







Bahasa
Halaman
Hukum


www.elsevier.com/locate/forsciint
Available online at www.sciencedirect.com
al 178 (2008) 192–198
Forensic Science InternationOff-line HPLC method combined to LC–MS for the determination
of sildenafil and its active metabolite in post-mortem human
blood according to confirmation criteria
Constantinos Pistos *, Ioannis Papoutsis, Artemis Dona, Maria Stefanidou,Sotiris Athanaselis, Constantinos Maravelias, Chara Spiliopoulou
University of Athens, Medical School, Laboratory of Forensic Medicine and Toxicology, 75M. Asias Str., Goudi 11527, Athens, Greece
Received 17 November 2007; received in revised form 2 March 2008; accepted 24 March 2008
Available online 16 May 2008
Abstract
A simple HPLC method has been validated for the determination of sildenafil and its active metabolite (N-desmethylsildenafil) in human blood,
using an octadecyl silica (ODS) hypersil column. The chromatographic run time is less than 25 min using a mobile phase of 35:65 (v/v)
acetonitrile–0.015 M disodium hydrogen phosphate (Na2HPO4), triethylamine 0.1%, pH 7.4 at 1 mL/min flow rate and UV–vis detection at
230 nm. The method is linear in the concentration range of 10–500 ng/mL (r > 0.999, n = 5) for each analyte, with relative standard deviation
(R.S.D.) less than 5.05%. Interday and intraday errors were found to be �11.94%. The limits of detection and quantitation for both analytes were
5.0 ng/mL (s/n > 3) and 10.0 ng/mL (s/n > 10), respectively. The method was applied in two post-mortem human blood samples, concerning two
fatal cases from sildenafil citrate use, reported for the first time in Greece, and the results were further confirmed with LC–MS. The method is
proposed as supplementary to LC–MS when inadequate mass fragmentation does not provide information appropriate to meet confirmation
criteria.
# 2008 Elsevier Ireland Ltd. All rights reserved.
Keywords: HPLC UV–vis; Confirmation criteria; Sildenafil; N-Desmethylsildenafil
1. Introduction
Sildenafil (S) (1-[4-ethoxy-3-(6,7-dihydro-1-methyl-7-oxo-
3-propyl-1H-pyrazolo-[4,3-d]pyrimidin-5-yl) phenylsulpho-
nyl]-4-methylpiperazine) (Fig. 1a) has been widely prescribed
for erectile dysfunction [1–4] and its bioavailabilty, metabo-
lism, elimination route and pharmacokinetics have been
extended reported [3,5]. Mean maximum sildenafil plasma
concentrations measured after a single oral dose of 100 mg to
healthy male volunteers is 450 ng/mL plasma. The lower
therapeutic concentrations in human plasma after a 25 mg
single oral dose are approximately 7 ng/mL [5].
Many analytical methods, using high performance liquid
chromatography (HPLC), have been published for quantification
of the parent drug sildenafil in plasma and not for its active
* Corresponding author. Tel.: +30 210 7462433; fax: +30 210 7716098.
E-mail address: [email protected] (C. Pistos).
0379-0738/$ – see front matter # 2008 Elsevier Ireland Ltd. All rights reserved.
doi:10.1016/j.forsciint.2008.03.018
metabolite, using ultraviolet visible (UV–vis) detector [6,7], or a
liquid chromatography system combined with a triple quadru-
pole mass spectrometry detector [8], as well as in oral fluids using
a liquid chromatography single mass spectrometry system [9].
