
β-CD conjugated dendrimer
117
Transcript of β-CD conjugated dendrimer






I

II
Preface
Dendrimers are a new class of synthetic polymers based on a well-defined
cascade motif. These macromolecules may be synthesized to reach the size of
nanoobjects having dimensions similar to proteins. Dendrimers are regarded as
ideal candidates for use in biomedical applications. Dendrimers containing
cyclodextrin exhibited that the -CD-grafted biodegradable dendrimers is a
potential candidate as an efficient drug carrier systems due to its relative
stability in aqueous solution and its drug encapsulation and release properties.
Cyclodextrins and their complexes constitute a fascinating subject that, due to
its specific chemical structure. This great attention makes today’s CD science
and technology extremely involved in a large number of important areas of
research covering both basic and applied science in chemistry and biology.
Their ability to modify the behavior of trapped guests is being used for example,
in chemistry, biology, food, pharmacy/medicine and agriculture.
This book reflects the height of the latest knowledge of these sweet nanocavities
to host different kinds of guests on the drug delivery systems.
I would like to thank the University of Tabriz and Tabriz University of medical
science, RCPN that enabled my research at faculty of chemistry and leading to
my deeper understanding of supramolecular complexes. Finally, I hope this
volume will be beneficial to readers of this book who like dendrimer and
cyclodextrin chemistry to enter the fascinating field of supramolecular
chemistry.
Prof. Hassan Namazi
Yousef Toomari
University of Tabriz, Iran, July 2012

III
Table of Contents
Preface ................................................................................................................. II
Table of Contents ............................................................................................. III
List of Figures .....................................................................................................V
List of Tables ................................................................................................. VIII
List of Symbols and Abbreviations ................................................................ IX
Chapter 1: Background and Literature Review .............................................. 1
1.1. Dendrimers ................................................................................................. 11.1.1. Introduction .......................................................................................... 11.1.2. History and Progress of Dendrimer Research ...................................... 21.1.3. Molecular Structure of Dendrimers ..................................................... 31.1.4. Synthesis Methods ............................................................................... 31.1.5. Properties of Dendrimers ..................................................................... 51.1.6. Application of Dendrimer in Drug Delivery Systems ....................... 141.1.7. Mechanism of Drug Delivery Using Dendrimers .............................. 151.1.8. Release of Guest Molecules ............................................................... 221.1.9. Modification of Periphery Groups of Dendrimer .............................. 231.1.10. Aim of Work .................................................................................... 25
Chapter 2: Experimental Section .................................................................... 27
2.1. Materials and Methods ............................................................................. 272. 1.1. Materials ............................................................................................ 272.1.2. Instrumental Measurement ................................................................. 28
2.2. Methods .................................................................................................... 292.2.1. Preparation of , -di(chlorocarbonylmethylene)poly(ethyleneglycol) (ClOC-PEG-COCl) .................................................... 292.2.2. Preparation of G1 Using Thionylchloride ......................................... 292.2.3. Preparation of G2 Using DCC ........................................................... 302.2.4. Preparation of G3 Using DCC ........................................................... 312.2.5. Synthesis of G1- -CD Using DCC .................................................... 322.2.6. Synthesis of G2- -CD Using DCC .................................................... 332.2.7. Synthesis of G3- -CD Using DCC .................................................... 34

IV
2.2.8. Synthesis of G1-NH- -CD Using DCC ............................................. 352.2.9. Synthesis of G2-NH- -CD Using DCC ............................................. 372.2.10. Synthesis of G3-NH- -CD Using DCC ........................................... 382.2.11. Preparation of Gn- -CD (n=1–2) and Gn-NH- -CD (n=1–2)/NLXComplexes .................................................................................................... 402.2.12. In Vitro Release Studies ................................................................... 402.2.13. Particle Size Analysis ....................................................................... 41
Chapter 3: Results and Discussion .................................................................. 42
3.1. Investigation of Dendrimer Synthesized Based on Citric acid ................ 423.1.1. Synthesis of G1 Compound ............................................................... 423.1.2. Synthesis of G2 Compound ............................................................... 453.1.3. Synthesis of G3 Compound ............................................................... 473.1.4. Synthesis of G1- -CD Compound ..................................................... 513.1.5. Synthesis of G2- -CD Compound ..................................................... 553.1.6. Synthesis of G3- -CD Compound ..................................................... 593.1.7. Synthesis of G1-NH- -CD Compound .............................................. 623.1.8. Synthesis of G2-NH- -CD Compound .............................................. 693.1.9. Synthesis of G3-NH- -CD Compound .............................................. 73
3.2. Investigation of Nanocarriers Size ........................................................... 773.3. In Vitro Release Studies ........................................................................... 80
3.3.1. Loading of NLX Drug Molecule into the Dendrimers ...................... 803.3.2. Calculating the Amount of the Trapped and Released Drug Molecule ...................................................................................................................... 803.3.3. Studying the Controlled Release of NLX from the Nanocarriers...... 82
Chapter 4: Conclusions .................................................................................... 87
Chapter 5: References ...................................................................................... 88

V
List of Figures
Figure 1. Typical architecture of dendrimer. ........................................................ 3
Figure 2. Schematic drawing showing the divergent method for synthesis of
dendrimers. ............................................................................................................ 4
Figure 3. Schematic drawing showing the convergent method for synthesis of
dendrimers. ............................................................................................................ 5
Figure 4. The close dimensional size (nm) of selected proteins to respective
generations of PAMAM dendrimers [12]. ............................................................ 8
Figure 5. Schematic illustration of the result of back-folding in an increased
molecular density in the interior of a dendrimer...................................................9
Figure 6. The effect of the solvent on the conformational state of a dendrimer. 12
Figure 7. Drawing of a dendrimer carrier encapsulating hydrophobic drug
molecules in the dendrimer’s voids to increase their aqueous solubility and
control their release rate. ..................................................................................... 17
Figure 8. Schematic drawing showing a dendrimer-drug conjugate where the
drug molecules (red ovals) are either directly coupled (solid lines) to
dendrimer’s surface groups or via a pH-sensitive linkage (blue rectangle). ...... 22
Figure 9. Chemical structure of naltrexone. ....................................................... 27
Figure 10. 1H NMR spectrum of G1 in DMSO-d6. ............................................ 44
Figure 11. FT-IR spectrum of G1. ...................................................................... 44
Figure 12. 1H NMR spectrum of G2 in DMSO-d6. ............................................ 46
Figure 13. FT-IR spectrum of G2. ...................................................................... 47
Figure 14. 1H NMR spectrum of G3 in DMSO-d6. ............................................ 50
Figure 15. 1H NMR spectrum of G3 in acetone deuterium. ............................... 50
Figure 16. FT-IR spectrum of G3. ...................................................................... 51
Figure 17. 1H NMR spectrum of G1- -CD in DMSO-d6. .................................. 54
Figure 18. FT-IR spectrum of G1- -CD. ............................................................ 54
Figure 19. 1H NMR spectrum of G2- -CD in DMSO-d6. .................................. 58

VI
Figure 20. FT-IR spectrum of G2- -CD. ............................................................ 58
Figure 21. FT-IR spectrum of G3- -CD. ............................................................ 62
Figure 22. 1H NMR spectrum of -CD-Tos in DMSO-d6. ................................. 65
Figure 23. 1H NMR spectrum of G1-NH- -CD in DMSO-d6. ........................... 68
Figure 24. FT-IR spectrum of G1-NH- -CD. ..................................................... 68
Figure 25. 13C NMR spectrum of G1-NH- -CD in DMSO-d6. .......................... 69
Figure 26. 1H NMR spectrum of G2-NH- -CD in DMSO-d6. ........................... 72
Figure 27. FT-IR spectrum of G2-NH- -CD. ..................................................... 72
Figure 28. 13C NMR spectrum of G2-NH- -CD in DMSO-d6. .......................... 73
Figure 29. 1H NMR spectrum of G3-NH- -CD in DMSO-d6. ........................... 76
Figure 30. FT-IR spectrum of G3-NH- -CD. ..................................................... 76
Figure 31. 13C NMR spectrum of G3-NH- -CD in DMSO-d6. .......................... 77
Figure 32. The size distribution profiles of G1- -CD and G2- -CD dendrimers
estimated by DLS. ............................................................................................... 78
Figure 33. Particle Diameter of G1-NH- -CD by using LDPSA. ...................... 79
Figure 34. Particle Diameter of G2-NH- -CD by using LDPSA. ...................... 79
Figure 35. Particle Diameter of G3-NH- -CD by using LDPSA. ...................... 80
Figure 36. Release of NLX from Gn (n=1-3)/NLX complexes as a function of time
and pH (pH 7.4, 37 ˚C). ...................................................................................... 84
Figure 37. Release of NLX from Gn (n=1-3)-NH- -CD/complexes as a function of
time and pH (pH 7.4, 37 ˚C). .............................................................................. 84
Figure 38. Release of NLX from Gn (n=1-2)- -CD/complexes as a function of time
and pH (pH 7.4, 37 ˚C). ...................................................................................... 85
Figure 39. Release of NLX from G3, G3- -CD and G3-NH- -CD/complexes as a
function of time and pH (pH 1, 37 ˚C). .............................................................. 85
Figure 40. Release of NLX from G3-NH- -CD/complexes as a function of time
and pH (pH 1, 7.4 and 10 and 37 ˚C). ................................................................. 86
Figure 41. Release of NLX from G2- -CD/complexes as a function of time and
pH (pH 1, 7.4 and 10 and 37 ˚C)......................................................................... 86

VII
List of Schemes
Scheme 1. Synthesis of G1. ................................................................................ 43
Scheme 2. Synthesis of G2. ................................................................................ 46
Scheme 3. Synthesis of G3. ................................................................................ 49
Scheme 4. Synthesis of G1- -CD. ...................................................................... 53
Scheme 5. . Synthesis of G2- -CD. .................................................................... 57
Scheme 6. Synthesis of G3- -CD. ...................................................................... 61
Scheme 7. Hierarchical synthesis of -CD-NH2. ............................................... 64
Scheme 8. Synthesis of G1-NH- -CD. ............................................................... 67
Scheme 9. Synthesis of G2-NH- -CD. ............................................................... 71
Scheme 10. Synthesis of G3-NH- -CD. ............................................................. 75

VIII
List of Tables
Table 1. Physical characteristics of PAMAM dendrimers. .................................. 7
Table 2. Diameter of nanocarriers obtained using laser diffraction particle size
analyzer. .............................................................................................................. 78
Table 3. Drug loading efficiency of Gn(n=1-3), Gn(n=1-3)- -CD and Gn(n=1-3)-NH- -
CD/NLX complexes. ........................................................................................... 81

IX
List of Symbols and Abbreviations
13C NMR carbon-13 nuclear magnetic resonance spectroscopy 1H NMR proton nuclear magnetic resonance spectroscopy
% percentage(s)
% w/v percentage weight per volume
vibration modes
chemical shift
extinction coefficient
celsius temperature or centigrade scale
aq aqueous
br broad
-CD -cyclodextrin
-CD-NH2 6-Deoxy-6-amino- -cyclodextrin
-CDTos 6-Deoxy-6-(p-toluenesulfonyl)- -cyclodextrin
-CD-N3 6-Deoxy-6-azido- -cyclodextrin
CD cyclodextrin
CDs cyclodextrins
cm-1 wavenumbers
Co company
D2O deuterium Oxide
DDS drug delivery systems
DCC dicyclohexylcarbodiimide
DLS dynamic light scattering
DMF N, N-dimethylformamide
DMSO dimethyl sulfoxide
DMSO-d6: perdeuterated dimethyl sulfoxide
e.g. for example
FT-IR fourier transform infrared spectroscopy

X
Fig. figure
G1- -CD Gn(n=1)- -cyclodextrin
G2- -CD Gn(n=2)- -cyclodextrin
G3- -CD Gn(n=3)- -cyclodextrin
G1-NH- -CD Gn(n=1)-NH- -cyclodextrin
G2-NH- -CD Gn(n=2)-NH- -cyclodextrin
G3-NH- -CD Gn(n=3)-NH- -cyclodextrin
G generation
g gram
m medium/multiplet
MHz mega Hertz
mol mole
MS mass spectrometry
nm nanometer(s)
NLX naltrexone
PAMAM poly(amidoamine)
PD pharmacodynamics
pH -log of proton concentration
PK pharmacokinetic
PPI poly(propylene imine)
ppm parts per million
PEG polyethylene glycol
PEO poly(ethylene oxide)
PEG–COCl , -di(chlorocarbonylmethylene) poly(ethyleneglycol)
Rf retention Factor
s strong/singlet
SEC size-exclusion chromatography
t triplet
TEM transmission electron microscopy

XI
THF tetrahydrofuran
TLC thin layer chromatography
TSA -toluenesulfonic acid
UV-vis ultraviolet-visible
v:v volume/volume

1
Chapter 1: Background and Literature Review
1.1. Dendrimers
1.1.1. Introduction
Dendrimers are a family of nanosized and perfect monodisperse polymers with
a regular and highly branched three-dimensional macromolecules and compact
spherical geometry in solution. Their name comes from the Greek word
“dendron”, meaning “tree” and meros meaning “part”. In comparison to the
traditional polymers, dendrimers are relative newcomers. Dendrimers differ
from many traditional polymers, such as linear and branched polymers, because
they are well defined size, with a high degree of molecular uniformity and
monodispersity, in addition to a highly functional surface [1-6]. Generally, low
generation dendrimers have an open structure, but as the generation increases
the structure becomes more spherical and dense. Dendrimers offer a plenty of
advantages compared to other architectural forms of polymers that have been
used in drug-delivery systems (DDS). Compared with classical linear polymers,
dendrimers offer several featured advantages as drug carrier applicants. These
advantages consist of: (1) high density and reactivity of functional groups on the
outside of dendrimers [7, 8]; (2) well-defined globular structure, predictable
molecule weight and monodispersity [9]; (3) controllable size [10-12]; (4) high
penetration abilities of dendrimers through the cell membrane [7, 13]; (5)
enhanced penetration and retention effect of dendrimers offers preferential
uptake of the materials by cancer tissues [8]; (6) programmed release of drugs
from the matrixes leads to reduced toxicity, increased bioavailability and
simplified dosing schedule [14, 15]. Commonly, the size, shape, and surface
properties of the polymeric carriers greatly influence the pharmacodynamic
(PD) and pharmacokinetic (PK) behaviors of drugs encapsulated in/complexed

2
to/conjugated to the carrier [16]. In the case of dendrimers, the loading ability of
drug molecules and other bioactive agents can be altered by varying dendrimer
generations, the water solubility, biodistribution, circulation time in blood and
therapeutic efficiency of drugs in dendrimer-based formulations can be tuned by
varying dendrimer surface components, the release of drugs from dendrimer
scaffolds can be controlled by using different degradable linkers between
dendrimers and drugs, and the specific accumulation of the dendrimer-based
therapeutics can be achieved by further modifying the dendrimers with targeting
moieties [16]. These properties together prove dendrimer perfect candidates in
the design of new DDS. This thesis will give an overview of the specific use of
modified dendrimers with -CD in drug delivery and how it relates to the
specific properties of these materials.
1.1.2. History and Progress of Dendrimer Research
The successful laboratory synthesis of dendrimers initially reported by Vögtle
and his group at the end of the 1970’s [17] with the so-called “cascade
molecules”. The concept of repetitive growth with branching was first reported
in 1978 by Vögtle. The result was a new core-shell macromolecular
architecture, now recognized as dendrimers. This was followed closely by the
Tomalia group [2], the first paper showing in great detail the preparation of
poly(amidoamine) (PAMAM) dendrimers appeared in 1985, the same year a
communication reported the synthesis of arborols by Newkome et al. [18], and
others to give rise to the larger dendritic structures [19, 20]. These
hyperbranched molecules were called “dendrimers” or “arborols” (from the
Latin “arbor” also meaning tree).

