







Bahasa
Halaman
Hukum


http://informahealthcare.com/drdISSN: 1071-7544 (print), 1521-0464 (electronic)
Drug Deliv, Early Online: 1–10! 2014 Informa Healthcare USA, Inc. DOI: 10.3109/10717544.2013.879753
ORIGINAL PAPER
Solid self-nanoemulsifying drug delivery system (S-SNEDDS) fororal delivery of glimepiride: development and antidiabetic activityin albino rabbits
Abdul Bari Mohd1, Krishna Sanka1, Srikanth Bandi1, Prakash V. Diwan1, and Nalini Shastri2
1Department of Pharmaceutics, School of Pharmacy, Anurag Group of Institutions, Hyderabad, Andhra Pradesh, India and 2Department of
Pharmaceutics, National Institute of Pharmaceutical Education and Research (NIPER), Hyderabad, Andhra Pradesh, India
Abstract
Context: This study presents novel self-nanoemulsifying drug delivery system potential of oraldelivering which leads poorly aqueous soluble drug glimepiride.Objective: The objective of this study was to prepare solid self-nanoemulsifying drug deliverysystem (S-SNEDDS) for the improved oral delivery of glimepiride and to evaluate its therapeuticefficacy in albino rabbits.Results and discussion: The droplet size analyses revealed a droplet size of less than 200 nm. Thesolid state characterization of S-SNEDDS by scanning electron microscopy (SEM), X-ray powderdiffraction and differential scanning calorimetry (DSC) revealed the absence of crystallineglimepiride in the S-SNEDDS. The in vitro dissolution studies revealed that the significantimprovement in glimepiride release characteristics. The effect of S-SNEDDS on therapeuticefficacy of glimepride was assessed in albino rabbits by monitoring blood glucose levels andcompared with free drug suspension, L-SNEDDS. The S-SNEDDS showed significant (p50.05)increase in in vitro drug release and therapeutic efficacy as compared with free drug.Conclusion: This study demonstrated that S-SNEDDS is a promising novel drug delivery systemof glimepride to enhance oral delivery.
Keywords
Aerosol 200, dissolution, glimepiride, oraldelivery, solid carrier, S-SNEDDS
History
Received 12 November 2013Revised 29 December 2013Accepted 29 December 2013
Introduction
The oral delivery of many new drug molecules is recurrently
associated with complications of low water solubility or high
lipophilicity which leads to poor and highly variable oral
bioavailability and the absence of dose proportionality
(Kyatanwar et al., 2010). This class of drug compounds can
be classified as low solubility and high permeability Bio
Pharmaceutical Classification System (BCS) class II drugs. In
these drugs, dissolution of the drug is the rate limiting step in
the absorption process. To triumph over these obstacles,
numbers of formulation approaches are reported including the
use of surfactants (Allaboun et al., 2003; Balakrishnan et al.,
2004; Chakraborty et al., 2009), lipids (Yeap et al., 2013),
permeation enhancers (Burcin et al., 2010; Beg et al., 2011),
formation of salt (Li et al., 2005; Serajuddin, 2007),
co-crystallization (Shan & Zaworotko, 2008; Qiao et al.,
2011; Chadha et al., 2012), solid dispersions (Serajuddin,
1999), inclusion complexes with cyclodextrins and modified
cyclodextrins (Miyake et al., 2000; Veiga et al., 2000; Wang
et al., 2000; Bannwart et al., 2001; Carrier et al., 2007;
Gamsiz et al., 2010a,b; Gamsiz et al., 2011; Badr-Eldin et al.,
2008; Kumar et al., 2013), nanosuspensions (Patravale et al.,
2004), and colloidal vesicles like liposomes (Nazzal et al.,
2002a; Manconi et al., 2013; Yang et al., 2013), and niosomes
(Khazaeli et al., 2007; Bayindir & Yuksel, 2010; Sezgin-
Bayindir et al., 2013; Jin et al., 2013)
In modern years, self-nanoemulsifying drug delivery
systems (SNEDDS) are the most popular and commercially
feasible lipid-based formulation approach for improving oral
bioavailability of poorly water soluble and lipophilic drugs
(Pouton, 2006; Date, 2007; Shweta et al., 2011). SNEDDS are
precisely defined as an isotropic multi-component drug
delivery systems composed of a synthetic or natural oil,
surfactant, and co-surfactant that have a unique ability of
forming fine oil in water micro- or nano-emulsion upon mild
agitation followed by dilution in aqueous media such as
gastro-intestinal fluid. As SNEDDS self-emulsifies in the
stomach and presents the drug in minute droplets of oil
(55 mm), it improves drug dissolution through presenting a
large interfacial area for partitioning of the drug between
the oil and the GIT fluid. The other advantages include
increased stability of drug molecules and ease of administer-
ing the final formulation as gelatin capsules (soft gelatin
capsules in the case of liquid self-nanoemulsifying drug
delivery system (L-SNEDDS) and hard gelatin capsules in
the case of solid self-nanoemulsifying drug delivery system
(S-SNEDDS)).
Address for correspondence: Dr. Nalini Shastri, Department ofPharmaceutics, National Institute of Pharmaceutical Education andResearch (NIPER), Hyderabad 500 037, AP, India. Tel: +40 23073741.Fax: +40 23073751. Email: [email protected]
Dru
g D
eliv
ery
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Gla
sgow
Cal
edon
ian
Uni
vers
ity o
n 02
/01/
14Fo
r pe
rson
al u
se o
nly.

Glimepiride,1-[[p-[2-(3-ethyl-4-methyl-2-oxo-3-pyrroline-
1-carboxami-do) ethyl] phenyl] sulphonyl]-3-(trans-4-methyl-
cyclohexyl) urea is the first third-generation sulphonyl urea.
It is a very potent sulphonyl urea employed for concomitant use
with insulin for the treatment of non-insulin-dependent (type
II) diabetes mellitus. It produces hypoglycemia by stimulating
release of insulin from pancreatic b cells and by increasing the
sensitivity of peripheral tissue to insulin. It also supports the
movement of sugar from the blood into the cells that need it.
