
A pre-emptive strike against malaria's stealthy hepatic forms
11
OPINION A pre-emptive strike against malaria’s stealthy hepatic forms Dominique Mazier, Laurent Rénia & Georges Snounou Abstract | Almost all the drugs that are widely used today against Plasmodium spp., the causative agent of malaria, target the asexual blood stages of the parasite. Widespread drug resistance severely restricts our ability to control malaria and makes it necessary to seek novel antimalarial compounds. Here, we advocate the development of true causal chemoprophylactic drugs that will fully inhibit the obligate short-lived hepatic forms that precede blood infections. Such drugs will prevent pathology and interrupt transmission, and could therefore have an important role in the control of malaria and its eventual eradication. Since the early twentieth century, thousands of compounds have been screened for anti- malarial activity, yet only a small number of drugs have been widely deployed against malaria (TABLE 1). Although at times some have been used for mass prophylaxis, today almost all of the antimalarial drugs in use are administered primarily to treat clinical malaria — that is, to inhibit the pathogenic, asexual blood stage parasite. All malaria parasites that infect humans (Plasmodium falciparum and Plasmodium vivax, which are responsible for most of the global burden of disease and mortality, Plasmodium ovale and Plasmodium malariae) must go through a multiplication phase in the liver before the pathogenic infection of red blood cells is initiated. The liver stage parasites of all species can be targeted by various drugs, but only one drug, primaquine, is currently used against liver stage parasites. This is because primaquine can kill the persistent, non- dividing hepatocytic forms — hypnozoites — that cause relapse on reactivation. To date, only P. vivax and P. ovale are known to form hypnozoites. In the 1990s, the spread of multidrug- resistant P. falciparum parasites became global 1 , leading to a re-appraisal of combina- tion therapy for malaria 2 . Artemisinin-based combination therapies (ACTs) are currently the mainstay of global malaria control pro- grammes 3 . However, the recent discovery of parasites with reduced susceptibility to artemisinins is a matter of considerable concern 4 . Until now, the search for novel drugs that are active against the liver stages of the parasite (referred to here as anti-liver stage drugs) has not received much attention, and efforts to find compounds with the activity of primaquine but without its disadvantages (BOX 1) have been limited. These efforts have been spurred by the emergence of P. vivax strains that are resistant to chloroquine 5,6 and reasonably tolerant to primaquine 7,8 , and by the rediscovery of P. vivax as an important contributor to the global malaria burden 9–11 . Given the increased commitment to global malaria control over the past few years and the recent re-adoption of eradica- tion as the ultimate goal of these activities, the development of drugs against liver stage parasites deserves serious consider- ation. Among the life cycle stages of the Plasmodium malaria parasites of mammals (FIG. 1), the short hepatic phase invariably precedes the cyclical multiplication of the parasite in red blood cells, which is responsible for the symptoms, the morbidity and the mortality of malaria infections. Therefore, full inhibition of liver stage para- site development will lead to true causal prophylaxis. Transmission will also be inter- rupted because it depends on gametocytes that mature in red blood cells, after the liver stage. Furthermore, the low numbers of hepatic parasites and their limited multipli- cation substantially reduce the likelihood for drug-resistant forms to emerge. Failure to develop a drug with true causal prophylactic activity against malaria is mostly related to the biology of the Plasmodium spp. liver stages and the inherent technical difficulties in studying them. Here, we review these factors and the prospects for developing novel drugs against liver stage parasites. The Plasmodium spp. liver stages The development of the malaria parasite in hepatocytes is an obligatory step that dominates the pre-erythrocytic phase of the infection (FIG. 1), but was not discovered until 68 years after the initial description of the malaria parasites in the blood 12,13 . A further 32 years elapsed before the latent hepato- cytic forms of the parasite, which cause the disease’s characteristic late relapses, were observed in primates 14 . Compared with the erythrocytic phase of the infection, the pre-erythrocytic stage is poorly understood. The cost and ethical considerations associated with obtaining sufficient quantities of infected cells to study hepatic infection restrict detailed investigations of liver stage Plasmodium spp. primarily to studies in rodents. Ultimately, two biological parameters are relevant to the development of drugs against hepatic parasites. In contrast to erythrocytic parasites, which cyclically multiply every 24, 48 or 72 days depending on the species, liver stage development ends after a unique multiplication step of highly variable duration between Plasmodium species 15 . The minimum time from infection with sporozoites to the first wave of merozoites that reach the blood- stream is shortest for rodent parasites 16,17 (~48 hours) and longest for P. malariae in humans 18 (~15 days). The liver stage development of P. falciparum and P. vivax lasts 6 and 8 days, respectively 19–22 , which is similar to many other parasite species that infect primates. There is also considerable variation in the time required for liver stage matura- tion within each Plasmodium species. In humans and other hosts, direct obser- vations of liver stage parasites in liver biopsies showed that the maturation of hepatic parasites is asynchronous 23,24 , which suggests that merozoite release from hepatocytes that were infected after one infective bite occurs over many days. Furthermore, observations of sporozoite-induced infection in malaria- naive volunteers indicated that the time of peak merozoite release from the liver can vary from one person to another by a few weeks 25 . In the case of P. vivax and P. ovale, the time to hypnozoite activation that leads to new blood infections can also vary, from a few weeks to many months after infection with sporozoites 15 . Full inhibition of liver stage parasite development will lead to true causal prophylaxis. PERSPECTIVES 854 | NOVEMBER 2009 | VOLUME 8 www.nature.com/reviews/drugdisc © 2009 Macmillan Publishers Limited. All rights reserved
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of A pre-emptive strike against malaria's stealthy hepatic forms

O P I N I O N
A pre-emptive strike against malaria’s stealthy hepatic formsDominique Mazier, Laurent Rénia & Georges Snounou
Abstract | Almost all the drugs that are widely used today against Plasmodium spp., the causative agent of malaria, target the asexual blood stages of the parasite. Widespread drug resistance severely restricts our ability to control malaria and makes it necessary to seek novel antimalarial compounds. Here, we advocate the development of true causal chemoprophylactic drugs that will fully inhibit the obligate short-lived hepatic forms that precede blood infections. Such drugs will prevent pathology and interrupt transmission, and could therefore have an important role in the control of malaria and its eventual eradication.
Since the early twentieth century, thousands of compounds have been screened for anti-malarial activity, yet only a small number of drugs have been widely deployed against malaria (Table 1). Although at times some have been used for mass prophylaxis, today almost all of the antimalarial drugs in use are administered primarily to treat clinical malaria — that is, to inhibit the pathogenic, asexual blood stage parasite. All malaria parasites that infect humans (Plasmodium falciparum and Plasmodium vivax, which are responsible for most of the global burden of disease and mortality, Plasmodium ovale and Plasmodium malariae) must go through a multiplication phase in the liver before the pathogenic infection of red blood cells is initiated. The liver stage parasites of all species can be targeted by various drugs, but only one drug, primaquine, is currently used against liver stage parasites. This is because primaquine can kill the persistent, non-dividing hepatocytic forms — hypnozoites — that cause relapse on reactivation. To date, only P. vivax and P. ovale are known to form hypnozoites.
In the 1990s, the spread of multidrug-resistant P. falciparum parasites became global1, leading to a re-appraisal of combina-tion therapy for malaria2. Artemisinin-based combination therapies (ACTs) are currently the mainstay of global malaria control pro-grammes3. However, the recent discovery of parasites with reduced susceptibility to artemisinins is a matter of considerable concern4.
Until now, the search for novel drugs that are active against the liver stages of the parasite (referred to here as anti-liver stage drugs) has not received much attention, and efforts to find compounds with the activity
of primaquine but without its disadvantages (bOX 1) have been limited. These efforts have been spurred by the emergence of P. vivax strains that are resistant to chloroquine5,6 and reasonably tolerant to primaquine7,8, and by the rediscovery of P. vivax as an important contributor to the global malaria burden9–11.
Given the increased commitment to global malaria control over the past few years and the recent re-adoption of eradica-tion as the ultimate goal of these activities, the development of drugs against liver stage parasites deserves serious consider-ation. Among the life cycle stages of the Plasmodium malaria parasites of mammals (FIG. 1), the short hepatic phase invariably precedes the cyclical multiplication of the parasite in red blood cells, which is responsible for the symptoms, the morbidity and the mortality of malaria infections. Therefore, full inhibition of liver stage para-site development will lead to true causal prophylaxis. Transmission will also be inter-rupted because it depends on gametocytes that mature in red blood cells, after the liver stage. Furthermore, the low numbers of hepatic parasites and their limited multi pli-cation substantially reduce the likelihood for drug-resistant forms to emerge.
Failure to develop a drug with true causal prophylactic activity against malaria is mostly related to the biology of the Plasmodium spp. liver stages and the inherent technical difficulties in studying
them. Here, we review these factors and the prospects for developing novel drugs against liver stage parasites.
the Plasmodium spp. liver stagesThe development of the malaria parasite in hepatocytes is an obligatory step that dominates the pre-erythrocytic phase of the infection (FIG. 1), but was not discovered until 68 years after the initial description of the malaria parasites in the blood12,13. A further 32 years elapsed before the latent hepato-cytic forms of the parasite, which cause the disease’s characteristic late relapses, were observed in primates14.
Compared with the erythrocytic phase of the infection, the pre-erythrocytic stage is poorly understood. The cost and ethical considerations associated with obtaining sufficient quantities of infected cells to study hepatic infection restrict detailed investigations of liver stage Plasmodium spp. primarily to studies in rodents.
Ultimately, two biological parameters are relevant to the development of drugs against hepatic parasites. In contrast to erythrocytic parasites, which cyclically multiply every 24, 48 or 72 days depending on the species, liver stage development ends after a unique multiplication step of highly variable duration between Plasmodium species15. The minimum time from infection with sporozoites to the first wave of merozoites that reach the blood-stream is shortest for rodent parasites16,17 (~48 hours) and longest for P. malariae in humans18 (~15 days). The liver stage development of P. falciparum and P. vivax lasts 6 and 8 days, respectively19–22, which is similar to many other parasite species that infect primates.
