







Bahasa
Halaman
Hukum


Molecular Cell
Short Article
TORC2 Signaling PathwayGuarantees Genome Stabilityin the Face of DNA Strand BreaksKenji Shimada,1,5 Ireos Filipuzzi,2,5 Michael Stahl,3 Stephen B. Helliwell,2 Christian Studer,2 Dominic Hoepfner,2
Andrew Seeber,1,4 Robbie Loewith,3 N. Rao Movva,2 and Susan M. Gasser1,4,*1Friedrich Miescher Institute for Biomedical Research, Maulbeerstrasse 66, 4058 Basel, Switzerland2Novartis Institutes for Biomedical Research, Novartis Pharma AG, Fabrikstrasse 22, 4056 Basel, Switzerland3Department of Molecular Biology, University of Geneva, rue Ernest-Ansermet 30, 1211 Geneva, Switzerland4Faculty of Natural Sciences, University of Basel, Klingelbergstrasse 70, 4056 Basel, Switzerland5These authors contributed equally to this work
*Correspondence: [email protected]://dx.doi.org/10.1016/j.molcel.2013.08.019
SUMMARY
A chemicogenetic screen was performed in buddingyeast mutants that have a weakened replicationstress response. This identified an inhibitor of targetof rapamycin (TOR) complexes 1 and 2 that selec-tively enhances the sensitivity of sgs1D cells tohydroxyurea and camptothecin. More importantly,the inhibitor has strong synthetic lethality in combi-nation with either the break-inducing antibiotic Zeo-cin or ionizing radiation, independent of the strainbackground. Lethality correlates with a rapid frag-mentation of chromosomes that occurs only whenTORC2, but not TORC1, is repressed. Genetic inhibi-tion of Tor2 kinase, or its downstream effectorkinases Ypk1/Ypk2, conferred similar synergisticeffects in the presence of Zeocin. Given that Ypk1/Ypk2 controls the actin cytoskeleton, we testedthe effects of actin modulators latrunculin A andjasplakinolide. These phenocopy TORC2 inhibitionon Zeocin, althoughmodulation of calcineurin-sensi-tive transcription does not. These results implicateTORC2-mediated actin filament regulation in the sur-vival of low levels of DNA damage.
INTRODUCTION
The maintenance of genome stability is essential for all living
organisms, as cells are continuously challenged by endogenous
and exogenous sources of DNA damage. Inappropriate repair of
lesions can lead to genetic aberration, including chromosome
loss, translocations, deletions, and point mutations. Cells are
particularly sensitive to genomic insult during DNA replication,
and inadequate protection of replisome stability engenders
gross chromosomal rearrangements (Aguilera and Gomez-
Gonzalez, 2008). Intriguingly, oncogene activation itself triggers
replication fork-associated stress and activates a low-level
Molecu
checkpoint response (Bartkova et al., 2006; Di Micco et al.,
2006). Two proteins that contribute synergistically to ensure
genomic stability at stressed replication forks are the ataxia tel-
angiectasia and Rad3-related (ATR) checkpoint kinase (Mec1 in
yeast), and the Bloom’s syndrome RecQ helicase (Sgs1 in yeast)
(Cobb et al., 2005; Errico and Costanzo, 2012). Consistently,
both of these are tumor suppressor genes in mammalian cells
(Bartek et al., 2007; Bernstein et al., 2010).
DNA damage and replication checkpoint pathways are
conserved from yeast to man. The budding yeast Tel1 and
Mec1 kinases (ataxia telangiectasia mutated [ATM] and ATR in
mammals, respectively) are members of the phosphoinositide
3-kinase-related family, and both have central roles in the check-
point signaling cascade (Lovejoy and Cortez, 2009). A further
PIKK family member is the cytoplasmic kinase TOR (target of
rapamycin), which does not control the checkpoint response,
but instead regulates cell growth in response to nutritional stress
(Heitman et al., 1991a). In mammalian cells, a single TOR kinase
subunit, mammalian TOR (mTOR), takes part in two distinct TOR
complexes, whereas in budding yeast there are two genes
encoding for related catalytic subunits. Yeast Tor1 serves only
in TOR complex 1 (TORC1), whereas Tor2 can function as the
catalytic subunit in either TORC1 or TORC2. The TORC1 com-
plex additionally contains Kog1, Tco89, and Lst8, while TORC2
includes Avo1, Avo2, Avo3/Tsc11, Bit61, Bit2, and Lst8 (Loewith
et al., 2002; Reinke et al., 2004). Both complexes are essential
and also widely conserved across eukaryotic species (Loewith
and Hall, 2011). Indeed, the immunosuppressant rapamycin
binds FKBP12, a cis-trans prolyl isomerase, to form a prodrug
complex that specifically inhibits TORC1 in both yeast and man.
The downstream effectors of TORC1 activation include the
AGC-family kinase Sch9 and a subset of type 2A protein phos-
phatases, in complex with a regulatory subunit, Tap42. TORC1
promotes an increase in cellular protein mass by simultaneously
stimulating anabolic processes, such as translation initiation and
ribosome biogenesis, and inhibiting catabolic processes (Loe-
with and Hall, 2011). The major effectors of TORC2 are Ypk1
and Ypk2, a redundant pair of AGC-family kinases related to
the mammalian kinase SGK1 (Kamada et al., 2005; Niles et al.,
2012). Through the Ypk1/Ypk2 kinases, TORC2 regulates actin
lar Cell 51, 829–839, September 26, 2013 ª2013 Elsevier Inc. 829
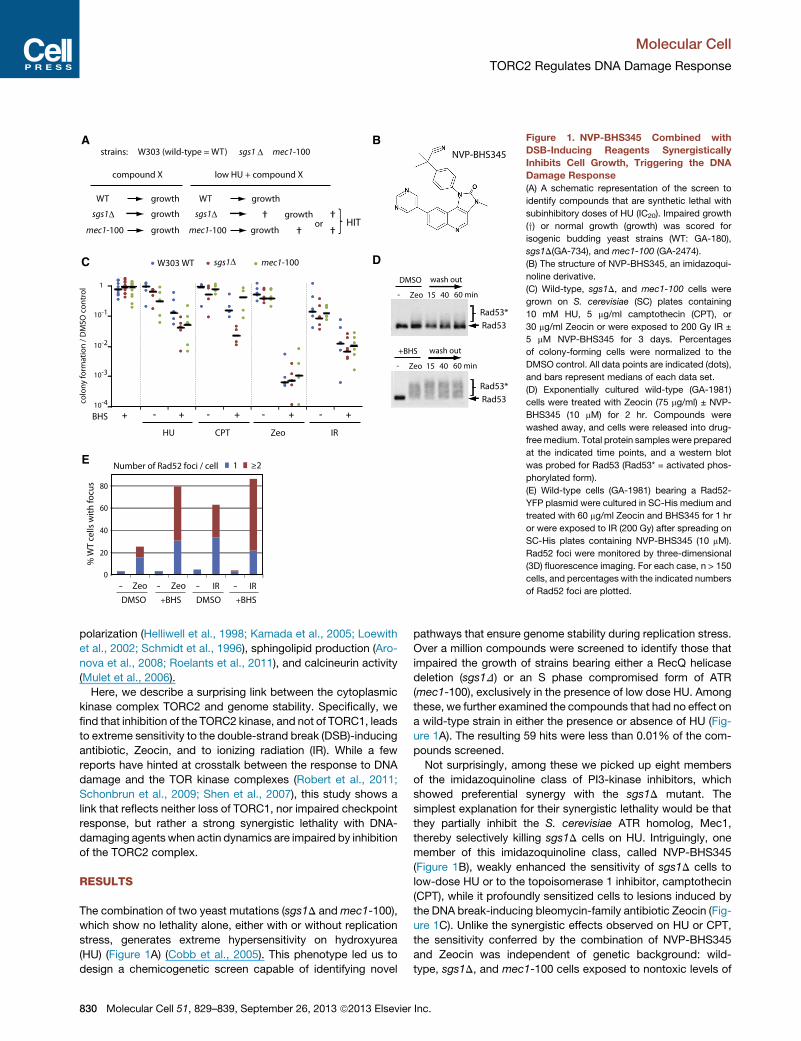
A
C
E
B
D
Figure 1. NVP-BHS345 Combined with
DSB-Inducing Reagents Synergistically
Inhibits Cell Growth, Triggering the DNA
Damage Response
(A) A schematic representation of the screen to
identify compounds that are synthetic lethal with
subinhibitory doses of HU (IC20). Impaired growth
(y) or normal growth (growth) was scored for
isogenic budding yeast strains (WT: GA-180),
sgs1D(GA-734), and mec1-100 (GA-2474).
