







Bahasa
Halaman
Hukum


ORIGINAL PAPER
Ammoxidation of ethylene to acetonitrile over chromium or cobaltalumina catalysts prepared by sol–gel method
F. Ayari Æ M. Mhamdi Æ G. Delahay ÆA. Ghorbel
Received: 19 May 2008 / Accepted: 12 November 2008 / Published online: 4 December 2008
� Springer Science+Business Media, LLC 2008
Abstract A series of Cr/Al2O3 and Co/Al2O3 catalysts were
tested in the selective ammoxidation of ethylene to acetoni-
trile. Catalysts were prepared either by sol–gel method or by
impregnation with chromium or cobalt acetylacetonate salts.
Physicochemical properties of catalysts were accomplished
by several techniques such as chemical analysis, physisorp-
tion of N2, X-ray diffraction (XRD), 27Al MAS NMR, UV–
Visible diffuse reflectance (DRS) and Raman spectroscopy
and temperature programmed reduction of H2 (H2–TPR).
Textural analysis reveals that mesoporous materials with
pronounced surface areas were obtained using sol–gel pro-
cedure while impregnation of the support produces a moderate
decrease of its surface area and pore volume. XRD analysis
confirms the presence of highly dispersed metal species which
reside essentially on the surface and measure less than 4 nm.
Furthermore, 27Al MAS NMR shows that for xerogels, part of
metal species occupies sites on/in A12O3 in close vicinity of
octahedral 27Al. This, apparently, is not the case for aerogels.
For Cr/Al2O3 catalysts, isolated Cr6?, mono and polychro-
mate species were identified using DRS, Raman Spectroscopy
and H2–TPR which seem to play a key role in the ammoxi-
dation of ethylene. Furthermore, for cobalt doped catalysts,
CoAl2O4 was identified as active phase on the basis of DRS
and H2–TPR results. From the supercritical drying, it results
generally better catalysts than catalysts calcined by ordinary
procedure which leads to inactive agglomerated Co3O4 and
CoO–Al2O3 phase.
Keywords Cobalt � Chromium � Alumina � Sol–gel �Ammoxidation � Acetonitrile
Abbreviation
ATB Aluminum-tri-sec-butoxide
SB Sec-butanol
TCD Thermal conductivity detector
1 Introduction
Sol–gel processing designates a type of highly divided
solid materials synthesis procedure, performed at low
temperature (typically T \ 100 �C) in a homogeneous
solution that contains both the metal and support precur-
sors. In general, sol–gel can allows the obtention of
materials such as glasses and ceramics with a metastable
character and very attractive physical and chemical prop-
erties not achievable by other means of low temperature
chemical synthesis [1]. Sol–gel solids have characteristics
which make them good materials for applications in many
catalytic reactions such as selective oxidation [2], selective
reduction [3], polymerization [4] and nitroxidation of ole-
fins [5], paraffins [6] and aromatics [7–10]. Catalytic
reactions can be of the acid–base or of the redox type. In
the second case redox catalytic sites are due to transition
metal atoms (Cu, Fe, Mo, V, Cr, Co) able to change their
valence state [11].
It is well known that alumina constitutes a family of
materials whose importance in the field of heterogeneous
catalysis is enormous and constantly growing. In fact,
F. Ayari (&) � M. Mhamdi � A. Ghorbel
Laboratoire de Chimie des Materiaux et Catalyse, Departement
de Chimie, Faculte des Sciences de Tunis, Campus Universitaire
Tunis ElManar, 2092 Tunis, Tunisie
e-mail: [email protected]
G. Delahay
Institut Charles Gerhardt Montpellier, UMR 5253, CNRS-UM2-
ENSCM-UM1, Eq. ‘‘Materiaux Avances pour la Catalyse et la
Sante’’ ENSCM (MACS - Site la Galera), 8, rue Ecole Normale,
34296 Montpellier Cedex 5, France
123
J Sol-Gel Sci Technol (2009) 49:170–179
DOI 10.1007/s10971-008-1860-7

Al2O3 has been extensively used as support in many cat-
alyst formulations, mainly due to their low cost, particular
texture, good thermal stability, and the important interac-
tion that it exhibits with deposited transition metals [12].
Cr/Al2O3 catalysts are active in the catalytic dehydro-
genation of alkanes. The main interest was devoted to the
dehydrogenation of propane and isobutane. In fact, propene
is used like precursor for the synthesis of high-purity
polypropylene, whereas, isobutene is needed for the syn-
thesis of some gasoline additives (like MTBE and ETBE)
[13–15]. Co/Al2O3 catalysts have important application in
Fischer-Tropsch synthesis [16–18] which is a promising
process on the conversion from natural gas and coal to oil
products, and exhibited in particular appreciable capacity
for the selective catalytic reduction (SCR) of NO with
propane or propene in the presence of oxygen [19–21].
Some works [19, 20, 22, 23] reveal that an optimum of NOx
SCR activity was observed in the case of Co/Al2O3 cata-
lysts prepared by sol-gel method.
Selective ammoxidation of light hydrocarbons (C2) to
acetonitrile is of a prime practical signification as it could
substitute the source for synthesis of a valuable nitrile cur-
rently produced from hazardous, high cost, and less-selective
processes. First, Li and Armor [24, 25], lately Wichterlova
and co-workers [26], and recently Ghorbel and co-workers
[27] reported that ethane or ethylene can be efficiently con-
verted to acetonitrile in the presence of ammonia and oxygen
over cobalt exchanged zeolite catalysts.
During the course of this work, a series of Cr/Al2O3 and
Co/Al2O3 catalysts were prepared using either the sol–gel
or impregnation methods, and then tested in the ammoxi-
dation of ethylene to acetonitrile. Impregnation method
was performed using aerogel support in the order to syn-
thesise reproducible, homogeneous and thermally resistant
materials [28]. The texture of catalysts was studied by
physical adsorption of N2 at liquid nitrogen temperature to
determine total surface areas and porosity. XRD and 27Al
MAS NMR were used to monitor the structure of catalysts,
while DRS, Raman and H2–TPR were used to elucidate the
metal coordination and the oxidation state. Catalytic
behavior of solids in the ammoxidation reaction is then
reported. This includes selectivity and activity results
versus the reaction temperature, the nature of the metal
incorporated, and the method of catalyst preparation.