There is also considerable variation in the time required for liver stage matura-tion within each Plasmodium species. In humans and other hosts, direct obser-vations of liver stage parasites in liver biopsies showed that the maturation of hepatic parasites is asynchronous23,24, which suggests that merozoite release from hepatocytes that were infected after one infective bite occurs over many days. Furthermore, observations of sporozoite-induced infection in malaria-naive volunteers indicated that the time of peak merozoite release from the liver can vary from one person to another by a few weeks25. In the case of P. vivax and P. ovale, the time to hypnozoite activation that leads to new blood infections can also vary, from a few weeks to many months after infection with sporozoites15.
Full inhibition of liver stage parasite development will lead to true causal prophylaxis.
P e r s P e c t i v e s
854 | NoVEMBER 2009 | VoLUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved
Screening for activitywith the exception of primaquine, anti-malarial drugs have mainly targeted the pathogenic, asexual blood stage parasite. Until the development of widely accessible methods of in vitro cultivation of P. falci-parum in erythrocytes in 1976 (ReFs 26,27), researchers could only screen large numbers of compounds through lengthy
testing in animal models. The automation of these methods has opened the way to cost-effective, high-throughput screening of millions compounds in chemical libraries. Unfortunately, the technically challenging nature of the in vitro cultivation methods that have been developed for the hepatic stages precludes such screening strategies for compounds that are active against liver stage
parasites. Thus, there remain a number of hurdles in the path to discover a compound that merits testing in clinical trials (FIG. 2).
Production of infective sporozoites. Any investigation of the pre-erythrocytic stages of Plasmodium spp. depends on the avail-ability of infective sporozoites. This requires mosquitoes in which complete parasite
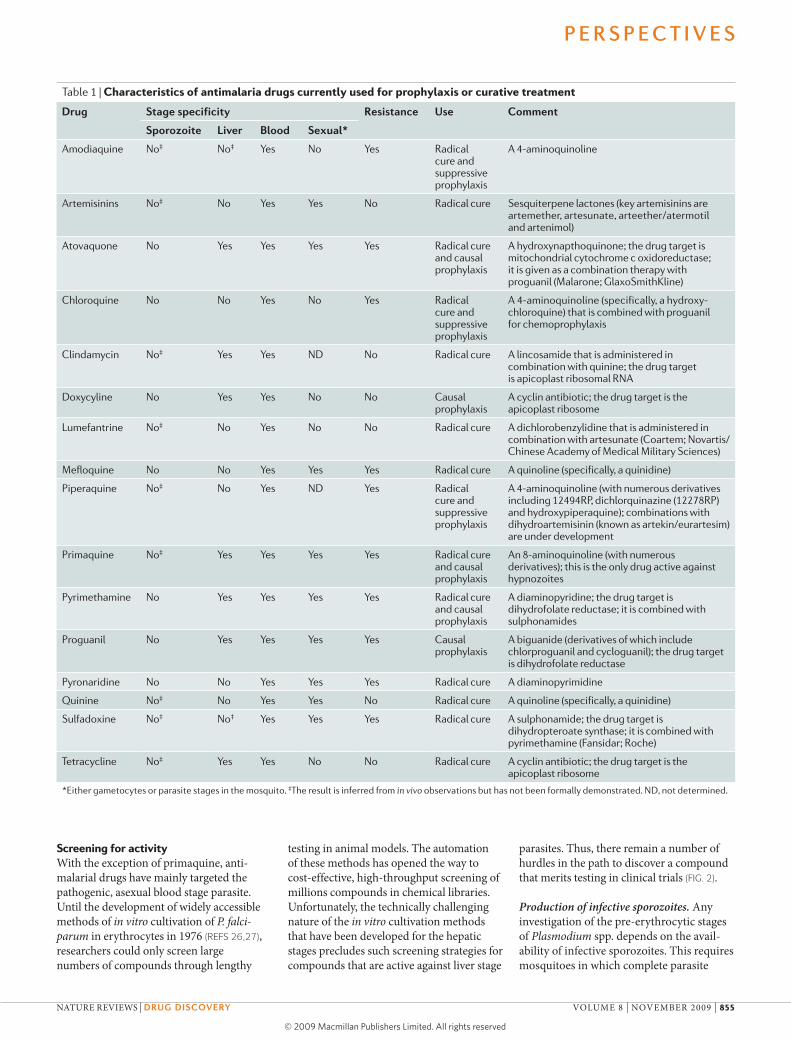
Table 1 | Characteristics of antimalaria drugs currently used for prophylaxis or curative treatment
Drug stage specificity resistance use comment
sporozoite liver Blood sexual*
Amodiaquine No‡ No‡ Yes No Yes Radical cure and suppressive prophylaxis
A 4-aminoquinoline
Artemisinins No‡ No Yes Yes No Radical cure Sesquiterpene lactones (key artemisinins are artemether, artesunate, arteether/atermotil and artenimol)
Atovaquone No Yes Yes Yes Yes Radical cure and causal prophylaxis
A hydroxynapthoquinone; the drug target is mitochondrial cytochrome c oxidoreductase; it is given as a combination therapy with proguanil (Malarone; GlaxoSmithKline)
chloroquine No No Yes No Yes Radical cure and suppressive prophylaxis
A 4-aminoquinoline (specifically, a hydroxy- chloroquine) that is combined with proguanil for chemoprophylaxis
clindamycin No‡ Yes Yes ND No Radical cure A lincosamide that is administered in combination with quinine; the drug target is apicoplast ribosomal RNA
Doxycyline No Yes Yes No No causal prophylaxis
A cyclin antibiotic; the drug target is the apicoplast ribosome
Lumefantrine No‡ No Yes No No Radical cure A dichlorobenzylidine that is administered in combination with artesunate (coartem; Novartis/chinese Academy of Medical Military Sciences)
Mefloquine No No Yes Yes Yes Radical cure A quinoline (specifically, a quinidine)
Piperaquine No‡ No Yes ND Yes Radical cure and suppressive prophylaxis
A 4-aminoquinoline (with numerous derivatives including 12494RP, dichlorquinazine (12278RP) and hydroxypiperaquine); combinations with dihydroartemisinin (known as artekin/eurartesim) are under development
Primaquine No‡ Yes Yes Yes Yes Radical cure and causal prophylaxis
An 8-aminoquinoline (with numerous derivatives); this is the only drug active against hypnozoites
Pyrimethamine No Yes Yes Yes Yes Radical cure and causal prophylaxis
A diaminopyridine; the drug target is dihydrofolate reductase; it is combined with sulphonamides
Proguanil No Yes Yes Yes Yes causal prophylaxis
A biguanide (derivatives of which include chlorproguanil and cycloguanil); the drug target is dihydrofolate reductase
Pyronaridine No No Yes Yes Yes Radical cure A diaminopyrimidine
Quinine No‡ No Yes Yes No Radical cure A quinoline (specifically, a quinidine)
Sulfadoxine No‡ No‡ Yes Yes Yes Radical cure A sulphonamide; the drug target is dihydropteroate synthase; it is combined with pyrimethamine (Fansidar; Roche)
Tetracycline No‡ Yes Yes No No Radical cure A cyclin antibiotic; the drug target is the apicoplast ribosome
*either gametocytes or parasite stages in the mosquito. ‡The result is inferred from in vivo observations but has not been formally demonstrated. ND, not determined.
P e r s P e c t i v e s
NATURE REVIEwS | Drug Discovery VoLUME 8 | NoVEMBER 2009 | 855
© 2009 Macmillan Publishers Limited. All rights reserved

development can be achieved and a supply of blood that contains infectious gameto-cytes. This is not a trivial undertaking as it necessitates substantial investments in laboratory space, equipment and staff. Few anopheline vectors are available for the large-scale laboratory breeding that is needed for drug screening programmes, and only some species can be used for infecting laboratory rodents. Infectious gametocytes are easily obtained for parasites that infect rodent models, and P. falciparum can be readily induced to produce gametocytes in culture. However, the availability of gameto-cytes from other species, such as P. vivax, is limited to laboratories with regular access to blood from gametocytaemic patients or to experimentally infected monkeys. Finally, the infectiousness of sporozoites can vary considerably among batches, and standardi-zation of each sporozoite batch for infectivity is costly and time consuming.
In vitro production of sporozoites was recently achieved for P. berghei28 and P. yoelii29. However, mass production of axenic (free from other microbes) sporo-zoites for parasites that infect humans and primates is unlikely to become available in the near future. In this context, scientists are devoting substantial efforts to develop protocols for sporozoite cryopreservation, which is a crucial objective if attenuated sporozoites are to be used for mass malaria vaccination campaigns30.
In vivo screening. Until the late 1950s, almost all of the initial screening of com-pounds was performed using avian malaria parasites31. However, investigations of the hepatic phase were restricted to experi-mental infections of humans and primates, because the Plasmodium parasites that infect birds differ in many respects from those that infect mammals. In birds, sporo-zoites invade and develop in endothelial cells and macrophages (for example, Kupffer cells) but not in hepatocytes, and the resulting merozoites can infect either other endothelial cells or nucleated red blood cells. The merozoites from an infected red blood cell are capable of infecting not only other red blood cells but also macrophages and endothelial cells13. The discovery of Plasmodium species in African thicket rats that could also infect laboratory rodents and
be transmitted by laboratory-bred anophe-lines (P. berghei, P. vinckei, P. chabaudi and P. yoelii)32–36 led researchers to adopt them as the standard experimental models for drug investigations37. This also made it pos-sible to investigate the liver stages without having to rely on experimental infections of primates or humans. The hepatic stages of Plasmodium species in laboratory rodents mature in 48–68 hours16. Furthermore, the duration of their erythrocytic cycle is short — 21 hours for P. berghei and 18 hours for P. yoelii strains — making it easy to obtain a readout from experimental infections. The two most widely used species for preclinical drug testing are P. berghei and P. yoelii in the mouse. with respect to the liver stage forms, one of the main advantages of the rodent model is that it is possible to directly quantify the efficacy of anti-liver stage treatment through microscopic examina-tion or molecular techniques. None of the Plasmodium species of rodents produces latent hepatic stages in laboratory animals, and so they are of little use for the assessment of anti-hypnozoite activity.