Lewis and Johnson [10] reported the detection of both sildenafil
and N-desmethylsildenafil in post-mortem fluids and tissues,
while Weinmann et al. [11] reported their detection in urine and
tissue samples using liquid chromatography tandem mass
spectrometry (LC–MS/MS) system. Al-Ghazawi et al. [12]
developed a method for the determination of both analytes in
plasma using electrochemical detection, Cooper et al. [13] in
plasma using UV–vis detector and Saisho et al. [14] in human
hair by GC–MS. Although most of these methods are sensitive
and accurate, none of them describe how to overcome the
inadequate LC–MS fragmentation of both sildenafil and its active
metabolite in human blood. According to our experiments, the
proposed methods in the literature for the determination of both
compounds in plasma appear to be problematic when they are
applied in human blood. Considering that at a number of cases,
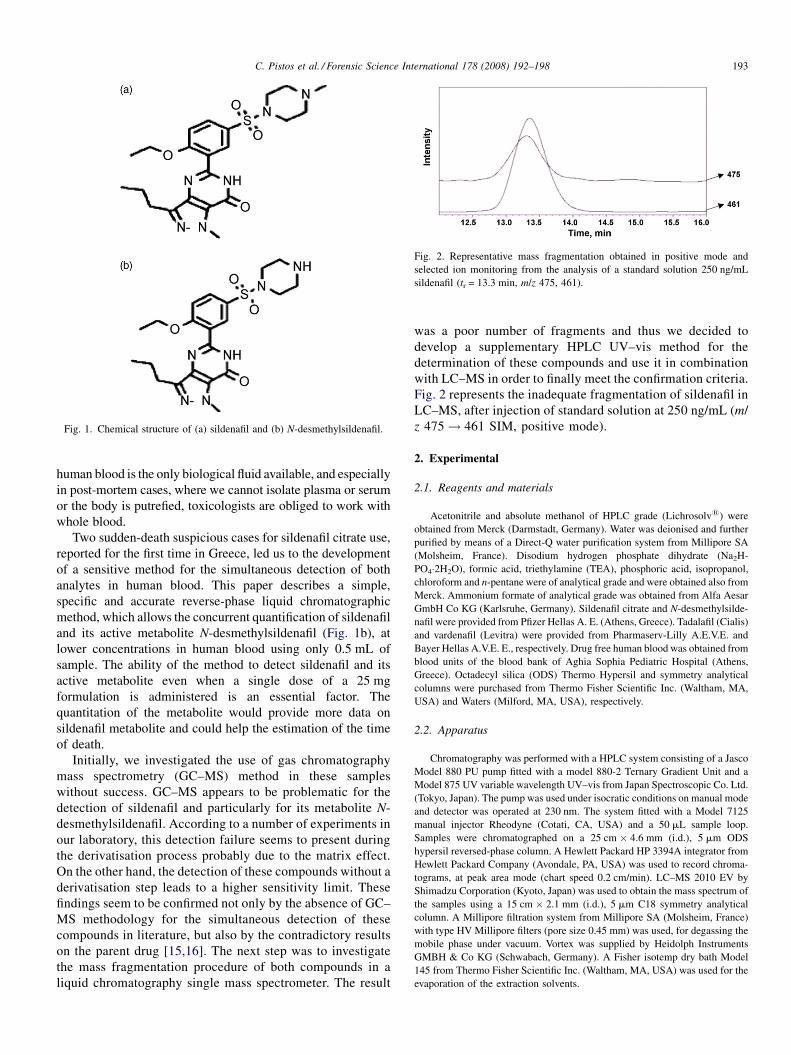
Fig. 1. Chemical structure of (a) sildenafil and (b) N-desmethylsildenafil.
Fig. 2. Representative mass fragmentation obtained in positive mode and
selected ion monitoring from the analysis of a standard solution 250 ng/mL
sildenafil (tr = 13.3 min, m/z 475, 461).
C. Pistos et al. / Forensic Science International 178 (2008) 192–198 193
human blood is the only biological fluid available, and especially
in post-mortem cases, where we cannot isolate plasma or serum
or the body is putrefied, toxicologists are obliged to work with
whole blood.
Two sudden-death suspicious cases for sildenafil citrate use,
reported for the first time in Greece, led us to the development
of a sensitive method for the simultaneous detection of both
analytes in human blood. This paper describes a simple,
specific and accurate reverse-phase liquid chromatographic
method, which allows the concurrent quantification of sildenafil
and its active metabolite N-desmethylsildenafil (Fig. 1b), at
lower concentrations in human blood using only 0.5 mL of
sample. The ability of the method to detect sildenafil and its
active metabolite even when a single dose of a 25 mg
formulation is administered is an essential factor. The
quantitation of the metabolite would provide more data on
sildenafil metabolite and could help the estimation of the time
of death.