Glimepiride shows low, pH-dependent solubility. It exhibits
very poor solubility at 37 �C (50.004 mg/ml) in acidic and
neutral aqueous media and it belongs to ‘‘BCS Class II’’ drugs
(Lobenberg & Amidon, 2000). It is likely to show low and
irregular bioavailability following oral administration due to
the low water solubility (Amidon et al., 1995; Grunenberg
et al., 1995). Hence administering glimepiride by oral appears
as a tough challenge due to its poor absorption pattern and
rapid and unpredictable hepatic first pass metabolism.
The present study deals with formulation of an Aerosol�
200 based SNEDDS of a poorly water soluble drug
(glimepride). The main objective of this study was to
investigate solid self-nanoemulsifying drug delivery system,
as a potential drug delivery system for glimepiride. S-
SNEDDS (consisting of Tween� 80/PEG and 400/Mygliol�
812) was characterized with regard to morphological analysis,
solid state characterization as well as its in vitro drug release
and therapeutic efficacy in albino rabbits.
Materials
Glimepiride was kindly gifted by Dr Reddy’s Labs, Hyderabad,
India. Miglyol� 812 (Capric Triglyceride), Cotton seed oil,
Aerosol� 200 (Dioxosilane), and Cremophor� RH 40
(Macrogolglycerol hydroxystearate) were gift samples from
Bari’s Pharmaceuticals, Hyderabad, India. Tween� 80
(Polyoxyethylene sorbitan monooleate), PEG 400 (polyethyl-
ene glycol 400), Span� 20 (Sorbitan monolaurate), oleic acid,
soya bean oil, ethyl alcohol, and HPLC grade methanol were
purchased from Merck Specialties Pvt. Ltd., Mumbai, India.
Span� 80 (Sorbitan monooleate), propylene glycol, and
potassium dihydrogen phosphate were obtained from SD-
Fine Chemicals Ltd., Mumbai, India. All other chemicals and
materials were of analytical grade.
Methods
Operating conditions of HPLC for glimepiride analysis
Glimepiride was determined by high-pressure liquid chroma-
tography (HPLC) using HPLC system (UFLC prominence
HPLC, Shimadzu, Kyoto, Japan) with a PDA detector
(SPD20A) at a wavelength of 228 nm and a manual injector
with injection volume setting at 20 ml. The mobile phase
consists of Acetonitrile: 0.2 M phosphate buffer (pH 7.4) was
pumped at a flow rate of 1 ml/min. The drug was separated by
Gemini C18, 100� 4.6 mm (ID), and 5 mm column at ambient
temperature.
Solubility studies
The solubility of glimepiride was determined in oils
(Miglyol� 812, castor oil, oleic acid, cotton seed oil, and
soya bean oil), surfactants, and co-surfactants (Tween� 80,
PEG 400, propylene glycol, Cremophor� RH 40, Span� 80,
and Span� 20). An excess amount (500 mg) of drug was
added to each 1.5 ml eppendorf tubes consisting of 1 ml of
each vehicle and the contents were mixed using a vortex
mixer (REMI CM 101DX, REMI Equipment, Mumbai, India)
to facilitate uniform mixing of drug for 15 min. The
Eppendorf tubes were kept at 25 ± 1.0 �C in an orbital
shaker (CL 24, Remi Electrotech Ltd., Mumbai, India) for
48 h to reach equilibrium (Wang et al., 2009). The tubes were
centrifuged at 5000 rpm using micro centrifuge (RM 12C,
REMI Equipment, Mumbai, India) for 20 min. The super-
natant was filtered through a 0.45-mm syringe filter and an
aliquot of 0.1 ml from the filtered supernatant was collected
and diluted with methanol. The quantification of glimepiride
was performed by HPLC (Prominence HPLC, Shimadzu,
Japan) at 228 nm using a PDA detector.
Selection of oil
The selection of oil was based on solubility of glimepiride in
the oils. Higher the solubility of the drug, higher will be the
drug loading potential (Pouton, 2000, 2006). In this study, the
drug was significantly more soluble in Miglyol� 812. Thus,
the same was chosen as the oil phase for formulating the
SNEDDS system.
Selection of surfactant
The screening of surfactant was based on both the ability to
solubilize the drug in it and its ability to emulsify the selected
oil phase. The solubility of all the surfactants include Tween�
80, Span� 20, Span� 80, and Cremophor� RH 40 was
determined from solubility studies. Then to examine their
emulsification ability, 20 ml of surfactant and 20 ml of the
above selected oil phase were mixed together thoroughly in an
Eppendorf tube. From this, 25 ml of mixture was added to
25 ml of distilled water in a standard flask. The ease of
emulsification was checked by the number of inversions of
standard flask enough to produce to uniform emulsion. The
formed emulsions were allowed to stand for 2 h. Then the
transmittance was determined using a double beam UV-
visible spectrophotometer (UV-3500, Labindia, Mumbai,
India) at 638.2 nm against distilled water as the blank (Date
& Nagarsenker, 2007; Shweta et al., 2011).
Selection of co-surfactant
The screening of co-surfactants was based on both the ability
to solubilize the drug in it and their efficacy to improve the
nanoemulsification ability of the selected surfactants. The
solubility of the drug in the co-surfactants namely PEG 400,
propylene glycol was determined from the solubility studies
same as in the case of oils and surfactants. Then to screen
the efficacy of co-surfactants, 40 ml of the selected surfactant
and 20 ml of co-surfactant to be screened and 60 ml of
selected oil phase were mixed together thoroughly in
an Eppendorf tube. From this mixture, 25 ml was added to
25 ml of distilled water in a standard flask. The ease of
emulsification was checked in a similar fashion as described
above.
2 A. B. Mohd et al. Drug Deliv, Early Online: 1–10
Dru
g D
eliv
ery
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Gla
sgow
Cal
edon
ian
Uni
vers
ity o
n 02
/01/
14Fo
r pe
rson
al u
se o
nly.

Construction of ternary phase diagram
For the construction of ternary phase diagrams, the screened
surfactant and co-surfactants were mixed (Smix) in three
different weight ratios (1:1, 2:1, and 3:1). These Smix ratios
were chosen to show increasing surfactant concentrations
with respect to co-surfactants. The oil phase and each Smix
ratios (a total of 51 ratios) were blended thoroughly in 17
different weight ratios from 1:9 to 9:1 (1:1, 1:2, 1:3, 1:4, 1:5,
1:6, 1:7, 1:8, 1:9, 9:1, 8:1, 7:1, 6:1, 5:1, 4:1, 3:1, and 2:1).
