







Bahasa
Halaman
Hukum


MolecularBioSystems
This article was published as part of the
Computational and Systems Biology themed issue
Please take a look at the full table of contents to access the other papers in this issue.
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online / Journal Homepage / Table of Contents for this issue

Molecular modeling and docking studies of human 5-hydroxytryptamine
2A (5-HT2A) receptor for the identification of hotspots for ligand
bindingwzKaruppiah Kanagarajadurai,y Manoharan Malini,y Aditi Bhattacharya,Mitradas M. Panicker and Ramanathan Sowdhamini*
Received 31st March 2009, Accepted 12th August 2009
First published as an Advance Article on the web 8th September 2009
DOI: 10.1039/b906391a
The serotonergic system has been implicated in emotional and cognitive function. In particular,
5-HT2A (5-hydroxytrytamine receptor 2A) is attributed to a number of disorders like
schizophrenia, depression, eating disorders and anxiety. 5-HT2A, being a GPCR (G-protein
coupled receptor), is important in the pharmaceutical industry as a proven target for these
disorders. Despite their extensive clinical importance, the structural studies of this protein is
lacking due to difficulties in determining its crystal structure.
We have performed sequence analysis and molecular modeling of 5-HT2A that has revealed a set of
conserved residues and motifs considered to play an important role in maintaining structural
integrity and function of the receptor. The analysis also revealed a set of residues specific to the
receptor which distinguishes them from other members of the subclass and their orthologs. Further,
starting from the model structure of human 5-HT2A receptor, docking studies were attempted to
envisage how it might interact with eight of its ligands (such as serotonin, dopamine, DOI, LSD,
haloperidol, ketanserin, risperidone and clozapine). The binding studies of dopamine to 5-HT2A
receptor can bring up better understanding in the etiology of a number of neurological disorders
involving both these two receptors. Our sequence analysis and study of interactions of this receptor
with other ligands reveal additional residue hotspots such as Asn 363 and Tyr 370. The function of
these residues can be further analyzed by rational design of site-directed mutagenesis. Two distinct
binding sites are identified which could play important roles in ligand binding and signaling.
Introduction
G-protein coupled receptors (GPCRs) and their downstream
signaling partners constitute one of the largest class of molecular
targets contributing to any human disease. This fact is
emphasized in the central nervous system, wherein GCPRs that
bind neurotransmitters, apart from ion channels, are the key signal
transducers and are involved in almost every aspect of neural
function. Perhaps, the most extensive of all neurotransmitter-
mediated GPCR is the serotonergic system (serotonin or
5-hyroxytryptamine and its cognate receptors), which has been
implicated in the etiology of many mental disorders such as
depression, anxiety, schizophrenia, eating disorders, obsessive
compulsive disorder (OCD), migraine and panic disorder.
GPCRs have been exhaustively characterized in terms of
their pharmacology and downstream signaling cascades.
However, structural information about these integral
membrane proteins is still elusive. The GPCR transmembrane
systems in general represent a most challenging task for
structure determination, as their ‘greasy’ hydrophobic surfaces
do not readily make the regular intermolecular contacts
required for crystal formation. Despite these technical
challenges, a few GPCR crystal structures have been solved
after the initial bovine rhodopsin structure,1 two adrenergic
receptor structures,2–5 and most recently the adenosine 2A
structure.6 These have been crystallized as engineered or
bound receptors to limit their flexibility while retaining
functionality. Due to the extensive biomedical importance of
GPCRs, there have been attempts to model7–9 the three-
dimensional structure of these receptors, like angiotensin
receptors,10 vasopressin receptor,11 5-HT2C receptor,12 and
of their interactions with their ligands, for instance.
During the past 20 years or so, multiple 5-HT receptor
subtypes have been characterized ranging from 5-HT1 to
5-HT7.13 In particular, the 5-HT2 receptor family currently
comprises of three receptor subtypes, 5-HT2A, 5-HT2B and
5-HT2C receptors, which are similar in terms of their molecular
structure, pharmacology and signal transduction pathways.
The amino acid sequences of the 5-HT2 receptor family have a
high degree of homology within the seven transmembrane
domains but they are structurally distinct from other 5-HT
receptors.14 Of all the 5-HT receptor subtypes found in cortex,
National Centre for Biological Sciences (TIFR), GKVK Campus,Bellary Road, Bangalore-560065, India. E-mail: [email protected],[email protected], [email protected], [email protected],[email protected] This article is part of a Molecular BioSystems themed issue onComputational and Systems Biology.z Electronic supplementary information (ESI) available: Multiplesequence alignments, Ramachandran plot, structural alignment onconserved residues and LIGPLOT representations. See DOI:10.1039/b906391ay Both have equally contributed to this work.
This journal is �c The Royal Society of Chemistry 2009 Mol. BioSyst., 2009, 5, 1877–1888 | 1877
PAPER www.rsc.org/molecularbiosystems | Molecular BioSystems
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online

the G-protein coupled 5-HT2A receptor has received extensive
attention in both physiological and pharmacological
experiments.15 This particular receptor has been found to bind
to a number of agonists, antagonists and inverse agonists
which in turn regulate the signaling via G-proteins. It has also
been shown that dopamine, considered a separate neuro-
transmitter with its own set of cognate GPCRs, can interact
with and activate the receptor initiating other effector path-
ways than the normal ones carried out by serotonin.16 In this
particular study, we have carried out docking of 5-HT2A
receptor with eight of its ligands, which are classified under
four categories, such as endogenous ligands (serotonin and
dopamine), synthetic agonists (DOI and LSD), antagonists
(haloperidol and ketanserin) and inverse agonists (clozapine
and risperidone). Two of the most used synthetic agonists to
the 5-HT2 receptor sub-family are DOI (4-iodo-2,5-dimethoxy-
phenylisopropylamine) and LSD (lysergic acid diethylamide),
a semi-synthetic psychedelic drug from the ergoline family.
Both are known to have hallucinogenic properties. In
addition, many commonly prescribed anti-psychotic drugs
serve as antagonists or inverse agonists to this receptor class.
The two antagonists used in this study include Haloperidol
4-[4-(4-chlorophenyl)-4-hydroxy-1-piperidyl]-1-(4-fluorophenyl)-
butan-1-one) an older typical antipsychotic used in the treatment
of schizophrenia and Ketanserin (3-{2-[4-(4-fluorobenzoyl)-
piperidin-1-yl]ethyl}quinazoline-2,4(1H,3H)-dione) an anti-
hypersensitive drug which binds with highest affinity to the
5-HT2A receptor and also used as a specific radioactive ligand
of the receptor in binding studies. Two other ligands which we
have used are the inverse agonists (also called as ‘atypical
antagonists’) of the receptor Clozapine 8-chloro-11-(4-methyl-
1-piperazinyl)-5H-dibenzo(b,e)(1,4)diazepine which is atypical
antipsychotic, most effective in the treatment of schizophrenia
and Risperidone 4-[2-[4-(6-fluorobenzo[d]2,4isoxazol-3-yl)-1-
piperidyl]ethyl]-3-methyl-2,6-diazabicyclo[4.4.0]deca-1,3-
dien-5-one which is also an atypical antipsychotic drug used in
the treatment of schizophrenia and bipolar disorder in young
people.
