
53rd National Conference of Indian Society of Hematology ...
66
ABSTRACTS 53rd National Conference of Indian Society of Hematology & Blood Transfusion (ISHBT) 2012, 9–11 November 2012, Puri, India Ó Indian Society of Haematology & Transfusion Medicine 2012 Acute Leukemias Abstract P 001 Anti-Cancer Study of a Popular NSAID, Lornoxicam on Human Leukemic Cell Line and Human Hepatocellular Carcinoma Cell Line Subhadeep Roy 1 , Sayantan Dey 1 , Moumita Ray 1 , Nilanjana Deb 1 , Aparna Gomes 1 , Shila Elizabeth Besra 1 1 Drug Development/Diagnostic & Biotechnology Division, Indian Institute of Chemical Biology (CSIR), 4 Raja S.C. Mullick Road, Kolkata-700032, West Bengal, India Objective: The aim of the present study was intended on cancer because they are the most responsible disease for causing signifi- cant morbidity, mortality, and incurring healthcare costs worldwide. Recent data have expanded the concept that many cancers arise from sites of infection, chronic irritation and inflammation. These observations imply that anti-inflammatory agents should have a potential in both the prevention and treatment of cancer. These drugs can also be used as adjuvant to the currently available chemotherapy and radiotherapy. Lornoxicam is used for the treatment of various types of pain, especially resulting from inflammatory diseases of the joints, osteoarthritis, surgery, sciatica, and other inflammations but on cancer no work done so far. We have studied the anti-proliferative, cytotoxic and apoptotic activity of Lornoxicam on human leukemic cell line and human hepato- cellular liver carcinoma cell line. Method: We studied cell viability by Trypan blue exclusion, cytotoxicity study by MTT assay, morphological study by fluorescence microscopy and DNA frag- mentation was studied by gel electrophoresis on U937 cell line and HepG-2 cell line. Result: Lornoxicam significantly inhibited the cell viability and cytotoxicity (MTT) in a time and concentration dependent manner. The fluorescence showed characteristic features of membrane blabbing, chromatin condensation the sign of early and late apoptotic changes in the cells after treatment with Lornoxicam. Gel electrophoresis study showed fragmented DNA in the form of ladder. Conclusion: The present study reveals that the Lornoxicam possesses potent anti-leukemic activity. Studies are in progress to identify the mechanism of anti-leukemic activity of Lornoxicam. Abstract P 002 Studies with the Different Extracts of Ruellia tuberosa Leaves Against AML Patients’ Cells and Normal Human Lymphocytes Sayantan Dey 1 , Subhadeep Roy 1 , Moumita Ray 1 , Nilanjana Deb 1 , Aparna Gomes 1 , Chinmay Chowdhury 2 , Prithvish Banerjee 3 , Santanu Bose 3 , Shila Elizabeth Besra 1 1 Drug Development/Diagnostic & Biotechnology Division, Indian Institute of Chemical Biology (CSIR), 4 Raja S.C. Mullick Road, Kolkata-700032, India; 2 Department of Chemistry, Indian Institute of Chemical Biology, Council of Scientific and Industrial Research, 4 Raja S. C. Mullick Road, Jadavpur, Kolkata 700 032, India; 3 Employee State Insurance Hospital Sealdah, 301/3, A.P.C. Road, Kolkata-700009 Objective: In the present scenario, the demand for herbal products is growing exponentially throughout the world. This enthusiasm seems to be a result of people all over the world looking to various alternative systems of medicine, especially herbal drugs which are claimed to be safe, equally effective in comparison to allopathic drugs and which provide some answer to some of the chronic dis- eases like cancer. Ruellia tuberose leaf has a wide range of biological activities which includes anti-diabetic, anti-inflammatory, antipyretic, analgesic etc. Therefore, we have studied the anti-leu- kemic activity on AML patient’s cell and compared with normal healthy human leukocytes with methanol and methanol–water extracts of Ruellia tuberosa leaves. Methods: Collection, identifi- cation and extraction of Ruellia tuberosa leaves and designated as RTLE. AML PBMNCs was isolated by Histopaque (Sigma), cell viability by Trypan blue exclusion and cytotoxicity study by MTT assay in both the cells. Morphological study was determined by Fluorescence microscopy and gel electrophoresis of fragmented DNA was observed. Results: The cell viability and MTT assay showed the methanol and methanol–water extracts of RTLE sig- nificantly inhibit the PBMNCs of AML patients in a time and concentration dependant manner but no toxicity towards normal lymphocytes after 24 h treatment. The Fluorescence microscopic images show early and late apoptogenic changes in the leukemic cells than control, treated with IC50 doses. DNA bands confirmed apoptosis with both the treatment of RTLEs. Conclusion: The present study reveals that both the extracts of Ruellia tuberosa leaf have potent antileukemic activity. 123 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 DOI 10.1007/s12288-012-0199-y
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of 53rd National Conference of Indian Society of Hematology ...

ABSTRACTS
53rd National Conference of Indian Society of Hematology &Blood Transfusion (ISHBT) 2012, 9–11 November 2012, Puri,India
� Indian Society of Haematology & Transfusion Medicine 2012
Acute Leukemias
Abstract P 001
Anti-Cancer Study of a Popular NSAID, Lornoxicam on HumanLeukemic Cell Line and Human Hepatocellular Carcinoma CellLine
Subhadeep Roy1, Sayantan Dey1, Moumita Ray1, Nilanjana Deb1,Aparna Gomes1, Shila Elizabeth Besra1
1Drug Development/Diagnostic & Biotechnology Division, Indian
Institute of Chemical Biology (CSIR), 4 Raja S.C. Mullick Road,
Kolkata-700032, West Bengal, India
Objective: The aim of the present study was intended on cancer
because they are the most responsible disease for causing signifi-
cant morbidity, mortality, and incurring healthcare costs worldwide.
Recent data have expanded the concept that many cancers arise
from sites of infection, chronic irritation and inflammation. These
observations imply that anti-inflammatory agents should have a
potential in both the prevention and treatment of cancer. These
drugs can also be used as adjuvant to the currently available
chemotherapy and radiotherapy. Lornoxicam is used for the
treatment of various types of pain, especially resulting from
inflammatory diseases of the joints, osteoarthritis, surgery, sciatica,
and other inflammations but on cancer no work done so far. We
have studied the anti-proliferative, cytotoxic and apoptotic activity
of Lornoxicam on human leukemic cell line and human hepato-
cellular liver carcinoma cell line. Method: We studied cell viability
by Trypan blue exclusion, cytotoxicity study by MTT assay,
morphological study by fluorescence microscopy and DNA frag-
mentation was studied by gel electrophoresis on U937 cell line and
HepG-2 cell line. Result: Lornoxicam significantly inhibited the
cell viability and cytotoxicity (MTT) in a time and concentration
dependent manner. The fluorescence showed characteristic features
of membrane blabbing, chromatin condensation the sign of early
and late apoptotic changes in the cells after treatment with
Lornoxicam. Gel electrophoresis study showed fragmented DNA in
the form of ladder. Conclusion: The present study reveals that the
Lornoxicam possesses potent anti-leukemic activity. Studies are in
progress to identify the mechanism of anti-leukemic activity of
Lornoxicam.
Abstract P 002
Studies with the Different Extracts of Ruellia tuberosa LeavesAgainst AML Patients’ Cells and Normal Human Lymphocytes
Sayantan Dey1, Subhadeep Roy1, Moumita Ray1, Nilanjana Deb1,Aparna Gomes1, Chinmay Chowdhury2, Prithvish Banerjee3,Santanu Bose3, Shila Elizabeth Besra1
1Drug Development/Diagnostic & Biotechnology Division, Indian
Institute of Chemical Biology (CSIR), 4 Raja S.C. Mullick Road,
Kolkata-700032, India; 2Department of Chemistry, Indian Institute
of Chemical Biology, Council of Scientific and Industrial Research,
4 Raja S. C. Mullick Road, Jadavpur, Kolkata 700 032, India;3Employee State Insurance Hospital Sealdah, 301/3, A.P.C. Road,
Kolkata-700009
Objective: In the present scenario, the demand for herbal products
is growing exponentially throughout the world. This enthusiasm
seems to be a result of people all over the world looking to various
alternative systems of medicine, especially herbal drugs which are
claimed to be safe, equally effective in comparison to allopathic
drugs and which provide some answer to some of the chronic dis-
eases like cancer. Ruellia tuberose leaf has a wide range of
biological activities which includes anti-diabetic, anti-inflammatory,
antipyretic, analgesic etc. Therefore, we have studied the anti-leu-
kemic activity on AML patient’s cell and compared with normal
healthy human leukocytes with methanol and methanol–water
extracts of Ruellia tuberosa leaves. Methods: Collection, identifi-
cation and extraction of Ruellia tuberosa leaves and designated as
RTLE. AML PBMNCs was isolated by Histopaque (Sigma), cell
viability by Trypan blue exclusion and cytotoxicity study by MTT
assay in both the cells. Morphological study was determined by
Fluorescence microscopy and gel electrophoresis of fragmented
DNA was observed. Results: The cell viability and MTT assay
showed the methanol and methanol–water extracts of RTLE sig-
nificantly inhibit the PBMNCs of AML patients in a time and
concentration dependant manner but no toxicity towards normal
lymphocytes after 24 h treatment. The Fluorescence microscopic
images show early and late apoptogenic changes in the leukemic
cells than control, treated with IC50 doses. DNA bands confirmed
apoptosis with both the treatment of RTLEs. Conclusion: The
present study reveals that both the extracts of Ruellia tuberosa leaf
have potent antileukemic activity.
123
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
DOI 10.1007/s12288-012-0199-y

Abstract P 003
Apoptotogenic Activity of Sulfonoquinovosyldiacylglyceride(SQDG): A Constituent of Azadirachta indica (Leaves) AgainstPBMNCs of AML and CML Patients
Shila Elizabeth Besra1, Moumita Ray1, Subhadeep Roy1,Sayantan Dey1, Aparna Gomes1, Sukdeb Banerjee2, Nirup BikasMondol2, Prithvish Banerjee3, Santanu Bose3
1Drug Development/Diagnostic & Biotechnology Division, Indian
Institute of Chemical Biology (CSIR), 4 Raja S.C. Mullick Road,
Kolkata-700032, West Bengal, India; 2Chemistry Division, Indian
Institute of Chemical Biology (CSIR), 4 Raja S.C. Mullick Road,
Kolkata-700032, West Bengal, India; 3Employee State Insurance
Hospital Sealdah, 301/3, A.P.C. Road, Kolkata-700009
Objective: Cancer, a dreadful disease characterized by uncontrolled
growth and spread of abnormal cells without apoptosis, is a leading
cause of death worldwide. Elimination of cancer cells through
apoptosis is the key target of cancer therapy. A sulfonoglycolipid
identified as a sulfonoquinovosyldiacylglyceride (SQDG) isolated
from leaves of Azadirachta indica showed significant anti- leukemic
activity in U937 and K562 human leukemic cell lines. Therefore, we
studied the apoptogenic activity of SQDG against PBMNC of ALL
and AML patients. Methods: In vitro cell proliferation assay were
done by MTT assay, morphological determination of the cells
undergoing apoptosis by Fluorescent dye staining, DNA fragmenta-
tion by gel electrophoresis, quantization of apoptosis by flow
cytometric analysis and PBMNCs isolated by lymphocyte separating
fluid Histopaque (Sigma). Results: After treatment, SQDG signifi-
cantly inhibited the metabolically active cell growth and cytotoxicity
of both the AML and CML patient’s cells. The effect of SQDG on
normal human peripheral blood mononuclear was studied by cell
viability and cytotoxicity and was found to be lower than that on
AML and CML cells, indicating its specificity towards cancer cells.
The morphological study showed the characteristic features of
apoptotic changes in the treated cells than control cells after 24 h. The
induction of apoptosis was confirmed by using Annexin-FITC/PI
staining by flow cytometric analysis and fragmented DNA was found
in the form of ladder after treatment with SQDG. Conclusion: Study
reveals that, sulfonoquinovosyldiacylglyceride (SQDG) may be used
as novel chemotherapeutic agent in future for better treatment of
cancer without systemic toxicity.
Abstract P 004
Treatment Related Complications in Patients with AcuteLymphoblastic Leukemia
Rajesh Kashyap, Mukul Agarwal, Pradeep Kumar, GarimaAgarwal
Department of Hematology, SGPGIMS, Lucknow, UP
Aim: To study the different non-haematological complications
occurring during the late phase (2 weeks of induction therapy to
remission) treatment of ALL patients. Materials and Methods:Patients diagnosed with ALL and being treated at department of
haematology, SGPGIMS were subject of the study. The diagnosis of
ALL was made based on complete hemogram, bone marrow exami-
nation with cytochemistry and immunophenotyping by flow
cytometry. The clinical records of the patients were analysed for
occurrence of non-haematological complications occurring 2 weeks
after the start of therapy. The patients were treated as per the BFM
protocol (BFM 90 of BFM 95 protocol). Result: One hundred and one
patients had 147 events of non-hematological complications. Gas-
trointestinal toxicity was the most frequent complication and occurred
predominantly as hepatitis. Hyperglycemia was seen in 25 cases
(24.7 %) and was most frequent in patients above the age of 20 years.
Neurological complication was predominantly seen in patients below
the age of 20 years (76.2 %) and seizures was the most common
presentation. Osteonecrosis involving the hip joints was seen two
adult young males. Conclusion: Non-hematological complications
are quite frequent during the late phase of treatment of ALL patients.
The Gastrointestinal tract, nervous system and endocrine system most
frequently affected. Many of these events are missed because of low
levels of clinical suspicion. This study highlights the incidence of
these complications as they are associated with high morbidity and
mortality.
Abstract P 005: Poster Presentation
Acute Erythroid Leukaemia: A Clinico-Hematological Reviewof 5 Case Series
Nidhi Rai, M Deepak Nayak1, Chethan Manohar, Sushma VBelurkar
Department of Pathology, Kasturba Medical College, Manipal;1Department of Pathology, Melaka Manipal Medical College,
Manipal
Introduction: Acute erythroid-leukaemia is a rare form of acute
myeloid leukaemia (AML), characterized by abnormal proliferation
of erythroid precursors (proerythroblasts and basophilic erythro-
blasts). It comprises \5 % of AML cases. Objective: To review the
clinico-hematological features of erythroleukaemia cases with review
of literature. Materials and Methods: We report 5 such cases seen in
our institution over a period of 1 year. 4 out of these 5 cases presented
with pallor, easy fatigability and hepatosplenomegaly, while one case
reported with non-classical symptoms. Peripheral smear examination
and complete haemogram revealed pancytopenia with circulating
blasts ([20 %) in all five cases. Bone marrow study yielded [50 %
proerythroblasts and basophilic erythroblasts and dyspoiesis in other
lineages. Results: Final diagnosis of Erythroleukaemia (erythroid/
myeloid) was made in four cases and one case with non-classical
presentation was typed morphologically as Pure Erythroleukaemia.
Conclusion: Erythroleukaemia is an uncommon hematopoietic neo-
plasm. Among these, pure erythroleukaemia is seldom reported in
literature and is known to have poor response to standard chemo-
therapy. In the present case series, clinical presentation did not differ
from other types of acute myeloid leukaemia cases. These five cases
presented in the most unobtrusive manner. Thus awareness of its
hematological and morphological features is necessary to avoid a
diagnostic dilemma since treatment protocols and prognosis are
varied.
Abstract P 006
Clinical, Hematological, Cytogenetic & Molecular Profileof Acute Myeloblastic Leukemia
J Latha Fathima, S Sitalakshmi, Parimala Puttaiah, PoornimaD Rao, A Vanamala, AM Shanthala Devi, Karuna Ramesh Kumar
Department of Clinical Pathology, St. John’s Medical College
Hospital, Bangalore;
192 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Introduction: Acute myeloid leukemia is a disease resulting from the
clonal expansion of myeloid blasts in the peripheral blood, bone
marrow or in other tissue. It is a clinically, morphologically, geneti-
cally and prognostically heterogeneous disease. In India the incidence
of AML is 2.8 to 3.5 cases per 100,000 population per year. The
course and prognosis of the disease in the western countries and India
differs. Objectives: 1. To study the clinical and hematological fea-
tures of AML. 2. To study the cytogenetic and molecular
abnormalities of AML. 3. To compare the course and prognosis of the
disease with western countries. Methods: This retrospective study
was done from the year 2010 to July 2012 for a period of 30 months
in the department of Clinical Pathology, St.John’s Medical College
Hospital, Bangalore. The clinical details were retrieved from the
patient records in the medical records department. The laboratory
parameters were obtained from the department of Clinical Pathology.
Result: The total number of cases were 131. The age ranged from 2 to
84 years. The peak incidence was in fourth decade. The most com-
mon presentation was fever. Morphologically AML-M2 was the
common subtype followed by acute promyelocytic leukemia.
Immunophenotypically, CD 13 was the most commonly expressed
(62 %) myeloid marker. CD 7 was aberrantly expressed in 28 %.
PML-RARA translocation t(15:17) was the common cytogenetic
abnormality. One case of therapy related AML was also encountered.
Conclusion: The common subtype is AML M2 followed by APML.
The commonest cytogenetic abnormality noted is PML-RARA
translocation.
Keywords Acute myeloid leukemia, Acute promyelocytic leukemia,
Cytogenetics
Abstract P 007
Extensive Extramedullary Involvement in a Child with AcuteMyeloid Leukemia
Anand Prakash1, Athira Ramakrishnan2, Balasubramaniam2,Suravi Mohanty3, Anuradha Ananthamurthy3
1St Johns Medical College Hospital, Department of Pediatrics,
Bangalore, India; 2St Johns Medical College Hospital, Department
of Otorhinolaryngology, Bangalore, India; 3St Johns Medical College
Hospital, Department of Pathology, Bangalore, India
Purpose: Extramedullary involvement can occasionally be the pre-
senting feature of Acute Myeloid Leukemia in children. We report a child
who presented with features of extensive paranasal, orbital, lymph node
and dural involvement of chloromas with facial nerve paralysis. Method:Case Report: A 12 year old girl presented with fever, loss of hearing,
proptosis and headache of 2 months duration. Clinical examination
revealed mild left orbital proptosis with painless cervical lymphade-
nopathy. She had no hepatosplenomegaly. Cranial imaging showed dural
enhancement in the left frontoparietal region with extensive maxillary,
ethmoid and sphenoid sinus obliteration. There were minimal deposits in
both retro-orbital areas. Peripheral blood film and bone marrow aspirate
revealed myeloblasts of 10 %. A biopsy of the paranasal mass showed
features of a granulocytic sarcoma. Immunohistochemistry was strongly
positive for Myeloperoxidase and CD117 and was negative for CD20 and
CD3. The Ki 67 index was about 50 %. CSF was negative for malignant
cells. Conventional Cytogenetics was attempted but unsuccessful.
Results: The patient has completed chemotherapy (induction with cyt-
arabine and daunorubicin and consolidation with three cycles of high
dose cytarabine). She had a complete resolution of her proptosis, facial
palsy and the paranasal sinus mass. She is currently well and in remission.
Conclusion: Extensive extramedullary manifestations of AML can
present with paranasal sinus extension and cranial nerve palsy.
Abstract P 008: Poster Presentation
CD133 and MLL in the Same Cup
G Smeeta1, Anita Chopra1, C Jagan1, Sameer Bakhshi2,Sunu Lazar Cyriac2, Rajive Kumar1
1Laboratory Oncology Unit, 2Department of Medical Oncology,
Dr. B.R.A Institute Rotary Cancer Hospital, All India Institute
of Medical Sciences, Ansari Nagar, New Delhi
Introduction: CD133 positivity has been described in, but is rarely
ever emphasized as a facet of mixed lineage leukemia (MLL)+ pro-B
acute lymphoblastic leukemia (ALL). Nuclear cupping is a feature
that hardly ever finds mention outside acute myeloid leukemia
(AML). We present two cases where these coexisted. Case Sum-mary: The first patient, a 5-year old girl, diagnosed B-lineage ALL
4 years back and treated on UK-MRC-ALL-2003 protocol till 2010,
and symptom-free for 2 years, presented to our institution in June
2012 with fever and lymphadenopathy. Peripheral blood smear
showed 80 % blasts; with over 80 % having prominent cup-like
nuclear invagination. The second patient was a 10-month old male,
presented with fever and splenomegaly. Peripheral blood smear
showed 90 % blasts, 20 % exhibiting cup-like morphology. In both
patients, blasts were positive for CD34, CD45, CD19, HLA DR,
CD133, CD38, CD15, cCD79a, CD117dim and negative for CD10,
cCD22, CD20, CD13, CD33, CD56, CD2, CD4, CD64, CD14, cCD3
and cMPO. NG2 was positive in 10 and 100 % blasts in first and
second case, respectively. Both the patients were diagnosed as pro-B
ALL. MLL gene rearrangement was identified in both by fluorescent
in situ hybridization using a dual color, break apart rearrangement
probe. Conclusion: We believe ours is the first case showing florid
nuclear cupping in association with CD133 positivity and MLLrearrangement in a setting of pro-B phenotype. We conclude that
nuclear cupping, even when present in nearly every blast cell, may not
necessarily mean AML and may be a pointer toward CD133 positive
MLL translocated pro-B ALL. CD133 should be evaluated as part of a
B-lineage ALL flow cytometric panel with a view to define its role as
a reliable indicator of MLL rearrangement.
Keywords Cupping, CD133, MLLL
Abstract P 009: Oral Presentation
Acute Promyelocytic Leukemia—from Morphology to MolecularDiagnosis
Tathagata Chatterjee1, Srishti Gupta2, Ajay Sharma2
1Armed Forces Medical College, Pune-40; 2Army Hospital (R&R),
Delhi Cantt-10
Introduction: Acute promyelocytic leukemia (APL) is a distinct
subtype of acute myeloid leukemia (AML) with typical clinico-
hematological features. Cytogenetically, it is predominantly charac-
terized by balanced reciprocal translocation between chromosome 15
and 17 which results in fusion between promyelocytic leukemia
(PML) gene and Retinoic acid receptor a (RARa) gene. There are 3
possible isoforms caused by these translocations. The breakpoint in
chromosome 17 is consistently found in intron 2, but varies in
chromosome 15. The 3 breakpoints on the PML gene can occur at
intron 3 (L-long form), intron 6 (S-short form), and exon 6 (V form).
The study on molecular characterization was undertaken due to lack
of sufficient data in Indian patients. Aims and Objectives: (1) To
study the clinic-hematological and morphological profile in APL
patients. (2) To study molecular characterization of BCR subtypes in
Indian APL patients. Materials and Methods: A prospective study of
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 193
123

fifteen APL patients presenting to Army Hospital (R&R) were taken
for study between November 2010 to March 2012. The clinical fea-
tures, hematological parameters and morphology were analyzed.
Peripheral blood and bone marrow aspirate were stained with
Leishman-Giemsa, myeloperoxidase (MPO) and chloroacetate ester-
ase (CAE) and non specific esterase (NSE) using Merck’s Diagnostic
reagents. Flowcytometric evaluation was done on bone marrow
aspirate/peripheral blood using Beckman Coulter FC 500, 4 color
flow cytometer using standard lyse wash technique. For molecular
studies, peripheral blood sample was collected and Real Time PCR
was performed using Fusion Quant� kits for bcr-1, bcr-2 and bcr-3
and Rotor GeneTM 3000 software. Results: Patients presented with
fever, loss of appetite, bleeding manifestations (petechial, conjunctiva
hemorrhage, gum bleed, vaginal bleed and bleeding per rectum) and
pallor. Median age was 42 years, TLC—4,500/mm3, Hb—7.5 g/dl
and platelet—35,000/mm3. Three patients were micro granular vari-
ants and rest were hyper granular variants. All cases stained for MPO
and CAE and 33.33 % for NSE. Flowcytometric analysis revealed
classic high SSC with dim CD45 scatter with hyper granular variants
and low SSC with dim CD45 with micro granular variants. Molecular
analysis revealed BCR1 in 6/15 (40 %), BCR2 in 3/15 (20 %) and
BCR 3 in 6/15 patients (40 %). Conclusions: In our study no cor-
relation was found between age, sex, TLC, hemoglobin and platelet
counts with different BCR isoforms. There was no significant increase
in particular isoform in Indian population unlike published data.
Morphological, cytochemical, flow cytometry and molecular diag-
nosis were achieved within 48 h of admission.
Abstract P 010
Trisomy Chromosome 6 as a Sole Cytogenetic Abnormalityin Acute Myeloid Leukemia
Monika Gupta, Nita Radhakrishnan, Manoranjan Mahapatra,Renu Saxena
Department of Haematology, All India Institute of Medical Sciences,
New Delhi, India
Abstract: Identification of cytogenetic abnormalities plays an impor-
tant role in the diagnosis and prognosis of leukemias. Isolated trisomy 6
is a rare abnormality, the prognostic significance of which is not well
established. We report one case of acute myeloid leukemia (AML M5
variant) with trisomy 6 as the sole cytogenetic abnormality. Previously,
trisomy 6 has been reported in aplastic anaemia, myelodysplastic syn-
drome and AML and they are usually associated with hypocellular
marrow. However our patient had a very short history and a hypercel-
lular marrow infiltrated with blasts. We report this case due to the rarity
of the condition. More studies are required to ascertain the role of
trisomy 6 in the development of leukemia as well as in prognosis.
Abstract P 011
Clinical and Immunophenotypic Characterisation of Early T CellPrecursor Acute Lymphoblastic Leukemia: a First Studyfrom India
G Smeeta1, Anita Chopra1, C Jagan1, Lalit Kumar2,Atul Sharma2, Sameer Bakhshi2, Rachna Seth3, Ajay Gogia2,RM Pandey4, Rajive Kumar1
1Laboratory Oncology Unit, 2Department of Medical Oncology,
Dr. B.R.A. Institute Rotary Cancer Hospital, 3Department of
Paediatrics, 4Bioststistics, All India Institute of Medical Sciences,
Ansari Nagar, New Delhi
Introduction: Early T cell precursor acute lymphoblastic leukemia
(ETP-ALL), a newly identified subtype of T-ALL, is characterized by a
strikingly distinct immature immunophenotype: CD5-, CD1a, CD8-
and expression of C1 myeloid or stem cell markers (CD117, CD34,
HLA-DR, CD13, CD33, CD11b, and/or CD65) on C25 % of lympho-
blasts. It has been reported to be associated with poor prognosis. A very
few studies, none from India, have evaluated the clinical and prognostic
significance of ETP-ALL cases. Objective: To determine the incidence
and clinical significance of ETP-ALL. Methods: Fifty five cases of
T-ALL were retrieved from the records of Laboratory Oncology,
AIIMS. The clinical and immunophenotypic characteristics of all cases
were noted. The immunophenotypic markers used were: CD3, CD45,
CD5, CD8, CD1a, CD13, CD33, CD117, HLA-DR, CD34, CD65 and
CD11b. The cases were subclassified based on expression of CD5:
CD5+ and CD5-. These were further subclassified into CD8+/CD1a+
and CD8-/CD1a- subgroups. In all these groups, presence of myeloid
and/or stem cell markers were also noted. The remission rate, relapse
rate and overall survival of these groups were compared. Results: CD5
was positive in 48 and negative in 7 cases. In the CD5+ group, 31 cases
were CD8+/CD1a+ (myeloid/stem cell markers: positive 19 and nega-
tive 12) and 17 were CD8-/CD1a- (myeloid/stem cell markers:
positive 16 and negative 1). All CD5- cases (n = 7) were CD8-/
CD1a- and myeloid/stem cell markers positive. All CD5- cases
(12.7 %) fulfilled the criteria for ETP-ALL. ETP-ALL cases had a poor
remission rate as compared to non- ETP-ALL cases (40 vs. 86.7 %;
p = 0.042). Death rate was also higher in ETP-ALL group (28.6 vs.
6.25 %; p = 0.02). Conclusion: ETP-ALL is a distinct, high risk
subtype of T-ALL, that must be identified by correctly designed im-
munophenotyping panels, so that therapy appropriate to the disease and
distinct from the non-ETP- type T-ALL can be instituted. Recognition
of the disease provides a fresh and meaningful perspective on the
nebulous concept of lineage infidelity of acute leukemias.
Abstract P 012
Biphenotypic Acute Leukemia: A Study of Clinical,Hematological & Immunophenotypic Profile
Man Updesh Singh Sachdeva1, Manupriya1, Neelam Varma1,Subhash Varma2, RK Marwaha3
1Department of Hematology, 2Department of Internal Medicine,3Department of Paediatrics, Postgraduate Institute of Medical
Education & Research, Chandigarh
Background: Acute biphenotypic leukemia (BAL) is a rare neoplasm
comprising of blasts showing more than one lineage on multi-colour
flow cytometry. This prospective study was designed to identify cases
of biphenotypic acute leukemia and study their clinical and hemato-
logical profiles. Methodology: EDTA anticoagulated bone marrow
aspirate/peripheral blood samples of patients diagnosed as acute
leukemia on the basis of morphology were utilized for immunophe-
notyping. A comprehensive panel of fluorochrome labeled monoclonal
antibodies was used to identify the lineage of leukemic cells on flow
cytometry. The patients diagnosed to have BAL, on basis of WHO 2008
classification, were selected for analyses of their clinical, hematological
and immunophenotypic profile. Results: BAL represented 2.99 % (15/
501) of all cases of acute leukemia over 2 years. 47 % (7/15) were
children, all males, mean age of 5 years. 53 % (8/15) were adults,
M:F = 6:2, mean age of 21.4 years. 53 % (8/15) were diagnosed as
B/Myeloid and 47 % (7/15) were T/Myeloid. No correlation was
observed between age and immunophenotype of BAL. Fever and
194 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

lymphadenopathy were more frequently observed in BAL than ALL
and AML, respectively. Thrombocytopenia was less frequent as com-
pared to ALL or AML. On morphology, (73 %) 11/15 were diagnosed
as AML and 27 % (4/15) were diagnosed as ALL. Conclusions: BAL is
a rare type of acute leukemia (2.99 % of all our AL cases). Most of the
clinical, hematological and morphological parameters fail to clearly
predict its occurrence and a comprehensive panel of antibodies should
be used to identify this neoplasm known to have a poor outcome.
Abstract P 013
Isolated CNS Relapse of in a Case of AML-M2 MimickingPosterior Fossa Intracranial Haemorrhage—a Case Reportand Brief Review of Literature
K Kishore, UK Nath, SS Roy, P Chakrabarty, U Chaudhuri
Institute of Hematology and Transfusion medicine, Kolkata
Objective: This report is intended to stress the importance of looking
for subtle clinical signs of CNS RELAPSE during follow-up visits even
when the blood counts are normal in AML patients. Methods: This 17/f
a case of AML-M2, 46XX, NPM1-FLT3-ITD/Asp835 Negative patient
achieved CR after one 7 + 3 and was further treated with 3 cycles of
HIDAC. She was in CR post consolidation. During her follow up visit,
in spite of normal counts, she complained of mild headache and nausea.
Her CT scan of the brain revealed increased attenuation in the midline
posterior fossa with compressed IV ventricle and a provisional diag-
nosis of intracranial haemorrhage was given. In view of normal
coagulation parameters we reviewed the plates again and went in for
MRI and CSF analysis. Results: MRI showed an heterogeneous area in
the posterior fossa in superior vermis region suggestive of a mitotic
mass lesion. CSF also shows blasts confirming an isolated CNS relapse
as bone marrow is also in remission now. Conclusion: The patients who
were treated with only local therapy (intrathecal chemotherapy with or
without radiation therapy) had an overall survival rate of 31.5 %
compared to 21.4 % in patients treated with systemic therapy. The
AML subtype, CNS-1 status at diagnosis, age at relapse, cytogenetic
characteristics and the initial CT picture makes the present case unique.
Abstract P 014
Erythroleukemia (Erythroid/Myeloid): A Case Report
T Santosh, RK Bhola, A Choudhary, AK Bal, MK Patro, J Naik,B behera, S Paradhan
Department of Pathology, MKCG Medical College, Berhampur,
Odisha
Background: Acute erythroid leukemia is a rare form of acute
myeloid leukemia (AML) with predominant erythroid lineage pro-
liferation. It is a heterogeneous entity amongst AML that can occur at
any age, including childhood, and comprises less than 5 % of AML.
It’s defined as ‘‘a proliferation of more than 50 % erythroblast and
[20 % myeloblasts within nonerythroid cells.’’ AML-M6 is a het-
erogeneous disease with poor response to standard chemotherapy that
carries a poor prognosis. The new WHO classification subdivides
acute erythroid leukemia into erythroleukemia (erythroid/myeloid)
and pure erythroid leukemia. Allogeneic bone marrow transplant
should be considered upfront for appropriate candidates once remis-
sion is achieved in AML-M6, as the risk of relapse and mortality is
very high with this disease. Case Report: A 36 year Hindu female
presented with fatigue, generalized body aches, pain since last
6 weeks. The patient’s peripheral blood showed a leukoerythroblastic
blood picture with Hb of 3.4 g/dL, TLC 6,400/mm3, and Platelet
count of 89,000/mm3. Bone marrow aspirate and biopsy revealed a
markedly hyper cellular bone marrow replaced by erythroid precur-
sors, representing approximately 53 % of the marrow cells and
Myeloblasts representing 49 % of non erythroid cells. The erythroid
precursors displayed dysplastic morphology, including megaloblastic
features, multinucleation, nuclear lobation and budding, and they
were periodic acid-Schiff (PAS)–positive. So a diagnosis of acute
erythroid leukemia (erythroid/myeloid) was made.
Keywords Erythroid leukemia, Erythroid/myeloid
Abstract P 015
Study of Clinico-Hematological and Immunophenotypic Profilein Patients with Acute Lymphoblastic Leukemia in SCB MedicalCollege & Hospital, Cuttack, Odisha
Bhattacharyya Debmalya1, Das Sidhartha1, RK Jena2
1Department of Medicine, SCBMCH, 2Department of Clinical
Hematology, SCBMCH
Aims and Objectives: To study clinical, hematological and immuno-
phenotypic profile of patients with Acute Lymphoblastic Leukemia (ALL)
and their association with mortality and remission. Methods: 60 naıve
consecutive cases of adult ALL (C15 years) were taken for study. Sec-
ondary and relapse ALL cases were excluded. Detailed history taking,
clinical examination, hematological parameters, bone marrow study and
Immunophenotyping using bone marrow sample or peripheral blood by
Flow cytometer (BD FACS CALIBUR) were done in all cases. Patients
who survived were treated with MCP-841 protocol and hematological
remission was evaluated after completion of induction phase (1 month).
Results: 68.3 % cases were suffering from B-ALL and 31.7 % from
T-ALL. 58.3 % of cases were young adults (between 15 and 24 years of
age). Bleeding manifestation was significantly associated with B-ALL
(p\0.01) and lymphadenopathy was associated with T-ALL (p\0.01).
FAB-L1 and FAB-L2 morphology in bone marrow was found in 38.3 and
61.7 % of cases respectively. In patients with B-ALL, cCD79a was
positive in 100 % of cases, followed by CD19 (80.49 %), CD10
(85.37 %), CD34 (43.9 %). In patients with T-ALL, cCD3 was positive in
100 % cases, followed by CD7 (94.73 %), CD5 (63.16 %) and CD34
(21.05 %) 0.21.67 % of cases had myeloid co-expression, of which CD13
was the most common (53.84 %), followed by CD33 (38.46 %) and
CD117 (7.69 %) 0.24.4 % of B-ALL and 15.8 % cases of T-ALL had
myeloid co-expressions. 25 % of cases died before completion of induc-
tion phase and 46.7 % cases achieved complete remission (CR) after
4 weeks of induction phase. 21.7 % cases had partial remission and only
6.7 % cases had no remission. 60.97 % of B-ALL cases could achieve CR
whereas only 15.78 % of T-ALL cases could achieve CR (p\0.002).
Conclusion: B-ALL is more prevalent than T-ALL. For Immunophe-
notyping a panel of CD markers is required. Prognosis of B-ALL is better
than T-ALL.
Abstract P 016
Pattern of Occurrence of Childhood Leukemias in a TertiaryCentre in Lucknow: A Eleven Year Study
R Kushwaha, A Kumar, US Singh, K Archana
King George Medical University. Lucknow
Introduction: Pattern of childhood leukemia is known to vary through
out the world. We present clinico-pathological profile of children
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 195
123

presenting with Leukemia in our centre during a span of 10 years from
January 2001 to August 2012. This reflects the leukemia pattern in
Eastern U.P from where no such data is published till date. Materialsand Methods: A retrospective and prospective study of children pre-
senting with Leukemia was done from January 2001 to August 2012.
Clinical details like demographic data, age, sex and presenting symp-
toms were noted. Peripheral smear was made and bone marrow
aspiration was done. Smears were stained with Giemsa stain and were
studied for morphology of cells. FAB classification was used to classify
Leukemias based on morphological basis. Results: A total of 888 cases
of Leukemia were studied. Distribution of cases males and females
showed that it was more common in males (72.86 %). Youngest patient
of the series was 2 month child presenting with Chronic Myeloprolif-
erative Disorder, which is very rare in this age group. Acute Leukemia
cases were 89.86 %. Chronic Leukemia was very rare (10.13 %).
Among Acute Leukemia 64.53 % cases were of Acule Lymphoblastic
Leukemia, 32.20 % cases were of Acute Myeloid Leukemia and 3.25 %
case were of Acute Undifferentiated Leukemia. Cases of Acute
Leukemia were equally distributed in two age groups, i.e. 0–5 years and
6–10 years. ALL-L2 was the commonest type of ALL (59.7 %).
ALL-L3 was seen only in 3.37 % of cases. In acute myeloid leukemia
AML-M2 was more common. Conclusion: There is geographic vari-
ation in the incidence of leukemia. In our series we have observed that
ALL-L2 is the most common type of childhood leukemia. Cases were
equally distributed from 0 to 10 years.
Abstract P 017
A Rare Clinical Antithesis of Hope and Despair: A Case Report
P Pujari Ganesh, LS Raut, VV Bohara, GV Badarkhe, SS Ray
Institute of Hematology & Transfusion Medicine, Kolkata
Abstract: Myeloid sarcoma is an extramedullary tumour of immature
cells of granulocytic series, generally occurring in approximately 2 %
of patients with acute myeloid leukaemia. Myeloid sarcoma occurs
mostly in adults aged 45–55 years, and it has a predilection for the
bone, soft tissue, and skin. We here report a 28 year old female,
presenting with fever, menorrhagia and gum hypertrophy. She was
diagnosed as Acute Myeloid Leukaemia (M4) and was treated with
7 + 3 induction therapy. On day 14 post-induction, multiple nodular
skin lesions developed gradually over both lower extremities. The
biopsy from the lesions revealed Leukemia cutis. Post-induction day
14 marrow revealed no evidence of disease and medullary remission
was documented on day 28. However, the skin lesions increased.
With high dose Arabinoside and Mitoxantrone (HAM) the lesions
completely disappeared after 10 days. However, patient succumbed to
septic shock on day 18 of HAM. This case highlights the extremely
unusual concurrent medullary remission and extramedullary relapse
in a case of acute myeloid leukemia treated on induction therapy.
Keywords Myeloid sarcoma, Induction, Failure, Leukemia cutis
Abstract P 018
Presence of FLAER Negative Population in Acute Leukemia
Kotteeswari Kathirvel, Bargavi Balakrishnan, HarikrishnanBabu, Ansu Abu Alex, Rayaz Ahmed, Aby Abraham, AuroViswabandya, Biju George, Vikram Mathews, Alok Srivastava
Department of Haematology, Christian Medical College, Vellore
Background: Fluorescent-labeled bacterial aerolysin (FLAER) is the
single most reliable marker to monitor small PNH clones especially in
conditions such as myelodysplastic syndromes when traditional GPI-
linked surface marker expression can be significantly altered. Studies
with FLAER have described a sensitivity ranging between 0.5 and
1 % in identifying GPI-negative WBCs in samples from aplastic
anemia patients (Cytometry B Clin Cyto, 2007). Also it has been
shown that 5-15 % of PNH patients develop leukocyte dyscrasias
which invariably are acute myelogenous leukemia (Leuk Lymphoma.
1999). Objective: To detect a FLAER negative population in acute
leukemia samples by a single tube multiparameter flowcytometric
assay. Methods: Peripheral blood and bone marrow samples of
patients with newly diagnosed acute leukemia received for immu-
nophenotyping are taken for this study. In a single tube assay, we
have combined FLAER with CD33, CD14 and CD45 for the detection
of PNH clone on blast and neutrophils of patient samples and neu-
trophils of healthy control samples simultaneously. 20,000 events
were acquired and analyzed for all the cases using FACS Calibur.
Blasts and neutrophils were gated by CD45 versus side scatter (R1)
and CD33 versus side scatter (R2) as shown in Fig. 1a. Results: We
have analyzed 31 newly diagnosed acute leukemia among which 13
cases of AML and ALL each (41.9 %) and 5 cases of APL (16.12 %).
The median age of AML, APL and ALL cohorts are 29 years (range:
13–73), 27 years (range: 12–37) and 3 years (range: 0–28) respec-
tively. The healthy control reference range for FLAER expression on
neutrophils was 99.98 % (range: 99.79–100). The FLAER negative
clone was detected on both blasts and neutrophils of patient samples.
In AML the median range of FLAER-ve cells on blasts is 74.4 (range:
0.92–90.18) which is significantly higher than in ALL of 13.56
(range: 4.13–91.22) (p = 0.006) (Fig. 1b) whereas on neutrophils are
11.31 (range: 1–59.06) and 0.62 (range: 0–8.65) respectively
(p = 0.0003) (Fig. 1c). Conclusion: In our study FLAER negative
population were more common in acute leukemia and significantly
higher in AML than ALL. This preliminary observation warrants a
more detailed study on the biology, relevance and prognostic impact
of this population on treatment outcomes.
Abstract P 019
Study of Incidence of Recurrent Genetic Translocations in AdultAcute Myeloid Leukemia
Shano Naseem1, Neelam Varma1, Prateek Bhatia2, JogeshwarBinota1, Subhash Varma3, Pankaj Malhotra3
1Departments of Hematology, 2Paediatrics, 3Internal Medicine;
PGIMER, Chandigarh
Background: World Health Organisation (WHO) classification of
acute myeloid leukemia (AML) incorporates morphologic, immuno-
phenotypic, genetic and clinical features in its classification scheme
and is believed to be of more clinical relevance then the FAB clas-
sification as it defines entities that are biologically homogeneous and
prognostically relevant. The latest 2008 WHO classification
includes a separate category of AML with balanced translocation/
inversion, which comprises of 2 provisional and 7 categories of
characteristic genetic abnormalities, including: (i) t(8;21);RUNX1-
RUNX1T1, (ii) t(15;17);PML-RARA, (iii) inv(16);CBFB-MYH11,
(iv) t(9;11);MLLT3;-MLL, (v) t(6;9);DEK-NUP214, (vi) inv3;RPN1-
EVI1 and (vii) t(1;22);RBM15-MKL1. Of these, t(8;21);RUNX1-
RUNX1T1, t(15;17);PML-RARA and inv(16) CBFB-MYH11 have
been reported in higher frequency than other translocations. Western
literature quotes the incidence of fusion transcripts to be around
40–45 % in AML. However the data from Indian sub-continent is
scarce. We, therefore planned this study to detect the incidence of
common translocation/chimeric fusion transcripts in adult patients
with AML for t(8;21);RUNX1-RUNX1T1, t(15;17);PML-RARA, and
196 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

(inv16); CBFB-MYH11 using a single multiplex RT-PCR assay.
Materials and Methods: The present study was carried in the
department of Hematology, PGIMER, Chandigarh from May 2010 to
May 2012. Cases diagnosed as AML by bone marrow morphology,
cytochemistry and flow cytometry immunophenotyping were enrolled
in the study. For detection of fusion transcript, a single multiplex
RT-PCR assay was carried out using primers specific to the transcript
being tested. Results: During the study period 125 cases (male:
female ratio = 1.2:1) of AML were tested for above mentioned
fusion transcripts. Of these, 86 (68.8 %) cases were negative and 39
(31.2 %) cases were positive for any of the fusion transcript.
t(15;17);PML-RARA was most common seen in 19/125 (15.2 %)
cases, followed by t(8;21);RUNX1-RUNX1T1 in 17/125 (13.6 %)
cases and inv(16);CBFB-MYH11 in 3/125 (2.4 %) cases. Conclu-sion: This study evaluated the incidence of common recurrent genetic
translocations/chimeric fusion transcript in 125 cases of adult AML.
The incidence of various fusion transcripts was 31.2 %, with
t(15;17);PML-RARA and t(8;21);RUNX1-RUNX1T1 being more
frequent, seen at a frequency of 15.2 and 13.6 % cases respectively.
inv(16);CBFB-MYH11 was less common. Identification of these
transcripts provides therapeutically and prognostically relevant clin-
ical information.
Abstract P 020
A Study of Hematological and Genetic Profile of PrecursorB Lymphoblastic Leukemia/Lymphoma Harbouring BCR-ABLRecurrent Genetic Abnormality
Anita Tahlan1, N Varma1, S Naseem1, J Binota1, D Bansal2,P Malhotra3, RK Marwaha2, S Varma3
Departments of Hematology1, 2Pediatric Hemato-Oncology and3Internal Medicine, PGIMER, Chandigarh
Introduction: Approximately 3 % of children and 25–30 % of adults
with B lymphoblastic leukemia/lymphoma (B-ALL) are reported to
harbour t(9;22)(q34;q11) and/or BCR-ABL positivity. This subset of
patients is considered a poor risk subgroup. Aim: To determine the
hematological and genetic profile of precursor B-ALL with
BCR-ABL positivity. Materials and Methods: The study was con-
ducted from June 2010 to June 2012, at PGIMER, Chandigarh.
Peripheral blood and bone marrow examination and immunopheno-
typing were done. Molecular analysis by reverse transcriptase PCR
(RT-PCR) for BCR-ABL hybrid transcripts was performed on bone
marrow or peripheral blood samples. Results: A total of 302 patients
were diagnosed to have ALL, of which 257 were B-ALL. Seventeen
patients (6.6 %) positive for BCR-ABL transcripts were included in
the study. Median age was 26 years (range 1 year to 49 years). 13 out
of 17 patients were males (76.4 %) and four females (23.5 %). Seven
patients (41 %) were below 18 years of age. The total leucocyte count
ranged from 38.6 to 404.9 9 109/L (median 49.5 9 109/L). Bone
marrow morphology showed a heterogeneous population of immature
cells in 13 cases with prominent nucleoli, irregular to indented
nuclear membrane and cytoplasmic blebbing/vacuolations. The
immunophenotypic profile was positive for the lymphoid markers in
all cases. In addition five cases showed positivity for myeloid markers
CD13 and CD33, One case showed positivity for CD13, CD33 and
CD 117. RT-PCR for BCR-ABL transcript showed e1a2 in 12 cases
(70.5 %), b2a2 in 3 cases and b3a2 in 2 cases (29.4 %). Conclusions:The overall prognosis improves with the addition of tyrosine kinase
inhibitors, in the treatment protocols of BCR-ABL positive B-ALL. In
a resource constraint setting, an attempt should be made to evaluate
BCR-ABL if the morphology is heterogenous, cytoplasmic blebbing/
vacuolations are noted, along with aberrant positivity for myeloid
markers CD13 and CD33.
Keywords B-ALL. BCR-ABL transcripts
Abstract P 021
The Clinical Outcome of Children with Philadelphia chromosome(Ph) Positive Acute Lymphoblastic Leukemia (ALL) in a TertiaryCare Centre
KG Srinivas, L Appaji, Aruna Kumari, KC Lakshmaiah
Department of Medical and Pediatric Oncology, Kidwai Memorial
Institute of Oncology, Bangalore, Karnataka
Objective: To evaluate the clinical outcome of children with
Ph positive ALL who received chemotherapy without tyrosine kinase
inhibitors (TKIs) from 2004 to 2009. In the current era of TKIs this
study serves as historical reference to evaluate the therapeutic impact
of TKIs on the outcome of Ph positive ALL. Methods: 28 children,
age less than 15 years with Ph positive ALL, registered at Kidwai
Institute of Oncology, from 2004 to 2009, who received standard
chemotherapy alone, according to MCP 841 protocol were analysed
retrospectively. Outcomes were compared with 28 Ph negative ALL
children who have taken same treatment during the same period.
Matching was done for appropriate parameters. Philadelphia chro-
mosome positivity was confirmed by translocation t(9:22) by
cytogenetics. Results: In our study. 19 (67.8 %) were males and 9
(32.2 %) were females (M:F 2.1:1). 21 (75 %) were less than 9 years
and 7 (25 %) were between 9 and 15 years. Complete remission was
attained in 82.1 % of children after induction treatment (Day 28). 10
(35.7 %) had early relapse, among them 7 (25 %) had early bone
marrow relapse, 2 (7.1 %) had early CNS relapse, 1 (3.5 %) had early
testicular relapse. 3 (10.7 %) had late bone marrow relapse. There
were three deaths during induction due to sepsis. The Event free
survival (EFS) and Overall survival (OS) at 3 years was 42.8 and
52 % respectively. Median OS was 38 months. In Ph negative ALL,
EFS and OS at 3 years was 73 and 80 % respectively. The median
duration of follow up is 37 months (range 2 months to 90 months).
Fig. 1 Analysis of FLAER-ve subset in newly diagnosed acute
leukemias. a Representative gating strategy to quantify GPI deficient
populations in AML. b FLAER-ve (%) on CD45dim population in
newly diagnosed acute leukemias. c FLAER-ve (%) on neutrophils in
newly diagnosed acute leukemias
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 197
123