3
1.1.3. Molecular Structure of Dendrimers
In contrast to traditional polymers, dendrimers are unique core–shell structures
possessing three basic structural components (Fig. 1): a core (central point
known as the initiator) (I), branched units (II), and end groups (the outer shell or
periphery) (III).
Figure 1. Typical architecture of dendrimer.
1.1.4. Synthesis Methods
1.1.4.1. Divergent Approach
In the divergent synthesis strategy, the construction of the dendrimer takes place
in a stepwise manner starting from the core and building up the molecule
towards the outside using two basic procedures (1) coupling of the monomer
and (2) deprotection or conversion of the monomer end-group to create a new
reactive surface functionality and then coupling of a new monomer. The process

4
is repeated for several generations and a dendrimer is built layer after layer. The
divergent synthesis was initially applied extensively in the synthesis of
poly(propylene imine) (PPI) and PAMAM dendrimers, but has also found wide
use in the synthesis of dendrimers having other structural designs [20, 21]. The
divergent technique is successful for the construction of large quantities of
dendrimers. Problems happen from side reactions and imperfect reactions of the
ending groups that lead to structure defects. To avoid side reactions and to force
reactions to achievement large excess of reagents is required. It causes some
difficulties in the purification of the ultimate product [22-24] (Fig. 2).
Figure 2. Schematic drawing showing the divergent method for synthesis of
dendrimers.
1.1.4.2. Convergent Approach
Fréchet and Hawker first proposed an alternative approach to dendrimer
syntheses, known as the convergent method. The convergent synthesis strategy
was developed as a comeback to the limitations of the divergent synthesis [24,
25]. The convergent method, proceeds in the opposite direction, from the
periphery to the core that is from the outside inwards. When the growing
branched polymeric arms, called dendrons, are large enough, dendrons are
attached to a multifunctional core molecule. The convergent growth technique

5
has several benefits. In this method the number of reactive sites during the
creation process remains minimal leading to faster reaction rates and yields.
Another benefit of this approach is the large “molecular difference” between the
reactant molecule and the product, facilitating the separation of the reactants
from the product during the purification process (Fig. 3).
Figure 3. Schematic drawing showing the convergent method for synthesis of
dendrimers.
1.1.5. Properties of Dendrimers
1.1.5.1. Nanoscale Monodispersity
Dendrimers can be constructed with a well-defined molecular structure
(monodisperse), dissimilar to linear polymers. The monodispersity of
dendrimers has been studied broadly by size-exclusion chromatography (SEC),
mass spectrometry (MS), gel electrophoresis and transmission electron
microscopy (TEM) [26]. The level of monodispersity is controlled by the ability
of the synthetic chemist, as well as the isolation and purification techniques
utilized. Generally, convergent technique create the most closely monodisperse
dendrimers as determined by mass spectrometry. This is because the convergent
method allows purification at each step of the synthesis and removes cumulative
effects because of unsuccessful couplings [27]. However, mass spectrometry

6
has shown that PAMAM dendrimers produced by the divergent technique are
remarkably monodisperse for earlier generation (G= 0–5) [28].
1.1.5.2. Functionalization of Dendrimer Surface Groups
The periphery groups of dendrimers within a generational series can be
estimated at least three different styles, specifically as a flexible, semiflexible,
or inflexible functionalized scaffolding. Based on mathematically defined
dendritic branching rules, the several exterior exhibitions become more crowded
and inflexible as a function of growing generation level. Additionally,
dendrimers may be viewed as adaptable and can be surface-functionalized with
a great range of chemical and application features. The capacity to engineer and
control these factors provides a limitless list of possibilities for utilizing
dendrimers as modules for nanodevice design [28-31].
1.1.5.3. Nanoscale Dimensions and Shapes that Mimic Proteins
Considering the extraordinary structure control and nanoscale dimensions
observed for dendrimers, it is not amazing to find extensive interest in their use
as globular protein mimics (Fig. 4) [32]. Based on their systematic, size-scaling
properties, electrophoretic and hydrodynamic [33] behavior, dendrimers are
referred to as synthetic proteins [34, 35]. Substantial effort has been focused
recently on the use of dendrimers for site-isolation mimicry of proteins [1], drug
delivery [35], surface engineering [36], and light harvesting [37]. These
fundamental properties have in fact led to their commercial use as globular
protein replacements for gene therapy, immunodiagnostics [38], and a variety of
other biological applications. Dendrimers are generally produced in an
interactive sequence of reaction steps, in that each additional repetition leads to
higher generation dendrimer. Each of the new layer creates a novel ‘generation’

7
along with doubling the number of terminal groups (or active sites) and almost
double the molecular weight of the previous generation, too (Table 1) [1].
Generation Number of surface groups Molecular weight a Diameter (nm)b
0 4 517 1.5
1 8 1430 2.2
2 16 3256 2.9
3 32 6909 3.6
4 64 14215 4.5
5 128 28826 5.4
6 256 58048 6.7
7 512 116493 8.1
8 1024 233383 9.7
9 2048 467162 11.4
10 4096 934720 13.5
a Molecular weight is based on defect-free, ideal-structure, amine terminated
dendrimers. b Molecular dimensions determined by size-exclusion chromatography.
Table 1. Physical characteristics of PAMAM dendrimers.

8
Figure 4. The close dimensional size (nm) of selected proteins to respective
generations of PAMAM dendrimers [12].
1.1.5.4. Physicochemical Properties of Dendrimers
As the dendrimer grows, the various parts of the dendritic architecture (the
dendrimer structure may be divided into three parts: a: The surface of dendrimer
b: the outer shell c: the core) begin to show different features which are
improved with increasing generation. In the subsequent section
physicochemical properties of dendrimers will therefore be presented in more
general terms.
1.1.5.4.1. Dendrimers and the Effect of Molecular Growth
The conformational behaviors of a dendrimer upon growing to upper
generations are defined by (1) the molecular dimensions of the monomers (2)
the flexibility of the dendrons and (3) the capability of the interactions of end-
groups with each other (by hydrogen bonding creating a dense outer shell).
In 1983 de Gennes and Hervet presented limits to the growing of branched
molecules, paying due attention to the effect of steric hindrance [39]. Their
studies determined that upon growth, the outside of the dendrimer becomes

9
gradually crowded while the molecular density of the core section remains low
during the molecular growth. As no back-folding (dendrons folding into the
inside of the dendrimer) is taken into account, the increasing molecular
crowding in the outside shell will give a limitation on the generation number
that a starburst dendrimer can grow to.
Dendrimers with amine surface groups have a relatively high mobility due to
the absence of binding interactions between both the dendrimer arms and the
functional groups at the superficial. This larger mobility enables the dendrons to
back-folding the dendrimer interior as a significance of entropy [40]. Thus, the
structural behavior of the dendrimer upon growing to higher generations is
estimated by the ability of the surface functionalities to form a network with
each other through, e.g. hydrogen bonding or ion pairing thus consolidating a
dense outer shell [41]. Also, study based on molecular dynamics indicate that
low generation dendrimers have conformations with low degree of back-folding
(“density overlap”) compared to higher generations [42] (Fig. 5).
Figure 5. Schematic illustration of the result of back-folding in an increased
molecular density in the interior of a dendrimer.

10
1.1.5.4.2. Dendrimers and the Effect of pH
PAMAM and PPI dendrimers with amino terminal groups have basic exterior
groups as well as a basic inner. For these types of dendrimers with interiors
containing tertiary amines, the low pH area commonly leads to extended
conformations because of electrostatic repulsion between the positively charged
ammonium groups. At this pH (pH 4), the interior is getting increasingly
“hollow” as the generation number rises because of repulsion between the
positively charged amines both at the dendrimer exterior and the tertiary amines
in the internal.
At neutral pH, back-folding occurs which may be a consequence of hydrogen
bonding between the uncharged tertiary amines in the internal and the positively
charged exterior amines. At higher pH (pH 10) the dendrimer contract as the
charge of the molecule becomes neutral, obtaining a more globular structure
based on a loose dense network, where the repulsive forces between the
dendrimer arms and between the surface groups reaches a minimum. At this pH,
the conformation has a higher degree of back-folding as a significance of the
weak “inter-dendron” repulsive forces.
PPI dendrimers having carboxylic acid end-groups at pH 2 has the most
extended conformation because of the electrostatic repulsion between the
positively charged protonated tertiary amines, leading to a large radius of the
core, while at pH 6 radius of the dendrimer because of equals of positively
charged amines with amount of negatively charged carboxylic groups is
decreased. At pH 11 the electrostatic repulsion between the negative charged
forces the surface groups apart to give a more extended conformation with a
highly expanded exterior area [43].

11
1.1.5.4.3. Dendrimers and the Effect of Salt
In general molecular simulations in the PPI dendrimers display that high
concentration of salts has a strong influence on charged of PPI dendrimers and
lead to compact conformation of dendrimers, with a high degree of back-folding
[44, 45]. At low concentration of salts, the repulsive forces between the charged
dendrimer fragments resulted in an extended conformation to minimize charge
repulsion in the construction.
1.1.5.4.4. Dendrimers and the Effect of Concentration
The conformation of dendrimers with flexible constructions is not only affected
by small molecules like solvents, salts or protons, but may also be sensitive to
other dendrimers or surfaces which can have a great effect on the molecular
density and conformation of the dendrimer. Conformation of PPI dendrimers
(G4, G5) in a polar solvent like methanol achieved with small angle X-ray
scattering (SAXS) experiments show that upon increasing concentration
becomes increasingly contracted. This molecular contraction may diminish the
repulsive forces between the dendrimer molecules and growth the ability of the
dendrimers to display a more tight intermolecular packing [46].
1.1.5.4.5. Dendrimers and the Effect of Solvent
The role of the solvent to solvate the dendrimer structure is a very significant
factor in the conformational state of a dendrimer. Investigations with molecular
dynamics show that the dendrimer conformation depends on dendrimer
generation in different solvents [42]. Generally all generations of dendrimers
show extend of back-folding with decreasing solvation. However, the low
generation dendrimers show the highest affinity towards back-folding because
of poor solvation compared to the higher generation dendrimers.

12
NMR studies performed on PPI dendrimers indicate that an apolar solvent,
poorly solvates the dendrons favoring intramolecular interactions between the
dendrimer sections and back-folding. However, a weakly acidic solvent like
chloroform can act as a hydrogen donor for the interior amines in a basic
dendrimer like PPI, leading to an extended conformation of the dendrimer due
to extensive hydrogen bonding between the solvent and the dendrimer amines
[47]. Both experimental as well as theoretical studies on amino-terminated PPI
and PAMAM dendrimers display the trend that apolar aprotic (“poor”) solvents
induce higher molecular densities in the core area as a result of back-folding,
however polar (“good”) solvents solvate the dendrimer arms and induce a
higher molecular density on the dendrimer surface (Fig. 6) [48].
Figure 6. The effect of the solvent on the conformational state of a dendrimer.
1.1.5.5. Biocompatibility of Dendrimers
In order to incorporate dendrimers as biological agents, they have to accomplish
definite biological requests. The dendrimer should be:

13
non-toxic;
non-immunogenic (except for vaccines);
able to cross biobarriers (biopermeable) for example, the intestines, the
blood–tissue barriers, cell membranes, etc.;
able to stay in circulation in the biological system for the time needed to
have the desired clinical effect;
able to target to particular structures
Biological properties like the toxicity or the immunogenicity profile of a
dendrimer is to a large extent governed by the size of the dendrimer and by the
exterior groups existing on the particular dendrimer. Dendrimers have to show
low toxicity and be non-immunogenic in order to be broadly used in biomedical
applications. Till now, the cytotoxicity of dendrimers has been chiefly
investigated in vitro, though; a few in vivo studies have been reported. Duncan
and co-workers have studied the relationship between structure and
biocompatibility of PAMAM, PPI, poly(ethylene oxide) (PEO) grafted
carbosilane dendrimers with cationic (NH2-terminated) and anionic (COONa-
terminated) dendrimers in vitro. They have reported that, regardless of structure,
cationic dendrimers were usually more hemolytic and cytotoxic effect at even
relatively low concentration in relationship to anionic dendrimers. Furthermore,
the cytotoxicity was found to be generation-dependent, with higher generation
dendrimers being the most toxic [49]. One more study reported that anionic
PAMAM dendrimers have displayed a significantly lower cytotoxicity than
cationic dendrimer using Caco-2 cells [50]. One way to decrease the
cytotoxicity of cationic dendrimers may reside in partial surface derivatization
with chemically inert functionalities such as PEG or fatty acids. Hydroxy- or
methoxy-terminated polyester dendrimers have been reported to have low
toxicity in both in vitro and in vivo studies. During in vitro study, at high
concentration, these dendrimers have induced some inhibition of cell growth but

14
no increase in cell death was observed. The results make these new dendritic
motifs promising candidates for drug-delivery devices [51].
Immunogenicity is one of the vital biological properties of the dendrimers. In
brief, dendrimers have varying degrees of immunostimulatory properties
depending on their surface functionalities. Generally, the immunogenicity of
unmodified amino dendrimers is low, but seems to increase with increasing
generation of the dendrimer. Initial reports on the immunogenicity of
dendrimers showed low or modest immunogenicity of unmodified amino
terminated PAMAM dendrimers (G3–G7). However, later investigations
showed some immunogenicity of these dendrimers and found that modification
of amino-terminated PAMAM dendrimers with polyethylene glycol (PEG)
chains decreases immunogenicity and gives longer life-time in the blood stream
in comparison to unmodified dendrimers [52].
1.1.6. Application of Dendrimer in Drug Delivery Systems
Drug delivery is a significant feature in the design of a drug because the suitable
choice of delivery system can control the bioavailability, concentration profile
and unwanted side effects (by targeted delivery). Dendrimers and polymers
have some properties in common, when considering drug delivery. The chief
problem related with using polymers for drug delivery is the wide molecular
weight dispersal often found in polymers, which in terms can lead to
irreproducible pharmacokinetic behavior. Dendrimers are in general useful as a
part of the molecular toolbox for drug delivery. Toxicity and biodistribution of
dendrimers are significant in medical use of any drug, and for dendrimers this is
highly dependent on the actual structure of the dendrimer [49, 53].
General principles are being established for designing dendrimer structures as
delivery systems which contain: 1) negatively-charged and neutral dendrimers
are commonly biocompatible, whereas positively-charged species display

15
varying degrees of toxicity; 2) PEGylation growths water solubility and
dendrimer size, and can lead to progressed retention and biodistribution
characteristics; 3) therapeutic agents can be internalized into the void space
between the edge and core, or covalently attached to functionalized exterior
groups; 4) targeting moieties bound to the dendrimer surface can be used to
specially treat cancer cells with definite over-expressed receptor targets. As
dendrimer structures have become more specialized, improved efficiency in in
vitro and in vivo models is being realized. The applications of such systems
have been several-fold, including the use of dendrimers to increase drug
solubility and bioavailability, and to act as release modifiers and platforms for
drug targeting.
1.1.7. Mechanism of Drug Delivery Using Dendrimers
Drug delivery with dendrimers can basically take place by two different types of
mechanisms: (1) by in vivo degradation of a drug–dendrimer conjugate, where
the drug is covalently bound to the dendrimer, or (2) by utilizing host–guest
chemistry where the drug is existing as a guest in the dendrimer, and is released
because of changes in the physical environment such as pH, temperature or
simply released by diffusion out of the dendrimer. Two other significant issues
in drug delivery are targeted delivery to a specific type of cell or controlled
release from a depot, which may be present in circulation or imbedded in some
suitable tissue. Host–guest based systems are treated first in the following
sections, since they are the most studied dendrimer-based drug delivery
systems.
The ideal dendrimer carrier should show high aqueous solubility and drug-
loading ability, biodegradability, low toxicity, favorable retention and
biodistribution characteristics, specificity, and suitable bioavailability. In
dendrimer-based drug delivery, a drug is either non-covalently encapsulated in

16
the inside of the dendrimer or covalently conjugated to form macromolecular
prodrugs.
An area that has attracted great interest is the interaction between drugs and
dendrimers. Numerous types of interactions have been discovered, which can be
generally subdivided into the entrapment of drugs within the dendritic
architecture (involving electrostatic, hydrophobic and hydrogen bond
interactions) and the interaction between a drug and the surface of a dendrimer
(electrostatic and covalent interactions). In this overview a range of drug/model
compound–dendrimer interactions will be considered together.
1.1.7.1. Noncovalent Encapsulation of Drugs
The nature of drug encapsulation inside a dendrimer may be simple physical
entrapment, or can involve non-bonding interactions with particular structures
within the dendrimer. Much of the work in this field has been based on the
hypothesis that dendrimers have a hollow core and a dense shell [7], and in fact,
much of the text based on drug encapsulation supports this theory (Fig. 7).
Primary studies of dendrimers as potential delivery systems focused on their use
as unimolecular micelles and ‘dendritic boxes’ for the noncovalent
encapsulation of drug molecules. For example, hydrophobic drugs and dye
compounds were incorporated into several dendrimer cores [54-57]. An
advantage of using dendritic unimolecular micelles rather than conventional
polymeric micelles is that the micelles structure is maintained at all
concentrations because the hydrophobic pieces are covalently attached.
Although the introduction of stabilizing PEO chains on the dendrimer outside
has expanded the scope of dendritic unimolecular micelles to incorporate
anticancer drugs such as 5-fluorouracil [58], methotrexate [15] and doxorubicin
[15] and can slow the drug release rates in these systems to some extent, this
method has yet to be demonstrated as a general strategy.

17
The encapsulation efficiency of drug molecules is influenced by dendrimer size,
exterior structure, solvent pH and functionality of the drug molecule. Generally,
higher generation dendrimers explain improved capability for drug
solubilization compared to lower generation dendrimers because of the larger
void volume available for drug encapsulation [59], and enhanced electrostatic
interactions with increasing dendrimer generation [60-63].
The encapsulation of guest molecules which is based on non-covalent
interactions (hydrophobic interactions, physical entrapment, hydrogen bonding,
electrostatic bonding, or a combination of these methods) will be discussed in
more detail below with the help of examples.
Figure 7. Drawing of a dendrimer carrier encapsulating hydrophobic drug
molecules in the dendrimer’s voids to increase their aqueous solubility and
control their release rate.

