
Whole-genome DNA methylation patterns and complex associations with gene structure and expression...
14
Whole-genome DNA methylation patterns and complex associations with gene structure and expression during flower development in Arabidopsis Hongxing Yang 1,2,† , Fang Chang 1, * ,† , Chenjiang You 1,3 , Jie Cui 1 , Genfeng Zhu 1 , Lei Wang 1,3 , Yu Zheng 4 , Ji Qi 1, * and Hong Ma 1,3, * 1 State Key Laboratory of Genetic Engineering and Collaborative Innovation Center for Genetics and Development, Institute of Plant Biology, Center for Evolutionary Biology, School of Life Sciences, Fudan University, Shanghai 200433, China, 2 Shanghai Chenshan Plant Science Research Center, Shanghai Institutes for Biological Sciences, Shanghai Chenshan Botanical Garden, Chinese Academy of Sciences, Shanghai 201602, China, 3 Ministry of Education Key Laboratory for Biodiversity Science and Ecological Engineering, Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China, and 4 New England Biolabs Inc., 240 County Road, Ipswich, MA 01938, USA Received 24 September 2014; revised 3 November 2014; accepted 6 November 2014. *For correspondence (e-mails [email protected], [email protected] and [email protected]). † These authors contributed equally to this work. SUMMARY Flower development is a complex process requiring proper spatiotemporal expression of numerous genes. Accumulating evidence indicates that epigenetic mechanisms, including DNA methylation, play essential roles in modulating gene expression. However, few studies have examined the relationship between DNA methylation and floral gene expression on a genomic scale. Here we present detailed analyses of DNA meth- ylomes at single-base resolution for three Arabidopsis floral periods: meristems, early flowers and late flow- ers. We detected 1.5 million methylcytosines, and estimated the methylation levels for 24 035 genes. We found that many cytosine sites were methylated de novo from the meristem to the early flower stage, and many sites were demethylated from early to late flowers. A comparison of the transcriptome data of the same three periods revealed that the methylation and demethylation processes were correlated with expression changes of >3000 genes, many of which are important for normal flower development. We also found different methylation patterns for three sequence contexts ( m CG, m CHG and m CHH) and in different genic regions, potentially with different roles in gene expression. Keywords: cytosine methylation, Arabidopsis DNA methylome, MspJI, RNA-seq, flower development, gene expression. INTRODUCTION Flowers are angiosperm reproductive structures, and develop from the floral meristem. The cells derived from the floral meristem undergo division and differentiation to form four types of flower organs, including the reproductive organs, stamens and pistil. Meiosis generates haploid spores that then develop into pollen grains and embryo sacs for fertilization and seed production. Flower development requires the normal function of receptor-like protein kinases and ligands, transcription factors, enzymes, and other mole- cules (Ma, 2005; Ge et al., 2010; Chang et al., 2011). Epige- netic mechanisms play essential roles by modulating the expression of numerous genes through histone modifica- tion, chromatin remodeling, microRNA-mediated mRNA degradation and DNA methylation. Studies over the past decade have revealed that these epigenetic pathways are important for normal gene expression, while DNA methyla- tion is also known to be involved in genome stability (Chan et al., 2005; Law and Jacobsen, 2010; Gan et al., 2013). Although conserved across eukaryotes, DNA methyla- tion in plants has several unique features with regard to the pattern of methylation, the methylation machinery, and demethylation enzymes in non-dividing cells (Chan et al., 2005). For example, mammalian DNA methylation occurs mostly at CG sites by the DNA methyltransferase DNMT1 and homologs. In contrast, plant DNA methylation occurs at CG, CHG and CHH sites, where H = A, T or C; in Arabid- © 2014 The Authors The Plant Journal © 2014 John Wiley & Sons Ltd 1 The Plant Journal (2014) doi: 10.1111/tpj.12726
Transcript of Whole-genome DNA methylation patterns and complex associations with gene structure and expression...

Whole-genome DNA methylation patterns and complexassociations with gene structure and expression duringflower development in Arabidopsis
Hongxing Yang1,2,†, Fang Chang1,*,†, Chenjiang You1,3, Jie Cui1, Genfeng Zhu1, Lei Wang1,3, Yu Zheng4, Ji Qi1,* and
Hong Ma1,3,*1State Key Laboratory of Genetic Engineering and Collaborative Innovation Center for Genetics and Development, Institute
of Plant Biology, Center for Evolutionary Biology, School of Life Sciences, Fudan University, Shanghai 200433, China,2Shanghai Chenshan Plant Science Research Center, Shanghai Institutes for Biological Sciences, Shanghai Chenshan
Botanical Garden, Chinese Academy of Sciences, Shanghai 201602, China,3Ministry of Education Key Laboratory for Biodiversity Science and Ecological Engineering, Institutes of Biomedical Sciences,
Fudan University, Shanghai 200032, China, and4New England Biolabs Inc., 240 County Road, Ipswich, MA 01938, USA
Received 24 September 2014; revised 3 November 2014; accepted 6 November 2014.
*For correspondence (e-mails [email protected], [email protected] and [email protected]).†These authors contributed equally to this work.
SUMMARY
Flower development is a complex process requiring proper spatiotemporal expression of numerous genes.
Accumulating evidence indicates that epigenetic mechanisms, including DNA methylation, play essential
roles in modulating gene expression. However, few studies have examined the relationship between DNA
methylation and floral gene expression on a genomic scale. Here we present detailed analyses of DNA meth-
ylomes at single-base resolution for three Arabidopsis floral periods: meristems, early flowers and late flow-
ers. We detected 1.5 million methylcytosines, and estimated the methylation levels for 24 035 genes. We
found that many cytosine sites were methylated de novo from the meristem to the early flower stage, and
many sites were demethylated from early to late flowers. A comparison of the transcriptome data of the
same three periods revealed that the methylation and demethylation processes were correlated with
expression changes of >3000 genes, many of which are important for normal flower development. We also
found different methylation patterns for three sequence contexts (mCG, mCHG and mCHH) and in different
genic regions, potentially with different roles in gene expression.
Keywords: cytosine methylation, Arabidopsis DNA methylome, MspJI, RNA-seq, flower development, gene
expression.
INTRODUCTION
Flowers are angiosperm reproductive structures, and
develop from the floral meristem. The cells derived from the
floral meristem undergo division and differentiation to form
four types of flower organs, including the reproductive
organs, stamens and pistil. Meiosis generates haploid
spores that then develop into pollen grains and embryo sacs
for fertilization and seed production. Flower development
requires the normal function of receptor-like protein kinases
and ligands, transcription factors, enzymes, and other mole-
cules (Ma, 2005; Ge et al., 2010; Chang et al., 2011). Epige-
netic mechanisms play essential roles by modulating the
expression of numerous genes through histone modifica-
tion, chromatin remodeling, microRNA-mediated mRNA
degradation and DNA methylation. Studies over the past
decade have revealed that these epigenetic pathways are
important for normal gene expression, while DNA methyla-
tion is also known to be involved in genome stability (Chan
et al., 2005; Law and Jacobsen, 2010; Gan et al., 2013).
Although conserved across eukaryotes, DNA methyla-
tion in plants has several unique features with regard to
the pattern of methylation, the methylation machinery, and
demethylation enzymes in non-dividing cells (Chan et al.,
2005). For example, mammalian DNA methylation occurs
mostly at CG sites by the DNA methyltransferase DNMT1
and homologs. In contrast, plant DNA methylation occurs
at CG, CHG and CHH sites, where H = A, T or C; in Arabid-
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd
1
The Plant Journal (2014) doi: 10.1111/tpj.12726

opsis thaliana, methylation at these three types of sites is
performed by DNA METHYLTRANSFERASE 1 (MET1), the
plant-specific CHROMOMETHYLASE 3 (CMT3), and
DOMAINS REARRANGED METHYLTRANSFERASEs
(DRMs), respectively (Chan et al., 2005). Each of the three
types of DNA methylation is crucial for development and
responses to environmental stresses (Bird, 2002; Chan
et al., 2005; Goll and Bestor, 2005; He et al., 2011; Jullien
et al., 2012; Song et al., 2013).
Consistent with the role of DNA methylation in genome
stability, a substantial proportion of the methylated cyto-
sines in Arabidopsis are found in genomic regions compris-
ing repetitive sequences such as transposable elements
(TEs) (Arabidopsis Genome Initiative, 2000; Martienssen
and Colot, 2001; Lippman et al., 2004; Chan et al., 2005).
Exogenous repetitive sequences such as transgenes may
also be methylated and induce methylation and consequent
transcriptional silencing of homologous sequences in trans-
genic lines (Mette et al., 2000; Soppe et al., 2000; Zilberman
et al., 2004; Chan et al., 2005). Case studies and genome-
wide analysis in Arabidopsis indicated that DNA methyla-
tion in promoter regions is often associated with transcrip-
tional gene silencing (Park et al., 1996; Stam et al., 1998;
Jones et al., 1999; Zhang et al., 2006; Zilberman et al.,
2007). Although genome-wide DNA methylation has been
investigated for vegetative tissues, little is known about the
patterns of DNA methylation and their association with
gene expression at various stages in plant development.
More specifically, a relationship between DNA methylation
and gene expression during flower development has not
been reported.
Whole-genome bisulfite sequencing as a gold standard
method has been applied in many studies of DNA methylo-
mes (Feng et al., 2010; Zemach et al., 2010), including Ara-
bidopsis (Cokus et al., 2008; Lister et al., 2008) and tomato
(Solanum lycopersicum) (Zhong et al., 2013). It requires
deep sequencing coverage for confident DNA methylation
calling, and thus is not cost-efficient. The recently identified
methylation-dependent endonuclease MspJI has both low
specificity in recognition sites and fixed cut distances: it rec-
ognizes 5-methylcytosine or 5-hydroxymethylcytosine in
the context of CNN(G/A), and cleaves both strands at fixed
distances (N12/N16–17) away from the modified cytosine on
the 30 side; these properties not only increase the number
of detectable methylcytosines (mCs), but also enable
identification of mCs at single-base resolution (Zheng et al.,
2010; Cohen-Karni et al., 2011; Horton et al., 2012; Huang
et al., 2013). In Arabidopsis, 48.8% of all cytosines and gua-
nines are part of CNNR/YNNG sites, of which 90.2% of
methylated sites were detected by methylation-dependent
MspJI DNA digestion combined with high-throughput
sequencing (MspJI-seq).
