







Bahasa
Halaman
Hukum


www.elsevier.com/locate/ynbdi
Neurobiology of Disease 23 (2006) 653–662Deposition of mouse amyloid β in human APP/PS1 double and singleAD model transgenic mice
Thomas van Groen,a,b,c,⁎ Amanda J. Kiliaan,d and Inga Kadishc
aDepartment of Neuroscience and Neurology, University of Kuopio, FinlandbDepartment of Neurology, Kuopio University Hospital, FinlandcDepartment of Cell Biology, University of Alabama at Birmingham, Birmingham, AL 35294-0006, USAdDepartment of Anatomy, Radboud University Nijmegen Medical Centre, 6525 EZ Nijmegen, The Netherlands
Received 21 February 2006; revised 9 May 2006; accepted 18 May 2006Available online 10 July 2006
The deposition of amyloid β (Aβ) peptides and neurofibrillary tanglesare the two characteristic pathological features of Alzheimer's disease(AD). To investigate the relation between amyloid precursor protein(APP) production, amyloid β deposition and the type of Aβ indeposits, i.e., human and/or mouse, we performed a histopathologicalanalysis, using mouse and human specific antibodies, of the neocortexand hippocampus in 6, 12 and 19 months old APP/PS1 double andAPP and PS1 single transgenic mice. There was a significantcorrelation between the human amyloid β deposits and the intrinsicrodent amyloid β deposits, that is, all plaques contained both humanand mouse Aβ, and the diffuse amyloid β deposits also colocalizedhuman and mouse Aβ. Furthermore, some blood vessels (mainlyleptomeningeal vessels) show labeling with human Aβ, and most ofthese vessels also label with mouse Aβ. Our findings demonstrate thatthe human amyloid deposits in APP/PS1 transgenic mice are closelyassociated with mouse Aβ, however, they do not precisely overlap. Forinstance, the core of plaques consists of primarily human Aβ, whereasthe rim of the plaque contains both human and mouse amyloid β,similarly, human and mouse Aβ are differentially localized in theblood vessel wall. Finally, as early as amyloid β deposits can bedetected, they show the presence of both human and mouse Aβ.Together, these data indicate that mouse Aβ is formed and deposited insignificant amounts in the AD mouse brain and that it is depositedtogether with the human Aβ.© 2006 Elsevier Inc. All rights reserved.
Keywords: Alzheimer's disease; Transgenic mice; Amyloid precursorprotein; Hippocampus; Amyloid deposition
⁎ Corresponding author. Current address: Dept. Cell Biology, Universityof Alabama at Birmingham, 1900 University Boulevard, THT 912,Birmingham, AL 35294-0006, USA. Fax: +1 205 934 7029.
E-mail address: [email protected] (T. van Groen).Available online on ScienceDirect (www.sciencedirect.com).
0969-9961/$ - see front matter © 2006 Elsevier Inc. All rights reserved.doi:10.1016/j.nbd.2006.05.010
Introduction
Alzheimer's disease (AD) is characterized pathologically by thepresence of large numbers of neuritic plaques and neurofibrillarytangles (e.g., Braak and Braak, 1991, 1998). Whereas tanglesconsist of the peptide tau, neuritic plaques primarily consist ofdeposits of amyloid β (Aβ), a 39–43 amino acid peptide that isderived through the processing of amyloid precursor protein (APP;e.g., Selkoe, 2004).
Plaques are formed by dense, aggregated deposits of Aβpeptides, but diffuse amyloid deposits have also been shown to bepresent in the brain (Braak and Braak, 1991; Dickson and Vickers,2001; Van Groen et al., 2003). It is still uncertain how this amyloidneuropathology and the development of AD are related to the change(s) in the processing of APP in AD patients. Transgenic miceexpressing mutated human genes associated with familial forms ofAD offer a goodmodel to study the role of Aβ in AD pathology (e.g.,Janus and Westaway, 2001; German and Eisch, 2004). The presentstudy employs the double transgenic mice expressing both thehuman APPswe and PS1-A246E mutations (AP/PS mice; Borcheltet al., 1996b) and the single mutant parental lines, i.e., either theAPPswe or the PS1-A246E mutation, and the APPswe/PS1ΔE9 line(Jankowsky et al., 2001). TheAP/PSmice develop elevated levels ofthe highly fibrillogenic Aβ42 peptide with age and show amyloid βplaques starting around the age of 9 months (Borchelt et al., 1996b;Van Groen et al., 2003; Wang et al., 2003).
In these mice, early in the development of pathology, there arepredominantly dense plaques, whereas diffuse deposits developlater (Van Groen et al., 2003). We have noted that plaques showdeposition of both human and mouse APP (Van Groen et al.,2003). Therefore, we hypothesized that mouse amyloid β wouldalso be present in most amyloid deposits, and we stained the brainsof our transgenic mice with a new, mouse Aβ specific antibody.Data show that, in our AP/PS mice, all plaques that consist ofhuman amyloid are surrounded by mouse amyloid β and thatdiffuse deposits consist of intermingled human and mouse Aβ.Furthermore, both human and mouse amyloid β are deposited in

Table 1Antibodies used for staining deposits of Aβ
Antibody Specificity Concentration
#9151 (Signet) Mouse Aβ1–16 1:50004G8 (Senetek) Human and mouse Aβ17–24 + APP 1:10006E10 (Senetek) Human Aβ3–8 + APP 1:1000W0–2 (T. Hartmann) Human Aβ4–10 + APP 1:20,000BAM10 (Sigma) Human Aβ1–12 + APP 1:2000A8842 (Sigma) Human and mouse APP
(KPI domain)1.2000
22C11 (Chemicon) Human and mouse APP (aa 60–100) 1:5000FCA18
(Oncogene)Human and mouse Aβ1–x 1:33,000
APLP1(Calbiochem)
APLP1 1:20,000
APLP2(Calbiochem)
APLP2 1:100,000
654 T. van Groen et al. / Neurobiology of Disease 23 (2006) 653–662
the wall of some blood vessels, predominantly in meningealarteries and arterioles. Finally, at the earliest time point thatdeposits can be detected, they contain both human and mouse Aβ.
Materials and methods
Animals
Male and female APP and PS1 single (APP and PS1,respectively) and double (AP/PS and AP/PSΔ) transgenic micewere used in the present study. The AP/PSmice were generated frommatings between APPswe transgenic mice and HuPS1-A246Etransgenic mice, the AP/PSΔ are bi-transgenic. These mice wereoriginally produced at Johns Hopkins University (Borchelt et al.,1996a; Jankowsky et al., 2001; Baltimore, MD, USA) and are nowbred locally. Throughout the experiments, the animals were housedindividually in a controlled environment (temperature 22°C,humidity 50–60%, light from 07:00 to 19:00), food and water wereavailable ad libitum. The experiments were conducted according tothe Council of Europe (Directive 86/609) and Finnish guidelines andapproved by the State Provincial Office of Eastern Finland.