18
1.1.7.1.1. Hydrophobic Interactions
The first method, which is most broadly applied, is based on the nature of the
micelles core: Hydrophobic cores interact favorably with hydrophobic guest
molecules [57, 64, 65]. Dendrimers consisting of an apolar core and polar shell
have been referred to as “unimolecular micelles”. Unlike usual micelles, the
dendritic structure is independent of dendrimer concentration [56, 65-68], i.e.
the dendrimers did not have a critical micelle concentration. The unimolecular
micelles structure of dendrimers was reported by Newkome et al. [56, 69]. The
ability of dendritic micelles to solubilize hydrophobic guest molecules in
aqueous media through hydrophobic interactions was first proved by Newkome
et al [70]. The location of the guest is not controlled nor is it known. The design
of dendrimers with well-defined and localized cavities or binding sites can be
realized by the specific incorporation of a hydrophobic core, hydrogen-bonding
moiety, or a metal ion coordination site.
PEG has been used to alter dendrimers in the design of solubilizing and drug
delivery systems [15, 58, 59, 71-75]. PEG is naturally conjugated to the outward
of a dendrimer to supply a hydrophilic shell around a hydrophobic dendritic
core to form a unimolecular micelle. PEG is of specific attention in the design
of dendrimer systems for pharmaceutical applications due to its high water
solubility, biocompatibility and ability to modify the biodistribution of carriers
[76]. Water-soluble dendritic unimolecular micelles of G3 dendrimer with
surface shell of PEG chains was synthesized by Liu et al. [69, 71] and the drug
loading of indomethacin was examined. The drug-loaded dendrimer supplied
sustained release of indomethacin over a period of approximately 30 h.

19
1.1.7.1.2. Physical Entrapment
Meijer et al. [54, 55] developed the famous ‘dendritic box’ idea, whereby
hydrophobic guest molecules are physically entrapped inside the internal
cavities of the dendrimers. During the synthetic process guest molecules could
be entrapped inside the cavities of the dendritic boxes, with a dense exterior
shell preventing dispersion from the structures, even after solvent extraction,
prolonged heating or sonication. The guest molecule becomes physically
trapped inside the core and can be released only when the ‘dense shell’ is
disrupted. The synthesis of dendritic boxes based on PPI dendrimers with
primary amine end groups were synthesized by Jansen et al [54, 77]. The
inflexible shell end group was obtained through modification with a bulky
amino acid derivative to yield a dense and rigid chiral shell with solid-phase
properties and a flexible core capable of entrapping molecules. A number of dye
molecules were encapsulated in the dendritic box, for example, up to 4
molecules of Bengal Rose could be encapsulated per dendrimer.
1.1.7.1.3. Hydrogen Bonding Interactions
Hydrogen Bonding interactions are possible between guest molecules and the
core or the exterior of dendritic micelles if appropriate functional groups are
existing on both components [78].
1.1.7.1.4. Electrostatic Interactions
The presence of large numbers of ionizable groups on the periphery and interior
of dendrimers supplies an interesting prospect for electrostatic attachment of
many ionizable drugs, providing the resulting complex retains adequate water
solubility [79-81]. For example, full generation PAMAM dendrimers have
primary amine ending groups (–NH2) on the periphery and tertiary amine

20
groups ( N–) positioned at the branching positions in the core both are
titratable having pKa values of 10.7 and 6.5, respectively [82].
The non-steroidal anti-inflammatory drug such as ibuprofen drug has been most
widely complexed to full generation of PAMAM dendrimers. Electrostatic
interaction can occur between the carboxyl groups of this poorly acidic drug and
the amine groups of the dendrimers. It has been estimated that approximately 40
ibuprofen molecules interact with G4 PAMAM dendrimer at pH 10.5 causing a
significant improvement of drug solubility [83].
The solubility improvement of the poorly water-soluble drug indomethacin in
the presence of a series of G4 and G4.5 PAMAM dendrimers and also G4
dendrimers with exterior hydroxyl groups was examined by Chauhan et al. [84].
The increasing solubility of indomethacin with G4 dendrimer was explained on
the basis of electrostatic interaction between the carboxyl group of
indomethacin and the amino groups of the dendrimer. As no electrostatic
interaction can be expected between the G4.5 and drug, the increased solubility
was thought to be a result of molecular encapsulation resulting from non-
specific, non-covalent interactions. Possible association of weak hydrogen
bonding was also thought to be effective in causing an increase of solubility in
the attendance of the hydroxyl-terminated G4 dendrimer. All three types of
dendrimer were shown to be efficient in increasing the change of indomethacin
crossways skin in both in vitro and in vivo experiments.
1.1.7.1.2. Covalent Dendrimer–drug Conjugates
The covalent binding of drugs to the exterior groups of dendrimers through
hydrolysable or biodegradable connections offers the opportunity for a greater
control over drug release than can be succeed by electrostatic complexation of
drugs to the dendrimers (Fig. 8). An alternative methodology to the
development of dendrimers as anticancer drug carriers is to exploit their well-

21
defined multivalency for the covalent attachment of drug molecules to the
dendrimer periphery. The drug loading can be altered by varying the generation
number of the dendrimer, and release of the drug can be controlled by
incorporating degradable linkages between the drug and dendrimer. For
example, Duncan and co-workers [85] have prepared conjugates of PAMAM
dendrimers with cisplatin, a potent anticancer drug with nonspecific toxicity and
poor water solubility. The conjugates display increased solubility, decreased
systemic toxicity and selective accumulation in solid tumors.
The preparation of multivalent folic acid conjugates of dendrimers [86] has
important suggestions for targeting to tumor cells, and the multivalent character
of dendrimers facilitates the attachment of several payloads, including targeting,
diagnostic and therapeutic molecules, as well as combinations of these agents.
Because expression of the folate receptor is improved in several human cancers
and restricted in most normal tissues [87], folic acid is an interesting candidate
for the active targeting of dendrimer–drug conjugates to tumors. Inspired by the
concepts of Esfand and Tomalia [35], and the multivalent dendrimer-folate and
dendrimer-methotrexate conjugates of Fréchet and co-workers [86], Quintana et
al. [88] prepared analogous PAMAM dendrimers with methotrexate conjugated
to their outside via either a stable amide or an ester linkage that could be
hydrolyzed under biological states. As expected, the introduction of folic acid
into these conjugates was found to increase their cellular uptake, resulting in an
increase in cytotoxicity of the methotrexate ester conjugate relative to that of the
free drug in vitro.

22
Figure 8. Schematic drawing showing a dendrimer-drug conjugate where the
drug molecules (red ovals) are either directly coupled (solid lines) to
dendrimer’s surface groups or via a pH-sensitive linkage (blue rectangle).
1.1.8. Release of Guest Molecules
Dendrimers have been used as devices for the release of hydrophobic
compounds [89]. Investigations have shown that these compounds structures are
able to release hydrophobic drugs in a sustained mode, as release has been
found at times to continue for several hours [89]. Release of hydrophobic
molecules from dendritic structures is dependent on both the construction of the
host and the size and shape of the guest molecules. The release of hydrophobic
compounds from amphiphilic dendritic polymers can also be affected by the
type of host-guest interactions involved. While weaker host-guest interactions
like hydrophobic interactions allow the almost complete release of the
encapsulated molecules from the micelle, partial release of the compounds has

23
been found to occur when stronger interactions (hydrogen and ionic bonding)
are involved. Release studies of lidocaine from a dendritic micelle have been
achieved in an aqueous medium [90]. At pH 7 lidocaine is hydrophobic and the
interactions between the core of the micelle and the drug are hydrophobic. The
release profile displays sustained character and it is detected that almost all the
drug molecules encapsulated are released after about 30 hours.
Encapsulation studies of ibuprofen in PAMAM dendrimers end-functionalized
with -NH2 groups showed two types of host-guest interactions: hydrophobic
interactions between the drug molecules and the hydrophobic core, and ionic
interactions between the carboxylic group of the drug and the amino termination
groups of the PAMAM dendrimer [91]. It was observed that the release of the
drug from the dendrimer was considerably slower, with 70% and 80% of the
drug being released from G3 and G4 dendrimers, respectively, after 8-9 hours. It
was demonstrated that because of the small size of the G3 dendrimer, ibuprofen
molecules were not solubilized in the hydrophobic internal of the micelle but
rather formed complexes through ionic interactions with the peripheral amino
groups of the dendrimer. In the G4 micelle, however, a small fraction of
ibuprofen (~ 18%) was also encapsulated within the hydrophobic voids of the
dendritic core, leading to the higher release of the drug from the G4 dendritic
micelles. In methanol and deionized water, less than 15% of the drug was
released from the G4 dendritic micelle even after 9 hours. This suggests that the
stronger the drug-dendrimer complex, the lower the fraction of drug released.
1.1.9. Modification of Periphery Groups of Dendrimer
Because of their perfect structure and the large number of chain ends present, it
is convenient to functionalize dendrimers at their periphery. Dendrimers with
hydrophobic cores derived from hexatricontaalchohol and poly(aryl ether)s,
were functionalized by Newkome et al. [56] and Hawker et al. [57] with polar

24
carboxylic acid groups, respectively. In this work, we conjugated -CD groups
on the surface of the dendrimers and investigated how the guest molecules get
trapped in the dendritic compounds in detail.
Cyclodextrins (CDs) are cyclic oligosaccharides created with 6, 7 or 8
glucopyranose units, respectively named as -, - or -cyclodextrin. Their
hydrophobic apolar interior (cavity) allows to form inclusion complexes with
organic compounds through formation of non-covalent inclusion complexes
[92]. The cavity size of -CD is insufficient for many drugs and -CD is
expensive. In general, -CD has weaker complex forming ability than
conventional CDs. -CD has been widely used in the early stages of
pharmaceutical applications because of its ready availability and cavity size
suitable for the widest range of drugs. But the low aqueous solubility and
nephrotoxicity limited the use of -CD especially in parenteral drug delivery.
Chemically modified CD derivatives have been prepared with a view to extend
the physicochemical properties and inclusion capacity of parent CDs. Several
amorphous, non-crystallizable CD derivatives with enhanced aqueous
solubility, physical and microbiological stability, and reduced parenteral
toxicity have been developed by chemical modification of parenteral CDs [93,
94].
-CD have a polar exterior and an apolar interior cavity [95] and is well known
for its capability to form host–guest inclusion complexes with hydrophobic
drugs [96]. Encapsulation ability of CDs has found numerous applications in
fields such as drug delivery systems. -CD is the most positive to be solubilized
of molecules but the solubilization capability of -CD is restricted by its poor
solubility in water (1.85 g/100 ml at 20 °C) [97] and almost unsolvable
complexes are often achieved with greatly a polar molecules [92]. Diverse
chemical alterations have been so far projected for improvement of solubility of
-CD and its complexes in aqueous medium, such as preparation of -CD
conjugates with biocompatible polar artificial polymers [98, 99]. In recent

25
times, it has been publicized that the merging of CDs into polymeric
nanoparticles can enhance the drug loading and modifies drug release [100-
102].
1.1.10. Aim of Work
Literature shows that due to their multivalency and globular shape dendrimers
are regarded as ideal candidates for use in biomedical applications. A number of
challenging targets remain in the design and synthesis of dendrimers for this
purpose. In this thesis, the supramolecular encapsulation of guest molecules in
the interior of a dendrimer is evaluated by using specific interactions
(electrostatic, hydrophobic and hydrogen bonding).
In this work, the synthesis of a new class of dendrimers is described that can be
applied in drug delivery systems (DDS). For this purpose, were described the
synthesis and characterization of multivalent citric acid based dendrimers and
modified citric acid dendrimers. Modified citric acid dendrimers are synthesized
via ester and amide linkage with -cyclodextrin ( -CD) and 6-Deoxy-6-amino-
-cyclodextrin ( -CD-NH2) for non-toxicity and enhanced water-solubility.
Cyclodextrins (CDs), including hydrophobic apolar interior (cavity) allows to
form inclusion complexes with organic compounds through formation of non-
covalent inclusion complexes
First, second and third generation (G) of citric acid based dendrimers are
prepared and the modification of synthesized dendrimers has been investigated
in detail. This thesis reports on the preparation of a citric acid-based dendrimers
bearing -CD as grafted side-chains by using dicyclohexyl carbodiimide (DCC)
as a coupling agent and an evaluation of its ability for the encapsulation of a
Naltrexone (NLX) and a biocompatible surface. The use of these dendrimers as
pH sensitive carriers for NLX has also been studied. Dendrimers were loaded
with NLX and the release pattern of the drug was investigated. Influences of the

26
dendrimer generation and number of grafted -CD with dendrimers on their
encapsulation ability have been described.

27
Chapter 2: Experimental Section
2.1. Materials and Methods
2. 1.1. Materials
Poly(ethylene glycol) 600 diacid (acid number 175, 96–98%, from Fluka) was
dried over Na2SO4 (Merck Chemical Co., Germany). -cyclodextrin (from
Fluka) dried in oven at 90 ˚C for 8 h. Citric acid and pyridine (purified with
refluxing over NaOH for 2 h and subsequent distillation) were obtained from
Merck Chemical Co. (Germany). -Toluenesulfonic acid (TSA) were purified
by conventional methods before use. Thionyl chloride (Merck Chemical Co.,
Germany) was refluxed on linseed oil for 2 h. Sodium azide was purchased
from Fluka. Dicyclohexyl carbodiimide (DCC) was purchased from Merck
Chemical Co. (Germany). N, N-dimethylformamide (DMF) (from Fluka) was
dried and distilled under reduced pressure. Dialysis membrane D7884 was
purchased from Sigma-Aldrich Co. (Steinhein, Germany) (retains molecular
weights greater than 2000 and releases smaller than 1200). Naltrexone (NLX)
hydrochloride was obtained from Sigma (St. Louis, MO, USA) and neutralized
with NaOH 0.25M. Fig. 9 shows the structure of NLX. Other reagents and
solvents purchased from Merck Chemical Co. (Germany).
Figure 9. Chemical structure of naltrexone.

28
2.1.2. Instrumental Measurement
FT–IR spectra were recorded on a Bruker Model Tensor-27 spectrometer.
Under appropriate conditions in a magnetic field, a sample can absorb
electromagnetic radiation in the radio frequency region at frequencies governed
by the characteristics of the sample. A plot of frequencies of the absorption
peaks versus peaks intensities constitutes a nuclear magnetic resonance
spectroscopy (NMR) spectrum. Thus, NMR is a very useful technique in
obtaining detailed information about molecular structure. In this dissertation,
NMR is used to identify the formation of specific products of reactions.1H and13C NMR spectra were recorded with a Bruker Advance 400 spectrometer
operating at 400.132 MHz (1H) and 100.623 (13C) using DMSO-d6, acetone
deuterium and D2O as a solvent. Analytical ultraviolet-Visible (UV-vis)
absorption spectroscopy is described by Beer’s Law (equation 2.1), where the
absorbance (A) of a molecule is a function of the molar absorptivity ( ), the path
length (l), and the concentration (c).
A = lc (2.1)
The UV absorption spectra were recorded using 1700 Shimadzu
spectrophotometer. Particle sizes were determined with dynamic light scattering
by commercially available equipment Zetasizer Nano ZS from Malvern using a
4-mW He–Ne laser (633 nm wavelength) with a fixed detector angle of 173°
and Laser Diffraction Particle Size Analyzer with model of SALD-2101 from
Shimadzu.

29
2.2. Methods
2.2.1. Preparation of , -di(chlorocarbonylmethylene) poly(ethyleneglycol)
(ClOC-PEG-COCl)
The diacyl halide poly(ethyleneglycol) was prepared by literature method [103].
In this procedure, dry poly-(ethyleneglycol) 600 diacid (PEG-A) was
chlorinated with refluxing in thionylchloride and ClOC–PEG–COCl obtained as
the light yellow oil, yield 100%.
2.2.2. Preparation of G1 Using Thionylchloride
A solution of citric acid (10.27 g, 5.83 10-2 mol) in 25 ml dry (THF) was
placed in a round-bottom flask equipped with a reflux condenser, dropping
funnel, argon inlet and magnetic stirrer. Dry pyridine (0.2 ml, 2.48 10-3 mol)
was added to this solution at 15 min through dropping funnel and mixture was
stirred for 20 min. A solution of ClOC–PEG–COCl (15.52 g, 2.43 10-2 mol) in
15 ml dry THF was added at for 30 min. The mixture was stirred at 0 for 1
h then at room temperature for 3 h and finally at 50 for additional 6 h (all
steps of reaction was carried out under argon) then was cooled and filtered off
and was precipitated in diethylether. The product was washed using
dichloromethane, cooled acetone, toluene and dissolved in 10 ml THF and
reprecipitated in diethylether and n-hexane for several times. The mixture was
poured in 5 ml of water at 25 . The mixture was conducted into cellophane
membrane dialysis bag. The bag was closed and transferred into a flask
containing 100 ml of water maintained at 25 . The external water was
continuously stirred for two days. The external water was removed after 24 h
and added 100 ml fresh water, then the product was removed from dialysis bag
and dried under vacuum at 50 as the reddish oil, yield 73% [20, 104].

