
A comparative study of Pt/CeO 2 catalysts for catalytic partial oxidation of methane to syngas for...
12
Applied Catalysis A: General 243 (2003) 135–146 A comparative study of Pt/CeO 2 catalysts for catalytic partial oxidation of methane to syngas for application in fuel cell electric vehicles Lidia Pino a,∗ , Antonio Vita a , Manuela Cordaro a , Vincenzo Recupero a , Manajanatha Subraya Hegde b a Institute CNR-ITAE, Via Salita S. Lucia sopra Contesse n. 5, 98126 S. Lucia, Messina, Italy b Solid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore 560012, India Received 6 August 2002; received in revised form 1 October 2002; accepted 8 October 2002 Abstract In the framework of a project aimed at developing a reliable hydrogen generator for mobile polymer electrolyte fuel cells (PEFCs), particular emphasis has been addressed to the analysis of catalysts able to assure high activity and stability in transient operations (frequent start-up and shut-down cycles). In this paper, the catalytic performance of 1 at.% Pt/ceria samples prepared by coprecipitation, impregnation and combustion, has been evaluated in the partial oxidation of methane. Methane conversion and hydrogen selectivity of 96 and 99%, respectively, associated with high stability during 100h of reaction under operative conditions (start-up and shut-down cycles), have been obtained. © 2002 Elsevier Science B.V. All rights reserved. Keywords: Catalytic partial oxidation of methane; Ceria-supported platinum catalyst 1. Introduction Fuel cells, directly converting the chemical energy of a fuel into electricity, may help to reduce the depen- dence from fossil fuels and, operating without combus- tion, can contribute to reduce environmental impacts. The different types of fuel cells have different po- tential applications such as stationary, transportation or portable power; particularly, the polymer electrolyte fuel cells (PEFCs), that operate at relatively low tem- perature (around 80 ◦ C), have high power density, can start instantly, and can be quickly adjusted to variable ∗ Corresponding author. E-mail address: [email protected] (L. Pino). power demands; thus, they have been proposed as an alternative to internal combustion engines [1]. In absence of a hydrogen refuelling infrastructure, these systems will require hydrogen produced from available fuels such as hydrocarbons and/or alcohols: the onboard conversion of a fuel into hydrogen is con- sidered as a promising method of supplying hydrogen for vehicle fuel cells [2]. Processes based on steam re- forming, partial oxidation and auto-thermal reforming are widely investigated [3–5]. Steam reforming, in- volving the reaction of steam with the fuel in presence of a catalyst, is endothermic [6]; some of the fuel must be burned and the heat transferred to the reformer via heat exchangers [7–9]. The resulting system is quite cumbersome and inadequate for frequent start-ups and shut-downs or transient conditions, connected with the 0926-860X/02/$ – see front matter © 2002 Elsevier Science B.V. All rights reserved. PII:S0926-860X(02)00542-2
Transcript of A comparative study of Pt/CeO 2 catalysts for catalytic partial oxidation of methane to syngas for...

Applied Catalysis A: General 243 (2003) 135–146
A comparative study of Pt/CeO2 catalysts for catalytic partialoxidation of methane to syngas for application in fuel
cell electric vehicles
Lidia Pinoa,∗, Antonio Vitaa, Manuela Cordaroa, Vincenzo Recuperoa,Manajanatha Subraya Hegdeb
a Institute CNR-ITAE, Via Salita S. Lucia sopra Contesse n. 5, 98126 S. Lucia, Messina, Italyb Solid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore 560012, India
Received 6 August 2002; received in revised form 1 October 2002; accepted 8 October 2002
Abstract
In the framework of a project aimed at developing a reliable hydrogen generator for mobile polymer electrolyte fuelcells (PEFCs), particular emphasis has been addressed to the analysis of catalysts able to assure high activity and stabilityin transient operations (frequent start-up and shut-down cycles). In this paper, the catalytic performance of 1 at.% Pt/ceriasamples prepared by coprecipitation, impregnation and combustion, has been evaluated in the partial oxidation of methane.Methane conversion and hydrogen selectivity of 96 and 99%, respectively, associated with high stability during 100 h ofreaction under operative conditions (start-up and shut-down cycles), have been obtained.© 2002 Elsevier Science B.V. All rights reserved.
Keywords: Catalytic partial oxidation of methane; Ceria-supported platinum catalyst
1. Introduction
Fuel cells, directly converting the chemical energyof a fuel into electricity, may help to reduce the depen-dence from fossil fuels and, operating without combus-tion, can contribute to reduce environmental impacts.
The different types of fuel cells have different po-tential applications such as stationary, transportationor portable power; particularly, the polymer electrolytefuel cells (PEFCs), that operate at relatively low tem-perature (around 80◦C), have high power density, canstart instantly, and can be quickly adjusted to variable
∗ Corresponding author.E-mail address: [email protected] (L. Pino).
power demands; thus, they have been proposed as analternative to internal combustion engines[1].
In absence of a hydrogen refuelling infrastructure,these systems will require hydrogen produced fromavailable fuels such as hydrocarbons and/or alcohols:the onboard conversion of a fuel into hydrogen is con-sidered as a promising method of supplying hydrogenfor vehicle fuel cells[2]. Processes based on steam re-forming, partial oxidation and auto-thermal reformingare widely investigated[3–5]. Steam reforming, in-volving the reaction of steam with the fuel in presenceof a catalyst, is endothermic[6]; some of the fuel mustbe burned and the heat transferred to the reformer viaheat exchangers[7–9]. The resulting system is quitecumbersome and inadequate for frequent start-ups andshut-downs or transient conditions, connected with the
0926-860X/02/$ – see front matter © 2002 Elsevier Science B.V. All rights reserved.PII: S0926-860X(02)00542-2

136 L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146
changes in the power demand. Alternatively, catalyticpartial oxidation is a more rapid process, with a muchhigher reaction rate than the steam reforming[10,11].A fuel processor based on partial oxidation of hydro-carbons[12] or alcohols could provide a low cost andcompact system, with fast start-up and capability tofollow load variations, more adequate for fuel cellselectric vehicles (PEFCVs).
The key to realize a successful onboard partial ox-idation reformer for light hydrocarbons (i.e. CH4) isthe development of an active and stable catalyst; in-deed, when the power demand rapidly decreases, thecatalyst can overheat, causing sintering which in turnresults in loss of activity. Besides, for repeated start-upand shut-down cycles, carbon deposition phenomenaeasily occur.
The catalytic partial oxidation of methane (POX) tosynthesis gas has been extensively studied. Supportedtransition (Ni, Co, Fe)[13–15] and noble metal cat-alysts (Pt, Pd, Rh, Ir)[16–18], as well as pyrochloreand perovoskite-type oxides[19,20]have demonstrateto give high conversion and selectivity to H2 and CO.In particular, nickel-based catalysts deactivate with thetime on stream; in order to reduce carbon depositionphenomena supports and mixed oxides[20,21] havebeen recently investigated.
