
The Molecular Basis of Enzymatic Catalysis - Projects at Harvard
12
e Molecular Basis of Enzymatic Catalysis 7 In earlier chapters we learned about the factors that influence the rates of chemical reactions, and we saw that many reactions that are essential for life occur extremely slowly. Consequently, living systems require catalysts to accelerate the rates of biochemical reactions so that they occur on a timescale that is consistent with life. We will see numerous examples of biological catalysts, or enzymes, including DNA polymerase, RNA polymerase, and kinases in later chapters. Nearly all enzymes are proteins, but some, as we will see in Chapter 13, are made of RNA instead. We will explore the arrow pushing mechanisms used by some enzymes (such as the kinase mechanism), but we first have to establish how enzymes are able to accelerate reaction rates. In this chapter we will explore the molecular basis of enzymatic catalysis to understand how and why certain reactions occur faster with the help of a catalyst. Enzymes accelerate reactions by lowering ΔG ‡ Enzymes do not affect ΔG° rxn ; instead, they increase reaction rates by lowering the activation energy, ΔG ‡ . As we saw in Chapter 4, an amount of free energy equal to ΔG ‡ must be added to substrates in order to form the transition state, which then proceeds to form products. e difference between a fast reaction and one that occurs slowly is simply the value of ΔG ‡ ; the larger the activation energy barrier, the more energy must be infused into the substrates before they can become products, and therefore the slower the reaction. Conversely, reactions with low ΔG ‡ values proceed quickly. • why enzymes are necessary for life. • how enzymes lower ΔG ‡ by stabilizing transition states. • how enzymes lower ΔG ‡ by using proximity and orientation effects. • how enzymes enhance the nucleophilicity and electrophilicity of reactants. Aſter this chapter, you should be able to explain: To understand how enzymes accelerate the rates of chemi- cal reactions. Objectives Goal
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of The Molecular Basis of Enzymatic Catalysis - Projects at Harvard

The Molecular Basisof Enzymatic Catalysis
7
In earlier chapters we learned about the factors that influence the rates of chemical reactions, and we saw that many reactions that are essential for life occur extremely slowly. Consequently, living systems require catalysts to accelerate the rates of biochemical reactions so that they occur on a timescale that is consistent with life. We will see numerous examples of biological catalysts, or enzymes, including DNA polymerase, RNA polymerase, and kinases in later chapters. Nearly all enzymes are proteins, but some, as we will see in Chapter 13, are made of RNA instead. We will explore the arrow pushing mechanisms used by some enzymes (such as the kinase mechanism), but we first have to establish how enzymes are able to accelerate reaction rates. In this chapter we will explore the molecular basis of enzymatic catalysis to understand how and why certain reactions occur faster with the help of a catalyst.
Enzymes accelerate reactions by lowering ΔG‡
Enzymes do not affect ΔG°rxn; instead, they increase reaction rates by lowering the activation energy, ΔG‡. As we saw in Chapter 4, an amount of free energy equal to ΔG‡ must be added to substrates in order to form the transition state, which then proceeds to form products. The difference between a fast reaction and one that occurs slowly is simply the value of ΔG‡; the larger the activation energy barrier, the more energy must be infused into the substrates before they can become products, and therefore the slower the reaction. Conversely, reactions with low ΔG‡ values proceed quickly.
• why enzymes are necessary for life.
• how enzymes lower ΔG‡ by stabilizing transition states.
• how enzymes lower ΔG‡ by using proximity and orientation effects.
• how enzymes enhance the nucleophilicity and electrophilicity of reactants.
After this chapter, you should be able to explain:
To understand how enzymes accelerate the rates of chemi-cal reactions.
Objectives
Goal

The Molecular Basis of Enzymatic CatalysisChapter 7 2
Enzymes lower ΔG‡ by allowing reactions to proceed via an alternate reaction mechanism that has a lower ΔG‡ than the uncatalyzed reaction. The substrates and products, and therefore their free energy values, are the same for both the catalyzed and uncatalyzed reactions, thus ΔG°rxn is the same for both reactions. Since the catalyzed and uncatalyzed reactions proceed via different mechanisms, however, the structures of the transition states are not the same for both reactions. Therefore, the free energies of the transition states are not the same either. The transition state of the catalyzed reaction is more stable; therefore, the catalyzed reaction has a smaller ΔG‡ and proceeds faster.
