
A one-pot synthesis of disiloxy, dihalo and dithiocyanato dioxo complexes bearing a bidentate...
11
A one-pot synthesis of disiloxy, dihalo and dithiocyanato dioxo complexes bearing a bidentate N-ligand from sodium or potassium molybdate tungstate and perrhenate Henri Arzoumanian Robert Bakhtchadjian Giuseppe Agrifoglio Reinaldo Atencio Alexander Bricen ˜o Received: 14 May 2008 / Accepted: 17 June 2008 / Published online: 20 September 2008 Ó Springer Science+Business Media B.V. 2008 Abstract Disiloxy, dichloro, dibromo and dithiocyanato molybdenum, tungsten and rhenium-oxo complexes bear- ing bidendate N-ligands have been synthesized in a one-pot process and fully characterized spectroscopically. The reaction, in acetonitrile, is significantly simpler to run than previously reported methods for the synthesis of analogous compounds and appears to be general in character. Introduction Transition metal-oxo species have aroused great interest in recent years due to their presence either as precursors or as real active species in many catalytic reactions. Function- alizing metal-oxo compounds in order to achieve judicious modifications of the coordination sphere of the metal has, thus, received considerable attention. One method exten- sively used has been the reaction with chlorosilanes, resulting either as chlorination or siloxylation. As a general rule, chlorination is obtained when trimethylchlorosilane is used, whereas the siloxy compound is formed when a bulkier trialkyl or triphenylchlorosilane is used. LMO 3 þ 2Me 3 SiCl ! LMO 2 Cl 2 þ Me 3 SiOSiMe 3 ½MO n 2 þ 2Ph 3 SiCl ! MO n2 ðOSiPh 3 Þ 2 þ 2Cl As an illustration, trimethylchlorosilane leads readily to mono or dichloro compounds with many metal-oxo complexes bearing various ligands such as: Me 3 ReO 3 [1], (g 5 -C 5 Me 5 ) 2 WO [2], (g 5 -C 5 Me 5 ) MoO 3 [3], (g 5 -C 5 Me 5 )ReO 3 [1], bis(diethyldithiocarbamate)MoO 2 [4], bis(dithiolene) WO 2 [5], bis(N-pyrrolidinedithiocarbamate)MoO 2 [6]. A few exceptions do exist, however, to this general trend such as for AgReO 4 and AgTcO 4 [3]. When a methyl group in Me 3 SiCl is replaced by a bulkier group such as tert-butyl or when triphenylchlorosilane is used, the reaction with a metal- oxo yields stable siloxy compounds. For example, Ag 2 MoO 4 [7], (g 5 -C 5 Me 5 ) WO 3 [7] bis(diethyldithiocarbamate)MoO 2 [4, 8], AgVO 3 [7], Ag 2 MoO 4 [8], bis(dithiolene) 2 WO [5]. In most cases, however, obtaining a metal-oxo species bearing a chloro or a siloxy group as well as a ligand necessitates at least two separate steps, either the metal-oxo bearing a ligand is synthesized and then treated with the chlorosilane [1–6] or the chloro or siloxy compound sepa- rately prepared is then complexed with the desired ligand [8]. More recently, a more direct and simpler procedure was reported for the silylation of potassium tungstate in the presence of a bidentate ligand [9]. The use of acetonitrile as a solvent was apparently the necessary medium for such an improvement. We have now extended this reaction mode to potassium and sodium molybdate and also showed that it can be applied to the chlorination, bromination and thio- cyanation of molybdate and tungstate ions in the presence of various ligands as depicted below. ½MoO 4 2 þ L þ 2Ph 3 SiCl ! LMoO 2 ðOSiPh 3 Þ 2 þ 2Cl ½MO 4 2 þ L þ 4Me 3 SiX ! LMO 2 X 2 þ 2Me 3 SiOSiMe 3 þ 2X M=Mo, W; X=Cl, Br, NCS; L = 4,4 0 -substituted-2,2 0 - bipyridine, bis(3,5-dimethylpyrazol-1-yl)methane. It was further tested with potassium perrhenate. H. Arzoumanian (&) R. Bakhtchadjian Chirosciences UMR 6263 CNRS ISM2, Universite ´ Paul Ce ´zanne, Faculte ´ des Sciences, St. Je ´ro ˆme, Marseille, France e-mail: [email protected] G. Agrifoglio R. Atencio A. Bricen ˜o Centro de Quı ´mica, Instituto Venezolano de Investigaciones Cientı ´ficas (IVIC), Apartado 21827, Caracas 1020, Venezuela 123 Transition Met Chem (2008) 33:941–951 DOI 10.1007/s11243-008-9123-6
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of A one-pot synthesis of disiloxy, dihalo and dithiocyanato dioxo complexes bearing a bidentate...

A one-pot synthesis of disiloxy, dihalo and dithiocyanato dioxocomplexes bearing a bidentate N-ligand from sodiumor potassium molybdate tungstate and perrhenate
Henri Arzoumanian Æ Robert Bakhtchadjian ÆGiuseppe Agrifoglio Æ Reinaldo Atencio ÆAlexander Briceno
Received: 14 May 2008 / Accepted: 17 June 2008 / Published online: 20 September 2008
� Springer Science+Business Media B.V. 2008
Abstract Disiloxy, dichloro, dibromo and dithiocyanato
molybdenum, tungsten and rhenium-oxo complexes bear-
ing bidendate N-ligands have been synthesized in a one-pot
process and fully characterized spectroscopically. The
reaction, in acetonitrile, is significantly simpler to run than
previously reported methods for the synthesis of analogous
compounds and appears to be general in character.
Introduction
Transition metal-oxo species have aroused great interest in
recent years due to their presence either as precursors or as
real active species in many catalytic reactions. Function-
alizing metal-oxo compounds in order to achieve judicious
modifications of the coordination sphere of the metal has,
thus, received considerable attention. One method exten-
sively used has been the reaction with chlorosilanes,
resulting either as chlorination or siloxylation. As a general
rule, chlorination is obtained when trimethylchlorosilane is
used, whereas the siloxy compound is formed when a
bulkier trialkyl or triphenylchlorosilane is used.