(B) The structure of NVP-BHS345, an imidazoqui-
noline derivative.
(C) Wild-type, sgs1D, and mec1-100 cells were
grown on S. cerevisiae (SC) plates containing
10 mM HU, 5 mg/ml camptothecin (CPT), or
30 mg/ml Zeocin or were exposed to 200 Gy IR ±
5 mM NVP-BHS345 for 3 days. Percentages
of colony-forming cells were normalized to the
DMSO control. All data points are indicated (dots),
and bars represent medians of each data set.
(D) Exponentially cultured wild-type (GA-1981)
cells were treated with Zeocin (75 mg/ml) ± NVP-
BHS345 (10 mM) for 2 hr. Compounds were
washed away, and cells were released into drug-
freemedium. Total protein samples were prepared
at the indicated time points, and a western blot
was probed for Rad53 (Rad53* = activated phos-
phorylated form).
(E) Wild-type cells (GA-1981) bearing a Rad52-
YFP plasmid were cultured in SC-His medium and
treated with 60 mg/ml Zeocin and BHS345 for 1 hr
or were exposed to IR (200 Gy) after spreading on
SC-His plates containing NVP-BHS345 (10 mM).
Rad52 foci were monitored by three-dimensional
(3D) fluorescence imaging. For each case, n > 150
cells, and percentages with the indicated numbers
of Rad52 foci are plotted.
Molecular Cell
TORC2 Regulates DNA Damage Response
polarization (Helliwell et al., 1998; Kamada et al., 2005; Loewith
et al., 2002; Schmidt et al., 1996), sphingolipid production (Aro-
nova et al., 2008; Roelants et al., 2011), and calcineurin activity
(Mulet et al., 2006).
Here, we describe a surprising link between the cytoplasmic
kinase complex TORC2 and genome stability. Specifically, we
find that inhibition of the TORC2 kinase, and not of TORC1, leads
to extreme sensitivity to the double-strand break (DSB)-inducing
antibiotic, Zeocin, and to ionizing radiation (IR). While a few
reports have hinted at crosstalk between the response to DNA
damage and the TOR kinase complexes (Robert et al., 2011;
Schonbrun et al., 2009; Shen et al., 2007), this study shows a
link that reflects neither loss of TORC1, nor impaired checkpoint
response, but rather a strong synergistic lethality with DNA-
damaging agents when actin dynamics are impaired by inhibition
of the TORC2 complex.
RESULTS
The combination of two yeast mutations (sgs1D andmec1-100),
which show no lethality alone, either with or without replication
stress, generates extreme hypersensitivity on hydroxyurea
(HU) (Figure 1A) (Cobb et al., 2005). This phenotype led us to
design a chemicogenetic screen capable of identifying novel
830 Molecular Cell 51, 829–839, September 26, 2013 ª2013 Elsevier
pathways that ensure genome stability during replication stress.
Over a million compounds were screened to identify those that
impaired the growth of strains bearing either a RecQ helicase
deletion (sgs1D) or an S phase compromised form of ATR
(mec1-100), exclusively in the presence of low dose HU. Among
these, we further examined the compounds that had no effect on
a wild-type strain in either the presence or absence of HU (Fig-
ure 1A). The resulting 59 hits were less than 0.01% of the com-
pounds screened.
Not surprisingly, among these we picked up eight members
of the imidazoquinoline class of PI3-kinase inhibitors, which
showed preferential synergy with the sgs1D mutant. The
simplest explanation for their synergistic lethality would be that
they partially inhibit the S. cerevisiae ATR homolog, Mec1,
thereby selectively killing sgs1D cells on HU. Intriguingly, one
member of this imidazoquinoline class, called NVP-BHS345
(Figure 1B), weakly enhanced the sensitivity of sgs1D cells to
low-dose HU or to the topoisomerase 1 inhibitor, camptothecin
(CPT), while it profoundly sensitized cells to lesions induced by
the DNA break-inducing bleomycin-family antibiotic Zeocin (Fig-
ure 1C). Unlike the synergistic effects observed on HU or CPT,
the sensitivity conferred by the combination of NVP-BHS345
and Zeocin was independent of genetic background: wild-
type, sgs1D, and mec1-100 cells exposed to nontoxic levels of
Inc.

Molecular Cell
TORC2 Regulates DNA Damage Response
NVP-BHS345 all became roughly 1,000-fold more sensitive to
nonlethal levels of Zeocin (30 mg/ml; Figure 1C). This compound
did not impair the DNA damage checkpoint activation: the
checkpoint effector kinase Rad53, a target of Mec1 and Tel1,
showed persistent hyperphosphorylation during and after treat-
ment with Zeocin in the presence of NVP-BHS345 (Figure 1D).
While the lethality could theoretically stem from enhanced Zeo-
cin uptake, this seemed an unlikely explanation, because similar
effects were observed when NVP-BHS345 was combined with
sublethal doses of IR (Figure 1C).
Both Zeocin and IR cause single- and double-strand breaks
(DSBs) and generate other adducts due to ionizing effects
(Povirk, 1996; Povirk et al., 1977). To monitor the number of
DSBs arising from the combined treatment of NVP-BHS345
with either Zeocin or IR, we scored for the appearance of
Rad52 foci by fluorescence microscopy. Such foci are formed
during the process of DSB repair by homologous recombination
(HR). Remarkably, after 1 hr of exposure to NVP-BHS345 in com-
bination with Zeocin, 79.6% of the cells had Rad52 foci, and
48.2% of cells had multiple Rad52 foci (versus 9.8% on Zeocin
alone; Figure 1E). Similar increases were seen in the presence
of NVP-BHS345 and IR (71.2% of cells had multiple foci). This
suggests that the presence of NVP-BHS345 either modifies the
damage induced by Zeocin or IR or strongly inhibits repair,
generating long-lasting Rad52 foci. The damage incurred by
the combined treatment was poorly reversed by plating in the
absence of reagents: less than 1% of the cells treated for
90 min with Zeocin and NVP-BHS345 were able to form colonies
when replated on rich media in the absence of drug, while >60%
of cells survived the same level of Zeocin in the absence of NVP-
BHS345 (Figure S1 available online).
NVP-BHS345 Induces Chromosomal FragmentationIndependent of Cell-Cycle ProgressionZeocin and IR, the two agents that synergize with NVP-BHS345,
albeit to different extents, induce single-strand and double-
strand breaks, and other types of oxidative damage. The ratio
of single-strand nicks to DSBs is roughly 9:1 for both Zeocin
and its related antibiotic, Bleomycin (Povirk, 1996; Povirk et al.,
1977). Nonetheless, yeast cells readily survive and repair
the damage provoked by Zeocin at concentrations of up to
75 mg/ml. To understand the type of damage induced during
the combined treatment of NVP-BHS345 and Zeocin, we moni-
tored the integrity of the yeast genome after exposure to either
compound alone or the two combined, by separating the 16
yeast chromosomes on a nondenaturing pulsed-field agarose
gel (clamped homogenous electric fields, CHEF). We found
that an incubation of yeast cells with 50 mg/ml Zeocin and
DMSO caused no visible loss of chromosome integrity, and the
chromosomes separated into discrete bands of expected size
(Figure 2A). However, incubation of the cells in as little as 5 mM
NVP-BHS345 together with 50 mg/ml Zeocin caused a dramatic
fragmentation of the chromosomes into pieces of roughly 150
kbp (Figure 2A). NVP-BHS345 alone at maximum dose had no
effect on chromosome integrity, suggesting that only the combi-
nation treatment leads to chromosome fragmentation.
We asked whether passage through the cell cycle was impor-
tant for this chromosome shearing, but this was not the case;
Molecu
cells arrested in G1 and exposed to the two drugs in a-factor
showed an identical chromosome effect (Figure 2B). Finally,
we made sure that the fragmentation did not arise during sample
preparation for CHEF gel electrophoresis by omitting the incuba-
tion of agarose-embedded spheroplasts at 50�C; gentler prepa-rations yielded similar results (Figure S1). The visible increase in
Rad52 foci in live cells exposed to NVP-BHS345 and either IR or
Zeocin further confirmed that breaks occur in these intact cells
(Figure 1D). We further rule out that Zeocin and the imidazoqui-
noline act on each other by showing that ionizing irradiation
in the presence of NVP-BHS345 also generates a significant
degree of chromosome fragmentation (Figure S2).