At the molecular level, an enzyme-catalyzed reaction unfolds as shown in Figure 1. First the enzyme randomly encounters the substrate in solution. Occasionally such an encounter will take place in a manner that allows the enzyme to bind to the substrate, forming an enzyme-substrate complex. When bound to the enzyme, the substrate experiences a precisely tailored environment that facilitates the substrate’s transformation into the transition state of the reaction. Rather than simply binding to the active site of the enzyme, the substrate also rearranges itself upon binding to more closely resemble the transition state. Similarly, the enzyme also undergoes slight conformational changes to better accommodate the substrate. This creates more favorable interactions between the substrate and active site and is called the induced fit model of enzyme catalysis. The energy released by forming these favorable interactions contributes to lowering the ΔG‡ in enzyme-catalyzed reactions.
The enzyme-stabilized transition state can then undergo additional changes to become enzyme-bound product. Since the substrate already more closely resembles the transition state, less energy is needed to push
substratebinding
catalyzedreaction
productrelease
free enzyme
product
substrate
[TS]‡
enzyme-substratecomplex
enzyme-productcomplex
Figure 1 Enzymes bind to their substrates to catalyze chemical re-actionsIn order for an enzyme to catalyze a chemical reaction it must first bind to its substrate to form an enzyme-substrate complex. The enzyme stabilizes the reaction’s transition state, making it easier for the bound substrate to form the transition state and convert to product. The resulting enzyme-product complex then dissociates, releasing free product and regenerating the free enzyme, which can carry out subsequent rounds of catalysis.
The Molecular Basis of Enzymatic CatalysisChapter 7 3
the substrate to product. The enzyme-product complex then dissociates into free product and free enzyme. The released enzyme is then ready to catalyze the conversion of another molecule of substrate into product. It is important to note that the enzyme is not changed by the reaction; when the reaction is complete, the enzyme is in the same state as before the reaction began.
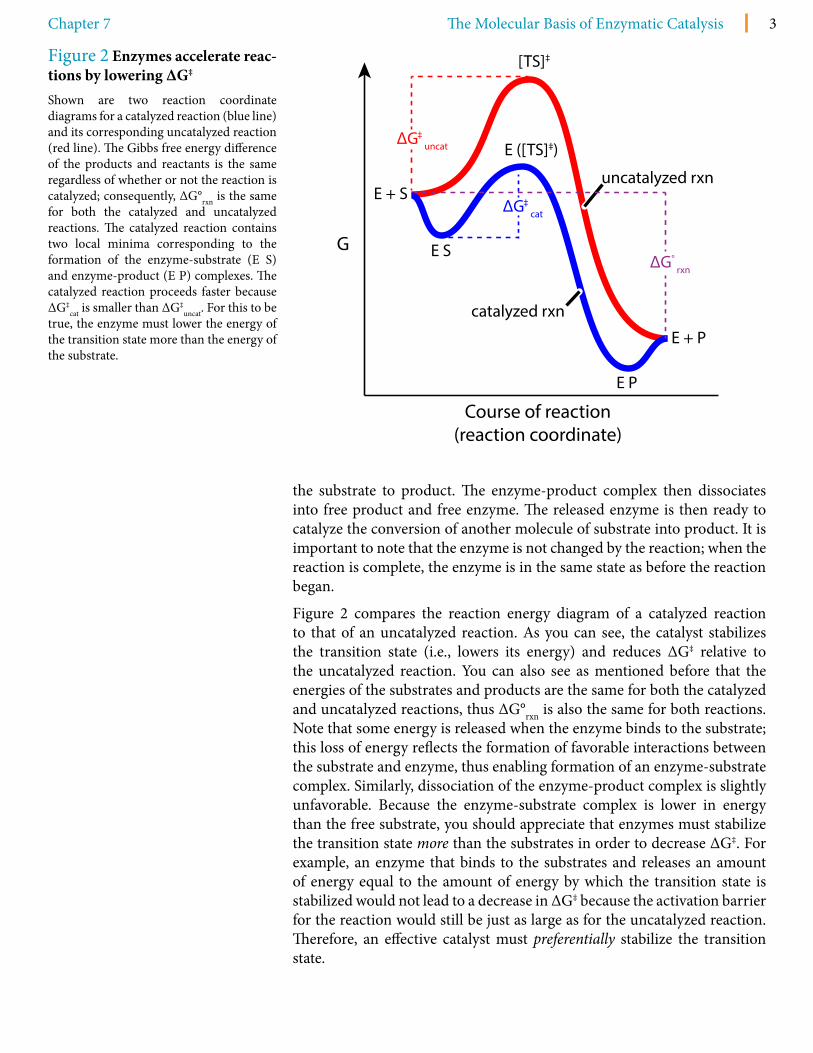
Figure 2 compares the reaction energy diagram of a catalyzed reaction to that of an uncatalyzed reaction. As you can see, the catalyst stabilizes the transition state (i.e., lowers its energy) and reduces ΔG‡ relative to the uncatalyzed reaction. You can also see as mentioned before that the energies of the substrates and products are the same for both the catalyzed and uncatalyzed reactions, thus ΔG°rxn is also the same for both reactions. Note that some energy is released when the enzyme binds to the substrate; this loss of energy reflects the formation of favorable interactions between the substrate and enzyme, thus enabling formation of an enzyme-substrate complex. Similarly, dissociation of the enzyme-product complex