LMO3 þ 2Me3SiCl! LMO2Cl2 þMe3SiOSiMe3
½MOn��2 þ 2Ph3SiCl! MOn�2ðOSiPh3Þ2 þ 2Cl�
As an illustration, trimethylchlorosilane leads readily
to mono or dichloro compounds with many metal-oxo
complexes bearing various ligands such as: Me3ReO3 [1],
(g5-C5Me5)2WO [2], (g5-C5Me5) MoO3 [3], (g5-C5Me5)ReO3
[1], bis(diethyldithiocarbamate)MoO2 [4], bis(dithiolene)
WO2 [5], bis(N-pyrrolidinedithiocarbamate)MoO2 [6]. A
few exceptions do exist, however, to this general trend such
as for AgReO4 and AgTcO4 [3]. When a methyl group in
Me3SiCl is replaced by a bulkier group such as tert-butyl or
when triphenylchlorosilane is used, the reaction with a metal-
oxo yields stable siloxy compounds. For example, Ag2MoO4
[7], (g5-C5Me5) WO3 [7] bis(diethyldithiocarbamate)MoO2
[4, 8], AgVO3 [7], Ag2MoO4 [8], bis(dithiolene)2WO [5].
In most cases, however, obtaining a metal-oxo species
bearing a chloro or a siloxy group as well as a ligand
necessitates at least two separate steps, either the metal-oxo
bearing a ligand is synthesized and then treated with the
chlorosilane [1–6] or the chloro or siloxy compound sepa-
rately prepared is then complexed with the desired ligand [8].
More recently, a more direct and simpler procedure was
reported for the silylation of potassium tungstate in the
presence of a bidentate ligand [9]. The use of acetonitrile as
a solvent was apparently the necessary medium for such an
improvement. We have now extended this reaction mode to
potassium and sodium molybdate and also showed that it
can be applied to the chlorination, bromination and thio-
cyanation of molybdate and tungstate ions in the presence
of various ligands as depicted below.
½MoO4��2 þ Lþ 2Ph3SiCl! LMoO2ðOSiPh3Þ2 þ 2Cl�
½MO4��2 þ Lþ 4Me3SiX! LMO2X2 þ 2Me3SiOSiMe3
þ 2X�
M=Mo, W; X=Cl, Br, NCS; L = 4,40-substituted-2,20-bipyridine, bis(3,5-dimethylpyrazol-1-yl)methane.
It was further tested with potassium perrhenate.
H. Arzoumanian (&) � R. Bakhtchadjian
Chirosciences UMR 6263 CNRS ISM2, Universite Paul
Cezanne, Faculte des Sciences, St. Jerome, Marseille, France
e-mail: [email protected]
G. Agrifoglio � R. Atencio � A. Briceno
Centro de Quımica, Instituto Venezolano de Investigaciones
Cientıficas (IVIC), Apartado 21827, Caracas 1020, Venezuela
123
Transition Met Chem (2008) 33:941–951
DOI 10.1007/s11243-008-9123-6

Experimental
General material and procedures
All materials were commercial and used without further
purification unless otherwise noted. All solvents were dis-
tilled, acetonitrile over CaH2, ether and THF over sodium
wire, stored over 4 A molecular sieves and thoroughly
degassed prior to use. All experiments were carried out
under inert atmosphere (dry nitrogen or argon) using
modified Schlenk techniques. 4,40-di-tert-butyl-2,20-bipyridine, 4,40-dimethoxy-2,20-bipyridine and bis(3,5-
dimethylpyrazol-1-yl)methane were synthesized using lit-
erature methods. NMR spectra were recorded on a Bruker
AVANCE 500 MHz spectrometer. IR spectra were recor-
ded on a Perkin Elmer 1720 XFT spectrometer.
Crystal structure determination
Intensity data for the compounds 1, 6 and 7 were recorded at
room temperature on a Rigaku AFC-7S diffractometer using
monochromated Mo(Ka) radiation (k = 0.71073 A).
Experimental details on unit cell and intensity measurements
can be found in the CIF files deposited with the Cambridge
Crystallographic Data Centre (68693, 68694 and 68695).
Crystal data, intensity data collection parameters and final
refinement results are summarized in Table 1. The structures
were solved by Direct Methods and refined by full-matrix
least-squares on F2. The H-atoms on C were placed in cal-
culated positions using a riding atom model with fixed C–H
distances [0.93 A for C(sp2), 0.96 A for C(sp3, CH3) and
0.97 A for C(sp3, CH2)]. All the H atoms were refined with
isotropic displacement parameters set to 1.2 9 Ueq for
Table 1 Crystal data, intensity
data collection parameters and
final refinement results
Compound 1 6 7
CCDC deposit no.
Crystal data
Formula C54H54MoN2O4Si � CH2Cl2 C11H16Cl2MoN4O2 C11H16Br2MoN4O2
Mw 1032.4 403.12 492.04
Specimen size (mm) 0.38 9 0.14 9 0.12 0.60 9 0.48 9 0.20 0.46 9 0.22 9 0.17
T (K) 293 (2) 293 (2) 293 (2)
a (A) 11.222 (6) 10.2723 (14) 10.500 (2)
b (A) 17.645 (6) 14.2462 (12) 14.307 (3)
c (A) 26.376 (6) 10.9094 (12) 11.103 (2)
b (�) 96.60 (3) 107.019 (9) 106.47 (3)
V (A3) 5188 (3) 1526.6 (3) 1595.5 (3)
Crystal system Monoclinic Monoclinic Monoclinic
Space group P21/n P21/n P21/n
Z 4 4 4
Dc (c cm-3) 1.321 1.754 2.043
l (mm-1) 0.45 1.22 5.82
Number of ref. for cell 20 25 2,372
Data collection
H Range (�) 26.0–26.8 22.6–25.3 2.3–28.0
h 0, 13 0, 12 -9, 12
k 0, 20 0, 16 -16, 16
l -31, 31 -12, 12 -13, 14
Number of ref. measured 9,603 2,487 18,255
Number of ref. unique 9,102 2,275 3,078
Ref. I [ 2r(I) 4,933 2,493 2,686
Rint 0.051 0.083 0.031
Refinement
R[F2 [ 2r(F2)] 0.069 0.027 0.035
wR(F2) 0.165 0.076 0.087
S 1.00 1.11 1.11
No. param. Refin. 600 185 185
(D/r) max. 0.001 0.001 0.001
Dq (min., max) (eA-3) -0.64, -0.48 -0.78, 0.73 -0.86, 0.44
942 Transition Met Chem (2008) 33:941–951
123

C(sp2) and 1.5 for C(sp3) of the attached atom. In the struc-
ture 1, the dichloromethane molecule was found to be
disordered. This disorder was modelled over three orienta-
tions with restraints in the C–Cl and Cl–Cl distances and
complementary occupancies. The occupational parameters
were refined and fixed during the final refinement cycle in
40:40:20. Only these atoms were refined with isotropic dis-
placement parameters. Data reduction for 1 and 6 were
performed using teXsan [10], whereas 7 was made using the
Crystal clear [11] crystallographic software packages,
respectively. All the refinement calculations were made
using SHELXTL-NT [12].
