leu2�0/leu2�0 Brachmann et al. (1998)
LYS2/lys2�0 MET15/met15�0 ura3�0/ura3�0Haploida As BY4742, orf�::kanMX4 Winzeler et al. (1999)Heterozygousa As BY4743, orf�::kanMX4/ORF Winzeler et al. (1999)Homozygousa As BY4743, orf�::kanMX4/orf�::kanMX4 Winzeler et al. (1999)SEY6210 MAT� leu2-3,112 ura3-52 his3-�200 Robinson et al. (1988)
trp1-�901 lys2-801 suc2-�9HAB880 As SEY6210 except mnn9::kanMX2 Shahinian et al. (1998)HAB900b As SEY6210 except f ks1::GFP-HIS3 Ketela et al. (1999)
a Indicates mutants obtained from the Saccharomyces Genome Deletion Consortium.b Haploid derived from TK103 strain.
of mutants tested (100–600 mutants/batch). Approximately Cell wall composition analysis: Total �-glucan analysis: Hap-loid strains used for alkali-insoluble �-glucan determinations1 � 106 cells were resuspended in 100 �l of sterile water, of
which 5 �l was used to inoculate 5 ml of molten YPD agar were MAT� sla1� and big1�, respectively, obtained or derivedfrom the Saccharomyces Genome Deletion Consortium (seemedium (1% agar, 0.001% methylene blue, and 1� Halvorson
buffered at pH 4.7) held at 45�. Sorbitol was supplemented Strains and media above) and compared to wild-type strainBY4742, while mnn9� (HAB880) and fks1� (HAB900) werefor big1 and pkc1 mutants, and for a wild-type control, as
described above. This medium was quickly poured into 60- � compared to parental strain SEY6210. Crude cell walls wereisolated and the levels of alkali-insoluble �-1,3-glucan and15-mm petri dishes and allowed to cool for 1 hr at room
temperature. Then 5 �l K1 killer toxin (1000� stock diluted �-1,6-glucan quantified as previously described (Dijkgraaf etal. 2002). The big1� mutant and the corresponding wild type1:10; Brown et al. 1994) was spotted on the surface of the
solidified medium. The plates were incubated overnight at were grown in medium containing 0.6 m sorbitol to provideosmotic support.18� followed by 24 hr at 30� (48 hr for slow growth mutants).
For each mutant showing a “killing” or “death” zone different Alkali-soluble �-1,6- and �-1,3-glucan analysis: Alkali-soluble�-1,6- and �-1,3-glucan immunodetection was performed asfrom wild type, a picture comparing the mutant and appro-
priate control was taken with the IS-500 Digital Imaging Sys- described by Lussier et al. (1998) and summarized here. Yeastwere pregrown on YPD/G418 for 18 hr at 30�, grown fortem, version 2.02 (Alpha-Innotech). Two measurements of
the killing zone were made with PhotoShop 4.0 and the aver- 24 hr at 30� in 10 ml YPD liquid, and harvested by a 10-mincentrifugation at 1860 � g. Cell pellets were washed with 5 mlage was saved in a database (FileMaker Pro 5.0) together with
the picture. Mutants with a killing zone �90% or �110% were of water and resuspended in 100 �l of water plus 100 �l ofglass beads. The cells were then subjected to five cycles ofretested up to four times to confirm the observed phenotype.
These percentages were determined as [(mutant killing zone vortexing for 30 sec, interspersed with 30-sec incubations onice. Total cellular protein of the lysate was determined withdiameter)/(wild-type killing zone diameter) � 100]. A subset
of mutants showing killing zones �75% or �115% was selected the Bradford assay (Bradford 1976; Bio-Rad, Mississauga,ON, Canada) prior to alkali extraction (1.5 n NaOH, 1 hr,for further characterization.
K1 toxin survival assay: To determine cell survival after 75�). A set of 1:2 serial dilutions of the alkali-soluble fractionswere then spotted on Hybond-C nitrocellulose membranetoxin treatment, 200 �l of a cell culture grown to log phase
in YPD pH 4.7 and adjusted to OD600 0.5 was incubated with (Amersham, Oakville, ON, Canada). The immunoblotting wasperformed in Tris-buffered saline Tween containing 5% non-50 �l toxin (1000� stock diluted up to 1:25) for 3 hr at 18�
on a labquake. Percentage of surviving cells was calculated fat dried milk powder using either a 2000-fold dilution ofaffinity-purified rabbit anti-�-1,6-glucan primary antibodyfollowing plating onto YPD agar after incubation with toxin
and counting colonies after 2 days at 30�. (Lussier et al. 1998) or a 1000-fold dilution of anti-�-1,3-glucan primary antibody (Biosupplies Australia Pty, Victoria,Drug phenotype assay: Drug sensitivity was determined by
spotting diluted cultures on plates containing various drugs Australia), both with a 2000-fold dilution of horseradish perox-idase goat anti-rabbit secondary antibody (Amersham). Theas described (Ram et al. 1994; Lussier et al. 1997). Briefly, 5
ml of liquid YPD medium, inoculated with freshly grown cells membranes were developed with a chemiluminescence detec-tion kit (Amersham). Dot blots were scanned with a UMAXon YPD/G418, was incubated overnight at 30�. The cell density
of these exponentially growing cultures was standardized with Astra 1220s scanner and signals were quantitated with AdobePhotoshop software, using the histogram function. The levelwater at an OD600 of between 0.485 and 0.515, and 2 �l of a
set of 10-fold serial dilutions were spotted on YPD supple- of �-1,6- and �-1,3-glucan for each mutant was estimated bya comparison with a wild-type dilution series, with mutantsmented with calcofluor white, hygromycin B, or SDS (seeclassified by ranges of 20–35% (see footnote in Table 5).Strains and media for drug concentrations). Hypersensitivity
or resistance was monitored for each drug after 48 and 72 hrgrowth at 30�. The cells were also spotted on a control plate(YPD without drug), which allowed a comparison with the RESULTSgrowth rate of the mutants after 24 hr growth at 30�. Pictures
K1 toxin sensitivity of deletion mutants: The toxinof all conditions tested were downloaded into a FileMaker 5.0database (see above for details). sensitivities of deletion mutants for 5718 genes were

877K1 Toxin Phenotypes of Yeast Genes
compared to those of the parental strain. The screen Glucan synthesis: The yeast cell wall is made princi-pally of four components: mannoproteins, chitin, �-1,3-was performed on haploid (MAT�) and homozygous
diploid mutants, with toxin sensitivity being almost iden- glucan, and �-1,6-glucan (Orlean 1997; Lipke andOvalle 1998). Protein mannosylation and �-1,6-glucantical in both backgrounds. The heterozygous diploid
collection was also tested, with the finding that 42 genes synthesis defects are known to lead to toxin resistanceby altering the cell wall receptor for the toxin (see Shah-have a haploinsufficient toxin phenotype. The individ-
ual deletion of most genes has no effect on toxin sensitiv- inian and Bussey 2000 for a review; Breinig et al. 2002).Many mutations resulting in resistance to the K1 toxinity. These aphenotypic mutations include genes with a
wide range of other phenotypes, such as slow growth have a reduced amount of �-1,6-glucan in the cell walland show slow growth or inviability depending on theand respiratory deficiency, and provide an important
control for trivial cellular alterations that might affect severity of the defect, and we anticipated finding newgenes affecting these processes. A complex pattern ofthe killing zone phenotype. For mutants in almost all
genes, despite some affliction, the killer phenotype is glucan phenotypes was found among the mutants exam-ined for alkali-extractable �-1,6- and �-1,3-glucan levels,wild type. Mutants in 268 genes (4.7%) have a pheno-
type distinct from wild-type toxin sensitivity, with 15 of with reduced or elevated amounts of one or both poly-mers found (Table 5). Of mutants in 63 genes withthese genes previously known to have such a phenotype
(Shahinian and Bussey 2000; de Groot et al. 2001). glucan phenotypes, 55 had effects on �-1,6-glucan levels,with the remaining 8 having �-1,3-glucan-specific alter-Tables 2–4 list these mutants in functional groupings.
A given gene is listed just once although some could ations. Of the 55 with �-1,6-glucan phenotypes, 40 alsohad some �-1,3-glucan phenotype, with 15 showing abe included in more than one category. Although the
phenotypes are significant and reproducible, most null �-1,6-glucan-specific phenotype. Principal findings areoutlined below.mutants have partial phenotypes. For example, among
155 haploid-resistant mutants, only 30 are fully resistant �-1,6-Glucan reduced: Mutants for five genes showingpartial toxin resistance had specific but partial alkali-at the toxin concentration used (Table 2). Toxin sensi-
tivity can be suppressed or enhanced in mutants, leading soluble �-1,6-glucan reductions. Among these was the�-1,3-glucan synthesis-associated gene FKS1, and thisto resistance or hypersensitivity. Toxin resistance, which
was always found as a recessive phenotype, is likely mutant also had reduced levels of alkali-insoluble �-1,3-glucan (Figure 1B; Table 5). The involvement of Fks1pcaused by a loss of function of some component needed
for toxin action. In hypersensitive mutants the mutation in both �-1,3- and �-1,6-glucan biogenesis has been stud-ied further (Dijkgraaf et al. 2002). Mutants in CNE1synthetically enhances toxin lethality and can be func-
tionally informative. Among the mutations resulting in encoding yeast calnexin have less �-1,6-glucan (Shahi-nian et al. 1998), and this is also a mutant phenotypea toxin phenotype, 42 were in uncharacterized open
reading frames (ORFs) of unknown function. Of these, of the uncharacterized gene YKL037W encoding a smallintegral membrane protein.3 were given a KRE (k iller toxin resistant) number, and
8 genes with hypersensitive mutants were called FYV �-1,6-Glucan reduced with altered �-1,3-glucan: big1mutants had greatly reduced levels of �-1,6-glucan and(function required for yeast v iability upon toxin expo-
sure) and given a number (Tables 2–4). an increase in �-1,3-glucan. BIG1 is a conditional essen-tial gene retaining partial viability on medium with os-Mutants for 118 genes with toxin phenotypes were
examined for altered sensitivity to SDS, calcofluor white, motic support (Bickle et al. 1998). Heterozygous big1/BIG1 diploids showed haploinsufficient toxin resistanceand hygromycin B as hypersensitivity or resistance to
these compounds is indicative of cell surface defects (Table 4), and haploid mutant cells grew very slowlyon medium containing 0.6 m sorbitol and were toxin(Lussier et al. 1997; Ross-Macdonald et al. 1999). Mu-
tants in 88 of these genes showed some additional phe- resistant (Figure 1A). Determination of the amount ofalkali-insoluble glucan in the cell wall of a big1 mutantnotype (Tables 2–4), independently suggesting that they
have some cell surface perturbation. As �-1,6-glucan is showed that the �-1,6-glucan was 5% of wild-type levels(Figure 1B). The amount of �-1,3-glucan in big1 mutantsthe primary component of the cell wall receptor for the
toxin, mutants in 144 genes with toxin phenotypes were increased, possibly through some wall compensatorymechanism. Elsewhere, we have extended work on theexamined for alkali-soluble �-1,6- and �-1,3-glucan levels
(Table 5) with 63 showing an altered level of one or both role of Big1p in �-1,6-glucan biogenesis (Azuma et al.2002).polymers. Genes previously identified as killer resistant
provide positive controls for this global screen (Tables Mutants for 13 genes had reductions in both alkali-soluble �-1,6- and �-1,3-glucan (Table 5), and three are2 and 4). A number of characterized genes not known
to have altered toxin sensitivity were found, suggesting described briefly below. smi1/knr4 mutants are resistantboth to the K1 toxin and to the K9 toxin from Hansenulathat they have roles in cell wall or surface organization.
Most mutants fall into a limited set of functional classes mrakii and have wall glucan defects and a reduced in vitroglucan synthase activity (Hong et al. 1994a,b). Smi1pand define specific areas of cellular biology, some of
which are described below (see also Tables 2 and 3). localized to cytoplasmic patches near the presumptive

