
Thermooxidative degradation of polyolefins in the solid state: Part 5. Kinetics of functional group...
23
ELSEVIER Polymer Degradation and Stuhility 55 (I 997) 21-43 0 lYY6 Elsevier Science Limited Printed in Northern Ireland. All rights reserved PII: SO141.3910(96)00109-7 0141-3910/97/$17.0(3 Thermooxidative degradation of polyolefins in the solid state: Part 5. Kinetics of functional group formation in PE-HD and PE-LLD F. Gugumus Ciha-Geigy AC, Busef, Swit~erlund (Received 4 March 1996; accepted 10 April 1996) The experimental kinetics of oxidation product formation on thermal oxida- tion of PE-LLD and PE-HD do not correspond to the rate laws reported in the literature. Instead of a quadratic or biquadratic formation of carbonyl groups with oxidation time, the rate law seems to be more complicated. There is an exponential-type increase in the early stages of the oxidation process, followed by a linear increase in the later stages. As previously seen with PE-LD, the discrepancy with the results in the literature is attributed on the one hand to the fact that the conclusions were based on a very limited amount of experimental results, and on the other hand, to the use of fitting to power laws. The heterogeneous oxidation model and the corresponding kinetics developed for PE-LD also explain the formation of free hydroperoxides, associated hydroperoxides and carbonyl groups in PE-LLD and PE-HD. Hence, they seem to be valid for polyethylene in general. The oxidation spreading rates are affected by polyethylene density as well as by the chemical nature of the catalyst residues. There is usually some similarity between PE-LLDs, although they are different from PE-LDs. There is also some similarity between different Ziegler-type PE-HDs. However, they behave differently from both Phillips-type PE-HDs and PE-LLDs. 0 1996 Elsevier Science Limited 1 INTRODUCTION In preceding publications in this series (Parts 1-4),‘-4 different aspects of thermal oxidation of polyolefins have been examined. After re- examination of the experimental kinetics of functional group formation in PE-LD, a model for heterogeneous oxidation of semi-crystalline polymers was proposed (Part 3). Then, the consequences of the heterogeneity of the polymer for the kinetics of oxidation were examined in detail (Part 4). The experimental verifications of the model were limited to conventional high pressure polyethylene (PE- LD). This was to avoid possible complications caused by the presence of transition metal catalyst residues in the other polyethylene types. In this work, the previous findings will be applied to linear low density polyethylene (PE-LLD) and high density polyethylene (PE-HD). 2 EXPERIMENTAL The polymers used for sample preparation were unstabilized commercial resins stored in a refrigerated room to minimize oxidation. Tables 1 and 2 give a few characteristics of the polymers used. PE-LD and PE-LLD films with an approximate thickness of 500 pm were prepared by compression molding at 170°C for 6 min. The films were immediately quenched in cold water (1OOC). The PE-HD films were compression molded at 21O”C, for 6 min and also quenched in cold water. Thermal oxidation was performed in forced draft air ovens (HORO, Model 080 V/EL) mainly at a temperature of 80°C. Some experiments involved slightly higher tempera- tures, 90 and 100°C. Functional group formation was measured by IR spectrophotometry with an unexposed sample
Transcript of Thermooxidative degradation of polyolefins in the solid state: Part 5. Kinetics of functional group...

ELSEVIER
Polymer Degradation and Stuhility 55 (I 997) 21-43 0 lYY6 Elsevier Science Limited
Printed in Northern Ireland. All rights reserved
PII: SO141.3910(96)00109-7 0141-3910/97/$17.0(3
Thermooxidative degradation of polyolefins in the solid state: Part 5. Kinetics of functional
group formation in PE-HD and PE-LLD
F. Gugumus Ciha-Geigy AC, Busef, Swit~erlund
(Received 4 March 1996; accepted 10 April 1996)
The experimental kinetics of oxidation product formation on thermal oxida- tion of PE-LLD and PE-HD do not correspond to the rate laws reported in the literature. Instead of a quadratic or biquadratic formation of carbonyl groups with oxidation time, the rate law seems to be more complicated. There is an exponential-type increase in the early stages of the oxidation process, followed by a linear increase in the later stages. As previously seen with PE-LD, the discrepancy with the results in the literature is attributed on the one hand to the fact that the conclusions were based on a very limited amount of experimental results, and on the other hand, to the use of fitting to power laws. The heterogeneous oxidation model and the corresponding kinetics developed for PE-LD also explain the formation of free hydroperoxides, associated hydroperoxides and carbonyl groups in PE-LLD and PE-HD. Hence, they seem to be valid for polyethylene in general. The oxidation spreading rates are affected by polyethylene density as well as by the chemical nature of the catalyst residues. There is usually some similarity between PE-LLDs, although they are different from PE-LDs. There is also some similarity between different Ziegler-type PE-HDs. However, they behave differently from both Phillips-type PE-HDs and PE-LLDs. 0 1996 Elsevier Science Limited
1 INTRODUCTION
In preceding publications in this series (Parts 1-4),‘-4 different aspects of thermal oxidation of polyolefins have been examined. After re- examination of the experimental kinetics of functional group formation in PE-LD, a model for heterogeneous oxidation of semi-crystalline polymers was proposed (Part 3). Then, the consequences of the heterogeneity of the polymer for the kinetics of oxidation were examined in detail (Part 4). The experimental verifications of the model were limited to conventional high pressure polyethylene (PE- LD). This was to avoid possible complications caused by the presence of transition metal catalyst residues in the other polyethylene types. In this work, the previous findings will be applied to linear low density polyethylene (PE-LLD) and high density polyethylene (PE-HD).
2 EXPERIMENTAL
The polymers used for sample preparation were unstabilized commercial resins stored in a refrigerated room to minimize oxidation. Tables 1 and 2 give a few characteristics of the polymers used. PE-LD and PE-LLD films with an approximate thickness of 500 pm were prepared by compression molding at 170°C for 6 min. The films were immediately quenched in cold water (1OOC). The PE-HD films were compression molded at 21O”C, for 6 min and also quenched in cold water.
Thermal oxidation was performed in forced draft air ovens (HORO, Model 080 V/EL) mainly at a temperature of 80°C. Some experiments involved slightly higher tempera- tures, 90 and 100°C.
Functional group formation was measured by IR spectrophotometry with an unexposed sample

22 F. Gugumus
Table 1. Characteristics of the polyethylene types used in the first series
Polyethylene
PE-LLD-A (butene) PE-LLD-B (butene) PE-HD-A (Ziegler) PE-HD-B (Ziegler) PE-HD-C (Phillips) PE-LD-C (comparison)
Density Vinylidene (g/cm’)
Vinyl (mol/l) X l@’ (mol/l) x 10‘
0.920 1.9 6.7 0.921 7.4 29.8 0.944 2.6 8.9 0.955 5.0 6.4 0.965 n.d. 57.2 0.918 18.9 8.2
Catalyst residues (mgkg)
3 (Ti) 2 (Ti) 9 (Ti)
39 (Ti) 2 (Cr) none
in the reference beam. The data are mostly expressed as the absorbance at the wavelength corresponding to the maximum of the absorption band, e.g. E 3410 for the absorbance of the associated hydroperoxides whose maximum is usually between 3400 and 3420 cm- ’ . The spectrophotometers used were Perkin-Elmer models 781.782 and FT-IR 1710.
3 EXPERIMENTAL KINETICS
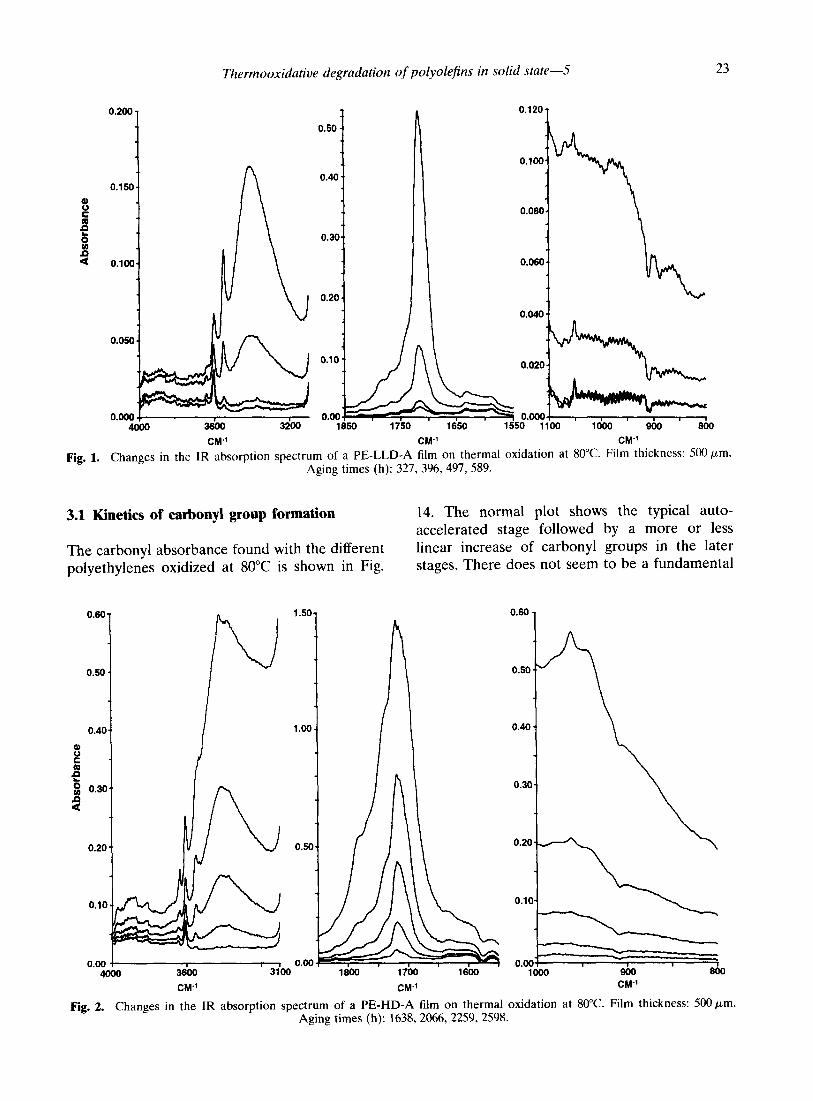
The changes in the IR spectrum of a typical PE-LLD film on thermal oxidation in a forced air oven are shown in Fig. 1. They are similar to those observed in PE-LD (Part l), with a considerable increase in the hydroxyl and carbonyl absorbance regions. At the same time there are important changes with respect to the hydrogen deformation peaks of the carbon- carbon double bonds and in the carbon-oxygen single bond absorption region. The changes observed on thermal oxidation of PE-HD films are roughly the same as shown in Figs 2 and 3 for Ziegler-type PE-HD (Ti-catalyst) and Phillips- type PE-HD (Cr-catalyst), respectively. The differences are mainly qualitative in nature. As with PE-LD, carbonyl groups and hydroxyl groups account for most of the oxidation products. Besides carbonyl groups with an IR absorption maximum at about 1715 cm- ‘, free
and associated hydroperoxides absorbing close to 3550 and 3410cm-‘, respectively, were used to monitor thermal oxidative degradation.
The plots of the variation of the absorbance of the carbonyl, free and associated hydroperoxide groups for the different polyethylene types examined and oxidized at 80°C are shown in Figs 4-8. It can be seen, that with the PE-LLD samples (Figs 4 and 5) as previously seen for PE-LD (Part l), the absorbance of the hydroperoxides is relatively important in com- parison with the absorbance of the carbonyl groups. This is much less pronounced with the high density polyethylenes (Figs 7 and 8). The oxidation time needed for some amount of oxidation depends both on the polyethylene type and the particular sample.
The results of oxidation at higher temperatures than 80°C are overall the same as those observed at 80°C. This can be seen in Figs 9-13 for the same polyethylene samples as above, oxidized either at 90°C (PE-LLD-A, PE-LLD-B and PE-HD-C) or at 100°C (PE-HD-A and PE-HD- B). The main visible difference from the corresponding results at 80°C consists in a shortened induction time.
In the following, carbonyl and hydroperoxide concentrations will be used to evaluate the advancement of thermal oxidation of PE-LLD and PE-HD.
Table 2. Characteristics of the polyethylene types used in the second series
Polyethylene Density (g/cm’)
Vinylidene (mol/l) X 10’
Vinyl (mol/l) X 10’
Catalyst residues (mgkg)
~.~. PE-LLD-A’ (butene) PE-LLD-C’ (butene) PE-HD-A’ (Ziegler) PE-HD-B’ (Ziegler) PE-HD-C’ (Phillips) PE-LD-C’ (comparison)
0.920 3.6 7.2 3 (Ti) 0.922 3.2 4.6 26 (Ti) 0.944 2.2 11.0 9.5 (Ti) 0.955 4.5 6.8 38 (Ti) 0.965 n.d. 64.4 2 (Cr) 0.918 18.9 8.2 none
Thermooxidative degradation of polyolejins in solid state-5 23
0.200-
0.150.
