
The study of genetically-tractable model organisms has advanced our knowledge of the molecular...
16
OLADOLAPO BELLO The study of genetically-tractable model organisms has advanced our knowledge of the molecular control of cellular function and these two disciplines underpin our understanding of cancer. Background Cancer is used to describe a large group of diseases affecting any part of the body which are capable of enabling the rapid proliferation of abnormal cells (malignant tumours) which exceed their boundaries and may invade other parts of the body. Genetically-tractable animal models of diseases have been of immense importance in the understanding of a number of human disease mechanisms (including cancer) through the elucidation of the genes, molecules and pathways involved in cellular function. Some of the models that have been employed specifically in cancer research include the Fruit-fly ( Drosophila melanogaster ), Zebrafish ( Danio rerio ), and the mouse ( Mus musculus ). Why Genetically-tractable animal models? In addition to characteristics such as high rates of fecundity and ease of manipulation and storage, these models have specific features that make them suitable for use in research. The development of genetic techniques that allow quick and easy identification and characterisation of genes involved in the formation and development of tumours make Drosophila a very good model organism for the study of cancer development and there is a high level of conservation of genes and pathways between the fruit-fly and humans (Potter et al ., 2000). Fish are also able to develop tumours which bear similarities with cancers, and genetic (forward & reverse) and small molecule screens, construction of transgenic models and disruption of specific genes can all be carried out in 1
Transcript of The study of genetically-tractable model organisms has advanced our knowledge of the molecular...

OLADOLAPO BELLO
The study of genetically-tractable model organisms has advanced our
knowledge of the molecular control of cellular function and these
two disciplines underpin our understanding of cancer.
Background
Cancer is used to describe a large group of diseases affecting any
part of the body which are capable of enabling the rapid
proliferation of abnormal cells (malignant tumours) which exceed
their boundaries and may invade other parts of the body.
Genetically-tractable animal models of diseases have been of immense
importance in the understanding of a number of human disease
mechanisms (including cancer) through the elucidation of the genes,
molecules and pathways involved in cellular function. Some of the
models that have been employed specifically in cancer research
include the Fruit-fly (Drosophila melanogaster), Zebrafish (Danio rerio),
and the mouse (Mus musculus).
Why Genetically-tractable animal models?
In addition to characteristics such as high rates of fecundity and
ease of manipulation and storage, these models have specific
features that make them suitable for use in research. The
development of genetic techniques that allow quick and easy
identification and characterisation of genes involved in the
formation and development of tumours make Drosophila a very good model
organism for the study of cancer development and there is a high
level of conservation of genes and pathways between the fruit-fly
and humans (Potter et al., 2000). Fish are also able to develop
tumours which bear similarities with cancers, and genetic (forward &
reverse) and small molecule screens, construction of transgenic
models and disruption of specific genes can all be carried out in
1
the fish, which make it an important and promising system in cancer
research (Stern & Zon, 2003). Also, it is possible to genetically
engineer mice with chemicals or retroviruses for different specific
cancer models, manipulate the levels and patterns of expression of
genes and even knockout genes in mice (Ma, 2004).