is slightly unfavorable. Because the enzyme-substrate complex is lower in energy than the free substrate, you should appreciate that enzymes must stabilize the transition state more than the substrates in order to decrease ΔG‡. For example, an enzyme that binds to the substrates and releases an amount of energy equal to the amount of energy by which the transition state is stabilized would not lead to a decrease in ΔG‡ because the activation barrier for the reaction would still be just as large as for the uncatalyzed reaction. Therefore, an effective catalyst must preferentially stabilize the transition state.
Figure 2 Enzymes accelerate reac-tions by lowering ΔG‡
Shown are two reaction coordinate diagrams for a catalyzed reaction (blue line) and its corresponding uncatalyzed reaction (red line). The Gibbs free energy difference of the products and reactants is the same regardless of whether or not the reaction is catalyzed; consequently, ΔG°rxn is the same for both the catalyzed and uncatalyzed reactions. The catalyzed reaction contains two local minima corresponding to the formation of the enzyme-substrate (E S) and enzyme-product (E P) complexes. The catalyzed reaction proceeds faster because ΔG‡
cat is smaller than ΔG‡uncat. For this to be
true, the enzyme must lower the energy of the transition state more than the energy of the substrate.
∆G‡ cat
Course of reaction(reaction coordinate)
G
E + S
E + P
E P
E S
[TS]‡
E ([TS]‡)
catalyzed rxn
uncatalyzed rxn
∆G‡ uncat
∆G° rxn

The Molecular Basis of Enzymatic CatalysisChapter 7 4
ΔG‡ can be expressed in terms of ΔH‡ and ΔS‡
To understand enzymatic catalysis we need to remind ourselves of the factors that influence reaction rate. As we have learned, the rate of a reaction is equal to the concentration of starting material multiplied by the rate constant (k). The rate constant accounts for all factors that influences rate except for reactant concentration; therefore, the rate constant accounts for molecular velocity, reaction cross-section, and the probability that molecules will collide with enough energy and in the correct orientation to react (Figure 3). Enzymes increase reaction rate by increasing the value of the rate constant, which is related to ΔG‡ by the Arrhenius equation that we saw in Chapter 4:
We can also express ΔG‡ in terms of ΔH‡ and ΔS‡ (ΔG‡ = ΔH‡ - TΔS‡). The ΔH‡ and ΔS‡ terms represent the changes in enthalpy and entropy, respectively, between the substrates and transition state (e.g., ΔH‡ = HTS – Hsubstrates). The ΔS‡ term relates to the reaction cross-section and the probability that molecules will collide in the correct orientation to react. Enzymes affect ΔS‡ by reducing the number of conformations the substrate can adopt. The ΔH‡ term relates to the probability that molecules collide with enough energy to react. Temperature, as we have discussed already, affects molecular velocity and the probability that molecules collide with enough energy to react. As we will see, enzymes can decrease ΔG‡ by affecting ΔH‡ and ΔS‡.