Conclusion: Children with Ph positive ALL have inferior outcome
with respect to EFS and OS when compared to Ph negative ALL
when given standard chemotherapy alone. Due to limited resources,
none of our children could afford transplantation. With the
encouraging results of combining Imatinib (TKI) with standard che-
motherapy in recent studies, these high risk children should be offered
combined modality of treatment.
Abstract P 022
Novel NPM Mutation in the 30-Untranslated Region Identifiedin a Patient with Acute Myeloid Leukemia
Vinodhini Kumaraswamy, Ajay Abraham, Ashok KumarJeyavelu, M Sathya, Vivi M. Srivastava, Biju George, AlokSrivastava, Vikram Mathews, Poonkuzhali Balasubramanian
Department of Haematology, Christian Medical College, Vellore
Abstract: Mutations in nucleophosmin (NPM) gene are known to
occur most frequently in normal karyotype acute myeloid leukemia
(NK-AML). The frequency of exon 12 NPM mutations in adult AML
patients’ ranges between 25 and 35 %, accounting for more than 50 % in
adult NK-AML. NPM is a nucleo-cytoplasmic shuttling protein initially
known to be involved in rearrangements in leukemia and lymphoma. The
NPM exon 12 mutations are mostly 4 bp insertions, which disrupts the
nucleolar localization signal (NoLS) at the C terminus of the NPM pro-
tein causing cytoplasmic accumulation of truncated NPM protein. The
most common among NPM mutation is type A, with the insertion of
tetranucleotide TCTG in the exon 12 seen in 75 to 80 % of cases. Type B
and D mutations (CATG and CCTG insertions respectively) are observed
in about 10 and 5 % of NPM mutated AML, other NPM mutations are
very rare. We report here a novel deletion detected in exon 12 of NPM
gene identified from a 41 year old male diagnosed with AML. Peripheral
blood smear of this patient revealed total WBC count of 1,200/mm3 with
7 % blasts. There was anemia (Hb 8.5 mg%) and thrombocytopenia
(Platelet counts 42,000/mm3). Bone marrow examination revealed FAB
AML-M2 based on morphology and immunophenotyping. He had tri-
somy 8 on cytogenetic analysis which placed him in the intermediate risk
category of AML. FLT3 ITD and NPM mutation detection was done
using genomic DNA sample at diagnosis by PCR followed by GeneScan
analysis. This patient was FLT3 ITD negative but there was a 5 bp
deletion seen in the NPM exon 12 based on GeneScan electropherogram
(Fig. 1). For further characterization of this mutation, the same sample
was PCR amplified and subjected to automated sequencing using ABI
genetic analyzer. DNA Sequencing revealed the presence of a novel 5
base pair (ATTTC) deletion in the exon 12 (Fig. 2). Unlike other exon 12
mutations, which are mostly insertions, this deletion is in the 30
un-translated region (30UTR) region, 45 bp downstream to the stop codon
(TAA) and hence will not result in the formation of a truncated protein.
This mutation was further confirmed by cDNA sequencing as well and
is in the position 930 from translational start site of the cDNA. NPM
RNA expression for this patient was checked by RQPCR and was found
not different from that of representative NPM type A, type B, type D and
NPM wild type subjects. Flowcytometric evaluation of the diagnostic
marrow showed 95 % positivity for CD34, which is against the previous
reports suggesting NPM mutation is associated with low CD34
expression. Bioinformatic analysis (http://www.microrna.org/) of NPM
30UTR region, where the mutation occurred, revealed potential loss of
binding site for miR208 due this 5 bp deletion. This patient underwent
conventional chemotherapy with daunorubicin and cytosine and
achieved CR1, received consolidation and is in complete remission
now. He did not develop any toxicity. This study reports a novel 30UTR
NPM mutation, which unlike the common NPM exon 12 mutations,
does not disrupt open reading frame but possibly affects the stability of
mRNA. Further characterization of this mutation is needed before we
comment on its clinical significance. The study also highlights the
importance of NPM gene sequencing in addition to GeneScan analysis
in order to identify the specific NPM mutation.
Abstract P 023
Impact of Cytogenetics on Outcomes of Pediatric Acute MyeloidLeukemia: Our Experience from a Tertiary Care Cancer Centrein South India
KS Rachan Shetty1, B Guruprasad1, L Appaji2, KC Lakshmaiah3
1Department of Medical Oncology, 2Department of Paediatric
Oncology, 3Department of Medical Oncology, Kidwai Memorial
Institute of Oncology, Bangalore 560029, India
Aim: To study the prognostic factors of paediatric Acute Myeloid
Leukemia (AML) with chemotherapy. Materials and Methods: 37
patients who had pathologically proven diagnosis of AML and had
received treatment at our institute from January 2007 to December
2010 were included for analysis. Patients were diagnosed based on
morphology, cytochemistry, cytogenetics and immunophenotypic
studies, molecular risk stratification was not done. AML patients
received 7 + 3 induction with Cytarabine and Daunomycin, followed
by 4 cycles of high dose Cytarabine consolidation, except for APML
(acute promyelocytic leukaemia) patients who received All Trans
retinoic acid (ATRA) with Daunomycin induction followed by 2
cycles of Daunomycin consolidation and maintenance with ATRA,
Methotrexate and 6-mercaptopurine. All patients were analyzed for
oncological outcome and these outcomes were correlated with initial
total leukocyte count and cytogenetics. Results: Majority of patients
had favourable cytogenetic abnormality. At the median follow-up at
24 months, (range 4–46 months), induction death rate was 27 and
73 % achieved complete remission. The relapse rate was 30 % and
event free survival rate was 43 %. Two year survival was 66.7 % for
favourable cytogenetics, 50 % for patients with intermediate risk, 0 %
for those with unfavourable cytogenetics. High tumor burden with
intermediate and unfavourable cytogenetics had significant correla-
tion with outcome. Conclusion: Tumor burden measured by initial
total count and WHO cytogenetic risk marker are the most important
prognostic factors even in paediatric AML. Even with advent of
molecular risk stratification cytogenetics remains the most important
risk factor.
Keywords Acute myeloid leukaemia (AML), APML (acute pro-
myelocytic leukemia), world health organisation(WHO) cytogenetic
risk matter
198 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Abstract P 024
Coding Variants of DNA Repair Genes and Its Associationwith De Novo Acute Promyelocytic Leukemia
GC Gaur1,2, SK Hasan1,2, T Ottone1,2, V Mantovani3,D Centonze4, F Lo-Coco1,2
1Department of Biopathology, University of Rome ‘‘Tor Vergata’’,
Rome, Italy; 2Laboratorio di Neuro-Oncoematologia, Fondazione
Santa Lucia, Rome, Italy; 3Centre for applied Biomedical Research,
St. Orsola-Malpighi University Hospital, Bologna, Italy; 4Clinica
Neurologica, Department of Neuroscience, University of Rome
‘‘Tor Vergata’’, Rome, Italy
Background: DNA is essential to life, but it is subject to damage
from interaction with various endogenous and exogenous agents.
Double-strand DNA breaks are arguably the most serious form of
DNA damage and are predominantly repaired through either the
homologous recombination (HR) or non-homologous end-joining
(NHEJ) pathways. Single nucleotide polymorphisms (SNPs) in dou-
ble-strand break repair genes may alter DNA repair capacity and, in
turn, confer predisposition to leukemia. We analyzed polymorphic
variants of DNA repair and detoxification genes in patients with acute
promyelocytic leukemia (APL). Methods: Using MassARRAY high-
throughput DNA analysis with matrix-assisted laser desorption/ioni-
zation time-of-flight mass spectrometry, we initially genotyped 26
APL patients and 561 healthy blood donors for 210 SNPs of 22 genes
mostly involved in DNA repair and drug detoxification. Based on
results of initial screening, we further analysed BRCA2, XRCC5,
NBN (formerly NBS1) and LIG4 gene in another 40 de novo APL
patients using DNA sequencing. Results: Based on our complete
cohort analysis, we identified 2 genes which were significantly
associated with risk of development of APL. We observed a differ-
ence in risk allele frequency between APL and Healthy controls for
NBN (rs1063045, NG_008860.1:g.6881G[A): 32.5 % and 45.5 %,
p = 0.003 and LIG4 (rs1805386, NG_007396.1:g.10970T[C):
12.1 % and 18.36 %, p = 0.07. The association of homozygous
variants of NBN yielded higher risk of APL (OR: 3.042, p = 0.003),
whereas T allele of LIG4 was found to be a susceptible allele in APL
(OR: 1.630, p = 0.07). Conclusions: Susceptibility to develop APL
may be linked to genetic variations in DNA repair genes that result in
inefficient repair of chemical or radiation induced genetic damage.
SNP rs1063045 of NBN gene could be an important factor which
plays an important role in NHEJ pathway repair in case of APL.
Abstract P 025
Sub Categorisation of T Cell Acute Lymphoblastic LeukemiaUsing Who Criteria—A Retrospective Analysis
BK Karthik Bommannan, MUS Sachdeva, Praveen Bose,Jasmina Ahluwali, Reena Das, Neelam Varma
Department of Hematology, Post Graduate Institute of Medical
Education and Research, Chandigarh
Introduction: According to the 2008-WHO CLASSIFICATION,
T-Acute Lymphoblastic Leukemia (T-ALL) has been subclassified
into PRO T-, PRE T-, CORTICAL T- and MEDULLARY T-ALL
based on immunophenotyping. Some studies indicate prognostic
significance of these subcategories. Aim: This study attempts to
subcategorize T cell acute lymphoblastic leukemia cases to assess
utility of WHO criteria for the subcategorization. Methodology: All
cases diagnosed as acute leukemia over a period of 3 years in the
department of hematology were screened for T cell ALL cases. An
attempt was made to subcategorize all T-ALL cases according to
WHO criteria and also to assess expression of aberrant markers.
Results: Out of 1740 acute leukemia cases diagnosed in 3 years,
29.1 % (507 cases) were of B-ALL and 3.1 % (54 cases) were of
T-ALL. Among the 54 T-ALL cases, a complete T-cell panel was
done in 33 cases. On applying stringent WHO criteria, 54 %
(18 cases) were classifiable and 45 % (15 cases) were not classifiable.
Among the classifiable cases, Pro T-ALL were 11.11 % (2/18), Pre
and Medullary T-ALL were 5.5 % (1/18) each, and Cortical T-ALL
were 77.77 % (14/18). In addition, aberrant expression of other
lineage markers including CD79a (5.5 %), CD13 (7.4 %), CD33
(7.4 %), CD117 (11.11 %), CD 19 (5.5 %), CD10 (18.5 %) were seen
in variable combinations. In addition, analysis of T Cell Receptor
subtypes done in 36 cases, showed 13.8 % of cases expressing TCR
alpha/beta and 50 % cases expressing TCR gamma/delta subtypes.
Conclusion: Among the cases examined, only 54 % of cases could be
subclassified following the present WHO criteria, indicating its lim-
ited utility in routine diagnostic use.
Abstract P 026
Clinicopathologic Profile of Leukemias in Infants—an IndianScenario
S Munot1, PG Subramanian1, S Gujral1, Y Badrinath1,A Kumar1, S Shinde1, S Mahadik1, P Amare-Kadam2, B Arora3,S Banavali3
1Hematopathology Laboratory, 2Department of Cytogenetics,3Department of Medical Oncology, Tata Memorial Centre, Mumbai
Introduction: Acute and chronic leukemias are rare in infants world-
wide, with very limited Indian data on their clinical and hematological
profile. We studied 50 consecutive cases of infant leukemias and the
clinical, morphology, cytochemistry, immunophenotyping and cytoge-
netic data was correlated. Objective: To study the incidence and
clinicopathological profile of leukemias in infants. Methods: A retro-
spective study was undertaken in our tertiary care cancer institute. The
data of all newly diagnosed cases of leukemias in infants from the year
2005 to 2012 was obtained from electronic medical records. Cases were
diagnosed either on peripheral blood, bone marrow aspirate or biopsy.
Results: Of the total 8,223 cases of newly diagnosed acute leukemias in a
7 year period, 49 were diagnosed in infants (0.5 %) It included 26 cases of
acute lymphoblastic leukemia, 10 cases of acute myeloid leukemia, 3
cases of juvenile myelomonocytic leukemia and1 case ofchronic myeloid
leukemia. 9 cases could not be subtyped for want of additional samples.
Only 2/49 cases occurred in neonates. There was a male preponderance
(34/49). A detailed immunophenotypic and cytogenetic analysis will be
presented in the study. Conclusion: This study provides important
insights in the spectrum and biological behavior of infant leukemias.
Abstract P 027
Role of Nrf2 and its Downstream Target Genes in In VitroSensitivity to Arsenic Trioxide in non-M3 AML
Sreeja Karathedath, Ajay Abraham, Savitha Varatharajan,Biju George, Alok Srivastava, Vikram Mathews, PoonkuzhaliBalasubramanian
Department of Haematology, Christian Medical College, Vellore
Background: Conventional chemotherapy with Cytarabine (Ara-C)
and Daunorubicin (Dnr) can cure about 25 % of patients with
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 199
123

Acute Myeloid Leukemia (AML) but majority fail to achieve long
term remissions. Drug resistance and relapse are considered major
causes of treatment failure. The toxic side effects of chemothera-
peutic drugs make therapy intolerable and inefficacious especially
in older patients. This emphasizes the need of developing new
therapeutic strategies to improvise the treatment both in terms of
cost and efficacy. Chemotherapeutic agents like Arsenic Trioxide
(ATO) brings cell kill by inducing Reactive Oxygen Species
(ROS). ATO has proved very effective in inducing remissions in
de novo acute promyelocytic leukemia (AML M3), but has shown
to have reduced activity in other AML subtypes. The redox sen-
sitive transcription factor, Nrf2 (Nuclear Factor Erythroid 2 Related
Factor) regulates oxidative stress in normal cells is an important
candidate which determine ROS mediated apoptosis. NRF2 trans-
criptionally regulates the levels of antioxidant response genes
((NADPH Quinone Oxidoreductase (NQO1), Heme Oxygenase
(HO-1), Glutamate Cysteine ligase (GCL) and drug efflux trans-
porters like Multi drug resistance related protein-2 (MRP2)) and
averts ROS mediated cytotoxic effects. Pharmacological modulation
of Nrf2 by Nrf2 inhibitors could be well exploited to provide better
treatment opportunities. We hypothesize that ATO insensitivity in
non-M3 AML cells is due to increased Nrf2 levels which could be
modulated. Materials and Methods: In vitro cytotoxicity to ATO
was done in NB4 (AML-M3), U937 (non-AML-M3) and
KASUMI1 (AML cell line with t(8;21)) cell line and primary AML
cells (n = 32) using MTT Assay. This assay was extended to cell
lines pretreated with pharmacological concentration (10 lM) of
Nrf2 Inhibitor, Luteolin. Nuclear protein extraction was done using
NE-PER kit (Pierce). Nuclear Nrf2 protein level was detected using
Western blot and normalized to b-lamin. RNA extraction from
AML cell lines and primary AML samples at diagnosis (n = 32)
was done using Trizol method and the expression of the down-
stream target genes (NQO1, HO1, GCLC, GCLM, MRP2) of Nrf2
was done using qRT-PCR. Results and Discussion: Based on the
MTT results, U937 was resistant compared to NB4 and Kasumi1
cell lines to ATO (IC 50 values; Table 1). Nuclear expression of
Nrf2 protein by western blot showed U937 cell line to have higher
nuclear Nrf2 compared to NB4 cell line (Fig. 1). To further iden-
tify the functional role of Nrf2, qRT-PCR analysis was done for the
downstream Nrf2 target genes NQO1, HO1, GCLC and GCLM.
ATO resistant U937 cell line showed 1.3, 3, 3.3 and 2.4 fold
increase in the mRNA levels of Nrf2 downstream target genes
NQO1, HO1, GCLC and GCLM respectively compared to ATO
sensitive cell line NB4 (Fig. 2). Pharmacological inhibition of Nrf2
by Luteolin pretreatment efficiently reduced the IC50 of ATO
resistant U937 cell line (Fig. 3). Nrf2 downstream target genes-
NQO1, HO1, GCLC and GCLM mRNA levels were analyzed in
primary AML cells by qRT PCR. Patients with ATO IC50 more
than the median (n = 16, IC 50 [ 2.37) had higher mRNA
expression for the above mentioned Nrf2 target genes when com-
pared to ATO sensitive patients (n = 16, IC 50 \ 2.37) though not
reaching statistical significance, probably due to low sample num-
ber. Conclusion: This pilot study extends the possibility of using
pharmacological inhibitors of Nrf2 to modulate ATO resistance in
AML.
Abstract P 028
Synchronous Presentation of Squamous Cell Carcinomawith Acute Promyelocytic Leukemia: Report of a Rare Case
Ayushi Sahay, Meena Desai, Vijay Hirani2, Pankhi Dutta1
SevenHills Hospital, Mumbai, 2Modern Haematology and
Chemotherapy Centre, Kolhapur, 1Kokilaben Dhirubhai Ambani
Hospital, Mumbai
Objective: Though rare, squamous cell carcinoma (SCC) has been
seen to occur in acute promyelocytic leukemia (APML) patients as a
consequence of treatment with arsenic trioxide. However, there is no
case reported in literature of simultaneous occurrence of these two
malignancies. We report a highly rare case. Method—Case Report:A 47 year old male presented with ulceration of the left inner cheek.
There was no other significant past history or treatment history. There
was history of regular tobacco chewing. Examination revealed a
buccal ulcer which was non tender along with a palpable and hard,
left submandibular lymph node. A biopsy done from the cheek ulcer
revealed a moderately differentiated SCC with nests and sheets of
moderately large malignant squamous cells irregularly infiltrating the
submucosa and muscle layer. A routine hematological evaluation at
this time revealed pancytopenia with Hb: 7.9 mg%; WBC: 660 cells/
mm3; platelet count: 53,000/mm3. A bone marrow (BM) examination
was done and showed highly cellular aspirates with near total
replacement of the marrow by abnormal, hypergranular promyelo-
cytes along with classical faggot cells. The corresponding BM
trephine biopsy showed near total replacement by sheets of immature
myeloid cells. A diagnosis of APML was made. PML-RARA testing
by gel PCR was positive for the bcr1 isoform. The patient refused any
specific treatment and expired after 8 days at home. Conclusion: To
the best of our knowledge, this is the first case in literature of syn-
chronous presentation of SCC with APML and highlights the
importance of bone marrow examination in carcinoma patients with
pancytopenia.
Abstract P 029
Mixed Phenotype Acute Leukemia—a Growing Concern
Shelly Poddar, SM Sethy, P Mohanty, RK Jena
Department of clinical Hematology, Department of Pathology,
SCB Medical College
Aim of the study: To study the incidence of Mixed Phenotype Acute
Leukemia (MPAL) out of 253 cases of Acute Leukemia studied.
Objective: To correlate the haematoclinical presentation with
cytomorphology, cytochemistry, immunophenotyping and treatment
200 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

outcome. Materials and Method: We evaluated 253 cases presenting as
Acute Leukemia. Each of these were completely evaluated by a
complete blood count, bone marrow aspiration, MPO &PAS positiv-
ity, multiparametric flow cytometry. Then they were evaluated for the
treatment response. Observation: Out of 253 cases of Acute Leu-
kemia 5 cases came out to be of Mixed Phenotype Acute Leukemia
(MPAL). Of these 5 cases morphologically 3 were AML and 2 were
ALL. They received a treatment plan of 3 + 7 CT followed by
MCP841. In all of these cases outcome was poor, all patients died
either before or after induction therapy. Discussion: MPAL accounts
for about 2 % of acute leukemias and can occur in both children and
adults but more common in adults. It can present morphologically as
either AML or ALL. So to distinguish these group of patients we
need to go for a immunophenotyping. In our study we have seen that
there is 100 % mortality rate. We can conclude that this leukemia has
a poor prognosis.
Slno.
Age/sex
PSC BM Immuno-phenotyping
Treatment Outcome
1. 1.8 mn/M
[80 %blast AML-M6
MPAL MCP841 Expired duringinduction
2. 27 years/M
Acleukemia
ALL MPAL 3 + 7CT IncompleteCR expiredbeforenext CT
3. 70 years/F
[50 %blast AML-M5
(B + T)AntiMPO-ve
Oral CT Expiredduringinduction
4. 51 years/M
Acleukemia
AML-M1
MPAL Planned for3 + 7 CT
Expired beforeinduction
5. 32 years/F
Acleukemia
ALL MPAL Received3 + 7CTfollowedbyMCP841
Cr after 2ndCT butexpireddue toinfectionafter4 months
Abstract P 030
Immunophenotypic Analysis of Transient Abnormal Myelopoiesisin 5 Children with Down Syndrome
V Baloda1, S Gujral1, PG Subramanian1, Y Badrinath1,A Kumar1, P Amare Kadam2, S Banavali3, B Arora3
1Department of Hematopathology, 2Cytogenetics Department,3Department of Medical Oncology, Tata Memorial Hospital, Mumbai
Objective: To study the immunophenotypic profile of transient
abnormal myelopoiesis (TAM). Materials and Methods: Retro-
spective analysis of cases reported as TAM during 2007-2012.
Results: 5 children (3 females, 2 males) with Down Syndrome with
age between 5 and 45 days presented with a high WBC counts and
high percentage of blasts in peripheral blood and/or bone marrow.
Blasts were MPO negative. Immunophenotypic analysis using multi
color flow cytometry was performed. Table 1 delineates the expres-
sion pattern of various markers on the blast population. The blasts did
not express any of these B or T cell markers—CD19, CD10, CD20,
CD3 and CD2 (2/2). Blasts expressed CD4 in one case. Conclusion:Transient abnormal myelopoiesis presented in Down syndrome chil-
dren within 45 days of birth in our series. CD34, CD13, CD33,
CD117, CD41, CD61, CD7 and HLA-DR are useful markers for
characterization of blasts of TAM.
Abstract P 031
Volume, Conductivity, Scatter: Simply Ornaments or DiagnosticTools of Acute Leukaemias: A Retrospective Analysis of 108Cases of Acute Leukaemias in a Tertiary Oncology Centre
Monali Gupta, DK Mishra, Mammen Chandy
Department of Lab Hematology, Tata Medical Centre, Kolkata
Introduction: The Automated Hematology analyser Coulter LH-780
uses a combination of three measurements Volume, Conductivity, Scatter
(VCS) to identify WBCs in their near native state but it could take
advantage of these parameters to evaluate their morphologic changes.
Objectives: The aim of this study was to investigate whether VCS can
become a new high-throughput screening method not only for the detection
but also for the exact categorization of Acute Leukaemias. Materials andMethods: VCS parameters of 108 diagnosed cases of acute leukaemias
analysed by Beckman Coulter LH 780 were studied retrospectively. The
CBCs, scatterplots, flags were correlated with VCS to calculate their
sensitivity and specificity alone and in combination. VCS trends and
characteristic cluster patterns were identified in 67 cases of ALLs, 7 AP-
MLs, 34 Non APMLs, 6 HLA-DR negative AMLs, 3 cases each of AML
with myelodysplasia related changes and Acute leukaemia of ambiguous
lineage. Results: High MLV, SD volume and high MLS and SD scatter
was the characteristic VCS trend observed in 61 cases of ALLs while 5
cases deviated from the trend. AMLs showed high MNV and SD but low
mean and SD scatter. APMLs exhibited least mean and SD scatter values in
spite the hypergranularity of abnormal promyelocytes. Interestingly AML
M5 cases exhibited high Monocyte mean scatter and normal SD scatter
despite the fact that the monoblasts are agranular! A tight merging cluster
pattern was seen in all 3 cases of HLA-DR negative AMLs. The current
study also found ‘‘cluster patterns’’ in 66 cases of ALLs and their associ-
ation with presence of aberrant myeloid antigen marker on
immunophenotyping was statistically significant (p value \0.001).
Conclusions: VCS alone proved to be a good diagnostic tool in ALLs as
the sensitivity was 91.8 % and specificity was 80 % while in AMLs the
sensitivity was 94.1 %. There were no AML cases wherein peripheral
blood did not have blasts, so specificity could not be calculated.
Keywords Beckman coulter LH 780, VCS, Mean neutrophil volu-
me(MNV), Mean Lymphocyte volume (MLV), Mean lymphocyte
scatter(MLS)
Abstract P 032
Minimal Residual Disease in Paediatric B Lineage AcuteLymphoblastic Leukemia: Comparison of Mid—InductionPeripheral Blood and Bone Marrow Samples Using Six ColourFlow cytometry
BK Karthik Bommannan1, MUS Sachdeva1, Parveen Bose1,Neelam Varma1, Deepak Bansal2, RK Marwaha2
1Department of Hematology, 2Department of Paediatrics, Post
Graduate Institute of Medical Education and Research, Chandigarh
Introduction: There is strong correlation between MRD levels and
risk of relapse in childhood leukemias. Bone marrow aspirate sample
on day 15 of induction is commonly used in paediatric cases to assess
MRD levels for further management decisions. Peripheral blood
sample might be used for the same if proven to yield similar MRD
levels. Aim: To compare MRD levels in paired peripheral blood and bone
marrow samples of paediatric B-ALL patients on day 15 of induction
using six colour flow cytometry. Methodology: Five newly diagnosed,
CD19 and CD10 dual positive, paediatric B lineage ALL patients were
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 201
123

analyzed on day 15 of chemotherapy for peripheral blood and bone
marrow MRD levels. Lyse-stain-wash technique was used to process
single 6 colour tube (CD19PECy7/CD10APC/CD20PerCP/CD34PE/
CD45APCH7/Syto13). One million events were acquired on BDFACS
Canto II and analysed by BDFACS Diva software. Results: MRD levels
of[0.01 % (range 0.02–0.81 %) were seen in all 5 bone marrow sam-
ples. All but one peripheral blood samples showed presence of
MRD [ 0.01 % (range 0.04–0.36 %). Although, the levels of MRD vary
slightly between bone marrow and peripheral blood samples, presence of
peripheral blood MRD in all but one case, may favour using peripheral
blood on day 15 for MRD detection instead of invasive bone marrow
procedure. Conclusion: There is good concordance (4 out of 5 cases) of
MRD level detection between mid-induction samples of peripheral blood
and bone marrow aspirate for paediatric B-ALL cases. This result needs
to be supported by more number of cases.
Abstract P 033
Monosomy 7 in Acute Myeloid Leukemia in India
S Yuvarani1, Usha Sitaram3, Rayaz Ahmed2, Aby Abraham2,Auro Viswabandya2, Biju George2, Vikram Mathews2,Alok Srivastava2, Vivi M. Srivastava1
1Cytogenetic Unit, 2Department of Haematology, 3Department
of Transfusion Medicine and Immunohaematology
Background: Monosomy 7 is seen in 5–10 % of all acute myeloid
leukemia (AML) and is associated with a poor prognosis. We describe the
cytogenetic features of monosomy 7 in AML. Patients and Methods:G-banded karyotypes of all patients with AML and monosomy 7 seen in
the Department of Haematology, Christian Medical College, Vellore
between January 2003 and December 2009 were studied. Cytogenetic
findings were correlated with clinical and laboratory features. Results:Monosomy 7 was seen in 71 patients (5.4 % of 1314 patients with AML),
52 of whom were males (73 %). There were 57 adults (median age:
39 years, range: 1–72). The median hemoglobin was 7.95 g/dl (range:
2.5–15.8); median WBC count, 6.4 9 109/L (range: 0.6 -271); median
platelet count, 25.5 9 109/L (range: 3–401). The most common AML
subtypes were AML-M2 (27 %) and AML, not otherwise specified
(29 %). Dysplasia was seen in 46 % of cases while 10 % of these cases
were of secondary AML. Monosomy 7 was the solitary abnormality in 19
patients (27 %). Complex karyotypes (two or more additional abnor-
malities) were seen in 38 (53 %). These included monosomy 5 (15 %),
deletion 5q (8 %), trisomy 8 and monosomy 17 (7 %), and trisomy 21 and
inversion 3 (6 %). Follow-up data was available for 20 of these patients.
Only two of them survived more than 8 months. Conclusion: This is the
first report of a large series of AML patients with monosomy 7 from India.
While the incidence (5.4 vs. 3–10 %) and poor outcome are similar to
other reports in the literature, these patients are younger (median age: 39
vs. 45–55 years) and show a higher male preponderance (2.3:1 vs. 1.5).
Myelo proliferative Neoplasms
Abstract P 034
JAK2 (V617F) Negative Myeloproliferative Neoplasms—Prevalence and Points to Ponder
Puttaiah Parimala, Karuna Rameshkumar and Cecil Ross1
1Department of Clinical Pathology and Hematology, St John’s
Medical College Hospital, Bangalore
Introduction: Myeloproliferative neoplasms (MPN) are characterised
by clonal proliferation of one or more of myeloid lineages and it is
known mutations in JAK 2 signaling contribute to pathogenesis.
There is a significant group of patients who are JAK2 (V617F) neg-
ative. Aims and Objectives: To study prevalence, clinical and
laboratory features of Philadelphia chromosome negative and JAK2
(V617F) negative myeloproliferative neoplasms. Materials andMethods: From January 2011 to July 2012, patients who fulfilled
2001 WHO criteria of MPN were included. Clinical features and
laboratory profile including complete hemogram, bone marrow aspi-
ration and biopsy were analysed. FISH for Philadelphia chromosome
and JAK2 mutation by allele specific PCR were done for further
characterization. Results: Among 44 patients diagnosed as MPN, 27
patients were diagnosed as JAK2 (V617F) negative. Polycythemia
Vera was most frequent (59 %) followed by idiopathic myelofibrosis
(26 %) and essential thrombocythemia (15 %). Male predominance
was noted (92 %). JAK 2 positive patients were older and had higher
frequency of splenomegaly. In comparison, JAK2 negative patients
had a higher platelet count in ET (11.6 ± 2.4 9 109/L) and isolated
erythrocytosis (19.4 ± 1.38 g/dl) in 94 % of patients with PV. Two
of them had presented with thrombosis. Comment and Conclusions:Mutations at exon 12 associate with erythropoietin signaling have
been reported in JAK 2 (V617F) negative PV in literature. The
present study reinforces the findings of our previous study. In addi-
tion, it highlights need to look at other mutations among JAK2
(V617F) negative myeloproliferative neoplasms, which may be
involved in activation of other signaling pathways.
Abstract P 035
Myelofibrosis—an Unusual Manifestation of HIV Infectionin a Child
Rohini Gupta2, Gunjan Mahajan1, Jagdish Chandra2, SunitaSharma1, Anju Seth2, Bhavna Dhingra2, Praveen Kumar2
1Department of Pathology, 2Department of Paediatrics, Lady
Hardinge Medical College and associated Hospitals, Delhi
Introduction: Myelofibrosis is a myeloproliferative disorder in which
abnormal type of bone marrow stem cells cause fibrosis. Secondary
Myelofibrosis can be associated with haematological malignancies,
infections, endocrine and connective tissue disorders. However,
Myelofibrosis in HIV infected children is very rare. Case Details: A
female child admitted postnatal for umbilical cord bleeding was given
blood transfusion in 2004. Patient remained asymptomatic till 4 years
of age when she presented with anasarca, acute gastroenteritis and
sepsis. On examination, patient had severe anemia, hepatospleno-
megaly and bilateral crepitations on auscultation. On investigations,
patient had normocytic anemia (Hb-6.6 g/dl, MCV-84.6 fl), throm-
bocytopenia (platelet-61 9 103/ll) and normal total leucocyte count
(TLC-6.7 9 103/ll). X-ray chest showed bilateral infiltrates in lungs.
HIV serology was positive. A probable diagnosis of tuberculosis with
HIV was made. CD4 counts were 602/mm3 (CD4 % = 22 %).Patient
was put in HIV clinical stage 3 and started on ATT. On further follow
up, the anemia and thrombocytopenia worsened, leucopenia devel-
oped and CD4 counts reduced to 255/mm3 (15 %). ART was thus
started on 11/01/2011, but pancytopenia of patient did not improve.
Serum vitamin B12 and folate levels were normal. Bone marrow
aspiration was diluted. Bone marrow biopsy was hypocellular for age
and showed focal islands of hematopoetic cells. Megakaryocytes were
reduced in number. On reticulin stain bone marrow showed grade 3
fibrosis. Von Gieson stain showed focal areas of fibrosis. Thus a
diagnosis of Myelofibrosis secondary to HIV infection was made.
Patient was started on thalidomide and steroids for 3 months and then
continued only on steroids. The peripheral blood count improved (Hb-
12.7 g/dl, TLC-5.8 9 103/ll, platelet-141 9 103/ll). She remained
asymptomatic till date and did not require blood transfusion.
202 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Conclusion: Myelofibrosis as presenting feature in HIV infection is
rare. Although a few case reports of Myelofibrosis secondary to HIV
infection in adult patients are present in Indian literature, none has
been documented in children to the best of our knowledge.
Stem Cell Transplant
Abstract P 036: Oral Presentation
Stemming Role of Stem Cell Transplantation in MultipleMyeloma: Who, When, and What Type?
Deepak Bansal
Maharishi Markandeshhwar Institute of Medical Science & Research,
Mullana
Abstract: High dose chemotherapy with autologous stem cell trans-
plantation has shown improved progression-free survival (PFS) over
the conventional chemotherapy regimens. In the era of novel agents
for myeloma in conjunction with the evolution of hematopoietic stem
cell transplantation, many new questions arise. How to incorporate
these novels into the transplant paradigm? Should transplant be
delayed until relapse? In earlier eras, autologous trans- plantation was
generally reserved for patients younger than 60 or 65 years. In this
modern era, should there be an age limit for myeloma transplant?
Now that medicine is individualized for every patient, chronologic
age alone should not drive decisions regarding transplantation. What
type of transplant (i.e., autologous compared with reduced intensity
allogeneic transplant) is best? Several large international trials have
demonstrated conflicting results in regard to an overall survival (OS)
benefit with the allogeneic approach. The role of allogeneic transplant
remains under study especially in the high-risk population, which has
high relapse rates with traditional autologous approaches. Future
directions to reduce relapse include post-transplantation consolidation
and maintenance therapy with either approved agents or new agents
and immunotherapy, either vaccine based or natural killer (NK) and
T-cell based.
Abstract P 037
Effect of GSTA1*B Polymorphism on Pharmacokineticsof Intravenous Busulfanin Patients Undergoing HematopoieticStem Cell Transplantation
S Gopinath, M Ezhil Pavai, Biju George, Vikram Mathews,Alok Srivastava, B Poonkuzhali
Department of Haematology, Christian Medical College, Vellore
Background: Busulfan in combination with cyclophosphamide is
widely used conditioning regimen in Hematopoietic cell transplan-
tation (HSCT) for various malignant and non-malignant disorders.
Wide inter-patient variability in busulfan systemic exposure is
observed with oral busulfan due to unpredictable intestinal absorption
and differences in hepatic metabolism. To overcome the problems
associated with oral administration, intravenous (i.v) busulfan was
introduced in early 2000. Busulfan is mainly metabolized in the liver
by glutathione S-transferases (GSTs). We have shown that GSTA1*B
polymorphism explained the inter-patient variability in busulfan
exposure and clearance in homozygous beta thalassemia patients
receiving oral busulfan (ASH 2009, ASH 2010). There is no report on
pharmacokinetics of i.v busulfan from Indian patients undergoing
HSCT. The aim of the present study is to document the PK of i.v
busulfan and to compare the first dose AUC and clearance in with the
GSTA1*B polymorphism. Methods: Sixty patients receiving i.vbusulfan once daily (n = 44) or four times daily (n = 16) as part of
their conditioning regimen before transplantation between June 2010
and July 2012 at the department of hematology, Christian Medical
College, Vellore were included in this study. Genomic DNA was
isolated from the peripheral blood MNCs before transplantation and
GSTA1*B polymorphism was analysed using PCR–RFLP method.
Peripheral blood samples were collected at different time points (0, 3,
4, 5 and 7 h for Q24h and 0, 1, 2, 3 and 4 h for Q6h) after busulfan
infusion. Busulfan plasma levels were measured using previously
reported LC–MS/MS method. The area under the concentration ver-
sus time curve (AUC) and clearance of busulfan after the first dose
was calculated as reported previously (Desire et al., IJMR, in press).
Based on the AUC of the first day, subsequent doses were adjusted to
achieve the target AUC. The AUC and clearance of busulfan were
compared with the GSTA1*B genotype using Mann–Whitney U test.
Results: Patient demographics and busulfan AUC and GSTA1*B
genotypes are listed in Table. There are contradictory reports on the
influence of GSTA1*B polymorphisms on i.v busulfan AUC and
clearance in adult patients as well as in children. We report here that
GST polymorphism does not significantly influence the first dose
AUC or the clearance of i.v busulfan, though in a small number of
patients. This is the first report from Indian population comparing
GST polymorphism and pharmacokinetics of i.v busulfan. We con-
clude that, along with GST polymorphisms, there may be other
genetic variants explaining the inter-individual variability in busulfan
systemic exposure and clearance, which needs to be evaluated.
Table 1 Immunophenotypic profile of blasts
Case 1 Case 2 Case 3 Case 4 Case 5
Age at
presentation
5 days 45 days 11 days Neonate 40 days
Sex M M F F M
Blast % 80 (BM) 57 (BM)
77 (PB) 59
(PB)
20 (PB)
MPO - - - - -
CD 34 + + + + +
CD 13 - - + - -
CD 33 + + + + +
CD 117 + + + - +
Anti MPO ND - + ND -
CD 41 + - + WK + Inconclusive
CD 61 + - + WK + Inconclusive
CD 14 - ND - ND ND
CD 235a - - ND ND ND
CD 7 - + + - +
CD 56 + - + - -
HLA DR - + - - -
M male, F female, ND not done, WK+ weak positive, BM bone
marrow, PB peripheral blood; +, positive; -, negative
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 203
123

Diagnosis N = 60 Bu
dosing
GST
A1*B
AUC (lmol) Clearance
(L/h/kg)
AML 19 Q24H (n = 44)
Median age—32 years
(1–58 years)
Variants—19
Median = 4,026(1,713–10,456)
Median—0.186(0.108–0.510)CML 9
CMML 1
MDS 8
PreB-ALL
1 Wildtype—21
Median = 3,554(1,990–10,450)
Median—0.240(0.097–0.494)
Severe AA 1 p value 0.3313 0.2361
PNH 2
Ph +ALL
1
JMML 1
SCID 1 Q6H(n = 16)
Median age—4 years
(2–7 years)
Variants—07
Median = 875(285–1,456)
Median—0.245(0.163–0.444)
THAL 16 Wildtype—09
Median = 670
(353–1,059)
Median—0.383(0.189–1.869)
p value 0.4079 0.2105
Abstract P 038
Targeted Dose Adjustment of i.v Busulfan in Patients UndergoingHematopoietic Cell Transplantation—CMC Vellore Experience
Ezhilpavai Mohanan, Poonkuzhali Balasubramanian, SalamunDesire, S Gopinath, Mammen Chandy, Vikram Mathews, AlokSrivastava, Biju George
Department of Haematology, Christian Medical College, Vellore
Background: Busulfan is a bi-functional alkylating agent used in
combination with cyclophosphamide or fludarabine as a conditioning
regimen prior to hematopoietic stem cell transplantation (HSCT).
Intravenous (i.v) busulfan is preferred over oral busulfan due to reduced
inter-individual variation and better predictability in pharmacokinetics.
Busulfan has a narrow therapeutic window, with toxic side effects such
as sinusoidal obstruction syndrome (SOS) occurring at high systemic
exposure and graft rejection or relapse at low exposure, thereby sig-
nificantly influencing clinical outcome. We share our experience in
targeted dose adjustment of i.v busulfan in patients undergoing HSCT
for various hematological diseases in the past 2 years. Patients andMethods: Sixty patients diagnosed with various haematological dis-
orders receiving high dose i.v busulfan as combination chemotherapy
between June 2010 and July 2012, were included in this study. Forty
three patients received single daily (Q24H) whereas 17 had 6 hourly
dose of (Q6H) busulfan. Peripheral blood samples were collected at
different time points and plasma were immediately separated. Busulfan
levels were analyzed using a rapid and sensitive LC-ESI MS/MS
method with deuterated internal standard (d8-Bu) and quantified using
Multiple Reaction Monitoring (MRM). The results are interpreted as
ng/ml. Inter and intra-day variability of the standards and controls was
less than 10 %.An AUC of 5,000–5,500 lmol (cumulative AUC target:
20,000–22,000 lmol) in Q24H or 900–1,350 lmol in Q6H were tar-
geted. Results: Diagnosis of the patients, busulfan dosing interval and
plasma AUC on day 1 and 3 is listed in Table. Figure shows the first dose
and dose adjusted AUCs after Q24H and Q6H respectively. Five
patients (8 %) achieved targeted levels after first dose and required no
further dose adjustment, while 39 (65 %) patients achieved targeted
levels after first dose adjustment. However, in 16 (27 %) patients the
dose had to be further adjusted on day 3. Eight (13 %) of these patients
did not achieve the targeted AUC even after increasing the dose the
second time and in 8 patients (13 %) the dose had to be reduced from the
initial rise. However, the dose was not increased beyond 20 % at any
given time. When we analyzed the regimen related toxicity in these
patients, 13 (22 %) developed mucositis Grade III-IV; 7 developed
SOS. Three patients rejected the grafts and all of the three had primary
graft failure. Conclusion: The target level busulfan for Indian patients
undergoing HSCT with various hematological conditions has not been
studied. There is also no documented pharmacokinetics (PK) of i.vbusulfan in our population. From our experience with targeted dose
adjustment of i.v busulfan, we observed that majority of these patients
required dose escalation, which is very different from previous reports.
The reason for this needs further PK and pharmacogenetic studies in our
population.
Abstract P 039
Hematopoietic Stem Cell Transplantation in Acute Leukemias:Long Term Results, Impact of Conditioning Regimensand the Role of Prognostic Factors from a Tertiary CancerCenter in India
Jayant Gawande1, Ravi Thippeswamy1, Bhausaheb Bagal1,Sadhana Kannan2, Manju Sengar1, Hari Menon1, Reena Nair1,Navin Khattry1
1BMT Unit, Department of Medical Oncology, 2Department
of Biostatistics, ACTREC, Tata Memorial Centre, Mumbai
Objectives: Hematopoietic Stem Cell Transplantation (HSCT) is the
most curative option in acute leukemias. We retrospectively ana-
lyzed our data for survival outcomes, impact of conditioning
regimens on outcomes and role of possible prognostic factors.
Methods: One hundred and fourteen patients (101 allogeneic, 13
autologous) who underwent transplant for acute myeloid leukemia
(AML-81) and acute lymphoblastic leukemia (ALL-33) between
March 1983 and August 2011 were evaluated. Poor risk cytogenetics
were seen in 84 and 38 % patients of ALL and AML respectively.
Seventy nine patients (69 %) received full intensity (FI; cyclo-
phosphamide-total body irradiation Cy-TBI and busulphan-
cyclophosphamide Bu-Cy) and 21 patients (18 %) received fludar-
abine based reduced intensity (RI) conditioning. Prognostic factors
evaluated for overall survival (OS) and relapse free survival (RFS)
were WBC count at presentation, baseline cytogenetics, disease
status at transplant, dose of cytarabine consolidation (18 vs. \18 g/
m2) in AML, conditioning regimen (Cy-TBI vs. Bu-Cy and FI vs.
RI), time from diagnosis to transplant, duration of relapse free
interval (RFI) pretransplant and chronic Graft versus Host disease
(cGVHD). Univariate and multivariate comparisons of survival
times for potential prognostic factors were made using log-rank test
and cox regression analysis respectively. Results: Median age at
Diagnosis N=60 Bu dosing AUC (µMoles) Dose adjustment
AML 19
Q24H (n= 44)Median age : 32 yrs
(1-58 yrs)
Day 1Median = 3776(2407-10456)
Increase : 30Median – 15%(4% to 47%)
CML 9 Decrease : 09Median – 25%
(5%-38%)CMML 1
MDS 8Pre B-ALL 1
Day 3Median = 4986
(3220-7661)
Severe AA 1PNH 2 No Change : 05
Ph+ ALL 1
JMML 1SCID 1
Q6H (n= 16) Median age : 4 yrs
(2-7 yrs)
Day 1Median = 671
(285-1456)
Increase : 13Median – 19%(2% to 39%)
THAL 16 Day 3Median = 995
(557-1712)
Decrease : 03Median – 8%
(4%-13%)
Q24H
Day 1
AUC
Day 3
AUC0
5000
10000
15000P = 0.0397
AU
C (
uM
ole
s/m
in)
Q6H
Day 1
AUC
Day 3
AUC0
500
1000
1500
2000P = 0.0138
AU
C (
uM
ole
s/m
in)
204 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

transplant was 26 years. Median time from diagnosis to transplant
was 7 months. At the time of transplant, 59 % were in first remis-
sion (CR-1), 26 % in second remission (CR-2), 10 % in refractory
state and in 5 % status at transplant was not known. Median follow
up time was 46 months. The incidence of acute and chronic GVHD
was 46 and 36 % respectively. The transplant related mortality
(TRM) was 30 %. The TRM was significantly higher (P = 0.001) in
the pre 2007 period (42 %) compared to the post 2007 period
(10 %). The cumulative probabilities of OS and RFS at 8 years were
37 and 35 % for the whole group, 35 and 32 % for AML, 29 and
20 % for ALL respectively. On multivariate analysis, Cy-TBI con-
ditioning (P = 0.01; P = 0.012)) and chronic GVHD (P = 0.009;
P = 0.004) each were prognostic factors for both better OS and
RFS. On univariate analysis, CR at transplant (P = 0.029) and
RFI [ 9 months pre transplant (P = 0.017) were also associated
with better OS and RFS respectively while RI conditioning showed
a trend towards better OS (P = 0.087). Conclusion: Our long term
results suggest that at least one-third of patients with acute leukemia
are cured with HSCT. Patients with cGVHD and those receiving
Cy-TBI had better survival. Reduced intensity conditioning is an
option in patients with acute leukemias with at least comparable
results.TRM has reduced significantly over recent years probably
due to better supportive care.
Abstract P 040
Bacterial Contamination of Peripheral Blood Stem Cell Harvest:Incidence, Risk Factor and Clinical Outcomes
Bhausaheb Bagal1, Sumathi SH2, Rajib De1, Vijay Patil1, JayantGawande1, Shashank Ojha2, Vivek Bhat3, Preeti Chavan4, RohiniKelkar3, Navin Khattry1
1BMT Unit, Department of Medical Oncology, 2Department
of Transfusion Medicine, 3Department of Microbiology, 4Department
of Laboratory Medicine, Tata Memorial Centre, Mumbai, India
Introduction: Microbial contamination is a potentially serious
complication of peripheral blood stem cell (PBSC) harvest. Data on
incidence, organism type, outcomes after infusion and risk factors
is scarce. Methods: We studied medical records of 195 patients
(autologous) and donors (allogeneic). Bacterial cultures of PBSC
harvest were obtained immediately after PBSC collections, prior to
freezing and after thawing of cryo-preserved PBSC. Risk factors
analyzed for positive culture were type of transplant, method of
mobilization, use of CVC versus peripheral venous access for
apheresis, number of days of collections and total blood volume
processed. Results: 155 subjects underwent apheresis yielding 334
PBSC bags (112 from healthy donors and 222 patients). Apheresis
of adequate PBSC required median of 2 days (range 1–5). Median
volume of blood processed per collection was 10.7 l. The incidence
of microbial contamination was 9.8 %. Microorganism cultured
were skin commensals (n = 19), gram negative pathogens (E. coli-4, Kleibsiella-8 and Proteus mirabilis-2). Seven post thaw samples
grew an organism, all of which were skin commensals. Only one
patient receiving culture positive PBSC harvest developed fever
with rigors during infusion. No microorganism present in the PBSC
was recovered from blood cultures collected in post-transplantation
period. Total blood volume processed greater than 10.7 liters was
associated with higher likelihood of positive culture in PBSC
harvest (p = 0.01). Conclusion: Our experience suggests infusion
of contaminated PBSC graft does not play a significant role as a
source for infections in the HSCT setting and higher blood vol-
umes processed during apheresis increases the risk of bacterial
contamination.
Abstract P 041
Breast Cancer Stem Cells (BCSCs): An Implication to Therapyin Chemoresistant Breast Cancer
Preetha Bhadra, S Gangopadhyay, S Sengupta, A Mukhopadhyay
Netaji Subhas Chandra Bose Cancer Research Institute, 16A Park
Lane, Kolkata-700016, West Bengal, India
Objective: Invasive and mesenchymal property of BCSCs with
CD44+/CD24low/ALDH1+ phenotype has made them promising tar-
get for targeted treatment. Chemotherapy treatment uses medicine to
weaken and destroy cancer cells in body, including cells at original
cancer site and any cancer cells that may have spread to another part
of body. Chemotherapeutic drugs for breast cancer are not well
defined yet. Combination of drugs is also not fully known. Our
objective is to define chemotherapeutic drugs and its action in breast
cancer. Methods: Total of 20 early chemo-naıve patients with biopsy
proven triple-negative metastatic breast cancer in age group of
18–36 years were selected randomly and tested for CD44/CD24 cell
analysis. Isolated BCSCs were cultured for in vitro drug sensitivity
towards Platinum, Anthracyclin, Docetaxel, Rapamycin, Sunitinib,
Sorafenib, Gefitinib. Correlation was drawn between cell differenti-
ations, % of stem cells and drug response. Accordingly chemotherapy
was designed for particular patient. % of BCSCs in pre and post-
chemotherapeutic condition was further compared. Results: We have
detected BCSCs in 90 % of cases. Among positive sample shows
in vitro drug sensitivity 4 (20 %) to Rapamycin, 1 (5 %) to Sunitinib,
1 (5 %) to Sorafenib, 1 (5 %) to Gefitinib, 3 (15 %) to Platinum, 1
(5 %) to Anthracyclin, 1 (5 %) to Docetaxel and rest shows no sen-
sitivity to any drug. Conclusion: Thus primary aim to target BCSCs
at onset of tumors in breast cancer patients to control metastasis and
relapse of disease was somewhat obtained. We further plan to cor-
relate ratio of selected markers present in pre and post-
chemotherapeutic condition with time to recurrence, mortality, mor-
bidity and progression free survival. If no BCSCs prevail after
chemotherapy, patients would be kept under observation and if traces
are found, we would proceed to targeted therapy.
Abstract P 042
Infections in Hematopoietic Stem cell Transplant Recipients—an12 Year Single Centre Experience of 207 Cases
N Grover, Nair Velu, S Bhattacharya, A Sharma, AK Sahni,S Das, L Singh, GS Chopra, S Sharma, R Kapoor, V Behera
AFMC, Pune
Background: Infections are the main cause of mortality and mor-
bidity in haematopoietic stem cell transplant (HSCT) patients
despite prophylaxis and newer anti microbial drugs. Aim: To study
the various infections in HSCT recipients at a large tertiary care
hospital of India between Feb 1998 to March 2010. Study Design:Retrospective case series. Methods: Total 207 patients underwent
HSCT with proper sterile precautions. Infections were diagnosed
according to the results of clinical examination or specific diagnostic
procedures and were defined based on CDC criteria. Bacterial
infections were documented on positive cultures, fungal infections
documented as per CDC criteria and viral infections diagnosed by
RT-PCR results. Proper antibiotic, antiviral and antifungal therapy
given whenever infection was suspected/documented. Results:164/207 patients (79.2 %) had one episode of fever and 35/207
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 205
123

(16.9 %) had second episode of fever. Second episode of fever was
more frequent in patients who underwent allogenic (23.3 %) than
autologous (4.2 %) and who nursed under clean side room (18/28)
versus protected environment (17/179) with p \ 0.001. Total 267
documented infections occurred in 207 transplants in which 134
(48.9 %) were bacterial, 98 (35.7 %) viral, 38 (14 %) fungal and 1
had plasmodium falciparum. Pathogens identified were more in
allograft than autograft transplants. Gram negative infections were
identified more in which the most common was E. coli. CMV was
the most common viral and candida the common fungal infection
documented. Death occurred in 72 patients in which 50 were post
allogenic. Conclusion: Overall incidence of all infections and feb-
rile episodes was high and were comparable to both western and
Indian studies. Infections were commoner in allograft than in
autograft patients. Major causes of death included fungal pneumonia
and bacterial pneumonia.
Abstract P 043
Long Term Results of Autologous Transplants for Hodgkin’sLymphoma—a Retrospective Multi-Center Analysis from India
Reetu Jain1, Navin Khattry2, Ravi Thippeswamy3, AdwaitaGore4, Bharat Bhosale5, Nandish Jeevangi6, Sadhana Kannan7,Anjana Sainani8, Ganapathy Bhat9, Reena Nair10, TapanSaikia11, On Behalf of Indian Study Oncology Group Trust
1,8,9Department of Medical Oncology, Jaslok Hospital and Research
Centre, Mumbai; 2,3,5,6,7,10BMT Unit, Department of Medical
Oncology, ACTREC, Tata Memorial Center, Mumbai;4,11Department of Medical Oncology, Prince Aly Khan Hospital,
Mumbai; 1Dr.G.Deshmukh Marg, Mumbai 26, India
Introduction: Autologous transplantation is the standard of care for
patients of relapsed and refractory Hodgkin’s lymphoma (HL). We report
the results of transplants from three centers in Mumbai and role of possible
prognostic factors. Methods: One Hundred and three patients underwent
transplant for HL from August 1994-May 2011. All patients received 2–3
cycles of DHAP, ESHAP, MINE, Mini-BEAM, GDP or ICE with or
without rituximab as salvage chemotherapy. Cheson’s response criteria
was applied for assessing response pre and post transplant at day 100.
Forty-eight percent of patients received BEAM (carmustine, etoposide,
ara-c and melphalan), while 52 % received LACE (lomustine, ara-c,
cyclophosphamide and etoposide) regimen. Ninety-two percent received
peripheral blood stem cells. Prognostic factors evaluated for progression
free survival (PFS) and overall survival (OS) were stage at diagnosis,
presence of B symptoms at diagnosis, time from diagnosis to transplant,
number of lines of chemotherapy pretransplant, remission status at trans-
plant, conditioning regimen, pretransplant salvage chemotherapy and
serum albumin at transplant. Univariate and multivariate comparisons of
survival times for potential prognostic factors were made using log-rank
test and cox regression analysis respectively. Results: The median age at
transplant was 23 years. B symptoms were present in 47 % at diagnosis.
The median time from diagnosis to transplant was 1.9 years. Fifty-seven
percent of patients had stage III and IV disease at diagnosis. At transplant,
41 % were in complete remission (CR) Sixty percent of patients who
received platinum- based salvage achieved CR compared to 30 % of those
who received ifosfamide (P = 0.012). Median number of lines of che-
motherapy pretransplant was 2 and median serum albumin at time of
transplant was 4 g/dl. Transplant related mortality was 11 %.The cumu-
lative probability of OS and PFS at 3 years were 65 and 49 % respectively.
Serum albumin[4.0 g/dl (P = 0.03) and CR at transplant (P = 0.025)
were associated with better OS while CR at transplant was associated with
improved PFS in multivariate analysis. Conclusion: Our data suggest that
serum albumin and CR at transplant are important factors affecting
survival. Platinum based pre-transplant salvage regimens achieved better
CR rates than predominantly ifosfamide based regimens.
Abstract P 044
Role of Reduced Intensity Conditioning Allogeneic Stem CellTransplant for Young Adults with Acute Myeloid Leukemiain First Complete Remission
Chepsy C Philip1, Abhijeet Ganapule1, Rayaz Ahmed1,Aby Abraham1, Biju George1, Kavitha M Lakshmi1, AlokSrivastava1, Vikram Mathews1
1Department of Clinical Hematology; CMC Vellore
Introduction: Allogeneic hematopoietic stem cell transplantation
(HSCT) is an effective treatment for patients with Acute Myeloid Leu-
kemia (AML). In AML, graft versus leukemia contributes significantly to
durable remission. Reduced intensity conditioning (RIC) is frequently
indicated in the elderly or those with co-morbidities. There is limited data
on clinical outcomes in young adults with AML with a RIC allogeneic
HSCT. Methods: From 2005 we have been using reduced intensity
conditioning with Fludarabine and melphalan in patients with AML in
CR1. The reason to change from myeloablative conditioning was our
concern about the high treatment related mortality with our earlier my-
eloablative regime. We performed an analysis of 46 consecutive patients
who received RIC. Kaplan Meir method was used to generate event free
survival (EFS) and overall survival (OS). Results: The median age of the
recipients was 34 (11–63). All patients engrafted. Median time to
ANC C 500 and platelets C20,000 was 14 and 17 days respectively.
Acute GVHD was seen in 24 patients [Grade 2–4: 20 (43.4 %); Grade
3–4: 09 (19.5 %)] and Chronic GVHD in 28 recipients [Limited: 21
(45.6 %); Extensive: 07 (15.2 %)] 0.4 recipients (8.7 %) relapsed. There
were 12 deaths. Treatment related mortality was as follows
(B100 days = 6.5 %, n = 03; B1 year = 17.3 %, n = 08). The esti-
mated EFS was 68.6 % and OS was 71 % at 2 years with a mean follow
up of 51 and 53 months respectively. Conclusion: The use of reduced
intensity conditioning is a reasonable option to consider in young adults
with AML in CR1.
Aplastic Anemia
Abstract P 045: Oral presentation
Pancytopenia: A Clinico Hematological Study
Vijai Tilak1, Madhukar Rai2, Vikram Singh3
Department of Pathology, Institute of Medical Sciences, B.H.U.,
Varanasi; 2Department of Internal Medicine, Institute of Medical
Sciences, B.H.U., Varanasi; 3Department of Internal Medicine,
Vir Chandra Singh Garhwali Govt. Institute of Medical Sciences
& Research, Srinagar, Pauri Garhwal, Uttarakhand
Objectives: (1) To study the different causes of pancytopenia. (2) To
evaluate the incidence of various disorders causing pancytopenia in chil-
dren and adults. (3) To explore a cost effective and rapid test to differentiate
between Megaloblastic anemia from megaloblastosis of myelodysplastic
syndrome (MDS). (4) To compare our data with other studies on pancy-
topenia. Material and Methods: The present study was conducted in the
Division of Hematology, Department of Pathology, Institute of Medical
Sciences over a period of 45 months on 428 patients of pancytopenia.
Results: The age of the patients ranged from 3 to 73 years. There was male
206 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

preponderance. The overall most common cause of pancytopenia was
Aplastic anemia followed by Megaloblastic anemia. Aplastic anemia and
acute Leukemia often bleed whereas Megaloblastic anemia rarely bleed.
Patients with aplastic anemia and acute Leukemia were often children.
Conclusions: Childhood pancytopenia is a distinct entity with a grave
prognosis vis-a-vis adult and elderly pancytopenia. Serum LDH is a cost
effective and rapid test to differentiate megaloblastosis of vitamin B12 and/
or folate deficiency from MDS. Bone marrow examination can be deferred
in pancytopenic patients with hypersegmented neutrophils; it can be per-
formed later if there is no response to hematinics.
Abstract P 046: Oral Presentation
Chronic Benign Neutropenia—a Case Report
Rachana Garg
KMC Manipal
Introduction: Chronic benign neutropenia is defined as absolute
neutrophil count (ANC)\1,000 cells/mm3 in infants and\1,500 cells/
mm3 in children and adults, persistently for more than 3 months. CaseHistory: 1 year old male child presented with fever for 5 days and
upper respiratory tract infection. Clinical examination was normal. His
peripheral blood smear showed ANC of 120 cells/mm3 with eosino-
philia and monocytosis. Bone marrow aspiration showed myeloid
hyperplasia with increase in precursor cells. IgG was raised. Child was
evaluated and no acquired immunodeficiency was seen. He was given
3 doses of G-CSF following which neutrophil counts improved,
however counts done 4 days after G-CSF again showed ANC -252
cells/mm3. Conclusion: Chronic benign neutropenia is a rare disorder,
but commonest cause of neutropenia in children in age group
(3–30 months). Careful evaluation is required to avoid misdiagnosing
it as leukemia or other hematological malignancies. In these cases
close follow up of patient is needed to assess the course of the disease.
Abstract P 047
A Clinico-Pathological Study of Cases of Pancytopenia—aHospital Based Study
Abhijit Phukan, Jina Bhattacharyya, DK Saikia, PK Gogoi
GMCH
Objective: This study was conducted to evaluate. (1) The disease
patter in different age and sexes. (2) Their clinical presentation. (3)
Various causes of pancytopenia and their relative frequencies in this
region. (4) Haematological parameters and bone marrow finding in
various causes. Methods: The study was conducted in the Depart-
ment of Pathology, GMCH, Guwahati, extending over a period of one
year from 1st July 2011 to 30th June 2012. The criterion of selection
of the patients was: (1) Hb \ 9 g. (2) Total leucocyte count \4,000/
mm3. (3) Platelet count\1 lakh/mm3. All the patients were subjected
to PBS study, bone marrow aspiration and biopsy. We also did special
stains, flow cytometry and cytogenetics as and when necessary.
Results: From the study of 88 cases, various important results can be
summarised as (1) Maximum cases Pancytopenia are observed in the
age group of 31–40 years, with male predominance (61 %). (2)
Common presenting features are generalized weakness (100 %),
bleeding (23 %) etc. (3) Among the various causes, Megaloblastic
anaemia is found to be the most prevalent (54 %). (4) Haematological
parameters vary according the causes. (5) Hypercellularity of marrow
is a feature in most of the cases (84 %). Conclusion: This study will
help in finding the disease scenario in this part of the world.
Abstract P 048
Antilymphocyte (ALG)/Antithymocyte Globulin (ATG)for Severe Aplastic Anemia—Single Centre Experience
Venkatesh S Ekbote, Biju George, Vikram Mathews,Aby Abraham, Riyaz Ahmed, M Kavitha, Alok Srivastava
Department of Hematology, Christian Medical College, Vellore,
Tamil Nadu
Objective: To analyze response to immunosuppressive therapy with
ATG/ALG in children and adults with severe and very severe aplastic
anemia treated between 1985 and 2012. Methodology: We conducted a
single institution retrospective analysis of severe and very severe aplastic
anaemia patients treated with ATG-based IST from 1989 to 2012.
Response, based on EBMTR guidelines, was determined at 3 and
6 months following ATG. Results: Two hundred ninety six patients with
Severe and Very Severe Aplastic Anaemia with median age of 38 years
(range 3–83 years) were studied, including 198 (66.9 %) males and 98
(33.2 %) females. The overall objective response (CR + PR) to ALG/
ATG CSA in this cohort was 50 %. Patients in younger age group
(1–15 years) had poorer response rate—34 % compared to adults who
had comparable response across all age groups; (16–29 years)—55 %,
(30–50 years)—56 % and more than 50 years—59 %. Also, 32
(10.8 %) patients died of infection within first month of ATG/ALG
treatment. On follow up, 14 patients (4.7 %) lost response while 13
(4.3 %) underwent clonal evolution to paroxysmal nocturnal haemo-
globinuria, myelodysplastic syndrome or acute myeloid leukemia. Seven
(2.4 %) patients underwent allogeneic stem cell transplantation. The
overall survival in this cohort of patients was 57 %. It was 51, 52, 59 and
71 % in age groups: 1–15, 16–29, 30–50 and more than 50 years,
respectively. Conclusion: This study suggests that ATG is a viable
option in adult Indian patients even more than 50 year old. The poorer
response seen in Indian children needs to be studied further.
Abstract P 049
Spectrum of Chromosomal Breakage Syndromes Observedin Patients with and Without Bone Marrow Failure Syndromes(BMFS), at PGIMER, Chandigarh
Priti, Anil Sood, KS Rana, N Varma
Department of Hematology, Post Graduate Institute of Medical
education and Research, Chandigarh
Introduction: Fanconi Anemia (FA) is the most common chromo-
somal breakage (CB) syndrome and is the prototype of inherited bone
marrow failure syndrome (BMFS). FA cases present during childhood
or at times later in adulthood. Other chromosomal instability syn-
dromes not characterized by BMFS include Nijmegen breakage
syndrome (NBS), Ataxia telangiectasia (AT), Bloom’s syndrome and
Xeroderma pigmentosa. Clinical heterogeneity of these disorders
often leads to an incorrect or missed diagnosis. Although clinical
suspicion index may be high for these disorders, definitive diagnoses
(many times) based upon chromosomal breakage studies (CBS) are
not well documented in India. This study was carried out to document
the spectrum of these syndromes observed in patients with and
without BMFS, at PGIMER, Chandigarh. Materials and Methods:The study included consecutive patients referred for work up of
chromosomal breakage syndromes, between Jan 2011 and August
2012. In our department, appropriate phytohemagglutinin (PHA)
stimulated peripheral blood lymphocyte cultures (PBLC) are set up
for patients and controls, with and without clastogens [1. diepoxy-
butane (DEB) and 2. mitomycin C (MMC) for Fanconi Anemia; 1.
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 207
123

