







Bahasa
Halaman
Hukum


Acce
pted M
anus
cript
1
© The Author 2014. Published by Oxford University Press on behalf of the Infectious Diseases Society of America. All rights reserved. For Permissions, please e-mail: [email protected].
Global dispersal pattern of HIV-1 CRF01_AE: A genetic trace of human
mobility related to heterosexual activities centralized in South-East Asia
Konstantinos Angelis1,†, Jan Albert2,3, Ioannis Mamais1,†, Gkikas Magiorkinis1,4,
Angelos Hatzakis1, Osamah Hamouda5, Daniel Struck6, Jurgen Vercauteren7,
Annemarie M.J. Wensing8, Ivailo Alexiev9, Birgitta Åsjö10, Claudia Balotta11, Ricardo J.
Camacho12, Suzie Coughlan13, Algirdas Griskevicius14, Zehava Grossman15, Andrzej
Horban16, Leondios G. Kostrikis17, Snjezana Lepej18, Kirsi Liitsola19, Marek Linka20,
Claus Nielsen21, Dan Otelea22, Roger Paredes23, Mario Poljak24, Elisabeth Puchhammer-
Stöckl26, Jean-Claude Schmit6, Anders Sönnerborg3,26, Danica Staneková27, Maja
Stanojevic28, Charles A.B. Boucher29, Lauren Kaplan3, Anne-Mieke Vandamme7,12 and
Dimitrios Paraskevis1,*
1Department of Hygiene, Epidemiology and Medical Statistics, Medical School, University
of Athens, Greece
2Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Stockholm,
Sweden
3Department of Clinical Microbiology, Karolinska University Hospital, Stockholm, Sweden
4Department of Zoology, University of Oxford, United Kingdom
5Robert Koch-Institut, Berlin, Germany
6Centre de Recherche Public de la Sante, Luxembourg, Luxembourg
7Clinical and Epidemiological Virology, Rega Institute for Medical Research, Department of
Microbiology and Immunology, KU Leuven, Leuven, Belgium
8Department of Virology, University Medical Center, Utrecht, The Netherlands
9National Center of Infectious and Parasitic Diseases, Sofia, Bulgaria
10University of Bergen, Bergen, Norway
Journal of Infectious Diseases Advance Access published December 15, 2014 by guest on D
ecember 18, 2014
http://jid.oxfordjournals.org/D
ownloaded from

Acce
pted M
anus
cript
2
11University of Milan, Milan, Italy
12Centro de Malária e OutrasDoenças Tropicais and Unidade de Microbiologia, Instituto de
Higiene e Medicina Tropical, Universidade Nova de Lisboa, Lisbon, Portugal
13University College Dublin, Dublin, Ireland
14Lithuanian AIDS Center, Vilnius, Lithuania
15Tel Aviv University, Tel Aviv, Israel
16Hospital of Infectious Diseases, Warsaw, Poland
17University of Cyprus, Nicosia, Cyprus
18Department of Molecular Diagnostics and Flow Cytometry, University Hospital for
Infectious Diseases "Dr. F. Mihaljevic", Zagreb, Croatia
19National Institute of health and welfare, Helsinki, Finland
20National Reference Laboratory of AIDS, National Institute of Health, Prague, Czech
Republic
21Statens Serum Institute, Copenhagen, Denmark
22National Institute for Infectious Diseases “Prof. Dr. Matei Bals”, Bucharest, Romania
23IrsiCaixa Foundation, Badalona, Spain
24Slovenian HIV/AIDS Reference Centre, University of Ljubljana, Faculty of Medicine,
Ljubljana, Slovenia
25University of Vienna, Vienna, Austria
26Divisions of Infectious Diseases and Clinical Virology, Karolinska Institute, Stockholm,
Sweden
27Slovak Medical University, Bratislava, Slovakia
28University of Belgrade Faculty of Medicine, Belgrade, Serbia
29Erasmus MC, University Medical Center, Rotterdam, The Netherlands
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
3
*Correspondence: Dimitrios Paraskevis, Department of Hygiene, Epidemiology and Medical
Statistics, Medical School, University of Athens, 75 Mikras Asias Street, 115 27 Athens,
Greece, Phone: +30 210 7462119, e-mail: [email protected]
†Contributed equally to the work
Abstract
Background: HIV-1 subtype CRF01_AE originated in Africa and then passed to Thailand
where it established a major epidemic. Despite the global presence of CRF01_AE little is
known about its subsequent dispersal pattern.
Methods: We assembled a global dataset of 2,736 CRF01_AE sequences by pooling
sequences from public databases and patient-cohort studies. We estimated viral dispersal
patterns using statistical phylogeography run over bootstrap trees estimated by the maximum
likelihood (ML) method.
Results: We show that Thailand has been the source of viral dispersal to most areas
worldwide, including 17 out of 20 sampled countries in Europe. Japan, Singapore, Vietnam
and other Asian countries have played a secondary role in the viral dissemination. In contrast,
China and Taiwan have mainly imported infections from neighbouring Asian countries,
North America and Africa without any significant exporting transmissions.
Discussion: The central role of Thailand in the global spread of CRF01_AE can be probably
explained by the popularity of Thailand as a vacation destination characterized by sexual
tourism and by Thai emigration to the Western world. Our study highlights the unique case of
CRF01_AE, the only globally distributed non-B clade for which its global dispersal was not
originated in Africa.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
4
Background
HIV-1 strains are divided into four major genetic groups M, O, N and P which represent four
separate transmissions of simian strains to humans [1]. Among them, group M is by far the
most prevalent and has established a global epidemic, while the other three are mainly
restricted to West and Central Africa. Group M is further classified into nine major genetic
subtypes named A-D, F-H, J and K and several recombinant forms [2, 3]. If a unique
recombinant form (URF) succeeds in being transmitted to several individuals, the URF is
called circulating recombinant form (CRF). One of the most prevalent CRFs is the
CRF01_AE, which is considered to have originated by recombination of a subtype A variant
and a putative extinct subtype E ancestor [4-6].