0)
2
d
i
9 0.100.
0.000 4ooo 3600 3200
CM-’ CM-’ CM-’
Fig. 1. Changes in the IR absorption spectrum of a PE-LLD-A film on thermal oxidation at 80°C. Film thickness: 500 pm. Aging times (h): 327, 396. 497, 589.
0.040.
3.1 Kinetics of carbonyl group formation
The carbonyl absorbance found with the different polyethylenes oxidized at 80°C is shown in Fig.
14. The normal plot shows the typical auto- accelerated stage followed by a more or less linear increase of carbonyl groups in the later stages. There does not seem to be a fundamental
0.00 4060 3600 3100 1600 1700 1600
0.60 1
Fig. 2. Changes in the IR absorption spectrum of a PE-HD-A film on thermal oxidation at 80°C. Film thickness: .5001_~m. Aging times (h): 1638,2066,2259,2598.
24 F. Gugumus
Fig.
1.501 0.225,
0.200 1
o%i'~~"~40g50 1800 1700 1800 -0.025 1OOO 950 soo 850 800 CM-' CM-' CM-'
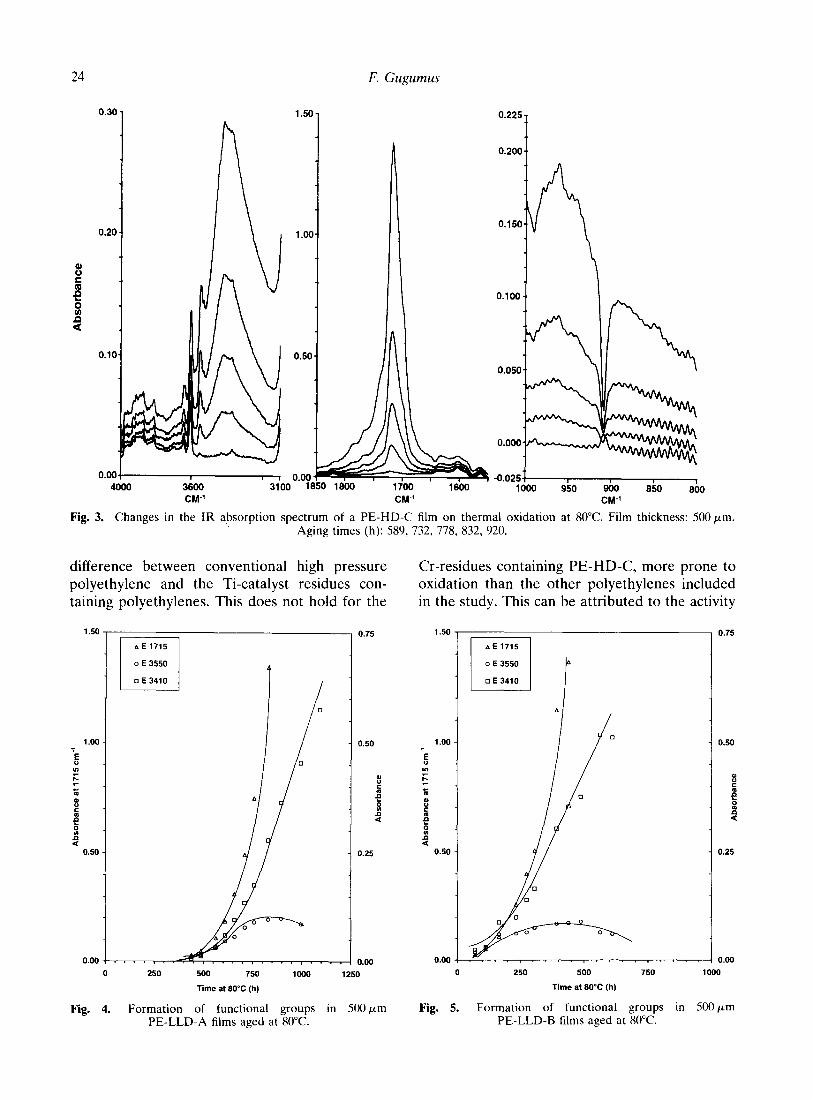
3. Changes in the IR absorption spectrum of a PE-HD-C film on thermal oxidation at 80°C. Film thickness: 500 pm. Aging times (h): 589, 732, 778, 832, 920.
difference between conventional high pressure Cr-residues containing PE-HD-C, more prone to polyethylene and the Ti-catalyst residues con- oxidation than the other polyethylenes included taining polyethylenes. This does not hold for the in the study. This can be attributed to the activity
1.50 1.75
A E 1715
L---l 0 E 3550
0 E 3410
- I I.75
0.50
0.25 0.25
0 250 500 750
Time at 60°C (h)
0.00
1000 1250 500
Time at 80% (h)
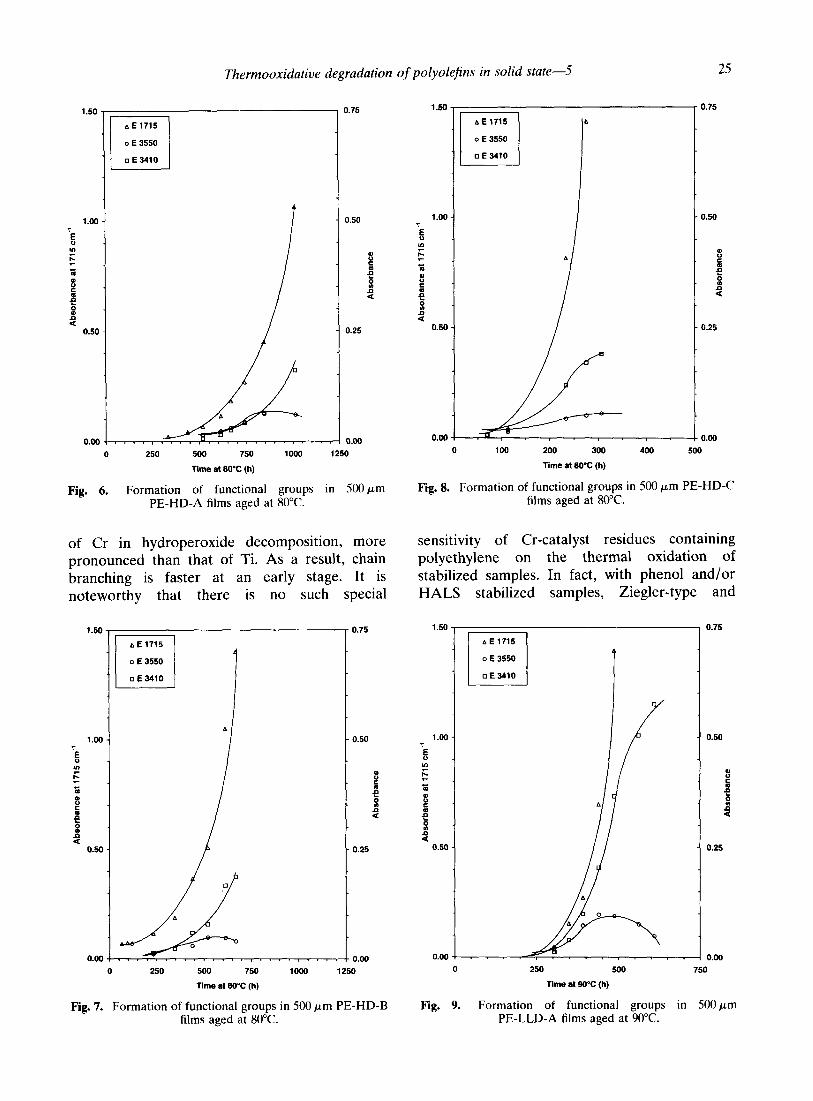
Fig. 4. Formation of functional groups in 500 pm Fig. 5. Formation of functional groups in 500 pm PE-LLD-A films aged at 80°C. PE-LLD-B films aged at 80°C.
Thermooxidative degradation of polyolefins in solid state-5
1 q E3410 1
4
- (
,
1.75
0.50
0.25
0.W
0 250 500 750 loo0 1250
Time at 80°C (h)
Fig. 6. Formation of functional groups in 5OOprn PE-HD-A films aged at 80°C.
of Cr in hydroperoxide decomposition, more pronounced than that of Ti. As a result, chain branching is faster at an early stage. It is noteworthy that there is no such special
1.50 -I
JII 0 oE3410 A E E 1715 3550 fi
1.00 -
Tg
z $
1
8 5 e P
9 0.50 -
0.00
0 250 500 750 loo0 1250
Time at 80% (h)
D.75
0.50
0.25
Fig. 7. Formation of functional groups in 500 pm PE-HD-B Fig. 9. Formation of functional groups in SOOpm films aged at 80°C. PE-LLD-A films aged at WC.
0.00. , . I . 1 . I . -r
0 100 200 300 400 SW
Time at 80°C (h)
25
0.75
0.50
8
s b d a
0.25
0.00
Fig. 8. Formation of functional groups in 500 pm PE-HD-C films aged at 80°C.
sensitivity of Cr-catalyst residues containing polyethylene on the thermal oxidation of stabilized samples. In fact, with phenol and/or HALS stabilized samples, Ziegler-type and
?
0.75
0.50
0.25
250 500 750
Time at 90°C (h)
26 F. Gugumus
2.00
1 so
rg
z $
z 1.00
e
B
B
9
0.50
0.00
1.00
0.75
0.50
0.25
0.00 0 100 200 300
Time at 90-C (h)
4W 500 0 50 100
Time at 1 W’C (h)
150
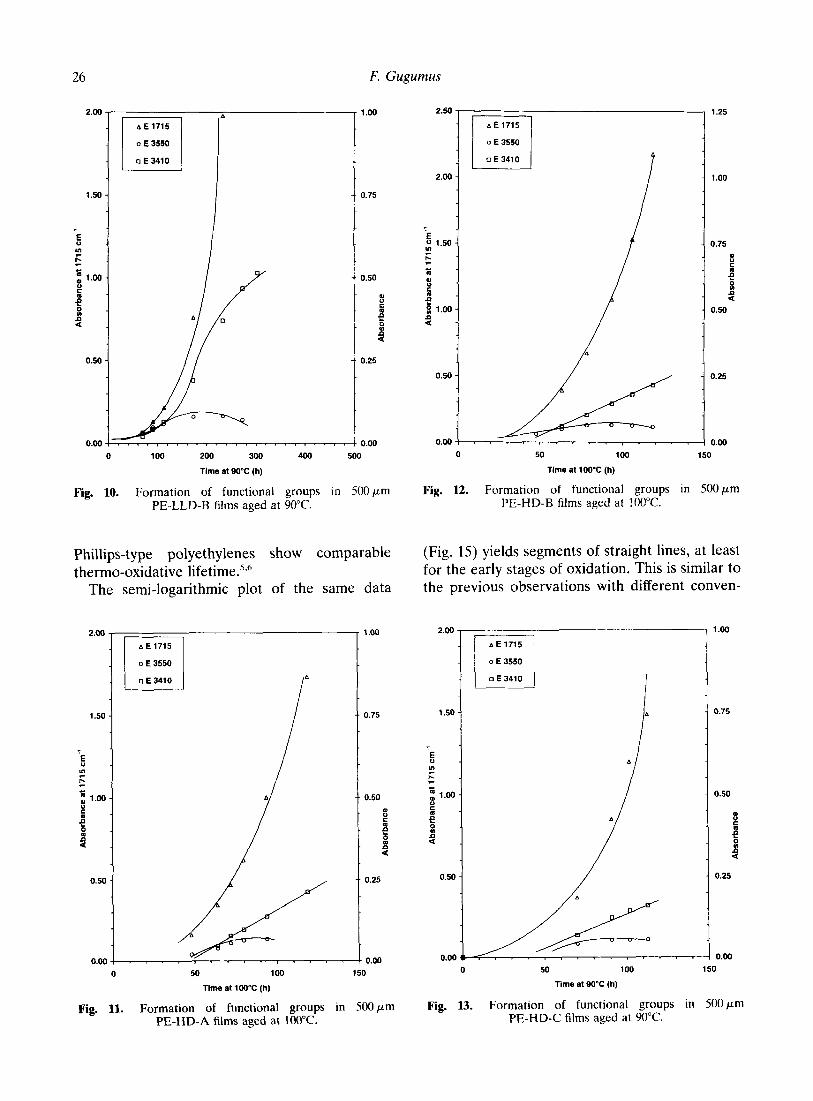
Fig. 10. Formation of functional groups in SOOpm Fig. 12. Formation of functional groups in SOO~m PE-LLD-B films aged at 90°C. PE-HD-B films aged at 100°C.