Here, we present detailed analyses of DNA methylomes
during Arabidopsis flower development, using MspJI-seq.
We analyzed three periods during flower development -
the meristem, early flower development (organogenesis),
and late flower development (maturation) - and detected
many more methylated cytosines in the second period
than either the first or third periods, suggesting de novo
methylation at many sites during organogenesis, followed
by demethylation at many sites. These developmental
stage-dependent methylation and demethylation activities
are correlated with changes in the expression levels of
over 3000 genes, including many genes that are important
for flower development. Moreover, the methylation pat-
terns and the potential influences on transcription vary sig-
nificantly across sequence contexts and genic regions. Our
study provides valuable insights into the possible func-
tions of DNA methylation during floral development.
RESULTS
The DNA methylation landscape during Arabidopsis floral
development
To survey the DNA methylation and gene expression dur-
ing flower development, we sampled wild-type early flow-
ers of stages 1–9 (E), wild-type late flowers of stages 10–12
(L), and meristems (M) from ap1 cal double mutant plants,
as this mutant is arrested at the inflorescence meristem
stage (Bowman et al., 1993; Ferrandiz et al., 2000), and has
been used previously as a source of meristems for tran-
scriptomic analyses (Gomez-Mena et al., 2005; Wellmer
et al., 2006; Kaufmann et al., 2010). Using high-throughput
sequencing of DNA fragments obtained by MspJI digestion
(Table S1), we identified 1 565 127 cytosines that were
potentially methylated during flower development. We ran-
domly selected nine regions of eight genes that contained
identified mC sites to perform real-time PCR-based valida-
tion, and the results confirmed the methylation status
detected by MspJI-seq (Figure S1 and Table S2). To further
validate the technical reproducibility of our MspJI-seq data,
we obtained methylation data for 44 randomly selected
genes, including the same eight genes mentioned above,
and 39 randomly selected genomic regions, by performing
bisulfite sequencing experiments on the same tissues; we
observed good consistency between MspJI-seq and bisul-
fite sequencing results (Figure S2).
Among the mC sites we identified, 453 066 were in the
CG dinucleotide context, including 207 115 that were previ-
ously detected in Arabidopsis seedlings (Cokus et al.,
2008). In addition, we detected 425 428 mCHG and 685 100mCHH sites (Figure 1a); these mC sites accounted for
17.5%, 13.7% and 5.2%, respectively, of the three types of
cytosine sites (in the CNNR context) that are potentially
digested and detected by MspJI-seq. Among detected
methylation sites, approximately 73% (1 141 758) were in
exons, 3% (46 208) were within introns, 8% (125 325) were
in putative promoter regions (1 kb region upstream of tran-
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
2 Hongxing Yang et al.
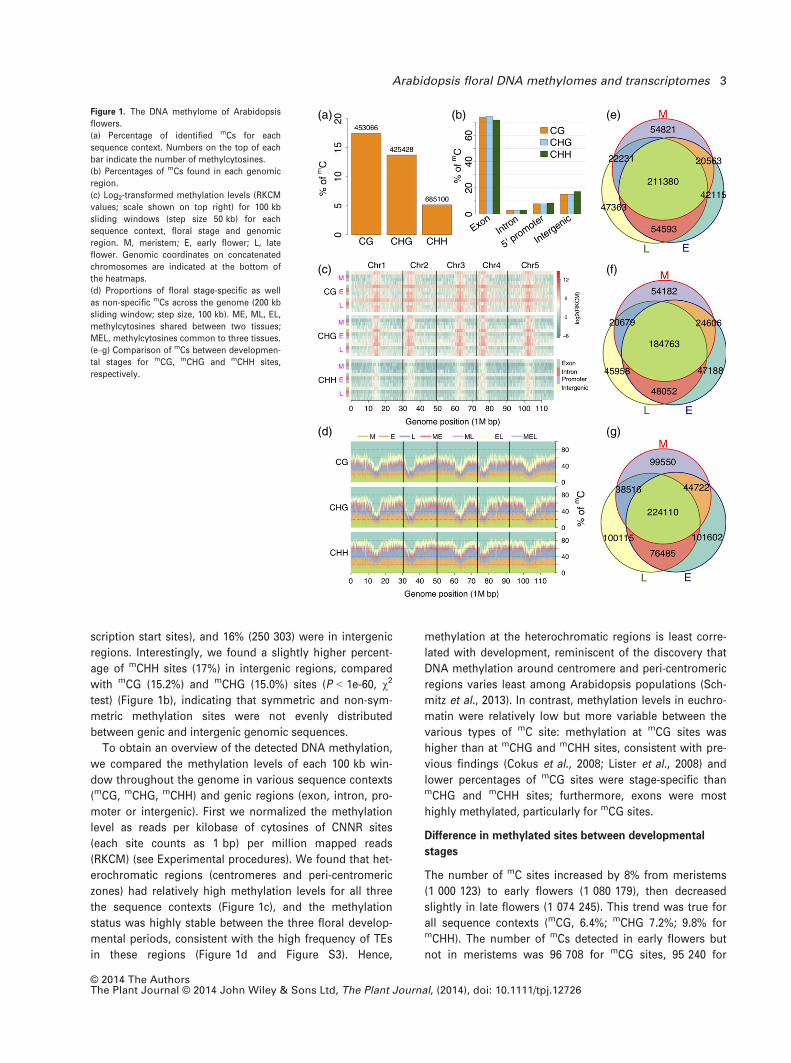
scription start sites), and 16% (250 303) were in intergenic
regions. Interestingly, we found a slightly higher percent-
age of mCHH sites (17%) in intergenic regions, compared
with mCG (15.2%) and mCHG (15.0%) sites (P < 1e-60, v2
test) (Figure 1b), indicating that symmetric and non-sym-
metric methylation sites were not evenly distributed
between genic and intergenic genomic sequences.
To obtain an overview of the detected DNA methylation,
we compared the methylation levels of each 100 kb win-
dow throughout the genome in various sequence contexts
(mCG, mCHG, mCHH) and genic regions (exon, intron, pro-
moter or intergenic). First we normalized the methylation
level as reads per kilobase of cytosines of CNNR sites
(each site counts as 1 bp) per million mapped reads
(RKCM) (see Experimental procedures). We found that het-
erochromatic regions (centromeres and peri-centromeric
zones) had relatively high methylation levels for all three
the sequence contexts (Figure 1c), and the methylation
status was highly stable between the three floral develop-
mental periods, consistent with the high frequency of TEs
in these regions (Figure 1d and Figure S3). Hence,
methylation at the heterochromatic regions is least corre-
lated with development, reminiscent of the discovery that
DNA methylation around centromere and peri-centromeric
regions varies least among Arabidopsis populations (Sch-
mitz et al., 2013). In contrast, methylation levels in euchro-
matin were relatively low but more variable between the
various types of mC site: methylation at mCG sites was
higher than at mCHG and mCHH sites, consistent with pre-
vious findings (Cokus et al., 2008; Lister et al., 2008) and
lower percentages of mCG sites were stage-specific thanmCHG and mCHH sites; furthermore, exons were most
highly methylated, particularly for mCG sites.
Difference in methylated sites between developmental
stages
The number of mC sites increased by 8% from meristems
(1 000 123) to early flowers (1 080 179), then decreased
slightly in late flowers (1 074 245). This trend was true for
all sequence contexts (mCG, 6.4%; mCHG 7.2%; 9.8% formCHH). The number of mCs detected in early flowers but
not in meristems was 96 708 for mCG sites, 95 240 for
(a)
(c)
(b) (e)
(f)
(d) (g)
Figure 1. The DNA methylome of Arabidopsis
flowers.
(a) Percentage of identified mCs for each
sequence context. Numbers on the top of each
bar indicate the number of methylcytosines.
(b) Percentages of mCs found in each genomic
region.
(c) Log2-transformed methylation levels (RKCM
values; scale shown on top right) for 100 kb
sliding windows (step size 50 kb) for each
sequence context, floral stage and genomic
region. M, meristem; E, early flower; L, late
flower. Genomic coordinates on concatenated
chromosomes are indicated at the bottom of
the heatmaps.
(d) Proportions of floral stage-specific as well
as non-specific mCs across the genome (200 kb
sliding window; step size, 100 kb). ME, ML, EL,
methylcytosines shared between two tissues;
MEL, methylcytosines common to three tissues.
(e–g) Comparison of mCs between developmen-
tal stages for mCG, mCHG and mCHH sites,
respectively.
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
Arabidopsis floral DNA methylomes and transcriptomes 3

mCHG sites, and 178 087 for mCHH sites, significantly out-
numbering mCs found in floral meristems but not in early
flowers (77 052 for mCG sites, 74 861 for mCHG sites, and
138 066 for mCHH sites) (Figure 1e–g), indicating extensive
new methylation during early flower development. The
newly methylated sites in early flowers are associated with
a large number of genes, of which 6570 genes containedmCG sites, 4570 contained mCHG sites, and 5602 containedmCHH sites (results supported by five reads or more). From
early to late flowers, only the number of mCG sites was
slightly increased (by 2.1%), whereas the numbers of mCHG
and mCHH sites were both slightly decreased by 1.7% (Fig-
ure 1e–g).
The proportion of tissue-specific mC sites also appeared
to vary across sequence contexts. In particular, 144 299mCG sites (31.8%), 147 328 mCHG sites (34.6%), and
301 267 mCHH sites (44.0%) were specific to one of the
three floral tissues. On the other hand, 32.7% of mCHH sites
were methylated in all three tissues, compared with 43.4%
and 46.7% of mCHG and mCG sites, respectively, observed
for all tissues. Therefore, we found more between-tissue
variations in number of mCHH sites than the number ofmCHG sites, which in turn was slightly more variable than
the number of mCG sites (Figure 1e–g).
Distinct gene methylation patterns during floral
development
To investigate the relationship between gene expression
and DNA methylation during floral development, we deter-
mined the positions of mC sites relative to individual genes.
In total, we found 24 035 genes with at least one mC site
supported by five reads or more (Table S3). With regard to
the three sequence contexts, we found 20 569, 17 409 and
18 746 genes containing at least one mCG, mCHG and mCHH
site, respectively. The overall methylation at gene level in
Arabidopsis flowers was compared with recently published
DNA methylome data (Schmitz et al., 2013) from mixed
stages of Arabidopsis flowers, for which >20 000 genes
were found to have at least one mC site. Over 83% of these
genes were also found in our list of methylated genes, rep-
resenting a high level of consistency even though different
ecotypes were used in these two studies (Col-0 for Schmitz
et al. and Landsberg erecta for this analysis).