Primates experimentally infected with Plasmodium spp. remain the gold standard for drug efficacy testing, mainly because they most accurately approximate human physiology. However, experimentation in primates is costly and tightly regulated.
one host–parasite combination, P. cynomolgi in Rhesus monkeys, was specifically devel-oped in the 1960s for assessing drug efficacy against the P. vivax hypnozoite. This model was chosen because sporozoite-induced infections of Macaca mulatta resemble those of the tropical strains of P. vivax in humans with respect to the duration of liver stage maturation and the relapse patterns38. This model is still used today to validate the most promising lead compounds before clinical trials. Physiological differences between humans and lower primates limit the accu-racy with which the rhesus monkey model can be predictive of success in humans39. Some South American monkeys that are susceptible to infection by Plasmodium spe-cies that infect humans (P. falciparum and P. vivax)40–42 are particularly useful. However, for some of these animals, splenectomy is needed for challenge infections to become evident in the blood.
In recent years, the development of so-called humanized mice has provided a valuable model that adds biological relevance. Immunodeficient mice (non-obese diabetic–severe combined immuno deficient and BXN) can be grafted with human red blood cells in which P. falciparum can develop and multiply. Early models, although limited by low and unpredictable receptivity to laboratory
Box 1 | Primaquine — missed opportunities
Primaquine is one of the oldest of the currently used antimalarial drugs and is the least well understood. This drug was first deployed to prevent Plasmodium vivax relapses in US soldiers returning to the United States from World War II and the Korean War98, and its primary role for the past 56 years has been the prevention of P. vivax and Plasmodium ovale relapses. An excellent and detailed review of the chemotherapeutic and chemoprophylactic uses of this long-serving drug has recently been published99. Primaquine also destroys the normal liver forms of all Plasmodium species, but toxicity considerations preclude its widespread use in the control of malaria. The long-held perception that primaquine is poorly tolerated and associated with unacceptable secondary effects has recently been challenged100. However, primaquine can precipitate acute and dangerous intravascular haemolysis in subjects with glucose-6-phosphate dehydrogenase (G6PD) deficiencies, who are most frequently found in malaria-endemic countries.
The US army launched a search for other compounds that could replace primaquine, when the protection provided by the chloroquine–primaquine tablet waned101. Compound WR-238605, now referred to as tafenoquine, underwent its first clinical trials in humans in 1998 (ReF. 102). Researchers in India are also developing another derivative of primaquine, bulaquine, and initial clinical trials are encouraging103–105. Tafenoquine’s long half-life would make it suitable for causal prophylaxis. However, like primaquine, it is not suitable for subjects with G6PD deficiencies. Surprisingly, bulaquine, which is converted to primaquine in vivo, seems to be better tolerated than primaquine in G6PD-deficient subjects105. Both of these compounds have activity against blood stage parasites — a characteristic that makes them less suitable for large-scale causal prophylaxis in malaria-endemic areas. The efficacy of primaquine against hypnozoites depends on co-administration with quinine or chloroquine106 and is a function of the total dose rather than the duration of treatment107. Moreover, P. vivax strains differ naturally in their susceptibility to primaquine108, and fully resistant lines can readily be experimentally selected in humans109. Ultimately, investigations of the mechanisms of action of primaquine against hypnozoites and gametocytes are likely to guide the development of better replacement drugs.
Anti malarial drugs have mainly targeted the pathogenic, asexual blood stage parasite.
P e r s P e c t i v e s
856 | NoVEMBER 2009 | VoLUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

Normal hepatic phase leading to primary infectionb Hepatic phase c Erythrocytic phasea Infective mosquito bite
Sporozoite
Blood vessel
Hepatocyte
Kupffer cell
Schizont
Schizont
Gametocytes
Red blood cell
Merozoite
Protracted hepatic phase leading to relapse infections from activated hypnozoites
Blood vessel
Hepatocyte
Kupffer cell
Hypnozoite
Weeks
Months to years
Schizont
Nature Reviews | Drug Discovery
Primary infection
Relapseinfections
parasite lines, were useful in immunological and chemotherapeutic investigations43,44. Now, animal models that can usually sustain the growth of any P. falciparum isolate, including those collected from patients, are available45,46. Furthermore, a mouse line with a liver defect that leads to endogenous hepatocyte destruction47,48 has been used to develop a model in which engrafted primary human hepatocytes
persist and remain susceptible to infection by P. falciparum sporozoites, which then develop normally49,50. This model may therefore allow the full life cycle of P. falci-parum to be investigated. The difficulties in obtaining regular supplies of human primary hepatocytes for engraftment have been addressed by the finding that grafted human hepatocytes can be serially sub-inoculated into new recipient animals51.
The rodents’ livers could eventually be repopulated by cultured cells lines engineered to be highly permissive for Plasmodium liver stage maturation, should these become available. The innate prolifera-tive capacity of such cells would need to be controlled to avoid liver tumour formation. These advances could also make it possible to obtain liver stage forms of P. vivax, including hypnozoites, in humanized mice.
Figure 1 | The Plasmodium life cycle. a | Sporozoites, Plasmodium cells that develop in the mosquito’s salivary glands, enter the skin of the host as the female mosquito probes for blood. b | Sporozoites reach the liver through the blood vasculature and the lymphatic system. The minimum time taken by the parasite to reach the liver after sporozoite deposition in the skin is usually in the order of minutes, but it can extend to hours. in the liver, the parasite traverses different cells, including Kupffer cells and hepato-cytes, before settling and developing in a hepatocyte. Here, it undergoes a process of asexual replication to give rise to a schizont. The obligatory hepatic phase is the only time in the life cycle of the parasite in which it invades and develops in a nucleated host cell. Maturation of the hepatic parasite can be completed in a few days or a few weeks, depending on the parasite species, and occurs asynchronously such that the release of merozoites (the invasive forms that result from the parasite’s asexual repro-duction) into the bloodstream occurs over many days (top panel) leading to
the primary infection. Some Plasmodium vivax and Plasmodium ovale sporozoites are predestined to develop into hypnozoites (non-dividing hepatocytic forms), which can remain latent in the liver for months to years before resuming development that leads to relapse infections (lower panel). The triggers that activate the maturation of hypnozoites are unknown. Potential anti-liver stage drugs could target sporozoite motility, hepatocyte invasion, maturation in hepatocytes or the egress of merozoites from the mature hepatic schizont. c | Merozoites invade red blood cells and divide mitotically to form an erythrocytic schizont. These merozoites can reinfect fresh erythrocytes, giving rise to a cyclical blood stage infection with a cycle length of 48–72 hours, depending on the Plasmodium species. As yet unknown factors trigger a subset of developing merozoites to differentiate into male and female gametocytes. Sporozoites are the culmination of the sexual phase of the life cycle that is initiated when the mosquito ingests gametocytes with the blood meal.
P e r s P e c t i v e s
NATURE REVIEwS | Drug Discovery VoLUME 8 | NoVEMBER 2009 | 857
© 2009 Macmillan Publishers Limited. All rights reserved

Nature Reviews | Drug Discovery
P. berghei and P. yoelii inprimary hepatocytesor hepatocyte cell lines
P. berghei and P. yoelii in normal mice
P. falciparum and P. vivax in humanized mice
P. cynomolgi inMacaca mulatta
Clinical trials
Medicinal chemistryLead optimization
P. falciparum, P. vivax and P. cynomolgi in primary hepatocytes or hepatocyte cell lines
• Efficacy• Specificity• Toxicity
• Efficacy• Pharmacokinetics• Pharmacodynamics• Toxicology
In vitro testing
In vivo testing
In vitro screening. The main impediment to in vitro culture of liver stage parasites is that they undergo a single round of division. Therefore, unlike cultures of erythrocytic parasites, those of liver stage forms cannot be maintained continuously. It was only in the 1980s that invasion and full maturation of liver stage parasites was reported in cultured host cells. Maturation of P. berghei pre-erythrocytic stages was first achieved in vitro using wI38, a human embryonic lung cell line52, and then in a clone line derived from the human hepatoma cell lines HepG2 (ReF. 53). At the same time, in vitro cultures of P. yoelii were obtained using primary
hepatocytes from laboratory rats54,55, from the natural host Thamnomys gazellae56, and eventually from laboratory mice57. Subsequently, the complete in vitro devel-opment of hepatic forms of P. falciparum and P. vivax, and the partial development of hepatic forms of P. ovale, was achieved in human primary hepatocytes58–61, and that of P. malariae was achieved in chim-panzee and Aotus primary hepatocytes62. In vitro development of other Plasmodium species that infect primates has also been possible in Macaca mulatta primary hepa-tocytes63,64. The HepG2 cell line was found to be suitable for the liver stage of P. berghei and P. vivax, but not for those of P. yoelii
and P. falciparum. Recently, other human hepatoma cell lines, HHS-102 (ReF. 65) and HC-04 (ReF. 66), were reported to sustain full development of P. falciparum and P. vivax.
