
A Mutational Analysis of Killer Toxin Resistance in ... - NCBI
13
Copyright Q 1993 by the Genetics Society of America A Mutational Analysis of Killer Toxin Resistance in Saccharomyces cereuisiae Identifies New Genes Involved in Cell Wall (1 + G)=@-Glucan Synthesis Jeffrey L. Brown, Zuzana Kossaczka,' Bo Jiang and Howard Bussey Biology Department, McGill University, Montreal, Quebec, Canada H3A IBI Manuscript received November 2, 1992 Accepted for publication December 14, 1992 ABSTRACT Recessive mutations leading to killer resistance identify the KRE9, KRElO and KREll genes. Mutations in both the KRE9 and KREl I genes lead to reduced levels of (1 + 6)-@-glucan in the yeast cell wall. The KREll gene encodes a putative 63-kD cytoplasmic protein,and disruption of the KREl I locus leads to a 50% reduced level of cell wall (1 + 6)-glucan. Structural analysis of the (1 + 6)-@-glucan remaining in a krell mutant indicates a polymer smaller in size than wild type, but containing a similar proportion of (1 -+ 6)- and (1 -+ 3)-linkages. Genetic interactions among cells harboring mutations at the KREII, KRE6 and KREl loci indicate lethality of krell kre6 double mutants and that krel I is epistatic to krel, with both gene products required to produce the mature glucan polymer at wild-type levels. Analysis of these KRE genes should extend knowledge of the B- glucan biosynthetic pathway, and of cell wall synthesis in yeast. T HE cellwall constitutes a massive extracellular organelle in fungi and plants. A major class of cell wall extracellular matrix polysaccharides are the large glucose-derived polymers, termed P-glucans (FLEET and PHAFF 198 1; KATO 1981 ; WESSELS and SIETSMA 1981). The yeast cell wall glucan contains a major (1 + 3)-P-glucan, and a minor (1 + 6)-P-linked polymer (MANNERS, MASSON and PATTERSON 1973a,b). The (1 + 3)-P-~-glucan contains up to 1500 glucose residues and has a small number of (1 + 6)- branch points and (1 + 3)-linked branches. The (1 --j 6)-P-~-glucan is smaller with 150-200 glucose resi- dues and is highly branched with (1 + 3)-branch points and (1 + 6)-linked side chains. These polymers are found extensively cross-linked to chitin and man- noprotein, and are alkali insoluble (VAN RINSUM, KLIS and VAN DEN ENDE 1991). A 0-glucan fraction is, however, alkali-soluble, and has classically been viewed as a distinct polymer class. Genetic results have simplified this view, as in a mutant defective in chitin synthase 3, all yeast P-glucan is alkali-soluble (RON- CERO et al. 1988).Chitin synthase 3appearstobe responsible for synthesis of cell wall chitin not found in the primary septum (SHAW et al. 1991). These findings suggest that the alkali-insoluble and alkali- soluble fractions differ only in the extent to which they are cross-linked to chitin in the cell wall. Understanding the synthesis of P-glucan polymers and their orderly assembly into the cell wall during growth, addresses some intrinsically interesting cell ' Present address: Institute of Chemistry, Slovak Academy of Sciences, Dubravska Cesta 9, 842 38 Bratislava, Slovakia. Genetics 133: 837-849 (April, 1993) biological questions. Glucan synthesis also has broad practical applications, ranging from polymer biotech- nology to the searchfor specific antifungalagents. The membrane-associated vectorial synthesis of P- glucans has been difficult to analyze biochemically. A genetic analysis to identify components involved in P- glucan biosynthesis circumvents this biochemical com- plexity, and once genes have been found, analysis of their function may be possible. Important insights have come from a genetic analysis of components involved in making a variety of cell wall polysaccha- rides, including cellulose (WONC et al. 1990) and chitin (BULAWA et al. 1986; SHAW et al. 1991; VALDIVISEO et al. 1991; BULAWA 1992). A number of studies have examined mutants in both 0-glucan synthesis and hydrolysis in fungi, but despitethe tractability of such systems forgenetic analysis, the P-glucan area remains relatively unex- plored. In S. cerevisiae, mutantsaffecting sensitivity (SHIOTA et al. 1985) or resistance, (JAMAS, RHA and SINSKEY 1986) to (1 + 3)-P-glucanases have been re- ported. Yeast genes whose products have P-glucanase activ- ity have been defined and examined (KLEBL and TAN- NER 1989; VASQUEZ DE ALDANA et al. 1991), but their biological role remains unclear. While these genes may be involved in P-glucan degradation, recentwork in Candida albicans suggests that they could also func- tion biosynthetically as transglycosylases (HARTLAND, EMERSON and SULLIVAN 1991). In Schizosaccharomyces pombe, two genes have been identified that affect P- glucan synthesis (RIBAS et al. 1991). Mutations in these
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of A Mutational Analysis of Killer Toxin Resistance in ... - NCBI

Copyright Q 1993 by the Genetics Society of America
A Mutational Analysis of Killer Toxin Resistance in Saccharomyces cereuisiae Identifies New Genes Involved in Cell Wall
(1 + G)=@-Glucan Synthesis
Jeffrey L. Brown, Zuzana Kossaczka,' Bo Jiang and Howard Bussey
Biology Department, McGill University, Montreal, Quebec, Canada H3A IBI Manuscript received November 2, 1992
Accepted for publication December 14, 1992
ABSTRACT Recessive mutations leading to killer resistance identify the KRE9, KRElO and K R E l l genes.
Mutations in both the KRE9 and K R E l I genes lead to reduced levels of ( 1 + 6)-@-glucan in the yeast cell wall. The K R E l l gene encodes a putative 63-kD cytoplasmic protein, and disruption of the K R E l I locus leads to a 50% reduced level of cell wall (1 + 6)-glucan. Structural analysis of the (1 + 6)-@-glucan remaining in a krell mutant indicates a polymer smaller in size than wild type, but
containing a similar proportion of (1 -+ 6)- and (1 -+ 3)-linkages. Genetic interactions among cells harboring mutations at the KREII , KRE6 and K R E l loci indicate lethality of krell kre6 double mutants and that krel I is epistatic to krel, with both gene products required to produce the mature glucan polymer at wild-type levels. Analysis of these KRE genes should extend knowledge of the B- glucan biosynthetic pathway, and of cell wall synthesis in yeast.
T HE cell wall constitutes a massive extracellular organelle in fungi and plants. A major class of
cell wall extracellular matrix polysaccharides are the large glucose-derived polymers, termed P-glucans (FLEET and PHAFF 198 1; KATO 1981 ; WESSELS and SIETSMA 1981). The yeast cell wall glucan contains a major (1 + 3)-P-glucan, and a minor (1 + 6)-P-linked polymer (MANNERS, MASSON and PATTERSON 1973a,b). The (1 + 3)-P-~-glucan contains up to 1500 glucose residues and has a small number of ( 1 + 6)- branch points and (1 + 3)-linked branches. The (1 --j 6)-P-~-glucan is smaller with 150-200 glucose resi-
dues and is highly branched with (1 + 3)-branch points and (1 + 6)-linked side chains. These polymers are found extensively cross-linked to chitin and man- noprotein, and are alkali insoluble (VAN RINSUM, KLIS and VAN DEN ENDE 1991). A 0-glucan fraction is, however, alkali-soluble, and has classically been viewed as a distinct polymer class. Genetic results have simplified this view, as in a mutant defective in chitin synthase 3, all yeast P-glucan is alkali-soluble (RON- CERO et al. 1988). Chitin synthase 3 appears to be responsible for synthesis of cell wall chitin not found in the primary septum (SHAW et al. 1991). These findings suggest that the alkali-insoluble and alkali- soluble fractions differ only in the extent to which they are cross-linked to chitin in the cell wall.
Understanding the synthesis of P-glucan polymers and their orderly assembly into the cell wall during growth, addresses some intrinsically interesting cell
' Present address: Institute of Chemistry, Slovak Academy of Sciences, Dubravska Cesta 9, 842 38 Bratislava, Slovakia.
Genetics 133: 837-849 (April, 1993)
biological questions. Glucan synthesis also has broad practical applications, ranging from polymer biotech- nology to the search for specific antifungal agents. The membrane-associated vectorial synthesis of P- glucans has been difficult to analyze biochemically. A genetic analysis to identify components involved in P- glucan biosynthesis circumvents this biochemical com- plexity, and once genes have been found, analysis of their function may be possible. Important insights have come from a genetic analysis of components involved in making a variety of cell wall polysaccha- rides, including cellulose (WONC et al. 1990) and chitin (BULAWA et al. 1986; SHAW et al. 1991; VALDIVISEO et al. 1991; BULAWA 1992).
A number of studies have examined mutants in both 0-glucan synthesis and hydrolysis in fungi, but despite the tractability of such systems for genetic analysis, the P-glucan area remains relatively unex- plored. In S. cerevisiae, mutants affecting sensitivity (SHIOTA et al. 1985) or resistance, (JAMAS, RHA and SINSKEY 1986) to (1 + 3)-P-glucanases have been re- ported.
Yeast genes whose products have P-glucanase activ- ity have been defined and examined (KLEBL and TAN- NER 1989; VASQUEZ DE ALDANA et al. 1991), but their biological role remains unclear. While these genes may be involved in P-glucan degradation, recent work in Candida albicans suggests that they could also func- tion biosynthetically as transglycosylases (HARTLAND,
EMERSON and SULLIVAN 199 1). In Schizosaccharomyces pombe, two genes have been identified that affect P- glucan synthesis (RIBAS et al. 1991). Mutations in these

838 J. L. Brown et al.
genes conferred temperature-sensitive lysis pheno- types that could be prevented in the presence of an osmotic stabilizer. Mutations in the C W G l gene gave conditional reduction in cell wall (1 + 3)-P-glucan in vivo, and in a membrane-associated (1 + 3)-P-glucan synthase activity in vitro. Defects in a second gene, CWG2, conferred a conditional pleiotropic phenotype with a reduced level of both a- and P-glucan in vivo, and a reduced activity of a soluble possible regulatory component in the in vitro P-glucan synthase assay.