Phillips-type polyethylenes show comparable (Fig. 15) yields segments of straight lines, at least thermo-oxidative lifetime.“.’ for the early stages of oxidation. This is similar to
The semi-logarithmic plot of the same data the previous observations with different conven-
1 .oo
-- 0.25
0 50 100 150
Time at 100°C (h)
0 50 100 150
Time at 90°C (h)
Fig. 11. Formation of functional groups in 500 pm Fig. 13. Formation of functional groups in 500 pm PE-HD-A films aged at 100°C. PE-HD-C films aged at WC.
-- 0.75
2.50
2.00
‘;E cJ 1.50 9 $
z
8 5 c 0 a 1.00
a
0.50
0.00
I
7
1.25
1 .oo
0.75
e d L B 4
0.50
0.25
0.00
/
2.00
1.50
rg
z $
t 1.00
K
: 01
9
0.50
4
l.w
A - 0.75
- 0.50
- 0.25
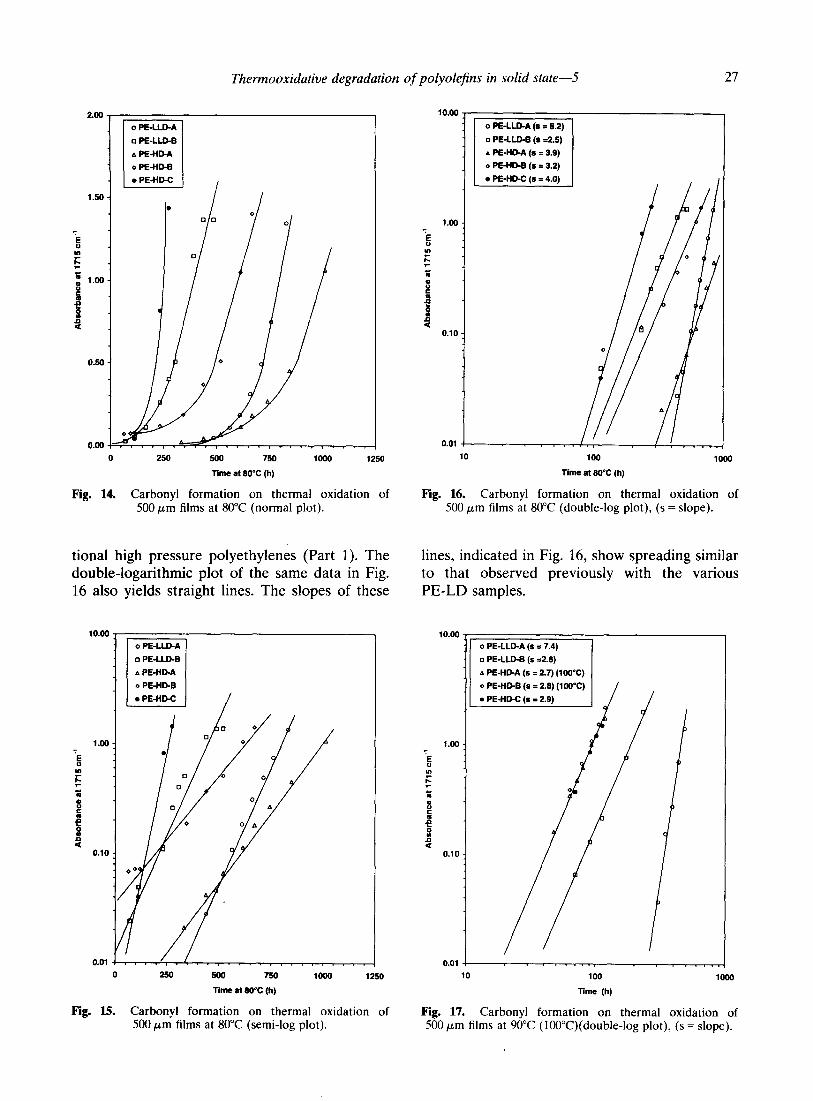
Thermooxidative degradation of polyolefins in solid state-5 27
1 so
0.00
0 250 500 750 1000 1250
Time at 80°C (h)
1.00
rE : K
G 8
c
9 0.10
0.01
10 loo
Time at 60% (h)
Fig. 14. Carbonyl formation on thermal oxidation of Fig. 16. Carbonyl formation on thermal oxidation of 500 pm films at 80°C (normal plot). 500 pm films at 80°C (double-log plot), (s = slope).
tional high pressure polyethylenes (Part 1). The lines, indicated in Fig. 16, show spreading similar double-logarithmic plot of the same data in Fig. to that observed previously with the various 16 also yields straight lines. The slopes of these PE-LD samples.
500 750
Time at 50°C (h)
4 1000 1250
Fig. 15. Carbonyl formation on thermal oxidation of Fig. 17. Carbonyl formation on thermal oxidation of 500 pm films at 80°C (semi-log plot). 500 pm films at 90°C (lOOT)(double-log plot), (s = slope).
q PE-LLD-B (s 12.5)
I.00 -
i : K
r
E $
$ 9
0.10 -
loo
Time (h)

28 F. Gugumus
These results are confirmed by the data obtained with the film samples from the same polymer series aged at 90°C (PE-LLD-A, PE-LLD-B and PE-HD-C) or at 100°C (PE-HD- A and PE-HD-B). The double-log plot of Fig. 17 again shows large variations in the values of the slopes. The results are also confirmed by a second testing series with different polyethylenes aged at 80°C. slopes of the and 9.9.
Therefore, quadratic or
It can be seen in Fig. 18 that the double-log plots vary between 3.6
carbonyl formation, according to biquadratic power laws, can be
excluded as much for PE-LLD and PE-HD as for conventional PE-LD. The different plots ex- amined point rather to an initial exponential-type increase of carbonyl groups followed by a more or less linear increase with oxidation time in the later stages.
3.2 Kinetics of hydroperoxide formation
The variation of the absorbance of free hydroperoxides in the different polyethylenes
q PE-LLD-6’ (s ~3.8)
A PE-HDA’ (s = 9.9)
-1
0.01 1
10 100 1000 10000
Time at 80°C (h)
Fig. 18. Carbonyl formation on thermal oxidation of 500 pm films at 80°C (second series), (double-log plot),
(s = slope).
0.15
0.10
‘g
5: s ;ii
2 d
P
9 0.05
0.00
L. PE-HD-c ]
500 750
Time at 90°C (h)
Fig. 19. Formation of free hydroperoxides on thermal oxidation of 500 pm films at 80°C (normal plot).
tested is shown in Fig. 19. It can be seen that, usually, after an initial autoaccelerated period, the concentration passes through a maximum and decreases. Although the maximum is not obvious for PE-HD-C in the plot shown, this has to be attributed to the particular sample and not to any special behavior of this polyethylene type. In fact, it has already been shown that in Phillips-type polyethylene also, the concentration of free hydroperoxides passes through a maxi- mum and decreases.’ Thus, PE-LLD and PE-HD show overall the same behavior as that observed with PE-LD.
The semi-logarithmic plot in Fig. 20 is linear in the initial stage of hydroperoxide formation, before the concentration starts to level off. This is again comparable to the observations made with PE-LD. The same is true for the double- logarithmic plot in Fig. 21. Again, the slopes of the segments of straight lines vary considerably. If the slope found with one of the samples, 1.9 for PE-HD-B, could be an indication of the quadratic dependency on oxidation time as reported in the literature (see Ref. 1 and references cited therein), the slopes for the other samples are either lower or significantly higher. The data generated with the same polymers at 90
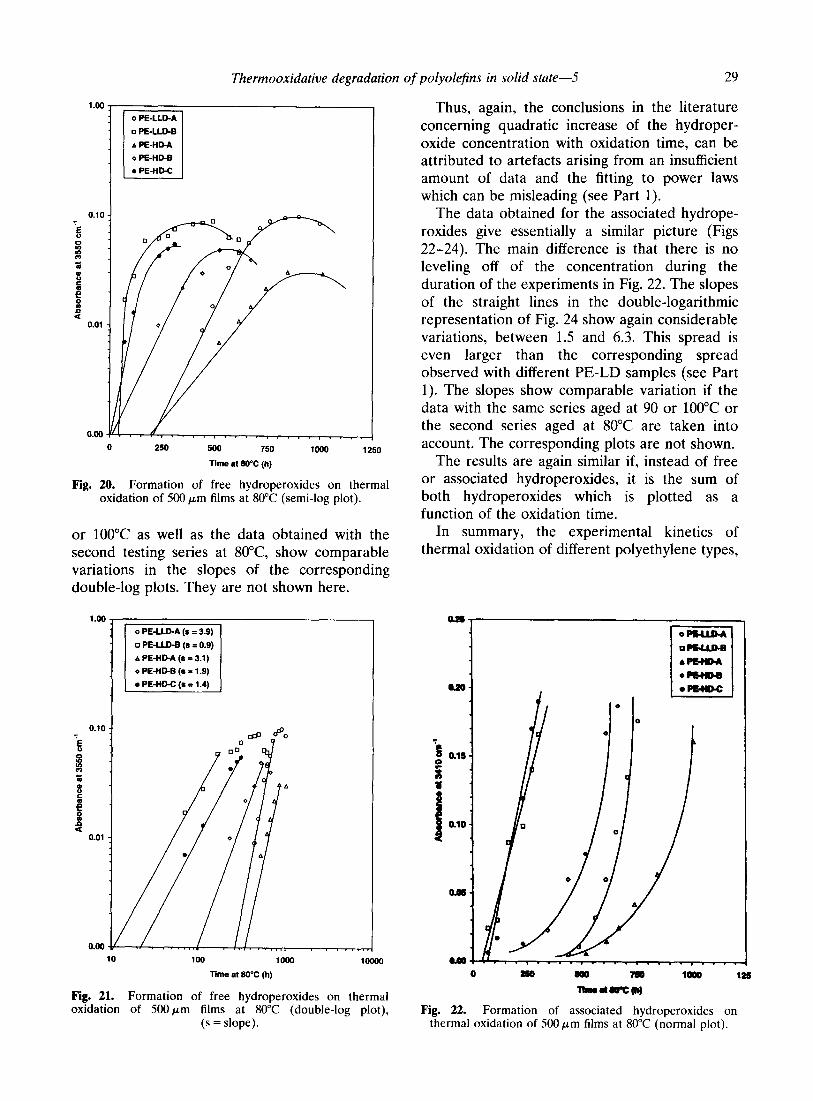
Thermooxidative degradation of polyolefins in solid state-5 29
1.00 -
0 250 500 750 1000 1250
Time at WC (h)
Fig. 20. Formation of free hydroperoxides on thermal oxidation of 500 pm films at 80°C (semi-log plot).
or 100°C as well as the data obtained with the second testing series at 8O”C, show comparable variations in the slopes of the corresponding double-log plots. They are not shown here.
l.o(
0.11
q PE-LLD-B (I = 0.9)
& PE-HDA (a = 3.1)
o PEHDB (s = 1.9)
0.00
10 100 1000 10000
Time at 90°C (h)
Fig. 21. Formation of free hydroperoxides on thermal oxidation of SOO~m films at 80°C (double-log plot),
(s = slope).
Thus, again, the conclusions in the literature concerning quadratic increase of the hydroper- oxide concentration with oxidation time, can be attributed to artefacts arising from an insufficient amount of data and the fitting to power laws which can be misleading (see Part 1).
The data obtained for the associated hydrope- roxides give essentially a similar picture (Figs 22-24). The main difference is that there is no leveling off of the concentration during the duration of the experiments in Fig. 22. The slopes of the straight lines in the double-logarithmic representation of Fig. 24 show again considerable variations, between 1.5 and 6.3. This spread is even larger than the corresponding spread observed with different PE-LD samples (see Part 1). The slopes show comparable variation if the data with the same series aged at 90 or 100°C or the second series aged at 80°C are taken into account. The corresponding plots are not shown.
The results are again similar if, instead of free or associated hydroperoxides, it is the sum of both hydroperoxides which is plotted as a function of the oxidation time.
In summary, the experimental kinetics of thermal oxidation of different polyethylene types,
0 x0 aa 700 1000 125
-a-(h)
Fig. 22. Formation of associated hydroperoxides on thermal oxidation of 500 pm films at 80°C (normal plot).