Moreover, ceria-based catalysts have been studiedbecause of their unique redox property, enhanced inthe presence of metal or metal oxide[22–24], associ-ated with a high oxygen storage capacity[25]. Theyhave been demonstrated to be active in total oxidationreactions such as CO oxidation[26–28], CH4 combus-tion [29,30], as promoter in water gas shift reactionat relatively low temperature[31,32] or in the directoxidation of CH4 to syngas, by the lattice oxygen, inabsence of gaseous oxidant[33–35].
The CeO2 reducibility determines a higher specificrate of the supported noble metal, with respect to theanalogous alumina and silica supported catalysts, par-ticularly in reactions such as water gas shift, steamreforming and CO2 reforming of CH4 [36,37].
The influence of the support reducibility/irreducibi-lity of the rhodium-based catalyst in the POX of CH4was reported by Ruckenstein and Wang[18]; the Au-thors suggest that the lower activity obtained whenrhodium was supported on the reducible oxides wasdue to the partial coverage of rhodium sites by migra-tion of partially reduced support.
Moreover, Pantu and Gavalas[38] pointed out, thatthe redox reactions near the interface of Pt/CeO2 cat-alyst could contribute to the higher methane conver-sion and H2/CO selectivity (at 600◦C) with respect toalumina-supported platinum.
Previously, we have studied POX of methane withundiluted air on a ceria-supported platinum catalystprepared by the solution-combustion method[39]. Inthis paper we have compared the catalytic activityand stability of 1 at.% Pt/CeO2 catalyst, obtainedby the solution-combustion method, with samplesprepared by impregnation and coprecipitation, in thepartial oxidation of CH4 to syngas at 800◦C andatmospheric pressure. Samples were characterizedby X-ray diffraction (XRD), transmission electronmicroscopy (TEM) and temperature-programmedreduction (TPR).
2. Experimental
2.1. Catalysts preparation
Samples containing 1 at.% Pt on ceria support wereprepared by different procedures: coprecipitation(sample A), impregnation (sample B) and combustionmethod (sample C).
Sample A was prepared by coprecipitation ofCe(NO3)3·6H2O and H2PtCl6·6H2O with ammoniaat 95◦C. The obtained slurry was continuously stirredat constant temperature (95◦C) for 22 h followed byfiltering and washing. The resulting catalyst was driedand calcined in an oven furnace at 800◦C for 2 h.
Sample B was prepared by impregnation of a CeO2support, (BET surface area= 106 m2/g), preparedby urea precipitation and calcined at 400◦C in airfor 2 h. The impregnation was carried out with anH2PtCl6·6H2O solution of appropriate concentration;the sample was dried and then calcined in an ovenfurnace at 800◦C for 2 h.
Sample C was obtained by the solution–combustion-method; the combustion mixture contained ceric am-monium nitrate [(NH4)2Ce(NO3)6], chloroplatinicacid (H2PtCl6) and oxalyldihydrazide (C2H6N4O2)in the molar ratios 0.99:0.01:2.33. Oxalyldihydrazidederived from diethyl oxalate, together with hydrazinehydrate was used as the fuel. Initially, 10 g of cericammonium nitrate (E-Merck India, 99.9%), 0.095 g
L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146 137
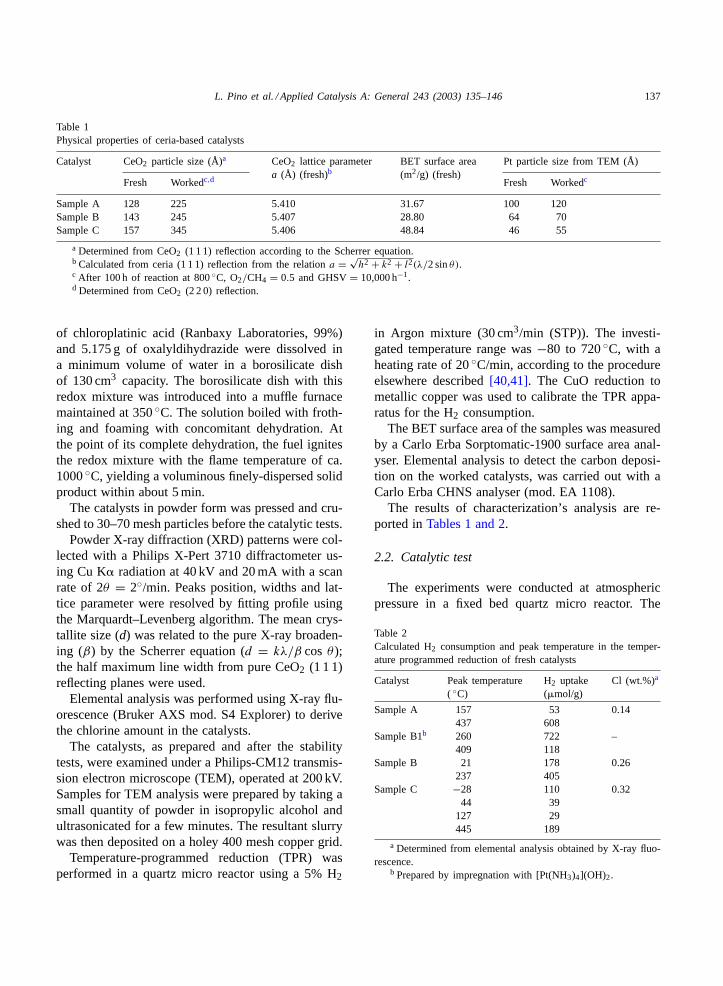
Table 1Physical properties of ceria-based catalysts
Catalyst CeO2 particle size (Å)a CeO2 lattice parametera (Å) (fresh)b
BET surface area(m2/g) (fresh)
Pt particle size from TEM (Å)
Fresh Workedc,d Fresh Workedc
Sample A 128 225 5.410 31.67 100 120Sample B 143 245 5.407 28.80 64 70Sample C 157 345 5.406 48.84 46 55
a Determined from CeO2 (1 1 1) reflection according to the Scherrer equation.b Calculated from ceria (1 1 1) reflection from the relationa = √
h2 + k2 + l2(λ/2 sinθ).c After 100 h of reaction at 800◦C, O2/CH4 = 0.5 and GHSV= 10,000 h−1.d Determined from CeO2 (2 2 0) reflection.
of chloroplatinic acid (Ranbaxy Laboratories, 99%)and 5.175 g of oxalyldihydrazide were dissolved ina minimum volume of water in a borosilicate dishof 130 cm3 capacity. The borosilicate dish with thisredox mixture was introduced into a muffle furnacemaintained at 350◦C. The solution boiled with froth-ing and foaming with concomitant dehydration. Atthe point of its complete dehydration, the fuel ignitesthe redox mixture with the flame temperature of ca.1000◦C, yielding a voluminous finely-dispersed solidproduct within about 5 min.
The catalysts in powder form was pressed and cru-shed to 30–70 mesh particles before the catalytic tests.