Yeast Chromosome Shattering Is Not Due to ImpairedHR, Impaired NHEJ, or ApoptosisThis observed massive chromosome fragmentation is reminis-
cent of the cancer-associated chromothripsis, or chromosome
shattering, deduced from the drastic reorganization of the
human genome in certain cancer cells (Stephens et al., 2011).
While we do not see efficient religation of the chromosomal frag-
ments during recovery from the combined drug stress, we do
observe enhanced levels of DSBs. Given this effect, we will refer
to the fragmentation observed here as yeast chromosome shat-
tering (YCS). An incubation of agarose-embedded YCS DNA
with T4 DNA ligase prior to migration in the pulsed-field gel
electrophoresis (PFGE) system failed to religate the fragments,
suggesting that the 30-hydroxy and the 50-phosphate termini
may not be intact after the dual drug treatment (data not shown).
YCS was not observed when either NVP-BHS345 or Zeocin
was added to cells alone, even at 4-fold-higher concentrations
(Figures 2A and S4F), nor did we observe chromosome fragmen-
tation when cells were treated with NVP-BHS345 and high levels
of HU, which stalls the replication fork (data not shown). We pro-
pose that NVP-BHS345 treatment leads to a selective change in
Zeocin- or IR-induced insults, generating DSBs.
To repair DSBs, cells use either nonhomologous end-joining
(NHEJ), which directly rejoins two broken ends, or HR, which
copies intact sequence information from a sister chromatid
or homologous chromosome (Symington and Gautier, 2011).
NHEJ occurs efficiently in G1 phase cells, while DSB resection
and recombination are preferred in S phase. We asked whether
NVP-BHS345 acts by inhibiting one of these repair pathways by
testing viable mutants that are deleted for genes essential for
DSB repair, namely LIG4 for NHEJ (Wilson et al., 1997) and
RAD51 for HR (Sung et al., 2003). Neither single nor double
deletions of these genes compromised chromosome integrity
in the presence of Zeocin (Figure 2C), and the NVP-BHS345-
Zeocin-induced fragmentation occurred in all repair mutants
tested.
We examined a time course of NVP-BHS345-Zeocin-induced
fragmentation and found that 30 min is sufficient for massive
YCS, which changes only slightly upon extending the incubation
to 90 min (Figure 2D). By monitoring membrane distribution with
the dye FM4-64 (Vida and Emr, 1995) and nuclear shape with
GFP-Nuc1, we could see that even 90 min in NVP-BHS345
and Zeocin did not alter nuclear or vacuolar ultrastructure (Fig-
ure 2E). Cells did not swell or become dramatically distorted,
as they do during H2O2-provoked yeast apoptosis (Figure 2E).
lar Cell 51, 829–839, September 26, 2013 ª2013 Elsevier Inc. 831

A B C
D E F
Figure 2. NVP-BHS345 Provokes Chromosome Breakdown in Cells Treated with Zeocin(A) Exponentially growingwild-type cells (GA-1981) in SCmediumwere treated for 1 hr with 50 mg/ml Zeocin (+) and the indicated concentrations of NVP-BHS345.
Ethidium bromide-stained CHEF gels show intact yeast chromosomes and fragmentation.
(B) Exponentially growing wild-type (GA-1981) cells were arrested in G1 by a factor, then treated in combination with NVP-BHS345 (10 mM) and Zeocin (75 mg/ml)
for 60 min as indicated. Genomic DNA was subjected to CHEF gel analysis. Cell-cycle arrest was examined by fluorescence-activated cell sorting (FACS)
(at right).
(C) Mutants in NHEJ or HR repair pathways do not mimic NVP-BHS345 treatment. Exponentially growing wild-type (GA-1981), lig4D (GA-6096), rad51D
(GA-6097), and lig4D rad51D (GA-6098) cells were cultured in SCmedium containing the following amounts of Zeocin (equivalent activity): 0 mg/ml (�), 12.5 mg/ml
(+), 50 mg/ml (++), or 12.5 mg/ml Zeocin with 10 mM NVP-BHS345, for 90 min. Genomic DNA was subjected to CHEF gel analysis.
(D)Wild-type (GA-1981) cells were incubated in Zeocin (50 mg/ml) ± NVP-BHS345 (BHS, 10 mM) in SCmedia for 30 or 90min, as indicated. Cells were prepared for
CHEF gel analysis (see A).
(E) Cells bearing Nup2-GFP (GA-6646) were treatedwith DMSO (control) or Zeocin (75 mg/ml) + NVP-BHS345 (10 mM) for 90min or with 0.6mMH2O2 for 24 hr. Cell
membranes are visualized by FM4-64-FIX (Invitrogen) and nuclear morphology by Nup2-GFP, using confocal microscopy. Bar = 5 mm.
(F) To rule out a role for apoptotic nuclease Nuc1 in chromosome fragmentation, exponential cultures of wild-type (GA-6696) and nuc1D (GA-6898) cells were
treated with Zeocin (75 mg/ml), NVP-BHS345 (10 mM), or both for 70 min. Chromosomal DNA was subjected to CHEF gel analysis as in (A).
Molecular Cell
TORC2 Regulates DNA Damage Response
832 Molecular Cell 51, 829–839, September 26, 2013 ª2013 Elsevier Inc.

A B
C
D E
Figure 3. NVP-BHS345 Inhibits TORC1 and
TORC2 in Budding Yeast
(A) Haploinsufficiency profiling (HIP) of NVP-
BHS345 was performed by growing heterozy-
gous deletion mutants covering 5,797 genes
(Open Biosystems) as a pool in the presence and
absence of subinhibitory concentrations of NVP-
BHS345 (30% overall growth inhibition). Relative
abundance of each individual deletion strain is
determined after 20 generations by comparing
growth ± NVP-BHS345. Sensitivity of the indi-
vidual deletion strains is given (y axis) and sup-
plemented by a statistical measure (selectivity,
x axis) to correct for frequent hitters. The most
sensitive and selective deletion strains are found
toward the bottom left. For complete hit list see
Table S1.
(B) Wild-type (GA-5743), TORC1 bypass (GA-
5744), TORC2 bypass (GA-5745), and TORC1 and
TORC2 bypass (GA-5746) strains (Table S2)
were grown for 2 days at 30�C on SC containing
20 nM rapamycin, 50 mM NVP-BHS345, or 1%
DMSO alone. Growth on NVP-BHS345 of TORC1
and TORC2 bypass strains suggests that NVP-
BHS345 inhibits these enzymes.
(C) MS113 (W303a URA3::HA-SCH9) cells were
treated with rapamycin (200 nM), wortmanin
(23 mM), and NVP-BHS345 as indicated. Proteins
separated on SDS gels were blotted for Sch9
phospho-T737 and HA-Sch9, or for Ypk1 phos-
pho-T662 and Ypk1 total. Phosphorylated sub-
strate was normalized to total protein in each lane
(P/Total).
(D) TORC1, TORC2, and their respective sub-
strates were TAP purified from yeast (see Experi-
mental Procedures). TORC1 and TORC2 sub-
strates (GST-tagged recombinant Sch9C-terminal
domain and kinase-dead Ypk2) were incubated
with the kinases in the presence of radiolabeled
ATP and increasing concentrations of NVP-
BHS345 (mM), as indicated. Reaction products
were separated on SDS gels, and incorporated
radioactivity was quantified by phosphoimager.
Each data point is an average of technical tripli-
cates normalized to the mock-treated samples.
Error bars = SE.
(E) Immunoprecipitated Mec1-Ddc2-GFP from
exponentially growing cells was incubated with substrate peptide Sgs1-404-604 (Hegnauer et al., 2012). Mec1 kinase activity was measured in vitro with the
ADP-Glo kit (Promega) in experimental triplicates. Kinase activity was normalized to mock-treated samples. Error bars = SE.
Molecular Cell
TORC2 Regulates DNA Damage Response
We tested whether YCS might nonetheless require elements of
the yeast apoptotic pathway (Carmona-Gutierrez et al., 2010),
yet strains lacking Nuc1, the major apoptosis-induced nuclease
(Buttner et al., 2007), underwent YCS on NVP-BHS345 and
Zeocin, just like wild-type cells (Figure 2F). Thus, we find nothing
that implicates yeast apoptosis in the rapid NVP-BHS345/Zeocin
induction of YCS.
