
$31.... - MSU Libraries - Michigan State University
239
ll] .. .uhuwvfiwmm .6. s 7.... .v . 7.21:0. «7:. o . .I..~¢»I . 2154‘s a $31.... « ‘0 :11! ' .ll-. .11. .4..U.._J.ua._..‘.na , . “'9 '«‘ 4\1 ‘10.- ..b‘.- n’!‘- z... «a... .o I... - or t.) .1 .11....) ... . . t IIJA. 0WD 5 “kw A"
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of $31.... - MSU Libraries - Michigan State University

ll]
...uhuwvfiwmm
.6.
s
7....
.v
.7.21:0.
«7:.
o.
.I..~¢»I
.2154‘s
a
H...
.
v.
4‘.
$31....
«
‘0:11! '
.ll-.
.11.
.4..U.._J.ua._..‘.na
, .
“'9
'«‘
4\1‘10.-
..b‘.-
n’!‘-
z...
«a...
.o
I...-or
t.)
.1.11....)
...
..
t
IIJA.
0WD
5
“kw
A"

’F—1m
SITY LIBRAARIE
llllllllll‘llllllllllllllllllllllllll I l lolelllll3 129300
This is to certify that the
dissertation entitled
MULTI ELECTRON PHOTOCHEMI STRY OF
QUADRUPLY BONDED BINUCLEAR COMPLEXES
presented by
Colleen Marie Partigianoni
has been accepted towards fulfillment
of the requirements for
PhoDo degreein ChemiStry
)000 [ADC/‘—Major professor
Date July 30, 1991
MSU is an Affirmative Action/Equal Opportunity Institution 0-12771 i
i i a w ,, ,s V, ,_ ._ ,_ _V , ,7 i , , 7 , _ _ fl ,7 7 _ fi_

PLACE IN RETURN BOX to remove this checkout from your record.
TO AVOID FINES return on or before date due.
DATE DUE DATE DUE DATE DUE
:l
j
Liv—'10:]MSU Is An Affirmative Action/Equal Opportunity Institution
chnS-nt

MULTIEIECTRONPHOTOCHEMISTRYOF QUADRUPLYBONDED
BINUCLEARCOMPLEXES
It
Colleen Marie Partigianoni
ADISSERTATION
submittedto
Michigan State University
in partial fulfillment ofthe requirement
for the degree of
DOCTOROFPHILOSOPHY
DepartmentofChemistry
1991

ABSTRACT
MULTIELECTRONPHOTOCHEMISTRYOF QUADRUPLYBONDED
BINUCLEARCOMPLEXES
by
ColleenMariePartigianoni
Guidelines for the development of multielectron excited state
chemistry of quadruply bonded binuclear complexes (M-LM) are rendered
from the study of a specific class of these complexes comprised of Mo(II)
and W(II) centers ligated by four chloride donors and either monodentate
(PR3) or bidentate (PP) ligands: M2014(PP)2 and M2C14(PR3)4. The mixed
valence character of the metal-to-metal charge transfer excited states of
these complexes, coupled with the structural flexibility of their ligation
sphere gives way to rich photophysics and photochemistry. Transient
absorption studies indicate that these complexes undergo structural
rearrangements upon metal—to—metal charge transfer excitation.
Specifically a bioctahedral distortion which ensures an octahedral
geometry about the oxidized metal center of the excited state is observed.
This rearrangement further provides cooperative stabilization and
coordinative unsaturation of the reduced metal center. These latter
features may be the crucial factors that enable the photochemical
multielectron transformations of the M-LM cores. Particularly intrigueing
is the direct addition of CH31 to the bimetallic core of electronically excited

ColleenMarie Partigianoni
W2014(dppm)2. This photochemistry is unique because photoproducts that
are a signature of overall multielectron transformations proceeding by
sequential one—electron transfer reactions are not observed for this system.
In short, the framework for the design of multielectron
photochemical schemes of quadruply bonded complexes to arise from these
studies directly parallels that established for the ground state reactivity of
square planar ML4 monomers containing d8 08(0), Ir(I) and Pt(II) metal
centers. Namely, low valent coordinatively unsaturated redox active metal
centers best facilitate oxidative addition of substrates. The subtle interplay
of chemical and electronic structure that appears to be required for
formation of photoinduced transients with these ideal features is presented.

To my family
forbeingwithmeeverystep oftheway

ACKNOWIEDGEMENTS
First and foremost I'd like to thank Dr. Daniel G. Nocera (how's that
for respect) for his support, guidance, and for taking the time to foster my
scientific growth during each and every of the step of my graduate career.
Dan's scientific training has taught me to look for the big picture, and his
support, enthusiasm and concern for students as individuals are qualities
to be emulated during my career. Most of all, I'd like thank him for being
one of the few research advisors who could tolerate me for five years.
I'd like to thank the present Nocera group for the memorable
celebrations and their support, especially during those stressful times,
when I needed it the most. Best wishes to all of you; and believe or not,
there is some truth to Dan's claim that "your graduate school years are the
best times of your life." I'd like to leave a special regard for my fellow
psychotic excited state chemist, Janice, but I can't find the appropriate
words to express my feelings (besides they wouldn't pass the censor.) I'm
so fortunate to have her little marks permanently documented in my lab
notebooks as a memoir. All who know her will agree, she's certainly one of
a kind; (could the world handle two??) I haven't forgotten the former
endeared "assholes" of the group, Bob Mussel], Randy King, I-Jy Chang,
Mark Newsham and Joel Dulebohn, who provided inspiration, training
and lots of life to the lab during my early years. The lab just wasn't the
same without them.
I am especially grateful to Claudia Turro for spending innumerable
days collecting that ”just one last" transient absorption spectrum over one
hundred times. I am greatly indebted to Claudia and the rest of the "rescue

squad", SuHane Chen, Jeong—a Yu, and Yeung Shin, for their help
during those last minute crunches.
The MSU College of Natural Science and Dow Chemicals are
recognized for their financial support. I was most impressed with the
outstanding moral support I have felt from the MSU Department of
Chemistry as a whole. In particular, I appreciate the support of Dr.
Dunbar, who not only served as my second reader, but shared numerous
helpful and insightful scientific discussions throughout my graduate
years. Of course, I can't forget the finest glassblowers in the US, Manfred
Langer, Scott Bankroff, and Keki Mistry, who definitely aim to please.
Nobody does it better.
Then there are those who have helped me maintain my sanity
through it all (or tried anyway). I'm grateful for the special and lasting
friendships I've made during the graduate study, especially those with Sue-
Jane, Janice, Brenda, Yeung, the Noceras, Claudia and all of my St.
John's family, especially Fr. Mark, Patrick Patterson, Dan Boyer, Chuck
Graff, Anne Curie, and Cindy Novak, who provided me with a home away
from home and many memories that will last forever.
Finally thanks a million to those back at home, whose contribution
extends beyond the past five years, esmcially Liz Kopp and the Radmores.
Needless to say, the once mentors and soon to be colleagues at good ol‘
Ithaca College Department of Chemistry have made a large contribution to
my career, (greater than I could have hoped for.) Thanks for having me
back on board, '63 a dream come true.
Most of all I want to thank Kelly, Jamie, Kathy, Pam, Mom, Dad and
Gram, for never expecting and wanting anything more from me than my
happiness. With your steadfast love support and love.............. WE DID IT!

TABLE OFCONTENTS
LIST OF TABLES ...................................................................
LIST OF FIGURES .................................................................
CHAPTER I INTRODUCTION ...........................................
CHAPTER II EXPERIMENTAL ...........................................
A. Solvent Purification ...................................
1. Solvents used for Synthesis ....................
2. Solvents used for Spectroscopy and
Photochemistry ....................................
B. Synthesis ..................................................
1. General Procedures ..............................
2. Synthesis of M2014(PR3)4 Complexes ...........
a. Precursors ......................................
i. W014 ............................................
ii. Mo(na-PhPMePhXPMePh2)3 ...........
iii. WCl4(PPh3)2 .................................
b. Dimolybedum Complexes ..................
i. D2d Isomer ....................................
ii. Green Isomer of M02014(PMePh2)4
c. Ditungsten Complexes .......................
d. MoWCl4(PMePh2)4 ............................
3. Synthesis of M2014(PP)2 (Dzh) Complexes

a. M02Cl4(dppm)2 and MegCl4(dmpm)2 34
b. W2Cl4(dppm)2 .................................. 34
c. MoWCl4(dppm)2 .............................. 35
4. Synthesis ofMZCI4(PR3)4X2 and
MzCl4(PP)2X2 Complexes ....................... 35
a. W2015(PR3)4 (PR3 = PEt3, PBu3) .......... 35
b. W2014(dppm)212 ............................... 35
c. W2015(dppm)2 .................................. 37
. Photochemistry ......................................... 37
1. General Procedures .............................. 37
2. Isolation and Photoproducts ................... 38
a. Photolysis of W2014(dppm)2 with CH3I 38
b. Photolysis ofW2014(PBu3)4 with PhSSPh 40
c . Photolysis ofW2Cl4(PBu3)4 with CH2012 40
. Electrochemistry ....................................... 40
1. General Procedures .............................. 4O
2. Preparation and Purification of Electrolytes 41
3. Bulk Electrolysis ofW204(PBu3)4 ........... 41
. Spectroscopic Instrumentation and Methods 42
1. Electronic Absorption Spectroscopy ............. 42
2. Steady-State Luminescence Experiments 43
3. Transient Absorption Spectroscopy .......... 44
4. Electron Paramagnetic Resonance .......... 44
5. Nuclear Magnetic Resonance ................. 44
6. Mass Spectrometry ................................ 44

CHAPTER III TRANSIENT ABSORPTION SPECTROSCOPY ..... 53
A. Background ............................................... 53
B. Results and Discussion ............................... 63
1. M2014(PP)2 (D211) Complexes ................... 63
a. Photochemically Inert Solutions ......... 63
b. Photochemically Active Solutions ........ 82
2. M2014(PR3)4 (ng) Complexes .................. 92
3. MozCl4(PMePh2)4 (D21, / Dzd) Isomers ...... 122
CHAPTER N PHOTOINDUCED REDOX CHEMISTRY ........... 127
A. Background ............................................... 128
B. Results ..................................................... 13)
1. Photo-oxidation Chemistry Accompanied
by Phosphine Displacement .................... 130
a. Photoreaction ofWzCl4(PBu3)4
with CH2012 ..................................... 130
b. Photoreaction of M02014(PBu3)4
with PhSSPh .................................... 142
2. Photo-oxidation Accompanied
by Disproportionation ............................. 149
3. On'dative Addition Reaction .................... 170
C. Discussion ................................................ 185
D. Conclusion ................................................ 188
CHAPTER V. FINAL REMARKS ........................................... 192
REFERENCES ........................................................................ 201

LIST OFTABLES
Table 1. Properties of the Luminescent 1(66*) State of M2X4(PR3)4
Table II. Comparison of Structural Properties of M2X4(PR3)4
Complexes and Lifetime of Nonluminescent Transient

LIST OF FIGURES
Latimer diagram for a transition metal complexes M, depicting
the relationship among the 0—0 transition energy (EM) and the
ground state (E0) and excited state (E') redox potentials described
in equations 1.1 and 1.2. ....................................................
Schematic diagram of a water splitting cycle utilizing
electronically excited Ru(bpy)32+ and relay molecules
methylviologen (MV)2"' and EDTA to transfer one—electron
equivalents to the Pt and Rqu catalysts. .............................
Reaction cycle for the conversion of isopropanol to acetone and
hydrogen with electronically excited Pt2(POP)4“ as a photo-
catalyst (Reference 62c). ....................................................
Relative energies of the d-derived molecular orbitals in a D4,,
binuclear M2L3 complex constructed from linear combination of
two ML4 fragments. ...........................................................
Relative energies of the lowest electronic states of M-LM
complexes as a function of torsional angle (t) or dxy orbital
overlap. The pictorial representation of the valence bond
description of these states as well as the corresponding
molecular orbital formalism is shown (Reference 137). ..........
xi

10.
Newman projection of M02X4(PP)2 complexes depicting the
torsional angle ¢. ...............................................................
(a) Electronic absorption spectrum of a finely ground state
sample of the edge—sharing bioctahedral complex W2016(PEt3)4.
(b) Electronic absorption spectral changes associated with the
conversion of W2016(PEt3)3 to the confacial bioctahedral complex
W2016(PEt3)3 in toluene solution at ambient temperature, In,”
nm (e M-1 cm-l) (a) W2016(PEt3)4: Am = 470 nm, (e = 1992 M-1
cm'l); 380 nm (1280 M-1 cm’l) (b) W2016(PEt3)3: Am = 510 nm,
(e = 2077 M-1 curl); 328 run (2078 M-1 curl). ........................
Electronic absorption spectrum of dichloromethane solutions of
the confacial bioctahedral complex W2016(PBu3)3, km“ = 500
nm, (e = 1970 M-1 cm-l); 315 run (2268 M-1 cm-l). .................
Positive fast atom bombardment mass spectrum of a product
from the thermal reaction of W2014(dppm)2 with 12. The
clusters centered at 1531, 1497 and 1405 amu are consistent with
(a) WzCl4(dppm)2lzt, (b) W2 C l 3( d p p m ) I 2 + and (c)
WZCl4(dppm)zl+, respectively. ............................................
Electronic absorption spectrum W2(II,III)C14(PBu3)4+ prepared
by bulk electrolysis in CH2012 in the (a) visible region and (b)
xii

11.
12.
13.
14.
15.
16.
NIR region. km, = 388 nm, (e = 2559 M-1 curl); 485 nm (949 M-1
cm’l); 1510 nm (2243 m-1 cm‘l). .........................................
(a) D26 structure of MZCl4(PR3)4 complexes; (1)) D21. structure of
MzCl4(PP)2 complexes where PP = bidentate phosphines. ......
Relative energies of the d—derived molecular orbitals in
M2014(PR3)4 complexes as a function of torsional angle (1)
(Reference 161). .................................................................
Electronic absorption spectra of dichloromethane solutions of
M2C14(dppm)2, where M = M0 ( ) and W (------) (a)
MozCl4(dppm)2: In,“ = 634 nm, (e = 2490 M‘1 cm‘l); 462 nm (900
M-1 cm-l); 364 run (2040 M-1 cm-l); 325 am (5600 M-1 cm-l) (b)
W1;Cl4(dppm)2:l.mIn = 710 nm, (e = 2585 M-1 cm-l); 500 nm (598
M’1 cm'l); 405 run (1923 M-1 curl); 325 run (2948 M‘1 cm'l). .....
Transient difference spectrum of M02014(dppm)2 in CH2012
recorded after 355 nm laser excitation. ...............................
Transient kinetics for W2014(dppm)2 in CsHs (1.95 x 10" M)
recorded at 440 nm following laser excitation at 532 nm. .......
Transient difference spectra of WZCI4(dppm)2 in CGHB recorded
recorded 100 ns and 4 us after 532 nm laser excitation (see
legend). ............................................................................
P889

17.
18.
19.
21.
Electronic absorption spectrum of dichloromethane solutions of
W2016(dppm)2, km“ = 822 nm, (e = 740 M‘1 cm‘l); 468 run (4800
m-1 curl); 387 an (3200 M-1 cm'l) (Reference 166). ..............
Electronic absorption spectrum of dichloromethane solutions of
M02016(dppm)2. .................................................................
Proposed edge—sharing bioctahedral distortion of the 1(n5“‘) (or
1(81t"')) excited state of the M2014(PP)2 complexes. Although the
former is designated in the diagram, the high energy metal
localized transitions of these complexes have not been
definitively assigned (see text). Transients are not observed from
the 1(86") excited state on the nanosecond time scale. The metal
centers which are oxidized and reduced in the transient species,
relative to the ground state, are denoted with + and —,
respectively. (See footnote 180 regarding M-«M notation). .......
Transient kinetics recorded at 440 am following 532 nm laser
excitation of (a) W2Cl4(dppm)2 (1.5 x 10" M) in 01131 (5.0
M)/C,H6. (b) W2014(dppm)2 (2.75 x 10“ M) in 011301121 (5.65
M)/CGH¢ (c) W2C14(dppm)g (3.0 x 10" M) in 011,012. ...............
Transient difl'erence spectrum of W2014(dppm)2 (1.5 x 10" M) in
CH31(5.0 MYCQHG in Cefie recorded 100 ns and 4 us after 532 nm
laser excitation (see legend). ...............................................
xiv
P88

. Transient difference spectrum of dichloromethane solutions of
W2014(dppm)2 (3.0 x 10‘11 M) collected 100 us after 532 nm laser
excitation. ........................................................................
. Transient difference spectrum upon 532 nm laser excitation of
W2C14(dppm)2 (2.75 x 10" M) in CH30H21 (5.65 M)/C,;H3 recorded
(a) 100 us after excitation (b) 10 us afier excitation. ................
. Emission of W2014(PMePh2)4 in toluene solution at ambient
temperature upon 691.5 nm excitation. ...............................
. Transient difference spectrum of W201‘(PHPh2)4 in CH2012 (~1.0
x 10'2 M) recorded 2 ps after 590 nm laser excitation. ...........
. Transient difi‘erence spectrum of W2014(PMePh2)4 in THF (~ 6.0
mmol) recorded 70 us after 683 nm laser excitation. ............
. Transient difference spectrum of WZCIJPBua)‘ in hexane (~ 6.0
mmol) recorded 70 us after 580 nm laser excitation. ............
. Transient difi'erence spectrum of W201‘(PEt3)4 in toluene (~ 6.0
mmol) recorded 70 us after 683 nm laser excitation. .............
the confacial bioctahedral complex WzCls(PEt3)3, In“ = 510 nm,
(e = 2077 M“ coo-1); 328 nm (2077 M-1 an“). ........................
XV
P820
101
KB
. Electronic absorption spectrum of a dichloromethane solution of _
109

30.
31.
Electronic absorption spectrum of the product obtained from
thermal reaction of one half molar equivalent of dichloroiodo—
benzene to angel, solutions 0fw2Cl4(P3113)4, An,“ = 500 nm, (e =
969 M-1 arr-1); 400 nm (588 M-1 coo-1); 330 on (3322 M-1 cm'l).
Proposed confacial bioctahedral distortion of the 1(88") excited
state for WZCl4(PR3)4 complexes. Transients are not observed
from the LMCT state on the nanosecond time scale. The metal
centers which are oxidized and reduced in the transient species,
relative to the ground state, are denoted with + and —,
respectively. (See footnote 180 regarding the M-«M notation).
Electronic absorption spectral changes accompanying addition
of dichloroiodobenzene to CH2C12 solutions of W2014(PBu3)4,
1...... =665 nm, (e = 3813 M-1 crn-l) in the (a) visible region and (b)
NIR region. The solid lines represent net addition of 0.5
equivalent and the dashed lines represent further addition.
Plot of phosphine v(CO) stetch vs cone angle of phosphines (a)
PBus, (b) PEta, (c) PMes, (d) PMCzPh, (e) PMOPhg, (0 PHth.
Photoinduced chemical intermediates are not observed from
M02014(PR3)4 where PR9 = PMe3 and PHth , which are marked
with *. ............................................................................
xvi
111
114
116

37.
Electronic absorption spectrum of THF solutions of D2 d
M02Cl4(PMePh2)4 (—) and green isomer of M02014(PMePh2)4 (--
--) that is proposed to have D2,, configuation of phosphines.
Transient difference spectrum of the green isomer of
M02014(PMePh2)4 in THF collected 1 us after 355 nm laser
excitation. .......................................................................
Electronic absorption spectral changes during photolysis (km >
375 nm) of deoxygenated dichloromethane solutions of
W2014(PBu3)‘ at 22°C in the (a) visible region and (b) NIR region.
The absorbance range in the NIR region at I > 800 nm is
expanded by a factor of two. The total time of photolysis was 20
minutes. ..........................................................................
X—band (9.598 GHz) EPR spectrum of a 2—MeTHF/CH2012 glass
ofphotolyzed CH2012 solutions ofW2C14(PBu3)4 at 77°K. ..........
Cyclic voltammograms of 0112012 solutions with 0.1M TBAPFG
of(a) Wzm4(PBu3)4 (b) w.,(:14(1>13113)4 and 3.2 x 10-2 M THACl (c)
photolyzed solutions of WZCI4(PBu3)4. ..................................
(a)1H and (b) 31P NMR smctra of products from photoreactions of
W204(PBu3)4 with 011,012. The spectra were recorded in CD202
solutions at -80 °C. ...........................................................
xvii

40.
41.
Electronic absorption spectral changes during thermal
reactions of benzene solutions of M02014(PBu3)4 containing a ten
fold excess of PhSSPh. The total reaction time at ambient
temperature was four hours. .............................................
(a) Electronic absorption spectral changes during photolysis
(1.,“ > 570 nm) of benzene solutions of M02014(PBu3)4 containing
a tenfold excess of PhSSPh. The total reaction time at ambient
temperature was 20 minutes. (b) Electronic absorption
spectrum of a photoproduct isolated from photolyzed solutions by
column chromography. The FABMS of this product is provided
in Figure 42. ....................................................................
Fast atom bombardment mass spectrum of a photoproduct
isolated from photolyzed solutions of M02014(PBu3)4 containing
PhSSPh. Selected assignments of the clusters in the spectrum
are: (a) M02014(PBu3)2(SPh)4t (b) M0201‘(PBu3)2(SPh)3+ (c)
M02C14(PBu3)(SPh)4+ (d) M02014(PBu3)2(SPh)4t. ...................
Electronic absorption spectral changes during photolysis (2.,“c >
570 nm) of dichloromethane solutions of M02014(PMe2Ph)‘
containing a ten fold excess of PhSSPh at 22°C. The total
reaction time at ambient temperature was 24 hours. ............
xviii
Pace
144
146
148

47.
49.
Fast atom bombardment mass spectrum of a photoproduct
isolated from photoreaction of MmCMPMezPh)‘ with PhSSPh.
The cluster is consistent with Mo,Clg(PMe2Ph)‘(SPh). ..........
Electronic absorption spectral changes during photolysis (1.“ >
435 nm) of acetone solution of Mogcl4(dppm)2 containing a forty
fold excess of TolSSTol at 22°C. ..........................................
Fast atom bombardment mass spectrum of photoproducts
isolated from photoreactions of Mogcl4(dppm)2 with TolSSTol.
Selected assignments of the clusters in the spectrum are (a)
[Mo]Cl4(STol)2t; (b) [MolCla(STol)2+; (e) [MolCls(STol)t; (d)
[Mo]Cl,(s'ro1)+; (e) [Mo]le where [Mo] = M02(dppm)2. .......
Electronic absorption spectral changes during photolysis (1.“ >
435 nm) of ethyl iodide solutions of W2014(dppm)2 at 0°C. The
total reaction time was 1.5 hours. .......................................
Fast atom bombardment mass spectrum of products isolated
fiom photolyzed ethyl iodide solutions of W2014(dppm)2. Selected
assignments of the clusters in the spectrum are (a) [WJCl4lzt;
(b) [W]Clsla"’; (c) [W]Cl;It; (d) may; where [W] =
W2(dppm)2. .......................................................................
Molecular ion cluster region of the fast atom bombardment mass
spectra of photolyzed (Ln. > 435 nm) solutions of
xix
161
163

51.
52.
M02014(dppm)2 in the presence of phenyl/tolyl disulfide
mixtures. Assignments of the clusters in the spectrum are (a)
MogCl4(dppm)2(SPh)2 (b) M02014(dppm)g(SPh)(STol) and (c)
Mogcl4(dppm)2(STol)g. ........................................................
Cyclic voltammograms of 1:1 CH3Utoluene solutions containing
0.1 M THAPF, of(a) wzolgdpprn)2 (h) waolgdmnn)2 (1.0 x 104)
and THAI (5.0 x 10-3 M) (c) WgCl4(dppm)2(I)2. .....................
Electronic absorption spectral changes during photolysis (km. >
435 nm) of methyl iodide solutions of WzCl4(dppm)2 at 0°C. The
wavelength scale in the near infrared region (3. = 900—1000 nm)
is twice that of the visible region (I < 900 nm). ......................
Fast atom bombardment mass spectra of (A) photolyzed (21,xc >
436 nm) solutions of W2014(dppm)2 and methyl iodide and of (B)
solutions of WZCI4(dppm)2 and methyl iodide refluxed in the
absence of light. Selected assignments of the clusters in the
spectrum are: (a) [W]Cl4CH3It; (b) [WIClsCH31+; (c)
(17101201135; ((1) [WlCltCH3+; (e) [W101412t; (t) [W1C1312+; (g)
[W]C151t; and (h) [WIC14I+ where [W] = W2(dppm)3. ...........
The relative isotopic distribution of the molecular ion cluster for
chl‘(dppm)2(CH3XI). The simulated relative abundances,
designated with solid lines, are superimposed on the observed
peaks. .............................................................................
XX
165
1%
172
174

57.
13C NMR of photoproduct from photolyzed solutions .of 1/1
“CI-1311126131 solutions ofW2C14(dppm)2, in 0112012 at -60°C.
Electronic absorption spectra of 01131 solutions of W2014(dppm)2
before (—) and alter (----) photolysis at A > 335 nm at 0°C. ......
Electronic absorption spectral changes observed upon refluxing
CHsl solutions of W2014(dppm)2. .........................................
Electronic absorption spectral changes during photolysis (1,“ >
405 nm) of CHal solutions of MoWCl4(dppm)2 at 0°C. No further
change in the spectra were noted alter an additional 0.5 hour of
irradiation. .....................................................................
Transient absorption kinetics recorded at 390 nm following 532
nm laser excitation of hexane solutions of displaying (a) the 120
ns transient (b) the initial rise and relative intensity of an
additional transient absorption (c) the rise and decay of the
additional long lived transient. ..........................................
Electronic absorption ofM02014(PBu3)‘ (—). M02C1¢(dppm)2 (~-
-) and irradiated solutions ofMo,Ch(PBu3)4 containing a 100 fold
excess of CH3N(PF2)2 (----- ). ................................................
xxi
179

CHAPTER I
INTRODUCTION
Both biological and chemical energy conversion processes typically
involve multiple oxidation-reduction transformations. Beyond an emcient
means of electron transport, the success of carrying out these
transformations is predicated on the ability to overcome the large kinetic
and/or thermodynamic barriers which confront these reactions. To this
end, electronically excited transition metal complexes are useful catalysts
in these transformations [1-3] because the increased driving force
garnered from an electronically excited state provides the impetus for
surmounting the large barriers confronting the corresponding ground
state species. Momover, electronically excited states are particularly useful
in redox reactions because the excited state is both a stronger oxidant and
redth than the ground state species.
The enhanced oxidation potential results from the promotion of an
electron to a higher energy orbital upon excitation. Likewise, owing to the
resultant hole produced in the orbital from which the electron was
promoted, the excited state is easier to reduce as well. Quantitative

2
comparision of the relative reduction potentials of the excited state and
ground state species are given by equations 1 and 2, where Eo_o(M—M*) is
defined as the spectroscopic energy of the 0—0 transition. The Latimer
diagram shown in Figure 1 illustrates the simple thermodynamic
relationship described by equations 1 and 2.
E*,ed(M*/M') = E°nd(M/M‘) + Eo-o(M/M*) (1.1)
13*” (M+/M*) = E°o,(M*/M) — Eo_o(M/M") (1.2)
Although numerous investigations during the past two decades have
proven single electron transfer to be an ubiquitous pathway of electronically
excited transition metal complexes [4—7], the capacity for multielectron
transfer which is ultimately required for energy conversion schemes is far
less common. Ingenious schemes have been designed to effect an overall
multielectron transformations by coupling successive excited state one-
electron transfers via relay molecules and catalysts [8-13], This is
exemplified by the classic water-splitting cycle of
tris(bipyridyl)ruthenium(II). Ru(bpy)32+, shown in Figure 2 [ll-13]. In this
scheme, electronically excited Ru(bpy)32"' ion transfers an electron to W“
which relays the electron to the platinum catalyst. The platinum catalyst
then couples the one-electron chemistry of MV"' to the two-electron
hydrogen production chemistry by effectively storing the reducing
equivalents of the viologen. Oxygen production is achieved from the
reducing equivalent of the photogenerated oxidant Ru(bpy)33+, which is
reduced back to mutiny);2+ by EDTA. The oxidized EDT/1+ reacts with H20
in the presence ofRu02 catalyst to produce oxygen.

Figure 1. Latimer diagram for a transition metal complexes M,
depicting the relationship among the 0—0 transition energy (EM) and the
ground state (E0) and excited state (E') redox potentials described in
equations 1.1 and 1.2.

Figune2. Schematic diagram of a water splitting cycle utilizing
electronically excited Ru(bpy)32"' and relay molecules methylviologen
(MV)2+ and EDTA to transfer one—electron equivalents to the Pt and Ruo2
catalysts.

hv
450nm
/\
Ru
2’
R2"
H20
EDTA
(130103
“(Willa
MV'
"2H2
114()2
EDTA.
Ru(bpy)33’
MV2+
H+
“@192

7
An alternative approach to multielectron chemistry predicated on
one—electron transfer is to mimic biological assemblies such as the
photosynthetic reaction center. The overall mechanism of the
photosynthetic assembly is not unlike that described for the Ru(bpy)32+
system in that electrons and holes resulting from the single electron
transfer reactions of the light harvester (i.e. porphyrin) are sequentially
transferred to and stored in catalytic centers which are capable of effecting
the overall multielectron transformation upon accumulation of sufficient
charge equivalents [14—22]. The primary difference in biological systems is
that the transport of electrons from the light harvester to the catalytic
center occurs through covalently bonded networks. Intramolecular
electron transport offers the potential for directional electron transport
which is difiicult to achieve in the intermolecular reactions of the relay
molecules used in the Ru(bpy)32+ scheme.
The required intricacy of designing covalently bonded networks
capable of efficient charge transport and storage is formidable. Research is
focused on successfully mimicking components of the overall system
including single electron transfer reactions of porphyrins [23-24], proteins
[25—27] and more generally covalently bonded organic networks [28—32].
Even the primary task of emcient separation of a single electron and hole is
challenging the imagination of several research groups. The inherent
difficulty in the seemingly simple charge separation process is that
recombination reaction of the hole and electron is thermodynamically
favored with respect to their continued evolution along the charge
separating network. In general, a highly exergonic electron transfer
between a donor (D) and acceptor (A) which is driven by the excess energy of
an excited state will yield D+ and A" which are unstable with respect to

8
back electron transfer owing to the predictions of Marcus inverted region
electron transfer [33]. The recombination of the hole and electron is less
favorable as they become spatially separated further due to the inverse
exponential distance dependence of the rate of electron transfer [33—34].
Thus a successful approach to efficient charge separation, which mimics
the photoreaction center [35—38], is the design of covalently bonded networks
which can transport electrons and holes away from the primary reaction
center to independent and far removed sites. ‘
This approach is exemplified by the cofacial diporphyrins [39], and
porphyrin based diads [40—43] and triads [44—45]. The latter consists of a
light harvesting porphyrin (P) and a covalently juxtaposed acceptor (A) and
donor (D). In this case charge separation is achieved by the primary
electron transfer from the electronically excited porphyrins to the acceptor
followed by subsequent electron transfer from the donor to the porphyrin, as
follows.
kfl kfzDP“A"
DP’A D+PA’ (1.3)
The photogenerated charge separated state D+PA’ may persist into the
microsecond range; the quantum yield however is usually relatively low
[46]. Efficient production of charge separated states requires forward
electron transfer rates (kfl, kfz) which are faster than the back electron
transfer rates. Clever ways to control these relative rates through efi'ects of
energy, distance and molecular structure are currently being explored [47-
48].

9
Yet the successful design of charge separating networks represents
only the first step towards the ultimate goal of multielectron
photochemistry. The initial electron]hole pair must then be stored at the
terminus of the network, and the overall process must be repeawd to build
up the necessary multielectron hole and electron equivalents. Finally, the
charge equivalents must then be coupled to a catalytic center capable of
promoting the overall multielectron process. Indeed, the synthesis of such
centers comprises a major research area in its own right with the design of
biomimetic models for nitrogenase [49], the oxygen-evolving complex [50—
51], copper-based proteins [52], cytochrome P—450 [53—55], hemerythrin [56—
57], hydrogenase enzymes [58], and a host of other metal based proteins and
enzymes [59-60].
An alternative approach to multielectron reactivity that does not rely
on independent catalytic centers employs electronically excited polynuclear
metal compounds (where polynuclear defines two or more metals in a
discrete complex). In these systems, transformations can be effected
directly at the electronically excited metal centers. Moreover, multielectron
capacity is offered by the combination of the one—electron chemistry of the
individual metal centers in the polynuclear core. Thus the polynuclear
core represents a self contained multielectron reaction center and therefore
the need to transfer, store and accumulate charge in independent catalytic
centers is obviated. Success with this approach is exemplified by the
plethora of exciwd state reactions of binuclear ds—d8 binuclear complexes
such as Pt2(P205H2)4" (P205H2 a POP) [61—63], Ir2(2,5-diisocyano-2,5-
dimethyl-hexaneh2+ [64] and [Ir(u-pyrazanole)-1,5-cyclooctadiene]2 [65],
which all ultimately effect the two-electron reduction of substrates. For
example, Pt2(POP)4“ photocatalytically converts isopropyl alcohol into

1 0
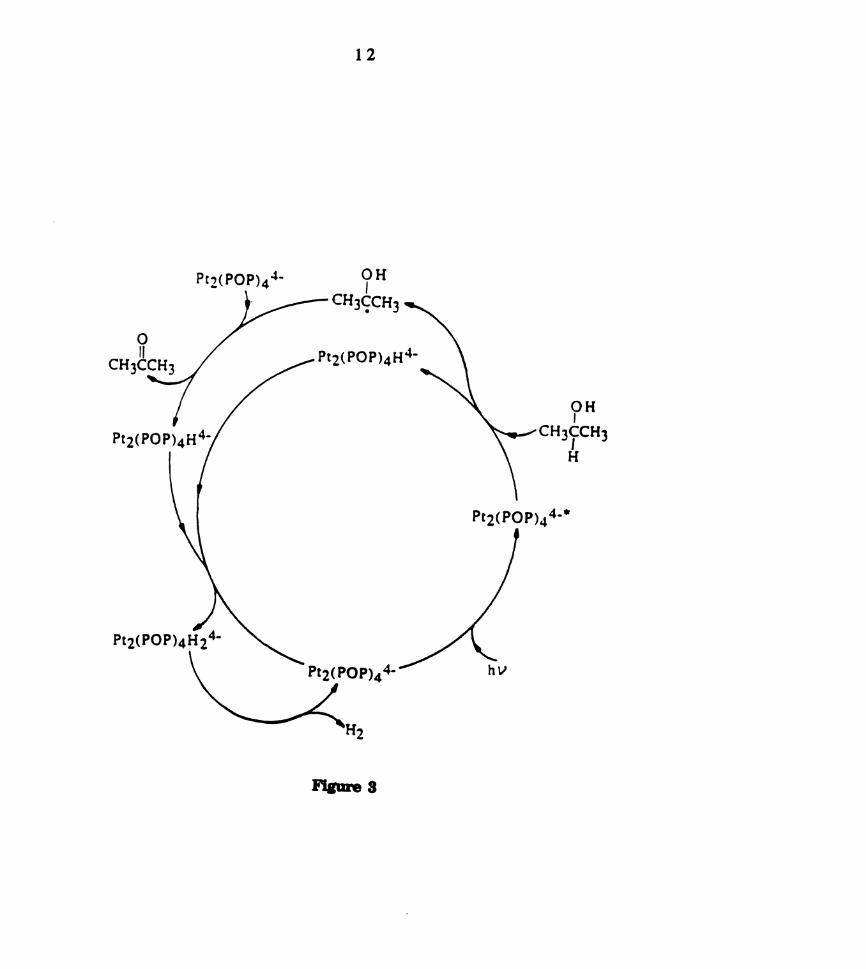
acetone and hydrogen by a pathway shown in Figure 3 [62s]. The primary
step in the photoreaction involves hydrogen atom abstraction by
electronically excited Pt2(POP)4" to give a Pt(II)Pt(II) mixed valence
intermediate Pt2(POP)4H" and the corresponding reactive (CH3)ZCOH
radical. Subsequent reaction of this reactive radical with another
equivalent of Pt2(POP)4" results in production of acetone as well as
Pt2(POP)4H" which can disproportionate to give Pt2(POP)4" and
Pt2(POP)4H24‘. Reductive elimination of H2 from the latter complex
completes the cycle.
The primary process of this reaction, namely hydrogen atom
abstraction, is exemplary of numerous other photoreactions of Pt2(POP)4"
with a wide variety of organic substrates R3EH for E = Sn, Ge, Si, C [62 b,c,
63]. Halide atom abstraction is an additional common reaction of the
electronically excited Pt2(POP)4"'. Recent transient absorption studies have
shown that this is the primary photoprocess in the reactions with
xcnzcnzx (X = Br, Cl) and aryl halides [62 b,d].
Each of the reactions of the electronically excited Pt2(POP)“‘
complexes described above involves an initial one-electron transformation
yielding a reactive organic or organometallic radical which can
subsequently be trapped by either the mixed valence intermediate or in
some cases the Pt2(POP)4"' complex to yield the two-electron reduced
substrate. This appears to be completely general for the photochemistry of
the binuclear da—d8 complexes [66]. Thus, even with the multiple redox
capacitance of the polynuclear core, multielectron transformations are still
confined to coupling one-electron reactions.
Our research is focused on a novel basic reaction type for electronic
excited states that difi‘ers from coupled one—electron schemes, namely a

11
Figure 3. Reaction cycle for the conversion of isopropanol to acetone and
hydrogen with electronically excited Pt2(POP)4" as a photo—catalyst
(Reference 62c).
12
Pr2(POP)44- 9”
' CH3C3CH3
i’ .CH3CCH3 PtthOP)4H
on
CH CCHPt2(POP)4H4‘
3) 3
P12(POP)44"'
Pt2(POP)4l-i24'
Pt2(P0p)44- hv
H2

13
discrete multielectron event. Concerted multielectron chemistry from an
excited state molecule is appealing from a practical standpoint because the
approach avoids competitive side reactions of one-electron intermediates,
including energy—wasting, back electron transfer. More fundamentally,
the ability to initiate a discrete multielectron process with a pulse of laser
light can contribute significantly to the understanding of the mechanisms
of multielectron processes, which are the least understood of any in
chemistry or biology. In particular, discrimination between a concerted
multielectron process and one that proceeds by sequential very rapid one-
electron transfers within a solvent cage is often problematic. This issue
can be addressed by opening the temporal window of multielectron
reactions from their heretofore conventional arena of study in the
millisecond range of stopped-flow kinetics to the picosecond and even
femtosecond range of laser kinetics. Thus the development of excited state
multielectron processes ofl‘ers the opportunity to explore and discover new
fundamental and practical aspects of oxidation—reduction chemistry.
Our initial approach to the development of discrete multielectron
transformations of electronically excited transition metal complexes uses a
photon to promote charge separation in a binuclear core to produce excited
states of the type M‘”—M"°". These excited states retain the two
important features contained in the binuclear ds—d8 complexes, multiple
redox equivalents of the polynuclear core and metal centers that can serve
as catalytic sites, but also includes charge transfer within the polynuclear
core. The latter feature may provide the final impetus required for the
novel discrete multielectron photoreactivity because the two electron mixed
valence character derived from charge separation should promote
multielectron redox reactivity; the M“ and M"+1 metal centers have the

1 4
capacity to serve as two electron donors and two electron acceptors,
respectively.
Two approaches to production of M‘m—M"l electronically excited
species are currently being pursued in our laboratory. The most
conceptually obvious one is the simple excitation of ground state species of
M““—M“'l character:
11 eM‘tl-M“ —"——— MN—Mx-1 (1.4)
To this end, the recently prepared Rh2(0,II) fluorophosphine
complexes are promising candidates [67]. Photophysical studies show that
they have long lived excited states which retain the mixed Rh2(0,II)
character and photochemical studies are currently being initiated. The
Rh2(0,II) complexes however represent a rare example of a
thermodynamically stable two-electron mixed valence species. A more
general approach to production of (MM—M“). excited states via MMCT
charge transfer in M—Mcomplexes:
M-M hv > w+1-w-1‘MMCT (1.5)
This latter approach has an advantage in that it allows for production of
less stable and more reactive M‘*l—M“’l transient species.
The preparation of localized charge-separation (M‘*1—M"1)‘ excited
states by optical excitation requires the transfer of electrons between weakly
coupled orbitals localized on the independent metal centers. Numerous
fundamental studies of MMCT excited states [68-72] have contributed

15
greatly to an understanding of the factors that govern the rate of electron
transfer such as the interconnecting bonding network [73—83], distance
[84—90], solvent [91-98], temperature [99-103] and free energy driving force
[104—110]. However few studies have centered on the photochemistry of
such excited states. The activation of 02 by the mixed valence anions,
[(CN)5M"(p—CN)Com(CN),J‘-, represents a relatively rare example [111].
Excitation of the metal-to-metal charge transfer (MMCT) induces an
intramolecular photoredox event to produce the corresponding
[(NC)5Mm(u-CN)Con(CN)5]6' (M = Ru, Os, Fe) mired valence species,
which undergoes subsequent dissociation to Mm(CN)33" and Con(CN)53'.
The coordinatively unsaturated intermediate is efficiently trapped by
oxygen to generate the peroxo dimer [(CN);Com(022') -Com(CN)5]6', which
submquently decomposes in acidic media to H202 and Com(CN)5(H20)2".
Whereas this is multielectron photochemistry, it reveals a
fundamental problem confi-onting MMCT photochemistry. Owing to the
weak coupling of the metal centers, population of the MMCT state results in
dissociation to monomeric species. Photodissociation of electronically
excited (M'M—M‘dr species can be circumvented by incorporating
bridging bidentate ligands. Alternatively multiply bonded M-LM binuclear
complexes have sufficiently strong metal—metal interactions to prevent
dissociation. Moreover, in the case of the quadruply bonded metal-metal
binuclear complexes (MA-M), the lowest energy transitions of are between
weakly coupled orbitals and hence the lowest energy excited states of these
species exhibit significant MMCT character. To this end, we became
interesmd in exploring the potential multielectron photochemistry ofMJ-M
binuclear complexes.

l 6
Since the initial discovery of 11620132' in 1964 [112], numerous other M
LM complexes comprised primarily of d‘ rhenium(III), chromium(II),
molybdenum(II) and tungsten(II) metal cores with an array of ligands
have been discovered, owing primarily to the efforts of Cotton and
coworkers [113]. The formulation of a quadruple bond was proposed to
account for the unusually short metal-metal bond distance, the
diamagnetic behavior and the eclipsed conformation of the two ML4 units
observed in these complexes. This proposal stimulated numerous
theoretical [114—120] and experimental [121-135] investigations and a self-
consistent description of their electronic structure has emerged in recent
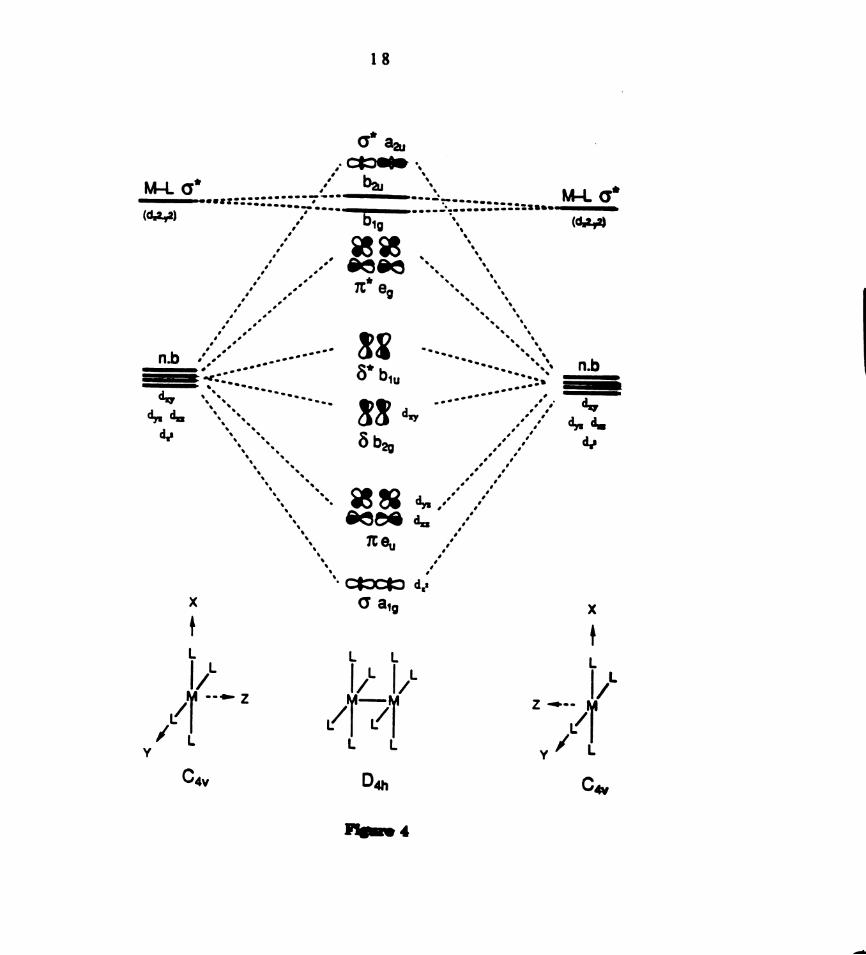
years. The general molecular orbital diagram for M-LM species depicted in
Figure 4 has evolved from these studies. It is constructed by taking the
linear combination of orbitals for two ML4 fragments. Each ML4 fragment
contains one highly destabilized M—L 0‘ molecular orbital, resulting from
interaction of the d,2.y2 orbital with the four ligands. Linear combination of
the M—L 6* orbital in the two M114 fragments gives rise to blg and b2u
molecular orbitals of 8 and 5* symmetry, respectively, which are nearly
degenerate. The M—M bonding interactions in the M-L-M binuclear
complexes arises from the linear combination of d.2, (du, dyg), and d,,
orbitals to form bonding and antibonding o, x, and 8 molecular orbitals,
respectively.
Numerous electronic absorption spectroscopic studies of various
M-‘-M complexes verify that the lowest energy absorption corresponds to the
spin and dipoled allowed 52a1(58’)(1A1‘-)1A2u) transition which is
predimd from this general molecular orbital scheme [136]. The energies of
these transitions are typically in the range of ~15,500 to 25,000 cm“.
Interestingly the 8—95’ transition typically red shifts by 10,000 cm"1 for the

l7
Figure4. Relative energies of the d—derived molecular orbitals in a D4},
binuclear M2L8 complex constructed from Linear combination of two ML4
fragments.
l8
0* am
e
\
M—LO’"
\
\
\be.
I
I
I
“no.
I.008:
M—LO’"
(dB-.1)(Gui-,2)
I'll-.4

l9
one—electron oxidized or reduced species [115]. The significant red shift
suggests that the 8/8‘ orbital splitting in Figure 4 is small owing to poor
overlap of the d” orbitals. This contention is further supporwd by estimates
of 10 kcal for 8 bond strength [115]. Owing to this relatively weak interaction
molecular orbital theory does not provide an accurate physical
representation of the states derived from population of the 8/8‘ orbital
manifold, and two electron exchange energies must be a significant
contributor to the 824'(85‘) transition ofM-LM species. Accordingly, states
derived from the population and depopulation of the 8 orbitals are more
accurately described within a valence bond framework [114—116].
The relative energies and pictorial representation of the valence bond
description of the four states derived from the purely atomic noninteracting
dry orbitals are shown in the right hand site of Figure 5 [137]. The lowest
energy degenerate states (1A13,3A2u) have one electron on each metal
center. These two states correspond to the 3(88’) and ‘(82) states in
molecular orbital formalism. At significantly higher energy are two
degenerate 1A“, 1A2“ states representing the positive and negative linear
combinations of electronic configurations where both electrons are on the
same metal center. These two states are far removed from the lA13,3A2u
states owing to the two-electron exchange energy associated with pairing of
two electrons in the (IU orbital [115]. The energies of these states are
perturbed as the overlap (S) of the d,y increases fiom 0 to the typical value of
0.1 for the M-LM species in the manner also shown in Figure 5. The ‘A13
state corresponding to 1(82) is stabilized and the A13 state corresponding to
1(8“) is destabilized. The energies of the 3A2“ (3(88‘))and 1A2“ (1(88'» states
are roughly invariant over this range.