From these each ratio, 0.1 ml of mixtures was transferred to
separate glass beakers. To these contents, 100 ml distilled
water was added gently agitated using a magnetic bar at
37 �C. The transmittance of the formed emulsions were
determined using an UV–visible double beam spectropho-
tometer (UV-3500, Labindia, Mumbai, India) at 638.2 nm
against distilled water as the blank. The resulted emulsions
were examined for clarity, phase separation, and coalescence
of oil droplets on standing for 2 h. When the oil droplets easily
spread out in water and formed a clear, transparent emulsion,
the emulsion was judged as ‘‘good’’ emulsion (% transmit-
tance 485), and when there was poor or no emulsion
formation with immediate coalescence of oil droplets, espe-
cially when stirring was stopped, the emulsion was judged as
‘‘bad’’ emulsion (% transmittance 585). The phase diagram
was constructed using Chemix school� version 3.50 software
(Arne Standnes, MN, USA) to identify the nanoemulsifying
region, using oil and Smix ratios which form ‘good’ emulsions
upon dilution with purified water. The nanoemulsion region in
the ternary phase diagram was represented as shaded area
(Figure 1).
Preparation of L-SNEDDS
Twelve L-SNEDDS of glimepiride were formulated by using
oil (5, 10, 20, and 30) and Smix in the ratios of 1:2.33, 1:4, 1:9,
and 1:19 as well as surfactant and co-surfactants in the ratios
of 1:1, 2:1, and 3:1. The drug loaded L-SNEDDS were
prepared by adding the oil phase, containing accurately
weighed quantity (20 mg) of glimepiride, drop wise into the
Smix with constant stirring for 30 min. It was equilibrated at
ambient temperature for 48 h and investigated for signs of
turbidity or phase separation. The compositions of prepared
L-SNEDDS were represented in Table 1.
Emulsion droplet size and PDI analysis
The globule size of the emulsion determines the rate and
extent of drug release (Tarr & Yalkowsky, 1989). Aliquot
(1 ml) of the optimized SNEDDS were diluted 100-fold
with purified water to assess the globule size using
Zetasizer (Zetasizer Nano ZS, Malvern Instruments,
Worcestershire, UK) at a wavelength of 635 nm with dynamic
light scattering angle of 90� at 25 �C. The Z-average diameter
was derived from cumulated analysis by the auto-measure
software.
Preparation of S-SNEDDS from L-SNEDDS
The simplest technique to convert L-SNEDDS to S-SNEDDS
is by adsorption onto the surface of carriers. In the present
study, Aerosol� 200 was used as an inert solid adsorption
Figure 1. Ternary phase diagram of theselected system (Miglyol, Tween� 80 andPEG 400) dispersed in water at 25 �C.
Table 1. Droplet size and PDI of liquid SNEDDS of glimepiride.
Formula Oil: Smix
Miglyol�
812%Tween�
80%PEG400%
Z-averagesize (d nm) PDI
F1 1:19 5.00 47.50 47.50 11 0.636F2 1:9 10.00 45.00 45.00 250 0.565F3 1:4 20.00 40.00 40.00 890 0.645F4 1:2.33 30.00 35.00 35.00 1406 0.919F5 1:19 5.00 63.33 31.66 270 0.820F6 1:9 10.00 60.00 30.00 432 0.991F7 1:4 20.00 53.33 26.66 630 0.830F8 1:2.33 30.00 46.66 23.33 1270 0.999F9 1:19 5.00 71.25 23.75 152 0.211F10 1:9 10.00 67.50 22.50 355 0.368F11 1:4 20.00 60.00 20.00 917 0.113F12 1:2.33 30.00 52.50 17.50 1126 0.688
DOI: 10.3109/10717544.2013.879753 S-SNEDDS for oral delivery of glimepiride 3
Dru
g D
eliv
ery
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Gla
sgow
Cal
edon
ian
Uni
vers
ity o
n 02
/01/
14Fo
r pe
rson
al u
se o
nly.

carrier. A fixed volume of formed L-SNEDDS equivalent to
dose was transferred to a China dish, and to this, the
Aerosol� 200 was added in increments with vigorous stirring
until a free flow powder was obtained. Then the dose
equivalent free flow powder was filled in hard gelatin
capsules.
In vitro drug release studies
Pure glimepiride (2 mg), marketed tablet (2 mg dose), dose
equivalent amount of glimepiride-loaded L-SNEDDS, and
S-SNEDDS were placed in a USP-II dissolution test apparatus
(DS 8000, LABINDIA, Mumbai, India). The dissolution test
was performed using 900 ml of phosphate buffer pH 7.4 as a
dissolution medium. Temperature and speed of the paddle
were adjusted to 35 ± 0.5 �C and 100 rpm, respectively.
At predetermined time intervals (0, 5, 10, 15, 30, 45, and
60 min) an aliquot (5 ml) of the samples was collected and
filtered through a membrane filter (0.4mm) at each time 5 ml
of fresh medium was added to dissolution medium. The
concentration of drug was determined by using HPLC
(Prominence HPLC, Shimadzu, Kyoto, Japan) with a PDA
detector (SPD-M20A) at 228 nm.
Solid state characterization of optimized S-SNEDDS
Morphological analysis of S-SNEDDS (SEM)
The morphological features of solid glimepiride-loaded
S-SNEDDS, pure glimepiride, and aerosol were observed
by SEM (S-4100, Hitachi, Shiga, Japan). The analysis
was performed by placing the samples on a brass stub using
a double-sided adhesive tape and was made electric-
ally conductive by coating in vacuum (6 pas) with platinum
using an ion sputter (E-1030) at 15 mA. The micro-
photographs were taken at an excitation voltage of 10 kV.
Differential scanning calorimetry (DSC)
The thermal investigations of glimepiride S-SNEDDS, pure
glimepiride, and pure Aerosol� 200 were performed by using
a DSC (SIIO, 6300, Tokyo, Japan) by placing about 3 mg of
each separated sample in sealed standard aluminum pans
before heating under a nitrogen flow (50 ml/min) and setting
the heat flow was from 0 �C to 300 �C.
X-ray powder diffraction (XRPD)
The powder crystallinity of the S-SNEDDS formulation, pure
glimepiride, and Aerosol� 200 were assessed with an X’Pert
PRO diffractometer (PW 1729, Philips, Amsterdam, The
Netherlands) at room temperature using monochromatic Cu-
Ka radiation, over a range of 2� angles from 2� to 50�, with
an angular increment of 0.02� per second.