2 Experimental
2.1 Catalysts preparation
Sol–gel derived materials were prepared as follow: ATB
(Acros 97%) was dissolved at room temperature in SB
(Acros 99%) for 1 hour. An appropriate amount of
chromium or cobalt metal, issued from chromium (III)
acetylacetonate (Acros 97%) or cobalt (II) acetylacetonate
(Merk 98%) respectively, was then added to the alcoholic
solution of aluminum alkoxide and the stirring was con-
tinued for a period of 1 h. A controlled amount of water
was added dropwise into the solution for hydrolysis. The
system gelled in a few minutes, resulting in a consistent
and coloured gel depending on the nature of the metal.
The molar ratios of reactants were SB/ATB = 1/0.1, H2O/
ATB = 0.15/0.1, Cr/ATB = 0.007/0.1, and Co/ATB =
0.006/0.1. Drying procedure is done either in supercritical
conditions of solvent or by evaporating under normal
conditions. In the first way, the gel was placed inside an
autoclave (Prolabo) that was sealed and heated to 275 �C,
resulting in pressure rise at 41 bar. The sample was
maintained for 0.5 h under supercritical conditions and the
aerogel (Ae) was obtained after solvent evacuation. Fur-
thermore, xerogel (Xe) is obtained after SB evaporation in
oven at 120 �C for 24 h. Aerogel and xerogel samples were
calcined in oxygen at 500 �C for 3 h (1.8 L/h, 2 �C/mn).
Wherein, this group of catalysts was referred to us by
AlAeMSG and AlXeMSG, where Al stands to alumina, M
to chromium or cobalt, and SG to sol–gel. For impregna-
tion, alumina aerogel was prepared according to the same
procedure described above but no metal salts were added to
the alcoholic solution of ATB before hydrolysis. The molar
ratios of reactants were kept the same mentioned above.
Before impregnation, a calcination of alumina aerogel
support was done in oxygen stream for 3 h at 500 �C.
Impregnation were performed by mixing alumina aerogel
support with chromium (III) or cobalt (II) acetylacetonate
in a minimum amount of SB for 0.5 h. Cr/ATB and
Co/ATB molar ratios were 0.007/0.1 and 0.006/0.1,
respectively, which correspond to 5wt% of metal. The
mixture was then dried at 120 �C for 24 h, heated in
helium atmosphere for 12 h at 500 �C (1.8 L/h, 2 �C/mn)
and finally calcined in oxygen stream for 3 h at 500 �C.
Impregnated catalysts and alumina support were labelled
as AlAeMI and AlAe500, respectively. CrO3–Al2O3 and
Cr2O3–Al2O3 catalysts were prepared by a simple
mechanical mixture of CrO3 (Merck 99%) and Cr2O3
(Aldrich 99.9%) with commercial grade alumina to serve
as a reference in catalysis.
2.2 Catalysts characterization
The analytical metal content of catalysts was determined
by atomic absorption spectroscopy using a Solar S4, PYE
UNICAM type spectrometer. Materials were dissolved in
HF, HCl and HNO3 at room temperature for 24 h and then
heated by mild heating. The remaining residue was dis-
solved in a minimum amount of HNO3 and made up to
standard volume. SBET and porosity were determined from
J Sol-Gel Sci Technol (2009) 49:170–179 171
123

adsorption–desorption isotherms measured at 77 K in an
automatic Micromeritics Asap 2000 Analyser. Before the
measurement, the samples were outgassed at 200 �C during
7 h. BET model was used to calculate the specific surface
area. The total pore volume was calculated by means of the
total amount of adsorbed gas at P/P� = 0.98. Furthermore,
desorption isotherms were used to calculate pore size dis-
tribution by applying the BJH method. XRD patterns of
catalysts were collected on a X’Pert Pro PANalytical dif-
fractometer with CuKa radiation, k = 1.54060 A at 40 kV
and 40 mA in the range 2h of 20–70�. The diffractometer
was operated at 1.0� diverging and 0.1� receiving slits at a
scan rate of 0.05�/mn. 27Al MAS NMR spectra were
recorded at 78.20609 MHz on a Bruker WB spectrometer
using AlClO3 � 6H2O as reference. An overall 4096 free
induction decays were accumulated. The excitation pulse
and recycle time were 6 ls and 0.063 s, respectively. DRS
spectra were recorded at room temperature in the range
200–900 nm on a Perkin Elmer Lambda 45 spectropho-
tometer equipped with a DR attachment. A Certified
Reflectance Standard (Labsphere) was used as the refer-
ence material for all catalysts and DR spectra were
converted to the Kubelka-Munk function in order to con-
vert reflectance data into pseudo-absorbance. Raman
measurements were carried out on a confocal Thermo
Scientific DXR Raman Microscopy system using the visi-
ble line at 532 nm. The maximum incident power at the
sample was approximately 10 mW. TPR in H2/Ar (3%,
temperature ramped from ambient to 1000 �C at 15�C/min)
was performed with 0.04 g of catalyst using a Micromer-
itics Autochem 2910 Analyser, in a Pyrex U-tube reactor
and with an on line TCD. The TCD, connected to a PC
computer, allowed studying the reduction profiles of H2
with the temperature. Catalytic experiments were per-
formed under atmospheric pressure using down flow
tubular glasses reactor. The temperature of the reactor was
controlled by a proportional band temperature controller,
using a Chromel–Alumel sensing thermocouple that was
directly attached to the outside of the reactor at the mid-
point of the catalyst bed. Measurements of the catalytic
activity of the various catalysts were performed at 450–
500 �C using an exact amount (0.1 g) of catalyst and a
reaction stream consisted of a mixture of 10 vol.% ethylene
(Air Liquid 99.995%), 10 vol.% ammonia (Air Liquid
99.96%), and 10 vol.% oxygen (Air Liquid 99.995%).The
total flow rate was then maintained at 0.1 L/mn by
appropriate adjustment of the remaining helium (Air
Liquid 99.998%) flow rate. The analysis of the outlet flow
was recorded on line by two chromatographic units, one
operated with a flame ionization detector while the other
was equipped with a TCD.