Discoveries arising from studies of genetically-tractable animal models
Shh Pathway
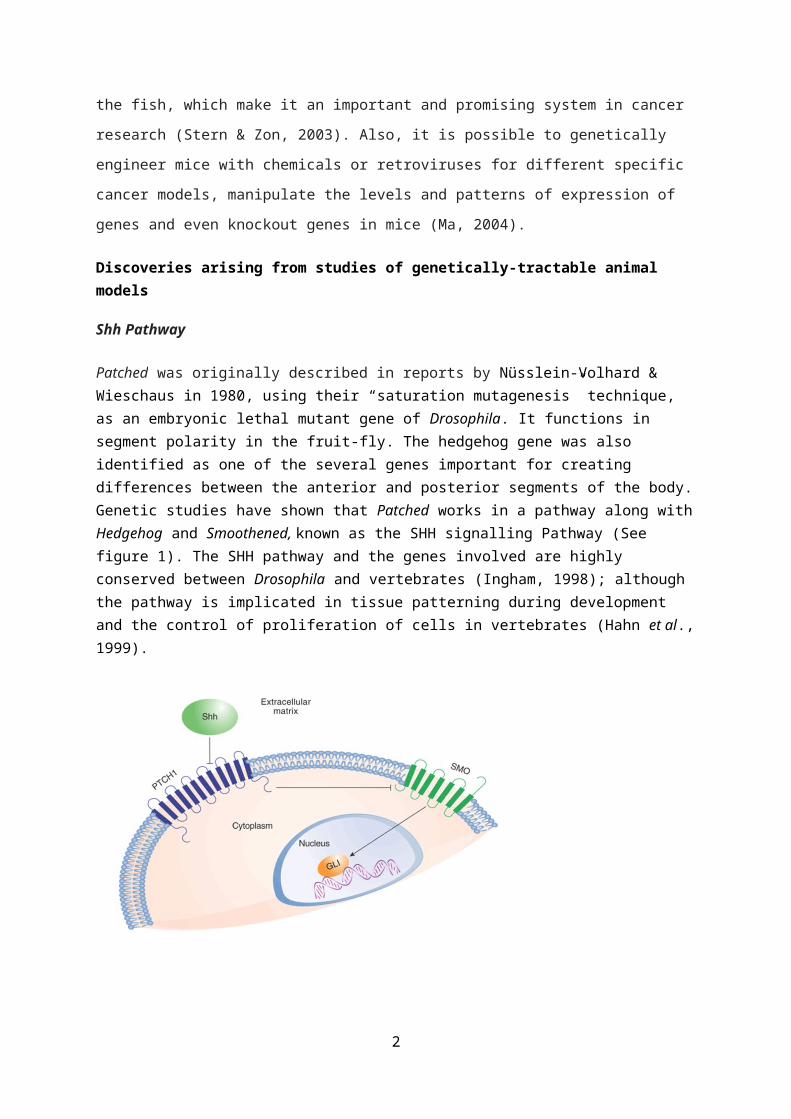
Patched was originally described in reports by Nüsslein-Volhard & Wieschaus in 1980, using their “saturation mutagenesis” technique, as an embryonic lethal mutant gene of Drosophila. It functions in segment polarity in the fruit-fly. The hedgehog gene was also identified as one of the several genes important for creating differences between the anterior and posterior segments of the body.Genetic studies have shown that Patched works in a pathway along withHedgehog and Smoothened, known as the SHH signalling Pathway (See figure 1). The SHH pathway and the genes involved are highly conserved between Drosophila and vertebrates (Ingham, 1998); although the pathway is implicated in tissue patterning during development and the control of proliferation of cells in vertebrates (Hahn et al.,1999).
2

Fig 1: The Sonic Hedgehog Signalling Pathway (Adapted from: Sheridan, 2009)
- In the Sonic hedgehog signalling pathway, binding of Shh to patched (a 12-span
transmembrane receptor protein) prevents the normal inhibition of smoothened (a 7-
span transmembrane protein) by patched. In the absence of the Shh ligand, the
pathway is turned off by Ptch which inhibits Smo, thus inhibiting the signalling of
downstream molecules (Villavicencio et al., 2000). The Ptch gene functions as a tumour
suppressor and activating Smo mutants function as oncogenes. Ptch and Smo gene
mutations have all been detected in basal cell carcinomas and medulloblastomas.