In vitro systems, although highly valuable for investigations of sporozoites and the biology of liver stage parasites67, suffer from practical difficulties that preclude their use for medium- to high-throughput drug screening. First, for all parasite species, the low efficiency of sporozoite invasion and maturation in cultured primary hepatocytes and hepatoma cell lines mean that only a small number of infected hepatocytes are obtained. For example, to obtain up to 2,000 infected hepatocytes in a typical well containing 80,000 primary hepatocytes, an inoculum of 30,000 sporozoites is needed (although fewer are needed for some host–parasite combinations).
Second, the sporozoites of some Plasmodium species, including P. falciparum and P. yoelii, are particularly inefficient at invading HepG2 cells, and the few forms that do invade do not develop to maturity68–70. For P. falciparum, higher infectivity and full maturation has been achieved mainly in human primary hepatocytes. This is a major limitation because primary hepatocytes, which do not multiply in vitro, can only be obtained after a patient’s informed consent and at the discretion of the surgeon treating them. The donor patients are usually admitted for various liver pathologies and in many cases are receiving drug treatment at the time of surgery. Consequently, the proportion of donated liver samples that yields primary hepatocytes of a sufficient quality for acceptable infection rates or optimal parasite development is variable and unpredictable.
There are as yet no validated in vitro systems for the production of P. vivax hypnozoites, owing to two main prob-lems. First, parasite liver stage cultures in hepatoma cell lines that divide every day become overgrown and damaged before truly latent parasite forms are observable. Second, the only way to confirm that the hepatic forms that are found in culture after an extended period are hypnozoites is to induce maturation and show that the resulting merozoites are infectious. For P. vivax this would require inoculation of hosts (primates or humans) with infected hepatoma cell cultures, which is ethically inadmissible. A potential solution to the first problem would be the arrest of hepatoma cell division by prior irradiation. In studies based on this approach71,72, uninucleate P. vivax forms were observed in P. vivax-infected
Figure 2 | Discovery path for drugs against the Plasmodium liver stages. A discovery programme will typically include in vitro screening of compounds for activity against the develop-ment of sporozoites from Plasmodium species that infect rodents in mouse primary hepatocytes or suitable hepatocyte cell lines. compounds with a half-maximal inhibitory concentration (ic
50) <20
μM will then be tested in vitro for activity against Plasmodium falciparum or Plasmodium cynomolgi liver stage development in cultured human or simian primary hepatocytes, or other adequate cell lines, should these become available. Only compounds that show both good efficacy and specificity against liver stage forms and low toxicity against hepatocytes will be selected for in vivo testing. Doses of 5–25 mg per kg or less will be administered to laboratory rodents that will be challenged with sporozoites, then to humanized mice inoculated with P. falciparum sporozoites. The tests could eventually be extended to primate models such as Plasmodium cynomolgi in Macaca mulatta. In vivo assays in rodents will include a test on day 10 for the detection of eventual overt parasitaemia, as well as a quantitative PcR assay for the evaluation of liver parasite burdens. initial in vitro and in vivo studies are directed towards lead compound identification. Medicinal chemistry and lead optimiza-tion are an essential and iterative component of the drug development pathway. Secondary in vivo tests are necessary for detailed compound evaluation, including dose ranging, tests to determine the onset of activity and recrudescence, tests for prophylaxis and screening for drug resistance, toxicity and preliminary pharmacokinetic and pharmacodynamic data.
P e r s P e c t i v e s
858 | NoVEMBER 2009 | VoLUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

irradiated HepG2-A16 cells a few days after the minimum time required for the normal liver stage maturation. The observed proportion of the non-dividing forms was consistent with the ratio of hypnozoites that was expected from the types of sporozoites used, leading the authors to conclude that these infected cells were indeed hypnozoites. Given the paucity of the data presented, and the fact that the observations did not extend beyond 15 days post-sporozoite inoculation, the precise nature of these slow-growing, non-dividing parasite forms remains unclear. It might have been possible to infer the presence of hypnozoites through observation of differential inhibition of development when cultures are treated with primaquine, which is active against hypnozoites, and atovaquone, which is not.
A possible means to achieve regular provision of human primary hepatocytes is through cryopreservation73,74 of the batches that have been shown to be suitable for P. falciparum liver stage invasion and matu-ration. However, cell lines that are highly susceptible to P. falciparum sporozoite inva-sion and liver stage development would be more useful, especially in the context of large-scale drug screening. one strategy to achieve this is to identify proteins that contribute to hepatocyte susceptibility or are damaging to the parasite, and then to modify a cell line accordingly. In a proof-of-concept study70, this approach was validated when HepG2 cells, which are not usually suscep-tible to sporozoite infection, were made receptive to P. yoelii invasion and maturation simply through transfection with CD81, a tetra spanin that is essential for the formation of a vacuole around the intracellular para-site70. The fact that the transfected HepG2 cells were still refractory to P. falciparum liver stage development70 indicated that an additional factor is needed by this parasite species or that there is a blocking mecha-nism specific to the different parasite species in HepG2 cells. Differential genome-wide transcriptome analyses of susceptible and non-susceptible cells might help to identify the other proteins and metabolic pathways that are linked to optimal P. falciparum liver stage development (J.-F. Franetich and D. Mazier, unpublished observations). Such data could then serve to develop easy-to-maintain cell lines for the investigation of the hepatic forms of P. falciparum.
Evaluation of hepatic stage inhibitionInhibition of the pre-erythrocytic phase in the mammalian host can be due to various causes: abrogation of hepatocyte invasion
by sporozoites, failure of the hepatic forms to mature, prevention of the egress of the merozoites from the mature liver schizont, or the production of defective merozoites that fail to invade erythrocytes or develop in them. There is no single assay by which to simultaneously distinguish between these possibilities. It is difficult to design assays that can measure reduction in hepatic mero-zoite egress and viability, as these events are not amenable to in vitro observation.
Identification of the compounds with potential anti-liver stage activity should begin in vitro; the rodent model and the CD81-transfected HepG2 cell line make undertaking this initial screen manageable70. Nonetheless, the high numbers of sporo-zoites (>10,000) that are needed per assay well restricts the number of compounds that can be screened. Some compounds have been found to have inhibitory activity only when primary hepatocytes are used (D. Mazier, unpublished observations) implying that, in some cases, potential lead compounds might be missed.
Evaluation of inhibitory activity depends on the ability to measure the number of liver stage parasites. This is usually performed ~48 hours after sporozoite inoculation for P. berghei and P. yoelii, and after ~5 days for P. falciparum, when parasites are large
enough to be easily observed under the microscope57,75,76. It should be noted that the period to reach maturity in vitro is longer than that observed in vivo for the species that infect rodents. Although microscopic examination is tedious and time-consuming, it provides an accurate measure of the numbers of sporozoites and the number and size of liver stage parasites. Recent developments in liver stage detection include automated microscopy77,78 and flow cyto metry assays using green fluorescent protein (GFP)-tagged parasites79, which will enable medium-throughput screening.
Another important parameter to assess in determining the suitability of a com-pound for further testing is its toxicity to the host cell. Hepatotoxicity can be measured through colorimetric assays in which mito-chondrial integrity is assessed or in which the specific release of lactate dehydrogenase is measured80. The observation that GFP expression correlated with cell viability in a HepG2-A6 cell line that expresses GFP has also been used to evaluate toxicity81.
The activity of compounds that are selected by in vitro screening needs to be reproduced in vivo before any preclinical investigations are considered. Ideally, an anti-liver stage drug will prevent the emergence of erythrocytic parasites. The
Box 2 | Selection of drug-resistant parasites
Parasites with reduced susceptibility to drugs are most efficiently selected when they are exposed to subcurative doses of a drug. Selection ultimately acts on random mutations that arise during DNA replication. The speed with which drug-resistant parasites emerge depends on the time over which drug pressure is applied, the number of parasites subjected to this pressure and the number of mutations needed to make the parasite resistant. The rate at which the resistant parasites spread is governed by other factors, including the fitness cost of the mutations, the transmission intensity, the extent of drug coverage in the community and the use and choice of drug combinations. Carefully designed drug deployment strategies could extend the useful life of a drug by decades.
If a causal prophylactic drug is to have a key role in the control of malaria, it will have to be deployed to all members of the community, who will have to take it at regular intervals for extended periods. Unless adherence is perfect, drug levels in many individuals are likely to fall below the minimum inhibitory concentration. If the drug used for causal prophylaxis has activity against blood stage parasites, then potentially large numbers of these parasites (often >109 per week in carriers) could be exposed to subcurative drug concentrations, which favours the development of resistance. By contrast, a maximum of 103 liver stage parasites per week are exposed to subcurative drug concentrations in subjects living in regions with high transmission intensities.
If a mutation that confers resistance to an anti-liver stage drug arises, the mutant parasite would have the potential to spread quickly in the community as the erythrocytic progeny, including gametocytes, would carry the mutation. The dissemination of parasites that are resistant to anti-liver stage drugs could be minimized by the adoption of drug combinations. It is interesting that the three experimental compounds known to specifically inhibit the liver stage parasite belong to distinct chemical classes89,110–112 (Table 2), suggesting that they differ in their mode of action. Furthermore, the drugs that are used most widely in the current anti-blood stage combination therapies act on different targets to anti-liver stage drugs, as they do not affect the hepatic parasite, which will further curtail the emergence and spread of parasites resistant to anti-liver stage drugs.
P e r s P e c t i v e s
NATURE REVIEwS | Drug Discovery VoLUME 8 | NoVEMBER 2009 | 859
© 2009 Macmillan Publishers Limited. All rights reserved

simplest way to evaluate this is to challenge mice with sporozoites while administering the test compound and then monitor the blood for the presence of parasites. For this data to be meaningful, the activity of the compound should be assessed in parallel on blood stage parasites. For compounds that efficiently inhibit the erythrocytic forms or that are only partially effective at killing the liver stage parasite, it is necessary to estimate the direct inhibitory activity on the infected liver. This has traditionally been performed through time-consuming microscopic enumeration of parasites in histological sections of infected mouse livers82,83, which necessitates challenge with about 500,000 sporozoites. Protocols based on PCR amplification84–87 provide a more practical alternative, as more accurate quantification of liver loads can be achieved more rapidly using animals inoculated with five to ten times fewer sporozoites. The disadvantage of PCR protocols is that they cannot differentiate between delayed maturation of liver stage parasites and their destruction. For example, DNA that has been purified from 10,000 hepatic parasites in which nuclear replication fails to occur would amplify in a PCR assay to the same extent as the DNA purified from a single, fully mature hepatic schizont that is ready to release 10,000 merozoites.