The conversion, selectivity and activity are defined as
follow:
Conversion of C2H4 : X ¼P
i yini
yEnE þP
i yini
Selectivity of product Pi Acetonitrileð Þ : Si ¼yiniPi yini
Activity of product Pi Acetonitrileð Þ : Ai ¼na � 190:97
mcatalyst
where yi and yE are the mole fractions of product Pi and
C2H4, respectively; ni and nE are the number of carbon
atoms in each molecule of product Pi and C2H4, respec-
tively, and na and mcatalyst are the mole number of produced
acetonitrile and catalyst weight, respectively. All the terms
were evaluated for the exit stream.
3 Results and discussion
3.1 Chemical analysis
The metal compositions of the different catalysts are pre-
sented in Table 1. As expected, the metal content is higher
for samples prepared by sol–gel method. Catalysts issued
from impregnation exhibit a low chromium or cobalt
retention since preparation steps result in a loss of metal.
The sublimation of metal acetylacetonate can explains both
the low Cr and Co content.
3.2 BET measurements
Nitrogen adsorption results indicate that adsorption–
desorption isotherms can be classified as type IV according
to the BDDT classification [29], and show additionally a
limited N2 uptake at low relative pressure, indicating the
presence of a mesoporous structure. The shape of the hys-
teresis as indicated by DeBoer classification [30] is similar to
the type ‘A’ hysteresis loop attributed to solids having
cylindrical pores with constant cross section. A pronounced
mesoporosity with a relatively mono-modal pore size dis-
tribution is observed for all samples, excepting AlXeCoSG
catalyst which has a bi-modal pore size distribution and point
out the presence of a second mesopore with a size close to the
Table 1 Metal content of catalysts
Catalysts Cr (wt%) Co (wt%)
AlAeCrSG 3.5 –
AlXeCrSG 3.9 –
AlAeCrI 2.1 –
AlAeCoSG – 4.2
AlXeCoSG – 3.9
AlAeCoI – 2.9
172 J Sol-Gel Sci Technol (2009) 49:170–179
123

macropore range. Alumina support exhibits the same type of
isotherm with a type ‘A’ hysteresis loop, indicating the
presence of the same network structure.
Table 2 summarises textural properties of synthesised
materials. It can be seen that SBET and Vp of support
(AlAe500) decrease after impregnation. On the other hand,
there is no noticeable change in the pore size distribution of
AlAe500 sample after impregnation, but Dp exhibits a
slight decrease after chromium doping providing the evi-
dence that some active phase is dispersed into the
intraparticle porosity of support [31]. Sol–gel derived cat-
alysts exhibit the highest SBET when compared to those of
impregnated catalysts. In fact, SBET values are in the range
of 346–430 m2/g while the higher are attributed to samples
which are dried in ordinary conditions. It can be also shown
that Vp and pore size of impregnated catalysts do not
undergo a significant change when compared to those of
sol–gel derived catalysts since the alumina support used is
also sol–gel derived.
Difference in textural properties for impregnated and
sol–gel derived catalysts can be explained as follow: the
SBET surface area of parent support undergoes a little
decrease after impregnation indicating an uniform spread-
ing of metal residues on the surface of alumina which leads
to pore blockage. The metal ions which most probably
exist at the pore entrance reduce the empty space available
for the adsorption of N2 [32, 33]. For sol–gel derived cat-
alysts, the direct inclusion of metal species to the solvated
alumina matrix during the sol–gel synthesis allowed the
formation of nanoscale primary particles which are con-
nected to form aggregates during gelation. After
calcination, these primary particles are bound together
strongly to form a solid network, and a large intraparticle
space with uniform nanoscale pore is formed and the larger
surface areas are expected [34].
3.3 XRD
Figure 1 shows the XRD patterns of Cr/Al2O3 and Co/
Al2O3 catalysts. The patterns are ascribed to an amorphous
phase in which the metal ions are dispersed at atomic level.
XRD patterns of aerogels show diffraction peaks at 2hvalues around 37, 46, and 67� which are attributed to cubic
c-Al2O3 [35]. No chromium or cobalt oxide peaks were
detected due to the high dispersion and their overlap with
those for c-Al2O3. This result suggests that metal species
measured less than 4 nm and reside essentially on the
surface. The crystallographic structure of prepared cata-
lysts is independent of the nature of metal but it largely
depends on drying method. It seems that drying in normal
conditions results in poorly crystallite phase but super-
critical drying reveals a nanosized crystallite state.
3.4 27Al MAS NMR
27Al MAS NMR spectra of Cr/Al2O3 and Co/Al2O3 cata-
lysts are shown in Figs. 2 and 3, respectively. As expected
from Fig. 2b and 3b, AlXeCrSG and AlXeCoSG NMR
spectra display a single peak at 2.31 and 2.87 ppm attrib-
uted to octahedral Al species [36–39]. Additional peak at
60 ppm, attributed to tetrahedral Al is observed for AlA-
eCrSG, AlAeCoSG and impregnated catalysts.
Most of peaks are asymmetric due to quadrupolar
interaction. It is known from the literature that paramag-
netic species (like Crn? with n \ 6 [40], or Co3O4 and CoO
[18]) quench 27Al lines in NMR [40–42] due to relaxation
phenomena [43]. The spectra of xerogels show that only
octahedral Al3? of Al2O3 is affected, suggesting that part
of paramagnetic species occupies sites on/in A12O3 in
Table 2 Textural properties of
support and catalysts
a By BET, using the equation:
Dp = 4 * Vp/SBET
b Calculated from the highest
partial-pressure adsorption pointc Most frequent pore size
Samples SBET (m2/g) Dpa (A) Vpb (cm3/g) Pore size distribution (A)
AlAe500 279 116 0.80 35–135 (52)c
AlAeCrI
AlAeCrSG
AlXeCrSG
270
385
423
112
94
87
0.75
0.90
0.90
30–140 (54)
20–160 (47)
20–140 (33)
AlAeCoI 243 122 0.75 30–150 (54)
AlAeCoSG
AlXeCoSG
346
430
112
108
0.96
1.15
20–170 (54)
20–70 (40); 100–450 (265)
Fig. 1 XRD patterns of Cr/Al2O3 and Co/Al2O3 catalysts
J Sol-Gel Sci Technol (2009) 49:170–179 173
123

close vicinity of octahedral 27Al. This, apparently, is not
the case for aerogels.