Initially, we investigated the use of gas chromatography
mass spectrometry (GC–MS) method in these samples
without success. GC–MS appears to be problematic for the
detection of sildenafil and particularly for its metabolite N-
desmethylsildenafil. According to a number of experiments in
our laboratory, this detection failure seems to present during
the derivatisation process probably due to the matrix effect.
On the other hand, the detection of these compounds without a
derivatisation step leads to a higher sensitivity limit. These
findings seem to be confirmed not only by the absence of GC–
MS methodology for the simultaneous detection of these
compounds in literature, but also by the contradictory results
on the parent drug [15,16]. The next step was to investigate
the mass fragmentation procedure of both compounds in a
liquid chromatography single mass spectrometer. The result
was a poor number of fragments and thus we decided to
develop a supplementary HPLC UV–vis method for the
determination of these compounds and use it in combination
with LC–MS in order to finally meet the confirmation criteria.
Fig. 2 represents the inadequate fragmentation of sildenafil in
LC–MS, after injection of standard solution at 250 ng/mL (m/
z 475! 461 SIM, positive mode).
2. Experimental
2.1. Reagents and materials
Acetonitrile and absolute methanol of HPLC grade (Lichrosolv1) were
obtained from Merck (Darmstadt, Germany). Water was deionised and further
purified by means of a Direct-Q water purification system from Millipore SA
(Molsheim, France). Disodium hydrogen phosphate dihydrate (Na2H-
PO4�2H2O), formic acid, triethylamine (TEA), phosphoric acid, isopropanol,
chloroform and n-pentane were of analytical grade and were obtained also from
Merck. Ammonium formate of analytical grade was obtained from Alfa Aesar
GmbH Co KG (Karlsruhe, Germany). Sildenafil citrate and N-desmethylsilde-
nafil were provided from Pfizer Hellas A. E. (Athens, Greece). Tadalafil (Cialis)
and vardenafil (Levitra) were provided from Pharmaserv-Lilly A.E.V.E. and
Bayer Hellas A.V.E. E., respectively. Drug free human blood was obtained from
blood units of the blood bank of Aghia Sophia Pediatric Hospital (Athens,
Greece). Octadecyl silica (ODS) Thermo Hypersil and symmetry analytical
columns were purchased from Thermo Fisher Scientific Inc. (Waltham, MA,
USA) and Waters (Milford, MA, USA), respectively.
2.2. Apparatus
Chromatography was performed with a HPLC system consisting of a Jasco
Model 880 PU pump fitted with a model 880-2 Ternary Gradient Unit and a
Model 875 UV variable wavelength UV–vis from Japan Spectroscopic Co. Ltd.
(Tokyo, Japan). The pump was used under isocratic conditions on manual mode
and detector was operated at 230 nm. The system fitted with a Model 7125
manual injector Rheodyne (Cotati, CA, USA) and a 50 mL sample loop.
Samples were chromatographed on a 25 cm � 4.6 mm (i.d.), 5 mm ODS
hypersil reversed-phase column. A Hewlett Packard HP 3394A integrator from
Hewlett Packard Company (Avondale, PA, USA) was used to record chroma-
tograms, at peak area mode (chart speed 0.2 cm/min). LC–MS 2010 EV by
Shimadzu Corporation (Kyoto, Japan) was used to obtain the mass spectrum of
the samples using a 15 cm � 2.1 mm (i.d.), 5 mm C18 symmetry analytical
column. A Millipore filtration system from Millipore SA (Molsheim, France)
with type HV Millipore filters (pore size 0.45 mm) was used, for degassing the
mobile phase under vacuum. Vortex was supplied by Heidolph Instruments
GMBH & Co KG (Schwabach, Germany). A Fisher isotemp dry bath Model
145 from Thermo Fisher Scientific Inc. (Waltham, MA, USA) was used for the
evaporation of the extraction solvents.