4,40-dichloro-2,20-bipyridine
A suspension of 4,40-dinitro-2,20-bipyridine-1,10-dioxide
[13] (1.5 g, 5.4 mmole) in 60 mL of acetyl chloride was
refluxed, under inert atmosphere, for 4 h. The reaction
mixture was cooled in an ice bath and to it was slowly
(15 min) added 15 mL of freshly distilled PCl3. The
resulting mixture was refluxed for an additional 4 h and the
volume reduced under vacuum to 20–30 mL. The solution
was cooled in an ice bath and slowly poured into 200 g of
crushed ice then made alkaline with NaOH. The precipitate
was filtered off and the product extracted with CH2Cl2,
dried over MgSO4 and recrystallized from ethanol/water.1H-NMR (CDCl3); d 7.39(dd), 8.40(d), 8.55(d).
Mo(O)2(4,40-t-butyl-2,20-bipy)(OSiPh3)2 � 1
To a 15 mL CH3CN solution of 4,40-di-tert-butyl-2,20-bipyridine (0.134 g, 0.5 mmol) was added 0.103 g
(0.5 mmol) of anhydrous Na2MoO4. The suspension was
stirred at 25 �C for 30 min before 0.265 g (0.9 mmol) of
triphenylchlorosilane was added. The resulting mixture
was heated to reflux for a period of 5 h and evaporated to
dryness. The white solid obtained was extracted with
CH3CN (3 9 10 mL). Removal of the solvent gave the
desired compound as a white crystalline solid. Yield
0.40 g, 93%. IR (KBr): m(Mo=O) 934(s), 910(sh); m(Si–O)
1,113(s) cm-1. 1H-NMR (CDCl3): d 1.41 (s), 7.1–7.3 (m),
7.43 [dd, J(H–H) = 6 Hz, 1.6 Hz], 7.75 [dd, J(H–H) =
6 Hz, 1.6 Hz], 9.03 [dd, J(H–H) = 6 Hz, 1.6 Hz]. Anal.
Calcd. for C54H54N2O4Si2Mo; C, 68.47; H, 5.75; N, 2.96.
Found; C, 68.58; H, 5.67; N, 2.87.
Mo(O)2 [bis(3,5-Me2-pyrazolylmethane)](OSiPh3)2 � 2
To a 25 mL CH3CN solution containing 0.296 g
(1.45 mmol) of bis-(3,5-Me2-pyrazolylmethane was added
0.299 g (1.45 mmol) of anhydrous Na2MoO4. The sus-
pension was stirred at 25 �C for 30 min and 0.740 g
(2.90 mmol) of triphenylchlorosilane was added. The
mixture was refluxed for 4 h and the resulting yellowish
suspension was evaporated to dryness, and extracted with
CH2Cl2 (2 9 50 mL). Evaporation gave a colourless
crystalline solid. Yield 1.16 g, 91%. IR(KBr): m(Mo=O)
929(m), 907(m) cm-1, m(Si–O) 1,118(s) cm-1. 1H-NMR
(CDCl3); d 2.40 (s), 2.65 (s), 5.95 (s), 6.50 (s), 7.1–7.3(m).
Anal. Calcd. for C47H46N4O4Si2Mo; C, 63.93; H, 5.25; N,
6.34. Found; C, 64.05; H, 5.28; N, 6.28.
Mo(O)2(4,40-t-butyl-2,20-bipy)Cl2 � 3
To a 15 mL CH3CN solution containing 134 mg (0.5 mmol)
of 4,40-di-t-butyl-2,20-bipyridine was added 103 mg
(0.5 mmol) of anhydrous Na2MoO4. The suspension was
stirred 30 min at room temperature and to it was added
216 mg (250 lL, 2 mmole) of trimethylchlorosilane. The
mixture was refluxed for 4 h and reduced to dryness under
vacuum to give a colourless solid residue. Recrystallization
from CH2Cl2/n-hexane (2/1) yielded a colourless crystalline
solid. Yield 0.22 g, 94%. IR (KBr); m(Mo=O), 939(s), 908(s)
cm-1. 1H-NMR (CDCl3) d 7.70 [dd, J(H–H) = 6 Hz,
1.6 Hz], 8.16 [d, J(H–H) = 6 Hz], 9.47 [d, J(H–H) =
6 Hz]. Anal. Calcd. for C18H24N2O2Cl2Mo; C, 46.27; H,
5.18; N, 5.99. Found; C, 46.12; H, 5.16; N, 6.12.
Mo(O)2(4,40-(OCH3)-2,20-bipy)Cl2 � 4
A 20 mL CH3CN suspension containing 21 mg (0.1 mmole)
of anhydrous Na2MoO4 and 22 mg (0.1 mmole) of 4,40-dimethoxy-2,20-bipyridine was stirred overnight at room
temperature. To the resulting white suspension was added
46.5 mg (53.5 lL, 0.42 mmole) of Me3SiCl; it resulted
immediately in the intensification of the white suspended
material. The mixture was refluxed for 4 h and evaporated
under vacuum to give a white solid. Treatment with CH2Cl2(2 9 30 mL) led to only partial extraction; it was slightly
improved by refluxing the solid in CH2Cl2 and filtering while
hot, but the overall yield remained modest (60%). Never-
theless, the complex obtained was quite pure. Yield 25 mg,
60%. IR (KBr); m(Mo=O) 937(s), 906(s) cm-1. 1H-NMR
(CD2Cl2); d 4.05(s), 7.20 [dd, J(H–H) = 6 Hz, 1.6 Hz], 7.67
[d, J(H–H) = 6 Hz], 9.32 [d, J(H–H) = 6 Hz]. Anal. Calcd.
for C12H12N2Cl2O4Mo; C, 41.87; H, 3.51; N, 8.14. Found; C,
41.79; H, 3.55; N, 8.20.