Histopathology
Mice were transcardially perfused with 0.9% saline followed by4% paraformaldehyde in 0.1 M Na–phosphate buffer. The brainswere removed and placed in the fixative for 2 h then transferred toa 30% sucrose solution, they were kept in that solution overnighton a shaker table. Six series of (1 in 6) coronal sections (35 μm)were cut through the brain using a sliding, freezing microtome. Thefirst series of sections was stained with cresyl violet, one half of thesecond series was immunohistochemically stained for human Aβusing the W0–2 antibody (mouse anti-human Aβ4–10; Dr. T.Hartmann, Heidelberg; Jensen et al., 2000). In a few cases, thisseries was stained for human Aβ with either BAM10 (human Aβ1–12; Sigma) or with FCA18 (human Aβ1–x; Oncogene; Barelli etal., 1997). The other half of the second series was immunohisto-chemically stained for mouse Aβ (rabbit anti-Aβ antibody; Signet,#9151). One half of the third series was stained with Congo Red, orthioflavine S, or thiazine red, the other half of this series wasstained with an antibody against APP (22C11; Chemicon). Onehalf of the fourth series was stained for GFAP (mouse anti-GFAP;Sigma), whereas the other half was stained for CD11b (rat anti-mouse CD11b; Serotec), a marker for microglia. The other twoseries were stored at −20°C in antifreeze for later analysis. In short,the sections were rinsed overnight in Tris-buffered saline (TBS),then the series of sections was transferred to a solution containingthe primary antibody (mouse anti-human Aβ at 1:20.000; rabbitanti-mouse Aβ at 1:5000, mouse anti-APP at 1:5000, mouse anti-GFAP at 1:1000; rat anti-mouse CD11b at 1:2000), this solutionconsists of TBS with 0.5% Triton X-100 added (TBS-T).Following incubation in this solution for 18 h on a shaker tableat room temperature (20°C) in the dark, the sections were rinsedthree times in TBS-T and transferred to the solution containing thesecondary antibody (goat anti-mouse*biotin, Sigma; goat anti-rabbit Ig*biotin, Chemicon; or sheep anti-rat Ig*biotin, Serotec) at1:500. After 2 h, the sections were rinsed three times with TBS-Tand transferred to a solution containing mouse ExtrAvidin®(Sigma), following another rinse, the sections were incubated forapproximately 3 min with Ni-enhanced DAB. All stained sectionswere mounted on slides and coverslipped with Depex (Table 1).
Double labeling
To analyze the colocalization of human Aβ (stained with eitherthe W0–2, the BAM 10 antibody or the FCA18 antibody) andmouse Aβ, secondary antibodies conjugated with alkalinephosphatase (AP; Abcam) were used, these were stained with acombination of either Fast Red (Sigma) and BCIP/NBT (Abcam)or Fast Red and Vector Blue (Vector). Similarly, the colocalizationof mouse Aβ and APP (22C11 antibody; Chemicon; C-terminalAPP, Synaptic Systems; Serotec) and mouse Aβ and APLP ([APPlike proteins], Calbiochem, Thinakaran) were examined with thesame procedure. All double-stained sections were mounted onslides, cleared in Neo-Clear (Merck) and coverslipped with Neo-Mount (Merck).
Images
The appropriate areas of the brain were digitized using aOlympus DP50 digital camera to avoid changes in lighting, allimages were acquired with the same camera setting. Furthermore,except rotation, clipping and sizing, and adjusting brightness andcontrast, no other manipulations were performed on theseimages.
Results
Specificity of the mouse Aβ antibody
For negative controls, sections containing amyloid β depositshave been processed for immunoreactivity with either: (1)omission of the primary antibody, (2) omission of the secondaryantibody; these control stainings are negative, i.e., a completeabsence of immunostaining (not illustrated). Furthermore, theselectivity of the anti-mouse Aβ antibody (Signet, #9151; Fig. 1A)has been characterized by: (1) preabsorption of the antibody withan excess of its immunizing peptide, (2) competitive binding assay,i.e., incubation of sections together with an excess of theimmunizing peptide, both treatments eliminate all immunolabeling(Fig. 1B). Furthermore, the Signet rodent Aβ antibody stains Aβdeposits in “normal” mice and PS1 mutant mice (i.e., mice that arenot expressing human APP) and in normal rats (Van Groen et al.,2005), and all of these deposits also stain with antibodies that are
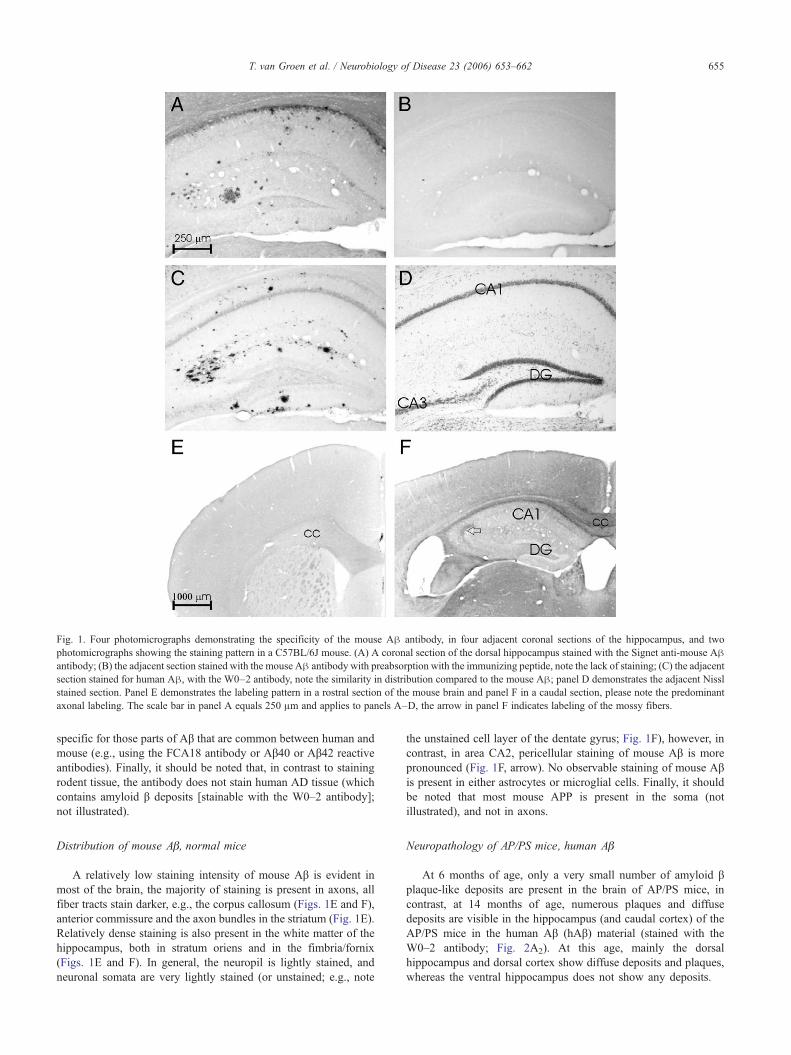
Fig. 1. Four photomicrographs demonstrating the specificity of the mouse Aβ antibody, in four adjacent coronal sections of the hippocampus, and twophotomicrographs showing the staining pattern in a C57BL/6J mouse. (A) A coronal section of the dorsal hippocampus stained with the Signet anti-mouse Aβantibody; (B) the adjacent section stained with the mouse Aβ antibody with preabsorption with the immunizing peptide, note the lack of staining; (C) the adjacentsection stained for human Aβ, with the W0–2 antibody, note the similarity in distribution compared to the mouse Aβ; panel D demonstrates the adjacent Nisslstained section. Panel E demonstrates the labeling pattern in a rostral section of the mouse brain and panel F in a caudal section, please note the predominantaxonal labeling. The scale bar in panel A equals 250 μm and applies to panels A–D, the arrow in panel F indicates labeling of the mossy fibers.