The CRF01_AE was first identified in female sex workers in northern Thailand in 1989
[7-9]. In the following years, phylogenetic studies showed that the CRF01_AE originated in
Central Africa, in the 1970s, and then migrated to Thailand, most likely through heterosexual
transmission [10]. Subsequently, CRF01_AE established a large-scale epidemic in South-
East Asia with a proportion of 79% among HIV-1 infections [11] and became one of the
successfully spreading recombinants [12, 13]. Currently, CRF01_AE has formed a nearly
global epidemic with viral strains reported in North America, Europe, Central and West
Africa, Asia and Australia and is estimated to represent approximately 5% of global HIV-1
infections [14]. The dispersal pattern after the transmission of CRF01_AE from Africa to
Thailand is largely unknown. Knowledge of viral dispersal pathways can inform the design of
interventions to prevent onward transmissions [15]. Previously published studies track the
mobility of the epidemic only to a local scale, within a country or to a small group of
neighbouring countries [16-21]. Larger studies, but still not on a global scale, have also been
reported [19]. To our knowledge no previous study has traced the dispersal pattern of
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
5
CRF01_AE on a global scale. Here, we use a global dataset of 2,736 CRF01_AE sequences
aiming to uncover global transmission routes using a statistical phylogeographic analysis.
Methods
Data assembly
We downloaded non-European sequences from the Los Alamos HIV sequence database [22].
Several sequences from the same patient might be available in the database, due to patient
inclusion in follow-up studies. To avoid potential bias due to driven viral evolution from
antiretroviral therapy, we kept only the oldest available sequence per patient. Duplicate
sequences from the same country were discarded using the ElimDupes [23] online tool. The
European sequences come from the SPREAD (Strategy to Control Spread of HIV Drug
Resistance) surveillance programme [24]. Information on this dataset has been previously
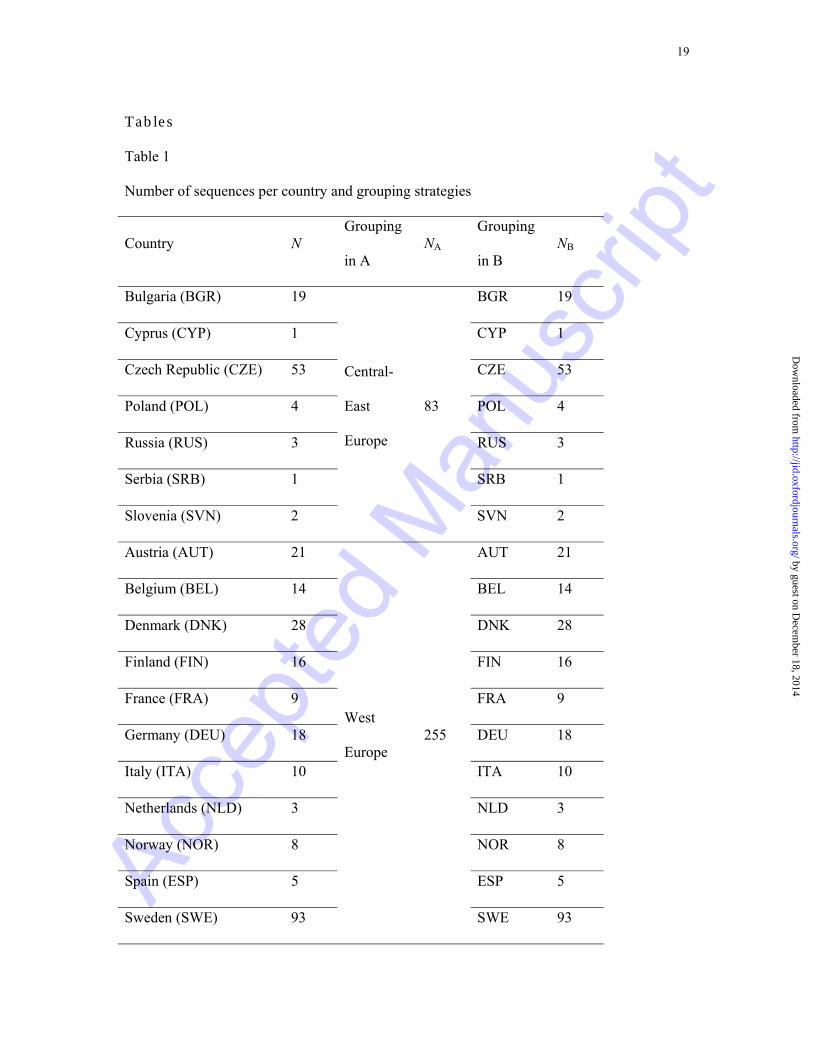
published [24, 25].The sequences come from most of the countries with an established
CRF01_AE epidemic as shown from Table 1. For the European sample some demographic
information (gender, transmission route and country of origin) obtained from standardized
questionnaires used in the SPREAD project, is reported in Table 2.
Alignment
The pol gene, protease (PR) and partial reverse transcriptase (RT) regions, of the HIV-1
genome was used, since it is among the most frequently sequenced genomic regions. We
aligned the sequences using the ClustalW algorithm incorporated into the MEGA5 program
[26] and manually corrected the alignment according to the encoded reading frame. We
discarded nucleotides corresponding to certain amino acid positions in order to avoid
potential biases due to convergent evolution caused by antiretroviral therapy (see
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
6
Supplementary Information). The final alignment was consisted of 2,736 sequences and 777
nucleotides.