C. Pistos et al. / Forensic Science International 178 (2008) 192–198194
2.3. Methods
2.3.1. Calibration curves
Two separate stock solutions containing 1.0 mg/mL of sildenafil and N-
desmethylsildenafil were prepared in absolute methanol and stored at �20 8C.
The stability of stock solutions was checked comparing the peak area of frozen
versus a fresh solution originated by a new weight of the reference standard. Stock
solutions were stable for up to 1 month. A combined working solution containing
both sildenafil and N-desmethylsildenafil was prepared in absolute methanol
containing 10 mg/mL from stock standard solutions of each compound. Addi-
tional dilutions were prepared containing 0.1, 0.2, 0.5, and 5.0 mg/mL of sildenafil
and N-desmethylsildenafil by appropriate dilutions of the reference solution with
absolute methanol. Blood standards for calibration curves were prepared by
spiking 0.9 mL aliquots of drug free human blood with 100 mL of the above-
mentioned working solutions, to make sildenafil and N-desmethylsildenafil blood
standards ranging from 10 to 500 ng/mL. Calibration graphs of the recovered
standards were prepared for each of the 5 days of analysis to establish linearity and
reproducibility of the HPLC system. Graphs were constructed of the peak area
against drug concentration according to European Union criteria [17].
2.3.2. Chromatographic conditions
Separation of the analytes was achieved on the ODS hypersil column with
the detector set at 230 nm and the column maintained at 30 � 1 8C temperature.
The mobile phase was a mixture of 35:65 (v/v) acetonitrile–0.015 M disodium
hydrogen phosphate buffer and 0.1% TEA. The pH of mobile phase was
adjusted at 7.4 using phosphoric acid 85% without prior dilution and the flow
rate was maintained at 1.0 mL/min, resulting in a pressure of about 70 bar.
Mobile phase was degassed by vacuum through filtration, after mixing. The
same ratios of solvents in mobile phase were used to apply the samples in LC–
MS. Thus, a mixture of 35:65 (v/v) acetonitrile–0.02 M ammonium formate
with the addition of 0.05% formic acid was prepared using a flow rate of
0.3 mL/min in SIM positive mode. The [M+H]+ ions used to create the MS
spectra were m/z 461 for N-desmethylsildenafil and m/z 475 for sildenafil,
respectively. Daughter ion for sildenafil was further obtained by the fragmenta-
tion of m/z 475 (m/z 475! 461).
2.3.3. Sample extraction
In 10 mL conical glass tube, 0.5 mL of human blood and 2.0 mL of
saturated solution of disodium hydrogen phosphate buffer (pH 9.0) were added
and mixed for 10 s on a vortex. Each sample was extracted with 4.0 mL of
chloroform: isopropanol: n-heptane (25:10:65, v/v/v) on vortex for 2 min at
speed 4. The sample was centrifuged for 10 min at 3000 rpm. The upper
(organic) layer was then transferred into a 10 mL conical glass tube and
evaporated to dryness at room temperature under a gentle stream of nitrogen.
The residue was reconstituted in 100 mL of mobile phase and an aliquot of
50 mL was injected onto the HPLC system.
2.3.4. Precision and accuracy
Within-run and between-run precision and accuracy were determined by
extracting blood supplemented with sildenafil and N-desmethylsildenafil to 35,
200 and 450 ng/mL (n = 3). Between-run assays were performed in three
different days.
2.3.5. Recovery and stability
Extraction recovery and stability were calculated at two levels of spiked
blood samples (35 and 450 ng/mL). Extraction recovery was determined by
comparing the peak areas from extracted samples with those obtained from a
direct injection of the corresponding unextracted standards dissolved in mobile
phase (n = 3 in three different days). Stability was investigated also for stock
and working solution at 35 and 450 ng/mL.
2.3.6. Selectivity
Selectivity of the method was examined for 10 different blood sources and
eight other drugs (Table 4), including two more selective inhibitors of (cGMP)-
specific phosphodiesterase type V, tadalafil and vardenafil. All compounds were
assayed independently by injecting standard solutions at 250 ng/mL in the HPLC
system using the same chromatographic conditions as the proposed method.