878 N. Page et al.
TA
BL
E2
Gen
esw
hose
dele
tion
caus
esre
sist
ance
toth
eK
1ki
ller
toxi
n(h
aplo
idor
hom
ozyg
ous
dipl
oid
deat
hzo
ne�
75%
ofth
ew
ildty
pe)
K1
kille
rto
xin
deat
hzo
ne
(%)
Cal
cofl
uor
Gen
en
ame
OR
FD
escr
ipti
onof
gen
epr
oduc
tH
aplo
idH
eter
ozyg
ous
Hom
ozyg
ous
wh
ite
Hyg
rom
ycin
BSD
S
1.K
inas
es,
phos
phat
ases
,si
gnal
tran
sduc
tion
(16
gen
es)
PHO
80YO
L00
1WC
yclin
that
inte
ract
sw
ith
Pho8
5ppr
otei
nki
nas
e48
8563
SS
wt
PHO
85YP
L03
1CC
yclin
-dep
ende
nt
prot
ein
kin
ase
56w
t37
RS
SPT
P3YE
R07
5CPr
otei
nty
rosi
ne
phos
phat
ase
65w
t63
Rw
tw
tC
KA
2YO
R06
1WC
asei
nki
nas
eII
�
chai
n65
wt
63R
wt
wt
SAC
7a
YDR
389W
GT
Pase
-act
ivat
ing
prot
ein
for
Rh
o1p
67w
t85
Sw
tw
tEL
M1
YKL
048C
ser/
thr-
spec
ific
kin
ase
69w
t58
Sw
tS
PTC
1YD
L00
6WPr
otei
nse
rin
e/th
reon
ine
phos
phat
ase
2c71
wt
64K
IN3
YAR
018C
ser/
thr
prot
ein
kin
ase
72w
t70
SAP1
55YF
R04
0WSi
t4p-
asso
ciat
edpr
otei
n73
wt
62SA
P190
YKR
028W
Sit4
p-as
soci
ated
prot
ein
74w
t75
GPA
2a
YER
020W
Gua
nin
en
ucle
otid
e-bi
ndi
ng
regu
lato
rypr
otei
n74
wt
83w
tw
tw
tR
RD
1YI
L15
3WPh
osph
otyr
osyl
phos
phat
ase
acti
vato
r75
wt
69M
KS1
YNL
076W
Neg
ativ
ere
gula
tor
ofR
as-c
AM
Ppa
thw
ay82
wt
83G
LC
8YM
R31
1CR
egul
ator
ysu
bun
itfo
rse
r/th
rph
osph
atas
eG
lc7p
83w
t69
PHO
86YJ
L11
7WIn
orga
nic
phos
phat
etr
ansp
orte
r83
wt
70SA
L6
YPL
179W
Phos
phop
rote
inph
osph
atas
e85
wt
67
2.T
ran
scri
ptio
n(2
1ge
nes
)SS
N6
YBR
112C
Gen
eral
repr
esso
rof
tran
scri
ptio
n0
810
TU
P1YC
R08
4CG
ener
altr
ansc
ript
ion
repr
esso
r0
830
CT
K3
YML
112W
CT
Dki
nas
e,ga
mm
asu
bun
it;
RN
APo
lII
regu
lati
on0
wt
0SN
F12
YNR
023W
Com
pon
ent
ofSW
I/SN
Fgl
obal
tran
scri
ptio
nac
tiva
tor
com
plex
0w
tN
AC
CR
4YA
L02
1CT
ran
scri
ptio
nal
regu
lato
r42
wt
57R
wt
wt
POP2
YNR
052C
Req
uire
dfo
rgl
ucos
ede
repr
essi
on60
wt
70EL
P4YP
L10
1WSu
bun
itof
RN
APo
lII
elon
gato
rco
mpl
ex68
wt
76S
wt
wt
AD
A2
YDR
448W
Gen
eral
tran
scri
ptio
nal
adap
tor
orco
acti
vato
r69
wt
63C
TK
1YK
L13
9WC
TD
kin
ase,
alph
asu
bun
it;
RN
APo
lII
regu
lati
on70
wt
58A
CE2
YLR
131C
Met
allo
thio
nei
nex
pres
sion
acti
vato
r71
wt
75R
Rw
tSI
R4
YDR
227W
Sile
nci
ng
regu
lato
ryan
dD
NA
-rep
air
prot
ein
72w
tN
Aw
tw
tw
tIK
13YL
R38
4CE
lon
gato
rco
mpl
ex;
RN
APo
lII
-ass
ocia
ted
prot
ein
73w
t75
SSN
3YP
L04
2CC
yclin
-dep
ende
nt
CT
Dki
nas
e73
wt
78SI
R1
YKR
101W
Sile
nci
ng
regu
lato
rypr
otei
n73
wt
80FU
N30
YAL
019W
Mem
ber
ofth
eSn
f2p
fam
ilyof
AT
P-de
pen
den
tD
NA
hel
icas
es76
wt
72EP
L6
YMR
312W
Subu
nit
ofR
NA
Pol
IIel
onga
tor
com
plex
79w
t58
ELP3
YPL
086C
His
ton
ean
dot
her
prot
ein
acet
yltr
ansf
eras
e80
wt
65SP
T10
YJL
127C
Tra
nsc
ript
ion
regu
lato
rypr
otei
n80
wt
70SA
P30
YMR
263W
Com
pon
ent
ofth
eR
pd3p
-Sin
3ph
isto
ne
deac
etyl
ase
com
plex
82w
t65
SIG
1YE
R06
8WT
ran
scri
ptio
nal
repr
esso
r83
wt
32S
SS
TA
F14
YPL
129W
TFI
IFsu
bun
it(t
ran
scri
ptio
nin
itia
tion
fact
or),
30kD
85w
t56
SS
S
(con
tinue
d)
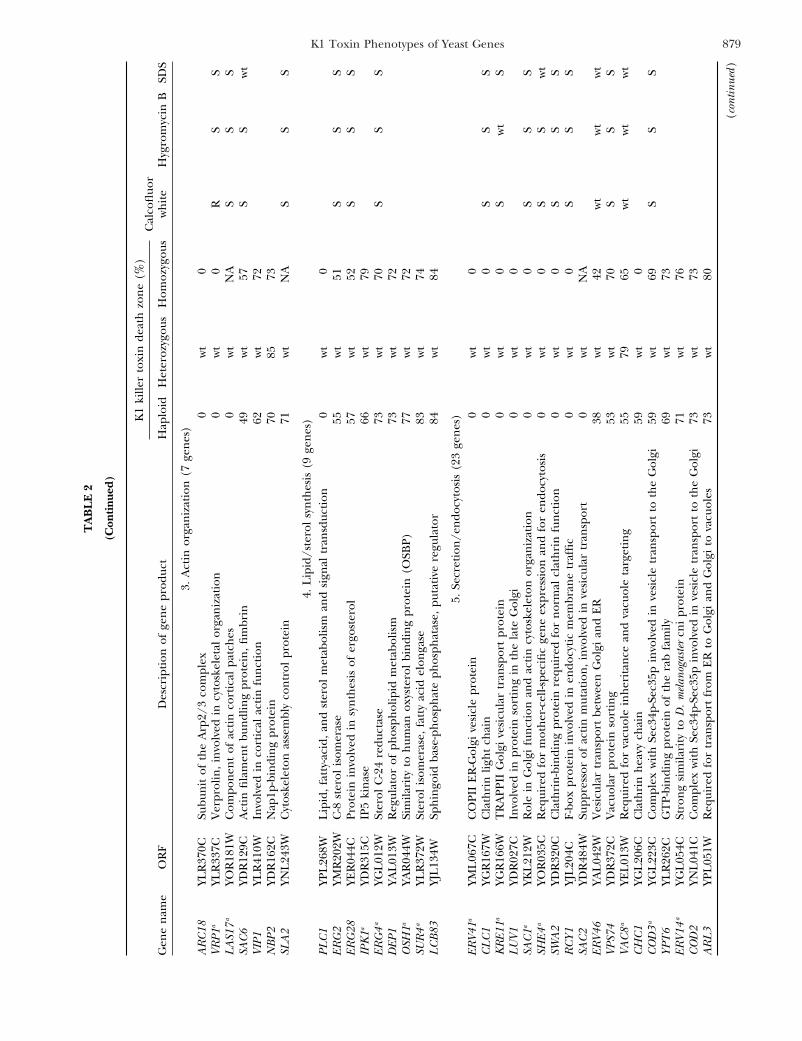
879K1 Toxin Phenotypes of Yeast Genes
TA
BL
E2
(Con
tinu
ed)
K1
kille
rto
xin
deat
hzo
ne
(%)
Cal
cofl
uor
Gen
en
ame
OR
FD
escr
ipti
onof
gen
epr
oduc
tH
aplo
idH
eter
ozyg
ous
Hom
ozyg
ous
wh
ite
Hyg
rom
ycin
BSD
S
3.A
ctin
orga
niz
atio
n(7
gen
es)
AR
C18
YLR
370C
Subu
nit
ofth
eA
rp2/
3co
mpl
ex0
wt
0VR
P1a
YLR
337C
Ver
prol
in,
invo
lved
incy
tosk
elet
alor
gan
izat
ion
0w
t0
RS
SL
AS1
7a
YOR
181W
Com
pon
ent
ofac
tin
cort
ical
patc
hes
0w
tN
AS
SS
SAC
6YD
R12
9CA
ctin
fila
men
tbu
ndl
ing
prot
ein
,fi
mbr
in49
wt
57S
Sw
tVI
P1YL
R41
0WIn
volv
edin
cort
ical
acti
nfu
nct
ion
62w
t72
NB
P2YD
R16
2CN
ap1p
-bin
din
gpr
otei
n70
8573
SLA
2YN
L24
3WC
ytos
kele
ton
asse
mbl
yco
ntr
olpr
otei
n71
wt
NA
SS
S
4.L
ipid
/ste
rol
syn
thes
is(9
gen
es)
PLC
1YP
L26
8WL
ipid
,fa
tty-
acid
,an
dst
erol
met
abol
ism
and
sign
altr
ansd
ucti
on0
wt
0ER
G2
YMR
202W
C-8
ster
olis
omer
ase
55w
t51
SS
SER
G28
YER
044C
Prot
ein
invo
lved
insy
nth
esis
ofer
gost
erol
57w
t52
SS
SIP
K1a
YDR
315C
IP5
kin
ase
66w
t79
ERG
4a
YGL
012W
Ster
olC
-24
redu
ctas
e73
wt
70S
SS
DEP
1YA
L01
3WR
egul
ator
ofph
osph
olip
idm
etab
olis
m73
wt
72O
SH1a
YAR
044W
Sim
ilari
tyto
hum
anox
yste
rol
bin
din
gpr
otei
n(O
SBP)
77w
t72
SUR
4a
YLR
372W
Ster
olis
omer
ase,
fatt
yac
idel
onga
se83
wt
74L
CB
83YJ
L13
4WSp
hin
goid
base
-ph
osph
ate
phos
phat
ase,
puta
tive
regu
lato
r84
wt
84
5.Se
cret
ion
/en
docy
tosi
s(2
3ge
nes
)ER
V41a
YML
067C
CO
PII
ER
-Gol
give
sicl
epr
otei
n0
wt
0C
LC
1YG
R16
7WC
lath
rin
ligh
tch
ain
0w
t0
SS
SK
RE1
1aYG
R16
6WT
RA
PPII
Gol
give
sicu
lar
tran
spor
tpr
otei
n0
wt
0S
wt
SL
UV1
YDR
027C
Invo
lved
inpr
otei
nso
rtin
gin
the
late
Gol
gi0
wt
0SA
C1a
YKL
212W
Rol
ein
Gol
gifu
nct
ion
and
acti
ncy
tosk
elet
onor
gan
izat
ion
0w
t0
SS
SSH
E4a
YOR
035C
Req
uire
dfo
rm
oth
er-c
ell-s
peci
fic
gen
eex
pres
sion
and
for
endo
cyto
sis
0w
t0
SS
wt
SWA
2YD
R32
0CC
lath
rin
-bin
din
gpr
otei
nre
quir
edfo
rn
orm
alcl
ath
rin
fun
ctio
n0
wt
0S
SS
RC
Y1YJ
L20
4CF-
box
prot
ein
invo
lved
inen
docy
tic
mem
bran
etr
affi
c0
wt
0S
SS
SAC
2YD
R48
4WSu
ppre
ssor
ofac
tin
mut
atio
n,
invo
lved
inve
sicu
lar
tran
spor
t0
wt
NA
ERV4
6YA
L04
2WV
esic
ular
tran
spor
tbe
twee
nG
olgi
and
ER
38w
t42
wt
wt
wt
VPS7
4YD
R37
2CV
acuo
lar
prot
ein
sort
ing
53w
t70
SS
SVA
C8
aYE
L01
3WR
equi
red
for
vacu
ole
inh
erit
ance
and
vacu
ole
targ
etin
g55
7965
wt
wt
wt
CH
C1
YGL
206C
Cla
thri
nh
eavy
chai
n59
wt
0C
OD
3a
YGL
223C
Com
plex
wit
hSe
c34p
-Sec
35p
invo
lved
inve
sicl
etr
ansp
ort
toth
eG
olgi
59w
t69
SS
SYP
T6
YLR
262C
GT
P-bi
ndi
ng
prot
ein
ofth
era
bfa
mily
69w
t73
ERV1
4a
YGL
054C
Stro
ng
sim
ilari
tyto
D.
mel
anog
aste
rcn
ipr
otei
n71
wt
76C
OD
2YN
L04
1CC
ompl
exw
ith
Sec3
4p-S
ec35
pin
volv
edin
vesi
cle
tran
spor
tto
the
Gol
gi73
wt
73A
RL
3YP
L05
1WR
equi
red
for
tran
spor
tfr
omE
Rto
Gol
gian
dG
olgi
tova
cuol
es73
wt
80
(con
tinue
d)