Powder X-ray diffraction (XRD) patterns were col-lected with a Philips X-Pert 3710 diffractometer us-ing Cu K� radiation at 40 kV and 20 mA with a scanrate of 2θ = 2◦/min. Peaks position, widths and lat-tice parameter were resolved by fitting profile usingthe Marquardt–Levenberg algorithm. The mean crys-tallite size (d) was related to the pure X-ray broaden-ing (β) by the Scherrer equation (d = kλ/β cos θ );the half maximum line width from pure CeO2 (1 1 1)reflecting planes were used.
Elemental analysis was performed using X-ray flu-orescence (Bruker AXS mod. S4 Explorer) to derivethe chlorine amount in the catalysts.
The catalysts, as prepared and after the stabilitytests, were examined under a Philips-CM12 transmis-sion electron microscope (TEM), operated at 200 kV.Samples for TEM analysis were prepared by taking asmall quantity of powder in isopropylic alcohol andultrasonicated for a few minutes. The resultant slurrywas then deposited on a holey 400 mesh copper grid.
Temperature-programmed reduction (TPR) wasperformed in a quartz micro reactor using a 5% H2
in Argon mixture (30 cm3/min (STP)). The investi-gated temperature range was−80 to 720◦C, with aheating rate of 20◦C/min, according to the procedureelsewhere described[40,41]. The CuO reduction tometallic copper was used to calibrate the TPR appa-ratus for the H2 consumption.
The BET surface area of the samples was measuredby a Carlo Erba Sorptomatic-1900 surface area anal-yser. Elemental analysis to detect the carbon deposi-tion on the worked catalysts, was carried out with aCarlo Erba CHNS analyser (mod. EA 1108).
The results of characterization’s analysis are re-ported inTables 1 and 2.
2.2. Catalytic test
The experiments were conducted at atmosphericpressure in a fixed bed quartz micro reactor. The
Table 2Calculated H2 consumption and peak temperature in the temper-ature programmed reduction of fresh catalysts
Catalyst Peak temperature( ◦C)
H2 uptake(�mol/g)
Cl (wt.%)a
Sample A 157 53 0.14437 608
Sample B1b 260 722 –409 118
Sample B 21 178 0.26237 405
Sample C −28 110 0.3244 39
127 29445 189
a Determined from elemental analysis obtained by X-ray fluo-rescence.
b Prepared by impregnation with [Pt(NH3)4](OH)2.

138 L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146
catalyst sample (about 25 mg diluted with quartzof the same size, 30–70 mesh, with a ratio ofcatalyst:diluent= 1:5) was placed between two quartzwool plugs in the centre of a quartz tube of 4 mminternal diameter and inserted into a furnace heatedto the reaction temperature. The reaction temperaturewas measured and controlled by two chromel-alumelthermocouples inserted into a thermowell. One ofthe thermocouples, which were centred within thecatalyst bed, was used to control and measure thereaction temperature while the other thermocouplewas kept just at the inlet of the catalytic bed. Highpurity gases CH4 (99.9999%), O2 (99.999%) and He(99.99%) were used in the experiments. The feedand products, subsequent to water removal from theproduct stream, were analyzed using a Dani-3800gas chromatograph equipped with a thermal conduc-tivity detector. A molecular sieve 5 Å column and aPorapak Q column in a series-by pass configurationwere used in the analysis. The reactants were dilutedwith He (90 vol.%, containing a small amount of N2for GC quantification) to minimize the axial tem-perature gradients. The catalytic tests were initiatedby heating the reactor, from ambient to the workingtemperature, under N2 flow (start-up). Subsequently,N2 flow was stopped and the reaction mixture (CH4,O2, He/N2) was allowed to flow through the reactor.Typical duration of a catalytic test was about 6 h. Thetemperature decrease, from working to ambient tem-perature (shut-down), was carried out after shuttingthe reaction mixture under N2 flow to purge the cat-alytic bed. For each test, the balance of oxygen andhydrogen, to account for the H2 and H2O produced,as well of carbon, conversion and selectivities werecalculated. The nitrogen peak was used for calibrationof mass balance. Conversion and selectivity (S) werecalculated from the following equations:
CH4conversion= �CH4
CH4,in= FCH4,in − FCH4,out
FCH4,in
SCO = FCO
�CH4
= FCO
FCO + FCO2 + 2(FC2H4 + FC2H6)
SH2 = 0.5FH2
�CH4
= FH2
FH2 + FH2O + 2FC2H4 + 3FC2H6
whereFi is the molar flow rate of speciesi.
The activity tests were carried out at 800◦C withgas hourly space velocities (GHSV) ranging between10,000 and 80,000 h−1. GHSV was defined as volumeper hour of the gaseous feed (including the He diluent)at 0◦C and 1 bar pressure per volume of the catalyticbed (including the inert particles). The variation in theGHSV was achieved by changing the catalyst weightand/or the total flow of the reagent at constant O2/CH4ratio. In all experiments the O2/CH4 molar ratio wasfixed at the stoichiometric value of 0.5.
The catalysts are tested without pre-reduction step.
3. Results and discussion
3.1. Catalysts characterization
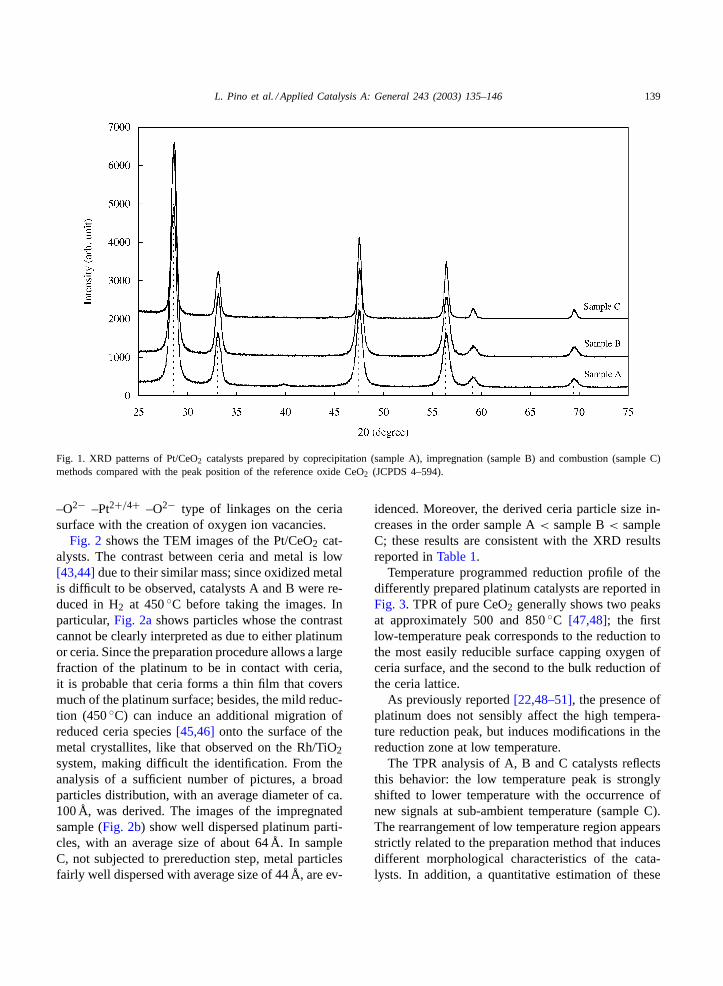
XRD diffraction patterns of 1 at.% Pt on CeO2 sam-ples, are shown inFig. 1. In the diffraction analysisonly the fluorite oxide type structure was identified;the related CeO2 particle size, derived by applicationof the Scherrer equation, range between 128 Å for thecoprecipitated sample to 157 Å for the sample C, asreported inTable 1. Samples B and C do not showthe presence of platinum metal or platinum oxides; inthe pattern of the co-precipitated catalyst pattern (sam-ple A), a broadening relative to the crystallographicdiffraction of the platinum (2θ = 40◦) is scarcely vis-ible. This not detectable presence of platinum may bedue to the low loading or can denote a high metaldispersion in the obtained catalysts. However, in allcatalysts the CeO2 reflections were slightly shifted tohigher degrees with respect to those of pure CeO2(JCPDS 4–594,a = 5.411 Å); the degree of the shiftincreases in the order sample A< sample B< sampleC. This observation can suggest the formation of a ox-ide solid solution, in agreement with previous results[42]. The existence of a solid solution by replacementof Ce4+ with Pt4+ or Pt2+, in the ceria lattice, candetermine a skrinkage of the lattice which accountsfor the evidence that the cation radius of Pt4+ (ionicradius= 0.625 Å) or Pt2+ (ionic radius= 0.80 Å) arelower than that of Ce4+ (ionic radius= 0.97 Å).