2.3.7. Application in post-mortem blood samples
The method was further applied in two sudden-death cases of two middle-
aged married men who were found dead after an extramarital sexual intercourse,
and they were suspected due to some investigation findings of taking sildenafil
citrate tablets.
3. Results and discussion
3.1. Chromatographic conditions optimization
During the preliminary experiments, several combinations
of mobile phase composition and mobile phase pH were
investigated, in order to obtain the optimum separation of
sildenafil and N-desmethylsildenafil. Thus, the pH of mobile
phase at 3, 4, 4.5, 5, and 7.4, the acetonitrile percentage at 15,
25, 35, 45 and 55% and the buffer concentrations at 0.015,
0.05, 0.10, 0.15 and 0.2 M disodium hydrogen phosphate
buffer were examined, respectively. It was observed that pH
affected the retention times of the analytes and their
separation. Decreasing the pH value resulted in an over-
lapping between the analyte peaks (sildenafil, N-desmethyl-
sildenafil) and matrix interferences, which consequently led to
a separation failure. Although low pH values were sufficient
for the separation of sildenafil from endogenous compounds,
it seemed not to be for N-desmethylsildenafil. Therefore, pH
7.4 was chosen as the optimum value. In addition, as the
percentage of acetonitrile decreased, the retention times for
the drugs were longer and the separation of the analytes
were better until 35%. At higher values, retention time of
sildenafil was too short and metabolite’s peak overlapped
with endogenous compounds. Thus, 35% of acetonitrile was
chosen as the optimum percentage of organic modifier.
Disodium hydrogen phosphate buffer concentration in the
mobile phase also affected retention time of the analytes. It
was found that a 0.015 M buffer concentration gave the best
separation in the shortest time since at higher concentrations
of the buffer; the endogenous blood peaks, particularly for the
metabolite, affected the analyte peaks. Consequently, the best
separation of sildenafil and N-desmethylsildenafil on the ODS
column was achieved using a mobile phase of 35:65 (v/v)
acetonitrile–0.015 M Na2HPO4�2H2O, TEA 0.1% with pH
7.4, to obtain retention times of 19.23 and 10.85 min,
respectively (Fig. 3b).
3.2. Sample extraction optimization
For the extraction of the analytes, liquid–liquid extraction
was selected comparing to solid phase extraction in order to
allow the optimization of a simple and rapid method with less
cost that would be applicable in routine analysis. Thus, an
alkaline liquid–liquid extraction method by Sheu et al. [18]
was applied with no success. However the replacement of
sodium hydroxide with disodium hydrogen phosphate buffer
appeared to facilitate the separation of the drug from the
protein complex providing a significant response. The
extraction efficiency of four different solvents or solvent
mixtures [diethylether, ethylacetate, ether/dichloromethane
(3:2, v/v), chloroform/isopropanol/n-heptane (25:10:65, v/v/

Fig. 3. Representative HPLC–UV–vis chromatograms obtained from the analysis of (a) blank blood sample, (b) spiked sample at 50 ng/mL with sildenafil
(tr = 19.23 min) and N-desmethylsildenafil (tr = 10.85 min) and (c) post-mortem blood sample.
C. Pistos et al. / Forensic Science International 178 (2008) 192–198 195
v)] were further investigated. The first three solvents provided
high matrix noise that overlapped with analytes’s peaks. The
optimum solvent mixture was chloroform/isopropanol/n-
heptane (25:10:65, v/v/v) that yielded a satisfactory baseline
and higher sensitivity.
Retention times of N-desmethylsildenafil and sildenafil, were
10.85 and 19.23 min, respectively. Fig. 3a shows a chromato-
gram obtained from an extracted drug free blood, while Fig. 3b
shows a chromatogram of extracted drug free plasma supple-
mented with N-desmethylsildenafil and sildenafil at 50.0 ng/mL.

Table 2
Recovery data for sildenafil and N-desmethylsildenafil in human blood (n = 9)
Concentration added (ng/mL) % Recovery (�S.D.) (ng/mL) %R.S.D.