Phylogenetic tree reconstruction
Phylogenetic reconstruction was performed under the general time reversible model of
nucleotide substitution with gamma-distributed rate heterogeneity (GTR + Γ4) among sites
[27, 28], using the maximum likelihood (ML) method as implemented in the RAxML
program [29]. To account for phylogenetic uncertainty 300 ML bootstrap trees were also
inferred using the rapid bootstrapping algorithm of RAxML [30] as implemented in the
CIPRES project cluster [31].
Inference of migration events
For each bootstrap tree we estimated CRF01_AE migration events using the approach
described by Slatkin and Maddison [32] as implemented in the PAUP 4.0 program [33]. All
tips of the bootstrap trees were assigned with a character (state) according to their geographic
origin (e.g. K, L, M for Austria, Belgium, Africa, respectively, etc.). The geographical
locations used in our study are described in Table 1. The algorithm reconstructs ancestral
states in the internal nodes of a tree using the parsimony criterion which minimizes the total
number of state changes.
When two branches from the same location (e.g. K and K) join each other the
reconstructed ancestral state is the same (e.g. K) and there is no migration. When the
branches come from different locations (e.g. K and L) the reconstructed state is ambiguous
and is assigned to be the union of the two characters [K, L] (Figure 4A, nodes 3, 6). This
implies a character change and thus a migration event is assigned. The ambiguity and thus the
direction of the migration may sometimes be resolved by the states of earlier nodes (Figure
4B, nodes 3, 6). In our analysis only unambiguous migration events were used.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
7
Migration matrices
Once the migration events between any two regions were estimated from all the 300
bootstrap trees, they were stored in 300 matrices (one matrix per bootstrap tree) with the rows
and columns to represent area of origin and destination, respectively. In this way a cell of a
matrix contains the number of estimated migration events from the area of the row to the area
of the column for a bootstrap tree. We summarized the medians of the events across the
bootstrap trees for all pathways in a single matrix (e.g. Supplementary Table1 1).
Steps of analysis
Migration events were estimated with two geographic grouping strategies. First
sequences were grouped into large geographic areas (analysis A, see Supplementary
Information)
At a next step (analysis B) we attempted to identify more specific dispersal routes
between countries, rather than between large geographic areas. Consequently, we grouped
together sequences from the same country, with the exception of African sequences which
formed a single group and sequences from the Asian countries Hong Kong, South Korea,
Afghanistan, Iran, Pakistan, Indonesia, Malaysia, Myanmar and Philippines, which formed
another group called "Rest Asia" (see Table 1). This was done because of the small number
of available sequences from these Asian countries. Although for some European countries
(e.g. Cyprus, Serbia,) the number of available sequences was small as well, we decided not to
merge them into a single group because transmission routes towards and from European
countries were of major interest.
Statistical phylogeography
In a random mixing population, an infected individual would have the same probability
to transmit the HIV-1 to any other healthy individual. Thus, a random shuffling of taxa at the
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
8
tips of any phylogenetic tree would simulate a tree inferred from such a population. To
estimate which transmission pathways of our analyses are significant, we performed a
random shuffling of taxa at the tips of the 300 bootstrap trees, and then inferred the number
of migration events between any two areas. We call these events "expected migration events",
as they are expected to occur under the assumption of random mixing, in contrast to the
"observed events" inferred from the bootstrap trees before shuffling. We then compared the
distribution of the events inferred from the two sets of trees (before and after shuffling) for
any two areas, using the one-sided Mann-Whitney test. The level of significance (α = 5%)
was adjusted according to Bonferroni correction for multiple comparisons. Pathways with
statistically significantly higher number of observed events than expected were identified as
significant. In case two pathways connecting the same two areas (i.e. K → L and L → K) are
both significant, a bidirectional relationship is defined between the two areas.
The ratio (r) of the observed versus the expected number of migration events provides
information on the amount of viral flow between different areas that is not attributed to
chance. For example, a ratio r = 1.20 for a pathway would indicate that the observed viral
flow across the pathway is 20% higher than what is expected from chance (random mixing
population). An estimate of this ratio for a pathway can be obtained by dividing the mean of
observed migration events by the mean of expected. Such ratios were estimated for all
pathways, except those having 0 mean of expected migration events. Ratios estimated in this
way are sensitive to the mean of expected events, as a mean of expected events very close to
0 may produce high ratio values (see Supplementary Information).
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
9
Results
Phylogenetic Analysis
The best-scoring maximum likelihood (ML) tree suggests an extensive pattern of viral
dispersal (Figure 1) with Thai sequences to be highly dispersed across the tree and positioned
basal to several monophyletic clusters. In contrast, the majority of Chinese and Vietnamese
sequences form large monophyletic clusters (four Chinese and two Vietnamese clusters,
Figure 1). This indicates that the CRF01_AE epidemic in those countries originated from a
limited number of viral introductions, which subsequently spread locally. Although Thai
sequences are highly dispersed across the tree, there are only a few Thai strains within the
Chinese and Vietnamese clusters suggesting limited interaction between Thailand and these
countries after the first main introductions.
European sequences are also dispersed in the tree and lie mainly next to Thai sequences,
indicating extensive interaction between European and Thai strains. The Japanese sequences
and those from North America and from other sampled Asian countries (denoted with
"Others" in Figure 1) are also dispersed across the tree, highly mixed with Thai strains. The
majority of African sequences seem to form a single cluster (top left corner of Figure 1).
However, no safe deduction can be made concerning the level of interaction between African
viral strains and strains from the rest of the world due to the small African sample size.
Overall, the above findings indicate a high level of phylogenetic mixing between Thai and
European, Japanese, North American strains and strains from sampled Asian countries
excluding China and Vietnam.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
10
Statistical Phylogeography
To statistically investigate the global mobility pattern of CRF01_AE, we estimated the
number of migration events between different countries and geographic areas. We then
compared them with the expected number of events under the null hypothesis of complete
geographic mixing, in order to infer significant dispersal pathways (see Methods).