Drug characteristics and benefitsCurrently, only compounds with activity against P. vivax hypnozoites are the focus of anti-liver stage drug develop-ment programmes. Ultimately, it will be
more profitable to focus development on compounds that are active specifically against the normal hepatic forms of all the Plasmodium species that infect humans. Taking into account that the development of anti-liver stage drugs is inherently expen-sive, we next consider the characteristics that such compounds should have and their advantages over currently available antimalarial drugs.
The window of opportunity for the destruction of hepatic parasites is relatively narrow compared with other parasite stages, making it necessary to maintain inhibi-tory concentrations over extended periods. Therefore, an effective anti-liver stage compound must have no adverse second-ary effects associated with long-term use. It is also crucial that the drug be a specific inhibitor of liver stage parasites, while having no activity against any of the blood stages, because community-wide intake of a drug with activity against blood stage parasites would probably lead to the selection and dis-semination of parasites that are resistant to this drug (bOX 2). Meeting these two criteria is less important if the anti-liver stage drug is used for radical and/or presumptive treat-ments, as is the case for primaquine or its eventual replacement, because drug exposure would be limited in duration and frequency.
Causal prophylaxis should supplement and not replace traditional control measures, such as the provision of rapid treatment or insecticide-treated bed nets. The main effect of causal prophylaxis is to provide a rapid and effective reduction in malaria transmission. Thus, causal prophyl-axis based on anti-liver stage drugs will be useful in all endemic areas, irrespective of endemicity. The benefits from causal prophylaxis will be evident even if the cover is partial. At the community level, a reduc-tion in the number of acquired infections will reduce the number of clinical cases and transmission. At the individual level, a par-tial reduction of the liver burden could lead to a decrease in the clinical severity of the ensuing blood stage infection, although this has yet to be confirmed88.
Discovery path for liver stage drugsExcluding the research concerning drugs that might replace primaquine, current antimalarial drug development strategies are strongly focused on compounds with activity against erythrocytic parasites. Thus, the traditional strategy is to discard compounds that have little activity against erythrocytic parasites from further study, thereby pre-cluding the identification of the compounds that are most suitable for causal prophylaxis. This represents a missed opportunity in light of the current interest in multidisciplinary programmes that aim to collect products from plants, fungi and marine organisms for drug discovery, which are helping to expand the vast private and public chemical libraries.
Table 2 | Compounds with activity directed exclusively against liver stage parasites
Drugs In vivo effects?
In vitro effects?
effect on normal liver stage parasites?
effect on hypnozoites?
Target molecule or pathways
references
Alkaloids
N-cyclopentyl-tazopsine PYY PF and PYY Yes NT Unknown 89
Anti-histaminic agents
cyproheptadine PYN PYY Yes NT Histamine H1 receptor 110,111
Ketotifen PYN PYY Yes NT Histamine H1 receptor 110,111
Imidazolidinediones
1 No PYY Yes NT Unknown 112
1a Pc and PYY PYY Yes Yes Unknown 112
1b PYY No Yes NT Unknown 112
1c No PYY Yes NT Unknown 112
1d PYY PYY Yes NT Unknown 112
1e PYY No Yes NT Unknown 112
Pc, Plasmodium cynomolgi; PF, Plasmodium falciparum; PYN, Plasmodium yoelii nigeriensis; PYY, Plasmodium yoelii yoelii; NT, not tested.
Ideally, an anti-liver stage drug will prevent the emergence of erythrocytic parasites.
P e r s P e c t i v e s
860 | NoVEMBER 2009 | VoLUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

A small subset of compounds with interesting activity against blood parasites are nonetheless occasionally tested against hepatic parasites, and some have shown promising activity (Table 2). Currently, anti-liver stage activity is considered to add value to drugs that are effective against blood parasites. However, this dual inhibi-tory activity substantially reduces their suitability for causal prophylaxis (bOX 2). of the many compounds with known anti-liver stage activity (see Supplementary information S1 (table)), only a few seem to be suitable for drug development (Table 2). No follow-up studies have been published for any of these compounds, and to our knowledge only the derivatives of tazopsine, a morphine-related compound extracted from a plant that is native to Madagascar89, are being investigated further.
The development of the parasite in the metabolically complex hepatocyte suggests two broad approaches to target selection: identifying parasite-specific molecules that are crucial to survival in the liver and/or maturation, or identifying host cell mol-ecules the synthesis or function of which must be specifically upregulated or activated to permit parasite development. Most host cell-specific inhibitors that affect normal hepatocyte processes are likely to have unac-ceptable toxicity, especially if the drug is to be ingested over an extended time frame, as would be the case for casual prophylactic drugs. The current list of possible path-ways that can be targeted (Table 3) is sparse because the biochemical and cellular pro-cesses in the liver stage parasite and its host cell are poorly understood. There is now hope that genomic and post-genomic meth-odologies will provide new information on the biology of the liver stage parasite by pro-ducing a whole-parasite transcriptome90–94 and eventually further proteome analyses94. The value of these studies has been substan-tially enhanced by the application of similar approaches to the host cell. For example, hepatocyte growth factor activation of the hepatocyte tyrosine kinase receptor MET has been proposed to have a role in the early liver infection stages, suggesting that tyrosine kinase inhibitors might be used to block parasite development95. The chemi-cals that are selected can also be tested using sophisticated bioinformatic approaches that could help reduce the numbers of compounds to be screened while increasing the probability of promising hits. Coupling quantitative structure–activity relationship studies with molecular topology analyses on a set of 127 active compounds that had
previously been tested against liver stage P. yoelii led to a model that predicted the in vitro anti-liver stage activity of novel compounds96. Application of this model to a large database of compounds with unknown activity identified molecules that proved highly active (half-maximal inhibitory con-centration (IC50) values in the nanomolar range) against liver stage parasites in vitro. one of these, an ionophore, was found to fully inhibit the development of both P. yoelii and P. falciparum in vitro, and to protect mice completely from a P. yoelii sporozoite challenge96.
Finally, it might be beneficial to prioritize the testing of drugs with known potent activity against other apicomplexan para-sites, as such parasites are likely to share some of the biological and metabolic path-ways of malaria parasites. Similarly, both experimental and established drugs that are used against other infectious pathogens, and libraries of compounds that have a specific target, such as kinases97, could be screened for anti-liver stage activity first.
It is our opinion that priority should be given to investigations of the biology and the metabolism of liver stage parasites because this knowledge will maximize the likelihood of uncovering promising leads while reducing the number of compounds to be screened.
ConclusionsSubstantial investments are likely to be needed to overcome the obstacles facing attempts to develop, and then successfully deploy, effective anti-liver stage drugs. It could be argued that development of new drugs should be directed only at treatment, whereas prevention should remain the pre-serve of vaccine developers and distributors of insecticide-treated bed nets. The history of the fight against malaria should have taught us that this epidemiologically com-plex disease is unlikely to be overcome by an attack on a few fronts, even if the tools avail-able to each are supremely efficient. The considerable efforts over the past three dec-ades that have been directed at developing a truly protective vaccine are yet to bear fruit.
Box 3 | Characteristics and deployment of anti-liver stage drugs
radical cureDrugs that provide a radical cure destroy all the liver stage parasites present in the body.
• Target population. Residents of malaria-endemic areas and travellers who present with clinical Plasmodium vivax or Plasmodium ovale. For some malaria-exposed subjects, treatment is sometimes provided presumptively.
• Drug characteristics. Exclusive specificity against liver stage parasites is not necessary. Considerations of cost and of minor adverse secondary effects are of reduced importance compared with drugs for causal prophylaxis because only a small fraction of the population is occasionally exposed to the drug for a short period.
• Contribution to malaria control. The primary benefit is to the individual. It is unclear whether provision of radical cure to patients with clinical P. vivax or P. ovale in malaria-endemic regions will substantially help to control or to eliminate these species. In endemic settings, asymptomatic carriers of P. vivax or P. ovale usually outnumber those with clinical symptoms. Thus, even if all the residents with symptoms of malaria were to seek and receive radical cure, most of the reservoir population will be unaffected.
causal prophylaxisDrugs that provide causal prophylaxis prevent the development of parasites that have been introduced into the body.
• Target population. For short-term cover (days to months), the target population comprises subjects who are transiently exposed to the parasite — at present, mainly travellers and the military. For long-term cover (months to year-long), the target population is residents of all malaria-endemic areas, including displaced populations.
• Drug characteristics. Lengthy exposure to the drug requires it to have minimal or no adverse secondary effects. Low cost is essential if the drug is to be deployed effectively in malaria-endemic populations, which tend to be economically disadvantaged. Exclusive specificity against liver stage parasites is a prerequisite to prevent the emergence of drug-resistant parasites.
• Contribution to malaria control. For travellers and the military, the benefit to the individual has little effect on malaria in the endemic areas visited. Judicious deployment of causal prophylaxis to residents in malaria-endemic regions, as a supplement to traditional control measures, would substantially help the elimination of malaria from any endemic setting.