Some functionally important residues, like D120 and D155,
were found to be important for the amine family of receptors
and are reported to be involved in binding of serotonin as well
as other ligands to its receptor 5-HT2A.17 Likewise, two
conserved tryptophans across the family (W336 and W367)
have been predicted to be involved in the binding of the
agonists of 5-HT2A.18–22 Mutation of conserved D155 was
reported to have lower affinity for serotonin, DOI, ketanserin,
mianserin, and spiperone, but not for LSD.17–21,23–25
It was initially proposed that at least one additional residue
in TM3 (S159) helps to anchor the charged terminal amine
moiety of serotonin and related ligands. Mutation of this Ser
(S159A) residue was reported to cause a 17.6-fold decrease
(1.2 kcal mol�1) in the receptor affinity to serotonin and
smaller changes in the affinities of N,N-9-dimethyl-5-HT
(bufotenine, 4-fold, 0.6 kcal mol�1) and LSD (1.4-fold,
0.15 kcal mol�1).25 It was also reported that the mutation of
Ser159 produces a 8–20 fold decrease in the binding of 5-HT,
5-MeOT, 4-HT.26 Detrimental effects on affinity, potency
and intrinsic activity were observed with Phe 339 mutant for
N-benzyl analogs, whereas N-substituted phenylalkylamines
and traditional agonists were only weakly affected and Phe
340 mutant had similar effects on almost all ligands.27 From
modelling studies, Ser 239 was found to act as a hydrogen
bond donor and Ser 242 acts as a hydrogen bond acceptor.
Ser 239 was predicted to engage oxygen substituents at either
4- or 5-posistion of tryptamine ligands and position-5 of
phenylalkyklamine ligands. Ser 242 seems to be engaged and
important for binding of polar ring substituted tryptamine
ligands.27 These serine residues, however, found to have little
effect in intrinsic activity.
Two independently derived molecular models have predicted
that a highly conserved tyrosine (Y370, TM7) has important,
but mechanistically distinct, roles in agonist binding.19 Tyr 370
has been proposed to have a direct role for DOM binding,23
and it was also suggested that Y370 actually stabilizes the
negative charge from Asp155.19 A mutation of Y370 (Y370A)
yielded a receptor with greatly diminished affinity for serotonin
and DOM (2,5-dimethoxy-4-methylamphetamine), but not for
a-methyl-5-HT or bufotenine.
Reports relating to intracellular signaling motifs present in
the 5-HT2A receptor are extensive and can indeed be classified
as an ‘interactome’.28 Relatively fewer attempts are reported
on modeling and docking of ligands with the extracellular
N-terminal domain. It may be surmised that given the wide
range of compounds that the receptor interacts with during
displacement studies, the ligand binding pocket might indeed
be dynamic and flexible. A number of mutagenesis studies
have been carried out, as discussed above, with respect to
binding site and the signaling of the 5-HT2A receptor.17–21,24,25
5-HT2A receptor is largely homologous and conserved across
several species. Still, a number of reports indicate that the
existence of small amino acid changes between isoforms are
responsible for significant changes in the function of the
receptor29 underlying the importance of detailed sequence
comparisons. In addition, modelling of human 5-HT2A receptor
and docking of known ligands were attempted due to its
important role implicated in a host of diseases, where future
drug design will be worthwhile. This article describes our
current examination of the sequence alignments and the
docked complexes of the 5-HT2A receptor in the process
of identification of structurally and functionally important
residues which would be further studied extensively by
mutagenesis in terms of ligand interaction, downstream
signaling and trafficking.
Results and discussion
Sequence analysis
Conservation of residues along the hierarchy of the GPCR
family with respect to 5-HT2A receptor was analyzed through
sequence comparison. The sequence alignments were performed
on human amine GPCR sequences (Fig. S1), human 5-HT
receptor sequences (Fig. S2) and 5-HT2A orthologous receptor
sequences (Fig. S3), which revealed a set of conserved residues
(Table 1) and motifs (Table 2). We have also compared the
human 5-HT2A sequence against the rat 5-HT2A sequence
(Fig. S4), and other rodent 5-HT2A sequences (Fig. S5).
1878 | Mol. BioSyst., 2009, 5, 1877–1888 This journal is �c The Royal Society of Chemistry 2009
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online
Similarly, human 5-HT2A sequence was compared with human
5-HT2B and human 5-HT2C sequences (Fig. S6).z
Human amine GPCR sequence alignment. The human amine
GPCR sequence alignment (42 sequences) includes sequences
from dopamine receptors, histamine receptors, adrenergic
receptors, muscarinic acetylcholine receptor, trace amine
associated receptors and 5-HT receptors. Sequences from 5-HT3
subtypes were not considered in this alignment, since they do
not belong to the GPCR class. The alignment was analyzed for
conservation of DRY and NPXXY motifs, and other con-
served residues. The analysis revealed the conservation of
three Tyr residues found at TM regions and the loop regions
at the cytoplasmic end of the receptor. The first important Tyr
residue, which is the part of the DRY motif of TM3, was
found to be conserved in all the sequences except the sequences
of TAAR2, TAAR3, DRD4 and HRH3 receptors (in which the
residue Tyr was replaced by Phe). The second Tyr residue was
found at the ICL2 region, a few residues away from the
DR[Y/F] motif and found to be conserved in all the human
amine GPCRs except in 5-HT2 receptor subtypes (where His
(H183 of 5-HT2A) replaced Tyr in 5-HT2A and 5-HT2B and
Ala replaced Tyr in 5-HT2C). The third Tyr (Y254 of 5-HT2A)
residue was observed to be conserved at the end of TM5 in all
amine GPCR sequences except HRH3 and HRH4 (where it is
replaced by Asn). The residues involved in the formation of
‘ionic lock’ in the intracellular region, implicated in one of the
major conformational changes between the inactive and active
form of bovine rhodopsin, were also found in the 5-HT2A
receptor sequence.30 Arg (R135 of bovine rhodopsin,
corresponds to R173 of 5-HT2A, part of DRY motif at TM3)
and Tyr (Y223 of bovine rhodopsin, corresponds toY254 of
Table 1 Amino acid conservation along the human 5-HT class of receptor sequences, human amine GPCR family of receptors and human GPCRsuperfamily
Secondary structureregion
Conserved residues from various set of alignments
5-HT receptor sequencealignment
Amine GPCR sequencealignment
Whole GPCR superfamily(literature reported)
TM1 N(92) Conserved ConservedTM2 N(110) — —
S(115) — —D(120) Conserved ConservedV(127) — —P(129) — —
ECL1 W(141) Conserved —TM3 C(148) Conserved —
D(155) Conserved —S(162) Conserved —I(163) — —I(169) — —D(172) Conserved —R(173) Conserved ConservedY(174) — —I(177) — —
TM4 W(200) Conserved ConservedTM5 F(243) Conserved —
P(246) Conserved ConservedY(254) — —
TM6 F(332) — —W(336) Conserved —P(338) Conserved ConservedF(339) — —F(340) — —
TM7 W(367) Conserved —G(369) — —Y(370) — —S(373) Conserved —N(376) Conserved —P(377) Conserved ConservedY(380) — —
Table 2 Conserved motifs obtained from various set of multiple sequence alignments
Structural region Reported GPCR motifs
Observed motifs in sequences
Human Amine GPCR Human 5-HT GPCR
TM2 — — V[M/L]PTM3 [D/E]RY DR[Y/F] TXSI, DRYTM5 — — F[Y/F]XPTM6 — WXP[F/Y] WXPFFTM7 [ND]PX2Y WX2[Y/W] SX2NPX2 [Y/H] WXGY, SX2NPX2Y
This journal is �c The Royal Society of Chemistry 2009 Mol. BioSyst., 2009, 5, 1877–1888 | 1879
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online

5-HT2A residue at TM5.31,32 The motif, WXP[F/Y] at the sixth
TM helix was found to be conserved throughout the alignment
(please see Fig. S1z). A glutamate residue (E318 of 5-HT2A)
was found to be conserved at the beginning of the sixth TM
region in all human amine receptors, except in TAAR2,
TAAR3, HRH4 and 5-HT6R receptors (where residues Asp
and Ala replace this residue). Similarly a motif WXX[Y/W] at
the beginning of seventh TM helix was found to be conserved
throughout the alignment. Likewise an Asn residue positioned
just before SXXNPXX[Y/H] motif at TM7 was found to be
conserved in all human amine GPCR sequences except in
5-HT2 subclass receptors (where it was replaced by either
Ser in 5-HT2A and 5-HT2B or Cys in 5-HT2C) (Table 2).