It was initially discovered that inhibition of the Hedgehog pathway
leads to cyclopia and holoprosencephaly (Chiang et al., 1996), then
it was later found that over-activation of the Hedgehog signalling
pathway promotes Basal Cell Carcinoma and that this overexpression
is caused by either a loss of function mutation in patched or an
activated smoothened mutation. Experiments were carried out using
transgenic mice using a promoter (K5) to drive smoothened mutation
and it was observed that non-keratinised and keratohyalin granules
were absent from the stratum granulosum (Xie et al., 1998), similarly
observed in earlier Mice BCC expression experiments which drove
overexpression of Shh in the skin (Oro, 1997). It was discovered
that Cyclopamine, a plant-derived compound, inhibits the activity of
HedgeHog and prevents abnormal cell growth (Taipale et al., 2000) and
Vismodegib, a drug closely related to Cyclopamine has now been
approved by the FDA for the treatment of advanced and metastatic
forms of Basal Cell carcinoma (Dubey et al., 2013)
Wnt Signalling
3

In 1982, Nusse & Varmus proposed that Mouse Mammary Tumour Virus
(MMTV) was capable of inducing tumours in mammary glands of mice by
activating the expression of a gene called Int1. Some researchers
also reported that DNA from carcinomas induced by MMTV further
induced carcinomas in other cells and that a DNA of human mammary
cancer cell line possessed a similar neoplastic transformation
potential. Nusse & Varmus hypothesized that the oncogenesis by MMTV
was dependent on the insertion of this proviral DNA in a specific
region of the host genome.
Fig 2: Insertion of MMTV into host genome leading to strong expression of
tumorigenic potential.
They tested their hypothesis by cloning integration sites from a new
provirus, isolating the uninterrupted MMTV int1 domain with the
integration site, and by using probes to detect rearrangements
within Int1 in different carcinomas. They found that although the
infection by proviral DNA of the normal mammary cells introduced the
proviral DNA into many sites in host genome, oncogenesis was more
likely to occur in cells that acquired a provirus within int1, and
4
they thus concluded that the integration of MMTV DNA in target cells
showed a certain regional specificity for the int1 domain.
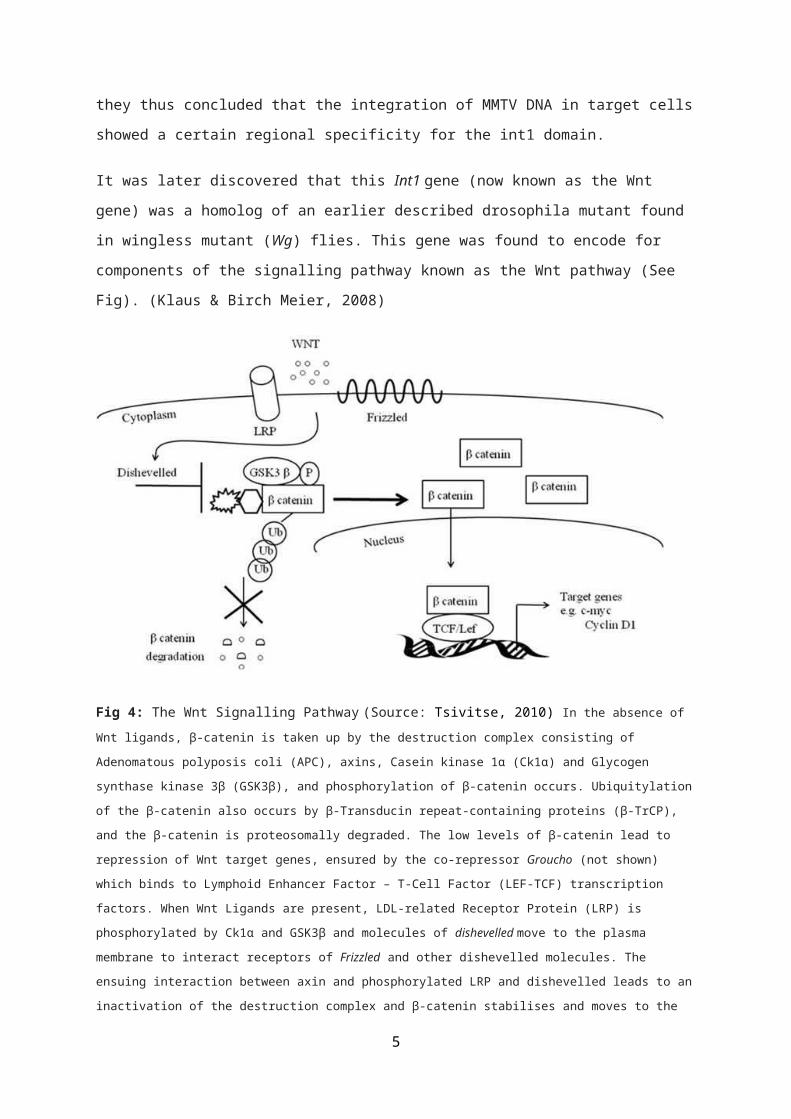
It was later discovered that this Int1 gene (now known as the Wnt
gene) was a homolog of an earlier described drosophila mutant found
in wingless mutant (Wg) flies. This gene was found to encode for
components of the signalling pathway known as the Wnt pathway (See
Fig). (Klaus & Birch Meier, 2008)
Fig 4: The Wnt Signalling Pathway (Source: Tsivitse, 2010) In the absence of Wnt ligands, β-catenin is taken up by the destruction complex consisting of
Adenomatous polyposis coli (APC), axins, Casein kinase 1α (Ck1α) and Glycogen