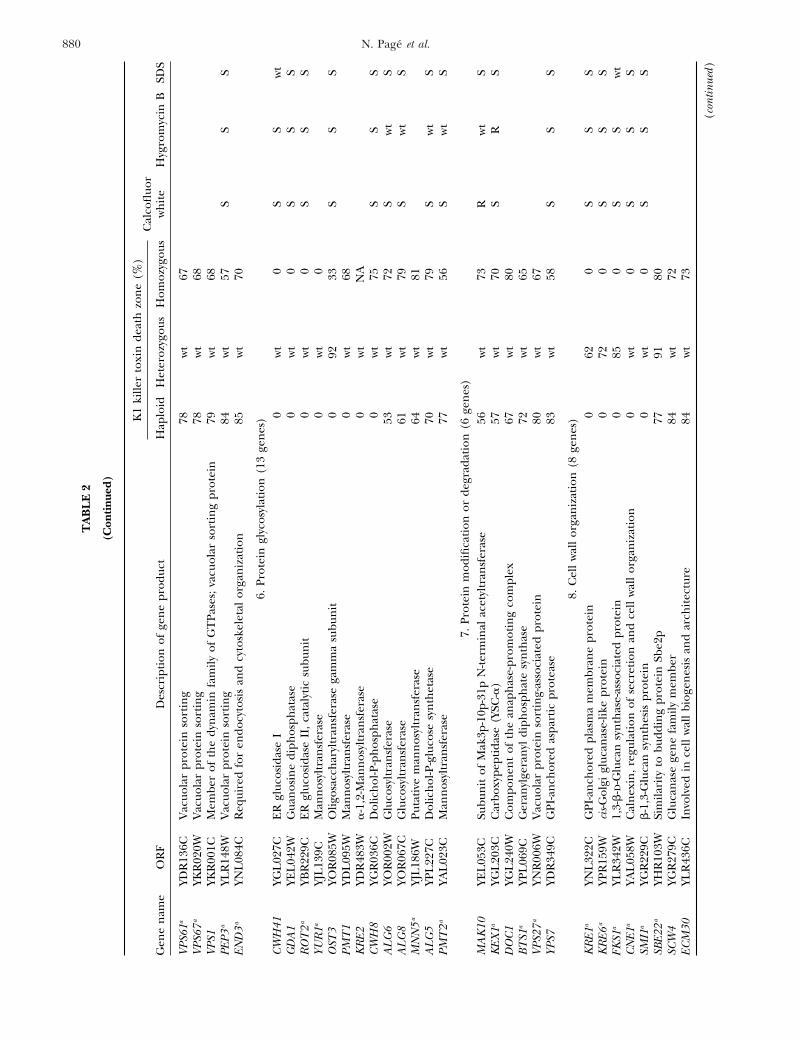
880 N. Page et al.
TA
BL
E2
(Con
tinu
ed)
K1
kille
rto
xin
deat
hzo
ne
(%)
Cal
cofl
uor
Gen
en
ame
OR
FD
escr
ipti
onof
gen
epr
oduc
tH
aplo
idH
eter
ozyg
ous
Hom
ozyg
ous
wh
ite
Hyg
rom
ycin
BSD
S
VPS6
1aYD
R13
6CV
acuo
lar
prot
ein
sort
ing
78w
t67
VPS6
7a
YKR
020W
Vac
uola
rpr
otei
nso
rtin
g78
wt
68VP
S1YK
R00
1CM
embe
rof
the
dyn
amin
fam
ilyof
GT
Pase
s;va
cuol
arso
rtin
gpr
otei
n79
wt
68PE
P3a
YLR
148W
Vac
uola
rpr
otei
nso
rtin
g84
wt
57S
SS
END
3a
YNL
084C
Req
uire
dfo
ren
docy
tosi
san
dcy
tosk
elet
alor
gan
izat
ion
85w
t70
6.Pr
otei
ngl
ycos
ylat
ion
(13
gen
es)
CW
H41
YGL
027C
ER
gluc
osid
ase
I0
wt
0S
Sw
tG
DA
1YE
L04
2WG
uan
osin
edi
phos
phat
ase
0w
t0
SS
SR
OT
2a
YBR
229C
ER
gluc
osid
ase
II,
cata
lyti
csu
bun
it0
wt
0S
SS
YUR
1aYJ
L13
9CM
ann
osyl
tran
sfer
ase
0w
t0
OST
3YO
R08
5WO
ligos
acch
aryl
tran
sfer
ase
gam
ma
subu
nit
092
33S
SS
PMT
1YD
L09
5WM
ann
osyl
tran
sfer
ase
0w
t68
KR
E2YD
R48
3W�
-1,2
-Man
nos
yltr
ansf
eras
e0
wt
NA
CW
H8
YGR
036C
Dol
ich
ol-P
-ph
osph
atas
e0
wt
75S
SS
AL
G6
YOR
002W
Glu
cosy
ltra
nsf
eras
e53
wt
72S
wt
SA
LG
8YO
R06
7CG
luco
sylt
ran
sfer
ase
61w
t79
Sw
tS
MN
N5
aYJ
L18
6WPu
tati
vem
ann
osyl
tran
sfer
ase
64w
t81
AL
G5
YPL
227C
Dol
ich
ol-P
-glu
cose
syn
thet
ase
70w
t79
Sw
tS
PMT
2a
YAL
023C
Man
nos
yltr
ansf
eras
e77
wt
56S
wt
S
7.Pr
otei
nm
odifi
cati
onor
degr
adat
ion
(6ge
nes
)M
AK
10YE
L05
3CSu
bun
itof
Mak
3p-1
0p-3
1pN
-term
inal
acet
yltr
ansf
eras
e56
wt
73R
wt
SK
EX1a
YGL
203C
Car
boxy
pept
idas
e(Y
SC-�
)57
wt
70S
RS
DO
C1
YGL
240W
Com
pon
ent
ofth
ean
aph
ase-
prom
otin
gco
mpl
ex67
wt
80B
TS1
aYP
L06
9CG
eran
ylge
ran
yldi
phos
phat
esy
nth
ase
72w
t65
VPS2
7a
YNR
006W
Vac
uola
rpr
otei
nso
rtin
g-as
soci
ated
prot
ein
80w
t67
YPS7
YDR
349C
GPI
-an
chor
edas
part
icpr
otea
se83
wt
58S
SS
8.C
ell
wal
lor
gan
izat
ion
(8ge
nes
)K
RE1
aYN
L32
2CG
PI-a
nch
ored
plas
ma
mem
bran
epr
otei
n0
620
SS
SK
RE6
aYP
R15
9Wci
s-Gol
gigl
ucan
ase-
like
prot
ein
072
0S
SS
FKS1
aYL
R34
2W1,
3-�
-d-G
luca
nsy
nth
ase-
asso
ciat
edpr
otei
n0
850
SS
wt
CN
E1a
YAL
058W
Cal
nex
in,
regu
lati
onof
secr
etio
nan
dce
llw
all
orga
niz
atio
n0
wt
0S
SS
SMI1
aYG
R22
9C�
-1,3
-Glu
can
syn
thes
ispr
otei
n0
wt
0S
SS
SBE2
2a
YHR
103W
Sim
ilari
tyto
budd
ing
prot
ein
Sbe2
p77
9180
SCW
4YG
R27
9CG
luca
nas
ege
ne
fam
ilym
embe
r84
wt
72EC
M30
YLR
436C
Invo
lved
ince
llw
all
biog
enes
isan
dar
chit
ectu
re84
wt
73
(con
tinue
d)
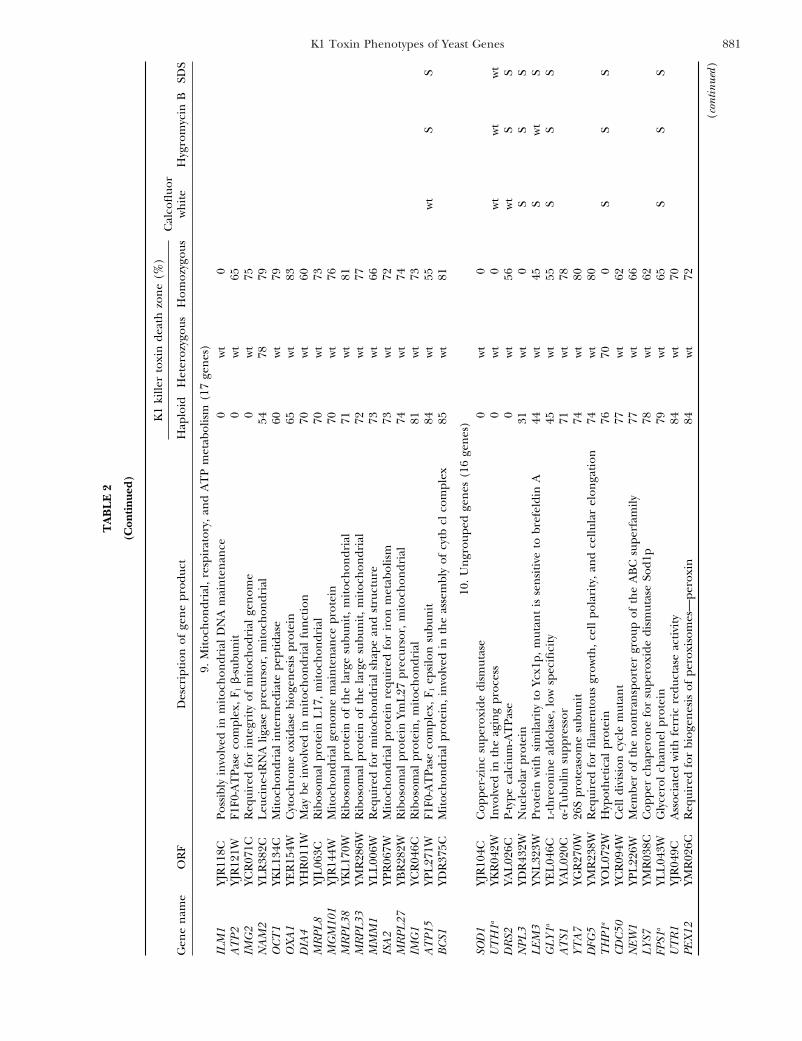
881K1 Toxin Phenotypes of Yeast Genes
TA
BL
E2
(Con
tinu
ed)
K1
kille
rto
xin
deat
hzo
ne
(%)
Cal
cofl
uor
Gen
en
ame
OR
FD
escr
ipti
onof
gen
epr
oduc
tH
aplo
idH
eter
ozyg
ous
Hom
ozyg
ous
wh
ite
Hyg
rom
ycin
BSD
S
9.M
itoc
hon
dria
l,re
spir
ator
y,an
dA
TP
met
abol
ism
(17
gen
es)
ILM
1YJ
R11
8CPo
ssib
lyin
volv
edin
mit
och
ondr
ial
DN
Am
ain
ten
ance
0w
t0
AT
P2YJ
R12
1WF1
F0-A
TPa
seco
mpl
ex,
F 1�
-sub
unit
0w
t65
IMG
2YC
R07
1CR
equi
red
for
inte
grit
yof
mit
och
odri
alge
nom
e0
wt
75N
AM
2YL
R38
2CL
euci
ne-
tRN
Alig
ase
prec
urso
r,m
itoc
hon
dria
l54
7879
OC
T1
YKL
134C
Mit
och
ondr
ial
inte
rmed
iate
pept
idas
e60
wt
79O
XA
1YE
R15
4WC
ytoc
hro
me
oxid
ase
biog
enes
ispr
otei
n65
wt
83D
IA4
YHR
011W
May
bein
volv
edin
mit
och
ondr
ial
fun
ctio
n70
wt
60M
RPL
8YJ
L06
3CR
ibos
omal
prot
ein
L17
,m
itoc
hon
dria
l70
wt
73M
GM
101
YJR
144W
Mit
och
ondr
ial
gen
ome
mai
nte
nan
cepr
otei
n70
wt
76M
RPL
38YK
L17
0WR
ibos
omal
prot
ein
ofth
ela
rge
subu
nit
,m
itoc
hon
dria
l71
wt
81M
RPL
33YM
R28
6WR
ibos
omal
prot
ein
ofth
ela
rge
subu
nit
,m
itoc
hon
dria
l72
wt
77M
MM
1YL
L00
6WR
equi
red
for
mit
och
ondr
ial
shap
ean
dst
ruct
ure
73w
t66
ISA
2YP
R06
7WM
itoc
hon
dria
lpr
otei
nre
quir
edfo
rir
onm
etab
olis
m73
wt
72M
RPL
27YB
R28
2WR
ibos
omal
prot
ein
YmL
27pr
ecur
sor,
mit
och
ondr
ial
74w
t74
IMG
1YC
R04
6CR
ibos
omal
prot
ein
,m
itoc
hon
dria
l81
wt
73A
TP1
5YP
L27
1WF1
F0-A
TPa
seco
mpl
ex,
F 1ep
silo
nsu
bun
it84
wt
55w
tS
SB
CS1
YDR
375C
Mit
och
ondr
ial
prot
ein
,in
volv
edin
the
asse
mbl
yof
cytb
clco
mpl
ex85
wt
81
10.
Un
grou
ped
gen
es(1
6ge
nes
)SO
D1
YJR
104C
Cop
per-
zin
csu
pero
xide
dism
utas
e0
wt
0U
TH
1aYK
R04
2WIn
volv
edin
the
agin
gpr
oces
s0
wt
0w
tw
tw
tD
RS2
YAL
026C
P-ty
peca
lciu
m-A
TPa
se0
wt
56w
tS
SN
PL3
YDR
432W
Nuc
leol
arpr
otei
n31
wt
0S
SS
LEM
3YN
L32
3WPr
otei
nw
ith
sim
ilari
tyto
Ycx1
p,m
utan
tis
sen
siti
veto
bref
eldi
nA
44w
t45
Sw
tS
GL
Y1a
YEL
046C
l-th
reon
ine
aldo
lase
,lo
wsp
ecifi
city
45w
t55
SS
SA
TS1
YAL
020C
�-T
ubul
insu
ppre
ssor
71w
t78
YTA
7YG
R27
0W26
Spr
otea
som
esu
bun
it74
wt
80D
FG5
YMR
238W
Req
uire
dfo
rfi
lam
ento
usgr
owth
,ce
llpo
lari
ty,
and
cellu
lar
elon
gati
on74
wt
80T
HP1
aYO
L07
2WH
ypot
het
ical
prot
ein
7670
0S
SS
CD
C50
YCR
094W
Cel
ldi
visi
oncy
cle
mut
ant
77w
t62
NEW
1YP
L22
6WM
embe
rof
the
non
tran
spor
ter
grou
pof
the
AB
Csu
perf
amily
77w
t66
LYS
7YM
R03
8CC
oppe
rch
aper
one
for
supe
roxi
dedi
smut
ase
Sod1
p78
wt
62FP
S1a
YLL
043W
Gly
cero
lch
ann
elpr
otei
n79
wt
65S
SS
UT
R1
YJR
049C
Ass
ocia
ted
wit
hfe
rric
redu
ctas
eac
tivi
ty84
wt
70PE
X12
YMR
026C
Req
uire
dfo
rbi
ogen
esis
ofpe
roxi
som
es—
pero
xin
84w
t72
(con
tinue
d)
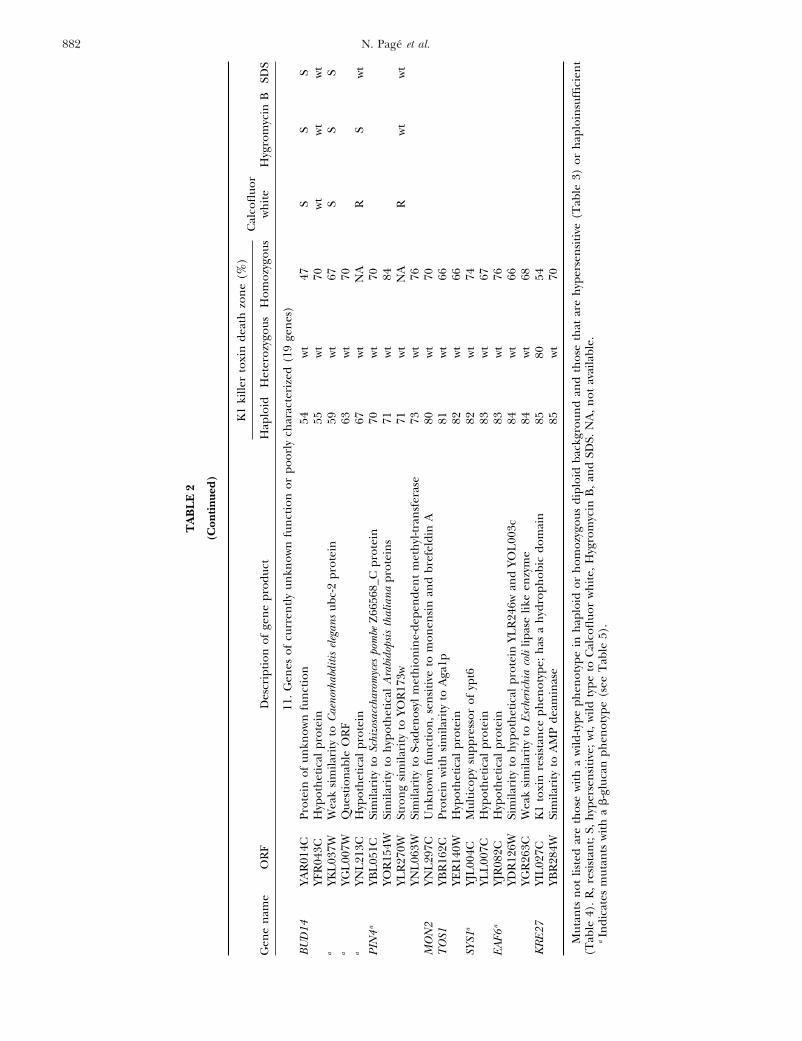
882 N. Page et al.
TA
BL
E2
(Con
tinu
ed)
K1
kille
rto
xin
deat
hzo
ne
(%)
Cal
cofl
uor
Gen
en
ame
OR
FD
escr
ipti
onof
gen
epr
oduc
tH
aplo
idH
eter
ozyg
ous
Hom
ozyg
ous
wh
ite
Hyg
rom
ycin
BSD
S
11.