The lowest and highest sequence identity between any two
sequences in this set was found to be between 19% and 80%,
respectively.
Human 5-HT receptor sequence alignment. 12 sequences
from human 5-HT1, 5-HT2, 5-HT4, 5-HT5, 5-HT6 and
5-HT7 receptors and their subtypes were referred to as human
5-HT receptor sequences. The conserved residues and motifs
found from this alignment are reported in Table 1 and Table 2,
respectively. The lowest and highest sequence identity between
any two sequences in this set was between 22% and 63%,
respectively.
Alignment of 5-HT2A ortholog sequences. Orthologs of
5-HT2A receptor sequences (14 sequences) were aligned to
identify amino acid exchanges (Fig. S3z). The lowest and
highest sequence identity between any two sequences in this
set was between 70% and 99.7% respectively. The major
differences among the ortholog sequences were found at the
N- and C-terminal ends.
On the whole, the residues observed to be conserved from
these alignments were found to be localized within the TM
region of the receptors with an exception of Trp 141 found in
the first extracellular loop (Table 1).
Comparison of human 5-HT2A sequence against rat 5-HT2A.
A close comparison of sequences between the human and rat
(Rattus norvegieus) 5-HT2A receptor was performed (Fig. S4z),owing to the fact that the rat is an extensively studied model
organism. The sequence identity between the human and rat
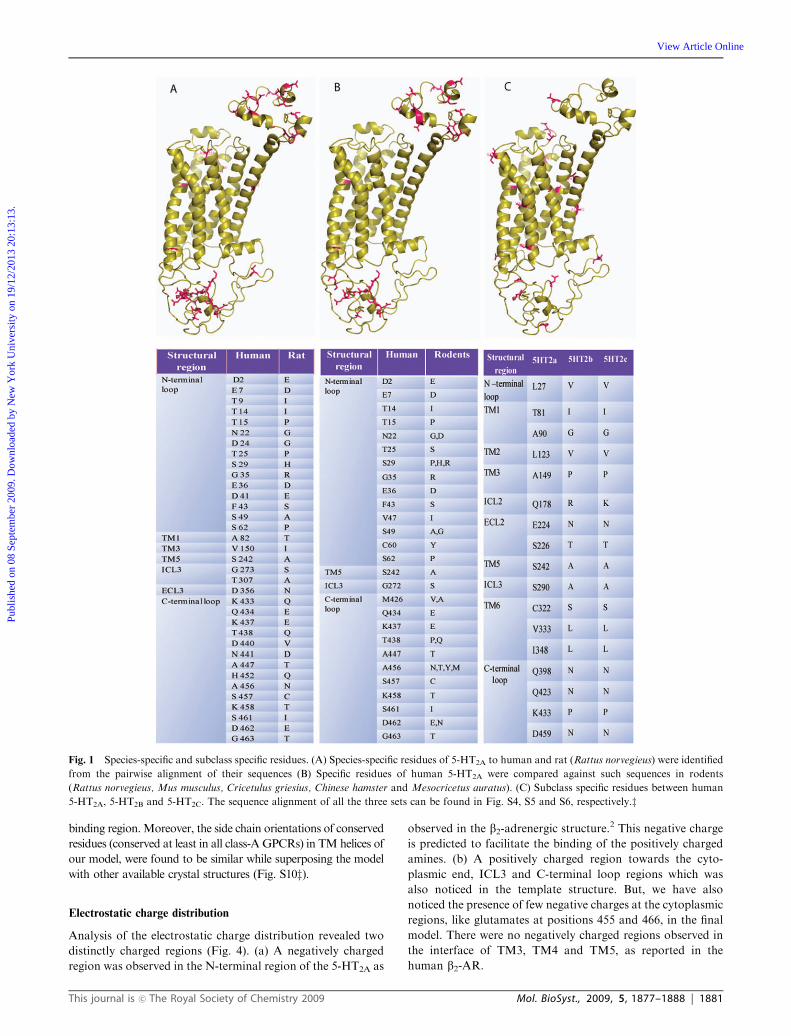
5-HT2A receptor is 91%. Species-specific residues (Fig. 1a)
were found to be distributed more in the loop regions (except
three residues, Ala 82, Val 150 and Ser 242, that were mapped
on TM1, TM4 and TM5, respectively). The substitution of
Ser 242 in the human 5-HT2A by alanine in the rat sequence,
had previously been shown to increase the affinity of the
receptor towards ergolines and tryptamines suggesting that
such changes could be pharmacologically important.33
Comparison of human 5-HT2A sequence against rodents 5-HT2A.
The 5-HT2A sequences of rodents such as Rattus norvegieus,
Mus musculus, Cricetulus griesius, Chinese hamster and
Mesocricetus auratus were compared against the human
5-HT2A (Fig. S5z). The sequence identity of human 5-HT2A
with any rodent 5-HT2A compared here lies between 90%
and 92%. The species-specific residues identified from this
alignment are reported in the Fig. 1b.
Comparison of human 5-HT2A sequence against human
5-HT2B and 5-HT2C sequences. Detailed comparison of amino
acid exchanges were also performed across the subclasses,
5-HT2A, 5-HT2B and 5-HT2C (Fig. S6z). The sequence identityof human 5-HT2A with human 5-HT2B and 5-HT2C is 43%
and 54% respectively suggesting that the human 5-HT2A is
closer to 5-HT2C than 5-HT2B. Subclass-specific residues were
found to be distributed across the transmembrane and loop
regions (Fig. 1c) suggesting that such exchanges could be
crucial in maintaining the specific function. Interestingly, one
residue amongst these was Ser 242 which is also specific to
Human 5-HT2A when compared to the rat 5-HT2A sequence.
The other residues stated in Fig. 1c may also play important
roles in function there by distinguishing the human 5-HT2A
from the other two members of its subclass.
Molecular modeling and validation
The human b2 Adrenergic receptor (b2-AR) crystal structure
was used as a template to model the human 5-HT2A receptor.
The human 5-HT2A receptor shares 34% sequence identity
with human b2-AR. The sequence alignment was analysed to
check the preservation of conserved residues throughout the
alignment and motifs, such as DR[Y/F] at the C-terminal of
TM3 and NPXXY at TM7 (Fig. 2). The sequence alignment
was compared with the alignment used for modelling by other
groups.34–37 and our alignment showed very few differences
(detailed comparison is addressed in the ESIz). The lengths ofthe TM helices were equivalent with the recently published
model by Bruno and co-workers,35 except that the TM5 of our
model has eight residues longer at the cytoplasmic end.
Cys residues at TM3 and ECL2, thought to be involved in
the formation of a disulfide bond, were conserved between the
template and the query. The sequence identity of individual
TM helices and loop regions can be obtained from
Supplementary Table 1.z TM helices were found to be highly
conserved between the two receptor sequences, but the loop
regions were more variable.
The model structure after loop building and energy
minimization through SYBYL (Fig. 3) was validated using
PROCHECK. The PROCHECK results for the model,
excluding the loop regions, shows more than 98% of the
residues are in allowed regions (92.4% in the strictly allowed
region and 6.4% in partially allowed region) (Fig. S7az). Thefull-length structure (model with loops) shows more than 96%
of the residues in the allowed region (strictly allowed was
84.7% and partially allowed region was 11.7%) of the
Ramachandran plot (Fig. S7b). The final model also retains
the disulfide bond formed between the Cys residues of TM3
and ELC2. Results of VERIFY3D38 showed that more than
90% residues are in allowed regions (Fig. S8z). There are fewresidues falling under negative regions of VERIFY3D plot
and were found to be in N-terminal (residue range: 18–20 and
60–80), ICL2 (residue range: 177–185) and ELC2 (residue
range: 351–356) loop regions. The observed variations with
both the PROCHECK and VERIFY3D in the disallowed and
negative regions were mainly due to the loop regions where
there was low sequence identity between the template and
query; however, not much variation was seen in the ligand
1880 | Mol. BioSyst., 2009, 5, 1877–1888 This journal is �c The Royal Society of Chemistry 2009
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online
binding region.Moreover, the side chain orientations of conserved
residues (conserved at least in all class-A GPCRs) in TM helices of
our model, were found to be similar while superposing the model
with other available crystal structures (Fig. S10z).