NVP-BHS345 Inhibits TORC1 and TORC2 in YeastImidazoquinolines similar to NVP-BHS345 have been character-
ized as inhibitors of mammalian PIKK and PI3K family kinases
(Maira et al., 2008). In order to determine the pathway targeted
by NVP-BHS345, we performed haploinsufficiency profiling
(HIP; Smith et al., 2010) with subinhibitory levels of NVP-
Molecu
BHS345. This assay scores a library of diploid strains (that are
heterozygous for deletions of all yeast open reading frames
[ORFs]) for reduced growth in the presence of the compound
(Hoepfner et al., 2012). Nine of the 20 most sensitive strains
were found to lack subunits of either the TORC1 or the TORC2
complex (including Tor1 and Tor2) or were targets of TORC2
(Figure 3A; Table S1). Consistently, strains lacking endogenous
downregulators of TORC1 or TORC1 complexes were resistant
to growth inhibition (Figure S3A). In a related assay called homo-
zygous profiling (HOP), which scores homozygous deletion
strains (Hoepfner et al., 2012), the loss of TORC1 or TORC2
inhibitors, or of factors that act downstream to repress TORC
responses, were also recovered as conferring resistance to
NVP-BHS345 (e.g., Tip41; Figure S3B). No component of the
lar Cell 51, 829–839, September 26, 2013 ª2013 Elsevier Inc. 833

D E
F G H I
A B C
(legend on next page)
Molecular Cell
TORC2 Regulates DNA Damage Response
834 Molecular Cell 51, 829–839, September 26, 2013 ª2013 Elsevier Inc.

Molecular Cell
TORC2 Regulates DNA Damage Response
DNA damage checkpoint pathway was detected in either assay.
Taken together, these results suggest that TORC1 and TORC2,
but not other members of the PIKK family (Mec1 and Tel1), are
targeted by NVP-BHS345.
In order to define whether TORC1, TORC2, or both complexes
are repressed by NVP-BHS345, we made use of bypass strains
that are insensitive to loss of one or the other complex (Huber
et al., 2009; Kamada et al., 2005). Because the two TOR com-
plexes have distinct downstream effectors, genetic modification
of specific downstream targets allows cells to grow in the
absence of the kinase itself. For instance, to bypass TORC1
lethality, TIP41 was deleted and SCH9 was replaced with
a constitutively active (SCH9DE) allele (Huber et al., 2009).
TORC2 function was bypassed by an overexpression of the
hyperactive YPK2D239A allele (Kamada et al., 2005). As a control
for complex specificity, we show that 20 nM rapamycin, which
inhibits only TORC1 (Loewith et al., 2002), does not impair
growth of the TORC1 bypass strain, although wild-type and
TORC2 bypass strains are blocked (Figure 3B). On plates con-
taining NVP-BHS345, strains that could bypass either TORC1
or TORC2 had partial resistance, and a strain that bypassed
both resisted best (Figure 3B). This argues that NVP-BHS345
most likely targets both TORC1 and TORC2.
We then confirmed that NVP-BHS345 directly inhibits the
kinase activities associated with both TOR complexes in vivo
by the use of phospho-specific antisera that recognize
TORC1- and TORC2-dependent phosphoepitopes on the tar-
gets Sch9 and Ypk1, respectively. In cells treated with NVP-
BHS345, we observed a rapid, dose-dependent loss of Sch9
and Ypk1 phosphorylation on western blots (Figure 3C). Positive
controls included wortmannin, a pleiotropic PI3K inhibitor that
inhibits both phosphorylation events and rapamycin. We note
that 25 mM NVP-BHS345 was needed to see a significant drop
in the phosphorylation status of these targets, while 2–5 mM is
sufficient to see synergy with Zeocin (see below). This difference
may reflect conditions of growth or strain background, as com-
Figure 4. Inhibition of TORC2/YPK Pathway or Actin Dynamics Synerg
(A) Tor2 active-site mutation confers resistance to NVP-BHS345. EMS-mutagen
trations of NVP-BHS345, and resistance conferringmutations in TOR2, V2126G, a
The mutations were introduced into the genomic TOR2 locus, and resulting strain
catalytic pocket of yeast Tor2 bound to NVP-BHS345, based on the mTOR/BEZ2
(B) TOR2 (GA-6148) and the tor2-V2126G mutant (GA-6150) cells were exponent
with the indicated concentrations of NVP-BHS345 for 80min. Chromosomal DNA
resistance.
(C) Rapamycin does not provoke YCS in combination with Zeocin. Cells (GA-198
(10 mM) for 80 min, and chromosomal DNA was subjected to the CHEF gel analy
(D) TORC2 inactivation causes YCS on Zeocin. Exponentially growing wild-type
containing Zeocin (75 mg/ml) with FK506 (1 mg/ml) or with NVP-BHS345 (10 mM) fo
mechanism of inactivation are described in Figures S4B–S4D.
(E) Inactivation of Ypk1/Ypk2 kinases phenocopies NVP-BHS345 effects on Zeo
sensitive) ypk2D (GA-5893) cells were treatedwith or without Zeocin (75 mg/ml) in t
for 80 min. Chromosome DNA was subjected to CHEF gel analysis. 1NM-PP1 in
(F) Cells expressing wild-type or constitutively active YPK2-D239A (Figure 3B, c
(75 mg/ml) with NVP-BHS345 (10 mM) for 80 min. Chromosomal DNA was subjec
(G) Exponentially growing cells (GA-4732) were cultured in SC media containing
a-amanitin (a-Am, 27 mM), LatA (50 mM), or nocodazole (Noc, 50 mM) for 90 min.
(H) GA-6148 cells were exponentially cultured in SC media and treated ± 6 mM ja
(50 mg/ml) and NVP-BHS345 (2 mM) for 80 min, and genomic DNA was subjected
(I) Deduced pathway of effects on genome stability provoked by NVP-BHS345 in
Molecu
pound uptake does vary among yeast strains. Importantly, it is
unlikely that Zeocin and NVP-BHS345 simply modify or enhance
each other’s action, since a comparison of haploinsufficiency
profiling (HIP) assays for Zeocin sensitivity and NVP-BHS345
sensitivity revealed absolutely no overlap in their profiles (Fig-
ure S3C). This would not be the case if the two acted on identical
pathways or on each other.
Kinase assays performed in vitro with purified TORC1 and
TORC2 complexes show that NVP-BHS345 also inhibits both
kinase activities in vitro on validated substrates at the half
maximal inhibitory concentration (IC50) = 1 mM (Figure 3D). How-
ever, in a similar range, the compound was inactive against
an immunoprecipitated Mec1-Ddc2 complex (Figure 3E). This
is consistent with the hyperactivated Rad53 checkpoint
observed on Zeocin and NVP-BHS345 in Figure 1. We conclude
that NVP-BHS345 is a potent, yeast cell-permeable compound
that selectively inhibits TORC1 and TORC2 without impairing
the activity of the checkpoint kinase Mec1, except at very high
concentrations (>100 mM).
TORC2 Inhibition Confers Zeocin-Induced ChromosomeFragmentationIt remained to be determined whether the relevant target of NVP-
BHS345 was the Tor1 or Tor2 catalytic subunit. We addressed
this by mutagenizing yeast with ethane methylsulfonate (EMS)
and selecting for survivors on NVP-BHS345-containing plates.
Of 96 resistant mutants, 3 were dominant. The sequencing of
these strains revealed mutations in the Tor2 catalytic site
(namely, V2126A, V2126G, and M2286T), but no mutations in
Tor1. We generated two of the mutations within the putative
active site of the Tor2 kinase by targeted mutagenesis of other-
wise wild-type cells and confirmed that these point mutations
alone confer resistance to NVP-BHS345 (Figure 4A). A modeling
of the active site of Tor2 bound to NVP-BHS345 was generated
based on the crystal structure of mTOR bound to NVP-BEZ235,
a related imidazoquinoline. Consistent with the resistance
izes with Zeocin to Provoke YCS
ized yeast cells were spread on plates containing growth-inhibitory concen-
ndM2286T were identified bywhole-genome sequencing of resistant colonies.
s were streaked on inhibitory concentrations of NVP-BHS345. A model of the
35 structure, shows likely contact of V2126 and M2286 in compound binding.
ially cultured in SC then treated with or without 50 mg/ml Zeocin in combination
samples were subjected to CHEF gel analysis. TOR2-M2286T conferred similar
1) were treated with 75 mg/ml Zeocin with rapamycin (100 nM) or NVP-BHS345
sis.