We then divided genes into classes according to their
coding potential, namely protein-coding, microRNA (miR-
NA), other non-coding RNA, pseudogenes and TE genes,
and observed distinct methylation patterns for the various
classes of genes. Approximately 70% of annotated protein-
coding genes (19 182 of 27 416) were methylated in one or
more of the floral tissues, forming the largest group of
methylated genes (79.81%) (Figure S4). Only 15% of these
protein-coding genes were methylated specifically in one
of the three tissues (meristem, 1518, approximately 7.9%;
early flower, 667, approximately 3.5%; late flower, 715,
approximately 3.7%) (Figure 2a,b). Furthermore, protein-
coding genes were more likely to be methylated at mCG
sites than at mCHG and mCHH sites (mCG: 16 187, approxi-
mately 59%, mCHG, 13 060, approximately 48%, mCHH,
14 438, approximately 53%; P < 1e-100, v2 test).Consistent with the role of DNA methylation in silencing
TEs (Zhang et al., 2006; Cokus et al., 2008; Law and Jacob-
sen, 2010), almost 92% of annotated TE genes (3588 of
3903) were methylated in one or more floral tissues, with
only approximately 2.4% (93) being specific to one of the
tissues (Figure 2a,b). We found no significant differences
in methylation at the different mC sequence contexts for TE
genes (P > 0.1 for all comparisons, v2 test). In addition to
TE genes, the Arabidopsis genome is annotated as pos-
sessing 31 118 TEs, most of which do not contain genes,
but may impair genome integrity after activation by trans-
posases or reverse transposases (Law and Jacobsen,
2010). We thus examined methylation of these TEs in floral
tissues. In contrast to the broad methylation of TE genes,
we found that only 24% of TEs (7516 of 31 188) were
methylated in meristems. Previous studies revealed strong
associations between TE methylation and actions of siR-
NAs (Lister et al., 2008; Ahmed et al., 2011). To check how
often siRNAs participated in the methylation of TEs in flo-
ral tissues, we obtained siRNA sequencing data for Arabid-
opsis seedlings (Chodavarapu et al., 2010), and found that
approximately 40% TEs were potential targets of siRNAs
(Figure S5). This relatively low hit ratio may be ascribed to
tissue-specific expression of some siRNA species, the pos-
sibility that a substantial proportion of TEs was not methy-
lated via siRNAs, or the possibility that silencing of TEs
may be primarily achieved via silencing of TE genes.
Genes of different classes also showed differences in the
patterns of methylation variation during floral develop-
ment. Significantly higher proportions of mCG-containing
protein-coding genes (42.1%) than mCHG-containing pro-
tein-coding genes (28.7%) or mCHH-containing protein-cod-
ing genes (23.2%) were differentially methylated between
meristems and early flowers. Similar patterns were
observed for pseudogenes, miRNA and non-coding RNA
genes, although to a lesser extent (data not shown), sug-
gesting similar DNA methylation-related regulatory mecha-
nisms that may reflect a common evolutionary origin and/
or functional constraints.
In contrast, more mCHH-containing TE genes showed
methylation variation than mCG or mCHG TE genes. A closer
look showed that, for TE genes, methylations at mCHH sites
primarily decreased during flower development, while
those at mCG sites were mainly increased (Figure 2c). Con-
cordantly, TEs mainly showed increased methylation,
which primarily occurred at mCG sites and mCHH sites,
although only 2124 TEs (6.8%) were differentially methylat-
ed between meristem and early/late flower development
stages (Figure S5). Taken together, TEs tended to become
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
4 Hongxing Yang et al.

hypermethylated as floral development proceeded, possi-
bly because of a greater need to protect the reproductive
cells against the mutagenic activities of TEs (Slotkin et al.,
2009; Yang et al., 2011). Furthermore, CG methylation may
play critical roles in the increased methylation of TEs as well
as TE genes. TEs may be classified into various families that
may have different methylation patterns during floral devel-
opment. Indeed, the LTR/Gypsy family of retrotransposons,
comprising 13.4% of all TEs in Arabidopsis, accounted for
36.0% of methylated TEs, whereas the RC/Helitron type of
DNA transposons, representing 41.5% of all TEs, consti-
tuted only 11.7% of methylated TEs in the meristem (Figure
S5), indicating that retrotransposons are more tightly con-
trolled by methylation than DNA transposons. Furthermore,
some TE families were more likely to be differentially
methylated than others. In brief, differentially methylated
retrotransposons mainly fall into the families of LTR/Copia,
LTR/Gypsy and Line/L1, with little preference observed with
respect to mC context class; differentially methylated DNA
transposons primarily came from the families of hAT and
DNA/others (Figure S6).
Finally, we examined relative methylation levels (RKCM)
across transcribed regions and the surrounding genomic
regions. We found similar profiles to those previously
(a)
(b)
(c)
(d)
(e)
(f)
Figure 2. Distinct methylation patterns for
genes in Arabidopsis flowers.
(a) Percentage of methylated genes for each tis-
sue. The colored squares at the bottom of each
vertical bar represent mC contexts. TE, trans-
posable element; ncRNA, non-coding RNA;
miRNA, microRNA.
(b) Between-tissue comparison of methylated
genes for each gene class. The colored bar at
the bottom of each vertical bar indicate mC con-
text, as defined in (a).
(c) Percentage of differentially methylated
genes for each class (ncRNA and miRNA com-
bined) and sequence context. The plus and
minus symbols indicate increased/decreased
methylation, respectively, between meristems
and early flowers.
(d–f) Normalized methylation levels at various
stages (M, E, and L) for protein-coding genes
(shades of green), pseudogenes (purple, red
and orange) and transposable element genes
(shades of blue). TSS, transcription start site;
TES, transcription termination site.
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
Arabidopsis floral DNA methylomes and transcriptomes 5

reported (Cokus et al., 2008; Feng et al., 2010; Zemach et
al., 2010), and these were also quite similar between floral
stages. For example, the methylation of protein-coding
genes mainly occurred at mCG sites, and was confined
within genic regions. The methylation of TE genes showed
similar patterns across sequence contexts, i.e. primarily
occurred within genic regions, dramatically decreased at
the transcription start site (TSS) and after the transcription
end site (TES), and increased again at adjacent regions fur-
ther upstream of the TSS and downstream of the TES (Fig-
ure 2d–f). Methylations in pseudogenes showed similar
patterns to those for TE genes, with levels intermediate
between TE genes and protein-coding genes, consistent
with previous observations (Cokus et al., 2008). These
results suggested a primary role of MET1 (for mCG) in the
methylation maintenance of protein-coding genes, and
comparable contributions of MET1, DRM1/2 and CMT2/3
(for mCHG and mCHH) to methylation of pseudogenes and
TE genes (Chan et al., 2005; Stroud et al., 2014).
DNA methylation at different genic regions differentially
correlates with gene expression
DNA methylation in promoter regions is often associated
with transcriptional silencing (Zhang et al., 2006), but
recent studies have revealed distinct relationships between
gene expression and methylation in different genic regions
(Brenet et al., 2011). To assess this relationship, we mea-
sured gene expression profiles by RNA sequencing using
the same three tissues used for DNA methylation studies.
Transcripts of 26 764 genes were detected in at least one
floral tissue, with 5768 of them significantly differentially
expressed between the three tissues (see Experimental
procedures). To examine the relationships between expres-
sion and methylation, genes were sorted into three equal-
sized groups according to expression levels or methylation
intensities at a specific genic region (exon, intron, 1 kb
upstream/downstream of the TSS). We found a general
trend of negative associations between expression levels
and methylation levels for all mC contexts and gene
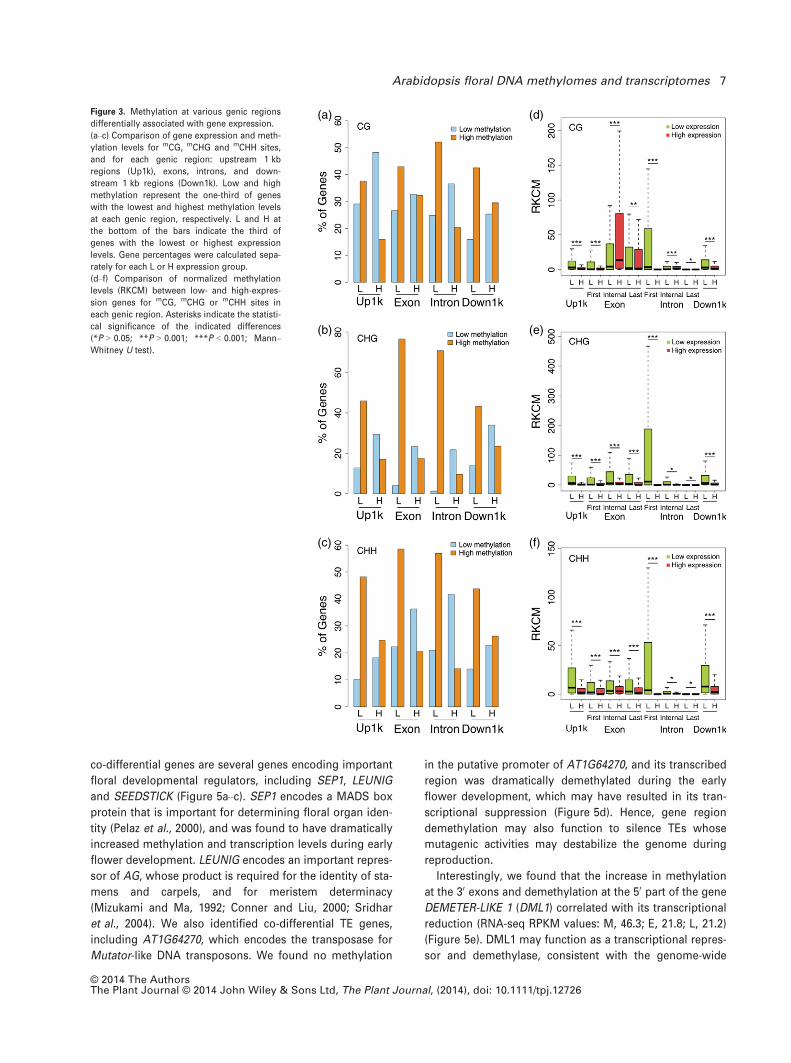
regions, especially gene body regions (Figure 3a–c).