Gen
esof
curr
entl
yun
know
nfu
nct
ion
orpo
orly
char
acte
rize
d(1
9ge
nes
)B
UD
14YA
R01
4CPr
otei
nof
unkn
own
fun
ctio
n54
wt
47S
SS
YFR
043C
Hyp
oth
etic
alpr
otei
n55
wt
70w
tw
tw
ta
YKL
037W
Wea
ksi
mila
rity
toC
aeno
rhab
ditis
eleg
ans
ubc-
2pr
otei
n59
wt
67S
SS
aYG
L00
7WQ
uest
ion
able
OR
F63
wt
70a
YNL
213C
Hyp
oth
etic
alpr
otei
n67
wt
NA
RS
wt
PIN
4a
YBL
051C
Sim
ilari
tyto
Schi
zosa
ccha
rom
yces
pom
beZ
6656
8_C
prot
ein
70w
t70
YOR
154W
Sim
ilari
tyto
hyp
oth
etic
alA
rabi
dops
isth
alia
napr
otei
ns
71w
t84
YLR
270W
Stro
ng
sim
ilari
tyto
YOR
173w
71w
tN
AR
wt
wt
YNL
063W
Sim
ilari
tyto
S-ad
enos
ylm
eth
ion
ine-
depe
nde
nt
met
hyl
-tran
sfer
ase
73w
t76
MO
N2
YNL
297C
Un
know
nfu
nct
ion
,se
nsi
tive
tom
onen
sin
and
bref
eldi
nA
80w
t70
TO
S1YB
R16
2CPr
otei
nw
ith
sim
ilari
tyto
Aga
1p81
wt
66YE
R14
0WH
ypot
het
ical
prot
ein
82w
t66
SYS1
aYJ
L00
4CM
ulti
copy
supp
ress
orof
ypt6
82w
t74
YLL
007C
Hyp
oth
etic
alpr
otei
n83
wt
67EA
F6a
YJR
082C
Hyp
oth
etic
alpr
otei
n83
wt
76YD
R12
6WSi
mila
rity
toh
ypot
het
ical
prot
ein
YLR
246w
and
YOL
003c
84w
t66
YGR
263C
Wea
ksi
mila
rity
toEs
cher
ichi
aco
lilip
ase
like
enzy
me
84w
t68
KR
E27
YIL
027C
K1
toxi
nre
sist
ance
phen
otyp
e;h
asa
hyd
roph
obic
dom
ain
8580
54YB
R28
4WSi
mila
rity
toA
MP
deam
inas
e85
wt
70
Mut
ants
not
liste
dar
eth
ose
wit
ha
wild
-type
phen
otyp
ein
hap
loid
orh
omoz
ygou
sdi
ploi
dba
ckgr
oun
dan
dth
ose
that
are
hyp
erse
nsi
tive
(Tab
le3)
orh
aplo
insu
ffici
ent
(Tab
le4)
.R
,re
sist
ant;
S,h
yper
sen
siti
ve;
wt,
wild
type
toC
alco
fluo
rw
hit
e,H
ygro
myc
inB
,an
dSD
S.N
A,
not
avai
labl
e.a
Indi
cate
sm
utan
tsw
ith
a�
-glu
can
phen
otyp
e(s
eeT
able
5).
883K1 Toxin Phenotypes of Yeast Genes
TA
BL
E3
Gen
esw
hose
dele
tion
caus
eshy
pers
ensi
tivi
tyto
the
K1
kille
rto
xin
(hap
loid
orho
moz
ygou
sdi
ploi
dde
ath
zone
�11
5%of
the
wild
type
)
K1
kille
rto
xin
deat
hzo
ne
(%)
Cal
cofl
uor
Gen
en
ame
OR
FD
escr
ipti
onof
gen
epr
oduc
tH
aplo
idH
eter
ozyg
ous
Hom
ozyg
ous
wh
ite
Hyg
rom
ycin
BSD
S
1.K
inas
es,
phos
phat
ases
,si
gnal
tran
sduc
tion
(8ge
nes
)H
OG
1YL
R11
3Wse
r/th
rpr
otei
nki
nas
eof
MA
PKfa
mily
196
wt
160
wt
wt
SPB
S2YJ
L12
8CT
yros
ine
prot
ein
kin
ase
ofth
eM
AP
kin
ase
kin
ase
fam
ily19
3w
t17
9SP
S1YD
R52
3Cse
r/th
rpr
otei
nki
nas
e15
2w
tN
AST
E11
YLR
362W
ser/
thr
prot
ein
kin
ase
ofth
eM
EK
Kfa
mily
139
wt
NA
wt
wt
SSS
K1
YLR
006C
Tw
o-co
mpo
nen
tsi
gnal
tran
sduc
er13
5w
t11
1R
wt
SSS
K2
YNR
031C
MA
Pki
nas
eki
nas
eki
nas
eof
the
HO
Gpa
thw
ay13
0w
t11
4T
PD3
YAL
016W
ser/
thr
prot
ein
phos
phat
ase
2A,
regu
lato
rych
ain
A12
2w
tN
AD
IG1
YPL
049C
Dow
nre
gula
tor
ofin
vasi
vegr
owth
and
mat
ing
113
wt
118
Rw
tw
t
2.T
ran
scri
ptio
n(7
gen
es)
MED
2YD
L00
5CT
ran
scri
ptio
nal
regu
lati
onm
edia
tor
166
wt
NA
GA
L11
YOL
051W
RN
APo
lII
hol
oen
zym
e(S
RB
)su
bcom
plex
subu
nit
158
wt
162
SS
SSR
B5
YGR
104C
RN
APo
lII
hol
oen
zym
e(S
RB
)su
bcom
plex
subu
nit
131
wt
124
SS
SSR
B2
YHR
041C
RN
APo
lII
hol
oen
zym
e(S
RB
)su
bcom
plex
subu
nit
124
wt
128
ITC
1YG
L13
3WSu
bun
itof
Isw
2ch
rom
atin
rem
odel
ing
com
plex
121
wt
125
UM
E6YD
R20
7CN
egat
ive
tran
scri
ptio
nal
regu
lato
r11
8w
t13
9S
wt
SSW
16YL
R18
2WT
ran
scri
ptio
nfa
ctor
118
wt
119
SS
S
3.R
NA
proc
essi
ng
(6ge
nes
)PR
P18
YGR
006W
U5
snR
NA
-ass
ocia
ted
prot
ein
148
wt
103
wt
wt
wt
CB
C2
YPL
178W
Smal
lsu
bun
itof
the
nuc
lear
cap-
bin
din
gpr
otei
nco
mpl
exC
BC
139
wt
119
Rw
tw
tC
DC
40YD
R36
4CR
equi
red
for
mR
NA
splic
ing
137
wt
NA
SS
SN
SR1
YGR
159C
Nuc
lear
loca
lizat
ion
sequ
ence
bin
din
gpr
otei
n12
2w
t11
6S
wt
wt
BR
R1
YPR
057W
Invo
lved
insn
RN
Pbi
ogen
esis
120
wt
116
wt
wt
wt
STO
1YM
R12
5WL
arge
subu
nit
ofth
en
ucle
arca
p-bi
ndi
ng
prot
ein
com
plex
CB
C11
9w
t12
4
4.R
ibos
omal
and
tran
slat
ion
init
iati
onpr
otei
ns
(18
2
gen
es)
RPS
16B
YDL
083C
Rib
osom
alpr
otei
nS1
6.e
157
wt
116
RPS
19B
YNL
302C
40S
smal
lsu
bun
itri
boso
mal
prot
ein
S19.
e14
9w
tN
AR
PS24
BYI
L06
9C40
Ssm
all
subu
nit
ribo
som
alpr
otei
nS2
4.e
149
wt
144
RPS
24A
YER
074W
40S
smal
lsu
bun
itri
boso
mal
prot
ein
S24.
e12
9w
t14
2w
tw
tw
tR
PS10
AYO
R29
3WR
ibos
omal
prot
ein
S10.
e12
811
211
8R
wt
wt
ASC
1YM
R11
6C40
Ssm
all
subu
nit
ribo
som
alpr
otei
n12
7w
t13
0R
PS17
AYM
L02
4WR
ibos
omal
prot
ein
S17.
e.A
127
wt
128
wt
wt
wt
RPS
11A
YDR
025W
Rib
osom
alpr
otei
nS1
1.e
126
wt
139
RPS
23B
YPR
132W
40S
smal
lsu
bun
itri
boso
mal
prot
ein
S23.
e12
6w
t12
3
(con
tinue
d)
884 N. Page et al.
TA
BL
E3
(Con
tinu
ed)
K1
kille
rto
xin
deat
hzo
ne
(%)
Cal
cofl
uor
Gen
en
ame
OR
FD
escr
ipti
onof
gen
epr
oduc
tH
aplo
idH
eter
ozyg
ous
Hom
ozyg
ous
wh
ite
Hyg
rom
ycin
BSD
S
RPL
13B
YMR
142C
60S
larg
esu
bun
itri
boso
mal
prot
ein
124
wt
130
RPS
16A
YMR
143W
Rib
osom
alpr
otei
nS1
6.e
121
wt
135
wt
wt
wt
RPS
0BYL
R04
8W40
Sri
boso
mal
prot
ein
p40
hom
olog
B12
0w
t12
5R
PS30
BYO
R18
2CSi
mila
rity
toh
uman
ubiq
uiti
n-li
kepr
otei
n/r
ibos
omal
prot
ein
S30
120
wt
120
RPL
2BYI
L01
8W60
Sla
rge
subu
nit
ribo
som
alpr
otei
nL
8.e
118
111
136
RPS
4BYH
R20
3CR
ibos
omal
prot
ein
S4.e
.c8
117
wt
114
RPS
30A
YLR
287C
-A40
Ssm
all
subu
nit
ribo
som
alpr
otei
n11
6w
t11
4R
PL14
AYK
L00
6WR
ibos
omal
prot
ein
115
wt
145
wt
wt
wt
RPS
6BYB
R18
1CR
ibos
omal
prot
ein
S6.e
114
115
126
TIF
3YP
R16
3CT
ran
slat
ion
init
iati
onfa
ctor
eIF4
B15
4w
t13
9w
tw
tS
YIF2
YAL
035W
Gen
eral
tran
slat
ion
fact
oreI
F2h
omol
og12
2w
tN
A
5.Pr
otei
nm
odifi
cati
onor
N-g
lyco
syla
tion
(5ge
nes
)M
NN
9a
YPL
050C
Req
uire
dfo
rco
mpl
exN
-gly
cosy
lati
on15
2w
t14
6S
SS
AN
P1a
YEL
036C
Req
uire
dfo
rpr
otei
ngl
ycos
ylat
ion
inth
eG
olgi
135
wt
129
SS
SM
AP1
aYL
R24
4CM
eth
ion
ine
amin
opep
tida
se,
isof
orm
113
4w
t13
0S
SS
MN
N10
aYD
R24
5WSu
bun
itof
�-1
,6-m
ann
osyl
tran
sfer
ase
com
plex
133
wt
136
SS
SL
AS2
1aYJ
L06
2WR
equi
red
for
side
-ch
ain
addi
tion
toG
PI11
4w
t11
9
6.C
ellu
lar
pola
rity
(5ge
nes
)B
UD
30a
YDL
151C
K1
toxi
nh
yper
sen
siti
vity
phen
otyp
e13
4w
t10
7B
UD
22YM
R01
4WPr
otei
nw
ith
poss
ible
role
inbu
dsi
tepo
lari
ty12
8w
t14
4S
Sw
tB
UD
25YE
R01
4C-A
Prot
ein
invo
lved
inbi
pola
rbu
ddin
g12
3w
t11
5S
SS
BEM
1YB
R20
0WB
udem
erge
nce
med
iato
r12
1w
t11
4S
SS
BU
D27
aYF
L02
3WK
1to
xin
hyp
erse
nsi
tivi
typh
enot
ype
115
wt
157
7.N
ewFY
Vge
nes
(8ge
nes
)FY
V1a
YDR
024W
K1
toxi
nh
yper
sen
siti
vity
phen
otyp
e14
1w
t13
8FY
V4YH
R05
9WK
1to
xin
hyp
erse
nsi
tivi
typh
enot
ype
127
wt
116
FYV5
aYC
L05
8CK
1to
xin
hyp
erse
nsi
tivi
typh
enot
ype
125
wt
119
FYV6
aYN
L13
3CK
1to
xin
hyp
erse
nsi
tivi
typh
enot
ype
123
wt
112
FYV7
aYL
R06
8WK
1to
xin
hyp
erse
nsi
tivi
typh
enot
ype
120
wt
116
wt
wt
wt
FYV8
YGR
196C
K1
toxi
nh
yper
sen
siti
vity
phen
otyp
e;si
mila
rity
toA
rp1p
119
wt
121
SS
SFY
V10
aYI
L09
7WK
1to
xin
hyp
erse
nsi
tivi
typh
enot
ype
116
wt
115
wt
SS
FYV1
2a
YOR
183W
K1
toxi
nh
yper
sen
siti
vity
phen
otyp
e11
4w
t12
3w
tS
wt
(con
tinue
d)

885K1 Toxin Phenotypes of Yeast Genes
bud site in unbudded cells and at the site of bud emer-gence (Martin et al. 1999) and may act in the polariza-tion of glucan synthetic components. CSF1 (YLR087C)encodes an integral membrane protein that may be aplasma membrane carrier. The null mutant is hypersen-sitive to K1 toxin, calcofluor white, SDS, and hygro-mycin; Tokai et al. (2000) showed the mutant to be saltand hydrogen peroxide sensitive with low temperaturedefects in growth and the uptake of glucose and leucine.LAS21 (YJL062W) participates in glycosylphosphatidylin-ositol (GPI) synthesis, adding an ethanolamine phos-phate to the �-1,6-linked mannose of the GPI mannosecore (Benachour et al. 1999). As this mannose core isthe site of attachment of the �-1,6-glucan moiety to GPI-linked cell wall proteins, altered levels of �-1,6-glucanmight be expected, although the basis of neither the�-1,3-glucan defect nor the mutant hypersensitivity toK1 toxin is evident, indicating a need for further work.