3.5 DRS
DRS spectra of Cr/Al2O3 catalysts are shown in Fig. 4; a
typical proposed deconvolution in Gaussian bands [44] for
AlAeCrI catalyst is shown in Fig. 5. Spectra consist of
absorption bands in the charge transfer (CT) region
(\500 nm) and monitor no d–d absorptions ([500 nm)
assigned to pseudo-octahedric Cr3? or (Cr2O3).
Such spectrum is typical for metallochromates [44, 45].
The bands are assigned to O2- ? Cr6? CT (see Table 3).
No dichromate band has been detected since dichromate
species occurs on acidic surface such as SiO2 (pHzc = 1–2)
[48]. Only chromate species are stable on Al2O3 surface
(pHzc is a neutral-to basic) [48]. On the other hand, the
appearance of 434–438 nm band may imply that chromium
oxide species are strongly anchored to the surface of alu-
mina [49] and are thermally stabilized since Cr(VI)
compounds are known to decompose to the trivalent state
at a temperature C400 �C [49, 50]. An esterification
reaction between chromate and hydroxyl groups of alumina
is proposed by Weckhuysen et al. [51] (Fig. 6).
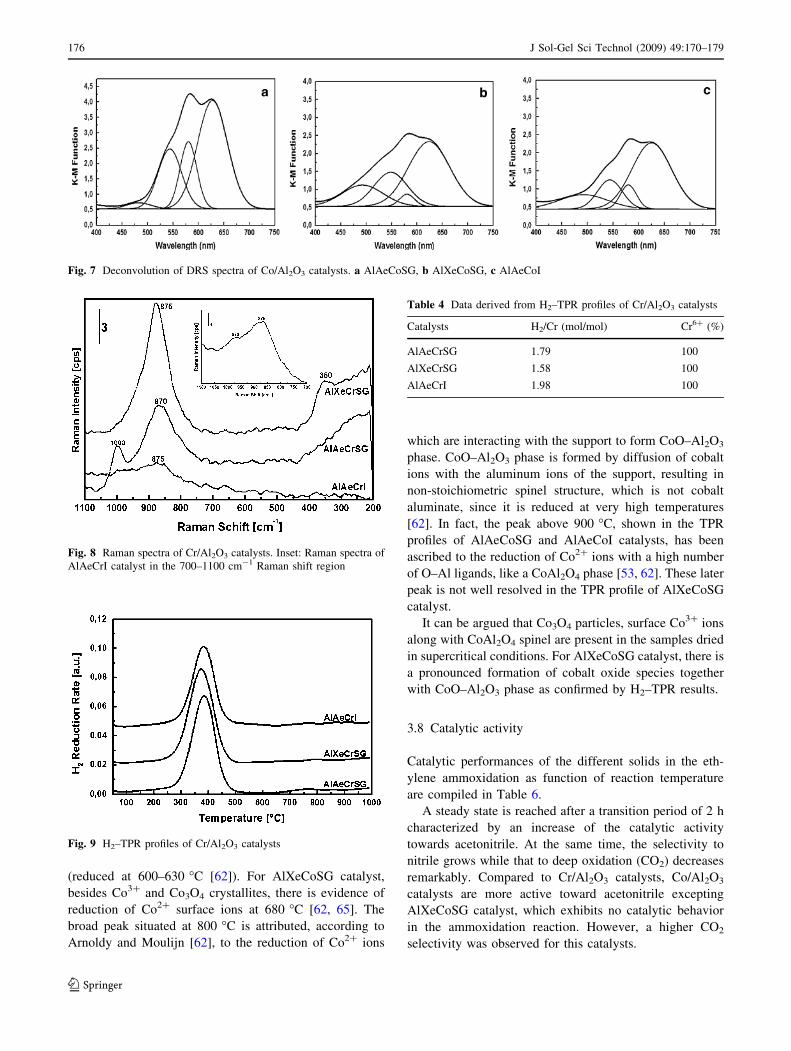
DRS spectra of Co/Al2O3 catalysts are shown in Fig. 7
with a proposed deconvolution. Since the UV–visible
spectra were collected under ambient conditions, surface
exposed cations are expected to adsorb gas molecules to
complete their coordination sphere. The spectra are char-
acterized by a triple band at around 627–623 nm (red
region), 580–579 nm (yellow orange region), and 548–
543 nm (green region), which gives rise to the blue col-
oration. This multiple bands are ascribed to the
[4A2(F) ? 4T1(P)] d–d transition of cobalt ions in
Fig. 2 27Al MAS NMR spectra of Cr/Al2O3 catalysts. a AlAeCrSG, b AlXeCrSG, c AlAeCrI
Fig. 3 27Al MAS NMR spectra of Co/Al2O3 catalysts. a AlAeCoSG, b AlXeCoSG, c AlAeCoI
Fig. 4 DRS spectra of Cr/Al2O3 catalysts (solid line) AlAeCrSG, (D)
AlXeCrSG, (h) AlAeCrI
174 J Sol-Gel Sci Technol (2009) 49:170–179
123

tetrahedral coordination as found in the spinel structure of
CoAl2O4 [52–56].
3.6 Raman
The Raman spectra of Cr/Al2O3 catalysts are presented in
Fig. 8. Alumina support does not shows any Raman fea-
tures in the 200–1100 cm-1 region and, therefore, all the
observed Raman bands are assigned to chromium species
vibrations. When we inspect Cr/Al2O3 catalysts spectra,
there is a complete absence of bands at 550 cm-1 (char-
acteristic of crystalline Cr2O3) or at 975 and 550 cm-1
(characteristic of crystalline CrO3). This result suggests
that chromium species are present as a two-dimensional
overlayer up to 5 wt% of metal [57]. The Raman shifts
observed at &875 and 350 cm-1 for AlXeCrSG catalyst
are assigned to the symmetric stretching and bending
modes for an isolated tetrahedral surface Cr(VI) oxide
species, respectively [51, 57–59]. The Raman shifts at 870–
875 and &970–1000 cm-1 for AlAeCrSG and AlAeCrI
catalysts are assigned to the symmetric stretching modes of
O–Cr–O for monomeric Cr(VI), and those of Cr=O bonds
for polymeric Cr(VI) oxide species, respectively [59, 60]. It
seems that ordinary drying leads essentially to the forma-
tion of isolated Cr6? species but mono and polychromates
are present when we use the autoclave method.