Enzymes use proximity and orientation effects to increase reaction rate
To accelerate chemical reactions, enzymes make use of a variety of molecular strategies, often with a remarkable effectiveness that is unmatched by analogous catalysts developed in the laboratory. A major strategy, called the
k = A e-∆G‡
R T
Figure 3 ΔH‡ and ΔS‡ relate to specific factors that affect reaction rateShown in the center row are the factors that affect reaction rate. All factors except for reactant concentration are included in the rate constant. Reaction cross-section and the probability that molecules collide in the right orientation determine ΔS‡, whereas the probability that molecules collide with enough energy to react determines ΔH‡.
rate constant(k)
ΔH‡ ΔS‡
ReactionRate
=Probability that
molecules collide inthe right orientation
Probability thatmolecules collide with
enough energy to reactx xx xReactant
concentrationMolecular
velocityReaction
cross-section

The Molecular Basis of Enzymatic CatalysisChapter 7 5
proximity effect, is to organize the substrates within the active site of the enzyme such that the reactants are much closer together than they would be in a typical solution. Enhancing the proximity of reactants increases their collision frequency, thus causing the reaction to proceed at a faster rate. The proximity effect effectively increases the local concentration of substrate (recall that rate is proportional to substrate concentration).
As an example of the proximity effect in catalysis, consider the rates of the two hypothetical reactions shown in Figure 4. The reaction at the top relies on the random collision between the two substrates to bring A and B close enough to react. In contrast, it is much more likely for A and B to encounter each other in the reaction at the bottom when they are already tethered together.
In addition to proximity effect, a related but distinct strategy used by enzymes to accelerate chemical reactions is to orient substrates into a maximally reactive conformation; this is known as the orientation effect. Simply confining two substrates close to one another does not guarantee a faster reaction rate because in order for two substrates to react they usually must achieve a specific relative orientation. This is similar to you finding your friend in a dining hall. Not only do you need to be in the same area at the same time, but you also need to be facing each other. In the example in Figure 5, it is not sufficient to simply tether A and B together; instead, a requirement of their reaction is to orient A and B properly. Many enzymes catalyze reactions not only by holding substrates close together, but also by forcing the substrates into an optimal orientation to lower the activation energy needed to reach the transition state.
Proximity and orientation effects lower the entropic barrier to forming the transition state (ΔS‡) because they pre-organize the substrates so that they lose less entropy during the formation of the transition state than the free substrates would. In other words, when enzymes bind to substrates they often reduce the entropy of the substrates by constraining them into reactive conformations, thus the favorability of binding between the enzyme and the substrates effectively “pre-pays” for the loss of entropy that is required to form the transition state.
We can quantify the proximity and orientation effects using the effective concentration of the reactants in the reactions. The effective concentration is defined as the ratio of the rate constant for the intramolecular reaction (with units of s-1) divided by the rate constant for the intermolecular
A + B A B
A B A B
increasing rxn rate,more favorable ΔS‡
Figure 4 The proximity effect in-creases collision frequencyThe top row shows an intermolecular reaction in which two reactants, A and B, form a new bond to each other. The bottom row shows an equivalent intramolecular reaction in which A and B are already connected. Intramolecular reactions are usually faster than intermolecular reactions because the reactants are held together in space (proximity effect) and collide more frequently as a result.

The Molecular Basis of Enzymatic CatalysisChapter 7 6
reaction (with units of s-1M-1). Effective concentration has units of molarity (M) and is a measure of the concentration of a reactant you would have to have in an intermolecular reaction to achieve the same rate as that in the intramolecular reaction with a substrate concentration of 1M. In the example shown in Figure 6, the rate constant for the intermolecular reaction (top) is kinter = 4 x 10-6 s-1M-1; in contrast, the rate constant of the intramolecular reaction (bottom) is kintra = 0.8 s-1. The effective concentration of A and B in the bottom case is kintra/kinter, or 200,000 M! In other words, A and B when tethered in close proximity and in the proper orientation by an enzyme react at the same rate as that of the untethered reaction when A or B is an impossibly high 200,000 M. Note that effective concentration takes into account both proximity and orientation effects; therefore, if the reactants were tethered close to each other but held firmly in the incorrect orientation for the reaction, the effective concentration would not be high. Enzymes use orientation and proximity effects, often with dramatic results, by confining reactants in the proper orientation within the relatively small active site of the enzyme.