Migration patterns with respect to countries
We grouped the sampled sequences with respect to country as described in Table 1
(analysis B). Results of this analysis revealed the striking role of Thailand and the secondary
of Japan to the global CRF01_AE viral epidemic (Figure 2).
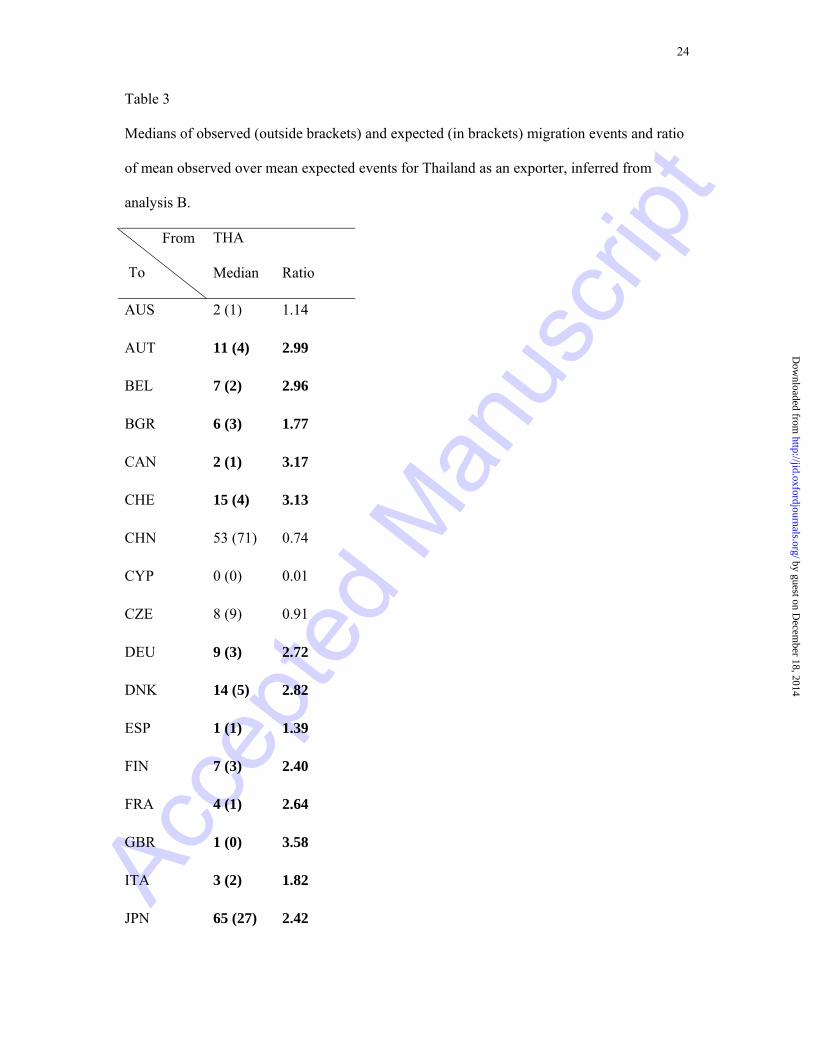
Specifically, Thailand has exported the epidemic to 17 of the 20 European countries
included in our sample, with the exception of Cyprus, Czech Republic and Serbia (Table 3,
Supplementary Table 1). United Kingdom, Slovenia, Switzerland and Austria are among the
countries with the highest ratios (3.58, 3.26, 3.13, 2.99, respectively) of importing
transmissions from Thailand (Table 3). In addition, Canada and the United States in North
America, as well as Japan, Singapore, and Taiwan in Asia, have constituted migratory targets
of Thai strains. Significant viral spread was also detected from Thailand to other Asian
countries, grouped and named "Rest Asia" in our analysis, including Hong Kong, South
Korea, Afghanistan, Iran, Pakistan, Indonesia, Malaysia, Myanmar and Philippines. Despite
the extensive viral exporting network inferred for Thailand, a single source of viral import
from Africa was detected in accordance to previous studies [10, 17, 19].
Japan also seems to have had an important role in viral dissemination. Japan has
exported the virus to several European countries (Austria, Denmark, Spain, Sweden and
Switzerland) as well as to Canada (Supplementary Table 1). These dispersal routes are
supported by high migration ratios (Supplementary Table 1). Singapore, Taiwan and "Rest
Asia" are also inferred as migratory targets for Japanese CRF01_AE strains. Our results also
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
11
suggest that Japan has imported viral strains from Spain, Singapore, "Rest Asia" and
Thailand (Supplementary Table 1). Consequently, Japan is a country with high bidirectional
transcontinental viral mobility.
Singapore, Vietnam and "Rest Asia" seem to have played a secondary role in viral
dissemination, as they interact (both as exporters or importers of the epidemic) with a smaller
number of countries mainly from Asia and Europe (Table 3, Supplementary Table 1). In
contrast, China and Taiwan have been migratory targets, having imported the epidemic
mainly from neighbouring Asian countries, and the United States and Africa, respectively,
without having any significant exporting pathways. Notably, we found significant migration
from Vietnam to China (Supplementary Table 1).
Concerning Africa it seems that it has been a major exporter, spreading the epidemic to
several European countries, such as Belgium, France, Italy, Sweden, Switzerland and Czech
Republic, to the United States as well as to Asian countries, such as Taiwan and Thailand
(Figure 2, Supplementary Table 1). Notably, no significant viral introduction was detected.
Viral mobility within Europe
Within Europe we found that Sweden and Finland seem to have played an important role
in disseminating the epidemic. Specifically, Sweden has exported viral strains to the
Netherlands, Norway, Russia and Finland (Supplementary Table 1). Notably, we found
significant export from Finland to Sweden as well, in accordance to previous studies [34, 35].