Some contraction of the CeO2 lattice parameter withsample B (a = 5.407 Å) and C (a = 5.406 Å) wasobserved. Probably the partial incorporation of plat-inum in the CeO2 structure, that determine the ob-served lattice shrinkage, resulting in a –O2− –Ce4+
L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146 139
Fig. 1. XRD patterns of Pt/CeO2 catalysts prepared by coprecipitation (sample A), impregnation (sample B) and combustion (sample C)methods compared with the peak position of the reference oxide CeO2 (JCPDS 4–594).
–O2− –Pt2+/4+ –O2− type of linkages on the ceriasurface with the creation of oxygen ion vacancies.
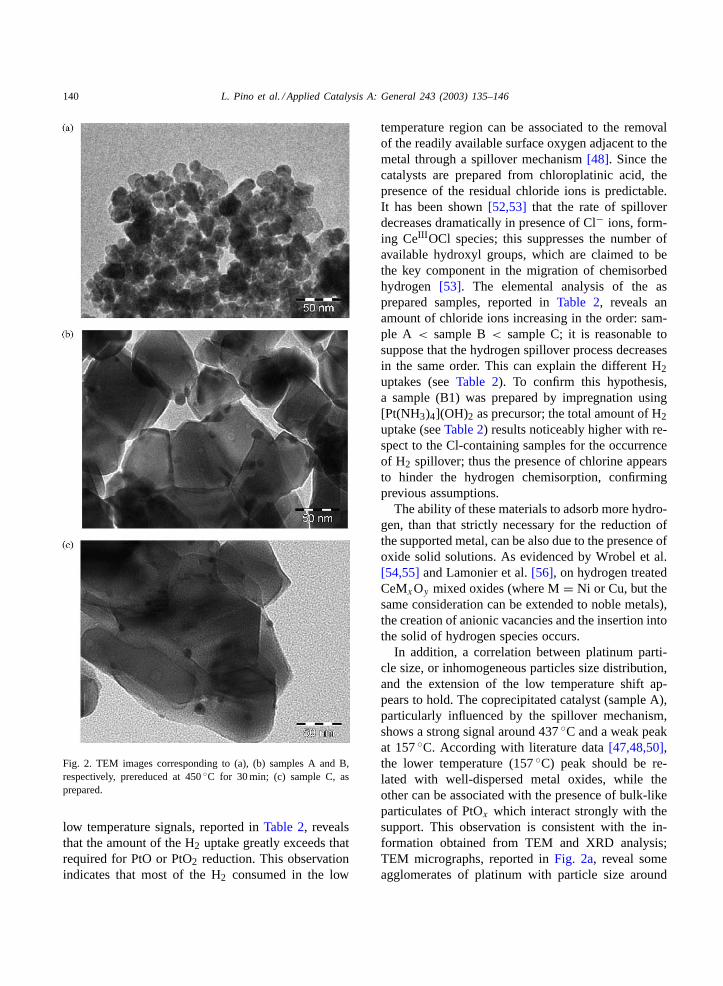
Fig. 2 shows the TEM images of the Pt/CeO2 cat-alysts. The contrast between ceria and metal is low[43,44]due to their similar mass; since oxidized metalis difficult to be observed, catalysts A and B were re-duced in H2 at 450◦C before taking the images. Inparticular,Fig. 2ashows particles whose the contrastcannot be clearly interpreted as due to either platinumor ceria. Since the preparation procedure allows a largefraction of the platinum to be in contact with ceria,it is probable that ceria forms a thin film that coversmuch of the platinum surface; besides, the mild reduc-tion (450◦C) can induce an additional migration ofreduced ceria species[45,46] onto the surface of themetal crystallites, like that observed on the Rh/TiO2system, making difficult the identification. From theanalysis of a sufficient number of pictures, a broadparticles distribution, with an average diameter of ca.100 Å, was derived. The images of the impregnatedsample (Fig. 2b) show well dispersed platinum parti-cles, with an average size of about 64 Å. In sampleC, not subjected to prereduction step, metal particlesfairly well dispersed with average size of 44 Å, are ev-
idenced. Moreover, the derived ceria particle size in-creases in the order sample A< sample B< sampleC; these results are consistent with the XRD resultsreported inTable 1.
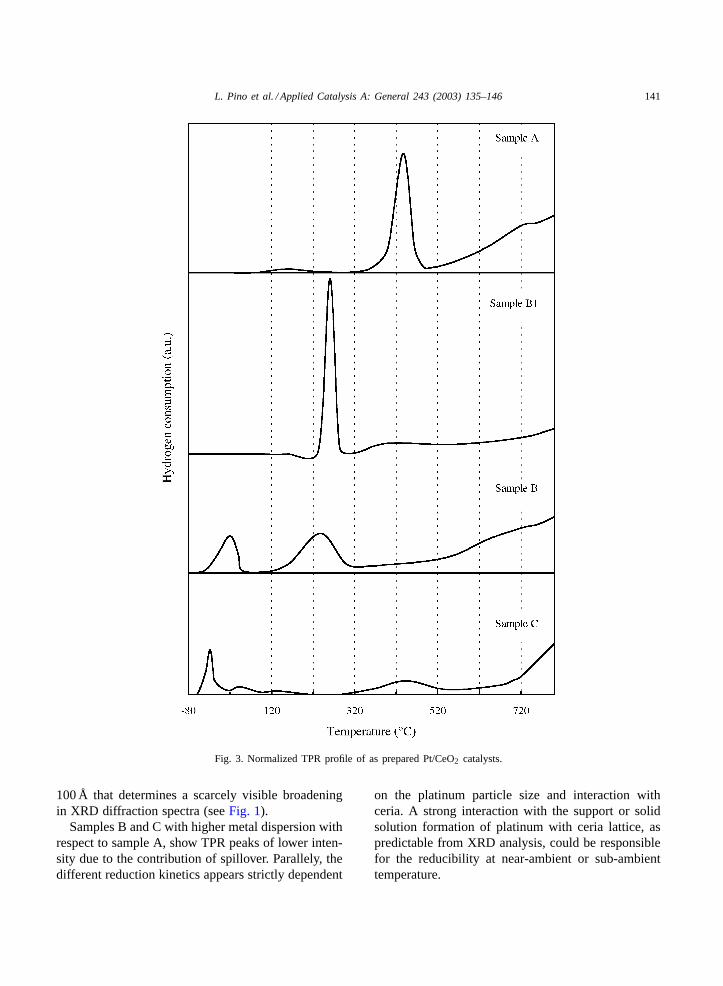
Temperature programmed reduction profile of thedifferently prepared platinum catalysts are reported inFig. 3. TPR of pure CeO2 generally shows two peaksat approximately 500 and 850◦C [47,48]; the firstlow-temperature peak corresponds to the reduction tothe most easily reducible surface capping oxygen ofceria surface, and the second to the bulk reduction ofthe ceria lattice.