We then compared the low- and high-expression genes
with respect to normalized methylation levels in different
genic regions, with exons and introns classified according
to their positions relative to TSS and TES sites. Consistent
with previous findings (Zhang et al., 2006; Zemach et al.,
2010), genes in the high-expression group were relatively
hypomethylated at all mC contexts in the 1 kb upstream and
downstream regions (Figure 3d–f), and first exons of genes
in the high-expression group were methylated at lower lev-
els than first exons of genes in the low-expression group,
for all mC contexts (Brenet et al., 2011; Chuang et al., 2012).
Notably, the methylation level in the first intron showed
even more significant negative correlation with expression
levels. Hence, regions near TSS may closely participate in
methylation-dependent transcriptional silencing. Further-
more, internal exons of genes with high expression tended
to be significantly highly methylated at mCG sites, but
methylated to low levels at mCHG and mCHH sites; internal
introns showed little or no association between expression
and methylation levels (Figure 3d–f). These observations
suggest that the influences of methylation on gene expres-
sion vary depending on genic region and sequence context.
DNA methylation in different sequence contexts is
differentially associated with gene expression levels
during floral development
By comparing raw read numbers and RKCM values for
each gene in each of the three sequence contexts (Figure
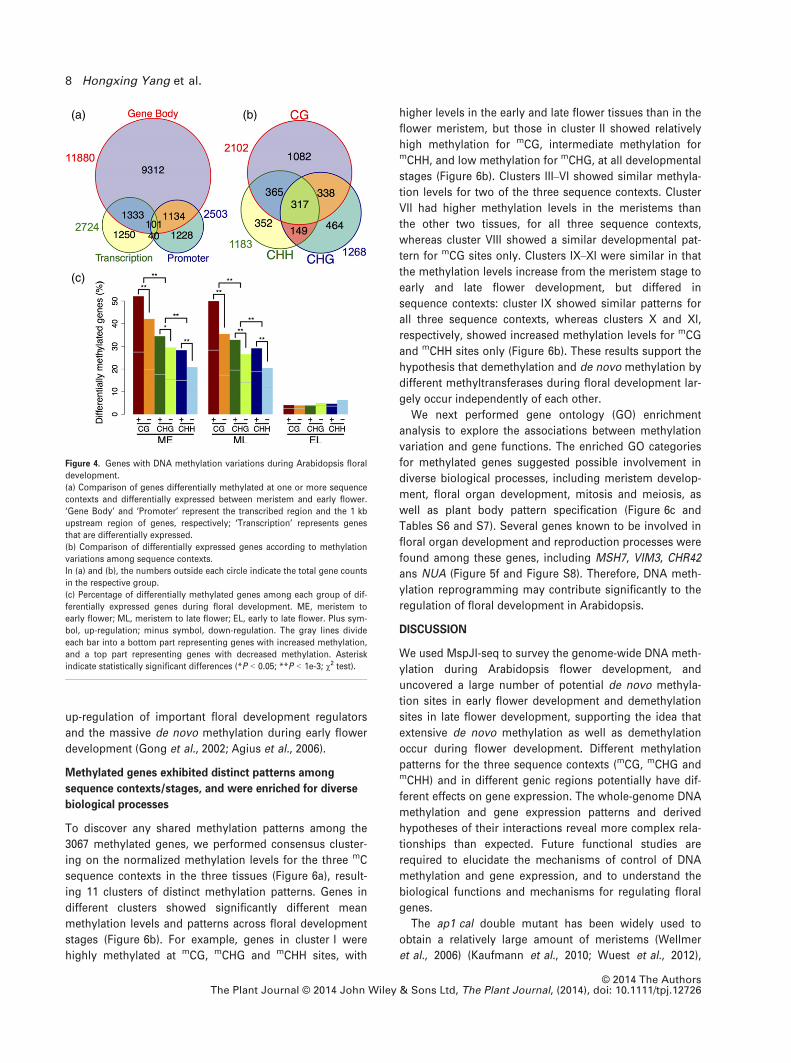
S7 and Table S3), we identified 11 880 genes with statisti-
cally significant methylation variations between meristem
and the early flower, indicating that DNA methylation in
the gene body (body methylation) is broadly regulated
across the genome (Figure 4a and Table S4). In addition,
2503 genes showed significant methylation variations in
their putative promoter regions during the same develop-
mental period, consistent with the relatively poor methyla-
tion of promoter regions (promoter methylation) (Zhang
et al., 2006). Further comparison revealed that only 1235
genes were simultaneously differentially methylated in
both body and promoter regions during early flower devel-
opment (Figure 4a), suggesting that body methylation and
promoter methylation are largely regulated separately,
possibly by independent mechanisms.
We identified 3067 genes that showed significant varia-
tions in both methylation and gene expression during floral
development (hereafter termed co-differential genes), with
only 10% (317 of 3067) being differentially methylated
across all the three sequence contexts (Figure 4b), suggest-
ing sequence context-dependent effects of methylation on
transcription. Moreover, among the 3986 genes differen-
tially expressed between meristems and early flowers, 2117
contained mCG sites, and 1048 (49.5%) of them were co-dif-
ferential at mCG sites, compared with 601 of 1744 mCHG-
containing genes (34.5%) and 509 of 1894 mCHH-containing
genes (26.9%) (Figure 4c and Table S5). We also observed
that a significantly higher proportion of transcriptionally
up-regulated genes were co-differential between meristem
and early or late flowers in comparison with genes that
were down-regulated at the same periods; this was consis-
tent across all mC sequence contexts (Figure 4c). Hence,
variation in DNA methylation levels may function as a sig-
nal affecting transcription during early flower development.
Body methylation variations may serve as important
transcriptional regulatory signals during floral
development
Our dataset provides clues to understanding how the varia-
tion in methylation level affects development. Among the
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
6 Hongxing Yang et al.
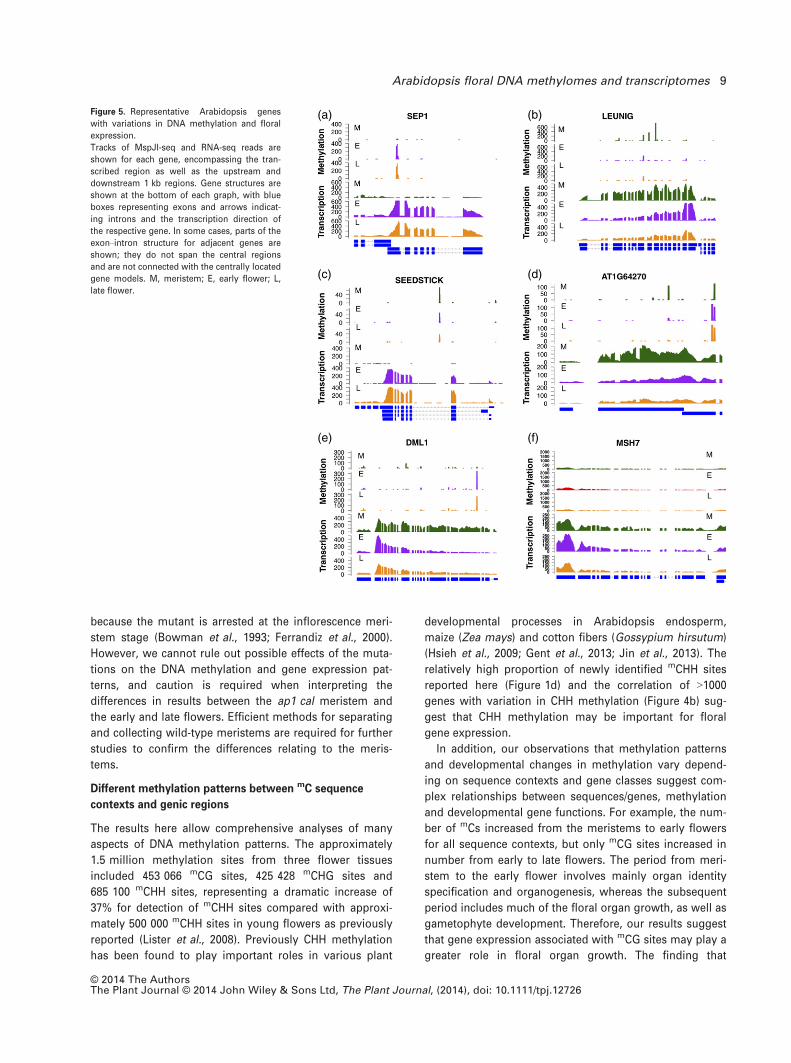
co-differential genes are several genes encoding important
floral developmental regulators, including SEP1, LEUNIG
and SEEDSTICK (Figure 5a–c). SEP1 encodes a MADS box
protein that is important for determining floral organ iden-
tity (Pelaz et al., 2000), and was found to have dramatically
increased methylation and transcription levels during early
flower development. LEUNIG encodes an important repres-
sor of AG, whose product is required for the identity of sta-
mens and carpels, and for meristem determinacy
(Mizukami and Ma, 1992; Conner and Liu, 2000; Sridhar
et al., 2004). We also identified co-differential TE genes,
including AT1G64270, which encodes the transposase for
Mutator-like DNA transposons. We found no methylation
in the putative promoter of AT1G64270, and its transcribed
region was dramatically demethylated during the early
flower development, which may have resulted in its tran-
scriptional suppression (Figure 5d). Hence, gene region
demethylation may also function to silence TEs whose
mutagenic activities may destabilize the genome during
reproduction.
Interestingly, we found that the increase in methylation
at the 30 exons and demethylation at the 50 part of the gene
DEMETER-LIKE 1 (DML1) correlated with its transcriptional
reduction (RNA-seq RPKM values: M, 46.3; E, 21.8; L, 21.2)
(Figure 5e). DML1 may function as a transcriptional repres-
sor and demethylase, consistent with the genome-wide
(a)
(b)
(c)
(d)
(e)
(f)
Figure 3. Methylation at various genic regions
differentially associated with gene expression.