synthase kinase 3β (GSK3β), and phosphorylation of β-catenin occurs. Ubiquitylation
of the β-catenin also occurs by β-Transducin repeat-containing proteins (β-TrCP),
and the β-catenin is proteosomally degraded. The low levels of β-catenin lead to
repression of Wnt target genes, ensured by the co-repressor Groucho (not shown)
which binds to Lymphoid Enhancer Factor – T-Cell Factor (LEF-TCF) transcription
factors. When Wnt Ligands are present, LDL-related Receptor Protein (LRP) is
phosphorylated by Ck1α and GSK3β and molecules of dishevelled move to the plasma
membrane to interact receptors of Frizzled and other dishevelled molecules. The
ensuing interaction between axin and phosphorylated LRP and dishevelled leads to an
inactivation of the destruction complex and β-catenin stabilises and moves to the
5

nucleus. In the nucleus, β-catenin binds to the LEF-TCF complex, displacing Groucho
and allowing transcription of Wnt target genes (Klaus & Birchmeier, 2008). Note: APC
is the unlabelled hexagonal structure next to β-catenin in the diagram.
It has now been shown that APC, a component of the Wnt pathway acts
as a tumour suppressor gene and it is commonly accepted that the
suppressor abilities of APC stem from its ability to destabilise
free B-catenin (Harada et al., 1999). It was also shown that APC loss
directly results in increased size of the proliferating crypt
compartment of the small intestine (Bienz and Clevers, 2000) and may
be implicated in about 85% of hereditary colorectal tumours (Kinzler
& Vogelstein, 1996) (See Fig). It is suggested that the
indiscriminate activation of TCF4 by stable B-catenin in the absence
of APC leads to this expansion of the crypt.
Fig 5: Adenomatous polyp formation in APC
mutant mice
A – Normal cells of the epithelium (crypt
cells in yellow; Differentiated villi
cells in blue)
B & C – Pouch outpocket formation in zone
of proliferation of crypt
D - F – Formation of microadenoma inside
a single villus G –
Microadenoma expansion into neighbouring
villus H – Formation of a
multi-villus polyp by the fusion of
multiple adenomas which are in the
process of rupturing through villus
6

epithelium to break into the lumen of the gut. Adapted from: Bienz & Clevers
(2000)
Zebrafish and Cancer
Cancer is commonly seen in wild fish and assays that involve
exposure to water-borne carcinogens show that fishes can develop a
wide variety of tumours (whether benign or malignant) in almost all
organs of their bodies (Spitsbergen et al., 2000). Also, fishes can
get tumours similar to human tumours and the cell cycle genes,
tumour suppressors and oncogenes in fish and humans are similarly
conserved. An advantage of this is that forward genetic screens can
be targeted to these highly conserved pathways and carcinogenesis
assays enable us test whether a mutation causing an embryonic
phenotype may induce predisposition to cancer in adults (Amatruda et
al., 2002).