655T. van Groen et al. / Neurobiology of Disease 23 (2006) 653–662
specific for those parts of Aβ that are common between human andmouse (e.g., using the FCA18 antibody or Aβ40 or Aβ42 reactiveantibodies). Finally, it should be noted that, in contrast to stainingrodent tissue, the antibody does not stain human AD tissue (whichcontains amyloid β deposits [stainable with the W0–2 antibody];not illustrated).
Distribution of mouse Aβ, normal mice
A relatively low staining intensity of mouse Aβ is evident inmost of the brain, the majority of staining is present in axons, allfiber tracts stain darker, e.g., the corpus callosum (Figs. 1E and F),anterior commissure and the axon bundles in the striatum (Fig. 1E).Relatively dense staining is also present in the white matter of thehippocampus, both in stratum oriens and in the fimbria/fornix(Figs. 1E and F). In general, the neuropil is lightly stained, andneuronal somata are very lightly stained (or unstained; e.g., note
the unstained cell layer of the dentate gyrus; Fig. 1F), however, incontrast, in area CA2, pericellular staining of mouse Aβ is morepronounced (Fig. 1F, arrow). No observable staining of mouse Aβis present in either astrocytes or microglial cells. Finally, it shouldbe noted that most mouse APP is present in the soma (notillustrated), and not in axons.
Neuropathology of AP/PS mice, human Aβ
At 6 months of age, only a very small number of amyloid βplaque-like deposits are present in the brain of AP/PS mice, incontrast, at 14 months of age, numerous plaques and diffusedeposits are visible in the hippocampus (and caudal cortex) of theAP/PS mice in the human Aβ (hAβ) material (stained with theW0–2 antibody; Fig. 2A2). At this age, mainly the dorsalhippocampus and dorsal cortex show diffuse deposits and plaques,whereas the ventral hippocampus does not show any deposits.
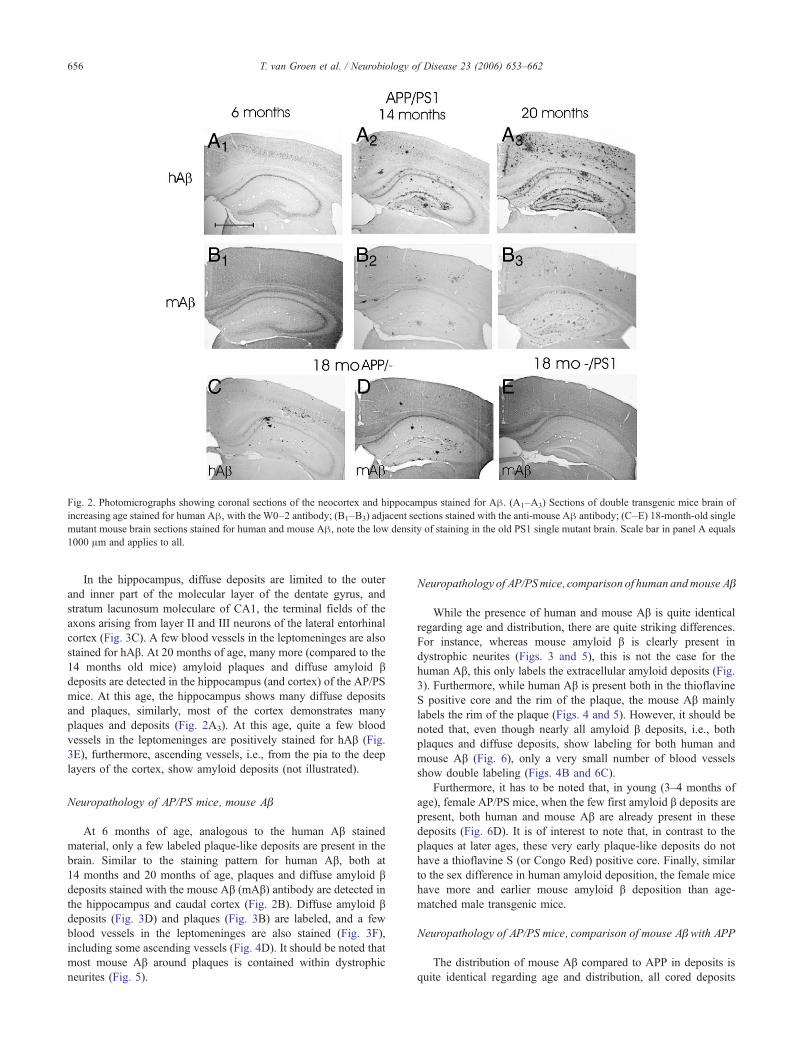
Fig. 2. Photomicrographs showing coronal sections of the neocortex and hippocampus stained for Aβ. (A1–A3) Sections of double transgenic mice brain ofincreasing age stained for human Aβ, with the W0–2 antibody; (B1–B3) adjacent sections stained with the anti-mouse Aβ antibody; (C–E) 18-month-old singlemutant mouse brain sections stained for human and mouse Aβ, note the low density of staining in the old PS1 single mutant brain. Scale bar in panel A equals1000 μm and applies to all.
656 T. van Groen et al. / Neurobiology of Disease 23 (2006) 653–662
In the hippocampus, diffuse deposits are limited to the outerand inner part of the molecular layer of the dentate gyrus, andstratum lacunosum moleculare of CA1, the terminal fields of theaxons arising from layer II and III neurons of the lateral entorhinalcortex (Fig. 3C). A few blood vessels in the leptomeninges are alsostained for hAβ. At 20 months of age, many more (compared to the14 months old mice) amyloid plaques and diffuse amyloid βdeposits are detected in the hippocampus (and cortex) of the AP/PSmice. At this age, the hippocampus shows many diffuse depositsand plaques, similarly, most of the cortex demonstrates manyplaques and deposits (Fig. 2A3). At this age, quite a few bloodvessels in the leptomeninges are positively stained for hAβ (Fig.3E), furthermore, ascending vessels, i.e., from the pia to the deeplayers of the cortex, show amyloid deposits (not illustrated).