(a–c) Comparison of gene expression and meth-
ylation levels for mCG, mCHG and mCHH sites,
and for each genic region: upstream 1 kb
regions (Up1k), exons, introns, and down-
stream 1 kb regions (Down1k). Low and high
methylation represent the one-third of genes
with the lowest and highest methylation levels
at each genic region, respectively. L and H at
the bottom of the bars indicate the third of
genes with the lowest or highest expression
levels. Gene percentages were calculated sepa-
rately for each L or H expression group.
(d–f) Comparison of normalized methylation
levels (RKCM) between low- and high-expres-
sion genes for mCG, mCHG or mCHH sites in
each genic region. Asterisks indicate the statisti-
cal significance of the indicated differences
(*P > 0.05; **P > 0.001; ***P < 0.001; Mann–Whitney U test).
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
Arabidopsis floral DNA methylomes and transcriptomes 7
up-regulation of important floral development regulators
and the massive de novo methylation during early flower
development (Gong et al., 2002; Agius et al., 2006).
Methylated genes exhibited distinct patterns among
sequence contexts/stages, and were enriched for diverse
biological processes
To discover any shared methylation patterns among the
3067 methylated genes, we performed consensus cluster-
ing on the normalized methylation levels for the three mC
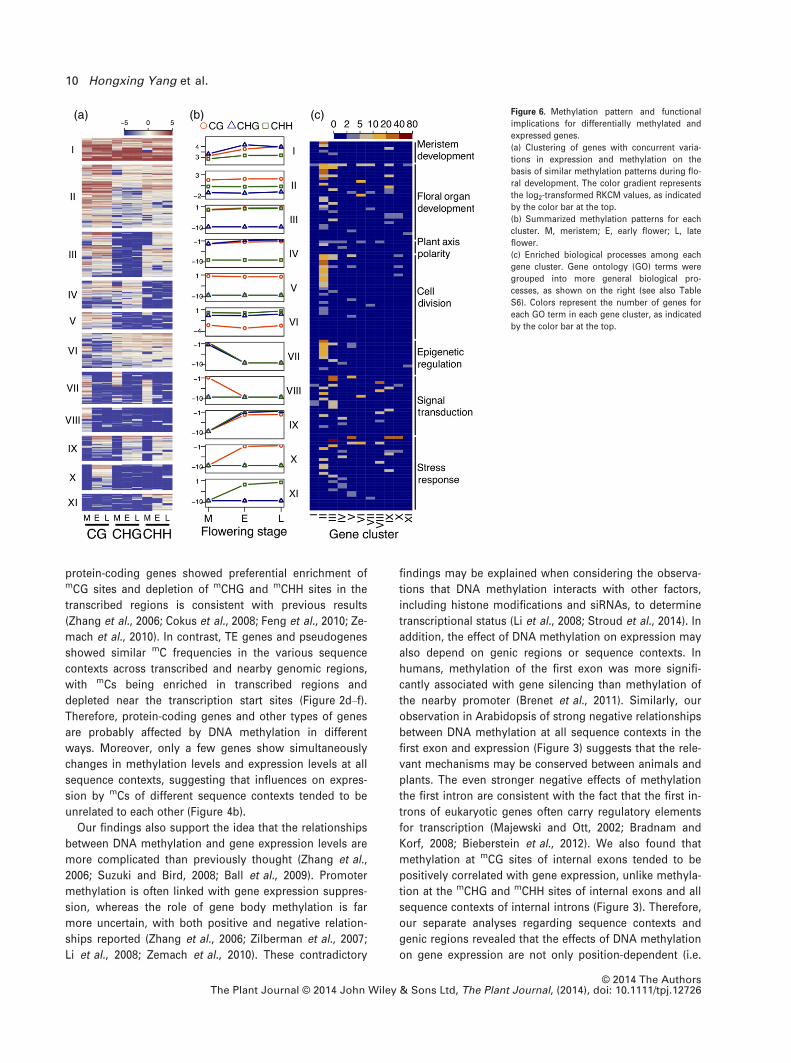
sequence contexts in the three tissues (Figure 6a), result-
ing 11 clusters of distinct methylation patterns. Genes in
different clusters showed significantly different mean
methylation levels and patterns across floral development
stages (Figure 6b). For example, genes in cluster I were
highly methylated at mCG, mCHG and mCHH sites, with
higher levels in the early and late flower tissues than in the
flower meristem, but those in cluster II showed relatively
high methylation for mCG, intermediate methylation formCHH, and low methylation for mCHG, at all developmental
stages (Figure 6b). Clusters III–VI showed similar methyla-
tion levels for two of the three sequence contexts. Cluster
VII had higher methylation levels in the meristems than
the other two tissues, for all three sequence contexts,
whereas cluster VIII showed a similar developmental pat-
tern for mCG sites only. Clusters IX–XI were similar in that
the methylation levels increase from the meristem stage to
early and late flower development, but differed in
sequence contexts: cluster IX showed similar patterns for
all three sequence contexts, whereas clusters X and XI,
respectively, showed increased methylation levels for mCG
and mCHH sites only (Figure 6b). These results support the
hypothesis that demethylation and de novo methylation by
different methyltransferases during floral development lar-
gely occur independently of each other.
We next performed gene ontology (GO) enrichment
analysis to explore the associations between methylation
variation and gene functions. The enriched GO categories
for methylated genes suggested possible involvement in
diverse biological processes, including meristem develop-
ment, floral organ development, mitosis and meiosis, as
well as plant body pattern specification (Figure 6c and
Tables S6 and S7). Several genes known to be involved in
floral organ development and reproduction processes were
found among these genes, including MSH7, VIM3, CHR42
ans NUA (Figure 5f and Figure S8). Therefore, DNA meth-
ylation reprogramming may contribute significantly to the
regulation of floral development in Arabidopsis.
DISCUSSION
We used MspJI-seq to survey the genome-wide DNA meth-
ylation during Arabidopsis flower development, and
uncovered a large number of potential de novo methyla-
tion sites in early flower development and demethylation
sites in late flower development, supporting the idea that
extensive de novo methylation as well as demethylation
occur during flower development. Different methylation
patterns for the three sequence contexts (mCG, mCHG andmCHH) and in different genic regions potentially have dif-
ferent effects on gene expression. The whole-genome DNA
methylation and gene expression patterns and derived
hypotheses of their interactions reveal more complex rela-
tionships than expected. Future functional studies are
required to elucidate the mechanisms of control of DNA
methylation and gene expression, and to understand the
biological functions and mechanisms for regulating floral
genes.
The ap1 cal double mutant has been widely used to
obtain a relatively large amount of meristems (Wellmer
et al., 2006) (Kaufmann et al., 2010; Wuest et al., 2012),
(a) (b)
(c)
Figure 4. Genes with DNA methylation variations during Arabidopsis floral
development.
(a) Comparison of genes differentially methylated at one or more sequence
contexts and differentially expressed between meristem and early flower.
‘Gene Body’ and ‘Promoter’ represent the transcribed region and the 1 kb
upstream region of genes, respectively; ‘Transcription’ represents genes
that are differentially expressed.
(b) Comparison of differentially expressed genes according to methylation
variations among sequence contexts.
In (a) and (b), the numbers outside each circle indicate the total gene counts
in the respective group.
(c) Percentage of differentially methylated genes among each group of dif-
ferentially expressed genes during floral development. ME, meristem to
early flower; ML, meristem to late flower; EL, early to late flower. Plus sym-
bol, up-regulation; minus symbol, down-regulation. The gray lines divide
each bar into a bottom part representing genes with increased methylation,
and a top part representing genes with decreased methylation. Asterisk
indicate statistically significant differences (*P < 0.05; **P < 1e-3; v2 test).
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
8 Hongxing Yang et al.
because the mutant is arrested at the inflorescence meri-
stem stage (Bowman et al., 1993; Ferrandiz et al., 2000).
However, we cannot rule out possible effects of the muta-
tions on the DNA methylation and gene expression pat-
terns, and caution is required when interpreting the
differences in results between the ap1 cal meristem and
the early and late flowers. Efficient methods for separating
and collecting wild-type meristems are required for further
studies to confirm the differences relating to the meris-
tems.
Different methylation patterns between mC sequence
contexts and genic regions
The results here allow comprehensive analyses of many
aspects of DNA methylation patterns. The approximately
1.5 million methylation sites from three flower tissues
included 453 066 mCG sites, 425 428 mCHG sites and
685 100 mCHH sites, representing a dramatic increase of
37% for detection of mCHH sites compared with approxi-
mately 500 000 mCHH sites in young flowers as previously
reported (Lister et al., 2008). Previously CHH methylation
has been found to play important roles in various plant
developmental processes in Arabidopsis endosperm,
maize (Zea mays) and cotton fibers (Gossypium hirsutum)
(Hsieh et al., 2009; Gent et al., 2013; Jin et al., 2013). The
relatively high proportion of newly identified mCHH sites
reported here (Figure 1d) and the correlation of >1000genes with variation in CHH methylation (Figure 4b) sug-
gest that CHH methylation may be important for floral
gene expression.
In addition, our observations that methylation patterns
and developmental changes in methylation vary depend-
ing on sequence contexts and gene classes suggest com-
plex relationships between sequences/genes, methylation
and developmental gene functions. For example, the num-
ber of mCs increased from the meristems to early flowers
for all sequence contexts, but only mCG sites increased in
number from early to late flowers. The period from meri-
stem to the early flower involves mainly organ identity
specification and organogenesis, whereas the subsequent
period includes much of the floral organ growth, as well as
gametophyte development. Therefore, our results suggest
that gene expression associated with mCG sites may play a
greater role in floral organ growth. The finding that
(a)
(c)
(e)
(b)
(d)
(f)
Figure 5. Representative Arabidopsis genes
with variations in DNA methylation and floral
expression.
Tracks of MspJI-seq and RNA-seq reads are
shown for each gene, encompassing the tran-
scribed region as well as the upstream and
downstream 1 kb regions. Gene structures are
shown at the bottom of each graph, with blue
boxes representing exons and arrows indicat-
ing introns and the transcription direction of
the respective gene. In some cases, parts of the
exon–intron structure for adjacent genes are
shown; they do not span the central regions
and are not connected with the centrally located
gene models. M, meristem; E, early flower; L,
late flower.
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
Arabidopsis floral DNA methylomes and transcriptomes 9
protein-coding genes showed preferential enrichment ofmCG sites and depletion of mCHG and mCHH sites in the
transcribed regions is consistent with previous results
(Zhang et al., 2006; Cokus et al., 2008; Feng et al., 2010; Ze-
mach et al., 2010). In contrast, TE genes and pseudogenes
showed similar mC frequencies in the various sequence
contexts across transcribed and nearby genomic regions,
with mCs being enriched in transcribed regions and
depleted near the transcription start sites (Figure 2d–f).