30
1H NMR (DMSO-d6) [400 MHz,δ,ppm]: 2.67-2.77 (citric acid CH2), 3.6-3.8 (-
OCH2CH2O) and 4.1-4.2 (-COCH2O-). IR (KBr,cm-1): 3439-2610 (COOH),
1738 (C=O), 1180 and 1106 (C-O).
2.2.3. Preparation of G2 Using DCC
A solution of G1 (12.8 g, 1.35 10-2 mol) in 20 ml dry THF was added to a
round-bottom flask equipped with reflux condenser, argon inlet, dropping
funnel and magnetic stirrer. Dry pyridine (0.5 ml) was added to this solution by
dropping funnel (15 min). The mixture was stirred vigorously for 10 min. A
solution of DCC (18.38 g, 9.72 10-2 mol) in 15 ml dry THF was added to
mixture at 0 by dropping funnel. The mixture was stirred for 30 min. Then a
solution of citric acid (17.12 g, 9.72 10-2 mol) in 15 ml THF was added
dropwise to this solution. The mixture was stirred at 0 for 1.5 h then at room
temperature for 24 h under argon. The solution was filtered off and was placed
at 5 for 24 h and again the solution was filtered off. The product was
precipitated in diethylether and was washed by acetone dichloromethane,
toluene then dissolved in THF and reprecipitated in diethylether for several
times. The product was dissolved in 5 ml water and stirred for 24 h at room
temperature. The solution was filtered off and poured in 5 ml of water at 25 .
The mixture was conducted into cellophane membrane dialysis bag. The bag
was closed and transferred into a flask containing 100 ml of water maintained at
25 . The external water was continuously stirred for two days. The external
water was removed after 24 h and added 100 ml fresh water. The product was
removed from dialysis bag and dried under vacuum at 50 as the amorphous
compound, yield 60% [20, 104].

31
1H NMR (DMSO-d6) [400 MHz,δ,ppm]: 2.6-2.8 (citric acid CH2), 3.57-3.8 (-
OCH2CH2O) and 4.1-4.2 (-COCH2O-). IR (KBr,cm-1): 3581-2620 (COOH),
1732 (C=O), 123180 and 1096 (C-O).
2.2.4. Preparation of G3 Using DCC
A solution of G2 (8.3 g, 4.16 10-3 mol) in 35 ml dry THF was added to a
round-bottom flask equipped with reflux condenser, argon inlet, dropping
funnel and magnetic stirrer. Dry pyridine (0.5 ml) was added to this solution by
dropping funnel at 20 min. The mixture was stirred vigorously for 20 min. A
solution of DCC (23.17 g, 1.12 10-1 mol) in 25 ml of dry THF was added to
mixture at 0 by dropping funnel. The mixture was stirred for 30 min. Then a
solution of citric acid (21.58 g, 1.12 10-1 mol) in 20 ml of THF was added
dropwise to this solution. The mixture was stirred at 0 for 2 h then for
additional 72 h under argon at room temperature. The solution was filtered off
and placed at 5 for 24 h and again the solution was filtered off. The product
was precipitated in diethylether and washed by acetone, dichloromethane (for
removing the excess of citric acid), and toluene then dissolved in THF and
reprecipitated in diethylether for several times. The product was dissolved in 5
ml water and stirred for 24 h at room temperature. The solution was filtered off
and poured in 5 ml of water at 25 . The mixture was conducted into
cellophane membrane dialysis bag. The bag was closed and transferred into a
flask containing 100 ml of water maintained at 25 . The external water was
continuously stirred for two days. The external water was removed after 24 h
and added 100 ml fresh water. The product was removed from dialysis bag and
dried under vacuum at 50 as the solid, yield 18% [20, 104]. 1H NMR (DMSO-d6) [400 MHz,δ,ppm]: 2.62-2.76 (citric acid CH2), 3.57-3.77
(-OCH2CH2O) and 4.1-4.2 (-COCH2O-). IR (KBr,cm-1): 3439-2610 (COOH),
1730 (C=O), 1260 and 1096 (C-O).

32
2.2.5. Synthesis of G1- -CD Using DCC
G1 was prepared as previously reported [20]. A solution of G1 (0.25 g, 2.6 × 10-3
mol) in 20 ml dry DMF was added to a round-bottom flask equipped with reflux
condenser, argon inlet, dropping funnel and magnetic stirrer. Dry pyridine (0.2
ml) was added to this solution by dropping funnel (15 min). The mixture was
stirred vigorously for 15 min. To this solution, DCC (0.385 g, 1.87 × 10-3 mol)
in 15 ml dry DMF was added as a coupling agent at 0 °C by dropping funnel.
The mixture was stirred for 30 min. Then a solution of -CD (1.795 g, 1.58 ×
10-3 mol) in 15 ml DMF was added dropwise to this solution. The mixture was
stirred at 0 °C for 3 h then at room temperature for 54 h and finally at 65-75 °C
for 5 h under argon. The solution was filtered off and was placed at 4 °C for 24
h and again the solution was filtered off and the solvent was removed under
vacuum. The product was precipitated in diethylether and was washed by
acetone. Then the product was dissolved in the methanol, and filtered off then
were precipitated in diethylether several times for removal of unreacted -CD.
The solution was filtered and the solvent was removed under vacuum. The
product was dissolved in 5 ml water and stirred for 24 h at room temperature.
The solution was filtered off and poured in 5 ml of water at 25 °C. The mixture
was conducted into cellophane membrane dialysis bag. The bag was closed and
transferred into a flask containing 100 ml of water maintained at 25 °C. The
external water was continuously stirred for two days. The external water was
removed after 24 h and added 100 ml fresh water. The product was removed
from dialysis bag and dried under vacuum at 50 °C and purified compound was
obtained as a light brown solid, yield 51%. 1H NMR (400 MHz, DMSO-d6): =2.8-2.9 (q, citric acid CH2), 3.26-3.8 (br, -
CD H2, H4, H5, H3, H6a, H6b and polyethylene glycol (PEG) -OCH2CH2O-), 3.92-
4.2 (br, PEG -COCH2O-), 4.2-4.3 (br, β-CD -CH2OCO-dendrimer), 4.57 (br, -
CD OH6), 4.81 (br, -CD H1), 5.62 (br, -CD OH3) and 5.79 (br, -CD OH2). IR

33
(KBr, cm-1): 3400 ( , OH β-CD and COOH dendrimer), 2928 and 2860 ( , C–
H), 1739 and 1704 ( , ester and acid C=O), 1151 ( , C-O-C), 1106 and 1033 ( ,
C-O).
2.2.6. Synthesis of G2- -CD Using DCC
G2 was prepared according to literature [20]. A solution of G2 (0.25 g, 1.25 ×
10-4 mol) in 20 ml dry DMF was added to a round-bottom flask equipped with
reflux condenser, argon inlet, dropping funnel and magnetic stirrer. Dry
pyridine (0.2 ml) was added to this solution by dropping funnel within 15 min.
The mixture was stirred vigorously for 20 min. To this solution, DCC (0.56 g,
2.7 × 10-3 mol) in 15 ml dry DMF was added as a coupling agent at 0 °C by
dropping funnel. The mixture was stirred for 30 min. Then a solution of -CD
(2.56 g, 1.29 × 10-3 mol) in 15 ml DMF was added dropwise to this solution.
The mixture was stirred at 0 °C for 3 h then at room temperature for 72 h and
finally at 65-75 °C for 6 h under argon. The solution was filtered off and was
placed at 4 °C for 24 h and again the solution was filtered off and the solvent
was removed under vacuum. The product was precipitated in diethylether and
was washed by acetone. Then the product was dissolved in the methanol, and
filtered off then were precipitated in diethylether several times for removal of
unreacted -CD. The solution was filtered and the solvent was removed under
vacuum. The product was dissolved in 5 ml water and stirred for 24 h at room
temperature. The solution was filtered off and poured in 5 ml of water at 25 °C.
The mixture was conducted into cellophane membrane dialysis bag. The bag
was closed and transferred into a flask containing 100 ml of water maintained at
25 °C. The external water was continuously stirred for two days. The external
water was removed after 24 h and added 100 ml fresh water. The product was
removed from dialysis bag and dried under vacuum at 50 °C and purified
compound was obtained as a brown solid, yield 32%.

34
1H NMR (400 MHz, DMSO-d6): =2.72-2.88 (q, citric acid CH2), 3.16-3.83 (br,
-CD H2, H4, H5, H3, H6a, H6b and PEG -OCH2CH2O-), 4-4.1 (br, PEG -
COCH2O-), 4.2-4.4 (br, β-CD -CH2OCO-dendrimer), 4.6 (br, -CD OH6), 4.83
(br, -CD H1), 5.6 (br, -CD OH3) and 5.73 (br, -CD OH2). IR (KBr, cm-1):
3423 ( , OH β-CD and COOH dendrimer), 2931 and 2859 ( , C–H), 1741 and
1705 ( , ester and acid C=O), 1151 ( , C-O-C), 1100 and 1031 ( , C-O).
2.2.7. Synthesis of G3- -CD Using DCC
G3 was prepared as previously reported [20]. A solution of G3 (0.25 g, 4.85 ×
10-5 mol) in 20 ml dry DMF was added to a round-bottom flask equipped with
reflux condenser, argon inlet, dropping funnel and magnetic stirrer. Dry
pyridine (0.2 ml) was added to this solution by dropping funnel within 15 min.
The mixture was stirred vigorously for 20 min. To this solution, DCC (0.65 g,
3.15 × 10-3 mol) in 15 ml dry DMF was added as a coupling agent at 0 °C by
dropping funnel. The mixture was stirred for 30 min. Then a solution of -CD
(2.99 g, 5.8 × 10-4 mol) in 15 ml DMF was added dropwise to this solution. The
mixture was stirred at 0 °C for 3 h then at room temperature for 96 h and finally
at 65-75 °C for 8 h under argon. The solution was filtered off and was placed at
4 °C for 24 h and again the solution was filtered off and the solvent was
removed under vacuum. The product was precipitated in diethylether and was
washed by acetone. Then the product was dissolved in the methanol, and
filtered off then were precipitated in diethylether several times for removal of
unreacted -CD. The solution was filtered and the solvent was removed under
vacuum. The product was dissolved in 5 ml water and stirred for 24 h at room
temperature. The solution was filtered off and poured in 5 ml of water at 25 °C.
The mixture was conducted into cellophane membrane dialysis bag. The bag
was closed and transferred into a flask containing 100 ml of water maintained at

35
25 °C. The external water was continuously stirred for two days. The external
water was removed after 24 h and added 100 ml fresh water. The product was
removed from dialysis bag and dried under vacuum at 50 °C and purified
compound was obtained as a brown solid, yield 25%.
1H NMR (400 MHz, DMSO-d6): =2.72-3.06 (q, citric acid CH2), 3.16 (m, -
CD H2), 3.3-3.9 (br, -CD H4, H5, H3, H6a, H6b and PEG -OCH2CH2O-), 4-4.3
(br, β-CD -CH2OCO-dendrimer), 4.57 (br, -CD OH6), 4.83 (br, -CD H1), 5.63
(br, -CD OH3) and 5.73 (br, -CD OH2). IR (KBr, cm-1): 3416 ( , OH β-CD
and COOH dendrimer), 2930 and 2858 ( , C–H), 1743 and 1705 ( , ester and
acid C=O), 1154 ( , C-O-C), 1186 and 1031( , C-O).
2.2.8. Synthesis of G1-NH- -CD Using DCC
In order to obtain a G1-NH- -CD, 6-Deoxy-6-amino- -cyclodextrin ( -CD-
NH2) was first synthesized. -CD-NH2 was prepared as previously reported
[98]. Briefly, -CD-NH2 was obtained through a three-step synthetic pathway.
In the first step 6-Deoxy-6-(p-toluenesulfonyl)- -cyclodextrin ( -CDTos) was
prepared. In this method, dry -CD dissolved in pyridine. The resulting solution
was cooled in an ice bath and reacted with a solution of p-toluenesulfonyl
chloride in pyridine. The mixture was stirred at room temperature under
nitrogen for 12 h. Then pyridine was evaporated under a reduced pressure at 40
°C to obtain a viscose material, then diethyl ether was added. The white
precipitate was recrystallized in distilled water three times [99]. Then -CD
converts to the 6-Deoxy-6-azido- -cyclodextrin ( -CD-N3). For this purpose -
CD-N3 was prepared by reacting at 85 °C an aqueous suspension of -CDTos
with sodium azide. The reaction was monitored by TLC on silica plates with the
mixture n-BuOH/EtOH/H2O 5:4:3 (v/v) as mobile phase until the complete
disappearance of the spot of -CD-Tos (Rf=0.45). The azide product was finally

36
recovered by precipitation in acetone as a white powder. Then -CD-N3 and
triphenylphosphine were dissolved in DMF, and to the solution was added
concentrated NH3 (aq). The mixture was stirred at room temperature for about 4
h, and the solution was poured into acetone, giving a crude product as a white
precipitate. The resulting product was purified and -CD-NH2 was obtained
[98]. The spectroscopic data show the complete agreement with the proposed
structures.
A solution of G1 (0.25 g, 2.6 × 10-3 mol) in 20 ml dry DMF was added to a
round-bottom flask equipped with reflux condenser, argon inlet, dropping
funnel and magnetic stirrer. Dry pyridine (0.2 ml) was added to this solution by
dropping funnel (15 min). The mixture was stirred vigorously for 20 min. To
this solution, DCC (0.385 g, 1.87 × 10-3 mol) in 15 ml dry DMF was added as a
coupling agent at 0 °C by dropping funnel. The mixture was stirred for 30 min.
Then a solution of -CD-NH2 (1.79 g, 1.89 × 10-3 mol) in 15 ml DMF was
added dropwise to this solution. The mixture was stirred at 0 °C for 3 h then at
room temperature for 54 h and finally at 45-55 °C for 5 h under argon. The
solution was filtered off and was placed at 4 °C for 24 h and again the solution
was filtered off and the solvent was removed under vacuum. The product was
precipitated in diethylether and was washed by acetone. Then the product was
dissolved in the methanol, and filtered off then were precipitated in diethylether
several times for removal of unreacted -CD-NH2. The solution was filtered and
the solvent was removed under vacuum. The product was dissolved in 5 ml
water and stirred for 24 h at room temperature. The solution was filtered off and
poured in 5 ml of water at 25 °C. The mixture was conducted into cellophane
membrane dialysis bag. The bag was closed and transferred into a flask
containing 100 ml of water maintained at 25 °C. The external water was
continuously stirred for two days. The external water was removed after 24 h
and added 100 ml fresh water. The product was removed from dialysis bag and

37
dried under vacuum at 50 °C and purified compound was obtained as a brown
solid, yield 43%.
1H NMR (400 MHz, DMSO-d6): = 2.06 (s, dendrimer -CH2CONH-), 2.7-2.92
(q, citric acid CH2), 3.11 (m, β-CD -CONHCH2-), 3.3-3.8 (br, -CD H2, H4, H5,
H3, H6a, H6b and PEG -OCH2CH2O-), 4.1-4.2 (br, PEG -COCH2O-), 4.57 (br, -
CD OH6), 4.81 (br, -CD H1), 5.62-5.64 (br, -CD OH3), 5.74-5.81 (br, -CD
OH2) and 7.97-8 (br, NH-C=O). 13C NMR (100 MHz, DMSO-d6): =31 and 70
(citric acid CH2), 50.22 ( -CD CH2-NH-CO), 60.49 ( -CD C6), 72.3-73.4 (β-
CD C2, C3 and C5) 70 and 72.3 (PEG carbons), 81 (β-CD C4), 101 (β-CD C1),
and 157, 162 and 167 (C=O). IR (KBr, cm-1): 3359 ( , OH β-CD and COOH
dendrimer), 2928 and 2862 ( , C–H), 1739 and 1705 ( , ester and acid C=O),
1653 ( , amide C=O), 1522 ( , CO–N–H), 1149 ( , C-O-C), 1109 and 1035 ( ,
C-O).
2.2.9. Synthesis of G2-NH- -CD Using DCC
A solution of G2 (0.27 g, 2.95 × 10-4 mol) in 20 ml dry DMF was added to a
round-bottom flask equipped with reflux condenser, argon inlet, dropping
funnel and magnetic stirrer. Dry pyridine (0.2 ml) was added to this solution by
dropping funnel within 15 min. The mixture was stirred vigorously for 20 min.
To this solution, DCC (0.58 g, 2.79 × 10-3 mol) in 15 ml dry DMF was added as
a coupling agent at 0 °C by dropping funnel. The mixture was stirred for 30
min. Then a solution of -CD-NH2 (2.65 g, 1.35 × 10-3 mol) in 15 ml DMF was
added dropwise to this solution. The mixture was stirred at 0 °C for 3 h then at
room temperature for 72 h and finally at 45-55 °C for 6 h under argon. The
solution was filtered off and was placed at 4 °C for 24 h and again the solution
was filtered off and the solvent was removed under vacuum. The product was
precipitated in diethylether and was washed by acetone. Then the product was