DEB induced CBS, 2. X-irradiation and 3. treatment with bleomycin
at G2 phase of cell cycle for Ataxia telangiectasia; and 1. PHA
stimulated PBLC: (a) spontaneous breakage (b) G-banding for
translocations, 2. DEB induced CBS, 3. MMC induced CBS 4.
X-irradiation and 5. treatment with bleomycin at G2 phase of cell
cycle for NBS]. Result: Out of 104 patients studied, 11 were diag-
nosed as Fanconi Anemia, 2 as Ataxia telangiectasia, whereas NBS
was diagnosed in just 1 patient. Median age of FA patients was
8 years (range 5–32 years). Both the AT patients were males. Patient
with Nijmegen breakage syndrome was an 18 months old boy. As
highlighted earlier, work up for chromosomal breakage syndromes is
extremely time consuming, though clinically very rewarding. Con-clusion: Approximately 130 cases of Ataxia telangiectasia have been
reported worldwide so far. Nijmegen breakage syndrome patient
included in this study is the second case reported in India. The exact
incidence of AT and NBS in India is not known. FA cases are being
diagnosed at few centers in India (including our department), however
the facilities to undertake specialized chromosomal breakage studies
for patients without BMFS need to be widely established. This is
required for appropriate genetic counseling and patient management.
Unless specific confirmatory tests using a battery of clastogens, are
performed, such cases will continue to remain under-diagnosed.
Chronic Leukemia
Abstract P 050
A Case of Early Ocular Manifestation of Maculopathyin a 37 year Old Male Patient of CLL
Sufia Khan, Syed Riaz Mehdi, Sharique Ahmad, Parul Gupta
Era’s Lucknow Medical College, Lucknow, UP
Abstract: We are presenting a case of 37 year old male patient
referred from medicine OPD for routine Complete Blood Count
(CBC). His Total Leukocyte Count (TLC) came out to be alarmingly
high .His general blood picture (GBP) and bone marrow examination
revealed it to be a case of Chronic Lymphocytic Leukemia (CLL).
Later on immunophenotyping was also performed. His cervical lymph
node biopsy was reported as a case of chronic lymphocytic leukemia/
small lymphocytic lymphoma. Subsequently, on his ophthalmic
examination maculopathy was detected. CLL in young male has not
yet been reported from India.
Abstract P 051
Clinico-Haematological Analysis of Haematological Malignancy,A Hospital Based Study
Rahul Varshney, Muktanjali Deka, Jina Bhattacharya, PK Gogoi
GMCH
Objective: Early diagnosis of haematological malignancy is important in
clinical practice to prevent mortality. This study was conducted to evaluate
their relative frequencies in different ages and sexes in this region.
Methods: The present study was conducted in the Department of
Pathology and Haematology, Gauhati Medical College & Hospital, Gu-
wahati from 1st July 2011 to 30th June 2012. Patients were examined and
blood tests including haemoglobin estimation, total and differential leu-
kocyte count, platelets count, reticulocyte count and PBS study, Bone
marrow aspiration and biopsy, special stains, flow cytometry and cyto-
genetics was done in each cases. Results: Out of 450 patient attending
Haematology OPD 236 were having malignancy. About 29.66 % patient
had chronic myeloid leukaemia while 1.69 % patient had chronic
lymphocytic leukemic. Among acute leukaemia, acute myeloid leukaemia
outnumbered acute lymphoblastic leukaemia, (21.19 %against 7.2 %).
Multiple myeloma was seen in 20.33 %. Non Hodgkin’s lymphoma was
seen in 10.59 % while Hodgkin’s disease 6.6 %, MDS 1.69 %, MPN and
LCH 1 %. Male to female ratio in haematological malignancies was
2.87:1. Maximum cases were observed in 51–60 years. Low grade fever,
progressive pallor, weakness and body aches were the commonest
symptoms (70 % cases) while pallor was the frequently observed sign.
Conclusion: The prevalence of haematological malignancy is
52.44 %.This study may help in finding out their relative distribution in
this region.
Abstract P 052
Monitoring of Response to Therapy in Chronic MyeloidLeukaemia Patients by Quantitative Real Time PCRon the International Scale: Our Experience in CML Patientsin a Tertiary Care Centre in India
J Kotwal, BK Chakrabarty, R Kapoor, V Nair, V Dutta
Armed Forces Medical College, Pune
Background: With advent of Tyrosine kinase inhibitors (TKIs),
treatment and prognosis of Chronic myeloid leukaemia (CML) has been
revolutionized. The patients go into complete haematological, cytolo-
genetic (CCyR) and major molecular remission (MMR) with therapy.
This is highly desirable monitoring reliability of RQ-PCR assay should
be optimized in different laboratories. The IRIS group has advised
cytogenetic remission but Adelaide group has favoured molecular
monitoring to be more effective as cytogenetic remission corresponds to
3 log reduction at molecular level. In our laboratory in 2005 Bcr-Abl
transcription was reported as copies/ml. Afterwards Bcr-Abl/Abl ratio
expressed as percentage was started using the M BCR fusion Quant kit
by Ipsogen in Rotor gene 3000. Base line value of Bcr-Abl/Abl ratio was
calculated by taking median of 30 newly diagnosed cases of CML. With
further improvements now Bcr-Abl Mbcr kit, compliant with updated
international recommendations and contains an IS-MMR calibrator,
aligned with international scale is used for converting NCN results to
the International scale and report MMR. Methods: RQ-PCR on Light
cycler 480 is used for monitoring of patients. WHO validated and
accredited reference reagent and IS MMR calibrator is used in each run
to calculate MMR on the IS scale. Results: Our experience related to 50
CML patients on therapy will be presented. Conclusion: Goals of
therapy are to reach major molecular remission. It seems desirable that
use of RQ-PCR for monitoring response to treatment for leukemia
should follow the same pattern to satisfy criteria for clinical use as
required by the appropriate international regulatory agencies.
Keywords Chronic myeloid leukemia, Complete molecular response,
Major molecular response, RQ-PCR, IS scale
Abstract P 053
Incidence of Different BCR-ABL Transcript Variants AmongstCML Patients: Their Correlation with Presenting Features,Relative Risk and Response to Treatment with Imatinib Mesylate
Pratik Deb, Prantar Chakrabarti, Utpal Chaudhuri,Shila Chakrabarti
Institute of Hematology and Transfusion Medicine, Medical College,
Kolkata
208 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Objective: To establish how different transcript variants of BCR-
ABL play their role in the disease process of Chronic Myeloid Leu-
kemia in our patient population. Method: RNA extraction, RTPCR,
Spectrophotometry and agarose gel electrophoresis, Statistical anal-
ysis. Results: Amongst 80 patients (56 male, 24 female) 56.25 %
patients showed the presence of b3a2 and 41.25 % patients showed
the presence of b2a2 transcript variant. The rest 2.5 % (numerically, 2
cases) showed the expression of rare e19b2 variety. Patients with
b2a2 transcript variant presented with higher Sokal, Hasford and
EUTOS score than the patients with b3a2 transcripts. Difference
between different parameters like Platelet count, TLC, Hemoglobin,
Organomegaly was minor. After receiving almost 18 months of I-
matinib therapy, patients with b2a2 reached hematological and
cytological remission more than their counterpart. Conclusion: CML
patients in our population with b2a2 variants tend to present with
higher risk scores but respond better to Imatinib therapy.
Abstract P 054
Standardisation of RTPCR for Confirmatory Diagnosisof Chronic Myeloid Leukemia: First Time in a Tertiary HealthCare and Teaching Hospital in West Bengal
Rajarshi Aich, Pratik Deb, Utpal Choudhuri, Shila Chakrabarty,Prantar Chakrabarti
Institute of Haematology and Transfusion Medicine, Kolkata
Objectives: Our challenge was to standardise a RTPCR protocol, for
the first time in a tertiary health care system of West Bengal, for the
confirmatory diagnosis of CML patients, and, at the same time, keeping
the procedure as costeffective as possible. Methods: Total 80 patients
were selected amongst which, 60 were diagnosed cases and 20 were
suspected cases of CML. Total RNA extraction was done from the
blood of all the patients and cDNA was made from it by reverse
transcriptase PCR using random primers. PCR amplification of the
BCR-ABL gene of cDNA was done using primers of standardized PCR
protocol of BIOMED1. The PCR products were subjected to 2 %
agarose gel electrophoresis and observed under UV light. Result: The
PCR reaction was able to successfully amplify the target products of
BCR-ABL. All 20 suspected cases of CML were diagnosed to be car-
rying BCR-ABL through our test and later 18 of those diagnosed cases
were confirmed by bone marrow cytogenetics (BMC). The 2 failed
cases were confirmed to be CML through FISH, thus establishing RT-
PCR’s higher sensitivity than BMC. All the established cases gave
positive results. Conclusion: Our present study has led us to the
development of a standardised RTPCR protocol for confirmatory
diagnosis of CML. It had also affirmed that the primer pair combination
used in Europe is properly working in our population.
Abstract P 055
Induction of Apoptosis in Chronic Myelogenous HumanLeukemic Cells K562 and U937 by Trichosanthes dioica LeafExtract
Nilanjana Deb1, Moumita Ray1, Biswajit Chakraborty2, ChinmayChowdhury2, Sugato Banerjee 3, Aparna Gomes1, Shila ElizabethBesra1
1Drug Development/Diagnostic & Biotechnology Division, Indian
Institute of Chemical Biology (CSIR), 4 Raja S.C. Mullick Road,
Kolkata-700032, West Bengal, India; 2Department of Chemistry,
Indian Institute of Chemical Biology, (C.S.I.R), 4 Raja S. C. Mullick
Road, Kolkata -700 032, West Bengal, India; 3Gupta College
of Technological Sciences, Ashram More, Asansol-713301
Objective: Plants have been an immense source of therapeutics. Cancer,
a dreadful disease is a leading cause of death worldwide. Synthetic anti-
cancer agents used in chemotherapy are generally toxic or immunosup-
pressive. But compounds from natural sources tend to have more
immunostimulants properties. We have studied the apoptosis on CML
cell lines along with the immunomodulatory effects of the leaf extracts of
Trichosanthes dioica. Method: We studied cell viability, cytotoxicity
and apoptosis on two CML cell lines. Cell morphology was determined
by Fluorescence and Confocal microscopy. DNA fragmentation was
studied by gel electrophoresis. Effect of TDLE on NO production was
studied in RAW264.7 cell line. Result: TDLE significantly inhibited the
cell viability and cytotoxicity (MTT) in a time and concentration
dependent manner. The fluorescence and Confocal study showed char-
acteristic features of membrane blabbing, chromatin condensation the
sign of early and late apoptotic changes in the cells after treatment with
TDLE. Gel electrophoresis study showed fragmented DNA in the form of
ladder. It was found that TDLE was able to induce NO production in
resting RAW264.7 cell line. But when TDLE was used in combination
with recombinant interferon-c, there was a marked cooperative induction
of NO production in RAW264.7 cells. Conclusion: The present study
reveals that the TDLE possesses potent anti-leukemic with immuno-
modulatory activity. Trichosanthes dioica constitute a major part of our
every day diet. The compounds from TDL can be used as immunother-
apeutic agents for cancer treatment. Studies are in progress to identify the
mechanism of anti-leukemic activity of TDLE.
Abstract P 056
Concordance Between ZAP-70 and IgVH Mutation Statusin Chronic Lymphocytic Leukemia
Ritu Gupta1, Lata Rani1, Nitin Mathur1, Atul Sharma2,Lalit Kumar2, Vinod Raina2
1Department of Laboratory Oncology, Dr. B.R.A. IRCH, AIIMS,
New Delhi, India-110029; 2Department of Medical Oncology,
Dr. B.R.A. IRCH, AIIMS, New Delhi, India-110029
Objective: Following gene expression profiling, ZAP-70 has emerged
as the most promising surrogate marker for the IgVH mutation status, a
robust prognostic marker in CLL. Western studies using flow cytom-
etry for ZAP-70 with 20 % cutoff value, showed an overall correlation
between ZAP-70 expression and IgVH mutational status ranging from
77 to 90 % concordance. However, there are no such data from Indian
population. We aimed to investigate the optimal cut off of ZAP-70
with IgVH mutational status and its concordance in CLL patients from
India. Methods: Peripheral blood samples from 100 B-CLL patients
were assessed for ZAP-70 and IgVH mutation status. ZAP-70
expression was analyzed on CD5+CD19+ CLL cells using multipara-
metric flow cytometry. For IgVH mutation status, RNA was extracted
from peripheral blood MNC, transcribed to cDNA which was ampli-
fied by PCR according to BIO-MED-2 protocol, using VH1-VH6
family FR1 primers and 30JH consensus primer. PCR products were
purified and sequenced with an automated DNA sequencer. Ig-BLAST
was used for comparing rearranged IgVH segments; cases with C98 %
identity to germline were classified as unmutated (UM) while cases
with\98 % identity were considered mutated (M). Results: Among a
total of 100 patients, the number of mutated and unmutated cases were
62 and 38 respectively. Using ROC analysis, the optimal cut-off value
for ZAP-70 was found to be C22 % (83 % sensitivity, 81 % speci-
ficity). Using this cut off value, 59 patients were ZAP-70 negative
while 41 patients were positive for ZAP-70. Out of 59 ZAP-70 neg-
ative, 52 (88.1 %) were mutated and 7 (11.8 %) were unmutated. Out
of 41 ZAP-70 positive patients, 31 (75.6 %) were unmutated and 10
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 209
123

(24.3 %) were mutated. Therefore, a total of 83 % cases were con-
cordant for ZAP-70 cut off and IgVH status (ZAP-70+UM, ZAP-
70-M) whereas 17 % cases were discordant (ZAP-70-UM, ZAP-70+
M). Conclusion: A cut off value of C22 % for ZAP-70 correctly
predicted the IgVH mutation status in 83 % cases in Indian population.
Abstract P 057
VEGF in CML: significance and Correlation with Phaseof Disease
Purvi Mathur1, Usha Rusia1, Meera Sikka1, RK Grover1
1Department of Pathology, University College of Medical Sciences
& GTB Hospital, Delhi-110095, India; 2Delhi State Cancer Institute
Background: Current data suggests that angiogenesis plays a significant
role in the progressive growth and metastasis of many tumors, including
hematological malignancies. Vascular endothelial growth factor (VEGF)
is a key cytokine involved in the angiogenic process in tumors leading to
acceleration of tumor growth rate, invasion and metastasis. The current
study focused on evaluating the serum levels and bone marrow expres-
sion of this cytokine in Chronic myeloid leukemia. Aims andObjectives: The present study aimed to document the serum level of
vascular endothelial growth factor (VEGF) in different stages of Chronic
myeloid leukemia (CML), to correlate the levels with clinicopathological
factors and to study the immunohistochemical expression of VEGF in
bone marrow in these patients. Materials and Methods: Serum levels of
VEGF were evaluated in 40 Philadelphia chromosome positive patients
of Chronic myeloid leukemia (CML) and 40 age and sex matched healthy
controls by using commercially available ELISA kits. Bone marrow
biopsies were obtained from 50 patients of CML. Biopsies from 10
patients suspected of lymphoma which were morphologically normal
were used as controls. Immunohistochemical staining of VEGF was
performed by the labeled streptavidin avidin biotin method to establish
it’s cellular distribution in bone marrow. Results: Serum VEGF levels
were found to be significantly (p \ 0.0001) elevated in patients as
compared to controls and in patients in blast and accelerated phase of
CML as compared to those in the chronic phase (p = 0.02).Correlation of
serum VEGF levels with established prognostic factors showed a positive
correlation between serum VEGF levels and total leucocyte counts
(p = 0.04), blast percentage (p = 0.006), Hasford score (p = 0.02) and
a positive trend with splenomegaly (p = 0.06). The cellular distribution
of VEGF in the bone marrow was found to be similar in both patients and
controls, being positive in megakaryocytes and negative in erythroid cells
and neutrophils. The myeloid precursors showed a variable positivity for
VEGF. Conclusion: Serum VEGF levels may play a role as an important
independent prognostic factor in patients of CML. The marked elevation
of VEGF levels in patients of blast crisis as compared to the chronic phase
indicates that raised VEGF levels could signify advanced disease stage.
The findings demonstrated in the study suggest that antiangiogenic
therapy could potentially be successful in treatment of these patients.
Abstract P 058: Oral Presentation
The Effect of Imatinib Mesylate Therapy on Bone MarrowMorphology in Chronic Myeloid Leukemia Patients—anImmunohistochemical Study
Kamal Preet1, Usha Rusia1, Meera Sikka1, RK Grover2
1Department of Pathology, University College of Medical Sciences
& GTB Hospital, Delhi, India; 2Delhi State Cancer Institute
Background: Chronic myeloid leukemia (CML) is the commonest
leukemia occurring in Indian adults. Imatinib Mesylate is now being
used in these patients as first line of treatment. There is scarcity of
data on the effect of this drug on bone marrow morphology from
Indian subcontinent. Moreover there is no study which has objectively
tried to measure the effect of Imatinib Mesylate on stem cells and
proliferation in Indian patients of CML. Aims: To evaluate the
morphological changes in the peripheral blood, bone marrow aspi-
rates and biopsies in patients of CML before and after treatment
(6 months) with Imatinib Mesylate and to study the effect of Imatinib
on proliferating marrow stem cells using CD34 and PCNA. Materialand Methods: Morphological features were studied on peripheral
blood, bone marrow aspirates and biopsies in 30 patients of CML at
presentation and at the end of six months of Imatinib therapy.
Results: The M:F ratio was 2.2:1 and the median age was 30 years.
Common complaints at presentation were splenomegaly (97.7 %),
anaemia (82 %), weakness (51 %), pain abdomen (53 %) and fever
(40 %). At the end of 6 months of therapy, 96 % of the patients
achieved complete hematological remission. The bone marrow aspi-
rates and biopsies showed reduction in overall cellularity, reduction in
the M:E ratio, reappearance of erythroid colonies and relative
decrease in myeloid precursors in all patients except one. Megakar-
yocytes showed normalization of morphology and numbers.
Myelofibrosis post therapy was seen in 48 %patients. Only one
patient had significant myelofibrosis (grade 2-3). A statistically sig-
nificant reduction in the number of CD34+ progenitors from 20 to 50
cells/hpf pre therapy to 3-35 cells/hpf post therapy was observed. Post
therapy PCNA counts were 42-126 cells/mm2 which were markedly
lower than the initial counts. Conclusion: Imatinib mesylate is an
effective drug as first line treatment of CML patients. Apart from
causing haematological remission, it also effectively acts on the
CD34+ stem cells and proliferating fraction in CML. Sequential bone
marrow biopsies can be useful in monitoring disease remission and
progression in CML patients, hence bone marrow biopsies need to be
an integral part in the follow up and management of CML patients.
Abstract P 059
Different Clinical and Haematological Features in CML PatientsDuring First Presentation—a Study of 166 Patients Seenat Referral Centre
Chetan Jain, Palash Kumar Mandal, Nirmal KumarBhattacharya, Swapan Kumar Sinha
Medical College, Kolkata
Introduction: Chronic myelogenous leukaemia (CML) is a common
myeloproliferative disorder (MPD) seen in our country and world
over. The condition has varied clinical presentations and haemato-
logical features but demonstration of Philadelphia chromosome is the
surest test for a confirmation at present.CML may be seen in one of
three stages-chronic phase (CP) or accelerated phase (AP) or blast
phase (BP), each having different features. Objectives: To analyze
the different clinical and hematological features at presentation of
patients diagnosed as chronic myelogenous leukaemia (CML). This
study was conducted in Medical College, Kolkata from July 2009 to
June 2012. A total of 166 patients of chronic myeloid leukaemia were
included in the study. The diagnosis was made on clinical history,
physical examination, complete haemogram, bone marrow examina-
tion and cytogenetic study. Results: Out of 166 CML patients, males
predominated, with a M:F ratio of 3:2. The youngest patient was
8 year old and the oldest patient was of 77 years old with a mean age
of 43 years. The peak age incidence was 53 years. Cytogenetic study
showed Ph chromosome to be positive in 160 cases while 6 patients
210 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

showed BCR-ABL1 positivity by RT-PCR. Symptoms of patients
included fatigue (79 %), low grade fever (18 %), body ache (38 %),
weight loss (40 %), arthralgia (17 %), bleeding manifestations
(10 %), dragging sensation in left loin (15 %) etc. Common
signs were splenomegaly (98 %), sternal tenderness (71 %),
lymphadenopathy (42 %), hepatomegaly (40 %) and purpura
(12 %).Haematological features were marked leucocytosis with
basophilia (2–17 %), thrombocytosis/thrombocytopenia (40,000–
8,00,000/lL), anemia (Hb6.1–10.4 g/dl). Marrow was hypercellular
in all, blasts varied from 1 to 7 %. Conclusion: Though CML may
have variety of clinical findings a correct approach to diagnosis with
proper management may prolong the life of these patients.
Keywords Chronicmyelogenousleukaemin, Clinical features,
Haematological features
Abstract P 060
Chronic Myeloid Leukemia in Children & Adolescents:An Experience from a Tertiary Centre
Lalit Raut, Vinay Bohara, Girish Badarkhe, Ganesh Pujari,Uttam Kumar, SS Ray, Prantar Chakrbarti, Utpal Chaudhuri
Institute of Haematology & Transfusion Medicine, Kolkata 700073
Aim: The aim of the study was to evaluate the characteristics at
presentation and the treatment outcome in CML in children and
adolescents age group. Methods: Retrospective analysis was carried
out at a single centre in India. Thirteen patients (B17 years) attending
CML outdoor from April 2008 to August 2012 were included in the
analysis. Results: CML-CP was the most common phase at presen-
tation. Maximum patients belonged to the 14–17 years age group.
Disease was common in male sex. Splenic discomfort and asthenia
were the most common symptoms and splenomegaly was the most
common sign. The median WBC count at presentation was 65 9 109/
L. In majority (69.23 %) of patients the WBC count was between
20 9 109/L to 99 9 109/L. The median hemoglobin at presentation
was 9.5 g/dL. The median platelet count at presentation was
462 9 109/L. CHR was documented in 11 out of 12 (91.66 %)
evaluable responses. At 18 months of treatment MMR was achieved
in 3 out of 5 evaluable patients. In one of the remaining two patients
MMR was documented at 24 months while in the other patient less
than MMR was documented at 12 months. Conclusion: The pre-
senting features of CML in the children and adolescent age group are
similar to that shown in other studies. The treatment with Imatinib
was effective and well tolerated.
Abstract P 061
Does the Concomitant Use of Antitubercular Drugs Alterthe Natural History of CML?
V Badarkhe Girish, Lalit Raut, Vinaykumar Bohra, GaneshPujari, Prantar Chakrabarti, SS Ray, Uttamkumar Nath, UtpalChaudhuri
Institute of Haematology & Transfusion Medicine Kolkata-73
Objective: We report the outcome of CML in five patients receiving
Imatinib and anti tubercular therapy. Methods: Four CML-CP patients
(C17 years) attending CML clinic developed tuberculosis (2 pulmon-
ary and 2 extra pulmonary) while on Imatinib and another one was
diagnosed with Pulmonary tuberculosis and CML upfront and started
on ATD and Imatinib concomitantly. All the four patients who devel-
oped tuberculosis later had achieved CCyR (Complete Cytogenetic
Response) prior to developing tuberculosis. Four drug ATT was started
under RNTCP. Result: Four out of five (80 %) patients progressed to
Accelerated Phase/Blast Crisis while on antitubercular therapy, while
IRIS data has showed that only 7 % progressed to Accelerated Phase/
Blast Crisis. The exact cause is not known, but drug interaction might
have played an important role in reducing the therapeutic efficacy of
Imatinib as the interaction of Imatinib with Rifampicin is well known.
Conclusion: The concomitant use of Imatinib and antitubercular
therapy may lead to disease progression in CML. A systematic registry
should be maintained to document the outcome of CML patients on
antitubercular therapy.
Abstract P 062
Clinical and Biological Features of Chronic LymphocyticLeukemia: A Single Tertiary Centre Experience
Punit Jain, Bargavi Balakrishnan, Ansu Abu Alex, Biju George,Aby Abraham, Rayaz Ahmed, Alok Srivastava, Vikram Mathews
Department of hematology, Christian Medical College, Vellore
Objective: To evaluate the clinical and biological features of chronic
lymphocytic leukemia (CLL) at presentation, indication for treatment
and comparison with reported data. Methods: We undertook a
descriptive retrospective study to evaluate cases diagnosed to have
CLL over the last 5 years at a tertiary care centre in India and review
its literature. Results: 125 cases were diagnosed to have CLL in this
period. The median age was 60 years (range; 34–85). 45 (36 %)
patients were \55 years (young CLL), while 3.2 % were \40 years.
We found a male to female ratio of 3:1. 52 % of the cases were
incidentally detected, while 29 % presented with swellings, followed
by anemia requiring transfusions in 5 % and fever in 1.6 % of cases.
Hepatomegaly was seen in 47.2 % and splenomegaly was seen in
40.8 %. 46.4 % patients had anaemia with autoimmune hemolytic
anaemia being seen in 3.2 % patients. Thrombocytopenia (\1 9 109/
L) was seen in 21.6 %. The median white cell count and the absolute
lymphocyte count were 40 9 109/L and 33 9 109/L, respectively.
Majority of patients presented in Rai stage II (25.6 %) followed by
stage IV (24 %), stage I (19.25 %), stage III (18.4 %) and lastly stage
0 (11.2 %). 68 (54 %) patients received treatment, out of which fol-
low up was available in 53 patients (range 3–45 months). Out of
these, 16 % had stable disease, 13.6 % had partial remission, 7.2 %
had progressive disease, while 5.6 % reached complete remission. In
our series, we also found CD 38 positivity in 42.4 % of the patients.
Of the patients treated, progressive disease was noted in 2.7 %
patients in CD38 negative patients as compared to 13.2 % patients in
CD 38 positive patients. Conclusions: Our findings are similar to data
reported internationally, except that we had a larger number of
younger patients. Most of our patients presented in an advanced stage.
Chlorambucil continues to be a widely used therapeutic option.
Abstract P 063
Profile of Chronic Lymphocytic Leukemia (CLL)—Single CentreStudy from Eastern India
Sandeep Saha, Basab Bagchi, Tuphan Kanti Dolai, PrakasKR Mandal, Ashutosh Panigrahi, Meet Kumar, MaitreyeeBhattacharyya, Shyamali Dutta, Rajiv De, Malay Ghosh
Hematology Department, NRS Medical College and Hospital,
Kolkata
Objective: To determined the demographic and clinical profile of CLL
patients from Eastern India. Methodology: Prospective demographic,
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 211
123

clinical, investigations including immunophenotype (IPT) data were
collected from patients attending hematology services of NRS Medical
College, Kolkata. Period: October 2009 to March 2012. Results:Among the 38 CLL, 29 were male and 9 female. Median age of pre-
sentation were 57 (range 37–78) years. Eight (21.05 %) patients were
asymptomatic at presentation and 73.68 % (28/38) with fever,
78.94 % (30/38) with pallor and 57.89 % (22/38) with infection (RTI).
On examination 28 (73.68 %) had generalized nontender lymphade-
nopathy, lymph node size average 2–4 cm and maximum 6 cm. Ten
(26.32 %) had no peripheral lymphadenopathy (clinically) which were
revealed by CECT or USG. Twenty six (68.42 %) patients presents
with both splenomegaly and hepatomegaly. One had extra nodal
involvement over skin (nodular lesion) below the right eye. Fifteen
(39.47 %) presents with TLC [50,000, 30 (78.94 %) with anemia
(normocytic normochromic anemia) and 23 (60.52 %) thrombocyto-
penia. Six (15.78 %) had positive Coombs test. BM involvement with
interstitial pattern was found in 17 (44.73 %), nodular in 8 (21.05 %),
mixed in 6 (15.78 %) and diffuse pattern in 7 (18.42 %). Myelofibrosis
was found in two cases. Immunophenotyping revealed CD5+ in all
cases, CD23+ and CD20+ in 36 (94.73 %), FMC7 negative in 21
(55.26 %) and sIgM positive in 23 (60.52 %) cases. Thirteen
(34.21 %) were CD 38 positive. Twenty two (57.89 %) patients had
advanced disease (Binet C) at diagnosis. Conclusions: (1) Majority of
CLL presents at fifth decades. (2) Poor prognostic feature (CD 38
positive) were found in 34.21 % subjects. (3) One third patients had
positive FMC7 and negative sIgM. (4) 57.89 % presents with
advanced stage of disease.
Abstract P 064
Spectrum of BCR-ABL Kinase Domain Mutations in Patientswith Chronic Myeloid Leukemia Receiving Imatinib MesylateTherapy
Senthamizh Selvi, Preetha Markose, C Ezhilarasi, EuniceS Edison, Sachin Jain, Biju George, Alok Srivastava, VikramMathews, B Poonkuzhali
Department of Haematology, Christian Medical College, Vellore
Abstract: Imatinib mesylate is the first line therapy for chronic
myeloid leukemia (CML) and induces durable complete haemato-
logical, cytogenetic responses. Despite the impressive responses, a
proportion of patients show resistance to imatinib. This has been
attributed to several mechanisms of which point mutations in the
BCR-ABL kinase domain is the most common mechanism. To date,
approximately 90 mutants in the BCR-ABL kinase domain conferring
different levels of resistance to imatinib have been identified. The aim
of the present study was to analyze the spectrum of BCR-ABL kinase
domain mutations in patients with clinical resistance to imatinib. A
total of 162 CML patients who failed to achieve major molecular
response (in the international scale—IS \ 0.1 %) were included in
the study. RNA was extracted from peripheral blood leucocytes,
cDNA was synthesized. BCR-ABL transcripts were quantified using
real time quantitative PCR (RQ-PCR) using ABL as control gene.
Mutations in BCR-ABL kinase domain was evaluated by nested PCR
followed by direct sequencing. Mutations were identified in 54 of 162
(33.33 %) cases. Nineteen were in chronic phase (CP), 26 in accel-
erated phase (AP) and 9 in blast crisis (BC). Twenty two different
mutations were identified and the distribution of these mutations is
shown in Figure. T315I mutation, which is associated with resistance
to first and second generation tyrosine kinase inhibitors, was the most
common mutation seen [11/54; 20.37 %] followed by the p-loop
mutation M244V [12.96 %]. Among the other reported mutations,
p-loop mutations (Q252H, G250E and Y253H), activation loop
mutation (L384M) and mutations in STI 571 contact site (F317L)
were also observed. In addition, one novel mutation (codon 246–273
indel) in p-loop was identified in two cases. The functional conse-
quence remains to be characterized. The median BCR-ABL IS ratio in
54 patients who had a mutation was 21.43 [0.57–82.42] compared to
16.32 [0.156–74.99] in patients who did not have a mutation which
was not statistically significant (p = 0.2). The frequency of ABL
kinase domain mutations seen in this study suggests that the emer-
gence of a mutation may not be the major mechanism of imatinib
resistance in this cohort. Other mechanisms of resistance including
pharmacogenetic variations, plasma sequestration of the drug or over
expression of BCR-ABL are being evaluated in an ongoing study.
Abstract P 065
A Study to Analyze the Hematological Parameters in NewlyDiagnosed CML Patients with b2a2 Type of BCR-ABLTranscripts
Jogeshwar Binota1, Shano Naseem1, Neelam Varma1,Prateek Bhatia1, Deepak Bansal1, S Varma2
1Department of Pediatrics, 2Department of Internal Medicine, Post
Graduate Institute of Medical Education and Research, Chandigarh
Introduction: Chronic myelogenous leukaemia (CML) is the com-
monest leukemia in Asia. However very few studies have been
published from India, documenting the frequency of BCR-ABL fusion
transcripts, possibly due to diagnostic techniques not being widely
available in our country. The sine qua non of CML, t(9;22)(q34;q11)
and/or BCR-ABL positivity must be detected in all cases of CML.
Major bcr (M-bcr) is almost always involved in CML patients, resulting
in b2a2 and b3a2 mRNA transcripts, p210 chimeric protein, and clas-
sical CML phenotype. Variable frequencies of BCR-ABL fusion
transcripts have been reported from different parts of the world. b3a2
and b2a2 type of BCR-ABL transcripts are detected in approximately
95 % of CML cases, b3a2 transcript being more common than b2a2
transcript. The aim of the present study was to analyze the hemato-
logical parameters at presentation in CML patients with b2a2 type of
transcripts. Materials and Method: A retrospective and prospective
analysis of CML cases was undertaken over a period of 3 years (Sep
2009–August 2012). Multiplex RT-PCR was performed to determine
the BCR-ABL transcripts in all the newly diagnosed CML cases. The
hematological parameters studied in CML cases with b2a2 transcript
were total leucocyte count (TLC), platelet count, absolute basophil
count and LAP score. Results: A total of 410 new CML cases were
0
2
4
6
8
10
12
Y2
53
F
Y2
53
H
T3
15
I
Q2
52
H
M3
51
T
M2
44
V
L3
87
M
L3
84
M
L2
73
M
L2
48
V
L 3
87
F
H3
96
R
F3
59
I
F3
17
L
F3
11
I
E3
55
G
E2
55
V
E2
55
K
D2
95
G
G2
50
E
T3
15
I a
nd Y
25
3F
Nov
el
inde
l
No
. of
pat
ien
ts
Mutations in ABL Kinase Domain
DISTRIBUTION OF ABL KINASE MUTATION
212 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

diagnosed over the study period. The incidence of b2a2 transcript was
26.3 % (108/410). The age ranged from 3.5 to 72 years with median age
of 36.9 years and M:F ratio was 1.6:1. The TLC ranged from 5.8 9 109
to 924.4 9 109/L with median of 160.2 9 109/L and 26 % of the cases
had a very high TLC of more than 200 9 109/L at presentation. Platelet
count ranged from 7 9 109 to 1644 9 109/L with a median
370.3 9 109/L. 5.6 % cases had a platelet count \100 9 109/L and
28.3 % [450 9 109/L. Absolute Basophil count ranged from
0.069 9 109 to 57.05 9 109/L. Severe basophilia ([1000 9 109/L)
was noted in 74 % cases. Discussion and Conclusion: The incidence of
b2a2 transcript in India has been reported to be 28–30 %. The present
study shows a slightly lower incidence of b2a2 transcript in CML cases
in our region. The median ranges for all the hematological parameters
are comparable to that described in western literature and slightly higher
than few of the studies reported from Indian subcontinent.
Keywords BCR-ABL transcripts, CML, Hematological parameters
Abstract P 066
Isolated Central Nervous System Blast Crisis in a Patientof Chronic Myeloid Leukemia on Imatinib Mesylate Therapy
Sanjeev, Ruchi Gupta, Soniya Nityanand
Department of hematology, Sanjay Gandhi Postgraduate Institute
of Medical Sciences, Lucknow
Objective: Imatinib mesylate is the treatment of choice in all phases of
chronic myeloid leukemia (CML). However, the drug or its active
metabolites have a poor penetration in the central nervous system (CNS).
Thus CNS acts as a sanctuary site for malignant cells in these patients. We
report an unusual case of CML on Imatinib therapy who presented with
an isolated CNS blast crisis and peripheral neuropathy. Case-Report: A
25 year old male patient, a known case of CML, presented to the
Department of Hematology, SGPGI, in Aug 2011 with complaints of
headache, double vision, vomiting and tingling sensation of both lower
limbs for past 3 months. He was on Imatinib therapy since February
2011. Salient examination findings were neck rigidity, bilateral flexor
plantar reflexes and absent deep tendon reflexes. The patient was in
hematological remission as confirmed by peripheral blood and bone
marrow examination. Nerve conduction study of lower limbs was sug-
gestive of axonal neuropathy. The CSF showed a turbid appearance with
protein—153 mg/dl, sugar—8 mg/dl, chloride—690 meq/l and TLC-
12,800/cmm. Giemsa staining of CSF smears showed 98 % blasts and
flow cytometry revealed a mixed phenotype blast crisis (B-Myeloid). The
BCR-ABL transcript was 51.4 %. Patient was started on Dasatinib.
Triple drug intra-thecal chemotherapy was given till three samples of
CSF were negative for blasts. The patient 4 months later developed a
systemic blast crisis and expired. Conclusion: Isolated CNS blast crisis
has been rarely reported in CML. Thus in a CML patient, who presents
with CNS symptoms like headache, vomiting, diplopia, etc., even if in
hematological remission, a suspicion of isolated CNS blast crisis should
arise and the patient should be investigated accordingly.
Abstract P 067
A Comparative Study of Sokal Versus Hasford Scoring Systemsin Chronic Myeloid Leukemia Patients in a Tertiary CareHospital in Eastern India
Lopamudra Chakravarty1, Prantar Chakrabarti1, Uttam KumarNath1, Siddhartha Sankar Ray1, Utpal Chaudhuri1
1Institute of Haematology & Transfusion Medicine (IHTM), 3rd floor,
MCH Building, Medical College, 88 College Street, Kolkata 700073, India
Introduction: Chronic myeloid leukaemia (CML) is one of the
commonest leukemias in India. Patients of CML are subjected to
risk stratification at the time of diagnosis according to the available
scoring systems, namely the Sokal and Hasford scores. Objective:Comparison of the Sokal and Hasford scoring systems for predicting
progression free survival and overall survival in patients with CML.
Methods: The data of 52 consecutive patients (n = 52) with diag-
nosis of chronic myeloid leukemia were analyzed retrospectively.
The Sokal score and Hasford score at diagnosis were compared in
terms of prediction of progression-free survival (PFS) and overall
survival (OS) at one year after diagnosis. Results: The median
patient age was 38 years (range 11–72). Thirty percent, 38 and 32 %
patients belonged to the Sokal low, intermediate and high-risk
groups respectively. Forty two percent, 35 and 23 % had low,
intermediate and high-risk Hasford scores, respectively. The PFS
was 90 % and OS was 94 % at one year after diagnosis. Both the
Sokal and Hasford scores predicted better PFS for low-risk patients.
However, there was no significant difference between the two scores
in predicting PFS or OS. Conclusion: In our study there was no
significant difference between the Sokal and Hasford scores in
predicting survival in CML. Late presentation of patients may be
one of the reasons for the higher prevalence of intermediate and
high-risk groups in the study.
Abstract P 068
Characterization of Genes Associated with Disease Progressionin Chronic Myeloid Leukemia by DNA Microarray: AIIMSExperience
Sudha Sazawal, Sunita Chhikara, Rekha Chaubey, RekhaKataria, Manoranjan Mahapatra, Pravas Mishra, Renu Saxena
Department of Hematology, All India Institute of Medical Sciences,
New Delhi-110029
Background: Imatinib resistance is associated with point mutation
in tyrosine kinase domain in 20–40 % of CML patients. The
studies on genetic events that cause the progression of chronic
phase to blast crisis CML in the remaining patients are very lim-
ited. In view of this, we explored gene expression profiling by
DNA microarray in CML patients. Objectives: To identify pre-
dictive genes that might prove informative for stage progression in
CML. Methods: Patients of CML in chronic, accelerated and blast
phase were the subjects of this study. RNA was isolated from
peripheral blood and QC checked using Agilent Bioanalyzer (Ag-
ilent Technologies, Palo Alto, CA). The samples were labelled,
hybridized, scanned and the data was extracted according to the
manufacturer protocol (Agilent Technologies). The data was ana-
lyzed using Agilent Genespring 12.0 software. Results: A total of
14 CML patients (M::8, F::6), median 40 years (range
20–60 years), chronic phase (n = 6), accelerated phase (n = 3),
blast crisis (n = 5) were analysed for changes in gene expression.
A set of genes involved in apoptosis (DAPK1, STAT protein, TNF
and PDCD1), cell adhesion (TNXB, VWF, CD6) and MAPK
pathway were significantly (p B 0.05) under expressed in all the
accelerated phase CML patients. Patients with blast crisis (n = 5)
showed significantly elevated expression of all the genes except
DAPK1. SOCS-2 showed elevated expression both in accelerated
(2/3) and blast phase (3/5) CML. The above genes showed no
expression in the chronic phase patients. The reported mutations
were not observed in any of our patients. Conclusion: Thus, under
expression or elevated expression of the above genes is associated
with the disease progression in CML.
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 213
123

Abstract P 069
Correlation of Multidrug Resistance Gene Polymorphism(T1236C) with Imatinib Response in Chronic Myeloid LeukemiaPatients: AIIMS Experience
Sunita Chhikara, Sudha Sazawal, Rekha Chaubey, RekhaKataria, Manoranjan Mahapatra, Pravas Mishra, Renu Saxena
Department of Hematology, All India Institute of Medical Sciences,
New Delhi-110029
Background: Imatinib mesylate (IM) is a selective tyrosine kinase
inhibitor used for treatment of patients with chronic myeloid leukemia
(CML), however; some patients develop resistance to it. Several gene
polymorphisms have been associated with the resistance and one such
polymorphism is MDR1. Its role has been reported differently among
different populations and different ethnic groups. So far there are no
reported studies from India. Aim of the study: To study the associa-
tion of MDR1 (T1236C) polymorphism in Indian CML patients and
to correlate with response to imatinib. Material and Method: CML,
chronic phase, patients treated with imatinib (400 mg/day) for
24 months were analyzed for T1236C polymorphism. DNA was
extracted from peripheral blood and subjected to PCR–RFLP using
restriction enzyme HaeIII. Results: A total of 100 CML chronic
phase, patients at presentation were included (M:F ratio 2:1) mean
age 40 (range 20–60 years). Of these, 66 patients showed response to
imatinib and 34 patients showed resistance. Among 66 responders the
frequency of three different alleles of T1236C locus was as follows;
CC-43 (86 %), CT-18 (69.3 %), TT-5 (21 %) and in non responders
(n = 34): CC-7 (14 %), CT-8 (30.7 %), and TT-19 (79 %). Resis-
tance to imatinib was higher in patients who were homozygous for the
TT allele in contrast to patients with CT/CC genotype (79 vs. 19.7 %,
P B 0.001). Thus, prevalence of TT genotype seems to be higher in
imatinib resistant patients compared to those who responded to the
drug. Conclusion: It was observed that homozygous TT allele was
associated with higher risk of imatinib resistance.
Abstract P 070
Effect of Long-Term Treatment with Imatinib on Bone Health:Single Institution Experience from Jaipur
Hemant Malhotra1, Bharti Malhotra2, Om Singh Rathore2, AjayYadav1, Shipra Bhargava2, Pratibha Sharma2, Ashwin Mathur1,Sandeep Jasuja1
1Division of Medical Oncology, Department of Medicine, 2Advanced
Research Laboratory, SMS Medical College & Hospital, Jaipur, India
Background: Standard treatment for newly diagnosed patients of
Chronic Myeloid Leukemia (CML) today is the bcr/abl tyrosine
kinase inhibitor, Imatinib. The drug has to be continued for several
years, possibly life-long. Long-term toxicity of the drug is not
established yet. In the present study, we report the effects of the
Imatinib on bone health as assessed by serum bone markers, serum
vitamin D3 levels and bone mineral density by the DEXA scan.
Design and Methods: We evaluated 104 CML patients who had
received Imatinib for more than 1 year at the Birla Cancer Center,
SMS Medical College Hospital, Jaipur. The serum calcium, magne-
sium and 25-hydroxyvitamin D3 levels were analyzed. The Bone
Mineralization Density (BMD) of AP spine in these patients was
measured using DEXA scan. Results: Eighty six percent (86 %,
n = 89) of the total patients (n = 104) received 400 mg IM daily
while 15 % (n = 11) of the patients received 600 mg IM. Eighty
percent (80 %) (n = 82) of the patients received IM for more than
3 year, 12 % (n = 13) received for more than 2 years while 8 %
(n = 9) received IM for more than one year. Eleven out of the 104
patients (11/104) (10 %) and 7/104 (6 %) patients were found to have
low serum calcium and low serum magnesium levels respectively.
The levels of 25-hydroxyvitamin D3 were found to be deficient in
24 % (n = 26) and insufficient in 39 % (n = 40) of the total patients.
Twenty three percent (23 %, n = 30) of the patients were found to be
osteopenic while 8 % (n = 11) were found to have osteoporosis
according to their T score values of AP spine as assessed by DEXA
scan. Conclusion: Our findings suggest that long-term Imatinib may
have an effect on bone health. Periodic monitoring for serum markers
of bone health (vitamin D3, calcium and magnesium) and for osteo-
porosis (by DEXA scan) should be considered in all patients on
long-term Imatinib, especially younger patients. Appropriate supple-
mentation with Vitamin D and/or calcium should be considered for
the deficient patient.
Abstract P 071
Detection of bcr/abl Kinase Domain Mutations by PCR–RFLPand Sequencing in Imatinib Resistant CML Patients
Hemant Malhotra1, Bharti Malhotra2, Shipra Bhargava2,Pratibha Sharma2, Om Singh Rathode2, Ashwin Mathur1,Sandeep Jasuja1
1Division of Medical Oncology, Department of Medicine, 2Advanced
Research Laboratory, SMS Medical College & Hospital, Jaipur, India
Background and Objective: BCR-ABL oncogene mutations are
responsible for the failure of Imatinib (bcr/abl tyrosine kinase
inhibitor (TKI)) treatment in patients with chronic myeloid leukemia
(CML). Moreover, the second generation TKI to be given after
Imatinib resistance depends on the type of mutation present. Patients
with the T315I mutation are resistant to second generation TKIs drugs
like Dasatinib and Nilotinib. Therefore it is important to routinely test
mutation in CML patients who have relapsed or have a sub-optimal
response to Imatinib. Sequencing has been widely used to screen for
bcr/abl kinase domain (KD) mutations in CML patient as a gold
standard but it is expensive and time consuming. PCR RFLP can be
used for early detection of the critical mutations like the T315I and
the technique can provide rapid and inexpensive detection of
important specific mutations. Our study was planned to compare PCR
RFLP and sequencing in CML patients who have relapsed after or are
resistant to Imatinib treatment. Methods: Forty CML patients who
have relapsed after or are resistant to standard Imatinib therapy after
one year were included in the study. PCR–RFLP of Thr315Ile
mutation was done using Dde1 restriction enzyme and ABL KD
mutations were screened by semi-nested reverse transcription
PCR followed by sequencing on ABI 3500 Dx Genetic Analyzer.
Results: Seven different point mutations were detected in the 7/40
(17.50 %) TKI resistant patients. These mutations were located at the
four hot spots of ABL kinase domain: one at the P-loop (Q260H), two
at the imatinib binding site (T315I, T317I), two at the catalytic
domain (M351, E355G) and two at the active A loop (V379E,
D381N). Dde1 RFLP showed wild-type pattern characterized by 2
fragments of 171 and 36 bp in thirty nine patients and one patient had
T315I mutation which showed 207 bp uncut band. Interpretation and
Conclusion: Screening of mutation by PCR RFLP helps in identi-
fying T315I mutation in non responders at an early stage; this is one
of the most important mutation as these patients do not respond to
dasatinib and nilotinib. However sequencing needs to be done in
214 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

patients not having this mutation to help in therapeutic decisions
regarding the choice of the second generation TKI.
MDS
Abstract P 072
Methylation Status of Fragile Histidine Triad (FHIT) Geneand its Impact on Prognosis of Patients with MyelodysplasticSyndrome-AIIMS Experience
Rekha Chaubey, Sudha Sazawal, Sunita Chikkara,Manoranjan Mahapatra, Renu Saxena
Department of Hematology, All India Institute of Medical Sciences,
New Delhi-29
Background: Myelodysplastic syndromes (MDSs) are clonal hema-
tologic disorders that frequently represent an intermediate disease stage
before progression to acute myeloid leukemia (AML). Although several
cytogenetic abnormalities have been associated with this heterogenous
disorder little is known about the genetic changes responsible for the
disease. Deregulated epigenetic mechanisms are likely involved in the
pathogenesis of MDS. Thus, we evaluated the FHIT methylation in
MDS patients and correlated with the prognosis. Aim of the Study: To
investigate the frequency of FHIT gene methylation and its correlation
with the prognosis in MDS patients. Methodology: DNA was extracted
from bone marrow/peripheral blood leucocytes of MDS patients.
Bisulphite modified DNA was amplified by methylation specific PCR.
All the baseline hematological and clinical parameters were correlated
with the FHIT methylation status. Results: A total of 100 MDS patients
were analyzed (M:F ratio 2:1) mean age 46 years (range
9–84 years).Forty three patients (43 %) showed methylation with sig-
nificantly high frequency in RAEB-2 subtypes (P \ 0.02). On
comparing the methylation status with other prognostic factors, FHIT
gene methylation was inversely associated with low WHO risk
(P \ 0.006). The overall survival (4 years) was significantly shorter in
patients with methylated FHIT gene when compared to the patients with
unmethylated FHIT gene (P \ 0.005). Conclusion: These results
suggested that aberrant methylation of the FHIT gene might be involved
in the disease progression and be an adverse prognostic factor in MDS.
Abstract P 073
A Rare Case of Transformation of MDS to AML
Abhisek Saini, Sima Chauhan, Sukumar Chakravarty,Raghumani Mohanty
Department of Pathology, Institute of Medical Science & SUM
Hospital, Bhubaneswar
Abstract: The MDS, represent a spectrum of stem cell malignancies
that manifest dysplastic and ineffective blood cell production in one
or more of the major myeloid cell lines compounded by a variable risk
of transformation to Acute Myeloid Leukemia (AML). MDS occurs
principally in older adults with a median age of 70 years, with a non-
age corrected annual incidence of 3–5/100,000 persons but rising to
[20/100,000 among those over the age of 70 years. MDS usually
arises denovo with the risk increasing proportionate to age. Although
progression to AML is the natural course in MDS, only 10–15 % of
MDS develop AML, and rest progress to BM failure. The percentage
of patient who progress to AML varies substantially in various sub-
types. A higher percentage of MDS with increased myeloblasts
transforms to AML, the biologic course in RA, RARS is prolonged
and indolent with a very low incidence of evolution to AML. A
50 year old female patient presented with Bicytopenia (anemia,
thrombocytopenia) and diagnosed to be a case of RCMD. After a span
of 4 months she transformed into AML which is quite rare.
Keywords MDS, AML, Myelodysplasia
Coagulation Abnormalities
Abstract P 074
Type 2 B Von Willebrand Disease—A Case Report
Sushma Belurkar, Chethan Manohar
KMC, Manipal
Introduction: Von Willebrand Disease (VWD) is the most common
human bleeding disorder. Most of the patients have simple quantitative
deficiency of von Willebrand factor (VWF) but around 20 % can have
qualitative defects in the VWF and are referred to as VWD type II. Type
II is further classified into IIA, B, M and N types. Type IIBVWD is a rare
type of VWD that is characterized by enhanced affinity of VWF to
aspecific platelet receptor, glycoprotein Ib-IX complex. The charac-
teristic functional abnormality in this variant is demonstrated by
ristocetin induced platelet aggregation (RIPA). In type II B VWD very
low doses of ristocetin, which have no effect on normal platelet rich
plasma result in full aggregation. Case report: A 7 year old child
presented with maculopapular rash on the body with bleeding tendency.
Patient also had history of prolonged bleeding following tooth fall.
There was no history of bleeding from any other sites. There was no
positive family or sibling history. Laboratory investigations revealed a
prolonged bleeding time ([15 min) and mildly prolonged APTT
(40.4 s). Platelets were mildly reduced in number. RIPA showed
[50 % aggregation with low dose ristocetin (0.5 mg/ml) and VWF
assay was also mildly reduced (38 %). Hence a diagnosis of type II-
bVWD was considered. Hyperaggregation with low dose ristocetin was
not corrected by adding normal platelets but was corrected by adding
normal plasma, hence platelet type of VWD was ruled out. Conclusion:Type IIBVWD is a rare subtype of VWD which can be easily distin-
guished from other subtypes of VWD due to its characteristic feature of
hyperaggregation with low dose ristocetin however it needs to be dif-
ferentiated from platelet type pfVWD by doing proper mixing studies.
Abstract P 075
Clinical and Haemostatic Profile in Suspected FemaleHaemophilia—A Carriers
S Kate1, A Saxena2, S Aggarwal1, N Gupta1
1Department of Medicine, MAMC, New Delhi; 2Department
of Biochemistry, MAMC, New Delhi
Background: Haemophilia A is an X-linked recessive bleeding disorder
resulting from deficiency of Factor VIII. Hence screening asymptomatic
women carriers is important for an effective informed decision at their
pregnancies. Objective: To study the hereditary transmission in families
with hemophilia A and to conduct screening coagulation tests amongst the
suspected carriers. Methods: Sixty one suspected carriers in 25 families of
haemophilia A were subjected to pedigree analysis and coagulation studies
including prothrombin time, activated partial thromboplastin time and
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 215
123