20
Figure 5. Relative energies of the lowest electronic states of M-i-M
complexes as a function of torsional angle ((0) or dxy orbital overlap. The
pictorial representation of the valence bond description of these states as
well as the corresponding molecular orbital formalism is shown (Reference
137).

(l_UJO) Afiieua
dxyOrbitalOverlap
L4M-ML4TorsionAngle———>
VB
MO
W5
21

22
The valence bond model is distinguished from simple molecular
prbital theory by the following predictions: a small energy gap between the
3A2“ and the ‘AIS ground state, a large energy gap between the 3A2u and the
1A“ state, and the lowest energy spin-dipoled electronic 1A lg—ilAzu
transition (correlating with the 8241(88") transition) involving conversion
from a ground state with a single electron on each metal center to an
excited state with both electrons on a single metal center is MMCT in
character. Each of these predictions of the valence bond model have been
experimentally verified in recent years. Evidence for the relative energy
ordering of states is provided by magnetic susceptibility measurements of
the torsionally strained M02Cl4(dmpe)2 complex [134]. Owing to the steric
contraints of the bridging bidentate dmpe, the two ML4 fragments in this
complex are nearly staggered (o= 40°) with respect to each other as depicted
in Figure 6, and thus the interaction of the d,, orbitals is very near the limit
of zero for the completely staggered configuration (¢= 45"). Indeed, as
predicted from Figure 5, magnetic measurements places the energy of the
thermally populated 3A2“ state only 400-500 cm“1 above the ground state,
whereas the lAlg—i1A2u transition is observed to be at 12,500 cm'l. From
these data and the observed 4700 cm'1 blue shift in the lAlg—ilAzu transition
of M02014(PMe3)4 complex, the 3142u state is predicted to be 5,200 cm'1 above
ground state. The increased energy for M02014(PMe3)4 is predicted owing to
maxium overlap of the dxy orbitals in an eclipsed conformation. Perhaps
the most compelling evidence supporting a valence bond model comes from
the studies of Winkler, Sutin, and their co-workers. Solvent dependence
1(88") luminescence reveal that the dipole moment of the 1A2“ (1(88'»
luminescent electronically excited state of M02014(PMe3)4 is 4.0 Debye (as
compared to the zero dipole moment of the ground state) [138]. Moreover,

23
Figure6. Newman projection of M02X4(PP)2 complexes depicting the
torsional angle (0.

25
time—resolved picosecond emission spectroscopy show the striking result
that the temporal evolution of 1(88”) luminescence occurs on the time-scale
of the microscopic solvent relaxation time [138]. These results closely show
that solvent is an important controlling factor in the dynamics of 1(88")
luminescence, and strongly corroborate a valence bond description of an
ionic excited state in which one metal center is on'dized and the other metal
center is reduced relative to the ground state.
The ionic character of 1(88*) state deems these complexes as logical
candidates for multielectron derived from charge separated excited states.
Beyond the mixed—valence character, additional assets of the 1(88") also
arises from the weak coupling of the dxy orbitals. Unlike the singlet excited
states of most transition metal complexes, the 1(88*) excited state of M-LM
binuclear complexes are sufficiently long-lived (1: ~ 100 ns) in some cases
[118d] to permit bimolecular reactivity [121d, 139—141]. The longevity of the
MMCT state most probably arises from the large 1(88"')-—3(88"') energy gap
which inhibits intersystem crossing. Furthermore, photodissociation of the
metal-metal bond is avoided upon population of 1(88$), since the 8 bond
contributes less than 10% to the overall M—M bond energy. The multiple
bonding in the M-LM complexes provides an array of excited states, and
although few studies have focused on excited states other than the 1(88"),
the valence bond description predicts mixed valence character in additional
states such as 1(1:8'") and 1(8x"') states [114]. These excited states formally
represent a one-electron charge separated state as compared with the
formal two—electron charge separation in the 1(88") excited state.
Irrespective of the formal electron counts, each of the MMCT transitions
yields partial reduction of one metal center and oxidation of the other metal
center relative to the ground state.

26
Studies described herein are focused on investigation of the
multielectron photochemistry of M-LM binuclear complexes M201‘(PR3)4
and M2014(PP)2, whose ligating spheres are comprised of chloride and
either monodentate (PR3) or bidentate (PP) phosphines in D2,, and D2}I
molecular geometries, respectively. A prerequisite for elucidating the
photochemistry of these binuclear complexes is a clear elucidation of the
physical and chemical properties in the excited states. Accordingly,
nanosecond transient absorption spectroscopic studies of the M2014(PR3)4
and M02Cl4(PP)2 complexes are described in Chapter III. These studies
reveal that the photophysics of MMCT states of these complexes is much
richer and more extensive than was heretofore realized. Efforts to develop
the multielectron photochemistry of these transients has centered on two-
electron oxidative addition reactions of mixed-valence excited states. The
observed photoreactions of the D2,! and D2}, complexes are described in
Chapter IV. Mechanisms of multielectron photoreactions of a M-LM
species with a variety of substrates are presented based on transient
absorption and electrochemical studies. Particularly intriguing is the
photoinduced oxidative addition of CH3I to electronically excited
W2Cl4(dppm)2. A framework for the rational design of variable discrete
multielectron schemes of M-LM complexes was formulated from these
studies and is briefly summarized in Chapter V.

CHAPTERII
EXPERIMENTAL
A. SolventPurification
LSolventsusedforSyntheses
All solvents were refluxed under N2 for no less than 8 h and freshly
distilled prior to use. Hexane, cyclohexane, diethyl ether, benzene, toluene
and tetrahydrofuran were refluxed over Na. In the case of the latter three
solvents, small amounts of benzophenone were added to form the blue or
purple ketyl radical anion or dianion, respectively, as an indicator. All
halogenated solvents were refluxed over P205. Methanol was refluxed over
Mg(OMe)2 prepared by initially refluxing 50 mL MeOH containing 5.0 g of
Mg turnings and 0.5 g of I2. Once the color of 12 disappeared, an additional
1 liter ofMeOH was added.
2. SolventsusedbrSpecuoecopyandPhotochemishy
With the exception of CH3I, all solvents used for electrochemical,
spectroscopic and photochemical experiments were spectroscopic grade
27

28
(Burdick and Jackson Laboratories). Each of the solvents was subject to
seven freeze—pump—thaw cycles (10'6 torr) and then vacuum distillamd into
storage flasks equipped with Kontes high-vacuum valves containing
various drying agents. Dichloromethane was stored over Linde 4A
molecular sieves, which had been activated under dynamic vacuum (10'6
torr) at 250 °C for 12 h. Hexane was stored over mixtures of sodium-
potassium alloy. Benzene, toluene, and THF was also stored over sodium-
potassium alloy except in these cases small amounts of benzophene were
used. Owing to the increased water content of THF, it was first vacuum
distilled and predried for two days over Na/benzophenone and finally
transferred into a storage flask containing the Na/K alloy and
benzophenone.
Methyl iodide (Baker Reagent Grade) used for photolysis was
refluxed over P205 and Cu for no less than 12 h. A Vigereux column was
used to further induce condensation of the very low boiling solvent (43 °C).
The doubly distilled solvent was collected in a round bottom flask containing
P205 and Cu, which was subsequently subject to seven freeze-pump—thaw
cycles (10"6 torr). Only freshly distilled solvent was used to avoid 12
impurties which will sublime at these pressures.
BSynthesis
LGeneralProcedures
Synthesis of all complexes were performed using standard Schlenk-
line techniques with rigorously deoxygenated and dried solvents; All
chemicals were reagent grade and were used as received unless otherwise

29
noted. The dppm (dppm = bis(diphenylphosphino)methane) and PBu3
phosphines were purchased from Aldrich Chemical Company, and the
remainder of the phosphines were obtained from Strem Chemicals. The
phosphines were stored under vacuum in flasks equipped with Kontes
high—vacuum valves. The highly reactive dmpm (dmpm a
bis(dimethylphosphino)methane) was dissolved in toluene (1.0 M in dmpm)
for storage and further use. Previously prepared compounds were
characterized by electronic absorption spectroscopy, cyclic voltammetry and
1H NMR spectroscopy.
2. SynthedsofMgCLa’Rggalmplems
aPrecur-sors
1. WCl4 [142]. A mixture ofWClg (9.9 g, 0.025 mol) (Aldrich Chemical
Co.) and W(CO)g (4.4 g, 0.0125 mol) (Aldrich Chemical Co.) in 50 ml of dry
chlorobenzene was refluxed until evolution of CO ceased (~ 12 h). The grey
product was filtered by suction filtration, washed with chlorobenzene, and
subsequently washed with dichloromethane to remove all traces of the
aromatic solvent. The solid was dried and stored under vacuum as
prolonged exposure (~ 1 h) to air results in decomposition.
ii. Mo(n'-PhPMePh)(PMePh,), [143]. This exceedingly moisture
sensitive complex was prepared by literature methods with slight
modifications to minimize the previously reported oligomerization. As
expected, formation of the brown—black oligomers was found to be
concentration and temperature dependent, and therefore reactions were
run under dilute conditions at 0 °C. To a solution of 50 mL of THF
containing 2.3 mL of PMePh2 (12 mmol), 0.65 g of MoC15 (2.38 mmol)

30
(Aldrich Chemical Co.) was dissolved. Untreated Grinard Mg turnings
(1.05 g, 43 mmol) (Mallinckrodt) were slowly added to the solution over a
period of 0.5 h. Numerous intermediates were observed during the
multiple reduction reaction. An initial orange precipitate observed after
the addition of Mg was complete, eventually redissolved with an ensuing
reaction to yield an olive green solution which finally turned brownish
orange. The last step was very exothermic and the heat induced
oligomerization when the temperature was not carefully controlled. Upon
completion of the reaction, the solution was allowed to slowly warm to room
temperature, stirred for an additional 0.5 h, and filtered to remove the Mg.
Complete removal of THF from the filtrate, as suggested in the literature,
was found to result in considerable oligomerization. Alternatively,
precipitation of the orange product from dilute solutions was achieved by
partial removal of the THF, addition of MeOH, and further removal of the
more volatile THF. Oligomer, if present, was removed by redissolving the
product in benzene, filtering and precipitating the pure product with
MeOH.
iii. WCI4(PPh,), [144]. This complex was prepared by reduction of
WClg (Aldrich Chemical Co.) with amalgamated mossy Zn (Baker
Chemical Co.). Mossy Zn (7.5g) was amalgamated by dissolving 0.2 g of red
HgO (Matheson, Coleman and Bell) in 2 mL of 12 M HCl (aq). This solution
was added dr0pwise to 80 mL of H20 containing the mossy Zn. The
amalgamated Zn was filtered, washed with H20 and acetone and dried in
the oven. The addition of 7.5 g of PPh3 (0.0325 mol) (Baker Chemical Co.) to
a saturated 60 mL solution of WCIG (3.75 g, 0.0095 mol) (Aldrich Chemical
Co.) containing 7.5 g Zn mossy (0.115 mol) resulted in immediate

31
precipitation of the yellowish-orange WCl4(PPh3)2. Afier shaking the
reaction mixture periodically for 15 min, the precipitate was filtered and
washed with small aliquots of CH2012. The mossy Zn was separated out
with forceps. Although the complex is quite stable in dry air, a blue
decomposition product is observed when moisture is present. Two triplets
at 8.0 and 8.4 ppm are observed in the 1H NMR of the WCMPPth complex
and the UV-visible spectrum exhibits a single absorption maximum at 420
nm.
h. DimolybedumComplemes
i. D“ learners. The entire series of M02014(PR3)4[145] complexes for
PR3 = PEt3, PBu3, PMezPh, PMeth, and PHth, were prepared by a
standard method from (NH‘)5M02C19-H20 [146]. A complete description of
the synthesis of (NH4)5M02C19-H20 is provided in the doctoral dissertation of
I—J. Chang [147]. Phosphine (4.5 mmol) was added to a suspension of 1
mmol of (NH4)5M02019-H20 in 50 mL of MeOH. The purple solution was
refluxed for 4 h to yield a blue microcrystalline product that was filtered by
suction filtration and washed with H20 to remove unreacted
(NH4)5M02019-H20, MeOH and diethyl ether (except for the soluble PEts and
PBu3 complexes). The complexes were purified by column chromatography
using Florisil as a solid support and CH2012 as the eluant. A blue band was
collected from the column, which retained yellow impurities at its top. The
purified complexes were precipitated by addition of MeOH. The complexes
exhibit very similar absorption profiles with intense absorptions at ~ 600 nm
(8 ~ 3200 M‘lcm‘l) and 330 nm (6 ~ 3000 M'lcm‘l) and a very weak
absorption at ~ 450 nm (8 ~ 250 M'lcm'l) [121 a].

32
11. Green Isomer of Mo,Cl4(PMePh,)4 [148]. The green isomer of
MozCl4(PMePh2)4 was prepared by the reaction of the very moisture
sensitive precursors MoCl,('l'HF)2 and Mo(n‘-PhPMePh)(PMePh2)3. A 30
mL THF slurry containing 400 mg of Mo(ne-PhPMePh)(PMePh2)3 (0.4
mmol) and 170 mg of McCl4(THF)2 (0.4 mmol) was stirred at ambient
temperature for a period of 10 h. The solution was concentrated to ~ 5 mL to
induce precipitation of unreacted MoCl4(THF)2, which was subsequently
filtered 011'. Complete removal of THF was found to result in production of
the blue isomer. The green isomer may be precipitated by addition of
cyclohexane to the filtrate followed by partial removal of the more volatile
THF. Residual of Mool,('rHr)2 and Mo(n“-PhPMePh)(PMePh2)3 can be
removed by column chromatography with Florisil and THF as an eluant.
The first green band was collected and precipitated with cyclohexane. The
UV-visible spectra of both the blue Dad isomer and the green isomer are
provided in Figure 34. The green isomer has a characteristic absorption at
450 run that is not observed in the blue D2,; isomer. Furthermore the ratio of
intensities of the absorption maxima at 600 and 330 nm vary for the two
isomers. It should be noted that the spectra of samples from different
preparations show different ratios of intensities of the absorption maxima
at 330 nm and 600 nm, as expected for varying mixtures of the green and
blue isomer. Furthermore, this ratio for a given sample decreased over a
period of hours indicating that isomerization from the green to blue isomer
might well occur in THF solution.
c. Ditungsten Complexes. The series ofW204(PR.3)4 complexes [142]
for PR3 = PEta, PBu3, PMe3, and PMeth, was prepared by Na reduction of
W014 [143]. Numerous attempts to prepare these complexes with

3 3
commercially available WC], (Aldrich Chemical Co.) were unsuccessful.
The synthesis described here is for the case of PR3 = PBu3. The PEta,
PMePh2 and PMes analogues were prepared in a similar manner, with the
slight variations previously reported in the literature: 2.5 mL of PBus (19
mmol) and 2.6 g ofW01. (8 mmol) were sequentially added to a 60 mL ofcold
THF (—78°C) containing a stoichiometric amount of 0.41% Na amalgam
(0.368 g Na (16 mmol)/ 90g Hg). Approximately 15 min after the reactants
were mixed, the reaction flask was put in an ice bath and vigorously stirred
and shaken periodically as it was allowed to slowly (~1.0 h) warm to room
temperature. The resultant blue green solution, obtained about 0.5 h after
the solution reached room temperature, was carefully decanted from the
amalgam. THF was then completely removed and the product was
extracted with hexane, and filtered to remove grey sideproduct. Complete
removal of hexane from the filtrate yielded a dark green-black oily residue.
The blue powder obtained upon addition of MeOH, was collected by suction
filtration and washed extensively with MeOH. The complex is stable
enough to be worked up in air, however prolonged exposure to air will
result in decomposition.
The complex was purified by column chromatography with Kieselgel
60 (EM Science) as a solid support and CH2012 as an eluant. The absorption
spectrum exhibits two intense maxima at 665 (e = 3813 M'lcm‘l) and 303
nm (e = 8933 M‘lcm’l) as well as a very weak absorption at 495 nm (e = 290
M‘lcm'l)
d. MoWCl,(PMePh,), [148]. A 15 mL 06H, solution of Mo(n“-
PhPMePh)(PMePh2)3 (100 mg, 0.1 mmol) was added dropwise over a 15 min
period to a suspension of WCl4(PPh3)2 (250 mg, 0.3 mmol) in 20 mL of CgHg.

34
After ~ 45 min, the green solution was concentrated to ~ 5 mL to induce
precipitation ofWCl‘(PPh3)2, and filtered to remove the latter.‘ The benzene
was entirely removed and a minimal amount (~ 3 mL) ofTHF was added to
yield a saturated solution followed by addition of 15 mL of MeOH. A
turquoise precipitate formed after 1 h at 0 °C was filtered and washed with
MeOH. The absorption spectrum exhibits maxima at 650 nm (e = 2609 M“
1cum-1), 460 nm (e = 394 M-lcm-l) and 320 nm (e a 7133 M-lcm-l). *(It should
be noted that the final olive green precipitate collected from the filtrate of
more dilute solutions contained significant amounts of WCl4(PPh3)2, as
determined by 1H NMR).
3. Synmsshormmmplms
a. MogCI..(dppm), and MogCLklmpm), [149-150]. The dimolybdenum
complexes, M02014(dppm)2 and M02014(dmpm)2 were prepared by the
method described for the M02014(PR3)4 complexes. Dichloromethane
solutions of M02014(dppm)2 exhibit two intense maxima at 634 nm (e = 2490
M‘lcm'l) and 325 nm (e = 5600 M'lcm'l) as well as a weak band at 462 nm
(e = 900 M'lcm‘l) . Two absorptions at 604 nm (e = 1730 M'lcm‘l) and 426
nm (e = 270 M’lcm'l) are present in the UV-visible spectrum of
M02014(dmpm)2.
b. W,Cl4(dppm), [151]. W2C14(PBu3)4 (0.5 g, 0.38 mmol) and dppm
(0.30 g, 0.788 mmol) were refluxed in a 1:3 mixture of toluene (10 mL) /
hexane (30 mL) for a period of 6 h. The resulting green crystalline solid (or
sometimes brown) was washed with hexane to remove traces of
W2014(PBu3)4. Benzene solutions of both the green and brown precipitates
dissolved in benzene are light brown, with identical absorption spectra with

3 5
maxima at 710 nm (e = 2585 M-lcm'l), 500 nm (e = 598 M-lcm-1)405 nm
(sh) (e = 1923 M-lcm-l) and 364 nm (e = 2948 M’lcm‘l) (Figure 14). The
change in color is attributed to the previous reported difi‘erences in the UV-
visible spectrum of W2014(dppm)2 in the solid state and solution [151]. The
W2014(dppm)2 complex is very reactive and exceedingly sensitive to oxygen,
and will decompose within seconds to yield green, blue, and purple
products. The complex will react thermally with CH2012 at ambient
temperature, as previously reported [152], as well as acetonitrile. These
reactions however do not result in major color changes but can be detected
by an increase in the 500 nm absorption band ofWZCI4(dppm)2.
c. MoWCleppm), Benzene solution of 0.15 g MoWCl4(PMePh2)4
(0.123 mmol) and 0.25 g (0.62 mmol) of dppm were stirred for 12 h at room
temperature (heating was found to result in decomposition). The green
precipitate was collected, redissolved in CH2C12 and filtered to remove
yellow-green insoluble material. The absorption spectrum exhibits
features at 665 nm (A = 0.5), 370 (sh) (A = 0.5), and 320 nm (A = 1.0).
4 aynmdmmAandma’PhxoCo-nflm
a. W,Cl.(PR.)4 (PR, I PEt., PBup) [142]. These complexes were
prepared by a method reported for the PM63 derivative, except that the
rather unstable precursor, chlg(THF)4, was prepared in situ by adding 2.6
g of WCl4 to 70 mL of THF containing 0.41% sodium amalgam (0.368 g Na
(16 mmol)/ 98 g Hg) at ambient temperature. After 1 h of vigorous shaking
and stirring, the resultant yellow-green solution of W2016(THF)4 was
decanted from the amalgam and filtered through Celite to remove a grey
sideproduct. The filtrate immediately turned red upon addition of

3 6
stoichiometric amounts of phosphine. The solution was stirred for 0.5
h,and then THF was completely removed in vacuo. Addition of MeOH
prompted the precipitation of a brown powder precipitate, which was
collected. Although reasonably good yields were obtained with the PEta
complex (61%), only 10% yield was obtained for the PBus complex. A UV—
visible spectrum of a finely ground powder of W2016(PEt3)4 is provided in
Figure 7a (1,“, = 770, A = 0.06; km, = 470, A = 0.53; In“ a 380 nm, A =
0.45). This complex has been shown to convert to confacial WzClg(PEt3)3 in
solution at ambient temperature [153]. The complete conversion requiring
~ 3 min was monitored by UV-visible spectroscopy (Figure 7b). The final
spectrum, with maxima at 510 nm (e = 2077 M-lcm-l) and 328 nm (e = 2080
M‘lcm’l), corresponds to W2016(PEt3)3. As reported for the PEta complex,
only signals attributable to W2016(PBu3)3 were observed in the ”P NMR
spectrum of a sample of W2013(PBu3)‘ dissolved in toluene at ambient
temperature: 8 = 421.17 (t), —33.9 (d), Jpp a 35 Hz. The UV—visible smctrum
of CH2012 solutions of W2013(PBu3)3, with maxima at 500 nm (e = 1970 M"
1cm‘l) and 315 (sh) (e = 2269 M'lcm’l), is similar to that of W2016(PEt3)3
(Figure 8). The FABMS ofWZClg(PBu3)3 exhibits a parent ion cluster at 1188
amu, as well as a fragment at 1150 amu corresponding to loss of Cl.
b. W,Cl4(dppm).lg. Equimolar amounts of 12 (0.03 g, 0.117 mmol) and
W2C14(dppm)2 (0.15 g, 0.117 mmol) in 20 mL ofbenzene were stirred for 0.5 h
at ambient temperature. The rose solution was passed through a Florisil
column with THF as the eluant and yielded a rose precipitate upon addition
of hexane. The solid was filtered, washed with benzene and hexane, and
dried under vacuo (10"6 torr) at 50 °C to remove excess I2. A parent ion
cluster at 1532 amu as well as two fragments at 1405 and 1497 amu,

3 7
corresponding to loss of I and Cl, respectively, are present in the FABMS
shown in Figure 9. The U'V-visible spectrum exhibits an intense
maximum at 500 nm (e = 6954 M’lcm'l) and a less intense shoulder at 390
nm (e = 3477 M’lcm‘l).
c. W3C1.(dppm),[152]. A CHzclz solution of W201‘(dppm)2 was
stirred for a period of two days. A precipitate of the air sensitive
W2C16(dppm)2 formed upon addition of hexane to the solution. The solid
was washed sequentially with benzene and pentane, and stored under
vacuum. The absorption spectrum exhibits two intense maxima at 468 nm
(e = 4800 M‘lcm'l) and 387 nm (e = 3200 M‘lcm‘l), as well as very weak
absorbances at 822 nm (e = 740 M‘lcm'l) and 641 nm (e = 285 M'lcm‘l).
GPhotochanish-y
LGeneralProcedures
Monitored photoreactions were carried out in a specially adapted
high vacuum UV-visible cell described in section E1. Bulk solutions were
also prepared under high vacuum conditions in quartz tubes. Sample
irradiations were performed by using a Hanovia 1000—W Hg/Xe high
pressure lamp. The beam was collimated and passed through a 10 cm
circulating water filter. The irradiation wavelength was selected with
Schott color glass high-energy cutoff filters which were placed in a glass
water circulating bath to avoid their heating. The collimated and filtered
beam was finally passed into the sample isolated in a separate water
circulating bath. Isolation of the sample in a separate compartment is
necessary to prevent previously encountered problems with reflected

3 8
unfiltered light reaching the sample. The sample temperatures were
thermostated with the water bath. An identical setup was used for
quantum yield determinations except that Oriel interference filters were
used in place of the cutofi‘ filters. Ferrioxalate actinometer [154, 155] was
used as the reference and described in detail in the dissertation of Dr. I-J.
Chang [147]. Measurements were made under optically dense conditions
(A > 2) at the exciting wavelengths. Photoproduct concentrations were
limimd to less than 10% to avoid inner filter effects. Conversions were
determined by monitoring the disappearance of the 8241(88‘) absorption of
the quadruply bonded binuclear complex.
2. IsolationofPhotoploduct:
a. Photolysis ofW,Cl4(dppm), with CHJ. The photoproduct ofthe
reaction of WzClg(dppm)2 with CH31 was precipitated upon addition of
hexane. There was no evidence of unprecipitated sideproducts in the
colorless mother liquor. The absence of sideproduct was further confirmed
by the fact the the UV-visible spectrum of the precipitated solid was
identical to the final spectrum of the photolysis. Elemental analysis of the
"crude" (i.e. nonrecrystallized) precipitate was performed at Galbraith
Laboratories. The photoproduct is not stable at ambient temperature and
photolyzed solutions were kept at S 0 °C at all times. Failure to do so
resulted in formation of W2014(dppm)2I2, as evidenced by a relative increase
in the absorption at 500 nm, and appearance of a parent ion cluster of the
diiodide complex in FABMS. Additionally, a decomposition product with
an absorption maximum at 470 nm was observed when the slightest trace of
moisture was present. Attempts to grow crystals layered from

3 9
CHzclzlhexane solutions at -20 °C were unsuccessful; only decomposition
products which were completely insoluble in CH2012 were obtained.
A 13C NMR spectrum of the photoproduct was obtained by
photolyzing W2014(dppm)2 in a 1:1 mixture of 12C and 13C enriched (99%,
Aldrich Chemical Co.) CHal. Upon completion of the photolysis, the CH3I
was completely removed under vacuum, and the photoproduct was further
subjected to dynamic vacuum (10“ torr) for 8 h. The product was
redissolved in CDzClz (Aldrich 99.6+%) and the 13C NMR spectrum was
recorded on a Bruker WM-250 NMR at -60 °C.
Analysis of ethane in the atmosphere above photolyzed CH31
solutions of W2C14(dppm)2 was performed by Toepler pumping a
quantitative volume V of the photolyzed solution containing a calculated
quantity (n) of the W2014(dppm)2(CH3)I photoproduct. The solutions were
kept at 0 °C to avoid decomposition of the photoproduct, but were not subject
to freeze-pump—thaw cycles at liquid N2 temperature.* The CH31 was
partially condensed in three successive traps at temperatures above the
boiling point of ethane (-78 °C). The collected gas was vacuum transferred
into a 0.5 ml tube equipped with a Kontes stopcock and an Ace high vacuum
septum. Identical procedures were carried out with two control solutions,
one was a blank sample of CH31 with volume V, and the other a sample of
CH3I with volume V containing n moles of ethane (control 2) These three
gas samples were analyzed with a Hewlett Packard 5985 GS/MS with an
open tubular 18" column at 50 °C. The relative intensities of the CHal peak
at 142.1 amu and the ethane peak at 30.1 amu were measured: (1142.1, 130:
Blank, 4824/5, control 2: 5000/105, sample 5464/3). *(Ethane was barely
detectable from control 2 solutions subject to freeze—pump—thaw cycles at
liquid N2 temperature.)

40
b. Photolysis ofMo,Clg(PBu3)4 with PhSSPh. A photoproduct Ohm,=
540 nm) from irradiated solutions of M02014(PBu3)4 with PhSSPh was
separated from a product (Inn: 460 nm) by column chromatography on
Florasil. The 540 nm product was selectively eluated from the column with
a 5% CH3CN and 95% CH2012 solvent mixture and the secondary product
was then removed by eluting with pure CHscN.
c. Photolysis ofW,CI.(PBu3)4 with CH,CI,. The photoproduct fiom
irradiated CH2C12 solution of W2014(PBu3)4 displayed absorption man'ma at
1430, 430, 404 and 335 nm. Attempts to isolate the photoproduct were
hindered by thermal decomposition at ambient temperature as evidenced by
the appearance of an absorption at 500 nm and disappearance of the NIR
absorption. The decomposition reaction is concentration dependent and
consequently forms immediately upon complete removal of solvent from
photolyzed solutions.
D.Electrochemish'y
LGeneralProeedures
Cyclic voltammograms were recorded with a Princeton Applied
Research (PAR) Model 173 potentiostat, Model 175 programmer, and a
Model 179 digital coulometer. The output of the digital coulometer was fed
directly into a Houston Instrument Model 2000 X-Y recorder. A Pt wire
gauze, a Pt button and a Ag wire were used as the counter, working and
reference electrodes, respectively. Potentials were referred to the SCE
reference scale by using the szFe+le2Fe couple of +0.31 V vs SCE as an

4 1
internal standard [156]. Typical concentrations were ~ 5 x 10" M in
quadruply bonded binuclear complexes with 0.1 M electrolyte. A two
compartment standard H cell was used in most cases except for
experiments involving the addition of halide anion to the sample
compartment. In this case, the reference electrode was separated from the
sample compartment by using a four compartment cell to avoid differences
in potential before and afizer the addition of halide. Bulk electrolysis
experiments were performed with the above apparatus in conjunction with
a PAR 179 coulometer. A standard three compartment H cell was used
with a platinum mesh electrode as the working electrode.
2. PreparationandPurificationofElecholytes
Tetrahexylammonium hexafluorophosphate (THAPFS) was prepared
by metathesis of tetrahexylammonium iodide (THAI, Fluka) and
ammonium hexafluorophosphate (NH4PF6 Aldrich) in 95 % EtOH [157].
The THAPFg precipitated immediately upon slow dropwise addition of a
saturated solution ofTHAI to a saturated to NH4PF6 solution. The THAPFG
was filtered and recrystallized from 95 % EtOH. Tetrabutylammonium
hexafluorophosphate (TBAPFG) was dissolved in ethyl acetate containing
MgSOb filtered, then recystallized by addition of ether. Both electrolytes
were dried under vacuum (10‘6 torr) at 90 °C for 10 h.
3. aulhalectrolysisorwpwsup,
Bulk electrolysis of a 4 mL CH2012 solution of W2Cl4(PBu3)4, (6.96 x
10" M) was performed in the N2 atmosphere of a glove box at an applied
potential of +0.1 V. The coulombs generated, 26.4 x 10’2, were consistent
with a one—electron oxidation process (theoretical = 26.8 x 10"2 coulombs).

42
Cyclic voltammograms obtained before and alter bulk electrolysis were
identical and showed no indication of side reactions accompanying the
electrolysis process. The UV—visible spectrum of the W2(II,III)C14(PBu3)4+
(1m, = 1510 nm, e = 2243 M-lcm-1,i.,,,,,x = 380 nm, e = 2240 M-‘cm-1.1m =
480 nm, e = 852 M‘lcm'l) species is provided in Figure 10. Although the
W2(II,III)Cl4(PBu3)4+ complex may be prepared chemically with NOBF4 as
an oxidant, the spectral changes indicate additional byproducts are also
formed.
E. Spectroscopic Instrumentation andMethods
1. ElectronicAbsorption Spectroscopy
Absorption spectra were recorded with Cary 17D or Cary 2300
spectrophotometers. In most cases solutions were prepared under high
vacuum conditions in a cell consisting of a 1 cm quartz cuvette and a 10 ml
side arm. The two chambers were separated by two Kontes high-vacuum
quick release teflon stopcocks. The samples were placed in the cuvette and
the appropriate solvent was transferred to the 10 ml side arm by bulb-to-
bulb vacuum distillation. After these subsequent freeze-pump—thaw cycles
were performed, the solvent was mixed with the sample. Extinction
coefficients were determined by standard procedures from solutions
prepared in a glove box. Three stock solutions with varying amounts of
weighed sample were first prepared. Appropriate dilutions of these stock
solutions provided seven solutions of known concentrations, with
absorbances varing from 0.20 to 0.90. Extinction coefficients were
calculated from Beer-Lambert plots.

43
asteady-StateLuminescenceExperiments
The emission spectra were recorded on an emission spectrometer
constructed at Michigan State University which is fully described in the
doctoral thesis of Dr. M. D. Newsham [158]. The R1104 Hamamatsu PMT
was used in the case of the dimolybdenum complexes and the R316
Hamamatsu PMT in the case of the ditungsten complexes. Absolute
emission quantum yields of optical dilute samples (A < 0.2) were measured
using M02014(PBu3)4 as a quantum yield standard (item = 0.013 in 2-Me-
pentane at 300 K) [121d]. The quantum yield was calculated from the
following equation [159]:
2Arm.) n_,_ g
4‘1.“ X [AthQ] X [111:2] X [Dr] (2.1)
where x and r designate the unknown and standard solutions, respectively,
n is the average refractive index of the solution, D is the integrated area
under the corrected emission spectrum, and A (2.) is the absorbance per
unit length (cm) of the solution at the exciting wavelength 2..
3. IramieritAbeorptionSpectroscopy
The nanosecond time resolved absorption spectroscopy of the
dimolybedum complexes was performed on an instrument described
elsewhere [160]. The same instrument was used for the ditungsten
transient spectra except that the signals were digitized on a Tetronix DSA
digitizing signal analyzer with a model 11A72 two-channel amplifier. The
solutions with absorptions of ~2 at the exciting wavelengths, except where

4 4
otherwise noted, were continually circulated through a flow cell with a 1
mm pathlength throughout the experiment. The solutions were prepared
in a glove box with freshly vacuum distilled high purity solvents.
4. Electron Paramagnetic Resonance
X band EPR spectra were obtained using a Bruker ER 200D
spectrometer. The magnetic field strength was measured with a Bruker
ER035M NMR Gaussmeter and a Hewlett-Packard 5245L frequency counter
equipped with a 3-12 GHz adapter was used to measure the microwave
frequencies.
5. Nuclear Magnetic Resonance
The phosphorus and proton NMR were recorded on a Varian VXR-
500 spectrometer. The 31PilH} chemical shifts were measured relative to
85% H3PO4. Deuterated methylene chloride (Aldrich, 99.6+%) and
deuterated chloroform (99.8%, Cambridge Isotope Laboratories) were dried
under high vacuum conditions as described in Section A2.
6. Mass Spectrometry
The fast atom bombardment mass spectra (FABMS) were performed
on a JEOL HX 110 double focusing mass spectrometer housed in National
Institute of Health/Michigan State University Mass Spectrometry Facility.
Samples were dissolved in 2-(octyloxy)nitrobenzene matrices.

45
Figure 7. (a) Electronic absorption spectrum of a finely ground state
sample of the edge—sharing bioctahedral complex W2016(PEt3)4. (b)
Electronic absorption spectral changes associated with the conversion of
W2C16(PEt3)3 to the confacial bioctahedral complex W2016(PEt3)3 in toluene
solution at ambient temperature, In,” nm (e M‘1 cm'l) (a) W2C16(PEt3)4:
4...... = 470 nm, (e = 1992 M’1 cm-l); 380 nm (1280 M-1 cm-l) (b) W2016(PEt3)3
: 1...... = 510 nm, (e = 2077 M-1 cm‘l); 328 nm (2078 M-1 cm-l).
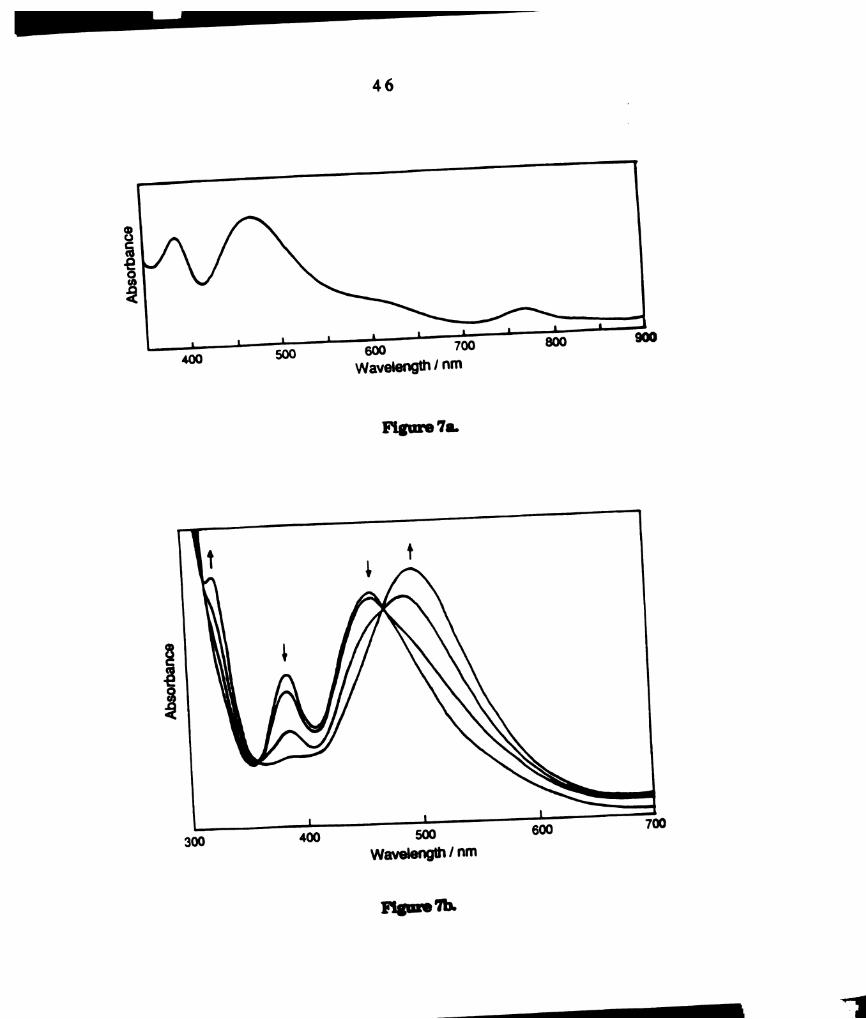
Absorbance
4 6
600700
WavelengthI nm
Figure'la.