Neuropathology of AP/PS mice, mouse Aβ
At 6 months of age, analogous to the human Aβ stainedmaterial, only a few labeled plaque-like deposits are present in thebrain. Similar to the staining pattern for human Aβ, both at14 months and 20 months of age, plaques and diffuse amyloid βdeposits stained with the mouse Aβ (mAβ) antibody are detected inthe hippocampus and caudal cortex (Fig. 2B). Diffuse amyloid βdeposits (Fig. 3D) and plaques (Fig. 3B) are labeled, and a fewblood vessels in the leptomeninges are also stained (Fig. 3F),including some ascending vessels (Fig. 4D). It should be noted thatmost mouse Aβ around plaques is contained within dystrophicneurites (Fig. 5).
Neuropathology of AP/PSmice, comparison of human andmouse Aβ
While the presence of human and mouse Aβ is quite identicalregarding age and distribution, there are quite striking differences.For instance, whereas mouse amyloid β is clearly present indystrophic neurites (Figs. 3 and 5), this is not the case for thehuman Aβ, this only labels the extracellular amyloid deposits (Fig.3). Furthermore, while human Aβ is present both in the thioflavineS positive core and the rim of the plaque, the mouse Aβ mainlylabels the rim of the plaque (Figs. 4 and 5). However, it should benoted that, even though nearly all amyloid β deposits, i.e., bothplaques and diffuse deposits, show labeling for both human andmouse Aβ (Fig. 6), only a very small number of blood vesselsshow double labeling (Figs. 4B and 6C).
Furthermore, it has to be noted that, in young (3–4 months ofage), female AP/PS mice, when the few first amyloid β deposits arepresent, both human and mouse Aβ are already present in thesedeposits (Fig. 6D). It is of interest to note that, in contrast to theplaques at later ages, these very early plaque-like deposits do nothave a thioflavine S (or Congo Red) positive core. Finally, similarto the sex difference in human amyloid deposition, the female micehave more and earlier mouse amyloid β deposition than age-matched male transgenic mice.
Neuropathology of AP/PS mice, comparison of mouse Aβ with APP
The distribution of mouse Aβ compared to APP in deposits isquite identical regarding age and distribution, all cored deposits
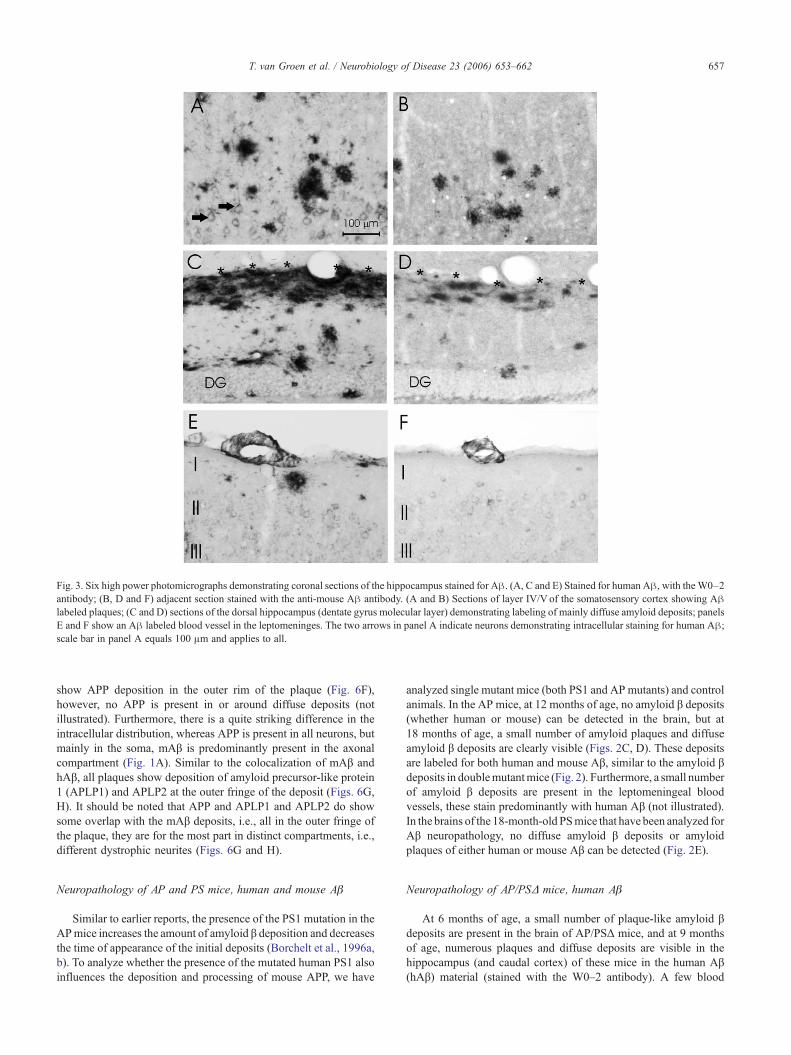
Fig. 3. Six high power photomicrographs demonstrating coronal sections of the hippocampus stained for Aβ. (A, C and E) Stained for human Aβ, with theW0–2antibody; (B, D and F) adjacent section stained with the anti-mouse Aβ antibody. (A and B) Sections of layer IV/V of the somatosensory cortex showing Aβlabeled plaques; (C and D) sections of the dorsal hippocampus (dentate gyrus molecular layer) demonstrating labeling of mainly diffuse amyloid deposits; panelsE and F show an Aβ labeled blood vessel in the leptomeninges. The two arrows in panel A indicate neurons demonstrating intracellular staining for human Aβ;scale bar in panel A equals 100 μm and applies to all.
657T. van Groen et al. / Neurobiology of Disease 23 (2006) 653–662
show APP deposition in the outer rim of the plaque (Fig. 6F),however, no APP is present in or around diffuse deposits (notillustrated). Furthermore, there is a quite striking difference in theintracellular distribution, whereas APP is present in all neurons, butmainly in the soma, mAβ is predominantly present in the axonalcompartment (Fig. 1A). Similar to the colocalization of mAβ andhAβ, all plaques show deposition of amyloid precursor-like protein1 (APLP1) and APLP2 at the outer fringe of the deposit (Figs. 6G,H). It should be noted that APP and APLP1 and APLP2 do showsome overlap with the mAβ deposits, i.e., all in the outer fringe ofthe plaque, they are for the most part in distinct compartments, i.e.,different dystrophic neurites (Figs. 6G and H).