It is important to note that in the Raman spectra of
cobalt samples, no bands were detected.
3.7 H2–TPR
TPR profiles of Cr/Al2O3 catalysts were presented in
Fig. 9. On the basis of redox behavior of reference samples
and literature [61], a single peak attributed to the reduction
of Cr6? to Cr3? is observed at around 380 �C.
Quantitative analysis of TPR results is reported in
Table 4. From H2/Cr molar ratio, it is possible to give an
evaluation of Cr6? fraction in the catalysts since a total
reduction of Cr(VI) to Cr(III) necessitates 1.5 mole H2.
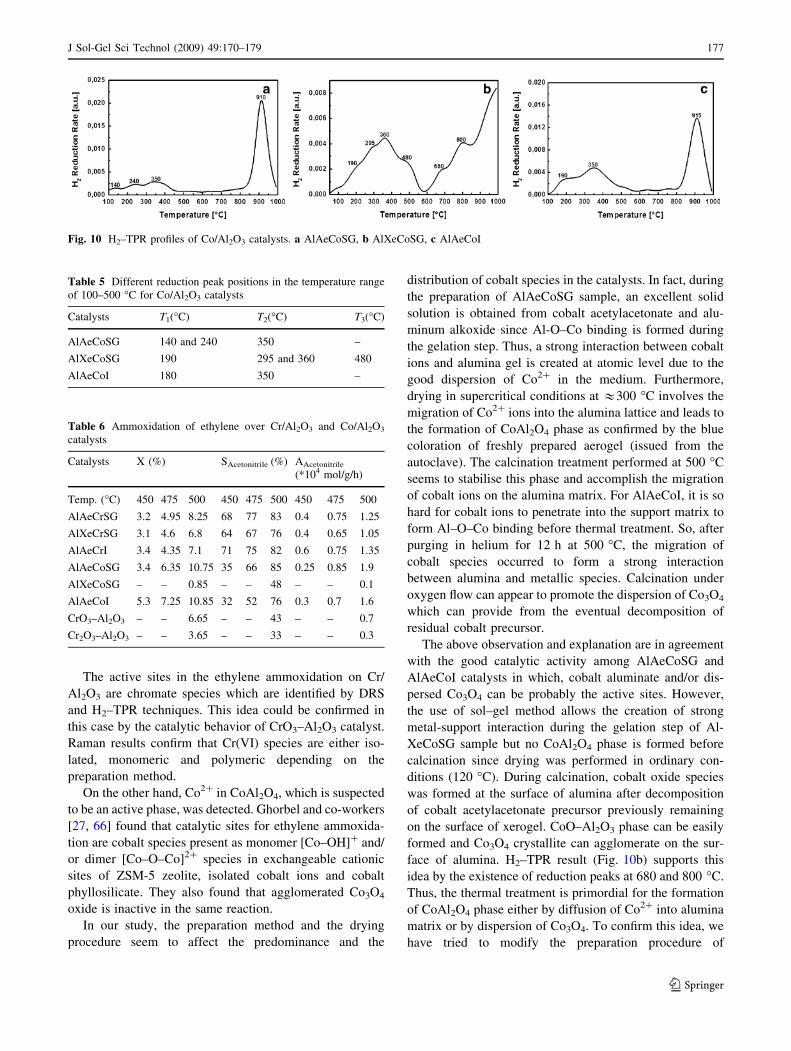
For Co/Al2O3 catalysts, TPR experiments up to 1000 �C
are illustrated in Fig. 10. Large reduction peak between
100 and 500 �C is observed for all catalysts. AlXeCoSG
catalyst exhibits in addition, a large reduction in the tem-
perature range of 600–850 �C (Fig. 10b). At relatively high
temperature (above 900 �C), a reduction peak is observed
in the TPR profiles of AlAeCoSG and AlAeCoI catalysts
(Fig. 10a, c). These later is not well resolved in the TPR
profile of AlXeCoSG catalyst.
The H2 consumption in the region of around 100–
500 �C has been attributed to the reduction of cobalt oxide,
as Co3O4 through two steps (Co3? ? Co2? ? Co0), along
with the reduction of Co3? surface ions [53, 62]. The
multiplicity of broad diffuse hydrogen consumption in this
region indicates that there are different populations of
cobalt species reducing at approximately the same tem-
perature. Table 5 illustrates the different peak positions in
the temperature range of 100–500 �C.
As mentioned in literature, reduction peak at T1 is
attributed to the reduction of a small fraction of cobalt
oxide with redox property similar to the Co3O4 bulk oxide
[53, 63, 64]. At T2, the reduction of Co3O4 weakly inter-
acted with alumina support takes place [62]. Furthermore,
Co3O4 crystallites interaction with alumina is responsible
of the apparition of reduction peak at T3 [59, 62]. Because
of the low calcination temperature, solid-state diffusion is
slow and the formation of crystalline Co3AlO6 is unlikely
Fig. 5 Deconvolution of the DRS spectrum of AlAeCrI catalyst
Table 3 DRS band positions and assignment of Cr/Al2O3 catalysts
Bands position (nm) Assignment
AlAeCrSG AlXeCrSG AlAeCrI
205 207 206 Support [45, 46]
257 249 257 (6t2 ? 2e)a [45, 47]
278 277 291 (1t1 ? 7t2)a [45, 47]
367 369 365 (1t1 ? 2e)a [45, 47]
434 434 438 (1t1 ? 2e)b [45, 47]
a Symmetry-allowedb Symmetry-forbidden
Fig. 6 Anchoring reaction of chromate on an alumina support
J Sol-Gel Sci Technol (2009) 49:170–179 175
123
(reduced at 600–630 �C [62]). For AlXeCoSG catalyst,
besides Co3? and Co3O4 crystallites, there is evidence of
reduction of Co2? surface ions at 680 �C [62, 65]. The
broad peak situated at 800 �C is attributed, according to
Arnoldy and Moulijn [62], to the reduction of Co2? ions
which are interacting with the support to form CoO–Al2O3
phase. CoO–Al2O3 phase is formed by diffusion of cobalt
ions with the aluminum ions of the support, resulting in
non-stoichiometric spinel structure, which is not cobalt
aluminate, since it is reduced at very high temperatures
[62]. In fact, the peak above 900 �C, shown in the TPR
profiles of AlAeCoSG and AlAeCoI catalysts, has been
ascribed to the reduction of Co2? ions with a high number
of O–Al ligands, like a CoAl2O4 phase [53, 62]. These later
peak is not well resolved in the TPR profile of AlXeCoSG
catalyst.