As previously reported[22,48–51], the presence ofplatinum does not sensibly affect the high tempera-ture reduction peak, but induces modifications in thereduction zone at low temperature.
The TPR analysis of A, B and C catalysts reflectsthis behavior: the low temperature peak is stronglyshifted to lower temperature with the occurrence ofnew signals at sub-ambient temperature (sample C).The rearrangement of low temperature region appearsstrictly related to the preparation method that inducesdifferent morphological characteristics of the cata-lysts. In addition, a quantitative estimation of these
140 L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146
Fig. 2. TEM images corresponding to (a), (b) samples A and B,respectively, prereduced at 450◦C for 30 min; (c) sample C, asprepared.
low temperature signals, reported inTable 2, revealsthat the amount of the H2 uptake greatly exceeds thatrequired for PtO or PtO2 reduction. This observationindicates that most of the H2 consumed in the low
temperature region can be associated to the removalof the readily available surface oxygen adjacent to themetal through a spillover mechanism[48]. Since thecatalysts are prepared from chloroplatinic acid, thepresence of the residual chloride ions is predictable.It has been shown[52,53] that the rate of spilloverdecreases dramatically in presence of Cl− ions, form-ing CeIII OCl species; this suppresses the number ofavailable hydroxyl groups, which are claimed to bethe key component in the migration of chemisorbedhydrogen [53]. The elemental analysis of the asprepared samples, reported inTable 2, reveals anamount of chloride ions increasing in the order: sam-ple A < sample B< sample C; it is reasonable tosuppose that the hydrogen spillover process decreasesin the same order. This can explain the different H2uptakes (seeTable 2). To confirm this hypothesis,a sample (B1) was prepared by impregnation using[Pt(NH3)4](OH)2 as precursor; the total amount of H2uptake (seeTable 2) results noticeably higher with re-spect to the Cl-containing samples for the occurrenceof H2 spillover; thus the presence of chlorine appearsto hinder the hydrogen chemisorption, confirmingprevious assumptions.
The ability of these materials to adsorb more hydro-gen, than that strictly necessary for the reduction ofthe supported metal, can be also due to the presence ofoxide solid solutions. As evidenced by Wrobel et al.[54,55] and Lamonier et al.[56], on hydrogen treatedCeMxOy mixed oxides (where M= Ni or Cu, but thesame consideration can be extended to noble metals),the creation of anionic vacancies and the insertion intothe solid of hydrogen species occurs.
In addition, a correlation between platinum parti-cle size, or inhomogeneous particles size distribution,and the extension of the low temperature shift ap-pears to hold. The coprecipitated catalyst (sample A),particularly influenced by the spillover mechanism,shows a strong signal around 437◦C and a weak peakat 157◦C. According with literature data[47,48,50],the lower temperature (157◦C) peak should be re-lated with well-dispersed metal oxides, while theother can be associated with the presence of bulk-likeparticulates of PtOx which interact strongly with thesupport. This observation is consistent with the in-formation obtained from TEM and XRD analysis;TEM micrographs, reported inFig. 2a, reveal someagglomerates of platinum with particle size around
L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146 141
Fig. 3. Normalized TPR profile of as prepared Pt/CeO2 catalysts.
100 Å that determines a scarcely visible broadeningin XRD diffraction spectra (seeFig. 1).
Samples B and C with higher metal dispersion withrespect to sample A, show TPR peaks of lower inten-sity due to the contribution of spillover. Parallely, thedifferent reduction kinetics appears strictly dependent
on the platinum particle size and interaction withceria. A strong interaction with the support or solidsolution formation of platinum with ceria lattice, aspredictable from XRD analysis, could be responsiblefor the reducibility at near-ambient or sub-ambienttemperature.
142 L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146
3.2. Catalytic activity
The catalytic partial oxidation of CH4 was carriedout with diluted feed gas of stoichiometric compo-sition (O2:CH4:He = 3.3:6.6:90 vol.%) at 800◦C
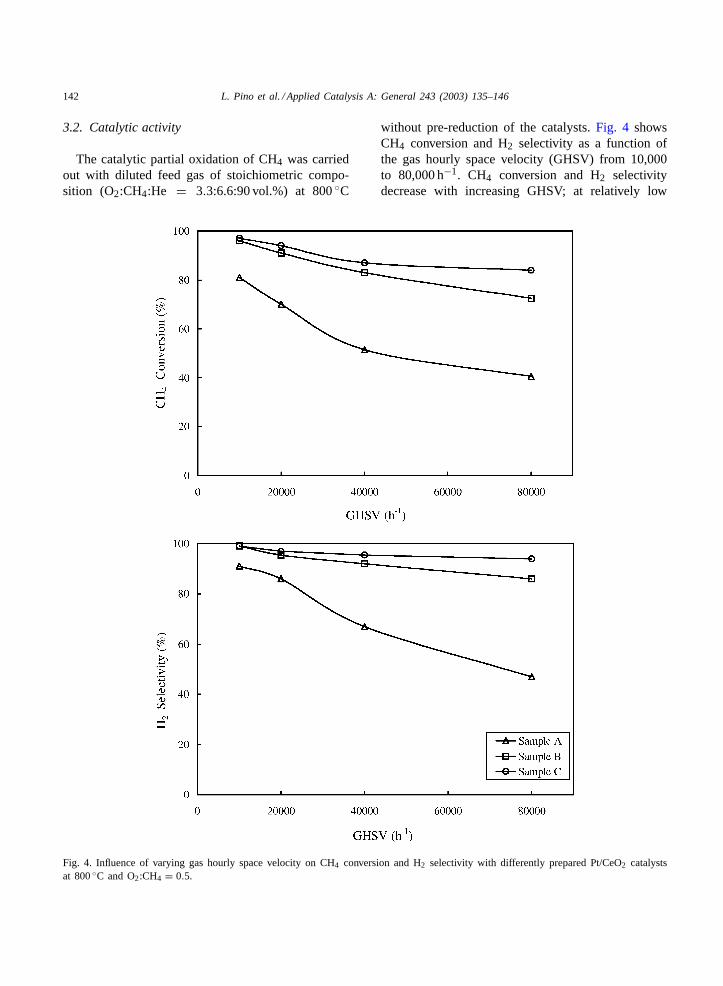
Fig. 4. Influence of varying gas hourly space velocity on CH4 conversion and H2 selectivity with differently prepared Pt/CeO2 catalystsat 800◦C and O2:CH4 = 0.5.