Neuropathology of AP and PS mice, human and mouse Aβ
Similar to earlier reports, the presence of the PS1 mutation in theAPmice increases the amount of amyloid β deposition and decreasesthe time of appearance of the initial deposits (Borchelt et al., 1996a,b). To analyze whether the presence of the mutated human PS1 alsoinfluences the deposition and processing of mouse APP, we have
analyzed single mutant mice (both PS1 and AP mutants) and controlanimals. In the AP mice, at 12 months of age, no amyloid β deposits(whether human or mouse) can be detected in the brain, but at18 months of age, a small number of amyloid plaques and diffuseamyloid β deposits are clearly visible (Figs. 2C, D). These depositsare labeled for both human and mouse Aβ, similar to the amyloid βdeposits in doublemutantmice (Fig. 2). Furthermore, a small numberof amyloid β deposits are present in the leptomeningeal bloodvessels, these stain predominantly with human Aβ (not illustrated).In the brains of the 18-month-old PSmice that have been analyzed forAβ neuropathology, no diffuse amyloid β deposits or amyloidplaques of either human or mouse Aβ can be detected (Fig. 2E).
Neuropathology of AP/PSΔ mice, human Aβ
At 6 months of age, a small number of plaque-like amyloid βdeposits are present in the brain of AP/PSΔ mice, and at 9 monthsof age, numerous plaques and diffuse deposits are visible in thehippocampus (and caudal cortex) of these mice in the human Aβ(hAβ) material (stained with the W0–2 antibody). A few blood
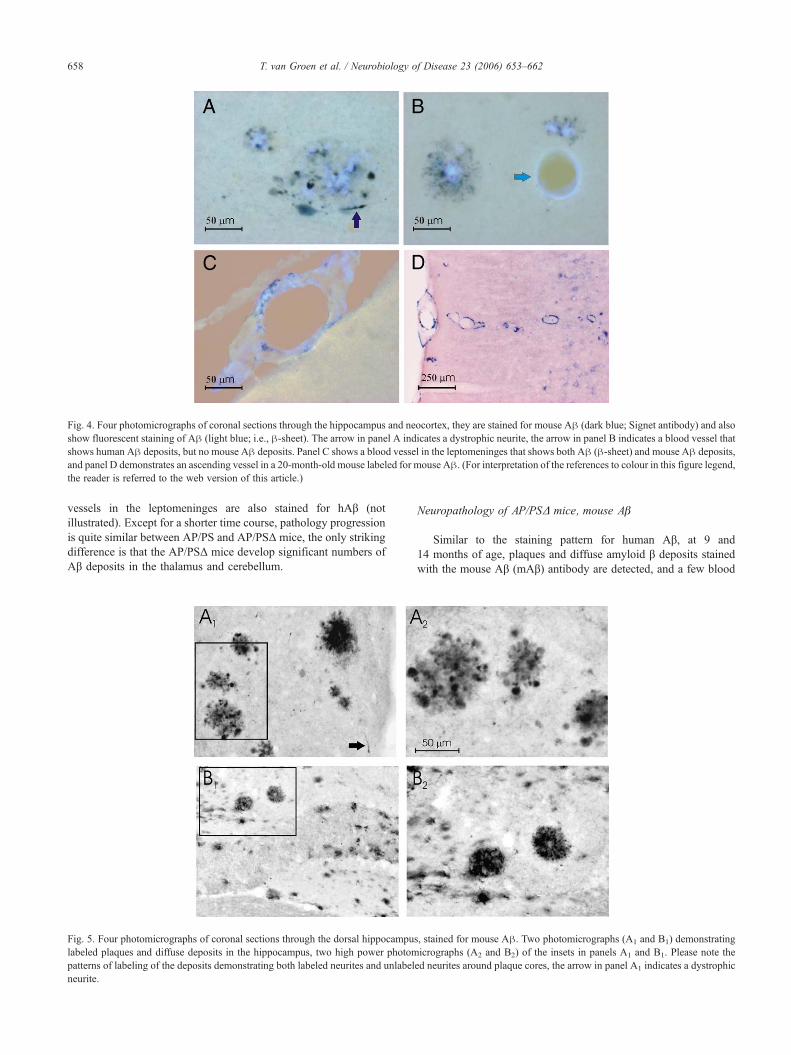
Fig. 4. Four photomicrographs of coronal sections through the hippocampus and neocortex, they are stained for mouse Aβ (dark blue; Signet antibody) and alsoshow fluorescent staining of Aβ (light blue; i.e., β-sheet). The arrow in panel A indicates a dystrophic neurite, the arrow in panel B indicates a blood vessel thatshows human Aβ deposits, but no mouse Aβ deposits. Panel C shows a blood vessel in the leptomeninges that shows both Aβ (β-sheet) and mouse Aβ deposits,and panel D demonstrates an ascending vessel in a 20-month-old mouse labeled for mouse Aβ. (For interpretation of the references to colour in this figure legend,the reader is referred to the web version of this article.)
658 T. van Groen et al. / Neurobiology of Disease 23 (2006) 653–662
vessels in the leptomeninges are also stained for hAβ (notillustrated). Except for a shorter time course, pathology progressionis quite similar between AP/PS and AP/PSΔ mice, the only strikingdifference is that the AP/PSΔ mice develop significant numbers ofAβ deposits in the thalamus and cerebellum.
Fig. 5. Four photomicrographs of coronal sections through the dorsal hippocampulabeled plaques and diffuse deposits in the hippocampus, two high power photompatterns of labeling of the deposits demonstrating both labeled neurites and unlabelneurite.