Enzymes use acid and base catalysis to affect ΔH‡
In addition to proximity and orientation affects, which affect ΔS‡, enzymes also catalyze reactions by providing proton donors (acids) and proton acceptors (bases) at precise locations in the active site. These strategies
A
B
A
B
A B
A B
A B
A B
reactive conformation
increasing rxn rate,more favorable ΔS‡
Figure 5 Substrates that are restrained into a reactive conformation react more rapidlyShown are three hypothetical substrates (top, middle, and bottom rows) in which the reactive regions, A and B, are connected by covalent bonds. The reactive conformation in which A and B are close together is drawn in the right-most column for each substrate. The substrate on the top row is the least constrained of the three and can adopt a large number of conformations; consequently, it must lose a large amount of entropy in order to adopt the reactive conformation, making ΔS‡ less favorable. In contrast, the substrate on the bottom row is already constrained into a reactive conformation; consequently, no loss of entropy is needed to adopt the reactive conformation, making ΔS‡ more favorable.

The Molecular Basis of Enzymatic CatalysisChapter 7 7
largely affect ΔH‡, accelerating the rate of breakage and formation of bonds. In the laboratory chemists can accelerate many reactions by adding acid or base to lower or raise the pH of the reaction solution. These pH changes typically accelerate reactions by altering the nucleophilicity or electrophilicity of the reactants.
As its name implies, nucleophilicity describes the effectiveness of a nucleophile and is a measure of how quickly an atom is able to form a covalent bond with an electron-deficient atom. Highly nucleophilic atoms are able to form bonds faster than weak nucleophiles. Nucleophilicity is a kinetic parameter because it reflects the rate of bond formation rather than the amount of energy released as a result of bond formation.
Nucleophiles can vary widely in their nucleophilicity, and as you would expect, the best nucleophiles tend to be atoms with an excess of negative charge. As a general rule of thumb, when comparing two atoms of similar size (such as among N, O, or F), the atom that is the strongest base (whose conjugate acid has the higher pKa) is the more nucleophilic atom. Therefore, a hydroxide anion (OH-), whose conjugate acid (H2O) has a pKa of 15.5, is a much better nucleophile than water, whose conjugate acid (H3O
+) has a pKa of -1.5 (Figure 7). Likewise, ammonia (NH3), whose conjugate acid (NH4
+) has a pKa of 10, is a better nucleophile than water.
Conversely, electrophilicity describes the effectiveness of an electrophile and measures how quickly an electron-deficient atom is able to form a covalent bond with an incoming nucleophile. Like nucleophilicity, electrophilicity is a kinetic parameter. As you might expect, those atoms attached to groups that can most readily accept electrons (including groups made of electronegative atoms) are the most electrophilic and make the best electrophiles. In the chemical reactions involved in life, electrophilic atoms are most often carbon or phosphorus atoms that are doubly bonded to oxygen atoms (Figure 8). If you take organic chemistry you will learn in much more detail the factors that determine electrophilicity, but for our purposes, we will focus on C=O and P=O groups as electrophiles.
A + B A B
A B A B
kinter = 4 x 10-6 M-1s-1
kintra = 0.8 s-1
4 x 10-6 M-1s-1
0.8 s-1
Ce� = kintra
kinter
= = 200,000 M
(A)
(B)
Figure 6 Effective concentration measures the rate enhancement of an intramolecular reaction rel-ative to a corresponding intermo-lecular reaction(A) Shown are two hypothetical reactions between reactants A and B. The top row shows an intermolecular reaction while the bottom row shows an otherwise equivalent intramolecular reaction. The rate constants of both reactions, kinter and kintra, are indicated. (B) Effective concentration (Ceff) equals the ratio of the intramolecular rate constant over the intermolecular rate constant. In this example Ceff equals 200,000 M, meaning that the reactants in the intermolecular reaction would have to be present at 200,000 M in order to match the rate of the intramolecular reaction when its reactants are present at 1 M.