Stability studies
The stability studies were conducted to determine the changes
in in vitro drug release studies, drug content, emulsion droplet
size, and PDI, on storage, according to ICH guidelines at
40 �C/75 ± 5% RH (Jain et al., 2005; Girish et al., 2007;
Srinivas et al., 2011) by storing the optimized S-SNEDDS for
3 months. After 3 months, the samples were collected and
analyzed for drug content, emulsion droplet size, PDI, and
in vitro release studies.
Assessment of therapeutic efficacy in albino rabbits
This study was with a single dose design using male albino
rabbits weighing 1–1.5 kg. The study was conducted prior
approval from institution animal ethical committee, School of
Pharmacy, Anurag Group of institutions, Hyderabad, India.
The animals were kept on a standard diet and fasted
overnight. Rabbits were divided into four groups, each of
six animals. Each animal of these first group received pure
glimepiride (1 mg/kg body weight), second group received
marketed formulation, third group received S-SNEDDS, and
forth group received L-SNEDDS in an amount equivalent to
calculated dose (1 mg/kg body weight). Blood samples were
withdrawn from the marginal ear vein of the animal. Fasting
blood glucose level was assessed using Semi-Auto analyzer
(Optimas, Labindia, Mumbai, India). Blood glucose level
(BGL) was measured at different time intervals (0, 1, 2, 3, 4,
5, 6, 8, 12, 18, and 24 h) up to 24 h. Each animal served as its
own control and hence, the hypoglycemic response was
evaluated as percentage decrease in blood glucose level. The
pharmacodynamic parameter of the area under percentage
decrease in BGL versus time curve (AUC0–24h) was calculated
by adopting the trapezoidal rule (Wagner, 1975; Ammar et al.,
2006; Sandya et al., 2012).
Results and discussion
Selection of oil
The determination of solubility of glimepiride in oils,
surfactants, and co-surfactants is the most important principle
for the selection of components for SNEDDS formulation.
The equilibrium solubility of glimepiride in different oils is
shown in Table 2. Among the all selected oils (Miglyol� 812,
Oleic acid, Sunflower oil, Soya bean oil, Isopropyl Myristate)
Miglyol� 812 and Oleic acid exhibited highest and lowest
solubilization capacity for glimepiride, respectively. As
Miglyol� 812 had good solubility for glimepiride compared
to all other oils, it was selected as oil phase for SNEDDS
formulation.
Selection of surfactant
Non-ionic surfactants are usually accepted for oral ingestion
and are considered safer than the ionic surfactants. The non-
ionic surfactants used were Tween� 80, Cremophor� RH 40,
Span� 20, and Span� 80. These surfactants were screened on
the basis of its ability to emulsify the selected oil as well as its
capacity to solubilize glimepiride. Tween� 80 showed highest
solubilization capacity for glimepiride (Table 3) and had
Table 2. Solubility of glimepiride in various oils.
Oil Solubility (mg/ml)
Oleic acid 0.16 ± 0.01Sunflower oil 16.2 ± 0.36Soya oil 15.23 ± 0.40Isopropyl Myristate 0.28 ± 0.02Miglyol� 812 18.3 ± 0.26
4 A. B. Mohd et al. Drug Deliv, Early Online: 1–10
Dru
g D
eliv
ery
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Gla
sgow
Cal
edon
ian
Uni
vers
ity o
n 02
/01/
14Fo
r pe
rson
al u
se o
nly.

ability to emulsify the selected oil, i.e., Miglyol� 812. The
emulsifying abilities were determined by measuring the %
transmittance of the resultant nanoemulsion. Since the use of
Tween� 80 and Cremophor� RH 40 was found to give
transmittance above 90% and as glimepiride was found to
have considerable solubility in these surfactants, they were
chosen for further investigations.
Selection of co-surfactant
The % transmittance obtained using PEG 400 and propylene
glycol as co-surfactants in combination with the above-
selected surfactants (namely Tween� 80 and Cremophor� RH
40) and Miglyol� 812 as the oil are given in Table 4. Among
the two co-surfactants examined, PEG-400 was found to
display highest % transmittance when used in combination
with Tween� 80 as a surfactant while propylene glycol was
found to show low % transmittance when combined with
Cremophor� RH 40 as the surfactant compared to PEG 400.
The co-surfactant was finally selected based on its ability to
solubilize the glimepiride and its ability to improve the
nanoemulsification efficiency of the selected surfactant.
Accordingly, PEG-400, which afforded high solubility of
glimepiride and which showed the highest transmittance
(98.55%), was selected as the co-surfactant for further
investigations.
Ternary phase diagram
The ternary phase diagram was constructed to identify the
nano-emulsifying region and to optimize the concentration of
the selected oil, surfactant, and co-surfactants (namely
Miglyol� 812, Tween� 80, and PEG 400, respectively). For
the development of a SNEDDS formulation, optimum ratios
of excipient concentrations established by means of phase
diagram studies provided the area of the monophasic region.
It is important to determine this area in order to confirm
successful aqueous dilution without breaking the nanoemul-
sion (Setthacheewakul et al., 2010). Figure 1 shows the
ternary phase diagram of the system containing selected oil,
surfactant, and co-surfactant. It was noted that incorporation
of the co-surfactant, PEG 400, within the self-emulsifying
region increased the spontaneity of the self-emulsification
process. The efficiency of emulsification was good when the
surfactant/co-surfactant concentration was more than 75% v/v
of the SNEDDS formulation. It was observed that spontan-
eous emulsion formation was not effective with less than 50%
v/v of the surfactant in the SNEDDS. In the present system,
the formulations surrounding the good self-emulsifying
region in the phase diagram exhibited a poor emulsion
forming ability. It has been reported that the drug
incorporated in the SNEDDS may have some effect on the
self-emulsifying performance (Oh et al., 2011). However, in
our study, no significant differences were found in the self-
emulsifying performance.
Optimization of glimepiride L-SNEDDS
The amount of oil, surfactant, and co-surfactant for the final
formulation of glimepiride SNEDDS was optimized on the
basis of Z-average size and PDI. The results are shown in
Table 1. Less than 200 nm Z-average size was observed with
two SNEDDS compositions namely 5:47.5:47.5 and
5:71.25:23.75 (oil:surfactant:co-surfactant, respectively). But
if the PDI value higher than 0.8, the systems were considered
as polydisperse. Hence the results obtained for SNEDDS
with oil:surfactant:co-surfactant of 5:71.25:23.75 was the
most consistent, it was considered as optimized L-SNEDDS
(F9) and composition was represented in Table 5.