A number of genes involved in P-glucan synthesis have also been identified through resistance to the S. cerevisiae K1 killer toxin, a protein that kills sensitive yeasts by forming lethal cation channels in the plasma membrane (MARTINAC et al. 1990; BUSSEY 1991). T h e toxin binds initially to yeast cells through a cell wall P-glucan receptor (HUTCHINS and BUSSEY 1983). Se- lection for mutants resistant to this toxin, has defined a series of genes required for P-glucan synthesis. T h e KREI and K R E 5 genes encode secretory pathway proteins necessary for (1 + 6)-glucan synthesis (BOONE et al. 1990; MEADEN et al. 1990). K R E 6 en- codes a membrane protein, possibly a glucan synthase, necessary to make normal levels of glucan (ROEMER and BUSSEY 1991). K R E 2 is a gene whose product is needed for protein glycosylation, and has recently been shown to encode a mannosyl transferase (HILL et al. 1992; HAUSLER and ROBBINS 1992). Mutations in K R E 2 may thus lead to defects in the cross-linking of glucans with mannoprotein in the cell wall.
Previous selections for killer resistant mutants were not exhaustive, (AL-AIDROOS and BUSSEY 1978; BOONE et al. 1990), and the present study aimed to more fully exploit the system.
MATERIALS AND METHODS
Yeast strains and culture conditions: Yeast strains used for this study are displayed in Table 1. Killer resistant mutants were generated in the SEY6210 background from S. D. EMIR, California Institute of Technology, as described below.
Yeast cells were grown under standard conditions, using media (YEPD, YNB and HALVORSON'S) as previously de- scribed (BUSSEY et al. 1982). Yeast genetics used standard procedures (SHERMAN, FINK and HICKS 1982). Routine transformations were by the lithium acetate procedure of ITO et al. (1983). Sorbitol-containing regeneration medium (SM2), was used for spheroplast transformations, and con- sisted of Halvorson's medium plus 2% agar, 1.2 M sorbitol and appropriate nutrilites.
Toxin preparation and seeded plate tests: K 1 killer toxin was prepared using strain T158C/S14a as previously de- scribed (BUSSEY et al. 1982), with the final product ap- proaching 1000-fold concentration. Yeast strains were as- sayed for resistance to killer toxin by resuspending approx- imately 1 X lo6 cells in 100 pI of sterile water. The slurry was used to inoculate 10 ml of 1 X Halvorson's buffered YEPD or minimal medium (pH 4.7, 1% agar at 45") that was quickly poured into a Petri dish and allowed to cool. Concentrated toxin (20 11) was spotted onto the surface of
the solidified medium, and placed at 18 O overnight. Follow- ing incubation at 30" for 24 h, either a zone of killed cells would appear when a strain was sensitive to the toxin or a confluent lawn of killer-resistant cells would emerge.
Selection for killer-resistant mutants: Some 1-2 x 10' cells from freshly grown colonies of strain SEY6210 were taken with a toothpick, mixed with 100 pl of YEPD pH 4.7 and 25 pl of sterile killer toxin and spread onto a YEPD 1.2 M sorbitol agar-containing Petri plate, and incubated at 18 O .
Thirty plates were prepared in this way, with a different colony being used for each plate to obtain independent spontaneous mutations. After 4-21 days, from 5-50 colo- nies of varying size grew up on each plate as survivors of the toxin treatment. From each plate up to four colonies of different size were taken for further analysis. These resistant mutants were crossed with strains bearing mutant alleles of the previously identified genes KREI, 2, 5, 6, and diploids tested for sensitivity to killer toxin. Forty-two mutants fell into one of the known complementation groups; these com- prised krel , 10; kre2, 15; kre5, 5; and kre6, 12. Of the remaining 35 mutants, 25 had a strong killer resistance phenotype. These mutants, presumed to identify new com- plementation groups, were crossed with SEY62 1 1, and all diploids were found to be toxin sensitive. Diploids were sporulated, and from 8 to 16 tetrads dissected and analyzed for killer resistance to establish if the resistance segregated as a single gene. Spore progeny harboring single gene mutations were subjected to complementation analysis among themselves. Pairs of mutations that gave resistant diploids were judged to be in the same complementation group.
Plasmids and recombinant DNA techniques: Yeast ge- nomic libraries constructed in either YCp50 (ROSE et al. 1987) or pRS3 16 vectors (from C. BOONE, University of Oregon, Eugene, Oregon), were used to identify isolate DNA fragments able to complement the kre9, and krell mutations. Both banks are based on single copy centromeric shuttle vectors, and contain the URA3 and Apr selectable markers.
Plasmids were isolated from yeast for transformation into Escherichia coli strain MC 106 1 according to the procedures of HOFFMAN and WINSTON (1987). Plasmids were purified from E. coli as described by SAMBROOK et al. (1989). Restric- tion endonucleases, Klenow and T4 DNA polymerases, and T4 DNA ligase were purchased from either Bethesda Re- search Laboratories, Inc. (Gaithersburg, MD), Pharmacia LKB Biotechnology (Piscataway, NJ), or New England Bio- labs (Beverly, MA), and were used according to the manu- facturers' instructions.
Subcloning and disruption of KREl I : The KREl I gene was originally isolated from a YCp50-based genomic library on a 12.5-kb insert. A restriction map of this region was generated and several subclonings into vector pRS316 lo- calized the complementing activity to a 4.2-kb HindIII- EcoRI fragment (MP53). Further subclonings of this frag- ment were incapable of complementing the k r e l l - 1 muta- tion. To create an insertional disruption of the KREll gene, the 4.2-kb HindIII-EcoRI insert from plasmid MP53 was cloned into YCp50, linearized at the unique XbaI site, and treated with the Klenow DNA polymerase to render the termini blunt. The complete HIS3 gene was excised from a PBSK:HIS3 plasmid as a 1.8-kb fragment by digestion with BamHI, also filled in with Klenow, and blunt ligated into the XbaI site to create MP195. A linear 4.8-kb PuuII-EcoRV fragment containing the HIS3 gene at this XbaI site was excised from MP195 and used to disrupt the KREl I locus (see below). Two deletion disruptions of the KREll locus were also performed, using the TRPl and URA3 auxotrophic
Yeast KRE Genes
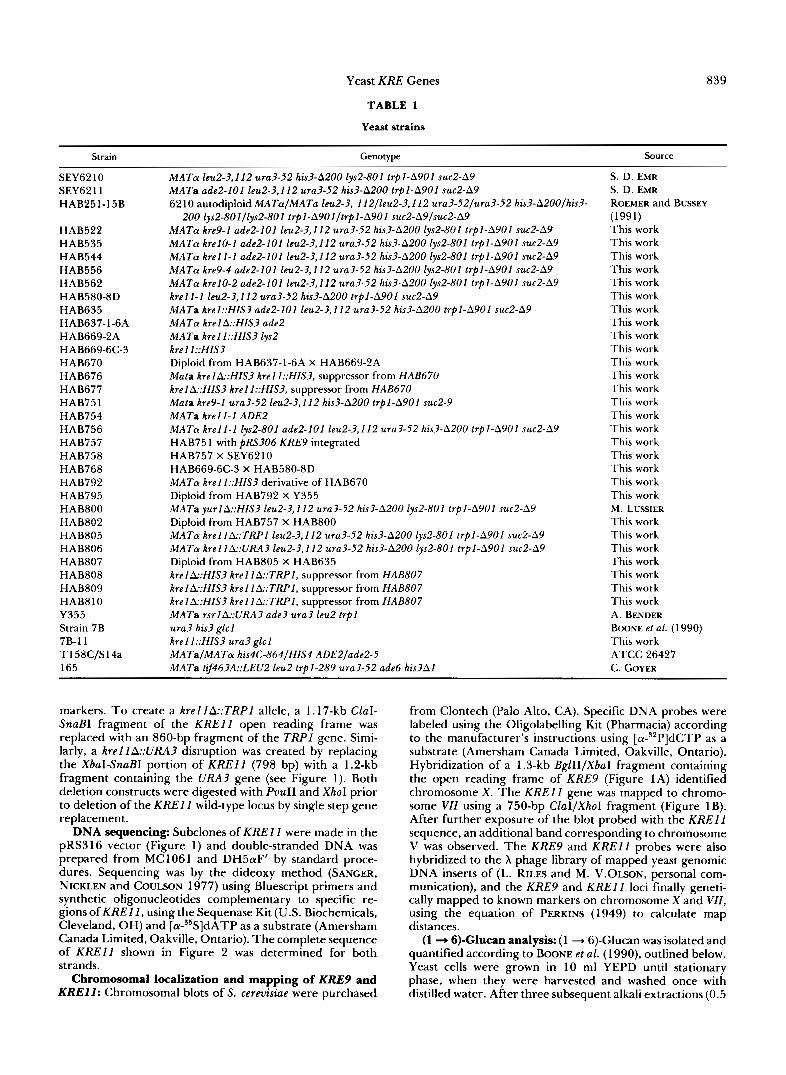
TABLE 1
Yeast strains
839
Strain
SEY62 10 SEY62 1 1 HAB251-15B
HAB522 HAB535 HAB544 HAB556 HAB562
HAB635 HAB637-1-6A
HAB580-8D
HAB669-2A HAB669-6C-3 HAB670 HAB676 HAB677 HAB751 HAB754 HAB756 HAB757 HAB758 HAB768 HAB792 HAB795 HAB800 HAB802 HAB805 HAB806 HAB807 HAB808 HAB809 HAB8 10 Y355 Strain 7B 7B- 1 1 T 158C/S 14a 165
Genotype
MATa leu2-3,112 ura3-52 his3-A200 lys2-801 trpl-A9Ol suc2-A9 MATa ade2-I01 leu2-3,112 ura3-52 his3-A200 trpl-A901 suc2-A9 6 2 10 autodiploid MATaIMATa leu2-3, 112/leu2-3,112 ura3-52/ura3-52 his3-A200/his3-