The Molecular Basis of Enzymatic CatalysisChapter 7 8
The protonation or deprotonation of a group can profoundly change its nucleophilicity or electrophilicity. As we have already seen in the case of water, removing a proton from a group typically increases its electron richness, and therefore its nucleophilicity. Conversely, adding a proton to a group typically makes that group more readily accept electrons, thereby increasing its electrophilicity. For example, the protonation of a carbonyl oxygen (Figure 9) results in a dramatic increase in the ability of the carbonyl carbon to accept an incoming nucleophile. This is because the protonation of the oxygen causes oxygen to become more positive because it loses one of its lone pairs and is not sharing those electrons with hydrogen. The positive charge on the oxygen causes the carbon-oxygen double bond to be even more polar than usual, with electrons in the bond being attracted to the positively charged oxygen. Since these electrons are more attracted to the protonated oxygen, the carbon atom becomes more positive and more electron-deficient, making it more electrophilic.
ConjugateAcid
Base HO
H
HO
H
H
15.5
HN
H
H
H NH
HH
9.25
HO
H
OH
-1.5pKa
Stronger base,better nucleophile
CO
O PO
OO
carbonyl phosphate
Figure 7 Stronger bases are often better nucleophilesFor atoms of similar size, stronger bases are generally more nucleophilic. Shown are three bases—water, ammonia, and hydroxide—arranged from left to right in order of increasing basicity and nucleophilicity. The conjugate acid of each base is also shown, along with the conjugate acid’s pKa. Stronger bases have conjugate acids with higher pKa values.
Figure 8 Carbonyl and phosphate groups are common electrophiles in living systemsShown are the structures of a carbonyl and phosphate group. The electrophilic atom is shown in red in both structures.
Figure 9 Protonation state affects nucleophilicity and electrophilici-tyDeprotonation by a base (top) increases an atom’s nucleophilicity by making it more electron-rich. Conversely, protonation by an acid (bottom) increases a functional group’s electrophilicity. In this example protonation of the carbonyl oxygen makes the C-O double bond more polar (electrons are more attracted to the positively charged oxygen), which in turn makes the carbon atom more electron-deficient.
HO
H
CO
base-H+
acid+H+
OHmore
electron-rich
CO
Hmore
electron-de�cient
better nucleophile
better electrophile

The Molecular Basis of Enzymatic CatalysisChapter 7 9
As an example, let us consider amide bond hydrolysis. You can now appreciate that neutral water is only weakly nucleophilic, whereas deprotonated water (the hydroxide anion) is a more effective nucleophile. A chemist might speed up the rate of amide bond hydrolysis in a test tube by adding base in order to deprotonate water and thereby convert it into a better nucleophile (Figure 10). Although enzymes in general cannot change the pH of an entire cell, they can use acid catalysis and base catalysis by positioning acidic or basic groups at just the right locations to protonate or deprotonate particular groups within a substrate. Acid and base catalysis lowers ΔH‡, thus decreasing ΔG‡ as well. Most commonly these acids and bases are the side chains of acidic and basic amino acids.
Even though we can draw reasonable transition state structures for the cases presented in Figure 10, it is not obvious simply from inspecting the structures of these transition states which would have a smaller ΔG‡ value. Instead, you should reason that the hydroxide anion is a better nucleophile than water; therefore, the hydroxide anion will more rapidly form a bond to the electrophile than water, and thus the faster rate will correspond to a lower ΔG‡ value.
Proteases catalyze peptide bond cleavage
As an example, proteases are enzymes that catalyze the cleavage of peptide bonds (Figure 11), which is chemically the same reaction as the amide hydrolysis example that we just examined. We saw the peptide bond cleavage reaction in Chapter 4 when we examined its arrow pushing mechanism. We also saw the active site of a protease called chymotrypsin in Chapter 5 as an example of how amino acids can function cooperatively. Your body
CO
NH
HO
H
OH
CO
NH
C NH
HO
H
O
δ+
δ−
C NH
OH
O
δ−
δ−
Water + Base
Weaker nucleophile:slower reaction,
higher ΔG‡
Stronger nucleophile:faster reaction,
lower ΔG‡
Figure 10 Base catalysis accelerates amide bond hydrolysis by increasing the nucleophilicity of waterShown is the example of amide bond hydrolysis in neutral water (top) and in a basic solution (bottom). The presence of base leads to the deprotonation of water, which increases its nucleophilicity. The stronger nucleophile leads to a faster reaction because the accompanying transition state that is lower in energy than the transition state in which water is used as a nucleophile.