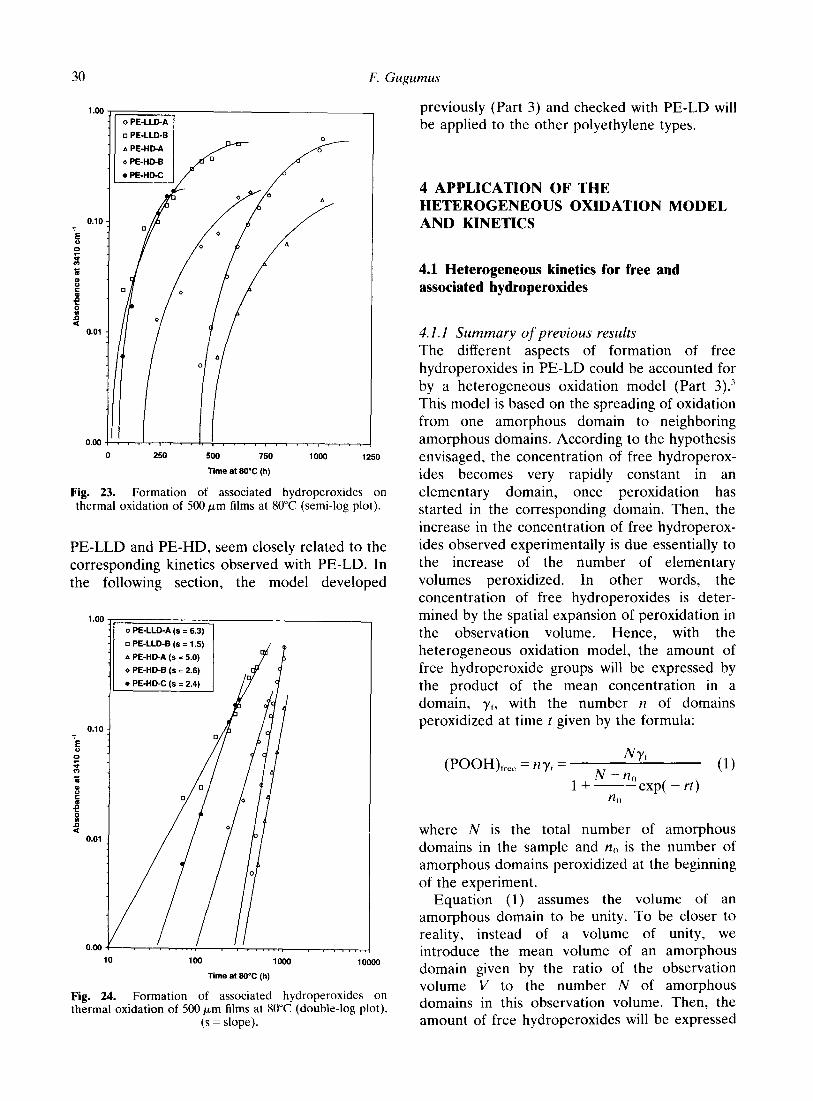
F. Gugumus
500 750
Time at WC (h)
1000 1250
Fig. 23. Formation of associated hydroperoxides on thermal oxidation of 500 pm films at 80°C (semi-log plot).
PE-LLD and PE-HD, seem closely related to the corresponding kinetics observed with PE-LD. In the following section, the model developed
q PE-LLPB (s = 1.5)
b PE-HDA (s = 5.0)
o PE-HDB (s = 2.6)
100 1000
Time at WC (h)
10000
Fig. 24. Formation of associated hydroperoxides on thermal oxidation of 500 Frn films at 80°C (double-log plot),
(s = slope).
previously (Part 3) and checked with PE-LD be applied to the other polyethylene types.
4 APPLICATION OF THE
will
HETEROGENEOUS OXIDATION MODEL AND KINETICS
4.1 Heterogeneous kinetics for free and associated hydroperoxides
4.1.1 Summary of previous results The different aspects of formation of free hydroperoxides in PE-LD could be accounted for by a heterogeneous oxidation model (Part 3).’ This model is based on the spreading of oxidation from one amorphous domain to neighboring amorphous domains. According to the hypothesis envisaged, the concentration of free hydroperox- ides becomes very rapidly constant in an elementary domain, once peroxidation has started in the corresponding domain. Then, the increase in the concentration of free hydroperox- ides observed experimentally is due essentially to the increase of the number of elementary volumes peroxidized. In other words, the concentration of free hydroperoxides is deter- mined by the spatial expansion of peroxidation in the observation volume. Hence, with the heterogeneous oxidation model, the amount of free hydroperoxide groups will be expressed by the product of the mean concentration in a domain, y,., with the number n of domains peroxidized at time t given by the formula:
(POOH),.,,, = ny, = NY,
N - no (1)
1+- exp( - rt) 6,
where N is the total number of amorphous domains in the sample and yl,) is the number of amorphous domains peroxidized at the beginning of the experiment.
Equation (1) assumes the volume of an amorphous domain to be unity. To be closer to reality, instead of a volume of unity, we introduce the mean volume of an amorphous domain given by the ratio of the observation volume V to the number N of amorphous domains in this observation volume. Then, the amount of free hydroperoxides will be expressed

Thermooxidative degradation of polyolefns in solid state-.5 31
by the product of mean volume, i.e.
V VYf
WOW,,, = n tg yf = N - no
1+- flo
exp( - rt)
the expression above with this
or, taking into account that only three para- meters are independent:
(POOWfree = A 1 + B exp( - rt)
(2)
where:
A = Vy,
N - no B=------ 4,
The model assumes that at the beginning of . .
oxidation, no amorphous domains are already peroxidized so that oxidation can spread from the first moment on. If this is not so, i.e. if the n,, domains are only slightly oxidized, it will mean that some time is required before they are fully oxidized and oxidation can spread out from them. Designating this ‘induction time’ by ti, the number of peroxidized domains will be given by the expression:
(POOWfrce = A
1 + B exp( - r[t - ti]) (3)
This equation is valid for oxidation times t 2 tie The problem is somewhat more complicated
with respect to the associated hydroperoxides. In Part 4 of this series,4 it was shown that the experimental results can be explained if the concentration of associated hydroperoxides in an amorphous domain is considered to consist of two parts: an initiating part comprising mainly associations hydroperoxide/hydroperoxide level- ing off as rapidly as the free hydroperoxides and a non-initiating part of associations hydro- peroxide/carbonyl groups:
(PO0H)a.i = (POOH),,,, + (POOH),,,,
where (POOH),,,i and (POOH),,,i are the con- centrations of the associations hydroperoxide/ hydroperoxide and of the associations hydroper- oxide/carbonyl groups, respectively. Designating the maximum values of these concentrations in the amorphous domains by yal and ya2, respectively, the maximum value of the con- centration of associated hydroperoxides will be Yal + Ya2. It has been assumed,” in a first
approximation, that in addition to the associa- tions hydroperoxide/hydroperoxide, the associa- tions hydroperoxide/carbonyl groups also level off rapidly after the beginning of peroxidation of a domain. Then, the total amount of associated hydroperoxides after an oxidation time t can be approximated by the product of this maximum concentration with the number of amorphous domains peroxidized at time t, i.e.
(POOH),,. = (~a, + ~a21 N
N - no (4)
l+--- n0
exp( - rt)
This equation is similar to the equation for the free hydroperoxides and, like the latter, it also assumes the volume to be unity for the amorphous domains. Introduction of the mean volume V/N of an amorphous domain and of the induction time yields the following equation:
(POOH),,, = ; N _“,[yai + ya23
1+---- n,, ‘)exp[ - r(t - fi)]
VYal + Ya21 = N-no
1+---- n,~ exP] - r(t - [i)]
or, reducing the parameters involved to a minimum:
(POOH),,, = A
1 + B exp[ - r(t - ti)] (5)
with:
A = V[Y,, + ~a21
N - no BE------
no
Equation (5) is formally the same as eqn (3) accounting for the free hydroperoxides. Howe- ver, the values of the parameters will usually be different, with the exception of B which has the same meaning in both equations.
4.1.2 Application to PE-LLD and PE-HD The data generated from the experimental
series already discussed above were fitted to the equations derived for the free and associated hydroperoxides.8 For comparison purposes, the data obtained from a typical PE-LD aged simultaneously were treated the same way. The plot of Fig. 25 shows the formation of free hydroperoxides with the various polyethylene samples aged at 80°C. Curve fitting to eqn (2) accounts well for the data, independently of the
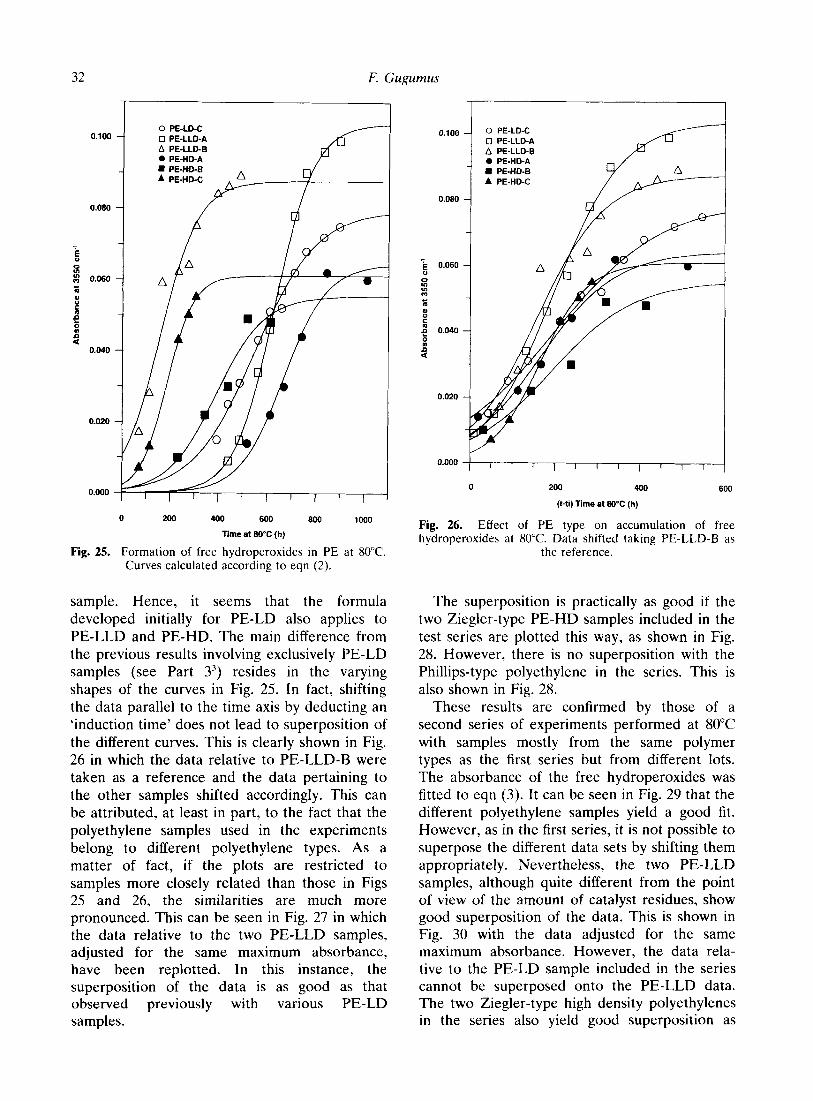
32 F. Gugumus
0.160 0 PE-LD-C
0 PE-LLD-A
A PE-LLD-8
0 PE-HD-A
0.080 0.080
i
% 4 0.060
I
8 5
c
::
9 0.040
0.020
0.000
k 0.060 0
P 4 z
8 s P 0.040
%
9
/
/
0.020
0.000
0 400 600
Time at 80°C (h)
Fig. 25. Formation of free hydroperoxides in PE at 80°C. Curves calculated according to eqn (2).
sample. Hence, it seems that the formula developed initially for PE-LD also applies to PE-LLD and PE-HD. The main difference from the previous results involving exclusively PE-LD samples (see Part 3’) resides in the varying shapes of the curves in Fig. 25. In fact, shifting the data parallel to the time axis by deducting an ‘induction time’ does not lead to superposition of the different curves. This is clearly shown in Fig. 26 in which the data relative to PE-LLD-B were taken as a reference and the data pertaining to the other samples shifted accordingly. This can be attributed, at least in part, to the fact that the polyethylene samples used in the experiments belong to different polyethylene types. As a matter of fact, if the plots are restricted to samples more closely related than those in Figs 25 and 26, the similarities are much more pronounced. This can be seen in Fig. 27 in which the data relative to the two PE-LLD samples, adjusted for the same maximum absorbance, have been replotted. In this instance, the superposition of the data is as good as that observed previously with various PE-LD samples.