200 lys2-801/lys2-801 trpI-A901/trpI-A901 suc2-A9/suc2-A9 MATa kre9-1 ade2-I01 leu2-3,112 ura3-52 his3-A200 lys2-801 trpl-A901 suc2-A9 MATa krel0-1 ade2-I01 leu2-3,I 12 ura3-52 his3-A200 lys2-801 trpI-A901 suc2-A9 MATa krell-1 ade2-I01 leu2-3,I 12 ura3-52 his3-A200 lys2-801 trpl-A90l suc2-A9 MATa kre9-4 ade2-I01 leu2-3,112 ura3-52 his3-A200 lys2-801 trpl-A901 suc2-A9 MATa krel0-2 ade2-101 leu2-3,112 ura3-52 his3-A200 lys2-801 trpl-A9Ol suc2-A9 krell-1 leu2-3,112 ura3-52 his3-A200 trpl-ASOl suc2-A9 MATa krel::HIS3 ade2-I01 leu2-3,112 ura3-52 his3-A200 trpl-A901 suc2-A9 MAT@ krel A::HIS3 ade2 MATa krelI::HIS3 lys2 krel I::HIS3 Diploid from HAB637-1-6A X HAB669-2A Mala krelA::HIS3 krel I::HIS3, suppressor from HAB670 krelk:HIS3 krel I::HIS3, suppressor from HAB670 Mata kre9-1 ura3-52 leu2-3,112 his3-A200 trpI-A901 suc2-9 MATa krell-I ADE2 MATa krell-1 lys2-801 ade2-I01 leu2-3,I 12 ura3-52 his?-A200 trpl-A90l suc2-A9 HAB75 1 with pRS306 KRE9 integrated HAB757 X SEY6210 HAB669-6C-3 X HAB580-8D MATa krell::HIS3 derivative of HAB670 Diploid from HAB792 X Y355 MATa yurlA::HIS3 leu2-3,112 ura3-52 his3-A200 lys2-801 trpl-A901 suc2-A9 Diploid from HAB757 X HAB800 MATa krel1k:TRPI leu2-3,112 ura3-52 his3-A200 lys2-801 trpl-A901 suc2-A9 MATa kreIlA::URA3 leu2-3,112 ura3-52 his3-A200 lys2-801 trpl-A901 suc2-A9 Diploid from HAB805 X HAB635 krelA::HIS3 krell A::TRPl, suppressor from HAB807 krelA::HIS3 krel I A::TRPI, suppressor from HAB807 krelA::HIS3 krel I A::TRPI, suppressor from HAB807 MATa rsrlA::URA3 ade3 ura3 leu2 trpl ura3 his3 glcl krel I::HIS3 ura3 glcl MATaIMATa his4C-864/HIS4 ADE2/ade2-5 MATa tif463A::LEU2 leu2 trpl-289 ura3-52 ade6 his3A1
Source
S. D. EMR S. D. EMR ROEMER and BUSSEY
This work This work This work This work This work This work This work This work This work This work This work This work This work This work This work This work This work This work This work This work This work M . LUSSIER This work This work This work This work This work This work This work A. BENDER BOONE et al. ( 1 990) This work ATCC 26427 C. COYER
(1991)
markers. T o create a kre1lA::TRPl allele, a 1.17-kb ClaI- SnaBI fragment of the KREll open reading frame was replaced with an 860-bp fragment of the TRPl gene. Simi- larly, a krel lA::URA3 disruption was created by replacing the XbaI-SnaBI portion of KREll (798 bp) with a 1.2-kb fragment containing the URA3 gene (see Figure 1). Both deletion constructs were digested with PuuII and Xhol prior to deletion of the KREll wild-type locus by single step gene replacement.
DNA sequencing: Subclones of KREll were made in the pRS316 vector (Figure 1) and double-stranded DNA was prepared from MC1061 and DH5aF’ by standard proce- dures. Sequencing was by the dideoxy method (SANGER, NICKLEN and COULSON 1977) using Bluescript primers and synthetic oligonucleotides complementary to specific re- gions of K R E l l , using the Sequenase Kit (U.S. Biochemicals, Cleveland, OH) and [a-35S]dATP as a substrate (Amersham Canada Limited, Oakville, Ontario). The complete sequence of KREll shown in Figure 2 was determined for both strands.
Chromosomal localization and mapping of KRE9 and K R E l l : Chromosomal blots of S. cerevisiae were purchased
from Clontech (Palo Alto, CA). Specific DNA probes were labeled using the Oligolabelling Kit (Pharmacia) according to the manufacturer’s instructions using [ C Y - ~ ~ P ] ~ C T P as a substrate (Amersham Canada Limited, Oakville, Ontario). Hybridization of a 1.3-kb BglII/XbaI fragment containing the open reading frame of KRE9 (Figure 1A) identified chromosome X. The KREl l gene was mapped to chromo- some VII using a 750-bp ClaI/XhoI fragment (Figure 1B). After further exposure of the blot probed with the KREll sequence, an additional band corresponding to chromosome V was observed. The KRE9 and KREll probes were also hybridized to the X phage library of mapped yeast genomic DNA inserts of (L. RILES and M. V.OLSON, personal com- munication), and the KRE9 and KREll loci finally geneti- cally mapped to known markers on chromosome X and VU, using the equation of PERKINS (1949) to calculate map distances.
(1 + B)-Glucan analysis: (1 ”* 6)-Glucan was isolated and quantified according to BOONE et al. (1 990), outlined below. Yeast cells were grown in 10 ml YEPD until stationary phase, when they were harvested and washed once with distilled water. After three subsequent alkali extractions (0.5

840 J. L. Brown et al.
ml of 3% NaOH at 75" for 1 hr), the residual cell wall glucans were neutralized by washing once with 1 ml of 100 r n M Tris-HCI, pH 7.5, and twice with 1 ml of 10 mM Tris- HCI, pH 7.5. The wall glucans were then resuspended in 1 rnl of 10 mM Tris-HCI, pH 7.5 and digested with 1 mg of the (1 + 3)-&glucanase, Zyrnolyase lOOT (ICN Biochemi- cals, Cleveland, OH) at 37" for 16 hr. After removing the residual insoluble material by centrifugation, the superna- tant was dialyzed against distilled water for 16 hr using Spectra/por tubing, with a 6,000-8,000-D pore size (Spec- trum Medical Industries, Los Angeles, CA). The carbohy- drate present prior to dialysis represents the total alkali- insoluble glucan (1 + 3)-@-linked plus (1 - 6)-linked, while the post-dialysis fraction represents (1 + 6)-&glucan. Car- bohydrate in each fraction was measured as hexose by the borosulfuric acid method (BADIN, JACKSON and SCHUBERT 1953).
For large scale isolation for structural analysis, the (1 + 6)-B-glucan was purified from cultures of 2 or 5 liters of
wild-type cells or yeast strains harboring disruptions at either the krelA or krellA loci, using the method of BOONE et al.
Gel filtration chromatography of glucans: A Sepharose CL-GB (Pharmacia) column with dimensions 109 X 1 cm was used to estimate the molecular weight distribution of glucan samples (-1 mg) by gel filtration chromatography. The eluant was 0.1 M NaOH, and 0.33-ml fractions were collected at a flow rate of 10 ml/hr. Molecular weights were estimated by comparison with dextran standards (Pharma- cia), with dextran blue used to determine the column void volume. The hexose content of each fraction was estimated by the borosulfuric acid method of BADIN, JACKSON and SCHUBERT (1953). "C NMR Glucan samples (50-100 mg) were dissolved
in -5 ml of D20, placed in 5-mm diameter tubes, and proton decoupled "C NMR spectra obtained using a Bruker WH 400 spectrometer (Bruker Instruments, Billerica, MA) operated in the Fourier-transform mode, at 100.61 5 MHz. Each spectrum represents approximately 10,000 scans with a sweep width of 12,500 Hz, and an acquisition time of 0.655 sec. The probe temperature was held at 18", and the reference peak was external dioxane at 67.4 ppm. ''C NMR spectra were generated for glucan samples isolated from two independent yeast strains, SEY6210 and 7B, differing slightly with respect to genetic background. Both wild-type and kreA disrupted representatives from each strain pro- duced an equivalent set of spectra.
Additional cell wall analyses: Alkali-soluble glucan plus mannan cell wall fractions of wild-type or mutant strains were estimated based on the procedure described by RON- CERO et al. (1988). Briefly, cells were extracted twice with 3% NaOH for 90 min at 80", and the alkali-soluble glucan plus mannan co-precipitated from the alkali extract with absolute ethanol. This material was subsequently digested overnight with 1 mg/ml Zymolyase lOOT in 100 mM Tris- HCI (pH 7.5) at 37", and one-half the volume dialyzed against distilled water using 6,000-8,000-D Spectra/por tubing. The total alkali-soluble glucan plus mannan fractions were estimated prior to dialysis as hexose by the borosulfuric acid method. The carbohydrate material remaining after dialysis was taken to represent a fraction of the cell wall mannan, and alkali-soluble (1 + 6)-glucan. The (1 + 3)$- glucan synthase activity was assayed from cell free extracts of wild-type or isogenic strains harboring a krelA::HIS3 or krel I::HIS3 disruption, according to the procedure of CABIB and KANC (1987). Cell wall chitin was observed by fluores- cence with Calcofluor White, and glycogen levels in cell colonies were qualitatively assessed using iodine vapors.