(GA-6053) and FK506-sensitive TORC2 (GA-6054) cells were cultured in SC
r 80 min. Chromosome DNA was subjected to CHEF gel analysis. Controls and
cin. Exponentially growing YPK1 ypk2D (GA-5892) and ypk1-as (ATP analog-
he presence of the ATP analog, 1NM-PP1 (1 mM), NVP-BHS345 (10 mM), or both
hibits the ypk1-as enzyme.
ontrol and TORC2 bypass strain, respectively) were cultured in SC ± Zeocin
ted to CHEF gel analysis.
1% DMSO and Zeocin (75 mg/ml) together with NVP-BHS345 (BHS, 10 mM),
Chromosome integrity was checked by CHEF gel analysis.
splakinolide for 40 min. Cells were further treated in combination with Zeocin
to CHEF gel analysis.
hibition of TORC2.
lar Cell 51, 829–839, September 26, 2013 ª2013 Elsevier Inc. 835

Molecular Cell
TORC2 Regulates DNA Damage Response
scored in vivo, TOR2-V2126G and TOR2-M2286T substitutions
are predicted to impair NVP-BHS345-Tor2 binding in the cata-
lytic pocket.
The generation of a NVP-BHS345-resistant form of Tor2
allowed us to test whether YCS stems exclusively from a loss
of Tor2 activity. To this end, we treated cells bearing TOR2-
V2126G with low levels of Zeocin and increasing concentrations
of NVP-BHS345. Cells bearing the Tor2 mutant that renders it
insensitive to NVP-BHS345 showed no YCS (Figure 4B). This
important result excludes a role for off-target effects or effects
that arise from inhibition of Tor1. It also identifies Tor2 as the rele-
vant target of NVP-BHS345with respect to both YCS and growth
inhibition.
Tor2 can serve in both TORC1 and TORC2 complexes, and to
date there is no small-molecule inhibitor specific for TORC2. To
determine which of the TOR complexes is involved in YCS, we
checked to see if rapamycin, a specific TORC1 inhibitor, induces
YCS in the presence of Zeocin. This was clearly not the case (Fig-
ure 4C). Thus, with respect to YCS, TORC2 appears to be the
relevant target of NVP-BHS345. Consistently, we observed chro-
mosome fragmentation in a tor2-21allele that only affects TORC2
function at a nonpermissive temperature (Helliwell et al., 1998).
YCS was detected in this strain at 37�C, but not 24�C, in the
presence, but not the absence, of Zeocin (Figure S4A).
To see whether inhibition of TORC2 complexes is sufficient to
enhance Zeocin-induced breaks, we developed a strain in which
TORC2 could be inactivated conditionally through the com-
pound FK506. Hsp90 family members are known to be required
for the maturation of many protein kinases, including Tor1 and
Tor2 (Takai et al., 2010). We rationalized that conditional recruit-
ment of Hsc82 (a yeast Hsp90 family member) to TORC2 might
lock it in an inactive conformation, thereby blocking TORC2
kinase activity (Figure 4D). To achieve this, we replaced the
endogenous AVO1 gene, which encodes an essential TORC2
subunit, with a construct expressingAVO1 fused toCNB1, which
encodes a regulatory subunit of the protein phosphatase
calcineurin. Additionally, we replaced HSC82 with a construct
expressing an Hsc82-Fpr1 fusion. Fpr1 is a proline isomerase
and receptor for FK506, which can be brought into proximity of
calcineurin by adding FK506 to the culture (Heitman et al.,
1991b). This is predicted to repress TORC2 selectively. Indeed,
in this strain background, but not in control strains lacking any
individual component, the addition of FK506 reduced TORC2
activity, as monitored by Ypk1 phosphorylation (Figure S4C).
Furthermore, Zeocin and FK506 treatment of TORC2-FK506-
sensitive cells, but not of control strains lacking either Hsc82-
Fpr1 or Avo1-Cnb1 proteins, triggered chromosome breakdown
in the presence of Zeocin, analogous to the effect of NVP-
BHS345 (Figures 4D and S4D). Importantly, there was no addi-
tive effect of NVP-BHS345 and FK506 in this strain background,
consistent with an epistatic mode of action (Figure 4D). Thus, the
selective inhibition of TORC2 is responsible and sufficient to
provoke catastrophic YCS in the presence of Zeocin.
We note that Crz1, a key transcription factor downstream of
TORC2, is activated by calcineurin-dependent dephosphoryla-
tion (Cyert, 2003). In the selective TORC2 inhibition protocol
used above, the calcineurin inhibitor FK506 is added. Incubation
with FK506, however, did not prevent YCS in response to Zeocin
836 Molecular Cell 51, 829–839, September 26, 2013 ª2013 Elsevier
and NVP-BHS345 (Figure S4D), allowing us to exclude the tran-
scriptional program driven by calcineurin-activated Crz1 in YCS
(Niles et al., 2012). Indeed, even a complete deletion ofCRZ1 did
not induce chromosome fragmentation in the presence of Zeocin
(Figure S4E), ruling out this pathway as being responsible for the
NVP-BHS345 effect.
The Inhibition of Ypk and Actin Dynamics ConfersZeocin-Induced YCSDownstream of TORC2 are two related kinases, Ypk1 and Ypk2,
which have partially redundant functions (Figure 4I) (Niles et al.,
2012). To test whether Ypk1/Ypk2 inhibition is the relevant
effector of YCS on Zeocin, we introduced an ATP analog-sensi-
tive YPK1 allele, ypk1-as (Bishop et al., 2001) in ypk2D cells (Fig-
ure 4E). We found that Ypk1/Ypk2 inhibition, triggered by adding
the ATP analog 1NM-PP1, resulted in YCS in the presence of
Zeocin (Figure 4E). Thus, inactivation of Ypk1/Ypk2 phenocopies
TORC2 inhibition by NVP-BHS345 with respect to enhanced
Zeocin sensitivity. Ypk1/Ypk2 inhibition was, moreover, epistatic
to NVP-BHS345 treatment (Figure 4E), arguing that loss of Ypk1/
Ypk2 kinase activity is the pathway through which NVP-BHS345
triggers YCS on Zeocin. Consistently, the expression of a hyper-
active form of Ypk2, Ypk2D239A, conferred resistance to com-
bined Zeocin and NVP-BHS345 treatment (Figure 4F). This
argues that NVP-BHS345 induces YCS and cell death in the
presence of Zeocin, at least in part by preventing Ypk1/Ypk2
kinase activation.
Ypk1/Ypk2 kinases regulate actin-targeted cell growth
(Schmelzle et al., 2002) as well as gene transcription. To see
whether regulation of the cytoskeleton, modulation of transcrip-
tion, or cell-cycle arrest mimics NVP-BHS345-dependent YCS,
we treated cells with either an inhibitor of RNA pol II transcription
(a-amanitin), the actin polymerization inhibitor, latrunculin A
(LatA), or the microtubule polymerization inhibitor nocodazole in
the presenceor absence of Zeocin. Remarkably, the combination
of Zeocin and LatA triggered efficient YCS, even though TORC2
and Ypk1/Ypk2 are active under these conditions (Figure 4G). In
contrast, the general inhibition of transcription and the depoly-
merization of microtubules by nocodazole had little or no effect.
None of the treatments led to YCS in the absence of Zeocin.
LatA binds monomeric actin (G-actin) and alters actin organi-
zation by preventing the formation of actin filament (F-actin).
Therefore, we next asked whether blocking actin dynamics
with jasplakinolide, a cyclic peptide that stabilizes F-actin and
locks actin dynamics (Ayscough, 2000; Bubb et al., 1994), would
influence chromosome integrity on Zeocin. Indeed, as with LatA,
the combination of Zeocin with jasplakinolide also resulted in
YCS, similar to the combined effects of NVP-BHS345 and
Zeocin (Figure 4H). These results suggest that the inhibition of
an actin-associated activity and/or actin dynamics, which are
under control of TORC2 and Ypk1/YPk2, triggers massive
chromosome fragmentation and cell death when the cells are
challenged with sublethal doses of Zeocin.
DISCUSSION
We have thoroughly documented an unexpected link
between TORC2 activity and a susceptibility to chromosome
Inc.