In 2005, Shepard et al., used forward genetic screens in Zebrafish
embryos to screen for cell cycle mutants using anti-PO4 Histone H3
antibodies which marked cells that were undergoing cellular
division. An increase in the number of cells was observed in a
mutant known as crb, where it was discovered that the cells were
blocked in the M-phase of mitosis. Homozygous crb mutants also
showed an increase in apoptosis. The associated gene was
positionally cloned and named b-myb. Crb mutants represented a loss of
function in the b-myb gene, which suggested that b-myb was a tumour
7

suppressor. As a follow-up to these experiments, Stern et al., (2005)
developed a chemical screen using zebrafish embryos to test for
small molecules that interact with the b-myb pathway. They crossed
crb heterozygotes (since the homozygous was embryonic lethal), and
arrayed them in multiwell plates and screened them with a 16,000
compound chemical library. It was observed that a previously unknown
compound, persynthamide, could rescue the phenotype, suppressing b-
myb mitotic defects.
SETDB1 & Melanoma (Zebrafish)
BRAF (V600E) is the most common mutation in Melanoma where it
activates BRAF and causes excessive activity in the Protein Kinase
pathway (Davies et al., 2002). However, it is also found in benign
naevi of melanocytes, which suggests that there are additional
genetic occurrences leading to melanoma Tumorigenesis. In 2011, Ceol
et al carried out experiments on zebrafish to test genes that were
recurrently amplified on a region of chromosome 1 for their
associative abilities with the BRAF (v600E) mutation in the
acceleration of melanoma using DNA sequence and gene expression
analyses. They used a transgenic fish where the oncogene,
BRAF(V600E) was expressed by the melanoma-specific promoter mitfa. In
a p53-/- background, this led to tumours but when the mitfa gene was
removed, the melanocytes died and the fish were normal. Using an
expression plasmid containing mitfa and several candidate genes which
was injected in the eggs of such fish lines, they discovered that
SETDB1, an enzyme that methylates histone on lysine9 (H3K9),
increased the rate of melanoma formation and identified SETDB1 as an
oncogene in melanoma in Zebrafish. Experiments were also carried out
using chromatin immuno-precipitation in conjunction with DNA
sequencing and analyses of gene expression and they some genes (such
as the HOX genes) were also discovered which are dsyregulated
8

transcriptionally, as a direct result of the increased levels of
SETDB1. These experiments show that SETDB1 is an oncogene in
melanoma formation in Zebrafish, and they also signify the
importance of chromatin factors in the regulation of Tumorigenesis.
Nodal signaling and Melanoma (Zebrafish)
Nodal ligands were initially identified by forward genetic screens
in both mice & Zebrafish. In 1986, researchers who were carrying out
a retroviral insertational mutagenesis screen in embryonic stem
cells of mice (Robertson et al., 1986). Through this screen, a mutant,
induced retrovirally, was discovered which showed defects in early
gastrulation and the locus that corresponded was discovered to be a
member of the superfamily of TGFβ ligands that often express in
mammalian nodes (Zhou et al., 1993). Ligands related to nodal were
also subsequently discovered in other species. Similarly, in 1998,
genetic screens in Zebrafish demonstrated the functions in
gastrulation of two nodal-related genes, Cyclops (cyc) and squint that
were isolated as loss-of-function alleles (Feldman et al., 1998).
In 2006, Topczewska et al., found that the nodal gene is expressed by
aggressive melanoma cells and may be highly functional in tumour
development. This was identified by studying the interactions of the
cancer cells with embryonic progenitors in zebrafish. Fluorescently-
labelled melanoma cells transplanted in zebrafish induced ectopic
structures while a control group did not. It was also observed that
the melanoma cells induced a notochord in these ectopic structures.
Again, they observed that many embryos had a duplicated embryonic
axis. Nodal protein was suggested to be implicated. The embryos were
then
9

stained for markers expressed in response to high and low levels of
nodal protein and it was found that the melanoma cells induced the
expression of both goosecoid (which is only expressed with high levels
of nodal) and notail. While the melanoma cells induced ectopic
structures, only a subset of the melanoma cells expressed nodal and
it was hypothesised that these were the actual cancer cells. To test
their observations, they tested several melanoma lines for the
expression of nodal. When two cell lines derived from the same
origin were used, it was observed that one was aggressive (MUM2B)
and the other was benign producing low levels of nodal. They
observed that nodal expression had a positive correlation with
melanoma tumour progression and there was more nodal protein in the
melanoma cancer cell metastases than in the primary tumours. It was
predicted that inhibiting the nodal pathway will lead to a decrease
in cell invasion and the reduced formation of the vasculogenic
network by downregulation of Keratin expression and VE cadherin.