For Sweden, we found significant viral import from Thailand, Japan and Africa. Almost all
significant migratory pathways of Sweden are supported by high ratios of observed versus
expected number of events (Supplementary Table 1). Significant viral dispersal was also
detected from Germany to Poland and from Bulgaria to Czech Republic. Transmission routes
within Europe are illustrated in Figure 3.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
12
To examine parameters potentially associated with the observed patterns of viral
dispersal we examined the demographic characteristics of patients infected with CRF01_AE
in Europe. Among those with a known country of origin, 29.7% and 11.6% were from
Thailand and Vietnam, respectively. Importantly, 70.7% of individuals reported heterosexual
contact as the main risk of HIV transmission (Table 2). In contrast, men having sex with men
(MSM) was reported as a transmission risk only for 15.7% of the patients.
Discussion
The origin of the HIV-1 CRF01_AE in Africa several years ago and its spread to
Thailand provided the first events for the current CRF01_AE global epidemic [4]. However,
the subsequent pattern of viral spread has remained largely unknown. Our study provides a
global phylogeographic analysis of the CRF01_AE epidemic using the largest dataset
currently available.
Concerning the transcontinental patterns of the epidemic spread we can note five
remarks. First, Africa has acted as a source of viral spread to several areas in Asia, Europe
and North America. Second, Thailand has shown the most extensive network with viral
dispersal to many countries in Europe, Asia and North America. Third, Japan and some other
Asian countries (e.g. Singapore, Vietnam) have had a secondary role in CRF01_AE
dissemination to Europe, Asia and North America. Fourth, China and Taiwan have had
monophyletic CRF01_AE epidemics. Fifth, Europe and North America have acted mainly as
sinks with import of CRF01_AE infections from several different countries.
Our results pinpoint the central role of Thailand in the global CRF01_AE epidemic. This
country has established an extensive network of within and transcontinental transmissions,
including neighbouring Asian countries as well many European countries and North America
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
13
(Table 3, Figure 2). Thailand experienced a dramatic increase in HIV-1 infections in early
nineties, where most of infections were linked to sexual transmission from sex workers [36].
The proportion of new infections due to CRF01_AE, named subtype E at that time, increased
rapidly from 2.6% in 1988-1989 to 4% in 1992-1993 [13]. The situation was alarming and
Thailand responded early by implementing the "100 percent condom" program that
successfully reduced the spread of HIV-1 and other sexually transmitted diseases among sex
workers. Specifically, between 1991 and 2001, the number of new HIV-1 infections in
Thailand dropped from 143,000 per year to less than 14,000 [14, 36]. However, the
generalized epidemic of CRF01_AE among heterosexuals and sex workers, the large
numbers of female sex workers operating in the country, the Thailand’s sexual tourism
industry [5] and human emigration to Europe and North America may explain the extensive
viral export from the country. In addition, Thailand is a popular touristic destination for many
Europeans and has an important geographical position as it is located in the crossroad of
Africa, Asia and Oceania attracting many travellers.
Considerable viral mobility was also indicated for Japan with export mainly to Europe,
North America and some neighbouring Asian countries and import mainly from Asian
countries. Trading connections of Japan with many Asian, European and American countries
as well as extensive tourism may have contributed to this pattern. Singapore and Vietnam
seem to have had a secondary role in viral dissemination. Interestingly, CRF01_AE was
introduced in those countries mainly by sexual transmission [37, 38].
In contrast to Thailand and Japan, our results suggest that China and Taiwan are sinks,
importing CRF01_AE strains mainly from other Asian countries. Interestingly, Thailand is
not one of them despite its extensive exporting viral network. This finding differs from the
results of Abubakar et al. (2013) who reported close phylogenetic relationships and
transmissions between Thai, Chinese and Vietnamese CRF01_AE strains [19]. Abubakar et
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
14
al. used a limited number of non-Thai, Chinese and Vietnamese sequences (83 out of 1957
sequences, 4.2%) and based their results on a single ML tree and a molecular clock analysis
using BEAST [39]. Here, we used a large number of sequences from countries other than
Thailand, China and Vietnam (736 out of 2736, 26.9%) and used a statistical
phylogeographic approach to estimate viral dispersal. It is likely that this difference between
findings is due to differences in sampling and phylogenetic methodology. On the other hand,
we detected significant viral dissemination from Vietnam and Singapore to China, in
accordance with Abubakar et al. [19].
Despite the high prevalence of the CRF01_AE in almost all South-East and East Asian
countries, those do not show the same pattern of viral dispersal. This may be explained by
differences in human migration, tourism, commerce and factors related to the characteristics
of local epidemics in Asia. For example in Thailand and Japan the CRF01_AE epidemics
were mainly driven by heterosexuals [5, 14] while in China, Vietnam and Taiwan by
heterosexuals, MSM and IDUs [17, 36, 37, 40, 41]. Furthermore, in our sample of HIV-1
infected individuals in Europe, we found 46 with Thai origin. Although it is unknown
whether these individuals became infected before emigrating to Europe, the low prevalence
of CRF01_AE in Europe and the high prevalence in Thailand suggests probably that most of
these infections took place in Thailand. These factors appear to be the most plausible
explanations for the different role of Thailand in the global CRF01_AE epidemic compared
to other Asian countries.
The European CRF01_AE sub-epidemic seems to be a result of viral transmissions
mainly from Thailand, Japan and Africa (Supplementary Table 1). International tourism and
extensive emigration into Europe around the turn of the century, might have contributed to
the observed transmission pattern. In particular, in Thailand the number of European tourists
visiting the country rose significantly in the past fifteen years, with travellers from United
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
15
Kingdom, France, Italy, Switzerland, Russia, Denmark and Finland being among the most
frequent in 2002 [16]. Extended commercial and tourist connections between West European
countries and Japan might also favour transmissions. Notably, all European viral movement
from Japan detected in our analysis concerned Western (and not Eastern) European countries
where commercial and tourist relationships with Japan are more intense (Supplementary
Table 1).