S
35 92.7 (�10.8) 11.6
450 92.6 (�1.8) 1.9
DS
35 86.4 (�2.6) 3.0
450 88.2 (�0.6) 0.6
Table 1
Intraday and interday accuracy and precision data for sildenafil (S) and N-desmethylsildenafil (DS) in spiked blood samples (3 days, n = 3)
Concentration added (ng/mL) S DS
Concentration found (ng/mL) %R.S.D. %Er Concentration found (ng/mL) %R.S.D. %Er
Intraday
35 35.0 � 1.7 4.8 0.1 31.9 � 0.4 1.1 �8.9
200 194.3 � 2.3 1.2 �2.9 178.6 � 2.6 1.5 �10.7
450 410.2 � 4.2 1.0 �8.8 502.8 � 5.0 1.0 11.7
Interday
35 35.2 � 1.8 5.1 0.5 30.9 � 0.4 1.1 �11.6
200 193.4 � 2.1 1.1 �3.3 177.9 � 2.6 1.5 �11.1
450 409.3 � 4.1 1.0 �9.1 503.8 � 5.0 1.0 11.9
C. Pistos et al. / Forensic Science International 178 (2008) 192–198196
3.3. Linearity
The peak areas were linearly related to blood concentrations
of sildenafil and N-desmethylsildenafil, respectively, from 10 to
at least 500 ng/mL. The average regression equation and slope of
five calibration curves of sildenafil in blood, prepared in five
different days over a period of 2 months is Ss = 0.0076
Cs + 0.0028, standard error (Sr) < 0.11, n = 5, r = 0.997. Ss,
corresponds to peak area of sildenafil while Cs, corresponds to
blood concentration of sildenafil (ng/mL). The correlation
coefficients square for each standard curve constructed
invariably exceeded 0.9991 and intercepts were all close to
zero and not statistically significant. %R.S.D. of the slopes
between the different calibration curves for sildenafil was 4.3.
The average regression equation and slope of five calibration
curves of N-desmethylsildenafil in blood, prepared in five
different days over a period of 2 months is SDS = 0.0081 �
Table 3
Stability data for sildenafil and N-desmethylsildenafil in human blood under vario
Storage conditions Time (%)Dev.a (35 ng/mL) R.S.D
S
Ambient temperature 6 h �7.3 6.1
6 8C 30 days �12.3 6.4
�20 8C 30 days �10.7 5.9
Freeze–thaw cycles; �20 8C 3 cycles �2.9 7.1
DS
Ambient temperature 6 h �3.7 6.4
6 8C 30 days �13.5 7.9
�20 8C 30 days �13.3 8.1
Freeze–thaw cycles; �20 8C 3 cycles �2.5 6.6
a Percentage of mean deviation from t = 0.b Percentage relative standard deviation, n = 3.
CDS � 0.0075, standard error (Sr) < 0.14, n = 5, r = 0.999. SDS,
corresponds to peak area of N-desmethylsildenafil and CDS,
corresponds to blood concentration of N-desmethylsildenafil
(ng/mL). The correlation coefficients for each standard curve
constructed invariably exceeded 0.9998 and intercepts were all
close to zero and not statistically significant. The lower limit of
detection was 5.0 ng/mL for sildenafil and N-desmethylsidenafil,
while the lower limit of quantification was 10.0 ng/mL for both
compounds based on S/N > 3 and S/N > 10, respectively.
%R.S.D. of the slopes between the different calibration curves
for N-desmethylsildenafil was 4.8.
3.4. Precision—accuracy
Interday precision and error in three different days were
found to be less than 5.1% and 9.1% for sildenafil, 1.5% and
11.9% for N-desmethylsildenafil, respectively (Table 1).
3.5. Recovery—stability
Extraction recovery and stability data, from blood samples
spiked with both analytes to 35 and 450 ng/mL are presented in
Tables 2 and 3, respectively.