P e r s P e c t i v e s
NATURE REVIEwS | Drug Discovery VoLUME 8 | NoVEMBER 2009 | 861
© 2009 Macmillan Publishers Limited. All rights reserved

However sufficient advances have been made in this field to maintain well-founded hope of success, and concerted searches for new antimalarial drugs have led to many useful new compounds. Despite this, we consider that drugs capable of providing efficient causal prophylaxis would make an important contribution to the control of malaria. The onus is on the decision makers to encourage researchers and pharmaceutical companies to combine their expertise and develop a potent inhibitor of liver stage parasites, which could then be added to the panoply of weapons aimed at the eradication of malaria.
Dominique Mazier and Georges Snounou are at the Institut National de la Santé et de la
Recherche Médicale U945, F-75013 Paris, France; and at Université Pierre et Marie Curie,
Faculté de Médecine Pitié-Salpêtrière, F-75013 Paris, France.
Dominique Mazier is also at the Assistance Publique Hôpitaux de Paris, Centre Hospitalo-Universitaire Pitié-
Salpêtrière, F-75013 Paris, France.
Laurent Rénia is at the Singapore Immunology Network, A*STAR, Biopolis, Singapore.
Laurent Rénia and Georges Snounou are also at the Department of Microbiology,
National University of Singapore, 5 Science Drive 2 Singapore 117597, Singapore.
All authors contributed equally to this work.
Correspondence to D.M. e-mail: [email protected]
doi:10.1038/nrd2960
1. White, N. J. et al. Averting a malaria disaster. Lancet 353, 1965–1967 (1999).
2. Newton, P. N. & White, N. J. Malaria: new developments in treatment and prevention. Ann. Rev. Med. 50, 179–192 (1999).
3. White, N. J. Antimalarial drug resistance and combination chemotherapy. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 354, 739–749 (1999).
4. Noedl, H. et al. Evidence of artemisinin-resistant malaria in western Cambodia. New Engl. J. Med. 359, 2619–2620 (2008).
5. Baird, J. K. et al. Resistance to chloroquine by Plasmodium vivax in Irian Jaya, Indonesia. Am. J. Trop. Med. Hyg. 44, 547–552 (1991).
6. Rieckmann, K. H., Davis, D. R. & Hutton, D. C. Plasmodium vivax resistance to chloroquine? Lancet 334, 1183–1184 (1989).
7. Bunnag, D. et al. High dose of primaquine in primaquine resistant vivax malaria. Trans. Royal Soc. Trop. Med. Hyg. 88, 218–219 (1994).
8. Luzzi, G. A., Warrell, D. A., Barnes, A. J. & Dunbar, E. M. Treatment of primaquine-resistant Plasmodium vivax malaria. Lancet 340, 310 (1992).
9. Mendis, K. N., Sina, B. J., Marchesini, P. & Carter, R. The neglected burden of Plasmodium vivax malaria. Am. J. Trop.Med. Hyg. 64, 97–106 (2001).
10. Rogerson, S. J. & Carter, R. Severe vivax malaria: newly recognised or rediscovered? PLoS Med. 5, e136 (2008).
11. Tjitra, E. et al. Multidrug-resistant Plasmodium vivax associated with severe and fatal malaria: a prospective study in Papua, Indonesia. PLoS Med. 5, e128 (2008).
12. Shortt, H. E. & Garnham, P. C. C. Pre-erythrocytic stage in mammalian malaria parasites. Nature 161, 126 (1948).
13. Bray, R. S. Studies on the exo-erythrocytic cycle in the genus Plasmodium (H. K. Lewis & Co., London, 1957).
14. Krotoski, W. A. et al. Relapses in primate malaria: discovery of two populations of exoerythrocytic stages. Preliminary note. BMJ 280, 153–154 (1980).
15. Garnham, P. C. C. Malaria Parasites and Other Haemosporidia (Blackwell Scientific Publications, Oxford, 1966).
16. Cox, F. E. G. in Malaria: Principles and Practice of Malariology (eds. Wernsdorfer, W. H. & McGregor, I.) 1503–1543 (Churchill Livingstone, London, 1988).
17. Landau, I. & Boulard, Y. in Rodent Malaria (eds. Killick-Kendrick, R. & Peters, W.) 53–84 (Academic Press, London, New York, San Francisco, 1978).
18. Lupascu, G. et al. The late primary exo-erythrocytic stages of Plasmodium malariae. Trans. Royal Soc.Trop. Med. Hyg. 61, 482–489 (1967).
Table 3 | Current potential targets for anti-pre-erythrocytic chemotherapy
Parasite stage Target location Target pathway Molecular target existing treatment Potential drugs references
Sporozoite cytosol Motility Kinases Aldolase
None Kinase inhibitors Aldolase inhibitors
113,114
cytosol, organelles and extracellular sites
Hepatocyte invasion
Kinases
cysteine protease proteins
None Protease inhibitors 115
Hypnozoites cytosol Unknown Unknown Primaquine (aminoquinolines)
Primaquine derivatives 116,117
Liver parasite Parasite cytosol Folate metabolism Dihydrofolate reductase Dihydropteroate synthase
Pyrimethamine Proguanil Sulphadoxine
New inhibitors 118–120
Parasite cytosol Polyamine biosynthetic pathway
Ornithine decarboxylase
None New inhibitors 121
Parasite proteasome Protein degradation
Proteases None Protease inhibitors 122
Parasite mitochondrion
electron transport cytochrome c oxidoreductase
Atovaquone New inhibitors 123
Parasite apicoplast Protein synthesis Apicoplast ribosome
Azythromycin Tetracyclines (such as doxycycline)
New cyclins 94,124,125
Parasite apicoplast DNA synthesis Topoisomerase None New quinolines 126
Parasite apicoplast DNA transcription RNA polymerase Rifampicin New inhibitors 94
Parasite apicoplast Type ii fatty-acid biosynthesis
FAB1 and FABG None Triclosan 94,127,128
Hepatocyte cytosol and mitochondrion
Hepatocyte apoptosis blockade
Unknown None inhibitors of apoptosis 129
Liver merozoite Hepatocyte cytosol and parasitophorous vacuole
Merozoite release Proteases None Protease inhibitors 130
P e r s P e c t i v e s
862 | NoVEMBER 2009 | VoLUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

19. Boyd, M. F. & Kitchen, S. F. Observations on induced falciparum malaria. Am. J. Trop. Med. 17, 213–235 (1937).
20. Boyd, M. F. & Stratman-Thomas, W. K. Studies on benign tertian malaria. 7. Some observations on inoculation and onset. Am. J. Hyg. 20, 488–495 (1934).
21. Bray, R. S. The exoerythrocytic phase of malaria parasites. Int. Rev. Trop. Med. 2, 41–74 (1963).
22. Fairley, N. H. Sidelights on malaria in man obtained by subinoculation experiments. Trans. Royal Soc. Trop. Med. Hyg. 40, 621–676 (1947).
23. Bray, R. S. The tissue phase of malaria parasites. J. Trop. Med. Hyg. 57, 41–45 (1954).
24. Vanderberg, J. P. Asynchronous maturation of Plasmodium berghei exo-erythrocytic forms in rats. Trans. Royal Soc. Trop. Med. Hyg. 76, 251–252 (1982).
25. Kitchen, S. F. in Malariology — A Comprehensive Survey of All Aspects of This Group of Diseases From a Global Standpoint (ed. Boyd, M. F.) 966–994 (W. B. Saunders Company, Philadelphia and London, 1949).
26. Haynes, J. D., Diggs, C. L., Hines, F. A. & Desjardins, R. E. Culture of human malaria parasites Plasmodium falciparum. Nature 263, 767–769 (1976).
27. Trager, W. & Jensen, J. B. Human malaria parasites in continuous culture. Science 193, 673–675 (1976).
28. Al-Olayan, E. M., Beetsma, A. L., Butcher, G. A., Sinden, R. E. & Hurd, H. Complete development of mosquito phases of the malaria parasite in vitro. Science 295, 677–679 (2002).
29. Porter-Kelley, J. M. et al. Plasmodium yoelii: axenic development of the parasite mosquito stages. Exp. Parasitol. 112, 99–108 (2006).
30. Luke, T. C. & Hoffman, S. L. Rationale and plans for developing a non-replicating, metabolically active, radiation-attenuated Plasmodium falciparum sporozoite vaccine. J. Exp. Biol. 206, 3803–3808 (2003).
31. Coatney, G. R. & Cooper, W. C. Symposium on exoerythrocytic forms of malarial parasites. III. The chemotherapy of malaria in relation to our knowledge of exoerythrocytic forms. J. Parasitol. 34, 275–289 (1948).
32. Landau, I. Description of Plasmodium chabaudi n. sp., parasite of African rodents. C. R. Hebd. Séances Acad. Sci. 260, 3758–3761 (1965) (in French).
33. Landau, I. & Chabaud, A. G. Natural infection by 2 plasmodia of the rodent Thamnomys rutilans in the Central African Republic. C. R. Hebd. Séances Acad. Sci. 261, 230–232 (1965) (in French).
34. Rodhain, J. Plasmodium vinckei n. sp.; second plasmodium parasite of wild rodents at Katange. Ann. Soc. Belge Méd. Trop. 32, 275–279 (1952) (in French).
35. Vincke, I. H. & Lips, M. A. H. Un nouveau plasmodium d’un rongeur sauvage du Congon, Plasmodium berghei n. sp. Ann. Soc. Belge Méd. Trop. 28, 97–104 (1948) (in French).
36. Yoeli, M. & Wall, W. J. Complete sporogonic development of Plasmodium berghei in experimentally infected Anopheles spp. Nature 168, 1078–1080 (1951).
37. Peters, W. Chemotherapy and drug resistance in malaria (Academic Press, London and New York, 1970).
38. Schmidt, L. H. Plasmodium cynomolgi infections in the rhesus monkey. Background studies. Am. J. Trop. Med. Hyg. 31, 609–611 (1982).
39. Schmidt, L. H. et al. Antimalarial activities and subacute toxicity of RC-12, a 4-amino-substituted pyrocatechol. Antimicrob. Agents Chemother. 28, 612–625 (1985).