Emulsion droplet size analysis
The particle size distribution is one of the important
parameters affecting the in vivo fate of emulsions.
The droplet size of the emulsion also defines the rate and
extent of drug release (Krutika et al., 2011). The smaller the
emulsion droplet size, larger the surface area provided for the
drug absorption. The Z-average size of the resulted nanoe-
mulsion at 100 times dilution was determined to be 152 nm
and the PDI value was low (0.211), representing that the
system had narrow size distribution (Figure 2).
Characterization of optimized S-SNEDDS ofglimepiride
Morphological analysis of S-SNEDDS (SEM)
The scanning electron microscopic pictures of glimepiride
powder, Aerosol� 200, and S-SNEDDS final formulation are
presented in Figure 3. The pure glimepiride powder
(Figure 3A) appeared as smooth surfaced, irregular shaped
crystals. Aerosol� 200 (Figure 3B) appeared as a rough
surface with porous particles. However, the S-SNEDDS
(Figure 3C) appeared as smooth surfaced particles without
Table 4. Emulsification efficiency with different co-surfactants andselected surfactants.
% Transmittance (at 638.2 nm)
Co-surfactantsSolubility(mg/ml) Tween� 80 Cremophor� RH 40
PEG 400 15.34 ± 0.21 98.55 ± 1.22 95.26 ± 1.33Propylene glycol 0.06 ± 0.005 62.61 ± 2.36 59.25 ± 0.82
Table 5. Composition of S-SNEDDS.
Formula Components in S-SNEDDS Proportions in mg
F 9 Glimepiride 20Miglyol� 812 50
Tween� 80 712.5PEG 400 237.5
Aerosol� 200 750
Table 3. Solubility of glimepiride in various non-ionic surfactants andtheir emulsification efficiency.
Surfactant HLB Solubility (mg/ml)% Transmittance
(at 638.2 nm)
Tween� 80 15 19.23 ± 0.30 91.3 ± 1.98Cremophor� RH 40 13 14.3 ± 0.4 90.1 ± 1.04Span� 20 8.6 4.41 ± 0.17 55.3 ± 0.28Span� 80 4.3 2.50 ± 0.15 54.4 ± 0.34
DOI: 10.3109/10717544.2013.879753 S-SNEDDS for oral delivery of glimepiride 5
Dru
g D
eliv
ery
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Gla
sgow
Cal
edon
ian
Uni
vers
ity o
n 02
/01/
14Fo
r pe
rson
al u
se o
nly.

any crystalline shape which indicates complete adsorption of
liquid NEDDS containing glimepiride inside the pores of
Aerosol� 200.
Differential scanning calorimetry (DSC)
The DSC thermograms of pure glimepiride, Aerosol� 200,
and S-SNEDDS are shown in Figure 4. Pure glimepiride
showed one sharp endothermic peak at 212.2 �C (Figure 4A),
corresponding to its melting point indicating its crystalline
nature. S-SNEDDS showed one broad endothermic peak with
reduced intensity at 153 �C (Figure 4C). It might be indicating
that the drug is present in molecularly dissolved and
amorphous state in S-SNEDDS (Nazzal et al., 2002b;
Balakrishnan et al., 2009; Wang et al., 2009; Shanmugam
et al., 2011).
X-ray powder diffraction (XRPD)
The XRPD patterns of pure glimepiride powder, Aerosol�
200, and S-SNEDDS are shown in Figure 5. XRPD was used
for further verification of the internal physical state of
glimepiride in the S-SNEDDS. Pure glimepiride powder
showed sharp peaks at the diffraction angles (2�), such as
10.9�, 12�, 16.5�, 17�, 18.8�, 21�, 23�, 28�, and 30�
(Figure 5A) indicating a typical crystalline pattern.
Aerosol� 200 showed no peaks at diffraction angles,
indicating an amorphous pattern (Figure 5B). No obvious
peaks representing crystals of glimepiride were observed for
the S-SNEDDS final formulation (Figure 5C), indicating the
absence of crystalline structure of glimepiride in the final
formulation.
In vitro drug release studies
The dissolution profiles of L-SNEDDS, S-SNEDDS, and
marketed formulation were compared with that of the pure
drug (Figure 6), using phosphate buffer pH 7 as a dissolution
medium. Both liquid and S-SNEDDS and marketed formu-
lation of glimepiride showed more than 85% drug within
15 min, whereas the pure drug showed less than 15% release
of the total amount of drug. Within 30 min, the drug is
completely released from the both solid and L-SNEDDS as a
result of the spontaneous emulsification and the resulted
nano-level droplet size. At the same time % drug release from
pure drug was found to be only 16% of overall drug release.
Figure 3. Scanning electron microscope (SEM) pictures of (A) pureglimepiride powder; (B) Aerosol� 200, and (C) S-SNEDDS formulation.
Figure 2. Z-average (nm) of liquid SNEDDSof glimepiride (F9).
6 A. B. Mohd et al. Drug Deliv, Early Online: 1–10
Dru
g D
eliv
ery
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Gla
sgow
Cal
edon
ian
Uni
vers
ity o
n 02
/01/
14Fo
r pe
rson
al u
se o
nly.

Figure 5. X-ray powder diffractograms of (A) pure glimepiride, (B) Aerosol� 200, and (C) S-SNEDDS formulation.
Figure 4. Differential scanning calorimetric thermograms of (A) pureglimepiride, (B) Aerosol� 200, and (C) S-SNEDDS formulation.
Figure 6. Comparative in vitro drug release profile of pure glimepiride,optimized L-SNEDDS, S-SNEDDS, and marketed formulation. Datarepresented are cumulative % drug release versus time (min) in terms ofmean ± SD (n¼ 3).
DOI: 10.3109/10717544.2013.879753 S-SNEDDS for oral delivery of glimepiride 7
Dru
g D
eliv
ery
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Gla
sgow
Cal
edon
ian
Uni
vers
ity o
n 02
/01/
14Fo
r pe
rson
al u
se o
nly.