Factor VIIIc levels. Ten families (10 patients and 31 suspected carriers) were
subjected toRFLPlinkageanalysisusingbcl1 (Intron18)and HindIII (Intron
19) markers. Results: On the basis of pedigree analysis, Fourteen suspected
carriers could be classified as Obligate (22.9 %) and 47 as Possible (77.0 %).
Six out of 61 suspected carriers (10 %) had an abnormal APTT correcting
with normal plasma whereas 53 (86.8 %) had an abnormal mean Factor
VIIIc activity with an average of 25.47 ± 16.3 %. None of the 8 suspected
carriers with normal FVIIIc was obligate carrier or had a history of bleeding.
Eight out of 25 families (32 %) had a family history of haemophilia. The
suspected carriers with a positive family history (inherited case) had a sig-
nificantly lower Factor VIIIc levels as compared to those without, i.e.
sporadic case (p = 0.04). Molecular genetic analysis in 10 families using
bcl1 and hind III RFLP markers gave noninformative results. Conclusions:Suspected women carriers of hemophilia are more likely to have lower
Factor VIIIc levels where there is a positive family history.
Abstract P 076
Genetic Diagnosis of Protein C Deficiency in Two UnrelatedFamilies from India
G Sankari Devi1, S Rajkumar 1, E Sumitha 1, P Shenbagapriya2,SC Nair#, V Mathews 1, GR Jayandharan1, A Srivastava1
Departments of 1Hematology, 2Immunohematology and Transfusion
Medicine, Christian Medical College, Vellore, Tamil Nadu, India
Introduction: Protein C (PC) is a vitamin K dependent coagulation
factor which regulates the activity of factor (F) V and FVIII. The
incidence of clinically significant protein C deficiency has been
estimated to be 1 in 20,000. Only *322 mutations in the protein C
gene (PROC) have been reported as being disease-causative in the
literature. Identification of additional mutations is important for
understanding the biology of protein C deficiency as well as offering
genetic testing to families affected by this disorder. Material andMethods: Two unrelated families diagnosed with hereditary PC
deficiency at Christian Medical College, Vellore were investigated.
PC activity and antigen levels were measured. Mutation analysis was
performed by amplification of the PROC gene using 9 pairs of
primers designed by Primer3 software. A direct DNA sequencing
analysis of the amplicons was carried out in an ABI 3130 genetic
analyzer. In silico analyses, including PolyPhen-2, SIFT, multiple
sequence alignment, splicing prediction were performed to predict the
consequences of each variant identified. Results and Discussion: In
family 1 the proband, a 21 year old female had lost two children due
to purpura fulminans and a spontaneous abortion. The Protein C
levels was 19.2 % (proband/mother) and 24.2 % (husband) reduced in
both the parents, consistent with a heterozygous phenotype and sug-
gesting a autosomal recessive pattern of inheritance. Mutation
analysis identified a p.Cys160Arg missense substitution in the epi-
dermal growth factor domain of Protein C n a heterozygous state in
both the parents. Cys160 is conserved across various species (Homosapiens, Bos Taurus, Canis lupus familiaris, Gallus gallus, Musmusculus, Pan troglodytes, Rattus norvegicus) and a mutation at this
site is predicted to affect the disulphide linkage between Cys160–
Cys147 and thus affect the tertiary structure of Protein C. This family
subsequently underwent antenatal diagnosis. The foetus was found to
be only a carrier of this mutation and the family continued with this
pregnancy. In the second family, a 6 day old, full-term neonate born
from a consanguineous marriage presented with progressive ischemia
and gangrene of multiple body parts. Excluding the common infective
causes, purpura fulminans secondary to a thrombotic cause or sepsis
was suspected. Protein C level in this patient was found to be 1 %. On
genetic analysis, a novel c.1048 A ? T transversion at codon 350 of
the PROC gene was identified in the proband in a homozygous state.
This nonsense mutation predicted premature termination of transla-
tion (Lys350Stop) in the catalytic domain of PROC to result in a
severe phenotype. Further studies confirmed that both the parents
were heterozygous carriers of Lys350Stop mutation. Subsequently,
the mother presented to us for a prenatal diagnosis during her next
pregnancy. The foetus was homozygous for the Lys350Stop mutation
in the PROC gene and thus affected by this disorder. Conclusions:This is the first report describing mutations in PROC gene from
southern India and the mutation data was helpful in offering genetic
diagnosis to families affected by this disorder.
Abstract P 077
Diagnostic Value of a Quantitative Immunoturbidimetricd-Dimer Assay in the Evaluation of Oncology Patients who havea Moderate Pretest Probability of Thrombosis
J Kotwal, MK Patra, A Kotwal, BN Kapoor, V Dutta, V Nair
Armed Forces Medical College, Pune
Objective: Malignancies may present with life-threatening compli-
cations like Deep vein thrombosis (DVT) and consequent pulmonary
embolism (PE). Unfortunately, diagnosis based on history and
examination alone is often unreliable. The d-dimer test has a high
sensitivity and low specificity in diagnosis of thrombosis. Earlier
evaluations studies were based on ELISA for d-dimer which is not
easily applicable for emergency evaluation of patients. The goal of
this study was to test the efficacy of automated quantitative immu-
noturbidimetric d-dimer assay on Coagulometer. Subjects andMethods: Patients at the oncology centre at a tertiary care centre with
moderate probability of DVT for 3 years were evaluated. 210 such
cases were studied. All (210) were in intermediate risk for DVT as per
Well’s score (score 2–4) and 105/210 (50 %) were in intermediate
score for PE as per the revised Geneva score. D-dimer was evaluated
by the automated immunoturbidimetric d-dimer assay (Lia test D-Di
from Stago diagnostica) on the STA compact. Results: 210 cancer
patients were evaluated. Taking the value of d-dimer \0.5 ml (FEU)
as negative, 50 patients were negative and 160 were positive. How-
ever, out of the 160 d-dimer positive cases only 16 were detected to
have DVT/PE. ROC curve was drawn and the max sensitivity and
specificity was at a cut off of 0.80 ml (Sens 75.4 %. Spec 73.8 % and
AUC 0.815). We observed that all the 16 cases of DVT/PE had
d-dimer [ 1.0 |ag/ml. Thus as per our study a d-dimer \1.0 fj.g/ml
would rule out DVT/PE in intermediate risk cancer patients. Con-clusion: The results seem to support the use of a quantitative d-dimer
assay on a coagulometer as a first-line test in evaluation for DVT/
pulmonary embolism in cancer patients when the clinical probability
of the presence of DVT/PE is intermediate.
Keywords d-dimer, Immunoturbidimetric, Coagulometry
Abstract P 078
Assessment of Protein C, Protein S and Prothrombin Levelin Gastrointestinal Carcinoma Patients with and WithoutMetastasis
Ashutosh Kumar1, Mohd. Yusuf1, MLB Bhatt2, Surya Kant3,Abhijit Chandra4
1Department of Pathology, 2Department of Radiation Oncology,3Department of Pulmonary Medicine, 4Department of Surgical
Gastroenterology, King Georg’s Medical University, Lucknow,
Uttar Pradesh, India
216 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Background: A hypercoagulable or prothrombotic state of malig-
nancy with metastasis occurs due to the ability of tumor cells to
activate the coagulation system. Material and Method: A total of 80
Patients with gastrointestinal carcinoma such as malignant carcinoma
rectum, malignant carcinoma oesophagus and malignant carcinoma
colon with and without metastasis were studied in order to evaluate
the presence and extent of hemostatic abnormalities in case of gas-
trointestinal carcinoma. Results: The mean average prothrombin time
in patients of gastrointestinal carcinoma was less than normal value.
Similarly, the average activated partial prothrombin time was also
less than the normal value. The mean level of Protein C ranged from
55 to 90 %. The mean level of Protein S ranged from 48 to 95 %.Out
of the total 80 patients, 7 were Protein C deficient and 5 were Protein
S deficient. However, 3 were Protein C and S both deficient. The
Protein C level was significantly lower (p \ 0.0001) in Protein C
deficient patients with metastasis compared to without metastasis.
Similarly, the Protein S level was significantly lower (p \ 0.0001) in
protein S deficient patients with metastasis as compared to without
metastasis. The Protein C and S levels were also lower in those who
were deficient with metastasis. Conclusion: Our study conclude that
APC resistance in Gastrointestinal Carcinoma with metastasis may
contribute thrombotic episodes in these patients. Cancer patients
including GI malignancy are at increased risk for the development of
thrombotic events that contribute significantly to the morbidity and
mortality of malignancy in these patients.
Abstract P 079
Changes in the Coagulation Profile During InductionChemotherapy in Childhood Acute Lymphoblastic Leukemia
Shivali Sehgal, Sunita Sharma, Jagdish Chandra1, Anita Nangia
Department of Pathology and Paediatrics1, Lady Hardinge Medical
College and Kalawati Saran Children’s Hospital
Introduction: Acute lymphoblastic leukemia (ALL) is the most
common childhood malignancy. The risk of thrombosis in children
with ALL varied from 1.1 to 36.7 % in literature. Two important
factors triggering hypercoagulability are tumor cells themselves and
chemotherapeutic drugs, primarily l-asparaginase. Aims and Objec-tives: To study the effect of induction chemotherapy on coagulation
parameters in paediatric ALL patients. Materials and Methods:Thirty seven newly diagnosed patients of ALL up to 18 years of age
were evaluated along with 30 age and sex matched controls. At the
time of diagnosis (day 0), various coagulation parameters (PT, APTT,
fibrinogen, d-dimer, Protein C, Protein S, Antithrombin, tPA, PAI-1)
were tested. These were sequentially analysed on day 14 (after the
completion of l-asparaginase doses) and on day 28 of therapy (after
the completion of induction). Results: No major change in PT and
APTT was observed during chemotherapy, however, fibrinogen levels
declined significantly (p = 0.04) following l-asparaginase treatment.
D-dimer levels were significantly raised at diagnosis and throughout
induction therapy. PC, PS and AT were reduced in the initial part of
induction followed by a rise in the second half of therapy, reaching
their respective baseline levels (p \ 0.05). The tPA levels were sig-
nificantly reduced in the patients at diagnosis and throughout therapy
(p \ 0.001). PAI-1 levels were comparable to controls at presentation
and showed a rising trend during therapy. Conclusion: The coagu-
lopathy in ALL is multifactorial. The thrombin activation during
induction therapy coupled with deficiency of anticoagulants and
decreased fibrinolysis shifts the balance towards hypercoagulability.
This may enhance the risk of thrombosis in these patients.
Abstract P 080
Hypobaric Hypoxia Alters Coagulation and FibrinolysisPathways
Neha Gupta1, Tathagata Chatterjee2, Srishti Gupta2, MohammadZ Ashraf1
1Genomics Group, Defence Institute of Physiology & Allied
Sciences, Lucknow Road, Timarpur, Delhi, 110054 India;2Department of Pathology, Army Hospital (R&R), Delhi Cantt,
New Delhi-110010 India
Introduction: Studies have indicated that high altitude facilitates the
occurrence of thromboembolic disorders (TED). However, the exact
mechanism still remains to be understood. The present study was
designed to understand the role of HH in TED using rat model of deep
vein thrombosis. Methods: Deep Vein thrombosis was induced by
flow restriction model via ligation of inferior vena cava just below the
renal veins. Hypoxic animals were exposed to simulated altitude of
15,000 ft (429 mmHg). Histopathological studies were conducted in
thrombus, lungs and heart tissue. Expression levels of fibrinolytic
phase protein including PAI-1, tPA were detected by western blotting.
Results: 24 h of hypobaric hypoxic exposure resulted in animal death
and there were indications of Disseminated Intravascular coagulation
(DIC) in HH exposed animals which led to excessive bleeding
resulting in animal death. Histopathological studies revealed the
presence of fibrin thrombi in lungs tissue of thrombotic animals on
exposure to HH. At a HH exposure of lower time point (6 h)
thrombus formation could be captured in IVC of rats. Moreover, the
presence of fibrin thrombi in coronary heart vessels suggests that
thrombus formed in the IVC would have detached and migrated to the
lungs via pulmonary circulation. Levels of fibrinolytic phase proteins;
PAI-1, tpA were found to be altered under hypoxic conditions.
Conclusion: The in vivo experiments conducted so far have sug-
gested that hypobaric hypoxia leads to the development of
hypercoagulation followed by hypofibrinolytic state which leads to
thrombo embolism and ultimately resulting in animal mortality.
Abstract P 081: Oral Presentation
Screening for Lupus Anticoagulant in Women with SpontaneousFetal Loss and its Association with Anticardiolipin Antibodies
Akanksha Rawat1, Meera Sikka1, Usha Rusia1, Kiran Guleria2
1Department of Pathology, 2Department of OBG, University College
of Medical Sciences and GTB Hospital, Delhi
Introduction: Recurrent pregnancy loss affects 1 % of pregnant
women. Several causes of RPL have been observed. However, the
potential cause remains unexplained in several cases which must be
accurately identified for appropriate treatment. Aims: To study the
prevalence of lupus anticoagulants (LA) and anticardiolipin (aCL) anti-
bodies in women with spontaneous fetal loss using a combination of tests,
and to confirm presence of LA by using a platelet neutralization proce-
dure. Methods: Hundred women with spontaneous/recurrent fetal loss
and fifty healthy controls were included in the study. The following tests
were done: complete blood counts (LH 500), PT, APTT, APTT using LA
sensitive reagent, TT, KCT, dRVVT (screening and confirmatory) for LA
detection and ELISA for IgG and IgMaCL antibodies. Results: PT and
TT were not prolonged in any patient or control. APTT of patients was
prolonged in 8 (8 %) patients. Use of LA sensitive reagent identified 13
additional patients suspected of having LA. dRVVT screening time was
found to be prolonged in 16 (16 %) patients and 15/16 patients showed
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 217
123

confirmed positivity. Thus, the prevalence of LA in women with recur-
rent fetal loss was 15 %.A combination of LA sensitive APTT and LA
screening test was found to be most sensitive and specific in identifying
LA. IgG and IgM anticardiolipin antibodies were estimated in 78 patients
and all controls. One (1.2 %) patient was positive for aCLIgG antibodies
and 3 (3.8 %) patients were positive for aCLIgMantibodies. Thus anti-
cardiolipin antibodies were detected in 4 (5.1 %) patients. Conclusions:Screening for antiphospholipid antibodies must be done in women with
spontaneous/recurrent fetal loss even in absence of other clinical mani-
festations. The use of sensitive tests like LA sensitive APTT and
confirmatory and specific test like dRVVT can diagnose a significant
number of these underdiagnosed patients.
Abstract P 082
Coagulation Aberration in HIV Infected Patients
Swapan Kumar Sinha, Soumit Mondal
Department of Pathology, Medical College, Kolkata
Objective: To study disorders of haemostasis in untreated cases of
HIV/AIDS. Total Platelet Count, prothrombin time (PT), activated
partial thromboplastin time (APTT) was performed on 53 diagnosed
patients of HIV. Methods: Citrated blood (1:9) samples were col-
lected for P.Time, APTT, FDPs and EDTA blood for Complete Blood
Count ART Centre of Medical College and School of Tropical
Medicine. PTimre and APPT was performed as per validated methods
with Internal Quality Control. Platelet count was done by making
blood smears of individual samples and counting under light micro-
scope. Results: Among them 19/53 pts. (35.84 %) had decreased plt.
count. Only 7/53 pts. (13.2 %) had elevated PTimer, 6/53 (11.32 %)
had elevated APTT and 9/53 (16.98 %) had elevated PT, APTT and
Low platelet. Conclusion: Thrombocytopenia may be due to
increased destruction commonly immune mediated, increased con-
sumption due to microangiopathy, TTP or decreased production.
Thromboplastin like substances, Factor X activation by angiopathy or
due to CMV infection may lead to activation of coagulation system.
Acquired Factor V Leiden may lead to hypercoagulable states
including APLAS. A detail study in HIV patients for coagulation
disorders will be more informative.
Abstract P 083
F8 Gene Mutation Analysis in Inversion Negative HaemophiliaA Patients—Identification of 29 Novel Mutations
Preethi Satish Nair, Shrimati Shetty, Kanjaksha Ghosh
Department of Haemostasis, National Institute
of Immunohaematology (I.C.M.R), K.E.M Hospital campus,
Parel, Mumbai-400012, India
Objective: Identification of causative mutations in HA is very
important for genetic diagnoses, assessing clinical manifestations and
establishing a mutation database, enabling an accurate genetic diag-
nosis in all affected families. Method: We analyzed 108 inversion
negative cases (37 severe, 36 moderate and 35 mild), by multiplex
PCRs and CSGE technique followed by DNA sequencing. The
pathogenicity of these mutations was assessed using various predic-
tion softwares and genotypic–phenotypic correlation was studied.
Results: We identified mutations in 82 patients with 56 unique
mutations (29 novel and 27 recurrent). 35 missense, 3 nonsense, 1
donor splice site, 10 deletions and 6 insertional mutations were
detected in the present study. A homozygous mutation in a classical
female hemophilic and heterozygous mutation in another female
bleeder was detected. Double mutations were detected in 2 cases (1
familial and 1 unrelated). 8 CRM (+) and 6 CRM reduced cases were
identified, p.R531C was observed to be associated with both CRM (+)
and reduced cases, whereas p.F309L was observed in 2 unrelated
CRM (+) cases. Conclusion: High heterogeneity of F8 gene muta-
tions, with unique mutations in most of the patients analyzed,
suggests the need for maintaining a mutation database in India.
Presence of double mutations gives an indication for a total gene scan,
even when one mutation is identified. Presence of a female bleeder
suggests that all carriers should be screened for FVIII levels, as;
occasionally the heterozygote carriers may have very low levels of
FVIII, most probably due to extreme lyonization.
Abstract P 084
Procoagulant Microparticles and Their Associationwith Pregnancy Loss
Rucha Patil, Kanjaksha Ghosh, Shrimati Shetty
National Institute of Immunohaematology (ICMR), Mumbai
Objective: To study and analyze the role played by the circulating
microparticles in women suffering from pregnancy loss (PL) with
both recurrent miscarriage (RM) and unexplained fetal loss (UFL) and
characterize their cellular origin. Methods: Twenty six women with
RM and 16 normal healthy control women were analyzed in the
present study. Methodology for analysis of microparticles has been
standardized on BD FACS Aria flow cytometer, using the ‘Megamix
beads’ by participating in the ‘‘Vascular Biology SSC workshop:
standardization of FCM-based PMP enumeration’’. Results: The total
annexin MP and activated endothelial (CD 62e) in patients with
recurrent miscarriages of all categories (early, late and early and late)
were higher than in women in the control group (P \ 0.05). Differ-
ences in platelet (CD41a), non activated endothelial (CD 146),
leukocyte (CD45) and erythrocyte (CD 235a) MP levels were not
statistically significant. Conclusion: Our findings suggest that mi-
croparticles may have a role in the pathogenesis of recurrent
miscarriages. One of the proposed causes of recurrent miscarriages is
uteroplacental thrombosis, and MPs may be associated with this. In
addition, presence of elevated total annexin and endothelial micro-
particles at a distance from the adverse pregnancy event suggest a
continued chronic endothelial damage or activation which might be
one of the causes of recurrent pregnancy loss. Although none of the
patients in this study had any thrombotic episode, it is possible that
this might become apparent in pregnancy.
Abstract P 085
A Study on the Environmental and Genetic Factors AffectingInhibitor Formation in Severe Hemophilia A Patients
Sachin David1, Vikram Mathews1, Shashikant Apte3, GiridharaR. Giridhara R. Jayandharan1, Sukesh C. Nair2, Dolly Daniel2,Suresh Kumar1, Ansu Abu Alex1, Saravanan Ganesan1, KannanSubramanian3, Rayaz Ahmed1, Aby Abraham1, AuroViswabandya1, Biju George1, Alok Srivastava1
Departments of 1Haematology, 2Immunohaematology and
Transfusion Medicine, Christian Medical College, Vellore,
Tamil Nadu; 3Department of Haematology, Sahyadri Speciality
Hospitals, Pune
218 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Introduction: Inhibitors or neutralizing antibodies against therapeu-
tic FVIII is a severe complication affecting the treatment of
hemophilia. A number of environmental and genetic factors have
been reported to be associated with the risk of inhibitor development.
We undertook a community based study to evaluate the prevalence of
inhibitors among patients with severe hemophilia A and to study the
predisposing environmental and genetic factors. Material andMethods: Samples from 312 patients were collected which fulfilled the
inclusion criteria (severe hemophilia A and at least 5 life time exposures
to factor replacement). A detailed questionnaire was administered to all
subjects to obtain demographic, clinical and treatment details. Genomic
DNA was used for PCR–RFLP’s (IL4Ra Ile50Val; Arg551Gln, IL4-
590C/T, IL5 746T[C, CTLA4 49G/A), allele specific PCR’s for
identifying cytokine polymorphisms (TNF a-308G/A, TGF b 1 codon
10T/C; codon 25C/G, IL6 -174C/G, IL10 -592A/C; -819C/T;
1082A/G, IFNc +874T/A) was done using a CYTGEN cytokine
genotyping trays from One Lambda (Canoga Park, CA, USA), VNTR’s
in IL5Ra 30UTR by gene scan. HLA Class II typing was done using
standard kit (AllSet+ SSP PCR kits. Dynal, Merseyside, UK). The
immunophenotype of the Treg population was analyzed by flow
cytometry using co-expression of the CD4+, CD25+ markers. Results:Sixty patients (19.2 %) had evidence of inhibitors. Of these 22 patients
(36.6 %) had high titre inhibitors (C5 BU). Table 1 compares the
demographic and laboratory variables among cases that had inhibitors
versus those that did not. We could not find a correlation with envi-
ronmental factors. While all patients in this study had FVIII levels
\1 %, patients with inhibitors had a significantly lower FVIII level than
those that did not (Table 1). Cases with inhibitors were significantly
more likely to have siblings with inhibitors (Table 1). Of the evaluated
immune-regulatory gene polymorphisms a significant protective asso-
ciation was noted with the heterozygous IL4-590 C/T allele and
development of inhibitors (RR = 0.22; 95 % CI 0.108–0.442:
P = 0.000). This protective effect was retained in the high titre subset
when low titre inhibitor positive cases were excluded from the analysis.
There was also a trend to decreased risk of inhibitor formation among
those that were HLA-DRB1-07 positive and this achieved statistical
significance among the inhibitor cases that were high titre positive
(RR = 0.24; 95 % CI 0.055–1.05: P = 0.047). It is recognized that
there is significant heterogeneity in allele distribution of DRB1-07 and
DRB1-13 in the Indian population (www.ebi.ac.uk/imgt/hla). There
was no significant association with T-reg levels and inhibitors. Con-clusions: In this series there was no apparent association with any of the
environmental parameters. There was a significant association with
IL4-590 C/T polymorphism, HLA-DRB1-13 and a trend with HLA-
DRB1-07. This along with the increased incidence of inhibitors in
siblings with hemophilia suggests that inherited parameters may have a
more significant pre-disposing effect than environmental factors in
inhibitor formation in this population.
Abstract P 086
Hematological Parameters with Perinatal Outcomein Preeclamptic Toxaemia—a Study in a Tertiary Care Hospitalof Eastern India
Koyela Mondal1, Swapan Kumar Sinha1, Palash Kumar Mandal1,Bandana Biswas2, Swati Bhattacharyaa3, SanghamitraChakraborty3
1Department of Pathology, Medical College, Kolkatta, Rabindra nath
Mandal, Dehanadapur, Hooghly. 2Department of Gynaecology
& Obstetrics, Medical College, Kolkatta, Rabindra nath Mandal,
Dehanadapur, Hooghly. 3Department of Biochemistry, Medical
College, Kolkatta, Rabindra nath Mandal, Dehanadapur, Hooghly.
Introduction: Preeclampsia and eclampsia are important causes
of morbidity and mortality in pregnant women in developing
countries. Hemolytic anaemia and related disorders like TTP,
HELLP and HUS are commonly seen during pregnancy associ-
ated with preeclampsia and eclampsia and may cause further
complications. Objectives: To study haematological parameters,
coagulation profile and liver enzymes with perinatal outcomes in
preeclasmpsia and eclampsia. Methods: Out of 10,782 number of
antenatal mothers attending the labour room during the period of
August 2011 to July 2012, 636 were found to have severe pre-
eclampsia and eclampsia. Physical examination of patients with
routine urine and blood examination was done in all the patients.
Complete hemogram with coagulation profile (PT, APTT, FDP)
and liver function tests of all 636 mothers were noted. Results:Patients were in age range of 21–38 years. The criteria for
severe preeclampsia included sustained blood pressure of at least
160/110 mmHg or higher with persistent proteinuria on urine
dipstick or elevated creatinine ([1.2 mg/dL). Out of 636, 305
(48 %) mothers were anaemic, fifty cases (8 %) of thrombocy-
topenia alone. Out of 305 anaemic mothers, 57 (19 %) presented
with HELLP syndrome. PT, APTT, FDP was raised in 19, 45
and 9 cases respectively while combined PT and APTT in 50
cases. Still birth was found in 31 cases (5 %) and 190 cases
(30 %) were of small for gestational age. Conclusion: Early
detection of toxaemia and also hemolytic disorders in antenatal
patients may in preventing morbidity and mortality of mothers
and newborns.
Abstract P 087
Genetic Determinants of Warfarin Dosage in Indian Patients
Tejasvita Gaikwad, Shrimati Shetty, Vrinda Kulkarni1,Cecil Ross2, Shanaz Khodaiji3, Kanjaksha Ghosh
National Institute of Immunohematology (ICMR), KEM Hospital,
Mumbai; 1Nair Hospital, Mumbai; 2St.John’s Hospital, Bangalore;3P.D Hinduja Hospital, Mahim
Introduction: Warfarin, a widely prescribed anticoagulant for
prevention of thrombosis, is associated with narrow therapeutic
window where even small variations in dosing may result in
hemorrhagic or thrombotic complications. Several polymorphisms
in VKORC1, CYP2C9, CYP4F2, EPHX1, GGCX, factor IX, factor
II and factor VII have been shown to be associated with warfarin
dose requirement. Material and Methods: 120 warfarin treated
patients were screened for CYP2C9*2, CYP2C9*3, VKORC1-
1639G/A, EPHX1 T113C (rs1051740) by PCR–RFLP and allele
specific PCR method. Results: See Table. (1) CYP2C9 and
VKORC1 promoter region polymorphisms: Overall 44.4 % studied
patient population was found to be carriers for any of the three
warfarin sensitive alleles, requiring low warfarin dose as compared
to wild type, i.e. 3.1 mg versus 6.25 mg (±1.3 mg) OD. Of this,
approximately 62 % patients manifested with bleeding problems
while on warfarin. (2) EPHX1 gene polymorphisms: Patients with
CC genotype require approximately 1 mg/day less warfarin than
patients with TT genotype. Conclusions: Approximately 45 % of
the patients carry any one of other 3 strongly linked warfarin
sensitive polymorphisms, suggesting the need for identifying the
warfarin genotype before starting the dose. The strongest deter-
minant of warfarin dosage was shown to be VKORC1 AA,
followed by cytochrome 3 and cytochrome 2 polymorphisms.
EPHX1 polymorphism showed a minor effect on warfarin dosage.
Keywords Warfarin genotype, VKORC1, CYP2C9, EPHX1,
Warfarin adverse effect
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 219
123

Association of different variants with warfarin dose requirement and
over anticoagulation
No. Mean warfa-
rin
dose
Patients
with
INR \ 3
Wild type allele carriers
VKORC1
GG + CYP2C9*1/
*1 + EPHX1 TT
31 6.2 7 (22.5 %)
One variant carriers
VKORC1 variant carriers
VKORC1
GA + CYP2C9*1/
*1 + EPHX1 TT
9 4.0 6 (66.6 %)
VKORC1
AA + CYP2C9*1/
*1 + EPHX1 TT
1 2.0 1 (100 %)
Cytochrome variant
carriers
VKORC1
GG + CYP2C9*1/
*2 + EPHX1 TT
3 4.41 1 (33.3 %)
VKORC1
GG + CYP2C9*1/
*3 + EPHX1 TT
5 3.36 3 (60 %)
VKORC1
GG + CYP2C9*3/
*3 + EPHX1 TT
1 2 1 (100 %)
VKORC1
GG + CYP2C9*2/
*3 + EPHX1 TT
1 3.5 1 (100 %)
EPHX1 variant carriers
VKORC1
GG + CYP2C9*1/
*1 + EPHX1 TC
30 5.19 8 (26.6 %)
VKORC1
GG + CYP2C9*1/
*1 + EPHX1 CC
10 5.02 3 (30 %)
Combined variant carriers
VKORC1
GG + CYP2C9*1/
*3 + EPHX1 TC
9 2.94 4 (44.4 %)
VKORC1
GG + CYP2C9*1/
*3 + EPHX1 CC
3 3.16 2 (66.6 %)
VKORC1
GA + CYP2C9*1/
*2 + EPHX1 TT
1 3.5 1 (100 %)
VKORC1
GA + CYP2C9*1/
*3 + EPHX1 TT
1 2.5 1 (100 %)
VKORC1
GA + CYP2C9*1/
*1 + EPHX1 CC
1 2.5 1 (100 %)
VKORC1
GA + CYP2C9*1/
*1 + EPHX1 TC
14 2.98 8 (57.14 %)
Abstract P 088
FXI Deficiency—a Rare Cause for APTT Prolongation
Archana Suman, SK Bose, Joseph, Alaknanda, C Batra,J Ahluwalia, S Varma1, N Varma
Department of Hematology and Internal Medicine1, Postgraduate
Institute of Medical Education and Research, Chandigarh
Introduction: FXI deficiency is a rare bleeding disorder character-
ized by a hemorrhagic syndrome of variable degree of severity. FXI is
involved in intrinsic coagulation pathway. Here we present a case
report of patient with FXI deficiency. Case Report: A 16 year old
Muslim girl, presented with an episode of hemochezia 2 years back
and epistaxis since 2 years. She had no family history of bleeding or
previous transfusion. She was born of a consanguineous marriage.
Laboratory tests revealed normal PT and prolonged APTT which was
corrected by normal pooled plasma. Mixing studies showed correc-
tion of APTT with Factor VIII and IX deficient plasma and no
correction with FXI deficient plasma. The platelet count was normal.
Factor XI assay revealed marked deficiency (1 %) in patient. Family
screening revealed low FXI activity in the mother and father, 40 and
39 % respectively. Her sister was asymptomatic and had normal FXI
levels. Conclusion: FXI deficiency is a rare cause of bleeding and
must be looked for in patients with isolated APTT prolongation. The
diagnosis is usually delayed in females with prolonged APTT, since a
commoner cause—Von Willebrand disease needs to be excluded.
This case reiterates the fact that as has been described earlier, the
factor levels do not coincide with symptom severity. Less than 150
cases of FXI deficiency have been reported so far in India.
Abstract P 089
The Laboratory Profile of the Thrombotic AntiphospholipidSyndrome
J Ahluwalia, Joseph, SK Bose
Department of Hematology, PGIMER Chandigarh
Objective: The antiphospholipid antibody syndrome (APS) is the
commonest acquired cause of thrombosis). We attempted to
determine the prevalence of APS in patients with thrombosis and to
study the pattern of positivity in the laboratory tests. Methods andPatients: Patients with thrombosis undergo 3 tests viz Lupus
anticoagulant testing (clot based screening and confirmation) and
anticardiolipin and anti beta 2 Glycoprotein 1 antibody assay (both
by EIA) as a part of workup of the APS. Laboratory data of
patients reporting for the investigation for thrombophilia for a
thrombotic event was reviewed and examined for the persistent
positivity of these tests at an interval of at least 3 months. Results:In the period of 3 years, 991 cases were tested for the APS. 889
cases were available for analysis. In 186 (20 %) of these any one
of the APS antibodies were positive at least once. In 30 (3.3 %) the
antibodies persisted for at least 12 weeks and they qualified for the
APS. Only 2 cases had persistent positivity for all 3 antibodies. In
22 cases only 1 antibody was positive. Anti beta 2 GP 1, Anti-
cardiolipin and LA antibodies were positive in 28, 9 and 4 cases
respectively. Fifteen of the 30 cases had CNS vascular obstruction,
whereas 6 had limb DVT. Conclusions: Positivity for all 3 anti-
bodies is rare. Anti beta2 GP 1 antibodies were the commonest
ones encountered. Testing for LA is less frequently positive pos-
sibly because of inability to reconfirm the initial positive test due
to start of anticoagulation.
220 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Abstract P 090
Molecular Basis of 19 Severe Factor XIII Deficient Cases
Sharda Shanbhag, Shrimati Shetty, Kanjaksha Ghosh
National Institute of Immunohaematology (ICMR), Mumbai
Introduction: Congenital Factor XIII (FXIII) deficiency is a rare
autosomal recessive disorder affecting 1 in 1–5 million individuals,
with a higher prevalence in countries where consanguineous mar-
riages are common. It is a serious bleeding diathesis, generally
manifested as umbilical stump bleeding, post-injury prolonged
bleeding, intra cranial bleeding, and spontaneous abortions in women.
FXIII deficiency is usually attributed to mutations in F13A gene, on
chromosome 6. Material and Methods: We analyzed 19 FXIII
deficient patients, diagnosed on the basis of their clinical history,
normal screening coagulation and clot solubility assay. Genomic
DNA was extracted by the phenol chloroform method, and mutations
were detected by direct DNA sequencing of F13A gene. 14 of 19
patients had history of primary consanguinity. Results: 17 mutations
were detected in 19 FXIII deficient patients, of which 8 were missense
(5 novel, 3 recurrent), 6 were nonsense (3 novel, 3 recurrent), and 2
patients showed a novel single base pair deletion; one patient showed
a splice site mutation in exon 14. A large deletion in exon 3 is sus-
pected in 2 unrelated patients because of repetitive failure of PCR
amplification of this exon. Seven polymorphisms were detected in
these patients, of which one is novel. Conclusions: We have identi-
fied F13A gene mutations in all the 19 FXIII deficient patients, and
observed a high heterogeneity in the mutation profile. The data
obtained would assist in establishing a National Mutation Database,
and enable an accurate carrier and antenatal diagnosis in affected
families by direct mutation analysis.
Abstract P 091
Variations of PT Reagent to FX Deficiency
Shenbagapriya, Ramya, Jennifer, Suresh, Vandana Kamath,Usha Sitaram, Sukesh C Nair
Department of Transfusion Medicine & Immunohaematology,
Christian Medical College, Vellore, India
Abstract: FX deficiency is a rare bleeding disorder which results in
variable bleeding tendency, but patients with severe FX deficiency
tend to have severe bleeding manifestations among the rare bleeding
disorders. Prothrombin time reagents may vary in sensitivity to FX
deficiency and congenital variants have been identified in which PT or
APTT being normal. We have a case of 31 year old female with
complaints of menorrhagia for the past 3 months, easy bruisability
and prolonged bleeding. Laboratory Findings: Plasma clotting test
and Global haemostatic test were done and the results are as follows:
Plasma Clotting test: PT PT (Innovin:Recombinant Thromboplastin):
Pt: [ 2 min Normal range: 10.0–12.5 s INR: [10.0;PT (Thromborel
S:Human Placental Thromboplastin): Pt: 28 s;aPTT: Pt:52.4 s Nor-
mal range: 25.0–34.8 s;� PT with � control plasma: 11.4 s;� PT
with � adsorbed plasma :23.1 s;� PT with � aged serum: 11.0 s;TT:
Pt:13.8 s Normal range: 12–16 s;FVIII: 367.6 % FIX:315.4 % FXI:
195.8 % FV: 167.0 %; FX: \1 % (Innovin:Recombinant Thrombo-
plastin); FX: 5.7 % (Thromborel S:Human Placental Thrombo-
plastin); Lupus Anticoagulant (DRVVT): Negative Pt: Screen: 64.5 s
Screen Mix: 40.2 s Control Screen: 37.2 s. Global Haemostatic test
also showed normal clotting parameters. Marked prolongation of PT
([2 min–132.5 s) to recombinant Thromboplastin which also resulted
in its assay system revealing severe deficiency of FX. This could be
either a laboratory phenomenon or FX variant.
Abstract P 092
The Epidemiology of FVIII Inhibitors in Indian HaemophiliaA Patients
Patricia Pinto, Preethi Nair, Priyanka Kasatkar, TejasvitaGaikwad, Shahnaz Ali, Anshul Jadli, Rucha Patil, BipinKulkarni, Kanjaksha Ghosh, Shrimati Shetty
Department of Haemostasis & Thrombosis, National Institute
of Immunohaematology, (Indian Council of Medical Research),
Mumbai, India
Objective: A serious complication of replacement therapy in patients
with bleeding disorders, is the development of specific antibodies or
‘inhibitors’ to the deficient coagulation factors, particularly in haemo-
philia A patients. This leads to an increase in the management cost,
morbidity and mortality, especially post-operatively in case of FVIII
inhibitors. The mechanism of FVIII inhibitor development is quite
complex and it is difficult to predict their development, but a prompt and
accurate diagnosis is critical as early therapy can save lives. The aim of
this study was to screen patients with bleeding disorders for inhibitors,
and analyse the incidence of inhibitors in different regions in India.
Methods: Samples were collected in sodium citrate vacutainers from
patients in different cities in India for coagulation and inhibitor screening
assays. The Bethesda assay was performed in inhibitor positive samples
for confirmation and quantification of the FVIII Inhibitor titre. Patient
Table 1 Comparison of cases with and without inhibitors
Demographic parameters Inhibitor posi-
tive: N = 60
N (%)/median
(range)
Inhibitor neg-
ative: n = 252
N (%)/median
(range)
P value
Age (years) 18,.5 (3–59) 18 (2–75) 0.647
Age at diagnosis of
hemophilia (years)
1.5 (0–55) 1.5 (0–32) 0.718
FVIII:C (%) 0.25 (0.04–0.99) 0.35
(0.10–0.60)
0.007
No: of exposures in first
year of life
0 (0–4) 0 (0–25) 0.696
Life time number of
exposures
30 (2–336) 23.5 (5–1,000) 0.164
FVIII (life time) total
factor replacement
used (IU)
13,200
(1,000–241,000) 9,200
(500–425,600) 0.133
Number of joints affected 2 (0–7) 3 (0–7) 0.505
Parental consanguinity 18 (30) 56 (22.2) 0.237
Familial hemophilia 38 (63) 152 (60.3) 0.769
Sporadic hemophilia 22 (36.7) 100 (39.7) 0.769
Siblings with inhibitors 4 (6.6) 5 (1.96) 0.016
Discordance between
siblings for inhibitors
7 (11.6) 12 (4.76) 0.044
Chronic Illness 6 (10) 19 (8) 0.596
Past history of surgical
procedure
7 (11.7) 52 (20.6) 0.142
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 221
123

details were recorded in a clinical proforma. A total of 800 patients with
bleeding disorders from different cities of India were screened for
‘Inhibitors’. Results: Out of the 800 samples screened, 710 were Hae-
mophilia A patients, out of which 51 were positive for ‘FVIII Inhibitors’.
Conclusion: The highest incidence of FVIII Inhibitors, i.e. 20.48 % was
seen among the Chennai samples, followed by Hyderabad, Mumbai and
Guwahati, which showed an incidence of 12.5, 11.02, and 8.51 % FVIII
Inhibitors respectively, with respect to the samples analysed. The other
regions showed an Inhibitor incidence\7 %. The overall FVIII Inhibitor
incidence in the haemophilia A samples studied was 7.18 %.
Keywords FVIII Inhibitors, epidemiology, haemophilia A
Abstract P 093
Analysis of FVIII Polymorphism Haplotypes as Risk Factorsfor FVIII Inhibitor Development in Indian Severe HaemophiliaA Patients
Patricia Pinto, Kanjaksha Ghosh, Shrimati Shetty
Department of Haemostasis & Thrombosis, National Institute
of Immunohaematology, (Indian Council of Medical Research),
Mumbai, India
Objective: ‘Factor VIII (FVIII) inhibitor’ development, a serious
complication of FVIII replacement therapy in haemophilia A patients,
leads to an increase in management cost, morbidity and mortality,
especially post-operatively. Recently, four non-synonymous single-
nucleotide polymorphisms (SNPs) in the F8 gene, G1679A [R484H],
A2554G [R776G], C3951G [D1241E], and A6940G [M2238V],
whose haplotypes encode six wild-type FVIII proteins (H1–H6), have
been implicated as risk factors for FVIII inhibitor development due to
mismatched FVIII transfusions. The aim of the study was to deter-
mine if mismatched FVIII transfusions could significantly contribute
to FVIII inhibitor development in Indian haemophiliacs. Methods:We analyzed the above four SNPs, in 60 Indian severe hemophilia A
patients, i.e. 30 inhibitor positive and 30 inhibitor negative control
patients, by direct DNA sequencing of the relevant regions of the F8gene. Results: The prevalence rates of the H1 and H2 haplotypes,
were found to be 1.00 and 0.00 among the inhibitor positive patients,
and 0.94 and 0.06 among the inhibitor negative patients, respectively.
These two haplotypes (H1 and H2) match the recombinant FVIII
products Kogenate (Bayer) and Recombinate (Baxter), used clini-
cally. Conclusion: The F8 polymorphism haplotypes are probably
not a risk factor for FVIII inhibitor development in Indian haemo-
philiacs, but could explain the lower inhibitor incidence. Further
studies, in a larger patient cohort, with regard to association with F8mutations and other genetic risk factors of inhibitor development,
could provide useful insights into the FVIII immune response.
Keywords FVIII Inhibitors, F8polymorphisms, Haemophilia A
Abstract P 094
Effect of Antitubercular Therapy on Haemostasis IncludingCoagulation Factor VIII in Patients with Tuberculosis
Aditya Kutiyal, Naresh Gupta, Sandeep Garg
Department of Medicine, Maulana Azad Medical College,
New Delhi-110002, India
Objective: To study the effect of antitubercular therapy on haemo-
static parameters including coagulation Factor VIII in patients with
different types of tuberculosis. Methodology: Adult patients over
12 years of either sex with newly diagnosed tuberculosis of different
sites categorized as pulmonary, CNS, disseminated and others were
included. Detailed clinical assessment included the haemostatic
parameters namely Prothrombin Time, Activated Partial Prothrombin
Time, Factor VIII, fibrinogen and D-dimers were done in all, before
and after 2 months of intensive standard antitubercular therapy.
Results: Mean age of the 128 subjects was 31.55 ± 15.03 years,
ranging 12–75 years. Pulmonary, CNS, disseminated and other types
of tuberculosis comprised 30.5, 28.9, 17.2, and 23.4 % patients
respectively. Abnormal PT and APPT at baseline was seen in 64
(50 %) and 23 (18 %) cases respectively whereas the mean coagu-
lation Factor VIII level was 125.08 % ± 72.83. Factor VIII was
deranged in 45 (35.15 %) cases, being high in 28.12 % and low in
7.03 %. Baseline fibrinogen levels were altered in 73 (57 %), with 62
being elevated beyond 400 mg%. D-dimers were high in 57.8 %
patients. Platelets were disturbed in 66 (51.5 %), with thrombocyto-
paenia in 47 and thrombocytosis exceeding 400 9 109/l in 19 cases.
Following 2 months ATT, the PT became normal in statistically
significant 32 (50 %) of 64 abnormal at baseline. Factor VIII levels
did not change significantly after ATT (125.08 ± 72.83 vs.
126.21 ± 37.65) whereas fibrinogen decreased significantly from
391.43 ± 167.34 to 272.33 ± 92.66 mg% (p \ 0.01). Cases with
elevated D-dimers came down to 12.5 %. Interestingly, all the platelet
disturbances reverted to normal within 2 months of ATT. Subgroup
analysis of different categories of tuberculosis mirrored these results
except the PT in disseminated group. Conclusions: Elevated fibrin-
ogen, D-dimers, and Factor VIII levels were observed in patients with
different types of tuberculosis, favouring a state of hypercoagulability
which improved with anti-tubercular therapy.
Keywords Tuberculosis, Haemostasis, Coagulation factor VIII,
Hypercoagulability
Hemoglobino Pathies
Abstract P 095
Evaluation of D-10 Hemoglobin Testing System for Diagnosisof Beta Thalassemia Carrier State
Debajyoti Singha Roy
Ramakrishna Mission Sevapratisthan and Vivekananda Institute of
Medical Science, 99 Sarat Bose Road, Kolkata-26
Objective: Prevention is the only way to reduce the burden of thal-
assemia through beta thalassemia carrier state detection. The most
reliable method is High Performance Liquid Chromatography
(HPLC). The conventional HPLC instrument (VARIANT, BIO-RAD)
is very expensive ([3 million).The present study examines the fea-
sibility of using a relatively less expensive HPLC instrument D-10
HEMOGLOBIN TESTING SYSTEM, BIO-RAD which was initially
launched for HbA1c estimation by HPLC. After a thorough search we
could not find any comparative study between the VARIANT and
D-10. Methods: Study population- Patients who are diagnosed as beta
thalassemia carrier by D-10 in RKMSP and VIMS. Exclusion criteria:
1. Children \ 6 months; 2. Patients with prior blood transfusion; 3.
Co existing hematinic deficiency and borderline HbA2 value. Data
collection: same blood samples of the cases diagnosed as beta thal-
assemia carrier by D-10 are tested in SYSMEX KX21 cell counter for
red cell indices and also in VARIANT, BIO-RAD. Some retained
samples are also run in D-10 on day 7. Analysis: statistical analysis
done to establish correlation between results obtained from D-10 and
VARIANT. Results: Sample size 96. (1) Between D-10 and
222 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

VARIANT. (2) HbF value- 99 % correlation. (3) HbA2 value—83 %
correlation. (4) Not a single case misdiagnosed. (5) 16 samples run on day
1 and 7 in D-10. (6) HbF—95 % correlation. (7) HbA2—97 % correla-
tion. Conclusions: After obtaining a good correlation value it can be
concluded that D-10 HEMOGLOBIN TESTING SYSTEM is a reliable,
low cost instrument for beta thalassemia carrier state detection.
Abstract P 096
Detection of Pattern of Hemoglobin in Patients AdvisedHPLC—A Hospital Based Study
Aishwarya Raj, Jina Bhattacharya, UC Dutta, PK Gogoi
Department of Hematology, Gauhati Medical College and Hospital,
Guwahati, Assam
Objective: Study was conducted to detect the pattern of hemoglobin
and relative distribution of different variants in patients attending the
Hematology department, GMCH. Methods: Present study was con-
ducted in Department of Haematology, Gauhati medical college and
hospital from 1st December 2010 to 31st July 2012. Geographical
distribution of cases predominantly included parts of Assam. Patients
presenting with clinical features and blood examinations indicating
haemoglobinopathies were subjected to HPLC. Further tests were
carried out in selected cases (sickling test, acid elution test, osmotic
fragility test etc.). Result: Out of total 783 patients subjected to HPLC,
394 cases (50.38 %) were having normal Hb A. Among the 49.62%
cases with abnormal hemoglobin, the most prevalent pattern was het-
erozygous Hb E (21.18 %) followed by homozygous Hb E (15.07 %).
Compound heterozygous Hb E-b was the next commonly observed
variant (6.13 %). 2.29 % had sickle cell disease; 0.26 % had sickle cell
trait, 0.38 % had Hb S-b trait, while two cases (0.24 %) with double
heterozygous Hb S-Hb E were observed. A single case (0.12 %) of
hereditary persistence of fetal hemoglobin was noted. 0.89 % cases
suffered from beta thalassemia major, while 3.06 % cases were het-
erozygous for beta thalassemia. Conclusion: Study shows that the
prevalence of abnormal hemoglobin pattern is 49.62 %, of which the
most common is noted in Hb E (36.25 %) in this part of India.
Abstract P 097
Trials of Hydroxyurea in Sickle Cell HemoglobinopathiesPatients of Eastern India
Prasanta Purohit, Dilip Kumar Patel1, Siris Patel, SnehadhiniDehury, Subasa C Bishwal, Satyabrata Meher, BidyapatiPradhan, Kishalaya Das
Sickle Cell Clinic & Molecular Biology Laboratory, VSS Medical
College, Burla, Sambalpur, Odisha; 1Department of Medicine, OSCP,
NRHM, V.S.S. Medical College, Burla, Odish
Introduction: Increased level of HbF (22.3 ± 6.9) was found to have
protective effect against painful crisis, osteo-necrosis, ACS and
splenic dysfunction in Sickle cell disease patients of Odisha (Mashon
RS, 2009). Low dose Hydroxyurea (10 mg/kg body-wt/day) effec-
tively increased the level of HbF in this patients (Patel DK, 2012).
Unfortunately, the use of HU is extremely limited in India, because of
its high cost and the apprehension of its toxicities. Objective: To
assess the clinical and haematological response of low dose
Hydroxyurea in patients of sickle cell hemoglobinopathies in eastern
India. Materials and Method: The study was undertaken at Sickle
Cell Clinic & Molecular Biology Laboratory, V.S.S. Medical College,
Burla, Odisha, India, from 2006 to 2012. 1887 patients were enrolled
including 1738 HbSS (35.7 % paediatrics and 64.3 % adults), 125
HbS-b thalassemia (33.6 % paediatrics and 66.4 % adults), 18 HbSD
and 6 HbSE. The indication were[3 VOC or[2 blood transfusion in
last 12 months of presentation. Detailed baseline studies, i.e. CBC,
HPLC, Bio-chemical, liver function test were done and cases were
followed up at three month intervals. Here we analysed the data of
364 cases under regular follow-up for more than 2 years of HU
therapy. Result: After 2 years HU therapy %HbF increased signifi-
cantly from 18.6 ± 6.9 to 22.5 ± 7.3 but the proportionate rise of
HbF varied from case to case. MCV, MCH and MCHC level also
increased significantly in all cases. The frequency of painful crises
reduced significantly after HU therapy. In paediatric cases response
rate was 71.5 % where as in adults it was 91.2 %. Following HU
therapy, about 95.0 % patients became transfusion independent.
Transient bone marrow suppression (Absolute Neutrophil Count
\2,500/lL, platelet count \80,000/lL) was occurs in 6.96 % cases.
Discussion: With a minimal dose (10 mg/kg body-wt/day) of HU,
most of the patients showed an impressive improvement in clinical
and haematological parameters. In resource poor country like India,
low dose HU therapy provides a suitable therapeutic option for a vast
number of untreated sickle cell hemoglobinopathies patients.
Abstract P 098
Clinical Hematological and Molecular Characterization of SickleCell HbDPunjab (HbSD) in Eastern India: The Largest seriesin world
Subasa C Bishwal, Dilip Kumar Patel1, Siris Patel, SnehadhiniDehury, Prasanta Purohit, Saytabrata Meher, Bidyapati Padhan,Kishalaya Das
Sickle Cell Clinic, VSS Medical College & Hospital, Burla,
Sambalpur, Odisha-768 017; 1Department of Medicine,
V.S.S. Medical College, Burla, Odisha
Introduction: Although HbDPunjab[b-121(GH4)Glu ? Gln] is the
commonest haemoglobinopathy in northern India, HbSD, the com-
pound heterozygote state with sickle gene is rare. Hereby we report
detailed profile of 36 cases Hb SD, the largest number hitherto reported
in world literature. Objectives: To describe the clinical, hematological
and molecular profile of 36 HbSD patients from Eastern India. Mate-rials and Methods: HbSD disease was diagnosed by HPLC. All cases
were subjected to detailed clinical, radiological, biochemical exami-
nation and CBC. HbS mutation was confirmed by ARMS-PCR,
HbDPunjab mutation was confirmed by EcoR1-RE digestion following
PCR, RFLP and Xmnl polymorphism were studied by analyzing six RE
sites as described by Orkin et.al (1982). a-Thalassaemia was diagnosed
by GAP-PCR. Results: The mean age of patients was
21.63 ± 11.7 years and majority belonged to a particular caste namely
the Agharia, which is a predominant caste of Western Odisha. Highest
number (30.55 %) of patients was from Sundergarh district. The
commonest clinical presentation was blood transfusion (in 69.44 % of
patients) followed by painful crisis (66.67 % of patients). 32.0 % of
patients had splenomegaly and hepatomegaly. 18.18 % had AVN of the
femur head, 12.0 % had cholelithiasis, whereas 8.0 % had splenic
atrophy. The mean Hb, MCV and MCH were 8.4 ± 2.59 g/dL,
88.54 ± 11.0 fl and 29.4 ± 4.34 pg respectively in the HbSD patients.
The mean HbF concentration was 19.05 ± 9.95 % and HbD was
41.46 ± 3.89 %. 81.18 % were heterozygote for Xmnl polymorphism
whereas 18.82 % were homozygotes and 5 cases (15.0 %) had dele-
tional a-thalassaemia. Conclusion: We report the detail profile of 36
cases of HbSD patients who were thoroughly investigated and their
detailed molecular characterization was accomplished.
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 223
123