(1 990).
RESULTS
Isolation of killer resistant mutants in SEY6210: To search for mutations in possible new genes re- quired for killer sensitivity, we selected for toxin resistant mutants, including those that grew slowly; in addition we selected in the presence of 1.2 M sorbitol, so that osmotically fragile mutants might survive. Sur- vivors of killer toxin treatment from a sensitive pop- ulation of 3-6 X 1 Os yeast, were selected as described in MATERIALS AND METHODS. Approximately 100 sur- viving colonies were taken and tested for toxin resist- ance on YEPD sorbitol seeded plates; 77 of these had a resistant phenotype. All grew on YEPD in the ab- sence of sorbitol, and thus none appeared to be os- motically fragile.
Assignment of three new complementation groups: T h e resistant mutants were tested for com- plementation of mutant alleles of previously identified KRE genes (see MATERIALS AND METHODS). Twenty- five mutants fell into no known complementation group and were tested for single gene defects and for new complementation groups (see MATERIALS AND METHODS). Three new complementation groups emerged; kre9, krelO and kre l l , with 6, 5 and 2 members, respectively.
Allelism was tested between the krel0-2 and krel l - I mutations. Strain HAB562 krel0-2 was crossed with HAB756 krel l -1 . T h e toxin-resistant phenotypes for these mutations segregated independently: parental ditypes (0), nonparental ditypes (2), tetratypes (lo), indicating that they were not allelic. In addition, the resistant krelO krell double mutants showed a more extreme slow growth phenotype than either the krel0- 2 or the krell-I mutants alone.
Mutants with a defect in kre9 grew very poorly, were found to be more sensitive to Zymolyase diges- tion than the wild type, and were difficult to work with. Cloning of the KRE9 gene indicated that it was distinct from K R E l l , see Figure 1 and below.
(1 + 6)-&Glucan levels in new mutants: T h e al- kali-insoluble (1 + 6)-glucan levels from kre9, 10 and 1 I mutants and the isogenic parental strain were compared to determine if the killer resistance pheno- types correlated with a reduction in cell wall glucan. Two kre9 alleles were tested for effects (Table 2) and both strains showed a reduction in the (1 "* 6)-glucan fraction of approximately 60% as compared to the wild-type. T h e krel0-2 mutation caused no significant change in glucan level, while the krell-1 allele re- duced the level of this polymer to about one half that of the SEY62 10 isogenic parental strain.
Cloning of KRE9: T h e KRE9 gene was cloned by functional complementation of the kre9-1 mutation in HAB751, as described by BOONE et al. (1990) using pRS316 and YCp50-based yeast genomic banks (BOONE et al. 1990; ROSE et al. 1987). Strains harbor-

Yeast KRE Genes 84 1
A ) KRE 9 COMPLEMENTATION
K p n l PvuII Bg111 Spel Xbal PvuII Kpnl
I I I I I I - + 1 I +/-
I I +/ - 8) KRE I t
I I -
1 I -
I kb
FIGURE 1 .-Restriction maps of KRE9 and K R E l 1 . Restriction maps of the (A) KRE9, and (B) K R E l l loci are shown, with black arrows indicating the size and positions of the open reading frames. Only selected restriction sites are shown. Subclones of each frag- ment were tested for their ability to complement the respective kre9-2 or kre l l -1 mutation based on sensitivity to toxin on seeded plate tests (+, full complementation; +/-, partial complementation; -, failure to complement).
ing kreY mutations failed to transform by the standard lithium acetate or electroporation techniques; so a modified spheroplast transformation procedure was employed. Approximately 20,000 Ura+ transformants were grown in sorbitol-containing selective regenera- tion medum (SM2), and the regeneration agar medum plus yeast colonies homogenized in a Waring-type blender. This slurry was re-plated onto YNB selective medum to isolate single colonies, and then replica- plated onto methylene blue-containing YEPD agar seeded with a diploid killer strain to test for toxin sensitivity. Cells with plasmids containing a wild-type copy of KREY should restore killer toxin sensitivity and be killed. Sensitivity in this test results in a white colony edged with a blue-stained ring of killed cells. Cells of toxin resistant colonies decolorize the meth- ylene blue dye, and remain white in this test. Of the 20,000 colonies screened, two different sized plasmids from the pRS3 16 bank, and three from the YCp50 bank were found to completely restore growth and toxin sensitivity to kreY-I strain HAB75 1, in a plasmid- dependent fashion. The smallest complementing frag- ment was a 7-kb insert from the pRS3 16 bank, and a restriction map of this fragment was generated. Sub- cloning experiments suggested KRE9 activity was lo- cated in a 3.8-kb KpnI fragment (Figure 1A). Addi- tional subclonings of this region revealed that two fragments overlapping by 1.3-kb were both able to
TABLE 2
Glucan levels of new kre mutants
Strain KRE allele @( 1 + B)-Glucan (pglmg dry wt)
SEY62 10 Wild type 36.3 f 3.7 HAB522 kre9-1 13.7 k 2.6 HAB556 kre9-4 14.7 f 0.6 HAB535 krel0-2 40.7 f 5.4 HAB544 kre l l -1 18.6 k 0.6
Cell wall (1 + 6)-glucan levels from representatives of the three new complementation groups are compared with the isogenic pa- rental strain, SEY6210. Total alkali-insoluble glucan was not signif- icantly different in any of these strains, with an average of 203 f 16 pg/mg dry weight. Error represents 1 SD.
partially complement the kreY-l mutation, suggesting the approximate location of this gene. The position of an open reading frame within this fragment has recently been determined (J. BROWN, unpublished results) and is shown.
KRE9 is allelic to kre9-I: To test if the 3.8-kb genomic KpnI complementing fragment was KRE9, the fragment was first cloned into the integrating vector pRS306. The resulting construct was linearized at a unique BglII site within the insert (Figure IA), and used to transform cells with a kreY-1 mutation. The Ura+ prototrophs selected should contain a tan- demly integrated URA3 gene at the kreY locus, as well as a restored wild-type copy of KREY. All transform- ants tested were sensitive to K1 toxin and grew at wild-type rates, suggesting the homologous recombi- nation event was capable of rescuing the kre9-1 mu- tation. One such transformant HAB757 was back- crossed to the isogenic wild-type parent, SEY62 10.
The resulting diploid HAB758 was sporulated and 24 tetrads analyzed. All 4 spores of each tetrad were toxin sensitive, with the Ura+ phenotype segregating 2:2. This result indicates that the complementing fragment directed integration to the kreY-1 locus and that the KRE9 gene had been cloned.
Mapping of KRE9: KRE9 was mapped to chromo- some X using a 1.3-kb BglII-XbaI fragment of the gene (Figure 1A) to probe a chromosome blot, and further to X clone 6699 of the X phage library of mapped yeast genomic DNA inserts of L. RILES and M. V. OLSON (personal communication). Clone 6699 lies between the cdcb and tzj2 genes, some 75-92 kb from the end of the left arm of the physical map of chro- mosome X (L. RILES, personal communication). We tested for linkage of KREY with y u r l , which is tightly linked physically to TIF2 in this region of chromosome X (MULLER, TRACHSEL and LINDER 1989; FOREMAN, DAVIS and SACHS 199 1). Tetrad analysis of spore progeny from diploid HAB802 YURl/yurl::HIS3 KRE9::URA3 kre9-IIKRE9 indicated a distance of 19 CM for the yurl-KRE9 interval (parental ditypes: 42, nonparental ditypes: 0, tetratypes: 25). This genetic

842 J. L. Brown et al.
distance is consistent with the physical mapping of KREY, approximately 50 kb centromere distal to t i p on the left arm of chromosome X .
Cloning of KREII : The wild-type K R E l 1 gene was cloned by functional complementation of the kre l l - I mutation in HAB754 as described above for KREY, using a YCp50-based yeast genomic bank. Approxi- mately 15,000 Ura+ colonies were screened for the killer-sensitive phenotype, and three different sized genomic inserts with a common fragment (Figure 1 B) were found to completely complement the kre l l -1 mutation in a plasmid-dependent fashion. The small- est complementing fragment contained a 12.5-kb in- sert, which was shown (see below) to contain the wild- type K R E l I locus.
Nucleotide sequence of KREII : The subcloning and disruption experiments indicated an XbaI restric- tion site in the functional region of K R E l 1 , and the DNA sequence in this region was obtained (Figure 2). This revealed a single, continuous open reading frame of 1680 nucleotides spanning the XbaI site (bold in Figure 2). The K R E l I sequence predicts that the gene encodes a protein, Krel lp, of 560 amino acids, with a predicted molecular mass of 63.3 kD (Figure 2). Structurally, Krel l p appears to be a soluble cyto- plasmic protein, and is devoid of any hydrophobic potential transmembrane or signal sequences. Com- parison of both the K R E l l nucleotide and deduced amino acid sequences with those in the EMBL and GenBank databases has been uninformative. Krel l p does however, contain the sequence RLGG, which conforms to the consensus (RXGG) found in the pro- posed UDP-glucose binding domains of yeast and rabbit glycogen synthases (FARKAS et al. 1990; MAH- RENHOLZ et al. 1988). The presence of several Asn- rich tracts located between amino acids 96-1 15, 343- 381 and 438-476 is also noteworthy.