Mo(O)2(4,40-Cl-2,20-bipy)Cl2 � 5
A 30 mL suspension containing 51.5 mg (0.25 mmole) of
anhydrous Na2MoO4 and 56.2 mg (0.25 mmole) of 4,40-dichloro-2,20 bipyridine was stirred overnight at room tem-
perature. To the resulting suspension was added 108 mg
(125 lL, 1 mmole) of Me3SiCl. The mixture was refluxed
for 4 h and evaporated to dryness under vacuum. Extraction
Transition Met Chem (2008) 33:941–951 943
123

of the desired product with CH2Cl2 was incomplete, hot
acetone was used instead to give a white solid upon evapo-
ration of the solvent. Yield calculated with two moles of
acetone, 78 mg, 65%. IR (KBr); m(Mo=O) 944(s), 915(s)
cm-1. 1H-NMR (d6 acetone) d 8.10 [dd, J(H–H) = 6 Hz,
1.6 Hz], 8.98 [d, J(H–H) = 6 Hz], 9.46 [d, J(H–H) =
6 Hz]. Anal. Calcd. for C10H6N2Cl4O2Mo + 2 (CH3)2CO;
C, 32.3; H, 2.5; N, 5.8. Found; C, 31.4; H, 2.3; N, 5.6.
Mo(O)2 [bis(3,5-Me2-pyrazolylmethane)]Cl 2 � 6
To a 25 mL CH3CN solution containing 0.296 g
(1.45 mmol) of bis-(3,5-Me2-pyrazolylmethane was added
0.299 g (1.45 mmol) of anhydrous Na2MoO4. After stirring
the suspension for 30 min, 0.630 g (733 lL, 5.8 mmol) of
trimethylchlorosilane was added and the mixture heated to
reflux for 4 h. Evaporation to dryness gave a white solid
(yield 0.54 g, 92%) which upon recrystallization from
CH2Cl2/n-hexane (2/1) gave the desired product as a col-
ourless crystalline solid. IR (KBr); m(Mo=O) 943(s), 910(s)
cm-1. 1H-NMR (CDCl3); d 2.45 (s), 2.75 (s), 6.01 (s), 6.56
(s). Anal. Calcd. for C11H16N4O2Cl2Mo; C, 32.77; H, 4.00;
N, 13.64. Found; C, 32.39; H, 3.86; N, 13.51.
Mo(O)2 [bis-(3,5-Me2-pyrazolylmethane)]Br2 � 7
A 25 mL CH3CN suspension containing 204 mg (1 mmol)
of bis-(3,5-Me2-pyrazolylmethane) and 206 mg (1 mmol)
of anhydrous Na2MoO4 was stirred for 30 min at room
temperature. To the resulting milky suspension was added
612 mg (430 lL, 4 mmol) of trimethylbromosilane and the
mixture refluxed for 4 h. Evaporation of the solvent gave a
yellow-green solid which was extracted with CH2Cl2(2 9 30 mL). Evaporation gave nearly pure product. Yield
0.47 g, 96%. Recrystallization from CH2Cl2/n-hexane (2/1)
gave an X-ray suitable material. IR (KBr) n(Mo=O), 941,
907 cm-1. 1H-NMR (CDCl3); d 6.53(s), 6.06(s), 2.73(s),
2.43(s). Anal. Calcd. for C11H16N4Br2Mo; C, 27.59; H,
3.24; N, 11.38. Found; C, 26.72; H, 3.26; N, 11.22.
Mo(O)2(t-butyl-bipy)(NCS)2 � 8
To a 15 mL CH3CN solution containing 134 mg (0.5 mmole)
of 4,40-di-t-butyl-2,20-bipyridine was added 103 mg
(0.5 mmole) of anhydrous Na2MoO4. The resulting suspen-
sion was stirred at room temperature for 30 min and to it was
added 263 mg (282 lL, 2 mmole) of Me3SiNCS. The mix-
ture became quickly yellow. It was refluxed for 4 h, turning
reddish. After evaporation to dryness, the resulting red solid
was extracted with CH2Cl2 (2 9 50 mL). Evaporation of the
solvent yielded nearly pure product. Yield 0.24 g, 95%. IR
(KBr): m(Mo=O) 935(s), 905(s), m(SCN) 2,014(vs) cm-1.1H-NMR (CDCl3) d 7.77 [dd, J(H–H) = 6 Hz, 1.6 Hz], 8.22
[d, J(H–H) = 6 Hz], 9.35 [d, J(H–H) = 6 Hz]. Anal. Calcd.
for C20H24N4S2O2Mo; C, 46.87; H, 4.72; N, 10.93; S, 12.51.
Found; C, 46.62; H, 4.69; N, 11.00; S, 12.46.
[Mo(O)2(t-butyl-bipy)(NCS)]2O � 9
A 25 mL CH3CN suspension containing 103 mg (0.5 mmole)
of anhydrous Na2MoO4 and 134 mg (0.5 mmole) of 4,40-di-t-
butyl-2,20-bipyridine was stirred overnight at room tempera-
ture. To the white milky suspension obtained was added
196 mg (210 mL, 1.5 mmole) of Me3SiNCS. The mixture
became quickly yellow, it was kept at room temperature for
1 h and then refluxed for 3 h. The yellow suspension was
evaporated under vacuum and the solid extracted with CH2Cl2(2 9 30 mL). Evaporation of the solvent afforded 220 mg of
pure product (94%). IR (KBr): m (Mo=O) 937(s), 907(s), m(Mo–O–Mo), 776(s). 1H-NMR identical with an authentic
sample [14]. 1H-NMR (CDCl3) d 1.36(s); 1.41(s); 1.43(s);
1.48(s); 7.45 [d, J(H–H) = 6 Hz], 7.57 [d, d, J(H–H) =
6 Hz, 1.6 Hz], 7.69 [t, J(H–H) = 6 Hz]; 7.98 [d, J(H–H) =
6 Hz]; 8.14 [d, J(H–H) = 1.6 Hz], 8.24 [d, J(H–H) = 6 Hz];
8.51 [d, J(H–H) = 6 Hz]; 8.78 [d, J(H–H) = 6 Hz]; 8.89
[d, J(H–H) = 6 Hz]; 9.36 [d, J(H–H) = 6 Hz], 9.406 [d,
J(H–H) = 6 Hz].
W(O)2(t-butyl-bipy)(NCS)2 � 11
To a 20 mL CH3CN solution containing 134 mg
(0.5 mmole) of 4,40-di-t-butyl-2,20-bipyridine was added
147 mg (0.5 mmole) of anhydrous Na2WO4. The suspen-
sion was stirred 30 min at room temperature and to it was
added 263 mg (282 lL, 2 mmole) of Me3SiNCS. The
mixture was refluxed for 4 h and the resulting suspension
evaporated to dryness. Extraction of the resulting solid with
CH2Cl2 (2 9 30 mL) followed by evaporation gave a
white solid. Yield 0.26 g, 87%. IR (KBr) m(W=O): 952(s),
912(s), m(SCN): 2,016 cm-1. 1H-NMR (CDCl3) d 1.50(s),
7.80 [dd, J(H–H) = 6 Hz, 1.6 Hz]; 8.20 [d, J(H–
H) = 6 Hz], 9.12 [d, J(H–H) = 6 Hz]. Anal. Calcd. for
C20H24N4S2O2W: C, 40.01; H, 4.03; N, 9.32; S, 10.68;
Found: C, 40.50; H, 4.15; N, 9.05; S, 10.65.