47
Figure 8. Electronic absorption spectrum of dichloromethane solutions
of the confacial bioctahedral complex W2C16(PBu3)3, In,“ = 500 nm, (e =
1970 M‘1 cm‘l); 315 nm (2268 M-1 cm'l).
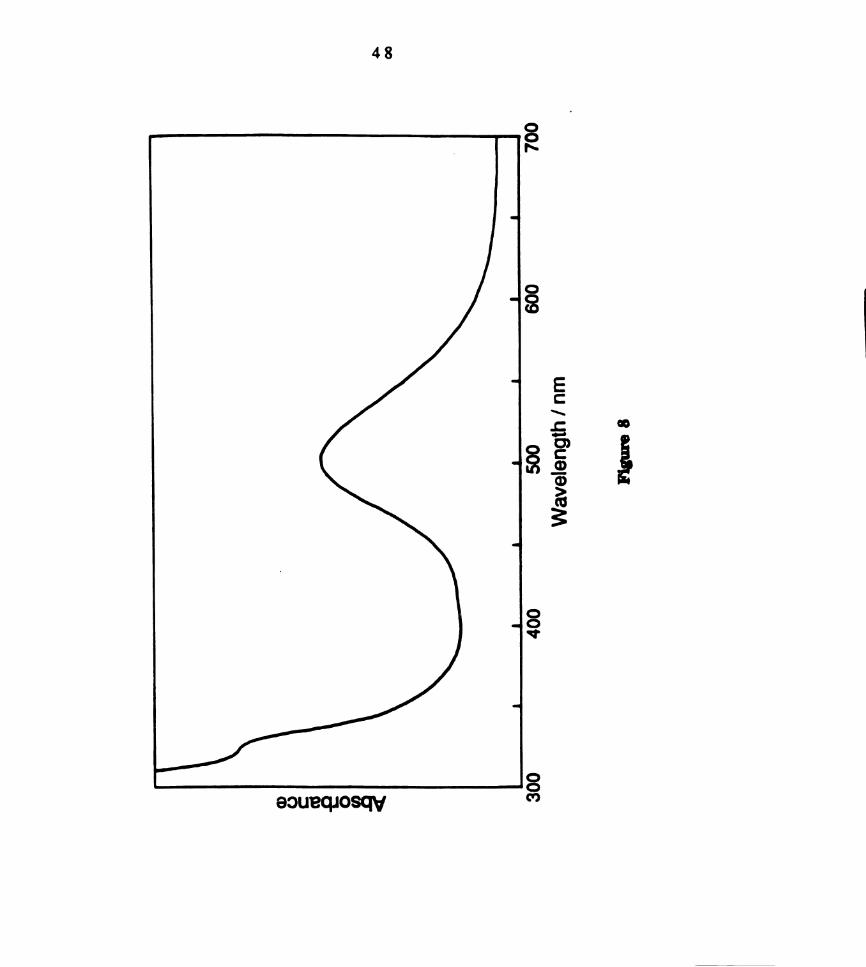
48
can
opus...—
E:
\56:235.;
com
00?
q
com
dd
eoueqiosqv

49
Figure 9. Positive fast atom bombardment mass spectrum of a product
from the thermal reaction of W2014(dppm)2 with 12. The clusters centered
at 1531, 1497 and 1405 amu are consistent with (a) W2014(dppm)212, (b)
W2013(dppm)I2 and (c)WZCI4(dppm)21, respectively.
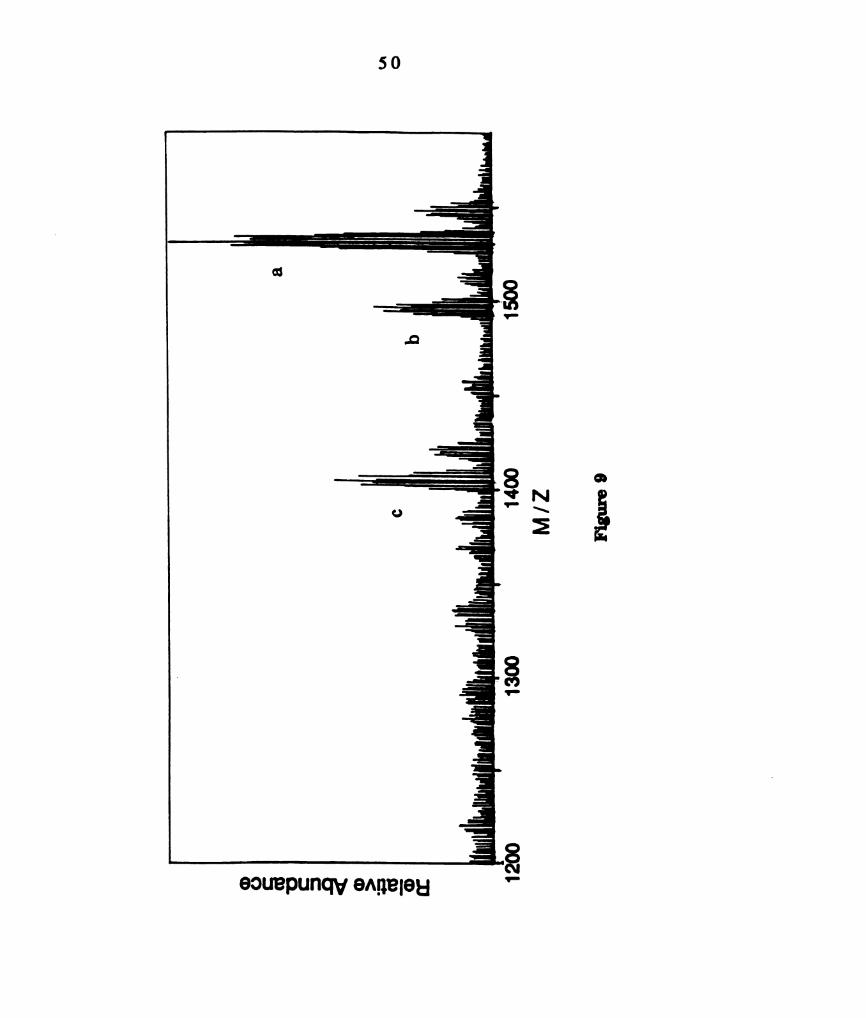
50
eouepunqv ennslea
M/Z
“m9

51
Figure 10. Electronic absorption spectrum W2(II,III)Cl4(PBu3)4+
prepared by bulk electrolysis in CH2C12. km” = 388 nm, (e = 2559 M‘1 cm‘l);
485 nm (949 M‘1 cm'l); 1510 nm (2243 M-1 cm-l).
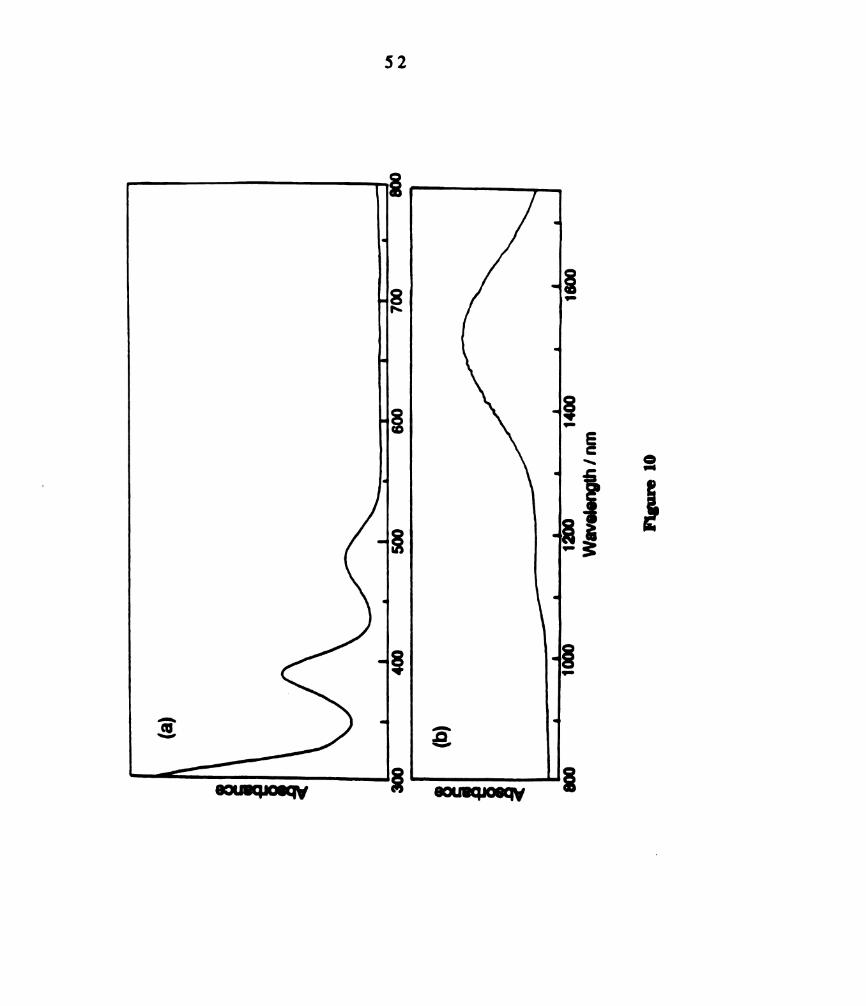
a.
52
3

CHAPTERIII
TRANSIENTABSORPTIONSPECTRIBCOPICSTUD-
A. BACKGROUND
The electronically excited molecule is the crucial reactant in a
photochemical transformation. Thus a clear understanding of the
photochemistry must begin with the elucidation of the electronic structure
of the photoreactant. The quadruply-bonded complexes whose electronic
structure is most intensively investigated are the M2014(PR3)4 complexes.
The lowest energy transition in the uv-visible absorption spectra of these
complexes, which adopt the D24 geometry depicted in Figure 11, is the
typical 8241(88") transition. Higher energy transitions in the electronic
absorption spectrum of M2Cl4(PMe3)4 have been assigned as well [121a]. A
weak transition (6 = 210 M’lcm‘l) centered at 441 nm is attributed to the
dipole-allowed 10:48") transition and a ligand—to—metal charge transfer
(LMCT) assignment has been made for the intense transition (6 = 3720 M"
1cm’l) centered at 324 nm based on its characteristic blue shift (11,,“ = 290
nm) for the ditungsten analogue.
53

54
Figure 11. (a) D2d structure of MZCI4(PR3)4 complexes; 0)) D2}1 structure of
MzCl4(PP)2 complexes where PP = bidentate phosphines.
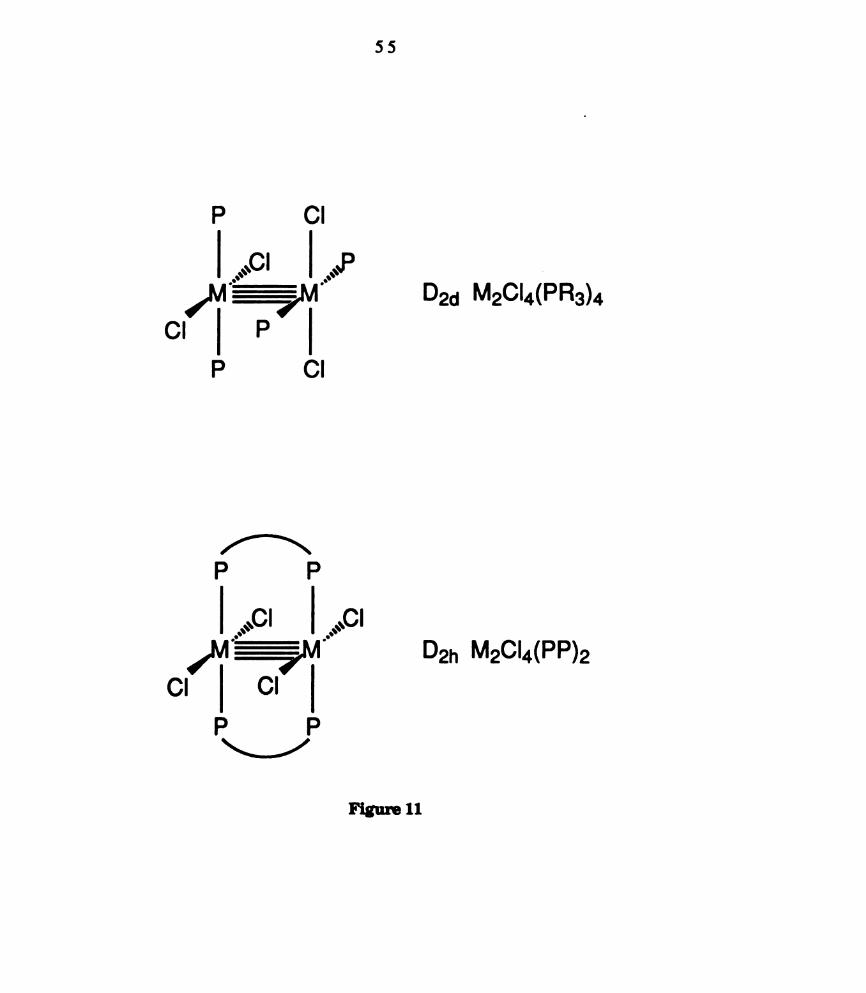
55
.0
£2
D2ci M20'4(PRa)4
/'\
P P
”on
(Cl\
e“
02h M20'4(PP)2
Cl I CI l
P PV
Figurell

56
In contrast to the excited state properties of the majority of
quadruply-bonded dimers, the 1(88”) state of these complexes is long-lived
and the luminescence is intense. For example, the 1(88") luminescent
lifetime of MozCl4(PBu3)4 in 2-methylpentane at ambient temperature is 16
ns and the emission quantum yield is 0.013 [121d]. The enhanced lifetime of
the 1(88*) excited state of M02014(PR3)4 complexes relative to other M02C14L4
complexes has been ascribed to the steric bulk of the coordinating
phosphines [139]. For complexes with sterically unhindered ligands such
as CH3CN and Cl’, free rotation about the metal-metal bond plays a
prominent role in the nonradiative decay of the 1(88") excited state due to the
annihilation of the 8 bond upon 82 -)1(88*) excitation. Indeed the broad and
weak emission and notable lack of a mirror image between absorption and
emission spectra of M02018“ [129] is consistent with the poor Franck—
Condon factors between the molecule in its staggered D“ excited state and
its eclipsed D41, ground state [139]. The lifetimes of the staggered excited
states are typically in the picowcond range [139]. This is not the case for the
M02014(PR3)4 complexes. The mirror image absorption and emission of the
l(88") excited state is explained by the steric constraint of the phosphines
which prevent rotation about the metal-metal bond. In effect, the excited
state is locked into the eclipsed geometry of the ground state. Nevertheless,
despite this simple excited state model, transition absorption studies of
M0201‘(PBu3)4 have identified multiple decay processes of the 1(88") excited
states. In addition to prompt decay from the luminescent 1(88") excited
state, a nonluminescent transient with a lifetime of 90 as is also observed
[139]. An even longer lived (1: = 46 us) nonluminescent transient is observed
with the M0201‘(CH30N)4 complex, though no analogous transient exists
for electronically excited M02013“. While definitive assignment of the

5 7
transients has not been achieved, the possibilities of a 3(88*) excited state,
3(1t8"‘) excited state or a distorted chemical intermediate have been
suggested. The longevity of this nonluminescent suggests that it is a
promising candidate for bimolecular photochemistry.
In principle, an eclipsed conformation about the quadruply bonded
core can be imposed not only by a bulky phosphine, but by a bidentate
phosphine spanning the bimetallic core as well. Despite the basic
similarities in the ligation spheres of the M2Cl4(PR3)4 and M2014(PP)2
complexes, a D2,, symmetry of the latter complexes is imposed by the trans
arrangement of the bridging bidentate phosphines depicted in Figure 11.
Although the electronic excited state properties of the MzCl4(PP)2 complexes
have not been investigated as extensively as those of MzCl4(PR3)4, there are
data that suggest this change in symmetry is not without consequences.
Indeed differences are predicted from analysis of the simple molecular
orbital diagram provided in Figure 12 [161]. The relative energetics of the
metal-metal based orbitals are displayed as a function of rotation of a
MClsz fragment about the metal-metal bond from 0° (D211) to 90° (Dzd): with
D2 symmetry occuring for all angles in between. The relative energies of
the 8 orbitals are a result of the variation of the d" overlap with the
torsional angle 0. Maximum overlap and hence the largest 8/8’ splitting is
achieved at o = 0° (Dzh) or 90° (D24) where the MClsz fragments are
eclipsed. The overlap and resultant 8/8' splitting decreases monotonically
with rotation away from the eclipsed conformation, until it reaches a
minimum of 0 at the completely staggered conformation (t = 45°). The
relative energetics of the x and xt orbitals vary with ¢~ In D24 symmetry, the
M—M it" and icy, orbitals are degenerate. However this degeneracy is
removed in D2,, symmetry. The (1,, metal orbitals, which are strongly

58
Figure 12. Relative energies of the d—derived molecular orbitals in
MZCl4(PR3)4 complexes as a function of torsional angle 0 (Reference 161).
59

60
metal-chlorine (M—Cl) x antibonding, are destabilized, and the d"
orbitals, which are strictly nonbonding with phosphine ligands, are
stabilized. The splitting of the it" and my, orbitals increases monotonically
with o. The in orbitals are perturbed in the same manner. Since theory
predicts 27% chlorine character in M—M x orbitals of M020134' [129], the
relative energetics of these orbitals are expected to be significantly affected
by this perturbation.
Experimental study of the torsional perurbation of the electronic
structure has become accessible with Cotton and coworkers synthesis of a
series of torsionally distorted D2 B—M2014(PP)2 complexes where a variation
in (t is induced by the steric restrictions of the bridging PP ligands. This
series of compounds reveal that the 8241(88') transition energies are
correlated with 00st where x = 0 or «I2 - 4) when t < 45 or t > 45, resmctively
[162, 163]. The electronic spectra of torsionally distorted complexes are far
more complex than those of their D24 cogeners. The optical activity of the
disymmetric D2 B-M2X4(S,S,—dppb)2 (S,S,-dppb = (28,38)-
bis(diphenylphosphino)butane) complexes provides a benchmark for the
assignment of these transitions based on their resultant circular dichroism
(CD) spectra [161]. As with the M,CI,(PR,), l)2d complexes, a ‘(x—is“)
transition was assigned to an electronic absorption band located
immediately to higher energy of 8241(88") absorption. However, the
assignment of the electric-dipole forbidden, magnetic-dipole allowed
1(rt-+8") transition of the B-M2X4(S,S,-dppb)2 complex was made for a
transition at considerably higher energy (1,“, = 362 nm) and much greater
intensity (2 > 3000 M‘lcm'l) than the similarly assigned electric-dipole
allowed transition centered at 440 nm (e = 200 M‘lcm’l) in the spectrum of
M02014(PMe3)4 [121a]. Also, as predicted from the model in Figure 12, the

61
nondegeneracy of the it orbitals potentially leads to two lbw—>8") transitions
in the case of the D2 dimers. In support of this contention, an absorption at
362 nm has been ascribed to the Tun—>8” component and a 340 nm feature
in the CD spectrum has been assigned to l(rim-+8"). The blue shift of the
l(rt—>8") transition upon a D2 to D2,, perturbation is opposite to that expected
from the model in Figure 12. As there was insuficient data in either case
for unequivocal assignment, it is not clear if either of these assignments is
correct. In general, the l(rt-48'") transition is difficult to distinguish from
the 1(8—-)rt"') which is predicted by theory to be energetically proximate. An
additional assignment of a 848x212 transition, where the 8x212 orbital is
primarily metal-ligand antibonding in character, was ascribed to an
absorption centered at 470 nm with approximately one-half the intensity of
the 8241(88") transition.
The monotonic increase in the run" splitting with increasing ¢ may
provide a basis for interpreting the luminescence intensity of M2014(PP)2
complexes. Previous studies described in the dissertation of Dr. I-J. Chang
show that the emission quantum yields of D2 complexes like M02014(dppe)2
(dppe = bis(diphenylphosphino)ethane) and M02014(dmpe)2 (dmpm =
bis(dimethylphosphino)ethane) are ~ 102 less than those of their respective
D25 analogues, M02014(PMePh2)4 and M02014(PMe3]4 [147]. An additional
102 attenuation is observed from the D2,, cogeners with dppm and dmpm
ligands. These relative emission quantum yields fit nicely with the model
in Figure 12. The energy gap between low lying 3(81'), 3(r:8"') states and the
1(88") state is smaller in the D21, complexes, accounting for the increased
nonradiative decay of their 1(88") states. The intermittent quantum yields of
emission from the D2 complexes are consistent with the predicted relative
energy gaps.

62
Despite these differences in their electronic excited state properties, a
recurrent theme in the redox chemistry common to both the M2X4(PP)2 and
M2014(PR3)4 complexes is that oxidation of the metal core is accompanied by
a major rearrangement of the ligation sphere. Two electron oxidized
M2X4(LL)2 species are stabilized by an edge-sharing bioctahedral
configuration of ligands [152, 164—167]. In the case of the more flexible
M2X4L4 complexes (M = Mo(II), W(II), Re(III); X = halide or pseudohalide;
and L = donor ligand including halide), which do not contain bridging
bidentate phosphines, the alternative confacial bioctahedral geometry is
sometimes observed upon oxidation [168-173] as well as the edge-sharing
bioctahedral structure [174—177]. Both the edge and confacial bioctahedral
arrangements ensure a stabilizing octahedral coordination geometry about
the on’dized metal center.
As discussed in Chapter I, the metal-to—metal charge transfer in
82—3 1(88*), 1(1:48") and l(8-nt"‘) transitions yield mixed valence
electronically excited states. Owing to the stabilization offered by confacial
or edge-bridging geometries about oxidized metal cores, the preparation of a
mixed valence excited states by light excitation should not be without
chemical consequences. To this end, these octahedrally based
rearrangements may presage a rich transient spectroscopy of D2,, and D24
complexes. Indeed the previous picosecond transient absorption studies of
M0201‘(PBu3)4 and M02Cl‘(CH3CN)4 indicate that nonluminescent
transients are generated upon 82-91(88"') absorption. These initial studies of
MozCl4L4 complexes (L= CH30N, PBu3, 01") are now elaborated by
nanosecond transient absorption studies of a complete series of
Mozcl‘(PR3)4 with phosphines of varying the basicity and lability. The
properties of 1(88") excited state of the ditungsten analogues are also

63
presented. Moreover, the electronic absorption spectroscopic results of the
D2 M2X4(PP)2 complexes suggest that studies should not be limited to the
1(88*) excited state, but rather extended to include the plethora of metal
localized excited states at higher energy. Accordingly, the deactivation
processes of the high energy states of both the M2014(PR3)4 D2,, and
M2X4(PP)2 D211 and complexes were probed. Transient absorption studies
reveal varying decay processes of the high energ states of the D2,, and Dzh
complexes. The M02014(PMePh2)4 complex provides an ideal test for the
model emerging from the studies of the MzCl4(PR3)4 D2d and M2X4(PP)2 D2,1
complexes because both the D2,, and D2d isomers can be prepared [148]. The
absorption spectra of the transient species generated in the photochemically
inert solutions are compared with those of photochemical intermediates
observed in halocarbon solutions.
B. RESULTSAND DISCUSSION
1. MgCMPP); (1);.)Complenes
a. PhotochemicallylnertSolutiom
Figure 13 displays the electronic absorption spectrum of
M02014(dppm)2, which is typical of D2}l complexes. The lowest energy
8241(88’) transition maximized at 634 nm (e = 2490 M’lcm‘l) is clearly
identified and a weaker transition at 462 nm (e = 900 M’lcm‘l) is
comparable in energy and intensity to the assigned 82—)1(88x2_y2) transition
in the spectrum of B—M2014(S,S,-dppb)2 [162]. At even higher energy (Inn =
325 nm) is a more intense transition (6 = 5600 M‘lcm'l) that displays a
shoulder at 364 nm. A similar spectral profile for the ditungsten analogue,

64
Figure 13. Electronic absorption spectra of dichloromethane solutions of
M2014(dppm)2, where M = M0 (——) and W (------ ) (a) MogCl4(dppm)2: km”
= 634 nm, (e = 2490 M"1 cm‘l); 462 nm (900 M’1 cm'l); 364 run (2040 M’1 cm’
1); 325 nm (5600 M'1 cm-l) (b) WzCl4(dppm)2: 1m = 710 nm, (e = 2585 M-1
cm-l); 500 nm (598 W1 cm“); 405 nm (1923 M-1 cm-l); 325 nm (2948 M-1
cm‘l).
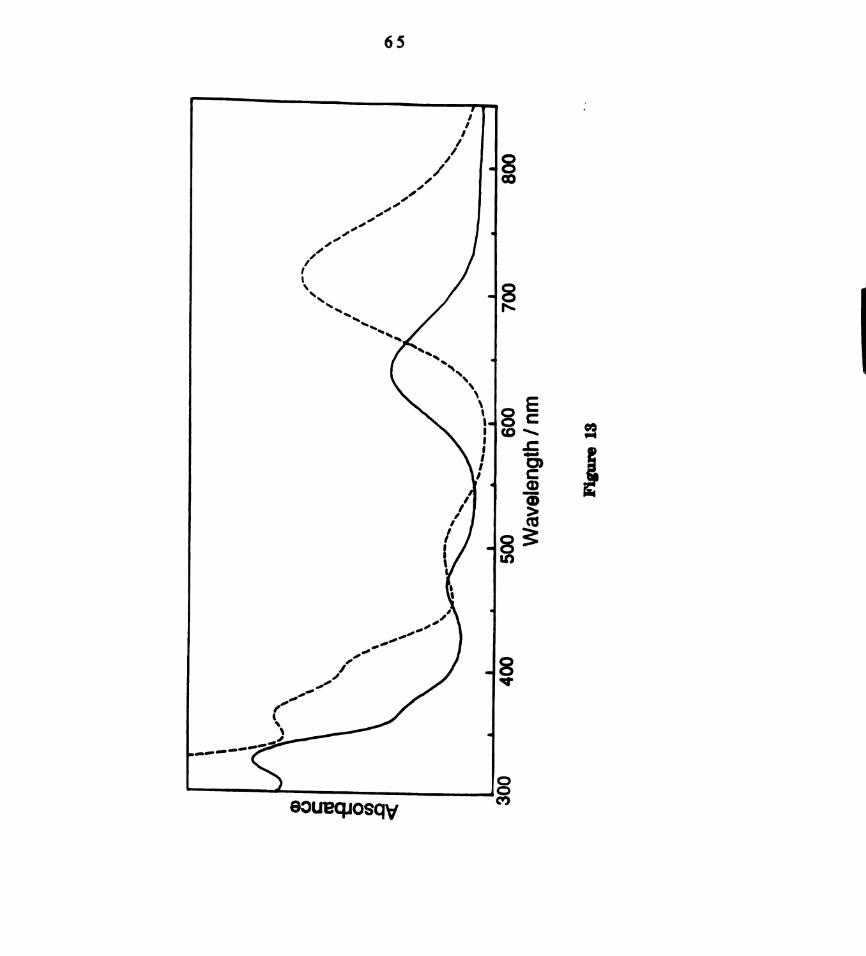
65
£23...—
Ec
\59.23.25
-
Id
d1
\lllll’ll
eoueqlosqv

6 6
shown in Figure 13, is red-shifted. This red shift of the higher energy
transitions is indicative of metal-localized transitions and is in clear
contrast to the expected blue shift of an LMCT transition, as observed for the
D2,, dimers (Imu(LMCT) = 324 nm and 290 nm for M02014(PMe3)4 and
W2Cl4(PMe3)4 complexes, respectively) [121a]. By analogy to the model of
Figure 12 for the torsionally distorted complexes, the energetically
proximate features at 325 and 364 nm absorptions of M02C14(dppm)2 are
consistent with the lacy—)5“) or 1(8--)rt,.,"') and 1(rim-98'") or l(8—)rt,,,"')
transitions, respectively. Irrespective of their specific assignment, the
intense high energy transitions are clearly metal-localized and are not of
LMCT character.
On the nanosecond time scale, no transients are observed with
excitation into 1(88*). Yet as reported in the doctoral dissertation of Dr. I—J.
Chang, short lived absorptions are observed for the 1(88*) excited state on
the picosecond time scale [147]. A more elaborate transient spectroscopy is
observed with high energy excitation. A long-lived (1 ~ 5 us)
nonluminescent transient, whose decay back to ground state is
monoexponential, is observed upon excitation of the higher energy metal
localized transitions (1.“ = 355 nm) of M02014(dmpm)2 in CHzclz. A
transient absorption (Figure 14) collected 1 us after excitation exhibits a
maximum at 520 nm. Unfortunately emission from an impurity in the free
ligand has prevented us from measuring the transient absorption
smctrum at wavelengths less than 460 nm [178].
High energy excitation (Am a 355 nm) of M2014(dppm)2 complexes (M
= M0, W) yields ligand based emission which exhibits a maximum at 460 ,
nm. The fluorescent lifetime of t = 2 us for free dppm [179] agrees with that
for the transient observed upon 355 nm excitation of M02014(dppm)2 as

67
Figure 14. Transient difference spectrum of MozCl4(dppm)2 in CH2012
recorded after 355 nm laser excitation.

A0.0.
68
0010— l
:1 iiiiif
0.....— E if}
' 450 500 550 M[1:100 700 750 '

69
reported in the dissertation of Dr. I-J. Chang. This ligand emission
precludes detection of nonluminescent transients of M02C14(dppm)2
complex arising from high energy metal localized excited states. However,
owing to the red-shifted spectral profile of the ditungsten analogue, ligand
based emission can be avoided with 532 nm excitation of W2014(dppm)2.
Accordingly the laser excitation of benzene solutions at 532 nm was
performed. A transient absorption which decays back to ground state
within 100 as is observed at 440 nm (Figure 15). The transient spectra
recorded 100 ns, 1 us and 4 us after excitation are provided in Figure 16.
Although slight variations are noted in these spectra, each of the profiles
exhibits common features at 390, 420 and 480 nm.
Both the ditungsten and dimolybdenum transient spectra exhibit
features which are comparable to those in the electronic absorption spectra
of the edge-sharing bioctahedral complexes. The feature centered at 480
nm in the ditungsten transient spectrum, and the 520 nm absorption of the
dimolybdenum transient spectrum, agree well with those at 465 and 500 nm
in the electronic absorption spectra of chlg(dppm)2 (Figure 17) [166] and
M02014dmpm)2 (Figure 18), respectively. Prominent absorption features in
the spectral range of 450 to 550 nm are ubiquitous to the spectra of
numerous dimolybdenum and ditungsten edge-sharing bioctahedral
complexes with M(III)—M(III) (d6) bimetallic cores. These include:
MozClg(dppm)2, In,“ = 530 nm [164b]; M02014(dppm)2Br2, km“ a 540 nm
[164a]; and M02015(dppm)2(SPh), In,“ a 540 nm. Similar features are
observed in the ditungsten complexes, although they are generally slightly
blue shifted relative to the dimolydenum complexes, (W2013(dppm)2, 1,,“ =
468 nm [163]; chl4(dppm)2I2, km“ = 500 nm; WZCl4(dppm)2(SPh)2, Am“ =
504 nm [164b]; W2015(dppm)2H, In“ = 464 nm [166]). Accordingly, the

70
Figure 15. Transient kinetics for WzCl4(dppm)2 in CSHG (1.95 x 10‘4 M)
recorded at 440 nm following laser excitation at 532 nm.
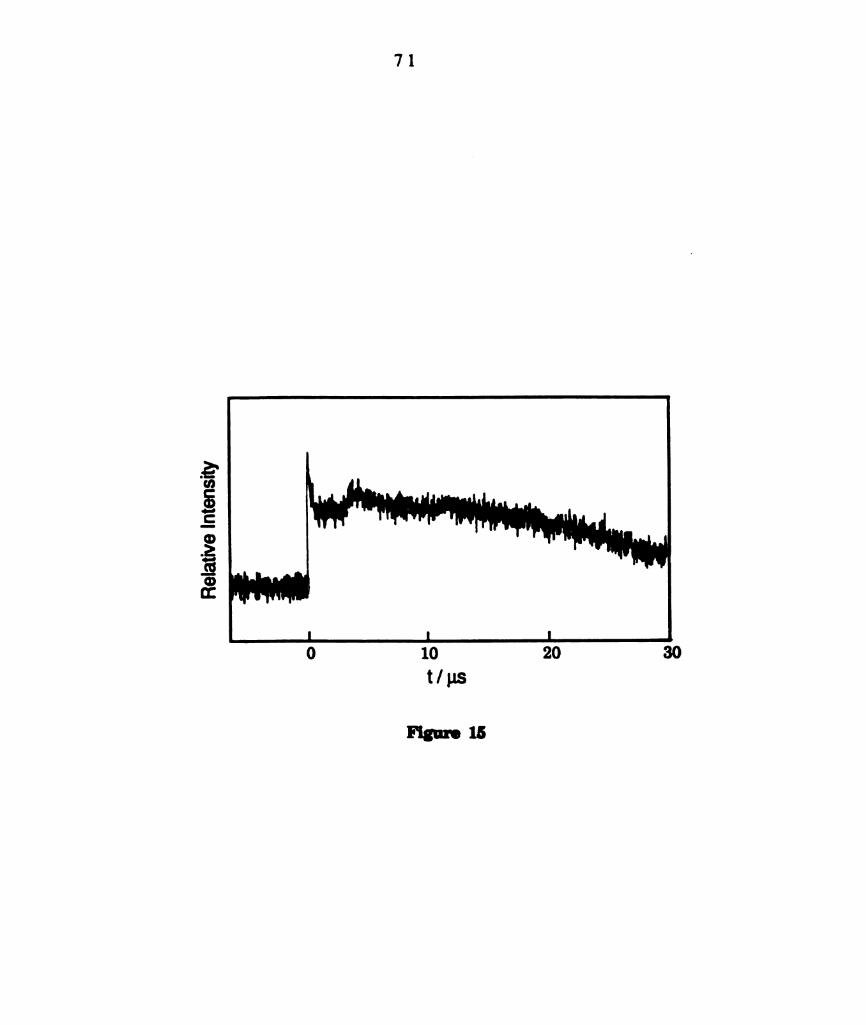
71
Emcee.peso:
2010
Hits
Figure“

72
Figure 16. Transient difference spectra of WZCI4(dppm)2 in C6H6 recorded
recorded 100 ns and 4 us after 532 nm laser excitation (see legend).
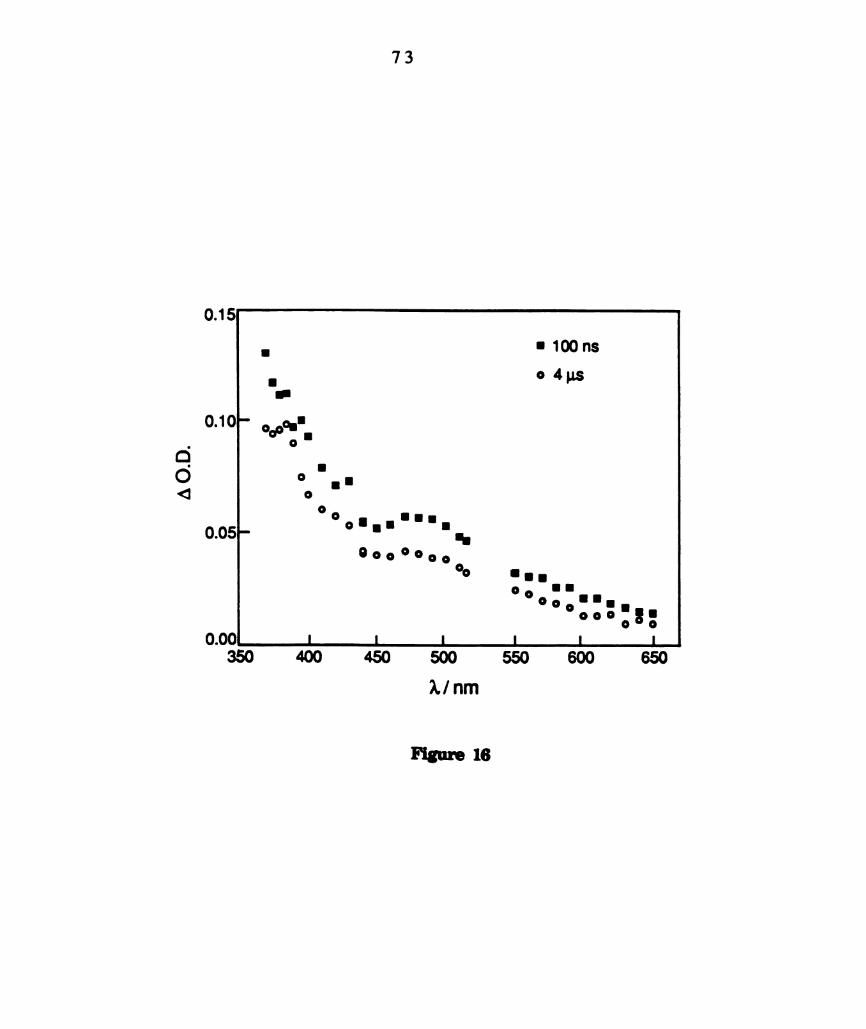
73
0J5
. -100ns
. o4us
\—
010)" coop-l.
o
Io ..
o
.0 II.
0.05i— "I" '..
Ooo°o°°
-II.-
°°¢°°II..
"as:and l l l l l 1
an “N «w an am (no fl”
Mnm
FlarreIB

74
Figure 17. Electronic absorption spectrum of dichloromethane solutions
of W2016(dppm)2, km“ = 822 nm, (e = 740 M”1 cm'l); 468 run (4800 M‘1 cm'
1); 387 nm (3200 M‘1 cm'l) (Reference 166).
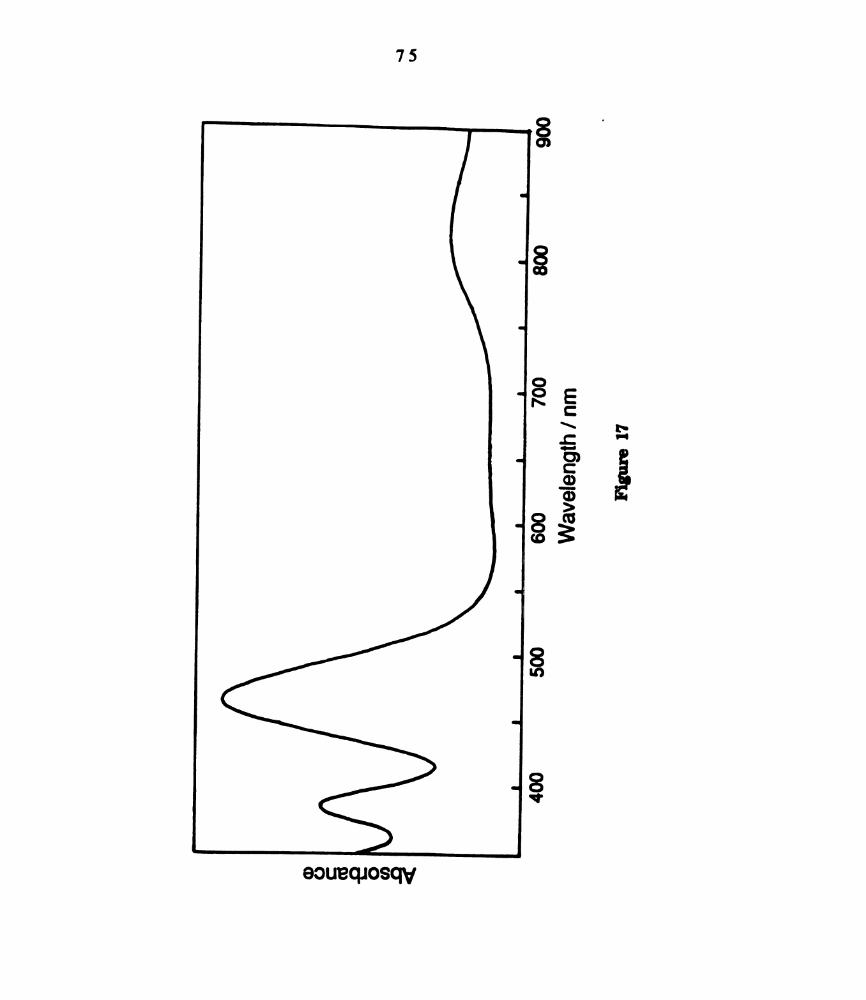
75
:23...—
E:
\£92353
com
com
d
eoueqiosqv

76
Figure 18. Electronic absorption spectrum of dichloromethane solutions
0f M02016(dppm)2.

77
com
39:55
E:
\£95.05;
com
com
oov
_a
_
aoueqiosqv

78
transient spectra of W2014(dppm)2 in benzene exhibits a distinct maxima at
480 nm, which is slightly blue shifted from that of the dimolybdenum
transient. Moreover, the ditungsten transient spectrum exhibits an
additional feature ~ 390 am that is typically observed in the spectra of
M(III)—M(III) edge-sharing bioctahedral complexes [166, 167a].
(Unfortunately, the dimolybdenum transient spectra could not be collected
in this region.) These data support the transient assignment of an edge-
sharing bioctahedral intermediate formed by the foldover of two chlorides to
the edge-bridging positions as depicted in Figure 19.
The relative intensities of the 390 and 480 absorptions of the proposed
edge—bridging bioctahedral transients differ formed from M-LM excitation
differ from those of the stable and known edge-sharing bioctahedral
complexes. This is not unexpecmd on the basis of the variation in electron
counts of the binuclear cores in the transient of MzCl4(PP)2 (ds) and the
M2016(PP)2 edge-sharing bioctahedral complexes (d6) [180]. Furthermore,
there are differences in the coordination numbers of the two species, with
the bioctahedral transient of M2014(PP)2 featuring two vacant coordination
sites (see Figure 19). It is known that the relative intensities of the
absorptions at ~ 390 and ~ 465 nm in the spectra of edge-sharing
bioctahedral complexes vary significantly with coordination sphere [166,
167a], as exemplified the relative intensities of these maxima in the
spectrum of W2015(dppm)2H (6464 = 460 M‘lcm‘l, 6336 = 3300 M'1cm‘1)
compared with those observed in the spectrum of W2013(dppm)2, (8463 = 4800
M’lcm’l 6336 = 3200 M‘1cm’l). Based on this dramatic difference observed in
replacing a chloride with a hydride, it is not surprising to find that the
relative intensities in the spectrum of the coordinatively unsaturated

79
\
Figure 19. Proposed edge—sharing bioctahedral distortion of 1(81t"‘) (or
1(rc8"‘)) excited state of the MzCl4(PP)2 complexes. Although the only the
former is designated in the diagram, the high energy metal localized
transitions of these complexes have not been definitively assigned (see text).
Transients are not observed from the 1(88*) excited state on the nanosecond
time scale. The metal centers which are oxidized and reduced in the
transient species, relative to the ground state, are denoted with + and -,
respectively. (See footnote 180 regarding the M-«M notation).