Conclusions
Our study describes for the first time the global dispersal pattern of CRF01_AE ,
highlighting the central role of Thailand as a major viral exporter to the Western world. The
key factor in this pattern might be Thailand’s popularity as a touristic destination, the
extensive network of commercial sex workers operating in the country and human migration.
The case of CRF01_AE is likely unique regarding endemicity and factors causing its global
dispersal pattern in contrast to other non-B infections mainly associated with immigration
from Africa. Africa provided the source for all globally prevalent non-B subtypes (A, C, D,
F) and CRFs. Currently, South Eastern Asia provides the only exception of a non-African
source for the global dissemination of CRF01_AE. Notably, this CRF spread beyond the area
of South Eastern Asia to distant western countries probably occurred through heterosexual
activities. Our study highlights the advance of phylogeographic analyses towards a better
understanding of infectious diseases epidemics and factors potentially associated with their
spread.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
16
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DP designed the analysis and contributed to writing the manuscript. IM and KA performed
the analysis. KA, DP, GM and JA prepared the manuscript. All co-authors reviewed the
manuscript and contributed to data provision and interpretation of the results.
Funding
This work was supported by the European Community’s Seventh Framework Programme
(FP7/2007-2013) under the project “Collaborative HIV and Anti-HIV Drug Resistance
Network (CHAIN)”. GM is supported by the Medical Research Council. This work was in
part supported by the AIDS Reference Laboratory of Leuven that receives support from the
Belgian Ministry of Social Affairs through a fund within the Health Insurance System; the
Fonds voor Wetenschappelijk Onderzoek – Flanders (FWO) [PDO/11 to K.T., G069214N].
Previously presented information
This study has been accepted as a late breaker presentation at the 20th International AIDS
conference in Melbourne.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
17
Figure legends
Figure 1. Best-scoring unrooted ML tree estimated from RAxML using 2,736 HIV-1
CRF01_AE sequences from 46 countries around the globe. Tree branches are coloured
according to sampling origin. Different colours were used for sequences from Europe, Africa,
China, Thailand, Vietnam, Japan and from all other regions.
Figure 2. Significant importing and exporting pathways around the globe. Countries and
geographic regions with at least one significant pathway are illustrated with different colours.
The same colour (light blue) was used for all European countries and significant pathways
within Europe are not reported for clarity of the figure. Asian countries clustered as "Rest
Asia" and Singapore are not reported for the same reason. The direction and thickness of the
arrows show the direction and level (based on the ratios of Supplementary Table 1) of
CRF01_AE migration, respectively, across the transmission routes.
Figure 3. Significant importing and exporting pathways within Europe. Sampling countries
are in blue. The direction and thickness of the arrows show the direction and level (based on
the ratios of Supplementary Table 1) of CRF01_AE migration, respectively, across the
transmission routes.
Figure 4. An example of how the parsimony algorithm is used to compute migration events
on an unrooted phylogeny of 8 taxa, sampled from 2 different geographic regions. Tips are
labeled with characters K or L which correspond to the geographic origin and internal nodes
are numbered. Ancestral states are recursively assigned to the internal nodes according to the
majority rule criterion. A: For example, the ancestral state at node 1 is K because the two
joined branches have the same character K. Similarly for node 2. When the branches come
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
18
from different regions, K, L, the ancestral state is set to be the union of the two characters [K,
L], as in node 3. This implies that a character change has occurred and thus a migration event
is assigned. However, because the state of the node is ambiguous (either K or L), the
direction of the migration is undetermined. The ambiguity and thus the direction of migration
may be resolved by the states of neighboring nodes using the parsimony criterion. B: In this
case, because the state of node 4 is [K] the ambiguity of node 3 is solved into [K], and thus
the migration is from region K to L (denoted by → in B). In this example, both ambiguities in
nodes 3 and 6 are resolved under the parsimony criterion into state K. However, not always
ambiguities can be resolved, especially in case the sampling of taxa is from many different
locations.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from
Acce
pted M
anus
cript
19
Tables
Table 1
Number of sequences per country and grouping strategies
Country N Grouping
in A NA
Grouping
in B NB
Bulgaria (BGR) 19
Central-
East
Europe
83
BGR 19
Cyprus (CYP) 1 CYP 1
Czech Republic (CZE) 53 CZE 53
Poland (POL) 4 POL 4
Russia (RUS) 3 RUS 3
Serbia (SRB) 1 SRB 1
Slovenia (SVN) 2 SVN 2
Austria (AUT) 21
West
Europe 255
AUT 21
Belgium (BEL) 14 BEL 14
Denmark (DNK) 28 DNK 28