Abstract P 99
Epidemiology of Beta Thalassemia Trait in Western Odisha
Satyabrata Meher, Dilip Kumar Patel1, Siris Patel, PrasantaPurohit, Snehadhini Dehury, Subasa C Bishwal, BidyapatiPadhan, Kishalaya Das
Sickle Cell Clinic, VSS Medical College & Hospital, Burla,
Sambalpur, Odisha-768 017; 1Department of Medicine & Project
coordinator, OSCP, NRHM, V.S.S. Medical College, Burla, Odisha
Introduction: Heterozygote state of Beta thalassaemia (Beta thalas-
saemia trait or BTT) is benign and asymptomatic. Detection of BTT
has been very important in the effort of prevention of the disease by
prenatal diagnosis. Microcytic hypochromic red cells and an elevated
HbA2 fraction is hallmark to detect BTT. Objective: To study the
epidemiology of BTT in Western Odisha from our Centre. Materialand Method: Total of 5690 blood samples were collected from cross
sectional study from different parts of Western Odisha were studied at
the Sickle Cell Clinic and Molecular Biology Laboratory, V.S.S.
Medical College, Burla, Odisha, India. Samples were subjected to
CBC and CE-HPLC to quantify haemoglobin fractions. Cases with
MCV \ 80 fl and MCH \ 27 pg were suspected for BTT (Colah
et al. 2007). Cases with HbA2 [ 3.5 % (Madan et al. 2010) and
HbF \ 3.0 % were considered as BTT. Results: From a total of 5690
cases, 213 (3.7 %) individuals were found with BTT. The mean age
of males and females were 32.1 ± 14.1 and 31.7 ± 14.6 years
respectively. 3 (1.16 %) cases of BTT were found among a total of
259 ANC cases screened. 97.65 % of BTT were Hindu by religion
and only 2.35 % were from other religions. Among Hindus, 44.6 %
fall under OBC and 24.9 % under SC category, whereas 30 cases each
were from ST and general category. While BTT was found prepon-
derant among individuals from various districts of Odisha; Cases of
BTT were also detected in individuals from West Bengal, Chhattis-
garh and Andhra Pradesh. Conclusion: The prevalence of beta
thalassemia gene in Western Odisha was 3.7 %. The finding that the
religious groups like Muslims and Christians have lower frequency of
BTT differs from the observation (Sur and Mukhopadhyay 2006)
from West Bengal. The trend observed in this study though is unique
in its representation of epidemiology of BTT from Eastern India; a
larger population-based investigation is required to map the spread of
b-thalassaemia gene in Eastern India.
Abstract P 100
Clinical and Molecular Characterization of 194 cases of Sickle-beta Thalassaemia in Western Odisha and Their Responseto Hydroxyurea Therapy
Snehadhini Dehury, Dilip Kumar Patel, Siris Patel, PrasantaPurohit, Subasa C Bishwal, Satyabrata Meher, Bidyapati Padhan,Kishalaya Das
Sickle Cell Clinic, VSS Medical College & Hospital, Burla,
Sambalpur, Odisha-768 017; 1 Department of Medicine, OSCP,
NRHM, V.S.S. Medical College, Burla, Odisha-768017
Introduction: Hemoglobin Sickle-b-thalassaemia is a variant form of
sickle cell disease, resulting from the inheritance of HbS and b-thalassemia
genes. Objective: To study the clinical, hematological and molecular
profile of sickle-b-thalassaemia double heterozygotes in western Odisha.
Materials and Methods: 194 cases of HbS-b-thalassemia were studied at
Sickle Cell Clinic, VSS Medical College Hospital, Burla, Odisha, India.
Cases were diagnosed by CE-HPLC (BioRad Variant II) and family study
and confirmation was done by ARMS-PCR. a-Thalassaemia was studied by
GAP-PCR. Cases with severe clinical features were treated with Hydroxy-
urea at a dose of 10 mg/kg body wt/day orally. Results: Three b-
thalassaemia mutations [IVS1-5(G-C), FS41/42(-CTTT), codon 15
(G[A)] accounted for 97 % of all HbS-b-thalassaemia cases. IVS1-5(G-
C) was the commonest mutation (184/194, 94.85 %). The mean %HbA2,
%HbF and %HbS were 5.02 ± 0.86, 19.04 ± 8.89 and 69.84 ± 9.91
respectively, whereas the mean RBC, MCV and MCH were found to be
3.90 ± 0.98 9 103/ll, 71.68 ± 9.48 fl and 23.72 ± 8.94 pg respectively.
Commonest clinical presentation was splenomegaly (39.69 %) followed by
hepatomegaly and AVN (23 and 7.2 % respectively). 57.21 % had severe
anaemia (Hb below 9 g/dL). Deletional a-thalassemia was found in 25 % of
cases. 64.43 % patients with VOC more than 3/year and BT more than
2/year were treated with hydroxyurea.Conclusion: IVS1-5(G-C) mutation
was the commonest and found in 94.85 % of cases. FS41/42(-CTTT) and
codon 15 (G[A) mutations were reported for the first time from this region.
a-Thalassaemia had no significant effect on the clinical and hematological
features except that HbF was significantly high in a-thalassaemia group.
Low dose Hydroxyurea had an ameliorating affect on the clinical and
hematological profile of HbS-b-thalassaemia patients in western Odisha.
Abstract P 101
The ‘Odisha Sickle Cell Project’—a New Horizon of Hopefor Sickle Cell Aggrieved Patients in Odisha
Siris Patel1, Dilip Kumar Patel, Prasanta Purohit, SnehadhiniDehury, Subasa C Bishwal, Satyabrata Meher, BidyapatiPradhan, Kishalaya Das
Sickle Cell Clinic & Molecular Biology Laboratory, VSS Medical
College, Burla, Sambalpur, Odisha; 1V.S.S. Medical College, Burla,
Odisha
Introduction: Sickle cell disease (SCD) is a serious health problem in
Odisha with significant morbidity and mortality. It is a huge burden for
the patients and his families and a serious challenge to the medical
fraternity. However the major constraint for management of this
problem is non-availability of health facility in remote areas and lack of
awareness amongst health care professionals about the various treat-
ment of this disease. In view of this, National Rural Health Mission
(NRHM), Govt. of Odisha sponsored Odisha Sickle Cell Project.
Objective: Screening of population for sickle cell haemoglobinopa-
thies and develop an infrastructure for investigation, registration,
treatment, follow-up and counselling of all patients with SCD of
Odisha. Materials and Method: The Odisha Sickle cell Project
(OSCP) was started in April-2010, under this project Sickle cell unit
were constructed at six DHHs of Western Odisha. Counselling and
diagnosis of sickle cell patients at peripheral hospitals is done by a field
worker and trained Laboratory technician. A state-of-art Molecular
Biology & Haematology Laboratory has been developed at the referral
centre, V.S.S. Medical College & Hospital, B urla. Cases referred from
periphery are examined, counselled, registered here. This laboratory
has facility for Hb electrophoresis, CBC, Biochemical test, CE-HPLC,
PCR & Flow Cytometry on a routine basis. Severe cases of SCD are
started Hydroxyurea therapy at a low dose (10 mg/kg body wt/day).
2073 individuals have been screened for sickle cell haemoglobinopa-
thies in 14 health camps organised in various districts of western
Odisha. Simultaneously 10 CMEs have been coordinated for creating
awareness among the doctors. Results: Till date the number of patients
examined, counselled, diagnosed to have sickle cell haemoglobinopa-
thies at VSS Medical College & Hospital, Burla are 28321, 13720 and
224 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123
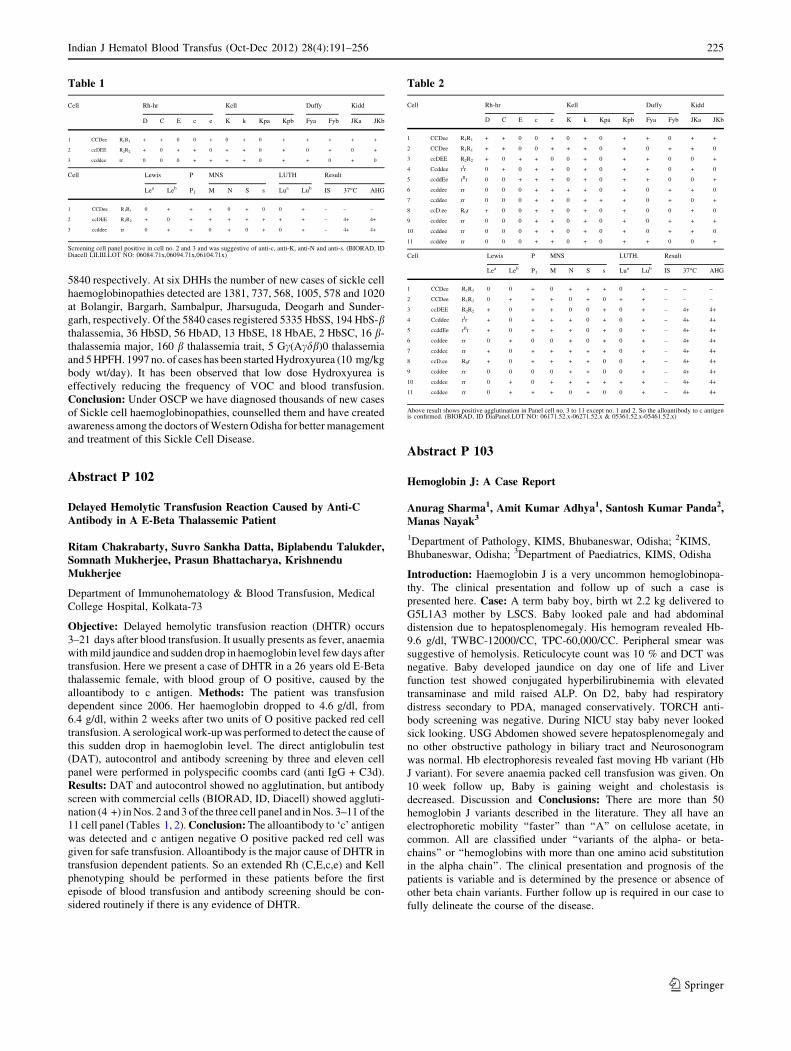
5840 respectively. At six DHHs the number of new cases of sickle cell
haemoglobinopathies detected are 1381, 737, 568, 1005, 578 and 1020
at Bolangir, Bargarh, Sambalpur, Jharsuguda, Deogarh and Sunder-
garh, respectively. Of the 5840 cases registered 5335 HbSS, 194 HbS-bthalassemia, 36 HbSD, 56 HbAD, 13 HbSE, 18 HbAE, 2 HbSC, 16 b-
thalassemia major, 160 b thalassemia trait, 5 Gc(Acdb)0 thalassemia
and 5 HPFH. 1997 no. of cases has been started Hydroxyurea (10 mg/kg
body wt/day). It has been observed that low dose Hydroxyurea is
effectively reducing the frequency of VOC and blood transfusion.
Conclusion: Under OSCP we have diagnosed thousands of new cases
of Sickle cell haemoglobinopathies, counselled them and have created
awareness among the doctors of Western Odisha for better management
and treatment of this Sickle Cell Disease.
Abstract P 102
Delayed Hemolytic Transfusion Reaction Caused by Anti-CAntibody in A E-Beta Thalassemic Patient
Ritam Chakrabarty, Suvro Sankha Datta, Biplabendu Talukder,Somnath Mukherjee, Prasun Bhattacharya, KrishnenduMukherjee
Department of Immunohematology & Blood Transfusion, Medical
College Hospital, Kolkata-73
Objective: Delayed hemolytic transfusion reaction (DHTR) occurs
3–21 days after blood transfusion. It usually presents as fever, anaemia
with mild jaundice and sudden drop in haemoglobin level few days after
transfusion. Here we present a case of DHTR in a 26 years old E-Beta
thalassemic female, with blood group of O positive, caused by the
alloantibody to c antigen. Methods: The patient was transfusion
dependent since 2006. Her haemoglobin dropped to 4.6 g/dl, from
6.4 g/dl, within 2 weeks after two units of O positive packed red cell
transfusion. A serological work-up was performed to detect the cause of
this sudden drop in haemoglobin level. The direct antiglobulin test
(DAT), autocontrol and antibody screening by three and eleven cell
panel were performed in polyspecific coombs card (anti IgG + C3d).
Results: DAT and autocontrol showed no agglutination, but antibody
screen with commercial cells (BIORAD, ID, Diacell) showed aggluti-
nation (4 +) in Nos. 2 and 3 of the three cell panel and in Nos. 3–11 of the
11 cell panel (Tables 1, 2). Conclusion: The alloantibody to ‘c’ antigen
was detected and c antigen negative O positive packed red cell was
given for safe transfusion. Alloantibody is the major cause of DHTR in
transfusion dependent patients. So an extended Rh (C,E,c,e) and Kell
phenotyping should be performed in these patients before the first
episode of blood transfusion and antibody screening should be con-
sidered routinely if there is any evidence of DHTR.
Abstract P 103
Hemoglobin J: A Case Report
Anurag Sharma1, Amit Kumar Adhya1, Santosh Kumar Panda2,Manas Nayak3
1Department of Pathology, KIMS, Bhubaneswar, Odisha; 2KIMS,
Bhubaneswar, Odisha; 3Department of Paediatrics, KIMS, Odisha
Introduction: Haemoglobin J is a very uncommon hemoglobinopa-
thy. The clinical presentation and follow up of such a case is
presented here. Case: A term baby boy, birth wt 2.2 kg delivered to
G5L1A3 mother by LSCS. Baby looked pale and had abdominal
distension due to hepatosplenomegaly. His hemogram revealed Hb-
9.6 g/dl, TWBC-12000/CC, TPC-60,000/CC. Peripheral smear was
suggestive of hemolysis. Reticulocyte count was 10 % and DCT was
negative. Baby developed jaundice on day one of life and Liver
function test showed conjugated hyperbilirubinemia with elevated
transaminase and mild raised ALP. On D2, baby had respiratory
distress secondary to PDA, managed conservatively. TORCH anti-
body screening was negative. During NICU stay baby never looked
sick looking. USG Abdomen showed severe hepatosplenomegaly and
no other obstructive pathology in biliary tract and Neurosonogram
was normal. Hb electrophoresis revealed fast moving Hb variant (Hb
J variant). For severe anaemia packed cell transfusion was given. On
10 week follow up, Baby is gaining weight and cholestasis is
decreased. Discussion and Conclusions: There are more than 50
hemoglobin J variants described in the literature. They all have an
electrophoretic mobility ‘‘faster’’ than ‘‘A’’ on cellulose acetate, in
common. All are classified under ‘‘variants of the alpha- or beta-
chains’’ or ‘‘hemoglobins with more than one amino acid substitution
in the alpha chain’’. The clinical presentation and prognosis of the
patients is variable and is determined by the presence or absence of
other beta chain variants. Further follow up is required in our case to
fully delineate the course of the disease.
Table 2
Cell Rh-hr Kell Duffy Kidd
D C E c e K k Kpa Kpb Fya Fyb JKa JKb
1 CCDee R1R1 + + 0 0 + 0 + 0 + + 0 + +
2 CCDee R1R1 + + 0 0 + + + 0 + 0 + + 0
3 ccDEE R2R2 + 0 + + 0 0 + 0 + + 0 0 +
4 Ccddee rIr 0 + 0 + + 0 + 0 + + 0 + 0
5 ccddEe rIIr 0 0 + + + 0 + 0 + + 0 0 +
6 ccddee rr 0 0 0 + + + + 0 + 0 + + 0
7 ccddee rr 0 0 0 + + 0 + + + 0 + 0 +
8 ccD.ee R0r + 0 0 + + 0 + 0 + 0 0 + 0
9 ccddee rr 0 0 0 + + 0 + 0 + 0 + + +
10 ccddee rr 0 0 0 + + 0 + 0 + 0 + + 0
11 ccddee rr 0 0 0 + + 0 + 0 + + 0 0 +
Cell Lewis P MNS LUTH. Result
Lea Leb P1 M N S s Lua Lub IS 37�C AHG
1 CCDee R1R1 0 0 + 0 + + + 0 + – – –
2 CCDee R1R1 0 + + + 0 + 0 + + – – –
3 ccDEE R2R2 + 0 + + 0 0 + 0 + – 4+ 4+
4 Ccddee rIr + 0 + + + 0 + 0 + – 4+ 4+
5 ccddEe rIIr + 0 + + + 0 + 0 + – 4+ 4+
6 ccddee rr 0 + 0 0 + 0 + 0 + – 4+ 4+
7 ccddee rr + 0 + + + + + 0 + – 4+ 4+
8 ccD.ee R0r + 0 + + + + 0 0 + – 4+ 4+
9 ccddee rr 0 0 0 0 + + 0 0 + – 4+ 4+
10 ccddee rr 0 + 0 + + + + + + – 4+ 4+
11 ccddee rr 0 + + + 0 + 0 0 + – 4+ 4+
Above result shows positive agglutination in Panel cell no. 3 to 11 except no. 1 and 2. So the alloantibody to c antigenis confirmed. (BIORAD, ID DiaPanel.LOT NO: 06171.52.x-06271.52.x & 05361.52.x-05461.52.x)
Table 1
Cell Rh-hr Kell Duffy Kidd
D C E c e K k Kpa Kpb Fya Fyb JKa JKb
1 CCDee R1R1 + + 0 0 + 0 + 0 + + + + +
2 ccDEE R2R2 + 0 + + 0 + + 0 + 0 + 0 +
3 ccddee rr 0 0 0 + + + + 0 + + 0 + 0
Cell Lewis P MNS LUTH Result
Lea Leb P1 M N S s Lua Lub IS 37�C AHG
1 CCDee R1R1 0 + + + 0 + 0 0 + – – –
2 ccDEE R2R2 + 0 + + + + + + + – 4+ 4+
3 ccddee rr 0 + + 0 + 0 + 0 + – 4+ 4+
Screening cell panel positive in cell no. 2 and 3 and was suggestive of anti-c, anti-K, anti-N and anti-s. (BIORAD, IDDiacell I,II,III.LOT NO: 06084.71x,06094.71x,06104.71x)
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 225
123

Abstract P 104
A Novel 9 bp Denovo Deletion [HBB: c.132_141del9bp] inb-Globin Gene Causing Heinz Body
K Neelakandan, Neeraj Arora, Sathish Kumar1, Usha Sitaram2,Alok Srivastava, Eunice Sindhuvi Edison, RV Shaji
Department of Haematology, Christian Medical College, Vellore;1Department of Child Health, Christian Medical College, Vellore;2Department of Transfusion Medicine & Immunohaematology,
Christian Medical College, Vellore
Abstract: The ‘‘Heinz-body anaemias’’ are a group of hemolytic
syndromes of diverse etiology with common morphologic charac-
teristics. One of the rare causes for Heinz-body anaemias is dominant
b-thalassaemia (b-thal). Dominant b-thal presents with moderate to
severe haemolytic anaemia with jaundice and splenomegaly and is
associated with a thalassemia intermedia phenotype in the heterozy-
gous state. We describe a case of dominant b-thal with severe
hemolytic anaemia caused by a novel denovo deletion in b-globin
gene. The patient was a 3 year old girl who presented with transfusion
dependent anaemia and hepatosplenomegaly (liver-4 cm; spleen-
8 cm). She had marked reticulocytosis (19 %) and elevated LDH
levels. Both direct and indirect coombs test were negative. The blood
picture and other common tests (G6PDH, autohaemolysis, sickling
preparation, Hams and sucrose lysis test, osmotic fragility) done for
haemolytic anaemia were unremarkable, but Heinz bodies test was
positive. Interestingly, peripheral blood smear stained with brilliant
cresyl blue showed numerous RBCs containing hemoglobin H (HbH)-
like inclusions. However, the eight common alpha (a)-globin dele-
tions prevalent in our population screened by a multiplex PCR were
absent. We performed sequencing of globin genes to identify possible
mutations that produce unstable haemoglobin causing haemolytic
anaemia. We identified a heterozygous state of a novel 9 bp deletion
(TCC TTT GGG) in the exon 2 of the beta globin gene (HBB:
c.132_141del9bp). This region of the b globin is situated in or around
the heme pocket and the mutations that destabilize heme binding has
been found to be associated with extremely unstable hemoglobins.
Both parents had normal HbF, HbA2, MCV and MCH values and had
normal b globin gene sequences. Most of the mutations that cause
dominant b-thalassaemia reside in the exon 3 of the b globin gene,
although rare cases with mutations in other exons 1 and 2 have also
been reported. Our case serves to underscore the importance of
considering dominant b thalassaemia as a cause of moderate to severe
hemolytic anaemia in children even without a significant family
history. DNA based diagnostics can readily detect and confirm these
uncommon b globin gene mutations, and help implement appropriate
treatment planning and genetic counselling.
Abstract P 105
Profile of Liver Fucntion Test in Sickle Cell Patients
Mitali Madhumita Rath, MK Panigrahi, Sudha Sethy, RajibNayak, Pranati Mohanty, Kalyani Prava Gouda, Sujata Pujari,SR Mohapatra, RK Jena
Department of Pathology & Clinical Hematology, S.C.B. Medical
College & Hospital, Cuttack
Introduction: Abnormal liver function tests are common in patients
with sickle cell anaemia, even in the absence of liver disease.It may be
hemolytic, cholestatic, hepatocellular and mixed pattern. There is a
paucity of data on liver function in sickle cell disease (SCD) from this
region. Aims and Objective: The present study aims at determinig the
prevalance and the profile of abnormal liver fuction test in sickle cell
patients. Material and Mathods: One hundred consucutive patients of
sickle cell disease (homozygous)attainding the out patients department
of hematology, S.C.B. Medical College from January 2012 to July 2012
were included in this study. Detailed clinical history was taken and
subjected for complete blood count, hemoglobin profile, liver function
tests, viral markers, ultrasonography of abdomen and pelvis. Results:The 100 patients included in the study ranged in age from 8 to 62 years
(mean 30.8 ± 9.11 years) and included 64 males and 36 females. Mean
episode of vaso-oclusive crisis was 2.65 ± 1.82 times and mean units of
blood transfusion was 3.74 ± 1.75. Mean hemoglobin concentration
was 8.2 ± 2.3 g/dl. Abnormality in liverfuction test was found in 86 %
of the study participants. The mean serum total bilirubin, AST, ALT,
ALP were 8.4 ± 6.5 mg/dl, 104 ± 43 U/L, 68 ± 27 U/L, 258 ± 76
U/L respectively. 49 % had hemolytic, 12 % had obstructive 25 % had
mixed pattern of LFTs. On ultrasonography 24 % had hepatomegaly,
46 % had spleenomegaly, 11 % cholelithiasis and 2 % had both cho-
lelithiasis and choledocholithiasis. Conclusion: Majority of sickle cell
homozygous patients had abnormal liver fuction. One fourth of the
patients had mixed pattern of LFTs that can not be explained by either
hemolytic or obstructive component.
Abstract P 106
Profile of Hb E/beta Thalassaemia Patients After Splenectomy—Experience of a Thalassemia Care Centre in Eastern India
Prakas Kumar Mandal, Basab Bagchi, Sandeep Saha, TuphanKanti Dolai, Sanjay Misra, Malay Kumar Ghosh, MaitreyeeBhattacharrya
Department of Hematology, N.R.S. Medical College and Hospital,
Kolkata-700014
Objective: The study was done to evaluate various complications
following splenectomy in HbE/beta thalassaemia (EBT). Methods:A total of 72 splenectomised EBT were studied retrospectively.
Detail history including age of splenectomy, transfusion require-
ment (before and after splenectomy) was taken. Physical
examination was done for facial deformities, pubertal growth.
Baseline Hb %, serum ferritin level, evidence of pulmonary
hypertension (PHTN), thrombo-embolic manifestations (VTE),
extramedullary hematopoiesis were noted. Results: Out of 1380
registered EBT patients, 72 (5.22 %) underwent splenectomy. Ten
(13.9 %) were diagnosed \1 year, 24 (33.3 %) 1–5 years, 23
(31.87 %) 5–15 years and 15 (20.83 %) [ 15 years. Thirty five
(48.57 %) started transfusion \ 5 years and one at 39 years. 19
(26.4 %) underwent splenectomy \ 10 years, 38 (52.7 %) between
10–20 years and 15 (20.8 %) [ 20 years of age. Mechanical dis-
comfort was the leading cause for splenectomy in 37 (51.39 %)
followed by increased transfusion requirement in 15 (20.83 %).
Mean baseline Hb level of 5.43 g/dl with 48 (66.6 %) pre-sple-
nectomy and majority had Hb % of 6.8 g/dl Post-splenectomy.
Mean transfusion requirement was remarkably reduced from
18.1 unit/year to 7.8 unit/year after splenectomy. Four (5.5 %)
received single transfusion till date. Mean serum ferritin level was
increased from 907.58 ng/ml (before) to 1091.6 ng/ml (after) ins
pite of reduced transfusion requirement after splenectomy. 48
226 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

(66.6 %) and 31 (43.05 %) patients had facial deformities and
delayed pubertal growth respectively. Four (5.5 %) had evidence
of extramedullary hematopoiesis, 15 (20.83 %) had VTE and 7
(9.7 %) developed PHTN. Though 48 (66.67 %) was started with
iron chelation therapy, majority 38 (52.7 %) stopped and 10
(13.89 %) are continuing till date. Conclusions: Though splenec-
tomy reduces requirement of transfusion; it does not prevent
skeletal abnormalities and delayed puberty. VTE are increased.
Patients should be encouraged for effective Iron chelation therapy.
Abstract P 107
Optimizing the Dose of Hydroxyurea in Thalassemia Intermedia
Vinaykumar V Bohara, Lalit Raut, Girish Badarkhe, GaneshPujari, P Chakrabarti, S Ray, U Nath, Utpal Chaudhuri
Institute of Hematology and Transfusion Medicine (IHTM), Kolkata
Introduction: The optimum dose of hydroxyurea in the manage-
ment of thalassemia patients is an area of gray zone. We report the
results of study carried out on patients of E-b thalassemia. Aims and
objectives: To determine the optimum dose of hydroxyurea in E-bthalassemia patients and to assess its efficacy and safety. Methods:This was a prospective, single center study in which E-b thalassemia
patients with no or minimum transfusion needs were enrolled and
randomised to receive 10 (group A) and 20 mg/kg/day (group B)
hydroxyurea. Complete response (CR) was defined as transfusion
independence or rise of haemoglobin (Hb) by [1.5 g%. Partial
response (PR) was rise in Hb by 0.5 to 1.5 g% or reduction in
transfusion needs. Anything less was defined as no response (NR).
Results: Median age in group A (n = 32) and B (n = 31) was 12.5
and 12 years, respectively (p [ 0.05). The median follow up was 9
and 6 months in groups A and B respectively (range 3–9 months).
In group A response was seen in 65.7 % (20 out of 32). Of this CR
and PR was seen in 32.3 % (n = 10) and 33.4 % (n = 11). The rise
in Hb in group A was statistically significant at 3 and 6 months. In
group B there was NR in 83.9 % (26 out of 31) of patients. There
was no CR and eight (25.8 %) patients discontinued hydroxyurea at
6 months due to adverse effects requiring intervention. High base-
line HbF was associated with higher response in group A but not in
group B. In Complete responders IVS 1-5(G-C) was the commonest
mutation. Conclusion: Hydoxyurea is effective and well tolerated at
10 mg/kg/d dose in E-b thalassemia (Thalassemia Intermedia) in
Eastern India.
Abstract P 108
Association of Genetic Modulators of b Thalassemia Linkedto Raised Fetal Hb and its Effect on Clinical Severity
Pooja Dabke, Roshan Colah, K.Ghosh, Anita Nadkarni
National institute of Immunohematology, 13th Floor NMS Building,
KEM hospital Campus, Parel, Mumbai-12
Objective: Study of genetic modifiers linked to raised HbF and its
effect on clinical severity of b thalassemia. Methods: Study group
consisted of 104 patients of hemoglobinopathies and 50 healthy
individuals. HbA2 and HbF levels were measured using HPLC System.
XmnI polymorphism, Ac-d intergenic haplotypes, (AT)x(T)y motif,
Pre G c haplotype, b globin LCR motifs were studied by PCR–RFLP,
Gene scan analysis and DNA sequencing. Results: Clinically 40
patients were classified as thalassemia major and 39 as thalassemia
intermedia. 13 different b thalassemia mutations were encountered.
IVS1nt5 (G ? C) was the prevalent mutation (56.96 %). 25 % of the
thalassemia intermedia chromosomes showed presence of (AT)9(T)5
motif. The Pre G c TAG haplotype associated with higher HbF levels
was present in homozygosity in 48.57 % of the thalassemia intermedia
cases as against 12.82 % of the thalassemia major cases. 80.55 % of
the milder patients showed presence of the Ac-d intergenic haplotype
T. All the 11 sickle cell anemia patients showed homozygosity for Pre
G c TAG and Ac-d intergenic T haplotypes showing their linkage with
Arab-Indian haplotype. In b LCR region the HS2 motif were well
spread among the patients. The HS3 motif did not show any change
with respect to the reference sequence. Conclusion: As 33.33 % of our
milder cases showed presence of 4 modifiers linked to higher HbF
(XmnI, (AT)9(T)5, TAG Pre G c, Ac-d intergenic T haplotype), it
appears that in the Indian scenario these secondary modifiers of bthalassemia seem to contribute to ameliorate the disease severity.
Abstract P 109
A Hospital Based Study of Thalassemia from Kolkata
Deboshree M. Bhattacharyya, Jayasri Basak,Soma Mukhopadhyay, Ashis Mukhopadhyay
Netaji Subhas Chandra Bose Cancer Research Institute, 16A Park
Lane, Kolkata-700016, West Bengal, India
Objective: The present study represents a population of Eastern
Indian state of West Bengal. This is a hospital based study where in the
last 3 years 242 individuals have been screened for their carrier status.
The main objective was to study the genotype-phenotype correlation
of b thalassemia cases, and finally to identify different b thalassemia
mutations prevalent in West Bengal. Method: After obtaining written
consent from each individual or parents of individuals under the age of
18 years, 3 ml of peripheral blood was drawn from each subject and
stored in EDTA vials. At first complete blood count was done followed
by High Performance Liquid Chromatography. DNA is then extracted
from the remaining blood following Millers method (1988). ARMS
PCR and GAP PCR were done to detect beta and alpha mutations
respectively. Results: Total of 242 subjects were studied which
included 22.31 % b thalassemia carriers, 2.1 % of b thalassemia major
1.24 % sickle beta thalassemia, 6.2 % Eb thalassemia, 8.3 % HbE
carriers and HbE 2.1 % homozygous. 10 Eb patients responded to
hydroxyurea treatment and required less blood transfusion whereas 5
Eb patients did not respond to the treatment and required regular blood
transfusion. The severity of the Eb mutations depends on the genotype
of the beta mutation. Apart from these we have identified families
having mutations for HPFH, Fanning Lubbock and a alpha thalasse-
mia. Conclusion: Genotype phenotype correlation plays a significant
role in determining the severity of thalassemia. Among the studied
samples the prevailing beta mutations are IVS 1-5, codon 15 and
codon 30. Compound heterozygosity of HbE with the above men-
tioned beta mutation produces mild to severe anemia. The above study
gives an idea of the thalassemia status of Kolkata.
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 227
123

Abstract P 110
Role of NESTROFT in Mass Screening of Thalassemia
Nabamita Pal, Deboshree M. Battacharyya, AshisMukhopadhyay, Abhijit Chakraborty, Swati Dasgupta,Priyabrata Das, Soma Mukhopadhyay, Jayasri Basak
Department of Molecular Biology, Netaji Subhas Chandra Bose
Cancer Research Institute, 16 A, Park Lane, Kolkata-700016,
West Bengal, India
Objective: Thalassemia represents a major health problem worldwide.
Present carrier status of Thalassemia in West Bengal according to Sur
D et al. is 20.47 %, which is comparatively higher than other states of
this country. Our main aim is to arrange Mass Awareness and
Screening camps among both urban and rural West Bengal and screen
people to know their thalassemia carrier status. Method: In the present
study from January 2009 to January 2011 screening was carried out
among various populations of West Bengal including tribal. With their
individual consent 3 ml of peripheral blood was collected in EDTA
vials. NESTROFT was done on spot using 0.36 % Saline Buffer
solution (Sodium chloride, Sodium dihydrogen phosphate, Disodium
hydrogen Phosphate). Complete Blood Count was performed within
24 h of collection. HPLC (High Performance Liquid Chromatography)
was done to identify the beta samples. Result: In this period total 6482
individuals have been screened. Among them 25 % were NESTROFT
positive and 50 % are negative. Rest of them were doubtful cases.
NESTROFT result was compatible with the respective HPLC reports.
Most of the NESTROFT positive were either carriers of beta thalas-
semia, sickle cell anemia, carrier of alpha thalassemia and victims of
Eb thalassemia. The doubtful cases of the test contributed to mostly E
carriers and individuals suffering from iron deficiency anemia. The
efficiency of the NESTROFT test is 71.29 % among the general
population of West Bengal. The specificity and sensitivity of the test
are 61.1 and 72 % respectively. Conclusion: Hence NESTROFT is
both cost-effective and cost-efficient test in Thalassemia Mass
Screening. It plays a significant role in thalassemia carrier detection
but HPLC is mandatory in the area where HbE is prevalent.
Abstract P 111
Role of Wheat Grass Juice as an Iron Chelator in IntermediateThalassemia Patients
Abhijit Chakraborty1, Priyabrata Das1, Manoj Kar2, SomaMukhopadhyay1, Suvra Mondol3, Ashis Mukhopadhyay1
1Netaji Subhas Chandra Bose Cancer Research Institute, 16A Park
Lane, Kolkata-700016, West Bengal, India; 2N.R.S Medical College
& Hospital, Kolkata-700014, West Bengal, India; 3National Research
Institute for Ayurvedic Drug Development, Kolkata-700091,
West Bengal, India
Objective: The aim of the study is to observe the effect of wheat grass
juice in reducing ferritin level and increasing the hemoglobin level in
intermediate Thalassemia patients. We will also study the biochemical
properties of wheat grass juice. Methods: We performed a study on
236 patients of intermediate thalassemia during the period from March
2009 to March 2012. Of these, 48 transfusion dependent intermediate
thalassemia patients whose mean age is 6.42 ± 1.71 were selected.
30 ml of fresh juice from 5-7-day-old wheat grass leaves including
stems was given daily to all 48 patients regularly for 6 months. Wheat
grass juice was analyzed by cation exchange column chromatography.
Deoxyribose degradation assay was performed to study the iron che-
lating activity of wheat grass. Result: Wheat grass juice is found to be
rich in active ingredients which play an important role in dietary
absorption of iron from intestine. It is also found to contain unique
active ingredients with iron chelating property. The mean serum fer-
ritin level of the patients before treatment was 2,250 (range
650–3,100), which reduced to 950 (range 68–1680) (p \ 0.0001) after
treatment. The mean levels of hemoglobin before starting wheat grass
juice were 6.2 g%. After 6 months of wheat grass therapy the mean
value for hemoglobin was 7.8 g% (p \ 0.005). The performance status
was improved from 60 to 80 % (Karnofsky) after wheat grass treat-
ment. The mean interval between transfusions was also found to
increase. Conclusions: Hence, wheat grass juice is an effective iron
chelator and it is an effective alternative of blood transfusion. Its use in
intermediate thalassemia patients should be encouraged.
Abstract P 112
Prevalence of Thalassemia in West Bengal with Special Referenceto Different Tribes
Jayasri Basak, Deboshree M Bhattacharyya, SomaMukhopadhyay, Swati Dasgupta, Abhijit Chakraborty,Priyabrata Das, Nabamita Pal, Sukanta Koner,Ashis Mukhopadhyay
Department of Molecular Biology, Netaji Subhash Chandra Bose
Cancer Research Institute, Kolkata
Introduction: Thalassemia is a hereditary anemia resulting from
defect in hemoglobin production and it is the most common genetic
disorder worldwide. Prevalence of Thalassemia (both alpha and beta)
is high in West Bengal, especially among the scheduled tribes. To
reduce the Thalassemia burden from West Bengal we have taken a
project for Thalassemia carrier screening through awareness and
screening program among the rural and urban populations. Our target
population were tribes, e.g., Toto, Rabha, Munda, Oraon, Kerketa,
Sardar etc. residing in remote rural areas. Materials and Methods:From January 2009 to December 2011, we have organized 73 camps
in different districts of West Bengal. After taking written consent,
2–3 ml peripheral blood samples were collected from each interested
people attending the mass awareness camp. NESTROF, CBC, HPLC
were done for all samples. To detect alpha and beta mutations GAP-
PCR and ARMS-PCR were performed following the standard pro-
tocol. Results: During the above mentioned period, we have screened
on total 8387 individuals (4,869 male; 3,518 female) for Thalassemia
carrier detection through camps, organized in 14 districts of West
Bengal. Out of 8,387 screened individuals, tribes were 2,885 and
general caste including scheduled castes was 5502. Among tribes
HbE carrier and homozygous percentage are 14.6 and 4.01 respec-
tively while percentage of sickle cell anemia and beta Thalassemia
carriers are 1.25 and 4.19 respectively. Total beta carrier percentage
among general caste and scheduled caste is 6.94. Our result also
revealed that alpha Thalassemia carrier among Oraon and Sardar
tribes is very high. Conclusion: Incidence of Thalassemia is high in
different rural populations especially among tribes. So, large scale
awareness and preventive program should be taken.
228 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Abstract P 113
Spectrum of Beta Globin Gene Mutations in North IndianPatients with Thalassemia Major: Application in PrenatalDiagnosis
Reena Das, Jasbir Kaur, Sanjeev Chabra, Jasmina Ahluwalia,Amita Trehan1, Subhash Saha2, Rashmi Bagga2, Deepak Bansal1,Inusha Panigrahi1, RK Marwaha1, G Garewal
Departments of Hematology, Pediatrics1 and Obstetrics
& Gynaecology2, PGIMER, Chandigarh 160 012, India
Objectives: Thalassemia major (TM) is an autosomal recessive dis-
order where the parents are asymptomatic beta thalassemia carriers
(bTT). The prevalence of bTT is 3.5 % in north Indians. We analyzed
the spectrum of mutations in 1500 TM alleles and its application to offer
prenatal diagnosis to couples who were both bTT. Method: ARMS-
PCR and beta globin gene sequencing from genomic DNA from the
peripheral blood was carried out. Prenatal diagnosis was offered to 570
pregnancies in the last 14 years using molecular analysis. Results:Common five Indian mutations were found in 86.8 % alleles; com-
monest being IVS 1,5 (G-C) [29.4 %], followed by Fr 8/9 (+G) [21 %],
619 bp deletion [17 %], IVS 1,1 (G-T) [8.5 %] and Fr 41/42 (-TTCT)
[10.8 %]. Codon 16 (-C) was seen in 4 % and uncommon b++ muta-
tions -88 (C-T) and Cap + 1 (A-C) was 2.6 and 2 % respectively. Rare
mutations constituted 4.5 % alleles and many were identified by beta
globin gene sequencing. Only 0.1 % alleles remained uncharacterized.
Results of the prenatal diagnosis of 570 pregnancies showed that in
25.7 % cases the fetuses were normal, 49.8 % cases were bTT, 23.2 %
were TM and in 1 % cases we were unable to distinguish between bTT
and TM. Seven cases had twin pregnancies and all except one case
showed similar normal/bTT/TM status. Conclusion: This application
has helped to prevent the birth of 132 children with thalassemia major in
the region and alleviated the anxiety of 430 pregnancies to have children
who were either normal or carriers.
Abstract P 114: Oral Presentation
Clinico-Hematological Analysis of Hb Variants and Spectrumof Hemoglobinopathies on HPLC: a 3 Year Study from a TertiaryCare Centre of North India
Poojan Agarwal, Snigdha Goyal, Aneesha Mohanpuria,Vijay Kumar, Sadhna Marwah, A.S.Nigam, Gurdeep Buxi
Department of Pathology, PGIMER, Dr. Ram Manohar Lohia
Hospital, New Delhi
Objective: To analyse and characterize various Hb variants and he-
moglobinopathies using high performance liquid chromatography
(HPLC). Methods: The present retrospective study was carried out at
PGIMER, Dr. RML Hospital, by analysing the data obtained from
September 2009 to August 2012. The clinical and haematological
records of patients suspected of hemoglobinopathies were retrieved
and studied with the HPLC pattern. Family studies carried out in
indeterminate cases were also reviewed. Results: Abnormal haemo-
globin fractions on HPLC were seen in 121 of the 1846 cases studied.
Of these, beta thalassemia trait was the predominant abnormality with
a total of 69 cases (57.02 %). HbS was the next most common Hb
variant, observed in 18 cases (14.8 %). Of these, four cases were
homozygous for sickle cell disease. Other hemoglobins observed were
Hb D Punjab (7 cases; 5.78 %), alpha thal (7 cases; 5.78 %) and Hb E
(5 cases; 4.13 %). Hb J Meerut, HPFH and combined Hb E/beta thal
inheritance had 3 cases each (i.e. 2.3 % each). 5 cases also showed
peak in the C window. 2 cases (1.6 %) were each of Hb Q India, Hb
Lepore trait and beta thal major. Conclusion: Our study highlights that
high performance liquid chromatography (HPLC) is a simple yet rapid
and reliable tool for the detection of haemoglobin variants.
Abstract P 115
Determination of Allelic Frequency of SNPs at Quantitative TraitLoci (QTL)s of Fetal Hemoglobin
Charu Batra1, Jasbir Kaur1, Amita Trehan2, Jasmina Ahluwalia1,Reena Das1
1Department of Hematology, 2Department of Pediatrics, Postgraduate
Institute of Medical Education and Research, Chandigarh
Introduction: Fetal Hemoglobin (HbF) is considered a ‘quantitative
trait’ (QT) wherein multiple genes together with a small environmental
component determine the value measured in any given individual. High
HbF levels are associated with milder disease progression and fewer
complications in patients with Sickle cell Disease (SCD) and b thal-
assemia. Recently conducted Genome-Wide Association Studies
(GWAS) in European and African American populations and patients
with hemoglobinopathies, have identified single-nucleotide polymor-
phisms (SNPs) from chromosomal loci that contribute to varying
expression levels of HbF and other clinical traits. They include the
Xmn1-Gc polymorphism and SNPs at BCL11A and HBSB1L-cMYBinter-region loci. To the best of our knowledge, the allelic frequency of
SNPs at BCL11A and HBS1L-MYB intergenic region in normal North
Indian population and their association with thalassemia in India has not
been studied. Objective: Determination of prevalence of polymor-
phism at SNP rs11886868 in BCL11A exon 2, SNP rs4895441 in the
HBSB1L-cMYB inter-region and SNP rs74822144 in b globin gene
cluster (The Xmn1-Gc polymorphism) in normal North Indian popula-
tion. Methods: Single-nucleotide polymorphism (SNP) analysis was
performed by using polymerase chain reaction (PCR)/restriction
enzymes on genomic DNA extracted from peripheral blood leukocytes
of fifty healthy normal children[3 years of age. Enzymatic digestion
was performed by XmnI, MboII, and RsaI for, rs74822144, rs11886868
and rs4895441 respectively. Results: Minor allele frequencies for
rs11886868 and rs4895441, rs74822144 were 0.35, 0.13 and 0.26 in
normal North Indian children. We plan to determine the allelic fre-
quency at these loci in children with thalassemia intermedia and to
correlate the SNPs with the clinical phenotypes
Abstract P 116
Non-Transfusion Dependent Thalassemia; Management Issues
Maitreyee Bhattacharyya, Asutosh Panigrahi
N.R.S Medical College, 138, A.J.C Bose Road, Kolkata 700014
Objectives: Non transfusion dependent thalassaemia (NTDT) usually
do not require regular RBC transfusions for survival but associated with a
variety of serious clinical complications that require proactive and
comprehensive management. Methods: We investigated clinical and
biochemical profile of non-transfusion dependent thalassemia patients
attending thalassemia clinic of N.R.S medical College, Kolkata. Results:Out of 179 patients studied, 158 were HbE b thalassemia and rest Sb or
Hb E disease or b thal intermedia. Mean age of diagnosis was 15 year
(range 1–48 years). Mean Hb % was 7.4 g/dl (range 4–10 g/dl). Mean
age of 1st BT was 17 years 20 (11 %) patients didn’t have splenomegaly,
47 (26 %) without skeletal deformity (ineffective erythropoiesis
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 229
123

indicator). Growth failure observed in 87 patients, 95 were with jaundice,
5 had delayed puberty. Predominant complications observed was Gall
stone in 1 patient, thyroid dysfunction in 5 patients VTE in 1. We didn’t
observe diabetes, pulmonary hypertension, in any patients. Serum ferritin
was above 1000 in 16 [with normal development in 4 (2 %) and normal
puberty in 6 (3 %)] and 122 patients had s.ferritin between 300 and 1,000.
Management provided were transfusion [required 98 (55 %), frequent in
7 (4 %) and not required in 81 (45 %)], splenectomy in 3 (1.6 %), hy-
droxylurea 2 (1 %), thromboprophylaxis in 1 (0.5 %) iron chelation in 17
(9 %) and monitoring. Conclusion: Results from our study shows that
effective management of NTDT patients in our genetic background can
give a far better quality of life.
Abstract P 117
Profile of Hemoglobinopathies in the State of Odisha
Anasuya Lenka, Sudha Sethy, Rajeeb Nayak, RK Jena, PranatiMohanty
Department of pathology and department of clinical hematology,
S.C.B. Medical College and Hospital, Cuttack
Objectives: To determine the clinical and hematological profile of
different hemoglobinopathies. Materials and Methods: Patients with
clinical features of chronic hemolytic anemia without history of blood
transfusion in last three months attending the department of clinical
hematology of SCB Medical College & Hospital, Cuttack from
November 2007 to February 2012 are included in the study. About 2 ml
of blood was collected from each patient which was investigated in fully
automated capillary zone electrophoresis. Screening tests like sickling
test and complete blood count by five part cell counter (sysmax 2000I)
were done in all cases. Results: Out of total 6,195 cases hemoglobin-
opathy was found in 3,263 cases. Beta-thalassemia constitutes 28.1 %
cases with 21.02 % of trait, 4.51 % thalassemia intermedia and 2.57 %
of thalassemia major cases. Sickle cell disorder was found in 27.03 %
cases with sickle cell trait in 23.17 % and sickle cell disease in 3.86 %
cases. Sicke-beta thal was 21.88 %. Hb E was found in 2.69 % cases and
E-beta thal in 9.65 % cases. Conclusion: Hemoglobinopathy is com-
mon in almost all parts of Odisha. They cause high degree of morbidity
and mortality along with socio- economic burden. In this study diag-
nosis could not be confirmed in 8.8 % of cases which needs further
investigations like parental evaluation and globin chain mutation
analysis.
Keywords Hemoglobinopathies, Sickle cell, Thalassemia, Hb-E,
Odisha state
Lymphomas
Abstract P 118
Plasmablastic Lymphoma of the Retroperitoneumin an HIV-Negative Patient
Nishad Dhakate1, Soniya Nityanand2
1S.G.P.G.I, Lucknow; 2Department of Hematology, SGPGI, Lucknow
Abstract: Plasmablastic lymphoma (PBL) is an aggressive non-
Hodgkin lymphoma classically occurring in individuals infected with
HIV. Plasmablastic lymphoma has a predilection for the oral cavity
and jaw. However, recent case reports have shown plasmablastic
lymphoma in the stomach, lung, nasal cavity, cervical lymph nodes
and jejunum in HIV negative individuals. We report what is, to the
best of our knowledge, the second case of plasmablastic lymphoma
occurring in the retroperitoneum of a HIV-negative man. Case
Report: A 57 years old male, known case of hypertension presented
with chief complaints of swelling in the right thigh since 3.5 months.
MRI pelvis revealed a soft tissue lesion in adductor compartment of
right thigh. FDG PET scan showed a soft tissue swelling in retro-
peritoneum extending into the pelvis along bilateral psoas muscle, on
the right extending into right inguinoscrotal region, inguinofemoral
region and in adductor compartment. Pathological examination
showed tumour cells with eccentrically placed nucleus and moderate
amount of eosinophilic cytoplasm consistent with plasmablastic
lymphoma. Immunohistochemistry was diffusely and strongly posi-
tive for vimentin and CD 138 and focally and weakly positive for
LCA, CD 38 and CD 79a consistent with plasmablastic lymphoma.
His LDH was 5864, creatinine 2.8 mg%, Na 132 meq, K 6.5 meq,
uric acid: [ 20.0 mg, phosphorus: 5.9 meq and calcium 8.8 mg%
suggestive of tumour lysis syndrome which required dialysis. He was
HIV and EBV negative. Serum protein electrophoresis was normal.
Serum free kappa light chains of 42.8 mg/L (3.3–19.40) and serum
free lambda light chains of 760 mg/L (5.71–26.30). Bone marrow
Aspiration and Biopsy showed no e/o myeloma or lymphoma. Our
case was started on standard CHOP (cyclophosphamide, doxorubicin,
vincristine, and prednisone) chemotherapy. In view of minimal
response he was shifted to salvage chemotherapy (ESHAP). But
patient expired due to neutropenic sepsis.
Abstract P 119
Primary Bone Marrow Lymphoma: A Diagnostic Dilemma
M Deepak Nayak1, Lakshmi Rao, Sushma V. Belurkar,Chethan Manohar
1Department of Pathology, Melaka Manipal Medical College,
Manipal University, Manipal; Department of Pathology, Kasturba
Medical College, Manipal University, Manipal
Introduction: Bone marrow involvement by non Hodgkin Lymphoma
(nHL) has conventionally been considered as a systemic dissemination
of a nodal malignancy. Primary Bone Marrow Lymphoma (PBML) is a
rare exception to this rule. This entity accounts for less than 50 cases in
published literature; thereby highlighting its diagnostic novelty and
clinical significance. Apart from the exclusion of a leukemia originating
in the bone marrow, the current criteria include ruling out a nodal or an
extranodal involvement (including the bone). We present one such
unique case in a 41 year old male patient presenting with isolated
thrombocytopenia. Case Report: The peripheral smear showed
abnormal large lymphoid cells; also reflecting in the bone marrow
aspirate. This led to consideration of alternative diagnoses such as acute
lymphoblastic leukemia and lymphoma. The clinico-radiological work
up showed an absence of organomegaly and lymphadenopathy; thus
necessitating a review of the bone marrow study. The trephine biopsy
revealed interstitial and paratrabecular infiltrates of abnormal lymphoid
cells with brisk mitotic figures. The immunohistochemistry panel for
lymphoma showed a strong positivity for CD10, CD 20 and BCL 2 in
the lymphoid cells. The other markers such as CD34, CD99 and BCL6
were negative. Considering all the above information, a diagnosis of
PBML was rendered. A 5 month follow up has thus far showed a
favourable response to chemotherapy. Conclusion: Primary Bone
Marrow Lymphoma is an uncommon lymphoid malignancy, often
overshadowed by its more common neoplastic counterparts. An
awareness of this entity is essential to establish an accurate diagnosis
and adequate therapy.
230 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Abstract P 120
Gastric Outlet Obstruction in a Child—a Rare Manifestationof Burkitts Lymphoma
Anand Prakash1, Kanishka Das2, Usha Kini3
1St Johns Medical College Hospital, Department of Pediatrics,
Bangalore, India; 2St Johns Medical College Hospital, Department
of Pediatric Surgery, Bangalore, India; 3St Johns Medical College
Hospital, Department of Pathology, Bangalore, India
Purpose: Burkitt’s lymphoma may arise in many atypical locations
which on rare occasions can include stomach. A case of one such
primary non-endemic gastric Burkitt’s lymphoma is described here in
a non-HIV child who presented with features of gastric outlet
obstruction and had a dramatic resolution of symptoms with che-
motherapy. Method: Case report: A 6 year old undernourished girl
with history of abdominal pain and persistent vomiting of 2 months
duration presented with a large, firm, irregular epigastric mass. The
CT-scan revealed a gastric mass with diffuse uniform thickening of its
wall involving the body and antrum with near complete obliteration of
the pyloric orifice. A pelvic lymphnodal mass lesion measuring
5.0 9 4.5 cm along with multiple perigastric and mesenteric lymph
nodes, were also seen. A tru-cut biopsy of the gastric mass showed a
high grade diffuse mature B cell lymphoma with positivity for CD10,
CD 20, CD 79a and Ki67 index of 100 % and negative expression for
Tdt, diagnostic of Burkitt’s lymphoma and Stage III as bone marrow
aspiration biopsy and CSF were negative for lymphoma cells.
Results: The patient was started on NHL-902 protocol. Within a
week of commencing therapy of cytoreduction with vincristine,
cyclophosphamide and prednisolone her symptoms of gastric outlet
obstruction resolved. She continued therapy as per protocol with
COPAD and COPADM and is currently well. Conclusion: The case
presented here highlights an uncommon presentation of a common
pediatric lymphoma and demonstrates the prompt resolution of gastric
outlet obstruction with chemotherapy.
Abstract P 121: Poster Presentation
A Case Series of Three Patients of Hematolymphoid Malignancywith Primary Presentation of Renomegaly
Bidish Kumar Patel, Pritinanda Mishra, Debdatta Basu, RakheeKar
Department of Pathology, JIPMER, Puducherry-6
Introduction: Extramedullary renal involvement in hematological
malignancies is a rare occurrence and may lead to diagnostic dilem-
mas. Report: We report a series of three patients, two children, a girl
aged seven and a boy of 11 years and an adult male aged 37 years, all
of whom on initial presentation had renomegaly and were investigated
for a suspected renal disease. Two of them underwent a renal biopsy.
Subsequently, based on further clinical evaluation, peripheral blood
and bone marrow examination and lymph node biopsy in one; all the
three patients were diagnosed with high-grade hematolymphoid
malignancies. Both the male patients had precursor B cell Acute
Lymphoblastic Leukemia with Renal infiltration while the girl was
diagnosed as a primary Renal T cell Acute Lymphoblastic Lymphoma
with infiltration of the bone marrow. Conclusion: This series of three
cases highlight the fact that hematolymphoid malignancies can at
times masquerade as renal disease. Leukemias and lymphomas can
have myriad clinical manifestations which can mislead the clinical
judgment necessitating a high index of clinical suspicion and a good
clinicopathological correlation to arrive at a final diagnosis.
Abstract P 122: Oral Presentation
Hodgkin Lymphoma: Immunohistochemical Featuresand Association with Epstein-Barr Virus
Debdatta Basu, S Muthu
Department of Pathology, JIPMER, Pondicherry
Background and Objectives: Hodgkin Lymphoma (HL) shows
variation in epidemiological, immunohistochemical features and
association with Epstein-Barr virus (EBV). Immunohistochemical
features as well as the EBV status of HL patients have an impact on
the prognosis. The objectives of this study were to classify HL based
on histomorphology and immunophenotype, to determine the asso-
ciation of EBV with HL and to compare the histological subtype,
proliferation index, and clinical features between EBV positive and
EBV negative cases. Materials and Methods: There were 82 patients
of HL diagnosed over a period of 5 years. The histomorphological
features were analyzed. Immunohistochemistry was done with a panel
of markers including CD3, CD20, CD15, CD30, LCA, EBV LMP-1
and PCNA. The clinical, histomorphological and immunohisto-
chemical parameters were correlated and statistical analysis were
performed using Fischers exact test and Chi-square test. Results:There were 80 cases of classical Hodgkin Lymphoma (CHL) and two
cases of Nodular Lymphocyte Predominant Hodgkin Lymphoma
(NLPHL). Nodular sclerosis was the most common subtype of CHL
(48 cases—60 %). CD30, CD15, CD20 were positive in 100, 83.8 and
10 % cases respectively. Group A immunophenotype (CD30+,
CD15+, CD20-) was the most common (75 %) followed by Group B
(CD30+, CD15-, CD20-). Both cases of NLPHL were CD20+,
CD45+, CD15-, CD30- and negative for EBV. EBV LMP-1 was
positive in 46 CHL cases (57.5 %). EBV association was more
common in children, males, mixed cellularity and lymphocyte
depleted subtypes, and in patients with bone marrow infiltration, high
clinical stage and B-symptoms. PCNA expression was high in almost
all cases irrespective of their EBV status. Conclusion: The immu-
nohistochemical features of HL in our study population was similar to
that of the Western countries. EBV positivity in HL was more often
associated with adverse prognostic factors. However the overall
impact of these differences on the clinical outcome of these patients
need to be studied in large scale clinical studies.
Abstract P 123: Poster Presentation
Diagnosis of Hematolymphoid Malignancies in SerousFluids—a Spectrum of Three Cases
S Prasath, NG Rajesh, Debdatta Basu
Department of Pathology, JIPMER, Pondicherry
Background: Involvement of serous fluids by hematolymphoid
malignancies is a common manifestation across the spectrum of
indolent as well as high grade lesions. At times, involvement of
serous cavities is the primary manifestation of lymphoma/leukemia
thus presenting a challenge in accurate diagnosis. Case Series: We
present a series of three cases presenting with pericardial, pleural
and peritoneal effusions. Our first patient, a 25 year old male pre-
sented with massive pericardial effusion to the casualty. His
pericardial fluid cytology, revealed numerous atypical lymphoid
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 231
123