80
01,, a on,'M:----- 1
Cl/ \Cl/
8*
—_
PllsMI/IIP
TIRMIIP
Cl/
Figure19

81
transient do not directly concur with those in the spectrum of
W2016(dppm)2.
Nonetheless, despite the differences in coordination number and
electronic configurations, the spectral profiles do exhibit common features.
The similarities in the spectral profiles of the d8 transient species and the d°
complexes can be explained by the near degeneracy of their highest
occupied molecular orbitals (HOMO), as indicated by theoretical [181, 167s]
and experimental [167d,e, 175a] studies. The small energy gap between the
HOMO's of the occupied manifold arises from a weak interaction of metal
orbitals of the 8 symmetry which are primarily M—L antibonding in
character. Transitions with this manifold are predicted to lie at very low
energy. Thus the high energy absorptions at ~ 470 and 390 nm of edge-
sharing bioctahedra, in the (16 complexes are likely to lie outside the HOMO
manifold. Thus the visible transitions (likely to be from electron promotion
from o and 1c orbitals, in the (1° and d8 edge—sharing bioctahedra might be
expected to be similar.
An edge-sharing ligand rearrangement of the mixed valence excited
state of M-LM complexes provides stabilization of the oxidized metal center
by achieving an octahedral coordination geometry and a depleted
coordination geometry of halides about the reduced metal center.
Moreover, owing to the assymmetry of the chemically distorted
intermediaite, the mixed valence character is enhanced by this
rearrangement. It should be noted that this structural distortion of
electronically exciwd MzCl4(PP)2 complexes does not occur from the lowest
energy MMCT 1(88") state, but rather from the higher energy l(rt8"') or
1(816") MMCT states. That edge-sharing bioctahedral distortions are not a
photophysical pathway of the 1(88*) excited state could simply be due to

82
insufficient energy. However, an additional feature of the l(8--)rt"') and
l(rt-48'“) excitation that may be important to the formation of the edge-
sharing bioctahedral intermediate is that the metal-metal x bonding
relative to the ground state is significantly weakened. This feature is
expected to enhance formation of a bioctahedral intermediate because
interactions of the metal dyx orbital with the bridging ligands are at the
expense of M—M x interactions. This may well explain why no transients
are formed from 82 -) 1(88") excitation since the M—M x interactions are
unperturbed in the 1(88") excited state relative to the ground state.
Thus a model for the proposed transient absorption studies of the D211
M2014(PP)2 complexes is summarized in Figure 19. Whereas long-lived
transients are not observed on the nanosecond time range upon 82 -) l(88$)
excitation, transients assigned to edge-sharing bioctahedral intermediates
are formed upon excitation of the higher energy 1(8—-)7c*) and l(rt—98'”)
transitions. In the absence of reactive substrates, this intermediate
converts to ground state.
hPhotochemicalb'ActiveSolutiom
Transient species with difi‘erent properties are observed upon 532 nm
excitation of W2014(dppm)2 in the presence of halocarbon solvent substrates.
(The photochemistry observed with these various substrates is described in
Chapter IV). The spectra of solutions containing CH3I and CchHzl were
only measured at wavelengths longer than those absorbed by the substrates
(1. > 430 nm). The initially observed transient spectra with each of the
various substrates are not that of the photoproduct. Moreover the transient
signal decreases with increasing photochemical conversion of the
quadruply bonded species, thereby establishing that the transient is from

83
an intermediate proceeding photochemical reaction. The transient
absorption decay profiles of WzCl4(dppm)2 in CHal (5.0 M)/ CsHs and
011301121 (5.65 M)/ CsHs and neat CH2012 solutions, provided in Figures 20a,
20b, and 20c, respectively, were each collected at 440 nm. In each case a
transient is observed which decays within approximately 1 us to a nonzero
absorption. In the case of CH3I substrate, the transient absorption is
similar to that observed in benzene. Namely, transient spectra recorded 100
ns and 4 us after excitation of the CH31/ CgHg solutions (Figure 21) each
display a dominant feature at 480 nm which is characterisitic of the edge-
sharing bioctahedral complexes. This is not true for the case of CH3CH2I
and CH2012 substrates. The transient spectrum in CH2C12 collected 100 ns
after excitation (Figure 22) exhibits an intense absorption at 380 nm with a
discrete shoulder at 420 nm. The transient spectra with CH2012 and CH31
substrates are most clearly difi'erentiable in the region between 450 and 500
nm. While a sharp increase in absorbance is observed in CHsl, the
transient absorption in CH2012 significantly decreases in this region to
nearly zero at 500 nm (Figure 22). The transient spectra in CH3CH21
solutions collected 100 us after excitation (Figure 23a) do not show any
notable features. At 10 us after excitation (Figure 23b) the absorption shows
a marked decrease between 450 and 500 nm, as observed in CH2012
solutions. Although present data is insufficient for assignment of the
transients in 011,012 and CHscHzl solutions, the spectra clearly do not
exhibit an absorption maximum at ~ 480 nm, which is characteristic of
edge-sharing bioctahedral complexes and they are not consistent with the
photoproduct. We believe that these transients in (311,012 and CH30H2I are
formed with one—electron oxidation of the bimetallic core (vide infra
Chapter IV).

84
Figure 20. Transient kinetics recorded at 440 nm following 532 nm laser
excitation of (a) W2Cl4(dppm)2 (1.5 x 10-4 M) in CH31 (5.0 M)/C,H6. (b)
WzCl4(dppm)2 (2.75 x 10-4 M) in CH30H21 (5.65 M)/C,;H6 (c) W2Cl4(dppm)2
(3.0 x 10-4 M) in CH2012 .
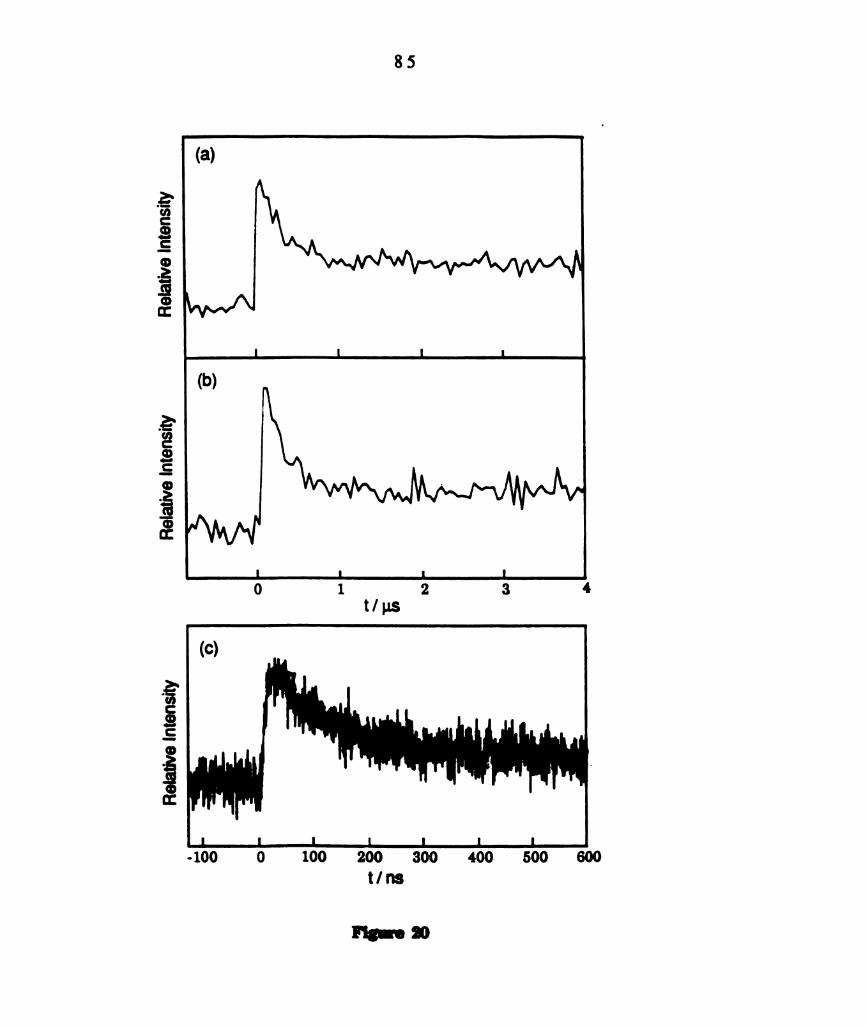
85
(a)
(b)
€325ozfiom
£2.25axiom
t/us
)not
arcs...cause:
100 200 300 400 5000
- 100
t/ns
Flo-'20

86
Figure 21. Transient difference spectrum of W2Cl4(dppm)2 (1.5 x 10" M)
in CH3I (5.0 M)/CGH6 in CSHG recorded 100 ns and 4 us after 532 nm laser
excitation (see legend).
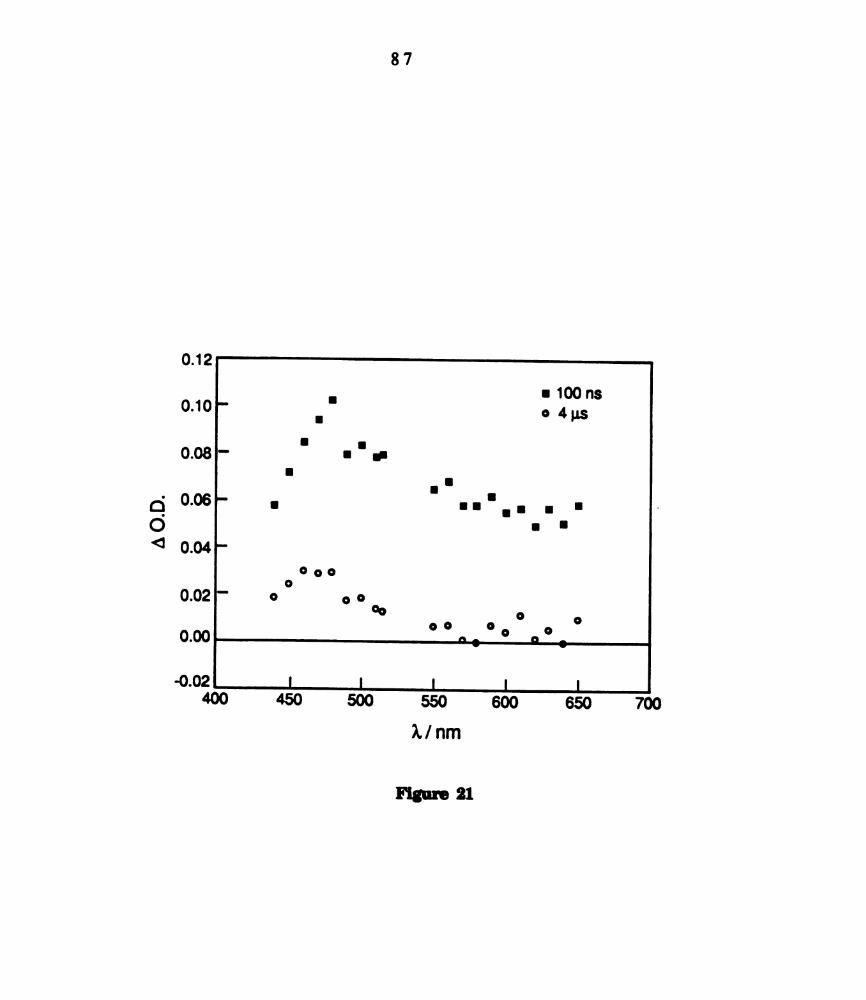
A0D.
87
0.12
. I 100 ns
0.10 I o 4115
0.08 . I '
l I
I
0.06- - . - . . .
. I
0.04
O o o
O
0.02 o 0
° oO 00 o O A O O 0
- 4—L—e—
°0.02 I I l l l
400 450 500 550 600 650 700

88
Figure 22. Transient difference spectrum of dichloromethane solutions of
W2Cl4(dppm)2 (3.0 x 10’11 M) collected 100 us after 532 nm laser excitation.
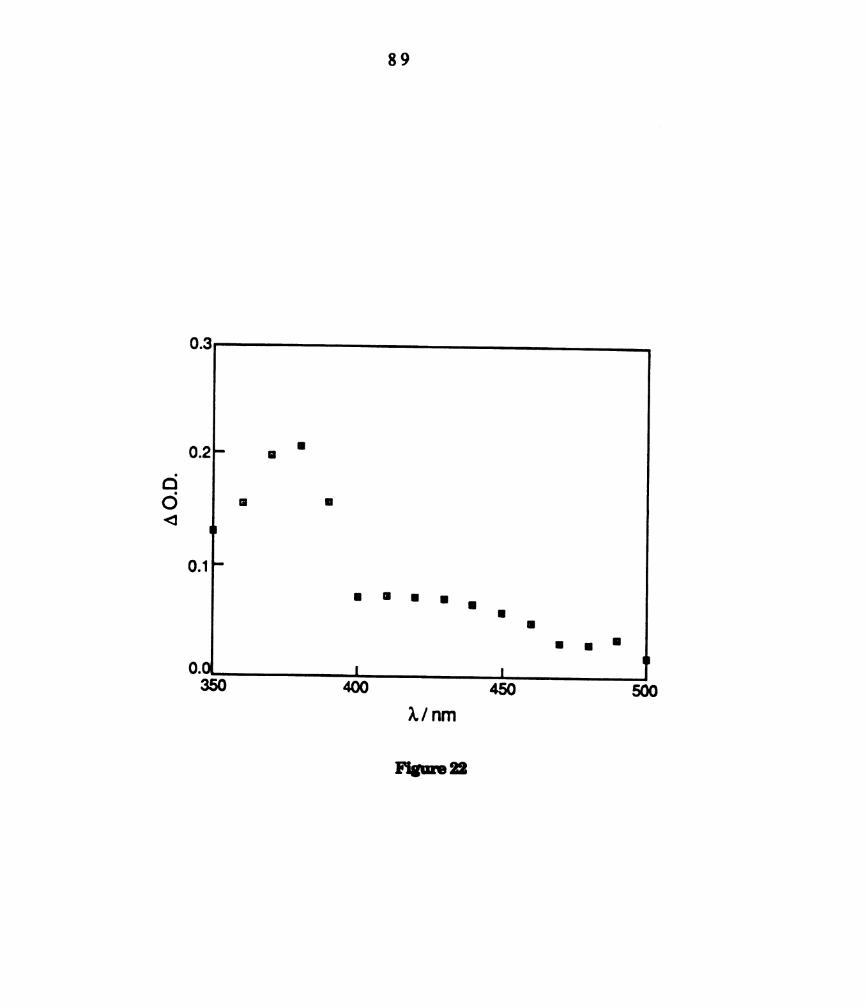
89
0.3
0.2L :1 .
A0.0.
u u

90
Figure23. Transient difference spectrum upon 532 nm laser excitation of
W2014(dppm)2 (2.75 X 10_4 M) in CH3CH21 (5.65 M)/C6H6 recorded (a) 100 us
after excitation (b) 10 us after excitation.

0.10
0.08
0.06
A0.0.
0.04
0.02
0.00
0.015
o“0' 0.010
0.005
0.000
91
(a)

92
2- mmiwccmflm
The transient spectrum of the 1(88"') excited state of M02014(PBu3)‘
has previously been obtained. As expected, the absorption maximum at 450
nm decays with a lifetime that is identical to that of the luminescent
lifetime of the complex. Table I lists the emission quantum yields and
lifetimes for the 1(88,") excited state previously reported for M02014(PR3)4
complexes PR3 = PMe3,PBu3,PEt3, PMezPh, PMePh2 and PHth. The
quantum yields for the series are quite similar except for the case where
PR3 = PMe3, which is an order of magnitude larger. This enhancement of
the luminescent properties of the PMe3 complex is also evidenced in the
lifetime of the 1(88") state. While the lifetime for the PMe3 complex is 135
ns, those of the remainder of the series are typically ~ 10 ns. We have found
that the 1(88") excited state of ditungsten analogues exhibit luminescence as
well. Consistent with the red shift in the 8241(88") absorption, the
ditungsten analogue exhibits an emission band centered at approximately
800 nm, as exemplified by the spectrum of W2014(PMePh2)4 provided in
Figure 24. The 1(88") lifetime and emissive quantum yield of the ditungsten
complex is slightly greater than those of the dimolybedum analogue;
W204(PMePh2)4 (t = 44.6 ns, 4),”In = 0.0437), M02014(PMePh2)4 (‘t = 11.4 ns, item
= 0.0114).
In addition to the luminescent 1(88") excited state, a nonluminescent
transient species has previously been observed upon 8241(88") absorption of
the MozCl4(PBu3)4 complex. We now extend these studies to include series
of dimolybedum and ditungsten MzCl4(PR3)4 complexes with varying with
PR3. The discussion described henceforth is restricted to the transient

93
Table I. Properties ofthe Luminescent 1(88") State ofM2X4(PR3)4‘
PR$ 1018, 300 K) 9cm
PMe3 135 0259
PEts 14 0.013
PBu3 21 0.013
PMePh2 11.4 0011
PHth 0.18 - (a) Data for PR3 = PMe3, PEta, and PBu3 taken fiom reference 118d.
Table 11. Comparison of Structural Properties ofM2X4(PR3), Complexes
and Lifetime of Nonluminescent Transient.
p33 vco (curl-1)ll 6 (deg)‘ M-M-Cl (deg)b M-M-P (deg)b t (ns)
PBu3 2060.3 132 12)
W3 2061.7 132 104.8(2) 1052 (2) 12)
”hp; 20641 118 1122(2) 1023 (1) '
PMegPh 20653 122 108.0(7) 1052(2) 12)
PMePhg 2067.0 136 so
PHPhg 20733 1% 111.0(1) 93.0(1) '
(a) From reference 169. (b) From reference 170. " transient not observed.

94
Figure24. Emission of W2Cl4(PMePh2)4 in toluene solution at ambient
temperature upon 691.5 nm excitation.
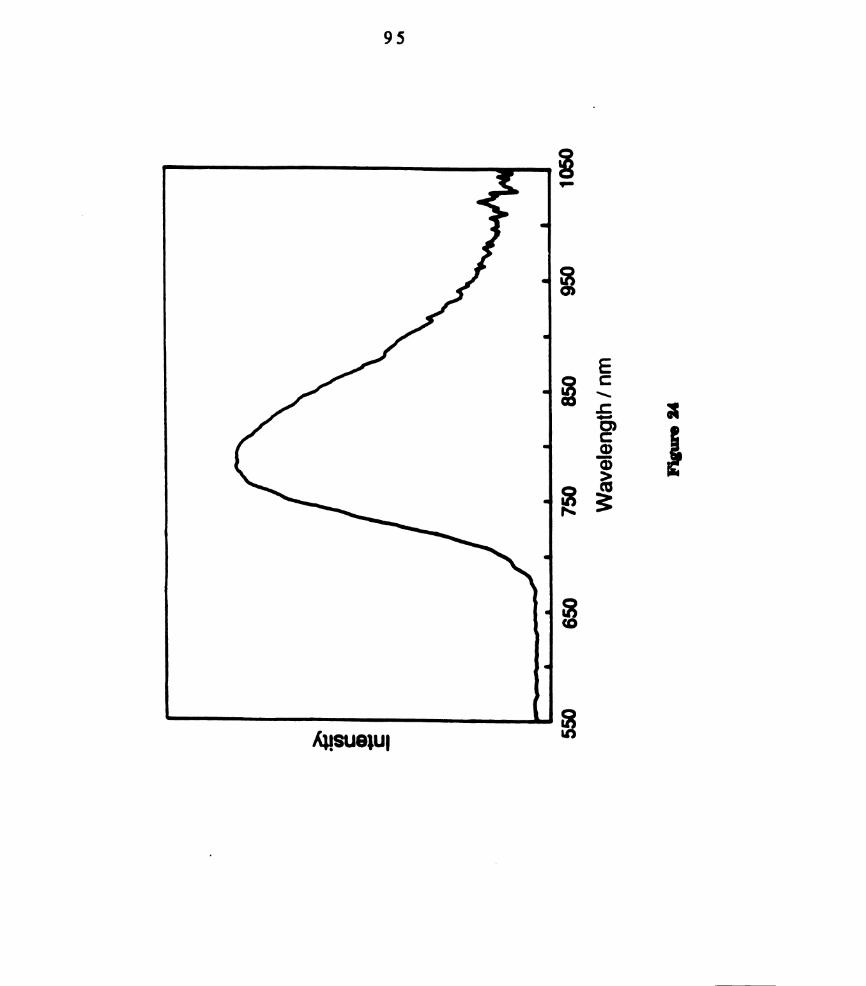
95
wag-8f.—
Ec
\586.353
85
one
Ausueiui

96
species other than the 1(88") excited state, which we will refer to as the
nonluminescent transient detected by nanosecond transient absorption.
Similar to the M02C14(PBu3)4 result, excitation within the 8241(88’)
absorption (Inc: 532 nm) of dichloromethane solutions of M02Cl4(PR3)4
complexes with PR3 = PMOPhg, PMezPh and PEta gives rise to a transient
which exhibits a monoexponential nonradiative decay back to ground state.
With the exception of M02014(PMePh2)4. (t = 80 ns) a lifetime of 120 us is
observed for the transient as indicated in Table II (pg 93) [182]. Each
transient, whose spectrum was recorded 60 as after excitation, exhibits a
maximum at ~ 400 am that is comparable to the reported 390 nm
maximum for M02014(PBu3)4 in CH30N (e = 1080 M'lcm’l). Although we
recorded spectra between 390 and 460 run, an additional very broad feature
(6 a 640 M'lcm‘l) centered at 740 nm and weak absorption at 470 nm (e = 341
M'lcm’l) have also been reported in the full spectrum of M02Cl‘(PBu3)4 in
CHscN. Of the complexes investigated, (i.e. M02C14(PMePh2)4 (4.3 x 10" M)
and M02014(PBu3)4 (3.8 x 10‘3 M), the lifetimes of the transient generated
from dichloromethane solutions do not change upon addition of excess
phosphine to concentrations as high as 0.50 M. Previous picosecond
transient absorption studies have shown that the 1(88") excited state of
M02014(PM63)4 directly decays back to ground state [139]. A nonluminescent
transient species is not observed. Similar results are observed with
picosecond transient absorption spectroscopic studies of M02014(PHPh2)4
upon 580 nm absorption. The only transient species detected is one that
monoexponentially decays back to ground state in 150 ps, whose absorption
spectrum (Figure 25) is consistent with the 1(88") excited state [139].
The W2014(PR3)4 (PR3 = PEta, PBua, PMeth, PM83) series also shows
a transient with properties comparable to that of the dimolybdenum series

97
Figure 25. Transient difference spectrum of WzCl4(PHPh2)4 in CH2012
(~1.0 x 10'2 M) recorded 2 ps after 590 nm laser excitation.
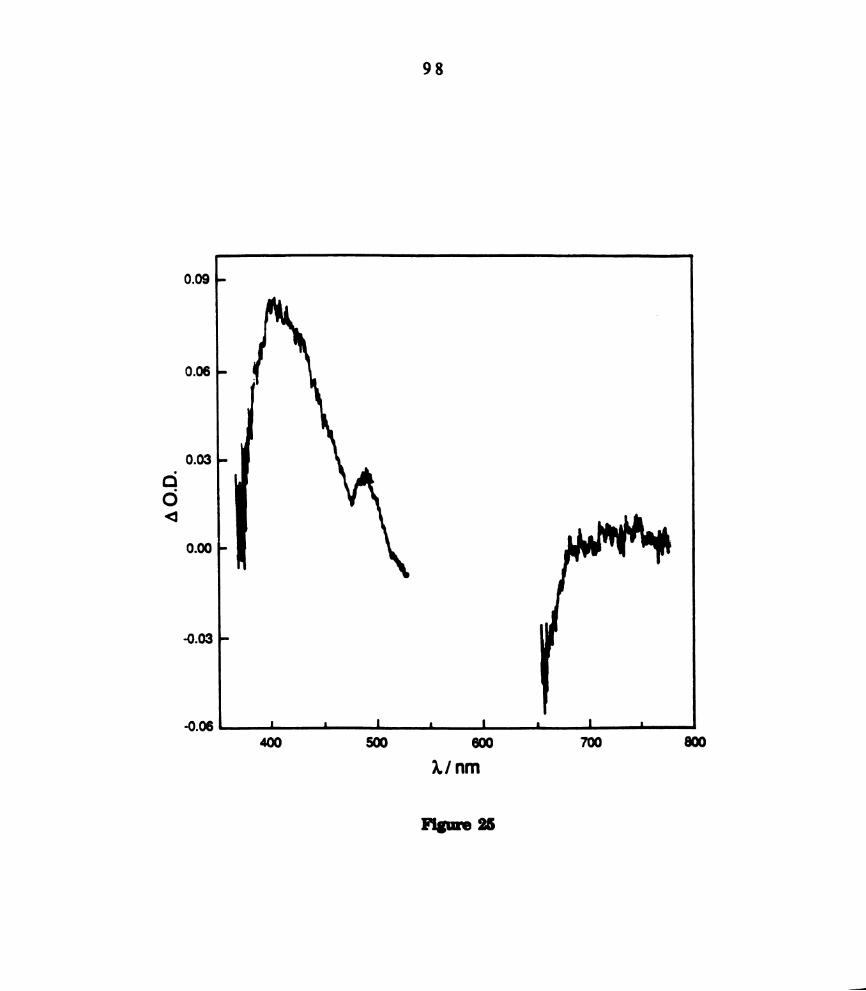
A0.0.
0.09
0.06
0.03
0.00
0.03
0.06
98
8r

99
upon excitations energetically coincident (Ina = 683 nm) with the 8241(88")
absorption. As was the case for dimolybdenum series, the transient decays
nonradiatively and monoexponentially back to the ground state with the
lifetimes that are virtually invariant to the nature of the phosphine.
However, a notable difference between the dimolybdenum and ditungsten
series is that a transient of the latter is observed when PR3 = PMes. The
transient spectra for PR3 = PMeth, PBus and PEts collected 70 ns after
excitation are provided in Figures 26—28, respectively. The spectra are of
varying quality, but all are similar in as much as an absorption in the UV
region is preeminent. Additionally, weaker absorption features in the
visible region of the spectra are noted. These are most clearly seen in the
spectrum of MozCl4(PMePh2)4 in THF solutions, which shows two weak
absorptions at 400 and ~ 475 nm. The latter may resolve into a double
maximum at 460 and 500 nm, but these features are within experimental
error limits, and in the cases of PEt3 and PBu3, are not resolved. A distinct
shoulder at 345 nm is observed on the rising UV absorption in all of the
spectra. This shoulder is most clearly evident in the spectrum of the PEta
complex, which was collected far into the UV region. For clarity, the
spectrum is expanded in an inset. Higher energy excitation of the D2,,
dimolybdenum or ditungsten complexes in hexane does lead to the
production of transients on the nanosecond time scale.
In the case of the dimolybdenum complexes, the occurrence of a
transient appears to be related to the nature of the phosphine. An
important result, and one that was previously observed, is the anomalous
behavior of the PMe3 complex. Excitation of Mo,Cl,(PMe,), produces l(2)15“)
luminescence which directly decays back to ground state. Only the 1(88")
excited state is observed with lifetime of 135 ns, and no additional transient

100
Figure26. Transient difference spectrum of WZCI4(PMePh2)4 in THF (~
6.0 mmol) recorded 70 ns after 683 nm laser excitation.
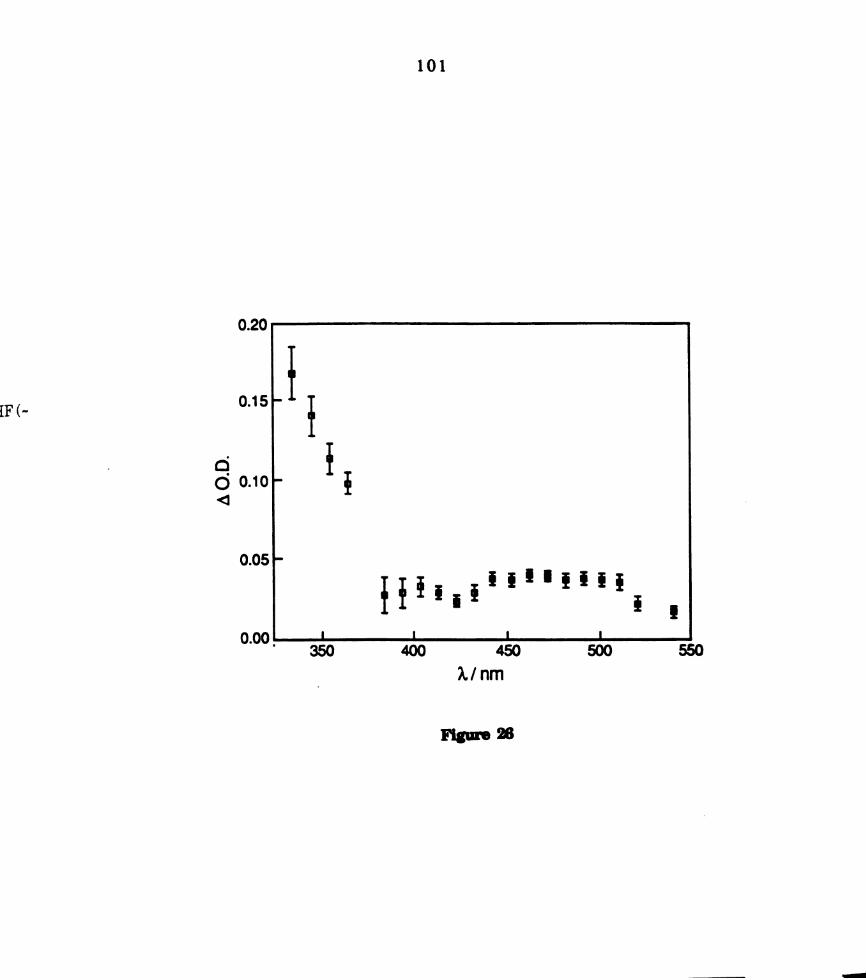
101
0.20
0.15-{i
o: l00.10- I
<1
0.05P !.
ii iii iii!
ii . t ,l l L l
0'00“ 350 400 450 500 550
I/nm

102
Figure 27. Transient difference spectrum of W2014(PBu3)4 in hexane (~ 6.0
mmol) recorded 70 ns after 580 nm laser excitation.
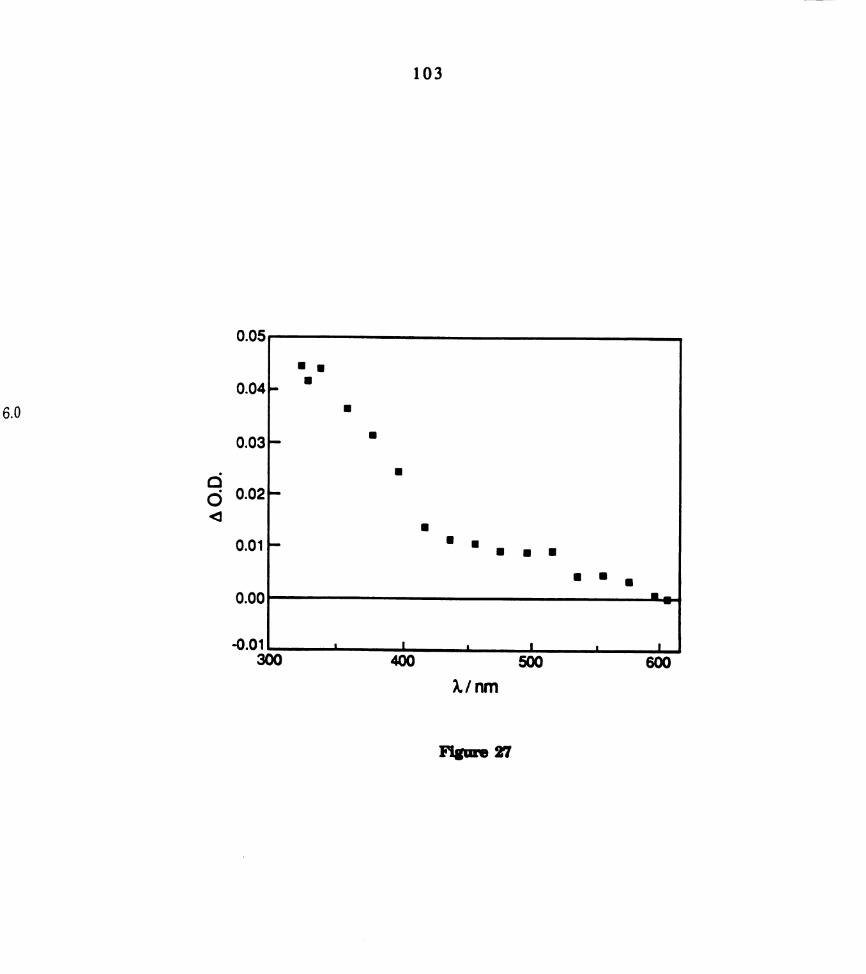
6.0
A0.0.
0.05
0.04
0.03
0.02
0.01
0.00
-0.01
103

104
Figure 28. Transient difference spectrum of WZCI4(PEt3)4 in toluene (~ 6.0
mmol) recorded 70 ns after 683 nm laser excitation.
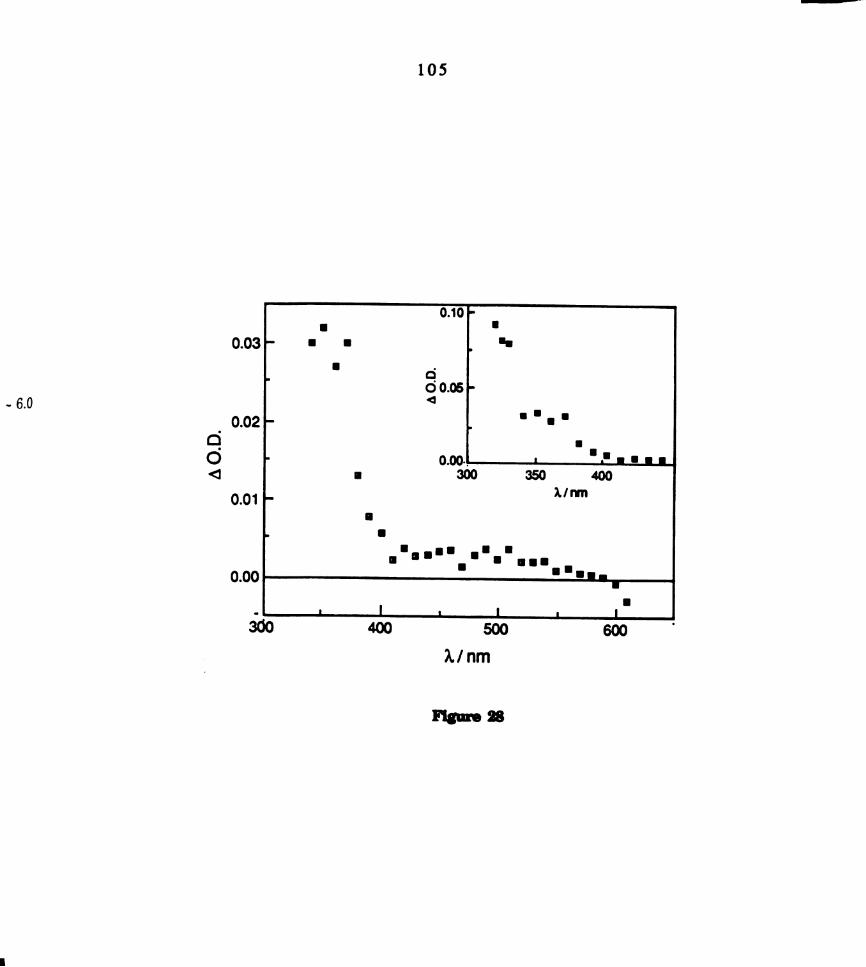
10 5
0.10
I
0.03 '-
O:
O 0.3 P
- 6.0 <
0.02 r ' ' - '
d u
d 0 -- . ' 4' ILL-.1
4 300 350 400
0.01 M"m
I
I I I
ul".u . u.. l
0,00 '4Ia.._.
. l 1 I 1 I .
300 400 500 600

1 0 6
is detected. The absence of a nonluminescent transient from M02C14(PMe3)4
is informative. First, it can account for the fact that the quantum yield of
emission (0.26) and lifetime (140 ns) of the 1(88*) excited state of
M02Cl4(PMe3)4 are each an order of magnitude larger than M02014(PR3)4
complexes from which nonluminescent transients are observed (see Table
I). Moreover, the anamolous behavior of M02014(PMe3)4 and
M02014(PHPh2)4 provides some insight into the electronic and chemical
nature of the transient species generated from the remainder of the series.
The previously proposed assignments of 3(88*) and 3(it8"') seem unlikely
since the slight perturbation upon replacing ethyl in M02014(PEt3)4 with
methyl in MozCl4(PMe3)4, or in replacing a methyl in M02Cl4(PMePh2)4 with
a hydride in M02C14(PHPh2)4 seem insufiicient to significantly alter the
intersystem crossing rate between the 1(88‘) state and these triplet states.
As was the case for the D21, complexes, the transient of dimolybedum
D2,, does exhibit spectral features which are similar to the cl6 edge—sharing
bioctahedral complexes. Most noted is the intense feature at 390 nm
reported of the dimolydenum transient does correlate with that reported for
d° edge-sharing bioctahedral complex M02016(PMePh2)4 at 400 nm (sh) (e =-
2400 M’lcm‘l). However, differences are noted in the lower energy
absorptions of the transient at 470 (e = 341 M‘lcm'l) and 740 nm (e = 640 M‘
lcm'l) relative to those of the d° bioctahedral complex at 526 nm (e = 800 M‘
lcm"1) and 650 nm (e = 640 M'lcm‘l). An alternative bioctahedral
geometry that is typically observed observed upon oxidation of the more
structurally flexible M2X4L4 complexes that do not contain bridging ligands
is a confacial bioctahedron. In fact the edge-sharing MOzCls(PR3)4
complexes have been shown to slowly disproportionate to confacial
M02016(PR3)3 complexes and monomers, indicating that the confacial

107
bioctahedral geometry is thermodynamically favored over the edge-sharing
bioctahedral geometry [175a]. This is also true with the“ ditungsten
analogues. While the edge-sharing W2016(PEt3)4 complex has been isolated
and characterized by X-ray crystallography, “P NMR spectra of crystalline
samples of this complex dissolved in toluene at ambient temperature
exhibit signals attributable to free phosphine and the confacial bioctahedral
WzClg(PEt3)3 complex [153].
Although the distinction between a confacial and edge-sharing
bioctahedral intermediate is difficult for the dimolybdenum complexes,
owing to the similarities in the spectral profiles of M(III)—M(III) confacial
and edge-sharing bioctahedral complexes [183],the spectra of edge-sharing
and confacial bioctahedral ditungsten complexes are distinguishable. The
W2(III,III)Clg(PEt3)4 (D2,!) edge-sharing bioctahedral complex exhibits an
absorption at 390 nm (Figure 7), as do the D211 W2(III,III)ClG(PP)2
analogues. But the d6 confacial bioctahedral complex, W2(III,III)C16(PEt3)3
has a distinct absorption at ~ 330 nm (Figure 29). The prominent feature at
~ 345 in the the d8 ditungsten transient spectrum is in good agreement with
the UV absorption of the d° confacial complex, and is distinguished from
the 390 absorption of the edge-sharing bioctahedral complexes. Moreover
the very weak absorptions of the transient at ~ 475 and 400 nm in the latter
are energetically similar to those of the confacial bioctahedral complex.
However discrepancies are noted in the relative intensities of the bands
which we believe may arise from the difi'erences in electron counts of the d8
transient as compared to the (1‘5 species. For instance the electronic
spectrum of a complex formed by addition of dichloroiodobenzene to
W2C14(PBu3)4 is shown in Figure 30. Although this complex has not been
definitively characterized, its EPR spectrum is consistent with a one-

108
Figure 29. Electronic absorption spectrum of a dichloromethane solution
of the confacial bioctahedral complex W2C16(PEt3)3, km” = 510 nm, (e = 2077
M-1 cm-l); 328 pm (2077 M"1 cm"1).

1 solution
(E = 2077
109
X8
1
600
700
l
500
Wavelength
/nm
300
eoueqiosqv
Figure29

110
Figure 30. Electronic absorption spectrum of the product obtained from
thermal reaction of one half molar equivalent of dichloroiodobenzene to
CH2012 solutions of WzCl4(PBu3)4, 1...... = 500 nm, (e = 969 M-1 cm‘l); 400
nm (588 M-1 cm-l); 330 nm (3322 M‘1 cm-l).

lll
Absorbance
1 l l l l
300 400 500 600
Wavelength / nm
Flames!)