It can be argued that Co3O4 particles, surface Co3? ions
along with CoAl2O4 spinel are present in the samples dried
in supercritical conditions. For AlXeCoSG catalyst, there is
a pronounced formation of cobalt oxide species together
with CoO–Al2O3 phase as confirmed by H2–TPR results.
3.8 Catalytic activity
Catalytic performances of the different solids in the eth-
ylene ammoxidation as function of reaction temperature
are compiled in Table 6.
A steady state is reached after a transition period of 2 h
characterized by an increase of the catalytic activity
towards acetonitrile. At the same time, the selectivity to
nitrile grows while that to deep oxidation (CO2) decreases
remarkably. Compared to Cr/Al2O3 catalysts, Co/Al2O3
catalysts are more active toward acetonitrile excepting
AlXeCoSG catalyst, which exhibits no catalytic behavior
in the ammoxidation reaction. However, a higher CO2
selectivity was observed for this catalysts.
Fig. 7 Deconvolution of DRS spectra of Co/Al2O3 catalysts. a AlAeCoSG, b AlXeCoSG, c AlAeCoI
Fig. 8 Raman spectra of Cr/Al2O3 catalysts. Inset: Raman spectra of
AlAeCrI catalyst in the 700–1100 cm-1 Raman shift region
Fig. 9 H2–TPR profiles of Cr/Al2O3 catalysts
Table 4 Data derived from H2–TPR profiles of Cr/Al2O3 catalysts
Catalysts H2/Cr (mol/mol) Cr6? (%)
AlAeCrSG 1.79 100
AlXeCrSG 1.58 100
AlAeCrI 1.98 100
176 J Sol-Gel Sci Technol (2009) 49:170–179
123
The active sites in the ethylene ammoxidation on Cr/
Al2O3 are chromate species which are identified by DRS
and H2–TPR techniques. This idea could be confirmed in
this case by the catalytic behavior of CrO3–Al2O3 catalyst.
Raman results confirm that Cr(VI) species are either iso-
lated, monomeric and polymeric depending on the
preparation method.
On the other hand, Co2? in CoAl2O4, which is suspected
to be an active phase, was detected. Ghorbel and co-workers
[27, 66] found that catalytic sites for ethylene ammoxida-
tion are cobalt species present as monomer [Co–OH]? and/
or dimer [Co–O–Co]2? species in exchangeable cationic
sites of ZSM-5 zeolite, isolated cobalt ions and cobalt
phyllosilicate. They also found that agglomerated Co3O4
oxide is inactive in the same reaction.
In our study, the preparation method and the drying
procedure seem to affect the predominance and the
distribution of cobalt species in the catalysts. In fact, during
the preparation of AlAeCoSG sample, an excellent solid
solution is obtained from cobalt acetylacetonate and alu-
minum alkoxide since Al-O–Co binding is formed during
the gelation step. Thus, a strong interaction between cobalt
ions and alumina gel is created at atomic level due to the
good dispersion of Co2? in the medium. Furthermore,
drying in supercritical conditions at &300 �C involves the
migration of Co2? ions into the alumina lattice and leads to
the formation of CoAl2O4 phase as confirmed by the blue
coloration of freshly prepared aerogel (issued from the
autoclave). The calcination treatment performed at 500 �C
seems to stabilise this phase and accomplish the migration
of cobalt ions on the alumina matrix. For AlAeCoI, it is so
hard for cobalt ions to penetrate into the support matrix to
form Al–O–Co binding before thermal treatment. So, after
purging in helium for 12 h at 500 �C, the migration of
cobalt species occurred to form a strong interaction
between alumina and metallic species. Calcination under
oxygen flow can appear to promote the dispersion of Co3O4
which can provide from the eventual decomposition of
residual cobalt precursor.
The above observation and explanation are in agreement
with the good catalytic activity among AlAeCoSG and
AlAeCoI catalysts in which, cobalt aluminate and/or dis-
persed Co3O4 can be probably the active sites. However,
the use of sol–gel method allows the creation of strong
metal-support interaction during the gelation step of Al-
XeCoSG sample but no CoAl2O4 phase is formed before
calcination since drying was performed in ordinary con-
ditions (120 �C). During calcination, cobalt oxide species
was formed at the surface of alumina after decomposition
of cobalt acetylacetonate precursor previously remaining
on the surface of xerogel. CoO–Al2O3 phase can be easily
formed and Co3O4 crystallite can agglomerate on the sur-
face of alumina. H2–TPR result (Fig. 10b) supports this
idea by the existence of reduction peaks at 680 and 800 �C.