They inhibited nodal expression using a morpholino and observed that
it resulted in the elimination of the axis-inducing properties of
the cell and it led to melanin formation in tumours and a reduction
in the volume of melanoma formed.
Fig 6: Graph showing reduction in tumour volume as a result of inhibition
of nodal signalling (Source: Topczewska et al., 2006)
10
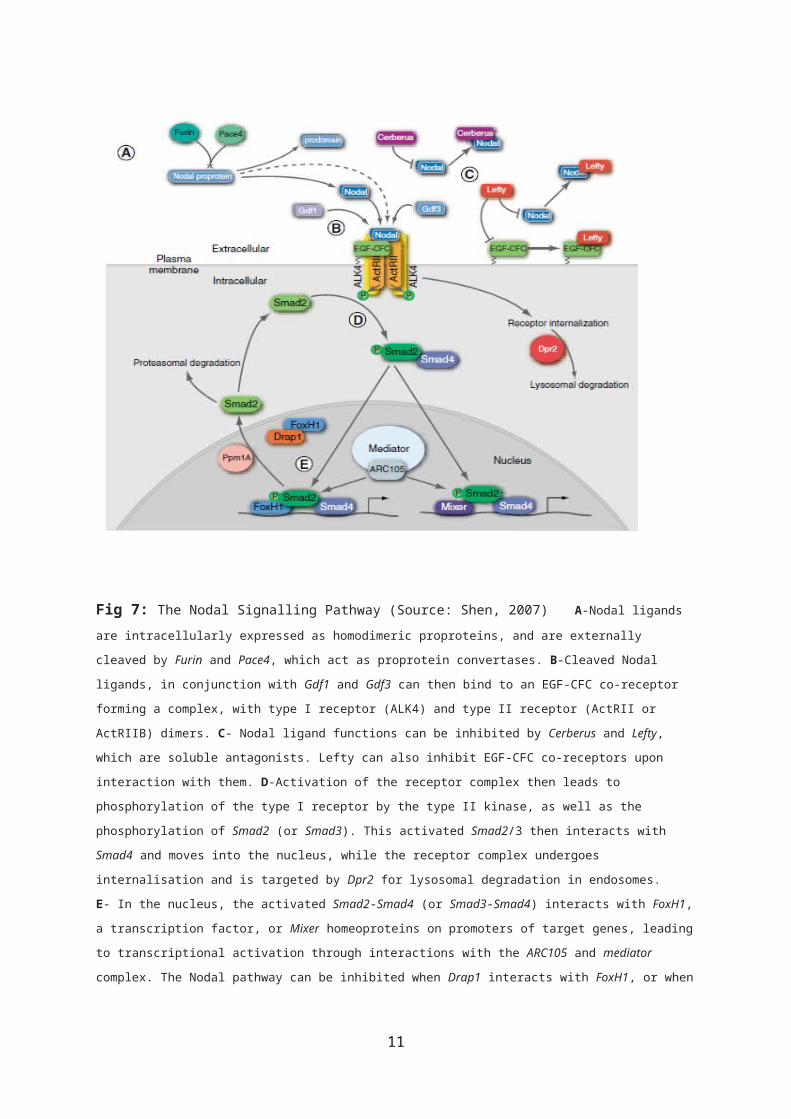
Fig 7: The Nodal Signalling Pathway (Source: Shen, 2007) A-Nodal ligands are intracellularly expressed as homodimeric proproteins, and are externally
cleaved by Furin and Pace4, which act as proprotein convertases. B-Cleaved Nodal
ligands, in conjunction with Gdf1 and Gdf3 can then bind to an EGF-CFC co-receptor
forming a complex, with type I receptor (ALK4) and type II receptor (ActRII or
ActRIIB) dimers. C- Nodal ligand functions can be inhibited by Cerberus and Lefty,
which are soluble antagonists. Lefty can also inhibit EGF-CFC co-receptors upon
interaction with them. D-Activation of the receptor complex then leads to
phosphorylation of the type I receptor by the type II kinase, as well as the
phosphorylation of Smad2 (or Smad3). This activated Smad2/3 then interacts with
Smad4 and moves into the nucleus, while the receptor complex undergoes
internalisation and is targeted by Dpr2 for lysosomal degradation in endosomes.