The solid carrier used for S-SNEDDS did not interfere with
the drug release. The in vitro dissolution studies of S-
SNEDDS revealed that initial glimepiride release from porous
carriers (Aerosol� 200) was slower compared with L-
SNEDDS, This could be because of additional steps involved
during dissolution such as disintegration of solid structure of
S-SNEDDS and desorption of L-SNEDDS from the voids of
porous carriers. The S-SNEDDS when exposed to dissolution
medium leads to desorption of the L-SNEDDS from the
Aerosol� 200 surface due to stronger interaction between
Aerosol� 200 and dissolution medium than those between
Aerosol� 200 and L-SNEDDS.
Stability studies
To determine the temperature sensitivity on dissolution
release profile, emulsion droplet size, PDI, and drug content
of optimized S-SNEDDS, the stability study was performed at
40 �C and 75% ± 5% RH for 3 months. The stability samples
were evaluated for dissolution release profile, emulsion
droplet size, PDI, and drug content. Samples withdrawn
after 3 months showed no significant changes in in vitro drug
release studies, drug content, emulsion droplet size, and PDI.
The similarity factor (f2) for the optimized S-SNEDDS was
found to be 94.51 (Table 6), which indicates good similarity
of dissolution release profile, prior and after stability studies.
No significant difference (p40.05) was observed in drug
content, emulsion droplet size, and PDI data, before and after
storage.
Assessment of therapeutic efficacy in albino rabbits
Blood glucose level (BGL) was measured at different time
intervals up to 24 h. Each animal served as its own control and
hence, the hypoglycemic response was evaluated as percent-
age decrease in blood glucose level. The pharmacodynamic
parameter of the area under percentage decrease in BGL
versus time curve (AUC0–24h) was calculated adopting the
trapezoidal rule
Decrease in BGL ð%Þ ¼ BGL at t ¼ 0� BGL at t
BGL at t ¼ 0� 100
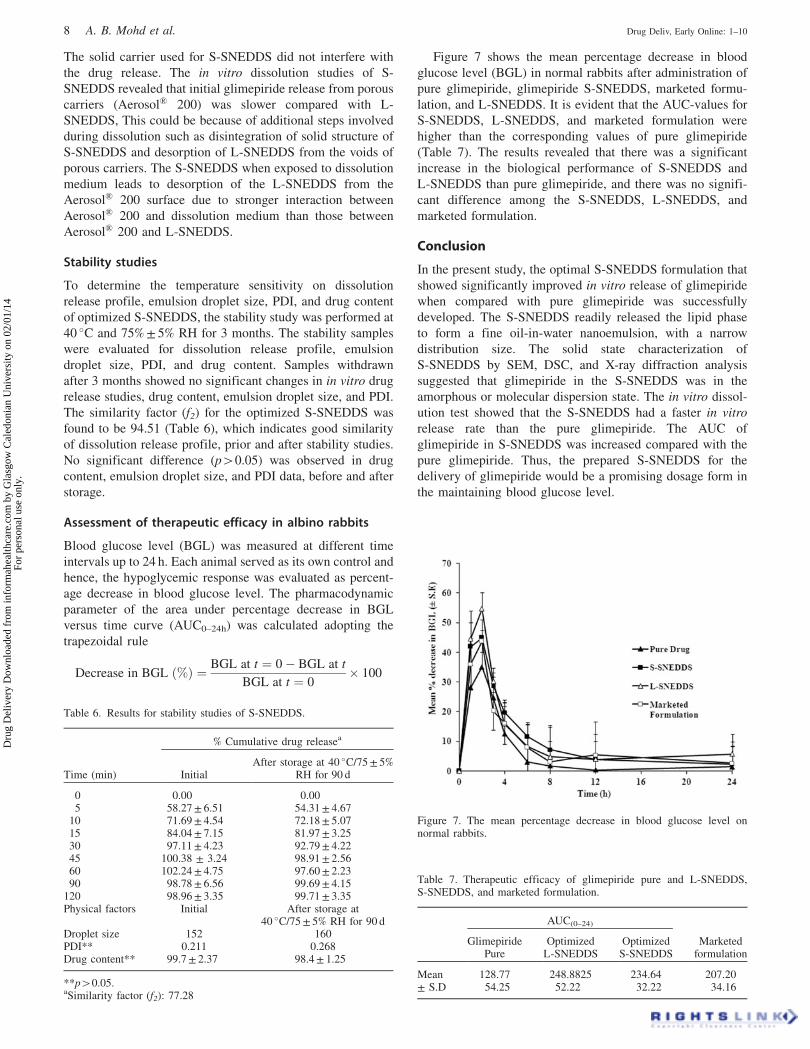
Figure 7 shows the mean percentage decrease in blood
glucose level (BGL) in normal rabbits after administration of
pure glimepiride, glimepiride S-SNEDDS, marketed formu-
lation, and L-SNEDDS. It is evident that the AUC-values for
S-SNEDDS, L-SNEDDS, and marketed formulation were
higher than the corresponding values of pure glimepiride
(Table 7). The results revealed that there was a significant
increase in the biological performance of S-SNEDDS and
L-SNEDDS than pure glimepiride, and there was no signifi-
cant difference among the S-SNEDDS, L-SNEDDS, and
marketed formulation.
Conclusion
In the present study, the optimal S-SNEDDS formulation that
showed significantly improved in vitro release of glimepiride
when compared with pure glimepiride was successfully
developed. The S-SNEDDS readily released the lipid phase
to form a fine oil-in-water nanoemulsion, with a narrow
distribution size. The solid state characterization of
S-SNEDDS by SEM, DSC, and X-ray diffraction analysis
suggested that glimepiride in the S-SNEDDS was in the
amorphous or molecular dispersion state. The in vitro dissol-
ution test showed that the S-SNEDDS had a faster in vitro
release rate than the pure glimepiride. The AUC of
glimepiride in S-SNEDDS was increased compared with the
pure glimepiride. Thus, the prepared S-SNEDDS for the
delivery of glimepiride would be a promising dosage form in
the maintaining blood glucose level.
Figure 7. The mean percentage decrease in blood glucose level onnormal rabbits.
Table 6. Results for stability studies of S-SNEDDS.