ReO3[bis-(3,5-Me2-pyrazolylmethane)]Cl � 12
A 40 mL CH3CN suspension containing 289 mg
(1 mmole) of KReO4 and 204 mg (1 mmole) of bis-(3,5-
dimethyl-pyrazol-1-yl)methane was stirred at room tem-
perature for 24 h. To the milky mixture obtained was added
217 mg (186 lL, 2 mmole) of trimethylchlorosilane and
refluxed for an additional 24 h. The resulting white sus-
pension was filtered. Evaporation of the filtrate under
vacuum yielded a low melting solid free of any unreacted
944 Transition Met Chem (2008) 33:941–951
123

ligand. Yield 0.37 g, 78%. IR (KBr) m(Re=O): 912 (s), 840
(m). 1H-NMR (CDCl3) d 6.39(s), 5.98(s), 2.47(s), 2.20(s).
Results and discussion
The one-pot procedure consists of reacting sodium
molybdate with the halosilane, in the appropriate stoichi-
ometry, in the presence of a bidentate ligand in acetonitrile.
Simple evaporation of the solvent at the end of the reaction
and separation from the sodium halide by-product, gives
the desired complex in high yield. The success of the
method is plausibly linked to the coordinating properties of
acetonitrile, since no other media allows the reaction to
work.
Siloxylation of Na2MoO4 with Ph3SiCl proceeds
in a manner analogous to the reported reaction
with K2WO4 [9]
With 4,40-di-t-butyl-2,20-bipyridine � 1
The acetonitrile solution containing bipyridine and triphe-
nylchlorosilane reacts readily with the molybdate salt in
suspension, to yield the colourless crystalline dioxo com-
plex upon straightforward work up. It shows two intense
Mo=O infrared bands at 956 and 905 cm-1 and one Si–O
stretch at 1,113 cm-1. The 1H-NMR spectrum is typical of
a species with a C2 symmetry with one single pattern for a
pyridyl ring (d 8.9, 7.6, 7.3) as well as a unique tert-butyl
resonance peak (d 1.34). As expected the complex exhibits
high stability towards moisture or air. Its structure was
confirmed by an X-ray spectroscopic analysis (Fig. 1).
With bis(3,5-dimethylpyrazol-1-yl)methane � 2
As in the case of the reaction with 4,40-t-butylbipyridine,
the reaction mixture in acetonitrile is a suspension and
yields the expected dioxo complex upon work up. The
yield is very much dependent on the dryness of the starting
molybdate salt. The infrared spectrum shows two intense
bands at 929 and 907 cm-1 for m Mo=O and one for m Si–O
at 1,118 cm-1. The 1H-NMR indicates again clearly a C2
symmetry with only one pyrazolyl pattern. Following an
identical procedure with both ligands is a good indication
of the general character of this method.
The simple one-pot procedure used for the silyloxylation
reaction with Na2MoO4 brought us to consider its exten-
sion to chlorination by the use of trimethylchlorosilane.
This would render easily accessible the synthesis of dioxo-
dichloro complexes and add a real versatile character to
this synthetic method. We tried, at first, the reaction with
molybdate ion in the presence of four different bidendate
ligands.
Chlorination of Na2MoO4 with Me3SiCl
With 4,40-di-t-butyl-2,20-bipyridine � 3
The procedure followed was analogous to the silylation
reaction with triphenylchlorosilane, except that four
equivalents of Me3SiCl were used instead of two. The
Fig. 1 Mo(O)2(4,40-t-butyl-
2,20-bipy)(OSiPh3)2. 1 The
displacement parameters are
drawn at 30% probability
Transition Met Chem (2008) 33:941–951 945
123

heterogeneous reaction mixture in acetonitrile after reflux
and work up yielded a colourless crystalline solid. It
showed in infrared spectroscopy two strong absorption
bands at 908 and 939 cm-1 attributed to m Mo=O, as well
as those corresponding to the bipyridyl moiety. No
absorption corresponding to m Si–O were observed. The1H-NMR spectrum displays one single set of signals for the
bipyridyl ligand at d 9.47, 8.16 and 7.70 well in accord
with a C2 symmetry. All these values are identical with
those obtained for the same dichloro-dioxoMo(VI) com-
plex prepared by another synthetic route [13]. Its X-ray
structure has been reported previously [13].
With 4,40-dimethoxy-2,20-bipyridine [15] � 4
The reaction was run with four equivalents of Me3SiCl
following an analogous procedure as for the 4,40-t-butyl-
2,20-bipyridine ligand. The white solid exhibited in IR
spectroscopy two absorption bands characteristic of mMo=O at 937 and 906 cm-1. The 1H-NMR in CD2Cl2,
showed a typical set of signals for a bipyridyl entity in
accord with a C2 symmetry at d 9.32, 7.67 and 7.20 for the
aromatic hydrogen and at d 4.05 for the methoxy group. It
should be noted that the synthesis of this same complex
was attempted with the multistep reported procedure [13],
yielding a mixture of the desired product contaminated
with a dimeric species, and thus, was not reported.
With 4,40-dichloro-2,20-bipyridine � 5
Under analogous reaction conditions and upon work up, a
white solid was obtained. The extraction with CH2Cl2 was
unsatisfactory, it was repeated with hot acetone which upon
evaporation gave a white solid. The infrared spectroscopy
(m Mo=O, 944,950 cm-1) as well as the 1H-NMR (d6
acetone) (d 9.50, 8.21, 7.77) were in accord with the pure
desired complex.
With bis(3,5-dimethylpyrazol-1-yl)methane � 6
As in the case of the 4,40-di-t-butyl-2,20-bipyridine ligand
the reaction proceeded as a heterogeneous suspension and
yielded upon work up a white crystalline product exhibit-
ing in infrared spectroscopy two sharp absorption bands at
943 and 910 cm-1 (m Mo=O). The 1H-NMR analysis
showed a pattern for the pyrazolyl ring corresponding to a
complex having a C2 symmetry at d 6.56, 6.01, 2.75 and
2.45. Interestingly, the downshift expected upon com-
plexation is less with this ligand than the downshift
observed for 4,40-di-t-butyl-2,20-bipyridine; this is in
agreement with the slight difference observed in infrared
spectroscopy. Its structure was confirmed by an X-ray
spectroscopic analysis (Fig. 2).