The Molecular Basis of Enzymatic CatalysisChapter 7 10
contains hundreds of different proteases. Some of the best studied examples of proteases are those involved in digestion, such as pepsin and trypsin. These proteases play key roles in allowing your body to make use of protein nutrients by breaking proteins down into smaller pieces. These protease digestion products are eventually processed into amino acids that can be used for many functions in the cell, including the synthesis of new proteins through the process of translation.
The reaction catalyzed by proteases, peptide bond cleavage, is highly favorable; therefore, its ΔG°rxn is negative. Although peptide bond hydrolysis is favorable, it does not proceed rapidly without a catalyst. In fact, this reaction is so slow that the half-life of a peptide bond in neutral water is on the timescale of years. Proteases, however, increase the rate of peptide bond hydrolysis by over a billion-fold, accelerating peptide bond hydrolysis to a timescale that is consistent with biological processes.
Summary
Many biological reactions do not occur on a timescale that is consistent with life. Living systems address this problem by using enzymes, which accelerate otherwise slow chemical reactions by lowering ΔG‡. In contrast, enzymes do not affect thermodynamics and do not change ΔG°rxn.
Enzymes catalyze reactions by first binding to free substrate to form an enzyme-substrate complex. In this complex, the enzyme stabilizes the reaction’s transition state, thereby lowering the overall energy and allowing substrate to convert more rapidly to product. The resulting enzyme-product complex then dissociates, releasing free product and regenerating the free enzyme, which can take part in subsequent rounds of catalysis. In order to decrease ΔG‡, the enzyme must preferentially stabilize the transition state relative to the substrate.
One strategy used by enzymes to lower ΔG‡ is the proximity effect, in which the enzyme constrains the substrates by holding reactive atoms in close proximity to each other, thereby increasing collision frequency and decreasing ΔS‡. Similarly, enzymes use the orientation effect, in which the enzyme constrains the substrates into specific reactive conformations, thereby increasing the probability that reactants will collide in the correct orientation to react, thus decreasing ΔS‡ as well.
Figure 11 Proteases catalyze peptide bond hydrolysisShown is the chemical reaction of the peptide bond hydrolysis reaction that is catalyzed by proteases. This reaction is thermodynamically favorable, but it proceeds slowly without a catalyst. Proteases greatly accelerate the reaction’s rate.
O
NHR
HN
R
OON
R
OH
HO
R
HN
OOHH
OH+ +
protein substrate carboxylic acidproduct
amineproduct

The Molecular Basis of Enzymatic CatalysisChapter 7 11
Enzymes also catalyze reactions using acid and base catalysis by providing proton donors (acids) and proton acceptors (bases) at precise locations in the active site. These strategies largely affect ΔH‡ and accelerate the rate of bond breakage and formation. In base catalysis, a base is used to deprotonate an atom, thereby giving it additional electron density and making it more nucleophilic. In acid catalysis, on the other hand, an acid is used to protonate an atom, thereby making it more positive, typically increasing the electrophilicity of atoms bonded to it. Acid/base catalysis can also accelerate the making/breaking of bonds by neutralizing high energy charges that develop during the course of a reaction. As we will see later, an example of this can be found in proteases, which are enzymes that accelerate the hydrolysis of peptide bonds.
Practice problems
1. Shown below is a reaction catalyzed by carbonic anhydrase, an enzyme present in red blood cells. The active site of the enzyme shown below, facilitates the generation of OH-. The three histidine residues on the left bind an important Zn2+ ion. Describe the different ways this Zn2+ ion catalyzes the reaction mechanism.

The Molecular Basis of Enzymatic CatalysisChapter 7 12
Solutions to practice problems
• Proximity and orientation effects: The Zn2+ ion positions the water molecule correctly oriented in the active site of the enzyme in close proximity of the histidine residue, which increases both the collision frequency of the reactants and the probability that reactants collide in the right orientation.
• Increasing electrophilicity: The Zn2+ ion places positive charge next to the oxygen of H2O, which makes the hydrogen atom a better electrophile.
• Stabilizing transition state: The positive charge of the Zn2+ ion also stabilizes the formation of the nega-tive charge on the oxygen of H2O and thus stabilizes the transition state, lowering the activation energy.
Question 1:




