40. Herrera, S., Perlaza, B. L., Bonelo, A. & Arévalo-Herrera, M. Aotus monkeys: their great value for anti-malaria vaccines and drug testing. Int. J. Parasitol. 32, 1625–1635 (2002).
41. Collins, W. E. Nonhuman primate models. II. Infection of Saimiri and Aotus monkeys with Plasmodium vivax. Methods Mol. Med. 72, 85–92 (2002).
42. Collins, W. E. Nonhuman primate models. I. Nonhuman primate host-parasite combinations. Methods Mol. Med. 72, 77–84 (2002).
43. Badell, E. et al. Human malaria in immunocompromised mice: an in vivo model to study defense mechanisms against Plasmodium falciparum. J. Exp. Med. 192, 1653–1660 (2000).
44. Moreno, A., Badell, E., Van Rooijen, N. & Druilhe, P. Human malaria in immunocompromised mice: new in vivo model for chemotherapy studies. Antimicrob. Agents Chemother. 45, 1847–1853 (2001).
45. Moreno, A. et al. The course of infections and pathology in immunomodulated NOD/LtSz-SCID mice inoculated with Plasmodium falciparum laboratory lines and clinical isolates. Int. J. Parasitol. 36, 361–369 (2006).
46. Moreno, A., Perignon, J. L., Morosan, S., Mazier, D. & Benito, A. Plasmodium falciparum-infected mice: more than a tour de force. Trends Parasitol. 23, 254–259 (2007).
47. Grompe, M. Mouse liver goes human: a new tool in experimental hepatology. Hepatology 33, 1005–1006 (2001).
48. Kneteman, N. M. & Mercer, D. F. Mice with chimeric human livers: who says supermodels have to be tall? Hepatology 41, 703–706 (2005).
49. Morosan, S. et al. Liver-stage development of Plasmodium falciparum, in a humanized mouse model. J. Infect. Dis. 193, 996–1004 (2006).
50. Sacci, J. B. Jr et al. Plasmodium falciparum infection and exoerythrocytic development in mice with chimeric human livers. Int. J. Parasitol. 36, 353–360 (2006).
51. Azuma, H. et al. Robust expansion of human hepatocytes in Fah–/–/Rag2–/–/Il2rg–/– mice. Nature Biotech. 25, 903–910 (2007).
52. Hollingdale, M. R., Leef, J. L., McCullough, M. & Beaudoin, R. L. In vitro cultivation of the exoerythrocytic stage of Plasmodium berghei from sporozoites. Science 213, 1021–1022 (1981).
53. Hollingdale, M. R., Leland, P. & Schwartz, A. L. In vitro cultivation of the exoerythrocytic stage of Plasmodium berghei in a hepatoma cell line. Am. J. Trop. Med. Hyg. 32, 682–684 (1983).
54. Lambiotte, M., Landau, I., Thierry, N. & Miltgen, F. Development of schizonts in cultured hepatocytes of adult rats after in vitro infection with Plasmodium yoelii sporoazoites. C. R. Hebd. Séances Acad. Sci. 293, 431–433 (1981) (in French).
55. Pirson, P. Culture of the exoerythrocytic liver stages of Plasmodium berghei sporozoites in rat hepatocytes. Trans. Royal Soc. Trop. Med. Hyg. 76, 422 (1982).
56. Mazier, D. et al. In vitro infection of adult Thamnomys hepatocytes by sporozoites of Plasmodium yoelii: development of schizonts and release of infective merozoites. Ann. Parasitol. Hum. Comp. 57, 99–100 (1982) (in French).
57. Rénia, L. et al. A malaria heat-shock-like determinant expressed on the infected hepatocyte surface is the target of antibody-dependent cell-mediated cytotoxic mechanisms by nonparenchymal liver cells. Eur. J. Immunol. 20, 1445–1449 (1990).
58. Mazier, D. et al. Complete development of hepatic stages of Plasmodium falciparum in vitro. Science 227, 440–442 (1985).
59. Mazier, D. et al. Cultivation of the liver forms of Plasmodium vivax in human hepatocytes. Nature 307, 367–369 (1984).
60. Mazier, D. et al. Plasmodium ovale: in vitro development of hepatic stages. Exp. Parasitol. 64, 393–400 (1987).
61. Smith, J. E., Meis, J. F., Ponnudurai, T., Verhave, J. P. & Moshage, H. J. In vitro culture of exoerythrocytic form of Plasmodium falciparum in adult human hepatocytes. Lancet 2, 757–758 (1984).
62. Millet, P. G., Collins, W. E., Fisk, T. L. & Nguyen-Dinh, P. In vitro cultivation of exoerythrocytic stages of the human malaria parasite Plasmodium malariae. Am. J. Trop. Med. Hyg. 38, 470–473 (1988).
63. Millet, P. G. et al. In vitro cultivation of Plasmodium cynomolgi bastianelli in hepatocytes of the Macaca rhesus. Ann. Parasitol. Hum. Comp. 62, 5–7 (1987) (in French).
64. Millet, P. G., Fisk, T. L., Collins, W. E., Broderson, J. R. & Nguyen-Dinh, P. Cultivation of exoerythrocytic stages of Plasmodium cynomolgi, P. knowlesi, P. coatneyi, and P. inui in Macaca mulatta hepatocytes. Am. J. Trop. Med. Hyg. 39, 529–534 (1988).
65. Karnasuta, C. et al. Complete development of the liver stage of Plasmodium falciparum in a human hepatoma cell line. Am. J. Trop. Med. Hyg. 53, 607–611 (1995).
66. Sattabongkot, J. et al. Establishment of a human hepatocyte line that supports in vitro development of the exo-erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax. Am. J. Trop. Med. Hyg. 74, 708–715 (2006).
67. Prudencio, M., Rodriguez, A. & Mota, M. M. The silent path to thousands of merozoites: the Plasmodium liver stage. Nature Rev. Microbiol. 4, 849–856 (2006).
68. Calvo-Calle, J. M., Moreno, A., Eling, W. M. C. & Nardin, E. H. In vitro development of infectious liver stages of P. yoelii and P. berghei malaria in human cell lines. Exp. Parasitol. 79, 362–373 (1994).
69. Hollingdale, M. R. Malaria and the liver. Hepatology 5, 327–335 (1985).
70. Silvie, O. et al. Expression of human CD81 differently affects host cell susceptibility to malaria sporozoites depending on the Plasmodium species. Cell. Microbiol. 8, 1134–1146 (2006).
71. Hollingdale, M. R., Collins, W. E., Campbell, C. C. & Schwartz, A. L. In vitro culture of two populations (dividing and nondividing) of exoerythrocytic parasites of Plasmodium vivax. Am. J. Trop. Med. Hyg. 34, 216–222 (1985).
72. Hollingdale, M. R., Collins, W. E. & Campbell, C. C. In vitro culture of exoerythrocytic parasites of the North Korean strain of Plasmodium vivax in hepatoma cells. Am. J. Trop. Med. Hyg. 35, 275–276 (1986).
73. Silvie, O. et al. A role for apical membrane antigen 1 during invasion of hepatocytes by Plasmodium falciparum sporozoites. J. Biol. Chem. 279, 9490–9406 (2004).
74. Meis, J. F. et al. Infection of cryopreserved adult human hepatocytes with Plasmodium falciparum sporozoites. Cell Biol. Int. Rep. 9, 976 (1985).
75. Rénia, L. et al. Malaria sporozoite penetration. A new approach by double staining. J. Immunol. Methods 112, 201–205 (1988).
76. Mazier, D. et al. Effect of antibodies to recombinant and synthetic peptides on P. falciparum sporozoites in vitro. Science 231, 156–159 (1986).
77. Gego, A. et al. New approach for high-throughput screening of drug activity on Plasmodium liver stages. Antimicrob. Agents Chemother. 50, 1586–1589 (2006).
78. Rodrigues, C. D. et al. Host scavenger receptor SR-BI plays a dual role in the establishment of malaria parasite liver infection. Cell Host Microbe 4, 271–282 (2008).
79. Prudencio, M., Rodrigues, C. D., Ataide, R. & Mota, M. M. Dissecting in vitro host cell infection by Plasmodium sporozoites using flow cytometry. Cell. Microbiol. 10, 218–224 (2008).
80. Jung, M. et al. Effects of hepatocellular iron imbalance on nitric oxide and reactive oxygen intermediates production in a model of sepsis. J. Hepatol. 33, 387–394 (2000).
81. Yalaoui, S. et al. Hepatocyte permissiveness to Plasmodium infection is conveyed by a short and structurally conserved region of the CD81 large extracellular domain. PLoS Pathogen 4, e1000010 (2008).
82. Most, H., Herman, R. H. & Schoenfeld, C. Chemotherapy of sporozoite- and blood-induced Plasmodium berghei infections with selected antimalarial agents. Am. J. Trop. Med. Hyg. 16, 572–575 (1967).
83. Fink, E. Assessment of causal prophylactic activity in Plasmodium berghei yoelii and its value for the development of new antimalarial drugs. Bull. World Health Organ. 50, 213–222 (1974).
84. Hulier, E. et al. A method for the quantitative assessment of malaria parasite development in organs of the mammalian host. Mol. Biochem. Parasitol. 77, 127–135 (1996).
85. Briones, M. R. S., Tsuji, M. & Nussenzweig, V. The large difference in infectivity for mice of Plasmodium berghei and Plasmodium yoelii sporozoites cannot be correlated with their ability to enter into hepatocytes. Mol. Biochem. Parasitol. 77, 7–17 (1996).
86. Bruña-Romero, O. et al. Detection of malaria liver-stages in mice infected through the bite of a single Anopheles mosquito using a highly sensitive real-time PCR. Int. J. Parasitol. 31, 1499–1502 (2001).