38
dissolved in the methanol, and filtered off then were precipitated in diethylether
several times for removal of unreacted -CD-NH2. The solution was filtered and
the solvent was removed under vacuum. The product was dissolved in 5 ml
water and stirred for 24 h at room temperature. The solution was filtered off and
poured in 5 ml of water at 25 °C. The mixture was conducted into cellophane
membrane dialysis bag. The bag was closed and transferred into a flask
containing 100 ml of water maintained at 25 °C. The external water was
continuously stirred for two days. The external water was removed after 24 h
and added 100 ml fresh water. The product was removed from dialysis bag and
dried under vacuum at 50 °C and purified compound was obtained as a brown
solid, yield 30%.
1H NMR (400 MHz, DMSO-d6): = 2.08 (s, dendrimer -CH2CONH-), 2.7-2.92
(q, citric acid CH2), 3.06 (m, -CD -CONHCH2-), 3.32-3.83 (br, -CD H2, H4,
H5, H3, H6a, H6b and PEG -OCH2CH2O-), 4.1-4.2 (br, PEG -COCH2O-), 4.6 (br,
-CD OH6), 4.82 (br, -CD H1), 5.62-5.64 (br, -CD OH3), 5.74-6 (br, -CD
OH2) and 8.1-8.2 (br, NH-C=O). 13C NMR (100 MHz, DMSO-d6): =31 and
69.8 (citric acid CH2), 49.53 ( -CD CH2-NH-CO), 60.2 ( -CD C6), 68-70 (β-
CD C2, C3 and C5) 72.3and 72.6 (PEG carbons), 81 (β-CD C4), 101 (β-CD C1),
and 156, 167 and 170 (C=O). IR (KBr, cm-1): 3388 ( , OH β-CD and COOH
dendrimer), 2930 and 2858 ( , C–H), 1743 and 1706( , ester and acid C=O),
1649 ( , amide C=O), 1561 ( , CO–N–H), 1223 ( , C-O-C), 1115 and 1032 ( ,
C-O).
2.2.10. Synthesis of G3-NH- -CD Using DCC
A solution of G3 (0.25 g, 4.85 × 10-5 mol) in 20 ml dry DMF was added to a
round-bottom flask equipped with reflux condenser, argon inlet, dropping
funnel and magnetic stirrer. Dry pyridine (0.2 ml) was added to this solution by

39
dropping funnel (15 min). The mixture was stirred vigorously for 20 min. To
this solution, DCC (0.65 g, 3.15 × 10-3 mol) in 15 ml dry DMF was added as a
coupling agent at 0 °C by dropping funnel. The mixture was stirred for 30 min.
Then a solution of -CD-NH2 (3.98 g, 5.8 × 10-4 mol) in 15 ml DMF was added
dropwise to this solution. The mixture was stirred at 0 °C for 3 h then at room
temperature for 96 h and finally at 45-55 °C for 8 h under argon. The solution
was filtered off and was placed at 4 °C for 24 h and again the solution was
filtered off and the solvent was removed under vacuum. The product was
precipitated in diethylether and was washed by acetone. Then the product was
dissolved in the methanol, and filtered off then were precipitated in diethylether
several times for removal of unreacted -CD-NH2. The solution was filtered and
the solvent was removed under vacuum. The product was dissolved in 5 ml
water and stirred for 24 h at room temperature. The solution was filtered off and
poured in 5 ml of water at 25 °C. The mixture was conducted into cellophane
membrane dialysis bag. The bag was closed and transferred into a flask
containing 100 ml of water maintained at 25 °C. The external water was
continuously stirred for two days. The external water was removed after 24 h
and added 100 ml fresh water. The product was removed from dialysis bag and
dried under vacuum at 50 °C and purified compound was obtained as a brown
solid, yield 24%.
1H NMR (400 MHz, DMSO-d6): =1.84 (s, dendrimer -CH2CONH-), 2.72-2.88
(q, citric acid CH2), 3.09 (m, -CD -CONHCH2-), 3.29-3.8 (br, -CD H2, H4,
H5, H3, H6a, H6b and (PEG) -OCH2CH2O-), 4.1-4.2 (br, PEG -COCH2O-), 4.6
(br, -CD OH6), 4.83 (br, -CD H1), 5.62-6 (br, -CD OH3 and OH2) 8.1-8.2 (br,
NH-C=O). 13C NMR (100 MHz, DMSO-d6): =30.8 and 69.8 (citric acid CH2),
50 ( -CD CH2-NH-CO), 64 ( -CD C6), 70-72.6 (β-CD C2, C3, C5 and PEG
carbons), 81 (β-CD C4), 106 (β-CD C1), and 156, 162 and 167 (C=O). IR (KBr,
cm-1): 3416 ( , OH β-CD and COOH dendrimer), 2930 and 2856 ( , C–H),

40
1740 and 1706( , ester and acid C=O), 1657 ( , amide C=O), 1564 ( , CO–N–
H), 1228 ( , C-O-C), 1156 and 1035 ( , C-O).
2.2.11. Preparation of Gn- -CD (n=1–2) and Gn-NH- -CD (n=1–2)/NLX
Complexes
For the preparation of Gn- -CD (n=1–2) and Gn-NH- -CD (n=1–2)/NLX complexes,
first the dendrimers were dissolved in 20 ml DMF and these solutions were
added to a round-bottom flask equipped with reflux condenser and magnetic
stirrer containing a solution of drug (excess of NLX) in 20 ml DMF. The
solutions were stirred for 2h at 35-45 ˚C. Complexes were precipitated in n-
hexane and then dissolved in water, filtered and precipitated in diethylether. The
sample obtained was dried at 35 ˚C for 3 h in a vacuum oven.
2.2.12. In Vitro Release Studies
In vitro release of drug–dendrimer complexes was carried out by the dialysis
method. A quantity of dried dendrimer/drug complexes (66 mg) were added to a
5 ml of aqueous buffered solution (pH 10, 7.4, and 1) at 37 °C. The resulting
solution was loaded into a cellophane membrane dialysis bag. The cellophane
bag was closed and transferred into glass beakers containing 25 ml of the equal
buffer solution maintained at 37 °C. The outer phase was stirred continuously
with a magnetic stirrer at 800 rev/min and a sampling (3 ml) was withdrawn at
regular selected intervals and replaced with 3 ml fresh buffer. The NLX released
in to the medium was analyzed through a UV spectrophotometer to characterize
the release of NLX and determined from the calibration curve achieved
previously under the same conditions. The absorbance of the outer phase was
monitored at 282 nm (pH=1 and 7.4) and 291 nm (pH=10) by a 1700 Shimadzu
UV spectrophotometer to characterize the concentration of NLX using a 1 cm

41
quartz cell. The results were presented in the terms of cumulative release as a
function of time.
2.2.13. Particle Size Analysis
Particle size of complexes were measured by dynamic light scattering (DLS)
using a Malvern Zeta-Sizer Nano ZS equipped with a 4-mW He–Ne laser (633
nm wavelength) with a fixed detector angle of 173°. Samples were dissolved in
distilled water, and Light scattering experiments were carried out at 25 °C and
were started 10 min after the cuvette was placed in the DLS apparatus to allow
the temperature to equilibrate. About 1 ml of the sample was transferred to a
special dust free light-scattering cell. The temperature was controlled to within
±0.02 °C. All samples were quietly inverted for homogenization before
measurement. The particle sizes were measured based on their average
diameters. Also, Particle sizes of resulted dendrimers were measured by Laser
Diffraction Particle Size Analyzer (SA-2101).

42
Chapter 3: Results and Discussion
3.1. Investigation of Dendrimer Synthesized Based on Citric acid
3.1.1. Synthesis of G1 Compound
The compound G1 was synthesized from the reaction of ClOC–PEG–COCl
with anhydrous citric acid and ClOC–PEG–COCl was prepared through
chlorination of diacid poly(ethyleneglycol) using thionylchloride in yield 100%
[20]. The general synthesis pathway is presented in Scheme 1. The condition of
reactions and the characterization of products are described in the
“Experimental section”. The structure of G1 was verified by 1H NMR and FT-
IR spectroscopy. Fig. 10 shows the 1H NMR spectrum of G1 compound. In Fig.
10 the 1H NMR of G1which shows a quartet at 2.67–2.77 ppm as a AB system
for the CH2 protons of citric acid, the protons of PEG at 3.6–3.8 ppm (–
OCH2CH2O–) and 4.1–4.2 ppm (–COCH2O–) can be recognized also the
chemical shifts at 2.5 ppm is related to the DMSO as the solvent. The integral
ratio of aliphatic protons of PEG to the citric acid part of the molecule is 6 (in
comparison to 6 as a theoretical calculation). This is evidence, which shows the
reaction has been accomplished completely and both of the acyl halide functions
of chlorinated PEG have been reacted with citric acid.
FT-IR spectrum of G1 (Fig. 11) shows a band around 1106 cm-1 can be
assigned as characteristic stretching vibration of C–O of G1. In the FTIR
spectrum the absorption band at 1738 cm-1 is attributed to the carbonyl groups
of ester. A strong and wide band at 3439 cm-1 is the absorption of -COOH
groups from dendrimer component. The FT-IR spectrum of G1 indicates a C–H
asymmetrical stretching vibration of the dendrimer segment at 2858-2860 cm-1.
The peak at 1180 cm-1 (C–O stretch) confirmed the presence of ether group of
PEG. Both these data confirmed the formation of ester bond between the acyl
group of ClOC–PEG–COCl and the hydroxyl group of citric acid.

43
Scheme 1. Synthesis of G1.

44
Figure 10. 1H NMR spectrum of G1 in DMSO-d6.
Figure 11. FT-IR spectrum of G1.

45
3.1.2. Synthesis of G2 Compound
The compound G2 was prepared via the reaction of G1 and dry citric acid in the
presence of DCC as a coupling agent. The general synthesis pathway is
presented in Scheme 2. The condition of reactions and the characterization of
products are described in the “Experimental section”. The structure of G2 was
verified by 1H NMR and FT-IR spectroscopy. Fig. 12 display 1H NMR of G2,
the chemical shifts at 2.6–2.8 ppm (CH2) of protons of citric acid as a quartet
(AB system), protons of PEG at 3.57–3.8 ppm (–OCH2CH2O–) and 4.1–4.2
ppm (–COCH2O–) can be recognized. In 1H NMR spectroscopy the comparison
of the proton numbers of CH2 of G2 shows that the number of protons of citric
acid versus number of protons of PEG is grown related to the G1, which
indicates the formation of dendrimer (G2). Also integral ratio of aliphatic
protons of PEG to citric acid is shows that the reaction was completed and the
growth of dendrimer is confirmed (G2). This is an evidence, which shows the
reaction has been accomplished completely and both of the between the
carboxylic group of G1 have been reacted with citric acid.
FT-IR spectrum of G2 (Fig. 13) shows a band around 1096 cm-1 can be
assigned as characteristic stretching vibration of C–O of G2. In the FTIR
spectrum the absorption band at 1734 cm-1 is attributed to the carbonyl groups
of ester. A strong and wide band at 3400 cm-1 is the absorption of -COOH
groups from dendrimer component. The peak at 1180 cm-1 (C–O stretch)
confirmed the presence of ether group of PEG. Both these data confirmed the
formation of ester bond between the between the carboxylic groups of G1 and
the hydroxyl group of citric acid.

46
Scheme 2. Synthesis of G2.
Figure 12. 1H NMR spectrum of G2 in DMSO-d6.

47
Figure 13. FT-IR spectrum of G2.
3.1.3. Synthesis of G3 Compound
The preparation of G3 was carried out via the reaction of G2 with dry citric acid
in the presence of DCC as a coupling agent and DMF as solvent. The general
synthesis pathway is presented in Scheme 3. The condition of reactions and the
characterization of products are described in the “Experimental section”. The
structure of G3 was verified by 1H NMR and FT-IR spectroscopy. Fig. 14 and
15 display 1H NMR of G3, the chemical shifts at 2.62–2.76 ppm (CH2) of
protons of citric acid as a quartet (AB system), protons of PEG at 3.57–3.77
ppm (–OCH2CH2O–) and 4.1–4.2 ppm (–COCH2O–) can be recognized. In 1H
NMR spectroscopy the comparison of the proton numbers of CH2 of G3 shows
that the number of protons of citric acid versus number of protons of PEG is
grown related to the G2, which indicates the formation of dendrimer (G3). The
number of protons of G3 in the same chemical shifts displays the growth of
citric acid part in comparison with the protons of PEG as a core and also in G2.

48
Also integral ratio of aliphatic protons of PEG to citric acid is 0.47 (in
comparison to 0.5 as a theoretical calculation) shows that the reaction was
completed and the growth of dendrimer is confirmed (G3). This is an evidence,
which shows the reaction has been accomplished completely and both of the
between the carboxylic group of G2 have been reacted with citric acid.
FT-IR spectrum of G3 (Fig. 16) shows a band around 1096 cm-1 can be assigned
as characteristic stretching vibration of C–O of G2. In the FTIR spectrum the
absorption band at 1730 cm-1 is attributed to the carbonyl groups of ester. A
strong and wide band at 3300 cm-1 is the absorption of -COOH groups from
dendrimer component. The FT-IR spectrum of G2 indicates a C–H
asymmetrical stretching vibration of the dendrimer segment at 2858-2860 cm-1.
The peak at 1260 cm-1 (C–O stretch) confirmed the presence of ether group of
PEG.

49
Scheme 3. Synthesis of G3.

50
Figure 14. 1H NMR spectrum of G3 in DMSO-d6.
Figure 15. 1H NMR spectrum of G3 in acetone deuterium.

51
Figure 16. FT-IR spectrum of G3.
3.1.4. Synthesis of G1- -CD Compound
Compound G1- -CD was synthesized from the conjugation of G1 with -CD by
‘‘one-step synthesis reaction’’, through the formation of an ester linkage in the
presence of pyridine after activation of acidic groups with DCC as a coupling
agent. The general synthesis pathway is presented in Scheme 4. The condition
of reactions and the characterization of products are described in the
“Experimental section”. The 1H NMR spectrum (Fig. 17) of the G1- -CD
conjugate shows signals originating from both G1 and -CD. 1H NMR data of
these compounds shows a quartet at 2.8–2.92 ppm as AB system for the CH2
protons of citric acid, the anomeric protons of -CD at 4.82 and CH2OCO-
dendrimer of β-CD at 4.2-4.4 can be recognized. All these chemical shifts were
in agreement with the projected structure of these compounds. The average
number of -CD in the G1- -CD dendrimers prepared was evaluated from their1H NMR spectrum. 1H-NMR spectrum of the resulting G1- -CD was measured
and the molar ratio of dendrimer and -CD was calculated from the peak areas

52
of anomeric protons of -CD and citric acid protons of the dendrimer. From the
integration ratio of the signal at 4.82ppm (corresponding to the anomeric
protons of -CD) to the signal at 2.8-2.92 ppm corresponding to the four
protons of citric acid, approximately 2 molecule of -CD were attached to each
molecule of G1.
FT-IR spectrum (Fig. 18) of this compound showed a band around 1033 cm-1
that could be assigned as characteristic stretching vibration of C–OH of -CD.
A strong and wide band at 3361cm-1 was the absorption of hydroxyl groups
from both -CD and dendrimer components. In the FT-IR spectrum of G1- -
CD, the strong peak at 1739 cm-1 could be assigned to the stretching vibration of
carbonyl group in which was not observed in -CD spectrum. The results
indicated that the carboxymethylation occurs at C6 position. The FT-IR
spectrum of G1- -CD indicated a C–H asymmetrical stretching vibration of the
dendrimer segment at 2926-2860 cm-1, which is not appeared in -CD. The
absorption at 1106 cm-1 (C–O stretch) confirmed the presence of ether group of
PEG. The results of FT-IR absorption and 1H NMR spectra confirmed the
successful synthesis of dendrimers.

53
Scheme 4. Synthesis of G1- -CD.

54
Figure 17. 1H NMR spectrum of G1- -CD in DMSO-d6.
Figure 18. FT-IR spectrum of G1- -CD.

55
3.1.5. Synthesis of G2- -CD Compound
Compound G2- -CD was synthesized from the conjugation of G2 with -CD by
‘‘one-step synthesis reaction’’, through the formation of an ester linkage in the
presence of pyridine after activation of acidic groups with DCC as a coupling
agent. The general synthesis pathway is presented in Scheme 5. The condition
of reactions and the characterization of products are described in the
“Experimental section”. The 1H NMR spectrum (Fig. 19) of the G2- -CD
conjugate shows signals originating from both G2and -CD. 1H NMR data of
these compounds shows a quartet at 2.72–3.04 ppm as AB system for the CH2
protons of citric acid, the anomeric protons of -CD at 4.83 and CH2OCO-
dendrimer of β-CD at 4-4.2 can be recognized. All these chemical shifts were in
agreement with the projected structure of these compounds. The average
number of -CD in the G2- -CD dendrimers prepared was evaluated from their1H NMR spectrum. 1H-NMR spectrum of the resulting G2- -CD was measured
and the molar ratio of dendrimer and -CD was calculated from the peak areas
of anomeric protons of -CD and citric acid protons of the dendrimer. From the
integration ratio of the signal at 4.83 ppm (corresponding to the anomeric
protons of -CD) to the signal at 2.72-3.04 ppm corresponding to the four
protons of citric acid, approximately 3 molecule of -CD were attached to each
molecule of G2.
FT-IR spectrum (Fig. 20) of this compound showed a band around 1033 cm-1
that could be assigned as characteristic stretching vibration of C–OH of -CD.
A strong and wide band at 3423 cm-1 was the absorption of hydroxyl groups
from both -CD and dendrimer components. In the FT-IR spectrum of G2- -
CD, the strong peak at 1741 cm-1 could be assigned to the stretching vibration of
carbonyl group in which was not observed in -CD spectrum. The results
indicated that the carboxymethylation occurs at C6 position. The FT-IR
spectrum of G2- -CD indicated a C–H asymmetrical stretching vibration of the

56
dendrimer segment at 2931-2859 cm-1, which is not appeared in -CD. The
absorption at 1151 cm-1 (C–O stretch) confirmed the presence of ether group of
PEG. The results of FT-IR absorption and 1H NMR spectra confirmed the
successful synthesis of dendrimers.