Mapping of KREII: A 750-bp ClaI/XhoI fragment present in the open reading frame of K R E l l (see Figure 1B) was used to probe a yeast chromosomal blot (see MATERIALS AND METHODS, and Figure 2), and hybridized to chromosome VZZ (data not shown). The K R E l l probe also indicated hybridization to X clone 3286 of the L. RILES and M. V. OLSON X phage library of yeast genomic inserts (see MATERIALS AND METH- ODS), which is an orphan and unassigned, and to cosmid clone 9943 which contains the X 3286 DNA and which was originally placed in the hip1 region of the right arm of chromosome VZZ, but could not be unambiguously assigned. In addition the gene TZF4631 hybridized to X 3286 as well as 6627, a X insert localized to the rsrl region of chromosome VZZ (L. RILES and C. COYER, personal communication). T o resolve the location of K R E l I, we tested for linkage with rsrl (BENDER and PRINGLE 1989), and TZF4631. Preliminary data indicated that tif4631 and
krel I were closely linked (parental ditypes: 14, non- parental ditypes: 0, tetratypes: 0). In a further cross, k r e l l linkage to rsrl and ade3 was examined, see Table 3. Close linkage was found of k r e l l to ade3, a marker close to hip1 on the right arm of chromosome V U , and based on linkage to rsrl , k re l l is centromere proximal to ade3.
Disruption of KREII : A 1.8-kb fragment contain- ing the HIS3 gene was inserted at the XbaI site in the K R E l I open reading frame, and a linear 4.8-kb PvuII l EcoRV fragment used to disrupt the wild-type K R E l I locus in an isogenic diploid strain, HAB 251-15B, (see below for proof that the cloned gene is KREI 1 ). Killer resistance co-segregated 2:2 with the His+ phenotype in 30 tetrads dissected. Resistant spore progeny from 5 tetrads had a 50% reduction in the cell wall (1 + 6)-glucan level, a representative example is shown in
Table 4. Two large deletions of the K R E l 1 locus were also created using the T R P l and URA3 selectable markers (see Materials and Methods). Deletion disrup- tions made in both haploid SEY6210 and in the iso- genic diploid strain, HAB 25 1-1 5B, gave growth and killer resistance phenotypes indistinguishable from the kreII::HIS3 insertion above, see Table 4. Correct integrations at the K R E l I locus were confirmed by genomic Southern blots (data not shown). A mutant strain harboring the larger of these deletions, HAB805 krel IA::TRPI, was assayed for cell wall (1 + 6)-glucan levels. Table 4 shows the kreI1A::TRPl
deletion has a similar effect on cell wall glucan, reduc- ing the in uiuo levels of the (1 + 6) polymer approxi- mately 50%. The reduction in (1 + 6)-glucan levels found with the krel1A::TRPI deletion was comparable to those found in the kreII::HIS3 insertion and the kre l l - I allele, suggesting all three mutations generate null alleles of the KREI I gene.
KREII is allelic to k r e l l - I : Since the insertion at the K R E l I locus did not severely impair cell growth it allowed an allelism test to be performed. Strain HAB 669-6C-3, krel I::HZS? harboring a disruption at the locus of the cloned gene, was crossed with HAB 580-8D, k r e l l - I , and the resulting toxin resistant diploid, HAB 768, sporulated and 27 meiotic tetrads analyzed. All tetrads segregated 4:O for killer resist- ance and 2:2 for His+; the HIS? insertion was, there- fore, closely linked to the krel l -1 mutation.
Analysis of glucan from a k r e l l mutant by gel filtration and "C NMR spectroscopy: The killer resistance phenotype and the reduction in the level of the (1 + 6)-glucan fraction, suggested a possible role for K R E l 1 in (1 += 6)-glucan biosynthesis. The resid- ual (1 + 6)-glucan from each of two strains, disrupted at the K R E I I locus, was purified for analysis. The (1 += 6)-glucan was isolated from genetic backgrounds SEY6210 and 7B, size fractionated by gel filtration, and analyzed by "C NMR spectroscopy, to examine

-535 -525 -450 -3 75 -300 -225 -150 - 75 1 1 58 20 115 39
1 72 58
229 77
286 96 343 115 400 134 457 153 51 4 172 5 71 191 62 8 21 0 685 229 742 248 799 267 856 286 91 3 3 05 970 324 1027 343
1084 3 62
1141 381 1198 400
1255 41 9
1312 438
1369 457
1426 4 76
1483 495
1540 51 4
1597 533
1654 552 1711
Yeast KRE Genes
ATAAGAATAC G A A G G T T A T A T A T G G T ~ G C T n : T ~ G G l Y X % ? i G A T C A C A A T A T ~ ~ A C T A A C G ~ G G T ~ G n ; G G 3 T A C C G T T A C G G T A G C D G 9 A A T A ~ T A A C A A G A A A G A C A G A A ~ ~ C T T T A A T A A A C ~ ~ TGGTATATATATAACCACTTTTTWCTTATGAGTGCTTl'CTTGTATATGCGTATATGTCGTCATATT~GCCA TTATTACTTGTACATAAAATATTCTACATAAATTTATATATCCCAlYX%?iGAGCCATGGTCACGlVZGGCACATT TCTn;4ATACTCGTTTGCAAGGCTTATTl l '~TC! l 'TTTCACA! l 'CTTTCTATTTTAGTG~GAAA~~C CTATGAAAAATTGGTAACAAAAATAATTAAMGTAAATGCTATAAAATTGlWXTAAATAAAAGTGGMiCAAGAG C n ; q G T T C A C A A C T C T A C A C T C T A T A ~ C A A T A T T ~ T T A A ~ C A A ~ T A A A ~ C A A C T C A T ~ C C ATG GAA TGT TTT GTA CCG CTG CGT TGC GAC TTA GAT GGA AGT AAT ATA GAA CAG TTA
M E C F V P L R C D L D G S N I E Q L CGT CAA TCA CAC TTA AGC CGT AAA TTT ATT ATA TTT GAT GAG CAA CTG AAT CTT TGG
R Q S H L S R K F I I F D E Q L N L W CTG Tcx: TTT CAA GGT AAT TCG CAA GAG AAC AAG AGA TTT GTA CTA CAG AAT ATG ATA
L W F Q G N S Q E N K R F V L Q N M I ATA TTA ATA AAT GAA GCG CAA GTT ACC A 0 9 ACA AGC ACT ATC GAT GAT TAT TTT ACC
I L I N E A Q V T R T S T I D D Y F T GU GTT GAG AAC AAT GAA AAT CTA TGG AGG TTG AAA AAC GAC TGC TGT TCG AAG ATT
Q V E N N E N L W R L K N D C C S K I CTT TTC AAA TCA AAT GTT GTT ATG AAT AAT GGT TAT AAT AAT CAG ATC AAA TTT GTC
L F K S N V V M N N G Y N N Q I K F V TTT GAA TAT AAA TCT GTG GAT GCA AAT TTC AAT AAC CAG GAT TCA TTA CAA GAT CCA
F E Y K S V D A N F N N Q D S L Q D P CAG GCA AAG TAT ACA TTA GAT AAG TAC TCT AGC GAG GAG ATT TTG CCA AGT TTT GAG
Q A K Y T L D K Y S S E E I L P S F E CCA GTT TAT TCC TGG TCT TCT GCA GCC ACC AAA TCA TCC AAA AAT ACT AAT AAT CAT
P V Y S W S S A A T K S S K N T N N H CTG GAG AAA AAT AAC AGG GCG ACT CAT CGA GTT AGT TCT AAA AAT AGC GAA GTC CAC
L E K N N R A T H R V S S K N S E V H GAA GCG GAC GTT !KT Mu AAT CCG AAT ACA TTT ACT CTT AAG CTC CAA TAC C c 4 A T A
E A D V S R N P N T F T L K L Q Y P I TTC TCA CTT TTG AAT ATG AGA Tl'G AGA AAC ATC TCC TTG AAA TCT GAG CAT TGC ATA
F S L L N M R L R N I S L K S E H C I TTA TCA TCG TTA GAC TTT CAA ACT TCT AAA GCA TCC GAA CAA CTG ACC AAA AAA TTT
L S S L D F Q T S K A S E Q L T K K F ATT TAT CCG CAA GAA CAC AAT TCT TTT CTC AAA CTG AAC TTT CAA GAA ATA TCG TAT
I Y P Q E H N S F L K L N F Q E I S Y AAA CTA ATC GAC Go4 ACC TCT CAA ATT GAA CTA GAT CCA ATC TGT CCT TTG AAA GTG
K L I D G T S Q I E L D P I C P L K V CCA CTC ACT GCA TTT TCA TAC GAT AGC ATT AGC GCT ACT TTT AAA CTG GTT CTG TTA
P L T A F S Y D S I S A T F K L V L L CCC AAA TCA ACT CAA CCA CAT CGT GTG AAA ATC ACC TTA GCG TAC GAA CTC GAG CTG
P K S T Q P H R V K I T L A Y E L E L CAT CCC AAT CTG AAG TTA CCT GTG AGA ACA TCA TGG GAA ACA GAA GTC ACA TTG AAG
H P N L K L P V R T S W E T E V T L K CGT TCT ATG CCA ATT TCC TCG ACA TCT TCT CAA TAC TCG AGT AAC AAC AAT AAT ACC
R S M P I S S T S S Q Y S S N N N N T AAT CAT AGC GCT TCT TTT AAT QZT GCG GCC AAC AAC GTT AAT TCT GGT GGT TTG GCC
N H S A S F N G A A N N V N S G G L A AAT CTA AGA TTA GGT GGG GTT TCC TCC T o 9 AGA TTT AGT CTT GGA GCT GCT TCG ACT
N L B L G G V S S S R F S L G A A S T ACA TCA TTG GTG AAT AGC AAA TTA AGC AAT GTA AAA TTC AAG TTT ATT AAT AGC AAT
T S L V N S K L S N V K F K F I N S N ATA AAA GTT ATT AAG GGC GAA AAG TTT ACT ATG AGG CTT CAG ATC ATT AAC TCG TCA
I K V I K G E K F T M R L Q I I N S S TCA TCC CCC CTG GAT CTT GTT GTG TAT TAT AAC AAT ACA ATA AAC CCA ATC CCT TCA
S S P L D L V V Y Y N N T I N P I P S GCT AAT AAC GTA CGT AAC AGC AAT GGT ATA AAC AAC TGT GGC ATG AAT AAT ACT
A N N V R N S N G I N N C G M N N G T ATC CCC AAT TCG CCC TTG ACA CIY: GAA AAT CAG TAC GU CTG CAT AAT AAA TAT AGA
I P N S P L T L E N Q Y Q L H N K Y R A m GCG GAG GGG ATT ATA CTA TTA TCC AAC GAT TAC AAA ATT CCA GTT GTA CCT
K I A E G I I L L S N D Y K I P V V P CCG A m GAA ACA TAC TTC GCG GAT TTA CGA TTT ATT GGT ATT ATG TCC GGA TAT TAT
P R E T Y F A D L R F I G I M S G Y Y GGc ACT CTC TCC Go4 CTT AAG GTA Tn: GAT TTA AAT ACA AAT GAA CTT ATA GTT
G T L S G L K V L D L N T N E L I E V GGA AAT GGC GCA TCT GTG TTA ATC CAG TAA GCGGUCAAGTATACATTTTATTAACATACTAC
G N G A S V L I Q S T O P A T A C T A A T C T G T A A C A T T T C T A T A A A A A A G T T A T T A T C ~ T
843
FIGURE 2.-The nucleotide and predicted amino acid sequence of KREZI. (GenBank accession number L10667) A, 1680-bp open reading frame is shown along with the pre- dicted amino acid sequence of Krel l p (560 amino acids). The XbaI restriction site used for subcloning and the insertional disruption of KREll is in bold. A possible UDP- glucose binding consensus sequence is underlined. Several Asn-rich re- gions of Krel l p can be seen between amino acids 96-115, 343-381 and 438-476.