Finland (FIN) 16 FIN 16
France (FRA) 9 FRA 9
Germany (DEU) 18 DEU 18
Italy (ITA) 10 ITA 10
Netherlands (NLD) 3 NLD 3
Norway (NOR) 8 NOR 8
Spain (ESP) 5 ESP 5
Sweden (SWE) 93 SWE 93
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
20
Switzerland (CHE) 28 CHE 28
United Kingdom (GBR) 2 GBR 2
United States (USA) 16 North
America 20
USA 16
Canada (CAN) 4 CAN 4
Australia (AUS) 8 AUS 8 AUS 8
Angola (ANG) 1
Africa 20 Africa 20
Cameroon (CMR) 6
Central African
Republic (CAF) 6
Chad (TCD) 1
Democratic Republic
of the Congo (COD) 3
Gabon (GAB) 1
Mali (MLI) 1
Senegal (SEN) 1
China (CHN) 723
East
Asia 927
CHN 723
Japan (JPN) 167 JPN 167
Taiwan (TWN) 33 TWN 33
Hong Kong (HKG) 1
Rest Asia 19
South Korea (KOR) 3
Afghanistan (AFG) 1 Central
Asia 3 Iran (IRN) 1
Pakistan (PAK) 1
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
21
Indonesia (IDN) 2
South-East
Asia 1420
Malaysia (MYS) 5
Myanmar (MMR) 1
Philippines (PHL) 4
Singapore (SGP) 131 SGP 131
Thailand (THA) 692 THA 692
Vietnam (VNM) 585 VNM 585
Total 2736 2736 2736
Note.− N is the number of available sequences per country in the dataset. NA and NB describe
the number of sequences in each group in analyses A and B, respectively. In each analysis the
grouping of countries was performed as described by the third and fifth columns. Text in
brackets refers to countries' 3-letter codes according to the United Nations.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
22
Table 2
Characteristics of European patients with known demographic record
Characteristic N %
Gender Male 100 64.10
Female 56 35.90
Country of origin
West Europe
Austria 13 8.39
Belgium 3 1.94
Denmark 8 5.16
Finland 13 8.39
Germany 9 5.81
Netherlands 1 0.65
Norway 1 0.65
Sweden 24 15.48
Central-East
Europe
Bulgaria 1 0.65
Czech Republic 4 2.58
Poland 2 1.29
Serbia 2 1.29
South America Colombia 1 0.65
Africa
Cameroon 1 0.65
Ethiopia 1 0.65
Nigeria 1 0.65
Tunisia 1 0.65
Asia China 2 1.29
Thailand 46 29.68
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
23
Vietnam 18 11.61
Myanmar 1 0.65
Indonesia 1 0.65
Iraq 1 0.65
Infection route
Patients with
any origin
Hete a 99 70.71
MSM b 22 15.71
IDU c 19 13.57
Patients with
Thai origin
Hete 36 78.27
MSM 6 13.04
IDU 1 2.17
Nwa d 3 6.52
Note.− Country of origin refers to the actual origin of the patient and not to the sampling
country. The infection route refers to the cause of infection, according to patients' responses. a
heterosexual, bmen having sex with men, c injection drug users, d no willing to answer.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from
Acce
pted M
anus
cript
24
Table 3
Medians of observed (outside brackets) and expected (in brackets) migration events and ratio
of mean observed over mean expected events for Thailand as an exporter, inferred from
analysis B.
From
To
THA
Median Ratio
AUS 2 (1) 1.14
AUT 11 (4) 2.99
BEL 7 (2) 2.96
BGR 6 (3) 1.77
CAN 2 (1) 3.17
CHE 15 (4) 3.13
CHN 53 (71) 0.74
CYP 0 (0) 0.01
CZE 8 (9) 0.91
DEU 9 (3) 2.72
DNK 14 (5) 2.82
ESP 1 (1) 1.39
FIN 7 (3) 2.40
FRA 4 (1) 2.64
GBR 1 (0) 3.58
ITA 3 (2) 1.82
JPN 65 (27) 2.42
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
25
NLD 1 (0) 2.66
NOR 3 (1) 2.51
POL 1 (1) 1.73
RUS 1 (0) 2.33
SGP 59 (21) 2.74
SVN 1 (0) 3.26
SRB 0 (0) 1.18
SWE 35 (15) 2.29
TWN 15 (6) 2.71
USA 8 (3) 2.75
VNM 22 (66) 0.33
Rest Asia 8 (3) 2.53
Africa 4 (3) 1.21
Note.− Cells in bold indicate statistically significant pathways under the null hypothesis of
random mixing population. Countries' codes are according to Table 1. This table is part of
Supplementary Table 1 and summarizes results for Thailand as an exporter.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
26
References
1. Sharp PM, Hahn BH. Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med
2011; 1:a006841.
2. Sharp PM, Hahn BH. The evolution of HIV-1 and the origin of AIDS. Philos Trans R Soc Lond B
Biol Sci 2010; 365:2487-94.
3. Vidal N, Peeters M, Mulanga-Kabeya C, et al. Unprecedented degree of human immunodeficiency
virus type 1 (HIV-1) group M genetic diversity in the Democratic Republic of Congo suggests that the
HIV-1 pandemic originated in Central Africa. J Virol 2000; 74:10498-507.
4. Gao F, Robertson DL, Morrison SG, et al. The heterosexual human immunodeficiency virus type 1
epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin. J Virol 1996;
70:7013-29.
5. WORLD HEALTH ORGANIZATION ROftWP. SEX WORK IN ASIA, July 2001.
6. Robertson DL, Sharp PM, McCutchan FE, Hahn BH. Recombination in HIV-1. Nature 1995;
374:124-6.
7. Carr JK, Salminen MO, Koch C, et al. Full-length sequence and mosaic structure of a human
immunodeficiency virus type 1 isolate from Thailand. J Virol 1996; 70:5935-43.
8. McCutchan FE, Hegerich PA, Brennan TP, et al. Genetic variants of HIV-1 in Thailand. AIDS Res
Hum Retroviruses 1992; 8:1887-95.
9. Nelson KE, Celentano DD, Suprasert S, et al. Risk factors for HIV infection among young adult
men in northern Thailand. JAMA 1993; 270:955-60.
10. McCutchan FE, Artenstein AW, Sanders-Buell E, et al. Diversity of the envelope glycoprotein
among human immunodeficiency virus type 1 isolates of clade E from Asia and Africa. J Virol 1996;
70:3331-8.
11. Hemelaar J, Gouws E, Ghys PD, Osmanov S, Isolation W-UNfH, Characterisation. Global trends
in molecular epidemiology of HIV-1 during 2000-2007. AIDS 2011; 25:679-89.