57
Scheme 5. . Synthesis of G2- -CD.

58
Figure 19. 1H NMR spectrum of G2- -CD in DMSO-d6.
Figure 20. FT-IR spectrum of G2- -CD.

59
3.1.6. Synthesis of G3- -CD Compound
Compound G3- -CD was synthesized from the conjugation of G2 with -CD by
‘‘one-step synthesis reaction’’, through the formation of an ester linkage in the
presence of pyridine after activation of acidic groups with DCC as a coupling
agent. The general synthesis pathway is presented in Scheme 6. The condition
of reactions and the characterization of products are described in the
“Experimental section”. The 1H NMR spectrum of the G3- -CD conjugate
shows signals originating from both G3 and -CD. 1H NMR data of these
compounds shows a quartet at 2.72–3.06 ppm as AB system for the CH2 protons
of citric acid, the anomeric protons of -CD at 4.83 and CH2OCO-dendrimer of
β-CD at 4.2-4.4 can be recognized. All these chemical shifts were in agreement
with the projected structure of these compounds. The average number of -CD
in the G3- -CD dendrimers prepared was evaluated from their 1H NMR
spectrum. 1H-NMR spectrum of the resulting G3- -CD was measured and the
molar ratio of dendrimer and -CD was calculated from the peak areas of
anomeric protons of -CD and citric acid protons of the dendrimer. From the
integration ratio of the signal at 4.83 ppm (corresponding to the anomeric
protons of -CD) to the signal at 2.72-3.06 ppm corresponding to the four
protons of citric acid, approximately 5 molecule of -CD were attached to each
molecule of G3.
FT-IR spectrum (Fig. 21) of this compound showed a band around 1031 cm-1
that could be assigned as characteristic stretching vibration of C–OH of -CD.
A strong and wide band at 3416 cm-1 was the absorption of hydroxyl groups
from both -CD and dendrimer components. In the FT-IR spectrum of G3- -
CD, the strong peak at 1743 cm-1 could be assigned to the stretching vibration of
carbonyl group in which was not observed in -CD spectrum. The results
indicated that the carboxymethylation occurs at C6 position. The FT-IR
spectrum of G3- -CD indicated a C–H asymmetrical stretching vibration of the

60
dendrimer segment at 2930-2859 cm-1, which is not appeared in -CD. The
absorption at 1154 cm-1 (C–O stretch) confirmed the presence of ether group of
PEG. The results of FT-IR absorption and 1H NMR spectra confirmed the
successful synthesis of dendrimers.

61
Scheme 6. Synthesis of G3- -CD.

62
Figure 21. FT-IR spectrum of G3- -CD.
3.1.7. Synthesis of G1-NH- -CD Compound
Also in this work, we describe the functionalization of -CD and its coupling to
synthesized dendrimers. Selective and mono-functionalization of -CD is a
most important challenge due to the existence of seven equivalent 6-primary
and 14 2, 3-secondary hydroxyl functions of the glucose units. In order to, -CD
was monofunctionalized into its amino derivative and then conjugated to the
carboxylic-side of dendrimers. For this purpose, we functionalized the -CD to
6-Deoxy-6-(p-toluenesulfonyl)- -cyclodextrin ( -CDTos) and 6-Deoxy-6-
amino- -cyclodextrin ( -CD-NH2) so as to selectivity and reactivity of mono
substitutions of -CD-NH2 that was prepared as previously reported. Then the
dendrimer Gn (n=1-3) covalently conjugated to the -CD-NH2, through the
formation of an amide band by Using DCC as a coupling agent.

63
3.1.7.1. Preparation of 6-Deoxy-6-amino- -cyclodextrin ( -CD-NH2)
-CD-NH2 was obtained through a three-step synthetic pathway. The first step
was the mono-tosylation of -CD [102]. This reaction was carried out in water,
in the presence of p-toluenesulfonyl chloride and sodium hydroxide, at 0 °C.
The 6- deoxy-6-(p-toluenesulfonyl)- -cyclodextrin ( -CD-Tos) was insoluble in
the reaction mixture and precipitated as a white solid during the process, thus
preventing further functionalization. The product was recovered by filtration
and washed with acetonitrile [105]. 6-Deoxy-6-azido- -cyclodextrin ( -CD-N3)
was prepared by reacting at 85 °C an aqueous suspension of -CD-Tos with
sodium azide. The reaction was monitored by TLC on silica plates with the
mixture isopropanol/ethyl acetate/water/NH4OH 5:2:3:1 as mobile phase until
the complete disappearance of the spot of -CD-Tos (Rf=0.52) and the presence,
besides a trace of unsubstituted -CD (Rf=0.28), of a single spot attributed to -
CD-N3 (Rf=0.40) [105]. -CD-N3 was finally recovered by precipitation in
acetone. Then crude -CD-NH2 was prepared by reacting mono-6-azido-6-
deoxy-CD with triphenyl phosphine in DMF at ambient conditions for 2 h.
After precipitation in acetone, filtering and washing with acetone, pure amine
was obtained as white solid [106]. All products were fully characterized by
NMR and FTIR. The spectroscopic data were shown in complete agreement
with the proposed structures. The general synthesis pathway is presented in
Scheme 7.
In the 1H NMR spectrum of -CD-Tos (Fig. 22) diagnostic peaks characteristic
of tosyl moiety were present, in particular the peaks at 7.4 and 7.7 ppm,
corresponding to the aromatic hydrogen’s of tosyl group, the peak at 4.27 ppm,
corresponding to the methylene adjacent to the sulfonyl moiety, and the
resonance peak at 2.41 ppm, corresponding to the methyl group of the tosyl
moiety. The anomeric protons of -CD appeared at 4.83 ppm. In the 1HNMR
spectrum of -CD-N3 the diagnostic peak of methylene group adjacent to the

64
azide moiety overlapped with those of the -CD hydrogen’s in position 3, 5 and
6 giving rise to a broad peak between 3.67 and 3.56. In the spectrum of -CD-
N3, all peaks relative to the tosyl moiety disappeared after reaction of with
sodium azide. The 1H NMR spectrum of -CD-NH2 lacked diagnostic peaks.
However, the completion of the reduction was as certained by the disappearance
in its FTIR spectrum of the diagnostic band at 2041 cm−1.
Scheme 7. Hierarchical synthesis of -CD-NH2.

65
Figure 22. 1H NMR spectrum of -CD-Tos in DMSO-d6.
3.1.7.2. Synthesis of G1-NH- -CD Compound
Compound G1-NH- -CD was synthesized from the conjugation of G1 with -
CD-NH2 through the formation of an amide linkage in the presence of pyridine
after activation of acidic groups with DCC as a coupling agent. The general
synthesis pathway is presented in Scheme 8. The condition of reactions and the
characterization of products are described in the “Experimental section”. The
obtained products were characterized using 1H NMR, 13C NMR and FTIR
spectroscopy. The 1H NMR spectrum of the G1-NH- -CD (Fig. 23) conjugate
shows signals originating from both G1 and -CD-NH2. 1H NMR data of these
compounds shows a quartet at 2.7–2.92 ppm as a AB system for the CH2
protons of citric acid, the anomeric protons of -CD at 4.81, the -CH2CONH-
dendrimer protons at 1.84-2.08 ppm and NH-C=O of β-CD at 7.97-8 ppm can
be recognized. All these chemical shifts were in accordance with the proposed
structure of these compounds. The average number of -CD in the G1-NH- -CD
dendrimers prepared was estimated from their 1H NMR spectra. 1H-NMR

66
spectrum of the resulting G1-NH- -CD was measured and the molar ratio of
dendrimer and -CD was calculated from the peak areas of anomeric protons of
-CD and citric acid protons of the dendrimer. From the integration ratio of the
signal at 4.81 ppm, which corresponds to the anomeric protons of -CD, to the
signal between 2.7 and 2.92 ppm corresponding to the four protons of citric
acid, approximately 2 molecule of -CD were attached to each molecule of G1-
NH- -CD.
FTIR spectrum of these compounds shows a strong and wide band at 3359 cm-1
is the absorption of hydroxyl groups from both -CD and dendrimer
components while peak at 1739 cm-1 confirmed the ester linkage. The peaks at
1653 and 1522 cm-1 were assigned to C=O stretching vibration of amide
carbonyl group and N–H bending vibration of a secondary amine, respectively.
The FTIR spectrum of these compounds indicates a C–H asymmetrical
stretching vibration of the dendrimer segment at 2928-2862 cm-1, which is not
appeared in -CD. Stretch vibration for ether band of PEG appeared at 1109-
1251 cm-1 (C–O stretch) (Fig. 24). The attachment of -CD to the chain end of
the dendrimer was further confirmed by 13C NMR spectrum for these
dendrimers. 13C NMR data of these compounds, shows signals at approximately
31, 70 and 72, 73 ppm are related to the carbon atoms of CH2 citric acid and
PEG respectively, also signals of C4 and C1 of β-CD carbons are appeared at 81
and 102 ppm, respectively. The signals of carbonyl groups are appeared at 157,
162 and 167 ppm for the carbon atoms of amide, ester and acid, respectively.
Thus, the 13C NMR resonances at 157 ppm confirmed the presence of the amide
band (Fig. 25). The results of FT-IR absorption, 1H NMR and 13C NMR spectra
confirmed the successful synthesis of these compounds.
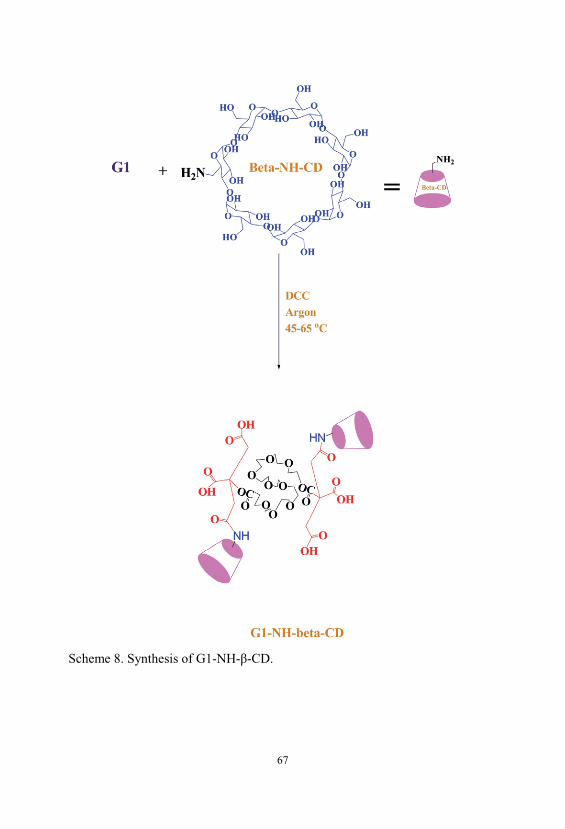
67
Scheme 8. Synthesis of G1-NH- -CD.

68
Figure 23. 1H NMR spectrum of G1-NH- -CD in DMSO-d6.
Figure 24. FT-IR spectrum of G1-NH- -CD.

69
Figure 25. 13C NMR spectrum of G1-NH- -CD in DMSO-d6.
3.1.8. Synthesis of G2-NH- -CD Compound
Compound G2-NH- -CD was synthesized from the conjugation of G2 with -
CD-NH2 through the formation of an amide linkage in the presence of pyridine
after activation of acidic groups with DCC as a coupling agent. The general
synthesis pathway is presented in Scheme 9. The condition of reactions and the
characterization of products are described in the “Experimental section”. The
obtained products were characterized using 1H NMR, 13C NMR and FTIR
spectroscopy. The 1H NMR spectrum of the G2-NH- -CD conjugate shows
signals originating from both G2 and -CD-NH2. 1H NMR data of these
compounds (Fig. 26) shows a quartet at 2.7–3 ppm as a AB system for the CH2
protons of citric acid, the anomeric protons of -CD at 4.82, the -CH2CONH-
dendrimer protons at 2.08 ppm and NH-C=O of β-CD at 8.1 ppm can be
recognized. All these chemical shifts were in accordance with the proposed
structure of these compounds. The average number of -CD in the G2-NH- -CD

70
dendrimers prepared was estimated from their 1H NMR spectra. 1H-NMR
spectrum of the resulting G2-NH- -CD was measured and the molar ratio of
dendrimer and -CD was calculated from the peak areas of anomeric protons of
-CD and citric acid protons of the dendrimer. From the integration ratio of the
signal at 4.82 ppm, this corresponds to the anomeric protons of -CD, to the
signal between 2.7 and 3 ppm corresponding to the four protons of citric acid,
approximately 4 molecule of -CD was attached to each molecule of G2-NH- -
CD.
FTIR spectrum of these compounds shows a strong and wide band at 3359–
3416 cm-1 is the absorption of hydroxyl groups from both -CD and dendrimer
components while peak at 1743 cm-1 confirmed the ester linkage. The peaks at
1649 and 1551-1649 cm-1 were assigned to C=O stretching vibration of amide
carbonyl group and N–H bending vibration of a secondary amine, respectively.
The FTIR spectrum of these compounds indicates a C–H asymmetrical
stretching vibration of the dendrimer segment at 2929-2858 cm-1, which is not
appeared in -CD. Stretch vibration for ether band of PEG appeared at 1150-
1228 cm-1 (C–O stretch) (Fig. 27). The attachment of -CD to the chain end of
the dendrimer was further confirmed by 13C NMR spectrum for these
dendrimers. 13C NMR data of these compounds, shows signals at approximately
31.7, 69.8 and 70.5, 70 ppm are related to the carbon atoms of CH2 citric acid
and PEG respectively, also signals of C4 and C1 of β-CD carbons are appeared
at 81 and 101 ppm, respectively. The signals of carbonyl groups are appeared at
156, 167 and 170 ppm for the carbon atoms of amide, ester and acid,
respectively. Thus, the 13C NMR resonances at 156 ppm confirmed the presence
of the amide band (Fig. 28). The results of FT-IR absorption, 1H NMR and 13C
NMR spectra confirmed the successful synthesis of these compounds.

71
Scheme 9. Synthesis of G2-NH- -CD.

72
Figure 26. 1H NMR spectrum of G2-NH- -CD in DMSO-d6.
Figure 27. FT-IR spectrum of G2-NH- -CD.

73
Figure 28. 13C NMR spectrum of G2-NH- -CD in DMSO-d6.
3.1.9. Synthesis of G3-NH- -CD Compound
Compound G3-NH- -CD was synthesized from the conjugation of G3 with -
CD-NH2 through the formation of an amide linkage in the presence of pyridine
after activation of acidic groups with DCC as a coupling agent. The general
synthesis pathway is presented in Scheme 10. The condition of reactions and the
characterization of products are described in the “Experimental section”. The
obtained products were characterized using 1H NMR, 13C NMR and FTIR
spectroscopy. The 1H NMR spectrum of the G3-NH- -CD conjugate shows
signals originating from both G3 and -CD-NH2. 1H NMR data of these
compounds shows a quartet at 2.72–2.88 ppm as a AB system for the CH2
protons of citric acid, the anomeric protons of -CD at 4.83, the -CH2CONH-
dendrimer protons at 1.84 ppm and NH-C=O of β-CD at 7.97-8 ppm can be
recognized. All these chemical shifts were in accordance with the proposed

74
structure of these compounds. The average number of -CD in the G3-NH- -CD
dendrimers prepared was estimated from their 1H NMR spectra. 1H-NMR
spectrum of the resulting G3-NH- -CD (Fig. 29) was measured and the molar
ratio of dendrimer and -CD was calculated from the peak areas of anomeric
protons of -CD and citric acid protons of the dendrimer. From the integration
ratio of the signal at 4.83 ppm, which corresponds to the anomeric protons of -
CD, to the signal between 2.72 and 2.88 ppm corresponding to the four protons
of citric acid, approximately 6 molecule of -CD were attached to each
molecule of G3-NH- -CD.
FTIR spectrum of these compounds shows a strong and wide band at 3416 cm-1
is the absorption of hydroxyl groups from both -CD and dendrimer
components while peak at 1740 cm-1 confirmed the ester linkage. The peaks at
1657 and 1564 cm-1 were assigned to C=O stretching vibration of amide
carbonyl group and N–H bending vibration of a secondary amine, respectively.
The FTIR spectrum of these compounds indicates a C–H asymmetrical
stretching vibration of the dendrimer segment at 2930-2858 cm-1, which is not
appeared in -CD. Stretch vibration for ether band of PEG appeared at 1154-
1224 cm-1 (C–O stretch) (Fig. 30). The attachment of -CD to the chain end of
the dendrimer was further confirmed by 13C NMR spectrum for these
dendrimers. 13C NMR data of these compounds (Fig. 31), shows signals at
approximately 31, 69 and 72, 72.4 ppm are related to the carbon atoms of CH2
citric acid and PEG respectively, also signals of C4 and C1 of β-CD carbons are
appeared at 81 and 106 ppm, respectively. The signals of carbonyl groups are
appeared at 157, 162 and 167 ppm for the carbon atoms of amide, ester and
acid, respectively. Thus, the 13C NMR resonances at 157 ppm confirmed the
presence of the amide band. The results of FT-IR absorption, 1H NMR and 13C
NMR spectra confirmed the successful synthesis of these compounds.