844 J. L. Brown et al.
TABLE 3
Mapping of KREl l
Tetrad type
Interval PD NPD TT (CM)
krel l -ade3 91 0 1 1 5 kre l l - r s r l 35 3 49 39 rsrl-ade3 28 4 72 52
Map distance
Diploid HAB795, isolated from crossing strains Y355 and HAB792, was sporulated and tetrad analysis was performed. Map distances were calculated using the equation of PERKINS (1949), and a graphical correction for longer map distances (MORTIMER and SCHILD (1980).
TABLE 4
Glucan levels in krel and krel I mutants
B(1 -P 6)-Glucan Strain KRE allele(s) (Pi+% dry wt)
SEY 62 10 Wild type 32.8 f 2.5 HAB 637 krelA::HIS3 14.3 f 0.8 HAB 669 krel l:HIS3 15.4 f 2.7 HAB 676 krelA::HIS3 8.4 f 1.5
HAB 677 krel&:HIS3 7.0 f 1.2
HAB 805 krell A::TRPl 15.0 rfr 0.6 HAB 808 krel&:HIS3 7.7 * 2.1
HAB 809 kre lk :HIS3 7.5 f 1.0
HAB 810 k r e l k : H l S 3 7.4 f 0.3
krell:HIS3
krel l:HIS3
kre l l A::TRPI
krell A::TRPl
krel1A::TRPl
The levels of (1 + G>glucan were analyzed in strains harboring disruptions at the h e 1 locus, the kre l l locus, or independently arising suppressors to krel krel l double mutants. Total alkali- insoluble glucan was an average of 145 f 23 ag/mg dry weight in all these strains. Error represents 1 SD.
the structure of the polymer. Gel filtration chroma- tography of (1 + 6)-glucan isolated from krell::HZS3 or wild-type strains (Figure 3), suggested the mutant glucan was somewhat smaller than that from the wild- type strain. The apparent weight average molecular mass of the wild-type glucan was approximately 40 kD (containing about 200 glucose residues), while the krell::HZS3 mutant glucan eluted with a profile cor- responding approximately to a 30-kD polymer.
13C NMR was performed on the kre l l mutant glu- can to assess the relative proportions of (1 ".* 3)-/3- and (1 + 6)-/3-linked residues in the polymer. The "C NMR spectrum of krel 1 (1 ".* 6)-glucan was similar to that of (1 + 6)-glucan purified from the isogenic wild- type strains SEY62 10 and 7B (Figure 4). These results suggest that disruption of the K R E l l gene leads to a (1 + 6)-glucan a little smaller in size than the wild- type polymer, but containing a similar proportion of (1 + 3)- and (1 + 6)-linkages.
The kre l l mutations affect (1 + 6)-glucan synthesis in a different manner than those in the K R E l gene (BOONE et al. 1990). Krelp is involved in the attach-
0.6
0.5
-
-- 0.2
- - 0 3
- - 0 4
"
0 1 - - m' i 8'
,..a' , 0 : '.- '. 06
0 .5
0.4
0 3
0 2
0. I
0
0 P v)
0 D 0 . 5
- '
0.4 - a
0 . 3 - '
0.2 -I
0.1 - -
kre I :: HIS3
100 120 140 160 180 200 220 2 4 0 260
Fractlon Number
FIGURE 3.--Gel filtration chromotagraphy of purified ( 1 + 6)- glucan isolated from kre mutant strains. A Sepharose CL-GB column was used to determine the average molecular weights of (1 + 6)- glucan isolated from strains: (A) SEY6210, (B) krel l::HlS3, (C) krelA::HlS3, (D) double mutant (krel&:HlS3 krell::HIS3), as de- scribed in the MATERIALS AND METHODS. Each glucan sample was purified from two independent strains and run two or three times. The elution volumes of dextran standards are indicated. The col- umn void volume was 30.5 ml, or fraction 94.
ment of (1 + 6)-/3-glucan side chains to a (1 + 3)- branched, (1 + 6)-glucan backbone. The (1 + 6)-glu- can from a kre lA mutant is smaller in size than wild type, but differs from the krel 1 mutant glucan above, in having a reduced proportion of (1 + 6)-linked res- idues in the mutant polymer. T o explore a possible

A) SEV8210
c - 1
Yeast KRE Genes
c-3
c-2 I
845
I / ' ' I I C-B(linked)
6) k r e l l HIS3
I
c) krel HIS3 I
FIGURE 4.-% NMR spectra of purified ( 1 * 6)-glucan isolated from kre mutant strains. Purified (1 + 6)-glucan samples were subject to I3C NMR spectroscopy to determine the relative proportions of ( 1 + 3)-/3 and (1 + 6)-/3 linkages present in the polymers. The predom- inant signals (A) C-1, C-3 (linked), C-3, C-5, C- 2, C-4, C-6 (linked) and C-6) characteristic of (1 + 6)-/3-linked glucan in S. cereuisiae are in- dicated as previously described by WNE et al. (1990). (A) SEY6210, (B) hrel l : :HIS3, (C) k r e l k : H I S 3 , (D) double mutant (krelA::HIS3 krel l : :HIS3).
1 I
D) k r e l l : . H I S 3
krel HIS3
I
100 90 80 70 80 PPn

846 J. L. Brown et al.
interaction of the K R E l and K R E l l gene products, we examined the phenotype of kre lA kre l l double mutants.
Phenotype of kre l l Krel double mutants: A krel A krell double disruption was created by mating strain HAB637- 1 -6A krel A::HIS3, to the isogenic strain HAB669-2A krel I::HIS3, and the resulting diploid HAB 670 sporulated, and tetrads dissected. Spore progeny harboring both the krelA krel I mutations showed a slow growth phenotype that was more severe than the modest growth reduction seen with either mutation alone. Indeed the double mutants gave mi- crocolonies that were so severely compromised for growth that they could not be tested directly. Spon- taneous second site suppressors of the slow growing double mutant phenotype arose at a frequency of approximately 1 X 1O"j. These suppressors partially restored growth but did not restore toxin sensitivity. The krelA krel I double mutants bearing the suppres- sor could be further characterized. The retention of disruptions at both the K R E l and K R E l I loci in these suppressed strains was confirmed by genomic South- ern blots. Previously, partial suppressors of the severe growth defect in a kre5A null mutant allowed the determination that the mutant lacked (1 + 6)-glucan (MEADEN et al. 1990), suggesting that suppression may occur through an alternative pathway. Glucan analysis on two independent suppressed variants of the kre lA kre l l double mutant, HAB 676 and HAB 677, re- vealed a 75-80% reduction in the (1 + 6)-glucan fraction, a reduction greater than that caused by either a kre lA or kre l l mutation alone, see Table 4. Similar results were also obtained in HAB808, HAB809 and HAB810, three independently derived suppressors of kre lA kre l IA double mutants gener- ated from crossing HAB637-1-6A krelA::HIS3, with the k r e l l A deletion mutant, HAB805 krel1A::TRPl (Table 4). Characterization of HAB677 kre lA kre l l glucan by gel filtration indicated an average molecular mass of 15 kD, similar to that of the kreIA::HIS3 glucan (Figure 3). The ''C NMR spectrum of the glucan from double mutant cells showed a structure with linkage ratios similar to those of a kre lA null mutant (Figure 4). These results suggest that both Kre l p and Kre 1 l p are required to produce the ma- ture glucan polymer at wild-type levels. KREII interacts with KRE6: As with krel I , mu-
tants with defects in the KRE6 gene show a reduced level of a wild-type (1 + 6)-glucan polymer (ROEMER and BUSSEY 1991). We tested whether mutants har- boring the kre l l -1 allele interacted with a kre6A::HZS3 containing mutant, by attempting to construct strains containing mutations in both genes. Diploid HAB6 13 heterozygous for kre l l -1 and kre6A::HIS3 was made, sporulated, and 13 tetrads dissected and analyzed. Three tetrads were parental ditypes for kre l l -1 and
kre6A::HIS3, with 4 killer-resistant spore progeny, 2 being His+. Nine tetrads were tetratypes and gave 3 viable spores, 1 sensitive His-, 2 killer resistant with one of these His+, and the fourth inviable spore di- vided 2-6 times before ceasing to grow. One tetrad was a nonparental ditype, and gave 2 viable, sensitive His- spore progeny. This segregation pattern is con- sistent with the kre l l - I kre6A::HIS3 double mutation leading to lethality.