Thus, the thermal treatment is primordial for the formation
of CoAl2O4 phase either by diffusion of Co2? into alumina
matrix or by dispersion of Co3O4. To confirm this idea, we
have tried to modify the preparation procedure of
Fig. 10 H2–TPR profiles of Co/Al2O3 catalysts. a AlAeCoSG, b AlXeCoSG, c AlAeCoI
Table 5 Different reduction peak positions in the temperature range
of 100–500 �C for Co/Al2O3 catalysts
Catalysts T1(�C) T2(�C) T3(�C)
AlAeCoSG 140 and 240 350 –
AlXeCoSG 190 295 and 360 480
AlAeCoI 180 350 –
Table 6 Ammoxidation of ethylene over Cr/Al2O3 and Co/Al2O3
catalysts
Catalysts X (%) SAcetonitrile (%) AAcetonitrile
(*104 mol/g/h)
Temp. (�C) 450 475 500 450 475 500 450 475 500
AlAeCrSG
AlXeCrSG
AlAeCrI
3.2
3.1
3.4
4.95
4.6
4.35
8.25
6.8
7.1
68
64
71
77
67
75
83
76
82
0.4
0.4
0.6
0.75
0.65
0.75
1.25
1.05
1.35
AlAeCoSG
AlXeCoSG
AlAeCoI
3.4
–
5.3
6.35
–
7.25
10.75
0.85
10.85
35
–
32
66
–
52
85
48
76
0.25
–
0.3
0.85
–
0.7
1.9
0.1
1.6
CrO3–Al2O3 – – 6.65 – – 43 – – 0.7
Cr2O3–Al2O3 – – 3.65 – – 33 – – 0.3
J Sol-Gel Sci Technol (2009) 49:170–179 177
123

AlXeCoSG catalyst. In this context, the xerogel was treated
in helium atmosphere for 3 h after being dried in an oven at
120 �C. An enhancement in the activity has been observed
(at 500 �C, X = 7.44%, SAcetonitrile = 86%, AAcetonitrile =
1.47 * 10-4 mol/g/h). Nevertheless, there is no activity
with the same catalyst after purging under oxygen stream
during the reaction. This result could be due to the
agglomeration of cobalt oxide on the surface of the catalyst
which favorises the hydrocarbon combustion.
4 Conclusion
Preparation method and operating conditions such as dry-
ing and thermal treatment are important parameters which
affect the nature and concentration of species present at the
surface of the alumina support. Chromium doped alumina
show similar activity in the ammoxidation of ethylene.
After calcination, most of chromium is stabilized as Cr6?
on the alumina surface. Ordinary drying leads essentially to
the formation of isolated Cr6? species but mono and
polychromates are present when we use the autoclave
method. Furthermore, catalytic behavior of cobalt doped
alumina depends on the preparation method and the drying
procedure. Agglomerated Co3O4 and CoO–Al2O3 phase are
present on the alumina upon drying in ordinary conditions.
When drying is performed under supercritical conditions,
the CoAl2O4 phase is easily formed and the catalytic
activity was largely improved. In summary, one could
expect that cobalt containing catalysts are more active in
the ethylene ammoxidation until we choosing the appro-
priate preparation method. Nevertheless, chromium doped
catalysts seems to be more selective.
References
1. Livage J, Henry M, Sanchez C (1988) Prog Solid State Chem
18:259. doi:10.1016/0079-6786(88)90005-2
2. Matis G, Juillet F, Teichner SJ (1976) Bull Soc Chim Fr 1633
3. Willey RJ, Lai H, Peri JB (1991) J Catal 130:319. doi:10.1016/
0021-9517(91)90116-L
4. Fanelli AJ, Burlew JV, Marsh GB (1988) J Catal 116:318.
doi:10.1016/0021-9517(89)90099-7
5. Zarrouk H, Ghorbel A, Pajonk GM, Teichner SJ (1984) Proc 9th
Ibero American Symposium on Catalysis, Lisbon, July 16–21,
p 339
6. Sayari A, Ghorbel A, Pajonk GM, Teichner SJ (1981) Bull Soc
Chim Fr 220
7. Zine S, Sayari A, Ghorbel A (1987) Can J Chem Eng 65:127
8. Zine S, Ghorbel A (1990) The 2nd International Symposium of
Heterogeneous Catalysis and Fine Chemicals, Poitiers, October
2–5, p 455
9. Kadri Y, Ghorbel A, Naccache C (1995) J Chim Phys 94:1993
10. Kadri Y, Ghorbel A, Rives A, Hubaut R (1998) J Chem Soc,
Faraday Trans 94:455. doi:10.1039/a705555b
11. Pierre AC, Pajonk GM (2002) Chem Rev 102:4243. doi:10.1021/
cr0101306
12. Sanchez-Valente J, Bokhimi X, Hernandez F (2003) Langmuir
19:3583. doi:10.1021/la020423?