75
Scheme 10. Synthesis of G3-NH- -CD.

76
Figure 29. 1H NMR spectrum of G3-NH- -CD in DMSO-d6.
Figure 30. FT-IR spectrum of G3-NH- -CD.

77
Figure 31. 13C NMR spectrum of G3-NH- -CD in DMSO-d6.
3.2. Investigation of Nanocarriers Size
Particle sizes were determined with DLS experiments in distilled water and
laser diffraction particle size analyzer. DLS results present that the average
diameters of G1- -CD and G2- -CD/NLX complexes were 50 and 70 nm,
respectively (Fig. 32). Typical examples of size distributions by laser diffraction
particle size analyzer (LDPSA) for -CD-modified dendrimers (Gn(n=1-3)-NH-
-CD) are presented in Fig. 33-35. These dendrimers exhibited a narrow size
distribution. Median diameter of G1-NH- -CD, G2-NH- -CD and G3-NH- -CD
were 63, 69 and 82 nm, respectively. It shows that have been increased with
increasing of generation of dendrimers. Size distribution profiles of these
complexes by laser diffraction particle size analyzer are also presented in Table
2.

78
Compound Diameter (nm)
G1-NH- -CD 63
G2-NH- -CD 69
G3-NH- -CD 82
Table 2. Diameter of nanocarriers obtained using laser diffraction particle size
analyzer.
Figure 32. The size distribution profiles of G1- -CD and G2- -CD dendrimers
estimated by DLS.

79
Figure 33. Particle Diameter of G1-NH- -CD by using LDPSA.
Figure 34. Particle Diameter of G2-NH- -CD by using LDPSA.

80
Figure 35. Particle Diameter of G3-NH- -CD by using LDPSA.
3.3. In Vitro Release Studies
3.3.1. Loading of NLX Drug Molecule into the Dendrimers
The resultant dendrimer-cyclodextrin conjugates revealed enormously high
water solubility. Generally, dendrimers serve as carriers of biologically active
agents by encapsulating them in the interior or, joining them on the exterior
edging of the dendrimers. When the guest molecules are drugs, the resulted
drug/dendrimer complexes could be used as ideal candidates for carriers and in
drug release. Herein, Gn(n=1-3), Gn(n=1-3)- -CD and Gn(n=1-3)-NH- -CD as
biocompatible compounds have been synthesized, and the encapsulation of
NLX drug, using these structures has been attempted. The measurements of UV
from complexes confirmed the presence of drug in the resulted complexes.
3.3.2. Calculating the Amount of the Trapped and Released Drug Molecule
For determining the amount of drug encapsulated in the complex, 66 mg of
dried drug/dendrimer complexes was dissolved in 100 ml of water. After
complete dissolution of the drug/dendrimer complexes, the amounts of drug in

81
drug/dendrimer complexes was measured by UV spectrometer and the amounts
of the trapped drug for each generation before and after the functionalization
with -CD are displayed in Table 3. The UV detection for to determine the
quantity of drug inside the complexes was measured at 282 nm.
NLX loading efficiency of the complexes was determined as fallowing:
Amount of trapped drug in the complexes (g) = (A/B)
Loading efficiency (%) = (C/A) × 100
where A, B and C are the primary feeding amount of drug (g), determined free
drug content (g) and amount of trapped drug in the complexes (g) respectively.
Compound Naltrexone
Loading (%)
G1/NLX 8
G2/NLX 22
G3/NLX 27
G1-β-CD/NLX 17
G2-β-CD/NLX 32
G3-β-CD/NLX 37
G1-NH-β-CD/NLX 22
G2- NH-β-CD/NLX 35
G3- NH-β-CD/NLX 39
Table 3. Drug loading efficiency of Gn(n=1-3), Gn(n=1-3)- -CD and Gn(n=1-3)-NH- -
CD/NLX complexes.
As shown in Table 3 the loading efficiency% of carried drug by dendrimers was
enhanced with the rising of dendrimer generations. The drug content and
percent encapsulation efficiency was varying between 8 and 39%. The

82
maximum encapsulation of NLX was observed with G3-NH-β-CD in distilled
water of about 39%. The increased entrapment was possibly due to increase the
number of acidic groups of dendrimers (for electroestatic interactions), cavities
(for trap of drugs) in higher generations and the number of grafted -CD cavity.
Coating with -CD further increased drug entrapment because of the
hydrophobic cavity of CDs is capable of including a variety of hydrophobic
compounds using host-guest complexation. Whenever, it seems the ionic
interaction between dendrimers and guest molecule and host–guest interactions
including hydrophobic interactions, hydrogen-bonding and Vander Waals
interactions influence the ability of dendrimers as drug carriers.
3.3.3. Studying the Controlled Release of NLX from the Nanocarriers
The controlled release of NLX from the nanocarriers was investigated in pH 1,
7.4, and 10.
In vitro release behavior of NLX from the NLX/dendrimer complexes were
examined in buffered solution at different pH values (pH=1 and pH=7.4
max=282, pH=10 max=291 and 37 °C). Hydrolysis was carried out in a
cellophane membrane bags permeable to low molecular weight of compounds
and the amount of the drug release was determined in different pH values (pH 1,
7.4, and 10 all at 37 °C). UV absorbance measurements were carried out for the
characterization of NLX concentration in the complex solution. In vitro release
rate of NLX from dendrimers having -CD and without -CD in end groups of
dendrimer is compared and subsequent results were obtained. The results
displayed that both the rate and the amount of the release of the drug from
dendrimer/drug complex is influenced by some factors such as the generation of
the host molecule, biodegradability of the dendrimers, functional groups on the
surface of the dendrimer, solubility of the drug, the interactions between the
drug and the dendrimer in the complex and pH and the effects of each factor is

83
considered as the following. The comparison of the data showed that in
different pH’s the NLX released in different rates. In pH 10 the rate of release
of NLX was slower than pH 1 and 7.4. Interestingly, in all cases after
approximately 600 min, the release of NLX from the complexes is
approximately completed and is changed to a very slow rate. Figs. 36, Fig. 37
and Fig. 38 displays the controlled release of NLX from Gn (n=1-3), Gn (n=1-2)- -CD
and Gn (n=1-3)-NH- -CD at pH 7.4, respectively. As shown, the rate of release and
the amount of released drugs in complexes (pH 7.4) were increased with the
increase of generations but the rate of release in Gn (n=1-3)-NH- -CD was found
to be less compared to their corresponding dendrimers (Gn (n=1-3)) and these
results is related the hydrophobic interactions between -CD cavity and NLX
that leading to stable complexes and steric hindrance preventing the drug release
from open structure. The comparison of the data was confirm that the rate of
release in Gn (n=1-3)-NH- -CD dendrimers slightly slow than Gn (n=1-3)- -CD.
Because amide linkage in Gn (n=1-3)-NH- -CD were comparatively stable against
hydrolysis, whereas the ester band showed pH dependent release and the degree
of release varied with pH. Also, Fig. 39 shows the release profiles of NLX from
G3, G3- -CD and G3-NH- -CD at 37 °C at pH 1. As shown, the rate of release
for G3 faster than to G3- -CD and G3-NH- -CD, respectivly. Control released in
this case is related to the stable complexes between drug and -CD cavity. The
Fig. 40 and Fig. 41 shows the rate of release of NLX from G3-NH- -CD and G2-
-CD at pH 1, 7.4 and 10. As shown, the rate of release in pH 1 is faster,
because the complexes in this pH are not stable and the esteric bonds are
hydrolyzed. Also, the results showed that the drug release rate from all kinds of
the complexes was faster in the initial period of time. This rapid release was due
to the free drug remaining at the surface that was not entrapped competently
inside the dendrimer matrix. After this initial burst, slow and constant release
rate is observed. The slow release after the initial burst release was as a result of
the low dispersion of NLX from complexes. Again as shown the rate of released

84
drug at pH 7.4 is higher than pH 10 and lower than the pH 1. This result could
be related to the relative stability of the complexes in the above different pH’s.
Figure 36. Release of NLX from Gn (n=1-3)/NLX complexes as a function of time
and pH (pH 7.4, 37 ˚C).
Figure 37. Release of NLX from Gn (n=1-3)-NH- -CD/complexes as a function of
time and pH (pH 7.4, 37 ˚C).
pH=7.4
0102030405060708090100
0 100 200 300 400 500 600 700
Time (min)
Cum
ulativerelease%
G1
G2G3
pH=7.4
0
20
40
60
80
100
0 100 200 300 400 500 600 700
Time (min)
Cum
ulativerelease%
G1-NH- -CD
G2-NH- -CD
G3-NH- -CD

85
Figure 38. Release of NLX from Gn (n=1-2)- -CD/complexes as a function of time
and pH (pH 7.4, 37 ˚C).
Figure 39. Release of NLX from G3, G3- -CD and G3-NH- -CD/complexes as a
function of time and pH (pH 1, 37 ˚C).
0
10
20
30
40
50
60
70
0 100 200 300 400 500 600 700
Cum
ulat
ive
rele
ase
%
Time (min)
pH=7.4
G1- -CD
G2- -CD
pH=1
0102030405060708090100
0 100 200 300 400 500 600 700
Time (min)
Cum
ulativerelease%
G3
G3- -CD
G3-NH- -CD

86
Figure 40. Release of NLX from G3-NH- -CD/complexes as a function of time
and pH (pH 1, 7.4 and 10 and 37 ˚C).
Figure 41. Release of NLX from G2- -CD/complexes as a function of time and
pH (pH 1, 7.4 and 10 and 37 ˚C).
G3-NH- -CD
01020
3040506070
8090100
0 100 200 300 400 500 600 700
Time (min)
Cum
ulativerelease%
PH=1
PH=7.4
PH=10

87
Chapter 4: Conclusions
In this study dendrimers of citric acid and poly (ethylene glycol) as a core
compound was synthesized through divergent method. Then the synthesized
dendrimers modified with -CD and or with -CD-NH2 groups through ester
and amide linkage using DCC as a coupling agent, as a novel drug carrier
having an interior for encapsulation of drugs and a biocompatible surface. The
results were shown that the numbers of conjugated -CD units to dendrimers
increased with modification of -CD into -CD-NH2. Also -CD-modified
dendrimers having a shell of amphiphilic residues in the peripheral moiety of
the dendrimer in order to increase their encapsulation ability designed.
Complexes of the synthesized dendrimers with NLX have been developed. Rate
of release of guest molecules shows that the release depends on interaction
between host and guest molecule, generation of linear–dendritic
macromolecules, biodegradability of the dendrimers, functional groups on the
surface of the dendrimer and pH. Also, findings show that the -CD-coated
dendrimers can be utilized for sustained release delivery of NLX. Thus, results
exhibited that the -CD-grafted biodegradable citric acid dendrimers is a
potential candidate as an efficient drug carrier systems due to its relative
stability in aqueous solution and its drug encapsulation and release properties.
-CD groups were conjugated onto the surface of the dendrimers and the size of
the nanocarriers in the distilled water was investigated using DLS and LDPSA
technique which was 50, 70 nm (Gn(n=1-2)- -CD) and 63, 69 and 82 nm for G1-
NH- -CD, G2-NH- -CD and G3-NH- -CD, respectively.

88
Chapter 5: References
[1] Tomalia DA, Naylor AM, Goddard WA. Starburst Dendrimers-Molecular-
Level Control of Size, Shape, Surface-Chemistry, Topology, and Flexibility
from Atoms to Macroscopic Matter. Angew Chem Int Ed Engl. 1990;29:138-
75.
[2] Tomalia DA, Baker H, Dewald JR, Hall M, Kallos G, Martin S, et al. A
New Class of Polymers: Starburst-Dendritic Macromolecules. Polym J.
1985;17:117-32.
[3] Haririan I, Alavidjeh MS, Khorramizadeh MR, Ghane ZZ, Ardestani MS,
Namazi H. Anionic linear-globular dendrimers: biocompatible hybrid materials
with potential uses in nanomedicine. J Mater Sci-Mater M. 2010;21:1121-33.
[4] Namazi H, Didehban K, Entezami AA. Triazine-based Dendrimers as Liquid
Crystals: Synthesis and Characterization. Iran Polym J. 2009;18:731-41.
[5] Namazi H, Didehban K, Entezami AA. Dendrimer-based hydrogen-bonded
liquid crystalline complexes: Synthesis and characterization. Eur Polym J.
2009;45:1836-44.
[6] Didehban K, Namazi H, Entezami AA. Non-covalent dendrimer-based
liquid crystalline complexes: Synthesis and characterization. Eur Polym J.
2010;46:1923-31.
[7] D'Emanuele A, Attwood D. Dendrimer-drug interactions. Adv Drug Deliv
Rev. 2005;57:2147-62.
[8] Gillies ER, Frechet JM. Dendrimers and dendritic polymers in drug delivery.
Drug Discov Today. 2005;10:35-43.
[9] Caminade AM, Laurent R, Majoral JP. Characterization of dendrimers. Adv
Drug Deliv Rev. 2005;57:2130-46.
[10] Svenson S, Tomalia DA. Commentary - Dendrimers in biomedical
applications - reflections on the field. Adv Drug Deliv Rev. 2005;57:2106-29.

89
[11] Lee CC, MacKay JA, Frechet JM, Szoka FC. Designing dendrimers for
biological applications. Nat Biotechnol. 2005;23:1517-26.
[12] Tomalia DA. Birth of a new macromolecular architecture: dendrimers as
quantized building blocks for nanoscale synthetic polymer chemistry. Prog
Polym Sci. 2005;30:294-324.
[13] Duncan R, Izzo L. Dendrimer biocompatibility and toxicity. Adv Drug
Deliv Rev. 2005;57:2215-37.
[14] Zhuo RX, Du B, Lu ZR. In vitro release of 5-fluorouracil with cyclic core
dendritic polymer. J Control Release. 1999;57:249-57.
[15] Kojima C, Kono K, Maruyama K, Takagishi T. Synthesis of
polyamidoamine dendrimers having poly(ethylene glycol) grafts and their
ability to encapsulate anticancer drugs. Bioconjug Chem. 2000;11:910-7.
[16] Cheng Y, Gao Y, Rao T, Li Y, Xu T. Dendrimer-based prodrugs: design,
synthesis, screening and biological evaluation. Comb Chem High Throughput
Screen. 2007;10:336-49.
[17] Buhler E, Wehner W, Vögtle F. Cascade-chain-like and nonskid-chain-like
synthesis of molecular cavity topologies. Synthesis. 1978:155-8.
[18] Newkome GR, Yao Z-Q, Baker GR, Gupta K. Micelles 1. Cascade
moleculess A new approach to micelless A [27]-Arborol. J Org Chem.
1985;50:2003-4.
[19] Namazi H, Adeli M. Solution proprieties of dendritic triazine/poly
(ethylene glycol)/dendritic triazine block copolymers. J Polym Sci Pol Chem.
2005;43:28-41.
[20] Namazi H, Adeli M. Novel linear–globular thermoreversible hydrogel
ABA type copolymers from dendritic citric acid as the A blocks and
poly(ethyleneglycol) as the B block. Eur Polym J. 2003;39:1491-500.
[21] Namazi H, Adeli M. Synthesis of barbell-like triblock copolymers,
dendritic triazine-block-poly(ethylene glycol)-block-dendritic triazine and
investigation of their solution behaviors. Polymer. 2005;46:10788-99.

90
[22] Klajnert B, Bryszewska M. Dendrimers: properties and applications. Acta
Biochim Pol. 2001;48:199-208.
[23] Buhleier E, Wehner W, Vögtle F. Cascade and Nonskid-Chain-like
Syntheses of Molecular Cavity Topologies. Synthesis. 1978:155-8.
[24] Aulenta F, Hayes W, Rannard S. Dendrimers: a new class of nanoscopic
containers and delivery devices. Eur Polym J. 2003;39:1741-71.
[25] Hawker CG, Fréchet JMJ. Preparation of polymers with controlled
molecular architecture. A new convergent approach to dendritic
macromolecules. J Am Chem Soc 1990;112:7638-47.
[26] Jackson CL, Chanzy HD, Booy FP, Drake BJ, Tomalia DA, Bauer BJ, et
al. Visualization of dendrimer molecules by transmission electron microscopy
(TEM): Staining methods and Cryo-TEM of vitrified solutions.
Macromolecules. 1998;31:6259-65.
[27] Fréchet JMJ, Tomalia DA. Dendrimers and other dendritic polymers.
Chichester ; New York: Wiley; 2001.
[28] Tomalia DA. Starburst Cascade Dendrimers - Fundamental Building-
Blocks for a New Nanoscopic Chemistry Set. Adv Mater. 1994;6:529-39.
[29] Tomalia DA, Brothers HM, 2nd, Piehler LT, Durst HD, Swanson DR.
Partial shell-filled core-shell tecto(dendrimers): a strategy to surface
differentiated nano-clefts and cusps. Proc Natl Acad Sci U S A. 2002;99:5081-
7.
[30] Tomalia DA, Majoros I. Dendrimeric supramolecular and
supramacromolecular assemblies (Reprinted from Supramolecular Polymers,
Marcel Dekker, Inc.: New York, pg 359-434, 2002). J Macromol Sci, Polym
Rev. 2003;C43:411-77.
[31] Namazi H, Adeli M. Synthesis of polyfunctional dendrimeric type
copolymer as the hydrogels and investigation of their thermoreversible
behaviours. Iran Polym J. 2005;14:942-52.