Therefore, protein-coding genes and other types of genes
are probably affected by DNA methylation in different
ways. Moreover, only a few genes show simultaneously
changes in methylation levels and expression levels at all
sequence contexts, suggesting that influences on expres-
sion by mCs of different sequence contexts tended to be
unrelated to each other (Figure 4b).
Our findings also support the idea that the relationships
between DNA methylation and gene expression levels are
more complicated than previously thought (Zhang et al.,
2006; Suzuki and Bird, 2008; Ball et al., 2009). Promoter
methylation is often linked with gene expression suppres-
sion, whereas the role of gene body methylation is far
more uncertain, with both positive and negative relation-
ships reported (Zhang et al., 2006; Zilberman et al., 2007;
Li et al., 2008; Zemach et al., 2010). These contradictory
findings may be explained when considering the observa-
tions that DNA methylation interacts with other factors,
including histone modifications and siRNAs, to determine
transcriptional status (Li et al., 2008; Stroud et al., 2014). In
addition, the effect of DNA methylation on expression may
also depend on genic regions or sequence contexts. In
humans, methylation of the first exon was more signifi-
cantly associated with gene silencing than methylation of
the nearby promoter (Brenet et al., 2011). Similarly, our
observation in Arabidopsis of strong negative relationships
between DNA methylation at all sequence contexts in the
first exon and expression (Figure 3) suggests that the rele-
vant mechanisms may be conserved between animals and
plants. The even stronger negative effects of methylation
the first intron are consistent with the fact that the first in-
trons of eukaryotic genes often carry regulatory elements
for transcription (Majewski and Ott, 2002; Bradnam and
Korf, 2008; Bieberstein et al., 2012). We also found that
methylation at mCG sites of internal exons tended to be
positively correlated with gene expression, unlike methyla-
tion at the mCHG and mCHH sites of internal exons and all
sequence contexts of internal introns (Figure 3). Therefore,
our separate analyses regarding sequence contexts and
genic regions revealed that the effects of DNA methylation
on gene expression are not only position-dependent (i.e.
(a) (b) (c) Figure 6. Methylation pattern and functional
implications for differentially methylated and
expressed genes.
(a) Clustering of genes with concurrent varia-
tions in expression and methylation on the
basis of similar methylation patterns during flo-
ral development. The color gradient represents
the log2-transformed RKCM values, as indicated
by the color bar at the top.
(b) Summarized methylation patterns for each
cluster. M, meristem; E, early flower; L, late
flower.
(c) Enriched biological processes among each
gene cluster. Gene ontology (GO) terms were
grouped into more general biological pro-
cesses, as shown on the right (see also Table
S6). Colors represent the number of genes for
each GO term in each gene cluster, as indicated
by the color bar at the top.
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
10 Hongxing Yang et al.

with respect to genic region), but also sequence context-
dependent, providing further insight into the relationships
between methylation and gene expression.
Implications for functional roles of DNA methylation in
flower development
Changes in genome-wide DNA methylation levels have
been associated with plant development, including global
demethylation in the endosperm of the developing seed
compared with the embryo (Gehring et al., 2009; Hsieh
et al., 2009) and lower methylation in the central cell of the
female gametophyte than in somatic cells (Jullien and Ber-
ger, 2010). Previous studies also suggested that the DNA
methylation level tends to increase during development
from seedling through vegetative stages to floral stages
(Ruiz-Garcia et al., 2005). Our analysis of differential meth-
ylation between three floral tissues revealed that more
sites are methylated de novo than demethylated from mer-
istem to early flowers, whereas similar numbers of sites
were methylated and demethylated from early to late
flower development (Figure 1e–g). These findings suggest
that plant DNA methylation is under critical control during
development, and proper re-patterning of DNA methyla-
tion is important for the developmental program.
The importance of DNA methylation in development has
been demonstrated by genetic studies of methyltransfer-
ase genes or genes encoding chromatin remodelers,
including MET1 and DDM1, with effects on embryogenesis,
meristem identity and flowering time (Finnegan et al.,
1996; Kakutani et al., 1996; Ronemus et al., 1996; Xiao
et al., 2006). The effects of DNA methylation occur at least
in part via modulating transcription of developmental reg-
ulators (Xiao et al., 2006; Li et al., 2008; Gehring et al.,
2009; Hsieh et al., 2009). In addition, DNA methylation reg-
ulates genes that play critical roles during flower develop-
ment; for example, hypermethylation of SUPERMAN and
AG phenocopies the corresponding mutants (Jacobsen
et al., 2000). Our results showing that DNA methylation is
associated with changes in the expression of over 3000
genes suggest that methylation affects genes with diverse
roles during flower development, such as regulation of
early flower development (33 genes) and pollen develop-
ment (21 genes) (Table S6). In addition, the expression of
regulatory genes is probably affected by methylation,
including genes for transcriptional control (201), chromatin
organization (29) and signal transduction (56).
In particular, changes in DNA methylation were linked to
expression changes for several key regulators. For
instance, the expression of floral regulators SEP1 and
SEEDSTICK was affected by DNA methylation (Figure 5a,
c). An effect of DNA methylation on the C function gene
AG was previously reported: hypermethylated epi-alleles
of AG were found in plants with reduced global methyla-
tion (Jacobsen et al., 2000). Our data suggest that DNA
methylation may indirectly influence the expression of AG
in wild-type plants, similar to the case for the FLC gene
(Finnegan et al., 2005). Decreased methylation in early and
late flowers may result in down-regulation of LEUNIG (Fig-
ure 5b), a negative regulator of AG (Sridhar et al., 2004). In
addition, methylation may also function to activate BLH9
and/or suppress PERIANTHIA (Figure S8a), both genes
encoding transcription factors that are required for proper
floral expression of AG (Bao et al., 2004; Das et al., 2009).
On the other hand, the methylcytosine-binding proteins
VIM1, VIM2 and VIM3, which are involved in hypermethy-
lation of the flowering-time gene FWA and its subsequent
suppression (Woo et al., 2008), were observed to have
high gene expression levels that may also be affected by
methylation (Figure S8b), suggesting deep involvement of
DNA methylation in flowering transition.
Our results also suggest that DNA methylation may
affect other cellular and developmental processes during
reproduction, such as pollen tube growth, mitosis and mei-
osis. For example, GO enrichment analysis revealed over-
representation of 21 genes associated with pollen develop-
ment in cluster VI of methylated genes (Figure 6 and Table
S7), and correlated changes in methylation and expression
between the three floral tissues/stages for genes participat-
ing in meiosis, including AtMSH7, AtMSH4, AtDMC1 and
AtSMC3 (Figure 5f and Table S4), suggesting that DNA
methylation may be involved in the floral development
program as well as embryogenesis and other aspects of
plant development. Our analyses provide insights into the
possible roles of DNA methylation in gene expression, and
important resources for further investigation of the genetic
pathway that regulates flower development.
EXPERIMENTAL PROCEDURES
Plant growth, tissues collection, and DNA isolation
Plants of the Arabidopsis thaliana Landsberg ecotype that werehomozygous for the erecta mutation (Ler) and ap1 cal mutantplants(also in the Ler background) were grown in soil in a plantgrowth room at 22°C under 16 h light/8 h dark cycles. The meris-tems of the ap1 cal mutant plants, wild-type early flowers (stages1–9) and late flowers (stages 10–12) were collected separately (Fig-ure S9). The materials collected for each sample were taken frommany individuals and in large amounts (2 g) to control the effect ofbiological variations. Genomic DNA was extracted using a DNeasyplant mini kit (Qiagen, http://www.qiagen.com), precipitated usingtwo volumes of EtOH with a one-tenth volume of 3 M NaAc (pH5.2), and resuspended in Tris/EDTA buffer (pH 8.0) to a final con-centration of 1 lg ll�1.
MspJI digestion and DNA recovery
A 5 lg aliquot of each DNA sample was added to 90 ll of well-mixed NE Buffer 4 (New England Biolabs, https://www.neb.com),BSA and nuclease-free digestion mix before addition of 20 unitsof MspJI enzyme (New England Biolabs, https://www.neb.com) foreffective digestion without obvious DNA degradation (FigureS10a), and incubated at 37°C for 16 h.
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
Arabidopsis floral DNA methylomes and transcriptomes 11

After digestion, the DNAs were separated in a 20% polyacryl-amide gel (arc/bis: 29:1; 50 mA, 2.5 h). The polyacrylamide gelpieces containing DNA of approximately 32 bp (Figure S9a) wereexcised from the polyacrylamide gel and crushed and transferredinto sterile microfuge tubes containing 300 ll of buffer comprising0.3 M sodium acetate, pH 7.5, with 0.1 mM EDTA), and shakenovernight at 37°C. Then the gel was pelleted by centrifugation for2 min at 14 000 g at room temperature, and the supernatant wascollected. DNA was precipitated by adding 2 ll glycogen and twovolumes of 100% ethanol to each tube, keeping the tubes at�80°C for 30 min, followed by centrifugation for 30 min at14 000 g at 4°C. The DNA pellets were washed with 70% ethanoltwice (for 2 min at 14 000 g at room temperature) before resuspen-sion in Tris/EDTA buffer (pH 8.0).
Construction of a DNA methylation fragment library for
sequencing
The recovered DNA samples were used to construct sequencinglibraries according to the fragment library preparation protocoldescribed in the SOLiDTM system library preparation guide (LifeTechnologies, http://www.lifetechnologies.com/) with some mod-ifications. Both ends of recovered DNA were repaired using ENDpolishing enzymes 1 and 2 from the SOLiDTM fragment libraryconstruction kit, and purified using a QIAquick nucleotideremoval kit (Qiagen) and spin columns from a MinElute� reac-tion clean-up kit (Qiagen). After purification, the P1 and P2 adap-tors from the SOLiDTM fragment library construction kit wereligated to the DNA fragments, followed by another purificationstep using the QIAquick nucleotide removal kit. Recovered DNAlibraries were nick-translated and amplified by PCR for tencycles, then purified using a QIAquick nucleotide removal kit.Purified DNA libraries were subjected to 4% agarose gel electro-phoresis, and bands of approximately 100 bp (Figure S10b) wererecovered using a QIAquick gel extraction kit (Qiagen). Theresulting libraries were analyzed using an Agilent bioanalyzer(http://www.agilent.com), and 0.5 pmol of each DNA library wereused to perform emulsion PCR reactions according to the tem-plated bead preparation guide from the SOLiDTM system. The35 bp sequence reads were obtained using the SOLiDTM 3.0 sys-tem, and were subsequently aligned against the Arabidopsis ref-erence genome sequences (TAIR10, http://www.arabidopsis.org)using BIOSCOPE software version 1.3 (Life Technologies) (seeMethods S1). The overall mapping rates were approximately61% (Table S1).