�-1,6-Glucan elevated: Killer mutants in 33 genes hadelevated levels of �-1,6-glucan (Table 5). A group of�-1,6-glucan overproducers are mutant in genes in-volved in assembly of the outer fungal-specific �-1,6-glucan chain of N-glycosyl chains (mnn9, mnn10, andanp1; see Table 5 and Figure 2). Mutants in these genesare hypersensitive to killer toxin and are described fur-ther in N-glycosylation below. A contrasting group of resis-tant mutants overproducing �-1,6-glucan (and to alesser extent, �-1,3-glucan) are in a subgroup of genesinvolved in cortical actin assembly and endocytosis (Ta-ble 2 and sla1 mutant in Figure 1). Our results areconsistent with work reporting thickened cell walls insome of these mutants (for a review see Pruyne andBretscher 2000). Cell wall synthesis is normally re-stricted to the growing bud, but in these mutants newmaterial is added inappropriately to the mother cell,resulting in a thickened wall (Li et al. 2002). It is surpris-ing that cells with thickened cell walls and more �-1,6-glucan can be killer toxin resistant, since resistance typi-cally arises through loss of cell wall �-1,6-glucan andless binding of the toxin. One explanation is that moretoxin is bound to the walls, reducing its effective concen-tration, a resistance mechanism proposed for the SMKTtoxin of Pichia farinosa (Suzuki and Shimma 1999). Asecond explanation is that the thickened cell wall blockstoxin access to the plasma membrane.
Mutants for other genes that specifically overproducealkali-soluble �-1,6-glucan have broadly acting geneproducts, with mutants expected to be pleiotropic andtheir effects indirect. These include MAP1 encodingone of an essential pair of methionine aminopeptidases;this mutant is killer toxin, calcofluor white, hygromycin,and SDS hypersensitive (Table 3) and has a randombudding pattern (Ni and Snyder 2001). ERG4 encodesan oxidoreductase required for ergosterol synthesis.This mutant is partially toxin resistant, hypersensitiveto calcofluor white, hygromycin, and SDS (Table 2),
TA
BL
E3
(Con
tinu
ed)
K1
kille
rto
xin
deat
hzo
ne
(%)
Cal
cofl
uor
Gen
en
ame
OR
FD
escr
ipti
onof
gen
epr
oduc
tH
aplo
idH
eter
ozyg
ous
Hom
ozyg
ous
wh
ite
Hyg
rom
ycin
BSD
S
8.U
ngr
oupe
dor
poor
lych
arac
teri
zed
gen
es(1
2ge
nes
)C
SF1a
YLR
087C
Req
uire
dfo
rn
orm
algr
owth
rate
and
resi
stan
ceto
NaC
lan
dH
2O2
140
wt
125
SS
SVI
D21
aYD
R35
9CM
utan
tim
pair
edin
fruc
tose
-1,6
-bis
phos
phat
ase
degr
adat
ion
139
wt
136
SS
SA
RV1
aYL
R24
2CL
ipid
and
ster
olm
etab
olis
m13
7w
t12
3S
SS
SEC
66a
YBR
171W
ER
prot
ein
-tran
sloc
atio
nco
mpl
exsu
bun
it13
3w
t14
4PF
D1
YJL
179W
Pref
oldi
nsu
bun
it1
133
wt
117
SS
SC
TF4
YPR
135W
DN
A-d
irec
ted
poly
mer
ase
�-b
indi
ng
prot
ein
127
wt
112
wt
wt
wt
SYG
1YI
L04
7CM
embe
rof
the
maj
orfa
cilit
ator
supe
rfam
ily12
6w
t14
5IW
R1
YDL
115C
Hyp
oth
etic
alpr
otei
n12
6w
t11
0YD
J1YN
L06
4CM
itoc
hon
dria
lan
dE
Rim
port
prot
ein
121
wt
130
APL
4YP
R02
9CG
amm
a-ad
apti
nof
clat
hri
n-a
ssoc
iate
dA
P-1
com
plex
116
wt
117
wt
Sw
tH
MO
1YD
R17
4WN
onh
isto
ne
prot
ein
115
wt
109
AD
K1
YDR
226W
Ade
nyl
ate
kin
ase,
cyto
solic
112
wt
127
SS
S
Mut
ants
wit
ha
wild
-type
phen
otyp
ein
hap
loid
orh
omoz
ygou
sdip
loid
back
grou
nd
and
thos
ew
ith
are
sist
antp
hen
otyp
ein
thes
eba
ckgr
oun
dsar
en
otlis
ted.
NA
,not
avai
labl
e.a
Mut
ants
wit
ha
�-g
luca
nph
enot
ype
(see
Tab
le5)
.
886 N. Page et al.
TA
BL
E4
Gen
esw
hose
dele
tion
resu
lts
ina
K1
kille
rto
xin
hapl
oins
uffi
cien
cyph
enot
ype
K1
kille
rto
xin
deat
hzo
ne
%C
alco
fluo
rG
ene
nam
eO
RF
Des
crip
tion
ofge
ne
prod
uct
het
eroz
ygou
sw
hit
eH
ygro
myc
inB
SDS
1.R
esis
tan
t(d
eath
zon
e�
90%
ofth
ew
ildty
pe;
28
3ge
nes
)R
SP5
YER
125W
Ubi
quit
in-p
rote
inlig
ase
59G
LC
7YE
R13
3Wse
r/th
rph
osph
opro
tein
phos
phat
ase
1,ca
taly
tic
chai
n69
PUP2
YGR
253C
20S
prot
easo
me
subu
nit
(�5)
70C
CT
4YD
L14
3WC
ompo
nen
tof
chap
eron
in-c
onta
inin
gT
-com
plex
72A
RC
35YN
R03
5CSu
bun
itof
the
Arp
2/3
com
plex
73C
CT
7YJ
L11
1WC
ompo
nen
tof
chap
eron
in-c
onta
inin
gT
-com
plex
78SM
D3
YLR
147C
Stro
ng
sim
ilari
tyto
smal
ln
ucle
arri
bon
ucle
opro
tein
D3
80w
tw
tw
tN
SA2
YER
126C
K1
toxi
nre
sist
ance
phen
otyp
e;n
ucle
arpr
otei
n81
AR
P3YJ
R06
5CA
ctin
-rel
ated
prot
ein
82A
RC
15YI
L06
2CSu
bun
itof
the
Arp
2/3
com
plex
83SF
H1
YLR
321C
Subu
nit
ofth
eR
SCco
mpl
ex83
wt
wt
wt
CC
T2
YIL
142W
Ch
aper
onin
ofth
eT
CP1
rin
gco
mpl
ex,
cyto
solic
84C
CT
5YJ
R06
4WT
-com
plex
prot
ein
1,ep
silo
nsu
bun
it84
TA
D3
YLR
316C
Subu
nit
oftR
NA
-spe
cifi
cad
enos
ine-
34de
amin
ase
85b
BIG
1aYH
R10
1CB
igce
llsph
enot
ype
85w
tw
tS
GC
D7
YLR
291C
Tra
nsl
atio
nin
itia
tion
fact
oreI
F2b,
43-k
Dsu
bun
it85
wt
wt
wt
SEC
53YF
L04
5CPh
osph
oman
nom
utas
e86
SSL
2YI
L14
3CD
NA
hel
icas
e86
AC
C1a
YNR
016C
Ace
tyl-C
oAca
rbox
ylas
e87
CD
C25
aYL
R31
0CG
DP/
GT
Pex
chan
gefa
ctor
for
Ras
1pan
dR
as2p
87w
tw
tw
tG
AA
1YL
R08
8WR
equi
red
for
atta
chm
ent
ofG
PIan
chor
onto
prot
ein
s87
wt
wt
wt
TA
F3YP
L01
1CC
ompo
nen
tof
the
TB
P-as
soci
ated
prot
ein
com
plex
87w
tw
tw
tT
IP20
YGL
145W
Req
uire
dfo
rE
R-to
-Gol
gitr
ansp
ort
87T
RS1
20YD
R40
7CW
eak
sim
ilari
tyto
Myo
1p87
wt
wt
wt
CEG
1YG
L13
0Wm
RN
Agu
anyl
yltr
ansf
eras
e(m
RN
Aca
ppin
gen
zym
e,�
-sub
unit
)88
MEX
67YP
L16
9CFa
ctor
for
nuc
lear
mR
NA
expo
rt88
wt
wt
wt
LSG
1YG
L09
9WR
equi
red
for
nor
mal
grow
th,
mor
phol
ogy,
mat
ing,
spor
ulat
ion
89ER
G27
YLR
100W
3-K
eto
ster
olre
duct
ase,
requ
ired
for
ergo
ster
olbi
osyn
thes
is89
wt
wt
wt
CD
C3
YLR
314C
Cel
ldi
visi
onco
ntr
olpr
otei
n89
wt
wt
wt
New
KR
Ege
nes
KR
E29
YER
038C
Tw
o-h
ybri
din
tera
ctio
nw
ith
Ym10
23p
and
Lys
14p
76K
RE3
3a
YNL
132W
Un
know
nfu
nct
ion
,A
mp1
p-in
tera
ctio
nco
mpl
ex(2
0m
embe
rs)
83
(con
tinue
d)

887K1 Toxin Phenotypes of Yeast Genes
and has a random budding pattern (Ni and Snyder2001). ERV14 (YGL054C) and ERV41 (YNL067C) encodeCOPII vesicle coat proteins involved in endoplasmicreticulum (ER)-to-Golgi trafficking (Otte et al. 2001),and both show toxin resistance. Mutants in four genesof unknown function also overproduce alkali-soluble�-1,6-glucan (Table 5). Two of these genes, BUD27(YFL023W) and BUD30 (YDL151C), have random bud-ding patterns when mutated (Ni and Snyder 2001), andboth are hypersensitive to killer toxin. FYV5 (YCL058C)encodes a predicted small integral membrane protein,with the mutant sensitive to sorbitol and low tempera-ture (Bianchi et al. 1999) and K1 toxin hypersensitive.Finally, the null mutant of YGL007C has partial killertoxin resistance (Table 2).
N-glycosylation: Defects in N-glucosylation and its pro-cessing can lead to partial toxin resistance and reducedlevels of �-1,6-glucan (Romero et al. 1997; Shahinianet al. 1998). Our results extend this finding to manyother genes whose products are involved in the biosyn-thesis and elaboration of the Glc3Man9GlcNAc2 oligosac-charide precursor of N-glycoproteins (Tables 2 and 3;Figure 2). If Golgi synthesis of the fungal-specific �-1,6-mannose outer arm of the N-chain is blocked by muta-tion in OCH1 or in MNN9, MNN10, or ANP1 of themannan polymerase complex, toxin hypersensitivity re-sults, concomitant with higher levels of �-1,6-glucan inthe cell wall (Tables 3 and 5; Figures 1 and 2; andsee Magnelli et al. 2002 for mnn9). The glucan levelsobserved in an och1 mutant were similar to those ob-tained in a mnn9 mutant (not shown). To explore thisfurther we determined the alkali-soluble glucan levelsfor other mutants in the mannan polymerase complexand the outer chain �-mannosyltransferases (Figure 2),irrespective of toxin phenotype. A mutant in mnn11,part of the �-1,6-mannose-synthesizing mannan poly-merase complex, also showed elevated glucan levels, asdid mnn2 encoding the major �-1,2-mannosyltransferasethat initiates mannose branching from the �-1,6-glucanbackbone. However, a mutant in mnn5, whose geneproduct extends the �-1,2-mannose branches from the�-1,6-glucan backbone, had reduced levels of both�-glucans. Previous work showed that a small amountof glucan is attached to the N-chain structure (Tkacz1984; van Rinsum et al. 1991; Kollar et al. 1997), anda genetic study by Shahinian et al. (1998) also suggestedthis possibility. Our results show that core N-chain pro-cessing is required for wild-type �-1,6-glucan levels,while absence of the outer �-1,6-linked mannose sidechain or its first �-1,2-mannose branch can result in anincrease in cell wall �-1,6-glucan. However, mutants inlater mannosylation steps in elaborating branches fromthe outer �-1,6-linked mannose side chain have no effector lead to reduced �-glucan levels.
Lipid and sterol synthesis and ion homeostasis: Mu-tants for 10 genes involved in the biosynthesis or regula-
TA
BL
E4
(Con
tinu
ed)
K1
kille
rto
xin
deat
hzo
ne
%C
alco
fluo
rG
ene
nam
eO
RF
Des
crip
tion
ofge
ne
prod
uct
het
eroz
ygou
sw
hit
eH
ygro
myc
inB
SDS
2.H
yper
sen
siti
ve(d
eath
zon
e�
110%
ofth
ew
ildty
pe;
11ge
nes
)T
UB
1YM
L08
5C�
-1tu
bulin
135
RPB
8YO
R22
4CD
NA
-dir
ecte
dR
NA
poly
mer
ase
I,II
,II
I16
kDsu
bun
it12
8S
wt
wt
RPS
13YD
R06
4WR
ibos
omal
prot
ein
122
wt
wt
wt
RPS
15YO
L04
0C40
Ssm
all
subu
nit
ribo
som
alpr
otei
n12
1w
tw
tw
tR
PB3
YIL
021W
DN
A-d
irec
ted
RN
A-p
olym
eras
eII
,45
kD11
8w
tw
tw
tR
PS3
YNL
178W
Rib
osom
alpr
otei
nS3
.e11
8w
tw
tw
tR
PB7
YDR
404C
DN
A-d
irec
ted
RN
Apo
lym
eras
eII
,19
-kD
subu
nit
117
wt
wt
wt
AU
T2
YNL
223W
Ess
enti
alfo
rau
toph
agy
116
Sw
tS
RPO
26YP
R18
7WD
NA
-dir
ecte
dR
NA
poly
mer
ase
I,II
,II
I18
-kD
subu
nit
116
wt
wt
wt
TSC
10YB
R26
5W3-
keto
sph
inga
nin
ere
duct
ase
115
Sw
tS
PKC
1YB
L10
5CR
egul
ates
MA
Pki
nas
eca
scad
ein
volv
edin
regu
lati
ng
cell
wal
lm
etab
olis
mw
tc
Th
ese
gen
ede
leti
onm
utan
tsar
eav
aila
ble
only
ash
eter
ozyg
otes
and
are
usua
llyes
sen
tial
.a
Mut
ants
wit
ha
�-g
luca
nph
enot
ype
(see
Tab
le5)
.bU
nde
rn
orm
alco
ndi
tion
sth
isge
ne
ises
sen
tial
,bu
th
aplo
idm
utan
tsca
ngr
owon
sorb
itol
and
are
toxi
nre
sist
ant.