without pre-reduction of the catalysts.Fig. 4 showsCH4 conversion and H2 selectivity as a function ofthe gas hourly space velocity (GHSV) from 10,000to 80,000 h−1. CH4 conversion and H2 selectivitydecrease with increasing GHSV; at relatively low
L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146 143
GHSV (10,000 h−1) sample B and C show CH4 con-versions of 96 and 95%, respectively; a low decay (13and 23%), was recorded by increasing the GHSV at80,000 h−1. H2 and CO selectivity (not shown) fol-low the same trend; in the whole GHSV interval theH2/CO ratio was almost constant (≈ 2.0). A lower cat-alytic activity was obtained for coprecipitated catalyst(sample A); the CH4 conversion decreased from 80%(SH2 = 91%; SCO = 88%) at GHSV= 10,000 h−1
to 58% (SH2 = 69%; SCO = 60%) at GHSV =80,000 h−1; parallels, the H2/CO ratio decreased fromthe stoichiometric value to 1.5. In all experiments theoxygen was completely consumed during the reaction:only at 80,000 h−1, an oxygen conversion of 95% wasobtained for sample A. The mass balance between re-actants and products was 100%± 2%: in particular,the carbon balance showed that no carbon deposition,neither formation of C2 products occurred.
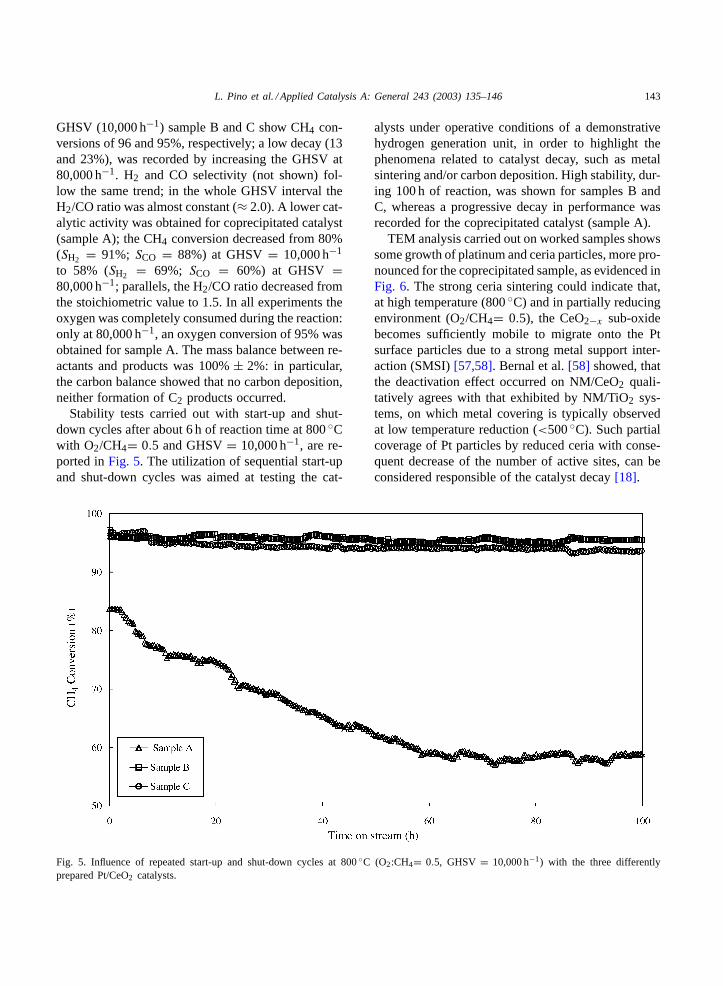
Stability tests carried out with start-up and shut-down cycles after about 6 h of reaction time at 800◦Cwith O2/CH4= 0.5 and GHSV= 10,000 h−1, are re-ported inFig. 5. The utilization of sequential start-upand shut-down cycles was aimed at testing the cat-
Fig. 5. Influence of repeated start-up and shut-down cycles at 800◦C (O2:CH4= 0.5, GHSV = 10,000 h−1) with the three differentlyprepared Pt/CeO2 catalysts.
alysts under operative conditions of a demonstrativehydrogen generation unit, in order to highlight thephenomena related to catalyst decay, such as metalsintering and/or carbon deposition. High stability, dur-ing 100 h of reaction, was shown for samples B andC, whereas a progressive decay in performance wasrecorded for the coprecipitated catalyst (sample A).
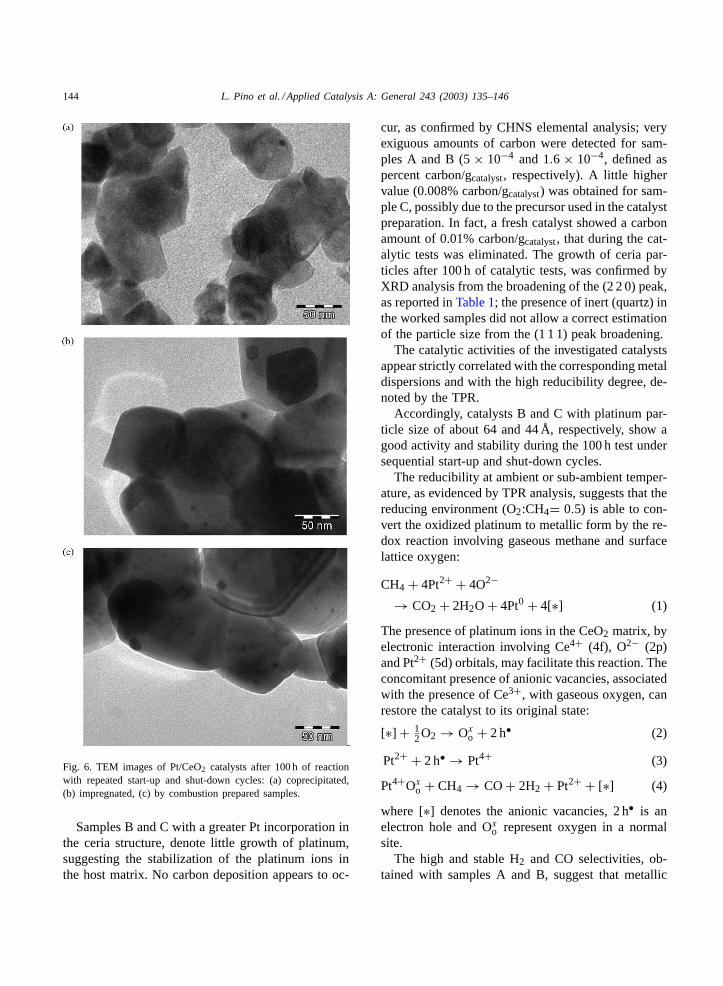
TEM analysis carried out on worked samples showssome growth of platinum and ceria particles, more pro-nounced for the coprecipitated sample, as evidenced inFig. 6. The strong ceria sintering could indicate that,at high temperature (800◦C) and in partially reducingenvironment (O2/CH4= 0.5), the CeO2−x sub-oxidebecomes sufficiently mobile to migrate onto the Ptsurface particles due to a strong metal support inter-action (SMSI)[57,58]. Bernal et al.[58] showed, thatthe deactivation effect occurred on NM/CeO2 quali-tatively agrees with that exhibited by NM/TiO2 sys-tems, on which metal covering is typically observedat low temperature reduction (<500◦C). Such partialcoverage of Pt particles by reduced ceria with conse-quent decrease of the number of active sites, can beconsidered responsible of the catalyst decay[18].