DNA methylome analysis
We developed an in-house program to identify SOLiD reads sup-porting methylcytosines. In brief, six cases of paired sequencepatterns were inferred based on the MspJI digestion properties,with each case resulting in DNA fragments that could be recov-ered (Figure S11 and Methods S2). Aligned reads that may beclassified into one of the six patterns were identified as methylreads, each supporting two potential mCs. MspJI cut double-stranded DNA at fixed distances downstream of the recognizedsequence (mCNNR), with the cleavage on the reverse strand wob-bling by one nucleotide (16 or 17) (Zheng et al., 2010). Methylreads were counted separately for each of the two cleavage pat-terns (N12/N16 or N12/N17), resulting in an N16/N17 ratio of approxi-mately 3:1 (Table S8). The number of reads supportingmethylcytosines was calculated for each genomic feature (genes,exons, etc.) and sequence context separately, and normalized asRKCM values. Fisher’s exact test was performed on the readcounts of a pair of tissues for each gene, and a gene was defined
as differentially methylated when the false discovery rate (calcu-lated by the Benjamini-Hochberg method) was < 0.001 and theratio of RKCM values was >2.
Gene ontology enrichment analysis was performed using the R
package topGO (Alexa and Rahnenfuhrer, 2010). The graphs inFigure 5 displaying the DNA methylation and gene transcriptionprofiles were produced using the R package Gviz (Hahne et al.,2013). Consensus clustering on the log2-transformed RKCM valuesof differentially methylated genes was performed using the R
package clusterCons (Simpson et al., 2010) with the algorithm ofpartitioning (clustering) of the data into ‘k’ clusters ‘around med-oids’ (PAM).
Total RNA isolation and transcriptome analysis
All three tissues were frozen in liquid nitrogen immediately aftercollection. Total RNAs were extracted using a Plant RNase mini kit(Qiagen). More than 2 lg of total RNA with an absorbance at 260/280 nm between 1.8 and 2.0 from each sample were used to cre-ate libraries that were deep-sequenced using the IlluminaTM Hi-seq2000 system (Illumina, http://www.illumina.com), to obtain 100 bppaired-end reads. All reads with fewer than two mismatches weremapped to the Arabidopsis genome (TAIR10) by TopHat (Trapnellet al., 2009). Calculation of expression values and differentialexpression analysis were performed using GFOLD (Feng et al.,2012) with default parameters.
ACKNOWLEDGEMENTS
We thank Zhiyi Sun from New England Biolabs for discus-
sion regarding computational identification of MspJI-
digested methylation sites. We appreciate the data kindly
provided by Steven Jacobsen and Matteo Pellegrini from
University of California, Los Angeles. This work was sup-
ported by the Ministry of Science and Technology of the
People’s Republic of China (MOST 2012CB910503), the
National Natural Science Foundation of China (31130006
and 31371330), and start-up funds from Fudan University
to F.C.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online ver-sion of this article.Figure S1. Validation of selected methylcytosine sites by PCRexperiments.
Figure S2. Methylation profiles determined by MspJI-seq andbisulfite sequencing were consistent for most of the randomlyselected genes and genomic regions.
Figure S3. Density of genes and TEs across Arabidopsis genome.
Figure S4. Percentages of genes of each class among all methylat-ed genes.
Figure S5. Methylation of TEs during Arabidopsis floral develop-ment.
Figure S6. Enrichment of TEs of various families in different mCsequence contexts.
Figure S7. Distribution of normalized methylation levels for eachmC context for genes of various classes.
Figure S8. Examples of genes with correlated variations in methyl-ation and expression levels.
Figure S9. Phenotypes of the three Arabidopsis floral stages usedfor experiments.
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
12 Hongxing Yang et al.

Figure S10. MspJI digestion and DNA library recovery.
Figure S11. Identification of mCs based on MspJI-seq.
Table S1. Summary of SOLiD reads sequenced and mappedagainst the Arabidopsis reference genome and reads that wereidentified as arising from MspJI digestion for each possible recog-nition site pattern.
Table S2. Primers used in PCR experiments for selected generegions digested by MspJI.
Table S3. DNA methylation and expression levels for genes in Ara-bidopsis flowers.
Table S4. Arabidopsis genes that were differentially methylatedand differentially expressed during floral development.
Table S5. Statistics for genes that were differentially expressedand methylated between flower meristems and early flowers.
Table S6. Significantly enriched biological processes for eachgene cluster in Figure 6.
Table S7. Number of genes for enriched GO terms for each genecluster in Figure 6.
Table S8. Relative frequencies of the wobbling cut positions ofMspJI.
Methods S1. Mapping of SOLiD short sequencing reads.
Methods S2. Identification of methylcytosines based on MspJI-seq.
REFERENCES
Agius, F., Kapoor, A. and Zhu, J.K. (2006) Role of the Arabidopsis DNA gly-
cosylase/lyase ROS1 in active DNA demethylation. Proc. Natl Acad. Sci.
USA, 103, 11796–11801.Ahmed, I., Sarazin, A., Bowler, C. et al. (2011) Genome-wide evidence for
local DNA methylation spreading from small RNA-targeted sequences in
Arabidopsis. Nucleic Acids Res. 39, 6919–6931.Alexa, A. and Rahnenfuhrer, J. (2010) topGO: Enrichment analysis for Gene
Ontology.
Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of
the flowering plant Arabidopsis thaliana. Nature, 408, 796–815.Ball, M.P., Li, J.B., Gao, Y. et al. (2009) Targeted and genome-scale strate-
gies reveal gene-body methylation signatures in human cells. Nat. Bio-
tech. 27, 361–368.Bao, X., Franks, R.G., Levin, J.Z. et al. (2004) Repression of AGAMOUS by
BELLRINGER in floral and inflorescence meristems. Plant Cell, 16, 1478–1489.
Bieberstein, N.I., Carrillo Oesterreich, F., Straube, K. et al. (2012) First exon
length controls active chromatin signatures and transcription. Cell Rep.
2, 62–68.Bird, A. (2002) DNA methylation patterns and epigenetic memory. Genes
Dev. 16, 6–21.Bowman, J.L., Alvarez, J., Weigel, D. et al. (1993) Control of flower develop-
ment in Arabidopsis thaliana by APETALA1 and interacting genes. Devel-
opment, 119, 721–743.Bradnam, K.R. and Korf, I. (2008) Longer first introns are a general property
of eukaryotic gene structure. PLoS One, 3, e3093.
Brenet, F., Moh, M., Funk, P. et al. (2011) DNA methylation of the first exon
is tightly linked to transcriptional silencing. PLoS One, 6, e14524.
Chan, S.W., Henderson, I.R. and Jacobsen, S.E. (2005) Gardening the genome:
DNAmethylation in Arabidopsis thaliana. Nat. Rev. Genet. 6, 351–360.Chang, F., Wang, Y., Wang, S. et al. (2011) Molecular control of microsporo-
genesis in Arabidopsis. Curr. Opin. Plant Biol. 14, 66–73.Chodavarapu, R.K., Feng, S., Bernatavichute, Y.V. et al. (2010) Relationship
between nucleosome positioning and DNA methylation. Nature, 466,
388–392.Chuang, T.J., Chen, F.C. and Chen, Y.Z. (2012) Position-dependent correla-
tions between DNA methylation and the evolutionary rates of mamma-
lian coding exons. Proc. Natl Acad. Sci. USA, 109, 15841–15846.Cohen-Karni, D., Xu, D., Apone, L. et al. (2011) The MspJI family of modifi-
cation-dependent restriction endonucleases for epigenetic studies. Proc.
Natl Acad. Sci. USA, 108, 11040–11045.
Cokus, S.J., Feng, S., Zhang, X. et al. (2008) Shotgun bisulphite sequencing
of the Arabidopsis genome reveals DNA methylation patterning. Nature,
452, 215–219.Conner, J. and Liu, Z. (2000) LEUNIG, a putative transcriptional corepressor
that regulates AGAMOUS expression during flower development. Proc.
Natl Acad. Sci. USA, 97, 12902–12907.Das, P., Ito, T., Wellmer, F. et al. (2009) Floral stem cell termination involves
the direct regulation of AGAMOUS by PERIANTHIA. Development, 136,
1605–1611.Feng, S., Cokus, S.J., Zhang, X. et al. (2010) Conservation and divergence
of methylation patterning in plants and animals. Proc. Natl Acad. Sci.
USA, 107, 8689–8694.Feng, J., Meyer, C.A., Wang, Q. et al. (2012) GFOLD: a generalized fold
change for ranking differentially expressed genes from RNA-seq data.
Bioinformatics, 28, 2782–2788.Ferrandiz, C., Gu, Q., Martienssen, R. et al. (2000) Redundant regulation of
meristem identity and plant architecture by FRUITFULL, APETALA1 and
CAULIFLOWER. Development, 127, 725–734.Finnegan, E.J., Peacock, W.J. and Dennis, E.S. (1996) Reduced DNA methyl-
ation in Arabidopsis thaliana results in abnormal plant development.
Proc. Natl Acad. Sci. USA, 93, 8449–8454.Finnegan, E.J., Kovac, K.A., Jaligot, E. et al. (2005) The downregulation of
FLOWERING LOCUS C (FLC) expression in plants with low levels of DNA
methylation and by vernalization occurs by distinct mechanisms. Plant J.
44, 420–432.Gan, E.-S., Huang, J. and Ito, T. (2013) Functional roles of histone modifica-
tion, chromatin remodeling and microRNAs in Arabidopsis flower devel-
opment. In International Review of Cell and Molecular Biology (Kwang,
W.J. ed). Waltham, MA: Academic Press, pp. 115–161.Ge, X., Chang, F. and Ma, H. (2010) Signaling and transcriptional control
of reproductive development in Arabidopsis. Curr. Biol. 20, R988–R997.