cells which were positive for PAS stain. His peripheral smear, bone
marrow aspirate and biopsy with immunohistochemical work up of
CD3, Tdt and CD 10 positive lymphoid cells confirmed the diag-
nosis of acute lymphoblastic leukemia. Our second patient presented
with history of ischemic heart disease and bilateral pleural effusion
and was suspected to have congestive cardiac failure. His pleural
fluid cytology revealed sheets of mature lymphoid cells and scat-
tered mesothelial cells. His peripheral smear and bone marrow
aspirate showed lymphocytosis (80 %) with CD20+, CD5+ and
CD23+ nodular to interstitial deposits of lymphoid cells in biopsy.
Our third patient a 5-year-old child presented with ascites and
ultrasound revealed retroperitoneal nodes. Ascitic fluid cytology
showed CD 10+, Tdt-, CD20+ lymphoid cells with basophilic
cytoplasm, prominent vacuolation and high mitotic activity. We
diagnosed Burkitt lymphoma and child is on treatment. Conclusion:Knowledge of various manifestations of hematolymphoid malig-
nancies in serous fluids is crucial in accurate diagnosis and timely
management.
Abstract P 124: Oral Presentation
Primary Extranodal non Hodgkin’s Lymphoma of the Headand Neck—a Study from a Tertiary Care Centre in South India
Pritinanda Mishra, Sreeya Das, Debdatta Basu, Rakhee Kar,Bhawana Badhe, Sajini E Jacob
Department of Pathology, Jawaharlal Institute of Postgraduate
Medical Education and Research, Pondicherry
Introduction: Primary extranodal non Hodgkin’s lymphoma (NHL)
of the head and neck accounts for 10–20 % of all cases of NHL. This
study was undertaken to ascertain the anatomic distribution, histo-
logical subtypes and sites of extranodal NHL presenting in the head
and neck region. Materials and Methods: This was a descriptive
study which included record review of the departmental archives over
a period of 3 years (2010—2012). Clinicopathologic features of 25
cases were studied in detail. Results: Of the 25 cases involving head
and neck 16 were males and nine were females. Mean age was
50 years. The most common sites of involvement were Waldeyer’s
ring, followed by nose, brain, orbit, palate and thyroid. Histologically
majority of the cases (80 %) were B cell origin of which diffuse large
B cell lymphoma was the commonest subtype (15 cases), followed by
plasmablastic lymphoma (2 cases) and a single case each of small
lymphocytic lymphoma, lymphoblastic lymphoma and low grade
marginal zone lymphoma of the mucosa associated lymphoid tissue
(MALT) type. T cell lymphomas constituted 20 % of the cases.
Conclusion: This retrospective study in a tertiary care centre in South
India illustrates the pattern of clinical and histological distribution of
various common and uncommon subtypes of extranodal NHL in the
head and neck region.
Abstract P 125
Gemcitabine Induced Skin Rash in a Boy with Hodgkin Disease
Vikas Dua1, Jai Bhagwan Sharma2
1Department of Pediatric Hematology Oncology, Action Cancer
Hospital, Delhi, India; 2Department of Medical Oncology, Action
Cancer Hospital, Delhi, India
Objective: Gemcitabine is used in various carcinomas like lung
cancer, pancreatic cancer, bladder cancer and breast cancer in adults.
It is considered to be a well-tolerated drug with little known side
effects. The reported toxic effects of gemcitabine include myelo-
suppression, altered liver function tests, flu-like syndrome,
bronchospasm, rash, itching, and fever. Gemcitabine has not been
frequently used in pediatric malignancies and to our knowledge
cutaneous reaction has never been reported in children. Method: A
8-year-old boy was admitted in our hospital because of fever and
multiple swellings on both sides of his neck in March 2012. On
examination multiple bilateral cervical lymph nodes were palpable.
Abdominal examination showed hepatosplenomegaly. Biopsy of
cervical lymph node suggested Hodgkin’s disease. Diagnosed as stage
III B Hodgkin’s disease, he was treated with ABVD based chemo-
therapy. Re evaluation following four cycles of chemotherapy
revealed progressive disease, so patient was put on ifosfamide,
gemcitabine, vinorelabine and prednisolone (IGV) based chemother-
apy. Results: On Day 3 of treatment child developed a
maculopapular, itchy skin rash. The rashes involved the neck, chest,
back, upper arms and abdominal wall. The rash subsided in severity
within 4-5 days with the use of oral antihistamine. However, it
reappeared again on Day 5 on repeat challenge with gemcitabine
during second cycle of chemotherapy. The skin lesions were again
easily managed with oral antihistamines. Conclusion: Doctors deal-
ing with chemotherapy drugs should be aware of this kind of side
effect in order to avoid a misdiagnosis and identify the cause of rash.
Abstract P 126
A Retrospective Analysis of Patterns of Distributionof Lymphomas in Elderly Patients in a Tertiary Care Hospitalin South India Over a Period of 5 years
Parimal Sarda1, Marie Therese Manipadam1, Sheila Nair1,Auro Viswabandya2
1Department of Pathology, Christian Medical College, Vellore;2Department of Hematology, Christian Medical College, Vellore
Introduction: This study aims to analyse the pattern of distribution of
lymphoid neoplasms in elderly patients (age more than 60 years) in a
single tertiary care centre in South India using WHO 2008 classifi-
cation. Material and Methods: This is a retrospective analysis of 359
cases diagnosed as lymphoma in elderly patients during the period of
Jan 2007 to Dec 2011. Results: A total number of 359 elderly patients
with diagnosis of lymphoma were included in this study. There were
245 males and 114 female patients. The mean age was 66.72 years.
Of these, non Hodgkin lymphomas (NHL) accounted for 89.97 %
(n = 323), Hodgkin lymphoma (HL) 9.75 % (n = 35) and post
transplant lymphoproliferative disorder 0.28 % (n = 1) respectively.
In NHLs, B cell neoplasms accounted for 90.09 % (n = 291) and T
cell neoplasms 9.91 % (n = 32). In B cell neoplasms, mature B cell
NHL accounted for 99.65 %. In mature B-NHL, indolent lymphomas
accounted for 43.10 % (n = 125) and aggressive lymphomas
56.90 % (n = 165). The subtypes of non Hodgkin lymphomas were
as follow: diffuse large B cell lymphoma (n = 161, 50 %), follicular
lymphoma (n = 33, 10 %), chronic lymphocytic leukemia/small
lymphocytic lymphoma (n = 17, 5.26 %). In T cell neoplasms,
mature T cell NHL accounted for 96.7 % and precursors T-LBL 3 %.
Among the mature T cell NHLs, the pattern of distribution was;
peripheral T cell lymphoma, not otherwise specified in 14 (4.34 %)
patients, angioimmunoblastic T cell lymphoma in 9 (2.78 %) and
anaplastic large cell lymphoma in 4 (1.23 %). Mixed cellularity was
the major subtype of classical Hodgkin lymphoma found in 13 (38 %)
where as Nodular lymphocyte predominant HL constituted 3 %
(n = 1). As compared to the distribution pattern of lymphomas
including all age groups in our institution the striking feature was that
232 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

B cell NHLs are more common in elderly patients. T-LBL and ALCL
are more common in the former group, but this included childhood
lymphoma also, hence when these are excluded overall frequency
becomes similar. Frequency of EBV + DLBCL of elderly will be also
presented. Conclusions: This is the first study from India showing the
pattern of distribution of lymphoid neoplasms in the elderly patients.
Abstract P 127
Lymphoma Involving Spleen and its Histopathological Patternin a Tertiary Care Hospital in South India: A Five Year Study
Subramaniam Kandasamy, Marie Therese Manipadam,Sheila Nair
Department of Pathology, Christian Medical College, Vellore
Introduction: Lymphoma involving spleen is heterogeneous array of
disease whose accurate diagnosis is essential in further management.
The aim of this study is to analyze the lymphoma involving spleen and
its histopathological pattern in a South India tertiary care centre using
WHO 2008 classification. Material and Methods: A retrospective
study of all splenic lymphomas diagnosed in Christian Medical Col-
lege Hospital, Vellore during a 5 year period (Jan 2007 to Dec 2011).
Results: This study includes 69 cases diagnosed as splenic lymphoma
from January 2007 to December 2011. This includes 50 males and 19
females. The mean age was 47 years (range from 7 to 74 years). Non-
Hodgkin lymphoma (NHL) was more common 88.4 % (n-69), than
Hodgkin lymphoma (HL) 11.6 % (n = 69). In NHL, B cell neoplasms
accounted for 73.8 % (n = 61) and T cell neoplasms 26.2 % (n = 61).
The subtypes of NHL were as follow; splenic marginal zone lym-
phoma (n = 13; 28.8 %), diffuse large B cell lymphoma (n = 12;
26.6 %), hairy cell leukemia (n = 6; 13.3 %), follicular lymphoma
(n = 4; 8.8 %), T cell/histiocyte-rich large B cell lymphoma (n = 2;
4.4 %) and one each of mantle cell lymphoma (2.2 %), lymphoplas-
macytic lymphoma (2.2 %) and chronic lymphocytic leukemia/small
lymphocyte lymphoma (2.2 %). 3 cases of low grade B cell lym-
phoma, unclassifiable. Among T cell NHL NK/T cell lymphoma
accounts for 37.5 % (n = 6/16) followed by hepatosplenic T cell
lymphoma (n = 5/16; 31.25), Peripheral T cell lymphoma (PTCL)
(n = 3/16; 18.25 %) and one case of T cell large granular lymphoma.
Splenic marginal zone and diffuse large B cell lymphomas were the
commonest types. As compared to western literature, follicular lym-
phomas were infrequent and peripheral T cell lymphomas including
Hepatosplenic T cell lymphomas and NK/T cell lymphomas were
more common. Conclusions: This study shows the frequency and
pattern of lymphomas involving spleen in a tertiary care centre.
Abstract P 128
Mature T/NK-Cell Lymphoproliferative Disorders: A TertiaryCentre Flow Cytometry Experience
Neeraj Arora, Bhargavi Balakrishnan, Ansu Abu Alex, RayazAhmed, Aby Abraham, Biju George, Alok Srivastava, VikramMathews
Department of Haematology, Christian Medical College and Hospital,
Vellore 632004
Abstract: The classification of the T cell malignancies is complex
and based on clinical features and laboratory investigations, which
include morphology, histology, immunophenotype, HTLV-I serology
and molecular genetics. Mature T/NK-cell lymphoproliferative dis-
orders are rare disorders with morphologic heterogeneity, and lack of
specific immunophenotypic markers for clonality. We retrospectively
analyzed immunophenotypic data from consecutive cases of mature
T/NK cell lymphoproliferative disorders diagnosed in our flow
cytometry laboratory from 2009 to August 2012. These cases were
diagnosed based on immunophenotyping of PB and BM aspirates and
morphology as per WHO 2008 guideline. The gating strategy used
included FSC/SSC, CD45/side scatter and CD3 gating. The initial
panels evaluated for acute leukemia or mature lymphoma depending
on the morphology. In all patients a T cell specific secondary panel
was used for further characterization using CD2, CD3, CD4, CD5,
CD7, CD8, CD10, CD16, CD19, CD25, CD34, CD56, CD57, TCR-
ab, TCR-cd, CD1a, HLA-DR and TdT (BD Biosciences, USA) in
most cases. Correlation was carried out with immunohistochemistry
on tissue (wherever available) such as BM, lymph node and skin
biopsies, serological tests for HTLV1 and other laboratory investi-
gations. Of the total 285 mature lymphoid neoplasms, T/NK-cell
lymphoproliferative disorders constituted 9.5 % (n = 27) and B cell
lymphoproliferative disorders including 12 cases of Burkitts lym-
phoma constituted 90.5 % (n = 258). Plasma cell disorders were not
included in the study. The commonest mature T/NK-cell lympho-
proliferative disorder in this study was hepatosplenic lymphoma
(HSL) (n = 10, 37.1 %) followed by T-large granular leukaemia (T-
LGL) (n = 4, 14.8 %), Adult T cell leukaemia/lymphoma (ATLL)
(n = 3, 11.1 %), Mycosis/Sezary syndrome (MF/SS) (n = 3, 11.1 %)
and T cell prolymphocytic leukaemia (T-PLL) (n = 1, 3.7 %).
Table 1 Immunophenotypic patterns in Mature T/NK cell lympho-
proliferative disorders
HSL (%) T-LGL (%) MF/SS (%) ATLL (%)
CD2 10 (100) 3/3 (100) 3 (100) 3 (100)
CD3+ 10 (100) 4 (100) 2 (66) 3 (100)
CD5+ 1 (10) 2/3 (67) 3 (100) 3 (100)
CD4+ CD8- 0 0 2 (66) 3 (100)
CD4- CD8+ 4 (40) 4 (100) 0 0
CD4- CD8- 6 (60) 0 0 0
CD4+ CD8+ 0 0 0 0
CD7+ 10 (100) 3/3 (100) 1 (33) 0
CD16+ 6 (85) 2 (50) 0 0
CD25+ 0 0 1 (33) 3 (100)
CD56+ 3 (30) 2/4 (50) 0/1 0
CD57 0/8 1/3 (33) ND 0
TCRab+ 1 (10) 3/3 (100) 1/1 (100) 2 (66)
TCRcd+ 9 (90) 0 0/1 0
Total cases 10 4 3 3
NKCLPD(%)
T-PLL(%)
AggressiveNK- cellleukaemia (%)
T-CLPDu (%)
CD2 1 (100) 1 (100) 2 3 (100)
CD3+ 0 1 (100) 0 3 (100)
CD5+ 0 1 (100) 0 3 (100)
CD4+ CD8- 0 1 (100) 0 2 (67)
CD4- CD8+ 1 (100) 0 1 (50) 1 (33)
CD4- CD8- 1 (100) 0 1 (50) 0
CD4+ CD8+ 0 0 0 0
CD7+ 1 (100) 1 (100) 1 (50) 2 (67)
CD16+ 1 (100) 0 2 (100) 0
CD25+ 0 0 0 1 (33)
CD56+ 0 1 (100) 2 (100) 0
CD57 0 0 1 (50) 0
TCRab+ 0 0 1 (50) 3 (100)
TCRcd+ 0 0 0 0
Total cases 1 1 2 3
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 233
123

Mature NK-cell disorders (n = 3, 11.1 %) included 2 cases of
aggressive NK-cell leukaemia and a case of chronic NK cell lym-
phoproliferative disorder (NK-CLPD). Because of the lack of clinical
details a definite diagnosis could not be made in 3 cases and these
cases were labeled as T cell lymphoproliferative disorders unclassi-
fiable (T-CLPD u).The different expression patterns of T cell markers
in various subtypes are summarized in Table 1. To conclude with an
appropriately selected panel, phenotypic analysis using FCM is very
useful for the diagnosis of T cell and NK-cell lymphoproliferative
disorders.
Abstract P 129
Result of Paediatric Non Hodgkin’s Lymphoma with IntensifiedShort Duration Chemotherapy
Prattusha Sengupta, P Gupta, S Biswas, A Mukhopadhyay
Netaji Subhas Chandra Bose Cancer Research Institute, 16A Park
Lane, Kolkata-700016, West Bengal, India
Objective:Aim of our study was to observe result of intensified
short duration chemotherapy in pediatric Non-Hodgkin’s Lympho-
mas (NHL). Methods: We included consecutive 240 pediatric NHL
patients in pediatric haematooncology department of Netaji Sub-
hash Chandra Bose Cancer Research Institute during period from
2000 to 2011. Inclusion criteria: patients \18 years of age with a
diagnosis of NHL. Exclusion criteria: Patients with [25 % blasts in
the bone marrow. Each patient received three cycles A and three
cycles B of MCP 842 protocol of INCTR. Response was assessed
at the completion of 2 cycles of chemotherapy (1 each of A and B)
and 6 cycles of chemotherapy. Result: 70 (29.17 %) patients-
Lymphoblastic Lymphoma (LL), 98 (40.83 %)-Burkitt Lymphoma.
60 (25 %)-diffuse Large B Cell Lymphoma (DLCL), 12 (5 %)-
Anaplastic Large Cell. The abdomen was the most common site in
80 (33.3 %) of involvement followed by mediastinum in 38
(15.83 %). 202 (84.17 %) patients achieved complete response after
2 cycles of therapy. 20 (8.33 %) achieved partial response, 10
(4.17 %) had no response, 10 (4.17 %) were not evaluable. With
median follow up of 4 years a total of 64 (26.67 %) patients (28
LL, 24 Burkitt Lymphoma, 8 DLCL and 4 ALCL) had died.
Causes of death were progressive disease in 48, infection in 12,
hepatitis in 2 and unknown 2. 176 (73.33 %) patients are alive and
disease free. Patients tolerated chemotherapy well. Grade-IV febrile
Neutropenia was seen in 52 patients. Conclusion: Result of short
duration chemotherapy MCP842 is promising. We intend to con-
tinue the same protocol for next few years.
Abstract P 130
Multiple CNS (Only) Relapse of a Case of Extra Nodal NonHodgkin’s Lymphoma with Repeated Complete Remissions
Deba Dulal Biswal, Dinesh Pendharkar, Akshay D. Shah,Suresh H. Advani
Asian Institute of Oncology, Mumbai
Abstract: Relapse of Non Hodgkin’s Lymphoma (NHL) in CNS
though occurs, is a rare phenomena. Relapse of extra nodal NHL in
CNS multiple times with complete response to conventional therapy
each time is very rare. We report a case of multiple CNS (Only)
relapse of a case of extra nodal Non Hodgkin’s Lymphoma with
repeated complete remissions. A 41 years male patient, who was a
known case of NHL, DLBCL, CD20 Positive, of Maxilla and Eth-
moid with mild extension to intracranial, but not involving the brain
parenchyma presented to TMH, Mumbai in 2003. This was an extra
nodal NHL in the face around nose, nearer to cranium. Following 6
cycles of CHOP chemotherapy and local Radiotherapy, he showed
complete response. About 3 years later again presented to the same
hospital with first time brain relapse only (ataxia, brain lesions on
MRI), received treatment as per CNS protocol. Post treatment scan
showed complete response and disappearance of lesion. Two years
later follow up MRI showed normal brain. Later he presented to our
hospital with multiple (two times) only CNS relapses at a gap of
4 years and 10 months respectively with complete response to
treatment.
Abstract P 131
Comparison of Efficacy and Safety of Rituximab (Mabthera�)and its Biosimilar (Reditux�) in Diffuse Large B Cell Lymphoma(DLBCL) Patients Treated with Chemo-Immunotherapy:A Retrospective Analysis
Partha Sarathi Roy, Shiji John, Sadhana Kannan, Sumeet Gujral,Jayant Gawande, Bhausaheb Bagal, Navin Khattry, ManjuSengar, Hari Menon, Reena Nair
Hematolymphoid Disease Management Group, Department
of Medical Oncology, Tata Memorial Centre, Mumbai, India
Objective: Rituximab with CHOP regimen (R-CHOP) has been the
standard of care for DLBCL. Reditux�, a biosimilar molecule of
Rituximab, was in use in India since 2007. The objective of this
retrospective audit is to compare the efficacy, safety and toxicity of
Mabthera� with Reditux�. Methods: Two hundred twenty-three
patients aged C18 years who received 4-8 cycles of R-CHOP
regimen between January 2004 to June 2010 were included.
Complete response (CR), Partial response (PR)] in both groups
were evaluated as per Cheson’s criteria and compared by Chi-
Square test. Overall survival (OS) and progression-free survival
(PFS) were estimated by Kaplan–Meier method and compared by
two-sided log rank test. All grade 3–4 hematological and non-
hematological toxicities were recorded. Results: One hundred one
patients received Mabthera�, 72 received Reditux�. In 17 patients,
the brand used was unknown and 33 patients received both.
Baseline characteristics at presentation were similar in both groups.
Median number of treatment cycle received was 6 in each group.
Observed CR rate was 75 % in Mabthera� group and 81 % in
Reditux� group. OS at 5 years was 84 % (Mabthera� 83 % and in
Reditux� 90 %; P = 0.214). PFS at 5 years was 67 % (Mabthera�
71 % and Reditux� 78 %; P = 0.579). We also evaluated patients
who had received C4 cycles of Mabthera� or Reditux� in their
respective groups, and found no differences in efficacy and out-
comes. Three patients died during treatment in each group. Grade
3–4 febrile neutropenia was observed in 23 % (Mabthera�) and
20 % (Reditux�); grade 3–4 oral mucositis and diarrhea was seen
in 27 % (Mabthera�) and 20 % (Reditux�). No difference in in-
fusional reactions was observed. Conclusion: In this retrospective
analysis we found Reditux� is as efficacious and safe as
Mabthera�.
234 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Abstract P 132
Utility of Flow Cytometry in Bone Marrow Staging of Lymphoma
K Ghodke, T Shet1, E Sridhar1, S Ghogale, Y Badrinath, S More,PG Subramanian, S Gujral
Department of Hematopathology, 1Department of Histotopathology,
Tata Memorial Centre, Mumbai
Introduction: Immunophenotypic (IPT) analysis is an established
tool in the diagnosis and classification of many hematolymphoid
disorders; however, the role of flow cytometry (FC) in detecting
bone marrow (BM) involvement for the staging of lymphoma is
yet to be defined. Objectives: To evaluate the role of FC im-
munophenotyping of BM in the staging of lymphoma and its
correlation with morphology on bone marrow aspirates (BMA)
and trephine biopsy (BM Bx) with immunohistochemistry.
Materials and Methods: A retrospective review was done of 193
consecutive staging BM over a period of 2 month (June–July
2012). Of these, FC analysis was asked for in 60 cases (as per
clinicians request). Morphologic and FC IPT analyses were per-
formed and results were compared with BM Bx findings. FC IPT
analysis was done by six color antibody panel on FACSCANTO
II. Results: As seen in following table, FC helped in detection of
BM involvement in 5 % (3/60) cases in which morphological
analyses on BM failed to detect the involvement. Conclusion: FC
IPT analysis on staging marrow contributed upstaging in 5 % of
cases.
Comparison of FC IPT analysis and morphological analysis in 60
cases
FC IPT
analysis
Morphological analysis Total
BMA (i)
BMBx
(i)
BMA (i)
BMBx
(ui)
BMA
(ui)
BMBx
(i)
BMA (ui)
BMBx
(ui)
i 14 0 7 3 24
ui 0 0 4 32 36
Total 14 0 11 35 60
i involved, ui uninvolved)
Abstract P 133: Poster Presentation
Flowcytometric Immunophenotypingof T Cell Leukemia/Lymphoma
Jyoti Garg, Poojan Agarwal, Vijay Kumar, Sadhna Marwah,AS Nigam, G Buxi
Department of Pathology, PGIMER & Dr. Ram Manohar Lohia
Hospital, New Delhi
Objective: To evaluate the role of flowcytometric immunophenotyping
in T cell leukemia/lymphoma. Methods: Flowcytometric analysis was
carried out on peripheral blood samples of 46 patients suspicious of
acute leukemia. Cases expressing T cell lineage markers were further
characterized with the help of a panel of antibodies comprising of TdT,
CD3, CD4, CD8, CD7, CD5, CD2, CD1a, CD56 and CD45 using BD
FACS Calibur. Results: Of the 10 cases expressing T cell lineage
markers, 8 cases were classified as T-lymphoblastic leukemia/lym-
phoma. All of these cases showed bright positivity for TdT, cCD3 and
CD7. Variable expression of CD1a, CD2, CD4, CD8 and CD5 was seen.
2 cases were classified as peripheral (mature) T cell leukemia/lym-
phoma. These cases showed bright positivity for CD45 and T lineage
markers with aberrant loss of some markers. Conclusion: Flowcy-
tometric immunophenotyping is a useful tool in identifying and
classifying T cell leukemia/lymphoma.
Plasma Cell Dyscrasias
Abstract P 134
sFLC Assay: Establishing National Reference Range—A Single Tertiary Care Centre Experience
Sarika Singh, Rajive Kumar, Vinod Raina, Lalit Kumar
All India Institute of Medical Sciences, New Delhi
Introduction: Serum free light chain (sFLC) assay measures levels of
free j&k immunoglobulin light chains. It is recommended to be used as
a screening test for all plasma cell dyscrasias (PCD).Therefore, inclu-
ded in both the guidelines for diagnosis as well as uniform response
criteria during therapy by International myeloma working group. To
conduct any study on sFLC we take a cue from reference range data
provided by west, due to non availability of Indian data. Aims andObjectives: This study was aimed to establish normal reference range
of sFLC in healthy donors and its comparison with other non myeloma
cohorts to validate the hypothesis that prevalent subclinical infections
and inflammation can result in higher j/k and to establish if any ethnic
variations exist in these cohorts compared to west. Materials andMethods: We studied 135 cases of healthy blood donors, 25 cases of
tuberculosis, autoimmune disorders, chronic renal failure (CRF) and
MM each during 2009–2010. sFLC assay was performed by nephelo-
metric bioassay. Results: j levels ranged from 6.8–70.7, 8.9–77.7, and
5.9–5420 mg/L in healthy donors, non myeloma cohort and MM cases
respectively. k levels were 3.3–66.7, 4.3–125, and 0.002–669.1 mg/L
levels in healthy donors, non myeloma and myeloma cohort respec-
tively. The j/k was 0.6–3.1 mg/L in healthy donors while was
0.4–1.1 mg/L in autoimmune disorder, 0.6–6.9 mg/L in CRF,
0.61–1.66 in TB patients. Conclusion: In contrast to west, all the
parameters were higher in Indian population, more so was the j/k. Since
this deviation is not observed in other non myeloma cohorts, this has a
bearing on the ethnicity rather than subclinical infections/inflammation
which was hypothesised to be the cause.
Abstract P 135: Oral Presentation
Immunohistochemical Profile and Bone Marrow Angiogenesisin Multiple Myeloma with Reference to its Clinical Significance
Sarah Grace Priyadarshini, Debdatta Basu, Rakhee Kar,TK Dutta1
Departments of Pathology and Medicine1, Jawaharlal Institute
of Postgraduate Medical Education and Research, Puducherry
Aims of the Study: To study the histomorphological features of bone
marrow in multiple myeloma and to evaluate the immunohisto-
chemical profile and angiogenesis using a panel of markers including
CD38, kappa and lambda light chain, CD56, Cyclin D1, Ki67 and CD
34 and to correlate immuno-hematological profile with various clin-
ical parameters. Materials and Methods: The study includes 48
cases of myeloma diagnosed over the period of 5 years. The
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 235
123

histomorphological features like plasma cell morphology, percentage
of plasma cells and pattern of infiltration were studied. Angiogenesis
was assessed by calculating the microvessel density (MVD) using
immunohistochemistry for CD34. Proliferation was assessed using
both Ki67 and CD38 highlighted cells. Immunohistochemistry for
CD56 and Cyclin D1 was also done. Results: A significant associa-
tion was seen between the plasma cell morphology and the pattern of
infiltration (p = 0.0067). The percentage of plasma cells showed a
significant association with the clinical staging. The MVD was sig-
nificantly associated with plasma cell morphology (p = 0.04) as well
as pattern of infiltration (p \ 0.001). The proliferation index was also
significantly associated with the plasma cell morphology (p = 0.003).
Both the Ki67 and MVD showed an increasing trend with clinical
staging and MVD was significantly associated with serum albumin
(p = 0.02). CD56 negativity was associated with circulating plasma
cells and also with higher clinical stage. Cyclin D1 positivity was not
seen in poorly differentiated morphology and the mean Ki67 was
lower in the cyclin D1 positive group. It was also found that the cases
with kappa light chain restriction had significantly less of poorly
differentiated morphology, diffuse pattern of infiltration, lower MVD
and Ki67. Conclusion: The study of bone marrow morphology and
angiogenesis with the aid of immunohistochemistry is useful for
prognosticating patients with multiple myeloma.
Abstract P 136
A Case of Hemophagocytic Syndrome with ReactivePlasmacytosis in a 16 Years Old Immunocompromised Patient
Prakriti Shukla, Syed Riaz Mehdi, Sharique Ahmad,Nishi Tandon
Era’s Lucknow Medical College and Hospital, Lucknow
Abstract: Hereditary and sporadic cases of hemophagocytic syndrome
(HPS) are primarily reported in children. We present a case of hemo-
phagocytic syndrome with reactive plasmacytosis in a 16 years young
male. The patient presented to us with persistent fever, hepatospleno-
megaly, lymphadenopathy, anaemia, leucopoenia and marked
thrombocytopenia with elevated triglycerides level. For all the above
complaints he was advised bone marrow examination which revealed
hypocellular smears with marked erythrophagocytosis by histiocytes
and his NK cell activity was reduced which was determined by Flow
cytometry. He was diagnosed as a case of hemophagocytic syndrome
but before any treatment could have been started, he expired. This
experience showed that HPS may be a life-threatening condition. Its
rarity combined with non-specific clinical features, such as fever and
lymphadenopathy, makes it a diagnosis that can be easily missed.
Keywords Hemophagocytosis, Plasma-cytosis, Immunocompromised
Abstract P 137
Prevalence of Autoimmune Hemolytic Anemia in MultipleMyeloma: A prospective study
Rajesh Kashyap1, Abhay Singh2, Pradeep Kumar1
1Department of Hematology, Sanjay Gandhi Postgraduate Institute
of medical Sciences, Lucknow, Uttar Pradesh, India; 2Department
of Transfusion Medicine, Sanjay Gandhi Postgraduate Institute
of medical Sciences, Lucknow, Uttar Pradesh, India
Objective: Autoimmune hemolytic anemia (AIHA) is often associ-
ated with B cell lymphoproliferative disorders. Patients with
malignant lymphoma often demonstrate a positive antiglobulin test,
but they rarely develop overt clinical symptoms of AIHA. AIHA
has rarely been documented in patients with multiple myeloma. We
conducted a prospective study to detect the presence of AIHA in
patients with multiple myeloma and its impact on clinical presen-
tation and outcome of disease. Study Design and Method: A total
of 66 patients diagnosed to have MM over a period of 18 months
were the subjects of the study. Patients with (a) severe anemia
(hemoglobin \ 6 g/dl), (b) requiring frequent blood transfusions and
(c) having clinical and laboratory features of hemolysis, were
screened for AIHA, by performing direct and indirect antiglobulin
(Coomb’s) test. Results: Of the total of 66 patients of multiple
myeloma, seventeen patients were screened for AIHA. Seven
(10.6 %) were found to be complicated with AIHA and carried
auto-antibodies in their sera. Five of these patients had denovo
multiple myeloma and two had multiple myeloma relapse. The IgG
subclass of the antibody binding to red cell membrane was com-
pared with that of M-protein and these findings were showing full
coincidence in all the seven patients. Six patients (85.7 %) had
stage IIIA and one patient (14.3 %) having stage IIIB disease. All
of these patients were positive for subtypes of IgG with IgG1
(n = 2), IgG2 (n = 4), IgG3 (n = 1) and IgG4 (n = 1). One patient
had simultaneous positivity for IgA and IgG2, with presence of cold
antibodies in the serum. Conclusion: We found that autoimmune
hemolysis is a frequent finding in patients with multiple myeloma
and failing to rule out this phenomenon may overburden the man-
agement protocol in form of multiple blood transfusion and related
complications.
Abstract P 138
A Case of Waldenstrom Macroglobulinemia Treatedwith Plasmapheresis
Suvro Sankha Datta, Biplabendu Talukder, Ritam Chakrabarty,Somnath Mukherjee, Prasun Bhattacharya, KrishnenduMukherjee
Department of Immunohematology & Blood transfusion, Medical
College Hospital, Kolkata-73
Objective: Therapeutic plasma exchange is a conjunctive modality
of treatment to decrease paraproteinemias associated with hyper-
viscosity. Here we narrate our experience in treating a diagnosed
case of Waldenstrom macroglobulinemia in 70 years old man with
moderate anaemia and severe features of hyperviscosity syndrome
by serial TPE and chemotherapy. Methods: Three therapeutic
plasma exchanges (TPE) were performed by intermittent cell sepa-
rator (Hemonetics, MCS+, USA). His first two exchanges were done
on alternate day followed by chemotherapy and third TPE was done
six weeks after the second TPE. Results: Initially his serum IgM
was 11.3 g/dl on serum protein electrophoresis. At the end of second
TPE the patient was relieved of his weakness and syncope. Before
third TPE his serum IgM again became 9.901 g/dl. After twenty
four hours of third TPE, his serum IgM level came down to 3.13
g/dl and the patient became asymptomatic. Conclusion: The rela-
tionship between blood viscosity and abnormal immunoglobulin
concentration is exponential, such that removal of relatively small
amount of immunoglobulin by TPE will result in a large reduction
in serum viscosity. So TPE must be used in conjunction with che-
motherapy for symptomatic relief in case of Waldenstrom
macroglobulinemia.
236 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Abstract P 139
Crystalline Inclusions in Myeloma Cells: A Case Report
Shweta Sushmita, Amit Kumar Adhya
Department of Pathology, KIMS, Bhubaneswar, Odisha, 751024
Introduction: The presence of crystalline inclusions in plasma cell
myeloma is a rare phenomenon and cases have been reported with
rod, needle, and rectangular shaped crystals. Here, we present a case
of plasma cell myeloma with needle and rhomboid shaped intracel-
lular crystalline inclusions and extracellular crystal depositions in the
bone marrow. Case: A 56 year old male patient presented with low
back pain. On investigation he was found to have multiple lytic bone
lesions. Serum M band was positive. Bone marrow study revealed
90 % myeloma cells with numerous intracellular and extracellular
needle and rhomboid shaped crystalline inclusions. The inclusions
were negative for MPO, SBB but positive for NSE. Discussion:Crystalline inclusions are believed to be due to accumulation of
cytoplasmic immunoglobulins secondary to a block in the protein
synthetic pathway Since rhomboid crystal depositions can be seen in
other clinical conditions such as pseudogout, this case invites con-
sideration of plasma cell myeloma in the differential diagnosis of
patients with rhomboid crystalline deposition in the bone marrow and
in sites/organs other than the bone marrow. This case and the review
of similar cases presented in the literature suggest that extracellular
rhomboid crystals in the bone marrow and extramedullary sites such
as articular spaces or renal parenchyma should raise suspicion of a
plasma cell neoplasm, yet other differential diagnoses such as gran-
ulocytic sarcoma or reactive processes should also be considered.
Abstract P 140
Multiple Myeloma Presenting as Proptosis: A DiagnosticDilemma
Pattanayak Lucy1, Padhi Sanjukta1, SN Senapati1, SamantaraySagarika2, Rout Niranjan2
1Department of Radiation Oncology, 2Oncopathology, AHRCC,
Cuttack
Background: Plasma cell neoplasms are characterized by monoclo-
nal proliferation of plasma cells or terminally differentiated B cells.
They may present as solitary plasmacytoma, which can affect bone or
soft tissues, or they can manifest with systemic involvement as
multiple myeloma. Extramedullary presentation of multiple myeloma
is uncommon and the orbit is involved in less than 0.25 % cases till
date. We describe an unusual case of multiple myeloma that presented
to us with proptosis. Case: A 45-year-old male presented with gradual
proptosis of the right eye for a period of 15 days. Local examination
revealed a forward and outward protrusion of the eyeball, restricted
movements and loss of vision in the right eye. On palpation there was
a firm mass 2.5 9 2.5 cm in size, nontender in the superior orbit of
the eye. Hb was 7 g% and ESR was 110 mm in the 1st hour. Scrape
cytology from the mass showed sheets of binucleate and multinu-
cleate plasma cells suggestive of plasmacytoma. Bone marrow
aspiration and cytology showed oval cells with pale basophilic
cytoplasm, perinuclear halo, nuclei with cartwheel chromatin sug-
gesting plasma cells which were [30 %. Serum Bens Jones protein
was positive and electrophoresis showed a sharp M band in the
gamma region confirming the diagnosis of Ig G multiple myeloma.
Skeletal survey was normal. Conclusion: Orbital myeloma with
negative skeletal survey is infrequently encountered. Cytological
picture from the lesion as well as bone marrow aspiration cytology is
confirmatory in the absence of bony lesions. Associated features like
anemia help in staging and prognostication.
Keywords Plasmacytoma, Multiple Myeloma, Orbit, Proptosis,
Abstract P 141
Primary Amyloidosis Associated with IgM k ProteinemiaPresented as Nephrotic Syndrome
Upasana Das, SM Sethy, RK Jena, P Mohanty
Department of Clinical haematology, Department of Pathology,
S.C.B. Medical College, Cuttack, Odisha
Introduction: Primary amyloidosis presents with systemic or localised
form without any evidence of preceding or coexisting disease, para-
proteinemia or plasma cell dyscrasias. The amyloid of primary
amyloidosis is made by plasma cells in bone marrow. The L chains are
secreted into the serum and they undergo partial lysosomal proteolysis
and get deposited as insoluble amyloid filament. Three fourth of
immunoglobulin light chains are of the k group. The organs most
commonly involved are the kidneys, heart, blood vessel walls. Renal
amyloidosis may manifests in many forms. Out of all presentations
nephritic syndrome is one of them. Usually the mean age is 43.9 years.
This is a case of presented in young male. Case Report: A 20 year male
boy presented with bilateral pitting pedal edema, puffiness of face and
loss of weight for last 6 months. Serum albumin was 1.5 g/dl, s. Protein
2.9 g/dl, urine albumin +++. The case was diagnosed to be nephrotic
syndrome. The capillary zone electrophoresis showed the presence of M
band, serum immunofixation revealed monoclonal IgG k type. Bone
marrow aspiration 5 % marrow plasma cells. The core needle biopsy of
kidney showed presence of glomerular and vascular deposition of
amyloid. Renal manifestation in amyloidosis carry a grave prognosis.
Keywords Primary Amycoidoses, Nephrotic Syndrome, 1Gms 2chain
Abstract P 142
Secondary Malignancy (AML) in Case of Post Chemotherapy(MPV) Multiple Myeloma
S Burma, P Bhuyan, S Sethi, RK Jena
Department of Pathology & Department of Clinical Hematology,
S C B Medical College, Cuttack, Odisha
Introduction: The multiple myeloma patients may develop AML or
MDS as a secondary malignancy either due to a complication of
Serial
no.
Pre-exchange
hematocrit
Total
blood
volume
processed
Replacement
fluid
1 37.10 % 2,139 ml *5 % albumin:850 ml
+ FFP:900 ml
+ ACD:220 ml
2 36.80 % 2,040 ml *5 % albumin:800 ml
+ FFP:750 ml
+ ACD:187 ml
3 35.30 2,087 ml *5 % albumin:900 ml
+ FFP:600 ml
+ ACD:260 ml
*Albumin is reconstituted with normal saline, FFP: Fresh Frozen
Plasma, ACD: Acid citrate dextrose
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 237
123

chemotherapy or may be in the absence of any chemotherapy. We
reported only two such cases out of 145 multiple myeloma receiving
MPV therapy from July 2010 to July 2012. Case Report: We have
reported two cases of multiple myeloma who developed AML 2 years
after the diagnosis. They had been treated with oral melphalan,
prednisone and vincristine (MPV) for MM. The patients were
achieved complete remission. But after 2 years later, one case pre-
sented with fever, generalized weakness. BM examination reveals
[50 % myeloblasts with[10 % maturation and diagnosed as AML-
M2. But the patient was died after 2 days of initiation of CT. Another
case, 1� years later, presented with fever, sternal tenderness and
mucocutaneous bleeding and diagnosed as AML-M4. The patient
expired due to thrombocytopenia before initiation of any chemo-
therapy. Conclusion: Incidence of secondary myeloid leukemia in the
setting of multiple myeloma is very uncommon. In our series it is
\2 %. Such leukemias are typically acute myeloid leukemia (AML).
The myelomonocytic subtype is particularly common.
Abstract P 143
Stomach Involvement in a Relapse Case of Multiple Myeloma
Priyadarshini Dehuri, Sudha Sethy, Rabindra Kumar Jena,Pranati Mohanty
Department of Clinical Haematology and Department of Pathology,
S.C.B Medical College, Cuttack
Introduction: Multiple myeloma is a neoplastic proliferation of
monoclonal plasma cells usually seen in the bone marrow. Relapse
cases with extramedullary presentation in the form of stomach lesion is
only rarely reported. Case Report: A 60 year female post chemother-
apy for multiple myeloma presented with painful swelling in abdomen,
fever and anaemia. The USG revealed a gastric mass involving fundus
and body. With a clinical suspicion of adenocarcinoma, biopsy was
advised. The biopsy reports were suggestive of either high grade lym-
phoma or undifferentiated carcinoma. Immunohistochemistry was
suggested for definite diagnosis. The IHC stains proved all lymphoma
markers to be negative. There was pan cytokeratin positivity. Most
importantly CD 56 positivity was noted. In the light chain analysis,
kappa to lambda ratio was 3:1. Thus all the investigations favoured a
diagnosis of a monoclonal plasma cell expansion. Conclusion: Relapse
cases of multiple myeloma with extramedullary presentation being very
rare, need attention for their further management.
Keywords Multiple Myeloma, Extramedullary, Stomach, CD56
Abstract P 144
Multiple Myeloma with CNS Involvement: A Rare Case Report
Surabhi, KL Tripathy, P Bhuyan, A Mishra, BP Das
Background: Involvement of CNS is extremely rare in multiple
myeloma, observed in approximately 1 % of the patients. CaseReport: A 35 year old female presented in Neurosurgery OPD with
rapidly progressive scalp swelling over left side of head since
4 months and weakness of right upper and lower limbs for last
1 month. On examination, the swelling measured 10 9 10 9 6 cm
and was tense with variegated consistency. X-ray skull showed a bony
erosion in left frontoparietal region with soft tissue swelling. CT scan
of brain showed a solid SOL in frontal lobe, 8 9 7 cm, with erosion
of left frontal bone and involvement of scalp. With differential diag-
nosis of meningioma with malignant transformation, lymphoma the
patient underwent debulking surgery. Result: The histopathological
study showed features of Plasmacytoma. On bone-marrow aspiration,
the case was diagnosed to be multiple myeloma supported by M-Band
in serum electrophoresis.
Platelet Disorders
Abstract P 145
Profile of Vascular Thrombotic Events in EssentialThrombocythemia
A Krishna Prasad, M Shetty, AMVR Narendra, VR Srinivasan
Nizam’s Institute of Medical Sciences, Hyderabad, AP
Introduction: Essential thrombocythemia (ET) is a chronic myelo-
proliferative disorder characterized by the clonal proliferation of
primarily megakaryocytes, resulting in thrombocythemia. Complica-
tions, often thrombotic, are more common in patients with high
platelet counts with an incidence of 84 %. Profile of vascular
thrombotic events in ET both at presentation and on follow up was
reported below. Aim: Study of vascular thrombotic events in ET
patients. Materials and Methods: Vascular thrombotic events
information, from the case records of ET patients attending General
Medicine department, was retrospectively analyzed. Period of col-
lection was 14 years. All cases satisfied the 2008 WHO criteria for
diagnosis of ET. Results: Total cases were 15. Males were 8 and
females 7. Mean age was 49 years (range of 24–75 years). Mean
platelet count at presentation 13.7 l/ll (range of 5–40 l/ll). All cases
presented with vascular events. Total thrombotic events were 25
(arterial 17 and venous 9). Mean platelet count in arterial thrombosis
was 11.1 l/ll. Arterial thrombosis noted in cerebral vessels (6), cor-
onaries (4), digital arteries (2), renal artery (1), anterior spinal artery
(1), aorto-iliac vessels (1), ulnar artery (1) and tibial artery (1). Mean
platelet count in venous thrombosis was 8.6 l/ll. Venous thrombosis
noted in portal veins (6), deep venous system of lower limbs (4),
spleenic vein (2) and superior mesenteric vein (2). Surgeries were done
in 7 cases [CABG (1), amputation of limbs (2), splenectomy (4)]. JAK2
positivity was noted in 3 out of 5 cases. Conclusions: Arterial events
were more than venous events. Recurrent vascular thrombosis was
noted in 11 cases due to poor control of the disease (twice in 9 and thrice
in 2 cases). Thrombotic complications affect the quality of life.
Abstract P 146
Study of the Cytokine Profile of Type 1 and Type 2 T-Cellsin Children with Acute Immune Thrombocytopenia (ITP)
Vanita Bhaskar1, Anita Nangia1, Sunita Sharma1, JagdishChandra2, Anju Seth2
Department of 1Pathology and 2Paediatrics, Lady Hardinge Medical
College and Associated Kalawati Saran Children’s Hospital,
New Delhi
Objectives: To study pretreatment and post treatment cytokine profile
of Type 1 (Th1 = IFNc, IL-2) and Type 2 (T2 = IL-4, IL-10) T-cells
in children with acute ITP after immunomodulatory treatment.
Method: 30 newly diagnosed ITP patients below 18 years of age,
were subjected to Complete Haemogram with platelet count, Periph-
eral smear and bone marrow aspirate (BMA). Serum cytokine level for
IFNc, IL-2 (Th1 subset) and IL-4, IL-10 (Th2 subset) were performed
238 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

in all using ELISA. Results: The mean values of Th1 cytokines (IFNy,
IL-2) were higher and Th2 cytokines (IL-4, IL-10) were lower in all
the patients pre treatment. Decrease in mean levels of Th1 and an
increase in mean values of Th2 cytokines post treatment were seen
with p value statistically significant. Conclusion: The present study
found an increase in Th1 cytokines (IFNc and IL-2) and decrease in
Th2 cytokines (IL-4 and IL-10), in all paediatric patients of acute ITP
at time of diagnosis emphasising the T cell dysregulation as the early
event in pathophysiology of acute ITP. The decrease in Th1 cytokines
and increase in Th2 cytokine post treatment with normalisation of Th1/
Th2 ratio is an excellent indicator of response to treatment.
Abstract P 147
T Regulatory Cells in Acute Immune ThrombocytopenicPurpura—Study from a Tertiary Centre
Pratibha Dhiman, Arnab Chattopadhyay, Utpal Chaudhuri
Institute of Haematology & Transfusion medicine (IHTM), Medical
College Kolkata
Introduction: Immune thrombocytopenic purpura (ITP) is charac-
terised by enhanced platelet destruction due to platelet autoantibodies.
There is an increasing evidence that regulatory T lymphocytes play an
important role in pathogenesis. Objective: To determine the fre-
quency of CD4+ C425+ CD127low T regulatory cells by flow
cytometry and to investigate the differences in their expression during
periods of disease activity and remission in acute ITP. Materials andMethods: A total of 30 patients with acute ITP (12 males and 18
females), aged 5–40 years were enrolled in the study. Diagnosis of
ITP was based on presence of thrombocytopenia only with no evi-
dence of red cell and white blood cell abnormalities, normal or
increased number of megakaryocytes in bone marrow smears and
exclusion of other causes of thrombocytopenia. Age and sex matched
30 control are also enrolled. All patients and controls are subjected to
clinical evaluation, complete blood count, direct coombs’ test, anti-
nuclear antibody detection, serology testing and morphological
examination of bone marrow samples (in case of patients only). T
regulatory cells were identified by flow cytometry after separation of
peripheral blood mononuclear cells using cell preparation tubes. T
regulatory cells are defined as CD4+ CD25+ CD127low lymphocytes.
Results: The frequency of T regulatory cells ranged from 1.4 to
3.4 % in acute ITP patients at the time of diagnosis. Patients who
achieved remission, the T regulatory cells were increased ranging
from 6.5 to 9 %. Among controls, the levels range from 7.2 to
10.5 %. Conclusion: The study imply one of the possible funda-
mental role of T regulatory cells in pathogenesis of ITP patients.
Abstract P 148
A-3 Haplotype of EPCR Gene: Is it a Risk Factorfor Recurrent Miscarriage in Indian Population
Amit Sharma, Ravi Ranjan, Suhail Akhter, Kamal Kishor,Hareram Panday, Renu Saxena
Department of Hematology, All India Institute of Medical Sciences,
New Delhi, India
Background: Recurrent miscarriage affects 1–5 % pregnancies and in
50 % of these women, the cause of the preceding miscarriages is
unknown. It is multi-factorial in origin and the role of inherited throm-
bophilia and gene polymorphisms of coagulation and anticoagulation
factors such as-thrombomodulin and endothelial protein C receptor
(EPCR) are involved in the pathogenesis of recurrent miscarriage. The
EPCR facilitates PC activation by the thrombin-thrombomodulin com-
plex. In plasma soluble form of EPCR (sEPCR) inhibits both
anticoagulant activity of activated protein C (APC) by blocking its
binding to phospholipids and protein C (PC) activation by competing for
PC with membrane-associated EPCR. sEPCR levels suggested under
genetic control. There are 13 known polymorphisms in EPCR gene which
are in complete linkage disequilibrium; these defined three haplotypes:
A-1, A-2, A-3. Out of them, A-3 was most studied and found strongly
associated with high sEPCR levels in deep vein thrombotic patients in
different population. However role of A3 haplotype of EPCR is a risk
factor for recurrent miscarriage remains controversial. Objective: The
aim of this study is to identify the role of EPCR haplotype (A3) in the
pathogenesis of recurrent miscarriage. Methods: 100 consecutive
patients with recurrent miscarriage and equal number of age and sex
matched healthy controls were the study subjects. All study subjects were
genotyped for A3 haplotype tagging SNP (A6936G) using PCR–RFLP.
Results: Homozygosity of A-3 genotype did not show any significant
association (P = 0.21, c2 = 1.55) while the allelic frequency of the
tagging SNP (A6936G) showed a trend of association (P = 0.07,
c2 = 3.10) with the recurrent miscarriage. Conclusion: A trend of
association in the allelic frequency of the tagging SNP (A6936G) reveals
that in a large sample size this A-3 Haplotype may be found associated
with the recurrent miscarriage in Indian population.
Abstract P 149
Immune Thrombocytopenic Purpura in a Girl with AcuteLymphoblastic Leukemia—an Unusual Association
Vikas Dua1, Jai Bhagwan Sharma2
1Department of Pediatric Hematology Oncology, Action Cancer
Hospital, Delhi, India; 2Department of Medical Oncology, Action
Cancer Hospital, Delhi, India
Objective: Lymphomas have been associated with Immune thrombocy-
topenic purpura (ITP) but it is rare to see ITP in patient with acute
lymphoblastic leukemia (ALL). Nine cases of ITP in children with ALL
have been reported so far, three of them were acute ITP, and rest were
chronic ITP. Only one such case has been reported from India. Method: A
15-year-old girl was admitted in our hospital because of fever, increasing
pallor and bleeding spots over the body in July 2010. Peripheral blood
smear showed 54 % blasts. The bone marrow was consistent with pre-B
ALL. Patient was started on chemotherapy using BFM-95 regimen. Bone
marrow showed remission on the 33rd day of treatment. After phases of
Protocol-I, Protocol-M, and Protocol-II, patient was started on mainte-
nance therapy including 6-mercaptopurine and methotrexate. After
20 months into therapy, the patient was noted to have a platelet count of
38,000/mm3. Results: Therapy was stopped and a suspicion of relapse was
kept and bone marrow was performed. An adequate number of megakar-
yocytes with findings of ALL in remission were detected. The patient was
started on prednisone at 2 mg/kg orally daily. After 3 weeks of prednis-
olone, she had improvement of her platelet count to 78,000/mm3. Her last
platelet count in July 2012 was 88,000/mm3.Secondary causes of ITP were
ruled out. Conclusion: The presence of thrombocytopenia in patient of
ALL does not always means relapse of ALL or as a result of chemotherapy
but the possibility of ITP should also be considered.
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 239
123