Neuropathology of AP/PSΔ mice, mouse Aβ
Similar to the staining pattern for human Aβ, at 9 and14 months of age, plaques and diffuse amyloid β deposits stainedwith the mouse Aβ (mAβ) antibody are detected, and a few blood
s, stained for mouse Aβ. Two photomicrographs (A1 and B1) demonstratingicrographs (A2 and B2) of the insets in panels A1 and B1. Please note theed neurites around plaque cores, the arrow in panel A1 indicates a dystrophic
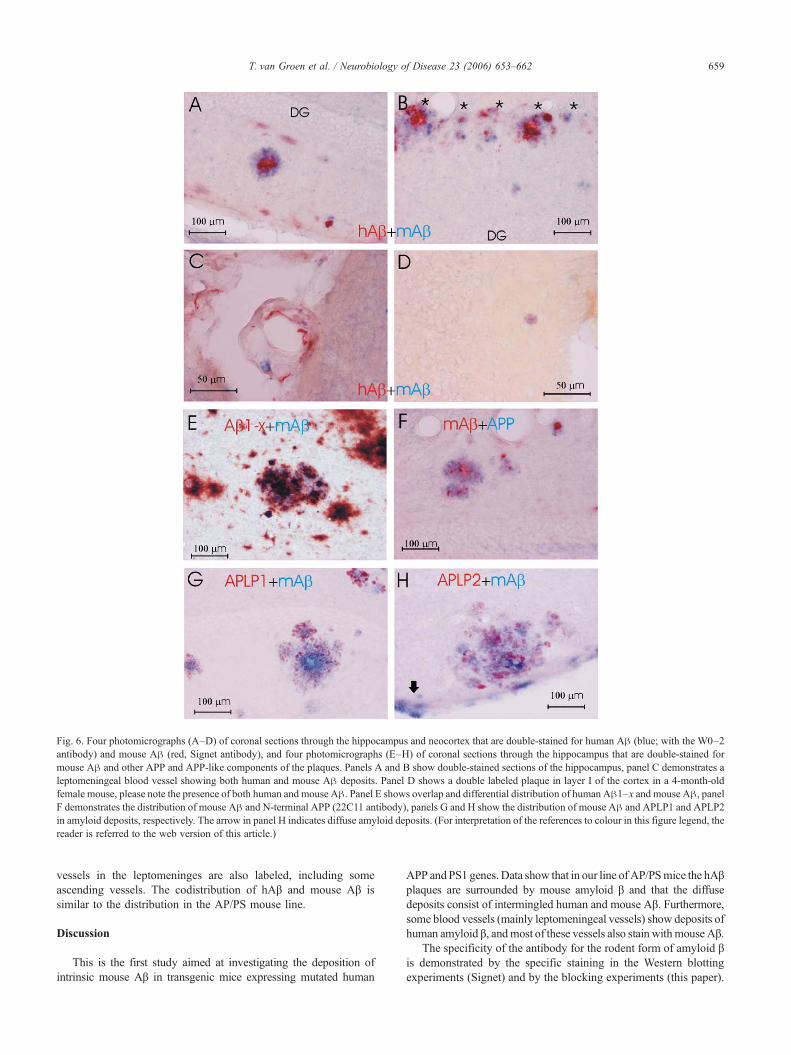
Fig. 6. Four photomicrographs (A–D) of coronal sections through the hippocampus and neocortex that are double-stained for human Aβ (blue; with the W0–2antibody) and mouse Aβ (red, Signet antibody), and four photomicrographs (E–H) of coronal sections through the hippocampus that are double-stained formouse Aβ and other APP and APP-like components of the plaques. Panels A and B show double-stained sections of the hippocampus, panel C demonstrates aleptomeningeal blood vessel showing both human and mouse Aβ deposits. Panel D shows a double labeled plaque in layer I of the cortex in a 4-month-oldfemale mouse, please note the presence of both human and mouse Aβ. Panel E shows overlap and differential distribution of human Aβ1–x and mouse Aβ, panelF demonstrates the distribution of mouse Aβ and N-terminal APP (22C11 antibody), panels G and H show the distribution of mouse Aβ and APLP1 and APLP2in amyloid deposits, respectively. The arrow in panel H indicates diffuse amyloid deposits. (For interpretation of the references to colour in this figure legend, thereader is referred to the web version of this article.)
659T. van Groen et al. / Neurobiology of Disease 23 (2006) 653–662
vessels in the leptomeninges are also labeled, including someascending vessels. The codistribution of hAβ and mouse Aβ issimilar to the distribution in the AP/PS mouse line.
Discussion
This is the first study aimed at investigating the deposition ofintrinsic mouse Aβ in transgenic mice expressing mutated human
APP andPS1 genes.Data show that in our line of AP/PSmice the hAβplaques are surrounded by mouse amyloid β and that the diffusedeposits consist of intermingled human and mouse Aβ. Furthermore,some blood vessels (mainly leptomeningeal vessels) show deposits ofhuman amyloid β, andmost of these vessels also stain withmouse Aβ.
The specificity of the antibody for the rodent form of amyloid βis demonstrated by the specific staining in the Western blottingexperiments (Signet) and by the blocking experiments (this paper).

660 T. van Groen et al. / Neurobiology of Disease 23 (2006) 653–662
Furthermore, there is a clearly differential distribution of the deposits,that is, exclusively mouse Aβ is found in dystrophic neurites.Finally, the antibody stains amyloid β deposits in non-transgenic ratsthat have sustained a period of ischemia (VanGroen et al., 2005), andit stains amyloid β deposits in injured axons in non-transgenic mice(unpublished data), furthermore, all these deposits also stain withantibodies that are specific for those parts of Aβ that are commonbetween human and mouse (e.g., the FCA18 antibody that stains thefirst aa of amyloid β).
The presence of mouse amyloid β in human amyloid β depositshas been shown before in transgenic mice expressing the LondonAPP mutation; Pype et al. (2003) using ELISAs demonstrated thatthe amyloid β deposits in these mice consisted of both forms ofamyloid. McGowan et al. (1999) also reported the presence ofmany isoforms of Aβ using both immunohistochemistry and massspectrometry in the cortex of Tg2576 mice that were crossed withTg mutant PS1 mice. Our report confirms these data in two lines ofdouble and single transgenic mice and shows the localization, anddifferential distribution, of these deposits.
Despite the fact that many studies have been done, therelationship between APP metabolism and amyloid β deposits isstill unclear. Discrepant results may be partly attributed to thedifferent time points studied and different approaches of APP/Aβdetection that were used (Iwatsubo et al., 1996). Moreover, speciesdifferences may also have an influence on APP metabolism, that is,amyloid β deposits in human AD patients differ from the Tg ADmouse amyloid β deposits (Saido et al., 1995; Iwatsubo et al., 1996;Kalback et al., 2002). Autosomal dominant early onset AD is causedby mutations of the amyloid precursor protein and the presenilin 1and 2 genes that all affect APP metabolism and Aβ proteinproduction (e.g., Czech et al., 1997; Price and Sisodia, 1998; Haassand De Strooper, 1999). Mutations in the APP and PS genes havebeen shown to alter APP processing resulting in an increase of themore amyloidogenic Aβ1–42 vs. Aβ1–40 (e.g., Borchelt et al.,1996b). Likely, this would affect the processing of the endogenousmouse APP, too, as has been demonstrated by Pype et al. (2003).
The extent of human Aβ deposition increases dramatically withage, female mice at 20 months of age can have an average Aβ loadof approximately 80% in areas of the hippocampus (Van Groen etal., 2003). Female AD mice have more Aβ deposits than male miceof the same age (Wang et al., 2003). Similarly, the mouse Aβ loadincreases with age, and, similarly, female mice have more depositsthan age-matched male mice, however, the mouse amyloid β doesnot seem to form β-sheet structures at any age point (Johnstone etal., 1991; Van Groen et al., 2005). It should be noted that, as earlyas Aβ deposits can be detected, they consist of both human andmouse Aβ. Based on the analysis of both the silver-stained and thematerial that was single and double-stained for Aβ, it appears thatboth dense core (plaques) and diffuse amyloid β deposits containhuman and mouse amyloid β. However, whereas the core,containing only human Aβ, has β-sheet staining properties, thatis, the core is Congo Red and thioflavine S positive, the outer rimof the plaque shows the presence of both β-amyloids and shows noβ-sheet staining with these agents. The diffuse amyloid β depositssimilarly are intermingled human and mouse Aβ, and they also donot have β-sheet staining characteristics (Van Groen et al., 2003).