cU
nde
rn
orm
alco
ndi
tion
sth
isge
ne
ises
sen
tial
,bu
th
aplo
idm
utan
tsca
ngr
owon
sorb
itol
and
are
toxi
nh
yper
sen
siti
ve.
tion of lipids or sterols show partial toxin resistance

888 N. Page et al.
TABLE 5TABLE 5
Genes whose deletion results in an altered alkali-soluble (Continued)�-glucan phenotype
Gene name ORF �-1,6-Glucan �-1,3-GlucanGene name ORF �-1,6-Glucan �-1,3-Glucan
Both reducedSMI1 YGR229C ��� ���-1,6-Glucan only affectedPIN4 YBL051C �� ��ElevatedCSF1 YLR087C � �MAP1 YLR244C wtLAS21 YJL062W � �ANP1 YEL036C wtCOD3 YGL223C (�) ��ERG4 YGL012W wtPMT2 YAL023C (�) �ERV14 YGL054C wtARV1 YLR242C (�) (�)ERV41 YML067C wtMNN5 YJL186W (�) (�)KEX1 YGL240W wtFYV10 YIL097W (�) (�)FYV5 YCL058C wtKRE33 YNL132W (�) (�)BUD30 YDL151C wtOSH1 YAR044W (�) (��)BUD27 YFL023W wtSBE22 YHR103W (�) (��)YGL007W wtFYV6 YNL133C (��) (��)
Reduced�-1,6-Glucan elevated and �-1,3-glucan reducedYKL037W �� wt
MNN10 YDR245W ��CNE1 YAL058W � wtMNN9 YPL050C �FKS1 YLR342W � wtYUR1 YJL139C () (�)KRE11 YGR166W � wtSHE4 YOR035C () (��)PEP3 YLR148W (�) wt
�-1,6-Glucan reduced and �-1,3-glucan elevated�-1,3-Glucan only affectedBIG1 YHR101C ��� ()ElevatedKRE1 YNL322C �� ()THP1 YOL072W wt KRE6 YPR159W �� ()YNL213C wt ROT2 YBR229C (�) ()BTS1 YPL069C wt ()
GPA2 YER020W wt ()Increase (I): , I � 100%; , 65 � I � 100; , 45 �FYV7 YLR068C wt ()
I � 65; (), 25 � I � 45; (), I � 25%. Decrease (D):���, 85 � D � 100; ��, 65 � D � 85; �, 45 � D � 65;Reduced(�), 25 � D � 45; (��), D � 25%.SEC66 YBR171W wt �
ACC1 YNR016C wt (�)SYS1 YJL004C wt (�)
cellular membrane potential leading to reduced toxin-�-1,6-Glucan and �-1,3-glucan affectedinduced ion permeability. Pertinently, defects in theBoth elevatedATP-dependent Drs2p and Atp2p membrane channelsEND3 YNL084C involved in cation and proton pumping confer toxinVRP1 YLR337C
SAC7 YDR389W resistance. The altered membrane composition in lipidLAS17 YOR181W () or sterol mutants could also affect secretory pathwayVAC8 YEL013W function, possibly linking their partial toxin resistanceFYV1 YDR024W phenotypes to those found in protein trafficking andIPK1 YDR315C ()
secretion (Table 2). For example, KES1 is implicated inFPS1 YLL043W ergosterol biology and can partially suppress the toxinSAC1 YKL212W resistance of a kre11-1 mutant, with Kre11p being in-VID21 YDR359C
EAF6 YJR082C volved in Golgi vesicular transport as a subunit of theCDC25 YLR310C () TRAPP II complex (Jiang et al. 1994; Sacher et al.GLY1 YEL046C () 2001).SUR4 YLR372W () High-osmolarity and stress response pathways: To sur-VPS61 YDR136C ()
vive hyperosmotic conditions, S. cerevisiae increases cel-UTH1 YKR042W () lular glycerol levels by activation of the high-osmolarityVPS27 YNR006W () glycerol (HOG) mitogen-activated protein kinase (MAPK)VPS67 YKR020W ()
FYV12 YOR183W () () pathway. Such activation leads to elevated transcriptionof genes required to cope with stress conditions, includ-(continued)ing the synthesis of glycerol with a resultant increase ininternal osmolarity (Posas et al. 1998; Rep et al. 2000).Mutants with an inactive HOG pathway are toxin hyper-(Table 2). These mutants have defects in membrane
structure, possibly affecting the efficiency of insertion sensitive, while deletion of protein phosphatases, suchas PTP3, PTC1, or PTC3, which act negatively on theof the toxin into the plasma membrane or altering the
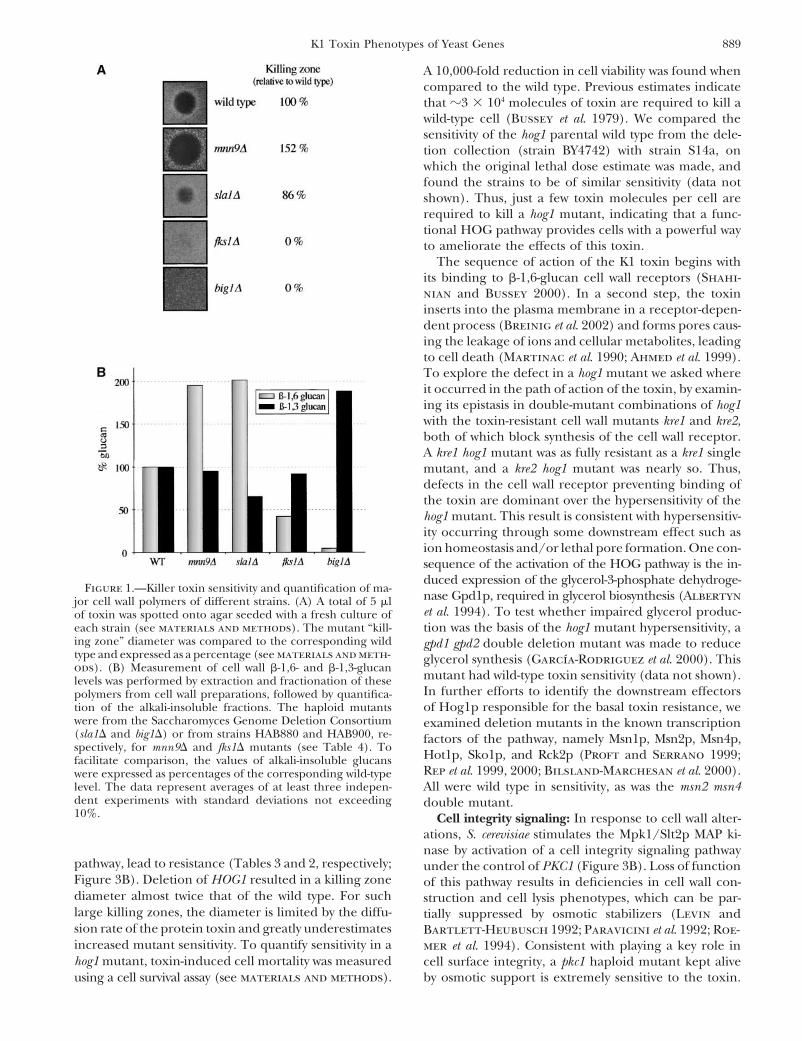
889K1 Toxin Phenotypes of Yeast Genes
A 10,000-fold reduction in cell viability was found whencompared to the wild type. Previous estimates indicatethat �3 � 104 molecules of toxin are required to kill awild-type cell (Bussey et al. 1979). We compared thesensitivity of the hog1 parental wild type from the dele-tion collection (strain BY4742) with strain S14a, onwhich the original lethal dose estimate was made, andfound the strains to be of similar sensitivity (data notshown). Thus, just a few toxin molecules per cell arerequired to kill a hog1 mutant, indicating that a func-tional HOG pathway provides cells with a powerful wayto ameliorate the effects of this toxin.
The sequence of action of the K1 toxin begins withits binding to �-1,6-glucan cell wall receptors (Shahi-nian and Bussey 2000). In a second step, the toxininserts into the plasma membrane in a receptor-depen-dent process (Breinig et al. 2002) and forms pores caus-ing the leakage of ions and cellular metabolites, leadingto cell death (Martinac et al. 1990; Ahmed et al. 1999).To explore the defect in a hog1 mutant we asked whereit occurred in the path of action of the toxin, by examin-ing its epistasis in double-mutant combinations of hog1with the toxin-resistant cell wall mutants kre1 and kre2,both of which block synthesis of the cell wall receptor.A kre1 hog1 mutant was as fully resistant as a kre1 singlemutant, and a kre2 hog1 mutant was nearly so. Thus,defects in the cell wall receptor preventing binding ofthe toxin are dominant over the hypersensitivity of thehog1 mutant. This result is consistent with hypersensitiv-ity occurring through some downstream effect such asion homeostasis and/or lethal pore formation. One con-sequence of the activation of the HOG pathway is the in-duced expression of the glycerol-3-phosphate dehydroge-
Figure 1.—Killer toxin sensitivity and quantification of ma-nase Gpd1p, required in glycerol biosynthesis (Albertynjor cell wall polymers of different strains. (A) A total of 5 �let al. 1994). To test whether impaired glycerol produc-of toxin was spotted onto agar seeded with a fresh culture of
each strain (see materials and methods). The mutant “kill- tion was the basis of the hog1 mutant hypersensitivity, aing zone” diameter was compared to the corresponding wild gpd1 gpd2 double deletion mutant was made to reducetype and expressed as a percentage (see materials and meth- glycerol synthesis (Garcıa-Rodriguez et al. 2000). Thisods). (B) Measurement of cell wall �-1,6- and �-1,3-glucan
mutant had wild-type toxin sensitivity (data not shown).levels was performed by extraction and fractionation of theseIn further efforts to identify the downstream effectorspolymers from cell wall preparations, followed by quantifica-
tion of the alkali-insoluble fractions. The haploid mutants of Hog1p responsible for the basal toxin resistance, wewere from the Saccharomyces Genome Deletion Consortium examined deletion mutants in the known transcription(sla1� and big1�) or from strains HAB880 and HAB900, re- factors of the pathway, namely Msn1p, Msn2p, Msn4p,spectively, for mnn9� and fks1� mutants (see Table 4). To
Hot1p, Sko1p, and Rck2p (Proft and Serrano 1999;facilitate comparison, the values of alkali-insoluble glucansRep et al. 1999, 2000; Bilsland-Marchesan et al. 2000).were expressed as percentages of the corresponding wild-type
level. The data represent averages of at least three indepen- All were wild type in sensitivity, as was the msn2 msn4dent experiments with standard deviations not exceeding double mutant.10%. Cell integrity signaling: In response to cell wall alter-
ations, S. cerevisiae stimulates the Mpk1/Slt2p MAP ki-nase by activation of a cell integrity signaling pathway
pathway, lead to resistance (Tables 3 and 2, respectively; under the control of PKC1 (Figure 3B). Loss of functionFigure 3B). Deletion of HOG1 resulted in a killing zone of this pathway results in deficiencies in cell wall con-diameter almost twice that of the wild type. For such struction and cell lysis phenotypes, which can be par-large killing zones, the diameter is limited by the diffu- tially suppressed by osmotic stabilizers (Levin andsion rate of the protein toxin and greatly underestimates Bartlett-Heubusch 1992; Paravicini et al. 1992; Roe-increased mutant sensitivity. To quantify sensitivity in a mer et al. 1994). Consistent with playing a key role inhog1 mutant, toxin-induced cell mortality was measured cell surface integrity, a pkc1 haploid mutant kept alive
by osmotic support is extremely sensitive to the toxin.using a cell survival assay (see materials and methods).

890 N. Page et al.
Figure 2.—Schematicsummary of N-glycan bio-synthesis in yeast. N-glycosylprecursor assembly is initi-ated in the endoplasmic re-ticulum. At the stage ofGlcNAc2Man9, three glu-cose residues are seriallytransferred from the Dol-P-Glc donor to the N-glycanby the glucosyltransferasesAlg6p, Alg8p, and Alg10p.Glucosylation is requiredfor efficient transfer of theN-glycan to target proteinsby a complex that includesOst3p. The glucose residuesare subsequently trimmedby the sequential action ofglucosidases I and II,Cwh41p and Rot2p, respec-tively. N-linked oligosaccha-rides undergo further matu-ration in the Golgi, whereaddition of the fungal-spe-cific “outer-chain” is initi-ated by Och1p and elabo-
rated by various enzymes, including the mannan polymerase complex (adapted from Orlean 1997; Shahinian and Bussey2000). Arrows indicate activation and bars indicate negative effects. (*) indicates essential genes; i.e., only heterozygous mutantswere tested. Genes whose deletion causes toxin hypersensitivity, red; resistance, blue; no phenotype, yellow; not tested, white.�-Glucans are shown as follows: �-1,6-glucan and �-1,3-glucan both reduced; �-1,6-glucan reduced and �-1,3-glucanwild type; �-1,6-glucan and �-1,3-glucan both wild type; �-1,6-glucan elevated and �-1,3-glucan wild type; �-1,6-glucan elevated and �-1,3-glucan reduced. Mnn2p, Alg10p, and Hoc1p are not listed in Tables 2 or 3; they are resistant orhypersensitive to K1 toxin, but fall outside of the chosen ranges.