Abstract P 150
Comparison of Platelet Counts by Sysmex XE 2100 and LH 750with the International Flow Reference Methodin Thrombocytopenic Patients
Tina Dadu, Sehgal Kunal, Shaikh Anjum, Khodaiji Shanaz
Department of Hematology, P.D.Hinduja Hospital & Medical
Research Centre, Mumbai
Background: There are several methods for counting platelets, of which
The International Flow Reference method is considered to be the gold
standard. We compared the platelet count given by this method to the
count given by automated analyzers using other methods. Aims: To
compare the platelet counts obtained by XE2100 (Impedance, optical
florescence and reported based on switching algorithm) and LH 750
(Impedance) with the international flow reference method in thrombo-
cytopenic blood samples. To calculate the sensitivity, specificity, PPV
and NPV of various technologies at the clinically relevant transfusion
thresholds of 10,000 and 20,000/ll. Materials and Methods: A total of
118 blood samples with platelet count of\50,000/ll were selected for the
study. Platelet counts of all samples were analyzed by all methods using
the Sysmex analyzer, LH750 and IRM in parallel within 6 h of collection.
Results and Conclusions: Sysmex-R had the least Bias and 95 %LA
range and thus correlated best with IRM values. LH-750 had a higher Bias
compared to Sysmex-O and Sysmex-R but a strikingly similar 95 %LA
ensures similar results in all three methods. In fact, in the oncology subset,
it had the narrowest 95 % LA which made it the best performer in this
subgroup. Of the three Sysmex results, Sysmex-I had the highest Bias,
widest 95 % LA, and highest potential risk of over transfusion. Hence
Sysmex-R and LH750 were found to be reliable tools for estimation of
platelet count in thrombocytopenic patients.
Abstract P 151
Evaluation of Various Laboratory Assays in Detection of HeparinInduced Thrombocytopenia in an Adult General ICU
Farhad N Kapadia1, Amruta S Ketkar2, Anand S Deshpande3,Vipla C Puri3, Shanaz J Khodaiji3
1Department of Intensive Care, P. D. Hinduja National Hospital and
MRC; 2Department of Research, P. D. Hinduja National Hospital and
MRC; 3Department of Laboratory Medicine, P. D. Hinduja National
Hospital and MRC
Objective: Heparin Induced Thrombocytopenia (HIT) is an immune
mediated complication of heparin therapy. Our objective was (1) to
compare various laboratory assays for HIT against clinical probability
(4-T score) and 14C-Serotonin Release Assay (SRA) which was the
composite gold standard and (2) to determine the incidence of HIT in the
ICU. Methods: The study Group (n = 217) consisted of consecutive
ICU patients with heparin exposure followed by thrombocytopenia.
The clinical probability (4-T score) was applied to the study group.
Enzyme Linked Immunosorbent Assay (ELISA), Particle Gel Immu-
noassay (PGIA), SRA and Platelet Aggregation Assay (PAA) were
performed. Results: The 4-T score showed that 1/217 patients had high
probability, 48 had intermediate probability and 168 had low proba-
bility for HIT. One patient was positive by SRA, 3 by PGIA and 33 by
ELISA. The incidence based on a combination of clinical features and
laboratory findings was 1.8 %. Conclusions: A greater number of false
positives were observed by ELISA than by PGIA when compared to a
composite gold standard of SRA and clinical probability. The incidence
of SRA positive HIT was 0.46 % (1/217).
Abstract P 152
Coexistence of Macro Thrombocytopenia and GlansmannThrombasthenia—a Case Report
Shenbagapriya, Ramya, Jennifer, Suresh, Vandana Kamath,Usha Sitaram Sukesh C. Nair
Department of Transfusion Medicine & Immunohaematology,
Christian Medical College, Vellore, India
Abstract: Congenital platelet disorders associated with thrombocytope-
nia often pose special diagnostic challenges to clinicians and diagnostic
laboratories. Glansmann thrombasthenia (GT) is a rare autosomal reces-
sive disorder characterized by mucocutaneous bleeding, normal platelet
counts, morphology, and prolonged bleeding time and absent clot retrac-
tion. Here we present a case of 10 year old female who presented to us with
the history of mucocutaneous bleeds requiring transfusions. Laboratory
investigations revealed mild thrombocytopenia with giant platelets (MPV:
15fL), prolonged bleeding time, good clot retraction, platelets failed to
aggregate with ADP, Collagen, AA and Epinephrine and aggregation seen
with Ristocetin. Flow cytometric analysis of GpII bIIIa also showed
findings consistent with GT. An unusual finding of macrothrombocy-
topenia with Glansmann thrombasthenia was identified.
Abstract P 153
Spectrum of Platelet Function Disorders Presenting with BleedingManifestations to a Tertiary Care Centre
Navanila Samanta, Prantar Chakrabarti, Siddhartha SankarRay, Uttam Kr. Nath, Utpal Chaudhuri
Institute of Haematology and Transfusion Medicine (IHTM), Medical
College, Kolkata
Introduction: Platelet aggregometry serves as an important tool for
evaluation of the patient with bleeding manifestations especially when
coagulation profile is normal. IHTM has been performing platelet
aggregometry as a clinical service for the last 10 years. Here, we are
presenting the data of 82 bleeders referred in the last 1 year for platelet
aggregometry using common platelet agonists. Aim: (1) Determination
of type of platelet dysfunction among patients with bleeding symptoms.
(2) To understand the correlation between bleeding time and platelet
aggregation. Method: The detailed clinical notes of the patients referred
for platelet aggregometry were analyzed. Tests were performed using
ADP, Collagen, Arachidonic acid and Ristocetin (standard and low dose)
by Chrono log Model-700. Results: Patients were referred because of
more skin-related bleeding symptoms rather than wet purpura. Using
platelet aggregometry, only 33 % demonstrated characteristic platelet
dysfunction. 7 patients had Glanzmann thrombaesthenia. Bleeding time
was prolonged ([7 min) in only 15 % patients who had platelet dys-
function on aggregometry. Impairment of aggregation with collagen and
arachidonic acid translates into prolonged bleeding time (p \ 0.05).
8.5 % patients showed hyperaggregation though they had bleeding
manifestations. Conclusion: Platelet aggregometry is a useful tool for
evaluation of a bleeding disorder but may remain inconclusive in 67 %
cases if not supplemented by other tests. Bleeding time is not a sensitive
test for detecting platelet dysfunction.
240 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Others
Abstract P 154: Poster Presentation
Systemic Mastocytosis—A Rare Entity
Ketan Mallya2, Bikash Singhania1, K Annamma2, B Sushma1,R Lakshmi1
1Kasturba Medical College, Manipal University, Manipal; 2Melaka
Manipal Medical College, Manipal
Introduction: Mastocytosis encompasses a rare and heterogeneous
group of clinical disorders characterised by abnormal growth and
accumulation of mast cells in various organs, commonly skin and bone
marrow. Here we report a case of systemic mastocytosis with bone
marrow involvement as the first manifestation. Specific histocytologi-
cal techniques were needed to establish the diagnosis. Case Report: A
52 year old lady presented with severe lower and upper back pain of
8 months duration. On local examination severe tenderness was elicited
over dorsal and lumbar spine. X-ray and MRI for the spine showed lytic
lesions at the L1 to L5 vertebrae. Bone scan was suggestive of osteo-
blastic changes. Gastroscopy demonstrated pangastritis. Metabolic and
endocrinological etiologies were excluded because laboratory evalua-
tion was unremarkable. Iliac crest bone marrow biopsy demonstrated
infiltration by mast cells, arranged in sheets and whorls. Extensive areas
of marrow fibrosis was seen along with bone destruction. Special stains:
AFB—Negative. Toluidine blue—positive in mast cells. Immunohis-
tochemistry: CD117—positive in mast cells. Conclusion: The
diagnosis of mastocytosis requires a great degree of clinical suspicion as
it is a rare disease that may go unrecognized or misdiagnosed because of
the non specificity of many of its symptoms, particularly in cases with
visceral involvement and absence of characteristic skin lesions of
urticaria pigmentosa. Bone involvement in mastocytosis is related to
infiltration of bone marrow by mast cells. Bone marrow biopsy and
histocytological techniques are the key for diagnosis.
Abstract P 155
A Template Based Reporting System for Peripheral SmearInterpretation
Shabnam Roohi1, Venkatesha Joshi2, Devendra Prasad2,Padam Kafle3
1Department of Haematology and Clinical Pathology,
Apollo Hospitals Bangalore; 1Apollo Hospitals Bangalore;3Akhil systems Pvt.Ltd
Objective: The peripheral smear examination and interpretation is an
important part of a pathologist’s day to day activity. A template based
reporting system was developed to help reduce the turnaround time
for preparation of an accurate, comprehensive and concise peripheral
smear report. Methods: The knowledge base of previously reported
cases, references from text books and the internet were used to
develop these templates. These templates were then uploaded onto to
the hospital information system using Microsoft visual studio 2005
and visual basic 6.0. A guide for appropriate template to be uploaded
based on the haematology analyzer output was developed. The
technicians were trained to upload the appropriate template based on
the analyzer output. The pathologist reviewed these for accuracy and
completeness after slide review. Results: Of 504 uploaded peripheral
smear templates screened for accuracy 242 were normal studies, 262
were abnormal, out of the abnormal studies 25 required to be modified
and there was no change in the normal study templates. Therefore the
error rate was 4.96 %. Implementation of this system reduced the
turnaround time for report preparation by 58 % and increased the
capacity of the lab by 30 %. Conclusion: A template based reporting
system for reporting peripheral smear interpretations is an effective
tool in reducing the turnaround time and the error rate of the lab for
reporting peripheral smears.
Abstract P 156
A Case of Hemophagocytic Syndrome with ReactivePlasmacytosis in a 16 years Old Immunocompromised Patient
Prakriti Shukla, Syed Riaz Mehdi, Sharique Ahmad,Nishi Tandon
Era’s Lucknow Medical College and Hospital, Lucknow
Abstract: Hereditary and sporadic cases of hemophagocytic syn-
drome (HPS) are primarily reported in children. We present a case of
hemophagocytic syndrome with reactive plasmacytosis in a 16 years
young male. The patient presented to us with persistent fever, hepa-
tosplenomegaly, lymphadenopathy, anaemia, leucopoenia and marked
thrombocytopenia with elevated triglycerides level. For all the above
complaints he was advised bone marrow examination which revealed
hypocellular smears with marked erythrophagocytosis by histiocytes
and his NK cell activity was reduced which was determined by Flow
cytometry. He was diagnosed as a case of hemophagocytic syndrome
but before any treatment could have been started, he expired. This
experience showed that HPS may be a life-threatening condition. Its
rarity combined with non-specific clinical features, such as fever and
lymphadenopathy, makes it a diagnosis that can be easily missed.
Keywords Hemophagocytosis, Plasmacytosis, Immunocompromised
Abstract P 157: Oral Presentation
VCS Parameters in Viral Infections
Mohammed Musheb, Chethan Manohar
Kasturba Medical College, Manipal, Udupi-576104
Objectives: To study the utility of WBC research population data
(RPD) provided as VCS parameters on the Coulter LH750 Hema-
tology analyzer for the screening of viral blood samples against
normal controls. Materials and Methods: The WBC differential
system of Coulter LH750 consists of a dedicated flow channel based
on the ‘Coulter principle’ & identifies the WBC types according to
three physical measurements: (1) Impedance method applied to direct
current-Cell volume (V). (2) Radiofrequency method—nuclear size
and density-conductivity (C). (3) Light scatter of a laser beam–
cytoplasmic granularity (S). Study groups: 1. 106 healthy blood
donors (controls). 2. 118 presumed viral infections—cases (with
bacterial cultures and serology negative, autoimmune serology neg-
ative). Samples were run on the Coulter LH750 on DC mode and their
VCS parameters, i.e. mean (M) and standard deviation (SD) of vol-
ume (V), conductivity (C) and scatter (S) for the different leukocyte
populations (N,L,M,E) were studied. Results: Studying the various
VCS parameters between 2 groups, it was found that mean and
standard deviation parameters for volume, conductivity and scatter for
neutrophils, lymphocytes and monocytes were significantly different
between the controls and cases. LSdV, NSdS in group 2 showed
significant difference and ROC curve analysis showed best AUCs for
mean monocyte volume-MMC (sensitivity 80% -specificity 57%
increasing to 74% for a sensitivity of 72% at a value of 166.3). The
scatter parameters proved to be non predictive. Conclusion: In
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 241
123

conclusion, the leukocyte VCS parameters is moderately predictive in
the detection of viral infections and further studies with serologically
proved viral infections would be useful.
Abstract P 158
Anti M Antibodies Complicating Transfusion Therapy: A Reportof Two Cases
Meenakshi Rao, Sangeeta Pahuja, P Lalita Jyotsna, Manjula Jain
Department of Pathology & Blood Bank, Lady Hardinge Medical
College, Delhi
Introduction: Anti M is a naturally occurring antibody usually active at
temperatures below 37�C and thus is of no clinical significance. However,
rarely the antibody agglutinates red cells at 37�C or at the antiglobulin
phase of testing and can lead to haemolytic transfusion reactions and
haemolytic disease of newborn. Case 1: A 3 year/F with no history of
blood transfusion (BT) presented with severe infection and anemia. For-
ward grouping showed AB positive; reverse grouping was inconclusive.
Blood grouping at different temperatures showed panagglutination at 22�Cwhich enhanced at 4�C and resolved at 37�C. DAT was negative. Antibody
identification using 11 cell panel suggested presence of anti-M antibodies.
DTT treatment of serum suggested presence of IgG + IgM type of anti-M
antibodies. Case 2: A 7 year/M with no history of BT presented with
severe anemia with impending CHF. Blood group (BG) was A positive.
Antibody screen was positive. Reverse BG at different temperatures
showed agglutination with all 3 cells at 22�C which enhanced at 4�C. Only
B cells showed reaction at 37�C. DAT was negative. Antibody identifi-
cation with 11 cell panel suggested presence of anti-M antibodies. DTT
treatment of serum suggested presence of IgM antibodies. Both children
were successfully transfused with M-negative compatible blood units
according to their antigen profile. Conclusion: Although considered
clinically insignificant, anti-M antibodies have been implicated in HDN
and delayed haemolytic transfusion reactions. They can assume signifi-
cance in cases of clinically induced hypothermia. Also, these are the cause
of incompatible cross-match results causing delay in blood transfusion.
Hence, it is extremely important to carefully interpret the results of blood
grouping and antibody screening.
Abstract P 159
Acute Megaloblastic Anemia Presenting as a Severe FulminatingPyrexial Illness. A Case Series
Vineet Behera1, Velu Nair2, Rajan Kapoor2, C Agrawal1
1Department of Medicine, AFMC Pune; 2Medicine & Hematology,
AFMC, Pune
Background: Megaloblastic anemia is a common illness known to have
diverse and protean manifestations. But megaloblastic anemia presenting
as a severe fulminating pyrexial illness is rarely reported. Objective: To
study the clinicopathological profile of megaloblastic anemia presenting as
an acute pyrexial illness. Materials and Methods: This is a retrospective
study in which the clinical, laboratory and therapeutic profile of patients of
megaloblastic anemia presenting as a acute febrile illness was studied.
Results: A total of 14 cases were included. The average age of patients was
31.8 years with 09 (64.28 %) vegetarians. All patients presented with an
acute febrile illness (mean duration 9.1 days) with 71 % having high grade
fever with chills or rigors. All had features of symptomatic anemia.
Jaundice was seen in 12 patients (85.71 %), splenomegaly in 10 (71.42 %),
hepatomegaly in 8 (57.14 %) and features of peripheral neuropathy in 4
(28.47 %) patients. Investigations revealed mean hemoglobin of 4.1 g/dl
(range 2.6–7.4), leucopenia and thrombocytopenia in all, mean ANC
1100/mm3 (ANC\500 in 4 patients) with mean MCV being 113 (max-
imum 132) with macrocytes and hypersegmented neutrophils seen in all.
The mean serum bilirubin was 3.88 mg/dl (unconjugated) with normal
enzymes, mean LDH 1733.42. Serum B12 levels were low (\140 pg/ml)
(mean—112) in all patients with folic acid levels low in 10 patients (71 %).
Bone marrow examination showed cellular reactive bone marrow with
megaloblastoid changes in all. All patients responded dramatically to
therapy with parenteral vitamin B12, folic acid and other supportive
therapy with gradual recovery and normalization of investigations. Con-clusion: Megaloblastic anemia should be considered as a differential
diagnosis of acute pyrexial illness in all patients having a similar presen-
tation to enable early diagnosis and treatment of this easily treatable
disorder.
Abstract P 160
High Incidence of Zidovudine Induced Anaemia in HIV InfectedPatients in Southern Odisha
Kaibalya Ranjan Dash, Lalit Kumar Meher, PK Hui, SPMohanty, SN Nayak
ART Center, Department of General Medicine, MKCG Medical
College & Hospital, Berhampur, Odisha
Aims and Objectives: This study was conducted with an aim to
determine prevalence of ZDV induced anaemia in HIV infected patients
initiated on ZDV containing ART regimen and also to find out corre-
lation with any factor for causing ZDV induced anaemia. Materialsand Methods: This is a retrospective study carried out in ART center,
MKCG MCH, Berhampur between 2009 Jan to 2011 Dec. HIV infected
patients registered at ART center were treated according to NACO
guidelines. Patients (n = 1221) with Hb [ 8 g/dl were prescribed ZDV
based ART regimen. Patients having anaemia (\8 g/dl) were excluded
from the study. Correlation of baseline characteristics (age, sex, weight,
Hb lebel, CD4 count, WHO clinical stage) with risk of developing
anaemia was also calculated. Results: 178 (14.6 %) patients on ZDV
regimen developed anaemia and 6.8 % (n = 83) of these developed
severe Anaemia (\6.5 g%). Patients with low CD4 count were more
prone to develop Anaemia. Age, sex, weight, WHO clinical stage had no
relation to development of Anaemia. Interpretation and Conclusion:Incidence of ZDV induced Anaemia is very High and patients having
low CD4 count were more susceptible to develop anaemia.
Abstract P 161: Poster Presentation
Validation of Automated ESR Machines with ModifiedWestergren Method
Asha Patil1, M Deepak Nayak2, Sushma V. Belurkar3,Seemitr Verma3
Manipal College of Allied Health Sciences, Manipal; 2Melaka
Manipal Medical College, Manipal; 3Kasturba Medical College,
Manipal
Aims and objectives: The study was conducted with the primary
objectives of comparing the following methods of determining ESR.
The new Ves MATIC cube 80TM instrument with modified Wester-
gren method. Ves MATIC cube 80TM with the Ves matic 20TM
instrument, which is currently used in our laboratory. Ves matic 20TM
with modified Westergren method. Materials and Methods: The Ves
MATIC cube 80TM and Ves matic 20TM ESR analyzer were compared
with modified Westergren method. A paired, simultaneously collected
venous blood sample was obtained from 200 random patients who had
a request for both CBC and ESR; that arrived at the sample collection
center, Kasturba Hospital, Manipal. All the samples were evaluated
242 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

using the three methods in our clinical laboratory as per the meth-
odology for each of the individual methods. Statistical Analysis:
Bland and Altman statistical analysis was applied for evaluating the
automated ESR analyser against the modified Westergren method.
Results: The analysis revealed a low degree of agreement between
manual and automated method especially for higher ESR values
([60 mm/h); whereas a reasonable concordance was observed
between values of 20–60 mm/h. When ESR values were\20 mm/h a
good concordance was seen with all the three methods. We went a
step further comparing Ves MATIC cube 80TM with Ves matic 20TM
and found that the discrepancy existed for higher values of ESR.
Conclusion: The fully automated Ves MATIC cube 80TM and Ves
matic 20TM for ESR measurement tend to underestimate the manual
ESR reading. Hence it is recommended that a correction factor should
be applied for a range of ESR values while using these machines.
Further studies & validation are needed.
Abstract P 162
Primary Diagnosis of an Unsuspected non-HaematologicMalignancy by Bone Marrow Biopsy Examination
Krishnendu Halder1, Palash Kumar Mandal1, Swapan KumarSinha1, Debdas Bose1, Debasis Banerjee2
1Department of Pathology Medical College; 2Department
of Pathology, Vivenkanda Institute of Medical Sciences, Kolkata
Objective: To diagnose the primary site of metastatic non-haematologic
malignancies by bone marrow trephine biopsy. Methods: A prospective
observational study was done at a tertiary care hospital of eastern India
from august 2009 to july 2012. Patients advised bone marrow biopsy
examination for causes like pancytopenia, anemia, leucoerythroblastic
blood picture on peripheral blood smears were reviewed during this period.
Patients with metastatic non-haematologic malignancies were selected out
of which only unsuspected malignancy with metastasis to bone marrow
formed the study group. Results: Sixteen cases of metastatic malignancy
with unknown primary site were noted. The age range of the patients was
10–65 years. The most common histologic subtype was adenocarcinoma
and the commonest site identified later was colon. Four cases remained
undiagnosed till the end. Conclusion: Bone marrow biopsy was fruitful
for initial diagnosis of a fair number of patients with occult malignancy. A
detailed history with ancillary investigations like endoscopy, advanced
imaging and PET scan could diagnose the primary site in most of the cases.
However prognosis remained poor due to disseminated malignancy.
Abstract P 163
Gelatinous Transformation of Bone Marrow: A ProspectiveTertiary Center Study, Indicating Varying Trendsin Epidemiology and Pathogenesis
S Singh1, R Sen1, M Gupta1, G Singh1, S Chabbra1, R Verma1,Abhinav2
1Department of Pathology, Pt.Bhagwat Dayal Sharma P.G.I.M.S,
Rohtak; 2Department of Hospital Administration, DCRUST, Murthal,
Sonipat
Introduction: Gelatinous transformation of bone marrow charac-
terized by fat cell atrophy, focal loss of hematopoietic cells and
deposition extracellular gelatinous material histochemically proven
to be mucopolysaccharides rich in hyaluronic acid. Objective: To
study the spectrum of causes of gelatinous transformation in bone
marrow. Materials and Methods: Bone marrow aspirates were
examined for a period of 1 year (1.09.2010 to 31.08.2012).
Gelatinous transformation was confirmed by Alcian blue stain (pH
2.5).This was correlated with the clinico-hematological profile of
the patient. Bone marrow cellularity and ratio of hematopoietic
cells: non hematopoietic cells (plasma cells, mast cells and stromal
cells), increased vascularity (capillary cores) was assessed. Results:A total of 732 samples of bone marrow aspirate were received
1 year period of which 35 showed gelatinous bone marrow
transformation (GBMT). Incidence was nearly 4.8 % Mal-
e:Female = 2:1 (23:12). Fourteen patients (40 %) were in pediatric
age group (\15 years), 13 (37 %) were adolescents and young
adults (15 to 40 years) 4 each (23 %) were in 5th and 6th decades
respectively. The bone marrow was hypocellular in 23 (66 %),
normocellular in 9 (25 %) and hypercellular in 3 (9 %) cases.
Conclusion: Capillary fronds in the bone marrow aspirates, irre-
spective of an increased number of lymphocytes, plasma cells,
mast cells and stromal cells was associated with specific infections
like TB, Leptospirosis and non-specific infectious conditions. 5
study cases (2 children) progressed to aplastic anaemia. The
presence of gelatinous transformation in bone marrow along with
relative increase in lymphoid, plasma, mast and stromal cells and
the paucity of capillary fronds could be a sign of impending
aplastic anaemia, moresoever in children.
Abstract P 164
Follicular Dendritic Cell Tumour in a Patient of Chronic MyeloidLeukemia: A Rare Association
Ragini Singh1, Rajeev Sen1, Nisha Marwah1, PS Gahlaut2,Soumik Chaudhuri3, Nisha Sharma1, Jatin Ahuja4, Kanika1
1Department of Pathology, PGIMS, Rohtak; 2Department of Medicine
& Haematology, PGIMS, Rohtak; 3Department of Haematology,
PGIMS, Rohtak; 4Department of Medicine, PGIMS, Rohtak
Abstract: Follicular dendritic cells, also known as dendritic retic-
ulum cells, are important components of the immune system and are
essential for functions of antigen presentation. Malignancies arising
from these cells are rare and the first such primary neoplasm arising
from follicular dendritic cells was reported only in 1986. The
commonest presence of these follicular dendritic cell sarcomas are
in the lymph nodes, especially of the cervical, axillary and medi-
astinal region but extra-nodal sites such as head and neck and
gastro-intestinal tract may be present in up to a third of patients. The
etiology remains an enigma with a possible link to the Epstein-Barr
virus. Immunohistochemical studies assume great importance in
differentiating this from its close congeners. We describe a case of
Follicular Dendritic Cell Sarcoma, arising in a patient of Chronic
Myeloid Leukemia (CML) on treatment with Imatinib mesylate for
the past 6 years. The Histopathological features of diffuse efface-
ment of the lymph nodes with spindle cells which were positive for
CD 23, CD 10, LCA and vimentin, showed the closest resemblance
to follicular dendritic cell sarcoma. This case deserves reporting due
to the rarity of the disease and hitherto unreported association with
CML. Furthermore, the pathological diagnosis is challenging and
requires a close-knit effort between the pathologist and the
haematologist.
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 243
123

Abstract P 165
Myelonecrosis—a Clinicopathological Study from a TertiaryCare Centre in South India
J Sree Rekha, Debdatta Basu, Rakhee Kar
Department of Pathology, Jawaharlal Institute of Postgraduate
Medical Education and Research (JIPMER), Pondicherry
Introduction: Myelonecrosis is a relatively uncommon clinicopatho-
logic entity. The etiology of myelonecrosis is diverse, and malignancy,
especially hematopoietic in origin, is the most common underlying dis-
ease. Objective: To describe the causes, diagnostic features and clinical
significance of myelonecrosis. Methods: 21/5679 bone marrow biopsies
showing myelonecrosis were identified from a retrospective review of
trephine biopsies done between January 2000 to July 2012. Clinical
findings, peripheral smear and bone marrow findings of these were
reviewed. Results: Among 21 patients, 16 (76.2 %) were males and five
(23.8 %) were female. Fever was the most common presenting complaint
seen in 12 cases (57.1 %). Pallor was the most common clinical sign seen
in 8 cases (38.1 %). Five patients were retropositive. Peripheral smear
examination showed pancytopenia in six cases (28.5 %), leucoery-
throblastic blood picture in six (28.5 %) and leukaemia in four (19.0 %).
Sickle cells were seen in peripheral smear in one case. Myelonecrosis was
identified on bone marrow aspiration smears in five cases. Among 21
cases 12 cases (57.1 %) were due to a neoplastic cause, four due to
tuberculous infection (three in retopositive patients), one due to sickle
cell disease. In three cases the cause could not be identified. Among the
neoplastic causes, acute leukemia was seen in 6 cases (4—ALL, 2—
AML), non-hodgkin lymphoma in two, metastatic adenocarcinoma in
two. Hodgkins disease and malignant melanoma accounted for one case
each. Conclusion: Though conditions associated with myelonecrosis are
varied, malignancy is the commonest cause. Haematological findings are
often present and may give clue to the underlying disorder.
Keywords Myelonecrosis, Bone marrow malignancy
Abstract P 166: Poster Presentation
Neutrophilic Cytoplasmic Inclusions and Their Utilityin Diagnosis of Diseases
Y Sharmila, Sajini Elizabeth Jacob, Debdatta Basu
Department of Pathology, JIPMER
Introduction: Qualitative changes in white blood cells have diag-
nostic significance in certain situations. These include changes like
cytoplasmic vacuolation, abnormal granulation, abnormalities in
nuclear segmentation etc. A detailed study of a peripheral smear,
stained by a standardized Romanowsky method can provide diag-
nostic clues which when correlated with the clinical findings can
clinch the diagnosis. Report: We report a series of eleven cases
where peripheral smear examination of neutrophils helped us to
diagnose varied clinical entities. There were five cases where the
leucocytes showed abnormal giant cytoplasmic granules. Correlating
with the clinical findings, a diagnosis of Chediak Higashi syndrome
was made. Examination of the peripheral smear in four cases of
neonatal sepsis showed neutrophils with golden yellow crystals in the
cytoplasm which are indicative of bilirubin crystals. One patient with
history of fever showed brownish pigmentation in neutrophils.
Detailed search of the smear picked up an occasional gametocyte of
Plasmodium falciparun. The last case was of a 2 year old male child
who presented with hepatomegaly and icthyosis. Peripheral smear
shows vacuolation in the neutrophils which on Sudan black staining
was optically clear. Oil red O staining shows reddish granules con-
firming Jordan anomaly. This finally was a case of Dorfman Chanarin
syndrome. Conclusion: Detailed study of neutrophils in an optimally
stained peripheral smear is a simple, though valuable diagnostic
procedure.
Abstract P 167: Oral Presentation
Tumors of non Haematopoietic Malignancies of Unknown Originin the Bone Marrow: A 5 Year Study from a Tertiary CareCentre in South India
Sreeya Das, Pritinanda Mishra, Rakhee Kar, Debdatta Basu,Sajini E Jacob
Department of Pathology, Jawaharlal Institute of Postgraduate
Medical Education and Research, Pondicherry
Introduction: Tumors of unknown origin represent a common presen-
tation of malignancy and account for 3 to 10 % of cancers. They are
infrequently reported and account for up to 12 % of all cancers detected
by bone marrow biopsy. The study was undertaken to assess the
involvement of bone marrow with tumors of unknown origin and its
significance in establishing primary diagnosis. Materials and Methods:This was a descriptive study which included record review of the
departmental archives for the last 5 years (January 2007 to December
2011).A bone marrow tumor of unknown origin was defined as a meta-
static cancer in the bone marrow in which a primary site was not apparent
at the time of clinical evaluation. A total 2,426 of bone marrow exami-
nation was done in this period of time, of which 15 cases were non
haematological malignancy of unknown malignancy. Results: Bone
marrow metastases of solid tumors were identified in 15 cases (0.6 %). Of
the 15 cases in 13 cases a definitive primary site could be identified by
correlating clinicoradiological findings with morphology and immuno-
histochemistry. In the pediatric population there was one case each of
neuroblastoma, rhabdomyosarcoma and metastatic small round cell
tumor. In the adult population adenocarcinoma of gastrointestinal tract
(41.7 %) was the commonest. Conclusion: Bone marrow metastasis can
masquerade a primary haematopoietic disorder; however its detection
has both therapeutic and prognostic significance. Immunohistochemistry
is an useful adjunct to morphology in reaching a definitive diagnosis.
Abstract P 168
Haematological Profile in HIV Patients Before and After Haart
PS Ghalaut, P Mehta, R Singh, S Dash, V Singh
Department of Clinical Haematology, Pathology & Biochemistry,
Pt. B.D. Shrama PGIMS, Rohtak
Introduction: A number of haematological abnormalities are repor-
ted in HIV patients such as anemia, neutropenia, thrombocytopenia.
Some of these are reversible with Highly active antiretroviral therapy
(HAART).The present study was undertaken to see the effect of
HAART in patients of HIV admitted at PGIMS, ROHTAK. Aims andObjectives: The present study was to evaluate the haematological
profile in HIV patients before and after HAART. Methods: A Total
newly diagnosed (by ELISA) 100 HIV positive patients, in age group
16-64 were taken for the study. 50 patients were put on HAART and
other 50 patients were not on HAART. Various haematological
parameters like Hb, TLC, DLC, APC, BT, CT, PTI, APTT, serum
ferritin, B12 level and serum folate level, CD4 levels and bone
marrow was done base line and after 3, 6 months post HAART level.
Statistical analysis was done by Chi square test. Observation: The
244 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

most frequent haematological abnormality was found to be normo-
cytic normochromic anaemia. (80 %) with mostly normo cellular
bone marrow. The observed value of mean Hb on HAART in study
group showed rise from 10.16 ± 2.30 g/dl to 10.96 ± 1.47 g/dl after
6 months which was statistically significant (p \ 0.01).in non HA-
ART group it show a fall in mean Hb from 9.84 ± 3.03, and to
9.42 ± 2.28 after 6 month follow up. The WBC count increased in
study group from 5110.64 ± 4401 to 6220 = -140 which was sig-
nificant (p \ 0.05), as compared to non significant rise from
5433 ± 4408 to 5888 ± 1547 in control group. The mean platelet
count in study group shows raised from 2.25 ± 0.737 to 2.52 ± 0.433
which was significant than the controlled group with non significant
rise from 2.19 ± 84 to 2.35 ± 6111. There were no significant
findings in other parameters. Conclusion: The study demonstrates
that normocytic normochromic anaemia was most common haema-
tological abnormality in HIV patient’s. There was significant
improvement in haemoglobin level, white blood cell level, platelet
level, absolute neutrophil count, and bone marrow picture in the
patients who were on HAART and rise was statistically significant.
Abstract P 169: Paper Presentation
Clinico-Haematological Analysis of Haematological Malignancy,a Hospital Based Study
Rahul Varshney, Muktanjali Deka, Jina Bhattacharya, PK Gogoi
Department of Haematology, GMCH
Objective: Early diagnosis of haematological malignancy is important
in clinical practice to prevent mortality. This study was conducted to
evaluate their relative frequencies in different ages and sexes in this
region. Methods: The present study was conducted in the Department
of Pathology and Haematology, Gauhati Medical College & Hospital,
Guwahati from 1st July 2011 to 30th June 2012. Patients were
examined and blood tests including haemoglobin estimation, total and
differential leukocyte count, platelets count, reticulocyte count and
PBS study, Bone marrow aspiration and biopsy, special stains, flow
cytometry and cytogenetics was done in each cases. Results: Out of
450 patient attending Haematology OPD 236 were having malignancy.
About 29.66 % patient had chronic myeloid leukaemia while 1.69 %
patient had chronic lymphocytic leukemic. Among acute leukaemia,
acute myeloid leukaemia outnumbered acute lymphoblastic leukae-
mia, (21.19 %against 7.2 %). Multiple myeloma was seen in
20.33 %. Non Hodgkin’s lymphoma was seen in 10.59 % while
Hodgkin’s disease 6.6 %, MDS 1.69 %, MPN and LCH 1 %. Male to
female ratio in haematological malignancies was 2.87:1. Maximum
cases were observed in 51-60 years. Low grade fever, progressive
pallor, weakness and body aches were the commonest symptoms
(70 % cases) while pallor was the frequently observed sign. Conclu-sion: The prevalence of haematological malignancy is 52.44 %.This
study may help in finding out their relative distribution in this region.
Abstract P 170
To Study Spectrum of Morphological Findings on Bone MarrowExamination in Cases of Visceral Leishmaniasis
Anshu Palta1, Sanjeev Garg1, Anita Tahlan1, Atul Sachdev2
1Department of Pathology, 2Department of Medicine, Govt. Medical
College and Hospital, Sector 32, Chandigarh
Objective: To evaluate various morphological findings on bone marrow
aspirate and trephine biopsies in cases of visceral leishmaniasis. Method:This is a retrospective analysis of 15 cases of visceral leishmaniasis
diagnosed in the Haematology section of Pathology Department,
GMCH-32B, Chandigarh over a seven year period (2004–2011). The
detailed clinical data, MGG stained slides of bone marrow aspirate and
H&E stained slides of trephine biopsies done in all the cases were
retrieved from archival material. Results: 15 cases diagnosed with vis-
ceral leishmaniasis were in the age group of 5–50 years with M:Fratio of
1.1:1. Sign and symptoms included fever (100 %), splenomegaly
(93.3 %), hepatomegaly (86.7 %) and lymphadenopathy (6.7 %). Pan-
cytopenia was noted in 73.3 % of cases while the remaining cases had
bicytopenia. Bone marrow hypercellularity was noticed in 53.3 % cases.
Main bone marrow aspirate findings were increase in plasma cells
(66.7 %), increase in histiocytes (66.7 %), increase in eosinophils with
precursors (40 %), hemophagocytosis (33.3 %), increased bone marrow
iron[ 2 + (40 %) and PelgerHuet cells (13.3 %) of cases. Additional
features noted in trephine biopsy were focal benign lymphoid aggregate
(6.7 %), gelatinous transformation (6.7 %), marrow fibrosis (13.3 %) of
cases.. The average parasite density in smear was 3+ and range of posi-
tivity was 1+ to 5+. Conclusions: A note of such aforementioned
observations together or individually should warn a pathologist to do
more vigorous search for L.D. bodies.
Abstract P 171: Oral Presentation
Endothelial Dysfunction in Young Patients of Coronary ArteryDisease in a Tertiary Care Setup
Shipra Garg1, Satendra Sharma1, Meera Sikka1, RajnishAvasthi2, Neelam Wadhwa1, Gopesh Mehrotra3
1Department of Pathology, University College of Medical Sciences
& Guru TegBahadur Hospital, Delhi; 2Department of Medicine,
University College of Medical Sciences & Guru TegBahadur
Hospital, Delhi; 3Department of Radiology & Imaging, University
College of Medical Sciences & Guru TegBahadur Hospital, Delhi
Aims and Objectives: A case-control study was planned to determine
the association between occurrence of CAD in young patients with
endothelial nitric oxide synthase polymorphism (eNOS) Glu298Asp
alone and in combination with TNF-a activity and Flow mediated dila-
tation (FMD) of brachial artery. Methods: The study enrolled 30
diagnosed young male patients of CAD below 40 years as cases and 30
healthy age matched male individuals without history of CAD as the
control group. Polymerase chain reaction- restriction fragment length
polymorphism (PCR–RFLP) analysis for detection of the eNOS-
Glu298 ? Asp (G894T) variant was carried out along with TNF-aactivity by ELISA technique in cases and controls. Endothelial dys-
function was also assessed by non-invasive brachial artery FMD through
ultrasonography. Results: The distribution of GG, GT genotypes in
young CAD was observed to be 73, 27 % and 80, 20 % in cases and
control subjects respectively with no mutant TT found in both study
groups. There was lack of significant association between T allele
inheritance (GT + TT vs GG) in young CAD patients (p = 0.76) with
similar ‘‘T’’ allele frequency in cases and controls. In premature CAD
patients increased TNF-a activity was found to be statistically significant
(p = 0.008) with strong association in young patients with eNOS
Glu298Asp polymorphism GT + TT (p = 0.004). Two-third of cases
with reduction in FMD brachial artery showed significant association
with eNOS Glu298Asp GT + TT genotype than GG (p = 0.02). Con-clusion: Although no significant association of eNOS Glu298Asp
polymorphism was found in young CAD, the observation of GT genotype
in almost one fourth cases of CAD may indicate a need to demonstrate
this polymorphism as a useful genetic marker with it’s possible role to
augment the risk of premature atherosclerosis and subsequent events in
younger patients with few conventional risk factors. The association of
elevated TNF-a activity and decreased FMD brachial artery in young
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 245
123

CAD with eNOS Glu298Asp polymorphism may indicate the probable
role of endothelial dysfunction and inflammation in pathophysiology of
atherosclerosis in young patients.
Abstract P 172
Role of Radiation in the management of Mycosis Fungoides
DK Parida
Department of Radiation Oncology, North Eastern Indira Gandhi
Regional Institute of Health & Medical Sciences (NEIGRIHMS),
Shillong
Abstract: Mycosis Fungoides is a low grade, chronic lymphoproliferative
disorder of the skin caused by abnormal proliferation of CD4+ T cells. The
clinical manifestation ranges from Premycotic, Macule, Plaques, Tumos
and Erythrodermic phase/stage. The leukemic variant is known as Sezary
Syndrome. In order to bring all the lymphoproliferative disorders of skin
the term ‘‘Cutaneous T-Cell Lymphoma’’ was introduced by Eldelson. The
overall incidence of mycosis fungoides is about 4 per 100,000 population.
Because of unavailability of diagnostic facilities many times there is
underreporting of the disease. Majority of the patients present between 40
and 60 years of age. From among the spectrum of treatment modalities,
radiotherapy treatment produces the best therapeutic response in terms of
cure and palliation. The important prognostic factors were (1) type of the
lesions, (2) extent of cutaneous involvement, (3) Involvement of lymph
node at the time of presentation and (4) Presence of lymphoma cells in
peripheral circulation. We treated 14 patients of mycosis fungoides patients
between the age of 27–82 years between 1985 and 1998. Total Skin
Electron Irradiation Treatment was given to the patients by Linear
Accelerator. The dose range was between 8 and 36 Gy. It was also
observed that the end response was directly proportional to the total dose of
radiation treatment delivered to the patient. Subsequently the treatment
techniques were innovated and modified. The further radiation treatment
was delivered by high dose rate mode. This innovation brought down the
treatment time from 2 h to 15 min, hence became very much patient as
well as machine compliant. By this method we could deliver total dose of
36 Gy to all the patients. Seven patients were treated between 1998 and
2000. The major radiation related toxicities were skin blisters, swelling of
joints, desquamation of skin etc. These adverse effects were also dose
dependant. Because of these problems the treatment has to be interrupted
and the total duration of treatment was stretching beyond 10-12 weeks of
time. The ultimate response of radiation treatment depends upon the (1)
total radiation dose and (2) total treatment duration. If the treatment
duration is unnecessarily prolonged, the effect of radiation on the disease
becomes poor resulting in either residual or quick recurrence of the disease.
We modified the treatment schedule from daily to alternate day radiation
treatment schedule beyond third week, which produced the desired result.
Four patients were treated by this method between 2000 and 2004. Skin
reactions were visibly less for which no treatment interruptions were
warranted. The total dose of 36 Gy could be delivered within a stipulated
time period which produced best of disease control. Out of 25 patients, 20
patients remained disease free at the end of 5 years, 5 patients had
recurrent disease. Two patients had progressive disease and 3 patients died.
Abstract P 173: Oral Presentation
Eosinophilia: An Approach to Management
Maya Gopalakrishnan, Karyampudi Arun, Kiran Kumar Matta,KK Nisar, TK Dutta
Jawaharlal Institute of Postgraduate Medical Education and Research
(JIPMER), Puducherry
Background: Peripheral blood eosinophilia is a common and
interesting presentation of various hematological and non-hemato-
logical disorders. Despite extensive evaluation, up to 50 % of
patients with eosinophilia remain undiagnosed. Objective: To
evaluate patients presenting with peripheral blood eosinophilia using
a diagnostic algorithm. Methods: We report a prospective case
series of patients presenting with peripheral blood eosinophilia. A
structured diagnostic algorithm was used, focusing on empiric
treatment of filarial and helminthic infections even after negative
test results owing to their high prevalence in south India. Results:Five patients presenting with different clinical features and absolute
eosinophil count ranging from 2,530 to 32,000 per mm3 were
diagnosed using the algorithm. A 47 year old businessman pre-
senting with low backache was diagnosed to have indolent systemic
mastocytosis. Another 54 year old shopkeeper with bleeding gums
was diagnosed as having myeloproliferative disorder. An asymp-
tomatic agricultural labourer aged 50 years who was referred for
eosinophilia improved with empiric antihelminthic therapy. A
48 year old labourer with wheeze for 2 months was found to have
tropical pulmonary eosinophilia. Whereas another 55 year old driver
presenting with dry cough and reticulonodular shadowing on X-ray
was ultimately diagnosed to have allergic bronchopulmonary
aspergillosis even after positive filarial serology. Conclusion: A
finding of eosinophilia is related to a wide range of disorders.
Therefore, a structured diagnostic algorithm suited to local preva-
lence of diseases may be useful in effective diagnosis and treatment
of patients with eosinophilia.
Abstract P 174
Anemia in Zidovudine Treated Patients in HIV/AIDS
Parna Bhaumik1, Swapan Kumar Sinha1, Pramit Ghosh1,Indranil Dhar2, Bibhuti Saha2, Mimi Gangopadhyay3, JyotirmoyPal3
1Medical College, Kolkata; 2School of Tropical Medicine, Kolkata;3North Bengal Medical College, SusrutaNagar, Darjeeling
Introduction: Anemia is common in HIV/AIDS patients primarily
due to HIV infection as well as due to secondary infections (CMV,
Parvo virus B19). Hemolytic anemia, anemia due to GI bleeding,
Vitamin B12 deficiency due to malabsorption, deranged Iron
metabolism anemia of chronic disease etc. are different types of
anaemia encountered in HIV/AIDS patients. HAART particularly
Zidovine (AZT) induced secondary anemia is commonly associated
with marrow toxicity. Inhibition of thymidine kinase and DNA
chain translocation may lead to anemia. However other lineages of
cells like neutrophils and platelets are affected. Aims and Objec-tives: The aim of the study was to find significant relation between
incidence of anemia and AZT therapy in Aids patients. Materialsand Method: 930 patients of HIV/AIDS were registered in Anti-
retroviral Centre, School of Tropical Medicine, Kolkata from July
2011 to June 2012 and 780 patients were registered in North
Bengal Medical college ART Centre. Out of these 1083 patients
were initiated with AZT and 325 patients developed anemia
(Hb \ 8.00 g/dl). EDTA blood samples were collected and CBC
was performed. Conclusion: The present study with limited num-
ber of patients show anaemia is commonly associated with AZT
HAART. Further study on pathophysiology of anemia including
types of anaemia e.g. Immunohaemolytic, Microangiopathic,
Nutritional, due to Marrow Suppression/dyserythropoietic anemia
may be necessary.
246 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

Abstract P 175
Bacterial Profile in Haematological Malignancies
Anjali Akhuj1, Lalit S. Raut2, Swagnik Roy1, Kalpana Karak1,Subrata Bhattacharya1, Prantar Chakrabarti2, UtpalChaudhuri2, Barun Saha Dalal1
1Department of Microbiology, KPC Medical College & Hospital,
Jadavpur, Kolkata; 2Institute of Haematology and Transfusion
Medicine, Medical College & Hospital, Kolkata
Introduction: The choice of empiric antimicrobial therapy in febrile
patients with haematological malignancies is based on susceptibility
pattern of locally prevalent pathogens. Aim: To review the bacterial
etiology and antimicrobial susceptibility pattern of blood culture isolates
in these patients. Method: In this one year retrospective study, blood
samples were processed by Rapid culture automated Bac T/Alert 3D
systems (Biomerieux). Results: Out of 458 blood samples, 18.34 % were
positive; gram negative organisms constituted 57.14 % and gram posi-
tive organisms 42.87 %. Gram-positive isolates were coagulase negativeStaphylococcus (CoNS) 77.77 %, Staphylococcus aureus 13.88 % and
Enterococci 8.33 %. There were 20 % Methicillin Resistant Staphylo-coccus aureus (MRSA). Methicillin sensitive CoNS were 64.28 %. All
gram positive isolates were sensitive to glycopeptides and linezolid. Most
common gram negative isolates were Pseudomonas aeruginosa 39.58 %
followed by Escherichia coli 22.99 %, Klebsiella pneumoniae 16.66 %,
Nonfermenting gram negative bacilli 8.33 % and Acinetobacter sp.
4.16 %. Ralstonia paucula, Providencea sp. and Cedeceae neteri were
isolated in 2, 1 and 1 cultures, respectively. In Enterobacteriaceae,
71.42 % were ESBL producers. Amongst the gram negative isolates
Fluoroquinolone resistance was noticed in 66.67 %. All gram negative
isolates were sensitive to Polymyxin and Colistin. Multidrug resistant
Pseudomonas aeruginosa sensitive only to Polymyxin and Colistin were
15.78 %. Conclusion: Due to emergence of multidrug resistant patho-
gens, monitoring antimicrobial resistance pattern locally is crucial in
making decisions of empirical therapy.
Abstract P 176
A Comparative Study of Cord Blood Hemoglobin, Iron, Ferritinand TIBC Level in Babies of Primigravida and Multigravidawith Relation to APGAR Score in a Tertiary Care Hospital
Madhumita Mondal1, Ranapratap Ojha1, Swapan Kumar Sinha1,Utpal Chowdhury2
1Department of Pathology, Medical College Kolkata; 1I.H.T.M.,
Medical College Kolkata
Objectives: (1) To compare the values of hemoglobin and iron profile
of newborns with APGAR score. (2)To establish relation between iron
profile and level of hemoglobin of newborn. Methods: We conducted a
prospective observational study over a period of 1 year. The study
population consisted of 48 pregnant women and their single live-birth
baby. Among the mothers 19 were primigravida and 29 were multi-
gravida. All the babies were delivered normally. Cord blood was
collected from all the cases in both EDTA and clotted vials. EDTA
blood was used for hemoglobin estimation and serum was used for
estimation of iron, ferritin and TIBC. APGAR score of all the newborns
at 5 min was measured. All the results were noted and analyzed by
slandered statistical methods. Results: APGAR score results showed 16
babies were in moderate to severe depression. Preterm babies were
proportionately more depressed than term ones. Hb concentration
varied from 13 to 18.7 g/dl. The iron profile of all the newborns were
within normal limit though case to case variation was present.
Conclusion: No statistical significant ‘p’ value was obtained from the
study. So there was no significant correlation between the cord blood
hemoglobin and iron profile with the APGAR score of the newborns.
Abstract P 177
Plasmapheresis in a Case of Severe Acute Hemolysis
Meenu Bajpai, Suman Lata1, Raju Singh
Department of Transfusion Medicine, 1Department of Nephrology,
Institute of Liver and Biliary Sciences, New Delhi
Introduction: Acute hemolysis is a difficult situation to treat and
usually leads to acute renal failure due to acute tubular necrosis. The
patient’s vascular system is loaded with free haemoglobin and toxic
effects of the same are seen. Here we present a case of acute hemolysis
of unknown origin treated successfully with plasmapheresis. Case: A
22 year old male presented at the emergency unit with fever, jaundice
for 2 days associated with progressive shortness of breath and altered
mutation for one day. The patient was labelled as Acute severe hemo-
lysis with jaundice and put on symptomatic therapy while awaiting
investigations. The patient was unresponsive to symptomatic therapy
and plasmapheresis was suggested as a therapeutic modality in con-
sultation with nephrology and transfusion medicine. Two sessions of
plasmapheresis were done on alternate days starting from day 2 of
admission in which one volume of plasma was exchanged using fresh
frozen plasma as replacement. Post plasmapheresis there was
improvement in the patient’s parameters. Investigation for all possible
causes of hemolysis was done. The patient was negative for all viral
hepatitis markers, leptospira, HIV, mycoplasma. Blood and urine cul-
ture were negative as were Indirect and direct Coombs test. The patient
gave no history of intake of any poisonous substance or animal bite. The
cause for the acute hemolysis remained an enigma. The patient suffered
from acute tubular necrosis of the kidneys from which he recovered and
was discharged. Conclusion: Therapeutic plasmapheresis was a life
saving intervention in the above case and should be considered when
routine therapies fail to bring about the desired outcome.
Abstract P 178
ABO Blood Groups Differentially Allow Plasmodium falciparumGrowth
Vrushali Pathak, Roshan Colah, Kanjaksha Ghosh
Objective: Plasmodium falciparum malaria has an impact on the
distribution of ABO blood groups in humans. The ratio of group O to
A is higher in geographic regions where malaria is currently or was
previously endemic. It is known that, during infection with P. falciparum,
group O offers a survival advantage, however the mechanism by
which the survival advantage is translated is not known. This study
was undertaken to correlate the P. falciparum parasitemia with the
ABO blood groups in an in vitro culture system. Methods: We used
an in vitro culture system that involved co-culturing of P. falciparum(strain 3D7) infected erythrocytes with uninfected different blood
group erythrocytes. A fixed number of parasites were allowed to grow
in fixed number of erythrocytes without addition of fresh cells for up
to 5 days. Mean percent parasitemias (Percent parasite-infected
erythrocytes) were calculated after every 24 h. Results: Percent
parasitemia (mean ± SD) in A, B and O group cultures on fourth day
were 15.4 ± 0.7, 13.3 ± 1.3 and 19.9 ± 3.2 respectively. Parasite-
mia was found to be significantly higher in O blood group when
compared to A and B blood groups (P \ 0.01). Conclusion: This
work demonstrated that blood groups differentially allow
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 247
123