Together, this might indicate that the deposition of mouse Aβcould interfere with the β-sheet formation by the human amyloid βand, thus, possibly the deposition of human amyloid β in thesemice. Studies have shown that “humanizing” the mouse APPsequence by replacing the three mouse amino acids in the amyloid
β part of APP changes the amyloidogenic properties of the peptide(De Strooper et al., 1995) such that it leads to increased productionof the Aβ peptide, through increased β-secretase activity. Data fromour single mutant mice overexpressing the humanized form of APP(Borchelt et al., 1996a) confirm this, that is, there is increasedhuman Aβ production and deposition. However, it should be notedthat in these mice there is also an increased deposition of mouse Aβat the sites of human Aβ deposition, and most of these amyloid βdeposits have a β-sheet positive core, that is, they are stainable withthioflavine S. In contrast, in our young double transgenic AP/PSmice, when only a few amyloid β deposits are present in the brain,the deposits consist of both human and mouse Aβ, and thesedeposits do not stain with thioflavine S, however, likely thesedeposits do not yet have a β-sheet core. The structure of thedeposits suggests that they are early “plaques”, whereas an “old”plaque has a densely stained core of human Aβ, with a“surrounding” of mouse Aβ deposits that are mainly present indystrophic neurites, these early, young plaques have a similarstructure, i.e., mouse Aβ in dystrophic neurites. Furthermore, densecore deposits, i.e., plaques with a β-sheet core, develop beforesignificant amounts of diffuse deposits are present, which consistof intermixed human and mouse Aβ (Van Groen et al., 2003).Thus, data do not indicate that the deposition of the mouse Aβinterferes with the deposition of human Aβ in plaques or with theformation of the β-sheet structure by amyloid β.
Data, however, do indicate that the presence of human Aβ leadsto concurrent deposition of mouse Aβ since none of the PS1transgenic mice, even at old age, shows any mouse Aβ deposits. Itis of interest to note that rodent Aβ deposits do form in rats thathave a long-term survival period following ischemic stroke (VanGroen et al., 2005). These deposits, however, are likely related tothe upregulated expression of APP in injured axons (e.g.,Stephenson et al., 1992; Van Groen et al., 2005). This would beconsistent with our observation that most rodent Aβ is observablein dystrophic neurites. Together, this might suggest that damageproduced by the human Aβ deposit leads to local upregulation ofthe mouse APP protein followed by Aβ production and deposition.
Our data indicate that the processing of APP is similar for boththe human and mouse forms (see above), however, the regulationof expression the APP protein is different, whereas mouse APP is“normally” regulated, the human APP is not under the control ofthe intrinsic APP promoter but is controlled by the prion proteinpromoter (Borchelt et al., 1996a). The APP 5′-untranslated regioncontains several regulatory elements, e.g., a functional heat shockelement, an AP1 site and a nuclear factor 6B element (Lahiri et al.,2005). These regulatory elements lead to different levels oftranscription in different cell types, within the brain most APPexpressed in neurons. Therefore, following cortical lesions in TgAD model mice, mouse APP expression is upregulated in damagedaxons, but not in astrocytes, whereas human APP is expressed inthe activated astrocytes surrounding the lesion and not in damagedaxons (Van Groen et al., 2005).
Dystrophic neurites surrounding plaques are also stainable withother markers such as APP (and APLP1 and APLP2), therefore wehave analyzed with double staining the colocalization of theseproteins with mouse Aβ. Surprisingly, all amyloid and amyloid-like proteins we analyzed with immunohistochemical doublestaining are found at similar but not totally overlapping distribu-tions at Aβ deposits. They are also present around plaque cores indystrophic neurites, where no overlap is found, suggesting thatthey are present in distinct neuronal compartments. It should be

661T. van Groen et al. / Neurobiology of Disease 23 (2006) 653–662
noted that a double protein deposition that is quite similar to thosefound in these mice has been observed in the Indiana kindred ofGerstmann–Sträussler–Scheinker disease (Bugiani et al., 1993).One of the affected members of this family carried PrP amyloiddeposits surrounded by Aβ. This double non-overlapping protei-nosis was been regarded as the effect of aging (i.e., Aβ)superimposed to, and enhanced by, the pathologic process (PrPamyloid deposition). Furthermore, uneven colocalization of Aβand other proteins (ApoE, ApoJ, etc.) is often observed inAlzheimer's disease (e.g., Aizawa et al., 1997).
In conclusion, we demonstrate that, in two lines of double andin single transgenic mice, both human and mouse Aβ aredeposited, and we show the localization of these deposits. HumanAβ and mouse Aβ are differently localized within these deposits,with human Aβ predominantly in the core of the plaque and mouseAβ preferentially located around the plaque, i.e., in the rim of theplaque. However, in diffuse amyloid β deposits, the amyloid βproteins are intermingled. Furthermore, these studies indicate that,in the mouse, without overexpression of the APP gene, over-production of Aβ can occur, similar to what is found in sporadicAlzheimer's disease.
Acknowledgments
We thank Dr. Egon von Schnier for his excellent comments onan earlier version of the manuscript and Pasi Miettinen for hisassistance with the histology. We thank J. Bertelsen (SignetLaboratories, Inc) for providing samples of the mouse Aβ antibody,and the mouse Aβ antigen protein was provided by Signet (SignetLaboratories, Inc., Dedham, MA, USA). This study was supportedby a travel grant from the Suomen Akatemia (#209716; TvG) and aresearch visit grant (#10-31-704; AK) from Zon-MW (TheNetherlands Organization for Health Research and Development).
References
Aizawa, Y., Fukatsu, R., Takamaru, Y., Tsuzuki, K., Chiba, H., Kobayashi,K., Fujii, N., Takahata, N., 1997. Amino-terminus truncated apolipo-protein E is the major species in amyloid deposits in Alzheimer'sdisease-affected brains: a possible role for apolipoprotein E inAlzheimer's disease. Brain Res. 768, 208–214.
Barelli, H., Lebeau, A., Vizzavona, J., Delaere, P., Chevallier, N., Drouot,C., Marambaud, P., Ancolio, K., Buxbaum, J.D., Khorkova, O., Heroux,J., Sahasrabudhe, S., Martinez, J., Warter, J.M., Mohr, M., Checler, F.,1997. Characterization of new polyclonal antibodies specific for 40 and42 amino acid-long amyloid beta peptides: their use to examine the cellbiology of presenilins and the immunohistochemistry of sporadicAlzheimer's disease and cerebral amyloid angiopathy cases. Mol.Med. 3, 695–707.
Borchelt, D.R., Davis, J., Fischer, M., Lee, M.K., Slunt, H.H., Ratovitsky,T., Regard, J., Copeland, N.G., Jenkins, N.A., Sisodia, S.S., Price, D.L.,1996a. A vector for expressing foreign genes in the brains and hearts oftransgenic mice. Genet. Anal. 13, 159–163.