However, most of the upstream activators of Pkc1p and duplicated gene mutants shows the phenotype (RPS0B,4B, 10A, 17A, 19B, 23B), suggesting that they have dis-all known downstream MAPK signaling components oftinct functions. Since some phenotypes were relativelythe cell integrity pathway show no toxin phenotype (seeweak (killing zone diameters �115% of the wild type),Figure 3B). The absence of phenotype for the upstreamnot all mutants are listed in Table 3. Of the 8 single-integral plasma membrane activators of the pathwaycopy genes of the small ribosomal subunit, heterozygousmay be explained by the functional redundancy of thedeletions in just 2 essential genes, RPS13 and RPS15,components (Verna et al. 1997; Ketela et al. 1999;gave toxin hypersensitivity (Table 4). The toxin hyper-Philip and Levin 2001). Rho1p, the GTP-binding pro-sensitivity phenotype was more prevalent among mu-tein involved in relaying the signal from the plasmatants in the small subunit (43%) than among those inmembrane to Pkc1p, is essential and the heterozygotethe large (16%). A total of 46 genes encode the largehas a wild-type phenotype. However, in the MAP kinaseribosomal subunit proteins, among which 35 are dupli-cascade downstream of Pkc1p, the kinase Bck1p andcated (Planta and Mager 1998); 12 of the duplicatedthe MAP kinase Mpk1p are unique and nonessentialgenes show toxin hypersensitivity when mutated (Tables(Levin and Errede 1995). The absence of a toxin phe-3 and 4).notype upon mutation of these components indicates
that hypersensitivity of a pkc1 mutant is not caused bythe absence of activation of the MPK1 MAP kinase path-
DISCUSSIONway, but in some other way (Figure 3B).Ribosomal subunit proteins: Defects in many ribo- The ability to directly establish a phenotype-to-gene
somal subunit proteins lead to toxin hypersensitivity. relationship is a great enabling strength of the mutantOf the 32 small ribosomal subunit genes, 8 are found collection. Moreover, since each gene can be examinedas single copy and 24 are duplicated, for a total of 56 simply by testing a mutant, partial or weak phenotypesORFs (Planta and Mager 1998). Toxin hypersensitivity can be readily analyzed (Bennett et al. 2001; Ni andis observed for mutants for 21 of the duplicated genes Snyder 2001). The collection allows comprehensive(Tables 3 and 4). A single deletion of either copy often screening and a knowledge of which genes have been
examined, overcoming many of the limitations of a clas-shows hypersensitivity. In some cases only one of the

891K1 Toxin Phenotypes of Yeast Genes
sical random mutant screen. Despite the extensive use lection used here neither the haploid MATa or MAT�nor the diploid heterozygous or homozygous deletionof random screens for toxin resistance these failed to
saturate the genome, as we have found mutants in many of this gene had a phenotype. Thus, in this strain back-ground Tok1p has no detectable role in toxin action,new genes. In addition, the mutant collection allows
one to know which genes remain to be tested and, im- indicating that despite the ability of the toxin to activateconductance of Tok1p, this channel protein cannot beportantly, which genes do not have phenotypes. Such
comprehensive testing can turn up the unexpected, as the only target for the K1 toxin and is not a significantin vivo target in this sensitive strain. Having mutants inillustrated by a few examples. The extent of the relation-
ships between cell wall polymers was unanticipated. Wall all cellular pathways allows the pursuit of phenotypethrough functional modules and has value in makingglucan work normally focuses on one or the other glu-
can synthetic pathway, and these are implicitly seen to such connections. Some specific examples are discussedbelow.be specific. Yet fks1 mutants, defective for a component
of the �-1,3-glucan synthase, are affected for both �-1,3- Functional clustering: The screen identified severalexamples of interactions that connect biological func-and �-1,6-glucan (Figure 1A), as are a large number
of other mutants (Table 5). These interactions likely tions into larger cellular processes, sometimes alreadyknown in detail. For example, toxin phenotypes traceindicate synthetic or regulatory links between these poly-
mers. The mnn9 mutation, which blocks synthesis of the relationship between almost every biosynthetic stepof the N-glycosyl moiety of glycoproteins. The cytoskele-the outer �-1,6-mannose arm of N-glycans, was assumed
specific and has been used to simplify structural analyses tal mutants provide an example of a less well-character-of glucomannoproteins in the cell wall (Van Rinsum et ized connectivity. Here a set of mutants in cytoskeletalal. 1991; Montijn et al. 1994). The fact that a mnn9 processes has a common toxin resistance phenotypemutation has other secondary effects that increase the that correlates with mother cells showing abnormal wallamount of glucan in the wall is an unexpected complica- proliferation. This wall phenotype, which is not a gen-tion, with the possibility that previous work analyzed eral one for all cytoskeletal defects, has been reportedstructures absent from wild-type cells. Electrophysiologi- for individual genes (see Pruyne and Bretscher 2000).cal work links the Tok1p potassium channel with toxin This functional cluster of genes, which may function inaction (Ahmed et al. 1999). In the deletion mutant col- limiting wall growth to daughter cells, offers insight into
a new facet of morphogenesis.
Figure 3.—Schematic summary of signal transduction path-ways involved in osmoadaptive responses and cell wall synthesisin yeast. (A) Exposure to high extracellular osmolarity triggersan adaptive response mediated by two pathways that convergeat Pbs2p. One arm of the pathway involves the binding ofPbs2p to plasma membrane protein Sho1p. Pbs2p is phosphor-ylated by the Ste11p MAPKKK, through a process requiringCdc42p, Ste50p, and Ste20p (Desmond et al. 2000). A secondpathway involves the two-component osmosensor moduleSln1p-Ypd1p-Ssk1p, which activates Pbs2p via a pair of relatedMAPKKK proteins, Ssk2p and Ssk22p. Activation of this MAPKcascade culminates at Hog1p with Hog1p-dependent activa-tion of the Rck2p protein kinase and activation and inactiva-tion of transcription factors. The model also outlines the ac-tion of some negative regulators of the pathway (Posas et al.1998; Rep et al. 1999, 2000; Bilsland-Marchesan et al. 2000;and references therein). (B) Environmental stresses causechanges in cell wall state, which are detected by the Wscproteins and Mid2p and Mtl1p. The information is transmittedto Rho1p by the guanine nucleotide exchange factors Rom1pand Rom2p. Tor2p is also an activator of Rho1p, whereasSac7p and Bem2p are GTPase-activating proteins for Rho1p.Activated, GTP-bound Rho1p interacts with a transcriptionfactor (Skn7p) and regulates the activity of proteins involvedin cytoskeleton assembly (Bni1p), cell wall synthesis (Fks1p),and signal transduction (Pkc1p). Pkc1p in turn activates thecell integrity MAP kinase pathway and independently on “an-other arm” effects Rap1p-dependent transcriptional repres-sion of ribosomal protein genes (Li et al. 2000; Philip andLevin 2001; and references therein). For the color-codingscheme, see Figure 2.

892 N. Page et al.
The HOG pathway buffers toxin action: Mutants in high osmolarity (Rep et al. 2000), ASC1 had a significanthypersensitivity (Table 3). ASC1 encodes a 40S smallhog1 are close to being maximally sensitive to the toxin,
dying at �1 molecule/cell, while in a HOG1 strain, four subunit ribosomal protein, one of many small ribosomalprotein encoding genes that, when mutated, show toxinorders of magnitude more toxin is needed to kill a cell.
How is this HOG1-dependent resistance achieved? One hypersensitivity (see Table 3 and below). Together,these observations suggest that, if the phenotype ob-possibility is that the HOG pathway is stress induced as
the toxin causes ion loss. Activation of this signaling served in a hog1 mutant results from a defect in expres-sion, it is not through a single gene but may originatepathway may result in changes in membrane conduc-
tance, intracellular osmotic pressure, or some other from a combined deficiency in more than one gene.Signaling components involved in toxin sensitivity:stress response, which can act to reduce the efficiency
of the toxin in promoting loss of cellular ions. Although Although the HOG pathway is the only MAP kinasecascade showing a toxin phenotype, two upstream acti-the toxin sensitivity of a gpd1 gpd2 double mutant is
similar to wild-type cells, the possible involvement of vators of MAPK pathways were identified in the screen:SSK1 and PKC1. The toxin hypersensitivity of an ssk1Hog1p-dependent osmoadaptation cannot be excluded.
Consistent with this scenario, Garcıa-Rodriguez et al. mutant is consistent with its place upstream of the HOGsignal transduction cascade. However, no toxin pheno-(2000) observed increased intracellular glycerol levels
after treatment with the cell-wall-perturbing agent cal- type is found for the components of the cell integrityMAPK pathway signaling downstream of PKC1, namely,cofluor white, independent of the action of GPD1 and
GPD2. An alternative explanation that there is some the sequentially acting kinases Bck1p, the redundantpair Mkk1p and Mkk2p, and the Mpk1p MAP kinaseconstitutive HOG1-dependent effect on cell wall synthe-
sis seems less likely on the basis of the following observa- (Figure 3B). This raises the question of how Pkc1p sig-nals in producing a normal response to the toxin. Previ-tions. Epistatic tests using kre1 hog1 and kre2 hog1 mu-
tants are consistent with the HOG pathway acting at the ous genetic analysis suggested a bifurcation of the signal-ing downstream of PKC1 (Errede and Levin 1993;membrane or intracellularly, as cell wall mutants are
epistatic to the hog1 defect and remain toxin resistant Helliwell et al. 1998). Our data are consistent withsuch a model since some “other arm” of the PKC path-in double mutants. Deficiencies in the HOG pathway
result in extreme toxin sensitivity, and we reasoned that way, distinct from the Bck1p-dependent arm, is responsi-ble for the toxin phenotype. Additional evidence for anmutations in genes regulated by this pathway might also
cause hypersensitivity. In looking for candidates, it is alternative pathway comes from studies on the coordina-tion of cell growth and ribosome synthesis, where astriking that some components specific to the RNA poly-
merase II complex (e.g., Gal11p, Med2p, Rpb4p, Rpb3p, block in protein secretion reduces ribosomal proteingene transcription (Mizuta and Warner 1994; Nier-Rpb7p, Srb5p, and Srb2p) or components shared be-
tween RNA polymerases I, II, and III (e.g., Rpb8p, ras and Warner 1999). This mechanism is: (i) depen-dent on Pkc1p activity; (ii) not mediated by the cellRpc10p, and Rpo26p) all display a strong toxin hyper-
sensitivity, similar to that of HOG pathway mutants (Ta- integrity pathway MAPK cascade (BCK1 or MPK1); and(iii) blocked by rap1-17, a silencing-defective allele ofbles 3 and 4). Is this response specific to the HOG
pathway? Among the MAPK pathways in yeast (Hunter RAP1 (Li et al. 2000). We found that a heterozygous rap1mutant exhibits haploinsufficient toxin hypersensitivityand Plowman 1997; Gustin et al. 1998), only the HOG
pathway exhibits toxin hypersensitivity. Mutants in (Figure 3B), providing additional support for Rap1pbeing an effector of Pkc1p.SMK1, MPK1, and YKL161c, which encode, respectively,
the MAP kinase of the sporulation pathway, the cell Ribosomal subunit mutants show toxin sensitivity:The coupling of protein secretion to ribosome synthesisintegrity pathway, and a putative uncharacterized path-
way, are not toxin hypersensitive. Similarly, a null muta- through the PKC pathway (Nierras and Warner 1999;Li et al. 2000) raises the possibility of regulation op-tion in the MAP kinase kinase encoding gene STE7,
which is involved in both the haploid mating and inva- erating in the reverse direction: that is, defects in pro-tein synthesis mediated predominantly through 40S ri-sive pathways, has no effect on toxin sensitivity. These
observations suggest a possible connection between the bosomal subunit proteins might affect protein secretionand cell wall synthesis. The binding of the rough ERsignaling elements of the HOG pathway and the activity
of the RNA polymerase II complex. To investigate which ribosomes to Sec61p of the signal recognition particleis through the 60S ribosomal subunit (Beckmann et al.potential target genes of Hog1p are responsible for the
hypersensitivity, we looked for toxin phenotypes re- 1997), and fewer mutants in 60S ribosomal proteinshave toxin phenotypes, arguing that the coupling stepsulting from mutations in genes known to be induced
by osmotic shock (Rep et al. 2000). None of these genes in itself is unlikely to be the primary site of any sucheffect. A more mundane alternative explanation is thathave an effect comparable to a hog1 mutant. Similar
results were obtained for genes whose mRNA level is nonessential defects in protein synthesis through lossof redundant ribosomal proteins have nonspecificaffected by a mutation of HOG1. However, among the
genes whose mRNA level is diminished after a shift to knock-on effects on protein secretion/cell wall synthesis

893K1 Toxin Phenotypes of Yeast Genes
F. Posas, 2000 Rck2 kinase is a substrate for the osmotic stress-through failure to make enough of a component re-activated mitogen-activated protein kinase Hog1. Mol. Cell. Biol.
quired for protein secretion. 20: 3887–3895.Brachmann, C. B., A. Davies, G. J. Cost, E. Caputo, J. Li et al., 1998Strength and limitations of comprehensive phenotyp-
Designer deletion strains derived from Saccharomyces cerevisiaeing with the collection: In addition to phenotypic cluster-S288C: a useful set of strains and plasmids for PCR-mediated
ing of genes, the simple discovery of biological roles for gene disruption and other applications. Yeast 14: 115–132.Bradford, M. M., 1976 A rapid and sensitive method for the quanti-genes through phenotype remains an important part
tation of microgram quantities of protein utilizing the principleof this screen. For example, a number of mutants inof protein-dye binding. Anal. Biochem. 72: 248–254.
poorly characterized genes have �-glucan phenotypes Breinig, F., D. J. Tipper and M. J. Schmitt, 2002 Kre1p, the plasmamembrane receptor for the yeast K1 viral toxin. Cell 108: 395–405.that warrant investigation. The yeast disruption mutant
Brown, J. L., T. Roemer, M. Lussier, A. M. Sdicu and H. Bussey,collection has limitations. Duplicated genes and gene1994 The K1 killer toxin: molecular and genetic applications
families having synthetic phenotypes but no phenotype to secretion and cell surface assembly, pp. 217–231 in MolecularGenetics of Yeast: A Practical Approach, edited by J. R. Johnston.when individually deleted will be overlooked. Also, theIRL Press/Oxford University Press, Oxford.1105 essential genes representing 18.7% of the yeast
Bussey, H., 1991 K1 killer toxin, a pore-forming protein from yeast.genome (Giaver et al. 2002) cannot be screened di- Mol. Microbiol. 5: 2339–2343.
Bussey, H., D. Saville, K. Hutchins and R. G. Palfree, 1979 Bind-rectly. Haploinsufficiency phenotypes in heterozygotesing of yeast killer toxin to a cell wall receptor on sensitive Saccharo-disrupted in one copy of an essential gene provide amyces cerevisiae. J. Bacteriol. 140: 888–892.
partial solution, as in the case of BIG1. In our screen de Groot, P. W. J., C. Ruiz, C. R. Vazquez de Aldana, E. Dueas,V. J. Cid et al., 2001 A genomic approach for the identificationsuch haploinsufficiency was found in the heterozygousand classification of genes involved in cell wall formation and itsmutants of 42 genes, but we still do not know the fullregulation in Saccharomyces cerevisiae. Comp. Funct. Genom. 2:
extent of the involvement of essential genes in cell sur- 124–142.Desmond, C., D. C. Raitt, F. Posas and H. Saito, 2000 Yeast Cdc42face biology. A set of conditional lethal mutants in all
GTPase and Ste20 PAK-like kinase regulate Sho1-dependent acti-essential genes would improve the value of the collec-vation of the Hog1 MAPK pathway. EMBO J. 19: 4623–4631.
tion for screening these genes. Dijkgraaf, G. J. P., M. Abe, Y. Ohya and H. Bussey, 2002 Mutationsin Fks1p affect the cell wall content of �-1,3- and �-1,6-glucan inWe thank Angela Chu and Ron Davis (Stanford University) andSaccharomyces cerevisiae. Yeast 19: 671–690.Sally Dow (Rosetta Inpharmatics) for providing strains and assistance.