Bromination of Na2MoO4 with Me3SiBr
With bis(3,5-dimethylpyrazol-1-yl)methane � 7
The use of trimethylbromosilane as a brominating agent for
metal-oxo compounds is less common, it was of great
interest to compare this halogenation method and extend it
to the bromination of sodium molybdate. The reaction
proceeds as a heterogeneous mixture in a manner analo-
gous to the chlorination reaction and yields upon work up a
light green solid exhibiting in infrared spectroscopy two
strong absorption bands at 907 and 941 cm-1 attributed to
m Mo=O. The 1H-NMR analysis showed resonance peaks
for the pyrazolyl ligand at 6.53, 6.06, 2.73 and 2.43 ppm.
The IR and NMR values are quite close to the chloro
analogue showing very little difference when varying the
halogen. This was already observed with the bidentate
ligand 4,40-t-butyl-2,20-bipyridine [13]. Its structure was
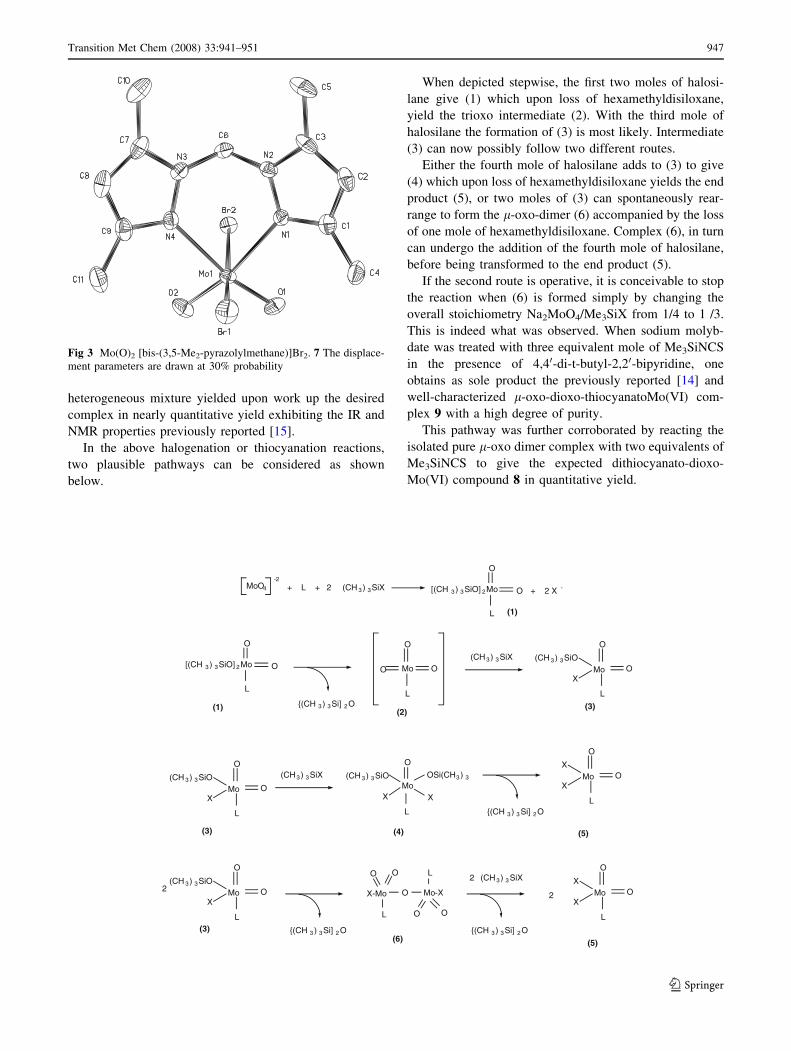
confirmed by an X-ray spectroscopic analysis (Fig. 3).
Thiocyanation of Na2MoO4 with Me3SiNCS
With 4,40-di-t-butyl-2,20-bipyridine � 8
Dithiocyanato-dioxo-4,40-di-t-butyl-2,20-bipyridine
Mo(VI) has been previously reported [15] and its synthesis
and reactivity as an oxo transfer agent largely studied. It
was of interest to check if it could be prepared by the
present more direct and easier method. Under analogous
reaction conditions as in the case of halogenation, the
Fig. 2 Mo(O)2 [bis(3,5-Me2-pyrazolylmethane)]Cl2. 6 The displace-
ment parameters are drawn at 30% probability
946 Transition Met Chem (2008) 33:941–951
123
heterogeneous mixture yielded upon work up the desired
complex in nearly quantitative yield exhibiting the IR and
NMR properties previously reported [15].
In the above halogenation or thiocyanation reactions,
two plausible pathways can be considered as shown
below.
When depicted stepwise, the first two moles of halosi-
lane give (1) which upon loss of hexamethyldisiloxane,
yield the trioxo intermediate (2). With the third mole of
halosilane the formation of (3) is most likely. Intermediate
(3) can now possibly follow two different routes.
Either the fourth mole of halosilane adds to (3) to give
(4) which upon loss of hexamethyldisiloxane yields the end
product (5), or two moles of (3) can spontaneously rear-
range to form the l-oxo-dimer (6) accompanied by the loss
of one mole of hexamethyldisiloxane. Complex (6), in turn
can undergo the addition of the fourth mole of halosilane,
before being transformed to the end product (5).
If the second route is operative, it is conceivable to stop
the reaction when (6) is formed simply by changing the
overall stoichiometry Na2MoO4/Me3SiX from 1/4 to 1 /3.
This is indeed what was observed. When sodium molyb-
date was treated with three equivalent mole of Me3SiNCS
in the presence of 4,40-di-t-butyl-2,20-bipyridine, one
obtains as sole product the previously reported [14] and
well-characterized l-oxo-dioxo-thiocyanatoMo(VI) com-
plex 9 with a high degree of purity.
This pathway was further corroborated by reacting the
isolated pure l-oxo dimer complex with two equivalents of
Me3SiNCS to give the expected dithiocyanato-dioxo-
Mo(VI) compound 8 in quantitative yield.