112
electron oxidized (17 species (vide infra, Chapter IV). The absorption
features at 500, 390 and 330 nm, which are energetically similar to those of
the confacial bioctahedral W2(III,III)Cls(PEt3)3 complex, indicate that it has
the typical confacial bioctahedral geometry observed upon oxidation of the
M2Cl4(PR3)4 complexes. The relative intensities of these absorptions of the
d7 complexes are markedly similar to those of the d8 transient species.
These data support the assignment of the transient species as a
confacial bioctahedrally distorted intermediate depicted in Figure 31. The
three chlorides assuming bridging positions and provide the stabilizing
octahedral coordination geometry about the oxidized metal center in the
MMCT excited state.
The similarities in the spectral features of the d8 transient with those
of the d7 and (16 complexes, despite the obvious differences differences in
electronic configuration are consistent with our observations that the
energies of the absorptions of confacial bioctahedral complexes appear to be
somewhat independent of oxidation state. This contention is based on the
spectral changes accompanying the addition of dichloroiodobenzene to
W2014(PBu3)4 shown in Figure 32. A decrease in the 82->1(88’) absorption of
W2Cl4(PBu3)4 and increases in NIR (I. = 1420 nm) and visible absorptions
with maxima at 335, 404, and 430 and 500 nm is observed. Four isosbestic
points are maintained during these changes until the 8241(88') absorption
maxima of the quadruply bonded W2(II,II) species have completely
disappeared. The solid line spectra correspond to the net addition of one
half molar equivalent of the oxidant. The presence of the isosbestic points is
consistent with smooth conversion of the quadruply bonded binuclear
complex to a W2(II,III) intermediate. Subsequent spectral changes upon
further addition of oxidant are denoted with dashed lines. Reaction of the

113
Figure 31. Proposed confacial bioctahedral distortion of the 1(88*) excited
state of WzCl4(PR3)4 complexes. See footnote 180 regarding M---M
designation.
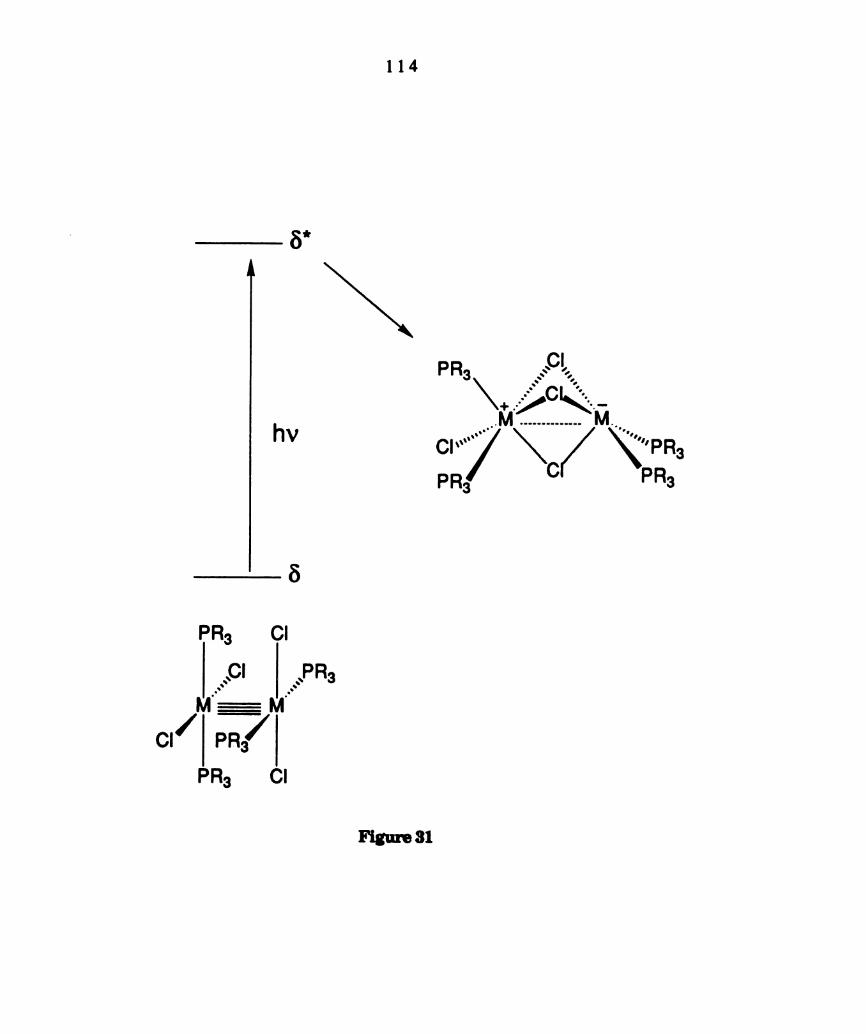
114
8*
“V Cl/\/\P’PZS
8
PR3 Cl
o0! “P123
M_______ M"
Cl/ I PR3
PR3 Cl
Figlne31

115
Figure32. Electronic absorption spectral changes accompanying addition
of dichloroiodobenzene to CH2C12 solutions of WzCl4(PBu3)4, km” =665 nm,
(e = 3813 M"1 cm’l). The solid lines represent net addition of 0.5 equivalent
and the dashed lines represent further addition.
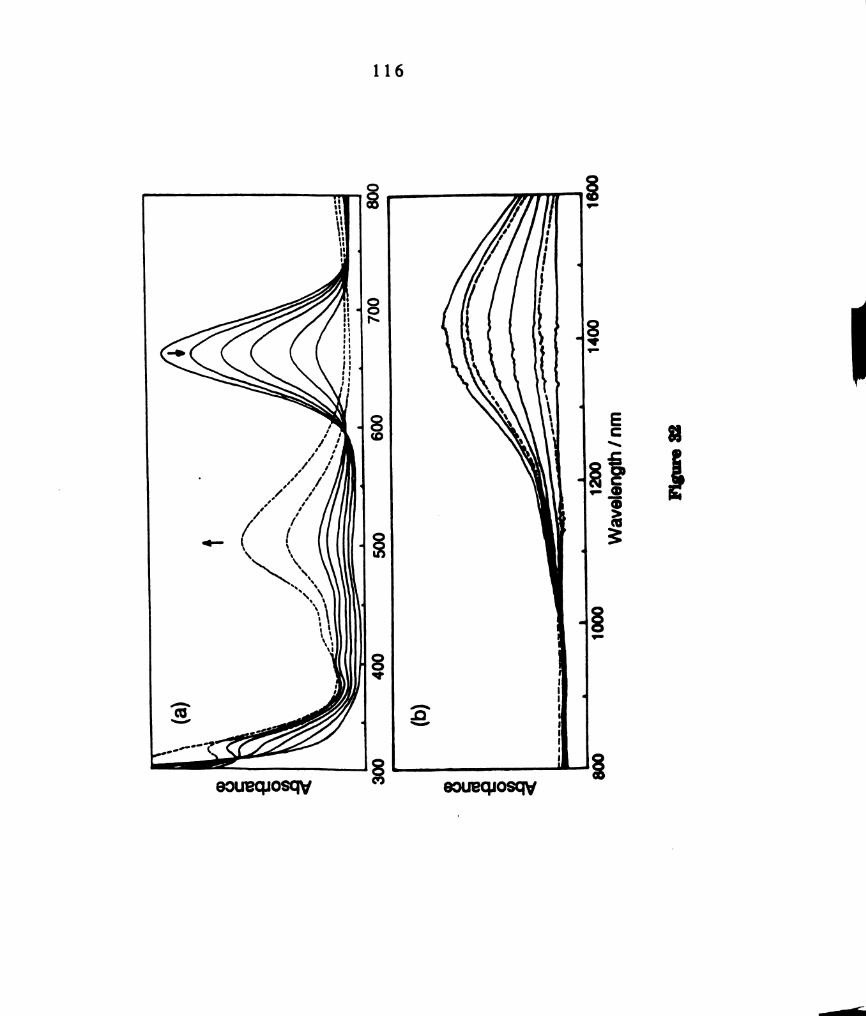
116
89
32.5..—
E:
\255.263
08
pcon
'il
‘
o8
I‘-
I-
int
-‘DII
I'l-
.
eoueqrosqv

117
W2(II,III) intermediate is evidenced by a decrease in the NIR absorption
and loss of all four isosbestic points. The final spectrum shows no NIR
absorption and the visible spectrum is similar to that of the independently
prepared W2(III,III) complex, W2(III,III)Clg(PBu3)3 (Figure 8). Despite the
change in the relative intensities of the visible transitions in the spectra, the
energies of the absorptions remain relatively unperturbed for the
conversion of the W2(II,III) to a W2(III,III) species.
Like the edge-bridging bioctahedra, the visible and UV transitions
are not predicted to involve orbitals within the HOMO—LUMO manifold
[184, 185]. Although the M—M bond in the (16 complexes may be formally
considered a triple bond, the extensive metal-ligand interactions in
complexes with bioctahedral geometries limits the metal-metal
interactions. Thus the energy gap between the HOMOs of (l6 and (18 species
is exceedingly small and transitions within this manifold are predicted to
lie in the NIR. As a point of reference, an absorption at 683 nm in the
spectrum of the M02013“ complex represents a transition from the al' (M—
M o) orbital to a metal-ligand antibonding orbital that lies well above the
LUMO level [185]. Thus the transitions between 330 and 550 nm of the d‘
and (17 complexes, which are comparable to those of the d8 transient species,
should lie well outside the manifold, thereby accounting for the similarities
of the spectra of confacial bioctahedra of difi'erent electron counts.
The transient absorption studies of the Dzd M2014(PR3)4 are
summarized in Figure 31. A bioctahedral intermediate is formed upon
excitation of the lowest energy MMCT 8241(88") transition. The spectra of
the ditungsten transient is consistent with a confacial bioctahedral
intermediate. The spectral similarities of the d6 edge-sharing and
confacial bioctahedral dimolybedum complexes precludes definitive

1 1 8
assignment of the dimolybedum transients, however the dependence of the
transient formation on the nature of the phosphine can be reconciled with
the assignment of either an edge-sharing or confacial bioctahedral
intermediate. Table II lists the cone angles of the phosphines and the CO
frequencies of Ni(CO)3(PR3) complexes which provides a relative measure of
the basicity of the phosphine [186]. Inspection of these data reveal that the
basicity of the phosphine is not an important controlling factor. Namely,
the complexes from which no nonluminescent transient is observed
M02014(PMe3)4 and MOZCI4(PHPh2)4, are ligated by one the most basic
phosphines, PMe3, and least basic phosphines, PHth. Similarily there is
no obvious correlation with cone angle of the phosphine. Although the cone
angles of PMe3, is the smallest in the series, the cone angle of PHPh2 is
larger that that of PMezPh. Moreover there is no interrelation between
these properties and transient formation. As discussed by Tolman, the
lability of the phosphines dependence can be correlated to both steric and
electronic effects on the basis of this plot [186]. Figure 33 which shows a plot
of cone angle vs CO stretching frequency of the Ni(CO)3(PR3) complexes.
The least labile phosphines located at the lower left hand corner of the plot
are strongly basic and sterically unhindered. The positions of PMe3 and
PHPh2 ligands on this plot indicate that phosphine dissociation is not a
controlling factor governing from formation of the transient. There is
however, a correlation of the transient with Mo—Mo—P and Mo—Mo—Cl
bond angles of the quadruply bonded dimers. Inspection of Table II reveals
that nonluminescent transients are not observed for complexes that have
the smallest Mo—Mo—P angle and the largest Mo—Mo—Cl angle. This
correlation may be rationalized in the context of a bioctahedral distortion
which involves the bend-over of chlorides to the bridging positions (is a

119
Figure33. Plot of phosphine v(CO) stetch vs cone angle of phosphines (a)
PBu3, (b) PEt3, (c) PMe3, (d) PMezPh, (e) PMeth, (0 PHth. Photoinduced
chemical intermediates are not observed from MOZCI4(PR3)4 where PR3 =
PMe3 and PHth , which are marked with *.
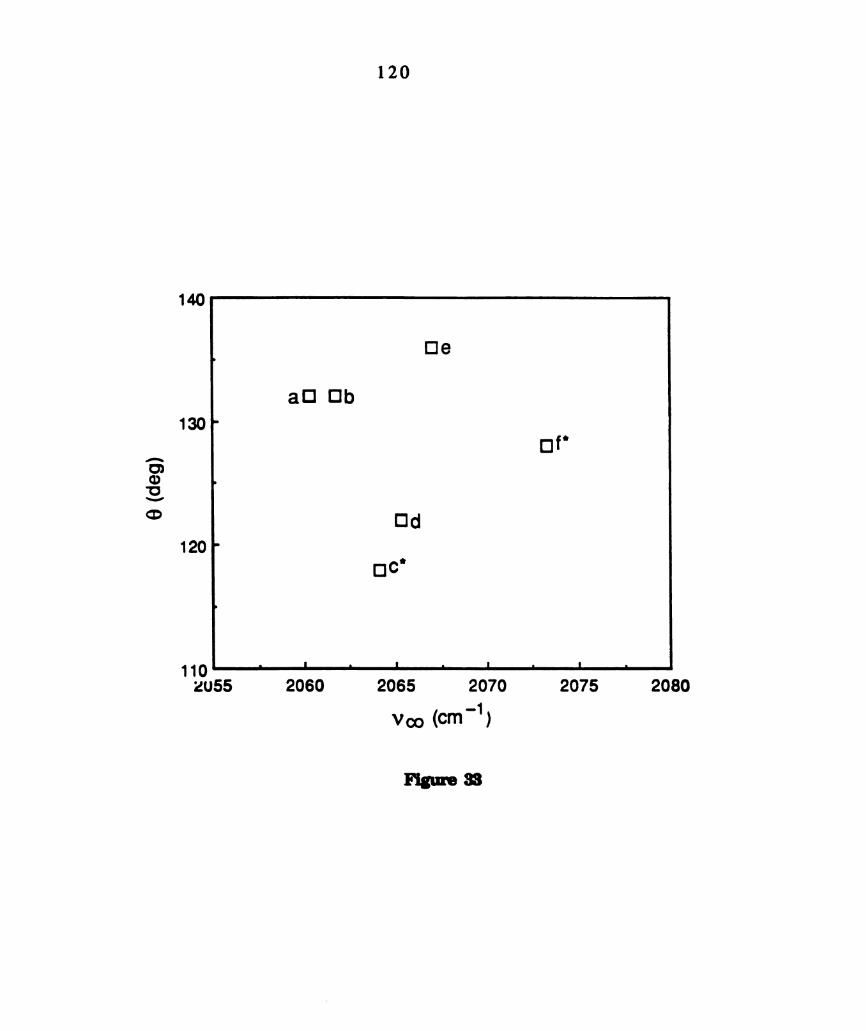
0(deg)
120
140
Be
an Elb
130i
Elf'
Eld
120 -
:10"
110 l a l x l a l
2055 2060 2065 2070 2075
V00 (cm")
W33
2080

121
decrease in the Mo—Mo—Cl angle) and concomitant increase in the Mo—
Mo—P angle. Namely, transients are not observed from complexes with
the greatest bond M—M—Cl angles and smallest M—M—P bond angles,
where a greater degree of structural distortion is predicted. No additional
transients species are observed on the nanosecond time scale with either
the dimolydenum nor the ditungsten D2,, M2014(PR3)4 complexes upon
higher energy excitation.
These results are in contrast to the transient absorption studies of the
MzCl4(PP)2 (D2).) complexes summarized in Figure 19. The differences in
the excited state properties of the D24 and D211 complexes can be accounted
for by the variation of their chemical and electronic structure. In the case
of the M2Cl4(PP)2 complexes, a confacial geometry is precluded by the
bridging bidentate phosphines. Thus the absence of transient species upon
8241(88’) absorption is consistent the assignment of a confacial
bioctahedral intermediate in the case of the D2,, M2014(PR3)4 complexes.
However, chemical structure cannot account for the differences observed in
the high energy states. Both edge-sharing MZCI4(PP)2X2 (D2).) and
MzCl4(PR3)4X2 (Dzd) complexes exist and thus the edge-sharing bioctahedral
intermediate could be achieved by simple foldover of chlorides to the
bridging position in both the electronically excited D2,, and D21, complexes.
That this distortion occurs upon high energy absorption of the the latter
complexes and not the D2,, complexes, can be explained by difl'erences in the
electronic nature of the excited states. Ligand-to-metal charge transfer
transitions are present in the high energy spectral region of the Dzd
complexes, while transitions at this energy in the spectra of the 132}.
complexes are clearly metal localized and are energetically coincident with
1(rt-48'") and 1(8-->x"') transitions. The charge transfer between metal

122
centers in the metal localized transitions of the D211 complexes yields a
mixed valence metal core. The observed edge-sharing bioctahedral
distortion provides cooperative stabilization ofboth the oxidized and reduced
metal centers in the mixed valence bimetallic core. In contrast, the net
one-electron reduction of the metal core which results from the LMCT
transition of the Dzd complexes does not facilitate formation of biOctahedral
type geometries that stabilize Mo(III) and W(III) centers. This model is
supported by the studies of the D2,, and D21, isomers of M02014(PMePh2)4
described below.
3. MW),(DalDu) learners
The green isomer of MozCl4(PMePh2)4 has been proposed to have a D2,,
configuration of phosphine ligands [148, 188]. This hypothesis is supported
by Figure 34 which displays the absorption spectrum ofTHF solutions of the
structurally characterized blue Dzd isomer M02C14(PMePh2)4 superimposed
over the spectrum of THF solutions containing the green isomer. While the
lowest energy 82 -) l(88") transition of both isomers is centered at 600 nm,
there are several differences in the spectra. First, the spectrum of the
solution containing the green isomer displays an absorption band at 450 nm
that is not observed in the spectrum of the Dad isomer. This distinguishing
absorption is comparable to those centered at 462 and 470 nm in the spectra
of M2014(dppm)2 and B—M2014(S,S,-dppb)2 respectively. This transition
appears to be characteristic of D2,, and D2 complexes and, as discussed, has
been tentatively assigned to 8 —) 8x2_y2. Second the relative intensities of the
82 —) l(88"') absorption band and the transition centered at 325 nm clearly
vary for the two solutions. As discussed, this transition has been assigned
as a LMCT in the case of the D24 complexes based on the characteristic blue

123
Figure 34. Electronic absorption spectrum of THF solutions of D2,,
MozCl4(PMePh2)4 (—) and green isomer of MozCl4(PMePh2)4 (-----) that is
proposed to have D21, configuation of phosphines.
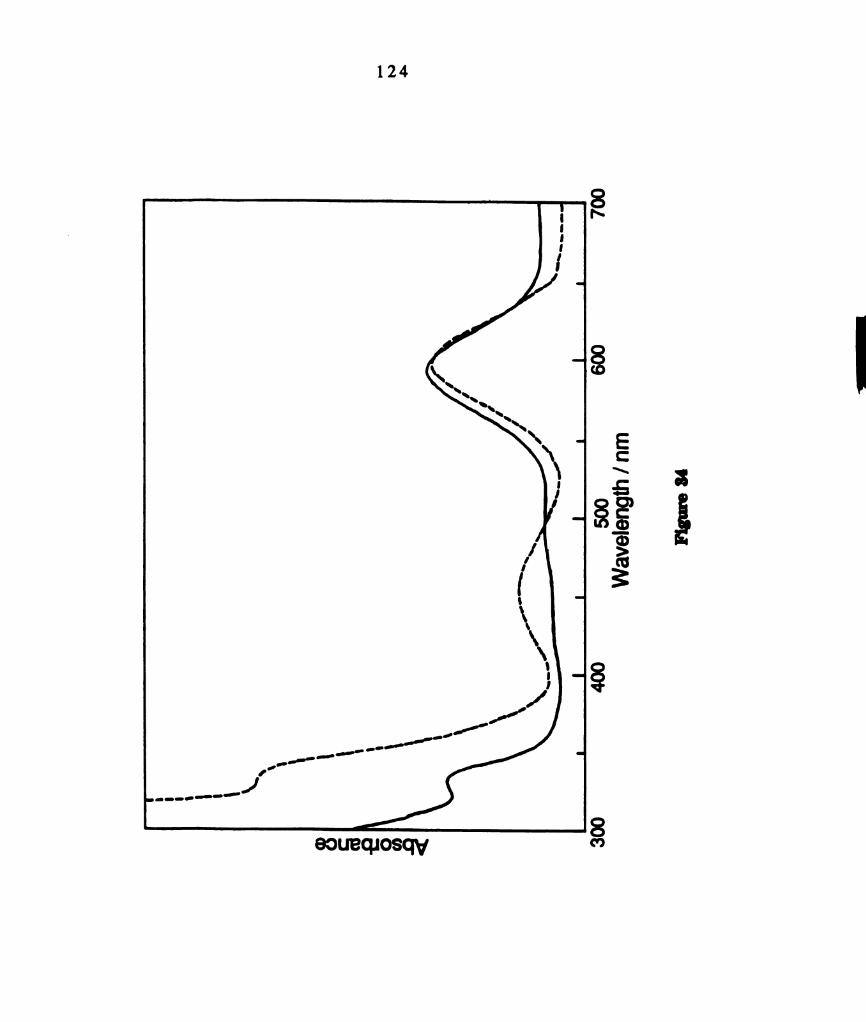
124
39:53
E:
\589935
com
b------’
eoueqiosqv

125
shift in the spectrum of the ditungsten analogue. The relative intensifies of
the absorption maximized at 330 nm (A = 2.1) and the 82 —) 1(88*) transition
(A = 1.0) in the spectrum of the green isomer are comparable to those of
analogous transitions at 325 nm (e = 5600 M'lcm“1) and 634 nm (e = 2490 M”
1cm“) in the spectrum of MozCl4(dppm)2 (Figure 15). The lower energy
transition in the spectrum of Mo2Cl4(dppm)2 is clearly the 82 —) 1(88*)
absorption band and, as discussed, the higher energy transition is metal
localized as well.
Excitation of a THF solution containing only the blue isomer, gives
rise to an emissive transient observed upon 355 nm whose energy (km, =
460 nm) is consistent with the reported emissive localized excited state of
the ligand [179]. No additional transient is observed. As expected, high
energy ligand-based emission is also observed upon excitation at 355 nm of
a THF solution containing the green isomer of M02014(PMePh2)4, however,
in contrast to solutions containing only the Dzd isomer, an additional non-
luminescent transient is also observed that decays monoexponentially back
to the ground state with a lifetime (1 = 3 us) and absorption (11,,“ = 520 nm)
provided in Figure 35 which is virtually identical to that of M02014(dmpm)2
transient. These results directly concur with those observed upon high
energy excitation of the D24 M201‘(PR3)4 complexes and the D2,, M2014(PP)2
complexes; namely, nonluminescent transients are only observed on the
nanosecond time scale with the D2,, complex and the transient spectrum is
consistent with an edge-sharing bioctahedral intermediate.

126
Figure35. Transient difference spectrum of green isomer of
MozCl4(PMePh2)4 in THF collected 1 us after 355 nm laser excitation
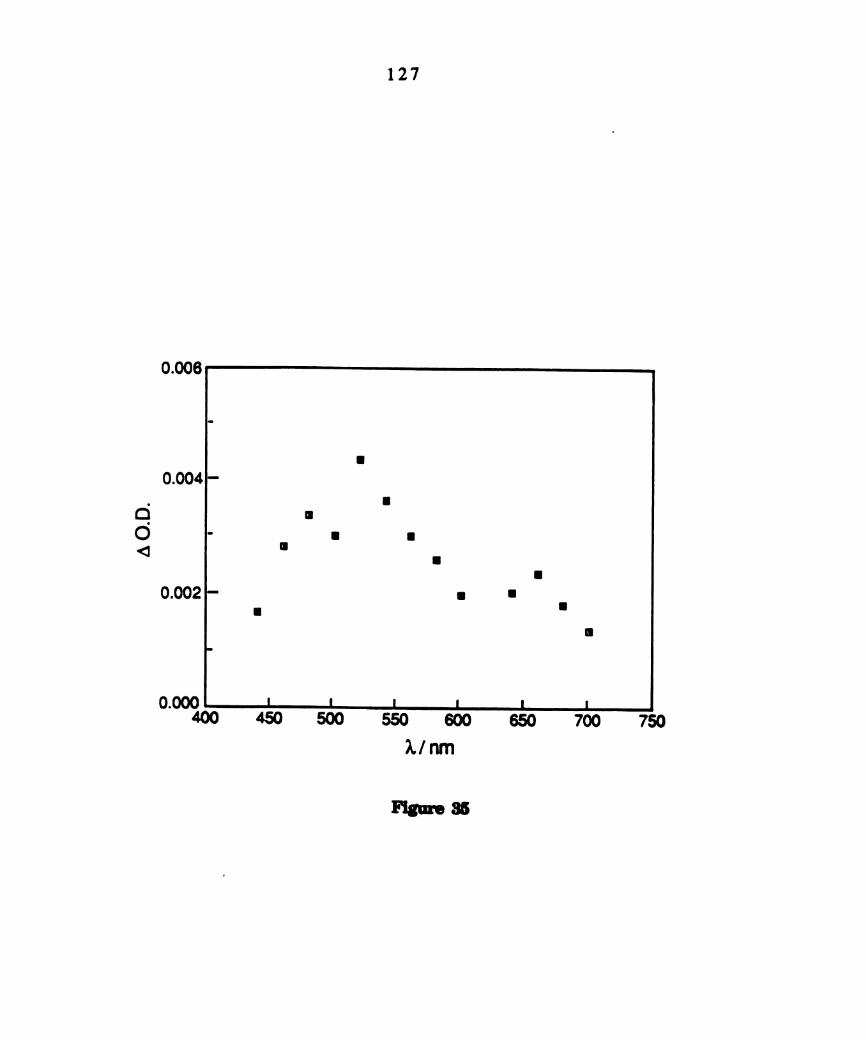
A0.0.
0.006
0.004
0.002
0.000
127
l l I - 550 600 650 700
Mnm
750

CHAPT‘ERIV
PHOTOINDUCEDREDOXCHEMISTRY
A. BACKGROUND
The photochemistry of M-LM complexes is generally characterized by
one—electron reactions promoted by ultraviolet irradiation. For instance,
despite long lived 1(88”) excited states for the M2Cl4(PR3)4 complexes, the
single photochemical reaction of these complexes reported to date requires
high energy excitation. Photolysis of CH2012 solutions of MozCl4(PEt3)4 at
254 nm yields products that are believed to be trichlorobridged binuclear
species [189]. Until very recently, the requirement for ultraviolet
irradiation in photochemistry of quadruply bonded metal-metal binuclear
complexes was completely general. Under these conditions, electronically
excited M02018" [1901,Mo2(so,),4- [191], M02(I-IP04)4" [192a], and Mo,(eq)‘*
(aq = H20) [190], in acidic media are capable of the two electron reduction of
protons yielding dihydrogen. The photochemistry of "M0203" has been
generalized with a study of M02(HP04)4“. The mechanism for H2
production involves two one-electron oxidations of independent bimetallic
cores. Detailed electronic absorption spectroscopy and action spectra
128

129
cores. Detailed electronic absorption spectroscopy and action spectra
identify the photoactivated state in this class of molecules'to be "(1trt*)"
[192a]. In this regard, the two electron reduction of dichloroethane to
ethylene effected upon 1(88*) irradiation of M02[02P(OCGH5)2]4 marks an
important development in the design of photochemical schemes of M-LM
complexes, because it represents the first example of multielectron
photochemistry accessible in the visible spectral region [191b].
Nevertheless, the overall transformation is again achieved by coupling the
redox function of independent metal cores. The final products are the
M02(II,III) mixed valence species, M02[(OCsH5)2]4Cl, and ethylene.
The plethora of thermal two electron oxidation M2014(PP)2 complexes
with strong oxidants such as halogens (Xz) [167] suggests that two electron
chemistry can be promoted at a discrete bimetallic core. The
thermodynamic accessibility of two—electron oxidation products may in
part from the structural rearrangement of the ligation sphere to a
bioctahedral geometry. Every oxidation reaction of the M2014(PP)2
complexes reported to date gives rise to an edge-sharing bioctahedron,
although in some cases products are obtained that do not correspond to
simple addition of substrate. For example, the edge-sharing bioctahedral
complex MozClg(dppm)2(SPh) has been structurally characterized from
reactions of MozCl4(dppm)2 with PhSSPh [167b]. The prevalence of
alternative pathways not involving simple addition of substrate is even
more prominent in the thermal oxidation chemistry of the M2014(PR3)4
complexes. Reaction of toluene solutions of M02I4(PMe3)4 with 12 results in
formation of the confacial bioctahedral complex M0217(PMe3)2‘, which is
deficient in phosphine [170b]. A similar product M02017(PMe3)2' is obtained
from reactions of MozCl4(PMe3)4 with PhIClz in CHzClz. In the case of the

1 3 0
thermal reaction of M02014(PEt3)4 with C014, all of phosphines are displaced
to yield M020193' [193]. Nonetheless, these reactions indicate that a
confacial bioctahedral geometry is important for stabilization of the metal
core upon its oxidation. This is most clearly demonstrated by a
comprehensive study of electrochemistry and photochemistry of Rezclaz'
[173]. One-electron oxidation of quadruply bonded Re2(III,III)Clsz' in the
presence of excess Cl‘ yields the confacial bioctahedral R02(III,N)0192-
complex. Similar chemistry may be affected photochemically upon
82-)1(88‘) absorption of Re20182'. One electron quenching of the 1(88‘)
excited state by acceptors TCNE (tetracyanoethylene) or DDQ (2,3-dichloro-
5,6-dicyano-1,4-benzoquinone) followed by trapping with a chloride anion
results in production of R82C192- [173].
The importance of bioctahedral geometries to the design of
multielectron pathways is presaged by the transient absorption
spectroscopy of MZCl4(PR3)4 and M2C14(PP)2 complexes presented in Chapter
III. Photoreactions of these structurally distorted bioctahedral
intermediates with various substrates is described in this chapter.
Independent electrochemical studies provide some insight into the
mechanisms of the photochemistry.
BRIBULTS
1. Photo-oxidationChemistryAccompeniedbyPhosphineDisplacement
a. “EDWINd W4CI4M4With@2013
Although dichloromethane solutions of W2014(PBu3)4 irradiated with
low energy light (A > 610 nm) are stable indefinitely, photolysis does proceed

131
when the LMCT transition is excited (I > 375 nm). The spectral changes
associated with the photochemistry are shown in Figure 36. A decrease in
the 8241(88‘) absorption maximum (8 = 3813 M‘1cm‘1) of the quadruply
bonded complex WZCl4(PBu3)4 is accompanied by the appearance of
absorptions exhibiting maxima at 1420 nm (e = 1628 M‘lcm‘l), 335 nm (e =
3164 M-‘cm-l), 404 nm (e = 744 M-1cm'1)and 430 nm (e = 725 M-‘cm-l).
Four isosbestic points are maintained throughout the course of the reaction
indicating that the reaction involves smooth conversion to a single product.
Although ensuing thermal decomposition of the photoproduct has
prevented definitive characterization of the photoproduct, its EPR,
electronic absorption, and NMR spectrum is informative in this regard.
The EPR spectrum of the photoproduct in a 2-MeTHF/CH2C12 glass at
77K (Figure 37) is consistent with a paramagnetic species in an axial
environment, g" = 2.020, g1: 1.835. The paramagnetism of the photoproduct
is indicative of a W2(II,III) species having one unpaired electron, since a
two-electron oxidized complex is diamagnetic. Whereas the photoproduct is
a W2(II,III) core, its electronic absorption spectrum varies significantly
from that of the mixed-valence species, W2(II,III)C14(PBu3)+, which retains
the D2,, structure of its quadruply bonded counterpart. The NIR transition
of the photoproduct (21,," = 1420 nm, e = 1628 M‘lcm‘l) is red-shifted and
less intense than that of W2(II,III)C14(PBu3)4'*' (Am, = 1510 nm, e = 2243 M‘
lcm“) whose spectrum is shown in Figure 10. Conversely, the spectral
features of the photoproduct are very similar to the tentatively assigned
confacial bioctahedral W2(II,III) intermediate formed by addition of
dichloroiodobenzene to WzCl4(PBu3)4 presented in Chapter III (Figure 29).
Further support for the production of a confacial bioctahedron comes
from electrochemical trapping studies. The cyclic voltammogram of

132
Figure36. Electronic absorption spectral changes during photolysis (Inc
> 375 nm) of deoxygenated dichloromethane solutions of WZCI4(PBu3)4 at
22°C. The total time of photolysis was 20 minutes.
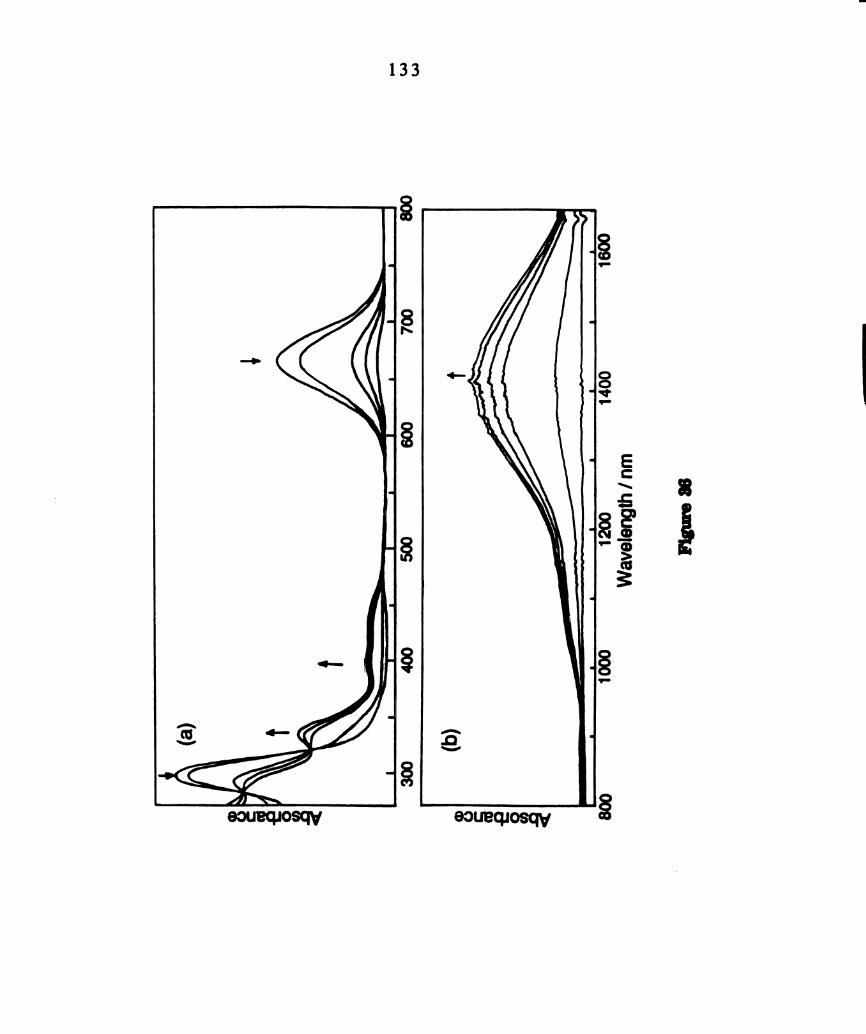
133
82.6...—
E:
\5929:;
8m—
83
eoueqiosqveoueqiosqv

134
Figure 37. X-band (9.598 GHz) EPR spectrum of a 2-MeTHF/CH2012 glass
of photolyzed CH2012 solutions ofW2014(PBu3)4 at 77°K.
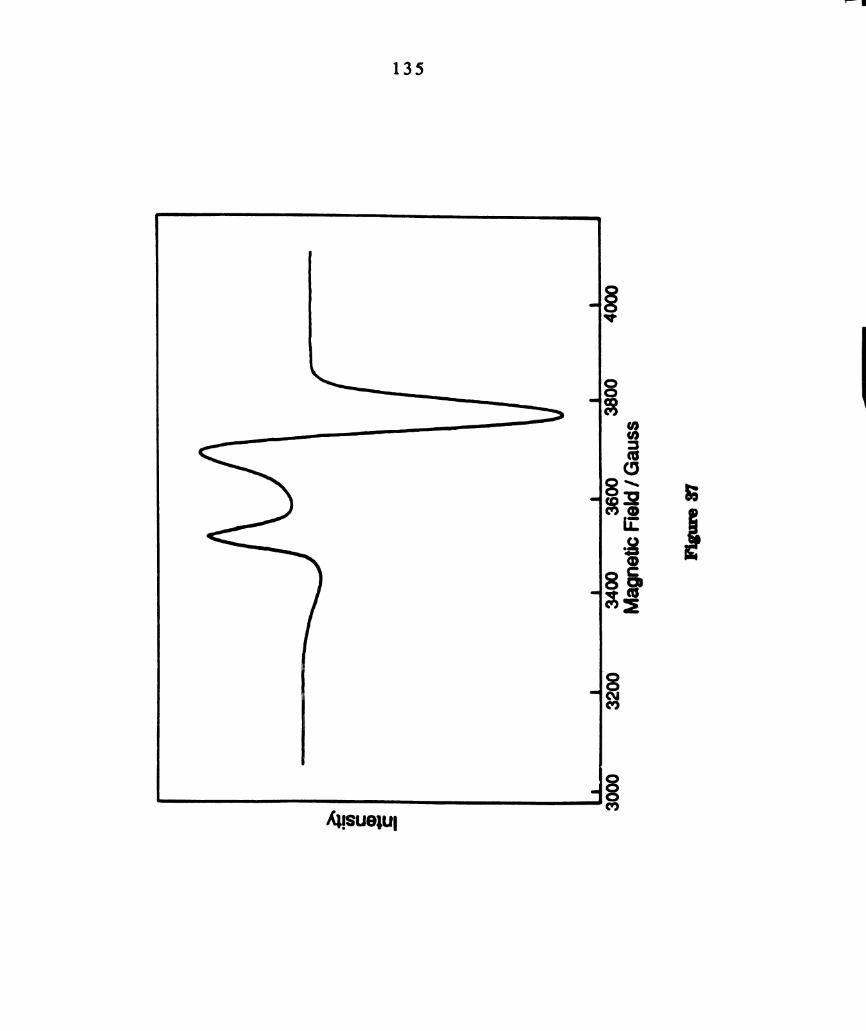
135
5958—:—
mmamo
\Romegages.
comm
Gown
—1
8mm
1
800
Ausueiui

136
photolyzed solution exhibits a reduction wave at -1.15 V and irreversible
oxidation waves at +0.14 V and +0.36 V (Figure 38c). This cyclic
voltammogram is similar to the that of a dichloromethane solution of
W2Cl4(PBu3)4 containing tetrabutylammonium chloride shown in Figure
38b. In the presence of C1‘ the cathodic component of the W2CI4(PBu3)4*’°
redox wave at —0.35 V (figure 38a) significantly decreases and new anodic
waves at +0.14 V and +0.36 V along with a reversible cathodic wave at -1.15
V are generated. Identical changes are observed in the cyclic
voltammogram when chloride anion is added to bulk electrolyzed solutions
of W2(II,III)C14(PBu3)‘+. Moreover, the UV-visible spectral profile of these
solutions show the 1420, 403 and 430 nm features of the photoproduct. The
observed electrochemistry is consistent with the thermal reaction studies of
W2014(PBu3)‘ in Chapter III. One electron oxidation of quadruply bonded
species in the presence of chloride typically yields confacial bioctahedra
species. For instance electrochemical oxidation of Re2(III,III)C182‘ in the
presence of 01-, yields the Re2(III,IV)0192- complex [173]. In the case of
M2Cl4(PR3)4 complexes, oxidation of the complex with subsequent trapping
by halide is usually accompanied by a concommitant displacement of
phosphine. For instance, the reaction of M02014(PMe3)4 with PhIC12 and
M021‘(PMe3)4 with 12 yield M02Cl1(PMe3)2' and M0217(PMe3)2', respectively
[170]. In these reactions simple halide addition to yield M2X5(PR3)4
confacial structures is supplanted by the addition of excess halide to the
core. Phosphine dissociation occurs to maintain the confacial bioctahedron
geometry. This also appears to be the case in the photooxidation reaction.
The spectral similarities of the electrochemical oxidation product
photoproduct and the product of the oxidation of W2014(PBu3)4 by P111012
suggest the photoproduct to be W2(II,III)C13(PBu3)3‘ or

137
Figure 38. Cyclic voltammograms of CH2C12 solutions with 0.1M TBAPF6
of (a) wzcmpsug)4 (b) W2Cl4(PBu3)4 and 3.2 x 10-2 M THACl (c) photolyzed
solutions of W2Cl4(PBu3)4.
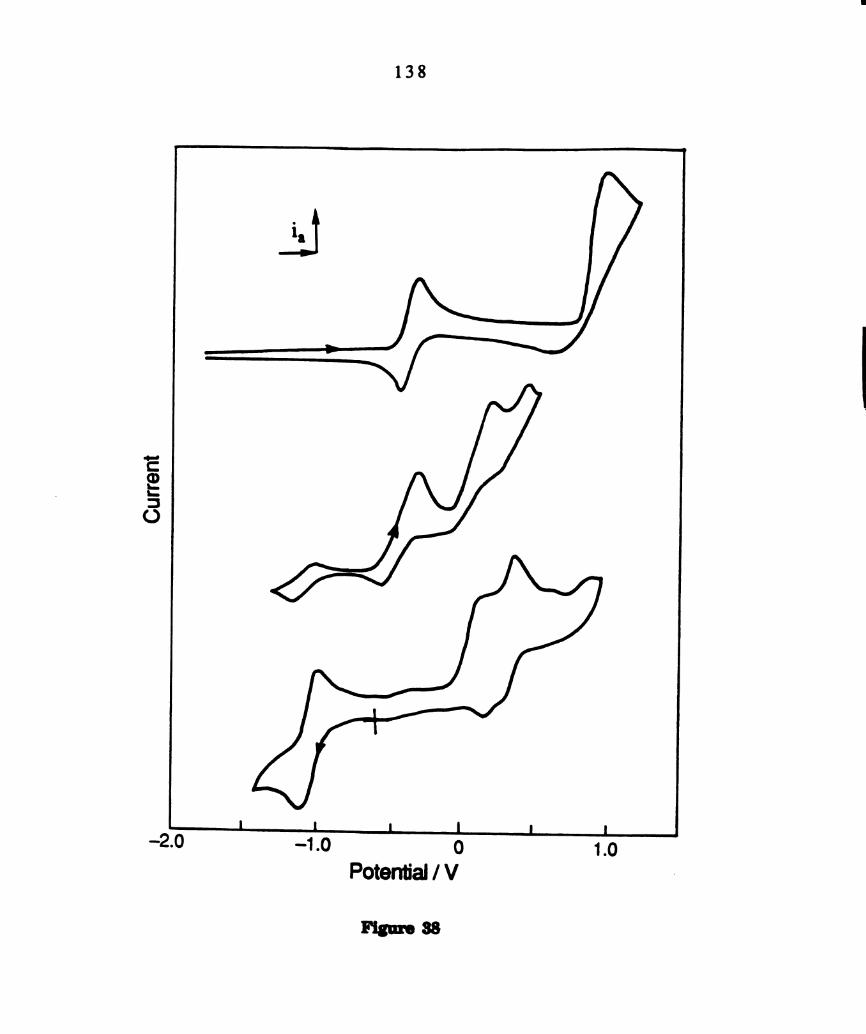
138
Current
—2.0 -1.0 0
Potential / V
“mass

139
W2(II,III)C17(PBu3)2’. In this case free phosphine should be observed in
the photoreaction mixture. Accordingly, NMR studies were undertaken.
Figure 39a shows the 1H NMR specrum of photolymd solutions. Very
weak and broad resonances of the protons of the phosphine ligands between
2.8 and 4.2 ppm are further evidence of a paramagnetic W2(II,III) center.
Similarly, very weak and broad 31P signals are observed for coordinated
phosphine (Figure 39b). Of greater interest is an intense peak at +34.4 ppm
(vs 85% H3PO4). This signal is comparable to that observed for the
chloromethyltrimethylphosphonium cation (5 = 433.3 ppm) [194].
Unfortunately, the tributyl derivative has not been reported, however it
appears that the chemical shift does not vary significantly with the
substituents on the phosphine, as exemplified by the resonances of various
diphenyl derivatives [thRPCHzClP which are all in a region ~30 ppm
comparable to that of the trimethyl analogue [195]. The intensity ofthe peak
relative to those of the other signals indicates that [Bu3P(3I-12Cl]+ exists free
in solution, and thus provides evidence for phosphine displacement during
the photoreaction.
Although it remains for the photoproduct to be definitively
characterized by X-ray analysis, the issue of primary interest to us is that
the photolysis involves one-electron oxidation of the bimetallic core. Indeed
the one-electron reduction of the core upon ligand-to-metal charge
transfer would facilitate such reactivity. This one-electron photochemistry
arising from LMCT absorption is in contrast to photoreactions of
M02014(PR3)4 complexes with PhSSPh which proceed from MMCT 5241(88’)
absorption.