Molecular Cell
TORC2 Regulates DNA Damage Response
fragmentation when cells are exposed to IR or the antibiotic Zeo-
cin. The Tor2 inhibitor NVP-BHS345 acts synergistically to pro-
voke cell death in the presence of sublethal doses of Zeocin or
IR by triggering a rapid and dramatic chromosome shattering,
YCS. Although the compound is shown to inhibit both TORC1
and TORC2 complexes, we show by mutagenesis, comparative
inhibitor studies, and the testing of specific mutations in the
absence of NVP-BHS345 that the critical target for preservation
of chromosomal integrity is TORC2. Downstream of TORC2, the
key regulators of Zeocin-induced YCS are the two effector
kinases, Ypk1/Ypk2, which are partially redundant homologs of
the mammalian SGK1 kinase (Loewith and Hall, 2011). Given
the conservation of these pathways, it is conceivable that such
inhibitors would be used to sensitize target cells to localized ra-
diation therapy or other DNA damage-inducing chemotherapies.
A previous study suggested that TORC2 in fission yeast regu-
lates telomere length on a pathway with ATR kinase and pro-
motes mitotic entry (Schonbrun et al., 2009). This is not the
case here, as the Mec1/Tel1 (ATR/ATM)-dependent checkpoint
pathway is hyperactivated when cells are treated with NVP-
BHS345 and Zeocin. Moreover, the ATR homolog Mec1 is not
significantly inhibited by NVP-BHS345 in vivo or in vitro. Finally,
there seems to be no crosstalk between NVP-BHS345 and
Zeocin; despite high levels of Zeocin, TORC2 activity on Ypk1
T662 is robust (Figure S4F). With this, we can rule out that
one chemical simply enhances the effect of the other. Rather,
we see that independent pathways intersect to provoke the
observed YCS.
TORC2 and Ypk1/Ypk2 kinases regulate polarized cell growth,
sphingolipid biogenesis, and the actin cytoskeleton (Loewith and
Hall, 2011). Whereaswe see no role for Crz1-regulated transcrip-
tion in Zeocin-induced YCS, the opposite is true for the modula-
tion of actin polymerization. Unexpectedly, both latrunculin A
and jasplakinolide, a cyclic peptide that stabilizes F-actin and
impairs actin dynamics, phenocopy the TORC2 inhibition with
respect to Zeocin hypersensitivity. We note that actin pools are
in continual flux between the globular (G) and filamentous (F)
forms of the molecule and that this exchange is essential for
cell survival. Our results argue that cells require this exchange
betweenG- and F-actin forms to prevent a still enigmatic conver-
sion of Zeocin-induced damage to frequent chromosomal
breaks.
Actin constantly shuttles to the nucleus in its G form in com-
plex with other proteins and is also selectively exported from
the nucleus (Stuven et al., 2003). For this reason, an impaired
exchange between forms could result in aberrantly high nuclear
actin levels. Nonfilamentous actin serves a range of roles within
the nucleus, including participation in nucleosome remodeler
complexes, interaction with RNA polymerase I, II, and III, and
coregulation of transcription factors (Vartiainen et al., 2007).
Thus, it is conceivable that an excess of nuclear globular (G)-
actin would inhibit a repair pathway or block a crucial enzyme
that allows cells to repair oxidative damage. It is well established
that actin binds ATP and DNA and inhibits prokaryotic DNaseI
(Kabsch et al., 1990), although the physiological counterpart of
these latter binding events is unknown. A recent report links
polymerized actin and its affinity for the repair factor Ku, to a
reduction in NHEJ (Andrin et al., 2012). Future studies will
Molecu
address both physiological and pathophysiological roles of actin
in the nucleus following DNA damage. In addition to a potential
impact of actin itself, actin-containing chromatin remodeling
complexes are known to influence DNA repair from yeast to
man, influencing nearly every type of repair, from nucleotide
and base excision, to NHEJ and HR.
YCS is not due to a block in the major DSB repair pathways
because the elimination of these does not generate chromo-
some fragmentation on Zeocin (Figure 2). We feel that the
more likely explanation of the observed chromosome fragmenta-
tion is that single-strand nicks or sugar-backbone adducts
deposited by Zeocin are converted to irreparable DSBs, possibly
because a crucial step in base or nucleotide excision repair is
blocked by TORC2 inhibition or by an imbalance of G- to F-actin.
Previous work in budding yeast showed that a lack of coordina-
tion between DNA polymerase d and the 50 flap endonuclease
Rad27/Fen1 with proliferating cell nuclear antigen (PCNA) can
result in the conversion of closely opposed methylated bases
to DSBs (Ma et al., 2009). Similarly, inhibition of apurinic/apyrimi-
dinic endonucleases or glycosylases could convert single-strand
nicks to DSBs. It remains to be seen how TORC2 inhibition
impacts these repair pathways.
Assays in a number of mammalian cancer cells have shown
that mTOR inhibition acts synergistically with various chemo-
therapeutic treatments, including gemcitabine (Awasthi et al.,
2012). Indeed, mTOR inhibitors that target both mTORC1 and
mTORC2 in human cells have been implicated in combined ther-
apeutic strategies against cancer (Maira et al., 2008; Tanaka
et al., 2011). Here, we find a pathway that is independent of
TORC1 function but links TORC2 inhibition to DNA damage
hypersensitivity. Hints that this may be therapeutically relevant
come from a study of glioblastoma in which TORC2 inhibition
through Rictor small hairpin RNA (shRNA) counteracted cisplatin
resistance and led to both 80% tumor shrinkage and 5-fold
increases in apoptosis in cells treated with cisplatin (Tanaka
et al., 2011). Our results in yeast predict such an outcome and
suggest that TORC2-controlled actin turnover itself could act
synergistically with DNA damaging agents to provoke cell death
through loss of genomic integrity.
EXPERIMENTAL PROCEDURES
Cell Culture, Strains, and Plasmids
Yeast strains used in this study are listed in Table S2. Yeast was cultured in SC
(synthetic complete + 2% glucose) medium at 30�C unless otherwise
indicated. Plasmid pWJ1213 bearing RAD52-YFP was a generous gift from
Dr. R. Rothstein.
Chemicogenetic Screening, HIP-HOP, and EMS Mutagenesis
Compound activity was scored for its effect on the relative growth of sgs1D
andmec1-100 yeast strains on low levels of HU, which is in each case normal-
ized to the growth of the WT strain. All compounds that selectively inhibited
either sgs1D or mec1-100 strains were further analyzed by dose-response
curves ± HU. HIP-HOP and EMS mutagenesis were performed as described
(Hoepfner et al., 2012).
X-Ray Exposure
Exposure to ionizing radiation was conducted on yeast cells spread on SC
agar plates with indicated doses by Torrex 120D (Astrophysics Research
Corporation).
lar Cell 51, 829–839, September 26, 2013 ª2013 Elsevier Inc. 837

Molecular Cell
TORC2 Regulates DNA Damage Response
Western Blots and Antibodies
Total cell extracts were prepared as described (Huber et al., 2009). Protein
extract was subjected to SDS-PAGE or NuPAGE (Invitrogen) and transferred
to nitrocellulose membranes for probing by anti-Rad53 (yc-19, Santa Cruz,
or Mab 11G3G6 custom made by GenScript), Mab anti-GFP (Roche), anti-
Ypk1 (SC-12051, Santa Cruz), anti-phospho-T662 Ypk1 (Berchtold et al.,
2012), anti-phospho-T737 Sch9 (Wanke et al., 2008), or anti-HA (12CA5).
CHEF Gel Analysis
Yeast genomic DNA was prepared in an agarose plug as described in the
instruction manual (Bio-Rad, CHEF-DR II), with slight modifications. After
washing yeast cells in ice-cold 50 mM EDTA-NaOH (pH 8.0), cell pellets
were suspended in Zymolyase buffer (50 mM Na-PO4 [pH 7.0], 50 mM
EDTA, 1 mM dithiothreitol [DTT]) and embedded in a 1% agarose plug.
Genomic DNA was prepared by treating the plug with 0.4 mg/ml Zymolyase
(20T, Seikagaku) in Zymolyase buffer at 37�C for 1 hr, followed by 1 mg/ml
proteinase K digestion in 10 mM Tris-HCl (pH 7.5), 50 mM EDTA, 1% sodium
N-lauroylsarcosinate at 50�C or 30�C for 15–20 hr. Chromosomal DNA was
migrated in 1% agarose/0.53 Tris-Borate-EDTA (TBE) on the CHEF-DR II
Pulsed Field Electrophoresis System (Bio-Rad) at 14�C, 6 V/cm, 60 s switch
time for 15 hr, then 90 s for 7 hr. Ethidium bromide-stained chromosomal
DNA was imaged on the ChemiDoc XRS system (Bio-Rad). Proteinase K treat-
ment at 30�C did not alter YCS (Figure S1).