144 L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146
Fig. 6. TEM images of Pt/CeO2 catalysts after 100 h of reactionwith repeated start-up and shut-down cycles: (a) coprecipitated,(b) impregnated, (c) by combustion prepared samples.
Samples B and C with a greater Pt incorporation inthe ceria structure, denote little growth of platinum,suggesting the stabilization of the platinum ions inthe host matrix. No carbon deposition appears to oc-
cur, as confirmed by CHNS elemental analysis; veryexiguous amounts of carbon were detected for sam-ples A and B (5× 10−4 and 1.6 × 10−4, defined aspercent carbon/gcatalyst, respectively). A little highervalue (0.008% carbon/gcatalyst) was obtained for sam-ple C, possibly due to the precursor used in the catalystpreparation. In fact, a fresh catalyst showed a carbonamount of 0.01% carbon/gcatalyst, that during the cat-alytic tests was eliminated. The growth of ceria par-ticles after 100 h of catalytic tests, was confirmed byXRD analysis from the broadening of the (2 2 0) peak,as reported inTable 1; the presence of inert (quartz) inthe worked samples did not allow a correct estimationof the particle size from the (1 1 1) peak broadening.
The catalytic activities of the investigated catalystsappear strictly correlated with the corresponding metaldispersions and with the high reducibility degree, de-noted by the TPR.
Accordingly, catalysts B and C with platinum par-ticle size of about 64 and 44 Å, respectively, show agood activity and stability during the 100 h test undersequential start-up and shut-down cycles.
The reducibility at ambient or sub-ambient temper-ature, as evidenced by TPR analysis, suggests that thereducing environment (O2:CH4= 0.5) is able to con-vert the oxidized platinum to metallic form by the re-dox reaction involving gaseous methane and surfacelattice oxygen:
CH4 + 4Pt2+ + 4O2−
→ CO2 + 2H2O + 4Pt0 + 4[∗] (1)
The presence of platinum ions in the CeO2 matrix, byelectronic interaction involving Ce4+ (4f), O2− (2p)and Pt2+ (5d) orbitals, may facilitate this reaction. Theconcomitant presence of anionic vacancies, associatedwith the presence of Ce3+, with gaseous oxygen, canrestore the catalyst to its original state:
[∗] + 12O2 → Ox
o + 2 h• (2)
Pt2+ + 2 h• → Pt4+ (3)
Pt4+Oxo + CH4 → CO+ 2H2 + Pt2+ + [∗] (4)
where [∗] denotes the anionic vacancies, 2 h• is anelectron hole and Oxo represent oxygen in a normalsite.
The high and stable H2 and CO selectivities, ob-tained with samples A and B, suggest that metallic

L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146 145
platinum (the active site for partial oxidation ofmethane) was generated according to reaction (1).Parallel reactions of the support near the metal–oxideinterface, where an oxygen-deficient compositionwas expected, can favour CO2 and H2O conversion(CeO2−x + H2O → CeO2 + H2; CeO2−x + CO2 →CeO2 + CO) according to[33,37]. The chemisorbedoxygen, largely influenced by the presence of oxy-gen vacancies, can evolve as lattice-oxygen, restoringthe reduction of Ce4+ to Ce3+ (2Ce4+ + Ox
o →2Ce3+ + 1
2O2(g) + [∗]) [59].In the coprecipitated sample, the predominant pres-
ence of bulk-like PtOx , probably induces the migrationof support sub-oxides over the partially reduced plat-inum particles. During time on stream, the sub-oxidesoxidation by gaseous oxygen occurs; the consequentdecrease in platinum active sites is thought to deter-mine the observed decrease in activity, with a progres-sive increase in the total oxidation products.
The catalytic activity was studied in a GHSV rangethat maximizes the H2 productivity, keeping a vol-ume of reactor consistent with the on-board utiliza-tion in PEFCVs. The H2 productivity is defined asmoles of hydrogen produced per volume of catalyticbed (including inert particles) per hour. If we con-sider the catalytic activity obtained with sample C,supposing that the activity would be kept constant dur-ing the reaction in undiluted air, an H2 productivityranging between 0.25 (at GHSV= 10,000 h−1) and1.68 moles H2/cm3 h (at GHSV= 80,000 h−1) can bederived.
To produce 5 Nm3/h of H2, related to a 5 kW fuelcell stack, from the above data, a POX catalytic bedvolume ranging between 0.88 and 0.13 l is derived; thispreliminary evaluation is consistent with an onboardutilization of the catalyst.
4. Conclusions
The evaluation of catalytic activity of Pt/CeO2 cat-alysts, differently prepared, for the partial oxidation ofmethane to syngas was reported. The results pointedout that the metal-ceria interaction leads to the for-mation of solid solution, which can contribute to theobserved activity and stability of the catalysts underoperative conditions of a demonstrative hydrogen gen-eration unit.
Acknowledgements
The authors thank Dr. L. Spadaro for the TPR mea-surements. The work was financial supported by theMIUR (Italian Ministry of University and Research).
References
[1] L. Carrette, K.A. Friedich, U. Stimming, Fuel Cells 1 (2001)5.
[2] D.L. Trimm, Z.I. Önsan, Catal. Rev. 43 (2001) 31.[3] Y. Jamal, M.L. Wyszynski, Int. J. Hydrogen Energy 19 (1994)
557.[4] J.C. Amphlett, K.A.M. Creber, J.M. Davis, R.F. Mann, B.A.
Peppley, D.M. Stokes, Int. J. Hydrogen Energy 19 (1994) 131.[5] S. Ahmed, J.P. Kopasz, B.J. Russel, H.L. Tomlinson, in:
Proceedings of the 3rd International Fuel Cell Conference,30 November–3 December 1999, Nagoya, Japan.
[6] J.R. Rostrup-Nielsen, Catalysis Science and Technology; J.R.Andersen, M. Boudart (Eds.), vol. 5, Springer-Verlag, NewYork, 1984, pp. 1–30.