Fig 3 Mo(O)2 [bis-(3,5-Me2-pyrazolylmethane)]Br2. 7 The displace-
ment parameters are drawn at 30% probability
Mo
OO
O
Mo
MoMoMo
Mo
(6) (5)
(5)(4)
(3)
(3)
(3)(2)(1)
(1)
2
X
X
L
O
O
{(CH 3 ) 3Si] 2 O
2 (CH 3) 3 SiX
{(CH 3) 3Si] 2O
L
L
Mo-XO
O
X-Mo2
X
(CH 3) 3SiO
L
O
O
X
X
L
O
O
{(CH 3) 3 Si] 2O
OSi(CH3) 3(CH 3) 3SiO
L
XX
O
(CH 3) 3SiX
X
(CH 3) 3SiO
L
O
O
X
(CH 3) 3SiO
L
O
O
(CH 3) 3SiX
(CH 3) 3SiX
L
O
O
O Mo
{(CH 3) 3Si] 2O
O
O
L
[(CH 3) 3SiO] 2Mo
+ 2 X -O
O
L
[(CH 3) 3SiO] 2Mo+ L + 2 -2
MoO4
Transition Met Chem (2008) 33:941–951 947
123

Although this last reaction, as such, is somewhat limited
from a practical point of view, it suggests, nevertheless, a
possible new synthetic route in which two different halo-
gens or pseudo halogens could be bonded to the dioxo-
Mo(VI) centre. Indeed, reacting a given l-oxo-dioxo
molybdenum(VI) dimer bearing one type of halogen with
trimethylsilane of another halogen, should plausibly lead to
a ‘‘mixed’’ dihalo-dioxo-Mo(VI) entity.
When [Mo(O)2(t-butyl-bipy)(NCS)]2O was reacted with
two equivalents of trimethylchlosilane, the product
obtained upon workup, showed in infrared spectroscopy
the absence of absorption band at 760 cm-1 indicating that
no l-oxo dimeric species remained. The rest of the spec-
trum was in accord with a typical molybdenum(VI) dioxo
complex with a C2 symmetry. The 1H-NMR analysis, on
the other hand, showed a pattern corresponding to three
distinct bipyridyl moieties integrating to approximately
0.5/1/0.5. The chemical shifts of the two minor species
correspond, respectively, to values for the dichloro and
dithiocyanato dioxo complexes, whereas the third bipydyl
set resonates at values just intermediate as is shown in
Fig. 4. This observation might be interpreted in terms of
the ‘‘mixed’’ complex 10 being indeed formed as the major
product but, unfortunately, accompanied by a scrambling
phenomenon via a probable equilibrium step [2], leading to
a mixture of dioxo-dichloro 3 and dioxo-dithiocyanato 8
species.
Chlorination of Na2WO4 with Me3SiNCS
With 4,40-di-t-butyl-2,20-bipyridine � 11
This one-pot halogenation and thiocyanation of molyb-
date ion has been extended to tungstate ions. We
reported recently [16] the chlorination of sodium tung-
state in the presence of 4,40-di-t-butyl-2,20-bipyridine.
The thiocyanation of sodium tungstate was shown also to
be effective by this simpler route using trimethylthiocy-
anatosilane to give as for the molybdate ion the
corresponding dithiocyanatodioxotungstate(VI) recently
reported [16].
ButBut
NNOO
NNOO
OCH3CN
+ 6 (CH3) 3SiNCS2 Na2MoO42+ Mo-NCSSCN-Mo
NN
O
OO
NNOO
2CH3CN
2 (CH3)3SiNCS+ (SCN)2Mo=O
NN
O Mo-NCSSCN-Mo
NN
O
OO
NNO
Mo=OY
X2
NN
+ 2 (CH3)3Si-YCH3CN
X-Mo Mo-XO
O
NN
948 Transition Met Chem (2008) 33:941–951
123

Chlorination of KReO4 with Me3SiCl
With bis(3,5-dimethylpyrazol-1-yl)methane � 12
Chlorination of the rhenium-oxo function with trimethyl-
chlorosilane is a well known reaction [1, 17–20]. It is
mainly encountered as an intermediate step in the synthesis
of alkyl rhenium-oxo species or halo rhenium-oxo com-
plexes bearing bidentate N-ligands. The most common
rhenium(VII) oxide used is Re2O7, although more recently
HReO4 was reported [20] as the source of rhenium-oxo for
the synthesis of a chloro rhenium trioxo complex stabilized
by two molecules of THF. It was of interest to test the
present simple, one-pot synthetic procedure with potassium
perrhenate. The reaction was performed in the presence of
the N-bidentate ligand bis(3,5-dimethylpyrazol-1-yl)meth-
ane, and shown to be as effective as with molybdate or
tungstate salts.
X-ray analysis
Figures 1–3 show an ORTEP representation of the
molecular structure of compounds 1, 6 and 7 in the solid
Fig. 4 1H-NMR spectrum:
3 = Cl2Mo(O)2-t-butyl-bipy,
10 = Cl(NCS)Mo(O)2-t-butyl-
bipy, 8 = (NCS)2Mo(O)2-t-
butyl-bipy
N
N
N
N
L =
L-ReO 3Cl + Me3SiOSiMe3 + KCl
CH3CNKReO4 + 2 Me3SiCl + L
ButBut
NNNCS
OOCH3CN
+ 4 (CH3) 3SiNCSNa2WO4 + SCN-W
NN
Transition Met Chem (2008) 33:941–951 949
123

state as determined by single-crystal X-ray diffraction
studies. The crystallographic data for the complexes are
given in Table 1 and selected bond lengths and angles are
in Table 2.
The molecular structure of 1 consists of a discrete
molecule where the central molybdenum atom binds to two
O atoms of the siloxy ligands, two nitrogen atoms of the
chelate substituted bipy and two oxygen atoms of oxo-type
and exhibits a distorted octahedral geometry. Thus, the
bond angles around the metal vary from 68� for the donor
atoms of bipy, 107� for the oxo ligands and 79� for the
siloxy groups. Meanwhile, in the distorted octahedral
geometry for the complexes 6 and 7, the bond angles vary
from 79� for the nitrogen atoms of the pyrazolyl chelate,
104� for the oxo ligands and 96� for the halogen ligands.
The bond lengths and angles of the MoO2(4,40-tBu-2,20-bipy)(OSiPh3)2 moiety do not vary significantly and are
comparable to those found in other molybdenum com-
pounds with the same units and geometry [21].
The two pyrazolyl rings have good planarity, with
maximum deviations from the mean planes of less than
0.009 A. The metallacycle C–N1–N2–N3–N4–Mo usually
has boat conformation, for the complex 6 and 7 we found
the boat conformation are very asymmetric, with the
dihedral angle: N1–N2–N3–N4 and N2–C6–N3 of 71.4�and N1–N2–N3–N4 with N1–Mo1–N4 of 16.2� for the
complex 6. For the complex 7 we have dihedral angle of
14.9� and 73.1�, respectively. Both complexes have a
quasi-sofa conformation [22]. For these compounds in
general, the values of bond lengths and angles are within
the expected values [23, 24].