TORC1 and TORC2 Activity Assays
TORC1 and TORC2 activity in vivo: a culture of MS113 (W303a URA3::HA-
SCH9) was exponentially cultured to optical density 600 (OD600) = 0.8. Proteins
were extracted under denaturing conditions after trichloroacetic acid (TCA;
6% g/v final) precipitation, and extracts were separated on SDS gels and
blotted with anti-Sch9 phosphorylated at T737 (Wanke et al., 2008) versus
anti-HA (12CA5) to detect total protein amount, while for TORC2 activity,
anti-Ypk1 (Santa Cruz, SC-12051) and anti-phospho-T662 Ypk1 antisera
were used (Berchtold et al., 2012).
For kinase assays in vitro, TORC1 and TORC2 were tandem affinity purified
(TAP) from yeast using RL170-2c for TORC1 and RL126-5d for TORC2, as pre-
viously described (Kamada et al., 2005; Urban et al., 2007). Substrates were a
glutathione S-transferase (GST)-tagged Sch9 C-terminal domain (TORC1) and
the kinase-dead allele of Ypk2 (TORC2; see Supplemental Experimental Pro-
cedures for details). Kinase and substrate were incubated with g-32P-labeled
ATP and increasing amounts of NVP-BHS345, and incorporated radioactivity
was quantified by SDS gel analysis with a phosphoimager. Kinase activity was
normalized to mock-treated samples.
Mec1-Ddc2 complex was isolated from exponentially growing cells
harboring endogenously tagged Ddc2-GFP (GA-7268). In vitro Mec1 kinase
assay was measured with ADP-Glo kit (Promega) using Sgs1-404-604 pep-
tide as a substrate (Hegnauer et al., 2012). Kinase activity was normalized
to the mock-treated samples, and Mec1 kinase specificity was confirmed
by comparing Ddc2-GFP immunoprecipitation (IP) samples from mec1D
cells.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures,
four figures, and two tables and can be found with this article online at
http://dx.doi.org/10.1016/j.molcel.2013.08.019.
ACKNOWLEDGMENTS
We thank the Novartis HIP-HOP team,W. Forrester (Novartis), and T. Schleker
and other Gasser laboratory members for helpful discussions and comments
on the manuscript. We thank G. Tevzadze, P. Schar, R. Rothstein, H.L. Klein,
K. Shirahige, and M. Hall for reagents. We thank N. Hustedt and Y. Bouza for
help with Mec1 assays and Ypk1 phosphorylation, respectively, and P. Furet
for modeling. Support was gratefully received from the Novartis Research
Foundation (S.M.G.), National Centers for Competence in Research ‘‘Frontiers
in Genetics’’ (S.M.G. and R.L.) and ‘‘Chemical Biology’’ (R.L.), the Canton of
838 Molecular Cell 51, 829–839, September 26, 2013 ª2013 Elsevier
Geneva (R.L.), European Research Council (R.L.), and the Swiss National
Science Foundation (S.M.G. and R.L.).
Received: March 30, 2013
Revised: July 5, 2013
Accepted: August 8, 2013
Published: September 12, 2013
REFERENCES
Aguilera, A., and Gomez-Gonzalez, B. (2008). Genome instability: a mecha-
nistic view of its causes and consequences. Nat. Rev. Genet. 9, 204–217.
Andrin, C., McDonald, D., Attwood, K.M., Rodrigue, A., Ghosh, S., Mirzayans,
R., Masson, J.Y., Dellaire, G., and Hendzel, M.J. (2012). A requirement for
polymerized actin in DNA double-strand break repair. Nucleus 3, 384–395.
Aronova, S., Wedaman, K., Aronov, P.A., Fontes, K., Ramos, K., Hammock,
B.D., and Powers, T. (2008). Regulation of ceramide biosynthesis by TOR
complex 2. Cell Metab. 7, 148–158.
Awasthi, N., Yen, P.L., Schwarz, M.A., and Schwarz, R.E. (2012). The efficacy
of a novel, dual PI3K/mTOR inhibitor NVP-BEZ235 to enhance chemotherapy
and antiangiogenic response in pancreatic cancer. J. Cell. Biochem. 113,
784–791.
Ayscough, K.R. (2000). Endocytosis and the development of cell polarity in
yeast require a dynamic F-actin cytoskeleton. Curr. Biol. 10, 1587–1590.
Bartek, J., Bartkova, J., and Lukas, J. (2007). DNA damage signalling guards
against activated oncogenes and tumour progression. Oncogene 26, 7773–
7779.
Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N.,
Vassiliou, L.V., Kolettas, E., Niforou, K., Zoumpourlis, V.C., et al. (2006).
Oncogene-induced senescence is part of the tumorigenesis barrier imposed
by DNA damage checkpoints. Nature 444, 633–637.
Berchtold, D., Piccolis, M., Chiaruttini, N., Riezman, I., Riezman, H., Roux, A.,
Walther, T.C., and Loewith, R. (2012). Plasmamembrane stress induces reloc-
alization of Slm proteins and activation of TORC2 to promote sphingolipid
synthesis. Nat. Cell Biol. 14, 542–547.
Bernstein, K.A., Gangloff, S., and Rothstein, R. (2010). The RecQ DNA heli-
cases in DNA repair. Annu. Rev. Genet. 44, 393–417.
Bishop, A.C., Buzko, O., and Shokat, K.M. (2001). Magic bullets for protein
kinases. Trends Cell Biol. 11, 167–172.
Bubb, M.R., Senderowicz, A.M., Sausville, E.A., Duncan, K.L., and Korn, E.D.
(1994). Jasplakinolide, a cytotoxic natural product, induces actin polymeriza-
tion and competitively inhibits the binding of phalloidin to F-actin. J. Biol.
Chem. 269, 14869–14871.
Buttner, S., Eisenberg, T., Carmona-Gutierrez, D., Ruli, D., Knauer, H.,
Ruckenstuhl, C., Sigrist, C., Wissing, S., Kollroser, M., Frohlich, K.U., et al.
(2007). Endonuclease G regulates budding yeast life and death. Mol. Cell 25,
233–246.
Carmona-Gutierrez, D., Eisenberg, T., Buttner, S., Meisinger, C., Kroemer, G.,
andMadeo, F. (2010). Apoptosis in yeast: triggers, pathways, subroutines. Cell
Death Differ. 17, 763–773.
Cobb, J.A., Schleker, T., Rojas, V., Bjergbaek, L., Tercero, J.A., and Gasser,
S.M. (2005). Replisome instability, fork collapse, and gross chromosomal rear-
rangements arise synergistically from Mec1 kinase and RecQ helicase muta-
tions. Genes Dev. 19, 3055–3069.
Cyert, M.S. (2003). Calcineurin signaling in Saccharomyces cerevisiae: how
yeast go crazy in response to stress. Biochem. Biophys. Res. Commun.
311, 1143–1150.
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C.,
Schurra, C., Garre’, M., Nuciforo, P.G., Bensimon, A., et al. (2006). Oncogene-
induced senescence is a DNA damage response triggered by DNA hyper-
replication. Nature 444, 638–642.
Errico, A., and Costanzo, V. (2012). Mechanisms of replication fork protection:
a safeguard for genome stability. Crit. Rev. Biochem. Mol. Biol. 47, 222–235.
Inc.

Molecular Cell
TORC2 Regulates DNA Damage Response
Hegnauer, A.M., Hustedt, N., Shimada, K., Pike, B.L., Vogel, M., Amsler, P.,
Rubin, S.M., van Leeuwen, F., Guenole, A., van Attikum, H., et al. (2012). An
N-terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53
kinase to stalled forks. EMBO J. 31, 3768–3783.
Heitman, J., Movva, N.R., and Hall, M.N. (1991a). Targets for cell cycle arrest
by the immunosuppressant rapamycin in yeast. Science 253, 905–909.
Heitman, J., Movva, N.R., Hiestand, P.C., and Hall, M.N. (1991b). FK 506-bind-
ing protein proline rotamase is a target for the immunosuppressive agent FK
506 in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 88, 1948–1952.
Helliwell, S.B., Howald, I., Barbet, N., and Hall, M.N. (1998). TOR2 is part of two
related signaling pathways coordinating cell growth in Saccharomyces cerevi-
siae. Genetics 148, 99–112.