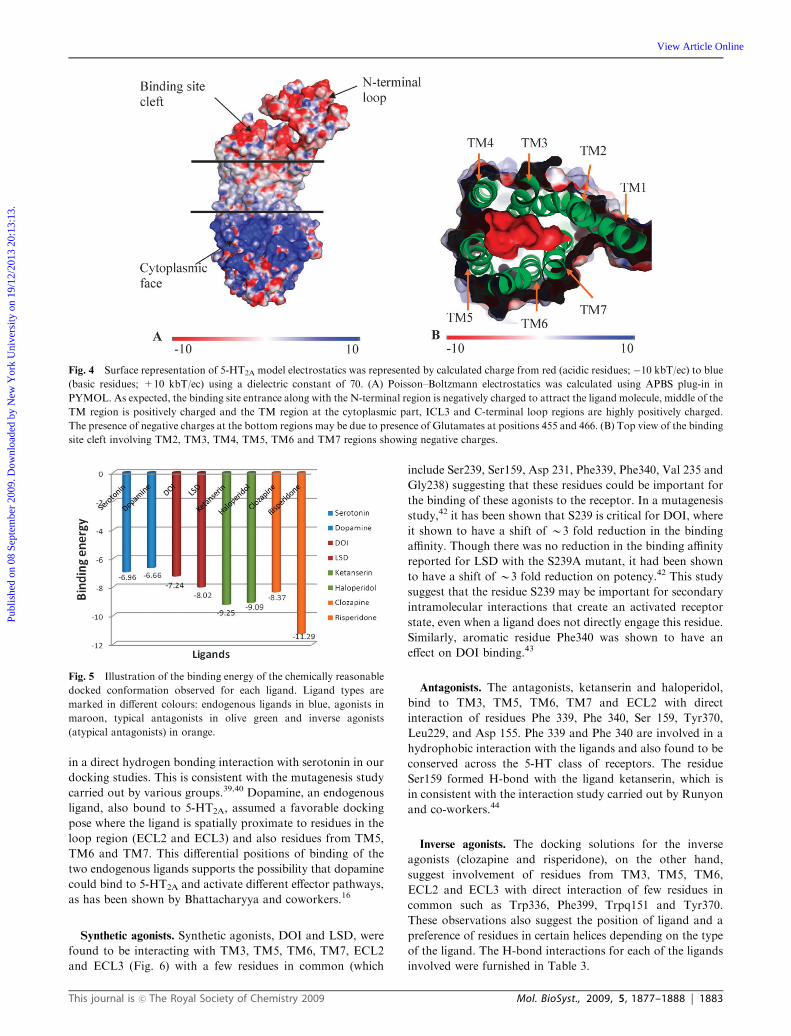
Electrostatic charge distribution
Analysis of the electrostatic charge distribution revealed two
distinctly charged regions (Fig. 4). (a) A negatively charged
region was observed in the N-terminal region of the 5-HT2A as
observed in the b2-adrenergic structure.2 This negative charge
is predicted to facilitate the binding of the positively charged
amines. (b) A positively charged region towards the cyto-
plasmic end, ICL3 and C-terminal loop regions which was
also noticed in the template structure. But, we have also
noticed the presence of few negative charges at the cytoplasmic
regions, like glutamates at positions 455 and 466, in the final
model. There were no negatively charged regions observed in
the interface of TM3, TM4 and TM5, as reported in the
human b2-AR.
Fig. 1 Species-specific and subclass specific residues. (A) Species-specific residues of 5-HT2A to human and rat (Rattus norvegieus) were identified
from the pairwise alignment of their sequences (B) Specific residues of human 5-HT2A were compared against such sequences in rodents
(Rattus norvegieus, Mus musculus, Cricetulus griesius, Chinese hamster and Mesocricetus auratus). (C) Subclass specific residues between human
5-HT2A, 5-HT2B and 5-HT2C. The sequence alignment of all the three sets can be found in Fig. S4, S5 and S6, respectively.z
This journal is �c The Royal Society of Chemistry 2009 Mol. BioSyst., 2009, 5, 1877–1888 | 1881
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online

Molecular docking
In order to identify residues potentially involved in ligand
binding and to analyze its functional importance, the receptor
was docked to various ligands which included two endogenous
ligands, two agonists, two antagonists and two inverse
agonists (atypical antipsychotics). The ligands were found to
dock with the receptor in various conformations and distinct
docking poses were realized after clustering of RMSD
structural deviation (2.0 A cutoff) each time from their initial
position. The number and binding energies of the generated
clusters (Fig. 5) varied with respect to the ligand. The extent of
energy differences of the various docking poses, measured as
standard deviation of the binding energy of the clusters were
not high (greater than 0.308 kcal mol�1). The highest standard
deviation was observed for haloperidol that also had the
highest number of clusters. Interestingly, DOI (the most
well-studied, synthetic, strong agonist for 5-HT2A) was found
to accommodate all the conformations into a single cluster.
Since the standard deviation of the binding energies of all the
ligands is within the standard deviation of the AutoDock force
field (2.5 kcal mol�1), the cluster which was more energetically
reasonable and encompassing a maximum number of
functionally important residues, already documented as
functional from the literature, was chosen for further analysis.