Additional cell wall analyses: The major cell wall components were examined in krel and krel I mutants to explore the possibility that defects in these genes led to pleiotropic effects in cell wall synthesis. The alkali-soluble glucan plus mannan fractions were de- termined from a wild-type strain, and isogenic kreIA::HIS3, krel1A::TRPI or krelA::HIS3 krel1A::TRPI double disruptant strains. The results indicated that total alkali-soluble glucan plus mannan fractions in the wild-type or mutant strains were not significantly different, with an average value of 167.1 f 3 &mg dry weight. After digestion with Zymolyase and dialysis to remove alkali-soluble (1 + 3)-glucan, no differences in the remaining fractions were seen, with average values of 72.8 f 4 pg/mg dry weight. Alkali-insoluble (1 + 3)-glucan levels were also meas- ured, and appeared normal in these strains (legend, Table 4), while the alkali-insoluble (1 + 6)-glucan fraction showed a reduction from the wild type in either single mutant, and a cumulative reduction (75- 80%) in the kreIA::HIS3 krel1A::TRP double mutants (Table 4). Cell wall chitin levels in wild-type and mutant strains were examined by Calcofluor White fluorescence. While no observable differences in flu- orescence were seen between the parental strain and either a krelA::HIS3 or kre1lA::TRPl single mutant, the krelA::HIS3 krel1A::TRP double mutants ap- peared to stain more intensely. Whether this apparent increase in cell wall chitin levels is the basis the suppression seen in the krel A::HIS3 krel I A::TRP dou- ble mutants has not presently been determined. A kreIA::HIS3, and a krelI::HIS3 mutant, along with the isogenic parental strain 7B, were assayed for (1 + 3)-/3-glucan synthase activity in vitro according to CABIB and KANC (1987). No significant difference in specific activity was seen between these mutants and the wild-type strain, with values for strain 7B; 97.4 f 13.6 nmol glucose incorporated/ mg protein/hr, krelA::HIS3; 109 * 25.5 nmol glucose incorporated/mg protein/hr, and krel l::HIS3; 133.1 2 49.2 nmol glucose incorporated/mg protein/hr. Finally, glycogen levels were tested by staining with iodine vapors, and appeared indistinguishable in wild type, kreIA::HIS3, krel IA::TRP, and kreIA::HIS3 krel1A::TRP double mutant strains. Cumulatively, these results suggest the cell wall defect observed in mutants harboring disruptions of K R E l or KREl I is

Yeast KRE Genes 847
A ) K r e l l p KreSp K r e 6 p K r e 1 p
Core + Matu re UDPG Glucan p( 1 +6) Glucan
p( 1 -+6) backbone p( 1 +6) side synthesls and chaln addlt lon p( 1-3) branchlng
K re9p K r e 1 Op
K r e l 1 p KreSp Kre6p K re 1 p
UDPG - Glucan I Core ~
UDPG - Core ~
Glucan 1 1 K r e 1 1 p- K r e l p
Ma tu re pC 1 + 6 ) Glucan Type I
Ma tu re PC 1 -+6) Glucan Type I I
Homolog? KreSp Kre6p
Kre9p K r e 1 Op
FIGURE 5.-Possible models for the functional role of K R E l I in the assembly of (1 -+ 6)-glucan. (A) (1 + 6)-Glucan is synthesized in a pathway from UDP-glucose, with Krelip, Kre6p and Krel Ip involved in making a glucan with (1 + 3)-branch points (Core Glucan), which is a substrate for Krelp-dependent side-chain addi- tion to elaborate the mature glucan polymer. (B) Depicts two independent pathways for (1 + 6)-glucan synthesis. Krel Ip would be involved only in the upper pathway, in making Core Glucan I, whose mature product is larger than type I1 glucan. A second pathway (lower part of B), uses a postulated functional homolog of Krel Ip to make core glucan 11, which is matured to the smaller type I1 glucan found in a K R E I l disrupted strain. The role of Kre9p and KrelOp in these processes is not known.
confined to a reduction in the (1 + 6)-P-glucan poly- mer.
DISCUSSION
This study extends genetic analysis of the yeast cell surface using killer toxin resistant mutants. The re- peated isolation of resistant alleles in seven genes indicates that further isolations using this method are unlikely to identify new genes. We have identified three genes implicated in cell wall synthesis. Mutations in KRE9 and KREI I show reduced levels of cell wall (1 + 6)-glucan. KREIO genetically interacts with
K R E l l , as cells with mutations in both genes grow more slowly than cells with a mutation in either gene alone. Disruption of the K R E l I gene and analysis of the resultant phenotype indicates that cell walls con- tain a reduced amount of a smaller glucan polymer. The exact structure of the (1 + 6)-glucan polymer remains unknown, but it is composed of a (1 -6)- linked core, with (1 - 3)-branch points and (1 + 6)- linked side chains. Failure to make normal amounts of the wild-type (1 + 6)-glucan structure likely leads to an impaired toxin receptor and killer resistance.
Functions and genetic interactions of the KREl , K R E l l and KRE6 genes involved in 8-glucan syn- thesis: The K R E l gene product is involved in (1 + 6)-side chain synthesis; the defect in strains with krel
mutations is consistent with a failure to extend (1 -+
6)-glucan sidechains to (1 -+ 3)-branchpoints on a (1 + 6)-glucan core (BOONE et al. 1990). The P-glucan phenotype in a k r e l l mutation appears to preserve the linkage arrangement of the wild-type polysaccha- ride in a polymer that is somewhat smaller in size. The cumulative reduction of the amount of the (1 + 6)-glucan polymer in the cell wall, and the slow
growth phenotype of the k r e l A k r e l I A double mutant strain argues that the defect in the polymer is more extreme than that caused by either a krel A or a krel I A mutation alone. The glucan structures and the mutant phenotypes imply that Krel l p precedes Krelp in a biosynthetic pathway, and that the glucan made in a krel I A mutant remains as a substrate for Kre 1 p. The finding that double mutations in K R E l 1 and KREG are lethal is consistent with these two gene products being required for (1 -+ 6)-glucan synthesis. Single mutants in these genes lead to reduced synthe- sis of a wild-type polymer. Thus both KREG and K R E l l appear to precede K R E l and together are essential for growth of this strain. In seeking to explain these results in an ordered glucan assembly pathway, there are a number of possible functions for Krel lp, outlined in Figure 5.
In a simple explanation (Figure 5A), Krel l p may be required in some way for efficient glucan synthesis, perhaps as a cytoplasmic component, with Kre6p as a membrane-associated synthase complex.
In a second model, Krel l p may be an essential component of one of several (1 + 6)-P-D-glucan syn- thases (Figure 5B) and involved in synthesis of a type I glucan. When Krel l p is absent, a redundant second synthase, possibly involving a Kre 1 1 p homolog, makes a lesser amount of a somewhat smaller glucan, called type I1 in Figure 5B. This model proposes two inde- pendent pathways for synthesis of the (1 -+ 6)-glucan (Figure 5B). The wild-type glucan would then be composed of a mixed population of the two types of mature (1 -+ 6)-glucans, which differ slightly in size. A formal, and more complex, possibility is that

848 J. L. Brown et al.
Krel l p is not involved in glucan synthesis but in preventing glucan degradation, and that in its absence wild-type (1 + 6)-glucan is made normally, but then partially degraded.
Pathway of (1 -+ 6)-glucan synthesis: The genes identified here augment the previous genetic model of (1 + 6)-glucan assembly in S. cerevisiae (MEADEN et al. 1990). While the roles of the Kre9p and KrelOp are unknown, analysis of glucan from krel l A dis- rupted cells suggests that the polymer is synthesized in a sequential manner involving several gene prod- ucts, some of which are in the yeast secretory pathway, and Krel l p which is likely cytoplasmic. Identification of these genes should allow access to their products, and eventually, through biochemistry and cell biology, to their functions. The search for further genes inter- acting with those described could usefully be contin- ued, as our present understanding remains incom- plete.
We thank TERRY ROEMER and ARTHUR PERLIN for helpful discussions, ANNE-MARIE SDICU for technical assistance, DIANE OKI for manuscript preparation, SILVI BILODEAU at the NMR Labora- tory, Universiti de MontrGaI, for NMR spectroscopy, SHAWN DE- LANEY for oligonucleotides, ALAN BENDER and CHARLE~ ~ Y E R for strains, and LINDA RILES for help with mapping. Supported by a Biotechnology Strategic Grant from The Natural Sciences and Engineering Research Council of Canada.
LITERATURE CITED
AL-AIDROOS, K., and H. BUSSEY, 1978 Chromosomal mutants of Saccharomyces cereuisiae affecting the cell wall binding site for killer factor. Can. J. Microbiol. 2 4 228-237.
BADIN, J., C. JACKSON and M. SCHUBERT, 1953 Improved method for determination of plasma polysaccharides with tryptophan. Proc. SOC. Exp. Biol. Med. 8 4 228-291.