E- In the nucleus, the activated Smad2-Smad4 (or Smad3-Smad4) interacts with FoxH1,
a transcription factor, or Mixer homeoproteins on promoters of target genes, leading
to transcriptional activation through interactions with the ARC105 and mediator
complex. The Nodal pathway can be inhibited when Drap1 interacts with FoxH1, or when
11

Ppm1A, a Smad phosphatase, promotes the nuclear export of Smad2, possibly targeting
it for proteasomal degradation (Shen, 2007).
Limitations of genetically-tractable animal models
While there have been significant advances in cancer research as a
result of broadened understanding of cellular function through the
experimental studies of genetically-tractable model organisms,
significant genetic, immunological and molecular differences exist
between animal and human systems and results gotten from animal
systems must be translated with caution. Also, animal models may
only show distinct processes or sets of processes within a disease
and may not display the entire array of changes in physiology that
take place in the disease being modelled (Hoyte et al., 2004).
12

References
Amatruda, J., Shepard, J., Stern, H. and Zon, L. (2002). Zebrafish as a cancer model system.Cancer Cell, 1(3), pp.229-231.
Bienz, M. and Clevers, H. (2000). Linking Colorectal Cancer to Wnt Signaling. Cell, 103(2), pp.311-320.
Chiang, C., Litingtung, Y., Lee, E., Young, K., Corden, J., Westphal, H. and Beachy, P. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature, 383(6599), pp.407-413.
Ceol, C., Houvras, Y., Jane-Valbuena, J., Bilodeau, S., Orlando, D.,Battisti, V., Fritsch, L., Lin, W., Hollmann, T., Ferré, F., Bourque, C., Burke, C., Turner, L., Uong, A., Johnson, L., Beroukhim, R., Mermel, C., Loda, M., Ait-Si-Ali, S., Garraway, L., Young, R. and Zon, L. (2011). The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature, 471(7339), pp.513-517.
Davies, H., Bignell, G., Cox, C., Stephens, P., Edkins, S., & Clegg,S. et al. (2002). Mutations of the BRAF gene in human cancer. Nature, 417(6892), 949-954. doi:10.1038/nature00766
Dubey, A., Dubey, S., Handu, S. and Qazi, M. (2013). Vismodegib: Thefirst drug approved for advanced and metastatic basal cell carcinoma. Journal of Postgraduate Medicine, 59(1), p.48.
Feldman, B., Gates, M. A., Egan, E. S., Dougan, S. T., Rennebeck, G., Sirotkin, H. I., ... & Talbot, W. S. (1998). Zebrafish organizer development and germ-layer formation require nodal-related signals. Nature, 395(6698), 181-185.
Hahn, H., Wojnowski, L., Miller, G. and Zimmer, A. (1999). The patched signaling pathway in tumorigenesis and development: lessons from animal models. Journal of Molecular Medicine, 77(6), pp.459-468.
Harada, N. (1999). Intestinal polyposis in mice with a dominant stable mutation of the beta -catenin gene. The EMBO Journal, 18(21), pp.5931-5942.
13

Hoyte, L., Kaur, J. and Buchan, A. (2004). Lost in translation: taking neuroprotection from animal models to clinical trials. Experimental Neurology, 188(2), pp.200-204.
Ingham, P. (1998). Transducing Hedgehog: the story so far. The EMBO Journal, 17(13), pp.3505-3511.
Kinzler, K., & Vogelstein, B. (1996). Lessons from Hereditary Colorectal Cancer. Cell, 87(2), 159-170. doi:10.1016/s0092-8674(00)81333-1
Klaus, A. and Birchmeier, W. (2008). Wnt signalling and its impact on development and cancer.Nat Rev Cancer, 8(5), pp.387-398.