[7] J.W. Jenkins US Patent 4,789,540 (1988).[8] J.W. Jenkins, E. Shutt, Platinum Metals Rev. 33 (1989) 118.[9] M. Krumpelt, T. Krause, J. Kopasz, D. Carter, S. Ahmed, in:
Proceedings of AICheE 2002 Spring Meeting, 10–14 March,New Orleans, LA, 2002.
[10] A.L. Dicks, J. Power Sources 61 (1996) 113.[11] A. Slagstern, H.M. Swaan, U. Olshye, I.M. Dahl, C.
Mirodatos, Catal. Today 46 (1998) 107.[12] V. Recupero, L. Pino, R. Di Leonardo, M. Laganà, G. Maggio,
J. Power Sources 71 (1998) 108.[13] D. Dissanayake, M.P. Rosynek, K.C.C. Kharas, J.H. Lunsford,
J. Catal. 132 (1991) 117.[14] Y.H. Hu, E. Ruckenstein, J. Catal. 158 (1996) 260.[15] T. Zhu, M. Flytzani-Stephanopoulos, Appl. Catal. A 208
(2001) 403.[16] D.A. Hichmann, E.A. Kaupfear, L.D. Schimdt, Catal. Lett.
17 (1993) 223.[17] K.H. Hofstad, A.O. Rokstad, A. Holmen, Catal. Lett. 36
(1996) 25.[18] E. Ruckenstein, H.Y. Wang, J. Catal. 187 (1999) 151.[19] P.D. Battle, J.B. Claridge, F.A. Copplestone, S.W. Carr, S.C.
Tsang, Appl. Catal. A 118 (1994) 217.[20] A.G. Andersen, T. Hayakawa, T. Tsunoda, H. Orita, M.
Shimizu, K. Takeira, Catal. Lett. 18 (1993) 37.[21] Q. Miao, G. Xiong, S. Seang, W. Cui, L. Xu, X. Guo, Appl.
Catal. A 154 (1997) 17.[22] H.C. Yao, Y.F. Yu Yao, J. Catal. 86 (1984) 254.[23] W. Liu, M. Flytzani-Stephanopoulos, Chem. Eng. J. 64 (1996)
283.[24] A. Trovarelli, G. Dolcetti, C. de Leitenburg, J. Kašpar, Stud.
Surf. Sci. Catal. 75 (1993) 2781.[25] A. Trovarelli, C. de Leitenburg, G. Dolcetti, Chemtech 32
(1997).[26] A. Trovarelli, Catal. Rev. Sci. Eng. 38 (1996) 439.

146 L. Pino et al. / Applied Catalysis A: General 243 (2003) 135–146
[27] W. Liu, M. Flytzani-Stephanopoulos, J. Catal. 153 (1995)304.
[28] W. Liu, M. Flytzani-Stephanopoulos, J. Catal. 153 (1995)317.
[29] L. Kundakovic, M. Flytzani-Stephanopoulos, J. Catal. 179(1998) 203.
[30] L. Kundakovic, M. Flytzani-Stephanopoulos, Appl. Catal. A171 (1998) 13.
[31] T. Bunluesing, R.J. Gorte, Appl. Catal. B 15 (1998) 107.[32] Y. Li, Q. Fu, M. Flytzani-Stephanopoulos, Appl. Catal. B 27
(2000) 179.[33] K. Otsuka, Y. Wang, E. Sunada, I. Yamanaka, J. Catal. 175
(1998) 152.[34] K. Otsuka, Y. Wang, M. Nakamura, Appl. Catal. A 183 (1999)
317.[35] P. Pantu, K. Kim, G.R. Gavalas, Appl. Catal. A 193 (2000)
203.[36] R. Cracium, B. Shreck, R.J. Gorte, Catal. Lett. 51 (1998) 149.[37] S. Sharma, S. Hilaire, J.M. Vohs, R.J. Gorte, H.W. Jen, J.
Catal. 190 (2000) 199.[38] P. Pantu, G.R. Gavalas, Appl. Catal. A 223 (2002) 253.[39] L. Pino, V. Recupero, S. Beninati, A.K. Shukla, M.S. Hegde,
P. Bera, Appl. Catal. A 225 (2002) 63.[40] F. Arena, F. Frusteri, A. Parmaliana, Appl. Catal. A 187
(1999) 127.[41] F. Frusteri, F. Arena, G. Calogero, T. Torre, A. Parmaliana,
Catalysis Comm. 2 (2001) 49.[42] P. Bera, K.C. Patil, V. Jayram, G.N. Subbanna, M.S. Hegde,
J. Catal. 196 (2000) 293.[43] M. Primet, M. El Azhar, R. Frety, M. Guenin, Appl. Catal.
A 59 (1990) 153.
[44] J.L. Duplan, H. Praliaud, Appl. Catal. A 67 (1991) 325.[45] A.K. Datye, D.S. Kalakkad, M.H. Yao, D.J. Smith, J. Catal.
155 (1995) 148.[46] S. Bernal, G. Blanco, J.M. Gatica, C. Larese, H. Vidal, J.
Catal. 200 (2001) 411.[47] P. Fornasiero, R. Di Monte, G.R. Rao, J. Kašpar, S. Meriani,
A. Trovarelli, M. Graziani, J. Catal. 151 (1995) 168.[48] C. de Leitenburg, A. Trovatelli, J. Kašpar, J. Catal. 166 (1997)
98.[49] B. Harrison, A.F. Diwell, C. Hallett, Platinum Met. Rev. 32
(1988) 73.[50] J.C. Vis, H.F.J. Van’t Blik, T. Huizinga, J. van Grondelle, R.
Prins, J. Catal. 95 (1985) 333.[51] S. Salasc, V. Perrichon, M. Primet, M. Chevrier, N.
Mouaddib-Moral, J. Catal. 189 (2000) 401.[52] D. Teschner, A. Wootsch, T. Röder, K. Matusek, Z. Paál,
Solid State Ionics 141 (2001) 709.[53] D.I. Kondarides, X.E. Verykios, J. Catal. 174 (1998) 52.[54] G. Wrobel, M.P. Sohier, A. D’Huysser, J.P. Bonnelle, J.P.
Marcq, Appl. Catal. A 101 (1993) 73.[55] G. Wrobel, C. Lamonier, A. Bennani, A. D’Huysser, A.
Aboukais, J. Chem. Soc., Faraday Trans. 92 (1996) 2001.[56] C. Lamonier, A. Ponchel, A. D’Huysser, L. Jalowiecki-
Duhamel, Catal. Today 50 (1999) 247.[57] S.J. Tauser, S.C. Fung, R.L. Garten, J. Am. Chem. Soc. 100
(1978) 170.[58] S. Bernal, J.J. Calvino, M.A. Cauqui, J.M. Gatica, C. Larese,
J.A. Pérez Omil, J.M. Pintado, Catal. Today 50 (1999)175.
[59] Y. Wu, T. Yu, B.S. Dou, C.X. Wang, X.F. Xie, Z.L. Yu, S.R.Fan, Z.R. Fan, L.C. Wang, J. Catal. 120 (1989) 88.




