P. falciparum growth. The resistance of O group hosts in vivo is not
due to low parasitemia some other mechanisms need to be explored.
Abstract P 179
Diagnosis of Malaria on the XT 2000i
Pankhi Dutta, Chitrangi Navadkar, Sharyu Awate1, SandhyaBastian2
Kokilaben Dhirubhai Ambani Hospital, Mumbai; 1SevenHills
Hospital Mumbai; 2Sysmex India Pvt Ltd
Objective: Detection of malaria on different fully automated 5-part
cell counters have been reported with varying sensitivities and
specificities. This study was conducted to evaluate the efficiency of
the XT 2000i (Sysmex) in diagnosing malaria. Method: The com-
plete blood counts including the various scattergrams of 446 febrile
patients suspected to have malaria were analyzed. Thick and thin
blood smears were examined by two observers and the cases were
divided into malaria positives and malaria negatives. In both groups,
abnormal scatter patterns were looked for in the WBC, WBC/Baso
and reticulocyte scattergrams respectively. Results: Malaria positive
cases were 276 while 170 were negative. In the malaria positive
group, additional cluster on the WBC scatter, complete voting out of
the differential, flag for ‘abnormal WBC scatter’, extended ‘ghost’
cluster in the WBC/Baso scatter and abnormal events in the reticu-
locyte scatter were seen in 93, 39.9, 27.5, 78.3 and 93.9 % of the
cases respectively while the same were seen in only 4.12, 0.6, 0.6, 0.6
and 7.6 % respectively, in the malaria negative group. In predicting
malaria positivity, the sensitivity and specificity of presence of an
additional cluster on WBC diff scatter were 92 and 96 % respectively
while it was 78 and 99 % respectively for an extended ghost cluster
on the WBC/Baso scatter. Presence of abnormal events in the retic
scatter had a sensitivity and specificity of 93.6 and 94 % respectively.
Conclusion: It is possible to detect malaria on the XT 2000i in an
automated manner with reasonable sensitivity and specificity.
Familiarization with the various abnormal scatter patterns will be
helpful in increasing the overall sensitivity of malaria diagnosis.
Abstract P 180
Haematological and Inflammatory Parameters in Patientswith Different Types of Tuberculosis and Their Responseto Antitubercular Therapy
Aditya Kutiyal, Sandeep Garg, and Naresh Gupta
Department of Medicine, Maulana Azad Medical College, New Delhi
Aim: To study the haematological parameters and markers of
inflammation in patients with tuberculosis involving different sites/
organs and to correlate these abnormalities following antitubercular
treatment. Methodology: Adult patients [12 years age of either sex,
newly diagnosed tuberculosis ATT naıve were studied. Four sub-
groups were analyzed stratified on the site of tuberculosis namely
pulmonary, CNS, disseminated and others. Haematological parame-
ters like Hb, WBC, platelet count, ESR, LDH, CRP and albumin were
assayed at baseline and 2 months after ATT. Results: Mean age
amongst the 128 patients was 31.55 years. Pulmonary, CNS, dis-
seminated and other types of tuberculosis comprised 30.5, 28.9, 17.2,
and 23.4 % patients respectively. At baseline, mean Hb, WBC,
platelet, ESR, LDH and albumin was 10.87 g% ± 2.17, 9.24 9 109/
l ± 4.27, 256 9 109/l ± 1.38, 61.43 mm ± 21.10, 753.92 iu ±
183.39 and 3.45 g% ± 0.65 respectively. Anaemia was present in
75.78 %, leukocytosis in 49.21 % and leukopenia in 3.9 %. Throm-
bocytosis and thrombocytopenia was encountered in 12.5 and
38.28 % respectively. ESR levels were raised in 98.43 %. Hypoal-
buminemia was seen in 75 % patients. CRP was elevated in 13
patients. Follow-up comparison after 2 months ATT revealed mean
Hb, WBC, platelet, ESR, LDH and albumin to be 11.72 g% ± 1.79,
7.93 v ± 1.4, 207 9 109/l ± 0.33, 8.34 mm ± 4.29, 205.70 iu ±
42.11 and 3.83 g% ± 0.38 respectively. The p values for change in
all the parameters were highly significant (\0.01). This was true
across the four subgroups of different tuberculosis. CRP was no
longer elevated in any patient after ATT. Conclusion: Tuberculosis
presents with varied haematological and inflammatory abnormalities
in majority which revert with 2 months of ATT. A normal ESR is
highly unlikely and anemia is very likely in tuberculosis at presen-
tation. Leucocytosis may occur in up to half of the cases.
Keywords Tuberculosis, Haematology, Inflammation, Acid phase
reactarts
Abstract P 181
Immature Granulocyte Percentage (IG%) in the Diagnosisof Sepsis
Pankhi Dutta1, Shashikala Shivaprakasha, Sharyu Awate,Monisha Sethi2
1Kokilaben Dhirubhai Ambani Hospital, Mumbai; SevenHills
Hospital, Mumbai; 2Sysmex India Pvt Ltd
Objective: Immature Granulocytes is a new, fully automated, FDA
approved parameter available on some cell counters from Sysmex,
Japan. We evaluated the role of IG% as an aid in the diagnosis of sepsis
as compared to the traditional parameters like absolute neutrophil count
(ANC) and total leukocyte count (TLC). Method- The complete blood
count (CBC) of 166 consecutive patients in whom sepsis was suspected
and for whom blood cultures were ordered, were analyzed on the XT
2000i for the IG%, TLC and ANC. Blood culture (BC) positivity was
considered diagnostic of sepsis. The counts in the BC positive group
versus the culture negative group were compared. The Mann–Whitney
test was applied to find the ‘p value’. The sensitivity and specificity of
these parameters in predicting sepsis were also calculated. Results: Of
166 patients (107-males, 59 females, aged 9–98 years), 62 were BC
positive and 104 were negative. In the BC positive group, the mean IG%
was 2.55 (range: 0–6.2 %) versus a mean of 1.46 (range: 0–3.35) in the
BC negative group (p = 0.0025). The BC positive group had higher
TLC and ANC values as compared to the negative group. The sensi-
tivity and specificity of IG% in predicting BC positivity was 61 and
59 % respectively while it was 87 and 29 % respectively for ANC and
84 and 38 % respectively for TLC. Conclusions: The IG % had a lesser
sensitivity but higher specificity versus ANC & TLC in predicting BC
positivity. It is automated, available at the time and cost of a routine
CBC, and can be a useful parameter in the diagnostic workup of sepsis.
Abstract P 182
Haematological Profile, Serum Iron and Ferritin Level in Anemiaof Inflammation: A Prospective study
Nibedita Sahoo, AK Dash, P Sahu, B Mishra, G Rath, SP Pradhan
Department of Pathology, MKCG Medical College, Berhampur
Introduction: Anemia of Chronic Disease (ACD) is the most common
anemia found in hospitalised patients and it is the SECOND most
248 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

prevalent after Iron Deficiency Anemia (IDA). ACD seen in patients
with chronic infections, malignancy, autoimmune disorders. Condition
also termed as ‘‘Anemia of Inflammation’’. Aim and Objective:Combined clinical history, haematological evaluation, serum iron and
ferritin estimation to reach diagnosis of ACD. Material and Methods:Patients of chronic infection, Autoimmune disorder or malignancy
admitted to different Department s of M.K.C.G.M.C & Hospital are
included in the study during period of 2010–2012. Routine haemato-
logical profile, ESR, serum iron, serum ferritin and bone marrow
aspiration with special emphasis to iron study was performed in 70
patients. Results: Prevalence of ACD was 71 %, IDA 13 % and non
ACD 16 %.Among the patients with ACD mild to moderate anemia
present in 74 % cases. On peripheral smear examination normochro-
mic normocytic anemia in 46 %, normocytic hypochromic 12 %,
dimorphic 16 %, microcytic hypochromic 26 % cases observed. Sig-
nificant correlation of degree of anemia with ESR was obtained. All
cases showed either increase or normal bone marrow iron store.
Conclusion: An increase serum Ferritin, increase Erythrocyte Sedi-
mentation Rate (ESR) and marrow iron staining results indicating
plentiful iron store distinguish people with ACD from IDA.
Keywords Anemia of inflammation (A1), Anemia of chronic disease
(ACD), Irondeficiency anemia (IDA)
Abstract P 183: Poster Presentation
Richter’s Transformation : A Case Report
Ipsita Dhal, Sudha Sethy, Rajeeb Nayak, Rabindra Kumar Jena
Department of pathology and Department of clinical haematology,
SCB Medical College, Cuttack
Introduction: Richter’s transformation or Richter’s syndrome is a
complication of B cell chronic lymphocytic leukemia (CLL) in which
the leukemia changes into a fast growing diffuse large Bcell lymphoma
(DLBCL). Richter’s syndrome affects nearly about 5 % of all CLL
patients at some point during their lives. Case History: A 56 year old
male presented with generalised lymphadenopathy and splenomegaly.
His haematological parameters were haemoglobin 7.5 g/dl, total leu-
cocyte count 50,000/cmm with 80 % atypical lymphocytes. He was
diagnosed as a case of CLL/Small lymphocytic leukemia and was put on
CVP regimen (4 cycles). He attended remission. After one and half
years he came with complaints of fever, myalgia and generalised
lymphadenopathy. His peripheral blood smear showed features of CLL.
Biopsy from cervical lymph nodes showed features of non Hodgkin’s
lymphoma (NHL), which was subsequently confirmed by immuno-
histochemical stains showing positivity for B lineage markers (CD 20,
CD 23). Thus the final impression was DLBCL. Conclusion: we report
this case because of its rarity.
Abstract P 184
Treatment Related Complications in Patients with AcuteLymphoblastic Leukemia
Rajesh Kashyap, Mukul Agarwal, Pradeep Kumar, GarimaAgarwal
Department of Hematology, Sanjay Gandhi Postgraduate Institute of
medical Sciences, Lucknow, Uttar Pradesh, India
Objective: To study the different non-haematological complications
occurring during the late phase (2 weeks of induction therapy to remis-
sion) treatment of ALL patients. Material and Methods: Patients
diagnosed with ALL and being treated at department of haematology,
SGPGIMS were subject of the study. The diagnosis of ALL was made
based on complete hemogram, bone marrow examination with cyto-
chemistry and immunophenotyping by flow cytometry. The clinical
records of the patients were analysed for occurrence of non-haemato-
logical complications occurring 2 weeks after the start of therapy. The
patients were treated as per the BFM protocol (BFM 90 of BFM 95
protocol). Result: One hundred and one patients had 147 events of non-
hematological complications. Gastrointestinal toxicity was the most
frequent complication and occurred predominantly as hepatitis. Hyper-
glycemia was seen in 25 cases (24.7 %) and was most frequent in patients
above the age of 20 years. Neurological complication was predominantly
seen in patients below the age of 20 years (76.2 %) and seizures was the
most common presentation. Osteonecrosis involving the hip joints was
seen two adult young males. Conclusion: Non-hematological compli-
cations are quite frequent during the late phase of treatment of ALL
patients. The Gastrointestinal tract, nervous system and endocrine system
most frequently affected. Many of these events are missed because of low
levels of clinical suspicion. This study highlights the incidence of these
complications as they are associated with high morbidity and mortality.
Abstract P 185
Diagnosis of Paroxysmal Nocturnal Hemoglobinuria (PNH)by Flow Cytometry, and Study of its Association with AplasticAnemia and MDS
Shivangi J. Harankhedkar, Sudha Sethy, Pranati Mahanty,BP Das, RK Jena, SR Mahapatra
SCB Medical College and Hospital, Cuttack
Background: PNH is a consequence of non malignant clonal
expansion of one or several hematopoietic stem cells due to a somatic
mutation in PIG-A gene. Resultant deficiency of glycosyl phospha-
tidyl inositol-anchored proteins (GPI-APs), CD55 and CD59,
accounts for the primary manifestation of intravascular hemolysis.
PNH may also arise in association with aplastic anemias and MDS.
Aims and Objectives: (1) Diagnosis of PNH by using multiparameter
flow cytometry with antibodies specific for GPI-APs, CD55 and
CD59, and assess severity of hemolysis. (2) Detect PNH clones in
patient of aplastic anemia and MDS, and correlate with therapy and
prognosis. Materials and Methods: 253 patients of pancytopenia and
refractory anemia (Coombs negative) were screened for PNH clones.
Using 2 ml of peripheral blood (with EDTA), CD55 (DAF) and CD59
(MIRL) deficient clones were detected on granulocytes by multipa-
rameter flow cytometry. Other supportive routine investigations were
done. Results: Out of 253 patients, classical PNH: 18. PNH clones in
50 aplastic anemia patients: 17. PNH clones in 5 MDS patients: 2. All
cases presented with anemia. Pancytopenia (80 %), hyperbilirubine-
mia (20 %), reticulocytosis and evidence of hemolysis (75 %).
Conclusion: (1) Flow cytometry is considered to be gold standard for
diagnosis of PNH. (2) Association of PNH with aplastic anemia and
MDS is identified as better prognostic indicator.
Abstract P 186
‘Bleeding’ and ‘transfusion Support’ in Acute Myeloid LeukemiaPatients: The Challenges
Rahul Chaurasia, Priti Elhence, Sonia Nityanand, AnupamVerma
Department of Hematology, Others Department of Transfusion
Medicine, SaNjay Gandhi PGIMS, Lucknow
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 249
123

Background: Bleeding due to severe thrombocytopenia in acute
myeloid leukemia (AML), may occur due to the disease process itself
or because of the chemotherapy. Prophylactic platelet transfusions are
given to these patients to prevent the occurrence of bleeding.
Occurrence of bleeding in these patients is therefore an indicator of
presence of factors whereby patient’s response to transfusion is sub-
optimal. Bleeding may be classified according to WHO grading
system and management of bleeding is done according to bleeding
grade. There is however insufficient data to validate this approach.
Objectives: To study the WHO bleeding grades and therapeutic
transfusion support in AML patients undergoing chemotherapy and
prophylactic platelet transfusions. Materials and Methods: A pro-
spective study of 19 patients suffering from AML was carried out at
our center. Patient details, clinical details, bleeding grades as per
WHO, the platelet transfusion data including the indication, trigger
dose and the response to the platelet transfusion were recorded and
analyzed. Results: 256 bleeding days was observed of the total 1302
patient days. WHO Bleeding grades observed were Grade
1 = 115 days, Grade 2 = 86 days, and Grade 3 = 55 days, no grade
4 bleeding was observed. GI bleeding (28.5 %) was the most common
site of the bleed followed by oropharyngeal bleed (19.1 %), epistaxis
(17.1 %), mucocutaneous bleeding (16.4 %), genitourinary (14.4 %)
and others (4.3 %). Mean pre transfusion Hb was 7.82 ± 1.32
(4.3–12.3) and platelet count was 17.54 ± 12.24 (6–46) for which
total 135 LPRBC units (mean 0.53 per episode) and 835 RDP units
(mean 3.26 per episode) was transfused. Clinically bleeding was
stopped after platelet transfusion. No bleeding related mortality was
noted. Conclusion: Despite the prophylactic platelet transfusion
19.6 % of observation days, majority of III bleeding episodes were in
patients who started as grade I or Grade II bleedings therefore this
management approach needs further improvement.
Abstract P 187
Sickle Cell Anaemia—Community Control Programme AmongstTribal Groups from Satpuda Hilly Ranges in Maharashtra, India
SL Kate, Gunvant H. Yeola, Prashant N. Dalvi,Girish T. Kulkarni, Yogesh S. Prabhune
Maharashtra Arogya Mandal, Hadapsar, Pune-411028
Abstract: Amongst all the tribal area of Maharashtra Sickle Cell
Disorder is very common amongst Bhill and Pawara tribal population
groups residing in the Satpuda hilly ranges (trait prevalence 1:5).
Considering counselling is the only alternative, to control the disease,
we have established a community control centre in high risk area
located between 3rd and 4th hilly ranges of Satpuda hills (at Roshmal
Budruk, Tal. Dhadgaon, Dist. Nandurbar Maharashtra state. We
provided accurate diagnosis, possible treatment, follow-up counsel-
ling. We have now 1501 sickle cell patients under our medical
supervision. Counselling is provided to patients, parents and public
from this area. Details will be described in the presentation.
Abstract P 188
Systemic Mastocytosis: A Rare Entity
M Ketan, Bikash Singhania, K Annamma, B Sushma, R Lakshmi
1Kasturba Medical College, Manipal University, Manipal; 2Melaka
Manipal Medical College, Manipal
Introduction: Mastocytosis encompasses a rare and heterogeneous
group of clinical disorders characterised by abnormal growth and
accumulation of mast cells in various organs, commonly skin and bone
marrow. Here we report a case of systemic mastocytosis with bone
marrow involvement as the first manifestation. Specific histocytological
techniques were needed to establish the diagnosis. Case Report: A
52 years old lady presented with severe lower and upper back pain of
8 months duration. On local examination severe tenderness was elicited
over dorsal and lumbar spine. X-Ray and MRI for the spine showed lytic
lesions at the L1 to L5 vertebrae. Bone scan was suggestive of osteo-
blastic changes. Gastroscopy demonstrated pangastritis. Metabolic and
endocrinological etiologies were excluded because laboratory evaluation
was unremarkable. Iliac crest bone marrow biopsy demonstrated infil-
tration by mast cells, arranged in sheets and whorls. Extensive areas of
marrow fibrosis was seen along with bone destruction. Special stains:
AFB—negative; Toluidine blue—positive in mast cells. Immunohisto-
chemistry: CD117—positive in mast cells. Conclusion: The diagnosis of
mastocytosis requires a great degree of clinical suspicion as it is a rare
disease that may go unrecognized or misdiagnosed because of the non
specificity of many of its symptoms, particularly in cases with visceral
involvement and absence of characteristic skin lesions of urticaria pig-
mentosa. Bone involvement in mastocytosis is related to infiltration of
bone marrow by mast cells. Bone marrow biopsy and histocytological
techniques are the key for diagnosis.
Abstract P 189
Flowcytometric Characterization of B-ALL: A 3 Year Studyfrom a Tertiary Care Centre
Nabhajit Mallik, Vijay Kumar, Anjali Sharma, Gurdeep Buxi
Department of Pathology, PGIMER & Dr. Ram Manohar Lohia
Hospital, New Delhi
Objective: To evaluate the expression of Common Acute Lympho-
blastic Leukemia Antigen (CD10), CD34, HLA-DR and myeloid
antigens in all cases of B-ALL over a period of 3 years in a tertiary
care centre in North India. Methods: The study was carried out at the
Department of Pathology, PGIMER & Dr. RML hospital, by ana-
lysing the flow cytometry data over a period of 3 years, from
September 2009 to August 2012. All cases of B-ALL were studied to
look for the expression of CD10, CD34, HLA-DR and myeloid
antigens (CD13, CD33). Results: A total of 36 cases of B-ALL were
retrieved, of which 27 (75 %) patients were male, and 9 (25 %) were
female. The youngest patient was 3 months old, while the oldest was
48 years. 29 cases (80.5 %) showed CALLA positivity, while 7
(19.5 %) were CALLA negative. CD34 positivity was seen in 32
cases (88.9 %) while 4 cases (11.1 %) were negative for CD34. Only
1 case (2.7 %) was negative for HLA-DR, and the remaining 35 cases
(97.3 %) were positive. A total of 13 cases (36.1 %) showed aberrant
expression of myeloid markers in the form of CD13 positivity.
Conclusion: The present study illustrates the role of flow cytometry
in immunophenotypic characterization of B-ALL, especially with
respect to markers that carry prognostic significance for the patient.
Abstract P 190: Poster Presentation
Hairy Cell Leukemia—A Rare Case Report
Sugatha Sahu, S Behera, SP Pradhan
Department of pathology, MKCG Medical college, Berhampur,
Odisha, India
Introduction: Hairy cell leukaemia (HCL) is a rare, clonal, chronic
lymphoproliferative disorder commonly seen in males in the middle years
of life. It represents 2 % of all leukemia’s. Pancytopaenia with moderate
250 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

to massive splenomegaly is the most common clinical presentation. The
diagnosis is based upon the recognition of the characteristic ‘‘hairy’’
nature of the leukemic lymphoid cells in the peripheral blood smears and
the typical appearance and pattern of infiltration in the bone marrow
biopsies in association with increased reticulin and eventual fibrosis of the
marrow. Case Report: We report a case of 54 year male who presented
with pallor, fever, abdominal distension. On physical examination
splenomegaly was detected. Peripheral blood examination showed pan-
cytopenia and lymphocytes with abundant cytoplasm which spread into
hair-like processes. The bone marrow aspirate was a dry tap. The trephine
biopsy has the characteristic features of a honey comb appearance. The
cytochemistry was positive with tartrate resistant acid phosphatase
(TRAP).Hence diagnosis of hairy cell leukemia was made. Conclusion:HCL is an easy diagnosis to make on the morphology of well made blood
and bone marrow smears. Although there are no specific markers for
HCL, cytochemistry and flow cytometry readily confirm the diagnosis.
Abstract P 191: Poster Presentation
Niemann-Pick Disease Presenting with Hepatosplenomegalyand Thrombocytopenia
Mitali Swain, SK Behera, SP Pradhan
Department of Pathology, MKCG Medical College, Berhampur,
Odisha, India
Introduction: Niemann-Pick disease (NPD) is an autosomal recessive
genetic disorder resulting in abnormal lipid metabolism. NPD is a clini-
cally and biochemically heterogeneous disorder, with four variants. Type
A and B have a generalized sphingomyelinase deficiency, whereas types C
and D have normal sphingomyelinase levels. Case Report: We report two
cases of Niemann-Pick disease in children, one aged 9 months and the
other of 2 years, complaining of abdominal distension and recurrent fever.
The older child also complained of epistaxis and melena. Both the children
had coarse facial features and growth retardation. The abdominal ultra-
sound confirmed the clinical findings of hepatosplenomegaly. Bone
marrow and liver biopsy examination revealed presence of foamy histio-
cytes, suggestive of Gauchers or Niemann-Pick. Confirmatory study by
lysosomal enzyme from leucocytes was normal for b-Glucosidase and
sphingomyelinase specific for Gauchers and Niemann Pick type A or B
respectively. So, a diagnosis of Niemann-Pick disease (types C or D) was
made. Conclusion: Niemann-Pick disease in children is very rare, with a
reported incidence of 1:1, 20,000 to 1:1, 50,000 live births. Prenatal
diagnosis with amniocentesis or chorionic villus sampling is available.
Keywords Niemann-Pick disease, Sphingomyelinase,
Hepatosplenomegaly
Abstract P 192
A Prospective Study to Test the Usefulnessof an Immunophenotype-Based Scoring System to DistinguishBetween T-ALL with Myeloid Markers [T-ALL(M)] and AMLWith T-Lymphoid Markers [AML(T)]
Vishal Mehrotra, Manisha Ramani, Janmejay Yadav, PallaviGujarathi, Santosh Parab, Archana Vazifdar and Amar DasGupta
Sections of Hematology and Flow Cytometry, SRL Limited, Mumbai
Introduction: From a retrospective analysis of 1874 cases of acute
leukemia we proposed a scoring system in 2010 to distinguish between
cases of AML(T) and T-ALL(M) on the basis of scores assigned for
expression of surface antigens that indicated myeloid phenotype of
blast cells because of their strong association with typical cases of
AML, e.g. CD5 negativity: score 2; CD10 negativity: 1; CD13 posi-
tivity: 1; CD117 positivity: 0.5 and HLA DR positivity: 1. Cases of
T-ALL(M) had a score\2.5 and those of AML(T) [ 3.5. In this pro-
spective study we tested the sensitivity and specificity of this scoring
system. Materials and Methods: Immunophenotyping was performed
in 1574 additional cases of acute leukemia from May 2010 to July 2012
using a primary panel consisting of antibodies against CD 3, 5, 7, 10,
13, 19, 20, 22, 33, 34, 45, 117 and HLA-DR. Additional markers such
as CD2, 4, 8, 14, 15, 41, 61, 64, 99 and glycophorin were tested as
indicated. The presence of cytoplasmic (c) myeloperoxidase, cCD79a
and cCD3 was examined in all cases with aberrant expression of CD
markers by blast cells. Results: 126/130 cases of AML(T) had a score
of C3.5 (sensitivity and specificity = 97 %) while 71/73 cases of
T-ALL(M) had a score B2.5 (sensitivity 97; specificity 98.5 %). There
was no case of true mixed (T and myeloid) leukemia. Conclusions: The
scoring system proposed by us allows accurate distinction between
AML(T) and T-ALL(M) in most cases at a much lower cost than that
involved in the use of expensive reagent panels, including cytoplasmic
markers, and advanced flow cytometry software.
Abstract P 193
CD200 Expression in B-Chronic Lymphocytic Leukemia(B-CLL)—it is not an all or None Phenomenon
Manisha Ramani, Vishal Mehrotra, Janmejay Yadav, PallaviGujarathi, Santosh Parab, Amar Das Gupta
Hematology & Flow Cytometry Sections, SRL Limited, Mumbai
Introduction: Immunophenotypic distinction between B-CLL and non-
CLL B-chronic lymphoproliferative disorders (B-CLPD) can be chal-
lenging and is sometimes impossible due to overlapping features among
these entities. However, a firm diagnosis is necessary for proper man-
agement of different types of CLPD. Recently CD200, a B-lymphoid
antigen has been shown to be useful in this regard by virtue of its close
(100 %) association with B-CLL and its absence in the lymphoma cells in
non-CLL B-CLPD, except hairy cell leukemia and B-lymphoblastic leu-
kemia/lymphoma. Hence CD200 has been proposed in recent studies as a
diagnostic marker for B-CLL. We wanted to verify this claim by checking
the specificity and sensitivity of this marker for the diagnosis of B-CLL.
Materials and Methods: Twenty eight cases of B-CLL and 28 cases of
other types of B-CLPD (splenic marginal zone lymphoma 7; mantle cell
lymphoma 10; ‘hairy cell’ leukemia 3; others 8) were immunophenotyped
between April and August 2012 using a large panel of antibodies against
CD3, 5, 10, 11c, 19, 20, 22, 23, 25, 38, 103, 200, FMC7, HLA-DR,
immunoglobulin light chains and surface immunoglobulins. Results:Lymphoma cells in all 28 cases of B-CLL were CD200 positive (sensitivity
100 %), while 9/28 cases of non-CLL B-CLPD (‘hairy cell leukemia: 3/3;
mantle cell lymphoma: 2/10; others: 4/8) also were CD200 positive indi-
cating a relatively low specificity (68 %). Conclusions: Our study
suggests that although CD200 is a highly sensitive marker for B-CLL, its
low specificity would limit its utility in the differential diagnosis between
this entity and the other types of B-CLPD.
Abstract P 194
Study of Treatment Outcome of Different Art Regimen Basingon CD4 Count
Vandana Raut, P Kote, RK Bhola, SK Behera, S Pradhan
Vandana Raut, MKCG Medical College, Brahmapur
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 251
123

Introduction: HIV is the leading infection in southern Orissa.
37.8 % of total HIV Population of Orissa is contributed by Ganjam
district alone, and the most probable reason being; unemployed
people from coastal districts like Ganjam migrate to industrial areas
where HIV cases are more. We have a functional ART centre since
2006 and mainly four different regimens are followed consisting of
two nucleoside reverse transcriptase inhibitors (NRTIs) and one
either a non-nucleoside reverse transcriptase inhibitor (NNRTI), or a
protease inhibitor (PI). Aims and Objectives: (1) To know the
efficacy of each of these regimen to restore the immune function (as
indicated by the CD4 cell count). (2) Compare the treatment out-
come of patients in relation to their base line cd4 count at onset of
ART. (3) Response to ART in HIV patient’s co infected with TB.
Materials and Method: The study included 464 HIV cases which
were followed over a period of 1 year. Efficacy of different ART
regimens is studied in terms of CD4 count. Result: Patients age
ranges from 15 to 60 years with mean age of 37.4 years and M:F is
1.4:1. Among 464 cases 220 patients are in regimen I(d4T, 3TC,
NVP), 60 patients are in regimen II(d4T, 3TC, EFV), 100 patients
are in regimen III(AZT, 3TC, NVP) and 84 patients are in regimen
IV(AZT, 3TC, EFV). % increase in CD4 count is maximum with
regimen I. Conclusion: From this we conclude that increase in CD4
count is best achieved with regimen I and presence of TB impairs
the response to ART.
Abstract P 195
Essential Thrombocythemia Transforming into Acute Leukemia:A Case Report
Shabnam Roohi1, Jyothsna Krishnappa2, PP Bapsy3, CN Patil3,Manish Chugh1
1Department of Haematology and Clinical Pathology; 2Department of
Internal Medicine; 3Department of Medical Oncology, Apollo
Hospitals, Bangalore
Introduction: The transformation of essential thrombocythemia into
myelofibrosis is a rare and late complication. The recent advances in
the management of this disease and its complications have raised a
hope for better control. In this case report we present a case of an
elderly patient having essential thrombocythemia with rapid trans-
formation to myelofibrosis and leukemia. Case Description: A
79 year old female case of essential thrombocythemia detected in
2007, on symptomatic treatment, presented to Apollo hospitals in
April 2012 with fever, low back ache, easy fatigability and loss of
appetite. She was thin, pale and had massive splenomegaly on
examination. Peripheral smear examination revealed a leukoery-
throblastic blood picture with severe thrombocytopenia. Bone
marrow aspirate was aparticulate with scattered immature myeloid
cells. The bone marrow biopsy revealed grade four fibrosis on a
scale of 0–4. A diagnosis of Essential Thrombocythemia trans-
forming to Myelofibrosis was made. Considering her high risk
status, regular follow-up with blood transfusion was initiated. In
august 2012 she presented with worsening general weakness and
breathlessness. Peripheral smear showed 24% blasts, suggestive of
leukemic transformation probably myeloid. Flow cytometry report is
awaited. Discussion: Progression of essential thrombocythemia to
Myelofibrosis is 2.8 and 2.3% for acute leukemia with a median of
11 years. In this case the progression to acute leukemia was within
5 years of the initial diagnosis. Conclusion: Transformation of
essential thrombocythemia to Myelofibrosis and acute leukemia can
be rapid. Therefore the use of targeted therapy even for high risk
patients needs to be explored.
Abstract P 196
Evaluation of ADVIA 2120 Haematology Analyzer
Shabnam Roohi, BC Sangamesh, LB Manigantan
Department of Haematology and Clinical Pathology, Apollo
Hospitals, Bangalore, Karnataka, India
Objective: To evaluate performance of Siemens ADVIA 2120 fully
automated haematology analyzer, compare it with the Beckman
Coulter ACT i 5 Diff and manual reference methods. Methods: The
precision, accuracy and Bias of the equipment were evaluated using
standard methods. The Linearity and Carryover of the equipment was
validated. The routine haematological parameters generated by the
ADVIA 2120 were compared to those obtained from ACTi 5 Diff.
The automated differential counts, platelet counts, n RBC and retic-
ulocyte percent were also compared to manual methods. Results: The
precision, accuracy and Bias of the equipment of the equipment were
within acceptable limits. Linearity and carryover were within the
limits established by the manufacturer. There was statistically sig-
nificant difference between the two equipments for platelet count and
eosinophil count, the difference for the other routine parameters was
not statistically significant. The leukocyte differential counts indi-
cated an excellent correlation with the manual reference method for
Neutrophils, Lymphocytes and Eosinophils, a good correlation for
Monocytes and a poor correlation for Basophils. The platelet count
and reticulocyte count indicated an excellent correlation with the
manual reference method, the enumeration of nucleated red blood
cells by the automated method showed moderate correlation with the
manual reference method. Conclusion: The results generated by the
ADVIA 2120 analyzer are comparable to the existing equipment and
the reference methods. The potential of new parameters generated by
the analyzer in extending the clinical role of haematological studies
needs to be explored.
Abstract P 197
Cytogenetic Abnormalities in Acute Leukemia Cases—SingleCentre Study from Eastern India
Basab Bagchi1,Tuphan Kanti Dolai1, Prakash KR Mondal1,Ashutosh Panigrahi1, Sandeep Saha1, Meet Kumar1, MaitreyeeBhattacharyya1, Shyamali Dutta1, Rajiv De1, Malay Ghosh1,Bani B Ganguli2
1Hematology Department, NRS Medical College and Hospital,
Kolkata; 2Genetics Centre, Navi Mumbai, Maharashtra
Objective: Cytogenetics has emerged as an effective tool in diag-
nosis, classification, risk stratification, prognostication and as a guide
for providing appropriate therapy in acute leukemia cases. However
there is not enough data from India. This study was conducted to
determine the incidence and common cytogenetic abnormalities in
AML and ALL. Methodology: Prospective collection of cytogenetics
data from patients attending hematology services of NRS Medical
College, Kolkata. Demographic data, diagnosis data collected, cyto-
genetics analysis done in all patients. Period: July 2009 to July 2012.
Results: Of the 235 patients 152 were males and 83 were females.
118 had ALL and 117 had AML. Among ALL(118) cases 86 patients
were \18 years of age and 32 patients were C18 years of age. Nor-
mal Karyotype was observed in 31/118 (26.3 %) patients.
Hyperdiploidy was the commonest cytogenetic abnormality observed
in 38/118 (32.2 %) of ALL patients, followed by t(9;22) detected in
15/118 (12.7 %) of patients. The next common abnormality was
t(4;11), detected in 9/118 (7.6 %) of ALL patients. Among the ph
252 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

positive patients 12/15 (80 %) were C18 years. Among AML 38/117
(32.5 %) had normal karyotype. t(15;17) was the commonest cyto-
genetic abnormality [13/117 (11.1 %)] followed by trisomy 8
(10.25 %) and t(8;21)9.4 %. Conclusions: 1. Cytogenetic abnor-
malities were present in 73.7 % of ALL and 67.5 % of AML. 2.
Hyperdiploidy was the most common cytogenetic abnormality in
ALL followed by t(9;22). 3. Among the Philadelphia positive patients
80 % were C18 years. 4. t(15;17) was the most common cytogenetic
abnormality in AML followed by trisomy 8 and t(8;21).
Abstract P 198
Coagulation Aberration in HIV Infected Patients
Swapan Kumar Sinha, Soumit Mondal
Department of Pathology, Medical College, Kolkata
Objective: To study disorders of haemostasis in untreated cases of
HIV/AIDS. Total Platelet Count, prothrombin time (PT), activated
partial thromboplastin time (APTT) was performed on 53 diagnosed
patients of HIV. Methods: Citrated blood (1:9) samples were col-
lected for P.Time, APTT, FDPs and EDTA blood for Complete Blood
Count ART Centre of Medical College and School of Tropical
Medicine. PTimre & APPT was performed as per validated methods
with Internal Quality Control. Platelet count was done by making
blood smears of individual samples and counting under light micro-
scope. Results: Among them 19/53 pts. (35.84 %) had decreased plt.
count. Only 7/53 pts. (13.2 %) had elevated PTimer, 6/53 (11.32 %)
had elevated APTT and 9/53 (16.98 %) had elevated PT, APTT and
Low platelet. Conclusion: Thrombocytopenia may be due to
increased destruction commonly immune mediated, increased con-
sumption due to microangiopathy, TTP or decreased production.
Thromboplastin like substances, Factor X activation by angiopathy or
due to CMV infection may lead to activation of coagulation system.
Acquired Factor V Leiden may lead to hypercoagulable states
including APLAS. A detail study in HIV patients for coagulation
disorders will be more informative.
Abstract P 199
Genotypic and Phenotypic Characterisation Bernard-SoulierSyndrome Patients from India
Objective: Molecular characterization of BSS patients from India.
Introduction: Bernard Soulier Syndrome (BSS) is a rare autosomal
recessive bleeding disorder caused by an absence of or defects in the
platelet membrane glycoprotein GP/IX/V complex. Methods: The diag-
nosis of 10 patients was based on low platelet count, presence of giant
platelets in the peripheral smear, platelet function and platelet receptor
study, flow cytometry study to assess the surface expression of GP1b/IX/V
complex showed reduced expression, genomic DNA was screened for
mutations in GP1BA, GP1BB and GP9 through direct sequencing.
Results: We studied 10 patients diagnosed with BSS with classical
bleeding symptoms. Bleeding diathesis measured by the World Health
Organisation bleeding scale (grade 0, no bleeding; grade 1, petechiae;
grade 2, mild blood loss; grade 3, gross blood loss; grade 4, debilitating
blood loss) ranged from 1 to 4. All subjects suffered from frequent
ecchymoses, epistaxis and gum bleeding, the three menstruated females
had severe menorrhagia that required treatment. All the patients had low
platelet count ranging from 6 9 109/L to 55 9 109/L determined auto-
mated cell counter (XT-2000i, Transasia Bio-Medical Ltd,
India).Thrombocytopenia is always more severe when platelets were
measured by a cell counter, because it does not recognises and enumerate
large platelets, as to mean platelet volume (MPV)/platelet size, in none of
the patients the instrument could report the MPV due to its platelet
abnormality. Ristocetin (normal and low dose) induced platelet aggrega-
tion was absent in all patients except one, which was unavailable for study
(BSS1.01), whereas it was normal in controls. Sequencing analysis of the
GPIba, GP1bb and GP9 gene revealed homozygous changes in the pro-
bands. Of the 6 mutations; 4 were found out to be novel mutations
including one nonsense changes in GP1bb (p.Cys32X) and two missense
changes in GP9 (p.Thr95Asp) and (p.Cys135Tyr) and one frameshift
change in GP1ba (p.Met338AsnfsX13). The known mutations found out to
be one missense change in GP9 (p.Cys24Arg) in one patient with severe
bleeding diathesis and one frameshift change in GP1ba (p.Val485Val-
fsX12). Conclusion: The molecular data presented here is a new data on
BSS patients from India apart from the existing data, adding significantly to
the mutation database of this condition and also useful for its genetic
diagnosis in India.
Abstract P 200
Comparative Evaluation of Bone Marrow Trephine Biopsieswith Bone Marrow Aspirates: A Tertiary Care InstitutionalExperience
Usha R Singh, Surbhi Goyal, Usha Rusia
University College of Medical Sciences & Guru Teg bahadur
Hospital, Delhi
Introduction: Bone marrow examination is an important diagnostic tool
to evaluate various neoplastic and non neoplastic hematological diseases.
Aims: To assess correlation between bone marrow biopsy and aspirate.
Correlation of lymphoma positivity with trephine biopsy length. Meth-ods: Diagnostic correlation was done between bone marrow aspirates &
bone marrow trephine biopsies in 518 cases received in the department of
pathology from Jan 2011 to Feb. 2012. Proportion of biopsy showing
marrow infiltration by lymphoma cells was studied in relation to total
trephine length. Biopsies were fixed in formalin & length measured. After
EDTA decalcification they were processed routinely and H&E stained.
Special stain & immunohistochemistry was done if needed. Results: We
found a positive correlation of 88.6 % for acute leukemias and plasma
cell dyscrasias, 60% for nonhematopoietic metastases, 53.7 % for NHL,
38.5 % for aplastic anemia, 5% for Hodgkin’s lymphoma, and 0 % for
granulomatous infections and myelofibrosis. The maximum proportion
of positivity in lymphoma was attained at a length of 1.3 cm. Conclu-sion: Granulomatous infections and myelofibrosis could only be
diagnosed on biopsy. Though both are complimentary, biopsy is more
sensitive and informative in detection of nonhematopoietic metastases,
assessing marrow involvement in lymphomas and plasma cell deposits in
multiple myeloma. In acute and chronic leukemias, immune thrombo-
cytopenic purpuras and nutritional anemias biopsy is a useful adjunct to
aspirate in diagnosis and follow up. A biopsy length of C1.3 cm was
adequate for assessing lymphoma infiltration.
Abstract P 201: Poster Presentation
Anticardiolipin Antibodies in Women with Spontaneous FetalLoss
Meera Sikka1, Akansha Rawat1, Usha Rusia1, Kiran Gulleria2
1Department of Pathology; 2Department of OBG, University College
of Medical Sciences & GTB Hospital, Delhi, India
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 253
123

Introduction: Spontaneous fetal loss is a frequent complication of
pregnancy. Recurrent pregnancy loss (RPL) i.e. occurrence of
three consecutive pregnancy losses during the first trimester
affects 1 % of pregnant women. Several causes of RPL have been
reported . Antiphospholipid syndrome, an autoimmune disorder
characterized by RPL, arterial and/or venous thrombosis and
elevated titers of antiphospholipid antibodies is a frequent cause
of RPL. Anticardiolipin antibodies (ACA) are one of the signifi-
cant antibodies detected in this syndrome. Aim: To ascertain the
prevalence of anticardiolipin antibodies in Indian women with
spontaneous/recurrent fetal loss. Methods: Seventy eight women
with spontaneous/ recurrent fetal loss in the first trimester were
included in the study. A detailed history and physical examination
was done. Anticardiolipin antibodies, IgG and IgM were deter-
mined by ELISA using commercially available kits. Results: IgG
antibodies were detected in 1 (1.2 %) woman and IgM in 3
(3.8 %). These women had no other clinical manifestation. Other
pregnancy related complications were significantly more in ACA
positive women as compared to ACA negative women. Conclu-sions: Presence of ACA defines a population at risk for fetal loss
and other pregnancy related complications. Women with RPL
must be screened for these antibodies even in the absence of other
clinical symptoms as on treatment these women have a better
chance of a subsequent successful pregnancy.
Abstract P 202
Non Immunoglobulin CMYC Translocation in High Grade B CellNHL: The Gray Zone Between Diffuse Large B Cell Lymphomaand Burkitt’s Lymphoma
M. Parihar1, A. Gupta1, A. Yadav1, S. R. Arun1, S. Bhave2,V.Radhakrishnan2, A. Chakrapani2, D. K. Mishra1, M. Chandy2
1Departments of Cytogenetics and Lab Hematology; 2Clinical
Hematology, Tata Medical Center, Kolkata, India
Background: High grade B cell non Hodgkins lymphoma (NHL) with
features between diffuse large B cell lymphoma (DLBCL) and Bur-
kitt’s lymphoma (BL) have been in the grey zone of diagnostic
uncertainty. Although immunoglobulin gene CMYC translocations
define BL, the CMYC gene may be involved in DLBCL as a secondary
hit to the BCL6 or the BCL2 gene translocations. We present a case of
non immunoglobulin gene (Ig) CMYC translocation,t(3;8)(q27;q24)
juxtaposing the CMYC gene to the BCL6 gene in a patient with features
of Burkitts lymphoma . Case report: Our patient a 29 year old male
presented with weakness and low backache following surgery for an
ileocaecal mass operated outside and diagnosed as DLBCL. Clinical
examination revealed no lymphadenopathy or hepatosplenomegaly. A
complete blood count showed low platelet counts with other parameters
being normal. The bone marrow aspirate showed 90 % L3 lympho-
blasts which on flowcytometry expressed CD43, CD10, CD19, CD38,
cCD79a and CD22 with surface immunoglobulin expression. The
trephine biopsy showed infiltration by sheets of abnormal cells with
grade 2 fibrosis. Immunohistochemistry showed positivity for
CD20,BCL6 and negativity for MUM-1 and BCL2 with 100 % Ki67
staining. Karyotyping showed 46,XY,t(3;8)(q27;q24)+2mar[20].
Metaphase FISH using breakapart CMYC probe confirmed the CMYC
gene rearrangement with the partner chromosome being 3q27.
Discussion: The non-IG-MYC translocations have been said to be
almost exclusively secondary events and rarely occur in typical BL.
From clinical point of view these cases with MYC rearrangements but
without an mBL signature are associated with a poor clinical outcome
and classified as intermediate BL/DLBCL by the WHO.
Abstract P 203
A Novel t(2;12) in Precursor T Cell ALL Involving the ETV6Gene
M. Parihar1, A. Gupta1, A. Yadav A1, S. R. Arun1,A. Bhattacharyya2, D. K. Mishra1, M. Chandy3
1Departments of Cytogenetics and Lab Hematology; 2 Paediatric
Oncology; 3Clinical Hematology, Tata Medical Center, Kolkata,
India
Background: Rearrangements of the short arm of chromosome 12 (12p),
that harbors the ETV6 and the CCND genes have been reported in acute
lymphoblastic leukemia (ALL) .Although the role of ETV6 in precursor
B cell ALL has been extensively studied only rare cases of ETV6 gene
involvement in paediatric T cell ALL have been described where the
CCND gene is more commonly involved. We report a case of early
precursor T cell ALL in a 13 year old male with t(2;12)(p13;q31)
involving the ETV6 gene. There have been no reports to date on t(2;12) in
ALL. Case Report: The patient presented with history of fatigue and
irregular fever for the past 6 months. On examination severe pallor,
generalized lymphadenopathy and hepatosplenomegaly were present.
Investigations revealed low hemoglobin, normal white blood cell count
and low platelet counts. Peripheral blood showed 14 % blasts. The bone
marrow showed 90 % L2 lymphoblasts with bright expression of cCD3
and CD7, and moderate expression of CD2, CD5, HLA DR and Tdt. The
karyotype showed 46,XY,t(2;12)(q31;p13) [20]. The involvement of
ETV6 gene was confirmed by metaphase FISH. The patient received the
UKALL 2003 Regimen B protocol. He was in complete remission (CR)
with negative MRD by flowcytometry on day 29 and remains to be in CR
post consolidation therapy. Discussion: The t(2;12)(p13;q31) results in
the relocation of the ETV6 present on 12p13 to 2q31 locus that harbors
the class 1 homeobox gene (HOX) cluster D. The homeobox genes are
expressed during early stages of T cell development and the exact
mechanism of activation of these HOX genes remains to be explored. Our
report highlights the importance of these genes in leukemegenesis of T
cell ALL by probable developmental arrest at specific T cell stages.
Abstract P 204
Cytogenetic Profile of Hematolymphoid Neoplasms in a NewlyOpened Tertiary Care Center: The First Year Experiencefrom East India
M. Parihar1, A. Gupta1, A. Yadav1, S. R. Arun1, S. Bhave2,V. Radhakrishnan2, A. Chakrapani2, A. Bhattacharyya3,D. K. Mishra1, M. Chandy2
1Departments of Cytogenetics and Lab Hematology; 2Clinical
Hematology; 3Paediatric Oncology, Tata Medical Center, Kolkata,
India
Introduction: Conventional cytogenetic analysis of hematolymphoid
neoplasms provides valuable diagnostic and prognostic information.
We describe the cytogenetic profile of hematolymphoid neoplasms
from East India. Material and methods: All patients that were
referred for karyotyping at Tata Medical Center, from August 2011 to
July 2012 were studied. The bone marrow findings and diagnosis was
recorded. G banded karyotypes were reported as per the ISCN.
Results: A total 221 bone marrow samples were studied. The turn-
over time ranged from 2 to 10 days with a median TAT of 5 days. The
disease distribution was as follows 108 patients with Acute Lym-
phoblastic leukemia (ALL),53 with acute myeloid leukemia
(AML),22 with myelodysplastic syndrome (MDS),16 with Non
hodgkins lymphomas(NHL), 10 with myeloproliferative neoplasms
(MPD), 9 with chronic myeloid lelukemia (CML), and 3 with
254 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123

multiple myeloma(MM). Metaphases were available in 212 cases
with a success rate of 96.5 %. Among the ALL patients 65 % had
abnormal karyotypes, hyperdiploidy being the commonest abnor-
mality in children (21/72, 30 %) and t(9;22) in adults(9/36, 25 %).
The other cytogenetic abnormalities seen were MLL gene rear-
rangements (3 %), hypodiploidy (2 %),t(1;19)(3 %), del6q(6 %) and
t(12;21)(2 %). The following cytogenetic abnormalities were seen in
patients with AML (58 % abnormal Karyotypes): t(15;17) in 7
patients, (8;21) in 5,trisomy 8 in 3,3q abnormalities in 3,monosomy 7
in 4,del 9q in 2, MLL gene rearrangements in 1 and complex
karyotype in three. Abnormal karyotypes were seen in 32, 62 and
30 % of patients with MDS,NHL and MM. Conclusion: The inci-
dence of hyperdiploidy in paediatric ALL patients is similar to what
has been reported in western literature and higher than some reports
from India. The incidence of t(9;22) in adult ALL,t(8;21) and t(15;17)
in AML is similar to what has been reported from India and west.
Abstract P 205
Jumping Translocations in AML: A Role in LeukemogenesisAlong with Tumor Progression
M. Parihar1, A. Gupta1, A. Yadav1, S. R. Arun1, A.Bhattacharyya2, D. K. Mishra1, M. Chandy3
1Departments of Cytogenetics and Lab Hematology; 2Paediatric
Oncology; 3Clinical Hematology, Tata Medical Center, Kolkata,
India
Background: Jumping translocations (JT) are rare cytogenetic phe-
nomenon resulting from translocation of the same segment of the
donor chromosome on to various recipient chromosomes creating
multiple related clones. Previous reports of JT in myeloid malig-
nancies have implicated its role in disease progression in patients with
myelodysplastic syndrome or myeloproliferative disorders that evolve
into acute myeloid leukemia (AML). We present a rare case of JT in a
1 year old female with de novo AML M5. Case report: The patient
presented with fever and purulent discharge from the left ear. On
examination pallor, facial edema, proptosis of the right eye, mass in
the maxillary region and hepatosplenomegaly were seen. Investiga-
tions revealed low hemoglobin, normal white blood cell count and
low platelet counts. There were no blasts in the peripheral blood. The
bone marrow was completely replaced by blasts having morphology
akin to monoblasts with expression of CD13, CD33, CD34, HLA-DR,
CD117, CD64, CD4 and MPO negativity on immunophenotyping.
Karytotyping showed a JT with the long arm of chromosome 1 (1q12)
as the donor fragment and short arms of chromosome 12 (12p13) and
13 (13p12) as the recipient chromosomes in 24 and 5 metaphases
respectively. The patient died within few days of admission. Dis-cussion: The most commonly involved chromosomal segment in JT
in myeloid malignancies is 1q and is associated with a poor prognosis.
However its presence here in a de novo pediatric AML indicates its
role in leukemogenesis unlike previous reports that have implicated
its role only in tumor progression.
Abstract P 206
Loss of Y Chromosome in Acute Lymphoblastic Leukemia:Constitutional or Neoplastic
S. R. Arun1, M. Parihar1, A. Gupta1, A. Yadav1,A. Bhattacharyya2, D. K. Mishra1, M. Chandy3
1Departments of Cytogenetics and Lab Hematology; 2Paediatric
Oncology; 3Clinical Hematology, Tata Medical Center, Kolkata,
India
Introduction: Loss of chromosome Y chromosome is a well estab-
lished cytogenetic abnormality in myeloid neoplasms and has been
reported with varying frequency ranging from 6.3 to 16.4 % in var-
ious hematological disorders. There have been no reports of a
neoplasia associated loss of Y in acute lymphoblastic leukemia (ALL)
and occasional reports in adult ALL have suggested it to be an age
related phenomenon rather than a neoplastic one. We report a case of
Philadelphia positive mixed phenotype ALL with loss of Y as a
secondary associated abnormality. Case Report: The patient a 4 year
old male child presented with a history of low back ache, bone pain,
and inability to walk. Systemic examination showed hepatospleno-
megaly. His blood counts revealed low hemoglobin, raised total
leucocyte count with 57 % blasts and low platelets. Bone marrow
examination showed 72 % L1 lymphoblasts. Immunophenotyping
showed mixed lineage phenotype with bright expression of CD10,
CD19, CD34, cCD22, and cCD3, TdT, HLA-DR and aberrant CD13.
Cytogenetic analysis (karyotype and FISH) showed two clones,one
showing t(9;22) with additional philadelphia chromosome (ph) and
loss of Y chromosome in 16 metaphases and the other showing t(9;22)
with t(1;19) in 4 metaphases. Phytohaemagglutinin stimulated
peripheral blood cultures showed a normal karyotype ruling out
constitutional loss of Y chromosome. Conclusion: Loss of Y chro-
mosome can be seen in ALL as a part of genetic evolution. The other
interesting findings were presence of additional Philadelphia chro-
mosome and t(1;19) as secondary abnormalities in genetic evolution
in a case of t(9;22) ALL.
Dr. J.C. Patel Award Session
Abstract P 207
RNA Expression of Genes Involved in Cytarabine Metabolismand Transport is an Independent Predictor of Ex-vivo CytarabineResponse in Acute Myeloid Leukemia
Ajay Abraham, Savitha Varatharajan, J Ashok Kumar, K Sreeja,Vivi M Srivastava, RV Shaji, Rayaz Ahmed, Aby Abraham, BijuGeorge, Mammen Chandy, Alok Srivastava, Vikram Mathewsand Poonkuzhali Balasubramanian
Department of Haematology, Christian Medical College, Vellore
Abstract: Wide inter-individual variation in terms of treatment
outcome exists among AML patients receiving chemotherapy with
cytarabine (Ara-C) and daunorubicin. The pre-requisite for the
cytotoxic action of Ara-C is its enzymatic conversion to active tri-
phosphorylated form Ara-CTP. The ex vivo cytotoxicity to Ara-C
in AML patients exhibited a wide inter individual variation, and
were stratified into 3 groups based on IC-50 values
(\5.4 lM, [5.4 lM and [80 lM of Ara-C respectively). DCK
and hENT1 RNA levels were significantly higher in Ara-C
ex vivo sensitive samples compared to those with intermediate
sensitivity and resistant patients. Ara-C resistance index (RI) was
proposed based on candidate gene expression data where,
RI = DCT (dCK 9 ENT1)/DCT CDA. RI values were signifi-
cantly higher in resistant and intermediately sensitive compared to
sensitive samples (median 5.428 and 5.278 vs. 3.475; p \ 0.0001).
The median RI for patients who presented at relapse was signif-
icantly higher compared to that of ex vivo sensitive and resistant
samples at diagnosis. Molecular markers including FLT3-ITD,
NPM mutation, AML-ETO and inv16, as well as MN1, BAALC,
ERG1 and CD34 expression, did not show any significant asso-
ciation with Ara-C RI or IC50. This study suggests RI as an
independent predictor of ex vivo Ara-C sensitivity irrespective of
cytogenetic and molecular risk factors.
Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256 255
123

Abstract P 208
Comparative Evaluation of the Coagulation Parameters Beforeand After Chemotherapy and/Radiotherapy in Solid Tumours
Supriya Dhar1, Swapan Kumar Sinha2, Pranab Kumar Biswas2,Shibashish Bhattacharya3
1Department of Transfusion Medicine, Tata Medical center, Kolkata;2Department of Pathology, Medical College, Kolkata; 3Department of
Oncology, Medical College, Kolkata
Objective: To evaluate the coagulation abnormalities in patients with
solid tumors and changes in their coagulation profile following che-
motherapy and or radiotherapy. Materials and Methods: A
prospective observational study was carried out in the Department of
Pathology, Medical College, and Kolkata. Forty one patients between
15 and 70 years with solid neoplasm were evaluated prior to therapy
and following treatment with radiotherapy and or chemotherapy
during the period between November 2010–November 2011. Results:In our study 14 % of cervical cancer patients showed prolonged PT
and APTT before therapy which normalized after therapy. Around
62 % of breast cancer patients showed prolonged PT and APTT
before therapy. After therapy, 37.5 % showed prolonged PT and 12 %
show shortening of APTT. Around 44 % lung cancer patients showed
pre therapy increased PT and APTT. After therapy both PT and APTT
were shortened in 33.3 and 44 % cases respectively. No coagulation
abnormality was seen in gastric cancer patients. Around 44 % lung
cancer patients had increased FDP levels before therapy which nor-
malised after therapy. In cervical cancer patients 14.2 % showed
increased FDP and all cases normalised after therapy. Plasma FDP
level was normal in all gastric cancer patients. All ovarian cancer
patients had increased pre therapy FDP and all cases normalized with
therapy. 62.5 % breast cancer patient had increased FDP which came
down to 12.5 % after therapy. Conclusions: Thus prolongation of PT
and APTT was seen in many untreated solid tumor patients, after
therapy few normalized, some showed shortening while others remain
prolonged. Pre therapy there was a 34 % increase in FDP which came
down to 2.4 % after therapy.
Abstract P 209
Indian MDS: Do Molecular Abnormalities Alter its Biology:AIIMS Experience
Rekha Chaubey, Sudha Sazawal, Manoranjan Mahapatra,Renu Saxena
Department of Hematology, All India Institute of Medical Sciences,
New Delhi-110029
Background: MDS in the Western countries are mainly found in the
elderly population but is being increasingly seen in young adults in
India. Also, the Indian patients are more severe at presentation and the
response to treatment in these patients is poorer than West. There may
be some molecular factors that may be responsible for the poor
prognosis of these patients. The present study is an attempt to look for
its effect on biology of Indian patients. Aim: To study the effect of
molecular factors on the biology of Indian MDS patients. Materialand Methods: Cytogentics, molecular mutations of RAS and FLT3
gene, hTERT expression, telomerase activity and methylation of
TSGs were analyzed in 100 MDS patients and correlated with disease
severity, progression and survival. Results: There were 67 % male
and 33 % female patients with Mean age at presentation 46 years and
median 48 years. The frequency of patients with age \60 years was
high as compared to the patients with age C60 years (75 vs. 25 %).
26/51 (51 %) patients had normal cytogenetics and 25/51 (49 %)
patients had chromosomal abnormalities. The frequency of N-RAS
mutation was 3 % and K-RAS mutation was 9 %. FLT3-LM and
FLT3-TKD-mutations were not observed at all. The increased telo-
merase activity (TA) was found in 17/100 (17 %) cases. hTERT
expression was present in 17/100 (17 %) cases. Forty patients (40 %)
had p15 INK4b gene methylation. Fifty three patients (53 %) had
SOCS-1 gene methylated. Forty three patients (43 %) had FHIT gene
methylated. Fifty eight patients (58 %) showed calcitonin gene
methylation. In multivariate analysis, of all the molecular factors
studied, only p15INK4b gene methylation was found as an important
predictor for progression of disease in MDS patients. Conclusion:The Indian patients showed some difference from the patients from
West in terms of molecular factors like rare chromosomal aberrations,
absence of FLT3 gene mutations, increased telomerase activity cor-
related with increased hTERT expression, and in methylation
pattern.
Abstract P 210: Oral Presentation
Immunohistochemical Profile and Bone Marrow Angiogenesisin Multiple Myeloma with Reference to its Clinical Significance
Sarah Grace Priyadarshini, Debdatta Basu, Rakhee Kar,TK Dutta1
Departments of Pathology and Medicine1, Jawaharlal Institute of
Postgraduate Medical Education and Research, Puducherry
Aims of the Study: To study the histomorphological features of bone
marrow in multiple myeloma and to evaluate the immunohisto-
chemical profile and angiogenesis using a panel of markers including
CD38, kappa and lambda light chain, CD56, Cyclin D1, Ki67 and CD
34 and to correlate immuno-hematological profile with various clin-
ical parameters. Materials and Methods: The study includes 48
cases of myeloma diagnosed over the period of 5 years. The histo-
morphological features like plasma cell morphology, percentage of
plasma cells and pattern of infiltration were studied. Angiogenesis
was assessed by calculating the microvessel density (MVD) using
immunohistochemistry for CD34. Proliferation was assessed using
both Ki67 and CD38 highlighted cells. Immunohistochemistry for
CD56 and Cyclin D1 was also done. Results: A significant associa-
tion was seen between the plasma cell morphology and the pattern of
infiltration (p = 0.0067). The percentage of plasma cells showed a
significant association with the clinical staging. The MVD was sig-
nificantly associated with plasma cell morphology (p = 0.04) as well
as pattern of infiltration (p \ 0.001). The proliferation index was also
significantly associated with the plasma cell morphology (p = 0.003).
Both the Ki67 and MVD showed an increasing trend with clinical
staging and MVD was significantly associated with serum albumin
(p = 0.02). CD56 negativity was associated with circulating plasma
cells and also with higher clinical stage. Cyclin D1 positivity was not
seen in poorly differentiated morphology and the mean Ki67 was
lower in the cyclin D1 positive group. It was also found that the cases
with kappa light chain restriction had significantly less of poorly
differentiated morphology, diffuse pattern of infiltration, lower MVD
and Ki67. Conclusion: The study of bone marrow morphology and
angiogenesis with the aid of immunohistochemistry is useful for
prognosticating patients with multiple myeloma.
256 Indian J Hematol Blood Transfus (Oct-Dec 2012) 28(4):191–256
123




