Ma, C. (2004). Animal Models of Disease. Modern Drug Discovery, 7(6), pp.30 -36.
Nusse, R. and Varmus, H. (1982). Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same regionof the host genome. Cell, 31(1), pp.99-109.
Nüsslein-Volhard, C. and Wieschaus, E. (1980). Mutations affecting segment number and polarity in Drosophila. Nature, 287(5785), pp.795-801.
Oro, A. (1997). Basal Cell Carcinomas in Mice Overexpressing Sonic Hedgehog. Science, 276(5313), pp.817-821.
Potter, C., Turenchalk, G. and Xu, T. (2000). Drosophila in cancer research. Trends in Genetics, 16(1), pp.33-39.
Robertson, E., Bradley, A., Kuehn, M., & Evans, M. (1986). Germ-linetransmission of genes introduced into cultured pluripotential cells by retroviral vector. Nature, 323(6087), 445-448. doi:10.1038/323445a0
Rudrapatna, V., Cagan, R. and Das, T. (2011). Drosophila cancer models. Developmental Dynamics, 241(1), pp.107-118.
Sheridan, C. (2009). Genentech obtains proof of concept for hedgehoginhibition. Nat Biotechnol, 27(11), 968-969. doi:10.1038/nbt1109-968
Shen, M. (2007). Nodal signaling: developmental roles and regulation. Development, 134(6), 1023-1034. doi:10.1242/dev.000166
14

Shepard, J., Amatruda, J., Stern, H., Subramanian, A., Finkelstein, D., & Ziai, J. et al. (2005). A zebrafish bmyb mutation causes genome instability and increased cancer susceptibility. Proceedings Of The National Academy Of Sciences, 102(37), 13194-13199. doi:10.1073/pnas.0506583102
Spitsbergen, J., Tsai, H., Reddy, A., Miller, T., Arbogast, D., Hendricks, J. and Bailey, G. (2000). Neoplasia in Zebrafish (Danio rerio) Treated with 7,12-Diniethylbenz[a]anthracene by Two Exposure Routes at Different Developmental Stages. Toxicologic Pathology, 28(5), pp.705-715.
Stern, H. and Zon, L. (2003). Cancer genetics and drug discovery in the zebrafish. Nat Rev Cancer, 3(7), pp.533-539.
Stern, H., Murphey, R., Shepard, J., Amatruda, J., Straub, C., & Pfaff, K. et al. (2005). Small molecules that delay S phase suppress azebrafish bmyb mutant. Nature Chemical Biology, 1(7), 366-370. doi:10.1038/nchembio749
Taipale, J., Chen, J., Beachy, P., Cooper, M., Wang, B., Mann, R., Milenkovic, L. and Scott, M. (2000). Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature, 406(6799), pp.1005-1009.
Topczewska, J., Postovit, L., Margaryan, N., Sam, A., Hess, A., Wheaton, W., Nickoloff, B., Topczewski, J. and Hendrix, M. (2006). Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nat Med, 12(8), pp.925-932.
Tsivitse, S. (2010). Notch and Wnt Signaling, Physiological Stimuli and Postnatal Myogenesis.International Journal of Biological Sciences, pp.268-281.
Villavicencio, E., Walterhouse, D., & Iannaccone, P. (2000). The Sonic Hedgehog–Patched–Gli Pathway in Human Development and Disease. The American Journal Of Human Genetics, 67(5), 1047-1054. doi:10.1016/s0002-9297(07)62934-6
Xie, J., de Sauvage, F., Murone, M., Luoh, S., Ryan, A., Gu, Q., Zhang, C., Bonifas, J., Lam, C., Hynes, M., Goddard, A., Rosenthal, A. and Jr, E. (1998). Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature, 391(6662), pp.90-92.
15

Zhou, X., Sasaki, H., Lowe, L., Hogan, B., & Kuehn, M. (1993). Nodalis a novel TGF-β-like gene expressed in the mouse node during gastrulation. Nature, 361(6412), 543-547. doi:10.1038/361543a0
16




