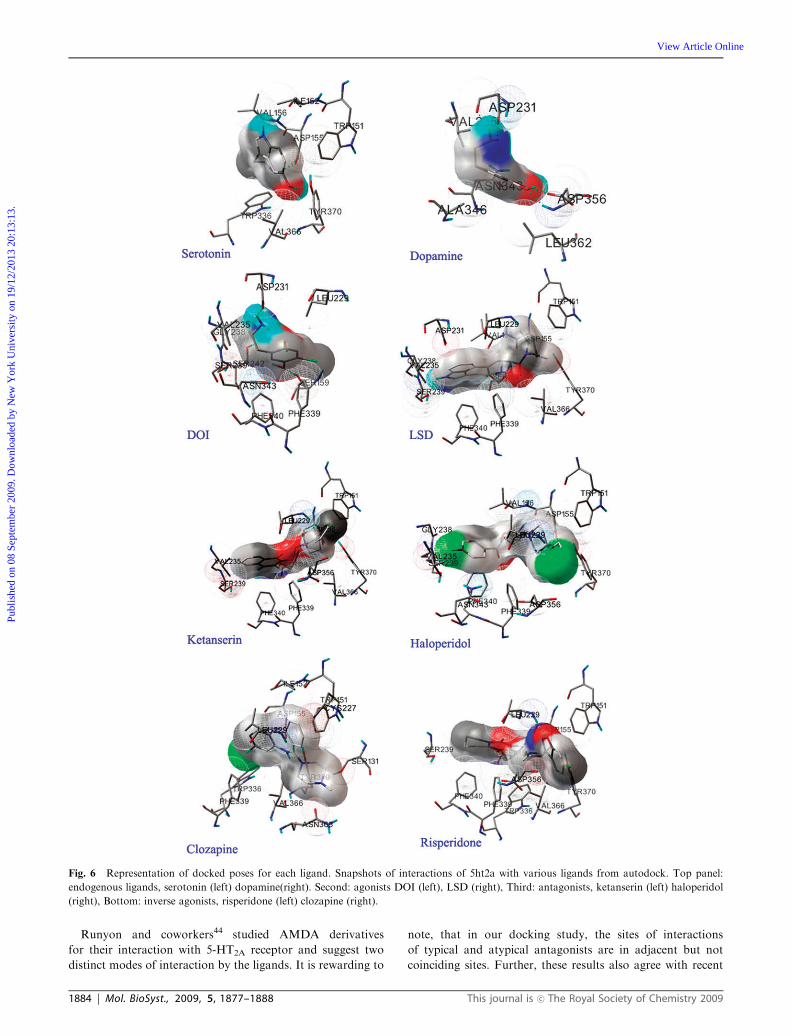
A number of residues reported to be involved in binding to
different ligands (Trp 151 in case of serotonin, Asp155 in case
of serotonin and ketanserin,39,40 Ser 159 in case of serotonin,25
Ser 207 in case of serotonin,41 Ser 239 in the case serotonin,
DOI and LSD,42 Phe 339 in case of serotonin and benzyl
analogs,42 Phe 340 in case of serotonin and other small classic
ligands,42,43 Trp 367 in case of all 5-HT2A ligands and Tyr 370
in case of serotonin, ketanserin and DOI) were observed close
to the docking poses (Fig. 6). The docking poses of these
ligands with their secondary structure is available in the
Fig. S11z). A few other residues were identified to interact
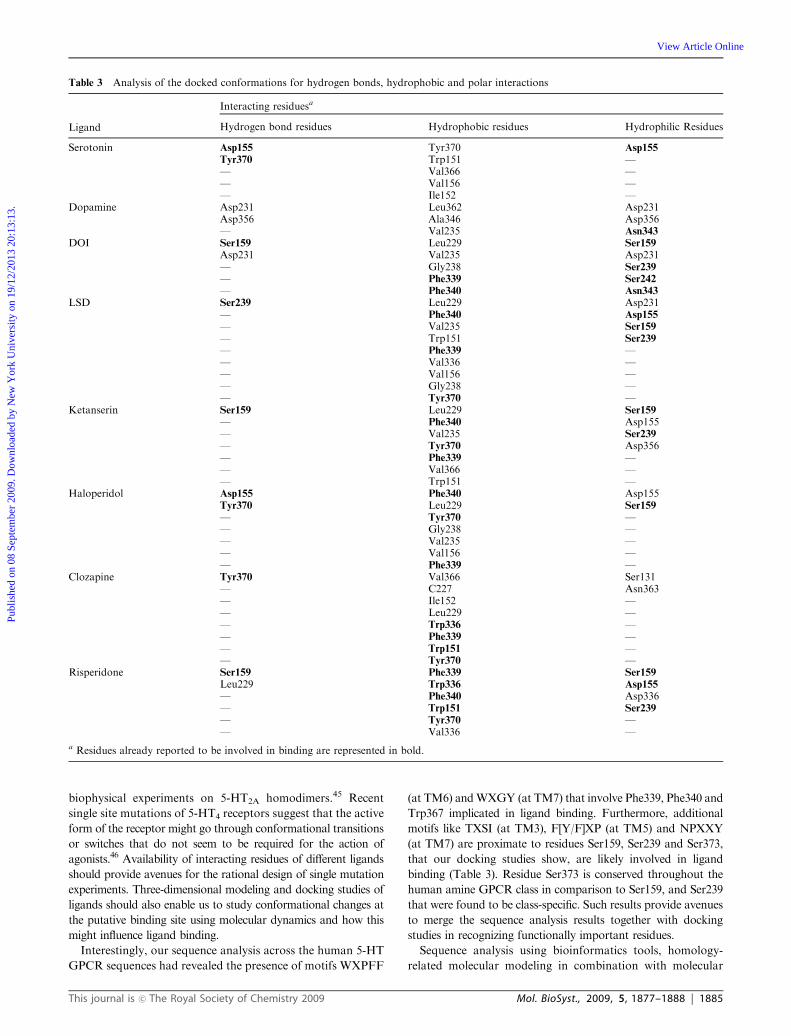
with the ligands from this study (Table 3).
Correlation of binding energy with experimentally determined
binding affinities. The binding energy obtained for each ligand
was found to correlate positively with the experimentally
determined binding affinities. The plots show a linear trend
with a correlation coefficient of �0.349 (Fig. S9) validating theapproach for docking of each of the ligands. Such correlation
coefficient values are accepted in the literature for training
docking algorithms and scoring schemes. Given the inherent
limitations in reproducing accurate docking poses from
computer algorithms and the heuristic approximations (such
as rigid ligand docking and the absence of lipids and solvents),
this correlation was merely to obtain confidence in our docking
protocol.
Endogenous ligands. In the case of endogenous ligands,
serotonin docked with slightly better binding energy
(�6.96 to �6.06) than dopamine (�6.66 to �5.76), as expected(data not shown). The synthetic agonists, antagonists and the
inverse agonists bind to the receptor with better binding
energy indicating better docking than the endogenous ligands.
The data therefore suggests them to be effective competitors
(Fig. 5).
The correlation of the residues in this study with the
literature-supported mutational and biochemical data also
validates the docking experiment. Serotonin binds to the
receptor forming hydrogen bonds with two residues Asp 155
and Tyr370 and is positioned within the transmembrane
region of TM3 and TM7. Tyr 370, which is already shown
to be conserved across the 5-HT class of receptors, is involved
Fig. 2 The final alignment used for modeling the 5-HT2A receptor.
The residues marked in green on each TM helix are conserved
throughout the GPCR family. The motifs (DRY and NPXXY),
conserved throughout the Class-A GPCR receptors, are covered by
green box. The motifs (TXSI, XFF, PFF and WXGY), covered by red
box, are conserved in all the 5-HT GPCR class except in 5-HT6
receptor. The dashed red line indicates the disulfide bridge formed
across the CYS residues in TM3 (C148) and ECL2 (C227).
Fig. 3 The energy minimized final structure of the 5-HT2A receptor.
TM helices are labeled for easy identification.
1882 | Mol. BioSyst., 2009, 5, 1877–1888 This journal is �c The Royal Society of Chemistry 2009
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online
in a direct hydrogen bonding interaction with serotonin in our
docking studies. This is consistent with the mutagenesis study
carried out by various groups.39,40 Dopamine, an endogenous
ligand, also bound to 5-HT2A, assumed a favorable docking
pose where the ligand is spatially proximate to residues in the
loop region (ECL2 and ECL3) and also residues from TM5,
TM6 and TM7. This differential positions of binding of the
two endogenous ligands supports the possibility that dopamine
could bind to 5-HT2A and activate different effector pathways,
as has been shown by Bhattacharyya and coworkers.16
Synthetic agonists. Synthetic agonists, DOI and LSD, were
found to be interacting with TM3, TM5, TM6, TM7, ECL2
and ECL3 (Fig. 6) with a few residues in common (which
include Ser239, Ser159, Asp 231, Phe339, Phe340, Val 235 and
Gly238) suggesting that these residues could be important for
the binding of these agonists to the receptor. In a mutagenesis
study,42 it has been shown that S239 is critical for DOI, where
it shown to have a shift of B3 fold reduction in the binding
affinity. Though there was no reduction in the binding affinity
reported for LSD with the S239A mutant, it had been shown
to have a shift of B3 fold reduction on potency.42 This study
suggest that the residue S239 may be important for secondary
intramolecular interactions that create an activated receptor
state, even when a ligand does not directly engage this residue.