% Cumulative drug releasea
Time (min) InitialAfter storage at 40 �C/75 ± 5%
RH for 90 d
0 0.00 0.005 58.27 ± 6.51 54.31 ± 4.67
10 71.69 ± 4.54 72.18 ± 5.0715 84.04 ± 7.15 81.97 ± 3.2530 97.11 ± 4.23 92.79 ± 4.2245 100.38 ± 3.24 98.91 ± 2.5660 102.24 ± 4.75 97.60 ± 2.2390 98.78 ± 6.56 99.69 ± 4.15
120 98.96 ± 3.35 99.71 ± 3.35Physical factors Initial After storage at
40 �C/75 ± 5% RH for 90 dDroplet size 152 160PDI** 0.211 0.268Drug content** 99.7 ± 2.37 98.4 ± 1.25
**p40.05.aSimilarity factor (f2): 77.28
Table 7. Therapeutic efficacy of glimepiride pure and L-SNEDDS,S-SNEDDS, and marketed formulation.
AUC(0–24)
GlimepiridePure
OptimizedL-SNEDDS
OptimizedS-SNEDDS
Marketedformulation
Mean 128.77 248.8825 234.64 207.20± S.D 54.25 52.22 32.22 34.16
8 A. B. Mohd et al. Drug Deliv, Early Online: 1–10
Dru
g D
eliv
ery
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Gla
sgow
Cal
edon
ian
Uni
vers
ity o
n 02
/01/
14Fo
r pe
rson
al u
se o
nly.

Declaration of interest
The authors report no conflicts of interest. The authors alone
are responsible for the content and writing of this article.
References
Allaboun H, Alkhamis KA, AlMomani WY. (2003). The application ofthe convective diffusion model and the film equilibrium model tosurfactant-facilitated dissolution of gliclazide. Eur J Pharm Sci 19:231–6.
Amidon GL, Lennernas H, Shah VP, Crison JR. (1995). A theoreticalbasis for a biopharmaceutic drug classification: the correlation ofin vitro drug product dissolution and in vivo bioavailability. PharmRes 12:413–20.
Ammar HO, Salama HA, Ghorab M, Mahmouda AA. (2006).Implication of inclusion complexation of glimepiride in cyclodex-trin–polymer systems on its dissolution, stability and therapeuticefficacy. Int J Pharm 320:53–7.
Badr-Eldin SM, Elkheshen SA, Ghorab MM. (2008). Inclusioncomplexes of tadalafil with natural and chemically modified beta-cyclodextrins. I: preparation and in-vitro evaluation. Eur J PharmBiopharm 70:819–27.
Balakrishnan A, Rege BD, Amidon GL, Polli JE. (2004). Surfactant-mediated dissolution: contributions of solubility enhancement andrelatively low micelle diffusivity. J Pharm Sci 93:2064–75.
Balakrishnan P, Lee BJ, Oh DH, et al. (2009). Enhanced oralbioavailability of dexibuprofen by a novel solid Self-emulsifyingdrug delivery system (SEDDS). Eur J Pharm Biopharm 72:539–45.
Bannwart B, Bertin P, Pehourcq F, et al. (2001). Piroxicam concentra-tions in plasma and synovial fluid after a single dose of piroxicam-beta-cyclodextrin. Int J Clin Pharmacol Ther 39:33–6.
Bayindir ZS, Yuksel N. (2010). Characterization of niosomes preparedwith various nonionic surfactants for paclitaxel oral delivery. J PharmSci 99:2049–60.
Beg S, Swain S, Rizwan Md, et al. (2011). Bioavailability enhancementstrategies: basics, formulation approaches and regulatory consider-ations. Curr Drug Deliv 8:1–12.
Burcin Y, Erem B, Imran V, Murat S. (2010). Alternative oralexemestane formulation: improved dissolution and permeation. Int JPharm 398:137–45.
Carrier RL, Miller LA, Ahmed I. (2007). The utility of cyclodextrins forenhancing oral bioavailability. J Control Release 123:78–99.
Chadha R, Saini A, Arora P, Bhandari S. (2012). Pharmaceuticalcocrystals: a novel approach for oral bioavailability enhancement ofdrugs. Crit Rev Ther Drug Carrier Syst 29:183–218.
Chakraborty S, Shukla D, Jain A, et al. (2009). Assessmentof solubilization characteristics of different surfactants forcarvedilol phosphate as a function of pH. J Colloid Interface Sci335:242–449.
Date AA, Nagarsenker MS. (2007). Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoximeproxetil. Int J Pharm 329:166–72.
Gamsiz ED, Miller L, Thombre AG, et al. (2010a). Modeling theinfluence of cyclodextrins on oral absorption of low solubility drugs:II. Experimental validation. Biotechnol Bioeng 105:421–30.
Gamsiz ED, Miller L, Thombre AG, et al. (2010b). Modeling theinfluence of cyclodextrins on oral absorption of low-solubility drugs:I. Model development. Biotechnol Bioeng 105:409–20.
Gamsiz ED, Thombre AG, Ahmed I, Carrier RL. (2011). Drug salts andsolubilization: modeling the influence of cyclodextrins on oralabsorption. Ann Biomed Eng 39:455–68.
Girish SS, Devendra KJ, Dhananjay MM. (2007). Preparation andin vitro evaluation of bilayer and floating bioadhesive tablets ofrosiglitazone maleate. Asian J Pharm Sci 2:161–9.
Grunenberg A, Keil B, Henck JO. (1995). Polymorphism in binarymixtures, as exemplified by nimodipine. Int J Pharm 118:11–21.
Jain SK, Awasthi AM, Jain NK, Agrawal GP. (2005). Calcium silicatebased microspheres of repaglinide for gastro retentive floating drugdelivery: preparation and in vitro characterization. J Control Release107:300–9.
Jin Y, Wen J, Garg S, et al. (2013). Development of a novel niosomalsystem for oral delivery of Ginkgo biloba extract. Int J Nanomed 8:421–30.
Khazaeli P, Pardakhty A, Shoorabi H. (2007). Caffeine-loaded niosomes:characterization and in vitro release studies. Drug Deliv 14:447–52.
Krutika K. Sawant, Shweta Guptha, Sandip Chavhan. (2011). Self-nanoemulsifying drug delivery system for Adefovir dipivoxil: design,characterization, in vitro and ex vivo evaluation. Colloids Surf A 392:145–55.
Kumar N, Shishu, Bansal G, et al. (2013). Preparation and cyclodextrinassisted dissolution rate enhancement of itraconazolium dinitrate salt.Drug Dev Ind Pharm 39:342–51.