Gehring, M., Bubb, K.L. and Henikoff, S. (2009) Extensive demethylation of
repetitive elements during seed development underlies gene imprinting.
Science, 324, 1447–1451.Gent, J.I., Ellis, N.A., Guo, L. et al. (2013) CHH islands: de novo DNA methyla-
tion in near-gene chromatin regulation in maize. Genome Res. 23, 628–637.Goll, M.G. and Bestor, T.H. (2005) Eukaryotic cytosine methyltransferases.
Annu. Rev. Biochem. 74, 481–514.Gomez-Mena, C., de Folter, S., Costa, M.M. et al. (2005) Transcriptional pro-
gram controlled by the floral homeotic gene AGAMOUS during early
organogenesis. Development, 132, 429–438.Gong, Z., Morales-Ruiz, T., Ariza, R.R. et al. (2002) ROS1, a repressor of
transcriptional gene silencing in Arabidopsis, encodes a DNA glycosy-
lase/lyase. Cell, 111, 803–814.Hahne, F., Durinck, S., Ivanek, R., Mueller, A., Lianoglou, S., Tan, G. and
Parsons, L. (2013) Gviz: Plotting data and annotation information along
genomic coordinates. R package version, 1(8), 4.
He, X.J., Chen, T. and Zhu, J.K. (2011) Regulation and function of DNA
methylation in plants and animals. Cell Res. 21, 442–465.Horton, J.R., Mabuchi, M.Y., Cohen-Karni, D. et al. (2012) Structure and
cleavage activity of the tetrameric MspJI DNA modification-dependent
restriction endonuclease. Nucleic Acids Res. 40, 9763–9773.Hsieh, T.F., Ibarra, C.A., Silva, P. et al. (2009) Genome-wide demethylation
of Arabidopsis endosperm. Science, 324, 1451–1454.Huang, X., Lu, H., Wang, J.W. et al. (2013) High-throughput sequencing of
methylated cytosine enriched by modification-dependent restriction
endonuclease MspJI. BMC Genet. 14, 56.
Jacobsen, S.E., Sakai, H., Finnegan, E.J. et al. (2000) Ectopic hyperme-
thylation of flower-specific genes in Arabidopsis. Curr. Biol. 10, 179–186.
Jin, X., Pang, Y., Jia, F. et al. (2013) A potential role for CHH DNA methyla-
tion in cotton fiber growth patterns. PLoS One, 8, e60547.
Jones, L., Hamilton, A.J., Voinnet, O. et al. (1999) RNA-DNA interactions
and DNA methylation in post-transcriptional gene silencing. Plant Cell,
11, 2291–2301.Jullien, P.E. and Berger, F. (2010) DNA methylation reprogramming during
plant sexual reproduction? Trends Genet. 26, 394–399.Jullien, P.E., Susaki, D., Yelagandula, R. et al. (2012) DNA methylation
dynamics during sexual reproduction in Arabidopsis thaliana. Curr. Biol.
22, 1825–1830.
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
Arabidopsis floral DNA methylomes and transcriptomes 13

Kakutani, T., Jeddeloh, J.A., Flowers, S.K. et al. (1996) Developmental
abnormalities and epimutations associated with DNA hypomethylation
mutations. Proc. Natl Acad. Sci. USA, 93, 12406–12411.Kaufmann, K., Wellmer, F., Muino, J.M. et al. (2010) Orchestration of floral
initiation by APETALA1. Science, 328, 85–89.Law, J.A. and Jacobsen, S.E. (2010) Establishing, maintaining and modify-
ing DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11,
204–220.Li, X., Wang, X., He, K. et al. (2008) High-resolution mapping of epigenetic
modifications of the rice genome uncovers interplay between DNA meth-
ylation, histone methylation, and gene expression. Plant Cell, 20, 259–276.Lippman, Z., Gendrel, A.V., Black, M. et al. (2004) Role of transposable ele-
ments in heterochromatin and epigenetic control. Nature, 430, 471–476.Lister, R., O’Malley, R.C., Tonti-Filippini, J. et al. (2008) Highly integrated
single-base resolution maps of the epigenome in Arabidopsis. Cell, 133,
523–536.Ma, H. (2005) Molecular genetic analyses of microsporogenesis and micro-
gametogenesis in flowering plants. Annu. Rev. Plant Biol. 56, 393–434.Majewski, J. and Ott, J. (2002) Distribution and characterization of regula-
tory elements in the human genome. Genome Res. 12, 1827–1836.Martienssen, R.A. and Colot, V. (2001) DNA methylation and epigenetic
inheritance in plants and filamentous fungi. Science, 293, 1070–1074.Mette, M.F., Aufsatz, W., van der Winden, J. et al. (2000) Transcriptional
silencing and promoter methylation triggered by double-stranded RNA.
EMBO J. 19, 5194–5201.Mizukami, Y. and Ma, H. (1992) Ectopic expression of the floral homeotic
gene AGAMOUS in transgenic Arabidopsis plants alters floral organ
identity. Cell, 71, 119–131.Park, Y.D., Papp, I., Moscone, E.A. et al. (1996) Gene silencing mediated by
promoter homology occurs at the level of transcription and results in
meiotically heritable alterations in methylation and gene activity. Plant J.
9, 183–194.Pelaz, S., Ditta, G.S., Baumann, E. et al. (2000) B and C floral organ identity
functions require SEPALLATA MADS-box genes. Nature, 405, 200–203.Ronemus, M.J., Galbiati, M., Ticknor, C. et al. (1996) Demethylation-induced
developmental pleiotropy in Arabidopsis. Science, 273, 654–657.Ruiz-Garcia, L., Cervera, M.T. and Martinez-Zapater, J.M. (2005) DNA meth-
ylation increases throughout Arabidopsis development. Planta, 222, 301–306.
Schmitz, R.J., Schultz, M.D., Urich, M.A. et al. (2013) Patterns of population
epigenomic diversity. Nature, 495, 193–198.Simpson, T.I., Armstrong, J.D. and Jarman, A.P. (2010) Merged consensus
clustering to assess and improve class discovery with microarray data.
BMC Bioinformatics, 11, 590.
Slotkin, R.K., Vaughn, M., Borges, F. et al. (2009) Epigenetic reprogramming
and small RNA silencing of transposable elements in pollen. Cell, 136,
461–472.Song, Y., Ma, K., Ci, D. et al. (2013) Sexual dimorphic floral development in
dioecious plants revealed by transcriptome, phytohormone, and DNA
methylation analysis in Populus tomentosa. Plant Mol. Biol. 83, 559–576.
Soppe, W.J., Jacobsen, S.E., Alonso-Blanco, C. et al. (2000) The late flower-
ing phenotype of fwa mutants is caused by gain-of-function epigenetic
alleles of a homeodomain gene. Mol. Cell, 6, 791–802.Sridhar, V.V., Surendrarao, A., Gonzalez, D. et al. (2004) Transcriptional
repression of target genes by LEUNIG and SEUSS, two interacting
regulatory proteins for Arabidopsis flower development. Proc. Natl Acad.
Sci. USA, 101, 11494–11499.Stam, M., Viterbo, A., Mol, J.N. et al. (1998) Position-dependent methyla-
tion and transcriptional silencing of transgenes in inverted T-DNA
repeats: implications for posttranscriptional silencing of homologous
host genes in plants. Mol. Cell. Biol. 18, 6165–6177.Stroud, H., Do, T., Du, J. et al. (2014) Non-CG methylation patterns shape
the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 21, 64–72.Suzuki, M.M. and Bird, A. (2008) DNA methylation landscapes: provocative
insights from epigenomics. Nat. Rev. Genet. 9, 465–476.Trapnell, C., Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice
junctions with RNA-Seq. Bioinformatics, 25, 1105–1111.Wellmer, F., Alves-Ferreira, M., Dubois, A. et al. (2006) Genome-wide analy-
sis of gene expression during early Arabidopsis flower development.
PLoS Genet. 2, e117.
Woo, H.R., Dittmer, T.A. and Richards, E.J. (2008) Three SRA-domain meth-
ylcytosine-binding proteins cooperate to maintain global CpG methyla-
tion and epigenetic silencing in Arabidopsis. PLoS Genet. 4, e1000156.
Wuest, S.E., O’Maoileidigh, D.S., Rae, L. et al. (2012) Molecular basis for the
specification of floral organs by APETALA3 and PISTILLATA. Proc. Natl
Acad. Sci. USA, 109, 13452–13457.Xiao, W., Custard, K.D., Brown, R.C. et al. (2006) DNA methylation is critical
for Arabidopsis embryogenesis and seed viability. Plant Cell, 18, 805–814.
Yang, H., Lu, P., Wang, Y. et al. (2011) The transcriptome landscape of Ara-
bidopsis male meiocytes from high-throughput sequencing: the com-
plexity and evolution of the meiotic process. Plant J. 65, 503–516.Zemach, A., McDaniel, I.E., Silva, P. et al. (2010) Genome-wide evolutionary
analysis of eukaryotic DNA methylation. Science, 328, 916–919.Zhang, X., Yazaki, J., Sundaresan, A. et al. (2006) Genome-wide high-reso-
lution mapping and functional analysis of DNA methylation in Arabidop-
sis. Cell, 126, 1189–1201.Zheng, Y., Cohen-Karni, D., Xu, D. et al. (2010) A unique family of Mrr-like
modification-dependent restriction endonucleases. Nucleic Acids Res.
38, 5527–5534.Zhong, S., Fei, Z., Chen, Y.R. et al. (2013) Single-base resolution methylo-
mes of tomato fruit development reveal epigenome modifications asso-
ciated with ripening. Nat. Biotechnol. 31, 154–159.Zilberman, D., Cao, X., Johansen, L.K. et al. (2004) Role of Arabidopsis ARG-
ONAUTE4 in RNA-directed DNA methylation triggered by inverted
repeats. Curr. Biol. 14, 1214–1220.Zilberman, D., Gehring, M., Tran, R. K. et al. (2007) Genome-wide analysis
of Arabidopsis thaliana DNA methylation uncovers an interdependence
between methylation and transcription. Nat. Genet. 39, 61–69.
© 2014 The AuthorsThe Plant Journal © 2014 John Wiley & Sons Ltd, The Plant Journal, (2014), doi: 10.1111/tpj.12726
14 Hongxing Yang et al.




