3.6. Selectivity
Blank samples from 10 different sources and all eight drugs
did not yield any interference at the elution time for the
us storage conditions
. (%)b (35 ng/mL) (%)Dev.a (450 ng/mL) R.S.D. (%)b (450 ng/mL)
�3.9 8.4
�11.3 7.6
�9.4 5.3
�8.9 6.0
�4.9 5.4
�8.6 7.6
�7.2 7.5
�5.6 8.3

Table 4
Compounds studied for interferences (C = 250 ng/mL)
Compound Retention time (min)
Tadalafil 13.2
Vardenafil 22.1
Lorazepam 28.4
Temazepam 25.8
Diazepam 25.6
Caffeine 4.5
Barbital 8.4
Butobarbital 14.3
Fig. 4. Total ion current mass chromatogram obtained positive mode and
selected ion monitoring from the analysis of a post-mortem blood sample
(sildenafil, tr = 13.3 min, m/z 475, 461 and N-desmethylsildenafil, tr = 4.2 min,
m/z 461.
C. Pistos et al. / Forensic Science International 178 (2008) 192–198 197
detection of sildenafil and N-desmethylsildenafil. Table 4
presents the retention times of the studied drugs.
3.7. Application in post-mortem blood samples
No interfering peaks were observed in the two case samples
investigated, concerning sudden death of two middle-aged men.
Sildenafil and N-desmethylsildenafil blood concentrations were
50.1 and 72.0 ng/mL in the first case and 52.46 and 38.66 ng/
mL in the second case, respectively. Fig. 3c presents the
chromatogram of a post-mortem blood sample, concerning a
middle-aged man, analyzed for sildenafil and N-desmethylsil-
denafil. The presence of both analytes in these samples was
further confirmed using the same extraction method in LC–MS.
Fig. 4, shows a TIC chromatogram of the autopsy sample in
which N-desmethylsildenafil and sildenafil are eluted at 4.2 and
13.3 min, respectively. The comparison of the UV–vis and MS
chromatograms from the same autospy sample, illustrates the
reliability of UV–vis to detect both analytes but also its
disadvantage of low selectivity, a problem that mass spectro-
meter overcomes. MS chromatogram presents the absence of
any interference in the background of the baseline.
4. Conclusion
Most laboratories analyze sildenafil and its active metabolite
in human plasma and not in whole blood. The fact that the
identification of sildenafil and more particularly N-desmethyl-
sildenafil in blood using GC–MS is problematic has led the
analysts to the application of LC–MS or LC–MS/MS
instrumentation. However, the need to combine a single LC–
MS methodology with a supplementary technique is often. In
this case and when human blood is the only biological sample
available, or plasma and serum cannot be separated as in most
post-mortem cases, HPLC–UV–vis method is considered the
method of choice for the simultaneous determination of both
sildenafil and N-desmethylsildenafil. Therefore, a simple
method for the quantification of both compounds in human
blood is proposed that allows quantitating small amounts of the
analytes even after the intake of the lowest dose of 25 mg tablet
of sildenafil citrate using a high performance liquid chromato-
graphy UV–vis system.
Acknowledgment
We would like to thank Dr. Leandros Arvanitakis from Pfizer
Hellas A. E. for providing us the reference standards.
References
[1] I. Goldstein, T.F. Lue, H. Padma-Nathan, R.C. Rosen, W.D. Steers, P.A.
Wicker, Oral sildenafil in the treatment of erectile dysfunction. Sildenafil
Study Group, N. Engl. J. Med. 338 (1998) 1397–1404.
[2] R.B. Moreland, I. Goldstein, A. Traish, Sildenafil, a novel inhibitor of
phosphodiesterase type 5 in human corpus cavernosum smooth muscle
cells, Life Sci. 62 (1998) PL309–PL318.
[3] M. Boolell, M.J. Allen, S.A. Ballard, S. Gepi-Attee, G.J. Muirhead, A.M.
Naylor, I.H. Osterloh, C. Gingell, Sildenafil: an orally active type 5 cyclic
GMP-specific phosphodiesterase inhibitor for the treatment of penile
erectile dysfunction, Int. J. Impot. Res. 8 (1996) 47–52.