140
Figure 39. (a) 1H and (b) 31P NMR spectra of products from photoreactions
of W2Cl4(PBu3)4 with CH2C12. The spectra were recorded in CD2012
solutions at —80 °C.
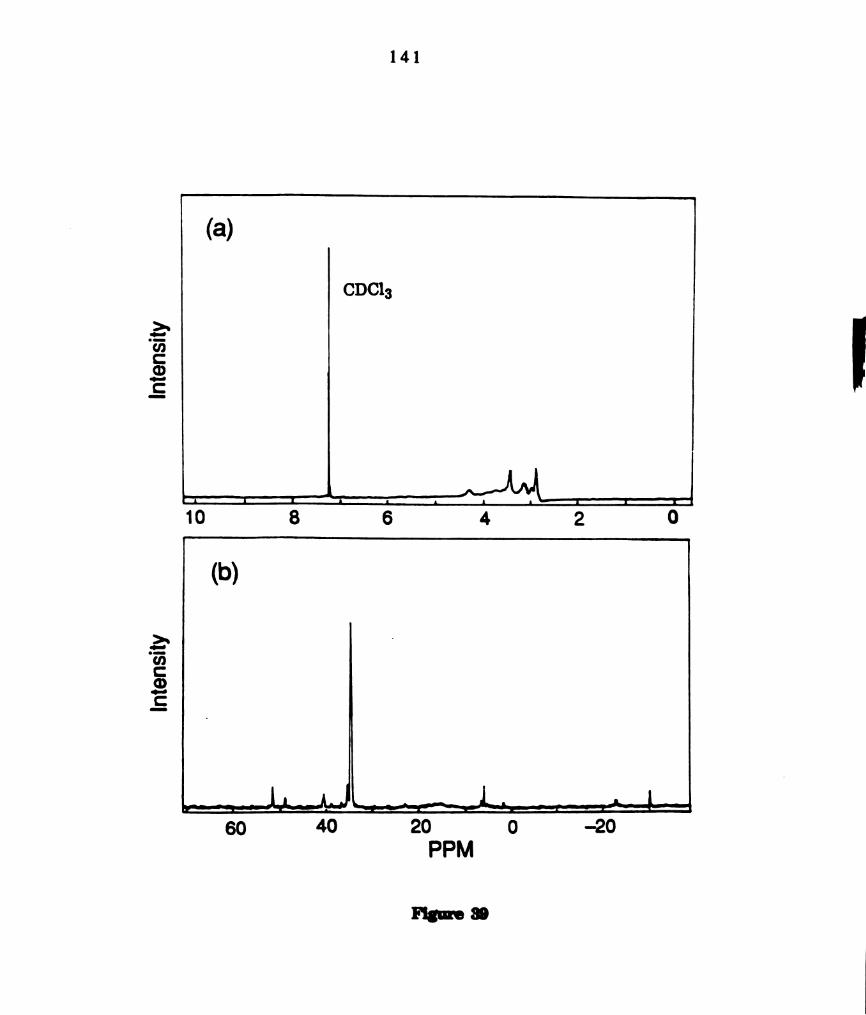
141
(a)
CDC13
26:25
10
(b)
33:25
40 20 -20
PPM
Flam 3
60

142
b. PhotmeacfionofMozChTBug4wifl1PhSSPh
Benzene solutions of M02C14(PR3)4 (PR3 = PBu3 and PMeth)
containing a ten-fold excess of phenyl disulfide (PhSSPh) at ambient
temperature are not indefinitely stable in the absence of light. Thermal
reaction is evidenced by the spectral changes shown in Figure 40 for the
case of PR3 = PBu3, where an absorption at 480 nm appears as the 82->1(56')
transition of M02C14(PBu3)4 decreases. Similar spectral changes are
observed with the MozCl4(PMePh2)4 complex. Reactions are accelerated by
irradiation of the 5241(88‘) transition (2. > 530 nm). More importantly, the
spectral changes associated with the photolysis differ from those of the
thermal reactions. An exemplary spectrum for the photolysis of
M02C14(PBu3)4 is provided in Figure 41a. The photolyzed solutions exhibit
absorption in the region between 500 and 550 nm which is not observed in
the spectra of the thermal reactions. Consistent with the lack of isosbestic
points, two products have been separated from photolyzed solutions of
M02Cl4(PBu3)4 by column chromatography. One product exhibits an
absorption maximum at 480 nm which is consistent with that of the
thermal product. Insuficient data precludes assignment of this product at
this time. Figure 41b shows the spectrum of an additional product which is
unique to the photoreaction with an absorption maximum at 540 nm. The
FABMS of this photoproduct reveals a parent ion cluster centered at 1173
amu, consistent with M02C14(PBu3)2(SPh)4, along with three fragments
corresponding respectively to loss of one SPh unit, one PBu3 and one of each
(Figure 42).
The M02014(PBu3)2(SPh)4 product corresponds to addition of two
equivalents of diphenylsulfide with accompanying displacement of two

143
Figure 40. Electronic absorption spectral changes during thermal
reactions of benzene solutions of MozCl4(PBu3)4 containing a ten fold excess
of PhSSPh. The total reaction time at ambient temperature was four hours.
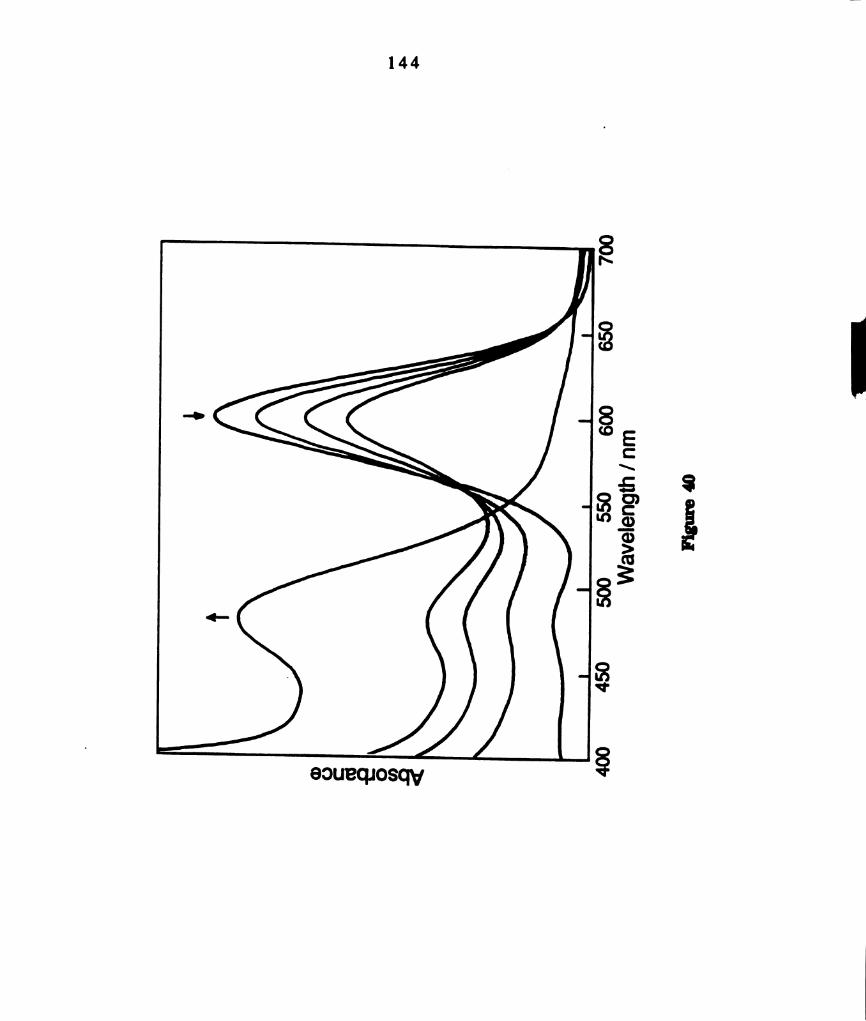
144
32.—BE
E:
\59.2963
can
own
coo
0mm
com
owe
8v
I_
q.
4_
\\.
eoueqmsqv

145
Figure 41. (a) Electronic absorption spectral changes during photolysis
(A.exc > 570 nm) of benzene solutions of M02C14(PBu3)4 containing a tenfold
excess of PhSSPh. The total reaction time at ambient temperature was 20
minutes. (b) Electronic absorption spectrum of a photoproduct isolated
from photolyzed solutions by column chromography. The FABMS of this
product is provided in Figure 42.
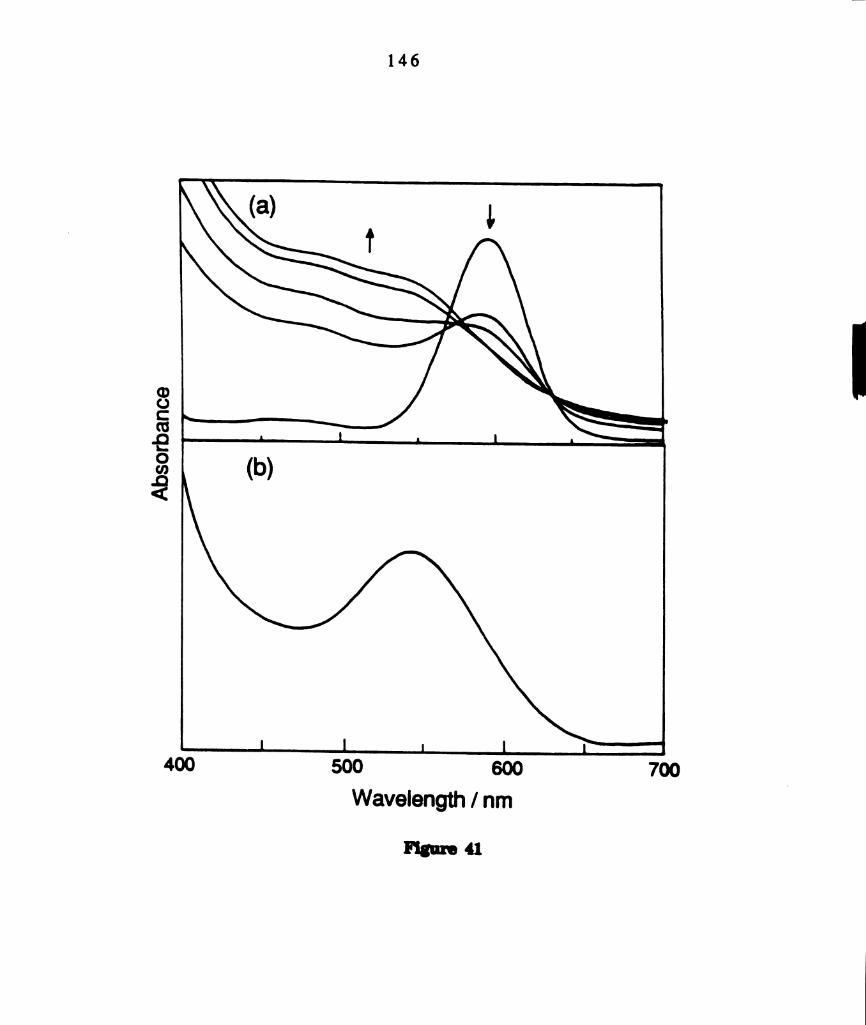
146
700
(a) l
1
s \ »c: M
‘
g . i . 1
g (b)<
l l 1 l L
400 500 600
Wavelength / nm
“M41

147
Figure42. Fast atom bombardment mass spectrum of a photoproduct
isolated from photolyzed solutions of M02C14(PBu3)4 containing PhSSPh.
Selected assignments of the clusters in the spectrum are: (a)
M02014“)BU3)2(SPh)4 (b) M02014(PB113)2(SP}1)3 (C) M02014(PBI13XSPh)4.
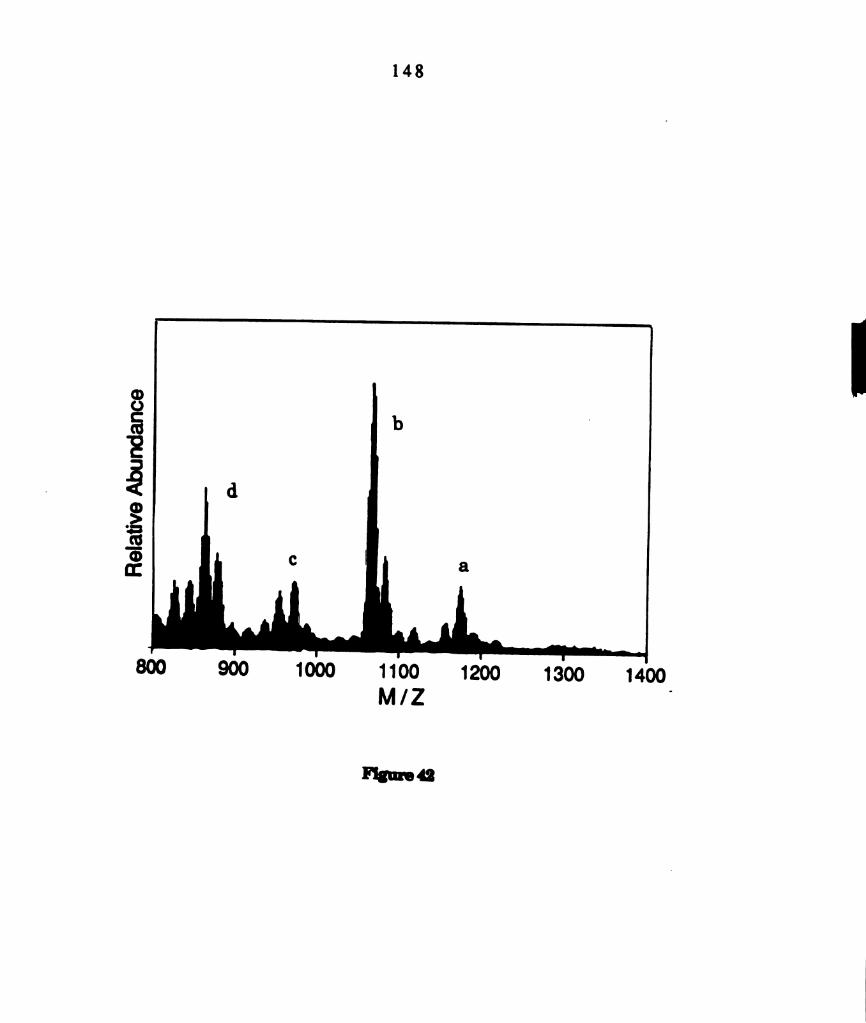
RelativeAbundance
800 900
148
1000 1100 1200
M/Z
Finn“!
1300
1400.

149
phosphine ligands. The observed phosphine displacement is not entirely
surprising based on independent studies which ' show that
thermodynamically unfavorable phosphine substitution reactions are
promoted by 8241(58') absorption of M02C14(PBu3)4 [196]. Nonetheless,
although quantitative oxidative addition has not been achieved, this
reaction with PhSSPh exemplifies the ability of MzCl4(PR3)4 complexes to
effect multielectron transformations upon 8241(85‘) absorption. However
these studies suggest that further pursuit of such photochemical schemes
should be restricted to M2Cl4(PR3)4 complexes with less labile phosphines
which are less easily displaced such as PMe3, and PMe2Ph and bidentate
phosphine ligands.
2. PhotooxidafionAmompaniedbyDiqropa-fionafion
On the basis of the results of Section 1b, the photolysis ofPhSSPh with
Mo2X4(PR3)‘ possessing less labile phosphines were undertaken.
Photoreactions are not observed within 10 h of irradiation (7L > 530 nm) of
M02C14(PMe3)4 complex in the presence of a tenfold excess of PhSSPh.
Photoreactions do proceed under the same conditions with the
M02C14(PMe2Ph)4 complex, however the photooxidation chemistry is
markedly different from that of the MozCl4(PBu3)4 complex. Furthermore
unlike the reactivity of the M02Cl4(PBu3)4 complex, thermal reactions of
PhSSPh with M02C14(PMe2Ph)4 do not proceed at ambient temperature.
The prompt reaction upon irradiation of the 82a1(86') absorption
M02C14(PMe2Ph)4 containing a ten-fold excess PhSSPh at A > 530 nm is
evidenced by the spectral changes shown in Figure 43. An isosbestic point
is maintained at 553 nm during the photolysis and two discrete maxima at

150
Figure43. Electronic absorption spectral changes during photolysis (Km.
> 570 nm) of dichloromethane solutions of MozCl4(PMe2Ph)4 containing a
ten fold excess of PhSSPh at 22°C. The total reaction time at ambient
temperature was 24 hours.
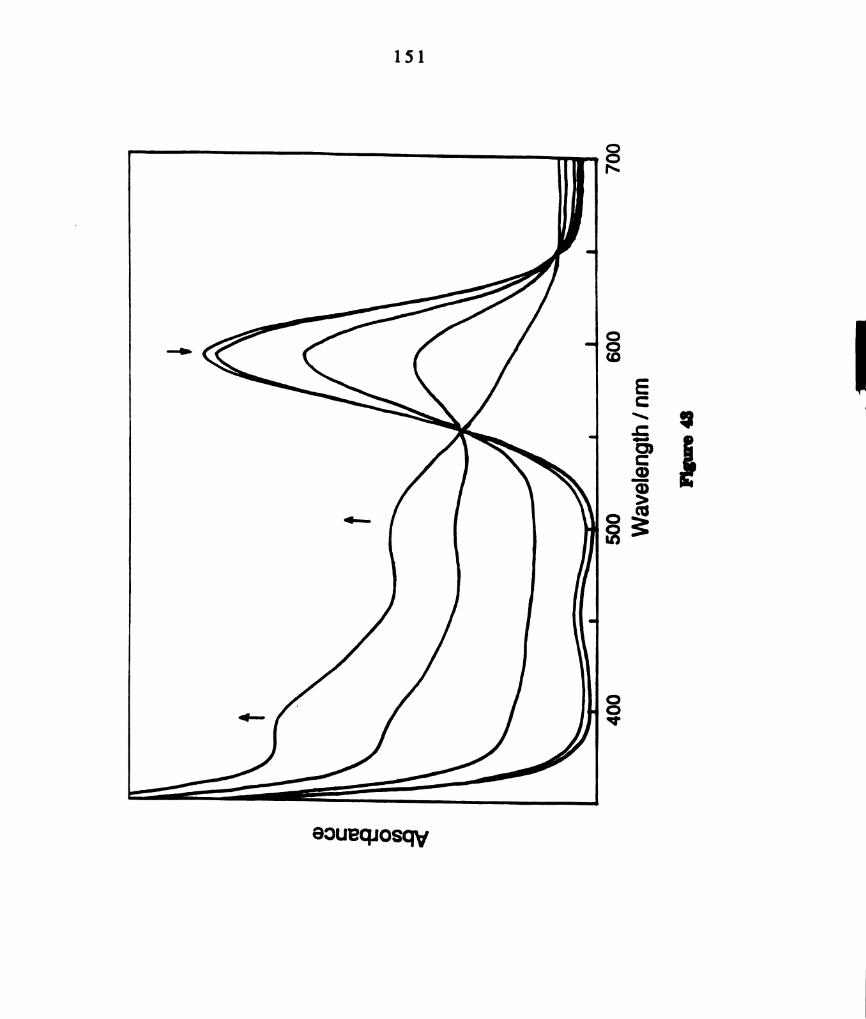
151
32.6...—
Ec
\26533.3
CON
cam
com
Gov
l».
..\
_a
(»
P
<)
aoueqiosqv

152
495 and 390 nm appear with the concommitant decrease of the 62—)‘(883
absorption. No NIR absorption is observed during the photolysis. The
FABMS of the photoproduct (Figure 44) isolated by addition of hexane,
reveals a parent ion cluster at 1128.8 amu consistent with
M02C15(PMe2Ph)4(SPh). Thus phosphine substitution is not observed with
the less labile PMezPh, but rather one observes addition of one Cl‘ and one
SPh‘ group to the bimetallic core.
Unlike the M02014(PMe2Ph)4 (Dzd) complex, acetone solutions of
M02C14(dppm)2 (D211) containing a forty-fold excess of PhSSPh are
photochemically inert upon irradiation at wavelengths coincident with the
82-41(88') absorption. However, photolysis does proceed upon higher energy
excitation (A. > 436 nm). The requirement for high energy irradiation is
completely general to all of the observed photoreactions of the D211
complexes, both bimolecular and pseudo first—order.
While solutions of M02C14(dppm)2 in the presence of a twenty fold
excess of tolyl disulfide (TolSSPhTol) are stable in the complete absence of
light, irradiation at x > 436 nm results in a concomitant decrease in the
8241(88’) absorption maximum at 634 nm with increases in absorption at
525 and 450 nm (Figure 45). No absorptions were observed in the NIR
region. Although isosbestic points are maintained throughout the reaction,
FABMS analysis of the solid precipitated with hexane with reveals the
presence of two products (Figure 46). One product, exhibiting a parent ion
cluster at 1260 amu, is the pentachloro product M02C15(dppm)2(STol), as is
the case for the photoreactions of M02014(PMe2Ph)4. However a second
product observed at 1348 amu is consistent with the oxidative addition
product, M02C14(dppm)2(STol)2. Similar reactions are observed with the
phenyl disulfide. The absorption spectrum of the major product is that of

153
Figure 44. Fast atom bombardment mass spectrum of a photoproduct
isolated from photoreaction of M02C14(PMe2Ph)4 with PhSSPh. The cluster
is consistent with MogCls(PMe2Ph)4(SPh).
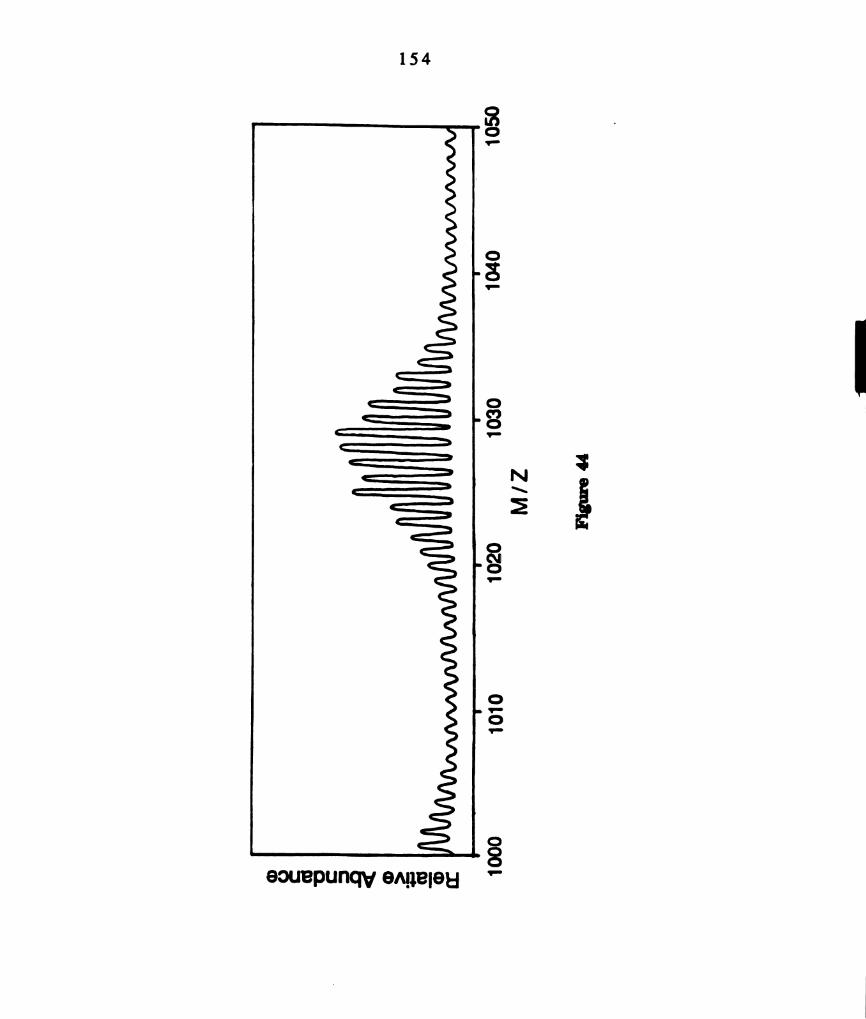
154
ome—
ovop
coo—
395:3
N\S_
omop
crop
oocp
C
eouepunqv engialea

155
Figure 45. Electronic absorption spectral changes during photolysis (2.exc
> 435 nm) of acetone solution of M02C14(dppm)2 containing a forty fold
excess of TolSSTol at 22°C.

156
anESH
E:
\£99962,
0::
com
com
com
4—
.q
d<
q
eoueqiosqv

157
Figure46. Fast atom bombardment mass spectrum of photoproducts
isolated from photoreactions of MozCl4(dppm)2 with TolSSTol. Selected
assignments of the clusters in the spectrum are (a) [Mo]Cl4(STol)2t; (b)
[MoJCl3(STol)2+; (c) [Mo]015(STol)-:; (d) [Mo]Cl4(STol)"'; (e) [Mo]C15"'
where [Mo] = M02C14.
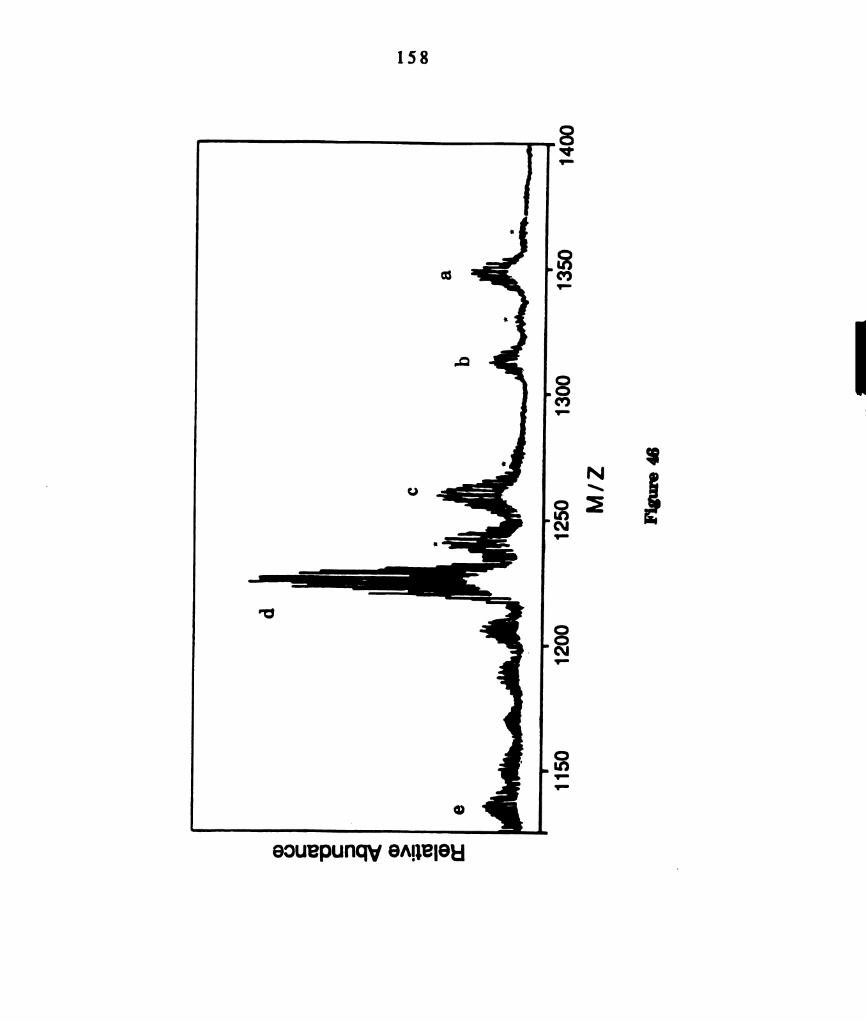
158
oovp
camp
avg
N\S_8w.
comp
amp.
eouepunqv eAgieleu

159
the pentachloro product M02015(dppm)2(SPh), (kmam = 525 and 450 nm),
whose identity has been confirmed by X-ray crystallography, and the minor
product, MozCl4(dppm)2(SPh)2, absorbs at 460 nm.
The pentachloroproduct is not restricted to reactions with RSSR but is
also observed from photoreactions with other substrates such as ethyl
iodide. Photolysis of CH3CH2I (EtI) solutions of WzCl4(dppm)2 at 0 °C
results in the spectral changes shown in Figure 47. The decrease in the
8241(55') transition of the quadruply-bonded binuclear complex is
accompanied by an increase in an absorption maximum at 500 nm (e = 6718
M’lcm’l). This absorption is consistent with that of independently
prepared W2Cl4(dppm)212 (6504 = 6954 M'lcm’l), as is the parent ion cluster
centered at 1531 amu in the mass spectrum of the solid sample isolated by
addition of hexane (Figure 48). However, an additional cluster centered at
1441 amu, not observed in the FABMS of WzCl4(dppm)212 (Figure 9), is also
present. The isotopic distribution of this cluster agrees well with that
theoretically calculated for the pentachloroproduct, W2015(dppm)2I,
comparable to that obtained from reactions of PhSSPh with the
dimolybdenum complexes.
The fact that direct addition of neither phenyl disulfide nor ethyl
iodide is effected in these photoreaction pathways is suggestive of a radical
mechanism. Compelling evidence for a radical mechanism in the disulfide
system is provided by performing photolysis of M02C14(dppm)2 in the
presence of equimolar mixtures of phenyl and tolyl disulfides. As Figure 49
shows, the molecular ion region reveals the presence of the the crossover
product MozCl4(dppm)2(STol)(SPh) in addition to the oxidative addition
products M02Cl4(dppm)2(SPh)2 and MozCl4(dppm)2(STol)2. Photolysis of

160
Figure 47. Electronic absorption spectral changes during photolysis (ken
> 435 nm) of ethyl iodide solutions of W2014(dppm)2 at 0°C. The total
reaction time was 1.5 hours.
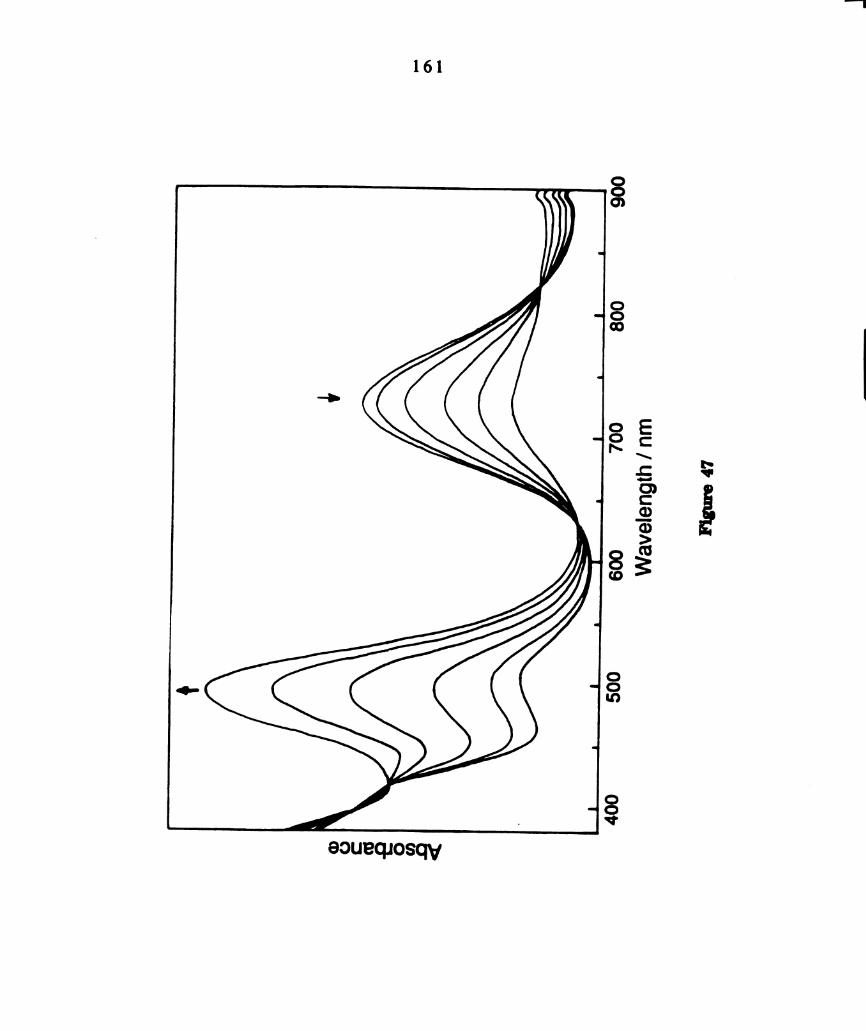
161
2.9:5:—
E:
\599963
8m
8»
as.
8o
--
com
.
cow
2
d
J
eoueqiosqv

162
Figure48. Fast atom bombardment mass spectrum of products isolated
from photolyzed ethyl iodide solutions of WZCI4(dppm)2. Selected
assignments of the clusters in the spectrum are (a) [W]Cl412t ; (b)
[W101312+; (c) [W]Cl51‘; (d) [W]CI4I"' where [W]: W2(dppm)2.
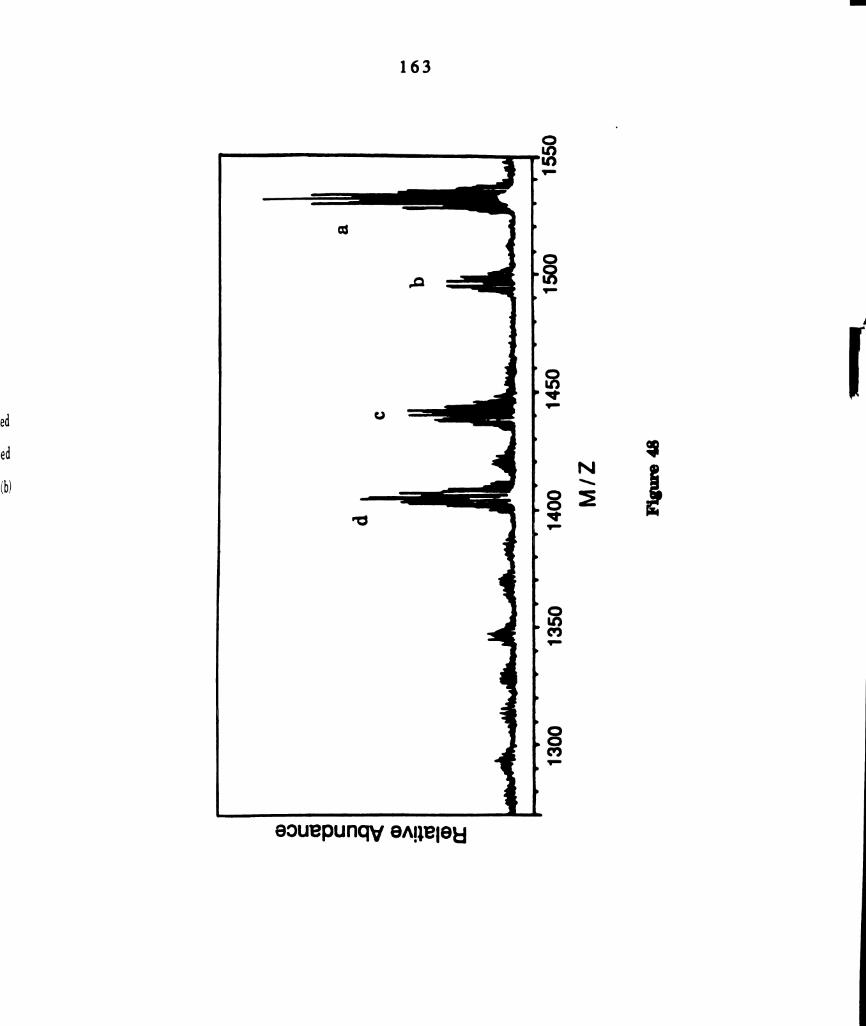
4
32.5:
N22
com.
on:
83
89
com.
c
163
ed
ed
(bl
eouepunqv eAneIea

164
Figure 49. Molecular ion cluster region of the fast atom bombardment
mass spectra of photolyzed 0.,“ > 435 nm) solutions of M02C14(dppm)2 in the
presence of phenyl/tolyl disulfide mixtures. Assignments of the clusters in
the spectrum are (a) MozCl4(dppm)2(SPh)2 (b) MozCl4(dppm)2(SPh)(STol)
and (c) MozCl4(dppm)2(STol)2 .

165
omm
P
9.953e—
N22
00%—
ompm_
ama—
lent
1the
5101)
min
eouapunqv 6A119|€H

1 6 6
mixtures of independently prepared M02014(dppm)2(SPh)2 and
M02014dppm)2(STol)2 show no evidence of exchange.
The photochemistry of the M2Cl4(dppm)2 complexes with phenyl
disulfide and ethyl iodide can be summarized by the following overall
reaction.
XY
M2Cl4(dppm)2 ——-> M2015(dppm)2x + M2Cl4(dppm2)Xg (4.1)
(X=I, SR; Y = Et, SR)
The production of M2C15(dppm)2(X) is suggestive of a free radical
mechanism. A radical mechanism that accounts for the formation of both
products, M2(III,III)C15(dppm)2(X) and M2(III,III)C14(dppm)2(X)2 is as
follows,
XY
M201, II)Cl4(dppm)2.T M2(II, III)Cl4(dppm)2X (4.2)
M2(II, III)C14(dppm)2X —>
étMsaII. III)CI.<dppm>sx + Meat. II)C13(dppm)2X] (4.3)
M201, III)Cl4(dppm)2X —->
{1504'er III)CI4(dppm)2X2 + M201. II)Clc(dppm)2] (4.4)
The primary step (4.2) involves one-electron oxidation of electronically
excited M2(II,II)Cl4dppm)2 by the substrate XY to yield the mixed-valence
M2(II,III)CI4(dppm)2(X) intermediate. Disproportionation upon chlorine
atom abstraction (eq 4.3) yields M2(III,III)Cls(dppm)2(X) and
M2(II,II)C13(dppm)2(X). Alternatively, disproportionation by X atom
transfer (eq 4.4) generates M2(III,III)C14(dppm)2(X)2 and

l 6 7
M2(II,II)CI4(dppm)2. A similar disproportionation reaction involving
halogen atom transfer has previously been proposed for reactions of
dinuclear platinum complexes with aryl halides [197].
This mechanism was investigated electrochemically. The
W2(II,III)CI4(dppm)2X mixed-valence intermediate can be produced by one-
electron oxidation of W2(II,II)CI4(dppm)2 in the presence of X‘. The cyclic
voltammogram of toluene/CH31 solutions of W2Cl4(dppm)2 reveal a one-
electron redox wave at +0.1 V (vs Ag wire reference electrode) (Figure 50a).
The cathodic component of this wave is not observed in the presence of I“
(Figure 50b) although a new wave at —0.82 V is present on the return scan.
This reduction wave is consistent with that of the independently prepared
W2(III,III)Cl4(dppm)2I2 shown in Figure 50c. These data are consistent
with the disproportionation mechanism shown in reactions 4.3 and 4.4.
The loss of the cathodic component of the WzCl4(dppm)2"'/° redox wave is
indicative of a trapping of W2(II,III)C14(dppm)2+ with I" to yield
W2(II,III)C14(dppm)2I. Disproportionation of the W2(II,III)C14(dppm)2I
intermediate is evidenced by the appearance of the reduction wave of
W2(III,III)Cl4(dppm)2I2. Unfortunately, we have not been able to prepare
and cleanly isolate the W2(III,III)C15(dppm)2I complex and thus its cyclic
voltammogram is not available for comparison. However the potential for
W 2(III,III)Clg(dppm)2 is only 0.13 V greater than that of
W2(III,III)CI4(dppm)212 and thus the wave at ~0.85 V could well be
reduction of both W2(III,III)C15(dppm)2I and W2(III,III)CI4(dppm)212.