87. Witney, A. A. et al. Determining liver stage parasite burden by real time quantitative PCR as a method for evaluating pre-erythrocytic malaria vaccine efficacy. Mol. Biochem. Parasitol. 118, 233–245 (2001).
88. Snounou, G., Grüner, A. C., Müller-Graf, C. D., Mazier, D. & Rénia, L. The Plasmodium sporozoite survives RTS,S vaccination. Trends Parasitol. 21, 456–461 (2005).
89. Carraz, M. et al. A plant-derived morphinan as a novel lead compound active against malaria liver stages. PLoS Medicine 3, e513 (2006).
90. Grüner, A. C. et al. Insights into the P. y. yoelii hepatic stage transcriptome reveal complex transcriptional patterns. Mol. Biochem. Parasitol. 142, 184–192 (2005).
P e r s P e c t i v e s
NATURE REVIEwS | Drug Discovery VoLUME 8 | NoVEMBER 2009 | 863
© 2009 Macmillan Publishers Limited. All rights reserved

91. Sacci, J. B. Jr, Aguiar, J. C., Lau, A. O. T. & Hoffman, S. L. Laser capture microdissection and molecular analysis of Plasmodium yoelii liver-stage parasites. Mol. Biochem. Parasitol. 119, 285–289 (2002).
92. Semblat, J.-P. et al. Laser capture microdissection of Plasmodium falciparum liver stages for mRNA analysis. Mol. Biochem. Parasitol. 121, 179–183 (2002).
93. Siau, A. et al. Temperature shift and host cell contact up-regulate sporozoite expression of Plasmodium falciparum genes involved in hepatocyte infection. PLoS Pathogen 4, e1000121 (2008).
94. Tarun, A. S. et al. A combined transcriptome and proteome survey of malaria parasite liver stages. Proc. Natl Acad. Sci. USA 105, 305–310 (2008).
95. Cunha-Rodrigues, M., Prudencio, M., Mota, M. M. & Haas, W. Antimalarial drugs — host targets (re)visited. Biotechnol. J. 1, 321–332 (2006).
96. Mahmoudi, N. et al. New active drugs against liver stages of Plasmodium predicted by molecular topology. Antimicrob. Agents Chemother. 52, 1215–1220 (2008).
97. Doerig, C. & Meijer, L. Antimalarial drug discovery: targeting protein kinases. Exp. Opin. Ther. Targets 11, 279–290 (2007).
98. Edgcomb, J. H., Arnold, J. D., Yount, E. H. Jr, Alving, A. S. & Eichelberger, L. Primaquine, SN 13272, a new curative agent in vivax malaria: a preliminary report. J. Natl Malaria Soc. 9, 285–292 (1950).
99. Hill, D. R. et al. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am. J. Trop. Med. Hyg. 75, 402–415 (2006).
100. Baird, J. K., Fryauff, D. J. & Hoffman, S. L. Primaquine for prevention of malaria in travelers. Clin. Infect. Dis. 37, 1659–1667 (2003).
101. Peters, W. The evolution of tafenoquine — antimalarial for a new millennium? J. Royal Soc. Med. 92, 345–352 (1999).
102. Brueckner, R. P., Coster, T., Wesche, D. L., Shmuklarsky, M. J. & Schuster, B. G. Prophylaxis of Plasmodium falciparum infection in a human challenge model with WR 238605, a new 8-aminoquinoline antimalarial. Antimicrob. Agents Chemother. 42, 1293–1294 (1998).
103. Dutta, G. P., Puri, S. K., Bhaduri, A. P. & Seth, M. Radical curative activity of a new 8-aminoquinoline derivative (CDRI 80/53) against Plasmodium cynomolgi B in monkeys. Am. J. Trop. Med. Hyg. 41, 635–637 (1989).
104. Krudsood, S. et al. Safety and tolerability of elubaquine (bulaquine, CDRI 80/53) for treatment of Plasmodium vivax malaria in Thailand. Korean J. Parasitol. 44, 221–228 (2006).
105. Valecha, N. et al. Comparative antirelapse efficacy of CDRI compound 90/53 (Bulaquine) vs primaquine in double blind clinical trials. Curr. Sci. 80, 561–563 (2001).
106. Alving, A. S. et al. Potentiation of the curative action of primaquine in vivax malaria by quinine and chloroquine. J. Lab. Clin. Med. 46, 301–306 (1955).
107. Schmidt, L. H., Fradkin, R., Vaughan, D. & Rasco, J. Radical cure of infections with Plasmodium cynomolgi: a function of total 8-aminoquinoline dose. Am. J. Trop. Med. Hyg. 26, 1116–1128 (1977).
108. Alving, A. S. et al. Korean vivax malaria. II. Curative treatment with pamaquine and primaquine. Am. J. Trop. Med. Hyg. 2, 970–976 (1953).
109. Arnold, J. D., Alving, A. S. & Clayman, C. B. Induced primaquine resistance in vivax malaria. Trans. Royal Soc. Trop. Med. Hyg. 55, 345–350 (1961).
110. Singh, N. & Puri, S. K. Causal prophylactic activity of antihistaminic agents against Plasmodium yoelii nigeriensis infection in Swiss mice. Acta Trop. 69, 255–260 (1998).
111. Singh, N. & Puri, S. K. Inhibition of the development of the hepatic stages of Plasmodium yoelii nigeriensis by antihistaminic agents. Ann. Trop. Med. Parasitol. 93, 419–422 (1999).
112. Zhang, Q. et al. Unambiguous synthesis and prophyl actic antimalarial activities of imidazoli dine-dione derivatives. J. Med. Chem. 48, 6472–6481 (2005).
113. Coppi, A. et al. Heparan sulfate proteoglycans provide a signal to Plasmodium sporozoites to stop migrating and productively invade host cells. Cell Host Microbe 2, 316–327 (2007).
114. Bosch, J. et al. Aldolase provides an unusual binding site for thrombospondin-related anonymous protein in the invasion machinery of the malaria parasite. Proc. Natl Acad. Sci. USA 104, 7015–7020 (2007).
115. Coppi, A., Pinzon-Ortiz, C., Hutter, C. & Sinnis, P. The Plasmodium circumsporozoite protein is proteolytically processed during cell invasion. J. Exp. Med. 201, 27–33 (2005).
116. Talati, S. M., Latham, M. R., Moore, E. G., Hargreaves, G. W. & DeWitt Blanton, C. Jr. Synthesis of potential antimalarials: primaquine analogs. J. Pharm. Sci. 59, 491–495 (1970).
117. Li, J. et al. Plasmodium berghei: quantitation of in vitro effects of antimalarial drugs on exoerythrocytic development by a ribosomal RNA probe. Exp. Parasitol. 72, 450–458 (1991).
118. Powell, R. D. & Brewer, G. J. Effects of pyrimethamine, chlorguanide, and primaquine against exoerythrocytic forms of a strain of chloroquine-resistant Plasmodium falciparum from Thailand. Am. J. Trop. Med. Hyg. 16, 693–698 (1967).
119. Nduati, E. et al. Effect of folate derivatives on the activity of antifolate drugs used against malaria and cancer. Parasitol. Res. 102, 1227–1234 (2008).
120. Gregory, K. G. & Peters, W. The chemotherapy of rodent malaria. IX. Causal prophylaxis. I. A method for demonstrating drug action on exo-erythrocytic stages. Ann. Trop. Med. Parasitol. 64, 15–24 (1970).
121. Hollingdale, M. R., McCann, P. P. & Sjoerdsma, A. Plasmodium berghei: inhibitors of ornithine decarboxylase block exoerythrocytic schizogony. Exp. Parasitol. 60, 111–117 (1985).
122. Gantt, S. M. et al. Proteasome inhibitors block development of Plasmodium spp. Antimicrob. Agents Chemother. 42, 2731–2738 (1998).
123. Davies, C. S., Pudney, M., Nicholas, J. C. & Sinden, R. E. The novel hydroxynaphthoquinone 566C80 inhibits the development of liver stages of Plasmodium berghei cultured in vitro. Parasitology 106, 1–6 (1993).
124. Andersen, S. L. et al. Efficacy of azithromycin as a causal prophylactic agent against murine malaria. Antimicrob. Agents Chemother. 38, 1862–1863 (1994).
125. Marussig, M. S. et al. Activity of doxycycline against preerythrocytic malaria. J. Infect. Dis. 168, 1603–1604 (1993).
126. Mahmoudi, N. et al. In vitro activities of 25 quinolones and fluoroquinolones against liver and blood stage Plasmodium spp. Antimicrob. Agents Chemother. 47, 2636–2639 (2003).
127. Yu, M. et al. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell Host Microbe 4, 567–578 (2008).
128. Vaughan, A. M. et al. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell. Microbiol. 11, 506–520 (2009).
129. Van De Sand, C. et al. The liver stage of Plasmodium berghei inhibits host cell apoptosis. Mol. Microbiol. 58, 731–742 (2005).
130. Sturm, A. et al. Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science 313, 1287–1290 (2006).
AcknowledgementsWe are grateful to A. C. Grüner for critical reading of the manuscript and many helpful and pertinent suggestions. D.M., L.R. and G.S. are currently part of an official collabora-tion between SIgN-/A*-STAR and INSERM (Laboratoire International Associé, INSERM).
DAtABASESUniProtKB: http://www.uniprot.orgG6PD | MeT
FuRtHER INFORMAtIONDominique Mazier’s homepage: http://www.inserm-u511.jussieu.fr/Laurent rénia’s homepage: http://www.sign.a-star.edu.sg
SuPPlEMENtARY INFORMAtIONsee online article: S1 (table)
All links Are AcTive in The online PDF
P e r s P e c t i v e s
864 | NoVEMBER 2009 | VoLUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved




