0 PE-LD-C
0 PE-LLD-A
A PE-LLD-6 0 PE-HD-A
1 PE-HD-6
A PE-HD-C
0 200 400
(t-ti) Time at 60% (h)
600
Fig. 26. Effect of PE type on accumulation of free hydroperoxides at 80°C. Data shifted taking PE-LLD-B as
the reference.
The superposition is practically as good if the two Ziegler-type PE-HD samples included in the test series are plotted this way, as shown in Fig. 28. However, there is no superposition with the Phillips-type polyethylene in the series. This is also shown in Fig. 28.
These results are confirmed by those of a second series of experiments performed at 80°C with samples mostly from the same polymer types as the first series but from different lots. The absorbance of the free hydroperoxides was fitted to eqn (3). It can be seen in Fig. 29 that the different polyethylene samples yield a good fit. However, as in the first series, it is not possible to superpose the different data sets by shifting them appropriately. Nevertheless, the two PE-LLD samples, although quite different from the point of view of the amount of catalyst residues, show good superposition of the data. This is shown in Fig. 30 with the data adjusted for the same maximum absorbance. However, the data rela- tive to the PE-LD sample included in the series cannot be superposed onto the PE-LLD data. The two Ziegler-type high density polyethylenes in the series also yield good superposition as
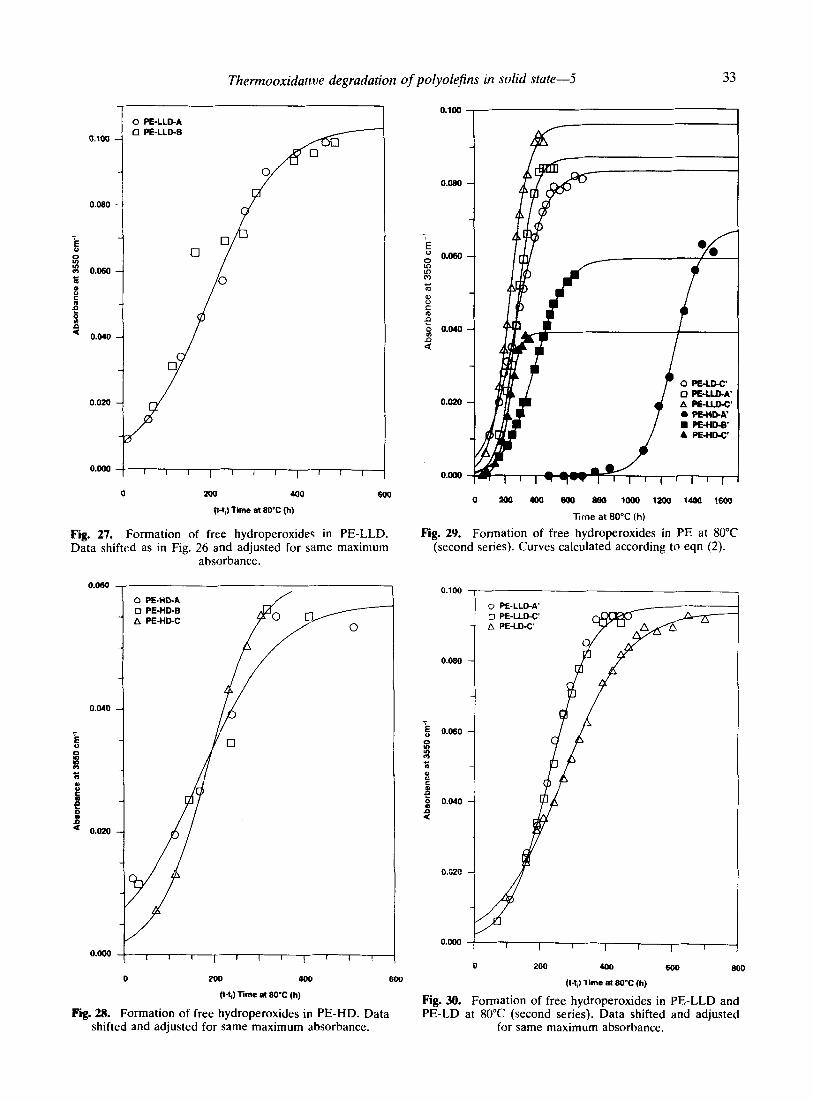
Thermooxidative degradation of polyolefins in solid state-5
0.100
0.080
7
0 PE-LLD-A cl PE-LLD-8
0 200 400
(t-t,) Time at 80°C (h)
600
Fig. 27. Formation of free hydroperoxides in PE-LLD. Data shifted as in Fig. 26 and adjusted for same maximum
absorbance.
0.060
0.040
f
% z 1 8
f
9 0.020
0.000
0 PE-HO-A 0 PE-HDB A PE-HDC
0 zoo wo
(t-t,) Time at 80°C (h)
600
Fig. 28. Formation of free hydroperoxides in PE-HD. Data shifted and adjusted for same maximum absorbance.
0.100
0.000
0.020
I 0 200400aJo 800 lmo 1200 14m IMX
prne at 80°C (h)
Fig. 29. Formation of free hydroperoxides in PE at 80°C (second series). Curves caklateh according to eqn (2).
0 200 400 Ml0 800
(t-ti) Time at 80% (h)
Fig. 30. Formation of free hydroperoxides in PE-LLD and PE-LD at 80°C (second series). Data shifted and adjusted
for same maximum absorbance.
34 F. Gugumus
0.060
g
5: 0.040
% 5
8 5 c 8
9
0.020
0.000
, 2
0 PE-HD-A’
q PE-HD-6’
A PE-HD-C’
0 200 400
(t-t,) Time at 60°C (h)
600
Fig. 31. Formation of free hydroperoxides in PE-HD at 80°C (second series). Data shifted and adjusted for same
maximum absorbance.
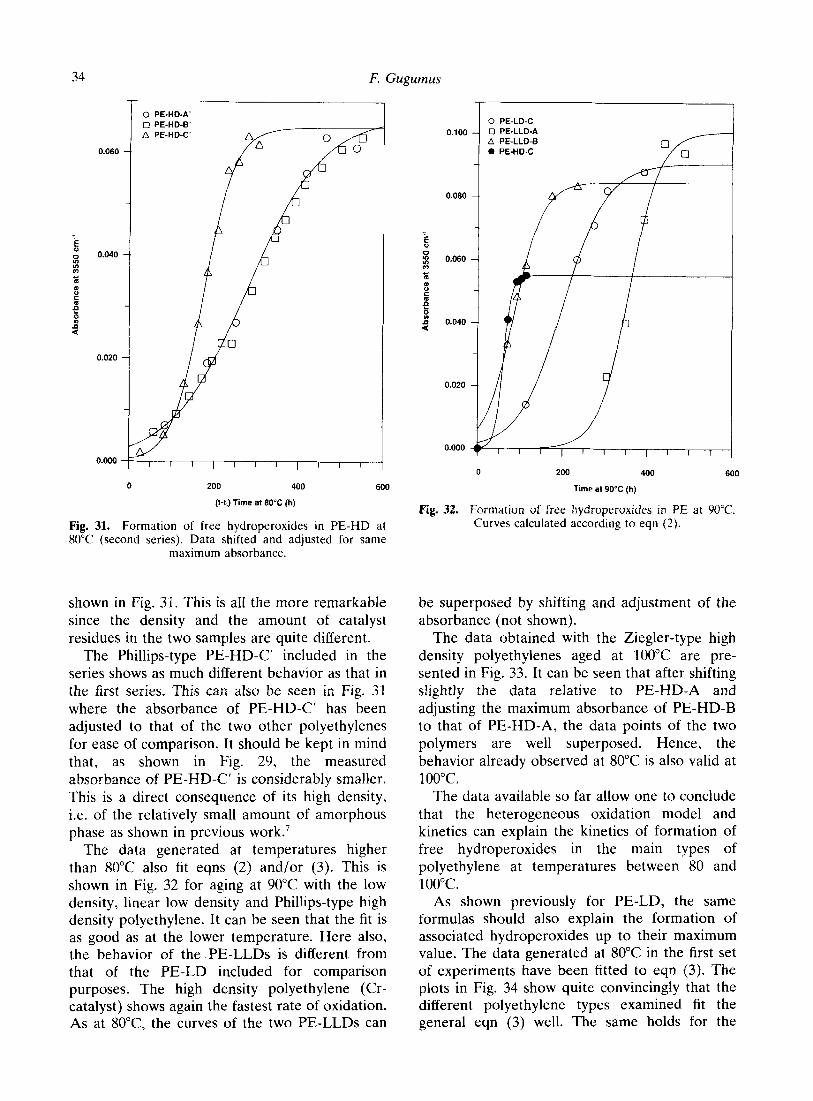
shown in Fig. 31. This is all the more remarkable since the density and the amount of catalyst residues in the two samples are quite different.
The Phillips-type PE-HD-C’ included in the series shows as much different behavior as that in the first series. This can also be seen in Fig. 31 where the absorbance of PE-HD-C’ has been adjusted to that of the two other polyethylenes for ease of comparison. It should be kept in mind that, as shown in Fig. 29, the measured absorbance of PE-HD-C’ is considerably smaller. This is a direct consequence of its high density, i.e. of the relatively small amount of amorphous phase as shown in previous work.’
The data generated at temperatures higher than 80°C also fit eqns (2) and/or (3). This is shown in Fig. 32 for aging at 90°C with the low density, linear low density and Phillips-type high density polyethylene. It can be seen that the fit is as good as at the lower temperature. Here also, the behavior of the PE-LLDs is different from that of the PE-LD included for comparison purposes. The high density polyethylene (Cr- catalyst) shows again the fastest rate of oxidation. As at 8O”C, the curves of the two PE-LLDs can
0.100
0.080
i
:: z 0.060
5
z c B
$ 8 0.040
0.020
0.000
Fig. 32.
-
/
0 PE-LD-C
0 PE-LLD-A
A PE-LLD-6
0 FE-HD-C
0 200 400 600
Time at 90°C (h)
Formation of free hydroperoxides in PE at YOT. Curves calculated according to eqn (2).
be superposed by shifting and adjustment of the absorbance (not shown).
The data obtained with the Ziegler-type high density polyethylenes aged at 100°C are pre- sented in Fig. 33. It can be seen that after shifting slightly the data relative to PE-HD-A and adjusting the maximum absorbance of PE-HD-B to that of PE-HD-A, the data points of the two polymers are well superposed. Hence, the behavior already observed at 80°C is also valid at 1oo”c.
The data available so far allow one to conclude that the heterogeneous oxidation model and kinetics can explain the kinetics of formation of free hydroperoxides in the main types of polyethylene at temperatures between 80 and 100°C.
As shown previously for PE-LD, the same formulas should also explain the formation of associated hydroperoxides up to their maximum value. The data generated at 80°C in the first set of experiments have been fitted to eqn (3). The plots in Fig. 34 show quite convincingly that the different polyethylene types examined fit the general eqn (3) well. The same holds for the
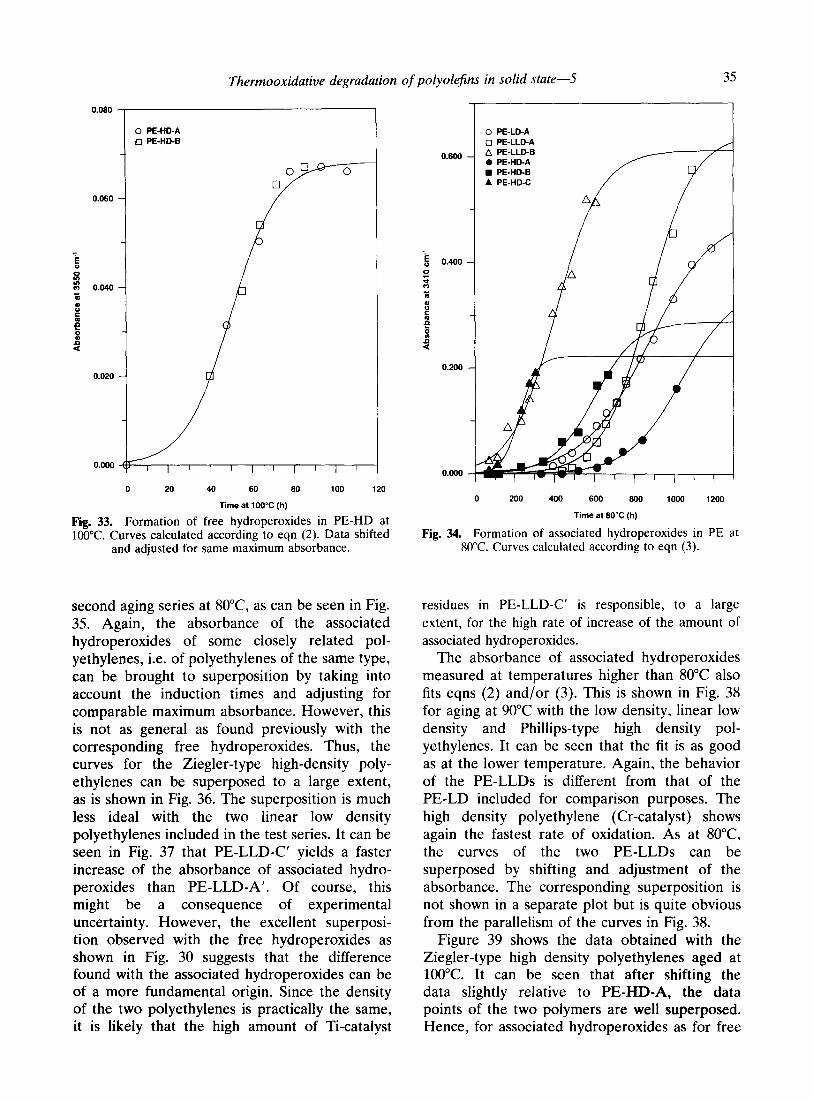
Thermooxidative degradation of polyolefins in solid state-5 35
0.060
0.060
i
5: 3 0.040 6 8 5 e 2 9
0.020
0 PE-HD-A
•I PE-HD-B
O.ooofT , , , , , , , , , , ,
0 20 40 60 60 100 120
Time at 100°C (h)
Fig. 33. Formation of free hydroperoxides in PE-HD at 100°C. Curves calculated according to eqn (2). Data shifted
and adjusted for same maximum absorbance.