168
Figure 50. Cyclic voltammograms of 1:1 CH3I/toluene solutions
containing 0.1 M THAPF6 of (a) WZCl4(dppm)2 (b) WzCl4(dppm)2 (1.0 x 10‘s)
and THAI (5.0 x 10-3 M) (c) W2Cl4(dppm)2(1)2
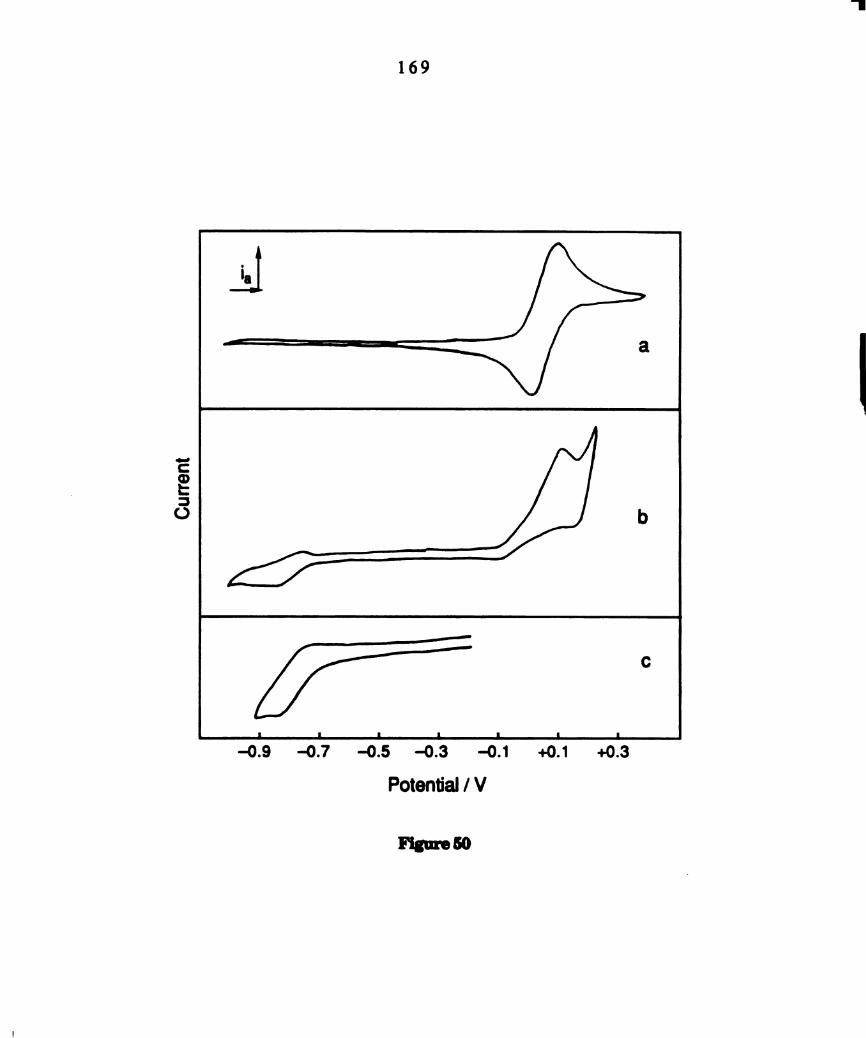
Current
169
& —
1
-o.9 -o.7 -o.5 -o.3 -o.1 +0.1 +0.3
Potential/ V
13m”

170
3. OxidafiveAddifionReacflon
The spectral changes upon irradiation at 71. > 436 nm of CH3I (MeI)
solutions of W2Cl4(dppm)2 clearly differ from those of the reactions with
ethyl iodide. In the case of CH3I (Figure 51) two absorption maxima at 490
(e = 2580 M‘lcm’l) and 582 nm (e = 1916 M‘lcm“) appear in the visible
region, as compared with the single absorption maximum at 500 nm in the
case of the reaction with ethyl iodide. Additionally, a distinguishing NIR
transition at 1090 nm (e = 646 M‘lcm'l) is observed from photolyzed CH3I
solutions. Two isosbestic points are maintained throughout the course of
the reaction with CH31. Consistent with the presence of these isosbestic
points, a single product is quantitatively obtained by addition of hexane to
photolyzed solutions. Elemental analysis of the isolated purple solid
corresponds to addition of CH31 to the tungsten-tungsten bond; Cald.
(Found) for WzCl4(dppm)2(CH3)(I): C, 43.1 (43.0); H, 3.32 (3.37); I, 8.93 (8.43);
P, 8.72 (8.54).
Further evidence that photolysis cleanly yields the simple oxidative-
addition product is provided by fast atom bombardment mass spectrometry
(FABMS). The molecular ion cluster in the FABMS spectrum of the
photoproduct (Figure 52), centered at 1418 amu, represents the molecular
ion, W2014(dppm)2(CH3)(I). Selective fragmentation of the parent ion
cluster at 1418 amu gives rise to the two major fragments centered at 1383
and 1291 amu also appearing in Figure 52, which corresponding to loss of
Cl and I, respectively. A simulation of the natural isotope distribution for
W2Cl4(dppm)2(CH3)(I) agrees well with the observed spectrum; the
simulated and observed relative abundances of the individual isotopic peaks
corresponding to the molecular ion are shown in Figure 53. The FABMS

171
Figure 51. Electronic absorption spectral changes during photolysis (Am,c
> 435 nm) of methyl iodide solutions of W2Cl4(dppm)2 at 0°C. The
wavelength scale in the near infrared region 0. = 900-1000 nm) is twice that
of the visible region (7. < 900 nm).
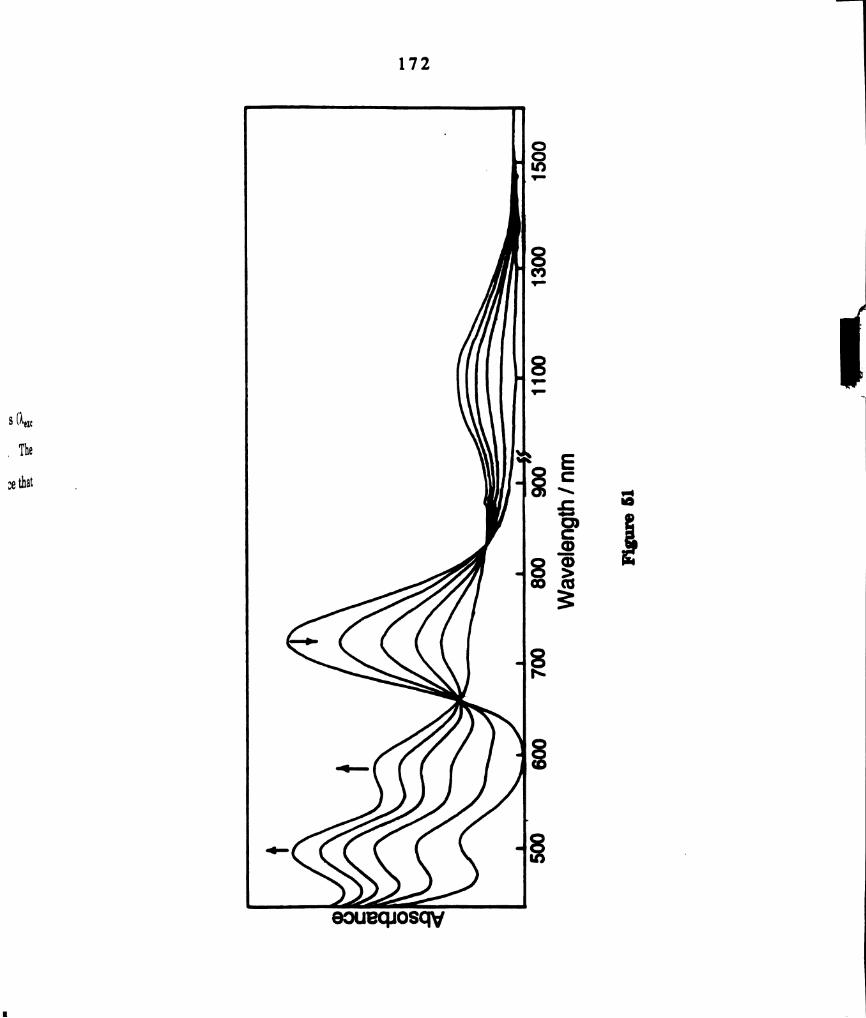
383...—
E:
\505.262,
83
82
8:
(realm
So
as.
8m
8m
lllilll
ll
v.
q.

173
Figure 52. Fast atom bombardment mass spectra of (A) photolyzed O.exc >
436 nm) solutions of WzCl4(dppm)2 and methyl iodide and of (B) solutions of
WzCl4(dppm)2 and methyl iodide refluxed in the absence of light. Selected
+
assignments of the clusters in the spectrum are: (a) [W]Cl4CH3I'; (b)
++
[WJCI3CH31+; (c) [WIClzCH31'; (d) [W]CI4CH3+; (e) [W]Cl412'; (f)
+
[W]Cl312+; (g) [W]ClsI’ ; and (h) [W]Cl4I+ where [W] = W2(dppm)2.
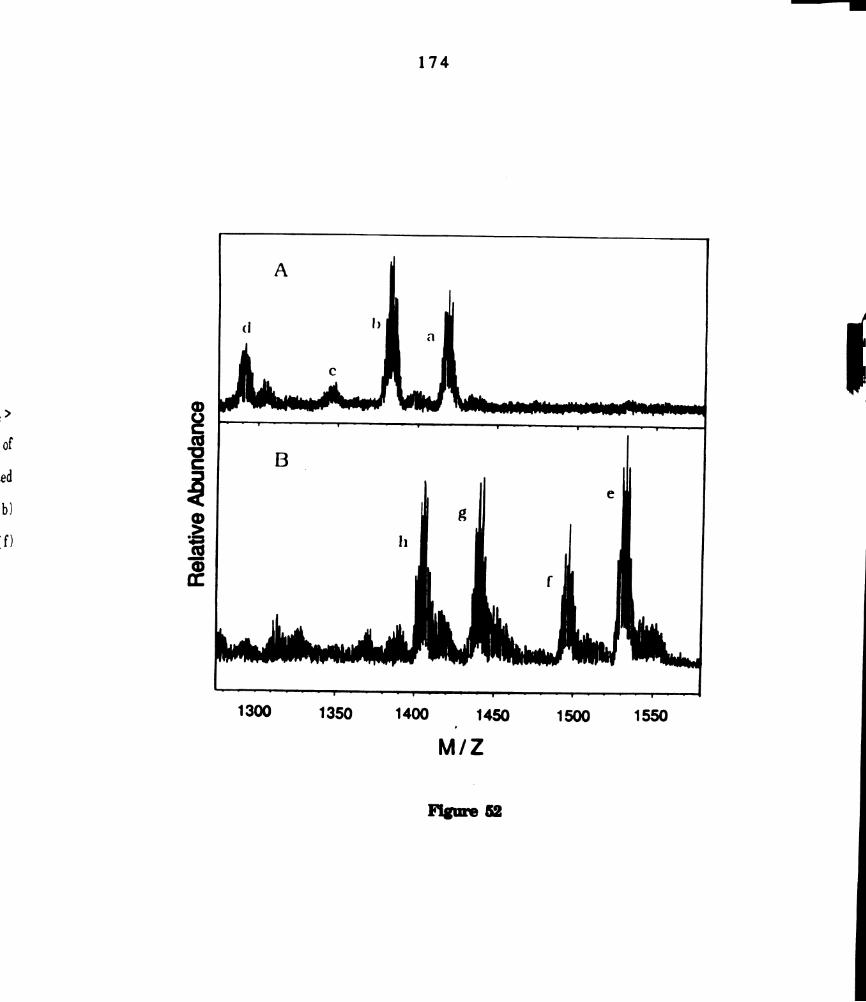
174.
('1
l)(l
1450 1500 1550'ircc' ',
M/Z
1300
8:352
028.6:
1350
“M52

175
Figure 53. The relative isotopic distribution of the molecular ion cluster
for WzCl4(dppm)2(CH3)(I) . The simulated relative abundances, designated
with solid lines, are superimposed on the observed peaks.
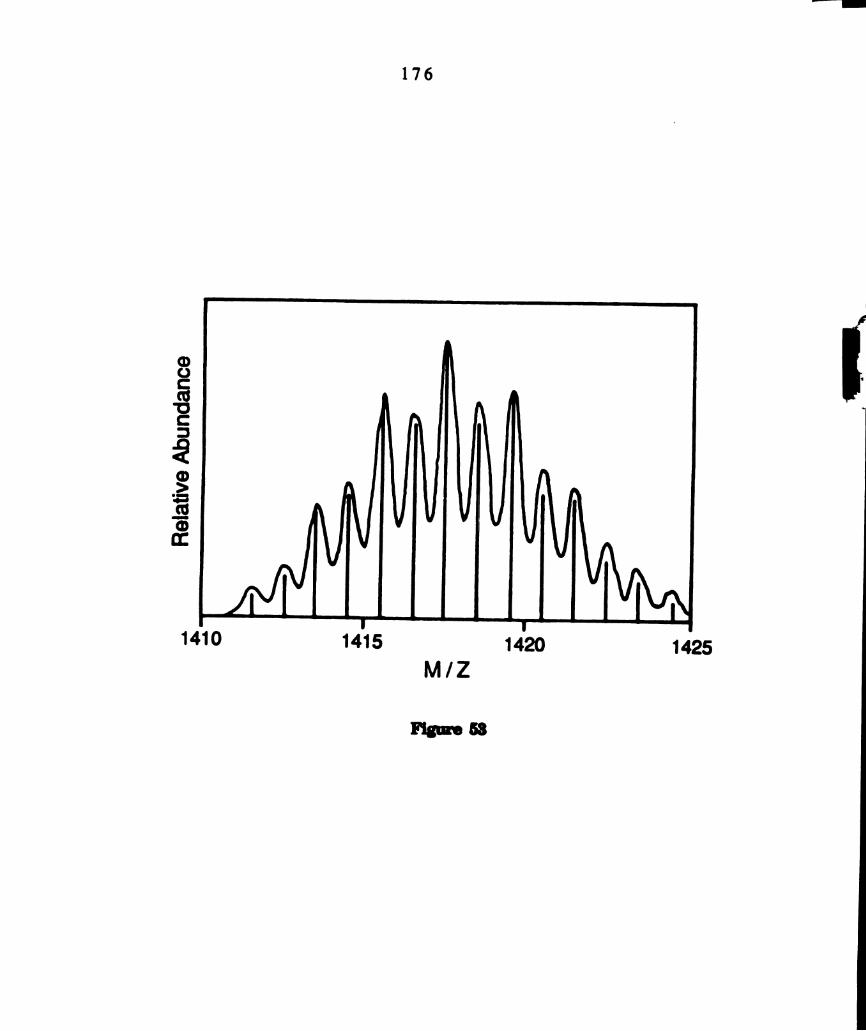
176
g '1 ll
33 u u U l ia:
‘ l l I I l J '1
1410 1415 1420 1425
M/Z

l 7 7
shows no evidence of the one-electron crossover products, W2014(dppm)212
and WzCl5(dppm)2I that arise from free radical pathways of Section 2. The
absence of free radical pathways is further supported by GC/MS analysis of
samples obtained by Toepler pumping photochemical reaction mixtures,
which gave no evidence of the production of ethane.
The coordination geometry about the metal-metal bond has not
unequivocally been established because we have not yet been able to obtain
single crystals suitable for X-ray diffraction. However, the absorption
spectrum is typical of the edge-sharing geometry which is observed from
every heretofore reported addition reaction of the MzCl4(PP)2 complexes. In
particular, close similarities are noted in the features of the photoproduct at
490, 582 and 1090 nm and a NIR transition with those of the edge-sharing
bioctahedral complex WzCl4(dppm)2(H)(I) at 464 nm, (e = 460 M'lcm‘l), 602
nm, (e = 450 M-lcm-l) and 1012 nm, (e = 340 M-‘cm-l), respectively. Insight
into the coordination position of the methyl group is provided by 13C nmr
spectroscopy. A single 13C resonance shifted 21 ppm upfield from TMS
(Figure 54) indicates that the methyl group is more likely to be in a terminal
rather than bridging coordination position, as the diamagnetic anisotropy
of the metal-metal quadruple bond would induce a downfield shift of the
resonance of a bridging ligand [198].
The quantum yield of the photoreaction is wavelength dependent @405
= 0.029(1), 4’436 = 0.011(2). ¢5lo = 0.001(3). t = 0 at A > 570 nm). These results
show that the photoactive state is not directly accessed by the 8241(55')
transition nor the absorption centered at 500 nm which has been assigned
as 8—95‘2_y2. The photoreaction is coincident with the 1(lt-i6')/1(6-m')
transition of M-LM species. Maximum quantum yields could not be
determined with increasing wavelength owing to absorption of the

178
Figure54. 13C NMR of photoproduct from photolyzed solutions of 1/1
13CH3I/12CH31 solutions of WZCI4(dppm)2, in CD2C12 at —60°C.
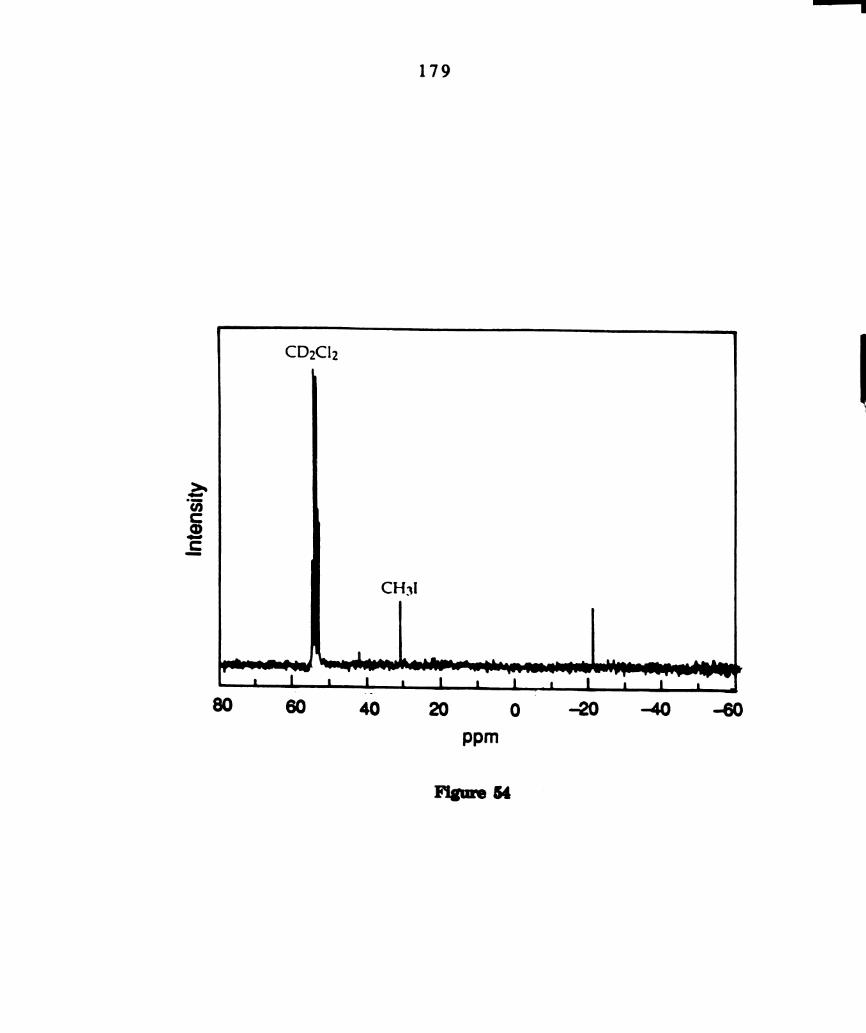
Intensity
179
CDzClz
CH3l

180
substrate at l. < 390 nm. The photochemistry is clearly not derived from
excitation of the substrate. Excitation at l. > 335 nm result in the spectral
changes shown in Figure 55. The characteristic absorption of
W2C14(dppm)2(CH3)(I) at 582 nm does not appear but rather a maxima at 500
nm, comparable to that observed from photoreactions with ethyl iodide,
appears. This result is not surprising, because it is well known that
photoexcitation of CH31 results in cleavage of the C—I bond yielding -CH3
and -I. Thus for this case radical mechanisms are initiated which would
result in production of ethane, W2Cl4(dppm)212 and W2015(dppm)21.
Furthermore photolytic cleavage of CH3I results in production of I2, which
we have shown independently will react with W2Cl4(dppm)2 to yield
WzCl4(dppm)2I2.
The photoproduct is unique. Thermal reactions occur at elevated
temperatures but the thermal reaction clearly differs from the
photochemical reaction as evidenced by the spectral changes observed upon
refluxing CH3I solutions ofW2Cl4(dppm)2 displayed in Figure 56. While the
shift in the 8241(88') transition is not understood, the absorption at 500 nm
is comparable to that of photolyzed EtI solutions. Indeed FABMS (Figure
52b) of these solutions reveal parent ion clusters at 1531 and 1441 amu,
consistent with WzCl4(dppm)2I2 and WzCl5(dppm)2I, respectively. Although
FABMS analysis indicates that W2014(dppm)2I2 is obtained in the thermal
decomposition of W2014(dppm)2(CH3XI) which proceeds even at ambient
temperature, W2015(dppm)2l is not observed. Thus the photochemical
pathway appears to be unique and corresponds to addition at a discrete
bimetallic core.

181
Figure 55. Electronic absorption spectra of CH3I solutions of
WZCl4(dppm)2 before ( ) and after(----- ) photolysis at A > 335 nm at 0°C.
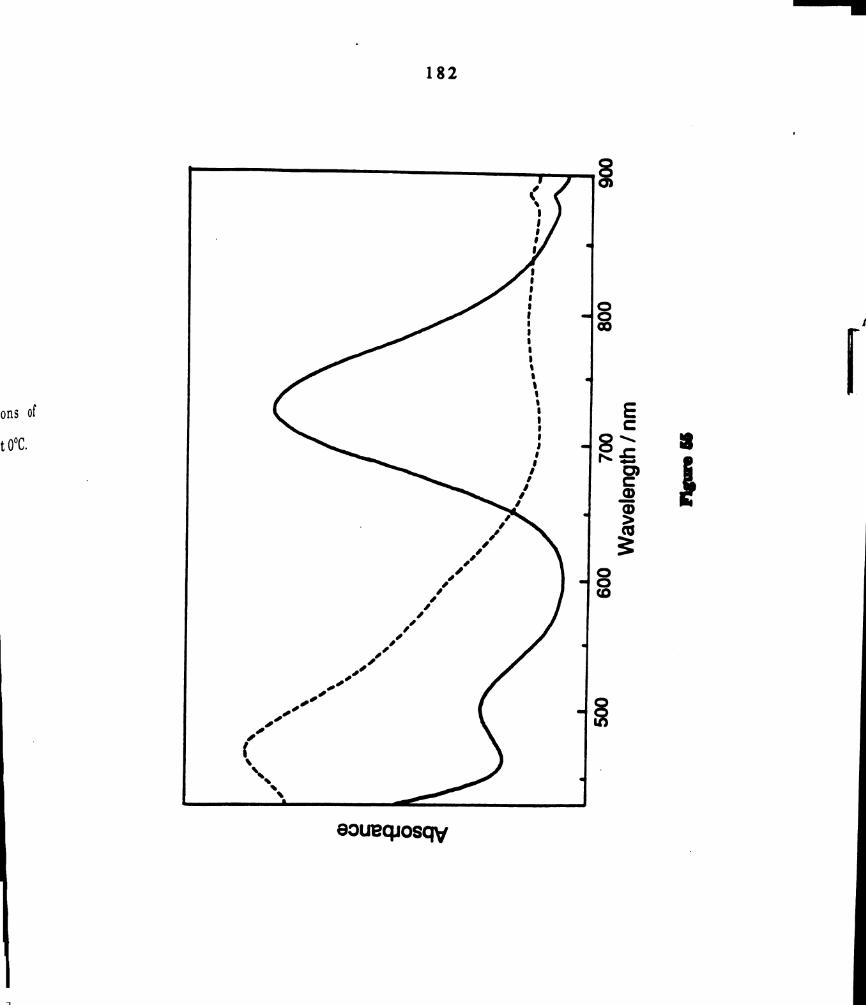
eoueqiosqv
ons of
t 0°C
moo
moo
woo
P
$55.96.:
\:3
mo:
39!...
182

183
Figln'e56. Electronic absorption spectral changes observed upon
refluxing CH3I solutions of W2Cl4(dppm)2.

184
com
8953..—
E:
\£95553
com
21:
8m
.d
q.
upon
eoueqlosqv

l 8 5
C. DISCUSSION
Transient absorption results of the Dzd M2C14(PR3)4 and D211
MzCl4(PP)2 complexes of Chapter III provide the underpinning to the
observed MMCT photochemistry of the M-LM complexes. First, a distinct
correlation is noted between the photochemical reactivity of the
dimolybdenum complexes with PhSSPh and the formation of the
bioctahedral intermediates. As summarized in Figure 19, structurally
distorted intermediates are not observed for 5241(85') excitation of the D2},
complexes, wheras an edge-sharing bioctahedral intermediate is formed
from the higher energy MMCT states. Accordingly, photoreactions of the
D211 complexes only proceed upon higher energy irradiation. Photoreactions
are however accessible upon 82* 1(551") absorption of the D24 complexes and
studies of Chapter III show that a bioctahedral intermediate is formed
under these conditions (Figure 31). A crucial role of the bioctahedral
intermediate is further implied by the relative photoreactivity within the
series of M02C14(PR3)4 complexes. Namely while photoreactivity is observed
for the cases of PR3 = PMeth, PBua, PMezPh, all of which form
bioctahedral intermediates, no photoreactivity is observed from
electronically excited M02C14(PMe3)4. The latter complex displays a
transient absorption for the 1(861") excited state with no distortion to a
bioctahedral intermediate.
Thus the emerging trend for the M-LM complexes is that light
initiated photoredox reactions predominates when bioctahedral
intermediates are formed. Indeed, results from Chapter III indicate that a
bioctahedral intermediate is responsible for the quantitative addition of
CH3I to the electronically excited bimetallic core of W2Cl4(dppm)2.

l 8 6
Specifically, the photochemical intermediate exhibits an absorption at 480
run that is characterisitic of an edge-sharing bioctahedral intermediate. A
photopathway of this reaction consistent with this data and with terminal
coordination of the methyl group is addition of the substrate at the open
axial position in the reduced metal center in the edge-sharing bioctahedral
intermediate as depimd in below
n A /\
P P P P
CI ClC1,, ,. Cl, , CL, 1 Cl,M'____ M‘ llv : /"M "M CH3! : /"M "M
Cl/ CI/ CI \Cl/ CI ’
P P P P P PV V \_/
This photoreaction with CH31 represents the first multielectron
transformation efl‘ected at a discrete electronically excited M-LM core. We
believe that the formation of the the edge-sharing bioctahedral intermediate
is central to this novel photoreactivity. The distortion enhances the mixed-
valence character and simultaneously provides two open coordination sites
at the reduced metal center. Indeed low valent, coordinatively unsaturated
redox active metal centers exhibit a propensity for oxidative addition of
substrates.
This pathway proposed for the photoreaction with methyl iodide is
directly analogous to that proposed for the concerted thermal oxidative
addition of H2 to (18 square planar complexes [199,200]. In these systems an
octahedral distortion of the square planar complex upon approach of the
substrate to yield a transition state that can undergo concertively addition
via a three center bond is proposed as depicted below.
CH3

187
””0... ..o\\\“\ L A
L/ M \L H2 i L/
An additional parallel between the photoreactions of the quadruply bonded
complexes and the thermal reactions of the d8 square square planar
complexes is noted in the varying mechanisms with ethyl iodide and
methyl iodide substrates. Whereas photoreactions of W2Cl4(dppm)2 with
MeI yield quantitative oxidative addition, the production of crossover
products W2Cl5(dppm)2I and W2Cl4(dppm)212 in the photoreactions with EtI
are consistent with pathways involving one-electron intermediates outside
a solvent cage. Similarly, a variation in the mechanisms of oxidative
addition reactions of the Vaska's complex, trans-IrCl(CO)(PR3)2 with EtI
and Mel has been noted [201]. Radical chain mechanisms have been
proposed for the the reactions with ethyl iodide, based on the attenuation of
the rate upon addition of radical inhibitors. The presence of these
inhibitors show no effect on the rate of reactions with CH3I. The solvent
dependence and large negative activation entropy in the reactions with
methyl iodide are consistent with a nucleophilic 8N2 mechanism, entailing
addition of the electropositive +CI-I3 to the metal center, followed by rapid
addition of the displaced iodide. However, a very short lived radical cage
mechanism cannot be ruled out.
Herein lies a common problem with definitive elucidation of
mechanistic details of thermal reactions, which do not offer temporal
resolution beyond the conventional arena of study in the millisecond range
of stopped-flow kinetics. We are now in a position, however, to investigate

188
the intimate mechanistic details of the multielectron transformation of
CHsl at the bimetach core of W2014(dppm)2. Smcifically, with' the ability to
initiate this reaction with a pulse of light, we can precisely monitor the
disappearance of photoreactant and formation of products with transient
absorption spectroscopy. The preliminary transient absorption studies of
Chapter III are promising in this regard. Whereas the photochemical
intermediate observed from methyl iodide solutions exhibits an absorption
at 480 nm, which is characteristic of an edge-sharing bioctahedral
intermediate, the transient generated in ethyl iodide solutions does not,
indicating that the one-electron intermediate in this photoreaction has a
distinct absorption profile. Thus the one-electron or two-electron
photochemical pathway of the CH3I system can be elucidated by further
defining the kinetics of both the ethyl iodide and methyl iodide systems on
the picosecond time scale.
D. CONCLUSION
The structural rearrangement to bioctahedral geometries has
important ramifications in the development of discrete multielectron
transformations of electronically excited quadruply bonded metal-metal
binuclear complexes. First it is noted that regardless of the overall
mechanism, each of the photoreactions of the MMCT states of the D2,,
M2014(PR3)4 and D2,, M2014(PP)2 complexes , results in two-electron
oxidation of the binuclear core. This is in contrast to the previously
observed redox reaction of M-‘-M complexes accessible by visible irradiation.
For instance, although a two electron reduction of 1,2 dichloroethane to
yield ethylene has been affected by the 1(88") excited state of

1 8 9
M02[02P(OC6H5)2]4, this reaction is achieved by coupling one-electron
changes at individual metal cores as follows,
Meiosmoccnchl. —h"—> Monomeric»: <45)
MoiOleOCgI-IM‘ + 01011,ch —>
Mo,[O,P(OC¢I-Ig)2LCl + - CHZCHZCI (4.6)
MOiOflOCM+ . CHchzcl ‘_’
MoiOngOCgthhCl + CH2CH2 (4.7)
The primary step involves one-electron transfer from electronically excited
M02[02P(OC6H5)2]4 to DCE yielding M02(II,III)[02P(OCGH5)2]4Cl and
°CHCH2C1. Ensueing thermal reaction of the reactive -CHCH2C1 with
M02(II,II)[02P(OCsH5)2]‘ results in the final formation of ethylene [192b].
Although the intimate mechanistic details of the primary step have not yet
been elucidated, the quantum yield was found to be severely attenuated in
solvents that coordinate in the axial positions, indicating that the primary
transformation is effected at this site. Indeed this is certainly expected,
owing to the encumbering ligation sphere that completely encases the
equatorial plane of the bimetallic core, as confirmed by computer generated
space filling molecular models.
We postulate these structural constraints as well as the structural
rigidity of the Mog[02P(OcsH5)2]4 complex govern the observed one-electron
oxidation of the binuclear complexes. First, two-electron oxidized M-LM
cores with structurally rigid ligation spheres are typically unstable. This is
evident in the case of the M02[02P(OCGH 5)2]4 complex by the
comproportionation of M02(III,III) and Moz(II,II) to yield M02(II,III)

190
smcies. Secondly, although axial attack of the substrate at a single metal
center is dictated by the steric congestion of the ligation sphere, oxidative
addition of the substrate at a single metal center is inhibited in these
structurally rigid "lantern" complexes containing four bidentate ligands
spanning the binuclear core. To date there is not a single report of a
lantern structure with two additional ligands in the equatorial positions at
a single metal center. Rather, oxidative addition typically results in
addition of a single ligand in the axial position at each metal center. Thus
two-electron reactions will rely on coupling one-electron changes at
individual metal centers. Because the steric congestion of the ligation
sphere of M02[02P(OCGH5)2]4 inhibits the ability of the substrate to
simaltaneously access both metal centers, a discrete two-electron
transformation at a single binuclear core will be difficult to achieve.
Rather, as discussed, a primary step involving a one-electron
transformation in the most easily accessed axial position of a single metal
center is postulated. For this case a net two-electron reaction, requiring
diffusion of the reactive organic radical intermediate to an additional metal
center, is likely to involve competitive reactions with other binuclear metal
cores in solution [202].
Thus the ramifications of a bioctahedral rearrangement upon
oxidation of the more structurally flexible M-LM complexes in multielectron
photochemical schemes is two fold. This rearrangement which ensures an
ocathedral coordination sphere about M(III) centers allows for two—
electron oxidation of the binuclear core. Moreover, the bioctahedral
distortion of the MMCT excited state provides open coordination sites at the
reduced metal center, which facilitates direct addition of substrate. Beyond
its apparent role in the quantitative oxidative addition of CH31 to

191
electronically excited WzCl4(dppm)2, the coordinative unsaturation of the
reduced metal in the bioctahedral intermediate may account for its
enhanced reactivity relative to the structurally undistorted l(65') excited
state. These studies suggest that oxidative addition reactions may be
accessible from the bioctahedral intermediates generated upon 1(88‘)
absorption of the Dad complexes are logical candidates for the future
development of multielectron photochemical schemes. However, future
pursuit of the multielectron photoreactivity of these complexes should be
limited to complexes from which nonluminscent transients are observed,
yet are ligated by "nonlabile" phosphines such as PMezPh and PEta [203].

CHAPTERV
FINALREMARKS
The aforementioned studies show that charge separation within a
polynuclear metal complex is a promising approach to discrete
multielectron transformations. However an additional hypothesis to arise
from these studies is that such reactivity in the case of the electronically
excited quadruply bonded dimers is facilitated by an additional feature
beyond the formal change in oxidation state of the metal centers induced by
MMCT. Transient absorption studies indicate that an edge-sharing
bioctahedral intermediate is responsible for the photoinduced quantitative
oxidative addition of CH3I to the bimetallic core of WzCl4(dppm)2. The
proposed pathway for this transformation based on transient absorption
studies is addition of the substrate at the two open coordination sites of the
reduced metal center in the edge-sharing bioctahedral intermediate.
Future investigations of the photoreaction of MoWCl4(dppm)2 with CH3I
provide an indirect test of this photopathway. Namely, based on the relative
redox properties of Mo and W, such a mechanism is expected to involve
production of Mo(I)——W(III) species, and addition of Mel should therefore
192

193
occur at the Mo center. Preliminary results are promising in this regard.
The spectral changes associated with the photolysis (l. > 405 nm)
proceeding from CH3I solutions of MoWC14(dppm)2 at 0°C are shown in
Figure 57. Two distinct absorptions at 520 and 655 nm. The latter is only
slightly shifted from the 8241(88‘) absorption of MoWCl4(dppm)2. No
further spectral changes were noted after an additional 45 minutes of
irradiation, indicating that the absorption corresponds to a photoproduct.
This absorption profile with two main features is comparable to that of
W2Cl4(dppm)2(CH3)(I), and is distinguished from the spectrum obtained
from the thermal reaction of MoWCl4(dppm)2 with I2, which exhibits a
single feature centered at 525 nm. The FABMS of a solid sample
precipitated by addition of hexane, reveals the presence of both
MoWCl4(dppm)2(CH3)(I) and MoWCl4(dppm)2I2 at 1330 and 1446,
respectively. The fact that a cluster corresponding to MoWC15(dppm)2I is
not observed suggests that the reaction may involve quantitative oxidative
addition to yield MoWCl4(dppm)2(CH3)(I), with ensuing thermal
decomposition to yield the observed MoWCl4(dppm)212. Indeed although the
W2Cl4(dppm)2(CH3XI) complex is stable at 0 °C, it will decompose at
ambient temperature to yield W2Cl4(dppm)212. That the
MoWCl4(dppm)2(CH3)(I) complex is apparently less stable than
W2014(dppm)2(CH3XI) is promising because Mo—CH3 bonds are less stable
than W—CHa bonds [204]. Future studies at lower temperatures which
may prevent decomposition of the MoWCl4(dppm)2(CH3)(I) photoproduct,
will be enlightening.
Because the reaction with MeI can be initiated with a pulse of light,
the intimate mechanistic details of the addition of substrate can be directly
addressed with picosecond laser spectroscopy. Thus investigations of this

194
Figm‘e 57. Electronic absorption spectral changes during photolysis (7texc
> 405 nm) of CH3I solutions of MoWC14(dppm)2 at 0°C. No further change
in the spectra were noted after an additional 0.5 hour of irradiation.
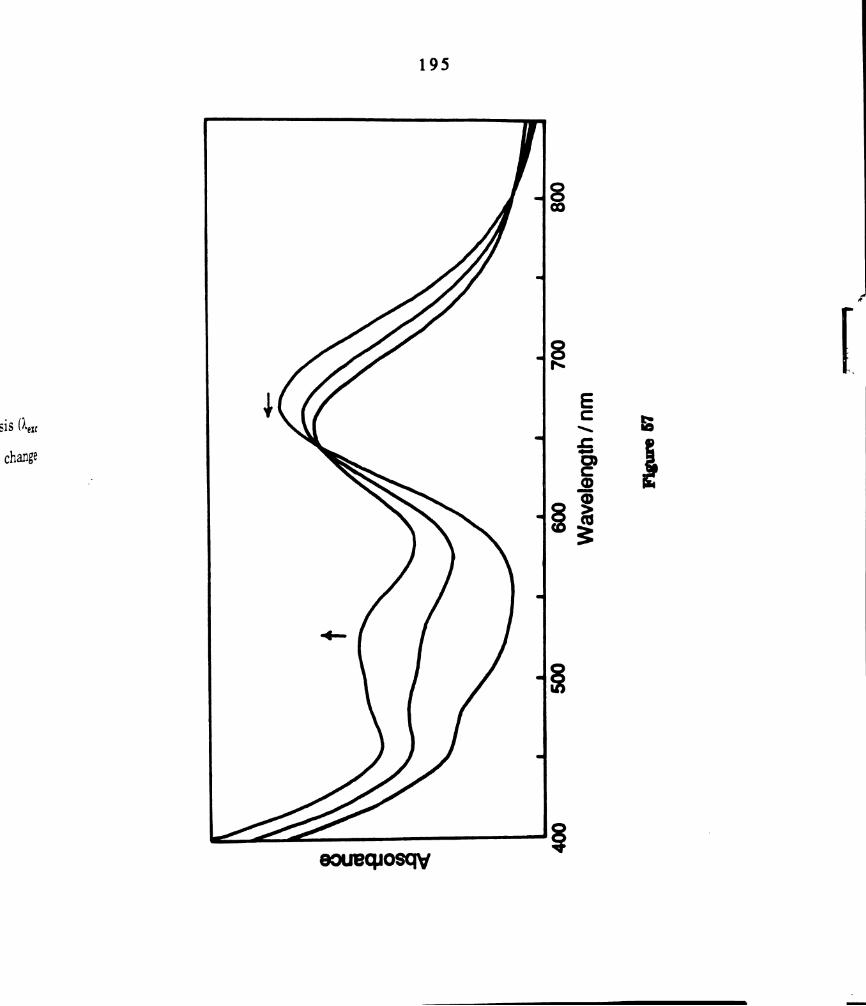
195
cosd
Bonfir—
E:
\coco-gm;
ooo
qd
lb 0 ex:
Change
eoueqlosqv

1 9 6
reaction can fundamentally lead to a better understanding of multielectron
reactivity and practically enable construction of a framework for design of
multielectron catalysts not only for excited state processes but for ground
state processes as well.
More generally, transient absorption spectroscopy opens up new
avenues in the deisgn of bimolecular photochemical schemes of M-LM
dimers. Although bimolecular photochemistry from numerous quadruply
bonded metal-metal dimers has heretofore remained unexplored owing to
the limited lifetime of the 1(85') state, studies presented here suggest that
long-lived l(88‘) excited states are as important for bimolecular
photochemistry as nonemissive structurally distorted intermediates. Thus
further pursuit of bimolecular multielectron photochemistry of quadruply
bonded metal-metal dimers may be significantly broadened to include the
majority of these complexes with shortlived nonemissive l(88’) excited
states.
In summary, the framework for the future design of photoinduced
multielectron small molecule activation schemes of MJ-M dimers to arise
from the studies described herein directly parallels that established for the
ground state reactions of square planar d8 Pd(II), Pt(II), and Ir(I)
mononuclear complexes. Namely, low valent, coordinately unsaturated
metal centers are ideal candidates for oxidative addition of substrates.
Moreover, the bioctahedral intermediate of the electronically excited
quadruply bonded dimers is directly analogous to an octahedral transition
that is believed to be important for concerted thermal oxidative addition
reactions of the mononuclear complexes. Electronically excited binuclear
complexes have the intiguing asset of mixed-valence character which
drives the formation of this ideal intermediate in the absence of substrate.