13. Weckhuysen BM, Schoonheydt RA (1999) Catal Today 51:223.
doi:10.1016/S0920-5861(99)00047-4
14. Poole CP, Maciver DS (1967) Adv Catal 17:223. doi:10.1016/
S0360-0564(08)60688-4
15. Kearby CP In: Emmet PH (ed) (1955) Catalysis, fundamental
principles. Reinhold, New York
16. Li JL, Zhan XD, Zhang YQ, Jacobs G, Das T, Davis BH (2002)
Appl Catal Gen 228:203. doi:10.1016/S0926-860X(01)00977-2
17. Zhang JL, Chen JG, Ren J, Sun YH (2003) Appl Catal Gen
243:121. doi:10.1016/S0926-860X(02)00541-0
18. Khodakov AY, Chu W, Fongarland P (2007) Chem Rev
107:1692. doi:10.1021/cr050972v
19. Yang YJ, Kung MC, Sachtler WMH, Kung HH (1997) J Catal
172:178. doi:10.1006/jcat.1997.1899
20. Horiuchi T, Fujiwara T, Chen L, Suzuki K, Mori T (2002) Catal
Lett 78:319. doi:10.1023/A:1014952400564
21. Inaba M, Kintaichi Y, Haneda M, Hamada H (1996) Catal Lett
39:269. doi:10.1007/BF00805594
22. Maunula T, Ahola J, Hamada H (2000) Appl Catal B 26:173.
doi:10.1016/S0926-3373(00)00118-1
23. Chen L, Horiuchi T, Mori T (2001) Catal Lett 72:71. doi:
10.1023/A:1009053614743
24. Li Y, Armor JN (1997) Chem Commun (Camb) 2013:214
25. Li Y, Armor JN (1998) J Catal 176:495. doi:10.1006/jcat.1998.
2089
26. Bulanek R, Novoveska K, Wichterlova B (2002) Appl Catal Gen
235:181. doi:10.1016/S0926-860X(02)00263-6
27. Mhamdi M, Khaddar-Zine S, Ghorbel A (2006) React Kinet Catal
Lett 88:149
28. Gonzalez RD, Lopez T, Gomez R (1997) Catal Today 35:293.
doi:10.1016/S0920-5861(96)00162-9
29. Brunauer S, Deming L, Deming W, Teller EJ (1940) J Am Chem
Soc 62:1723. doi:10.1021/ja01864a025
30. DeBoer JH, Lippens BC (1946) J Catal 3:38. doi:10.1016/0021-
9517(64)90090-9
31. Xiong H, Zhang Y, Wang S, Li J (2005) Catal Commun 6:512.
doi:10.1016/j.catcom.2005.04.018
32. Afzal M, Mahmood F (1993) Collect Czech Chem Commun
58:474. doi:10.1135/cccc19930474
33. Khattak AK, Afzal M, Saleem M, Yasmeen G, Ahmad R (2000)
Colloids Surf A Physicochem Eng Asp 162:99. doi:10.1016/
S0927-7757(99)00218-6
34. Buelna G, Lin YS (1999) Microporous Mesoporous Mater 303:59
35. Wefers K, Mirsa C (1987) In: Oxides and hydroxides of aluminum,
Alcoa Technical Paper No. 19 (Revised). Alcoa Laboratories,
Alcoa Center
36. Akitt JW (1987) In: Mason J (ed) Multinuclear NMR. Plenum,
New York
37. Bell AT, Pines A (1994) NMR techniques in catalysis. Dekker,
New York
38. Acosta S, Corria R, Leclercq D, Mutin PH, Vioux A (1994) Sol–
Gel Sci Technol 2:25. doi:10.1007/BF00486208
39. Jaymes Y, Douy A, Florian P, Massiot D, Loutures JP (1994)
Sol–Gel Sci Technol 2:367. doi:10.1007/BF00486272
40. Weckhuysen BM, De Ridder LM, Grobet RJ, Schoonheydt RA
(1995) J Phys Chem 99:320. doi:10.1021/j100001a048
41. Abragam A (1961) In: The principles of nuclear magnetism.
Clarendon Press, Oxford
42. Bloembergen N (1949) Physica 15:386. doi:10.1016/0031-8914
(49)90114-7
43. Carrington A, McLachlan AD (1967) In: Introduction to mag-
netic resonance. Harper & Row, New York
178 J Sol-Gel Sci Technol (2009) 49:170–179
123

44. Weckhuysen BM, De Ridder LM, Schoonheydt RA (1993) J Phys
Chem 97:4756. doi:10.1021/j100120a030
45. Weckhuysen BM, Verberckmoes AA, De Baets AR, Schoo-
nheydt RA (1997) J Catal 166:160–171. doi:10.1006/jcat.1997.
1518
46. Shi D, Zhao Z, Xu C, Duan A, Liu J, Dou T (2006) J Mol Catal
Chem 245:106. doi:10.1016/j.molcata.2005.09.031
47. Hillier JH, Saunders VR (1971) Chem Phys Lett 9:219.
doi:10.1016/0009-2614(71)85034-0
48. Parks GA (1965) Chem Rev 65:177. doi:10.1021/cr60234a002
49. Zaki MI, Mansour SAA, Taha F, Mekhemer GAH (1992)
Langmuir 8:727. doi:10.1021/la00038a069
50. Zaki MI, Fahim RB (1986) J Therm Anal 3:825. doi:10.1007/
BF01913553
51. Weckhuysen BM, Wachs IE, Schoonheydt RA (1996) Chem Rev
96:3327. doi:10.1021/cr940044o
52. Lever ABP (1984) Inorganic electronic spectroscopy, 2nd edn.
Elsevier, Amsterdam
53. Liotta LF, Pantaleo G, Macaluso A, Di Carlo G, Deganello G
(2003) Appl Catal Gen 245:167. doi:10.1016/S0926-860X
(02)00652-X
54. Van de Water LGA, Bezemer GL, Bergwerff JA, Versluijs-
Helder M, Weckhuysen BM, de Jong KP (2006) J Catal 242:287.
doi:10.1016/j.jcat.2006.06.004
55. Jacono RL, Cimino A, Schuit GCA (1973) Gazz Chim Ital
103:1281
56. Cava S, Tebcherani SM, Pianaro SA, Paskocimas CA, Longo E,
Varela JA (2006) Mater Chem Phys 97:102. doi:10.1016/
j.matchemphys.2005.07.057
57. Hardcastle FD, Wachs IE (1988) J Mol Catal 46:173. doi:
10.1016/0304-5102(88)85092-2
58. Cho DH, Yim SD, Cha GH, Lee JS, Kim YG, Chung JS, Nam I-S
(1998) J Phys Chem A 102:7913. doi:10.1021/jp981927c
59. Yim SD, Koh DJ, Nam I-S, Kim YG (2000) Catal Lett 64:201.
doi:10.1023/A:1019076112539
60. Gorriz OF, Cadus LE (1999) Appl Catal A 180:247. doi:10.1016/
S0926-860X(98)00344-5
61. Gaspar AB, Brito JLF, Dieguez LC (2003) J Mol Catal Chem
203:251. doi:10.1016/S1381-1169(03)00381-9
62. Arnoldy P, Moulijn JA (1985) J Catal 93:38. doi:10.1016/0021-
9517(85)90149-6
63. Van Steen E, Sewell GS, Makhothe RA, Micklethwaite C,
Manstein H, De Lange M, O’Connor CT (1996) J Catal 162:220.
doi:10.1006/jcat.1996.0279
64. Okamoto Y, Nagata K, Adachi T, Imanaka T, Inamura K, Takaya
T (1991) J Phys Chem 95:310. doi:10.1021/j100154a057
65. Simionato M, Assaf EM (2003) Mat Res 6 (short communication
No. 4)
66. Mhamdi M, Khaddar-Zine S, Ghorbel A (2008) Appl Catal Gen
337:39. doi:10.1016/j.apcata.2007.11.033
J Sol-Gel Sci Technol (2009) 49:170–179 179
123
Top Related
Copyright © 2022 FDOKUMEN