second aging series at 8O”C, as can be seen in Fig. 35. Again, the absorbance of the associated hydroperoxides of some closely related pol- yethylenes, i.e. of polyethylenes of the same type, can be brought to superposition by taking into account the induction times and adjusting for comparable maximum absorbance. However, this is not as general as found previously with the corresponding free hydroperoxides. Thus, the curves for the Ziegler-type high-density poly- ethylenes can be superposed to a large extent, as is shown in Fig. 36. The superposition is much less ideal with the two linear low density polyethylenes included in the test series. It can be seen in Fig. 37 that PE-LLD-C’ yields a faster increase of the absorbance of associated hydro- peroxides than PE-LLD-A’. Of course, this might be a consequence of experimental uncertainty. However, the excellent superposi- tion observed with the free hydroperoxides as shown in Fig. 30 suggests that the difference found with the associated hydroperoxides can be of a more fundamental origin. Since the density of the two polyethylenes is practically the same, it is likely that the high amount of Ti-catalyst
0.606
0 PE-LD-A
~1 PE-LLD-A
0 200 400 600 600 1000 1200
Time at 60°C (h)
Fig. 34. Formation of associated hydroperoxides in PE at 80°C. Curves calculated according to eqn (3).
residues in PE-LLD-C’ is responsible, to a large extent, for the high rate of increase of the amount of associated hydroperoxides.
The absorbance of associated hydroperoxides measured at temperatures higher than 80°C also fits eqns (2) and/or (3). This is shown in Fig. 38 for aging at 90°C with the low density, linear low density and Phillips-type high density pol- yethylenes. It can be seen that the fit is as good as at the lower temperature. Again, the behavior of the PE-LLDs is different from that of the PE-LD included for comparison purposes. The high density polyethylene (Cr-catalyst) shows again the fastest rate of oxidation. As at 8O”C, the curves of the two PE-LLDs can be superposed by shifting and adjustment of the absorbance. The corresponding superposition is not shown in a separate plot but is quite obvious from the parallelism of the curves in Fig. 38.
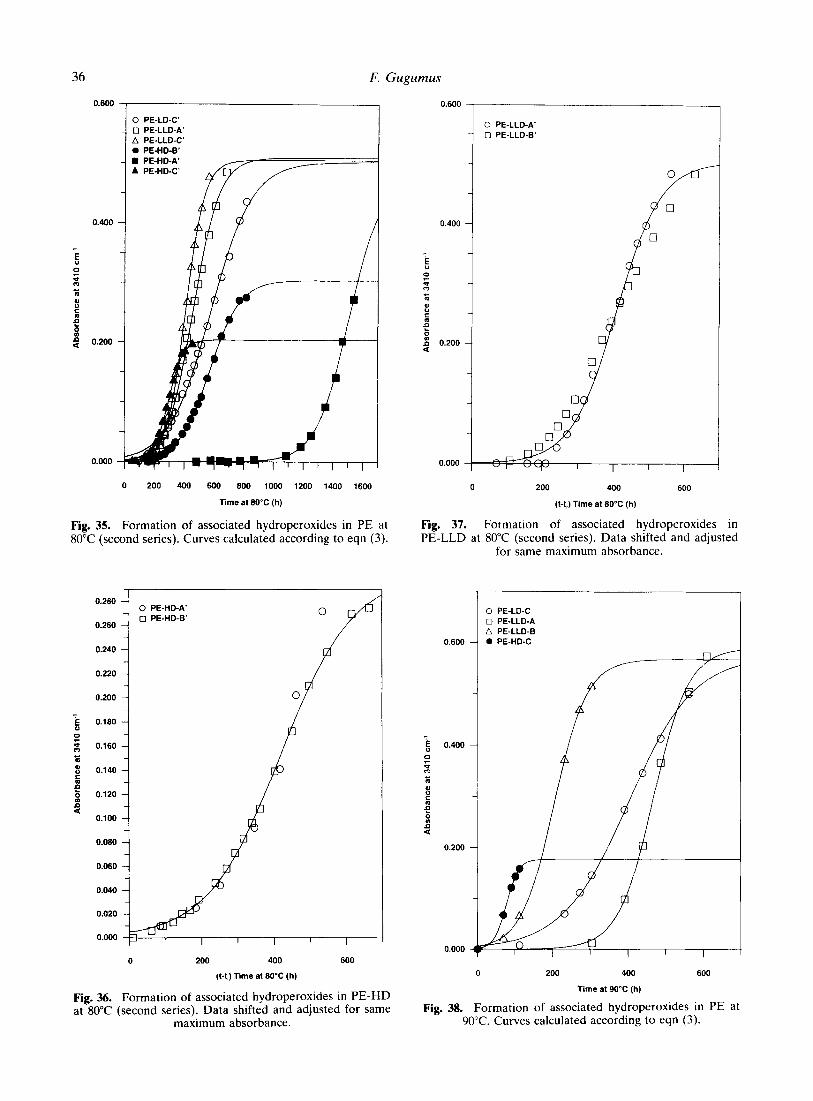
Figure 39 shows the data obtained with the Ziegler-type high density polyethylenes aged at 100°C. It can be seen that after shifting the data slightly relative to PE-HD-A, the data points of the two polymers are well superposed. Hence, for associated hydroperoxides as for free
36
0.600
0.400
g
z .z 6 :: 6 0
8 :: 0.200
F. Gugumus
/
0 PE-LD-C’
0 PE-LLD-A’
A PE-LLD-C’
l PE-HD-B’
0 200 400 600 600 1000 1200 1400 160C
Time at 80°C (h)
Fig. 35. Formation of associated hydroperoxides in PE at Fig. 37. Formation of associated hydroperoxides in 80°C (second series). Curves calculated according to eqn (3). PE-LLD at 80°C (second series). Data shifted and adjusted
for same maximum absorbance.
0.280
I
0 PE-HD-A’
0.260 III PE-HD-B’
0.240
0.220
0.180
0.080
P i
0 200 400 600
(t-t,) Time at 80°C (h)
Fig. 36. Formation of associated hydroperoxides in PE-HD at 80°C (second series). Data shifted and adjusted for same
maximum absorbance.
0.600
0 PE-LLD-A’
0 PE-LLD-B’
0.400
200 400 600
(t-tJ Time at 80°C (h)
0.600
0 PE-LD-C
0 PE-LLD-A
A PE-LLD-B
0 PE-HD-C
400
Time at 90°C (h)
600
Fig. 38. Formation of associated hydroperoxides in PE at 90°C. Curves calculated according to eqn (3).

Thermooxidative degradation of polyolefins in solid state-5 37
hydroperoxides, the behavior already observed at 80°C is also valid at 100°C.
The decrease of the concentration of as- sociated hydroperoxides after the maximum can be explained qualitatively the same way as previously for the associated hydroperoxides in PE-LD.4 This is caused by the fact that there is not only accumulation of non-initiating associa- tions hydroperoxide/carbonyl groups but also decomposition of such associations without formation of free radicals. Therefore, the rate of initiation is constantly decreasing whereas the rate of hydroperoxide decomposition is increas- ing with advancing oxidation. As a consequence, the overall concentration of hydroperoxides is decreasing more or less rapidly.
4.2 Heterogeneous oxidation kinetics for carbonyl groups
It is assumed that the chemical reactions occurring in polyethylene are summarized by the reactions shown in Scheme 1. It is thought that the reactions proceed in the individual amor- phous domains envisaged as more or less independent chemical reactors. As pointed out previously,4 the main difference between Scheme 1 and conventional homogeneous kinetics con-
k&W D=
d k :k
9a 96
kza + k,,
b#-‘W E=
J k >k (k2” + cl2
Yh
terns the initiation reactions: the bimolecular hydroperoxide decomposition reactions are re- placed by the pseudo-monomolecular decom- positions of the association complexes between a hydroperoxide group and a polyethylene segment or between two hydroperoxide groups. It is understood that the other reactions proceed under the same conditions. This means that the reacting groups are necessarily close enough spatially to be able to react. If this is not so, it is mandatory that at least one of the reacting groups is a low molecular mass fragment.
It has been shown4 that, according to the formal heterogeneous kinetics, the rate of formation of carbonyl groups in an amorphous domain becomes constant when the concentra- tion of initiating associated hydroperoxides becomes constant. Then, the amount of carbonyl groups in an amorphous domain can be expressed by the following equation:
(P=O)=Dt-E (6)
where the constants D and E are related to yf, the maximum value of the free hydroperoxide concentration in an amorphous domain, and the rate constants of the reactions in Scheme 1 by the expressions:
I
[k,,.(PH) + kal J k
90
:k Yh
k&b k&9, k :k
kjo(PH) + k,, + kg, + kw, 1
ksh(PH) 9a 9b
kzo + k,,
k,
k:,(PH)‘k :,
(k, + l:J’
9b
2[kO’W + &RI J k
90 :k
9h
ksh(PH) + 3 kzh +
[ kzcrk.v, kz,ks,
k,,(PH) + kv, + k,, + k9, I J k “:;,
k2, + k::
9h
+;k, J
k 211
k:,(PH)’ k “:; 9h
k9, + k9, (kz,, + k:)’
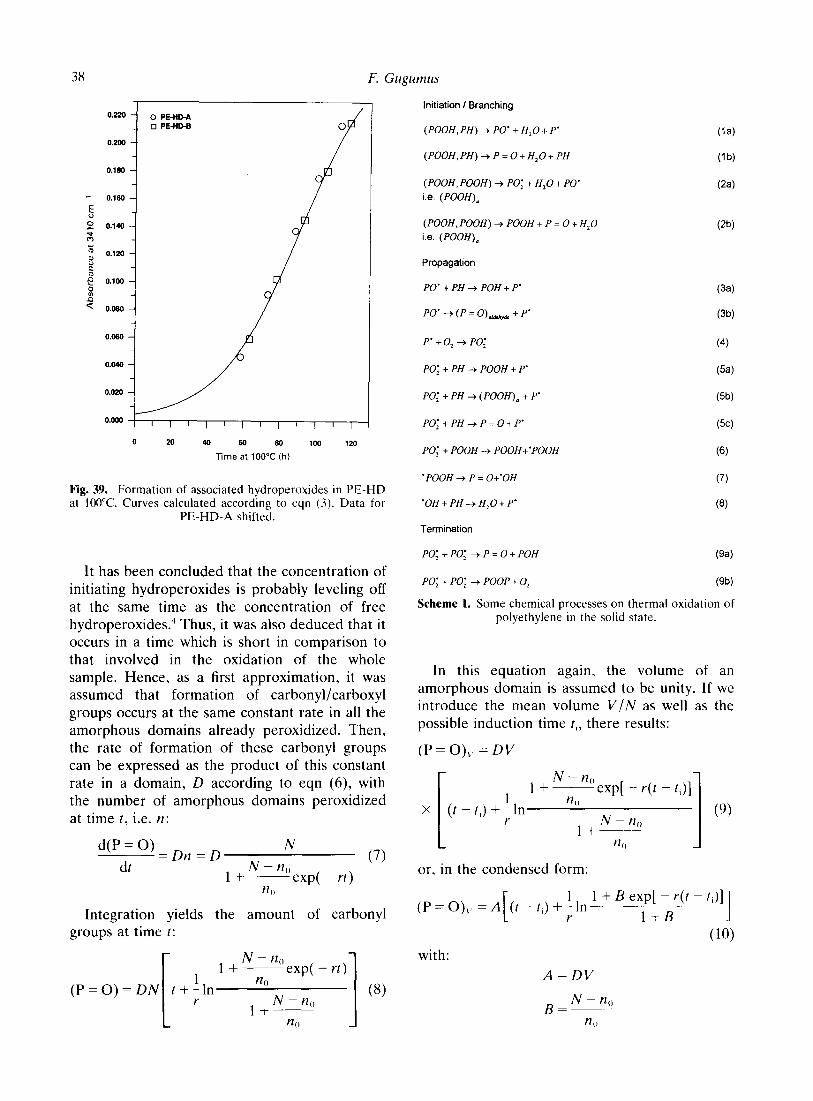
38 F. Gugumus
0 PE-HD-A 0 PE-HD-B
0 20 40 a0 a0 100 120
lime at 100°C (h)
Fig. 39. Formation of associated hydroperoxides in PE-HD at 100°C. Curves calculated according to eqn (3). Data for
PE-HD-A shifted.