Hoepfner, D., McNamara, C.W., Lim, C.S., Studer, C., Riedl, R., Aust, T.,
McCormack, S.L., Plouffe, D.M., Meister, S., Schuierer, S., et al. (2012).
Selective and specific inhibition of the plasmodium falciparum lysyl-tRNA syn-
thetase by the fungal secondary metabolite cladosporin. Cell Host Microbe 11,
654–663.
Huber, A., Bodenmiller, B., Uotila, A., Stahl, M., Wanka, S., Gerrits, B.,
Aebersold, R., and Loewith, R. (2009). Characterization of the rapamycin-sen-
sitive phosphoproteome reveals that Sch9 is a central coordinator of protein
synthesis. Genes Dev. 23, 1929–1943.
Kabsch, W., Mannherz, H.G., Suck, D., Pai, E.F., and Holmes, K.C. (1990).
Atomic structure of the actin:DNase I complex. Nature 347, 37–44.
Kamada, Y., Fujioka, Y., Suzuki, N.N., Inagaki, F., Wullschleger, S., Loewith,
R., Hall, M.N., and Ohsumi, Y. (2005). Tor2 directly phosphorylates the AGC
kinase Ypk2 to regulate actin polarization. Mol. Cell. Biol. 25, 7239–7248.
Loewith, R., and Hall, M.N. (2011). Target of rapamycin (TOR) in nutrient
signaling and growth control. Genetics 189, 1177–1201.
Loewith, R., Jacinto, E., Wullschleger, S., Lorberg, A., Crespo, J.L., Bonenfant,
D., Oppliger, W., Jenoe, P., and Hall, M.N. (2002). Two TOR complexes, only
one of which is rapamycin sensitive, have distinct roles in cell growth control.
Mol. Cell 10, 457–468.
Lovejoy, C.A., and Cortez, D. (2009). Common mechanisms of PIKK regula-
tion. DNA Repair (Amst.) 8, 1004–1008.
Ma, W., Panduri, V., Sterling, J.F., Van Houten, B., Gordenin, D.A., and
Resnick, M.A. (2009). The transition of closely opposed lesions to double-
strand breaks during long-patch base excision repair is prevented by the
coordinated action of DNA polymerase d and Rad27/Fen1. Mol. Cell. Biol.
29, 1212–1221.
Maira, S.M., Stauffer, F., Brueggen, J., Furet, P., Schnell, C., Fritsch, C.,
Brachmann, S., Chene, P., De Pover, A., Schoemaker, K., et al. (2008).
Identification and characterization of NVP-BEZ235, a new orally available
dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor
with potent in vivo antitumor activity. Mol. Cancer Ther. 7, 1851–1863.
Mulet, J.M., Martin, D.E., Loewith, R., and Hall, M.N. (2006). Mutual antago-
nism of target of rapamycin and calcineurin signaling. J. Biol. Chem. 281,
33000–33007.
Niles, B.J., Mogri, H., Hill, A., Vlahakis, A., and Powers, T. (2012). Plasmamem-
brane recruitment and activation of the AGC kinase Ypk1 is mediated by target
of rapamycin complex 2 (TORC2) and its effector proteins Slm1 and Slm2.
Proc. Natl. Acad. Sci. USA 109, 1536–1541.
Povirk, L.F. (1996). DNA damage and mutagenesis by radiomimetic DNA-
cleaving agents: bleomycin, neocarzinostatin and other enediynes. Mutat.
Res. 355, 71–89.
Povirk, L.F.,Wubter, W., Kohnlein, W., and Hutchinson, F. (1977). DNA double-
strand breaks and alkali-labile bonds produced by bleomycin. Nucleic Acids
Res. 4, 3573–3580.
Reinke, A., Anderson, S., McCaffery, J.M., Yates, J., 3rd, Aronova, S., Chu, S.,
Fairclough, S., Iverson, C., Wedaman, K.P., and Powers, T. (2004). TOR com-
Molecu
plex 1 includes a novel component, Tco89p (YPL180w), and cooperates with
Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J. Biol.
Chem. 279, 14752–14762.
Robert, T., Vanoli, F., Chiolo, I., Shubassi, G., Bernstein, K.A., Rothstein, R.,
Botrugno, O.A., Parazzoli, D., Oldani, A., Minucci, S., and Foiani, M. (2011).
HDACs link the DNA damage response, processing of double-strand breaks
and autophagy. Nature 471, 74–79.
Roelants, F.M., Breslow, D.K.,Muir, A.,Weissman, J.S., and Thorner, J. (2011).
Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to
control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc. Natl.
Acad. Sci. USA 108, 19222–19227.
Schmelzle, T., Helliwell, S.B., and Hall, M.N. (2002). Yeast protein kinases and
the RHO1 exchange factor TUS1 are novel components of the cell integrity
pathway in yeast. Mol. Cell. Biol. 22, 1329–1339.
Schmidt, A., Kunz, J., and Hall, M.N. (1996). TOR2 is required for organization
of the actin cytoskeleton in yeast. Proc. Natl. Acad. Sci. USA 93, 13780–13785.
Schonbrun, M., Laor, D., Lopez-Maury, L., Bahler, J., Kupiec, M., and
Weisman, R. (2009). TOR complex 2 controls gene silencing, telomere length
maintenance, and survival under DNA-damaging conditions. Mol. Cell. Biol.
29, 4584–4594.
Shen, C., Lancaster, C.S., Shi, B., Guo, H., Thimmaiah, P., and Bjornsti, M.A.
(2007). TOR signaling is a determinant of cell survival in response to DNA
damage. Mol. Cell. Biol. 27, 7007–7017.
Smith, A.M., Ammar, R., Nislow, C., and Giaever, G. (2010). A survey of yeast
genomic assays for drug and target discovery. Pharmacol. Ther. 127,
156–164.
Stephens, P.J., Greenman, C.D., Fu, B., Yang, F., Bignell, G.R., Mudie, L.J.,
Pleasance, E.D., Lau, K.W., Beare, D., Stebbings, L.A., et al. (2011). Massive
genomic rearrangement acquired in a single catastrophic event during cancer
development. Cell 144, 27–40.
Stuven, T., Hartmann, E., and Gorlich, D. (2003). Exportin 6: a novel nuclear
export receptor that is specific for profilin.actin complexes. EMBO J. 22,
5928–5940.
Sung, P., Krejci, L., Van Komen, S., and Sehorn, M.G. (2003). Rad51 recombi-
nase and recombination mediators. J. Biol. Chem. 278, 42729–42732.
Symington, L.S., and Gautier, J. (2011). Double-strand break end resection
and repair pathway choice. Annu. Rev. Genet. 45, 247–271.
Takai, H., Xie, Y., de Lange, T., and Pavletich, N.P. (2010). Tel2 structure and
function in the Hsp90-dependent maturation of mTOR and ATR complexes.
Genes Dev. 24, 2019–2030.
Tanaka, K., Babic, I., Nathanson, D., Akhavan, D., Guo, D., Gini, B., Dang, J.,
Zhu, S., Yang, H., De Jesus, J., et al. (2011). Oncogenic EGFR signaling acti-
vates an mTORC2-NF-kappaB pathway that promotes chemotherapy resis-
tance. Cancer discovery 1, 524–538.
Urban, J., Soulard, A., Huber, A., Lippman, S., Mukhopadhyay, D., Deloche,
O., Wanke, V., Anrather, D., Ammerer, G., Riezman, H., et al. (2007). Sch9 is
a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 26, 663–674.
Vartiainen, M.K., Guettler, S., Larijani, B., and Treisman, R. (2007). Nuclear
actin regulates dynamic subcellular localization and activity of the SRF
cofactor MAL. Science 316, 1749–1752.
Vida, T.A., and Emr, S.D. (1995). A new vital stain for visualizing vacuolar mem-
brane dynamics and endocytosis in yeast. J. Cell Biol. 128, 779–792.
Wanke, V., Cameroni, E., Uotila, A., Piccolis, M., Urban, J., Loewith, R., and De
Virgilio, C. (2008). Caffeine extends yeast lifespan by targeting TORC1. Mol.
Microbiol. 69, 277–285.
Wilson, T.E., Grawunder, U., and Lieber, M.R. (1997). Yeast DNA ligase IV
mediates non-homologous DNA end joining. Nature 388, 495–498.
lar Cell 51, 829–839, September 26, 2013 ª2013 Elsevier Inc. 839
Top Related
Copyright © 2022 FDOKUMEN