Borchelt, D.R., Thinakaran, G., Eckman, C.B., Lee, M.K., Davenport, F.,Ratovitsky, T., Prada, C.M., Kim, G., Seekins, S., Yager, D., Slunt, H.H.,Wang, R., Seeger, M., Levey, A.I., Gandy, S.E., Copeland, N.G.,Jenkins, N.A., Price, D.L., Younkin, S.G., Sisodia, S.S., 1996b. FamilialAlzheimer's disease-linked presenilin 1 variants elevate Aβ1–42/1–40ratio in vitro and in vivo. Neuron 17, 1005–1013.
Braak, H., Braak, E., 1991. Neuropathological stageing of Alzheimer-relatedchanges. Acta Neuropathol. 82, 239–259.
Braak, H., Braak, E., 1998. Evolution of neuronal changes in the course ofAlzheimer's disease. J. Neural Transm. 53, 127–140.
Bugiani, O., Giaccone, G., Verga, L., Pollo, B., Frangione, B., Farlow, M.R.,Tagliavini, F., Ghetti, B., 1993. Beta PP participates in PrP-amyloidplaques of Gerstmann–Straussler–Scheinker disease, Indiana kindred. J.Neuropathol. Exp. Neurol. 52, 64–67.
Czech, C., Delaere, P., Macq, A.F., Reibaud, M., Dreisler, S., Touchet, N.,Schombert, B., Mazadier, M., Mercken, L., Theisen, M., Pradier, L.,Octave, J.N., Beyreuther, K., Tremp, G., 1997. Proteolytical processingof mutated human amyloid precursor protein in transgenic mice. Mol.Brain Res. 47, 108–116.
De Strooper, B., Simons, M., Multhaup, G., Van Leuven, F., Beyreuther, K.,Dotti, C.G., 1995. Production of intracellular amyloid-containingfragments in hippocampal neurons expressing human amyloid precursorprotein and protection against amyloidogenesis by subtle amino acidsubstitutions in the rodent sequence. EMBO J. 14, 4932–4938.
Dickson, T.C., Vickers, J.C., 2001. The morphological phenotype of beta-amyloid plaques and associated neuritic changes in Alzheimer's disease.Neuroscience 105, 99–107.
German, D.C., Eisch, A.J., 2004. Mouse models of Alzheimer's disease:insight into treatment. Rev. Neurosci. 15, 353–369.
Haass, C., De Strooper, B., 1999. The presenilins in Alzheimer's disease—Proteolysis holds the key. Science 286, 916–919.
Iwatsubo, T., Saido, T.C., Mann, D.M., Lee, V.M., Trojanowski, J.Q., 1996.Full-length amyloid-beta (1–42(43)) and amino-terminally modified andtruncated amyloid-beta 42(43) deposit in diffuse plaques. Am. J. Pathol.149, 1823–1830.
Jankowsky, J.L., Slunt, H.H., Ratovitski, T., Jenkins, N.A., Copeland, N.G.,Borchelt, D.R., 2001. Co-expression of multiple transgenes in mouseCNS: a comparison of strategies. Biomol. Eng. 17, 157–165.
Janus, C., Westaway, D., 2001. Transgenic mouse models of Alzheimer'sdisease. Physiol. Behav. 73, 873–886.
Jensen, M., Hartmann, T., Engvall, B., Wang, R., Uljon, S.N., Sennvik,K., Naslund, J., Muehlhauser, F., Nordstedt, C., Beyeuther, K.,Lannfelt, L., 2000. Quantification of Alzheimer amyloid beta peptidesending at residues 40 and 42 by novel ELISA systems. Mol. Med. 6,291–302.
Johnstone, E.M., Chaney, M.O., Norris, F.H., Pascual, R., Little, S.P.,1991. Conservation of the sequence of the Alzheimer's diseaseamyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Mol. Brain Res. 10,299–305.
Kalback, W., Watson, M.D., Kokjohn, T.A., Kuo, Y.M., Weiss, N., Luehrs,D.C., Lopez, J., Brune, D., Sisodia, S.S., Staufenbiel, M., Emmerling,M., Roher, A.E., 2002. APP transgenic mice Tg2576 accumulate Abetapeptides that are distinct from the chemically modified and insolublepeptides deposited in Alzheimer's disease senile plaques. Biochemistry41, 922–928.
Lahiri, D.K., Ge, Y.W., Maloney, B., 2005. Characterization of the APPproximal promoter and 5′-untranslated regions: identification of celltype-specific domains and implications in APP gene expression andAlzheimer's disease. FASEB J. 19, 653–655.
McGowan, E., Sanders, S., Iwatsubo, T., Takeuchi, A., Saido, T., Zehr, C.,Yu, X., Uljon, S., Wang, R., Mann, D., Dickson, D., Duff, K., 1999.Amyloid phenotype characterization of transgenic mice overexpressingboth mutant amyloid precursor protein and mutant presenilin 1transgenes. Neurobiol. Dis. 6, 231–244.
Price, D.L., Sisodia, S.S., 1998. Mutant genes in familial Alzheimer'sdisease and transgenic models. Annu. Rev. Neurosci. 21, 479–505.
Pype, S., Moechars, D., Dillen, L., Mercken, M., 2003. Characterization ofamyloid beta peptides from brain extracts of transgenic mice over-expressing the London mutant of human amyloid precursor protein.J. Neurochem. 84, 602–609.
Saido, T.C., Iwatsubo, T., Mann, D.M., Shimada, H., Ihara, Y., Kawashima,S., 1995. Dominant and differential deposition of distinct beta-amyloidpeptide species, A beta N3(pE), in senile plaques. Neuron 14,457–466.
Selkoe, D.J., 2004. Alzheimer disease: mechanistic understanding predictsnovel therapies. Ann. Intern. Med. 140, 627–638.

662 T. van Groen et al. / Neurobiology of Disease 23 (2006) 653–662
Stephenson, D.T., Rash, K., Clemens, J.A., 1992. Amyloid precursor proteinaccumulates in regions of neurodegeneration following focal cerebralischemia in the rat. Brain Res. 593, 128–135.
Van Groen, T., Liu, L., Ikonen, S., Kadish, I., 2003. Diffuse amyloiddeposition, but not plaque number, is reduced in amyloid precursorprotein/presenilin 1 double-transgenic mice by pathway lesions.Neuroscience 119, 1185–1197.
Van Groen, T., Puurunen, K., Maki, H.M., Sivenius, J., Jolkkonen, J., 2005.Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion ofthe middle cerebral artery in rats. Stroke 36, 1551–1556.
Wang, J., Tanila, H., Puolivali, J., Kadish, I., van Groen, T., 2003. Genderdifferences in the amount and deposition of amyloidbeta in APPswe andPS1 double transgenic mice. Neurobiol. Dis. 14, 318–327.
Copyright © 2022 FDOKUMEN