It has been concluded that the concentration of initiating hydroperoxides is probably leveling off at the same time as the concentration of free hydroperoxides.’ Thus, it was also deduced that it occurs in a time which is short in comparison to that involved in the oxidation of the whole sample. Hence, as a first approximation, it was assumed that formation of carbonyl/carboxyl groups occurs at the same constant rate in all the amorphous domains already peroxidized. Then, the rate of formation of these carbonyl groups can be expressed as the product of this constant rate in a domain, D according to eqn (6), with the number of amorphous domains peroxidized at time t, i.e. n:
d(P = 0) N =Dn=D
dt N - no c-9
1+- exp( - rt) no
Integration yields the amount of carbonyl groups at time t:
, + N - no ~ exp( - rt) 4)
N - no 1 (8) 1+- no
Initiation / Branching
(POOH,PH)~PO'+H,O+P'
(POOH,PH)-tP=O+H,O+PH
(la)
(lb)
(POOH.POOH)-tPO; +H,O+PO'
i.e. (POOH), (2a)
(POOH,POOH)-+POOH+P=O+H,O i.e. (POOH),
Propagation
PO'+PH+POH+P'
PO' -r(P=O),,, +P'
(2b)
(W
(3b)
P'+O, jpo; (4)
PO;+PH_,POOH+P' (5a)
PO; iPH+(POOH), +P' (5b)
PO;+PH-tP=O+P' (5c)
PO;+POOH-+POOH+'POOH (6)
'POOH_,P=O+'OH
'OH+PH~H,O+P'
Termination
PO;+PO;+P=O+POH
(7)
(8)
(W
po;+po; ~PooP+o, (9b)
Scheme 1. Some chemical processes on thermal oxidation of polyethylene in the solid state.
In this equation again, the volume of an amorphous domain is assumed to be unity. If we introduce the mean volume V/N as well as the possible induction time t,, there results:
(P = O),, = DV
X
N - no 1+---
1 exp[ - r(t - 4)l
(t - t,) + -In 4
Y N - no 1+---
fl,l I (9)
or, in the condensed form:
(p = O),, = /+ - t,) + ; In l + B ex,Pi;+ - i)l]
with:
(10)
A=DV
B = N - no
4,
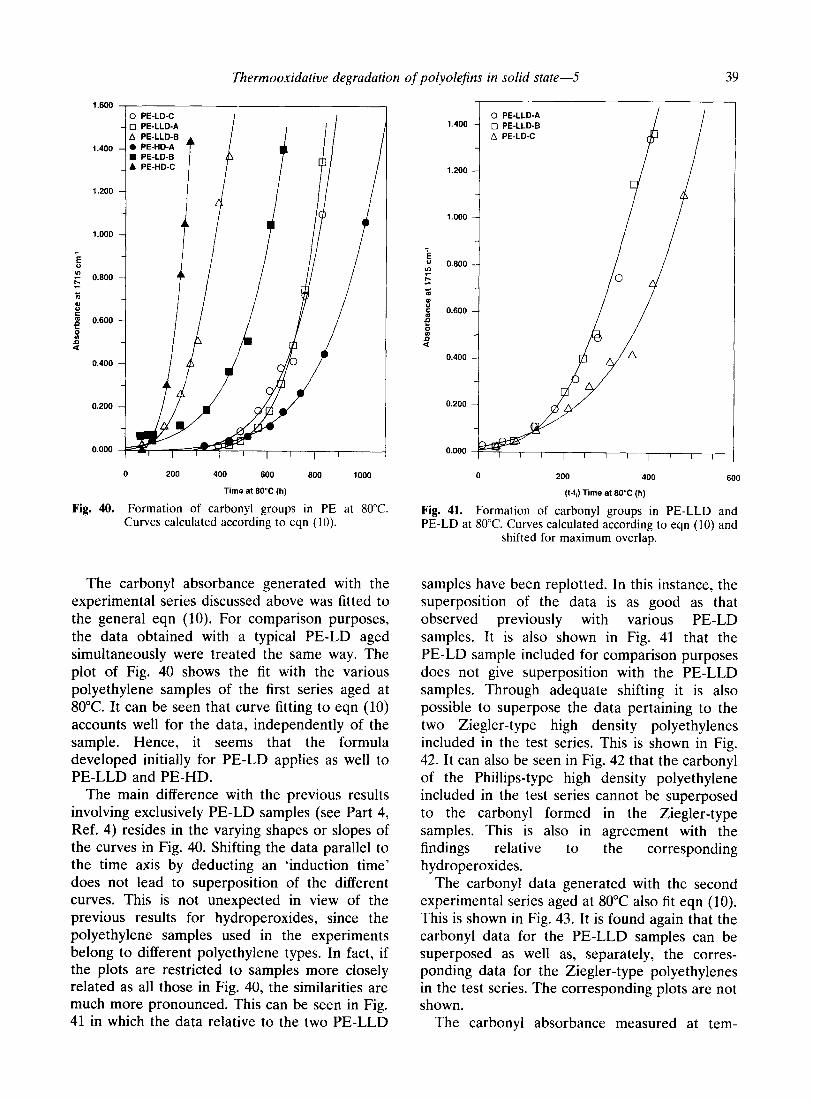
Thermooxidative degradation of polyolefins in solid state-5
1.600
0 200 400 600
Time at 60°C (h)
600 1000
Fig. 40. Formation of carbonyl groups in PE at 80°C. Curves calculated according to eqn (10).
The carbonyl absorbance generated with the experimental series discussed above was fitted to the general eqn (10). For comparison purposes, the data obtained with a typical PE-LD aged simultaneously were treated the same way. The plot of Fig. 40 shows the fit with the various polyethylene samples of the first series aged at 80°C. It can be seen that curve fitting to eqn (10) accounts well for the data, independently of the sample. Hence, it seems that the formula developed initially for PE-LD applies as well to PE-LLD and PE-HD.
The main difference with the previous results involving exclusively PE-LD samples (see Part 4, Ref. 4) resides in the varying shapes or slopes of the curves in Fig. 40. Shifting the data parallel to the time axis by deducting an ‘induction time’ does not lead to superposition of the different curves. This is not unexpected in view of the previous results for hydroperoxides, since the polyethylene samples used in the experiments belong to different polyethylene types. In fact, if the plots are restricted to samples more closely related as all those in Fig. 40, the similarities are much more pronounced. This can be seen in Fig. 41 in which the data relative to the two PE-LLD
0 PE-LLD-A
0 PE-LLD-B
A PE-LD-C
k : 0.600
E
%
z 5 0.600
e
%
8
0.400
0 200 400
(t-t,) Time at 60°C (h)
600
Fig. 41. Formation of carbonyl groups in PE-LLD and PE-LD at 80°C. Curves calculated according to eqn (10) and
shifted for maximum overlap.
samples have been replotted. In this instance, the superposition of the data is as good as that observed previously with various PE-LD samples. It is also shown in Fig. 41 that the PE-LD sample included for comparison purposes does not give superposition with the PE-LLD samples. Through adequate shifting it is also possible to superpose the data pertaining to the two Ziegler-type high density polyethylenes included in the test series. This is shown in Fig. 42. It can also be seen in Fig. 42 that the carbonyl of the Phillips-type high density polyethylene included in the test series cannot be superposed to the carbonyl formed in the Ziegler-type samples. This is also in agreement with the findings relative to the corresponding hydroperoxides.
The carbonyl data generated with the second experimental series aged at 80°C also fit eqn (10). This is shown in Fig. 43. It is found again that the carbonyl data for the PE-LLD samples can be superposed as well as, separately, the corres- ponding data for the Ziegler-type polyethylenes in the test series. The corresponding plots are not shown.
The carbonyl absorbance measured at tem-
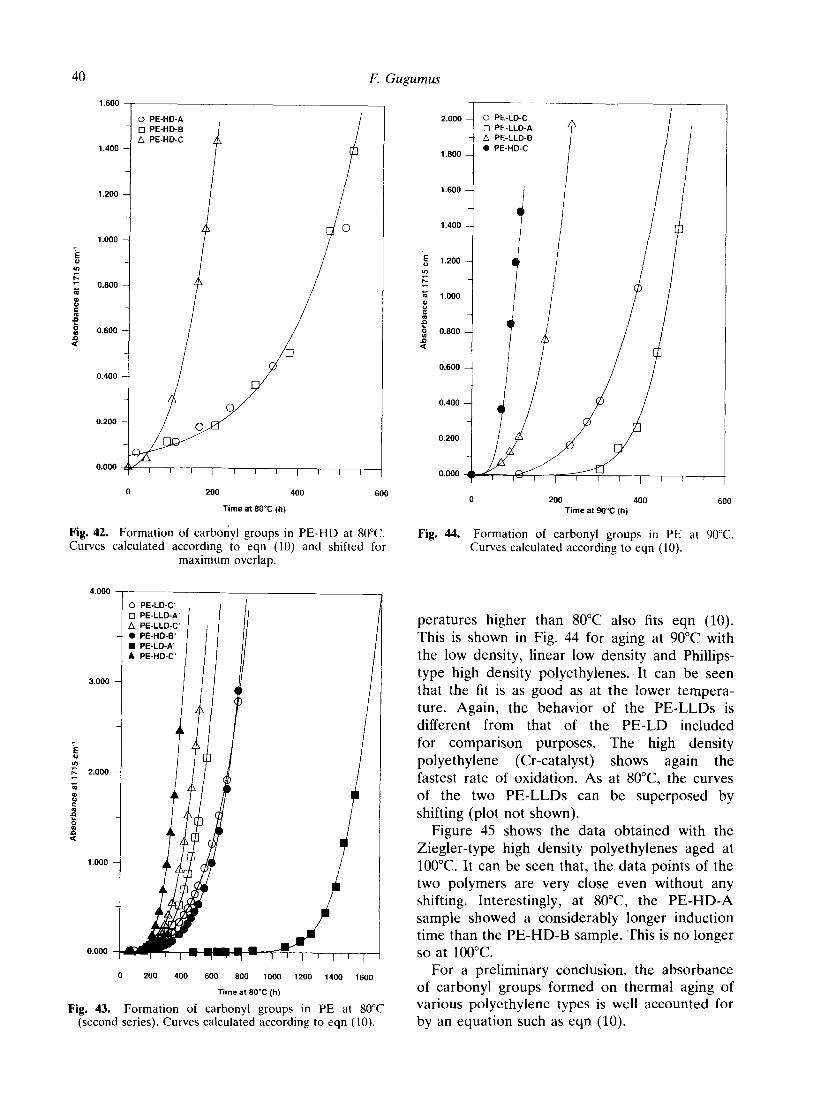
40
1.600
1.400
1.200
1.000
i
: $
z 0.600
E d
$ 0.600
9
0.400
0.200
0.000
F. Gugumus
0 200 400 600
Time at 80°C (h)
Fig. 42. Formation of carbonyl groups in PE-HD at 80°C. Curves calculated according to eqn (10) and shifted for
maximum overlap.