BENDER, A., AND J. R. PRINGLE, 1989 Multicopy suppression of the cdc24 budding defect in yeast by CDC42 and three newly identified genes including the ras-related RSRl. Proc. Natl. Acad. Sci. USA 8 6 9976-9980.
BOONE, C., S. S. SOMMER, A. HENSEL and H. BUSSEY, 1990 Yeast KRE genes provide evidence for a pathway of cell wall 8-glucan assembly. J. Cell Biol. 1 1 0 1833-1843.
BULAWA, C. E. , 1992 CSD2, CSD3, and CSD4 genes required for chitin synthesis in Saccharomyces cerevisiae: the CSD2 gene product is related to chitin synthases and to developmentally regulated proteins in Rhizobium species and Xenopus laevis. Mol. Cell. Biol. 12: 1764-1776.
BULAWA, C. E., M. L. SLATER, E. CABIB, J. Au-YOUNG, A. SBURLATI, W. L. ADAIR and P. W. ROBBINS, 1986 The S. cerevisiae structural gene for chitin synthase is not required for chitin synthesis in vivo. Cell 4 6 2 13-225.
BUSSEY, H., 1991 K l killer toxin, a pore-forming protein from yeast. Mol. Microbiol. 5: 2339-2343.
BUSSEY, H., W. SACKS, D. GALLEY and D. SAVILLE, 1982 Yeast killer plasmid mutations affecting toxin secretion and activity and toxin immunity function. Mol. Cell. Biol. 2: 346-354.
CABIB, E., and M. S. KANG, 1987 Fungal 1,3-@-glucan synthase. Methods Enzymol. 1 3 8 637-642.
FARKAS, I., T . A. HARDY, A. A. DEPAOLI-ROACH and P. J. ROACH, 1990 Isolation of the GSYl gene encoding yeast glycogen synthase and evidence for the existence of a second gene. J. Biol. Chem. 265: 20879-20886.
FLEET, G. H., and H. J. PHAFF, 1981 Fungal glucans-structure and metabolism, pp. 416-440 in Encyclopedia of Plant Physiol- ogy, New Series, Vol. 13b, Plant Carbohydrates 11, edited by W. TANNER and F. A. LOEWUS. Springer-Verlag, Berlin.
FOREMAN, P. K., R. W. DAVIS and A. B. SACHS, 1991 The Saccha- romyces cerevisiae RBP4 gene is tightly linked to the TIF2 gene. Nucleic Acids Res. 1 9 2781.
HARTLAND, R. P., G. W. EMERSON and P. A. SULLIVAN, 1991 A secreted @-glucan-branching enzyme from Candida albicans. Proc. R. SOC. Lond. B 246: 155-160.
HAUSLER, A., and P. W. ROBBINS, 1992 Glycosylation in Saccha- romyces cerevisiae: cloning and characterization of an a-1,2- mannosyltrans-ferase structural gene. Glycobiology 2: 77-84.
HILL, K . , C. BOONE, M. GOEBL, R. PUCCIA, A. M. SDICU and H. BUSSEY, 1992 Yeast KREP defines a new gene family encod- ing probable secretory proteins, and is required for the correct N-glycosylation of proteins. Genetics 1 3 0 273-283.
HOFFMAN, C. S. and F. WINSTON, 1987 A ten-minute DNA prep aration from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 57: 267-272.
HUTCHINS, K., and H. BUSSEY, 1983 Cell wall receptor for yeast killer toxin: involvement of (1 + 6)-@-~-ghcan. J. Bacteriol.
ITO, H., Y. FUKUDA, M. MURATA and A. KIMURA, 1983 Transformations of intact yeast cells with alkali cations. J. Bacteriol. 153: 163-168.
JAMAS, S., C. K. RHA and A. J. SINSKEY, 1986 Morphology of yeast cell wall as affected by genetic manipulation of p(1-6)- glycosidic linkage. Biotechnol. Bioeng. 28: 769-784.
KATO, K . , 1981 Ultrastructure of the plant cell wall: biochemical viewpoint, pp. 29-46 in Encyclopedia of Plant Physiology, New Series, Vol. 13b, Plant Carbohydrates 11, edited by W. TANNER and F. A. LOEWUS. Springer-Verlag, Berlin.
KLEBL, F., and W. TANNER, 4989 Molecular cloning ofa wall exo- 1,3-@-glucanase from Saccharomyces cerevisiae. J. Bacteriol. 171:
MANNERS, D. J., A. J. MASSON and J. C. PATTERSON, 1973a The structure of a p-(1 + 3)-D-ghCan from yeast cell walls. Biochem. J. 135: 19-30.
MANNERS, D. J., A. J. MASSON and J. C. PATTERSON, 1973b The structure of a p-(1 + 6)-~-ghcan from yeast cell walls. Biochem. J. 135: 3 1-36.
MARTINAC, B., H. ZHU, A. KUBALSKI, X. ZHOU, M. CULBERTSON, H. BUSSEY and C. KUNG, 1990 Yeast K1 killer toxin forms ion channels in sensitive yeast spheroplasts and in artificial liposomes. Proc. Natl. Acad. Sci. USA 87: 6228-6232.
MEADEN, P., K. HILL, J. WAGNER, D. SLIPETZ, S. S. SOMMER and H. BUSSEY, 1990 The yeast KRE5 gene encodes a probable endo-plasmic reticulum protein required for (1 + 6)@-~-glucan synthesis and normal cell growth. Mol. Cell. Biol. 1 0 3013- 30 19.
MORTIMER, R. K., and D. SCHILD, 1980 The genetic map of Saccharomyces cerevisiae. Microbiol. Rev. 44: 5 19-57 1.
MULLER, P. P., H. TRACHSEL and P. LINDER, 1989 Genetic local- ization of the Saccharomyces cerevisiae genes tiyland hf2. Curr. Genet. 16: 127-128.
PERKINS, D., 1949 Detection of linkage in tetrad analysis. Genetics
RIBAS, J. C., M. DIAZ, A. DURAN and P. PEREZ, 1991 Isolation and characterization of Schizosaccharomyces pombe mutants de- fective in cell wall (1-3)@-D-gluCan. J. Bacteriol. 173: 3456- 3462.
ROEMER, T., and H. BUSSEY, 1991 Yeast @-glucan synthesis: KRE6 encodes a predicted type I1 membrane protein required for glucan synthesis in vivo and for glucan synthase activity an vitro. Proc. Natl. Acad. Sci. USA 88: 11295-1 1299.
RONCERO, C., M. H. VALDIVIESO, J. C. RIBAS and A. DURAN, 1988 Isolation and characterization of Saccharomyces cereuisiae
154: 161-169.
6259-6264.
3 8 187-197.

Yeast KRE Genes a49
mutants resistant to calcofluor white. J. Bacteriol. 170: 1950- 1954.
ROSE, M. D., P. NOVICK, J. H. THOMAS, D. BOTSTEIN and G. R. FINK, 1987 A Saccharomyces cerevisiae genomic plasmid bank based on a centromeric-containing shuttle vector. Gene 6 0
SAMBROOK, J., E. F. FRITSCH and T. MANIATIS, 1989 Molecular Cloning: A Laboratory Manual, Ed. 2. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
SANGER, F., S. NICKLEN and A. R. COULSON, 1977 DNA sequenc- ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci.
SHAW, J. A., P. C. MOL, B. BOWERS, S. J. SILVERMAN, M. H. VALDIVIESO, A. DURAN and E. CABIB, 1991 The function of chitin synthases 2 and 3 in the Saccharomyces cerevisiae cell cycle. J. Cell Biol. 114 1 1 1-123.
SHERMAN, F., G. R. FINK and J. B. HICKS, 1986 Methods in Yeast Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
SHIOTA, M., T. NAKAJIMA, A. SATOHAN, M. SHIDA and K. MAT- SUDA, 1985 Comparison of @-glucan structures in a cell wall mutant of Saccharo-myces cerevisiae and the wild type. J. Biochem. 9 8 1301- 1307.
237-243.
USA 74: 5463-5467.
VALDIVIEW, M. H., P. C . MOL, J. A. SHAW, E. CABIB and A. DURAN, 1991 CALI , a gene required for activity of chitin synthase 3 in Saccharomyces cereuisiae. J. Cell Biol. 114 101-109.
VAN RINSUM, J., F. M. KLIS and H. VAN DEN ENDE, 1991 Cell wall glucomannoproteins of Saccharomyces cereuisiae mnn9. Yeast 7:
VASQUEZ DE ALDANA, C. R., J. CORREA, P. S. SEGUNDO, A. BUENO, A. R. NEBREDA, E. MENDEZ and F. DEL REY, 1991 Nucleotide sequence of the exo-l,3-B-glucanase-encoding gene, E X G l , of the yeast Saccharomyces cereuisiae. Gene 97: 173-182.
WESSELS, J. G. H., and J. H. SIETSMA, 1981 Fungal cell walls: a survey, pp 352-394 in Encyclopedia of Plant Physiology, New Series, Vol. 13b, Plant Carbohydrates 11, edited by W. TANNER and F. A. LOEWUS. Springer-Verlag, Berlin.
WONG, H. C., A. L. FEAR, R. D. CALHOON, G. H. EICHINGER, R. MAYER, D. AMIKAM, M. BENZIMAN, D. H. GELFAND, J. H. MEAD, A. W. EMERICK, R. BRUNER, A. BEN-BASSAT and R. TAL, 1990 Genetic organization of the cellulose synthase operon in Acetobacter xylinum. Proc. Natl. Acad. Sci. USA 87:
7 17-726.
8 130-8 134.
Communicating editor: E. W. JONES




