Similarly, aromatic residue Phe340 was shown to have an
effect on DOI binding.43
Antagonists. The antagonists, ketanserin and haloperidol,
bind to TM3, TM5, TM6, TM7 and ECL2 with direct
interaction of residues Phe 339, Phe 340, Ser 159, Tyr370,
Leu229, and Asp 155. Phe 339 and Phe 340 are involved in a
hydrophobic interaction with the ligands and also found to be
conserved across the 5-HT class of receptors. The residue
Ser159 formed H-bond with the ligand ketanserin, which is
in consistent with the interaction study carried out by Runyon
and co-workers.44
Inverse agonists. The docking solutions for the inverse
agonists (clozapine and risperidone), on the other hand,
suggest involvement of residues from TM3, TM5, TM6,
ECL2 and ECL3 with direct interaction of few residues in
common such as Trp336, Phe399, Trpq151 and Tyr370.
These observations also suggest the position of ligand and a
preference of residues in certain helices depending on the type
of the ligand. The H-bond interactions for each of the ligands
involved were furnished in Table 3.
Fig. 4 Surface representation of 5-HT2A model electrostatics was represented by calculated charge from red (acidic residues; �10 kbT/ec) to blue
(basic residues; +10 kbT/ec) using a dielectric constant of 70. (A) Poisson–Boltzmann electrostatics was calculated using APBS plug-in in
PYMOL. As expected, the binding site entrance along with the N-terminal region is negatively charged to attract the ligand molecule, middle of the
TM region is positively charged and the TM region at the cytoplasmic part, ICL3 and C-terminal loop regions are highly positively charged.
The presence of negative charges at the bottom regions may be due to presence of Glutamates at positions 455 and 466. (B) Top view of the binding
site cleft involving TM2, TM3, TM4, TM5, TM6 and TM7 regions showing negative charges.
Fig. 5 Illustration of the binding energy of the chemically reasonable
docked conformation observed for each ligand. Ligand types are
marked in different colours: endogenous ligands in blue, agonists in
maroon, typical antagonists in olive green and inverse agonists
(atypical antagonists) in orange.
This journal is �c The Royal Society of Chemistry 2009 Mol. BioSyst., 2009, 5, 1877–1888 | 1883
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online
Runyon and coworkers44 studied AMDA derivatives
for their interaction with 5-HT2A receptor and suggest two
distinct modes of interaction by the ligands. It is rewarding to
note, that in our docking study, the sites of interactions
of typical and atypical antagonists are in adjacent but not
coinciding sites. Further, these results also agree with recent
Fig. 6 Representation of docked poses for each ligand. Snapshots of interactions of 5ht2a with various ligands from autodock. Top panel:
endogenous ligands, serotonin (left) dopamine(right). Second: agonists DOI (left), LSD (right), Third: antagonists, ketanserin (left) haloperidol
(right), Bottom: inverse agonists, risperidone (left) clozapine (right).
1884 | Mol. BioSyst., 2009, 5, 1877–1888 This journal is �c The Royal Society of Chemistry 2009
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online
biophysical experiments on 5-HT2A homodimers.45 Recent
single site mutations of 5-HT4 receptors suggest that the active
form of the receptor might go through conformational transitions
or switches that do not seem to be required for the action of
agonists.46 Availability of interacting residues of different ligands
should provide avenues for the rational design of single mutation
experiments. Three-dimensional modeling and docking studies of
ligands should also enable us to study conformational changes at
the putative binding site using molecular dynamics and how this
might influence ligand binding.
Interestingly, our sequence analysis across the human 5-HT
GPCR sequences had revealed the presence of motifs WXPFF
(at TM6) andWXGY (at TM7) that involve Phe339, Phe340 and
Trp367 implicated in ligand binding. Furthermore, additional
motifs like TXSI (at TM3), F[Y/F]XP (at TM5) and NPXXY
(at TM7) are proximate to residues Ser159, Ser239 and Ser373,
that our docking studies show, are likely involved in ligand
binding (Table 3). Residue Ser373 is conserved throughout the
human amine GPCR class in comparison to Ser159, and Ser239
that were found to be class-specific. Such results provide avenues
to merge the sequence analysis results together with docking
studies in recognizing functionally important residues.
Sequence analysis using bioinformatics tools, homology-
related molecular modeling in combination with molecular
Table 3 Analysis of the docked conformations for hydrogen bonds, hydrophobic and polar interactions
Ligand
Interacting residuesa
Hydrogen bond residues Hydrophobic residues Hydrophilic Residues
Serotonin Asp155 Tyr370 Asp155
Tyr370 Trp151 —— Val366 —— Val156 —— Ile152 —
Dopamine Asp231 Leu362 Asp231Asp356 Ala346 Asp356— Val235 Asn343
DOI Ser159 Leu229 Ser159
Asp231 Val235 Asp231— Gly238 Ser239
— Phe339 Ser242
— Phe340 Asn343
LSD Ser239 Leu229 Asp231— Phe340 Asp155
— Val235 Ser159
— Trp151 Ser239
— Phe339 —— Val336 —— Val156 —— Gly238 —— Tyr370 —
Ketanserin Ser159 Leu229 Ser159
— Phe340 Asp155— Val235 Ser239
— Tyr370 Asp356— Phe339 —— Val366 —— Trp151 —
Haloperidol Asp155 Phe340 Asp155Tyr370 Leu229 Ser159
— Tyr370 —— Gly238 —— Val235 —— Val156 —— Phe339 —
Clozapine Tyr370 Val366 Ser131— C227 Asn363— Ile152 —— Leu229 —— Trp336 —— Phe339 —— Trp151 —— Tyr370 —
Risperidone Ser159 Phe339 Ser159
Leu229 Trp336 Asp155
— Phe340 Asp336— Trp151 Ser239
— Tyr370 —— Val336 —
a Residues already reported to be involved in binding are represented in bold.
This journal is �c The Royal Society of Chemistry 2009 Mol. BioSyst., 2009, 5, 1877–1888 | 1885
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online

dynamics simulation methodologies can aid and often
complement the experimentally derived data. The structural
data from these approaches can not only be used for the
purpose of rationalizing structure-activity data on low-molecular
weight agonists and antagonists within the context of a
protein binding pocket, but also for a better understanding
of mutagenesis experiments, thus qualifying GPCR structural
models as useful to provide rationale for biochemical experiments.
Such approaches would serve as important and valid
communication platforms to establish functional links
between molecular biology, biophysics, bioinformatics and
organic chemistry in a highly efficient manner.
Methodology
Sequence analysis
Sequence alignments were performed on sets of sequences to
identify conserved residues and motifs, which could have
structural and or functional implications in the biological
systems. All the sequences used in the analysis were downloaded
from the SWISSPROT47 and NCBI (http://www.ncbi.nlm.nih.gov/)
database including the human 5-HT2A sequence (P28223). The
alignment of the entire human GPCR dataset which includes
1426 entries was obtained from the GPCRDB database48
(http://www.gpcr.org/7tm/). The sequence alignments of the
human amine GPCR sequences include 42 GPCR sequences
which belong to the amine family of receptors, the human
5-HT receptor sequences include the different subtypes of
the 5-HT class of receptors and 5-HT2A ortholog receptor
sequences includes 14 5-HT2A sequences from other organisms.
The 5-HT3 class of receptors was excluded from the alignment
of the 5-HT class as they are not characterized as GPCRs and
they belong to the ion channel family of receptors. We have
also compared human 5-HT2A receptor sequences against
sequences, of (ii) rat 5-HT2A. (ii) rodents 5-HT2A and (iii) human
5-HT2B and 5-HT2C, to identify species-specific and subclass-
specific residues. The alignments were constructed using
CLUSTALW49 (Version�1.83) and the alignments were
manually-edited using JALVIEW50 (Version 2.4) to retain
high equivalence of conserved regions. These alignments were
analyzed for the identification of amino acid conservation and
for the identification of class-specific residues and motifs.
Throughout the text, the residues are numbered according to
human 5-HT2A receptor sequence.