Kyatanwar AU, Gajbhiye ND, Jadhav KR, Kadam VJ. (2010). Solidself emulsifying drug delivery system: a review. J Pharm Res 3:877–82.
Li S, Wong S, Sethia S, et al. (2005). Investigation of solubility anddissolution of a free base and two different salt forms as a function ofpH. Pharm Res:628–35.
Lobenberg R, Amidon GL. (2000). Modern bioavailability, bioequiva-lence and biopharmaceutics classification system. New scientificapproaches to international regulatory standards. Eur J PharmBiopharm 50:3–12.
Manconi M, Nacher A, Merino V, et al. (2013). Improving oralbioavailability and pharmacokinetics of liposomal metformin byglycerolphosphate-chitosan microcomplexation. AAPS Pharm SciTech 14:485–96.
Miyake K, Arima H, Hirayama F, et al. (2000). Improvement of solu-bility and oral bioavailability of rutin by complexationwith 2-hydroxypropyl-beta-cyclodextrin. Pharm Dev Technol 5:399–407.
Nazzal S, Guven N, Reddy IK, Khan MA. (2002a). Preparation andcharacterization of coenzyme QlO-Eudragit solid dispersion. DrugDev Ind Pharm 28:49–57.
Nazzal S, Smalyukh II, Lavrentovich OD, Khan MA. (2002b).Preparation and in vitro characterization of a eutectic based semisolidself nanoemulsified drug delivery system (SNEDDS) of ubiquinone:mechanism and progress of emulsion formation. Int J Pharm 235:247–65.
Oh DH, Kang JH, Kim DW, et al. (2011). Comparison of solid self-microemulsifying drug delivery system (solid SMEDDS) preparedwith hydrophilic and hydrophobic solid carrier. Int J Pharm 420:412–8.
Patravale VB, Date AA, Kulkarni RM. (2004). Nanosuspensions: apromising drug delivery strategy. J Pharm Pharmacol 56:827–40.
Pouton CW. (2000). Lipid formulations for oral administration of drugs:non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drugdelivery systems. Eur J Pharm Sci 11:S93–8.
Pouton CW. (2006). Formulation of poorly water-soluble drugs fororal administration: physicochemical and physiological issues andthe lipid formulation classification system. Eur J Pharm Sci 29:278–87.
Qiao N, Li M, Schlindwein W, et al. (2011). Pharmaceutical cocrystals:an overview. Int J Pharm 419:1–11.
Sandya RT, Sujatha S, Veeresham C. (2012). Pharmacokinetic andpharmacodynamic interaction of curcumin with glimepiride in normaland diabetic rats. Phcog Commn 2:14–21.
Serajuddin AT. (2007). Salt formation to improve drug solubility.Adv Drug Deliv Rev 59:603–16.
Serajuddin TM. (1999). Solid dispersion of poorly water-soluble drugs:early promises, subsequent problems, and recent breakthroughs.J Pharm Sci 88:1058–66.
Setthacheewakul S, Mahattanadul S, Phadoongsombut N, et al. (2010).Development and evaluation of self-microemulsifying liquid andpellet formulations of curcumin, and absorption studies in rats. Eur JPharm Biopharm 76:475–85.
Sezgin-Bayindir Z, Onay-Besikci A, Vural N, Yuksel N. (2013).Niosomes encapsulating paclitaxel for oral bioavailability enhance-ment: preparation, characterization, pharmacokinetics and biodistri-bution. J Microencapsul, [Epub ahead of print]. Pages 1–9.
Shan N, Zaworotko MJ. (2008). The role of cocrystals in pharmaceuticalscience. Drug Discov Today 13:440–6.
Shanmugam S, Baskaran R, Balakrishnan P, et al. (2011). Solidself-nanoemulsifying drug delivery system (S-SNEDDS) containingphosphatidylcholine for enhanced bioavailability of highly lipophilicbioactive carotenoid lutein. Eur J Pharm Biopharm 79:250–7.
Shweta G, Sandip C, Krutika KS. (2011). Self-nanoemulsifying drugdelivery system for adefovir dipivoxil: design, characterization,in vitro and ex vivo evaluation. Colloid Surf A 392:145–55.
DOI: 10.3109/10717544.2013.879753 S-SNEDDS for oral delivery of glimepiride 9
Dru
g D
eliv
ery
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Gla
sgow
Cal
edon
ian
Uni
vers
ity o
n 02
/01/
14Fo
r pe
rson
al u
se o
nly.

Srinivas M, Krishna S, Prabhakar RV. (2011). Design and evaluation ofhydrochlorthiazide gastroretentive floating drug delivery system.Asian J Pharm Sci 6:208–17.
Tarr BD, Yalkowsky SH. (1989). Enhanced intestinal absorption ofcyclosporine in rats through the reduction of emulsion droplet size.Pharm Res 6:40–3.
Veiga F, Fernandes C, Teixeira F. (2000). Oral bioavailability andhypoglycaemic activity of tolbutamide/cyclodextrin inclusion com-plexes. Int J Pharm 202:165–71.
Wagner SG. (1975). Fundamentals of clinical pharmacokinetics.Hamilton, IL: Drug Intelligence Publications Inc.
Wang D, Miller R, Zheng J, Hu C. (2000). Comparative populationpharmacokinetic-pharmacodynamic analysis for piroxicam-beta-cyclodextrin and piroxicam. J Clin Pharmacol 40:1257–66.
Wang L, Dong J, Chen J, et al. (2009). Design and optimization of a newself-nanoemulsifying drug delivery system. J Colloid Interface Sci330:443–8.
Yang Z, Lu A, Wong BC, et al. (2013). Effect of liposomes on theabsorption of water-soluble active pharmaceutical ingredients via oraladministration. Curr Pharm Des 19:6647–54.
Yeap YY, Trevaskis NL, Quach T, et al. (2013). Intestinal bile secretionpromotes drug absorption from lipid colloidal phases via induction ofsupersaturation. Mol Pharm 10:1874–89.
10 A. B. Mohd et al. Drug Deliv, Early Online: 1–10
Dru
g D
eliv
ery
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Gla
sgow
Cal
edon
ian
Uni
vers
ity o
n 02
/01/
14Fo
r pe
rson
al u
se o
nly.
Top Related
Copyright © 2022 FDOKUMEN