91
[32] Tomalia D, Henderson S, Diallo M. Dendrimers-an Enabling Synthetic
Science to Controlled Organic Nanostructures. Handbook of Nanoscience,
Engineering, and Technology, Second Edition: CRC Press; 2007. p. 24-1--47.
[33] Zhang C, Tomalia DA. Gel Electrophoretic Characterization of Dendritic
Polymers. Dendrimers and Other Dendritic Polymers: John Wiley & Sons, Ltd;
2002. p. 237-53.
[34] Tomalia DA, Huang B, Swanson DR, Brothers HM, Klimash JW. Structure
control within poly(amidoamine) dendrimers: size, shape and regio-chemical
mimicry of globular proteins. Tetrahedron. 2003;59:3799-813.
[35] Esfand R, Tomalia DA. Poly(amidoamine) (PAMAM) dendrimers: from
biomimicry to drug delivery and biomedical applications. Drug Discov Today.
2001;6:427-36.
[36] Tully DC, Fréchet JMJ. Chem Commun. 2001:1229-39.
[37] Adronov A, Frechet JMJ. Light-harvesting dendrimers. Chem Commun.
2000:1701-10.
[38] Singh P. Dendrimer-Based Biological Reagents: Preparation and
Applications in Diagnostics. Dendrimers and Other Dendritic Polymers: John
Wiley & Sons, Ltd; 2002. p. 463-84.
[39] DE Gennes PG, Hervet H. Statistics of « starburst » polymers. J Phys Lett.
1983;44:351-61.
[40] Ballauff M. Dendrimers III, Design, Dimension, Function. Top Curr Chem.
2001;177:212.
[41] Ballauff M, Likos CN. Dendrimers in solution: insight from theory and
simulation. Angew Chem Int Ed Engl. 2004;43:2998-3020.
[42] Murat M, Grest GS. Molecular Dynamics Study of Dendrimer Molecules
in Solvents of Varying Quality. Macromolecules. 1996;29:1278-85.
[43] Rietveld IB, Bouwman WG, Baars MWPL, Heenan RK. Location of the
outer shell and influence of pH on carboxylic-acid functionalised
poly(propylene imine) dendrimers. Macromolecules. 2001;34:8380.

92
[44] Welch P, Muthukumar M. Tuning the Density Profile of Dendritic
Polyelectrolytes. Macromolecules. 1998;31:5892-7.
[45] Ramzi A, Scherrenberg R, Joosten J, Lemstra P, Mortensen K.
Structure−Property Relations in Dendritic Polyelectrolyte Solutions at Different
Ionic Strength. Macromolecules. 2002;35:827-33.
[46] Topp A, Bauer BJ, Prosa TJ, Scherrenberg R, Amis EJ. Size Change of
Dendrimers in Concentrated Solution. Macromolecules. 1999;32:8923-31.
[47] Chai M, Niu Y, Youngs WJ, Rinaldi PL. Structure and Conformation of
DAB Dendrimers in Solution via Multidimensional NMR Techniques. J Am
Chem Soc. 2001;123:4670-8.
[48] Butler JE, Ni L, Nessler R, Joshi KS, Suter M, Rosenberg B, et al. The
physical and functional behavior of capture antibodies adsorbed on polystyrene.
J Immunol Methods. 1992;150:77-90.
[49] Malik N, Wiwattanapatapee R, Klopsch R, Lorenz K, Frey H, Weener JW,
et al. Dendrimers: Relationship between structure and biocompatibility in vitro,
and preliminary studies on the biodistribution of 125I-labelled polyamidoamine
dendrimers in vivo. J Controlled Release. 2000;65:133-48.
[50] Jevprasesphant R, Penny J, Jalal R, Attwood D, McKeown NB,
D’Emanuele A. The influence of surface modification on the cytotoxicity of
PAMAM dendrimers. Int J Pharm. 2003;252:263-6.
[51] Padilla De Jesus OL, Ihre HR, Gagne L, Frechet JM, Szoka FC, Jr.
Polyester dendritic systems for drug delivery applications: in vitro and in vivo
evaluation. Bioconjug Chem. 2002;13:453-61.
[52] Kobayashi H, Kawamoto S, Saga T, Sato N, Hiraga A, Ishimori T, et al.
Positive effects of polyethylene glycol conjugation to generation-4
polyamidoamine dendrimers as macromolecular MR contrast agents. Magn
Reson Med. 2001;46:781-8.
[53] Patri AK, Majoros IJ, Baker JR. Dendritic polymer macromolecular
carriers for drug delivery. Curr Opin Chem Biol. 2002;6:466-71.

93
[54] Jansen JFGA, Meijer EW, Debrabandervandenberg EMM. The Dendritic
Box-Shape-Selective Liberation of Encapsulated Guests. J Am Chem Soc.
1995;117:4417-8.
[55] Jansen JF, de Brabander-van den Berg EM, Meijer EW. Encapsulation of
Guest Molecules into a Dendritic Box. Science. 1994;266:1226-9.
[56] Newkome GR, Moorefield CN, Baker GR, Saunders MJ, Grossman SH.
Unimolecular Micelles. Angew Chem Int Ed Engl. 1991;30:1178-80.
[57] Hawker CJ, Wooley KL, Frechet JMJ. Unimolecular Micelles and
Globular Amphiphiles - Dendritic Macromolecules as Novel Recyclable
Solubilization Agents. J Chem Soc, Perkin Trans 1. 1993:1287-97.
[58] Bhadra D, Bhadra S, Jain S, Jain NK. A PEGylated dendritic
nanoparticulate carrier of fluorouracil. Int J Pharm. 2003;257:111-24.
[59] Namazi H, Adeli M. Dendrimers of citric acid and poly (ethylene glycol) as
the new drug-delivery agents. Biomaterials. 2005;26:1175-83.
[60] Cheng Y, Xu T. The effect of dendrimers on the pharmacodynamic and
pharmacokinetic behaviors of non-covalently or covalently attached drugs. Eur
J Med Chem. 2008;43:2291-7.
[61] Cheng Y, Li M, Xu T. Potential of poly(amidoamine) dendrimers as drug
carriers of camptothecin based on encapsulation studies. Eur J Med Chem.
2008;43:1791-5.
[62] Namazi H, Adeli M, Zarnegar Z, Jafari S, Dadkhah A, Shukla A.
Encapsulation of nanoparticles using linear-dendritic macromolecules. Colloid
Polym Sci. 2007;285:1527-33.
[63] Alavidjeh MS, Haririan I, Khorramizadeh MR, Ghane ZZ, Ardestani MS,
Namazi H. Anionic linear-globular dendrimers: biocompatible hybrid materials
with potential uses in nanomedicine. J Mater Sci - Mater Med. 2010;21:1121-
33.

94
[64] Newkome GR, Moorefield CN, Baker GR, Johnson AL, Behera RK.
Alkane Cascade Polymers Possessing Micellar Topology: Micellanoic Acid
Derivatives. Angew Chem Int Ed Engl. 1991;30:1176-8.
[65] Stevelmans S, Hest JCMv, Jansen JFGA, Van Boxtel DAFG, Brabander-
van den Berg EMM, Meijer EW. Synthesis, characterization, and guest–host
properties of inverted unimolecular dendritic micelles. J Am Chem Soc.
1996;118:7398-9.
[66] Newkome GR, Yao Z, Baker GR, Gupta VK. Casacade Molecules: A New
Approach to Micelles. J Org Chem. 1985;50:2003-4.
[67] Tomalia DA, Berry V, Hall M, Hedstrand DM. Starburst dendrimers: 4.
Covalently fixed unimolecular assemblages reminiscent of spheroidal micelles.
Macromolecules. 1987;20:1164–7.
[68] Hawker CJ, Wooley KL, Frechet JMJ. Unimolecular micelles and globular
amphiphiles: dendritic macromolecules as novel recyclable solubilization
agents. J Chem Soc, Perkin Trans 1. 1993:1287-97.
[69] Liu M, Kono K, Fréchet JMJ. Water-soluble dendritic unimolecular
micelles:: Their potential as drug delivery agents. J Controlled Release.
2000;65:121-31.
[70] Newkome GR, Moorefield CN, Baker GR, Johnson AL, Behera RK.
Chemistry of Micelles .11. Alkane Cascade Polymers Possessing Micellar
Topology-Micellanoic Acid-Derivatives. Angew Chem Int Ed Engl.
1991;30:1176-8.
[71] Liu M, Fréchet JMJ. Preparation of water-soluble dendritic unimolecular
micelles as potential drug delivery agents. Polym Mater Sci Eng. 1999;80:167–
8.
[72] Ooya T, Lee J, Park K. Effects of ethylene glycol-based graft, star-shaped,
and dendritic polymers on solubilization and controlled release of paclitaxel. J
Control Release. 2003;93:121-7.

95
[73] Yang H, Morris JJ, Lopina ST. Polyethylene glycol–polyamidoamine
dendritic micelle as solubility enhancer and the effect of the length of
polyethylene glycol arms on the solubility of pyrene in water. J Colloid
Interface Sci. 2004;273:148-54.
[74] Haba Y, Harada A, Takagishi T, Kono K. Synthesis of biocompatible
dendrimers with a peripheral network formed by linking of polymerizable
groups. Polymer. 2005;46:1813-20.
[75] Pan G, Lemmouchi Y, Akala EO, Bakare O. Studies on PEGylated and
Drug-Loaded PAMAM Dendrimers. J Bioact Compat Polym. 2005;20:113-28.
[76] Greenwald RB, Choe YH, McGuire J, Conover CD. Effective drug
delivery by PEGylated drug conjugates. Adv Drug Deliv Rev. 2003;55:217-50.
[77] Jansen JFGA, de Brabander-van den Berg EMM, Meijer EW.
Encapsulation of Guest Molecules into a Dendritic Box. Science.
1994;266:1226-9.
[78] Newkome GR, Woosley BD, He E, Moorefield CN, Guther R, Baker GR,
et al. Supramolecular chemistry of flexible, dendritic-based structures
employing molecular recognition. Chem Commun. 1996:2737-8.
[79] Ottaviani MF, Bossmann S, Turro NJ, Tomalia DA. Characterization of
Starburst Dendrimers by the Epr Technique .1. Copper-Complexes in Water
Solution. J Am Chem Soc. 1994;116:661-71.
[80] Ottaviani MF, Cossu E, Turro NJ, Tomalia DA. Characterization of
Starburst Dendrimers by Electron-Paramagnetic-Resonance .2. Positively
Charged Nitroxide Radicals of Variable Chain-Length Used as Spin Probes. J
Am Chem Soc. 1995;117:4387-98.
[81] Jockusch S, Turro NJ, Tomalia DA. Aggregation of Methylene-Blue
Adsorbed on Starburst Dendrimers. Macromolecules. 1995;28:7416-8.
[82] Milhem OM. PhD Thesis. University of Manchester. 2002.

96
[83] Milhem OM, Myles C, McKeown NB, Attwood D, D’Emanuele A.
Polyamidoamine Starburst® dendrimers as solubility enhancers. Int J Pharm.
2000;197:239-41.
[84] Chauhan AS, Sridevi S, Chalasani KB, Jain AK, Jain SK, Jain NK, et al.
Dendrimer-mediated transdermal delivery: enhanced bioavailability of
indomethacin. J Controlled Release. 2003;90:335-43.
[85] Malik N, Evagorou EG, Duncan R. Dendrimer-platinate: a novel approach
to cancer chemotherapy. Anticancer Drugs. 1999;10:767-76.
[86] Kono K, Liu M, Fréchet JMJ. Design of dendritic macromolecules
containing folate or methotrexate residues. Bioconjug Chem. 1999;10:1115-21.
[87] Sudimack J, Lee RJ. Targeted drug delivery via the folate receptor. Adv
Drug Deliv Rev. 2000;41:147-62.
[88] Quintana A, Raczka E, Piehler L, Lee I, Myc A, Majoros I, et al. Design
and function of a dendrimer-based therapeutic nanodevice targeted to tumor
cells through the folate receptor. Pharm Res. 2002;19:1310-6.
[89] Liu M, Kono K, Frechet JM. Water-soluble dendritic unimolecular
micelles: their potential as drug delivery agents. J Control Release.
2000;65:121-31.
[90] Liu H, Farrell S, Uhrich K. Drug release characteristics of unimolecular
polymeric micelles. J Control Release. 2000;68:167-74.
[91] Kolhe P, Misra E, Kannan RM, Kannan S, Lieh-Lai M. Drug
complexation, in vitro release and cellular entry of dendrimers and
hyperbranched polymers. Int J Pharm. 2003;259:143-60.
[92] Rekharsky MV, Inoue Y. Complexation Thermodynamics of
Cyclodextrins. Chem Rev. 1998;98:1875-918.
[93] Szente L, Szejtli J. Highly soluble cyclodextrin derivatives: chemistry,
properties, and trends in development. Adv Drug Deliv Rev. 1999;36:17-28.
[94] Matsuda H, Arima H. Cyclodextrins in transdermal and rectal delivery.
Adv Drug Deliv Rev. 1999;36:81-99.

97
[95] Szejtli J. Cyclodextrins and their inclusion complexes. Budapest:
Akadémiai Kiadó; 1982.
[96] Connors KA. The Stability of Cyclodextrin Complexes in Solution. Chem
Rev. 1997;97:1325-58.
[97] Szejtli J. Introduction and General Overview of Cyclodextrin Chemistry.
Chem Rev. 1998;98:1743-54.
[98] Salmaso S, Semenzato A, Caliceti P, Hoebeke J, Sonvico F, Dubernet C, et
al. Specific antitumor targetable beta-cyclodextrin-poly(ethylene glycol)-folic
acid drug delivery bioconjugate. Bioconjug Chem. 2004;15:997-1004.
[99] Giammona G, Cavallaro G, Maniscalco L, Craparo EF, Pitarresi G.
Synthesis and characterisation of novel chemical conjugates based on
alpha,beta-polyaspartylhydrazide and beta-cyclodextrins. Eur Polym J.
2006;42:2715-29.
[100] Boudad H, Legrand P, Lebas G, Cheron M, Duchene D, Ponchel G.
Combined hydroxypropyl-beta-cyclodextrin and poly(alkylcyanoacrylate)
nanoparticles intended for oral administration of saquinavir. Int J Pharm.
2001;218:113-24.
[101] Temtem M, Pompeu D, Jaraquemada G, Cabrita EJ, Casimiro T, Aguiar-
Ricardo A. Development of PMMA membranes functionalized with
hydroxypropyl-beta-cyclodextrins for controlled drug delivery using a
supercritical CO(2)-assisted technology. Int J Pharm. 2009;376:110-5.
[102] Namazi H, Kanani A. Investigation diffusion mechanism of beta-lactam
conjugated telechelic polymers of PEG and beta-cyclodextrin as the new
nanosized drug carrier devices. Carbohydr Polym. 2009;76:46-50.
[103] Geckeler KE, Arsalani N. Synthesis and Properties of Hydrophilic
Polymers. 4. Preparation and Characterization of Poly(Oxyethylene) Telechelics
with Different Aromatic Termini. J Macromol Sci A. 1996;33:1165-79.

98
[104] Adeli M, Namazi H. Synthesis of Nano-scale Dendritic Supramolecules
and Investigation of Their Applications as the Nanocarrier Agents. Iran- East-
Azarbaijan: University of Tabriz; 2005.
[105] Ferruti P, Ranucci E, Trotta F, Gianasi E, Evagorou EG, Wasil M, et al.
Synthesis, characterisation and antitumour activity of platinum(II) complexes of
novel functionalised poly(amido amine)s. Macromol Chem Phys.
1999;200:1644-54.
[106] Tang W, Muderawan IW, Ng SC, Chan HS. Synthesis and application of
mono-6-ammonium-6-deoxy-beta-cyclodextrin chloride as chiral selector for
capillary electrophoresis. J Chromatogr A. 2005;11:1-2.


Buy your books fast and straightforward online - at one of world’s
fastest growing online book stores! Environmentally sound due to
Print-on-Demand technologies.
Buy your books online at
www.get-morebooks.com
Kaufen Sie Ihre Bücher schnell und unkompliziert online – auf einer
der am schnellsten wachsenden Buchhandelsplattformen weltweit!
Dank Print-On-Demand umwelt- und ressourcenschonend produzi-
ert.
Bücher schneller online kaufen
www.morebooks.deVDM Verlagsservicegesellschaft mbH
Heinrich-Böcking-Str. 6-8 Telefon: +49 681 3720 174 [email protected] - 66121 Saarbrücken Telefax: +49 681 3720 1749 www.vdm-vsg.de





