Molecular modeling and validation
The crystal structure of the human b2 adrenergic receptor2
(PDB ID: 2rh1) was used as a template for the construction of
the model since it was found to be a more suitable template in
comparison to bovine rhodopsin51 (PDB ID: 1u19) in terms of
sequence. Like 5-HT2A, the b2-AR is also an amine GPCR,
which is also more closely related in terms of sequence identity
than the bovine rhodopsin and other available crystal
structures. The human 5-HT2A (ID: P28223) and the human b2adrenergic receptor (P07550) sequences was aligned using
CLUSTALW49 (Version�1.83) and edited using JALVIEW50
(Version 2.4). The TM regions were predicted for the human
5-HT2A receptor sequence using transmembrane prediction
servers, such as TMHMM,52 SOSUI,53 and HMMTOP,54 to
guide and improve the alignment in the TM regions.
The structure of the template was obtained from RSCB55
(http://www.rcsb.org/pdb). The coordinates corresponding to
1–28, 231–262, 343–365 segments were not available in the
template crystal structure due to poor electron density and
hence these residues were removed from the query sequence
before the alignment. The final alignment was used to
construct the model using the software MODELLER56
(Version 9.1). A set of 100 structures were generated, from
which the low energy structure was used for further processes.
The initial low energy model obtained from MODELLER
was validated by using PROCHECK57 server. The structure
was further energy minimized using the SYBYL software
package (Version 7.1) (Tripos Associates Inc.). Tripos force
field, using 500 iterations of steepest descent and 100 iterations
of conjugate gradient, with a distance dependent dielectric
constant equal to 1 and a non-bonded interaction cutoff
value of 8 and was terminated at a convergence of
0.05 kcal mol A�1. The energy minimized structure was further
utilized to build the N-terminal (1–70 residues), C-terminal
(396–471 residues) and the ICL3 (265–316 residues) regions
using pre-installed PRODAT database tool in SYBYL and
the structure was again energy minimized using the same
parameters successively after the construction of every loop
region. The final structure was validated again using the
PROCHECK57 and VERIFY3D38 server. We have also
analyzed the side chain orientation of conserved residues
(which are conserved at least in all class-A GPCR sequences),
by superposing our model against the available crystal
structures (bovine rhodopsin, turkey b1-AR, human b2-AR
and human A2A receptor) to analyze how good our model was
coinciding with other structures.
Electrostatic charge distribution
The Adaptive Poisson-Boltzmann Solver (APBS)61 program
was employed to calculate the electrostatic charge distribution
of 5-HT2A model structure. The consideration of a membrane
environment has been indirectly included by reducing the
dielectric constant to 70, as used in the b2 adrenergic structure2
to calculate the charges. The electrostatics were mapped on
the model using the molecular surface representation and was
compared with the electrostatic charge distribution of the
crystal structure of human b2-AR.
Molecular docking
Docking was performed using the program Autodock4.058
with the Lamarkian genetic algorithm to dock eight different
ligands which included two endogenous ligands (serotonin and
dopamine), two agonists (DOI59 and LSD25), two antagonists
(ketanserin60 and haloperidol62) and two inverse agonists
(clozapine62 and risperidone63). The binding cavity was
defined to cover the entire transmembrane region as a grid
of size 78*70*70 and a spacing of 0.375. The docking was
carried out, with flexible ligand, for 100 GA runs with a
population size of 150 with 25*105 evaluations and a maximum
of 27 000 generations. The conformations were analyzed based
on the generated clusters using the MGL Autodock tools
1886 | Mol. BioSyst., 2009, 5, 1877–1888 This journal is �c The Royal Society of Chemistry 2009
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online

(Version 1.5.2). The 3D structures of the ligands were
generated using the molecular design suite (MDS; VLife
Sciences Pvt. Technologies Ltd.) with their initial coordinates
from the PUBCHEM database. The correlation of binding
energy of the ligands with their experimental binding affinities,
obtained from the literature (as mentioned above), was studied
using scatter plot.
Acronyms
5-HT2A 5-hydroxytrytamine receptor 2A
GPCRs G-protein coupled receptors
DOI 4-iodo-2,5-dimethoxyphenylisopropylamine
DOM 2,5-Dimethoxy-4-methylamphetamine
ECL Extracellular loop
ICL Intracellular loop
LSD Lysergic acid diethylamide
TM Transmembrane region
Acknowledgements
R.S. was a Senior Research Fellow of Wellcome Trust, U.K.
K.K. is supported by University Grant Commission, India.
A.B. and M.M.P. would like to thank Department of
Biotechnology, India for financial support. M.M. is supported
by Career Enhancement Award of R.S. by Department of
Biotechnology, India. Both M.M.P. and R.S. thank NCBS
(TIFR) for financial and infrastructural support.
References
1 K. Palczewski, T. Kumasaka, T. Hori, C. A. Behnke,H. Motoshima, B. A. Fox, I. L. Trong, D. C. Teller, T. Okada,R. E. Stenkamp, M. Yamamoto and M. Miyano, Science, 2000,289, 739–745.
2 V. Cherezov, D. M. Rosenbaum, M. A. Hanson, S. G. F.Rasmussen, T. S. Foon, T. S. Kobilka, H.-J. Choi andR. C. Stevens, et al., Science, 2007, 318, 1258–1265.
3 S. G. F. Rasmussen, H.-J. Choi, D. M. Rosenbaum, T. S. Kobilka,F. S. Thian, P. C. Edwards, M. Burghammer and B. K. Kobilka,et al., Nature, 2007, 450, 383–387.
4 D. M. Rosenbaum, V. Cherezov, M. A. Hanson, S. G. Rasmussen,F. S. Thian, T. S. Kobilka, H. J. Choi, P. Kuhn, W. I. Weis,B. K. Kobilka and R. C. Stevens, Science, 2007, 318, 1258–65.
5 T. Warne, M. J. Serrano-Vega, J. G. Baker, R. Moukhametzianov,P. C. Edwards, R. Henderson, A. G. Leslie, C. G. Tate andG. F. Schertler, Nature, 2008, 454, 486–91.
6 V. P. Jaakola, M. T. Griffith, M. A. Hanson, V. Cherezov,E. Y. Chien, J. R. Lane, A. P. Ijzerman and R. C. Stevens, Science,2008, 322, 1211–7.
7 A. S. van Neuren, G. Muller, G. Klebe and L. Moroder, J. Recept.Signal Transduction, 1999, 19, 341–53.
8 E. Archer, B. Maigret, C. Escrieut, L. Pradayrol and D. Fourmy,Trends Pharmacol. Sci., 2003, 24, 36–40.
9 S. Dastmalchi, W. B. Church andM. B.Morris, BMCBioinformatics,2008, 9, S14.
10 T. Tuccinardi and A. Martinelli, Curr. Med. Chem., 2007, 14,3105–21.
11 C. Czaplewski, R. Kazmierkiewicz and J. Ciarkowski, J. Comput.-AidedMol. Des., 1998, 12, 275–87.
12 A. Farce, S. Dilly, S. Yous, P. Berthelot and P. Chavatte,J. Enzyme Inhib. Med. Chem., 2006, 21, 285–92.
13 D. Hoyer, D. E. Clarke, J. R. Fozard, P. R. Hartig, G. R. Martin,E. J. Mylecharane, P. R. Saxena and P. P. Humphrey, PharmacolRev., 1994, 46, 157–203.
14 G. Baxter, G. Kennett, F. Blaney and T. Blackburn, TrendsPharmacol. Sci., 1995, 16, 105–110.
15 V. Williams, G. R. Srinivas and S. G. Patrica, Journal ofNeuroscience, 2002, 22, 2843–2854.
16 S. Bhattacharyya, I. Raote, A. Bhattacharya, R. Miledi andM. M. Panicker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103,15428–15253.