Acknowledgements This work was done under the auspices of the
project ECOS-Nord France-Venezuela and Fundayacucho. Financial
support is kindly acknowledged.
References
1. Hermann WA (1988) Angew Chem Int Ed Engl 27:1297
2. Parkin G, Bercaw JE (1989) J Am Chem Soc 111:391
3. Nugent WA, Mayer JM (1983) Metal-ligand multiple bonds.
Wiley-Interscience, New York
4. Arzoumanian H, Krentzien H, Corao C, Lopez R, Agrifoglio G
(1995) Polyhedron 14:2887
5. Lorber C, Donahue JP, Goddard CA, Nordlander E, Holm RH
(1998) J Am Chem Soc 120:8102
6. Teruel H, Gorrin YC, Falvello LR (2001) Inorg Chim Acta 316:1
7. Huang M, DeKock CW (1993) Inorg Chem 32:2287
8. Thapper A, Donahue JP, Musgrave KB, Willer MW, Nordlander
E, Hedman B, Hodgson KO, Holm RH (1999) Inorg Chem
38:4104
9. Miao M, Willer MW, Holm RH (2000) Inorg Chem 39:2843
10. Molecular Structure Corporation (1999) TEXSAN. Single crystal
structure analysis software, version 1.10. MSC, 9009 New Trails
Drive, The Woodlands, TX 77381-5209, USA
11. Rigaku/MSC, Inc. (2000) CRYSTALCLEAR, Software users
guide, version 1.3.6, The Woodlands, TX, USA
12. Bruker (1998) SHELXTL-NT, version 5.1 Bruker AXS Inc.,
Madison, WI, USA
13. Arzoumanian H, Bakhtchadjian R, Agrifoglio G, Atencio R,
Briceno A (2006) Trans Met Chem 31:681
14. Arzoumanian H, Bakhtchadjian R, Agrifoglio G, Krentzien H,
Daran JC (1999) Eur Inorg Chem 2255
15. Arzoumanian H, Maurino L, Agrifoglio G (1997) J Mol Catal A
Chem 117:471
16. Arzoumanian H, Agrifoglio G, Capparelli M, Atencio R, Briceno
A, Alvarez-Larena A (2006) Inorg Chim Acta 359:81
17. Herrmann WA, Thiel WR, Herdtweck E (1990) Chem Ber
123:271
Table 2 Selected bond lengths
(A´
) and angles (�)Compound 1 Compound 6 Compound 7
Mo1–O1 1.695 (4) Mo1–O2 1.683 (2) Mo1–O2 1.684 (3)
Mo1–O2 1.696 (4) Mo1–O1 1.684 (2) Mo1–O1 1.686 (3)
Mo1–O3 1.921 (4) Mo1–Cl2 2.351 (8) Mo1–Br2 2.5067 (7)
Mo1–O4 1.927 (4) Mo1–Cl1 2.3773 (7) Mo1–Br1 2.5399 (8)
Mo1–N2 2.345 (5) Mo1–N1 2.356 (2) Mo1–N1 2.363 (3)
Mo1–N1 2.348 (5) N1–C1 1.337 (3) Mo1–N4 2.355 (3)
Si1–O4 1.600 (4) N2–C6 1.442 (3) Mo1–N1 2.363 (3)
Si2–O3 1.604 (4)
O1–Mo1–O2 107.2 (2) O2–Mo1–O1 103.72 (11) O2–Mo1–O1 103.61 (16)
O1–Mo1–O3 97.3 (2) O2–Mo1–N1 168.14 (10) O1–Mo1–N4 167.83 (14)
O2–Mo1–O3 97.3 (2) N1–Mo1–N4 78.99 (7) N4–Mo1–N1 79.32 (11)
O3–Mo1–O4 154.28 (18) Cl2–Mo1–Cl1 162.07 (3) Br2–Mo1–Br1 162.73 (2)
O1–Mo1–N2 92.5 (2) N2–N1–Mo1 125.32 (15) N2–N1–Mo1 126.0 (2)
O2–Mo1–N2 160.3 (2) O2–Mo1–Cl1 94.19 (9) O2–Mo1–Br2 96.84 (10)
Si1–O4–Mo1 179.7 (3) C1–N1–N2 105.5 (2) O2–Mo1–N4 88.51 (13)
Si2–O3–Mo1 175.3 (3)
950 Transition Met Chem (2008) 33:941–951
123

18. Herrmann WA, Kuhn FE, Romao CC, Kleine M, Mink J (1994)
Chem Ber 127:47
19. Kuhn FE, Haider JJ, Herdtweck E, Herrmann WA, Lopes AD,
Pillinger M, Romao CC (1998) Inorg Chim Acta 279:44
20. Noh W, Girolami GS (2007) J Chem Soc Dalton Trans 674
21. Thapper A, Donahue JP, Musgrave KB, Miller MW, Nordlander E,
Hedman B, Hodgson KO, Holm RH (1999) Inorg Chem 38:4104
22. Agrifoglio G, Capparelli M (2005) J Chem Crystallogr 35:95
23. Pettinari C, Pettinari R (2005) Coord Chem Rev 249:663
24. Santos AM, Kuhn FE, Bruns-Jensen K, Lucas I, Romao CC,
Herdtweck E (2001) Dalton Trans 1332
Transition Met Chem (2008) 33:941–951 951
123
![Thermal, oxidative and radiation stability of polyimides III. Polyimides based on N-[3-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)phenyl]acetamide and different diamines](https://static.fdokumen.com/doc/165x107/63448d5903a48733920af0ae/thermal-oxidative-and-radiation-stability-of-polyimides-iii-polyimides-based-on.jpg)


![Aqua(4,4'-bipyridine-[kappa]N)bis(1,4-dioxo-1 ... - ScienceOpen](https://static.fdokumen.com/doc/165x107/63262349e491bcb36c0aa51f/aqua44-bipyridine-kappanbis14-dioxo-1-scienceopen.jpg)













![N ′-[1-(2,4-Dioxo-3,4-dihydro-2 H -1-benzopyran-3-ylidene)ethyl]thiophene-2-carbohydrazide](https://static.fdokumen.com/doc/165x107/63252fe2c9c7f5721c01f37f/n-1-24-dioxo-34-dihydro-2-h-1-benzopyran-3-ylideneethylthiophene-2-carbohydrazide.jpg)