Appendix

197
Figure 58. Transient absorption kinetics recorded at 390 nm following 532
nm laser excitation of hexane solutions of displaying (a) the 120 ns
transient (b) the initial rise and relative intensity of an additional transient
absorption (c) the rise and decay of the additional long lived transient.
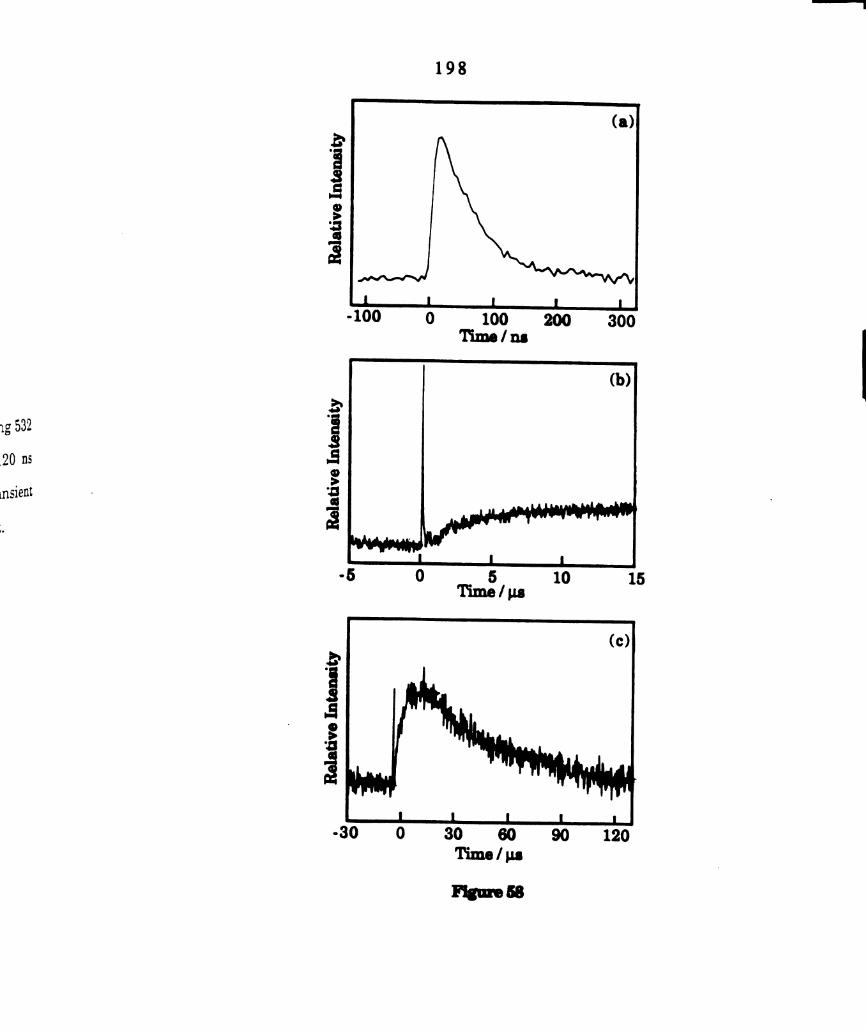
198
(s)
i
i
2'5
2
e1
1 l l l l
-100 O 100 200 300
Time/us
(b)
3'
lg 53?. g
8
.20 ns '5O
r
.>
mSIentg
a l I H
' l

199
Figure 59. Electronic absorption of M02C14(PBu3)4 (—), MozCl4(dppm)2
(-----) and irradiated solutions of M02Cl4(PBu3)4 containing a 100 fold excess
0f CH3N<PF2)2 (°°°°° ).
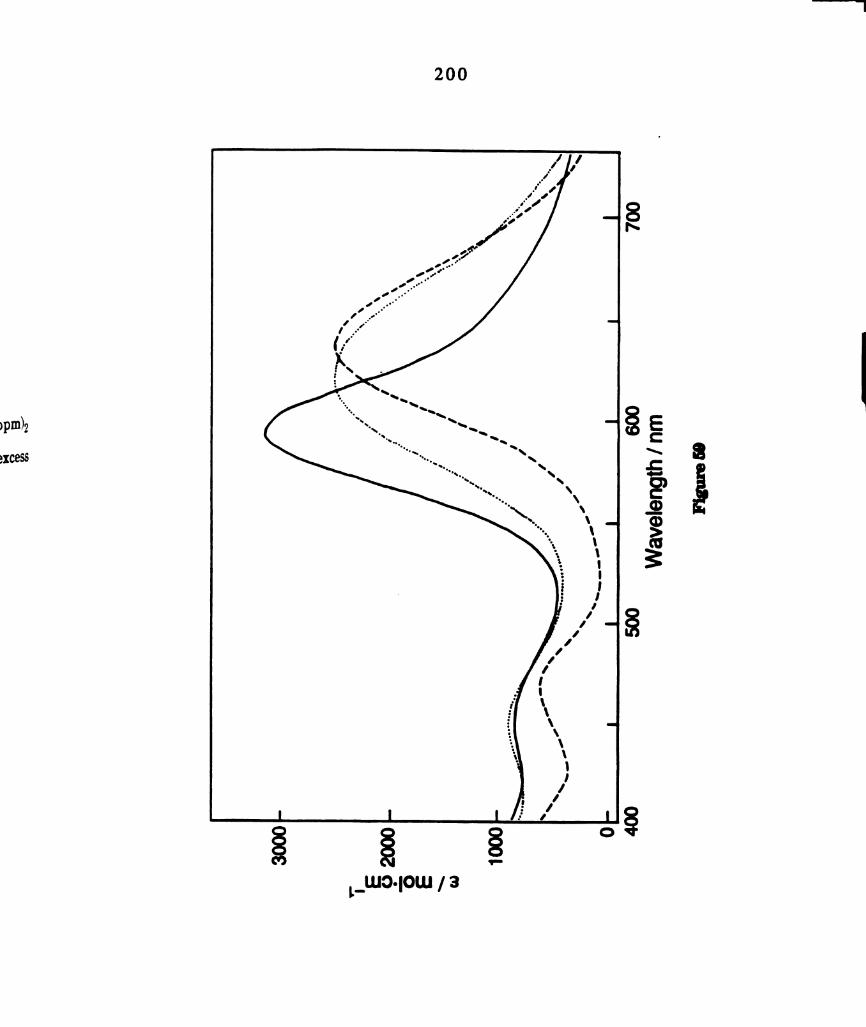
200
SE
E:
\£92925
__
_w
_.
lo
\s!isI:Is'5I
I
\\
II
I
\\
I
I/
s..
...
I\\
l...
.l..[I
\\
t.
II
‘\\
’a’oo”
\\
‘00
''\
I
’0'I....,
c.\
..
‘
0..
O
/\
..._.
*0
‘55
Axx
'
Is”...
\s
Ice-of
\\
oooS
3
I’M.-
‘\
sous
/
’aoccu
\u‘.
w
/...4
0
cs
w
|
a
s..
new
I..
.
Ia...
.\
w
I/
OS”
II
....a.
\.s
I.‘
IIIIVKI...
>pm)2
EXCESS

10.
11.
13.
Energy Resources through Photochemistry and Catalysis; Gratzel
M., Ed.; Academic Press: New York, 1983.
Gray, H. B.; Maverick, A. W. Science 1981, 214, 1201.
Photochemical Conversion and Storage of Solar Energy; Connolly, J.
8., Ed.; Academic Press: New York, 1981.
Photoinduced Electron Transfer; Fox, M. A., Chanon, M., Eds.;
Elsevier: Amsterdam, 1988; Part D.
Meyer, T. J. Acc. Chem. Res. 1989, 22, 163.
Kavarnos, G. J.; Turro, N. J. Chem. Rev. 1988, 86, 401.
Balzani, V.; Sabbatini, N.; Scandola, F. Chem. Rev. 1986, 86, 319.
Kalyanasundaram, K; Gratzel, M.; Pelizzetti, E. Coord. Chem. Rev.
1986, 6.9, 57 and references therein.
Gratzel, M. Heterogeneous Photochemical Electron Transfer; CRC
Press: Boca Raton, FL, 1989.
JuIis, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von
Zewelsky, A. Coord. Chem. Rev. 1988, 84, 85.
Sprintschnik, G.; Sprintschink, H. W.; Kirsh, P. P.; Whitten, D. G.
J. Am. Chem. Soc. 1976, 98, 2337
(a) Lehn, J. M.; Sauvage, J. P. Nouv. J. Chim. 1977, 1, 449; b) Lehn,
J. M. In Photochemical Conversion Storage of Solar Energy, Conolly
J. S. Ed; Academic Press; New York, 1981, Chapter 6.
Zameraev, K. I.; Parmon, V. N. In Energy Resources Through
Photochemistry and Catalysis, Gratzel, M. Ed; Academic Press; New
York, 1983, Chapter 5
201

14.
15.
16.
17.
18.
19.
21.
.5
202
Johnson, 8. G.; Tang, D.; Tankowiak, R. J. Phys. Chem. 1990, 94,
5849.
Lavi, B. G. Physics Today 1989, 42, 17.
Barber, J. Nature 1987, 325, 663.
(a) Renger, G. In Biopysics; Hoppe, W.; Lohmann, W.; Mark], H.;
Ziegler, H. Eds.; Springer-Verlag: Berlin, Heidelberg, 1983: Chapter
13, p 515. (b) Renger, G. Angew. Chem. Int. Ed. Engl. 1987, 26, 643.
Christou, G. Acc. Chem. Res. 1989, 22, 328.
Wieghard, K. Angew. Chem. Int. Ed. Engl. 1989, 28, 1153.
Pecoraro, V. L. Photoche. Photobiol. 1988, 49, 248.
Babcock, G. T. In New Comprehensive Biochemistry 15,
Photosynthesis; Amasz, J. Ed.; Elsevier: Amsterdam, 1987.
Kok, B.; Forbush, B.; McGloin, M. Photochem. Photobiol. 1970, 11,
457.
Wasielewski, M. R.; Niemczyk, M. P. ACS Symp. Ser. 1986, 321, 154.
(a) Cho, K. C.; Che, C. M.; Ng, K. M.; Choy, C. L. J. Phys. Chem.
1987, 91, 3690. (b) Cho, K. C.; Che, C. M.; Ng, K. M.; Choy, C. L. J.
Am. Chem. Soc. 1986, 108, 2814. (c) Cho, K. C.; Ng, K. M.; Choy, C.
L.; Che, C. M. Chem. Phys. Lett. 1986, 129, 521.
(a) McLendon, G. Acc. Chem. Res. 1988, 21, 160. (b) McLendon, G;
Miller, J. R.; Simolo, K.; Taylor, K.; Mauk, A. G.; English, A. M.
ACS Symp. Series 1986, 150.
(a) Gray, H. B. Chem. Soc. Rev. 1986, 15, 17. (b) Mayo, S. L.; Ellis, W.
R., Jr.; Crutchley, R. J.; Gray, H. B. Science 1986, 233, 948. (c) Scott,
R. A.; Mauk, A. G.; Gray, H. B. J. Chem. Ed. 1985, w, 932.
Moore, G. R.; Williams, R. J. P. Coor. Chem. Rev. 1976, 18, 125.
Isied, S. S. Prog. Inorg. Chem. 1984, 32, 443.
Hush, N. S.; Padden-Rowe, M. N.; Cotsaris, E.; Oevering, H.;
Verhoeven, J. W.; Heppener, M. Chem. Phys. Latt. 1985, 117, 8.

31.
32.
5:3
37.
41.
203
Warman, J. M.; deHass, M. P.; Padden-Rowe, M. N.; Cotsaris, E.;
Hush, N. S.; Oevering, H.; Verhoeven, J. W. Nature, 1986, 320, 615.
Close, G. L.; Calcaterra, L. T.; Green, N. J.; Penfield, K. W.; Mille, J.
R. J. Phys. Chem. 1986, 90, 3673.
Heitele, H.; Michel-Beyerle, M. E. J. Am. Chem. Soc. 1985, 107, 8286.
Marcus, R. A., Sutin, N. Biochem. Biophys. Acta 1985, 811, 265
Maurzerall, D. C. In Photoinduced Electron Transfer; Fox, M. A.,
Chanon, M., Eds.; Elsevier: Amsterdam, 1988; Part D, Chapter 6
Babcock, G. T.; Barry, B. A.; Debus, R. J.; Hoganson, C. W.;
Atamian, M.; Mcintosh, L.; Sithole, I.; Yocum, C. F. Biochem. 1989,
28,9557.
Friesner, R. A.; Won, Y. Biochim. Biophys. Acta, Rev. Bioenerg.
1989,99, 977.
Allen, J. P.; Feher, G.; Yeates, T. O.; Rees, D. C.; Deisenhofer, J.;
Michel, H.; Huber, R. Proc. Natl. Acad. Sci. USA 1986, 83, 8589.
McDowell, L. M.; Kirmaier, C.; Holten, D. J. Phys. Chem. 1991, 95,
3379.
Netzel, T. L.; Bergkamp, M. A.; Chang, C. K. J. Am. Chem. Soc.
1982, 104, 1952.
Siemiarczuk, A.; McIntosh, A. R.; Ho, T.-F.; Stillman, M. J.; Roach,
K. J.; Weedon, A. C.; Bolton, J. R.; Connolly, J. S. J. Am. Chem. Soc.
1983. 105, 7224.
Leland, B. A.; Joran, A. D.; Felker, P. M.; Hopfield, J. J.; Zewail, A.
H.; Dervan, P. B. J. Phys. Chem. 1985, 89, 5571.
Schmidt, J. A.; McIntosh, A. R.; Weedon, A. C.; Bolton, J. R.;
Connolly, J. 8.; Hurley, J. K; Wasielewski, M. R. J. Am. Chem. Soc.
1988, 110, 1733.
Osuka, A.; Maruyama, K.; Mataga, N.; Asahi, T.; Yamazaki, 1.;
Tamai, N. J. Am. Chem. Soc. 1990, 112, 4958.
(a) Gust, D.; Moore, T. A.; Moore, A. L. et. al. Science 1990, 248, 199.
(b) Gust, D.; Moore, T. A.; Moore, A. L.; Makings, L. R.; Seely, G. R.;

47.
49.
51.
52.
57.
204
Ma, X.; Trier, T. T.; Gao, F. J. Am. Chem. Soc. 1988, 110, 7567. (c)
Gust, D.; Moore, T.A. In Supramolecular Photochemistry; Balzani,
V., Ed.; Reidel: Boston, 1987.
(a) Wasielewski, M. R.; Minsek, D.W.; Niemczyk, M. P.; Svec, W. A.;
Yang, N. C. J. Am. Chem. Soc. 1990, 112, 2823. (b) Wasielewski, M.
R. In Photoinduced Electron Transfer; Fox, M. A., Chanon, M., Eds;
Elsevier: Amsterdam, 1988; Part A, p 161. (c) Wasielewski, M. R.;
Niemczyk, M. P.; Svec, W. A.; Pewitt, E. B. J. Am. Chem. Soc. 1985,
107, 5562.
Gust, D.; Moore, T. A.; Moore, A. L.; Barrett, D.; Harding, L. O.;
Makings, L. R.; Liddell, P. A.; De Schryver, F. C.; Van der
Auweraer, M.; Bensasson, R. V.; Rougée, M. J. Am. Chem. Soc.
1988, 110, 321.
Miller, J. R. Nouveau J. Chim. 1987, 11, 83.
Wasielewski, M. R. In Photoinduced Electron Transfer; Fox, M. A.,
Chanon, M., Eds.; Elsevier: Amsterdam, 1988; Part A, Chapter 4.
Helm, R. H.; Simhon, E. D. In Molybdenum Enzymes; Spiro, T., Ed.;
Wiley-Interscience: New York, 1985; p 1.
Christou, G. Acc. Chem. Res. 1989, 22, 328.
Wieghardt, K. Angew. Chem, Int. Ed. Engl. 1989, 28, 1153.
Tyeklar, Z.; Karlin, K. D. Acc. Chem. Res. 1989, 22, 241 and
references cited therein.
Groves, J. T.; Ann. N. Y. Acad. Sci. 1986, 47, 99.
Collman, J. P.; Hampton, P. D.; Brauman, J. I. J. Am. Chem. Soc.
1990, 112, 2977, 2986.
Panicucci, R.; Bruice, T. J. Am. Chem. Soc. 1990, 112, 6063.
(a) Menage, S.; Brennan, B. A.; Juarez-Garcia, C.; Munck, E.; Que,
L., Jr. J. Am. Chem. Soc. 1990, 112, 6423. (1)) Que, L., Jr.; True, A. E.
ng. Inorg. Chem. 1990, 38, 000.
Lippard, S. J. Angew. Chem., Int. Ed. Engl. 1988, 27, 344.
Lancaster, J. R., Jr., Ed. The Bioinorganic Chemistry of Nickel;
VCH: New York, 1988.

61..3
70.
71.
205
Que, L., Jr., Ed. In Metal Clusters in Pmteins; American Chemical
Society: Washington. DC, 1988; Series 372.
Sykes, A. G., Ed. Advances in Inorganic and Bioinorganic
Mechanisms; Academic Press: London, 1983.
Roundhill, D. M.; Gray, H. B.; Che, C.-M. Acc. Chem. Res. 1989, 22,
55.
(a) Roundhill, D. M.; Shen, Z.-P.; King, C.; Atherton, S. J. J. Phys.
Chem, 1988, 92, 4088. (b) Roundhill, D. M.; Dickson, M. K.; Atherton,
S. J. J. Organometallic Chem. 1987, 335, 413. (c) Roundhill, D. M.;
Atherton, S. J.; Shen, Z.-P. J. Am. Chem. Soc. 1987, 109, 6076. (d)
Roundhill, D. M.; Atherton, S. J. Inorg. Chem. 1986, 25, 4071. (e)
Roundhill, D. M. J. Am. Chem. Soc. 1985, 107, 4354.
(a) Harvey, E. L.; Stiegman, A. E.; Vlcek, A., Jr.; Gray, H. B. J. Am.
Chem. Soc. 1987, 109, 5233. (b) Vlcek, A., Jr.; Gray, H. B. Inorg.
Chem. 1987, 26, 1997. (c) Vleck, A.; Gray, H. B. J. Am. Chem. Soc.
1987, 109, 286
(a) Smith, D. C.; Gray, H. B. Coord. Chem. Rev. 1990, 100, 169. (b)
Smith, D. C.; Gray, H. B. ACS Symposium Series 1989, 394, 356.
(a) Marshall, J. L.; Stobart; S. R.; Gray, H. B. J. Am. Chem. Soc.
1984, 106, 3027. (b) Caspar, J. V.; Gray, H. B. J. Am. Chem. Soc. 1984,
106, 3029.
Marshall, J. L.; Stiegman, A. E.; Gray, H. B. ACS Symp. Ser. 1986,
307, 166 and references therein.
(a) Dulebohn, J. 1.; Ward, D. L.; Nocera, D. G. J. Am. Chem. Soc.
1990, 112, 2969. (b) Dulebohn, J. 1.; Nocera, D. G. J. Am. Chem. Soc.
1988, 110, 4054.
Hush, N. S. Coord. Chem. Rev. 1985, 64, 135.
Vogler, A.; Osman, A. H.; Kunkley, H. Coord. Chem. Rev. 1985, 64,
159.
Meyer, T. J. In Mixed Valence Compounds; Brown, D. B. Ed.; Reidel:
Boston, 1980; Volume 58, p 75.
Creutz, C. Prog. Inorg. Chem. 1983, 30, 1.

72.
73.
74.
75.
76.
77.
78.
79.
81.
82.
206
Newton, M. D.; Sutin, N. Ann. Rev. Phys. Chem. 1984, 35, 437.
Dong, T.-Y.; Hwang, M.-Y.; Hsu, T.-L.; Schei, C.-C.; Yeh, S.-K.
Inorg. Chem. 1990, 29, 80.
Doorn, S. K.; Hupp, J. T. J. Am. Chem. Soc. 1989, 111, 1142.
(a) Rendell, A. P. L.; Bacskay, G. B.; Hush, N. S. J. Am. Chem. Soc.
1988, 110, 8343. (b) Beattie, J. K.; Del Favero, P.; Hambley, T. W.;
Hush, N. S. Inorg. Chem. 1988, 27, 2000.
(a) Onuchic, J. N.; Beratan, D. N. J. Am. Chem. Soc. 1987, 109, 6771.
(b) Beratan, D. N.; Hopfield, J. J. J. Am. Chem. Soc. 1984, 106, 1584.
Haga, M.-a.; Matsumura-Inoue, T.; Yamabe, S. Inorg. Chem. 1987,
26,4148.
Bertrand, P. Chem. Phys. Lett. 1987, 140, 57.
Newton, M. D. J. Phys. Chem. 1986, 90, 3734.
Isied, S. S.; Vassilian, A.; Magnuson, R. H.; Schwarz, H. A. J. Am.
Chem. Soc. 1985, 107, 7432.
(a) Richardson, D. E.; Taube, H. J. Am. Chem. Soc. 1983, 105, 40. (b)
Krentzien, H.; Taube, H. Inorg. Chem. 1982, 21, 4001. (c) Dowling,
N.; Henry, P. M.; Lewis, N. A.; Taube, H. Inorg. Chem. 1981, 20,
2345. (d) Krentzien, H.; Taube, H. J. Am. Chem. Soc. 1976, 98, 6379.
(e) Tom, G. M.; Taube, H. J. Am. Chem. Soc. 1975, 97, 5310. (f) Tom,
G. M.; Creutz, C.; Taube, H. J. Am. Chem. Soc. 1974, 96, 7827.
(a) Stein, C. A.; Lewis, N. A.; Seitz, G.; Baker, A. D. Inorg. Chem.
1983, 22, 1124. (b) Stein, C. A.; Lewis, N. A.; Seitz, G. J. Am. Chem.
Soc. 1982, 104, 2596.
Callahan, R. W.; Brown, G. M.; Meyer, T. J. Inorg. Chem. 1975, 14,
1443.
Siddarth, P.; Marcus, R. A. J. Phys. Chem. 1990, 94, 2985.
Woitellier, S.; Launay, J. P.; Spangler, C. W. Inorg. Chem. 1989, 28,
758.

87.
58
EB
91.
207
Katz, N. E.; Creutz, C.; Sutin, N. Inorg. Chem. 1988, 27, 1687.
(a) Diril, H.; Chang, H.-R.; Nilges, M. J.; Zhang, X.; Potenza, J. A.;
Schugar, H. J.; Isied, S. S.; Hendrickson, D. N. J. Am. Chem. Soc.
1989, 111, 5102. (b) Isied, S. S.; Vassilian, A.; Wishart, J. F.; Creutz,
C.; Schwarz, H. A.; Sutin, N. J. Am. Chem. Soc. 1988, 110, 635. (c)
Isied, S. S.; Vassilian, A. J. Am. Chem. Soc. 1984, 106, 1732.
Geselowitz, D. A. Inorg. Chem. 1987, 26, 4135.
Haim, A. Pure Appl. Chem. 1983, 55, 89.
(a) Powers, M. J.; Meyer, T. J. J. Am. Chem. Soc. 1980, 102, 1289. (b)
Meyer, T. J. Acc. Chem. Res. 1977, 11, 94. (c) Callahan, R. W.;
Keene, F. R.; Meyer, T. J.; Salmon, D. J. J. Am. Chem. Soc. 1977, 99,
1064. (d) Powers, M. J.; Salmon, D. J.; Callahan, R. W.; Meyer, T. J. l
J. Am. Chem. Soc. 1976, 98, 6731.
(a) Blackbourn, R. L.; Hupp, J. T. J. Phys. Chem. 1990, 94, 1788. (b)
Hupp, J. T. J. Am. Chem. Soc. 1990, 112, 1563. (c) Blackboum, R. L.;
Hupp, J. T. J. Phys. Chem. 1988, 92, 2817. (d) Hupp, J. T.; Meyer, T.
J. J. Phys. Chem. 1987, 91 , 1001. (e) Hupp, J. T.; Meyer, T. J. Inorg.
Chem. 1987, 26, 2332.
(a) Lewis, N. A.; Obeng, Y. S.; Purcell, W. L. Inorg. Chem. 1989, 28,
3796. (b) Lewis, N. A.; Obeng, Y. S. J. Am. Chem. Soc. 1989, 111,
7624. (c) Lewis, N. A.; Obeng, Y. S. J. Am. Chem. Soc. 1988, 110,
2306.
McManis, G. E.; Gochev, A.; Nielson, R. M.; Weaver, M. J. J. Phys.
Chem. 1989, 93, 7733.
(a) Ennix, K. S.; McMahon, P. T.; de la Rosa, R.; Curtis, J. C. Inorg.
Chem. 1987, 26, 2660. (b) Chang, J. P.; Fung, E. Y.; Curtis, J. C.
Inorg. Chem. 1986, 25, 4233. (c) Curtis, J. C.; Sullivan, B. P.; Meyer,
T. J. Inorg. Chem. 1983, 22, 224.
Brunschwig, B. S.; Eherenson, S.; Sutin, N. J. Phys. Chem. 1986, 90,
3657.
(a) Kober, E. M.; Goldsby, K. A.; Narayana, D. N. S.; Meyer, T. J. J.
Am. Chem. Soc. 1983, 105, 4303. (b) Sullivan, B. P.; Curtis, J. C.;
Kober, E. M.; Meyer, T. J. Nouv. J. Chim. 1980, 4, 643. (c) Powers,

97..33
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
208
M. J.; Meyer, T. J. Inorg. Chem. 1978, 17, 1785. ((1) Powers, M. J.;
Meyer, T. J. J. Am. Chem. Soc. 1978, 100, 4393. (e) Powers, M. J.;
Callahan, R. W.; Salmon, D. J.; Meyer, T. J. Inorg. Chem. 1976, 15,
1457.
German, E. D. Chem. Phys. Lett. 1979, 64, 295.
Cannon, R. D. Chem. Phys. Lett. 1977, 49, 299.
Lewis, N. A.; Obeng, Y. S.; Taveras, D. V.; van Eldik, R. J. Am.
Chem. Soc. 1989, 111, 924.
Jernigan, J. C.; Surridge, N. A.; Zvanut, M. E.; Silver, M.; Murray,
R. W. J. Phys. Chem. 1989, 93, 4620.
Hammack, W. S.; Drickamer, H. G.; Lowery, M. D.; Hendrickson, D.
N. Inorg. Chem. 1988, 27, 1307.
Worl, L. A., Meyer, T. J. Chem. Phys. Lett. 1988, 143, 541.
Isied, S. S.; Vassilian, A. J. Am. Chem. Soc. 1984, 106, 1726.
Marcus, R. A. J. Phys. Chem. 1989, 93, 3078.
Lee, G.-H.; Ciana, L. D.; Haim, A. J. Am. Chem. Soc. 1989, 111, 2535.
(a) Curtis, J. C.; Blackbourn, R. L.; Ennix, K. S.; Hu, S.; Roberts, J.
A.; Hupp, J. T. Inorg. Chem. 1989, 28, 3791. (b) Hupp, J. T.; Weydert,
J. Inorg. Chem. 1987, 26, 2657.
Palaniappan, V.; Agarwala, U. C. Inorg. Chem. 1988, 27, 3568.
de la Rosa, R.; Chang, P. J.; Salaymeh, F.; Curtis, J. C. Inorg.
Chem. 1985, 24, 4229.
(a) Creutz, C. Inorg. Chem. 1978, 17, 3723. (b) Creutz, C.; Taube, H.
J. Am. Chem. Soc. 1969, 91 , 3988.
Callahan, R. W.; Brown, G. M.; Meyer, T. J. J. Am. Chem. Soc. 1974,
96, 7829. -
(a) Vogler, A.; Osman, A. H.; Kankely, H. Inorg. Chem. 1987, 26,
2337. (b) Vogler, A.; Kisslinger, J. Inorg. Chim. Acta. 1986, 115, 193.

112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
5.3.
E
209
Cotton, F. A.; Curtis, N. F.; Harris, C. B.; Johnson, B. F. G.;
Lippard, S. J.; Magus, J. T.; Robinson, W. R.; Wood, J. S. Science,
1964, 145, 1305. ’
(a) Cotton, F. A.; Walton, R. A. Multiple Bonds Between Metal
Atoms, Wiley-Intersience: New York, 1982 and references therein.
(b) Cotton, F. A.; Walton, R. A. Structure and Bonding, 1985, 62, 1
and references therein.
Hay, P. J. J. Am. Chem. Soc. 1982, 104, 7007.
Hopkins, M. D.; Gray, H. B.; Miskowski, V. M. Polyhedron, 1987, 6,
705.
Hall, M. B. Polyhedron, 1987, 6, 679.
Bursten, B. E.; Clark, D. L. Polyhedron, 1987, 6, 695.
(a) Ziegler, T.; Tschinke, V.; Beeke, A. Polyhedron, 1987, 6, 685. (b)
Ziegler, T. J. Am. Chem. Soc. 1985, 107, 4453. (c) Ziegler, T. J. Am.
Chem. Soc. 1984, 106, 5901.
Norman, J. G.; Kolan, H. J. J. Am. Chem. Soc. 1975, 97, 33.
Mortola, A. P.; Moskowitz, J. W.; Rosch, N. Int. J. Quantum Chem.,
Symp. No. 8, 1974, 161.
(a) Hopkins, M. D.; Miskowski, V. M.; Gray, H. B. J. Am. Chem.
Soc. 1988, 110, 1787. (b) Hopkins, M. D.; Schaefer, W. P.;
Bronikowski, M. J.; Woodruff, W. H.; Miskowski, V. M.; Dallinger,
R. F.; Gray, H. B. J. Am. Chem. Soc. 1987, 109, 408. (c) Hopkins, M.
D.; Gray, H. B.; Miskowski, V. M. Polyhedron, 1987, 6, 705. (d)
Zietlow, T. C.; Hopkins, M. D.; Gray, H. B. J. Solid State Chem. 1985,
57, 112.
Morris, D. E.; Sattelberger, A. P.; Woodruff, W. H. J. Am. Chem.
Soc. 1986, 108, 8270.
Dallinger, R. F. J. Am. Chem. Soc. 1985, 107, 7202.
(a) Fanwick, P. E. Inorg. Chem. 1985, 24, 258. (b) Fanwick, P. E.;
Bursten, B. E.; Kaufmann, G. B. Inorg. Chem. 1985, 24, 1165.

127.
EEl
131.
132.
133.
134.
135.
136.
137.
138.
210
(a) Huang, H.-W.; Martin, D. S. Inorg. Chem. 1985, 24, 96. (b)
Martin, D. S.; Huang, H.-W.; Newman, R. A. Inorg. Chem. 1984, 23,
699.
Robbins, G. A.; Martin, D. S. Inorg. Chem. 1984, 23, 2086.
Lichtenberger, D. L.; Blevins, C. H. J. Am. Chem. Soc. 1984, 106,
1636.
Clark, R. J. H.; Stead, M. J. Inorg. Chem. 1988, 22, 1214.
Fraser, I. F.; Peacock, R. D. Chem. Phys. Lett. 1983, 98, 620.
Martin, D. S.; Newman, R. A.; Fanwick, P. E. Inorg. Chem. 1982, 21 ,
3400.
(a) Manning, M. C.; Trogler, W. C. J. Am. Chem. Soc. 1983, 105,
5311. (b) Manning, M. C.; Trogler, W. C. Inorg. Chem. 1982, 21 , 2797.
Rice, S. F.; Wilson, R. B.; Solomon, E. I. Inorg. Chem. 1980, 19, 3425.
(a) Trogler, W. C.; Cowman, C. D.; Gray, H. B.; Cotton, F. A. J. Am.
Chem. Soc. 1977, 99, 2993. (b) Fanwick, P. E.; Martin, D. 8.; Cotton,
F. A.; Webb, T. R. Inorg. Chem. 1977, 16, 2103. (c) Cotton, F. A.;
Martin, D. S.; Fanwick, P. E.; Peters, T. J.; Webb, T. R. J. Am.
Chem. Soc. 1976, 98, 4681.
Trogler, W. C.; Gray, H. B. Acc. Chem. Res. 1978, 11, 232.
(a) Miskowski, V. M.; Goldbeck, R. A.; Kliger, D. 8.; Gray, H. B.
Inorg. Chem. 1979, 18, 86. (b) Cowman, C. D.; Trogler, W. C.; Gray,
H. B. Isr. J. Chem. 1977, 15, 308. (c) Cowman, C. D.; Gray, H. B. J.
Am. Chem. Soc. 1973, 95, 8177.
Reference 115 and 131(a) and references cited therein.
Hopkins, M. D.; ZieHow, T. C.; Miskowski, V. M.; Gray, H. B. J. Am.
Chem. Soc. 1985, 107, 511.
Zhang, X.; Kosik, M.; Sutin, N.; Winkler, J. R. ACS Symposium
Series, 1990, in press.

211
139. Winkler, J. R.; Nocera, D. G.; Netzel, T. L. J. Am. Chem. Soc. 1986,
108, 4451.
140. Hopkins, M. D.; Gray, H. B. J. Am. Chem. Soc. 1984, 106, 2468.
141. Miskowski, V. M.; Goldbeck, R. A.; Kliger, D. 8.; Gray, H. B. Inorg.
Chem. 1979, 18, 86.
142. Schrock, R. R.; Stargeofi‘, L. G.; Sharp, P. R. Inorg. Chem. 1983, 22,
2801.
143. Azizian, H.; Luck, R.; Morris, R. H.; Wong, H. J. Organomet. Chem.
1982, 238, C24.
144. Butcher, A. V.; Leigh, G. J.; Richards, P. L. J. Chem. Soc. Dalton.
Trans. 1972, 1064.
145. San Filippo. J. Jr. Inorg. Chem. 1972, 11, 3140.
146. Brencie, J. V.; Cotton, F. A. Inorg. Chem. 1970, 9, 346.
147. Chang, I-J. Ph. D. Dissertation, Michigan State University.
148. Luck, R. L.; Morris, R. H.; Sawger, J. F. Inorg. Chem. 1987 , 226,
2422.
149. Best, S. A.; Smith, T. J.; Walton, R. A. Inorg. Chem. 1978, 17, 99.
150. Taube, H.; Bowen, A. R. J. Am. Chem. Soc. 1971, 193, 323.
151. Canich, J. M.; Cotton, F. A. Inorg. Chim. Acta, 1988, 142, 69.
152. Canich, J. M.; Cotton, F. A.; Daniels, L. M.; Lewis, D. B. Inorg.
Chem. 1987, 26, 4046.
153. Chacon, S. T.; Chisholm, M. H.; Streib, W. E.; Van der Sluys, W.
Inorg. Chem. 1989, 28, 5.
154. Gordon, A. J.; Ford, R. A. The Chemist's Companions A Handbook
of Practical Data, Techniques, and References
155. Calvert, J. G.; Pitts, J. N. Photochemistry; Wiley-Interscience: New
York, 1966.
a.-
.‘

156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
212
Gordon, R. R.; Koval, C. A.; Lisensky, G. C. Inorg. Chem. 1980, 19,
2854.
Zoski, c. G.; Sweigart, D. A.; Stone, N. J.; Rieger, P. H. Mocellin, E.;
Mann, T. F.; Mann, D. R.; Gosser, D. K.; Doefi', M. M.; Bond, A. M.
J. Am. Chem. Soc. 1988, 110, 2109.
Newsham, M. D. Ph. D. Dissertation, Michigan State University.
Demas, N. J.; Crosby, G. A. J. Phys. Chem. 1971, 75, 991.
Jackson, J. A.; Turro, C.; Newsham, M. D.; Nocera, D. G. J. Phys.
Chem. 1990, 94, 4500.
Agaskar, P. A.; Cotton, F. A.; Fraser, 1. F.; Manojlovic, Muit, L.;
Mair, K. W.; Peacock, R. D. Inorg. Chem. 1986, 25, 2511.
Campell, F. L.; Cotton, F, A.; Powell, G. L. Inorg. Chem. 1985, 24,
177.
Cotton, F. A.; Powell, G. L. Inorg. Chem. 1983, 22, 1507.
For a review of coordination chemistry of dinuclear Mo(III) and
W(III) dimers see Chisholm, M. H. Acc. Chem. Res. 1990, 23, 419.
For a review of edge-sharing bioctahedral complexes see Cotton, F.
A. Polyhedron, 1987, 6, 667.
Fanwick, P. E.; Harwood, W. 8.; Walton, R. A. Inorg. Chem. 1987 ,
26, 242.
(a) Cotton, F. A.; Daniels, L. M.; Dunbar, K. R.; Falvello, L. R.;
O'Connor, C. J.; Price, A. C. Inorg. Chem. 1991, 30, 2509. (b) Canich,
J. M.; Cotton, F. A.; Dunbar, K. R, Falvello, L. R. Inorg. Chem. 1988,
27, 804. (c) Agaskar, P. A.; Cotton, F. A.; Dunbar, K. R.; Falvello, L.
R.; O'Connor, C. J. Inorg. Chem. 1987, 26, 4051. (d) Chakravarty, A.
R.; Cotton, F. A.; Diebold, M. P.; Lewis, D. B.; Roth, W. J. J. Am.
Chem. Soc. 1986, 108, 971. (e) Cotton, F. A.; Diebold, M. P.; O'Connor,
C. J.; Powell, G. L. J. Am. Chem. Soc. 1985, 107, 7438. (1') Cotton, F.
A.; Powell, G. L. J. Am. Chem. Soc. 1984, 106, 3371. (g) Cotton, F. A.;
Mott, G. N. J. Am. Chem. Soc. 1982, 104, 5978.

168.
169.
170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
213
Moynihan, K. J.; Gao, X.; Boorman, P. M.; Fait, J. F.; Freeman, G.
K. W.; Thornton, P.; Ironmonger, D. J. Inorg. Chem. 1990, 29, 1648.
Bott, S. G.; Clark, D. L.; Green, M. L. H.; Mountford, P. J. Chem.
Soc., Chem. Commun. 1989, 418.
(a) Cotton, F. A.; Luck, R. L. Inorg. Chem. 1989, 28, 182. (b) Cotton,
F. A.; Poli, R. Inorg. Chem. 1987, 26, 3310.
Bergs, D. J.; Chisholm, M. H.; Folting, K.; Humnan, J. C.; Stahl, K.
A. Inorg. Chem. 1988, 27, 2950.
Chisholm, M. H.; Eichhorn, B. W.; Folting, K.; Hufi‘mann, J. C.;
Ontiveros, C. D.; Streib, W. E.; Van Der Sluys, W. G. Inorg. Chem.
1987,26, 3182.
Nocera, D. G.; Gray, H. B. Inorg. Chem. 1984, 23, 3686.
Cotton, F. A.; Eglin, J. L.; Luck, R. L.; Son, K. Inorg. Chem. 1990, 29,
1802.
(a) Poli, R.; Mui, H. D. Inorg. Chem. 1990, 30, 2509. (b) Mui, H. D.;
Poli, R. Inorg. Chem. 1989, 28, 3609.
Schrock, R. R.; Sturgeofi', L. G.; Sharp, P. R. Inorg. Chem. 1983, 22,
2801.
Jackson, R. B.; Streib, W. E. Inorg. Chem. 1971, 10, 1760.
Although the impurity with an emission maximum at 420 am has
not yet been identified, it is noted that it is present in newly received
unopened samples of dmpm.
(a) Fife, D. J. J. Photochem. 1984, 24, 249. (b) Fife, D. J.; Moore, W.
M.; Morse, K. W. Inorg. Chem. 1984, 23, 1545.
The M—M bond order of the photoinduced transients are not
specified because the M—M bonding in complexes with bioctahedral
arrangements of ligands is not clearly defined. Furthermore the
mixed valence character of the bimetallic core will affect the M—M
bond order. A possible description of the M(I)—-M(III) bimetallic
core with the bioctahedra arrangements of ligands is a M—M triple

181.
182.
183.
184.
185.
186.
187.
188.
189.
190.
191.
192.
193.
214
bond with a lone pair localized at the MO) center. See references 181
and 183.
Shaik, 8.; Hofi‘man, R.; Fisel, R.; Summerville, R. H. J. Am. Chem.
Soc. 1980, 102, 4555.
Interestingly, an additional transient of much weaker intensity has
recently been observed concurrently with 1,“: 532 nm in these
species. Preliminary data appears in Figure 58 of Appendix I.
The spectral features of the edge-sharing bioctahedral complex
Mo¥C13(PMePh2)4 at 400 nm (sh) (c r 2400 M'lcm'l), 526 nm (e = 300
M“ cm“) and 650 nm (e = 640 M‘lcm‘l) are very similar to those of
the confacial bioctahedral analogue, MozCls(PMePh2)3 at 390 nm (sh)
(s = 1800 M-lcm-I), 515 nm (sh) (c = 540 M'lcm'l) and 590 (c = 800
M'lcm‘l).
Summerville, R. H.; Hoffman, R. J. Am. Chem. Soc. 1979, 101, 3821.
Trogler, W. C. Inorg. Chem. 1980, 19, 697.
Tolman, C. A. Chem. Rev. 1977, 77, 313.
Cotton, F. A.; Daniels, L. M.; Powell, G. L.; Kahaian, A. J.; Smith, T.
J.; Vogel, E. F. Inorg. Chim. Acta. 1988, 144, 109.
Carmona-Guzman, E.; Wilkinson, G. J. Chem. Soc. Dalton Trans.
1977, 1716.
Trogler, W. C.; Gray, H. B. Nouv. J. Chim. 1977, 1, 475.
Trogler, W. C.; Erwin, D. K; Geofi'rey, G. L.; Gray, H. B. J. Am.
Chem. Soc. 1978, 100, 1160.
Erwin, D. K; Geoffrey, D. K; Gray, H. B. J. Am. Chem. Soc. 1977, 99,
3620.
(a) Chang, I-J.; Nocera, D, G, J. Am. Chem. Soc. 1987, 109, 4901. (b)
Chang, I-J.; Nocera, D. G. Inorg. Chem. 1989, 28, 4309.
Glicksman, H. D.; Haamer, A. D.; Smith, T. J.; Walton, R. A. Inorg.
Chem. 1976, 15, 2205.

194.
195.
196.
197.
198.
199.
201.
202.
21 5
Karsch, H. H. Chem. Ber. 1982, 115, 823.
Appel, V. R.; Huppertz, M. Z. anorg. allg. Chem. 1979, 459, 7.
Irradiation of hexane solutions of M02014(PBu3)4 in the presence of a
hundred-fold excess of CH3N(PF2)2 yields a nonemissive product
whose uv-visible absorption spectrum shown in Figure 59 is similar
to MozCl4(dppm)2. The thermodynamic unfavorability of this reaction
is evidenced by the fact that M02C14(PBu3)4 is reformed within
minutes after the irradiation is discontinued, as confirmed by
absorption and emission spectroscopy. This thermal back reaction
has prevented us from isolating this product, which is believed to be
MozCl4[CH3N(PF2)2]2. However this reaction suggests that
phosphine substitution is likely to dominate the photochemical
pathways of this complex.
Roundhill, D. M. Inorg. Chem. 1986, 25, 4071.
Cotton, F. A.; Walton, R. A. Multiple Bonds Between Metal Atoms;
Wiley-Interscience: New York, 1982, p. 221.
Fundamental Transition Metal Organometallic Chemistry;
Lukehart, C. M. Ed.; Wadsworthg Inc: California, 1985, Chapter 10.
Stills, J. K, Lau, K S.Y. Acc. Chem. Res. 1977, 10, 434.
Labinger, J. A.; Osborn, J. A.; Coville, N. J. Inorg. Chem. 1980, 19,
3236.
Partigianoni, C. M.; Chang, I-J.; Nocera, D. G. Coor. Chem. Rev.
1990, 97, 105.
As previously noted these complexes exhibit an additional transient
species that has not yet been characterized, which may prove to be a
promising candidate for multielectron photoreactivity. Namely, the
equilibrium between confacial and edge-sharing bioctahedral
complexes may account for the presence of two transient species.

nICHIan STATE UNIV. LIBRARIES
lllllllllllllllllllllllllllllllllllllllllllllllllll31293009141106




