12. Vanichseni S, Kitayaporn D, Mastro TD, et al. Continued high HIV-1 incidence in a vaccine trial
preparatory cohort of injection drug users in Bangkok, Thailand. AIDS 2001; 15:397-405.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
27
13. Wasi C, Herring B, Raktham S, et al. Determination of HIV-1 subtypes in injecting drug users in
Bangkok, Thailand, using peptide-binding enzyme immunoassay and heteroduplex mobility assay:
evidence of increasing infection with HIV-1 subtype E. AIDS 1995; 9:843-9.
14. UNAIDS. Evaluation of the 100% Condom Programme in Thailand UNAIDS Case Study, 2000.
15. Cohen MS, Chen YQ, McCauley M, et al. Prevention of HIV-1 infection with early antiretroviral
therapy. N Engl J Med 2011; 365:493-505.
16. Tourism Authority of Thailand. Tourism Statistics. Available at:
http://www2.tat.or.th/stat/web/static_tsi_detail.php?L=&TsiID=1.
17. Liao H, Tee KK, Hase S, et al. Phylodynamic analysis of the dissemination of HIV-1 CRF01_AE
in Vietnam. Virology 2009; 391:51-6.
18. Ng KT, Ng KY, Khong WX, et al. Phylodynamic profile of HIV-1 subtype B, CRF01_AE and the
recently emerging CRF51_01B among men who have sex with men (MSM) in Singapore. PloS One
2013; 8:e80884.
19. Abubakar YF, Meng Z, Zhang X, Xu J. Multiple independent introductions of HIV-1 CRF01_AE
identified in China: what are the implications for prevention? PloS One 2013; 8:e80487.
20. Ng KT, Ong LY, Lim SH, Takebe Y, Kamarulzaman A, Tee KK. Evolutionary history of HIV-1
subtype B and CRF01_AE transmission clusters among men who have sex with men (MSM) in Kuala
Lumpur, Malaysia. PloS One 2013; 8:e67286.
21. Ye J, Xin R, Yu S, et al. Phylogenetic and temporal dynamics of human immunodeficiency virus
type 1 CRF01_AE in China. PloS One 2013; 8:e54238.
22. Los Alamos National Laboratory. HIV Databases. Available at:
http://www.hiv.lanl.gov/content/index.
23. Los Alamos National Laboratory. HCV sequence database. Available at:
http://hcv.lanl.gov/content/sequence/ELIMDUPES/elimdupes.html.
24. Wensing AMJ VJ, Van De Vijver DA, Albert J, Åsjö B, Balotta C, Camacho R, Coughlan S,
Grossman Z, Horban A, Kücherer C, Nielsen C, Paraskevis D, Loke WC, Poggensee G, Puchhammer-
Stöckl E, Riva C, Ruiz L, Schmit JC, Schuurman R, Salminen M, Sonnerborg A, Stanojevic M,
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
28
Struck D, Vandamme AM, Bouche, CAB, Spread programme. Transmission of drug-resistant HIV-1
in Europe remains limited to single classes. AIDS 2008; 22:625-35.
25. Paraskevis D, Pybus O, Magiorkinis G, et al. Tracing the HIV-1 subtype B mobility in Europe: a
phylogeographic approach. Retrovirology 2009; 6:49.
26. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary
genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony
methods. Mol Biol Evol 2011; 28:2731-9.
27. Yang Z. Estimating the pattern of nucleotide substitution. J Mol Evol 1994; 39:105-11.
28. Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates
over sites: approximate methods. J Mol Evol 1994; 39:306-14.
29. Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with
thousands of taxa and mixed models. Bioinformatics 2006; 22:2688-90.
30. Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers.
Syst Biol 2008; 57:758-71.
31. CIPRES Cyberinfrastructure for Phylogenetic Research. The CIPRES Science Gateway V. 3.3.
Available at: http://www.phylo.org/index.php/portal/.
32. Slatkin M, Maddison WP. A cladistic measure of gene flow inferred from the phylogenies of
alleles. Genetics 1989; 123:603-13.
33. Swofford D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version
4. Sinauer Associates 2003.
34. Skar H, Axelsson M, Berggren I, et al. Dynamics of two separate but linked HIV-1 CRF01_AE
outbreaks among injection drug users in Stockholm, Sweden, and Helsinki, Finland. J Virol 2011;
85:510-8.
35. Skar H, Sylvan S, Hansson HB, et al. Multiple HIV-1 introductions into the Swedish intravenous
drug user population. Infect Genet Evol 2008; 8:545-52.
36. World Health Organization. Thailand's new condom crusade. Bulletin of the World Health
Organization, 2010.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
29
37. Kalish ML, Korber BT, Pillai S, et al. The sequential introduction of HIV-1 subtype B and
CRF01AE in Singapore by sexual transmission: accelerated V3 region evolution in a subpopulation of
Asian CRF01 viruses. Virology 2002; 304:311-29.
38. Nerurkar VR, Nguyen HT, Dashwood WM, et al. HIV type 1 subtype E in commercial sex
workers and injection drug users in southern Vietnam. AIDS Res Hum Retroviruses 1996; 12:841-3.
39. Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC
Evol Biol 2007; 7:214.
40. Ou CY, Takebe Y, Luo CC, et al. Wide distribution of two subtypes of HIV-1 in Thailand. AIDS
Res Hum Retroviruses 1992; 8:1471-2.
41. Ubolyam S, Ruxrungtham, Sirivichayakul S, Okuda K, Phanuphak P. Evidence of three HIV-1
subtypes in subgroups of individuals in Thailand. Lancet 1994; 344:485-6.
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
31
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
32
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
33
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from

Acce
pted M
anus
cript
34
by guest on Decem
ber 18, 2014http://jid.oxfordjournals.org/
Dow
nloaded from
Top Related
Copyright © 2022 FDOKUMEN