[4] C.G. Stief, S. Uckert, A.J. Becker, M.C. Truss, U. Jonas, The effect of the
specific phosphodiesterase (PDE) inhibitors on human and rabbit caver-
nous tissue in vitro and in vivo, J. Urol. 159 (1998) 1390–1393.
[5] Viagra/Clinical Pharmacology/Pharmacokinetics and Metabolism, http://
www.rxlist.com/cgi/generic/viagra.htm, May 2007.
[6] M. Lee, D. Min, Determination of sildenafil citrate in plasma by high-
performance liquid chromatography and a case for the potential interac-
tion of grapefruit juice with sildenafil citrate, Ther. Drug Monit. 23 (1)
(2001) 21–26.
[7] M. Thau Sheu, A.B. Wu, Y.G. Cheng, A. Hsia, H. Ho, Development of a
liquid chromatographic method for bioanalytical applications with silde-
nafil, J. Chromatogr. B 791 (2003) 255–262.
[8] Y. Wang, J. Wang, Y. Cui, J.P. Fawcett, J. Gu, Liquid chromatographic-
tandem mass spectrometric method for the quantitation of sildenafil in
human plasma, J. Chromatogr. B 828 (2005) 118–121.
[9] A. Tracqui, B. Ludes, HPLC-MS for the determination of sildenafil citrate
(Viagra) in biological fluids. Application to the salivary excretion of
sildenafil after oral intake, J. Anal. Toxicol. 27 (2003) 88–94.
[10] J.R. Lewis, R.D. Johnson, L.C. Blank, Quantitative determination of silde-
nafil (Viagra) and its metabolite (UK-103,320) in fluid and tissue specimens
obtained from six aviation fatalities, J. Anal. Tox. 30 (2006) 14–20.
[11] W. Weinmann, M. Bohnert, A. Wiedemann, M. Renz, N. Lehmann, S.
Pollak, Post-mortem detection and identification of sildenafil (Viagra) and
its metabolites by LC/MS and LC/MS/MS, Int. J. Legal Med. 114 (2001)
252–258.
[12] M. Al-Ghazawi, M. Tutunji, S. Aburuz, Simultaneous determination of
sildenafil and N-desmethyl sildenafil in human plasma by high-perfor-
mance liquid chromatography method using electrochemical detection
with application to a pharmacokinetic study, J. Pharm. Biomed. Anal. 43
(2007) 613–618.
[13] J.D. Cooper, D.C. Muirhead, J.E. Taylor, P.R. Baker, Development of an
assay for the simultaneous determination of sildenafil (Viagra) and its
metabolite (UK-103,320) using automated sequential trace enrichment of
dialysates and high-performance liquid chromatography, J. Chromatogr. B
701 (1997) 87–95.

C. Pistos et al. / Forensic Science International 178 (2008) 192–198198
[14] K. Saisho, K.S. Scott, S. Morimoto, Y. Nakahara, Hair analysis for
pharmaceutical drugs. II. Effective extraction and determination of silde-
nafil (Viagra) and its N-desmethyl metabolite in rat and human hair by
GC–MS, Biol. Pharm. Bull. 24 (12) (2001) 1384–1388.
[15] S. Pagani, D. Mirtella, R. Mencarelli, D. Rodriquez, M. Cingolani,
Postmortem distribution of sildenafil in histological material, J. Anal.
Toxicol. 29 (2005) 254–257.
[16] P. Van Hee, H. Neels, W. Lambert, V. Coucke, M. Dedoncker, Comment
on: J. Anal. Toxicol. 29 (2005) 254–257: Postmortem distribution of
sildenafil in histological material, J. Anal. Toxicol. 30 (2006) 403–404;
author reply 404–405.
[17] Official Journal of the European Communities L221, 8-36. Commission
decision (2002/657/EC) of 12 August 2002 concerning the performance of
analytical methods and the interpretation of results. Brussels, Belgium,
2002.
[18] M.T. Sheu, A.B. Wu, G.C. Yeh, A. Hsia, H. Ho, Development of a liquid
chromatographic method for bioanalytical applications with sildenafil, J.
Chromatogr. B 791 (2003) 255–262.
Top Related
Copyright © 2022 FDOKUMEN