Errede, B., and D. E. Levin, 1993 A conserved kinase cascade forWe also thank Ashley Coughlin, Steeve Veronneau, Marc Lussier, and MAP kinase activation in yeast. Curr. Opin. Cell Biol. 5: 254–260.Terry Roemer for discussions and contributions and Robin Green Garcıa-Rodriguez, L. J., A. Duran and C. Roncero, 2000 Cal-and Federico Angioni for comments on the manuscript. Supported by cofluor antifungal action depends on chitin and a functionaloperating and CRD grants from the Natural Sciences and Engineering high-osmolarity glycerol response (HOG) pathway: evidence for
a physiological role of the Saccharomyces cerevisiae HOG pathwayResearch Council of Canada. N.P. was a predoctoral fellow of theunder noninducing conditions. J. Bacteriol. 182: 2428–2437.Fonds pour la Formation de Chercheurs et l’Aide a la Recherche
Giaver, G., A. M. Chu, L. Ni, C. Connelly, L. Riles et al., 2002(Quebec).Functional profiling of the Saccharomyces cerevisiae genome. Nature418: 387–391.
Gustin, M. C., J. Albertyn, M. Alexander and K. Davenport,1998 MAP kinase pathways in the yeast Saccharomyces cerevisiae.LITERATURE CITEDMicrobiol. Mol. Biol. Rev. 62: 1264–1300.
Helliwell, S. B., A. Schmidt, Y. Ohya and M. N. Hall, 1998 TheAhmed, A., F. Sesti, N. Ilan, T. M. Shih, S. L. Sturley et al., 1999A molecular target for viral killer toxin: TOK1 potassium chan- Rho1 effector of Pkc1, but not Bni1, mediates signaling from
Tor2 to the actin cytoskeleton. Curr. Biol. 8: 1211–1214.nels. Cell 99: 283–291.Albertyn, J., S. Hohmann, J. M. Thevelein and B. A. Prior, 1994 Hong, Z., P. Mann, N. H. Brown, L. E. Tran, K. J. Shaw et al., 1994a
Cloning and characterization of KNR4, a yeast gene involved inGPD1, which encodes glycerol-3-phosphate dehydrogenase, is es-sential for growth under osmotic stress in Saccharomyces cerevis- (1,3)-��glucan synthesis. Mol. Cell. Biol. 14: 1017–1025.
Hong, Z., P. Mann, K. J. Shaw and B. Didomenico, 1994b Analysisiae, and its expression is regulated by the high-osmolarity glycerolresponse pathway. Mol. Cell. Biol. 14: 4135–4144. of �-glucans and chitin in a Saccharomyces cerevisiae cell wall mutant
using high-performance liquid chromatography. Yeast 10: 1083–Azuma, M., N. Page, J. Levinson and H. Bussey, 2002 Saccharomycescerevisiae Big1p, a putative endoplasmic reticulum membrane pro- 1092.
Hunter, T., and G. D. Plowman, 1997 The protein kinases of bud-tein required for normal levels of cell wall �-1,6-glucan. Yeast 19:783–793. ding yeast: six score and more. Trends Biochem. Sci. 22: 18–22.
Jiang, B., J. L. Brown, J. Sheraton, N. Fortin and H. Bussey, 1994Beckmann, R., D. Bubeck, R. Grassucci, P. Penczek, A. Verschooret al., 1997 Alignment of conduits for the nascent polypeptide A new family of yeast genes implicated in ergosterol synthesis is
related to the human oxysterol binding protein. Yeast 10: 341–chain in the ribosome-Sec61 complex. Science 278: 2123–2126.Benachour, A., G. Spiros, I. Flury, F. Reggiori, E. Canivene- 353.
Ketela, T., R. Green and H. Bussey, 1999 Saccharomyces cerevisiaeGansel et al., 1999 Deletion of GPI7, a yeast gene required foraddition of a side chain to the glycosylphosphatidylinositol (GPI) Mid2p is a potential cell wall stress sensor and upstream activator
of the PKC1-MPK1 cell integrity pathway. J. Bacteriol. 181: 3330–core structure, affects GPI protein transport, remodelling, andcell wall integrity. J. Biol. Chem. 274: 15251–15261. 3340.
Kollar, R., B. B. Reinhold, E. Petrakova, H. J. Yeh, G. AshwellBennett, C. B., L. K. Lewis, G. Karthikeyan, K. S. Lobachev, Y. H.Jin et al., 2001 Genes required for ionizing radiation resistance et al., 1997 Architecture of the yeast cell wall. Beta (1→6)-glucan
interconnects mannoprotein, beta(1→3)-glucan, and chitin. J.in yeast. Nat. Genet. 29: 426–434.Bianchi, M. M., G. Sartori, M. Vandenbol, A. Kaniak, D. Uccel- Biol. Chem. 272: 17762–17775.
Levin, D. E., and E. Bartlett-Heubusch, 1992 Mutants in the S.letti et al., 1999 How to bring orphan genes into functionalfamilies. Yeast 15: 513–526. cerevisiae PKC1 gene display a cell cycle-specific osmotic stability
defect. J. Cell Biol. 116: 1221–1229.Bickle, M., P. A. Delley, A. Schmidt and M. N. Hall, 1998 Cellwall integrity modulates RHO1 activity via the exchange factor Levin, D. E., and B. Errede, 1995 The proliferation of MAP kinase
signaling pathways in yeast. Curr. Opin. Cell Biol. 7: 197–202.ROM2. EMBO J. 17: 2235–2245.Bilsland-Marchesan, E., J. Arino, H. Saito, P. Sunnerhagen and Li, H., N. Page and H. Bussey, 2002 Actin patch assembly proteins

894 N. Page et al.
Las17p and Sla1p restrict cell wall growth to daughter cells, and bZIP protein Sko1p confers HOG-dependent osmotic regulation.Mol. Cell. Biol. 19: 537–546.interact with the cis Golgi protein Kre6p. Yeast 19: 1097–1112.
Pruyne, D., and A. Bretscher, 2000 Polarization of cell growth inLi, Y., R. D. Moir, I. K. Sethy-Coraci, J. R. Warner and I. M. Willis,yeast. J. Cell Sci. 113: 571–585.2000 Repression of ribosome and tRNA synthesis in secretion-
Ram, A. F., A. Wolters, R. Ten Hoopen and F. M. Klis, 1994 Adefective cells is signaled by a novel branch of the cell integritynew approach for isolating cell wall mutants in Saccharomycespathway. Mol. Cell. Biol. 20: 3843–3851.cerevisiae by screening for hypersensitivity to calcofluor white.Lipke, P. N., and R. Ovalle, 1998 Cell wall architecture in yeast:Yeast 10: 1019–1030.new structure and new challenges. J. Bacteriol. 180: 3735–3740.
Rep, M., V. Reiser, U. Gartner, J. M. Thevelein, S. Hohmann et al.,Lussier, M., A. M. White, J. Sheraton, T. di Paolo, J. Treadwell1999 Osmotic stress-induced gene expression in Saccharomyceset al., 1997 Large scale identification of genes involved in cellcerevisiae requires Msn1p and the novel nuclear factor Hot1p.surface biosynthesis and architecture in Saccharomyces cerevisiae.Mol. Cell. Biol. 19: 5474–5485.Genetics 147: 435–450.
Rep, M., M. Krantz, J. M. Thevelein and S. Hohmann, 2000 TheLussier, M., A. M. Sdicu, S. Shahinian and H. Bussey, 1998 Thetranscriptional response of Saccharomyces cerevisiae to osmoticCandida albicans KRE9 gene is required for cell wall �-1, 6-glucanshock: Hot1p and Msn2p/Msn4p are required for the inductionsynthesis and is essential for growth on glucose. Proc. Natl. Acad.of subsets of HOG-dependent genes. J. Biol. Chem. 275: 8290–Sci. USA 95: 9825–9830.8300.Magnelli, P., J. F. Cipollo and C. Abeijon, 2002 A refined method
Robinson, J. S., D. J. Klionsky, L. M. Banta and S. D. Emr, 1988for the determination of Saccharomyces cerevisiae cell wall composi-Protein sorting in Saccharomyces cerevisiae : isolation of mutantstion and �-1,6-glucan fine structure. Anal. Biochem. 301: 136–defective in the delivery and processing of multiple vacuolar150.hydrolases. Mol. Cell. Biol. 8: 4936–4948.Martin, H., A. Dagkessamanskia, G. Satchanska, N. Dallies and
Roemer, T., G. Paravicini, M. A. Payton and H. Bussey,J. Francois, 1999 KNR4, a suppressor of Saccharomyces cerevisiae 1994 Characterization of the yeast (1→6)-�-glucan biosyntheticcwh mutants, is involved in the transcriptional control of chitin components, Kre6p and Skn1p, and genetic interactions betweensynthase genes. Microbiology 145: 249–258. the PKC1 pathway and extracellular matrix assembly. J. Cell Biol.Martinac, B., H. Zhu, A. Kubalski, X. Zhou, M. Culbertson et al., 127: 567–579.
1990 Yeast K1 killer toxin forms ion channels in sensitive yeast Romero, P. A., G. J. P. Dijkgraaf, S. Shahinian, A. Herscovics andspheroplasts and in artificial liposomes. Proc. Natl. Acad. Sci. H. Bussey, 1997 The yeast CWH41 gene encodes glucosidaseUSA 87: 6228–6232. I. Glycobiology 7: 997–1004.
Mizuta, K., and J. R. Warner, 1994 Continued functioning of the Ross-Macdonald, P., P. S. R. Coelho, T. Roemer, S. Agarwal, A.secretory pathway is essential for ribosome synthesis. Mol. Cell. Kumar et al., 1999 Large-scale analysis of the yeast genome byBiol. 14: 2493–2502. transposon tagging and gene disruption. Nature 402: 413–418.
Montijn, R. C., J. van Rinsum, F. A. van Schagen and F. M. Klis, Sacher, M., J. Barrowman, W. Wang, J. Horecka, Y. Zhang et al.,1994 Glucomannoproteins in the cell wall of Saccharomyces cere- 2001 TRAPP I implicated in the specificity of tethering in ER-visiae contain a novel type of carbohydrate side chain. J. Biol. to-Golgi transport. Mol. Cell 7: 433–442.Chem. 269: 19338–19342. Shahinian, S., and H. Bussey, 2000 �-1,6-glucan synthesis in Saccha-
Ni, L., and M. Snyder, 2001 A genomic study of the bipolar bud romyces cerevisiae. Mol. Microbiol. 35: 477–489.site selection pattern in Saccharomyces cerevisiae. Mol. Biol. Cell Shahinian, S., G. J. Dijkgraaf, A. M. Sdicu, D. Y. Thomas, C. A.12: 2147–2170. Jakob et al., 1998 Involvement of protein N-glycosyl chain gluco-
Nierras, C. R., and J. R. Warner, 1999 Protein kinase C enables the sylation and processing in the biosynthesis of cell wall �-1,6-glucanregulatory circuit that connects membrane synthesis to ribosome of Saccharomyces cerevisiae. Genetics 149: 843–856.synthesis in Saccharomyces cerevisiae. J. Biol. Chem. 274: 13235– Sherman, F., 1991 Getting started with yeast. Methods Enzymol.13241. 194: 3–21.
Orlean, P., 1997 Biogenesis of yeast wall and surface components, Suzuki, C., and Y. Shimma, 1999 P-type ATPase spf1 mutants showpp. 229–362 in Molecular and Cellular Biology of the Yeast Saccharo- a novel resistance mechanism for the killer toxin SMKT. Mol.myces, Vol. 3: Cell Cycle and Cell Biology, edited by J. R. Pringle, Microbiol. 32: 813–823.
Tkacz, J. S., 1984 In vivo synthesis of �-1,6-glucan in SaccharomycesJ. R. Broach and E. W. Jones. Cold Spring Harbor Laboratorycerevisiae, pp. 287–295 in Microbial Cell Wall Synthesis and Autolysis,Press, Cold Spring Harbor, NY.edited by C. Nombela. Elsevier, Amsterdam.Otte, S., W. J. Belden, M. Heidtman, J. Liu, O. N. Jensen et al.,
Tokai, M., H. Kawasaki, Y. Kikuchi and K. Ouchi, 2000 Cloning2001 Erv41p and Erv46p: new components of COP II vesiclesand characterization of the CSF1 gene of Saccharomyces cerevisiae,involved in transport between the ER and Golgi complex. J. Cellwhich is required for nutrient uptake at low temperature. J. Bacte-Biol. 152: 503–518.riol. 182: 2865–2868.Paravicini, G., M. Cooper, L. Friedli, D. J. Smith, J. L. Carpentier
Van Rinsum, J., F. M. Klis and H. van den Ende, 1991 Cell wallet al., 1992 The osmotic integrity of the yeast cell requires aglucomannoproteins of Saccharomyces cerevisiae mnn9. Yeast 7: 717–functional PKC1 gene product. Mol. Cell. Biol. 12: 4896–4905.726.Philip, B., and D. E. Levin, 2001 Wsc1 and Mid2 are cell surface
Verna, J., A. Lodder, K. Lee, A. Vagts and R. Ballester, 1997 Asensors for cell wall integrity signaling that act through Rom2,family of genes required for maintenance of cell wall integritya guanine nucleotide exchange factor for Rho1. Mol. Cell. Biol.and for the stress response in Saccharomyces cerevisiae. Proc. Natl.21: 271–280.Acad. Sci. USA 94: 13804–13809.Planta, R. J., and W. H. Mager, 1998 The list of cytoplasmic ribo-
Wickner, R. B., 1996 Microbiol. Rev. 60: 250–265.somal proteins of Saccharomyces cerevisiae. Yeast 14: 471–477.Winzeler, E. A., D. D. Shoemaker, A. Astromoff, H. Liang, K.Posas, F., M. Takekawa and H. Saito, 1998 Signal transduction by
Anderson et al., 1999 Functional characterization of the S. cere-MAP kinase cascades in budding yeast. Curr. Opin. Microbiol.visiae genome by gene deletion and parallel analysis. Science 285:1: 175–182. 901–906.
Proft, M., and R. Serrano, 1999 Repressors and upstream repress-ing sequences of the stress-regulated ENA1 gene in S. cerevisiae: Communicating editor: M. Johnston




