4.000
3.000
i
u) K 2.000
x 3 5 8 2 4
1.000
0.000
0 PE-LO-C’
0 PE-LLD-A’
A PE-LLD-C’
0 PE-HD-E’
n PE-LD-A’ A PE-HD-C’
0 200 400 600 600 1000 1200 1400 1600 For a preliminary conclusion, the absorbance
Time at 60°C (h) of carbonyl groups formed on thermal aging of
Fig. 43. Formation of carbonyl groups in PE at 80°C various polyethylene types is well accounted for (second series). Curves calculated according to eqn (10). by an equation such as eqn (10).
1.600 -
1.400 -
0.600 -
0.400 -
0.200 -
0 200 400 600 Time at 90°C (h)
Fig. 44. Formation of carbonyl groups in PE at 90°C. Curves calculated according to eqn (10).
peratures higher than 80°C also fits eqn (10). This is shown in Fig. 44 for aging at 90°C with
the low density, linear low density and Phillips- type high density polyethylenes. It can be seen that the fit is as good as at the lower tempera- ture. Again, the behavior of the PE-LLDs is different from that of the PE-LD included for comparison purposes. The high density polyethylene (Cr-catalyst) shows again the fastest rate of oxidation. As at 8O”C, the curves of the two PE-LLDs can be superposed by shifting (plot not shown).
Figure 45 shows the data obtained with the Ziegler-type high density polyethylenes aged at 100°C. It can be seen that, the data points of the two polymers are very close even without any shifting. Interestingly, at SO”C, the PE-HD-A sample showed a considerably longer induction time than the PE-HD-B sample. This is no longer so at 100°C.
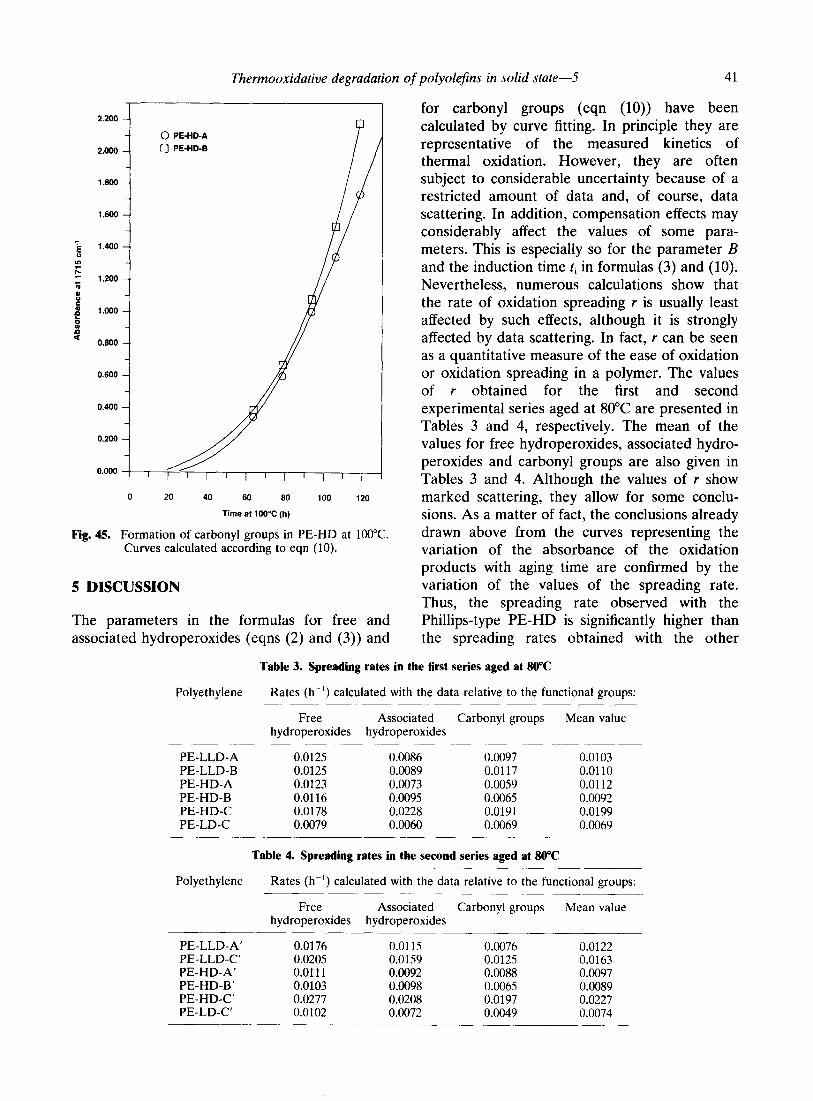
Thermooxidative degradation of polyolefins in solid state-5 41
0 PEHD-A
q PE-HD-B
0.600
0.000 1 , /, ( , , , , ,
0 20 40 60 60 100 120
Time at 100°C (h)
I
Fig. 45. Formation of carbonyl groups in PE-HD at 1OOT. Curves calculated according to eqn (10).
5 DISCUSSION
The parameters in the formulas for free and associated hydroperoxides (eqns (2) and (3)) and
for carbonyl groups (eqn (10)) have been calculated by curve fitting. In principle they are representative of the measured kinetics of thermal oxidation. However, they are often subject to considerable uncertainty because of a restricted amount of data and, of course, data scattering. In addition, compensation effects may considerably affect the values of some para- meters. This is especially so for the parameter B and the induction time ti in formulas (3) and (10). Nevertheless, numerous calculations show that the rate of oxidation spreading r is usually least affected by such effects, although it is strongly affected by data scattering. In fact, r can be seen as a quantitative measure of the ease of oxidation or oxidation spreading in a polymer. The values of r obtained for the first and second experimental series aged at 80°C are presented in Tables 3 and 4, respectively. The mean of the values for free hydroperoxides, associated hydro- peroxides and carbonyl groups are also given in Tables 3 and 4. Although the values of r show marked scattering, they allow for some conclu- sions. As a matter of fact, the conclusions already drawn above from the curves representing the variation of the absorbance of the oxidation products with aging time are confirmed by the variation of the values of the spreading rate. Thus, the spreading rate observed with the Phillips-type PE-HD is significantly higher than the spreading rates obtained with the other
Table 3. Spreading rates in the first series aged at WC
Polyethylene Rates (h-l) calculated with the data relative to the functional groups:
Free Associated Carbonyl groups Mean value hydroperoxides hydroperoxides
PE-LLD-A 0.0125 0.0086 0.0097 0.0103 PE-LLD-B 0.0125 0.0089 0.0117 0.0110 PE-HD-A 0.0123 0.0073 0.0059 0.0112 PE-HD-B 0.0116 0.0095 0.0065 0.0092 PE-HD-C 0.0178 0.0228 0.0191 0.0199 PE-LD-C 0.0079 0.0060 0.0069 0.0069
Table 4. Spreading rates in the second series aged at 80°C _ ~____
Polyethylene Rates (h-l) calculated with the data relative to the functional groups: -___
Free Associated Carbonyl groups Mean value hydroperoxides hydroperoxides
PE-LLD-A’ 0.0176 0.0115 0.0076 0.0122 PE-LLD-C’ 0.0205 0.0159 0.0125 0.0163 PE-HD-A’ 0.0111 0.0092 0.0088 0.0097 PE-HD-B’ 0.0103 0.0098 0.0065 0.0089 PE-HD-C’ 0.0277 0.0208 0.0197 0.0227 PE-LD-C’ 0.0102 0.0072 0.0049 0.0074

42 F. Gugumus
polyethylenes in the test series, including polyethylenes with comparable or lower crystalli- nity. The high spreading rate as well as the limited induction time observed even at 80°C are clearly indicative of the determining role of the Cr-catalyst residues in the ease of oxidation of this polymer type. The accelerating effect of Ti-catalyst residues on oxidation spreading is clearly visible from the comparison of PE-LLD with PE-LD. Although the polymer densities/crystallinities involved are very close, the spreading rate is systematically higher with PE-LLD than with PE-LD. This is valid for the first as well as for the second experimental series. This effect of Ti-catalyst residues can be seen although the amount of Ti is usually limited to 2-3 mg/kg. However, the sample with a much higher amount of Ti residues, PE-LLD-C’, also shows a much higher spreading rate than the other linear low density polyethylenes. In fact, it comes relatively close to the PE-HD-C’ sample containing Cr-catalyst residues.
The Ziegler-type PE-HDs show mean spread- ing rates comparable to or lower than those of the PE-LLD samples. This is all the more remarkable since the samples tested usually contain relatively high amounts of Ti-residues. It is concluded that the high crystallinity of these polyethylenes strongly reduces the oxidation spreading rate. This more than compensates for the relatively high Ti content.
The individual values of the oxidation spread- ing rate deduced from the data for the free hydroperoxides, associated hydroperoxides and carbonyl groups for a single polymer should be the same, within experimental error. It can be seen in Tables 3 and 4 that this is not so, even allowing for reasonable scattering. Furthermore, a close look at the data reveals some trends. Thus, the spreading rates Y deduced from the absorbance of the associated hydroperoxides are almost systematically lower than the correspond- ing values deduced from the absorbance of the free hydroperoxides. In fact, there is only one exception, PE-HD-C in Table 3. Very often, there is a further decrease of the values of Y on passing from the values deduced from the associated hydroperoxides to the values deduced from the carbonyl data. It can be seen in Table 4 that this decrease is systematic with the second experimental series.
It is not yet clear if the deviations have a chemical or a physical origin. As a matter of fact,
variations in the composition of the carbonyl groups and of the associated hydroperoxides might induce more or less strong deviations. With respect to the physical aspects, it is clear that imperfections of the model may have a significant impact on the results. Possible reasons for the deviations are currently being investigated.
The induction time of thermal oxidation depends heavily on the polymer and/or polymer sample. There are not many straightforward conclusions to be drawn from the data accumu- lated so far. There is, at least, an indication that polyethylenes with Cr-catalyst residues show relatively short induction periods. Similarly, with high amounts of Ti-residues, possibly inadequ- ately deactivated, the induction time also becomes short. However, PE-LLD with some Ti-residues may present an induction period which is longer than that of a typical PE-LD without any catalyst residues. One of the high density polyethylenes in the test series shows exceptionally long induction times. Of course, this could be caused by the presence of small amounts of stabilizers. However, no phenolic antioxidant could be detected by the usual analysis methods. It is obvious that a very low amount of hydroperoxide groups present initially in the polymer will also prolong the induction time. The same is certainly true if the Ti-catalyst residues have been well deactivated.
6 CONCLUSIONS
The experimental kinetics of oxidation product formation on thermal oxidation of PE-LLD and PE-HD do not correspond to the rate laws reported in the literature. Instead of quadratic or biquadratic formation of carbonyl groups with oxidation time, the rate law seems to be more complicated. As observed previously with PE- LD, there is an exponential-type increase in the early stages of the oxidation process, followed by a linear increase at the later stages.
The heterogeneous oxidation model and the corresponding kinetics developed for PE-LD are also valid for other polyethylenes. They include PE-LLDs as well as Ziegler and Phillips-type PE-HDs. The differences between the poly- ethylene types concern mainly the rate of oxidation, which seems to be most heavily affected by the nature of the polyethylene. In this respect, polymer density/crystallinity as well as

Thermooxidative degradation of polyolefins in solid state-5 43
the manufacturing process are important. Thus, oxidation model may need amendments. This will increasing density seems to lead to decreasing be the object of future work. rates of oxidation spreading. The catalyst residues appear to be even more important. If small amounts of Ti-catalyst residues have only a
REFERENCES
slightly accelerating effect, Cr-catalyst residues increase considerably the rate of spreading from
i. 3:
one amorphous domain to the next. The thermal oxidation data fit well to the
heterogeneous oxidation kinetics developed for PE-LD. However, a close look at the calculated values of the oxidation spreading rates shows the limits of the agreement with the theoretical model. Hence, some of the chemical and/or physical characteristics of the heterogeneous
Gugumus, F.. Polym. Degrad. Stab., 52 (1996) 131. Gugumus, F.. Polym. Degrad. Stab., 52 (1996) 145. Gugumus, F., Polym. Degrad. Stab., 52 (1996) 159. Gugumus, F.. Polym. Degrad. Stab., 53 (1996) 161. Gugumus, F., In Oxidation Inhibition in Organic Materials, eds P.P. Klemchuk & J. Pospisil. CRC Press Inc., Boca Raton, FL, 1990, Vol. 1, p. 61. Gugumus, F., In Plastics Additives, 3rd edn, eds R. Gaechter & H. Mueller. Hanser Publishers, Munich, 1990, p. 1. Gugumus, F.. Polym. Degrad. Stab., 49 (1995) 29. Leatherbarrow, R.J., GraFit Version 2.0. Erithacus Software Ltd, Staines, U.K., 1990.




