17 C. D. Wang, T. K. Gallaher and J. C. Shih,Mol. Pharmacol., 1993,43, 931–940.
18 M. F. Hibert, S. Trumpp-Kallmeyer, A. Bruinvels and J. Hoflack,Mol. Pharmacol., 1991, 40, 8–15.
19 R. B. Westkaemper and R. A. Glennon, Pharmacol. Biochem.Behav., 1991, 40, 1019–1031.
20 O. Edvardsen, I. Sylte and S. G. Dahl, Brain Res. Mol. Brain Res.,1992, 14, 166–78.
21 S. Trumpp-Kallmeyer, J. Hoflack, A. Bruinvels and M. Hibert,J. Med. Chem., 1992, 35, 3448–3462.
22 H. D. Holtje and U. K. Jendretzki, Arch. Pharm., 1995, 328,577–584.
23 S. C. Sealfon, L. Chi, B. J. Ebersole, V. Rodic, D. Zhang,J. A. Ballesteros and H. Weinstein, J. Biol. Chem., 1995, 270,16683–8.
24 M. S. Choudhary, N. Sachs, A. Uluer, R. A. Glennon,R. B. Westkaemper and B. L. Roth, Mol. Pharmacol., 1995, 47,450–457.
25 N. Almaula, B. J. Ebersole, D. Zhang, H. Weinstein andS. C. Sealfon, J. Biol. Chem., 1996, 271, 14672–14675.
26 B. J. Ebersole and S. C. Sealfon, Methods Enzymol., 2002, 343,123–136.
27 M. R. Braden, J. C. Parrish, J. C. Naylor and D. E. Nichols,Mol. Pharmacol., 2006, 70, 1956–1964.
28 J. Bockaert, P. Marin, A. Dumuis and L. Fagni, FEBS Lett., 2003,546, 65–72.
29 S. M. Miggin and B. T. Kinsella, J. Biol. Chem., 2002, 277,27053–27064.
30 D. A. Shapiro, K. Kristiansen, W. K. Kroeze, B. L. D. A. Shapiro,K. Kristiansen, W. K. Kroeze and B. L. Roth, Mol. Pharmlcol,2000, 58, 877–86.
31 J. H. Park, P. Scheerer, K. P. Hofmann, H. W. Choe andO. P. Ernst, Nature, 2008, 454(7201), 183–7.
32 D. M. Rosenbaum, S. G. Rasmussen and B. K. Kobilka, Nature,2009, 459(7245), 356.
33 M. P. Johnson, R. J. Loncharich, M. Baez and D. L. Nelson,Mol. Pharmacol., 1994, 45, 277–86.
34 J. Gonzalez-Maeso, R. L. Ang, T. Yuen, P. Chan,N. V. Weisstaub, J. F. Lopez-Gimenez, M. Zhou, Y. Okawa,L. F. Callado, G. Milligan, J. A. Gingrich, M. Filizola, J. J. Meanaand S. C. Sealfon, Nature, 2008, 452(7183), 93–7.
35 A. Bruno, A. E. Guadix and G. Costantino, J. Chem. Inf. Model.,2009, 49(6), 1602–1616.
36 J. Chambers and E. Nichols, J. Comput.-Aided Mol. Des., 2002,16(7), 511–520.
37 Evers Andreas, Hessler Gerhard, Matter Hans andKlabunde Thomas, J. Med. Chem., 2005, 48(17), 5448–65.
38 Roland Luthy, James U. Bowie and David Eisenberg, Nature,1992, 356, 83–85.
39 K. Kristiansen, W. K. Kroeze, D. L. Willins, E. I. Gelber,J. E. Savage, R. A. Glennon and B. L. Roth, J. Pharmacol. Exp.Ther., 2000, 293, 735–46.
40 H. A. Muntasir, M. Rashid, T. Komiyama, J. Kawakami andT. Nagatomo, J. Pharmacol. Sci., 2006, 102, 55–63.
41 B. R. Johnson and R. M. Harris-Warrick, J. Neurophysiol., 1997,78, 3210–21.
42 M. R. Braden and D. E. Nichols, Mol. Pharmacol., 2007, 72,1200–9.
43 A. Chaudhary, R. A. Lane, D. Woo and R. J. Herman,J. Pharmacol. Exp. Ther., 1993, 267, 1034–8.
44 S. P. Runyon, P. D. Mosier, B. L. Roth, R. A. Glennon andR. B. Westkaemper, J. Med. Chem., 2008, 51, 6808–6828.
45 J. Brea, M. Castro, J. Giraldo, J. F. Lopez-Gimenez, J. F. Padın,F. Quintian, M. I. Cadavid, M. T. Vilaro, G. Mengod, K. A. Berg,W. P. Clarke, J. Vilardaga, G. Milligan and M. I. Loza,Mol. Pharmacol., 2009, 75, 1380.
46 L. P. Pellissier, J. Sallander, M. Campillo, F. Gaven,E. Queffeulou, M. Pillot, A. Dumuis, S. Claeysen, J. Bockaertand L. Pardo, Mol. Pharmacol., 2009, in press.
This journal is �c The Royal Society of Chemistry 2009 Mol. BioSyst., 2009, 5, 1877–1888 | 1887
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online

47 A. Bairoch and B. Boeckmann, Nucleic Acids Res., 1997, 25,217–21.
48 F. Horn, E. Bettler, L. Oliveira, F. Campagne, F. E. Cohen andG. Vriend, Nucleic Acids Res., 2003, 31, 294–7.
49 D. Higgins, J. Thompson, T. Gibson, J. D. Thompson,D. G. Higgins and T. J. Gibson, Nucleic Acids Res., 1994, 22,4673–4680.
50 M. Clamp, J. Cuff, S. M. Searle and G. J. Barton, Bioinformatics,2004, 20, 426–7.
51 T. Okada, M. Sugihara, A. N. Bondar, M. Elstner, P. Entel andV. Buss, J. Mol. Biol., 2004, 342, 571–583.
52 A. Krogh, B. Larsson, G. Von Heijne and E. L. Sonnhammer,J. Mol. Biol., 2001, 305, 567–580.
53 T. Hirokawa, S. Boon-Chieng and S. Mitaku, Bioinformatics,1998, 14, 378–379.
54 G. E. Tusnady and I. Simon, Bioinformatics, 2001, 17, 849–850.55 H. M. Berman, T. N. Bhat, P. E. Bourne, Z. Feng, G. Gilliland,
H. Weissig and J. Westbrook, Nat. Struct. Biol., 2000, 7, 957–9.
56 A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815.57 R. A. Laskowski, M. W. MacArthur, D. S. Moss and
J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291.58 G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey,
W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem.,1998, 19, 1639–1662.
59 A. R. Knight, A. Misra, K. Quirk, K. Benwell, D. Revell,G. Kennett and M. Bickerdike, Naunyn-Schmiedebergs Arch.Pharmacol., 2004, 370, 114–123.
60 A. J. Sleight, N. J. Stam, V. Mutel and P. M. Vanderheyden,Biochem. Pharmacol., 1996, 51, 71–76.
61 R. S. Ostrom and P. A. Insel, Br, J. Pharmacol, 2004, 143(2),235–245.
62 A. Schotte, P. F. Janssen, W. Gommeren, W. H. Luyten, P.Van Gompel, A. S. Lesage, D. K. Loore and J. E. Leysen, Psycho-pharmacology, 1996, 124, 57–73.
63 C. T. Egan, K. Herrick-Davis and M. Teitler, J. Pharmacol. Exp.Ther., 1998, 286, 85–90.
1888 | Mol. BioSyst., 2009, 5, 1877–1888 This journal is �c The Royal Society of Chemistry 2009
Publ
ishe
d on
08
Sept
embe
r 20
09. D
ownl
oade
d by
New
Yor
k U
nive
rsity
on
19/1
2/20
13 2
0:13
:13.
View Article Online
Copyright © 2022 FDOKUMEN

